

Summary
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection causes severe COVID-19 in some patients and mild COVID-19 in others. Dysfunctional innate immune responses have been identified to contribute to COVID-19 severity, but the key regulators are still unknown. Here, we present an integrative single-cell multi-omics analysis of peripheral blood mononuclear cells from hospitalized and convalescent COVID-19 patients. In classical monocytes, we identified genes that were potentially regulated by differential chromatin accessibility. Then, sub-clustering and motif-enrichment analyses revealed disease condition-specific regulation by transcription factors and their targets, including an interaction between C/EBPs and a long-noncoding RNA LUCAT1, which we validated through loss-of-function experiments. Finally, we investigated genetic risk variants that exhibit allele-specific open chromatin (ASoC) in COVID-19 patients and identified a SNP rs6800484-C, which is associated with lower expression of CCR2 and may contribute to higher viral loads and higher risk of COVID-19 hospitalization. Altogether, our study highlights the diverse genetic and epigenetic regulators that contribute to COVID-19.
Keywords: COVID-19, genetic regulation, epigenetic regulation, scATAC-seq, scRNA-seq, allele-specific open chromatin
Graphical abstract
Highlights
-
•
Massive regulatory changes can be found in monocytes of COVID-19 patients
-
•
Hospitalization is marked by open chromatin of C/EBP motifs in classical monocytes
-
•
Interaction of C/EBPs with gene LUCAT1 highlighted the epigenetic regulator
-
•
Allele-specific open chromatin explains the regulatory role of genetic risk loci
With a single-cell multi-omics study, Zhang et al. linked both genetic and epigenetic regulation to transcriptional responses in COVID-19 patients. Their findings suggest the identification of underlying regulators of innate immune dysfunction in COVID-19 patients and the regulatory roles of known genetic risk loci in the COVID-19 pathogenesis.
Introduction
COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),1 and clinical symptoms of patients with SARS-CoV-2 infection range from asymptomatic to severe pneumonia and acute respiratory distress syndrome.2 Although vaccines reduce the risk of major illness and mortality, the molecular mechanisms underlying the heterogeneous outcome in disease presentation remain unclear.3
A number of studies have examined the complex interplay between peripheral blood leukocytes in COVID-19 and linked immune activation and specific cell subsets to disease severity.4,5 The adaptive immune system is clearly linked to disease presentation, because prominent lymphopenia is a hallmark of severe disease.6 Alterations in T cell function have also been observed, with T cells from severe patients showing increased signs of migration to inflamed site and apoptosis7 and excessive or suboptimal CD4+ and CD8+ T cell responses detected in severe disease.8 The innate immune system has also been reported to be dysregulated in severe disease, which is characterized by high neutrophil counts,9 likely contributing to tissue damage and hyperinflammation, dysfunctional monocytes with low expression of HLA-DR and interferon (IFN)-stimulated genes (ISGs),4 and functionally impaired NK cells.10 Additionally, long noncoding RNAs are reported to be involved in the regulation of antiviral immune responses in COVID-19 and subsequent disease states.11 These studies have shed light on the detrimental immune responses that contribute to immunopathology in severe COVID-19.
In addition to studies of molecular signatures, several genome-wide association studies (GWASs) of severe COVID-19 have been performed.12,13,14 These revealed the impact of genetic variations on disease severity, improving our understanding of COVID-19 pathology. Moreover, although an epigenetic study on individuals convalescing from COVID-19 revealed remodeling of the chromatin accessibility landscape that established immunological memory,15 a recent study shows the immune responses and cytokine production capacity generally recover without major sequelae after COVID-19.16 More importantly, because the majority of these risk factors were identified in noncoding regions, they are predicted to have functional effects on gene expression via transcription factor (TF) binding and interaction with regulatory elements.17 These regulatory effects are highly cell type specific18,19 and are not yet understood in relation to COVID-19 risk factors.
Bridging the existing gaps requires an integrative approach that connects genetic variations, epigenetic factors, and immune responses at the cellular level.20 For this reason, we captured both the transcriptome and epigenome of individual peripheral blood mononuclear cells (PBMCs), as well as genome-wide genotypes, from hospitalized and convalescent COVID-19 samples. We identified C/EBP-motif-enriched open chromatin profiles in classical monocytes and illustrated their interaction with the immune-regulatory LUCAT1 RNA locus using single-cell omics and loss-of-function experiments. Additionally, we demonstrate that COVID-19 GWAS risk variants contribute to the disease by regulating chromatin accessibility through allele-specific open chromatin (ASoC) effects. Our ASoC analysis reveals that the COVID-19 GWAS risk SNP rs6800484 is associated with the expression of CCR2 via chromatin accessibility of an enhancer in monocytes. Together, these data indicate that altered chromatin accessibility and ASoC both result in impaired epigenetic regulation that contributes to COVID-19 pathogenesis, while the complex co-action of these factors could lead to heterogeneous and individualized disease outcomes. Our study further provides a broad resource for exploring cell-type-specific genetic and epigenetic regulatory effects that contribute to COVID-19.
Results
Study overview and patient population
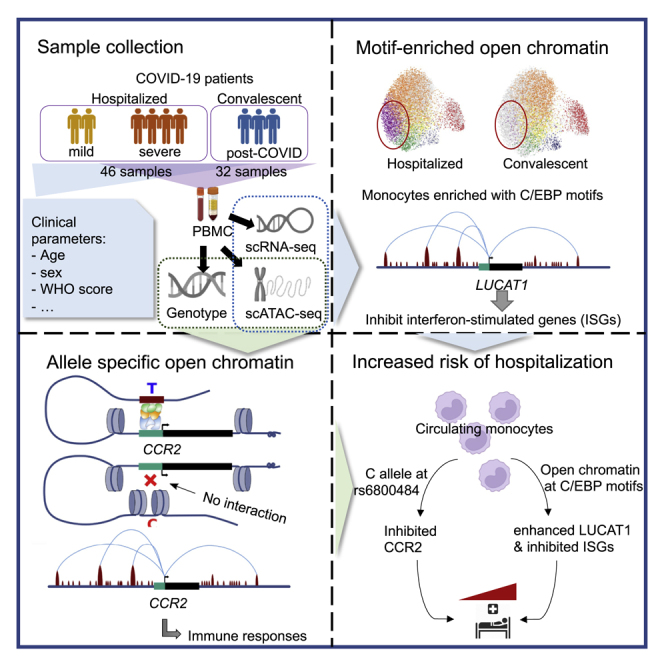
Using single-cell RNA sequencing (scRNA-seq), single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq), and genotype array, we examined the transcriptomics and epigenomics of PBMCs, as well as individual genotypes, across 46 hospitalized COVID-19 and 32 convalescent samples from 48 individuals, including 20 individuals from whom we have samples from multiple time points (Figures 1A, 1B, and S1). Hospitalized COVID-19 patients were further allocated to mild or severe patient categories using World Health Organization (WHO) scores (severe: 5–7, mild: 3–4). Clinical characteristics of all study participants are summarized in Table S1.
Figure 1.

Study overview and single-cell multi-omics
(A) Workflow of the study. Sample numbers in each data layer and disease condition are indicated.
(B) Schematic overview of all patients enrolled in the study. Sampling dataset, disease conditions, and convalescent days are indicated.
(C and D) UMAP showing the cell distribution of hospitalized and convalescent conditions in scRNA-seq (C) and in scATAC-seq (D); see also Figure S3 and Table S2 for annotation details.
(E) Boxplots showing cell proportion of hospitalized and convalescent samples in main cell types of scRNA-seq and scATAC-seq.
(F) Scatterplots showing the log-fold-change (log2FC) of DE-Gs identified in monocytes between the comparison of severe versus convalescent and the comparison of mild versus convalescent. More details on DE-Gs can be found in Figure S4 and Table S3.
(G) Scatterplots showing the log2FC of DE-Gs and log2FC of differentially accessible peaks (DAPs) identified in classical monocytes between comparison of severe versus convalescent and comparison of mild versus convalescent. Details of the matched DE-Gs and DAPs can be found in Table S4.
In total, after quality control, we obtained scRNA-seq data for 165,054 cells from 64 samples (n = 37 active, n = 27 convalescent samples) taken from 41 individuals and scATAC-seq for 46,690 cells from 49 samples (n = 25 hospitalized, n = 24 convalescent samples) taken from 39 individuals (Figure S2). We characterized these cells with unsupervised clustering and, based on the marker genes or gene activity scores in each cluster (Table S2), identified 10 major cell types in the scRNA-seq dataset and 8 major cell types in the scATAC-seq dataset (Figures 1C, 1D, and S3). The relative percentage of cell types in the PBMC fractions of each sample reveals a higher abundance of classical monocytes and a lower abundance of non-classical monocytes, as well as CD4+ and CD8+ T cells, in hospitalized COVID-19 compared with convalescent patients in both datasets (Figures 1E and S4A, Dirichlet regression test, false discovery rate [FDR] adjusted p < 0.05), in line with a recent publication.21 Additionally, a high proportion of CD163+ classical monocytes was found in five hospitalized COVID-19 patients (four severe and one mild) exclusively in the scRNA-seq dataset (Figures 1C and 1E).
Severe and mild COVID-19 patients show different magnitudes of transcriptional responses
Differential expression (DE) tests per cell type between hospitalized and convalescent samples revealed that a large number of differentially expressed genes (DE-Gs) were in NK cells, classical monocytes, and non-classical monocytes (Table S3; Figure S4B), suggesting that these cell types respond most prominently during COVID-19. Within these cell types, a large proportion of DE-Gs were shared between the mild versus convalescent and the severe versus convalescent comparison, especially for classical monocytes (Figure 1F). This suggests that similar transcriptional changes occur in mild and severe COVID-19, and that the difference between mild and severe COVID-19 is due to a difference in the magnitude of the response, rather than different transcriptional programs. For example, we observed several ISGs, such as IFI6, IFI27, IFI30, and IFI44L, to be significantly upregulated in both severe and mild samples compared with convalescent samples, whereas a significantly higher expression of IFI27 and IFITM3 was detected in mild patients compared with severe patients, reminiscent of the results of a previous study.4 Gene-enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database showed a clear upregulation of oxidative phosphorylation, across different immune cells, in both mild and severe COVID-19, as well as an enrichment of immune-related pathways, such as antigen processing and presentation and phagosome (Figure S4C).
