

Abstract
Vitamin E was identified almost a century ago as a botanical compound necessary for rodent reproduction. Decades of research since then established that of all members of the vitamin E family, α-tocopherol is selectively enriched in human tissues, and it is essential for human health. The major function of α-tocopherol is thought to be that of a lipid-soluble antioxidant that prevents oxidative damage to biological components. As such, α-tocopherol is necessary for numerous physiological processes such as permeability of lipid bilayers, cell adhesion, and gene expression. Inadequate levels of α-tocopherol interfere with cellular function and precipitate diseases, notably ones that affect the central nervous system. The extreme hydrophobicity of α-tocopherol poses a serious thermodynamic barrier for proper distribution of the vitamin to target tissues and cells. Although transport of the vitamin shares some steps with that of other lipids, selected tissues evolved dedicated transport mechanisms involving the α-tocopherol transfer protein (αTTP). The critical roles of this protein and its ligand are underscored by the debilitating pathologies that characterize human carriers of mutations in the TTPA gene.
Keywords: vitamin E, tocopherol, oxidative stress, neurodegeneration, ataxia
INTRODUCTION
Vitamin E was identified as an essential nutrient almost a century ago when Evans & Bishop (26) discovered a fat-soluble botanical that restored fertility to diet-restricted rats. The compound was termed “tocopherol” (Greek for “to bring offspring”), and its activity in enabling rodent reproduction is used as a measure of its activity to this day. We review here available data on the physiological functions of vitamin E and the mechanisms of action of αTTP. We focus the reader’s attention on the mechanisms that control vitamin E status in the central nervous system (CNS), the tissue most sensitive to malfunctions in vitamin E transport.
Vitamin E
The term vitamin E refers to a family of tocochromanols, neutral plant lipids that include tocopherols and tocotrienols. All family members possess a common hydroxylated chromanol ring and a hydrophobic 13-carbon phytyl side chain. Tocopherols have a saturated phytyl chain, whereas the side chain of tocotrienols contains three double bonds. The extent and location of chromanol ring methylation distinguish the various “vitamers” in the family, as shown in Figure 1. One of the striking features of vitamin E biology is that regardless of the tocopherol composition of dietary intake, the α-tocopherol form is selectively enriched in plasma and tissues of most organisms. Hence, although it has been recently proposed that other members of the vitamin E family display biological activities (e.g., 1), we focus this review on α-tocopherol, the only form that is actively retained in vivo.
Figure 1.
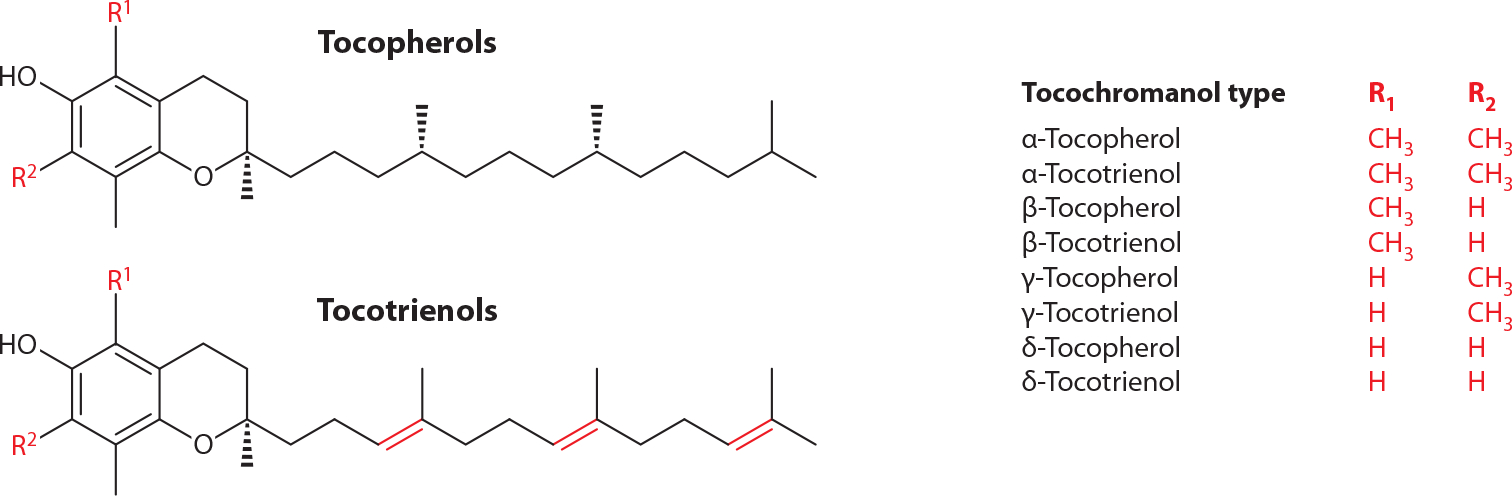
Molecular structures of vitamin E family members (the tocochromanols). Courtesy: J. Atkinson.
Antioxidant action.
Soon after its discovery, vitamin E’s ability to inhibit rancidity of stored lard was realized (57). During the following decades, chemical, biophysical, and physiological studies established that the major biological activity of α-tocopherol is to serve as an antioxidant that quenches free radicals formed by the oxidation of unsaturated lipids. In doing so, vitamin E breaks the chain reaction by which lipid peroxidation propagates, and it prevents oxidative damage to biological membranes. Consequently, vitamin E adequacy is critical for numerous biochemical reactions that rely on bilayer integrity (88). Chemical and spectroscopic approaches established that the antioxidant activity of vitamin E resides in the lone hydroxyl group on the chromanol ring. The quenching reaction, in which the vitamin donates its hydroxyl hydrogen to the lipid peroxyl radical, converts tocopherol to the more stable tocopheroxyl radical (14, 16). In turn, the tocopheroxyl radical either can be reduced back to the ground-state vitamin by other redox cyclers such as vitamin C (9, 66) or can be further oxidized to tocopherol quinones (74). At present, surprisingly little is known regarding the metabolic steps that regulate the formation and fate of these postreactive forms of α-tocopherol in vivo (76). Vitamin E’s low abundance in membranes (approximately one mole α-tocopherol to 1,000 moles phospholipid; 13), its slow turnover in some tissues, and the possible involvement of other antioxidants in its recycling place critical importance on its distribution in biological membranes and its orientation within the bilayer. Multiple lines of evidence suggest that tocopherol is primarily localized in microdomains enriched with polyunsaturated fatty acids that tend to be more curved and fluid than other membrane regions (cf. 5). X-ray diffraction and spectroscopic studies (28) led to a proposed structural model in which the hydrophobic tail portion of α-tocopherol inserts into one leaflet of the lipid bilayer, while the chromanol ring is situated near the phospholipid head groups, orienting the molecule for effective donation of its hydroxyl hydrogen to lipid radicals, as depicted in Figure 2.
Figure 2.
