

Abstract
The cilium evolved to provide the ancestral eukaryote with the ability to move and sense its environment. Acquiring these functions required the compartmentalization of a dynein‐based motility apparatus and signaling proteins within a discrete subcellular organelle contiguous with the cytosol. Here, we explore the potential molecular mechanisms for how the proximal‐most region of the cilium, termed transition zone (TZ), acts as a diffusion barrier for both membrane and soluble proteins and helps to ensure ciliary autonomy and homeostasis. These include a unique complement and spatial organization of proteins that span from the microtubule‐based axoneme to the ciliary membrane; a protein picket fence; a specialized lipid microdomain; differential membrane curvature and thickness; and lastly, a size‐selective molecular sieve. In addition, the TZ must be permissive for, and functionally integrates with, ciliary trafficking systems (including intraflagellar transport) that cross the barrier and make the ciliary compartment dynamic. The quest to understand the TZ continues and promises to not only illuminate essential aspects of human cell signaling, physiology, and development, but also to unravel how TZ dysfunction contributes to ciliopathies that affect multiple organ systems, including eyes, kidney, and brain.
Keywords: cilia, ciliary gate, ciliary trafficking, ciliopathies, transition zone
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membranes & Trafficking
The proximal‐most region of the cilium, termed transition zone, acts as a gate for membrane and soluble proteins. This review provides a comprehensive overview of the composition and assembly of the transition zone and discusses molecular mechanisms underlying its function as a selective diffusion barrier to maintain cilium homeostasis.

All organisms have the ability to sense and respond to environmental cues. Prokaryotes, protists, unicellular algae, and metazoans can all, for example, sense and move toward energy (food) sources. The signal transduction machinery that enables this process is normally polarized in the cell. In bacteria, receptor clustering creates a membrane domain that helps increase detection sensitivity and amplify downstream signaling (Watts et al, 2019). Such signaling cascades regulate cell motility via the flagellar apparatus, which is often situated on the opposite pole of the cell. Signal transduction molecules, including cyclic di‐GMP, modulate the dynamics of the rotary molecular motor and promote chemotaxis toward attractants or movement away from noxious substances (Jenal et al, 2017). Archaea employ similar mechanisms to regulate their flagella and, for example, promote chemotaxis or phototaxis (Quax et al, 2018).
During the emergence of eukaryotes some 2 billion years ago, the prokaryotic flagellar apparatus was lost and replaced by a substantially more complex organelle termed cilium (Mitchell, 2017; Khan & Scholey, 2018). Unlike prokaryotic flagella, motility apparatus and signal transduction machinery could be incorporated directly into the ciliary compartment. This remarkable evolutionary innovation required the adaptation of various pre‐existing cellular components, including tubulins (microtubules), kinesin and dynein molecular motors, membrane‐associated trafficking machinery, and at least initially, a sensory signaling system comprised of receptors and the cyclic nucleotides cAMP and cGMP (Johnson & Leroux, 2010; Carvalho‐Santos et al, 2011; Wickstead & Gull, 2011; Sung & Leroux, 2013). While eukaryotic motile cilia are often named flagella, we use the former terminology to avoid any possible confusion with the prokaryotic namesakes.
At their simplest, cilia are membrane‐encased cellular protrusions with microtubule “train tracks” that are served by a bidirectional transport system (termed intraflagellar transport or IFT) modified for ciliary, rather than vesicular, trafficking (Sung & Leroux, 2013; Fig 1A). In addition to supporting IFT retrograde (tip to base) transport, dynein machinery was co‐opted to power the whip‐like movement of the organelle (Lee & Ostrowski, 2020; Wallmeier et al, 2020). The need to compartmentalize not just the ciliary motility framework but also the signaling machinery was essential and required the formation of a multifunctional gate, or diffusion barrier. As we will discuss in detail, this functionality appears to be concentrated within the first subdomain of the ciliary axoneme, namely the transition zone (Fisch & Dupuis‐Williams, 2011; Reiter et al, 2012; Barker et al, 2014; Khanna, 2015; Takao & Verhey, 2016; Garcia‐Gonzalo & Reiter, 2017; Gonçalves & Pelletier, 2017; Jensen & Leroux, 2017; Mukhopadhyay et al, 2017; Garcia et al, 2018; Nachury, 2018).
Figure 1. Overview of evolutionarily conserved components and compartments of cilia, including the transition zone “ciliary gate”.

(A) All cilia possess a microtubule‐based axoneme that stems from a basal body foundation anchored to the plasma membrane via transition fibers. To build the cilium and make it dynamic, an intraflagellar transport (IFT) machinery powered by molecular motors (IFT‐Kinesin and IFT‐Dynein) docks at transition fibers and uses three functional modules (IFT‐A, IFT‐B and BBS protein complex or BBSome) to traffic ciliary proteins (cargoes) into and out of the organelle. Immediately distal to the basal body is a transition zone (TZ) region, widely believed to form a ciliary gate or diffusion barrier for membrane and soluble proteins. The TZ harbors typically Y‐shaped structures that span from the axoneme to the overlying membrane. In metazoans, the proximal segment includes a so‐called Inversin compartment that functionally interacts with the TZ. Most cilia also possess a distal segment with singlet microtubules. (i) TZ‐associated Y‐links are shown with arrowheads in cross‐section electron microscope images of motile cilia TZs from Chlamydomonas (modified from Craige et al, 2010) and rabbit oviduct (Anderson, 1974), as well as nonmotile cilia TZs from C. elegans (Jensen et al, 2018) and rat photoreceptor (Besharse et al, 1985; “Copyright 1985 Society for Neuroscience”). (ii) The ends of the TZ Y‐links, which connect to the membrane, likely form ciliary necklace “beads.” Shown is an electron micrograph of freeze‐fractured hamster respiratory airway cilia showing the ciliary necklace region, which contains 7–8 strands of membrane particles (arrowheads) and may form a spiral (arrows show start/end; modified from Heller & Gordon, 1986). Scale bars, 100 nm. (B) Rhodopsin (arrowheads) is highly concentrated in the distal ciliary region of the cilium (outer segment), and virtually absent from the connecting cilium (TZ) and near the basal body. Modified from ref. (Liu et al, 1999; “Copyright 1999 Society for Neuroscience”). Scale bar, 0.5 μm.