Differential open chromatin accessibility contributes to transcriptional differences between hospitalized and convalescent COVID-19 patients
To reveal epigenetic alterations at the level of chromatin accessibility in COVID-19, we explored open chromatin signatures of PBMCs across the different disease conditions. Among all 49 samples, 15% of the 157,330 reproducible peaks are in promoter regions, while 32% and 45% are located in intergenic and intronic regions, respectively. When comparing across cell types and conditions, we observed no general enrichment of cell-type- or condition-specific peaks in the promoter or enhancer region (Figure S5A). We noticed that among all cell types, the number of open chromatin peaks is highest in classical monocytes, where it is significantly higher in samples from hospitalized than from convalescent COVID-19 patients (chi-square test, p < 2.22 × 10−16). When comparing peaks from one cell type with all other cell types, a large number of cell-type-specific peaks were identified. This includes 17,105 peaks specific for classical and/or non-classical monocytes (FDR-adjusted p < 0.05 and log-fold-change [log2FC] > 0.5, compared with other cell types), which comprise 10.9% of all peaks in our data. Next, we investigated the condition-specific peaks by comparing open chromatin peaks between disease conditions (hospitalized versus convalescent, mild versus convalescent, and severe versus convalescent) within each cell type. The lack of genome-wide significant condition-specific peaks (FDR-adjusted p < 0.05) suggests that cell types contribute more than disease conditions to variation in open chromatin accessibility.
To test the regulatory impact of open chromatin marks on transcriptional responses, we integrated the significant DE-Gs (Bonferroni-corrected p < 0.05) described above with the nominal differential peaks (p < 0.05) through peak-to-gene linkages, i.e., correlating the gene expression from scRNA-seq and peak accessibility from scATAC-seq (Table S4; for details, see STAR Methods). Interestingly, we observed an enrichment of open chromatin peaks that associated DE-Gs in classical monocytes in both severe and mild patients (Fisher exact test, FDR-adjusted p = 6.03 × 10−4 and 4.08 × 10−4, respectively), which includes in total 977 out of 2,367 (41.3%) genes that are upregulated in either severe or mild comparing with convalescent patients (Figures 1G and S5B). These results illustrate that there is a large overlap of changes in chromatin accessibility and gene expression in the monocyte compartment during COVID-19, suggesting the underlying epigenetic regulation on transcriptional responses.
Motif enrichment reveals different transcriptional regulation between hospitalized and convalescent COVID-19 in classical monocytes
To further characterize the epigenetic regulation of gene expression in COVID-19, we performed TF motif-enrichment analysis for the open chromatin peaks identified in each cell type and condition. In total, we found 60 TFs with significantly enriched motifs among the identified peaks. This included SPI1 (PU.1), JUN/FOS, and C/EBP motifs, which were enriched in classical monocytes in both hospitalized and convalescent COVID-19 patients. Of note, C/EBP motifs (CEBPA, CEBPB, CEBPD, CEBPE, and CEBPG) are even more significantly enriched in hospitalized patients than in convalescent patients (Figure 2A). Given their important role in monocyte differentiation and pro-inflammatory activation,22,23 we further investigated the interaction between these TFs and their targets, which were identified based on the genes with motif-binding peaks in classical monocytes. In total, 4,681 genes were associated with peaks harboring either SPI1, JUN/FOS, or C/EBP motifs, of which 1,514 were also DE-Gs between hospitalized and convalescent COVID-19 in classical monocytes (Table S5).
Figure 2.

TF regulation via motifs in the open chromatin peaks
(A) Heatmap showing the significantly enriched TF motifs in the open chromatin peaks of each cell type and condition. Colors represent −log10 p value of enrichment. Rows are significantly enriched TF motifs.
(B) Track plots showing the peaks around the LUCAT1 gene. Blue lines indicate inferred linkages between peaks and LUCAT1 expression.
(C) Heatmap showing the expression correlation of LUCAT1, IFI27, and IFI30 and TF genes in classical monocytes of hospitalized patients with a dot plot showing the expression of these genes in different conditions.
(D) Boxplots showing the molecular responses after knockdown of SPI1 and LUCAT1 or inhibition of C/EBP proteins.
(E) Schematic plot summarizing the potential regulating program in LUCAT1, SPI1, and C/EBP, as well as ISG, and COVID-19 severity.
Interestingly, we found that the long noncoding RNA LUCAT1 was associated with monocyte-specific accessible peaks harboring SPI1, JUN/FOS, and C/EBP motifs (Figure 2B), suggesting a monocyte-specific influence of the SPI1, JUN/FOS, and C/EBP TFs on LUCAT1. Because LUCAT1 has previously been reported as a negative regulator of IFN responses,24 we determined the co-expression patterns of LUCAT1, the TFs, and the two highly expressed ISGs, IFI27 and IFI30, in classical monocytes. As shown in Figure 2C, the expression of LUCAT1 is positively correlated with the expression of SPI1, JUN/FOS, and CEBPD/CEBPE TFs but negatively correlated with IFI27 and IFI30 expression in active COVID-19 patients. A similar co-expression correlation was observed in convalescent individuals, although the correlations between LUCAT1 and CEBPE or IFI30 were no longer significant (Figure S5C). Furthermore, in the DE comparison between disease conditions, SPI1 and LUCAT1 showed significantly higher expression in severe and mild samples compared with convalescent samples in classical monocytes (Bonferroni-corrected p < 0.05; Table S3; Figure 2C), whereas CEBPD showed significantly higher expression in severe samples compared with both mild and convalescent ones (Bonferroni-corrected p = 3.64 × 10−10 [severe versus mild] and 4.15 × 10−43 [severe versus convalescent]). IFI27 and IFI30, as mentioned above, were significantly upregulated in mild samples compared with severe samples (Bonferroni-corrected p = 9.23 × 10−71 [IFI27] and 3.00 × 10−15 [IFI30]; Table S3; Figure 2C). Together, these findings indicate a complex interaction of these genes at the expression level through epigenetic regulation that results in their altered expression under different disease conditions.
To validate the interactions between LUCAT1 and C/EBPs in monocytes of COVID-19 patients, we measured the expression of LUCAT1 in isolated monocytes after inhibiting C/EBP using celastrol and betulinic acid (Figure 2D). In unstimulated monocytes, the C/EBP inhibitors enhanced LUCAT1 expression at lower doses but suppressed LUCAT1 expression at higher doses. In monocytes activated with a cocktail of interleukin (IL)-1α, IFNα, and 3p-hairpin RNA (viral mimic), both inhibitors suppressed LUCAT1 expression. In addition, in a stable LUCAT1 knockdown monocyte cell line, we observed increased CEBPE expression (Figure 2D), which indicates strong negative feedback of LUCAT1 to upstream regulatory C/EBP TFs. Because LUCAT1 was previously reported to suppress inflammatory and ISGs,24 and ISGs are suppressed in severe COVID-19 patients,4 we speculate that the interaction of LUCAT1, SPI1, and C/EBPs inhibits ISG responses, resulting in a more severe condition in COVID-19 patients (Figure 2E).
Single-cell RNA and ATAC profiles revealed altered C/EBP regulation in a monocyte subset associated with oxygen supply of COVID-19 patients
To further investigate the heterogeneity of gene regulation in the monocyte compartment of COVID-19 patients, we explored disease condition-specific subsets. Subsampling the monocytes and sub-clustering them revealed eight transcriptionally distinct cell clusters (R1–R8; Figure 3A), from which R3, R4, and R8 were largely contributed by hospitalized COVID-19 patients. Through the DE tests comparing expression of gene between one cluster with the rest of the clusters and visualization of selected marker gene expression by Uniform Manifold Approximation and Projection (UMAP) (Table S6; Figure 3B), we identified R1 as CD14−CD16+ non-classical monocytes and R2–R8 as CD14+CD16− classical monocytes. In the classical monocytes, CEBPD and SPI1 TFs were expressed similarly among all clusters. However, a more distinctive expression pattern was observed for the previously mentioned TF target genes. LUCAT1 was significantly higher expressed in R2 and R8 compared with the other clusters (Bonferroni-corrected p < 2.22 × 10−16; Table S6), while ISGs such as IFI27, IFI30, and IFITM3 showed expression predominantly in the R3 cluster (Bonferroni-corrected p < 2.22 × 10−16) (Figure 3B; Table S6).
Figure 3.

RNA and ATAC profiles in monocyte sub-clusters in hospitalized and convalescent COVID-19 patients
(A) UMAP showing the cell distribution of hospitalized and convalescent conditions in monocyte sub-clusters of scRNA-seq.
(B) Expression of marker genes in monocyte sub-clusters. See also Table S6 for all the markers.
(C) Violin plots showing the AUCell-based gene signature scores for each sub-cluster from HLA-DRloS100Ahi monocytes in PBMCs (Schulte-Schrepping et al.4) and infiltrating monocytes (FCN1-Mono) in bronchoalveolar lavage (BAL) fluid (Wendisch et al.25).
(D) UMAP showing the monocyte sub-clusters of scATAC-seq.
(E) Dot plots and heatmap showing the expression and imputed activity scores of shared marker genes identified in monocyte sub-clusters of scRNA-seq and scATAC-seq, respectively.
(F) Boxplots showing the cell proportion of severe, mild, and convalescent patients, as well as oxygen supply needed, not needed, and convalescent patients in each monocytes sub-cluster of scRNA-seq. See the cell distributions in Figure S8.
(G) Heatmap showing the significantly enriched TF motifs in the open chromatin peaks of each monocytes sub-cluster; TFs that were also enriched as regulon in R4 cluster cells by SCENIC (single-cell regulatory network inference and clustering) are marked with red asterisks.
See also Figures S6–S8 and Table S7.
To validate our findings, we compared our monocyte sub-clusters with the previously published transcriptional markers of monocytes from COVID-19 patients.4 By applying the AUCell scores based on the top 30 marker genes from the PBMC datasets, we confirmed that R1 is non-classical monocytes, whereas R2–R5 should be classical monocytes (Figure S6A). Additionally, we identified that the hospitalized COVID-19-specific R4 and R8 sub-clusters were similar to HLA-DRloS100Ahi monocytes (Figure 3C), which are previously found as dysfunctional CD14+ monocytes in severe COVID-19 patients. Moreover, the ISG-predominated R3 sub-cluster was also found similar to another reported severe COVID-19 patient-specific HLA-DRloCD163hi monocyte (Figure S6A).4 In response to SARS-CoV-2 infection, circulating monocytes could be recruited to the lung tissue and participate in tissue immune responses by further differentiating into macrophages.26 We therefore assessed the transcriptional similarity between the monocyte sub-clusters and monocytes/macrophages reported in bronchoalveolar lavage (BAL) fluid samples from COVID-19 patients.25 By applying AUCell scores again to the BAL dataset markers, we identified that the hospitalized COVID-19-specific R3 and R4 were similar to FCN1-Mono in BAL (Figure 3C), which was reported25 as infiltrating monocytes that would later differentiate toward macrophages, whereas the macrophages themselves were not identified in any of our clusters (Figure S6B). These together suggest that both R3 and R4 sub-clusters were associated with COVID-19 severity and play an important role in the immune responses by infiltrating to the patients’ lungs.