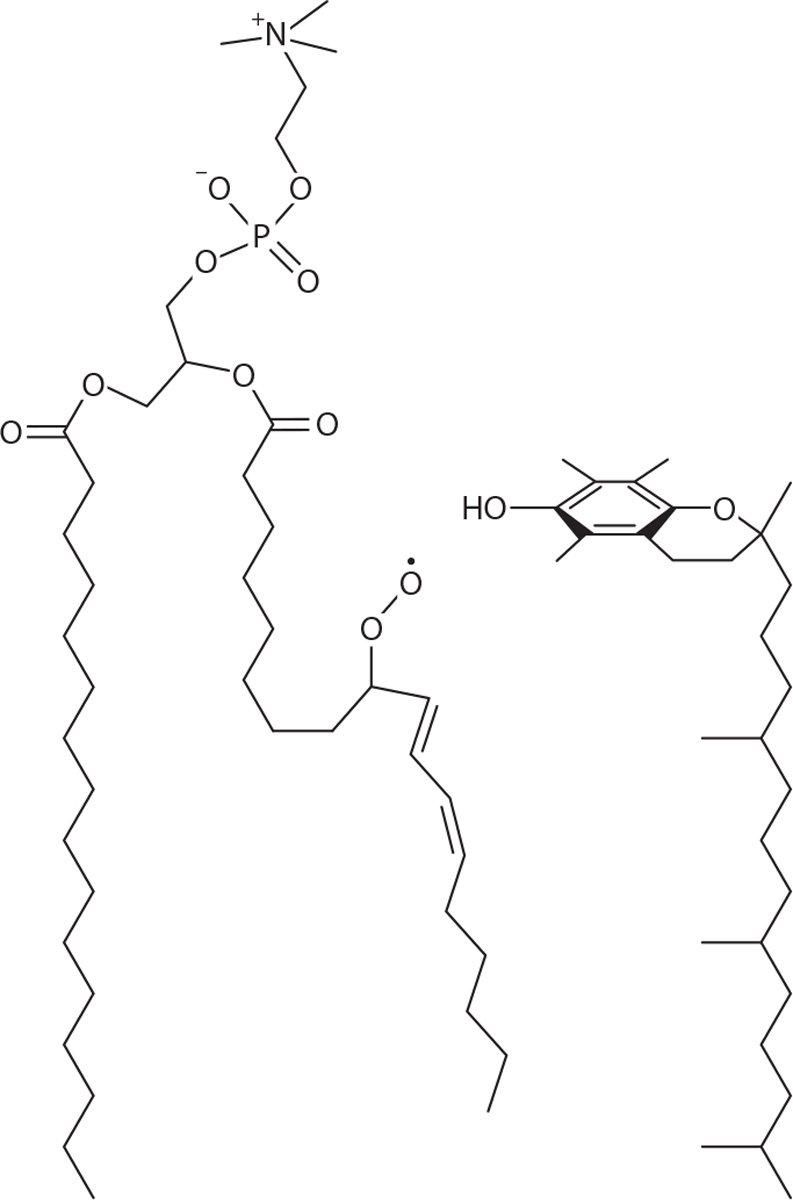
Conformational model of α-tocopherol within the lipid bilayer, in proximity to a radical product of unsaturated phospholipid peroxidation in phosphatidylcholine. Courtesy: J. Atkinson; drawn after (4).
Tocopherol functions as an efficient antioxidant that scavenges lipid peroxyl radicals some 200-fold faster than butylated hydroxytoluene, a synthetic antioxidant commonly used in the food industry (13). Accordingly, observations made during a century of research led to the designation of α-tocopherol as the major lipid-soluble antioxidant in humans (13, 15, 20, 99). We note that antioxidant-independent activities of vitamin E have also been proposed (cf. 108). However, the molecular mechanisms and physiological relevance of such possible activities are not well understood and remain controversial (88), and they are not discussed here.
VITAMIN E AND HUMAN HEALTH
The discovery of α-tocopherol was rooted in the vitamin’s ability to prevent pathological conditions and led to its assignment as an essential micronutrient. Indeed, deficiency in this nutrient precipitates a number of pathologies, and adequate intake of α-tocopherol is defined by the Institute of Medicine as critical for human health. It should be emphasized that although ill effects of supplementation with vitamin E have been reported (e.g., 50), these reports do not refute the recognition that α-tocopherol is an essential nutrient whose absence severely compromises human health. It is also noteworthy that the daily recommended intake of α-tocopherol (15 mg per day for adults) is rarely satisfied in the US population (27, 29).
Vitamin E Deficiency
Insights into α-tocopherol’s biological roles and its mechanisms of action can be obtained from studying the consequences of deficiency in this nutrient. Symptomatic dietary vitamin E deficiency is very rare, perhaps due to the abundance of α-tocopherol in plant oils. Metabolic vitamin E deficiencies are of two general types: (a) primary deficiency, arising from specific alteration in vitamin E status, and (b) secondary deficiency, in which low levels of vitamin E are secondary to other global perturbations such as disorders in lipid malabsorption or lipoprotein metabolism and transport. Commonly diagnosed primary and secondary vitamin E deficiencies manifest primarily in altered CNS function and present as neuropathological disorders. In all cases, high-dose supplementation with vitamin E effectively attenuates disease progression, and if started at an early stage, can reverse some of the neurological deficits. Unfortunately, because analytical measurement of vitamin E levels is not included in routine diagnostic protocols, deficiency is usually diagnosed at an advanced disease stage, when neurological dysfunctions are already debilitating and irreversible.
Primary Vitamin E Deficiency
Primary deficiency is seen in cases of the autosomal recessive disorder ataxia with vitamin E deficiency (AVED; OMIM #277460), which is caused by heritable mutations in TTPA, the gene that encodes α-tocopherol transfer protein (αTTP; 19, see discussion below). The disease is characterized by progressive spinocerebellar ataxia accompanied by very low plasma vitamin E levels. Clinical features include dysarthria, clumsiness of the hands, loss of proprioception, areflexia, dysdiadochokinesia, decreased visual acuity, and positive Babinski sign. A genetic mouse model in which the expression of the TtpA gene has been disrupted faithfully recapitulates the human disease. These animals display late-onset spinocerebellar ataxia, compromised electrophysiological responses, and neurodegeneration. In accordance with vitamin E’s function as an antioxidant, these pathophysiological presentations are accompanied by marked elevation in markers of oxidative stress (85, 101, 103).
Secondary Vitamin E Deficiency
Vitamin E deficiency is also observed in some disorders that affect metabolic pathways that are not specific to vitamin E processing. Interestingly, neurological symptoms of vitamin E deficiency, similar to those observed in patients with AVED, comprise a prime clinical signature of these disorders. Specifically, cerebellar ataxia is often one of the major clinical presentations of these diseases. Moreover, vitamin E supplementation, often at pharmacologic doses, is a beneficial intervention in these cases.
Secondary vitamin E deficiency in lipid malabsorption disorders.
Humans with compromised intestinal fat absorption diseases of various causes become vitamin E deficient. These diseases include cholestatic liver disease, short bowl syndrome, Crohn’s disease, and abetalipoproteinemia (62). Clinical presentations include ataxia, neuropathy, degeneration of the posterior column fibers, retinopathy, and compromised sensory conduction (61). In light of the hydrophobic character of α-tocopherol and the dependence of its absorption and transport on lipoprotein carriers, effective therapeutic intervention in patients with fat malabsorption often requires extreme vitamin E supplementation measures such as repeated injections of 10–100 mg α-tocopherol/kg (62). An interesting example is the case of cystic fibrosis (CF; OMIM #219700), one of the most common lethal autosomal diseases affecting Caucasians. Due to compromised pancreatic function, patients with CF present with severe fat malabsorption that leads to deficiency in fat-soluble vitamins, including vitamin E (77). Prior to the adoption of a regimented supplementation with pancreatic enzymes and fat-soluble vitamins, numerous cases were documented with neurological deficits and evidence of elevated lipid peroxidation. The new supplementation practices lead to significant improvement in vitamin E status, concomitant with significant amelioration of neurological deficits (47).
Secondary vitamin E deficiency in other disorders.
Vitamin E deficiency is also occasionally observed despite normal intestinal fat absorption owing to molecular defects that affect lipid transport and trafficking, such as in the two examples below.
Niemann-Pick disease type C (NPC; OMIM #257220 and #607625) is a lysosomal storage disorder caused by heritable mutations in the NPC1 or NPC2 transporters of the late endocytic compartment (12). NPC is characterized by accumulation and aggregation of lipids in multivesicular bodies of lysosomal origin and culminates in fatal neurodegeneration. Although the molecular culprit underlying NPC pathology was originally thought to be altered cholesterol status, it is now clear that other trapped lipids also play important roles (51). Indeed, vitamin E levels are altered in the brains and livers of Npc1−/− and Npc2−/− mouse models (92, 100). It is likely that, like cholesterol, vitamin E accumulates in the altered lysosomal compartment and is therefore sequestered in an unavailable state, causing the increased oxidative stress that is known to accompany NPC disease (105). It may also be postulated that unavailability of vitamin E and subsequent oxidative stress are important contributors to the spinocerebellar ataxia and neurodegeneration of Purkinje neurons presented by patients with NPC (30, 37). In support of this notion, vitamin E supplementation improves cognitive function in Npc1-null mice (7).
Tangier disease (OMIM #205400) is a rare autosomal recessive malady caused by mutations in the ATP-binding cassette-1 gene (ABCA1) encoding a membrane-bound lipid transporter (63). The disease is characterized by an absence of circulating high-density lipoprotein (HDL). Consequently, patients with Tangier disease accumulate cholesterol and lipids in many tissues, are at high risk for cardiovascular disease, and display progressive neuropathy. Since ABCA1 mediates the transport of α-tocopherol (64, 71, 75), it is reasonable to propose that this deficiency is a major contributor to the demyelination of peripheral neurons that characterizes Tangier disease. Surprisingly, vitamin E supplementation of affected humans or mouse models has not been reported.