Motile cilia with sensory capabilities persist to this day across most eukaryotes, with the exception that the organelle was secondarily lost in some lineages, for example, flowering plants and most fungi (Avidor‐Reiss et al, 2004; Li et al, 2004). The emergence of metazoans gave rise to a second major ciliary category, the so‐called “primary” cilia. These nonmotile organelles evolved to help support the diverse functional requirements of different cell types, making them specialized cellular antennae (Satir & Christensen, 2007; Goetz & Anderson, 2010; Guemez‐Gamboa et al, 2014; Malicki & Johnson, 2017; Wang & Dynlacht, 2018; Nachury & Mick, 2019; Sreekumar & Norris, 2019).
In humans, defects in motile cilia have long been associated with a genetic disorder (ciliopathy) known as primary ciliary dyskinesia (PCD), which is characterized by chronic respiratory problems, laterality defects, and infertility (Reiter & Leroux, 2017; Lee & Ostrowski, 2020; Wallmeier et al, 2020). More recently, primary cilia have attracted considerable attention, owing to their diverse roles in human physiology and development (Yoder, 2008; Valente et al, 2014; Falk et al, 2015; Braun & Hildebrandt, 2017; Mitchison & Valente, 2017; Anvarian et al, 2019; Engle et al, 2020; Gabriel et al, 2020). The clinical ailments arising from primary cilia anomalies include retinal degeneration, cystic kidneys, obesity, as well as skeletal and brain malformations (Reiter & Leroux, 2017). The multiorgan involvement of ciliopathies stems from the essential roles that primary cilia perform in signal transduction pathways essential for the functions and/or formation of most organs (Wheway et al, 2018; Anvarian et al, 2019; Kopinke et al, 2020). In all, over 400 genes are known to participate in distinct ciliary processes, including IFT‐dependent cilium formation (ciliogenesis) and maintenance, ciliary gating, motility, and/or cell signaling (Reiter & Leroux, 2017). Nearly 200 of these genes are linked to several types of ciliopathies, which often correlate with the specific ciliary functions being disrupted.
The focus of this review is to discuss the evolutionarily conserved components and organization of the transition zone and provide insights into how this ciliary domain functions molecularly as a membrane diffusion barrier to compartmentalize, in a manner made dynamic by trafficking machinery, both motile and nonmotile cilia.
Cilia are distinct cell compartments with specialized proteomes and membranes
Evolutionarily conserved ciliary structures
The notion that cilia represent discrete, specialized organelles has been held for some time. Early electron microscopy images of cilia from different organisms, including protists and metazoans, revealed that their basic structures are evolutionarily conserved (De Robertis, 1956; Tokuyasu & Yamada, 1959; AFZELIUS, 1961; Sorokin, 1962; Renaud & Swift, 1964; Reese, 1965; Dingle & Fulton, 1966; Ringo, 1967; Fig 1). All stem from the same foundation, a basal body (formed from a centriole) normally consisting of a ninefold arrangement of triplet microtubules (Fisch & Dupuis‐Williams, 2011). The basal body attaches to the plasma membrane via distal appendages that are often called transition fibers (Fisch & Dupuis‐Williams, 2011; Reiter et al, 2012). The first segment of the axoneme, termed transition zone (abbreviated TZ), contains typically Y‐shaped structures, termed Y‐links, that physically connect the doublet microtubules to the overlying ciliary membrane (Fisch & Dupuis‐Williams, 2011; Reiter et al, 2012; Barker et al, 2014; Fig 1A). The rest of the axoneme is often bipartite, harboring a proximal region with doublet microtubules, and a distal region comprised of singlet microtubules (Silverman & Leroux, 2009; Fisch & Dupuis‐Williams, 2011; Blacque & Sanders, 2014). This description fits typical nonmotile cilia, such as the ones that line the olfactory epithelium of vertebrates and are responsible for the sense of smell (Reese, 1965). Motile cilia almost invariably contain an additional central pair of microtubules, and accessory structures (including axonemal dynein motors) required for ciliary movement, as observed in human sperm and respiratory airway cilia (Lee & Ostrowski, 2020; Wallmeier et al, 2020).
Whereas most cilia are antenna‐ or whip‐shaped, some adopt comparatively more elaborate forms (Silverman & Leroux, 2009). In vertebrates, rod and cone photoreceptors represent the most obvious example, where the TZ (or “connecting cilium”) is followed by an enlarged outer segment containing numerous membranous stacks that harbor light‐sensing opsins and other signaling proteins (Roepman & Wolfrum, 2007; Bujakowska et al, 2017; Bachmann‐Gagescu & Neuhauss, 2019; Baehr et al, 2019; Fig 1B).
Aside from their conserved ultrastructures, cilia have several features relevant to the TZ that distinguish them as distinct cellular compartments. Below, we describe their autonomous nature, unique proteomes and lipid composition, requirement for characteristic gene expression patterns, and particular phylogenetic profiles.