Next, we performed sub-clustering on the monocytes from the scATAC-seq data. Unsupervised clustering revealed six epigenetically distinct cell clusters (C1–C6; Figures 3D, S7A, and S7B). Of these, the C4 cluster is specific to hospitalized COVID-19 patients, and the C2 cluster is specific to convalescent patients. Through a multi-omics alignment of the transcriptomic and epigenomic profiles across monocyte sub-clusters (see STAR Methods for details), we confidently matched C1 to R1 as non-classical monocytes, as well as C2 to R2 and C4 to R4 as classical monocytes (with >90% of aligned cells matched; Figures S7C and S7D). This can be confirmed by the shared pattern between gene expression levels of marker genes and estimated gene activity scores of the same marker genes based on peak data (Table S6; Figure 3E). When comparing the cell proportions across different disease conditions, the non-classical monocytes (C1/R1) had a higher abundance in convalescent COVID-19 patients (Figures 3F and S8A) that could be seen even before sub-clustering (Figure 1F). More interestingly, the remaining classical monocytes displayed high heterogeneity of cell proportions, with the C2/R2 and C4/R4 sub-clusters varying dramatically across disease conditions. C2/R2, which expresses LUCAT1 and harbors a strong antigen-presentation capacity with high expression of MHC class II components (including HLA-DQA and HLA-DPA), was largely contributed by convalescent patients (Figure 3F). In contrast, the C4/R4 cluster, which is annotated as HLA-DRloS100Ahi monocytes and shows a reverse expression pattern of MHC class II components and suppressed expression of ISGs, is mainly contributed by hospitalized COVID-19 patients (both mild and severe) and has a higher proportion in patients requiring oxygen supply than in those without (Figure 3F), suggesting a potential correlation between these monocytes and impaired lung function in patients.
To disclose the epigenetic regulation that underpins the transcriptional differences of these monocyte subsets, especially the condition-specific ones, we performed TF motif enrichment for marker peaks identified in each monocyte subset. The results demonstrate a distinct pattern of enriched motifs in different subsets. SPI1 is enriched in the convalescent-specific R2/C2 subset, together with RUNX1/2, IRF4, STAT2, and BCL11 A/B, whereas C/EBP motifs (CEBPA, CEBPB, CEBPD, CEBPG, and CEBPE) are enriched in the hospitalized patient-specific R4/C4 subset, together with an ATF4 motif (Figure 3G). From the scRNA-seq data, we found that among these TFs, CEBPD, CEBPB, and ATF4 were also widely expressed in R4 cells (Figure S8B). Through an independent TF regulon enrichment analysis in R4 cluster cells,27 we have confirmed that the identified C/EBPs and ATF4 were also high-confidently enriched TFs, together with IRF4, FOS, JUNB, JUND, BACH1, etc. (red asterisks in Figure 3G; see all enriched TFs in Table S7). This result indicates a shift of the regulatory elements between convalescent and hospitalized COVID-19 patients. Additionally, the suppressed expression of IFI27 and IFITM3 in R4 in comparison with R3 (Figure 3B) corresponds to its matched C4 cluster, which was enriched with C/EBP motifs. These results again suggest that altered TF motif accessibility may contribute to the dysregulation of IFN responses in COVID-19, and further indicate a potential correlation with the need for oxygen supply of COVID-19 patients.
COVID-19 GWAS variants are overrepresented in open chromatin regions of classical monocytes
Previous GWASs have revealed a number of genome regions associated with COVID-19 conditions. Therefore, we tested whether the identified GWAS hospitalization risk variants (“Hospitalized covid vs. population”, release 6)14 have an impact on open chromatin peaks in specific immune cell types. Our data reveal that these variants are significantly enriched in open chromatin peaks of classical monocytes from hospitalized COVID-19 patients (Fisher exact test, p = 2.98 × 10−12) and of CD4+ T cells from convalescent individuals (Fisher exact test, p = 2.68 × 10−6) compared with the other conditions and cell types. In classical monocytes, risk variants on chromosomes (chr) 3, 12, 17, and 21 were found to be located in several open chromatin peaks that were highly accessible in hospitalized patients (Figures 4A, 4B, and S9). When looking at the genes linked to the risk variant peaks mapped using the aforementioned method of peak-to-gene linkage (see STAR Methods), we identified significantly elevated expression (Wilcoxon rank-sum test, Bonferroni-corrected p < 0.05) of CCR1 and CCR2 (chr3), OAS3 (chr12), and IFNAR1 and IFNGR2 (chr21) in hospitalized patients compared with convalescent individuals in classical monocytes (Figure 4C). These results suggest that several GWAS risk variants may impact the expression of linked immune response genes through epigenetic regulation.
Figure 4.

GWAS risk variants associated with peaks and genes
(A) Schematic plot showing the potential regulatory role of a GWAS risk variant located in an open chromatin peak that is bound by the TF motif and associated with gene expression.
(B) Heatmap showing chromatin accessibility of peaks detected with hospitalized COVID-19 risk variants. More details can be found in Figure S9.
(C) Dot plots showing the expression of DE-Gs associated with peaks from (B).
ASoC analysis reveals epigenetic effects of genetic variants
To further investigate the epigenetic effects of genetic variants, we evaluated the ASoC, which represents the imbalance of chromatin accessibility between alleles, at heterozygous SNPs by integrating scATAC-seq and SNP data from the same individuals. In total, 292 and 86 ASoC SNPs were identified in hospitalized and convalescent COVID-19 individuals, respectively (FDR-adjusted p < 0.05; Figures 5A and S10A–S10C). Of these identified ASoC SNPs, about 5% were shared by hospitalized and convalescent conditions, which is in contrast with the fact that the majority of heterozygous SNPs (89.18%) available for testing the ASoC effect were shared by participants between conditions. This result suggests there is distinct allele-specific regulation in open chromatin regions between hospitalized and convalescent COVID-19 patients.
Figure 5.

ASoC analysis reveals the epigenetic effect of COVID-19 GWAS variants
(A) Venn diagram of ASoC SNPs identified in six cell types from hospitalized and convalescent participants. ASoC SNPs were merged per disease condition. ASoC SNPs identified in more than one cell type were counted once.
(B) Upset plot showing functional annotation of identified ASoC SNPs. Regulatory element annotations were determined based on 25-state models from the Roadmap Epigenomics Project. ASoC SNPs were assigned to eQTL genes and DE genes based on significant variant-gene pairs (GTEx V8) and positions (25 kbp up/downstream of ASoC SNPs), respectively. Numbers at the top of each bar indicate the exact number of ASoC SNPs belonging to the annotation or the gene group.
(C) Bar plot showing the enrichment of ASoCs assigned to our DE-Gs. The x axis represents cell types, and the y axis represents odds ratio that ASoCs are assigned to the DE-Gs (i.e., ASoC SNP is located in the promoter of the DEG). Color indicates disease conditions: red for hospitalized COVID-19 and blue for convalescent COVID-19. The numbers on the bar are FDR-adjusted p values and number of ASoC SNPs assigned to DEG out of the number of ASoC SNPs identified for the cell type.
(D) Heatmap of correlations between allelic imbalance and TF motif disruption. For each ASoC SNP, the allelic imbalance was represented by log2(reference read counts/alternative read counts), while the motif disruption was the difference between altScore and refScore by motifbreakR. Colors of the heatmap are Spearman’s rho, and multiplication symbols (×) indicate significant correlations (FDR-adjusted p < 0.05).
(E) Q-Q plot of COVID-19 GWAS p values for identified ASoC SNPs. The y axis represents observed GWAS p values (converted by −log10) of ASoC SNPs in cMono of hospitalized (red), convalescent COVID-19 (green) participants, and random selected SNPs (blue) with matched minor allele frequency.
(F) Allelic reads depth of ASoC SNP rs6800484 at the COVID-19-related CCR locus.
(G) Integration of gene-to-peak link, single-cell ATAC-seq, promoter capture Hi-C, eQTL SNPs, and COVID-19 GWAS SNPs around ASoC SNP rs6800484.
(H) Schematic plot showing the potential epigenetic and genetic regulating program at CCR2 locus under COVID-19 scenario.
(I) CCR2 expression in the differentiation trajectory of monocytes and macrophages of BAL fluid samples of COVID-19 patients (Wendisch et al.25).
<NA>, no valid estimation available. See also Figure S10.
As shown in Figure 5B, the majority of ASoC SNPs were located in enhancer (>25%) or promoter (>65%) regions, suggesting that epigenetic regulations occur in regulatory DNA sequences. In whole-blood samples, more than 55% of ASoC SNPs were reported to be associated with the expression of nearby genes (expression quantitative trait loci or eQTL),28 which indicates that these ASoC SNPs potentially affect gene expression by controlling allele-specific chromatin accessibility. In addition, in our scRNA-seq analysis for about 10% of ASoC SNPs, the nearby genes (of which promoters overlap with at least one ASoC SNP) were identified to be differentially expressed in at least one cell type (i.e., cell-type-dependent DE-Gs identified by comparisons between conditions). Further enrichment analysis revealed an over-representation of ASoC SNPs assigned to DE-Gs in COVID-19 patients (Fisher exact test, FDR-adjusted p < 0.05; Figure 5C), suggesting that the genetic risk variants have an impact on the transcriptional responses to SARS-CoV-2 infection through allele-specific chromatin accessibilities. Of note, we also observed that the ASoC SNPs were enriched in enhancer regions in hospitalized patients (Fisher exact test, p = 0.047), but not in the convalescent ones, showing the alteration of transcriptional profiles/activities in hospitalized COVID-19 patients compared with convalescent ones.
When zooming in on cell subsets, the ASoC SNPs we identified show significant over-representation in open chromatin regions (Figure S10D) and TF binding sites (TFBSs) (Figure S10E). Given that the genetic variants can perturb TF binding affinities by breaking the corresponding TF motifs, resulting in dysregulation of target genes,29 we calculated motif disruption scores (MDSs) for each ASoC SNP.30 We found that the allelic chromatin accessibilities were significantly correlated with MDSs for several TF motifs in classical monocytes from hospitalized COVID-19 patients (Spearman’s rank rho, FDR-adjusted p < 0.05; Figure 5D), suggesting that ASoC SNPs can play regulatory roles by disrupting TF motifs (i.e., affecting TF binding affinities).