Regardless of its particular molecular origins, vitamin E deficiency manifests primarily in neurological disorders, indicating that adequate levels of α-tocopherol are critical for CNS health. This notion is underscored by multiple findings documenting that α-tocopherol protects fragile neurons from oxidative stress–related damage (60), that oxidative stress and clinical severity of CNS dysfunction are correlated with vitamin E levels (e.g., 42, 54), and by therapeutic benefit of vitamin E supplementation in neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and dementia (e.g., 25). Moreover, studies in cultured cells and animal models demonstrated that vitamin E attenuates hypoxia-induced oxidative stress (53) and protects cerebellar neurons from oxidative stress–induced apoptosis (8). The association between adequate α-tocopherol status and neurological health led to widespread supplementation with vitamin E in Western cultures (83, 84).
VITAMIN E TRANSPORT
Many steps in vitamin E transport are shared with those of other dietary lipids. Absorbed tocopherols and tocotrienols are assembled in enterocytes into chylomicra particles that are secreted into the circulation and subsequently taken up by the liver. It is in the liver that a remarkable selection among vitamin E molecules takes place: α-Tocopherol is retained while other forms are degraded and destined to excretion (46). Following processing in the liver, vitamin E is delivered to nonhepatic tissues incorporated in lipoprotein complexes. Thus, although the vitamin E content of a typical US diet contains >70% γ-tocopherol, α-tocopherol constitutes >90% of the vitamin E in plasma and tissues. This selectivity is obtained by two independent hepatic activities: the αTTP (see below) that selectively retains α-tocopherol and a catabolizing cytochrome P-450 system that preferentially degrades all other forms of vitamin E (81).
Regulation of Systemic Vitamin E Status: The αTTP
The long-held view that tocopherol levels in blood and tissues simply reflect dietary vitamin E intake has been challenged following the realization that a specific hepatic protein, αTTP, regulates vitamin E levels and composition in vivo. Several lines of evidence established that αTTP regulates vitamin E status. (a) A number of groups identified a high-affinity, high-specificity α-tocopherol binding activity in liver cytosol (17, 18, 48). Moreover, the purified binding protein was shown to catalyze the transfer of α-tocopherol between lipid bilayers in vitro (73, 97). These criteria were used to purify αTTP from rat liver and to clone it from a human cDNA library (3, 72, 73). (b) Sokol and colleagues (80) reported that a subset of patients afflicted with familial ataxia exhibits extremely low levels of plasma vitamin E despite normal lipid absorption. This heritable disorder was initially named familial isolated vitamin E deficiency (FIVE) and later renamed ataxia with vitamin E deficiency (AVED; cf. 24). The genetic locus of the AVED mutations was mapped to the TTPA gene (36, 65). (c) Traber et al. (89, 90) noted that some patients with AVED are unable to discriminate between different forms of vitamin E, i.e., to retain α-tocopherol over other forms of the vitamin. These investigators hypothesized that the impairment may be caused by mutations in αTTP that affect its ligand selectivity. Subsequent studies revealed that αTTP is expressed at highest levels in parenchymal cells of the liver and to a smaller extent in the brain and the placenta. Low-level expression was also reported in the spleen, lung, and kidney (45, 55, 87). It may be postulated that while/whereas hepatic αTTP regulates α-tocopherol levels in blood and in blood-equilibrated tissues, its expression in other locations regulates distinct tissue-specific pools of the vitamin.
Activities of αTTP in Vitro
αTTP possesses two principal biochemical activities: (a) It binds α-tocopherol with a high affinity and selectivity and (b) it can facilitate the transfer of α-tocopherol between donor and acceptor membranes. It is usually assumed that these activities are at the basis of the protein’s ability to maintain vitamin E status in vivo. Hence, the preferential high-affinity binding to α-tocopherol enables the selective retention of α-tocopherol over other dietary forms of vitamin E, whereas facilitation of intermembrane transfer of the ligand may be at the basis of αTTP’s ability to stimulate intracellular trafficking of α-tocopherol. This notion is underscored by observations that many AVED-causing mutations in αTTP impair the ability of the protein to bind tocopherol and to facilitate its intermembrane transfer (59).
Activities of αTTP in Cultured Hepatocytes
Dietary vitamin E arrives at the liver incorporated in chylomicra or their remnants and is taken up by hepatocytes in a receptor-mediated process. There is no discrimination among the different forms of vitamin E during uptake by hepatocytes, and this step is αTTP independent (2, 71). Fluorescent microscopy studies using the analog nitrobenoxadiazyl-tocopherol enabled the study of vitamin E trafficking within hepatocytes at high spatial and temporal resolution. It was thus established that in hepatocytes, αTTP mediates transport of the vitamin from the late-endocytic compartment to specific sites at the plasma membrane, from which it is secreted to the extracellular milieu (70, 71; depicted in Figure 3).
Figure 3.
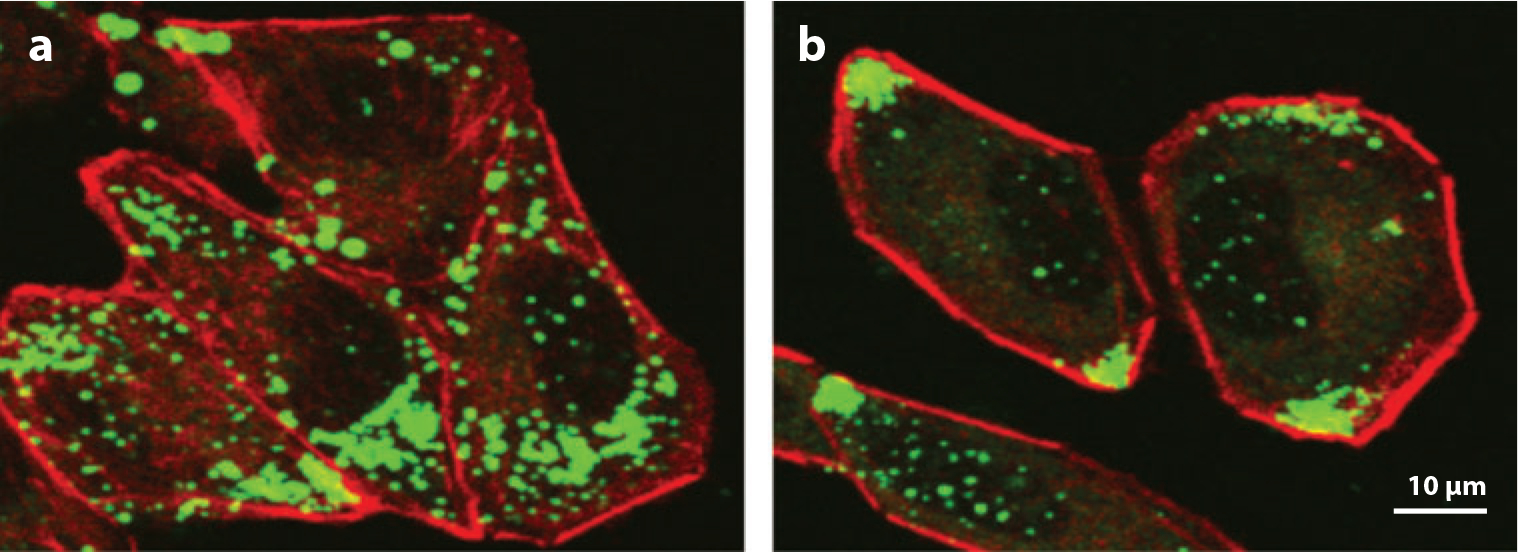
Facilitation of vitamin E secretion by αTTP. McA-RH7777 cells that express a control cDNA (a) or an αTTP expression vector (b) were “loaded” with nitrobenoxadiazyl-tocopherol, washed, and visualized using confocal fluorescence microscopy. Note that in αTTP-expressing cells, the fluorescent vitamin has translocated from perinuclear vesicles to the plasma membrane.
Although the exact path utilized for the αTTP-facilitated transport of α-tocopherol has not been characterized in detail, a number of key features of this process have been noted. (a) Intrahepatocyte trafficking of α-tocopherol relies on the tubulin cytoskeleton for vesicular movement (10, 71). (b) The α-tocopherol egress through the plasma membrane occurs via the membrane transporter ABCA1 (71, 75). (c) αTTP-mediated secretion of vitamin E is not affected by disruption of the Golgi apparatus, indicating that αTTP-mediated intracellular trafficking and secretion of α-tocopherol are independent of nascent very-low-density lipoprotein, i.e., the vitamin associates with lipoproteins following its secretion to the sinusoidal space. (d) Heritable mutations in αTTP that are found in AVED patients disrupt the transport process and result in retention of tocopherol in the lysosomal compartment (70). A model of vitamin E trafficking through the hepatocyte and the role(s) of α-TTP in this process are presented in Figure 4.