Ciliary shedding
Cilia from different organisms, including protists, algae, and metazoans/vertebrates, have the natural ability to be severed from the cell body and can be induced to do so through physical or chemical treatments (Quarmby, 2004). Such ciliary (flagellar) shedding, alternatively termed deciliation, deflagellation, or ciliary autotomy, represents a form of autonomy for the organelle. Notably, the site of cleavage occurs just distal to the TZ, and a recent study in Paramecium implicates several TZ‐localized proteins in this process (Gogendeau et al, 2020). Once released, cilia quickly grow back in an IFT‐dependent manner (Cole et al, 1998; Dentler, 2005; Marshall et al, 2005).
A second site of axoneme cleavage, found at the proximal end of the TZ, is engaged before ciliated cells enter mitosis (Parker et al, 2010). To free the basal body from the axoneme, so that it can act as a microtubule‐organizing center during cell division, resorption of the ciliary axoneme takes place; in addition, the microtubules between the basal body and TZ are severed, leaving behind a seemingly intact TZ. This discovery allowed for the isolation and proteomic analysis of the TZ in Chlamydomonas (Diener et al, 2015).
Ciliary proteomes
The ability to excise cilia enabled Piperno et al (1977) to perform 2D gel electrophoresis on excised motile cilia (minus the TZ) from Chlamydomonas and revealed the presence of over 130 proteins (Piperno et al, 1977; Dutcher, 1995). A subsequent, more comprehensive analysis of isolated Chlamydomonas cilia using mass spectrometry uncovered at least 350 proteins (Pazour et al, 2005). Among the proteins uniquely present within motile cilia (from both unicellular organisms and multiciliated vertebrate cells) are those required for movement, including inner and outer dynein arms, as well as dynein regulatory complex and radial spoke components (Smith et al, 2005; Subota et al, 2014; Lee & Ostrowski, 2020; Sim et al, 2020). Mass spectrometry of vertebrate cilia, including photoreceptors, has also provided insights into the compositions of different primary cilia (Ostrowski et al, 2002; Liu et al, 2007; Mayer et al, 2009; Ishikawa et al, 2012; Narita et al, 2012). Some proteomic studies have focused on specific ciliary components, for example, the membrane, or compared ciliary proteomes across basal species in the metazoan lineage (Yano et al, 2013; Sigg et al, 2017). A large proportion (although not all) of the identified proteins are unique to cilia, and there is growing evidence, which we discuss later, that the TZ ciliary gate helps regulate ciliary protein composition and homeostasis.
Ciliary lipid composition
In addition to having unique proteomes, ciliary compartments are ensheathed with a membrane whose lipid composition is substantially different from the cell body (Rohatgi & Snell, 2010; Garcia et al, 2018; Conduit & Vanhaesebroeck, 2020; Nechipurenko, 2020). This is evidenced from lipidomic approaches from protists and metazoan species. For instance, a Trypanosome study suggests that its ciliary membrane is selectively enriched in raft‐forming lipids (including phosphatidylserine, ceramide, and sphingomyelin), which may be important for signaling (Serricchio et al, 2015). Porcine olfactory cilia have reduced levels of phosphatidylethanolamine and more of a particular sphingolipid (Lobasso et al, 2010). The use of genetically encoded lipid reporters has also been helpful in identifying differences between ciliary and nonciliary membranes, including the presence of different species of phosphoinositide lipids (reviewed in ref. Conduit & Vanhaesebroeck, 2020).
Aside from an apparent ciliary enrichment of phosphatidylinositol 4,5‐bisphosphate (PIP2), whether there are evolutionarily conserved and biologically significant differences between cell body and ciliary membranes remains largely unexplored. Nevertheless, to maintain such a privileged lipid domain, cilia likely possess a membrane diffusion barrier, as mixing lipids between the cell body and ciliary membrane would otherwise occur rapidly. There is, however, limited direct experimental evidence for such a hypothesis. Furthermore, another potential mechanism, not mutually exclusive, is that some specific lipid species can be generated in situ. We discuss these scenarios, and PIP2 enrichment, in more detail later in the context of the TZ functional mechanism.
Ciliary expression signatures
Consistent with cilia being distinct cellular compartments, their construction requires coordinated expression of numerous specific genes. This phenomenon is apparent, for example, following deflagellation (ciliary excision) in Chlamydomonas (Stolc et al, 2005). Naegleria, which is normally amoeboid but can generate a basal body and cilium de novo, rapidly upregulates the expression of several hundred genes to support ciliary growth (Fritz‐Laylin & Cande, 2010). Despite the abundance of ciliary genes known to be co‐regulated in these and other protists, the transcription factor(s) responsible for this activity remain to be identified. In metazoans, ciliary gene expression is better understood (Hoh et al, 2012; Jensen et al, 2016; van Dam et al, 2019). A seminal study, published by Swoboda et al (2000), unveiled DAF‐19, an RFX transcription factor, as a master regulator for ciliary gene expression in C. elegans. The promoter regions of ciliary genes, including those associated with IFT and the TZ, possess a 14–15 bp consensus sequence termed X box (Blacque et al, 2005; Laurencon et al, 2007; Phirke et al, 2011). Loss of DAF‐19 predictably results in a lack of TZ or ciliary axoneme formation. In mammals, eight RFX transcription factors exist, several of which several (RFX1, 2, 3, 4, 7) collectively participate in ciliary gene expression (Ashique et al, 2009; Chu et al, 2010; Thomas et al, 2010; Narita et al, 2012; Manojlovic et al, 2014). To enable the further specialization of cilia within specific cell types, such as multiciliated cells, additional transcription factors are deployed, including p73 and FoxJ1 (Jackson & Attardi, 2016).