ASoC of COVID-19 GWAS variants
Next, we intersected our ASoC SNPs with the above-mentioned COVID-19 GWAS hospitalization risk variants.14 We found that ASoC SNPs identified in classical monocytes from hospitalized COVID-19 patients were also associated with COVID-19, compared with randomly selected SNPs with matched minor allele frequency (Figures 5E and S10G). Among them, rs6800484 (COVID-19 GWAS p = 6.58 × 10−9) showed an imbalance of chromatin accessibility in classical monocytes in hospitalized COVID-19 patients (binomial test, p < 0.05), but not in convalescent individuals (Figure 5F). Of note, this SNP is located in a classical monocyte-specific open chromatin peak that was annotated as an EnhA1 enhancer (Roadmap Epigenomics Project)31 close to the CCR gene family.
This observation led us to further explore this locus by combining our results (ASoC SNPs, scRNA-seq, and scATAC-seq) with publicly available data, including promoter capture Hi-C of monocytes (PCHiCs),32 eQTL of whole-blood samples,33 and COVID-19 GWAS summary statistics.14 As shown in Figure 5G, we illustrated a potential regulatory program showing the effect of this variant underlying the COVID-19 context. Specifically, the publicly available monocyte PCHiC data and our peak-to-gene linkage analysis (see STAR Methods) suggest that the expression of CCR2 is correlated with the regulatory elements pinpointed by rs6800484 in classical monocytes from hospitalized COVID-19 patients. In addition, rs6800484-C is significantly associated with both COVID-19 (p = 6.58 × 10−9) and decreased CCR2 expression (p = 4.29 × 10−13). Meanwhile, in classical monocytes, homozygous risk allele (C/C) carriers show significantly lower CCR2 levels compared with other COVID-19 patients (Wilcoxon test, Bonferroni-corrected p = 3.2 × 10−3; Figure S10H), validating the inhibiting role of the risk allele on CCR2 expression.
As summarized in Figure 5H, the COVID-19 risk allele rs6800484-C identified in hospitalized patients is associated with decreased chromatin accessibility of an enhancer at the locus, which further inhibits the CCR2 expression. Of note, a recent study using a mouse-adapted SARS-CoV-2 strain has shown that mice lacking Ccr2 demonstrate higher viral loads and increased lung viral dissemination.34 In addition, our scRNA-seq data of classical monocytes confirms the importance of CCR2 because it was significantly upregulated in classical monocytes both in hospitalized COVID-19 patients (compared with convalescent ones, Bonferroni-corrected p < 2.22 × 10−16) and the disease-related R4 sub-cluster (compared with other monocyte sub-clusters, Bonferroni-corrected p < 2.22 × 10−16). In the public BAL samples from COVID-19 patients,25 CCR2 was also highly expressed in the infiltrating monocytes that would later differentiate toward macrophages (Figure 5I).
Another interesting example of potential regulation programs for DPP9, a candidate gene for COVID-19 severity, is depicted in Figures S10I–S10K. The DPP9 gene harbors SNPs associated with COVID-19 (p < 5 × 10−8) and was prioritized as a candidate gene that is involved in host-driven inflammatory lung injury in severe patients.13 Also, this locus has been previously reported to be associated with fibrotic idiopathic interstitial pneumonias,35 which suggests the potential role of dipeptidylpeptidase 9 (the enzyme encoded by DPP9) in severe COVID-19 patients. Moreover, early studies reported the enzyme is involved in antiviral signaling pathways,36 antigen presentation,37 and the activation of inflammasome.38 Taken together, these data depict an epigenetic regulation effect of risk allele of DPP9 locus in severe COVID-19 patients.
Discussion
Both host response and genetic predisposition affect the course and outcome of COVID-19, although the interplay between the two is not yet fully understood. Our single-cell multi-omics study has revealed numerous insights into the (epi)genetic mechanisms that regulate immune cells in COVID-19. First, we observed that COVID-19 has a pronounced effect on the transcriptional signature of classical monocytes, which was shown to be epigenetically regulated, and that the difference between mild and severe COVID-19 is due to a difference in the magnitude of response rather than differing transcriptional programs. Second, we depicted the regulatory properties of the long noncoding RNA LUCAT1 on the C/EBPs TFs, linking it to COVID-19 severity, and we experimentally validated the regulatory relationships. Finally, we identified a number of ASoC SNPs with potential regulatory effects in hospitalized COVID-19 patients. Interestingly, among these ASoC SNPs, rs6800484-C was associated with COVID-19 risk and linked to decreased chromatin accessibility, as well as reduced expression of CCR2, specifically in classical monocytes from hospitalized COVID-19 patients. Together, these findings shed light on the genetic, epigenetic, and transcriptional regulation of immune cells in COVID-19 (Figure 6).
Figure 6.

Schematic plot summarizing the genetic and epigenetic dysregulation of innate immunity in COVID-19
In our study, we observed a number of changes in cell proportions between hospitalized and convalescent COVID-19 patients, but fewer between severe and mild patients. Lymphopenia has been linked to COVID-19, as is also observed in our data, with hospitalized COVID-19 patients having a lower percentage of CD4+ T cells. We also saw an increased proportion of classical monocytes in hospitalized COVID-19 patients, as also observed earlier.7 In addition, we observed an upregulation of type I IFN signaling, which is crucial for antiviral immunity, in various monocyte subsets in COVID-19 patients compared with recovered individuals, e.g., IFI27, ISG15, and IFI6, which was also observed earlier in monocytes from COVID-19 patients compared with healthy controls.7 Interestingly, most of the observed differences were shared between mild and severe COVID-19 patients compared with convalescent individuals. This suggests that the difference in immunity between mild and severe COVID-19 is a matter of degree rather than reflecting distinct transcriptional profiles. This is in line with a previous observation that there are no immunological endotypes within the spectrum of COVID-1939 like those seen, for example, in sepsis.40
With open chromatin profiles, we observed enrichment of C/EBP, JUN, FOS, and SPI1 motifs in classical monocytes, which are also reported as critical TFs to monocyte development in sepsis.23 Because a subgroup of severe COVID-19 patients also developed a sepsis-like syndrome,40,41,42 there could be some overlapping immune-regulatory mechanisms at play. Examining co-expression of genes and peak-to-gene linkages, we found that the long noncoding RNA LUCAT1 interacts with all these SPI1, JUN, FOS, CEBPD, and CEBPE TFs. We applied knockout and inhibitor experiments to decode the regulatory and feedback mechanism among these molecules. The sub-clustering of monocytes further illustrated C/EBP motif-enriched classical monocyte subsets specific to hospitalized COVID-19 patients. These results together led us to envision a dysregulated cascade where increased C/EBP regulation enhanced LUCAT1 expression and further suppressed IFN responses to SARS-CoV-2 infection, which finally led to dysfunctional immune responses of COVID-19. Activation of C/EBP TFs was also reported to license the differentiation of profibrotic macrophages and trigger lung fibrosis in COVID-19.25 In our study, we observed the enrichment of open chromatin regions with C/EBP motifs in an oxygen-supply-associated monocyte sub-cluster, suggesting the activation of C/EBP regulation programs in circulating monocytes may also be associated with lung fibrosis and contribute to the need for oxygen in COVID-19 patients.
Finally, we identified ASoC SNPs in regulatory elements that potentially disrupt regulation and consequently affect gene expression.43,44 Of note, we observed that the COVID-19 risk allele rs6800484-C corresponds to lower chromatin accessibility and lower expression of CCR2 in classical monocytes,13 suggesting the potential genetic and epigenetic regulatory function of rs6800484 in COVID-19 patients. The CCR2 gene encodes the chemokine receptor for monocyte chemoattractant protein-1 (MCP-1/CCL2), which promotes the migration of monocytes to sites of inflammation.45,46 MCP-1/CCL2 was reported to be enriched in BAL samples collected from severe COVID-19 patients, indicating active recruitment of CCR2+ monocytes and high inflammation in lung tissues.47 This suggests that the ASoC in the observed variant may impact monocyte recruitment to tissue by reducing CCR2 expression and thereby further influence the innate immune responses in COVID-19 patients. Although the down-regulation of CCR2 expression corresponds to higher viral loads and increased viral dissemination in animal models,34 we observed a significant down-regulation of CCR2 expression only in patients with C/C alleles compared with others, but not in the comparisons between hospitalized/severe COVID-19 patients and convalescent.
In summary, our data have improved the understanding of the genetic and transcriptional regulation of dysregulated immune responses in COVID-19 and identified LUCAT1 and CCR2 as key regulators of detrimental immunity. Both factors contribute to COVID-19 pathogenesis in a subset of patients, while the co-action of these factors could bring heterogeneous responses to the SARS-CoV-2 infection. These leads can be used as a starting point for the development of personalized host-directed therapy to treat COVID-19.