Figure 4.

A model depicting the trafficking of vitamin E through hepatocytes and the role of αTTP in this process. Following endocytosis-mediated uptake, dietary vitamin E arrives at the late endocytic compartment. In the presence of functional lysosomal transporters NPC1 and NPC2, α-tocopherol exits this compartment, and αTTP facilitates its vesicular transport to the plasma membrane. After ABCA1-dependent secretion to the extracellular space, α-tocopherol is complexed to lipoproteins and delivered to nonhepatic tissues. AVED-causing mutations in αTTP inhibit trans-hepatocyte trafficking and result in an accumulation of vitamin E in the hepatocytes and deficiency in nonhepatic tissues. Abbreviations: αTTP, α-tocopherol transfer protein; AVED, ataxia with vitamin E deficiency; NPC, Niemann-Pick disease type C; SRB1, scavenger receptor B1; TTP, tocopherol transfer protein.
Structure-Function Relationship in αTTP
The three-dimensional structure of αTTP (Figure 5) reveals a densely folded polypeptide characterized by the following structural features: (a) The protein is folded into a compact spherical shape, the surface of which is covered with charged residues (Figure 5a), accounting for the protein’s water solubility. (b) α-Tocopherol is bound within a deep cavity lined with hydrophobic residues (Figure 5b). (c) Access of the ligand to the binding pocket is limited by a channel, the entrance to which is gated by a flexible helix (lid). The surface of αTTP contains a hydrophobic prong proposed to enable association with membranes by penetrating one leaflet of the bilayer (Figure 5c).
Figure 5.

The three-dimensional structure of αTTP. (a) The electrostatic surface potential of the protein, emphasizing the abundance of basic (blue) and acidic (red) amino acids on the protein’s exterior surface. (b) The ligand binding pocket and the amphipathic lid that controls ligand access. Bound α-tocopherol is shown in red, and the lid is shown in the open, unligated conformation (green) and the closed, ligand-bound conformation (purple). (c) The association of αTTP with a lipid bilayer is shown based on a calculated model of free energies of binding to a model hydrophobic phase (cyan spheres) (52). The lid helix is colored purple. Courtesy: J. Atkinson & S. Chung.
The mechanism by which αTTP facilitates the transfer of α-tocopherol between membranes is poorly understood. Morley et al. (58) suggested that direct physical interaction between αTTP and membranes precedes extraction of the ligand into the protein’s binding pocket. Importantly, α-tocopherol reduces the affinity of αTTP to membranes, suggesting that it dissociates from membranes following acquisition of the ligand (58, 107). One possible mechanism involves active extraction of the ligand from the bilayer, using the flexible lid as a scoop, and then diffusion of the holo-protein through the aqueous phase to the acceptor membrane. Other possible scenarios include direct transfer of α-tocopherol from one vesicular membrane to another, e.g., from the lysosome to vesicles that are formed from the endocytic compartment.
The αTTP−/− Mouse
The roles of αTTP in regulating vitamin E homeostasis in vivo were critically evaluated using mouse models in which expression of the TtpA gene has been disrupted (cf. 55). The finding that TtpA gene dosage in the different mice (i.e., αTTP+/+, αTTP+/−, αTTP−/−) is proportional to plasma α-tocopherol levels reinforces the notion that αTTP determines plasma levels of the vitamin. TTP-null mice are characterized by vitamin E deficiency, oxidative stress, late-onset ataxia, and female infertility, all of which can be prevented by timely supplementation with α-tocopherol (43, 44, 85, 101). Although the first three phenotypes are analogous to the disease hallmarks observed in human patients with AVED, the relationship of αTTP and vitamin E to human fertility is presently unknown.
VITAMIN E AND THE CENTRAL NERVOUS SYSTEM
The half-life of α-tocopherol in the brain is one to two orders of magnitudes longer than in any other organ (11, 40), and unlike other tissues, the brain exclusively retains RRR-α-tocopherol at the expense of other tocols (21, 40, 69, 94). Notably, αTTP is expressed in the brain (38), and the rates of depletion and repletion of α-tocopherol in this organ are markedly slower in αTTP−/− mice (49). Moreover, expression levels of αTTP were shown to increase in patients afflicted with oxidative stress–related diseases (22). Taken together, these findings indicate that a separate pool of vitamin E is maintained in the CNS and that αTTP may serve to maintain and regulate this localized pool.
CNS Damage During Vitamin E Deficiency
Vitamin E deficiency manifests primarily as neurological and neuromuscular disorders (32, 60, 78, 79). The best-described disorder is vitamin E deficiency–induced axonalpathy, which affects sensory neurons and progresses from the periphery toward the head. In vitamin E–deficient rodents, monkeys, and humans, the primary affected regions are the medulla oblongata and the brain stem. At the microscopic level, injury progresses from gracile nuclei to the cuneate nuclei of the dorsal root ganglia sensory nerves. These regions are affected in a similar manner in multiple fat-malabsorption diseases, implicating vitamin E deficiency as a likely contributing factor to the associated neurological deficits. An important histological hallmark of vitamin E deficiency is the presence of axonal swellings, intracellular aggregates of proliferated endoplasmic reticulum, mitochondria, and neurofilaments (82). These are thought to present physical barriers for axonal transport, thereby disrupting neuron function. In support of this notion are findings of attenuated rates of axonal retrograde and anterograde transport and altered respiratory control in mitochondria isolated from brains of vitamin E–deficient rats (23, 86). An additional histological hallmark of vitamin E deficiency that parallels axonal dystrophy is the deposition of lipid peroxidation products in lipofuscin aggregates, indicating severe oxidative stress. The observed axonalpathy manifests also on a functional level. Electrophysiological studies in vitamin E–deficient rodents indicated compromised somatosensory-evoked potentials and visual-evoked potentials (33, 34, 101). Another neuropathological injury induced by vitamin E deficiency is reduced myelination in the spinal cord fibers. Transcriptome profiling analyses revealed that expression of key regulators of myelination, including retinoid orphan nuclear receptor-α, myelin proteolipid protein, and myelin basic protein, is attenuated in the CNS of vitamin E–deficient mice as compared to wild-type controls (31, 39).
Muscular lesions were also observed in vitamin E–deficient animals, in humans with malabsorption diseases, and in αTTP−/− mice. Histological observations revealed dense structures reminiscent of multivesicular bodies that reflect accumulation of aberrant lysosomal and autophagocytic vacuoles (68) that are accompanied by functional deficits observed in electromyographic recordings. However, muscular changes occur late during disease progression and likely represent consequences of neuronal degeneration rather than primary injury by vitamin E deficiency.
In accordance with the ataxic hallmark of vitamin E deficiency, mild cerebellar atrophy was reported in cholestasis-affected patients (78), and both humans and αTTP−/− mice display mild loss of cerebellar Purkinje neurons, the major coordinators of motor output from the CNS (101, 102). In accordance, cerebella of αTTP−/− mice display atrophy of Purkinje cell bodies and pronounced reduction in their dendritic branching (Figure 6).
Figure 6.

Diminished Purkinje cell branching and arborization in vitamin E–deficient mice. Shown are thin sections of Golgi-Cox stained cerebella from (a) 17-month-old αTTP−/− mice and (b) wild-type control animals.
From “Vitamin E Is Essential for Purkinje Neuron Integrity,” L. Ulatowski, G. Warrier, R. Sultana, D.A. Butterfield, R. Parker, & D. Manor, manuscript submitted.
Vitamin E deficiency precipitates both neuropathic and myopathic lesions, and both appear to precede the onset of ataxia. However, the root cause(s) and etiology of cerebellar injury are difficult to assess because of inconclusive longitudinal studies and difficulties in interpreting dietary depletion studies due to food auto-oxidation (32). The observation that the primary lesion that accompanies vitamin E deficiency is neuronal in humans but myopathic in rodents further confounds clear mechanistic understanding of the underlying impairment. Based on available data, we speculate that the initial events of vitamin E deficiency are intracellular aggregations that initiate axonal dystrophy. These events ultimately culminate in compromised Purkinje neuron function and diminished control of muscle coordination.