Phylogenetic signature
Although the cilium is widely conserved across eukaryotes, which points to the Last Eukaryotic Common Ancestor (LECA) as its origin, some lineages have since dispensed with the organelle entirely (Avidor‐Reiss et al, 2004; Li et al, 2004; Mitchell, 2017). For example, fungal phyla—Zygomycota, Basidiomycota, and Ascomycota such as S. cerevisiae—have lost virtually all known genes associated with cilia. By contrast, an “ancestral” fungal phylum, Chytridiomycota, together with its relatives Neocallimastigomycota and Blastocladiomycota, retained motile cilia to propel their spores (Hibbett et al, 2007). The wholesale loss of genes specifying a ciliary apparatus is again consistent with the cilium being a discrete organelle. In some cases, more selective gene losses occurred. For example, virtually all metazoans encode proteins required for ciliary motility, except roundworms, whose somatic cells only possess primary cilia, and which have nonciliated, amoeboid sperm (Fraire‐Zamora & Cardullo, 2010). Notably, TZ‐associated genes are present in nearly all ciliated organisms, suggesting that compartmentalization is integral to the function of most cilia (Barker et al, 2014).
In brief, cilia represent, much like the mitochondria or nuclei, discrete cellular compartments with specialized functions. However, unlike the mitochondrion, which is bounded by a double membrane, the cilium is encased in a membrane that is contiguous with the plasma membrane, and its internal space, or cilioplasm, is “open” to the cytosol. In this sense, cilia are more similar to nuclei, whose membranes contain pores and a transport system that facilitates regulated entry and exit (Schmidt & Görlich, 2016; Takao & Verhey, 2016; Beck & Hurt, 2017). So, by what mechanism do cilia maintain and regulate their internal composition, or homeostasis?
A ciliary gate at the transition zone
The idea of a structural domain separating the cell body from the ciliary compartment became apparent in early transmission electron microscopy (TEM) studies of vertebrate photoreceptors. In 1953, Fritiof Sjöstrand from the Karolinska Institute observed a specialized structure between the inner segment (cell body) and ciliary outer segment, which was bounded by its own membrane and contained a bundle of “fibrils” (SJOSTRAND, 1953). Three years later, Eduardo De Robertis recognized from more defined transverse TEM sections its relationship to cilia and coined the word “connecting cilium” for this region (De Robertis, 1956). Connections between microtubule doublets and the ciliary membrane, previously observed in mouse oviduct motile cilia (Fawcett & Porter, 1954), were also seen in the connecting cilium (Tokuyasu & Yamada, 1959). Such structures, which are often although not always Y‐shaped, would ultimately be observed in the proximal‐most domains of ciliary axonemes from a multitude of unicellular organisms, including Chlamydomonas and Naegleria, and different metazoan cell types (Fisch & Dupuis‐Williams, 2011; Barker et al, 2014; Fig 1A). As such, the connecting cilium is recognized as being analogous to the transition zone of other cilia (Rohlich, 1975).
Early evidence for a membrane diffusion barrier at the base of cilia
An influential study by Gilula and Satir in 1972 revealed in beautiful detail the ninefold symmetrical Y‐shaped structures present in the TZ of a nonmotile cilium from a mollusk (Gilula & Satir, 1972). Moreover, as seen by freeze‐etching, the Y‐links appear to organize intramembranous particles that look like a ciliary necklace, made up of rings of seemingly unconnected beads. Most necklaces have several rows, with photoreceptors typically sporting the most at over 20 (Rohlich, 1975; Fisch & Dupuis‐Williams, 2011; Fig 1A and Aii). The purpose of these rows of particles, which were observed one year prior by Noel Flower, was not obvious (Flower, 1971). Flower suggested a resemblance to particles within cell junctions, while Gilula and Satir proposed a prescient hypothesis: control of localized membrane permeability. Subsequent analysis of Chlamydomonas reinhardtii cilia by Weiss et al (1977) uncovered not only similar concentric rings within the TZ, but also a more proximal, continuous feature termed “bracelet” (Weiss et al, 1977). Although not apparently found in other species, the authors proposed that the bracelet “might act as a lateral diffusion barrier, restricting flagellar membrane proteins to the flagellum and denying any mobile proteins access to the flagellar compartment”; in other words, it may be a ciliary gate.
The molecular function of the TZ still remained untested. But in the 1980s, researchers working on photoreceptors were becoming aware that the light‐sensing G protein‐coupled receptor (GPCR), rhodopsin, becomes massively enriched within the outer segment (cilium) following its synthesis in the inner segment (cell body; Fig 1B). A barrier between the two compartments was suspected (Nir & Papermaster, 1983), and a fortuitous observation substantiated this notion. In dissociated photoreceptor cells, opsins remain concentrated within the outer segment. However, in happenstance cases where the outer segment fuses with the inner segment via their membranes, the outer segment opsins somehow bypass the connecting cilium (TZ) and diffuse into the inner segment (Spencer et al, 1988).
These findings provided strong evidence that a mammalian TZ acts as a membrane diffusion barrier. Two years earlier, this conclusion was independently arrived at using the green alga Chlamydomonas eugametos as a model system. In their study, Musgrave et al (1986) demonstrated a striking segregation of different cell body and ciliary markers, including an overall difference in membrane glycoprotein composition (Musgrave et al, 1986). They specifically attributed this to a membrane diffusion barrier at the TZ.
At least until the start of the century (see below), relatively little attention was seemingly paid to the emerging evidence of an evolutionarily conserved gating function for the TZ. For instance, a highly influential review article on intraflagellar transport (IFT) by Rosenbaum and Witman published in 2002 highlighted a “flagellar pore complex” that functions “as a gateway for the admission of specific proteins to the cilium,” but never mentions the TZ (Rosenbaum & Witman, 2002). Instead, the pore complex is proposed to be formed by the basal body transition fibers (Fig 1A). Notably, these fibers likely play a role in ciliary gating, which we discuss in Box 1, but their overall contribution to this process is independent from, and likely less prominent than, the TZ.