Limitations of the study
Despite our interesting findings, this study also has several limitations. First, we focused our analysis to monocytes in peripheral blood. This choice was motivated by an over-representation of significant differences in the myeloid cell compartment, which also corresponds to our previous results.4 Although our data provided limited observations in lymphocytes, the importance of their role in the immune response in COVID-19 should not be ignored. Considering the large diversity and complexity of T cell populations, it will be interesting to dissect the T cell subsets in COVID-19 through T cell enrichment combined with T cell receptor sequencing in future studies. Second, although we used computational methods to link cells across scRNA-seq and scATAC-seq, the sequencing libraries of the two platforms were constructed independently. Therefore, we were unable to simultaneously profile gene expression and open chromatin from the same cell, which limited our power to characterize the full regulatory programs for different cells. Finally, our ASoC analyses were limited by the number of heterozygous SNPs among our participants. A future study based on a large cohort or a cohort pre-selected to have heterozygous alleles along the COVID-19 GWAS risk variants would address the full picture of genetic regulators of immune responses in COVID-19.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human peripheral blood mononuclear cell | N/A | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 Medium | SIGMA | MDL# R0883-500ML |
| Fetal Bovine Serum | PAN BIOTECH | Cat# P30-5500 |
| Dulbecco’S Phosphate Buffered Saline | PAN BIOTECH | Cat# P04-36500 |
| Buffer EB | QIAGEN | Cat# 19086 |
| SPRIselect | Beckmann Coulter | Cat# B23318 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3′Regent Kits v3.1 | 10X | CG000315 Rev A |
| Chromium Next GEM Single Cell ATAC Regent Kits v1.1 | 10X | CG000209 Rev D |
| High sensitivity DNA kit | Agilent | Cat# 5067-4626 |
| Deposited data | ||
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium48 | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Human reference epigenomic annotations | Roadmap Epigenomics Consortium31 | https://egg2.wustl.edu/roadmap/web_portal/index.html |
| Bulk RNA-seq eQTL summary statistics (release 2019-12-11) | eQTL-Gen Consortium33 | https://www.eqtlgen.org |
| GTEx eQTL summary statistics (v8) | GTEx v828 | https://www.gtexportal.org/home/datasets |
| Promoter capture HiC results | Javierre et al.32 | https://osf.io/u8tzp/ |
| COVID-19 GWAS summary statistics (release round 6) | The COVID-19 Host Genetics initiative12 | https://www.covid19hg.org/results/r6 |
| snRNA-seq data | Wendisch et al.25 | EGAS00001004928; EGAS00001005634; https://nubes.helmholtz-berlin.de/s/XrM8igTzFTFSoio |
| snRNA-seq data | This paper | EGAS00001006559 |
| snATAC-seq data | This paper | EGAS00001006560 |
| Genotypes | This paper | EGAZ00001823187 |
| Software and algorithms | ||
| Plink v1.90b6.21 64-bit (19 Oct 2020) | Chang et al.49 | https://www.cog-genomics.org/plink/ |
| R 4.1 | R Core Team | https://www.r-project.org/ |
| R package Seurat version 3.2.2 | Stuart et al.50 | https://cran.r-project.org/web/packages/Seurat/index.html |
| R package ArchR version 1.0.1 | Granja et al.51 | https://www.archrproject.com/ |
| R package ggplot2 version 3.3.2 | Wickham et al.52 | https://cran.r-project.org/web/packages/ggplot2/index.html |
| R package Bioconductor version 3.12 | Gentleman et al.53 | https://bioconductor.org |
| R package clusterProfiler version 4.0.5 | Yu et al.54 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| R package ggpubr version 0.4.0 | Kassambara, The Comprehensive R Archive Network (CRAN) | https://cran.r-project.org/web/packages/ggpubr/index.html |
| R package pheatmap version 1.0.12 | Raivo, The Comprehensive R Archive Network (CRAN) | https://cran.r-project.org/web/packages/pheatmap/index.html |
| R package tidyverse version 1.3.0 | Wickham et al.52 | https://CRAN.R-project.org/package=tidyverse |
| R package org.Hs.eg.db version 3.12.0 | Carlson55 | https://bioconductor.org/packages/3.12/data/annotation/html/org.Hs.eg.db.html |
| R package ggrepel version 0.9.0 | Slowikowski56 | https://cran.r-project.org/web/packages/ggrepel/index.html |
| R package data.table version 1.14.0 | Dowle57 | https://cran.r-project.org/web/packages/data.table/index.html |
| R package GenomicInteractions version 1.30.0 | Harmston et al.58 | https://bioconductor.org/packages/release/bioc/html/GenomicInteractions.html |
| R package GViz version 1.20.1 | Hahne and Ivanek59 | https://bioconductor.org/packages/3.15/bioc/html/Gviz.html |
| R package motifbreakR version 2.10.0 | Coetzee, Coetzee, and Hazelett30 | https://bioconductor.org/packages/release/bioc/html/motifbreakR.html |
| Python version 3.9.6 | Van Rossum60 | https://www.python.org |
| Python package MACS2 version 2.2.7 | Gaspar61 | https://pypi.org/project/MACS2/ |
| Bowtie2 version 2.4.4 | Langmead and Salzberg62 | N/A |
| Python package matplotlib version 3.4 | Hunter63 | https://matplotlib.org |
| Python package pysam version 0.17.0 | Github pysam-developers | https://github.com/pysam-developers/pysam |
| BCFtools version 1.12 | Danecek et al.64 | http://www.htslib.org |
| SAMtools version 1.12 | Danecek et al.64 | http://www.htslib.org |
| Htslib version 1.12 | Danecek et al.64 | http://www.htslib.org |
| WASP pipeline version 0.3.4 | van de Geijn et al.65 | https://github.com/bmvdgeijn/WASP |
| GATK/ASEReadCounter version 4.2.0.0 | Castel et al.66 | https://gatk.broadinstitute.org |
| VEP (online) | McLaren et al.67 | https://www.ensembl.org/Tools/VEP |
| TOPMed and Michigan Imputation server | Fuchsberger, Abecasis, and Hinds; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium et al.; Das et al.68,69,70 |
https://imputation.biodatacatalyst.nhlbi.nih.gov; https://imputationserver.sph.umich.edu |
| Original codes and scripts | This paper | https://doi.org/10.5281/zenodo.7270242 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Yang Li (Yang.Li@helmholtz-hzi.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
We collected DNA and PBMCs from blood samples of COVID-19 patients. Samples taken on days when patients were hospitalized were considered hospitalized samples and samples taken from discharged patients were considered convalescent samples. Clinical information on age, sex, medication, active days, O2 supply, etc. is recorded for each sample and listed in Table S1. WHO scores were used to allocate the samples to mild (WHO 3–4) or severe (5–7) conditions according to the WHO clinical ordinal scale. The study was approved by the institutional review board at Hannover Medical School (#9001_BO_K2020) and informed consent was obtained from all patients.
Method details
Single-cell RNA-seq library preparation and sequencing
Cells were counted, and an equal number of cells from five or six different individuals were pooled together. In total, 16,000 cells in total were loaded into the 10X ChromiumTM Controller, and libraries were prepared based on the manufacturer’s instructions (Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 (Dual Index) User Guide, Rev A, CG000315 Rev A). Library quality per pool was examined using the Agilent Bioanalyzer High Sensitivity DNA kit. Sequencing was carried out on NovaSeq 6000 (Illumina), with a depth of 50,000 reads per cell.
Single-cell ATAC-seq library preparation and sequencing
Nuclei isolation was performed based on manufacturer’s instructions from 10X (CG000169 ⋅ Rev D). Briefly, cells were washed and lysed for 3 min on ice. After discarding the supernatant, lysed cells were diluted within 1× diluted nuclei buffer (10x Genomics) and counted using a Countess II FL Automated Cell Counter to validate lysis. An equal number of nuclei from five or six individuals were pooled and then loaded into the Chromium Next GEM Chip H based on the user guides from 10X genomics (Chromium Next GEM Single Cell ATAC Reagent Kits v1.1 User Guide, CG000209 Rev D). After breaking the emulsion, the barcoded tagmented DNA was purified and amplified for sample indexing and generation of scATAC-seq libraries. The final library was quantified using the Agilent Bioanalyzer High Sensitivity DNA kit. Sequencing was performed on NovaSeq 6000 (Illumina) with a depth of 25,000 reads per nuclei.
Genotyping
Genotyping of DNA samples isolated from subjects in the current study were performed using the GSA-MDv3 array (Infinium, Illumina) following the manufacturer’s instructions. In total, 725,875 variants of 48 individuals were called by Optical 7.0 with default settings.
Quantification and statistical analysis
Genotype imputation
Quality control (QC) for raw variants was performed using PLINK.71 In brief, no sample was excluded initially due to failure in sex-check (--check-sex). Then, low-quality variants and individuals were excluded by parameters --geno 0.1 --mind 0.1. Next, missingness of genetic information and rates of heterozygosity were filtered by --missing and --het, respectively. After QC, 719,942 variants from 48 individuals were retained for the imputation procedure. The clean raw variants were uploaded to the TOPMed Imputation Server and imputed against the TOPMed (Version R2 on GRC38) reference panel.70 The imputed variants (n = 290,971,705) were downloaded and filtered by BCFtools,64 excluding variants with R2 < 0.5, with 14,232,029 variants retained for the downstream analysis.
Additional QC and annotation was performed to obtain the genotypes that were used in the ASoC analysis. The variants were assigned reference SNP id (rs) by BCFtools against common variants (b151 GRCh38) downloaded from dbSNP. Subsequently, only variants that have rs numbers and that were heterozygous in at least three individuals were retained for the ASoC analysis.
Data pre-processing and demultiplex of 10x genomics Chromium scRNA-seq data
BCL files from each library were converted to FASTQ files using bcl2fastq Conversion Software (Illumina) using the respective sample sheet with the 10x barcodes utilized. The proprietary 10x Genomics CellRanger pipeline (v4.0.0) was used with default parameters. CellRanger was used to align read data to the reference genome provided by 10x Genomics (Human reference dataset refdata-cellranger-GRCh38–3.0.0) using the aligner STAR,72 and a digital gene expression matrix was generated to record the number of UMIs for each gene in each cell.
The single-cell transcriptome in each library was further demultiplexed by assigning cell barcodes to their donor. The pre-mapped bam files of each library were loaded to Souporcell (v1.3gb)73 for a genotype-free SNP-based demultiplex with default settings, where candidate variants were called for each library and cells from each library were clustered into different samples based on their allele patterns. SNPs called from each sample were then matched with known genotypes of donors to assign a donor ID to each sample. The demultiplex assignments were double-checked using the expression of Y chromosome genes (ZFY, RPS4Y1, EIF1AY, KDM5D, NLGN4Y, TMSB4Y, UTY, DDX3Y, and USP9Y) in male samples.
Data pre-processing and demultiplex of 10x genomics Chromium scATAC-seq data
BCL files from each library were converted to FASTQ files using bcl2fastq Conversion Software (Illumina) using the respective sample sheet with the 10x barcodes utilized. The proprietary 10x Genomics CellRanger-ATAC pipeline (v1.2.0) was used with default parameters. CellRanger-ATAC was used to align read data to the reference genome provided by 10x Genomics (Human reference dataset refdata-cellranger-atac-GRCh38–1.2.0) and a fragments matrix was generated to record the number of reads for each open chromatin region in each cell.
The cells in each library were further demultiplexed by assigning cell barcodes to their donor. The pre-mapped bam files of each library were loaded to Souporcell (v1.3gb) for genotype-free SNP-based demultiplexing. To call robust SNPs from the ATAC-seq samples, candidate variants were first called by freebayes74 with minimal mapping quality = 20, minimal base quality = 20, minimal coverage = 6, and minimal alternative allele = 2. Next, cell allele matrices from each library were generated with vartrix with minimal mapping quality = 20, and cells were clustered based on their allele patterns to identify different samples in one library. SNPs called from each sample were matched with known genotypes of donors to assign the donor IDs.
Independent sample set
In our cohort, some samples with different disease statuses came from the same donor. To obtain independent samples for pairwise comparison across conditions, we manually selected an independent sample set after QC for both scRNA-seq and scATAC-seq based on the following criteria: one sample per donor, early-stage for multiple stages, with samples shared by scRNA-seq and scATAC-seq data are preferred. The selected samples are marked in Table S1.
QC for scRNA-seq data
After the demultiplex, the expression matrix from PBMC was loaded to R/Seurat package (v3.2.2)50 for downstream analysis. To control the data quality, we first excluded cells with ambiguous assignments from Souporcell demultiplex. Next, we further excluded low-quality cells with >15% mitochondrial reads, <100 or >3,000 expressed genes, or <500 UMI counts (criteria were chosen according to the overall distribution of samples). In addition, genes expressed in less than three cells were also excluded from further analysis.