Transport of Vitamin E in the CNS
Limited information is available regarding the paths by which α-tocopherol travels through the CNS and the mechanisms that regulate this transport process. Uptake of HDL-complexed α-tocopherol across the blood-brain barrier was suggested to occur through a scavenger receptor class B1–mediated process (6, 35, 56). Within the murine CNS, apolipoprotein E (apoE)–containing particles are thought to transport α-tocopherol through the brain and in the cerebrospinal fluid (95, 96). Thus it appears that, similar to the situation in the liver, vitamin E utilizes transport routes in the CNS that are shared with other lipids. In situ hybridization revealed that αTTP mRNA is expressed in cerebellar cells that surround Purkinje neurons (38). Fluorescence microscopy approaches further show that αTTP is expressed in glial fibrillary acidic protein–positive cerebellar astrocytes but is excluded from β-tubulin-III-positive neurons (see Figure 7). These observations suggest that α-tocopherol-specific targeting in the brain may be directed by the localized expression of αTTP.
Figure 7.
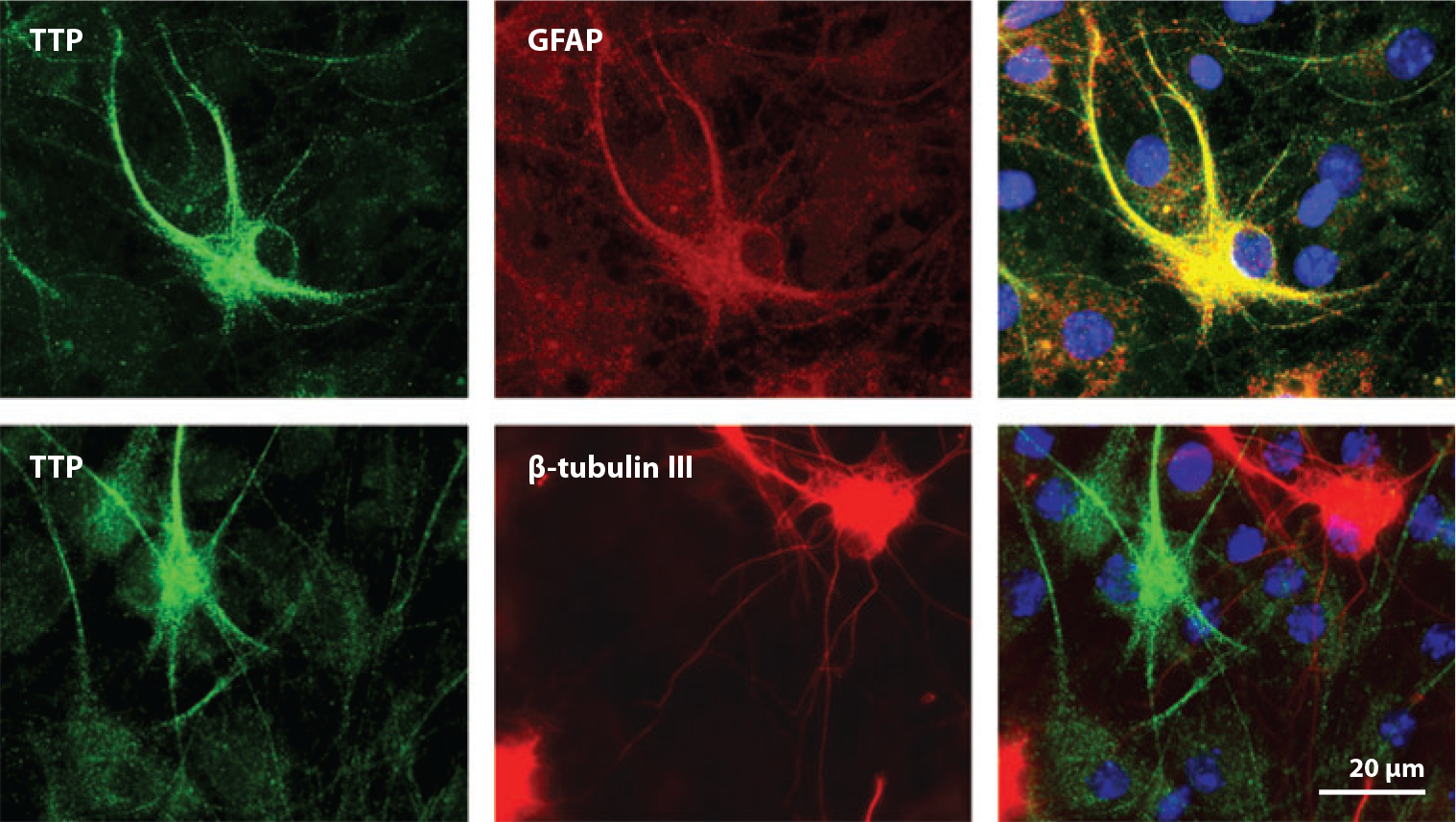
Astrocyte-specific expression of cerebellar αTTP. Primary cerebellar cells were stained with specific antibodies directed against αTTP (green) and the astrocyte marker glial fibrillary acidic protein (GFAP) (red, upper panel) or the neuronal marker β-tubulin III (red, lower panel). Note that αTTP and GFAP (yellow) localize to the same cells. From “Vitamin E Is Essential for Purkinje Neuron Integrity,” L. Ulatowski, G. Warrier, R. Sultana, D.A. Butterfield, R. Parker, & D. Manor, manuscript submitted.
Taken together with the known roles of astrocytes in metabolic support of neurons (104) and in apoE-mediated trafficking of cholesterol between astrocytes and neurons (67, 93), the astrocyte localization of αTTP suggests the working model for α-tocopherol trafficking in the cerebellum depicted in Figure 8. Under normal conditions, astrocytes store α-tocopherol in perinuclear vesicles, and αTTP regulates apoE-mediated egress of the vitamin to the neighboring neuron. Upon induction of oxidative stress, expression of αTTP in the astrocytes increases to facilitate delivery of α-tocopherol to the neurons, thereby protecting them from oxidative damage. Hence, modulation of the expression of the TTPA gene provides a homeostatic feedback response that protects Purkinje neurons from oxidative damage. Excessive oxidative stress, vitamin E deficiency, or αTTP mutations may undermine the ability of the system to prevent oxidative damage in Purkinje neurons, and ataxia ensues.
Figure 8.

Proposed model for trafficking of α-tocopherol between cerebellar astrocytes and neurons. α-Tocopherol is taken up by astrocytes and stored in perinuclear vesicles. αTTP facilitates egress of the vitamin via ABCA1 to nascent apolipoprotein E (ApoE)-containing lipoprotein particles. Particles are taken up by the neighboring neurons and distributed throughout its body, axon, and dendrites, where they quench lipid peroxidation. Since αTTP levels are the limiting factor in this transport chain, oxidative stress–induced increase in αTTP expression enhances vitamin E flux, thereby combating neuronal damage. Abbreviations: ABCA1, ATP-binding cassette transporter A1; Chol, cholesterol; LP, lipoprotein particle; PL, phospholipids; TTP, tocopherol transfer protein.