Box 1. The transition fibers as a flagellar (ciliary) pore complex and transport hub at the base of cilia.
Transition fibers are ninefold‐symmetric appendages present at the distal end of the basal body. Their most obvious function is to stably anchor the basal body to the base of the ciliary membrane (Wei et al, 2015; Garcia‐Gonzalo & Reiter, 2017). They are also docking sites for intraflagellar transport (IFT) particles and thus represent a primary hub (or “gateway”) for ciliary trafficking (Nachury et al, 2010; Reiter et al, 2012; Sung & Leroux, 2013; Wei et al, 2013). In addition, the pinwheel array of transition fibers is thought to form a “flagellar/ciliary pore complex” that impedes the entry of trafficked vesicles into cilia (Rosenbaum & Witman, 2002; Reiter et al, 2012; Wei et al, 2015; Garcia‐Gonzalo & Reiter, 2017). Rather than being transported into the organelle, vesicles fuse within the periciliary membrane region next to the transition fibers (Sung & Leroux, 2013; Mukhopadhyay et al, 2017). How important this latter function of transition fibers is cannot be readily ascertained, since perturbing their formation abrogates basal body docking and formation of the cilium in the first place (Schmidt et al, 2012; Tanos et al, 2013; Lu et al, 2015). Irrespective, it seems likely that the transition zone could fulfill a similar, redundant role, as it too creates a ninefold multispoke physical barrier that could prevent vesicle entry into cilia.
Another possibility for the function of transition fibers is that they act as scaffolds for membrane and/or soluble protein diffusion barrier(s). Cryo‐electron microscopy reconstructions of Tetrahymena ciliary bases reveal an elaborate complex at the level of transition fibers that spans from the membrane to the basal body cylinder and contains nine small, circular openings (Ounjai et al, 2013). However, the nature of the proteins forming this pore complex is unknown, and it has not been observed at the base of other cilia, making its significance unclear. Furthermore, there is no direct evidence that transition fiber components influence the membrane diffusion properties at the base of cilia. Yet, as described in the main text, at least some nucleoporins may localize at the level of the basal body, suggesting the presence of a nuclear pore‐like mesh barrier for soluble proteins at this level.
Molecular evidence for gating proteins at the transition zone
As far as we are aware, the first putative role for a TZ‐localized protein in ciliary gating was published by the Tiansen Li group in 2000. Using a mouse model to study RPGR, a protein implicated in retinitis pigmentosa in humans (RP3), they made two key findings (Hong et al, 2000). First, RPGR localizes specifically to the photoreceptor TZ. Second, its disruption results in reduced rhodopsin levels in the outer segment, concomitant with an ectopic redistribution of rod and cone opsins to the photoreceptor inner segments.
The discovery of evolutionarily conserved components within the TZ, and their molecular roles in forming a ciliary gate began in earnest in 2010. Using Chlamydomonas as a model system, the Witman laboratory found that loss of CEP290, which localizes to a proximal region within the TZ, causes a partial detachment of the TZ membrane from the axoneme, consistent with an apparent loss of Y‐shaped linkers (Craige et al, 2010). This perturbation coincided with differences in the composition of cilia, including increased levels of IFT and BBS proteins and decreased levels of a ciliopathy‐associated membrane protein, PKD2, which is mutated in the autosomal dominant polycystic kidney.
Three publications in 2011 greatly expanded on the number of proteins that localize to the TZ and help maintain a normal ciliary protein composition. Williams et al (2011) revealed roles for C. elegans orthologs of MKS1, B9D1/MKSR1, B9D2/MKSR2, TMEM67/MKS3, RPGRIP1L/MKS5, CC2D2A/MKS6, NPHP1, and NPHP4 in this process (Williams et al, 2011). Individually or in different combinations, disruption of these TZ‐localized proteins causes abnormal ciliary entry of two plasma membrane‐associated proteins, namely RP2 and TRAM1. In a separate study using mammalian cells, Chih et al (2011) demonstrated that knockdown of murine B9D1 and TMEM231 caused a reduction in the levels of cilium‐localized somatostatin and serotonin receptors (SSTR3 and HTR6, respectively; Chih et al, 2011). Moreover, two membrane‐associated proteins, GFP‐CEACAM1 and GPI‐anchored GFP, entered cilia more readily upon disruption of B9D1, TMEM231, TMEM17, and CC2D2A. Lastly, Garcia‐Gonzalo et al (2011) found that loss of the mammalian TZ proteins TCTN1, TCTN2, and CC2D2A prevented the ciliary enrichment of four proteins, namely membrane‐associated Adenylate Cyclase 3 (ADCY3), the small GTPase ARL13b, PKD2, and smoothened (hedgehog signaling protein).
Since then, studies have revealed the presence of additional TZ‐localized proteins—including TMEM80, TMEM107, TMEM138, TMEM218, TMEM237, and TCTN3—and confirmed in different systems the roles of these and others in regulating the composition of proteins in cilia (Awata et al, 2014; Basiri et al, 2014; Jensen et al, 2015; Lambacher et al, 2015; Li et al, 2016a; Lin et al, 2018; Datta et al, 2019; Okazaki et al, 2020; Wiegering et al, 2021). For example, a proteomic analysis of photoreceptors from a mouse RPGR mutant found both increased and decreased abundance of a variety of both membrane‐associated and soluble proteins (Rao et al, 2015). The number of reported TZ‐associated proteins is at least 28 (Reiter & Leroux, 2017), and a growing number of proteins are reported to depend on an intact TZ for their correct ciliary localization.