Dimensionality reduction and clustering for scRNA-seq data
After QC, we applied LogNormalization (Seurat function) to each cell, where original gene counts were normalized by total UMI counts, multiplied by 10,000 (TP10K), and then log-transformed by log10(TP10k+1). We then scaled the data, regressing for total UMI counts, and performed principal component analysis (PCA) based on the 2,000 most-variable features identified using the vst method implemented in Seurat. Subsequently, data from each sequencing batch was integrated, using the ‘harmony’ algorithm, based on the first 20 principal components to correct technical differences in the gene expression counts of different libraries. Cells were then clustered using the Louvain algorithm based on the first 20 ‘harmony’ dimensions with a resolution of 0.4. For visualization, we applied UMAP based on the first 20 dimensions of the ‘harmony’ reduction.
Annotation of scRNA-seq clusters
Clusters were annotated based on a double-checking strategy: 1) checking by automatic annotation with R/SingleR package75 and 2) manually checking the expression of cluster markers or known marker genes. Specifically, automatic annotation was applied with five pre-installed reference datasets in SingleR: HumanPrimaryCellAtlas data (HPCA),76 BlueprintEncode data,77,78 ImmuneCellExpression data,79 Novershtern Hematopoietic data,80 and MonacoImmune data.81 Cluster marker genes were identified by comparing gene expression of each cluster to all other clusters of the tested dataset using the FindAllMarkers function in Seurat with the Wilcoxon rank-sum test. Only upregulated genes with a log-fold change >0.25 and a Bonferroni-corrected p-value <0.05, and were expressed in at least 25% of cells were calculated for each cluster, and genes from each cluster of interest were ranked by their log-fold changes.
In addition, T and NK cell clusters were further characterized by expression of marker genes related to memory T cells (IL7R), naive/central memory (SELL, CCR7), cell cytotoxic (CD8A, CD8B, NKG7, GZMB), interferon responses (IFI6, ISG15), and other data-derived cluster markers (Table S2). Monocyte clusters were then characterized by classical and non-classical monocyte markers (CD14, FCGR3A) and pro-inflammatory cytokines (TNF, IL1B), and the data-derived cluster markers, such as CD163 (Table S2).
DE-Gs across Covid-19 conditions
For pairwise comparison between COVID-19 conditions, differential expression (DE) tests were performed using the FindMarkers functions in Seurat with the Wilcoxon rank-sum test. The non-parametric Wilcoxon rank-sum test is distribution-free, but the results may still be biased by age effects. We therefore also performed DE tests using MAST,82 where we fit a hurdle model to the expression of each gene consisting of a linear regression for age as supplementary results. In both tests, genes with a log-fold change >0.05 and a Bonferroni-corrected p-value <0.05, and were expressed in at least 10% of tested groups were regarded as significantly differentially expressed.
QC for scATAC-seq data
After alignment and demultiplex, we used ArchR,51 a full-featured scATAC-seq analysis package, with minor adaptation to analyze our scATAC-seq data. Briefly, we created an Arrow file for the CellRanger mapped fragments file from each single-cell library and annotated the cells with the Souporcell demultiplex assignments. For QC, we filtered out cells that had fewer than 1,000 unique fragments, a transcription start site enrichment <4, or potential doublets recognized by the ArchR package. We also excluded cells with ambiguous assignments from Souporcell demultiplex. Finally, an ArchRProject combining all of the Arrow files was created for downstream analysis.
Dimensionality reduction and clustering for scATAC-seq data
After QCl, we used the ArchR function ‘addIterativeLSI’ to process iterative latent semantic indexing using the top 25,000 variable features and top 30 dimensions. We then used the harmony algorithm to correct batch effects from different libraries and clustered cells based on the results with a resolution of 0.8. For visualization, we applied UMAP based on the dimensions of the ‘harmony’ reduction with nNeighbors = 30 and minDist = 0.5.
Annotation of scATAC-seq clusters
To annotate the scATAC-seq clusters, gene scores were calculated and imputed for each cell, and marker genes from each cluster were detected with functions in ArchR package. Briefly, we firstly use ‘addGeneScoreMatrix’ function to independently compute gene activity scores per cell, then applied ‘addImputeWeights’ to impute gene scores by smoothing the signal across nearby cells using the MAGIC algorithm. Next, we compared independent gene scores between cells from one cluster and all the other clusters using the Wilcoxon rank-sum test to detect cluster-specific genes. The ‘bias’ parameter in ‘getMarkerFeatures’ from ArchR was used to account for transcription start site enrichment scores and the number of unique fragments per cell during the comparison. Finally, we visualized these genes and other cell-type-specific marker genes used for our scRNA-seq data to assign an identity to each cluster.
Peaks calling and marker peaks detection
To generate a comparable peak matrix for cross-sample comparison of differential open chromatin accessibility, reproducible peaks were called based on the pseudo-bulk replicates for each condition and clustered using the ‘addReproduciblePeakSet’ functions with Macs2 algorithm.83
After adding a peak matrix based on the called reproducible peaks, we applied differential peak detection with the Wilcoxon rank-sum test, again accounting for transcription start site enrichment scores and the number of unique fragments per cell during the comparison. For marker peaks per cell type and disease conditions, peaks were compared between cells from the tested group and cells from all other groups, and peaks with FDR <0.05 were considered as significant cell type- and/or condition-specific peaks. For pairwise comparison between conditions within a cell population, peaks with p-value <0.05 were considered as nominal differential accessible peaks and used for integrative analysis with DE-Gs.
TF motif annotation and enrichment
After calling peaks, we looked for the motifs that are enriched in peaks that are openly accessible in different cell types and conditions. To do this, we first added motif annotation based on the “CIS-BP” database,84 then, we applied the ‘peakAnnoEnrichment’ function in ArchR to obtain overrepresented motifs in test peak sets.
Cross-platform linkage of scATAC-seq data with scRNA-seq data
To do an integrative analysis of scATAC-seq and scRNA-seq data, we performed a preliminary integration by aligning all cells from scATAC-seq with cells from scRNA-seq by comparing the above-mentioned scATAC-seq cell-independent gene score matrix with the scRNA-seq expression matrix using the ‘FindTransferAnchors’ function from the Seurat package and the ‘addGeneIntegrationMatrix’ function from the ArchR package. Based on the result of this initial integration and the cell type annotation, we filtered out undefined scATAC-seq clusters and clusters with <100 cells aligned to annotated scRNA-seq clusters. We then annotated remaining scATAC-seq clusters based on the aligned scRNA-seq clusters. Finally, we re-ran the integration process by aligning remaining scATAC-seq cells to cells from the aligned scRNA-seq clusters and created a gene-integration matrix by adding gene integration scores to each cell.
Peak-to-gene linkage
To find potential regulation from peaks to genes, we inferred a peak-to-gene linkage by calculating the correlation between peak accessibility and gene expression within the above-mentioned integrated scRNA-seq and scATAC-seq cells. A co-accessibility >0.45 and FDR-adjusted p < 0.05 were regarded as regulatory links.
TF footprinting with scATAC-seq data
To calculate the TF footprint for each motif, we first obtained all the positions from one TF motif. To profile the footprint, cells were grouped again by each condition and each cell type to create pseudo-bulk ATAC-seq profiles. To account for the insertion sequence bias of the Tn5 transposase, which can lead to misclassification of TF footprints, we used the “Substract” normalization method to subtract the Tn5 bias from the footprinting signal.
Sub-clustering of monocyte compartments in scRNA-seq
In the scRNA-seq dataset, the monocyte subpopulations were investigated by applying sub-clustering on the three monocyte clusters (cMono, CD163+ cMono, and ncMono) identified in PBMC. We first identified the 1,000 most-variable features again in monocytes using the vst method implemented in Seurat. Next, we scaled the data and performed PCA based on these 1,000 most-variable features. Subsequently, the cells were clustered using the Louvain algorithm based on the top-10 PCs with a resolution of 0.3. For visualization, we applied UMAP based on the top-10 PCs. The marker genes for each sub-cluster were calculated by the FindAllMarkers function in Seurat and a contaminated lymphocyte cluster with CD3 gene expression was identified and removed from further analyses.
AUCell-based gene signature scores were calculated using the AUCell method.27 We set the threshold for the calculation of the AUC to the top 3% of ranked genes and normalized the maximum possible AUC to 1. Top-30 marker genes reported from monocytes and macrophages reported in BAL fluid and PBMC4 were used to calculate AUC scores for each monocyte sub-clusters respectively. The resulting AUC values were subsequently visualized in violin plots.
Sub-clustering of monocyte compartments in scATAC-seq
In the scATAC-seq dataset, the two monocyte clusters (cMono and ncMono) identified in PBMC were extracted and investigated for sub-clustering analyses. Again, we used ArchR function ‘addIterativeLSI’ to process iterative latent semantic indexing using the top-25,000 variable features and top-30 dimensions. We then clustered cells based on the IterativeLSI reduced dimensions with a resolution of 0.8 and calculated UMAP with nNeighbors = 30 and minDist = 0.5. The resulting sub-clusters were aligned to scRNA-seq monocyte sub-clusters using the Cross-platform linkage method described above. Cells with a predicted linkage score >0.6 were regarded as aligned cells, and a scATAC-seq sub-cluster with a percentage of aligned cells >90% matched to the same scRNA-seq sub-cluster was regarded as the confidently matched sub-cluster.
TF gene expression and regulon enrichment analysis
In order to estimate the expression of the motif-enriched TFs in R4/C4 monocyte cluster, we ranked the genes based on their expressed percentages of R4 cells from hospitalized COVID-19 patients in scRNA-seq dataset and marked out TF genes among them. Next, we applied a regulon enrichment analysis across genes that were expressed at least 10% of R4 cells with SCENIC.27 Then, we intersected the enriched TFs, that were marked with 'high confidence' annotations by the algorithm, with the TF-motif enrichment results and marked the overlap TFs on the Heatmap.
Chromatin accessibility of COVID-19 risk variants
The genetic variants with a reported p-value <5 × 10−8 from the COVID-19 GWAS summary statistics (Hospitalized covid vs. population” release 6) by HGI14 were considered as risk variants of COVID-19. An open-chromatin peak was regarded to be associated with risk variants if its genomic location overlaps with at least one significant variant. The over-representation of “risk-variants-overlapping” peaks was estimated by the Fisher exact test comparing between peaks found in hospitalized patients and convalescent samples in each cell type, respectively.
Identification of ASoC SNPs
To estimate the allelic open chromatin for each identified cell type, the ATAC-seq reads of each subject were first split into individual BAM files per cell type using an in-house Python script according to the CB barcode which was added by CellRanger pipeline and error-corrected. The resulting BAM files were then calibrated using the WASP65 pipeline with Bowtie262 as aligner (-X 2000) to remove the mapping bias to reference allele at heterozygous sites. Afterward, the GATK/ASEReadCounter tool66 was used to count allelic reads at each heterozygous site with the default parameters. To detect the maximum allelic imbalance, the read counts from each subject were allelicly summed for each cell type at each heterozygous SNP, a pool approach that was justified in the previous study.43 Finally, only biallelic SNP sites with at least 20 read counts and at least 2 read counts for either allele were retained for downstream statistical analyses. To alleviate possible mapping bias to the reference allele, the WASP pipeline was applied, but no apparent bias effect was observed (Figure S10A).