CONCLUSIONS AND FUTURE DIRECTIONS
A century of research established the critical roles that vitamin E plays in maintaining optimal neurological health in humans. Despite these productive efforts, critical questions remain unanswered. Thus, the steps and entities involved in the transport of α-tocopherol between astrocytes and neurons, the exact role(s) of αTTP in this process, and the physiological factors that regulate the process need to be elucidated. Similarly, studies on the alterations in vitamin E status that accompany neurologic diseases await attention from bench scientists and clinicians alike. We see special urgency in examining the notion of sub-clinical α-tocopherol deficiency. Since severe deficiency is rare, levels of vitamin E are not routinely measured, and outcomes of mild deficits have not been considered. Rationale for addressing this notion is provided by recent reports demonstrating an association between common polymorphisms in αTTP and vitamin E status (91, 98, 106). It is hoped that future multidisciplinary efforts will address these questions, lead to novel insights, and, in turn, benefit public health.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Aggarwal B, Nesaretnam K. 2012. Vitamin E tocotrienols: life beyond tocopherols. Genes Nutr. 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arita M, Nomura K, Arai H, Inoue K. 1997. α-Tocopherol transfer protein stimulates the secretion of α-tocopherol from a cultured liver cell line through a brefeldin A-insensitive pathway. Proc. Natl. Acad. Sci. USA 94:12437–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arita M, Sato Y, Miyata A, Tanabe T, Takahashi E, et al. 1995. Human α-tocopherol transfer protein: cDNA cloning, expression and chromosomal localization. Biochem. J. 306:437–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkinson J, Epand RF, Epand RM. 2008. Tocopherols and tocotrienols in membranes: a critical review. Free Radic. Biol. Med. 44:739–64 [DOI] [PubMed] [Google Scholar]
- 5.Atkinson J, Harroun T, Wassall SR, Stillwell W, Katsaras J. 2010. The location and behavior of α-tocopherol in membranes. Mol. Nutr. Food Res. 54:641–51 [DOI] [PubMed] [Google Scholar]
- 6.Balazs Z, Panzenboeck U, Hammer A, Sovic A, Quehenberger O, et al. 2004. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated α-tocopherol by an in vitro blood-brain barrier model. J. Neurochem. 89:939–50 [DOI] [PubMed] [Google Scholar]
- 7.Bascunan-Castillo EC, Erickson RP, Howison CM, Hunter RJ, Heidenreich RH, et al. 2004. Tamoxifen and vitamin E treatments delay symptoms in the mouse model of Niemann-Pick C. J. Appl. Genet. 45:461–67 [PubMed] [Google Scholar]
- 8.Behl C 2000. Vitamin E protects neurons against oxidative cell death in vitro more effectively than 17-beta estradiol and induces the activity of the transcription factor NF-kappaB. J. Neural. Transm. 107:393–407 [DOI] [PubMed] [Google Scholar]
- 9.Bendich A, D’Apolito P, Gabriel E, Machlin LJ. 1984. Interaction of dietary vitamin C and vitamin E on guinea pig immune responses to mitogens. J. Nutr. 114:1588–93 [DOI] [PubMed] [Google Scholar]
- 10.Bjorneboe A, Bjorneboe GE, Hagen BF, Nossen JO, Drevon CA. 1987. Secretion of α-tocopherol from cultured rat hepatocytes. Biochim. Biophys. Acta 922:199–205 [DOI] [PubMed] [Google Scholar]
- 11.Bourre JM, Clément M. 1991. Kinetics of rat peripheral nerve, forebrain and cerebellum α-tocopherol depletion: comparison with different organs. J. Nutr. 121:1204–7 [DOI] [PubMed] [Google Scholar]
- 12.Brady RO, Carstea ED, Pentchev PG. 1997. The Niemann-Pick diseases group. In The Molecular and Genetic Basis of Neurological Disease, ed. Rosenberg RN, Pruisner SB, DiMauro S, Barchi RL, pp. 387–403. Boston: Butterworth-Heinmann [Google Scholar]
- 13.Burton GW, Cheeseman KH, Doba T, Ingold KU, Slater TF. 1983. Vitamin E as an antioxidant in vitro and in vivo. Ciba Found. Symp. 101:4–18 [DOI] [PubMed] [Google Scholar]
- 14.Burton GW, Ingold KU, Cheeseman KH, Slater TF. 1990. Application of deuterated α-tocopherols to the biokinetics and bioavailability of vitamin E. Free Radic. Res. Commun. 11:99–107 [DOI] [PubMed] [Google Scholar]
- 15.Burton GW, Joyce A, Ingold KU. 1982. First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 2:327. [DOI] [PubMed] [Google Scholar]
- 16.Burton GW, Traber MG. 1990. Vitamin E: antioxidant activity, biokinetics, and bioavailability. Annu. Rev. Nutr. 10:357–82 [DOI] [PubMed] [Google Scholar]
- 17.Catignani GL. 1975. An α-tocopherol binding protein in rat liver cytoplasm. Biochem. Biophys. Res. Commun. 67:66–72 [DOI] [PubMed] [Google Scholar]
- 18.Catignani GL, Bieri JG. 1977. Rat liver α-tocopherol binding protein. Biochim. Biophys. Acta 497:349–57 [DOI] [PubMed] [Google Scholar]
- 19.Cavalier L, Ouahchi K, Kayden HJ, Di Donato S, Reutenauer L, et al. 1998. Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am. J. Hum. Genet. 62:301–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheeseman KH, Burton GW, Ingold KU, Slater TF. 1984. Lipid peroxidation and lipid antioxidants in normal and tumor cells. Toxicol. Pathol. 12:235–39 [DOI] [PubMed] [Google Scholar]
- 21.Clément M, Dinh L, Bourre JM. 1995. Uptake of dietary RRR-alpha- and RRR-gamma-tocopherol by nervous tissues, liver and muscle in vitamin-E-deficient rats. Biochim. Biophys. Acta 1256:175–80 [DOI] [PubMed] [Google Scholar]
- 22.Copp RP, Wisniewski T, Hentati F, Larnaout A, Ben Hamida M, Kayden HJ. 1999. Localization of α-tocopherol transfer protein in the brains of patients with ataxia with vitamin E deficiency and other oxidative stress related neurodegenerative disorders. Brain Res. 822:80–87 [DOI] [PubMed] [Google Scholar]
- 23.Cuddihy SL, Ali SS, Musiek ES, Lucero J, Kopp SJ, et al. 2008. Prolonged α-tocopherol deficiency decreases oxidative stress and unmasks α-tocopherol-dependent regulation of mitochondrial function in the brain. J. Biol. Chem. 283:6915–24 [DOI] [PubMed] [Google Scholar]
- 24.Di Donato I, Bianchi S, Federico A. 2010. Ataxia with vitamin E deficiency: update of molecular diagnosis. Neurol. Sci. 31:511–15 [DOI] [PubMed] [Google Scholar]
- 25.Etminan M, Gill SS, Samii A. 2005. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol. 4:362–65 [DOI] [PubMed] [Google Scholar]
- 26.Evans HM, Bishop KS. 1922. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 56:650–51 [DOI] [PubMed] [Google Scholar]
- 27.Ford ES, Sowell A. 1999. Serum α-tocopherol status in the United States population: findings from the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 150:290–300 [DOI] [PubMed] [Google Scholar]
- 28.Fukuzawa K, Ikebata W, Sohmi K. 1993. Location, antioxidant and recycling dynamics of α-tocopherol in liposome membranes. J. Nutr. Sci. Vitaminol. (Tokyo) 39(Suppl.):S9–22 [DOI] [PubMed] [Google Scholar]
- 29.Fulgoni VL 3rd, Keast DR, Bailey RL, Dwyer J. 2011. Foods, fortificants, and supplements: Where do Americans get their nutrients? J. Nutr. 141:1847–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.German DC, Quintero EM, Liang CL, Ng B, Punia S, et al. 2001. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J. Comp. Neurol. 433:415–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gohil K, Schock BC, Chakraborty AA, Terasawa Y, Raber J, et al. 2003. Gene expression profile of oxidant stress and neurodegeneration in transgenic mice deficient in α-tocopherol transfer protein. Free Radic. Biol. Med. 35:1343–54 [DOI] [PubMed] [Google Scholar]
- 32.Gohil K, Vasu VT, Cross CE. 2010. Dietary α-tocopherol and neuromuscular health: Search for optimal dose and molecular mechanisms continues! Mol. Nutr. Food Res. 54:693–709 [DOI] [PubMed] [Google Scholar]