Another property of the TZ also deserves attention. In C. elegans, ciliary membrane‐associated signaling proteins, such as GPCRs and ARL13, are largely excluded from the TZ (Cevik et al, 2013; Jensen et al, 2015; Li et al, 2016a). This observation parallels the relative absence of opsins from the connecting cilium in vertebrate photoreceptors (Nir & Papermaster, 1983; Hong et al, 2000). Removal of TZ ultrastructure (Y‐links) in the RPGRIP1L/MKS5 or CEP290 C. elegans mutant—or removing RPGR from mouse photoreceptors—allows ciliary proteins to “fill” this region at the base of cilia (Hong et al, 2000; Cevik et al, 2013; Jensen et al, 2015; Li et al, 2016a). This finding suggests that the TZ has a composition and physical properties that make it uniquely adapted for excluding at least some proteins and thus, carrying out its gating function. Indeed, a variety of genetic, cell biology, biochemistry, and other evidence, which we explore below, are beginning to piece together the physical and functional connections between a growing number of TZ proteins and their roles in ciliary gating.
Components and functional modules of the transition zone
To date, at least 15 proteins have been localized to the TZ in a variety of divergent organisms, including Chlamydomonas, Trypanosoma, C. elegans, Drosophila, and vertebrates. The majority, mentioned above and shown in Fig 2 and Table EV1, have been found to support a gating function for the TZ.
Figure 2. Genetic, physical, and functional interaction network of transition zone proteins.

Interactions within the ring include among the most evolutionarily conserved transition zone (TZ) proteins, and interactions with other selected TZ proteins are shown outside of the ring in gray dotted boxes. Genetic interactions between two genes encoding TZ proteins (circles) and/or TZ protein localization dependency (arrows point to dependent protein) are shown on gray connecting lines and colored according to the model organism (Chlamydomonas, purple; Drosophila, blue, C. elegans, green, and vertebrates, red). Direct physical interactions and co‐precipitation of proteins are represented by yellow dotted and solid connections, respectively (wine‐colored dotted lines depict both types of interactions). Tentative grouping of proteins into MKS, NPHP or core scaffolding modules are colored purple, blue and green, respectively; uncertain grouping is shown as gray. Proteins are shown alongside their ciliopathy associations (MKS, Meckel syndrome; JBTS, Joubert syndrome; BBS, Bardet–Biedl syndrome; NPHP, Nephronophthisis; OFD, Orofaciodigital syndrome; COACH, COACH syndrome; SLSN, Senior–Løken syndrome; LCA, Leber congenital amaurosis) as well as domain structures (C2 and related B9 domains; TM, transmembrane; CC, coiled coil; β‐prop, β‐propeller; SH3, SRC homology 3; CYS, cysteine‐rich; MSP, major sperm protein). See text for additional details. All interaction data are presented in Table EV1.
To understand the molecular activities of individual TZ proteins, efforts have been made to assign them to functional categories, or modules. Together with genetic and cell biology approaches, five complementary experimental methods, discussed below, have been informative. These are genetic interaction studies, co‐dependent localization experiments, proteomic (physical interaction) approaches, phylogenetic groupings, and uncovering links to ciliopathies.
Genetic interactions
In 2005, the Yoder and Barr laboratories discovered that mutations in the C. elegans protein orthologs linked to Nephronophthisis (NPHP1 and NPHP4), present at the ciliary base, resulted in sensory and behavioral phenotypes but not overt ciliogenesis defects (Jauregui & Barr, 2005; Winkelbauer et al, 2005). Similarly, disrupting the C. elegans orthologs of TZ‐localized proteins associated with Meckel syndrome (MKS1 and related proteins B9D1/MKSR1 and B9D2/MKSR2) was later shown not to cause evident cilia ultrastructure anomalies (Williams et al, 2008; Bialas et al, 2009). However, any combination of MKS/MKSR and NPHP gene mutations (e.g., nphp‐4/NPHP4 and mksr‐1/B9D1) resulted in significant cilium formation defects, including the TZ itself. This was first suggested by a dye‐filling assay which queries for the correct exposure of full‐length cilia to the external environment (Williams et al, 2008) and later confirmed by electron microscopy observations (Williams et al, 2011).
These genetic data suggested the existence of two distinct functional modules, designated MKS and NPHP. Further genetic interaction studies carried out with additional C. elegans TZ‐associated gene mutants (disrupting MKS‐2/TMEM216, MKS‐3/TMEM67, MKS‐6/CC2D2A, TMEM‐17/TMEM17, TMEM‐107/TMEM107, TMEM‐218/TMEM218, and TMEM‐237/TMEM237) reinforced the existence of two functional modules which are redundant for ciliogenesis, with two proteins (NPHP1/NPHP4) affiliating with the NPHP module, and the rest with the MKS module (Huang et al, 2011; Williams et al, 2011; Warburton‐Pitt et al, 2012; Jensen et al, 2015; Lambacher et al, 2015; Li et al, 2016a). One interesting case is CEP‐290/CEP290, which genetically fits the description of an MKS module protein (Schouteden et al, 2015; Li et al, 2016a), but as discussed below, appears to occupy a more central “scaffolding” role in assembling the entire MKS module. An extensive map of known genetic interactions between TZ‐associated genes is shown in Fig 2.
Thus far, comparatively less evidence is available for genetic redundancy between the NPHP and MKS modules in other model systems. One notable exception is that in both C. elegans and mice, disrupting TCTN1 together with an NPHP mutant (NPHP1 or NPHP4) results in more severe ciliogenesis and ciliopathy (exencephaly and polydactyly) phenotypes, respectively (Yee et al, 2015). This suggests that Tectonic proteins (TCTN1/2/3 in mammals) are associated with the MKS module (Fig 2).