Binomial p values were calculated for the allelic read counts per SNP per cell-type by the R function binom.test(), with the alternative read counts as success trials and all read counts as total trials. Next, the R function p.adjust() was exploited to perform a multiple testing correction using the “fdr” method, and an FDR-adjusted p < 0.05 was considered the significant threshold. No obvious mapping bias to reference alleles was observed by visualizing the volcano plot of -log10(p values) and allelic read counts ratio.
Annotation and function enrichment of ASoC SNPs
To understand the function of the identified allelic imbalances, the ASoC SNPs were annotated against the GRCh38 reference genome using the online version of vep.67 Next, epigenomic annotations from RoadMap epigenomics projects85 were assigned to each identified ASoC SNPs based on their physical position and cell type. These epigenomic annotations were further grouped into promoters (including “TssA”, “PromU”, “PromD1”, and “PromD2”) and enhancers (including “TxReg”, “TxEnh5”, “TxEnh3”, “TxEnhW”, “EnhA1”, “EnhA2”, “EnhAF”, “EnhW1”, “EnhW2”, “EnhAc”, and “DNase”). To evaluate the effects of the identified ASoC SNPs, significant variant-gene pairs of whole blood tissue were downloaded from the GTEx Portal (V8) and allocated to the corresponding ASoC SNPs. Further, the DE genes identified by scRNA-seq between each pair of conditions in the current study were also attached to ASoC SNPs if the transcription start site of the gene is located in a 50Kbp-window of the ASoC SNPs. In addition, ASoC SNPs were allocated to TFs if the corresponding TF motifs were identified by chromVAR in scATAC-seq analysis of the current study. Finally, all the enrichment estimations were performed by R function fisher.test() while the adjustment of p values from multiple tests were done by p.adjust() using the “fdr” method except for those indicated in the context.
Correlation between allelic imbalance and motif disruptions
To test the effects of ASoC SNPs, i.e. a genetic perturbation, on TF binding motifs, we calulated motif break scores at each ASoC SNP using R package motifbreakR.30 Concretely, for each cell type, we first compiled a set of ASoC SNPs that are in the TF binding footprints identified in the TF binding footprints analysis. Subsequently, the disruptiveness of ASoC SNPs on TFBS were evaluated using motifbreakR() function with parameters: threshold = 1 × 10−4, method = "log", bkg = c(A = 0.25, C = 0.25, G = 0.25, T = 0.25). Next, only SNPs with a “strong” effect were retained, and motif break scores were represented by alleleDiff which is calculated by the difference between the scoreAlt and scoreRef in the motifbreakR results. Then, for each cell type of each condition, we estimated the correlation between motif break scores and allelic imbalance for each TF motif using Spearman’s rank correlation using cor.test() R function. The allelic imbalance was evaluated by the log2-transform ratio between alternative and reference ATAC-seq read counts per ASoC SNP. Finally, the correlations measured by Spearman’s rho were plotted as a heatmap using the ggplot2 package.
Multi-omics integration from the public resource and the current study
The functions of ASoC SNPs were also evaluated in scenarios of multi-omics integration. We downloaded publicly available GWAS/omics data, including COVID-19 GWAS summary statistics by HGI,14 whole blood eQTL summary statistics from the meta-analysis by eQTLGen,33 and promoter capture Hi-C data from Javierre and colleagues’ study.32 After integrating with scATAC-seq read depth, peak-to-gene links, and ASoC SNPs from the current study, the cross-omics results were visualized by the R/Gviz package59 along the genomic coordinates to show the ASoC SNP examples. Specifically, the track for promoter capture Hi-C and peak-to-gene links were visualized by R/GenomicInteractions.58
CRISPR- and inhibitor-experiments
Cells deficient in LUCAT1 were generated using a lentiviral CRISPR interference vector (Addgene #71237). A gRNA inserts targeting the transcriptional start site of LUCAT1 was cloned into the vector followed by lentiviral particle production. To this end, HEK293T cells were transfected with the lentiviral vector, a VSVG pseudotyping plasmid (pVSVG) and a helper-plasmid (psPAX2), using lipofectamine 2000 reagent. Viral particles were collected by passing transfected cell supernatants through a 0.45 μm filter, followed by ultracentrifugation. For transduction, the viral pellet was resuspended in PBS and transferred to THP1 cells, followed by centrifugation at 37°C and 800 g for 2 h. Transduced cells were enriched using an Aria III cell sorter (BD), based on GFP-expression from the lentiviral backbone.
THP1 and Hek293T cells were cultivated in RPMI 1640 medium (Thermo Fisher), supplemented with 10% FCS (Biochrom) and 1% penicillin/streptomycin solution (Thermo Fisher). Primary monocytes were isolated from Buffy coats (deidentified prior to use) using Lymphoprep gradient centrifugation and CD14-micoboeads (Miltenyi) and cultivated in X-vivo 15 medium (Lonza). All cells were kept in a 37°C incubator with a humidified atmosphere containing 5% CO2. For inhibitor experiments, cells were pre-incubated with the respective inhibitor for 2 h, followed by further stimulations. IL1α, IFNα were purchased from Preprotech and 3p-hairpin-RNA from Invivogen. Stimulations were carried out for 4 h (100 ng of each factor). RNA was extracted with Trizol reagent and qRT-PCR was done using the High Capacity cDNA Reverse Transcription kit (Thermo Fisher), LUNA Universal qPCR master mix (NEB) and a Quantstudio 3 instrument. Fold-changes based on CT values were calculated using the 2ˆ-DDCT method.
Acknowledgments
The authors thank all volunteers from the Hannover Medical School (MHH) for participation in the study. This study was supported by the COFONI (COVID-19 Research Network of the State of Lower Saxony) with funding from the Ministry of Science and Culture of Lower Saxony, Germany (14-76403-184) to Y.L., R.F., T.I., and J.H.; the Network Universities of Medicine (NUM) CODEX+ fund from the Federal Ministry of Education and Research (BMBF) to Y.L.; the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under Germany’s Excellence Strategy—EXC 2155 project number 390874280 to Y.L.; and the Helmholtz Centre for Infection Research network fund (NASAVIR) to Y.L. and J.H. Y.L. was also supported by a European Research Council (ERC) Starting Grant (948207) and the Radboud University Medical Center Hypatia Grant (2018) for Scientific Research. Z.Z. was supported by a joint scholarship by the University of Groningen and China Scholarship Consortium (CSC201706350277) and Singh-Chhatwal-Postdoctoral Fellowship at the Helmholtz Centre for Infection Research. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), National Heart, Lung, and Blood Institute (NHLBI), National Institute on Drug Abuse (NIDA), National Institute of Mental Health (NIMH), and National Institute of Neurological Disorders and Stroke (NINDS). The data used for the analyses described in this manuscript were obtained from the GTEx Portal on May 28, 2021. A.-E.S. acknowledges FOR-COVID (Bayerisches Staatsministerium für Wissenschaft und Kunst) and the Helmholtz Association for support.
Author contributions
Conceptualization and study design: Y.L.; data analysis and investigation: B.Z., Z.Z., V.A.C.M.K., S.K., A.V., and R.G.; loss-of-function experiments: L.N.S.; discussion and interpretation: B.Z., Z.Z., V.A.C.M.K., Y.L., C.-J.X., S.K., M.C., L.N.S., L.E.S., A.V., U.O., and J.H.; sample collection and biospecimen resources: M.C., L.N.S., R.F., T.I., U.O., H.-C.T., Z.L., C.F.S., B.B., and R.G.; writing – original manuscript: B.Z., Z.Z., and V.A.C.M.K.; review and editing manuscript: all authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100232.
Supplemental information
Data and code availability
All raw sequencing and genotypes generated during this study are deposited at the European Genome-phenome Archive (EGA) under the accession numbers EGA: EGAZ00001823187, EGAS00001006559, and EGAS00001006560, which are hosted by the EBI and the CRG. Processed data as Seurat objects with scRNA-seq count matrices and ArchR objects with scATAC-seq peak matrices have been deposited via Nubes: https://nubes.helmholtz-berlin.de/s/wqg6tmX4fW7pci5. Original codes and scripts used for the analyses are available at GitHub: https://github.com/CiiM-Bioinformatics-group/MHH50_COVID19_code and Zenodo: https://doi.org/10.5281/zenodo.7270242.