- 33.Goss-Sampson MA, Kriss A, Muddle JR, Thomas PK, Muller DP. 1988. Lumbar and cortical somatosensory evoked potentials in rats with vitamin E deficiency. J. Neurol. Neurosurg. Psychiatry 51:432–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goss-Sampson MA, Kriss A, Muller DP. 1990. A longitudinal study of somatosensory, brainstem auditory and peripheral sensory-motor conduction during vitamin E deficiency in the rat. J. Neurol. Sci. 100:79–84 [DOI] [PubMed] [Google Scholar]
- 35.Goti D, Hammer A, Galla HJ, Malle E, Sattler W. 2000. Uptake of lipoprotein-associated α-tocopherol by primary porcine brain capillary endothelial cells. J. Neurochem. 74:1374–83 [DOI] [PubMed] [Google Scholar]
- 36.Gotoda T, Arita M, Arai H, Inoue K, Yokota T, et al. 1995. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the α-tocopherol-transfer protein. N. Engl. J. Med. 333:1313–18 [DOI] [PubMed] [Google Scholar]
- 37.Higashi Y, Murayama S, Pentchev PG, Suzuki K. 1993. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 85:175–84 [DOI] [PubMed] [Google Scholar]
- 38.Hosomi A, Goto K, Kondo H, Iwatsubo T, Yokota T, et al. 1998. Localization of α-tocopherol transfer protein in rat brain. Neurosci. Lett. 256:159–62 [DOI] [PubMed] [Google Scholar]
- 39.Hyland S, Muller D, Hayton S, Stoecklin E, Barella L. 2006. Cortical gene expression in the vitamin E-deficient rat: possible mechanisms for the electrophysiological abnormalities of visual and neural function. Ann. Nutr. Metab. 50:433–41 [DOI] [PubMed] [Google Scholar]
- 40.Ingold KU, Burton GW, Foster DO, Hughes L, Lindsay DA, Webb A. 1987. Biokinetics of and discrimination between dietary RRR- and SRR-α-tocopherols in the male rat. Lipids 22:163–72 [DOI] [PubMed] [Google Scholar]
- 41.Inst. Med. 2000. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington, DC: Natl. Acad. Sci. [PubMed] [Google Scholar]
- 42.Jimenez-Jimenez FJ, de Bustos F, Molina JA, Benito-Leon J, Tallon-Barranco A, et al. 1997. Cerebrospinal fluid levels of α-tocopherol (vitamin E) in Alzheimer’s disease. J. Neural Transm. 104:703–10 [DOI] [PubMed] [Google Scholar]
- 43.Jishage K, Arita M, Igarashi K, Iwata T, Watanabe M, et al. 2001. α-Tocopherol transfer protein is important for the normal development of placental labyrinthine trophoblasts in mice. J. Biol. Chem. 276:1669–72 [DOI] [PubMed] [Google Scholar]
- 44.Jishage K, Tachibe T, Ito T, Shibata N, Suzuki S, et al. 2005. Vitamin E is essential for mouse placentation but not for embryonic development itself. Biol. Reprod. 73:983–87 [DOI] [PubMed] [Google Scholar]
- 45.Kaempf-Rotzoll DE, Traber MG, Arai H. 2003. Vitamin E and transfer proteins. Curr. Opin. Lipidol. 14:249–54 [DOI] [PubMed] [Google Scholar]
- 46.Kayden HJ, Traber MG. 1993. Absorption, lipoprotein transport, and regulation of plasma concentrations of vitamin E in humans. J. Lipid Res. 34:343–58 [PubMed] [Google Scholar]
- 47.Koscik RL, Farrell PM, Kosorok MR, Zaremba KM, Laxova A, et al. 2004. Cognitive function of children with cystic fibrosis: deleterious effect of early malnutrition. Pediatrics 113:1549–58 [DOI] [PubMed] [Google Scholar]
- 48.Kuhlenkamp J, Ronk M, Yusin M, Stolz A, Kaplowitz N. 1993. Identification and purification of a human liver cytosolic tocopherol binding protein. Protein Expr. Purif. 4:382–89 [DOI] [PubMed] [Google Scholar]
- 49.Leonard SW, Terasawa Y, Farese RV Jr, Traber MG. 2002. Incorporation of deuterated RRR- or all-rac-α-tocopherol in plasma and tissues of α-tocopherol transfer protein–null mice. Am. J. Clin. Nutr. 75:555–60 [DOI] [PubMed] [Google Scholar]
- 50.Lim WS, Liscic R, Xiong C, Morris JC. 2005. High-dosage vitamin E supplementation and all-cause mortality. Ann. Intern. Med. 143:152; author reply 156–58 [DOI] [PubMed] [Google Scholar]
- 51.Lloyd-Evans E, Platt FM. 2010. Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic 11:419–28 [DOI] [PubMed] [Google Scholar]
- 52.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. 2006. OPM: orientations of proteins in membranes database. Bioinformatics 22:623–25 [DOI] [PubMed] [Google Scholar]
- 53.Magalhaes J, Ferreira R, Neuparth MJ, Oliveira PJ, Marques F, Ascensao A. 2007. Vitamin E prevents hypobaric hypoxia-induced mitochondrial dysfunction in skeletal muscle. Clin. Sci. (Lond.) 113:459–66 [DOI] [PubMed] [Google Scholar]
- 54.Mangialasche F, Kivipelto M, Mecocci P, Rizzuto D, Palmer K, et al. 2010. High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J. Alzheimer’s Dis. 20:1029–37 [DOI] [PubMed] [Google Scholar]
- 55.Manor D, Morley S. 2007. The α-tocopherol transfer protein. Vitam. Horm. 76:45–65 [DOI] [PubMed] [Google Scholar]
- 56.Mardones P, Strobel P, Miranda S, Leighton F, Quinones V, et al. 2002. α-Tocopherol metabolism is abnormal in scavenger receptor class B type I (SR-BI)-deficient mice. J. Nutr. 132:443–49 [DOI] [PubMed] [Google Scholar]
- 57.Mattill HA. 1947. Antioxidants. Annu. Rev. Biochem. 16:177–92 [DOI] [PubMed] [Google Scholar]
- 58.Morley S, Cecchini M, Zhang W, Virgulti A, Noy N, et al. 2008. Mechanisms of ligand transfer by the hepatic tocopherol transfer protein. J. Biol. Chem. 283:17797–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morley S, Panagabko C, Shineman D, Mani B, Stocker A, et al. 2004. Molecular determinants of heritable vitamin E deficiency. Biochemistry 43:4143–49 [DOI] [PubMed] [Google Scholar]
- 60.Muller DP. 2010. Vitamin E and neurological function. Mol. Nutr. Food Res. 54:710–18 [DOI] [PubMed] [Google Scholar]
- 61.Muller DP, Lloyd JK, Wolff OH. 1983. Vitamin E and neurological function. Lancet 1:225–28 [DOI] [PubMed] [Google Scholar]
- 62.Muller DP, Lloyd JK, Wolff OH. 1985. The role of vitamin E in the treatment of the neurological features of abetalipoproteinaemia and other disorders of fat absorption. J. Inherit. Metab. Dis. 8(Suppl. 1):88–92 [DOI] [PubMed] [Google Scholar]
- 63.Oram JF. 2002. ATP-binding cassette transporter A1 and cholesterol trafficking. Curr. Opin. Lipidol. 13:373–81 [DOI] [PubMed] [Google Scholar]
- 64.Oram JF, Vaughan AM, Stocker R. 2001. ATP-binding cassette transporter A1 mediates cellular secretion of α-tocopherol. J. Biol. Chem. 276:39898–902 [DOI] [PubMed] [Google Scholar]
- 65.Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, et al. 1995. Ataxia with isolated vitamin E deficiency is caused by mutations in the α-tocopherol transfer protein. Nat. Genet. 9:141–45 [DOI] [PubMed] [Google Scholar]
- 66.Packer JE, Slater TF, Willson RL. 1979. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 278:737–38 [DOI] [PubMed] [Google Scholar]
- 67.Pfrieger FW. 2003. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol. Life Sci. 60:1158–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pillai SR, Traber MG, Kayden HJ, Cox NR, Toivio-Kinnucan M, et al. 1994. Concomitant brainstem axonal dystrophy and necrotizing myopathy in vitamin E-deficient rats. J. Neurol. Sci. 123:64–73 [DOI] [PubMed] [Google Scholar]
- 69.Podda M, Weber C, Traber MG, Packer L. 1996. Simultaneous determination of tissue tocopherols, tocotrienols, ubiquinols, and ubiquinones. J. Lipid Res. 37:893–901 [PubMed] [Google Scholar]
- 70.Qian J, Atkinson J, Manor D. 2006. Biochemical consequences of heritable mutations in the α-tocopherol transfer protein. Biochemistry 45:8236–42 [DOI] [PubMed] [Google Scholar]
- 71.Qian J, Morley S, Wilson K, Nava P, Atkinson J, Manor D. 2005. Intracellular trafficking of vitamin E in hepatocytes: role of tocopherol transfer protein. J. Lipid Res. 46:2072–82 [DOI] [PubMed] [Google Scholar]
- 72.Sato Y, Arai H, Miyata A, Tokita S, Yamamoto K, et al. 1993. Primary structure of α-tocopherol transfer protein from rat liver. Homology with cellular retinaldehyde-binding protein. J. Biol. Chem. 268:17705–10 [PubMed] [Google Scholar]
- 73.Sato Y, Hagiwara K, Arai H, Inoue K. 1991. Purification and characterization of the α-tocopherol transfer protein from rat liver. FEBS Lett. 288:41–45 [DOI] [PubMed] [Google Scholar]