The reason behind the MKS‐NPHP functional redundancy is unclear. Studies by Jensen et al (2015) suggest that at least in C. elegans, the MKS module may operate specifically at the TZ, while NPHP proteins act both at the TZ and within the basal body (transition fibers), which represent the two major membrane attachment points that ensure ciliary integrity. Hence, in C. elegans, ciliary integrity is maintained when either an NPHP protein or MKS protein is lost, but when both are disrupted, basal body‐TZ membrane connections are severely disrupted. More work will need to be done to assess potential genetic redundancy and its mechanism in other model systems, including mammals.
Co‐dependent localization
Experiments probing the co‐dependency of TZ protein localization have provided complementary insights into the existence of separate functional module(s) (Winkelbauer et al, 2005; Williams et al, 2008; Bialas et al, 2009; Chih et al, 2011; Garcia‐Gonzalo et al, 2011; Huang et al, 2011; Williams et al, 2011; Roberson et al, 2015; Yee et al, 2015; Pratt et al, 2016; Li et al, 2016a). In Fig 2, we show a compilation of such results, juxtaposed with the genetic interaction data. In sum, disruption of MKS module proteins can lead other MKS proteins to become delocalized, but not NPHP proteins. The converse is also true; loss of NPHP4 causes the mislocalization of NPHP1, but does not influence MKS protein enrichment at the TZ.
Loss of an NPHP or MKS module component may have no effect, or may perturb the localization of one or more, TZ protein(s). This led to the notion of “core” subunits of the TZ modules that are responsible for the localization of “peripheral” subunits (Li et al, 2016a). For example, loss of C. elegans NPHP‐4 delocalizes NPHP‐1, but loss of NPHP‐1 has no apparent effect on NPHP‐4 (Winkelbauer et al, 2005). The evidence for a hierarchical assembly of TZ proteins is compelling within a given model system, such as C. elegans, but may not always parallel the situation in others, for example, Chlamydomonas, Drosophila, or mammalian cells. At least for C. elegans MKS module proteins, MKS‐1/MKS1, MKSR‐1/B9D1, MKSR‐2/B9D2, MKS‐2/TMEM216, and TMEM‐231/TMEM231 largely behave like “core” components, whereas TMEM‐17, TMEM‐218, TMEM‐237 and the sole tectonic protein appear more “peripheral” (Huang et al, 2011; Williams et al, 2011; Jensen et al, 2015; Roberson et al, 2015; Li et al, 2016a).
In C. elegans, abrogation of CEP‐290 is specifically associated with the delocalization of MKS module components, but not NPHP module proteins (Schouteden et al, 2015; Li et al, 2016a). By contrast, disrupting the MKS module itself does not interfere with CEP290 from localizing to the TZ (Li et al, 2016a). The Drosophila ortholog of CEP290 is similarly required for the assembly of the MKS module (Basiri et al, 2014). This suggests that CEP290 is a “core” scaffold for the MKS module. Ostensibly, it likely fulfills such a central role by supporting the formation of TZ Y‐link structures (Craige et al, 2010; Li et al, 2016a). Interestingly, another evolutionarily conserved TZ protein, TMEM‐138/TMEM138, is not delocalized in either NPHP or MKS module mutants in C. elegans—but requires CEP‐290, as well as MKS‐5/RPGRIP1L for assembly at the TZ (Li et al, 2016a). As such, it cannot be readily classified as an NPHP or MKS module component, but appears specifically associated with CEP‐290 (Fig 2).
Orthologs of RPGRIP1L (MKS5), present alone, or together with its paralog RPGRIP1 in vertebrates, appear to be even more pivotal as central organizers of TZ proteins. Disruption of the single MKS‐5 protein in C. elegans results in no observable TZ localization of MKS and NPHP module proteins, or of CEP‐290 (Jensen et al, 2015; Li et al, 2016a). By contrast, loss of MKS or NPHP module proteins, or CEP‐290, does not perturb MKS‐5 TZ localization. Similarly, mutation of mouse RPGRIP1L or RPGRIP1, alone or in combination depending on the cell type, similarly displaces MKS, NPHP, and CEP290 proteins from the TZ (Patil et al, 2012; Shi et al, 2017; Wiegering et al, 2018, 2021). Consistent with these findings, mutations in the C. elegans and mammalian RPGRIP1/RPGRIP1L counterparts lead to an observable loss in Y‐link structures, suggesting the complete absence of a TZ (Patil et al, 2012; Jensen et al, 2015; Wiegering et al, 2018).
Based on the above, a potential hierarchical organization of the TZ is one where RPGRIP1L/RPGRIP1 acts as a central scaffold for the NPHP module, as well as for CEP290, which itself is a platform for the MKS module in C. elegans (Li et al, 2016a). However, as alluded to above, there are apparent differences in the hierarchical organization of TZ proteins when comparing across different model systems. This could be ascribed to differences in specific contacts and strength of interactions between TZ proteins, and the presence or absence of certain TZ proteins (including paralogs) within species. For example, TMEM80 is a paralog of TMEM17/TMEM216 that is part of the MKS module but only emerged recently, in tetrapods (Huang et al, 2011; Li et al, 2016a).