References
- 1.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verity R., Okell L.C., Dorigatti I., Winskill P., Whittaker C., Imai N., Cuomo-Dannenburg G., Thompson H., Walker P.G.T., Fu H., et al. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect. Dis. 2020;20:669–677. doi: 10.1016/S1473-3099(20)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184:1895–1913.e19. doi: 10.1016/j.cell.2021.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J.-Y., Wang X.-M., Xing X., Xu Z., Zhang C., Song J.-W., Fan X., Xia P., Fu J.-L., Wang S.-Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z., John Wherry E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020;20:529–536. doi: 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reusch N., De Domenico E., Bonaguro L., Schulte-Schrepping J., Baßler K., Schultze J.L., Aschenbrenner A.C. Neutrophils in COVID-19. Front. Immunol. 2021;12:652470. doi: 10.3389/fimmu.2021.652470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krämer B., Knoll R., Bonaguro L., ToVinh M., Raabe J., Astaburuaga-García R., Schulte-Schrepping J., Kaiser K.M., Rieke G.J., Bischoff J., et al. Early IFN-α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity. 2021;54:2650–2669.e14. doi: 10.1016/j.immuni.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Q., Lin F., Wang Y., Zeng M., Luo M. Long noncoding RNAs as emerging regulators of COVID-19. Front. Immunol. 2021;12:700184. doi: 10.3389/fimmu.2021.700184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Severe Covid-19 GWAS Group. Ellinghaus D., Degenhardt F., Bujanda L., Buti M., Albillos A., Invernizzi P., Fernández J., Prati D., Baselli G., et al. Genomewide association study of severe covid-19 with respiratory failure. N. Engl. J. Med. 2020;383:1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pairo-Castineira E., Clohisey S., Klaric L., Bretherick A.D., Rawlik K., Pasko D., Walker S., Parkinson N., Fourman M.H., Russell C.D., et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 14.Callaway E. Mapping the human genetic architecture of COVID-19. Nature. 2021;596:472–473. doi: 10.1038/s41586-021-03767-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.You M., Chen L., Zhang D., Zhao P., Chen Z., Qin E.-Q., Gao Y., Davis M.M., Yang P. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol. 2021;23:620–630. doi: 10.1038/s41556-021-00690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z., Kilic G., Li W., Bulut O., Gupta M.K., Zhang B., Qi C., Peng H., Tsay H.-C., Soon C.F., et al. Multi-omics integration reveals only minor long-term molecular and functional sequelae in immune cells of individuals recovered from COVID-19. Front. Immunol. 2022;13:838132. doi: 10.3389/fimmu.2022.838132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallagher M.D., Chen-Plotkin A.S. The post-GWAS era: from association to function. Am. J. Hum. Genet. 2018;102:717–730. doi: 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nott A., Holtman I.R., Coufal N.G., Schlachetzki J.C.M., Yu M., Hu R., Han C.Z., Pena M., Xiao J., Wu Y., et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134–1139. doi: 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corces M.R., Shcherbina A., Kundu S., Gloudemans M.J., Frésard L., Granja J.M., Louie B.H., Eulalio T., Shams S., Bagdatli S.T., et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020;52:1158–1168. doi: 10.1038/s41588-020-00721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu X., Zhang B., Koeken V.A.C.M., Gupta M.K., Li Y. Multi-omics approaches in immunological research. Front. Immunol. 2021;12:668045. doi: 10.3389/fimmu.2021.668045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson B.K., Francisco E.B., Yogendra R., Long E., Pise A., Rodrigues H., Hall E., Herrara M., Parikh P., Guevara-Coto J., et al. Persistence of SARS CoV-2 S1 protein in CD16+ monocytes in post-acute sequelae of COVID-19 (PASC) up to 15 Months post-infection. Immunology. 2021;12:746021. doi: 10.1101/2021.06.25.449905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenbauer F., Tenen D.G. Transcription factors in myeloid development: balancing differentiation with transformation. Nat. Rev. Immunol. 2007;7:105–117. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- 23.Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal S., Vierbuchen T., Ghosh S., Chan J., Jiang Z., Kandasamy R.K., et al. The long non-coding RNA LUCAT1 is a negative feedback regulator of interferon responses in humans. Nat. Commun. 2020;11:6348. doi: 10.1038/s41467-020-20165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wendisch D., Dietrich O., Mari T., von Stillfried S., Ibarra I.L., Mittermaier M., Mache C., Chua R.L., Knoll R., Timm S., et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. 2021;184:6243–6261.e27. doi: 10.1016/j.cell.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charo I.F., Ransohoff R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 27.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.GTEx Consortium. Laboratory, Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA. Biospecimen Collection Source Site—NDRI Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abramov S., Boytsov A., Bykova D., Penzar D.D., Yevshin I., Kolmykov S.K., Fridman M.V., Favorov A.V., Vorontsov I.E., Baulin E., et al. Landscape of allele-specific transcription factor binding in the human genome. Nat. Commun. 2021;12:2751. doi: 10.1038/s41467-021-23007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coetzee S.G., Coetzee G.A., Hazelett D.J. motifbreakR : an R/Bioconductor package for predicting variant effects at transcription factor binding sites: fig. 1. Bioinformatics. 2015;31:3847–3849. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R., et al. The NIH Roadmap epigenomics mapping Consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Hill S.M., Sewitz S., Cairns J., Wingett S.W., Várnai C., Thiecke M.J., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–1384.e19. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S., et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanderheiden A., Thomas J., Soung A.L., Davis-Gardner M.E., Floyd K., Jin F., Cowan D.A., Pellegrini K., Shi P.Y., Grakoui A., et al. CCR2 signaling restricts SARS-CoV-2 infection. mBio. 2021;12 doi: 10.1128/mBio.02749-21. e0274921-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fingerlin T.E., Murphy E., Zhang W., Peljto A.L., Brown K.K., Steele M.P., Loyd J.E., Cosgrove G.P., Lynch D., Groshong S., et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013;45:613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H., Maqsudi S., Rainczuk A., Duffield N., Lawrence J., Keane F.M., Justa-Schuch D., Geiss-Friedlander R., Gorrell M.D., Stephens A.N. Identification of novel dipeptidyl peptidase 9 substrates by two-dimensional differential in-gel electrophoresis. FEBS J. 2015;282:3737–3757. doi: 10.1111/febs.13371. [DOI] [PubMed] [Google Scholar]
- 37.Geiss-Friedlander R., Parmentier N., Möller U., Urlaub H., Van den Eynde B.J., Melchior F. The cytoplasmic peptidase DPP9 is rate-limiting for degradation of proline-containing peptides. J. Biol. Chem. 2009;284:27211–27219. doi: 10.1074/jbc.M109.041871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griswold A.R., Ball D.P., Bhattacharjee A., Chui A.J., Rao S.D., Taabazuing C.Y., Bachovchin D.A. DPP9’s enzymatic activity and not its binding to CARD8 inhibits inflammasome activation. ACS Chem. Biol. 2019;14:2424–2429. doi: 10.1021/acschembio.9b00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen N.A.F., Grondman I., de Nooijer A.H., Boahen C.K., Koeken V.A.C.M., Matzaraki V., Kumar V., He X., Kox M., Koenen H.J.P.M., et al. Dysregulated innate and adaptive immune responses discriminate disease severity in COVID-19. J. Infect. Dis. 2021;223:1322–1333. doi: 10.1093/infdis/jiab065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reyes M., Filbin M.R., Bhattacharyya R.P., Sonny A., Mehta A., Billman K., Kays K.R., Pinilla-Vera M., Benson M.E., Cosimi L.A., et al. Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro. Sci. Transl. Med. 2021;13:eabe9599. doi: 10.1126/scitranslmed.abe9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams T.S., Schupp J.C., Poli S., Ayaub E.A., Neumark N., Ahangari F., Chu S.G., Raby B.A., DeIuliis G., Januszyk M., et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webb B.J., Peltan I.D., Jensen P., Hoda D., Hunter B., Silver A., Starr N., Buckel W., Grisel N., Hummel E., et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: a cohort study. Lancet. Rheumatol. 2020;2:e754–e763. doi: 10.1016/S2665-9913(20)30343-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang S., Zhang H., Zhou Y., Qiao M., Zhao S., Kozlova A., Shi J., Sanders A.R., Wang G., Luo K., et al. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science. 2020;369:561–565. doi: 10.1126/science.aay3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atak Z.K., Taskiran I.I., Demeulemeester J., Flerin C., Mauduit D., Minnoye L., Hulselmans G., Christiaens V., Ghanem G.-E., Wouters J., Aerts S. Interpretation of allele-specific chromatin accessibility using cell state–aware deep learning. Genome Res. 2021;31:1082–1096. doi: 10.1101/gr.260851.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ginhoux F., Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 46.Serbina N.V., Pamer E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27:883–890.e2. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider V.A., Graves-Lindsay T., Howe K., Bouk N., Chen H.-C., Kitts P.A., Murphy T.D., Pruitt K.D., Thibaud-Nissen F., Albracht D., et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27:849–864. doi: 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the tidyverse. J. Open Source Softw. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 53.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. 2004. Bioconductor: Open Software Development for Computational Biology and Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R Package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlson M. 2017. org.Hs.eg.db: Genome Wide Annotation for Human. R package version 3.12.0. [DOI] [Google Scholar]
- 56.Slowikowski K., Schep A., Hughes S., Dang T.K., Lukauskas S., Irisson J.-O., Kamvar Z.N., Thompson R., Christophe D., Hiroaki Y., et al. CRAN; 2020. R package ggrepel version 0.9.0. [Google Scholar]
- 57.Dowle M., Srinivasan A., Gorecki J., Chirico M., Stetsenko P., Short T., Lianoglou S., Antonyan E., Bonsch M., Parsonage H., et al. CRAN; 2021. R package data.table version 1.14.0. [Google Scholar]
- 58.Harmston N., Ing-Simmons E., Perry M., Barešić A., Lenhard B. GenomicInteractions: an R/Bioconductor package for manipulating and investigating chromatin interaction data. BMC Genom. 2015;16:963. doi: 10.1186/s12864-015-2140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hahne F., Ivanek R. In: Statistical Genomics Methods in Molecular Biology. Mathé E., Davis S., editors. Springer New York; 2016. Visualizing genomic data using Gviz and bioconductor; pp. 335–351. [DOI] [PubMed] [Google Scholar]
- 60.Van Rossum G. Python Software Foundation; 2009. Python version 3.9.6. [Google Scholar]
- 61.Gaspar J.M. Improved peak-calling with MACS2. Bioinformatics. 2018 doi: 10.1101/496521. [DOI] [Google Scholar]
- 62.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunter J.D. Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering. 2007;9:90–95. [Google Scholar]
- 64.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van de Geijn B., McVicker G., Gilad Y., Pritchard J.K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castel S.E., Levy-Moonshine A., Mohammadi P., Banks E., Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015;16:195. doi: 10.1186/s13059-015-0762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuchsberger C., Abecasis G.R., Hinds D.A. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–784. doi: 10.1093/bioinformatics/btu704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.NHLBI Trans-Omics for Precision Medicine. TOPMed) Consortium. Taliun D., Harris D.N., Kessler M.D., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., Pitsillides A.N., et al. Sequencing of 53, 831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heaton H., Talman A.M., Knights A., Imaz M., Gaffney D.J., Durbin R., Hemberg M., Lawniczak M.K.N. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods. 2020;17:615–620. doi: 10.1038/s41592-020-0820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012 doi: 10.48550/ARXIV.1207.3907. Preprint at. [DOI] [Google Scholar]
- 75.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mabbott N.A., Baillie J.K., Brown H., Freeman T.C., Hume D.A. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genom. 2013;14:632. doi: 10.1186/1471-2164-14-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martens J.H.A., Stunnenberg H.G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 2013;98:1487–1489. doi: 10.3324/haematol.2013.094243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G., et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018;175:1701–1715.e16. doi: 10.1016/j.cell.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Novershtern N., Subramanian A., Lawton L.N., Mak R.H., Haining W.N., McConkey M.E., Habib N., Yosef N., Chang C.Y., Shay T., et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26:1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing and genotypes generated during this study are deposited at the European Genome-phenome Archive (EGA) under the accession numbers EGA: EGAZ00001823187, EGAS00001006559, and EGAS00001006560, which are hosted by the EBI and the CRG. Processed data as Seurat objects with scRNA-seq count matrices and ArchR objects with scATAC-seq peak matrices have been deposited via Nubes: https://nubes.helmholtz-berlin.de/s/wqg6tmX4fW7pci5. Original codes and scripts used for the analyses are available at GitHub: https://github.com/CiiM-Bioinformatics-group/MHH50_COVID19_code and Zenodo: https://doi.org/10.5281/zenodo.7270242.