- 74.Shi H, Noguchi N, Niki E. 1999.Comparative study on dynamics of antioxidative action of α-tocopheryl hydroquinone, ubiquinol, and α-tocopherol against lipid peroxidation. Free Radic. Biol. Med. 27:334–46 [DOI] [PubMed] [Google Scholar]
- 75.Shichiri M, Takanezawa Y, Rotzoll DE, Yoshida Y, Kokubu T, et al. 2010. ATP-binding cassette transporter A1 is involved in hepatic α-tocopherol secretion. J. Nutr. Biochem. 21:451–56 [DOI] [PubMed] [Google Scholar]
- 76.Shrader WD, Amagata A, Barnes A, Hinman A, Jankowski O, et al. 2012. Towards a modern definition of vitamin E—evidence for a quinone hypothesis. Bioorg. Med. Chem. Lett. 22:391–95 [DOI] [PubMed] [Google Scholar]
- 77.Sitrin MD, Lieberman F, Jensen WE, Noronha A, Milburn C, Addington W. 1987. Vitamin E deficiency and neurologic disease in adults with cystic fibrosis. Ann. Intern. Med. 107:51–54 [DOI] [PubMed] [Google Scholar]
- 78.Sokol RJ. 1988. Vitamin E deficiency and neurologic disease. Annu. Rev. Nutr. 8:351–73 [DOI] [PubMed] [Google Scholar]
- 79.Sokol RJ. 1990. Vitamin E and neurologic deficits. Adv. Pediatr. 37:119–48 [PubMed] [Google Scholar]
- 80.Sokol RJ, Kayden HJ, Bettis DB, Traber MG, Neville H, et al. 1988. Isolated vitamin E deficiency in the absence of fat malabsorption—familial and sporadic cases: characterization and investigation of causes. J. Lab. Clin. Med. 111:548–59 [PubMed] [Google Scholar]
- 81.Sontag TJ, Parker RS. 2002. Cytochrome P450 omega-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J. Biol. Chem. 277:25290–96 [DOI] [PubMed] [Google Scholar]
- 82.Southam E, Thomas PK, King RH, Goss-Sampson MA, Muller DP. 1991. Experimental vitamin E deficiency in rats. Morphological and functional evidence of abnormal axonal transport secondary to free radical damage. Brain 114(Part 2):915–36 [DOI] [PubMed] [Google Scholar]
- 83.Stampfer MJ, Hennekens CH, Manson JE, Colditz GA, Rosner B, Willett WC. 1993. Vitamin E consumption and the risk of coronary disease in women. N. Engl. J. Med. 328:1444–49 [DOI] [PubMed] [Google Scholar]
- 84.Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. 1996. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 347:781–86 [DOI] [PubMed] [Google Scholar]
- 85.Terasawa Y, Ladha Z, Leonard SW, Morrow JD, Newland D, et al. 2000. Increased atherosclerosis in hyperlipidemic mice deficient in α-tocopherol transfer protein and vitamin E. Proc. Natl. Acad. Sci. USA 97:13830–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomas PK, Cooper JM, King RH, Workman JM, Schapira AH, et al. 1993. Myopathy in vitamin E deficient rats: muscle fibre necrosis associated with disturbances of mitochondrial function. J. Anat. 183(Part 3):451–61 [PMC free article] [PubMed] [Google Scholar]
- 87.Traber MG, Arai H. 1999. Molecular mechanisms of vitamin E transport. Annu. Rev. Nutr. 19:343–55 [DOI] [PubMed] [Google Scholar]
- 88.Traber MG, Atkinson J. 2007. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 43:4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Traber MG, Burton GW, Hughes L, Ingold KU, Hidaka H, et al. 1992. Discrimination between forms of vitamin E by humans with and without genetic abnormalities of lipoprotein metabolism. J. Lipid Res. 33:1171–82 [PubMed] [Google Scholar]
- 90.Traber MG, Sokol RJ, Kohlschutter A, Yokota T, Muller DP, et al. 1993. Impaired discrimination between stereoisomers of α-tocopherol in patients with familial isolated vitamin E deficiency. J. Lipid Res. 34:201–10 [PubMed] [Google Scholar]
- 91.Ulatowski L, Dreussi C, Noy N, Barnholtz-Sloan J, Klein E, Manor D. 2012. Expression of the alpha-tocopherol transfer protein gene is regulated by oxidative stress and common single-nucleotide polymorphisms. Free Radic. Biol. Med. 53:2318–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ulatowski L, Parker R, Davidson C, Yanjanin N, Kelley TJ, et al. 2011. Altered vitamin E status in Niemann-Pick type C disease. J. Lipid Res. 52:1400–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vance JE, Hayashi H. 2010. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim. Biophys. Acta 1801:806–18 [DOI] [PubMed] [Google Scholar]
- 94.Vatassery GT, Angerhofer CK, Knox CA. 1984. Effect of age on vitamin E concentrations in various regions of the brain and a few selected peripheral tissues of the rat, and on the uptake of radioactive vitamin E by various regions of rat brain. J. Neurochem. 43:409–12 [DOI] [PubMed] [Google Scholar]
- 95.Vatassery GT, Lam C, Smith WE, Quach HT. 2006. Apolipoprotein E exerts selective and differential control over vitamin E concentrations in different areas of mammalian brain. J. Neurosci. Res. 84:1335–42 [DOI] [PubMed] [Google Scholar]
- 96.Vatassery GT, Quach HT, Smith WE, Santacruz KS, Roy S. 2007. Apolipoprotein E deficiency leads to altered brain uptake of α-tocopherol injected into lateral cerebral ventricles. Biochim. Biophys. Acta 1772:797–803 [DOI] [PubMed] [Google Scholar]
- 97.Verdon CP, Blumberg JB. 1988. Influence of dietary vitamin E on the intermembrane transfer of α-tocopherol as mediated by α-tocopherol binding protein. Proc. Soc. Exp. Biol. Med. 189:52–60 [DOI] [PubMed] [Google Scholar]
- 98.Wright ME, Peters U, Gunter MJ, Moore SC, Lawson KA, et al. 2009. Association of variants in two vitamin E transport genes with circulating vitamin E concentrations and prostate cancer risk. Cancer Res. 69:1429–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yau TM, Weisel RD, Mickle DA, Burton GW, Ingold KU, et al. 1994. Vitamin E for coronary bypass operations. A prospective, double-blind, randomized trial. J. Thorac. Cardiovasc. Surg. 108:302–10 [PubMed] [Google Scholar]
- 100.Yevenes LF, Klein A, Castro JF, Marin T, Leal N, et al. 2012. Lysosomal vitamin E accumulation in Niemann-Pick type C disease. Biochim. Biophys. Acta 1822:150–60 [DOI] [PubMed] [Google Scholar]
- 101.Yokota T, Igarashi K, Uchihara T, Jishage K, Tomita H, et al. 2001. Delayed-onset ataxia in mice lacking α-tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA 98:15185–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yokota T, Uchihara T, Kumagai J, Shiojiri T, Pang JJ, et al. 2000. Postmortem study of ataxia with retinitis pigmentosa by mutation of the α-tocopherol transfer protein gene. J. Neurol. Neurosurg. Psychiatry 68:521–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yoshida Y, Itoh N, Hayakawa M, Habuchi Y, Saito Y, et al. 2010. The role of α-tocopherol in motor hypofunction with aging in α-tocopherol transfer protein knockout mice as assessed by oxidative stress biomarkers. J. Nutr. Biochem. 21:66–76 [DOI] [PubMed] [Google Scholar]
- 104.Yu C, Youmans KL, LaDu MJ. 2010. Proposed mechanism for lipoprotein remodelling in the brain. Biochim. Biophys. Acta 1801:819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zampieri S, Mellon SH, Butters TD, Nevyjel M, Covey DF, et al. 2009. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J. Cell Mol. Med. 13:3786–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zanon-Moreno V, Asensio-Marquez EM, Ciancotti-Oliver L, Garcia-Medina JJ, Sanz P, et al. 2013. Effects of polymorphisms in vitamin E-, vitamin C-, and glutathione peroxidase-related genes on serum biomarkers and associations with glaucoma. Mol. Vis. 19:231–42 [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang WX, Thakur V, Lomize A, Pogozheva I, Panagabko C, et al. 2011. The contribution of surface residues to membrane binding and ligand transfer by the α-tocopherol transfer protein (α-TTP). J. Mol. Biol. 405:972–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zingg JM, Azzi A. 2004. Non-antioxidant activities of vitamin E. Curr. Med. Chem. 11:1113–33 [DOI] [PubMed] [Google Scholar]