Physical interactions
The co‐dependent localization of proteins at the TZ suggests either direct or indirect physical connections between subunits of the MKS and NPHP modules, and the scaffolding proteins RPGRIP1L/MKS5 and CEP290. An influential study by the Jackson laboratory in 2011 provided the first evidence for networks of interacting TZ proteins, obtained by a series of pull‐down and mass spectrometry experiments (Sang et al, 2011). Later studies, also encompassing other approaches such as proximity ligation, expanded or refined this protein‐protein interaction network to include the majority of known TZ proteins, as well as TZ protein candidates, and potential connections to other ciliary proteins not specifically localized to the TZ (Chih et al, 2011; Remans et al, 2014; Gupta et al, 2015; Dean et al, 2016; see also review articles and meta‐analyses: Roepman & Wolfrum, 2007; Gonçalves & Pelletier, 2017; Arslanhan et al, 2020; Karunakaran et al, 2020).
Satisfyingly, the results from these interaction studies, shown amalgamated with the genetic and localization networks (Fig 2), are largely consistent with the existence of separate modules harboring MKS or NPHP proteins. For example, precipitation of the MKS module proteins MKSR1/B9D1 or MKSR2/B9D2 uncovered most of the other evolutionarily conserved MKS module proteins, but not NPHP proteins, RPGRIP1L, or CEP290 (Chih et al, 2011). Interestingly, mammalian interaction studies suggest the presence of a third member of the NPHP module, namely RPGRIP1L (NPHP8; Sang et al, 2011). Indeed, an association between MKS5/RPGRIP1L and NPHP4 is supported in C. elegans (Jensen et al, 2015), and the paralog RPGRIP1 also interacts with NPHP4 (Roepman et al, 2005). However, as mentioned above and further discussed below, the characteristics of RPGRIP1L deviate substantially from those of an NPHP module component and align better with its being part of a separate, core scaffolding module.
Phylogenetic distribution of the MKS and NPHP modules
The comparison of genome sequences from a wide range of ciliated and nonciliated organisms represents a powerful means of identifying cilium‐associated proteins (Avidor‐Reiss et al, 2004; Li et al, 2004, 2014). The different phylogenetic distribution and thus evolutionary histories of ciliary proteins could also point, in theory, to their roles in distinct functional modules.
TZ proteins are widely conserved across ciliated organisms, indicating that the ancestral eukaryote possessed, in addition to an elaborate IFT‐BBS trafficking system, a functional ciliary gate (Hodges et al, 2010; van Dam et al, 2013; Barker et al, 2014). However, the TZ is not universally conserved among ciliated organisms. It is absent from some protists, including Giardia intestinalis, Toxoplasma gondii, and Plasmodium falciparum, as well as mosses such as Physcomitrella patens (Barker et al, 2014). Why the TZ has been lost in some ciliated species is unclear. A potentially telling feature of protists devoid of a TZ is that cilium formation occurs within the cytosol, meaning that the basal body is not directly associated with a ciliary membrane and the cilioplasm is not compartmentalized; however, in Physcomitrella, ciliogenesis does take place within a membrane‐encased compartment, like in most eukaryotes (Avidor‐Reiss & Leroux, 2015). It is suggested that in Giardia, the IFT particles present at the so‐called flagellar pore complex, found where the cytosolic axoneme becomes encased in a membrane, could be “analogous” to the TZ (McInally et al, 2019). But the notion that IFT proteins provide TZ‐like gating functionality is speculative and unlikely (the periciliary membrane region, where transition fibers normally attach, does act as a transport hub for the IFT machinery; see Box 1).
Based on the distribution of TZ proteins across eukaryotes, Barker et al (2014) proposed that the MKS module likely evolved before the NPHP module. Yet, while MKS module proteins are more prevalent in ciliated eukaryotes, the NPHP module proteins (NPHP1 and NPHP4) are widely distributed in Opisthokonta (including ciliated fungi such as B. dendrobatidis) and NPHP4 is found in essentially all eukaryotic clades that possess cilia, including Plantae (e.g., Chlamydomonas), Chromalveolata (e.g., Tetrahymena), Rhizaria (B. natans), and potentially Excavata (T. brucei; Barker et al, 2014). This suggests that at least one “core” NPHP module protein (NPHP4) was present in LECA and that the NPHP module has been secondarily lost in certain lineages. Indeed, this loss can even occur even in metazoans: in the ecdysozoan lineage, nematodes encode NPHP1 and NPHP4, but insects such as Drosophila have neither. Furthermore, the core scaffolding protein RPGRIP1L (MKS5) is also absent in Drosophila and other arthropods, whereas another scaffolding protein, CEP290, is still present together with the MKS module.
Altogether, these phylogenetic observations are consistent with potentially separable functions for the MKS and NPHP modules, the close affiliation of CEP290 with MKS module proteins, and the physical connection observed between RPGRIP1L and NPHP proteins.
Disease connections
Further support for the notion of two TZ functional modules (NPHP and MKS) can be gleaned from ciliopathy associations (Reiter & Leroux, 2017; Fig 2). On the one hand, the NPHP module proteins NPHP1 and NPHP4 are linked to Nephronophthisis (NPHP), a kidney disorder, but not the more severe Meckel syndrome (MKS) which includes central nervous system and skeletal anomalies. They are also linked to Senior–Løken syndrome (SLSN), which is characterized by both NPHP and retinal dystrophy phenotypes. On the other hand, nearly all MKS module proteins, namely MKS1, B9D1/MKSR1, B9D2/MKSR2, TMEM216/MKS2, CC2D2A/MKS6, TMEM107/MKS13, TCTN1/JBTS13, TCTN2/MKS8, TCTN3/JBTS18, TMEM218/JBTS39, TMEM231/MKS11, and TMEM237/JBTS14, are associated with MKS and/or Joubert Syndrome (JBTS), but not NPHP or SLSN. By contrast, CEP290 (linked to MKS, NPHP, JBTS and SLSN) is known to be important for the correct TZ localization of MKS module proteins in several model systems (including C. elegans, Drosophila, and mammals), as well as NPHP module proteins in mammals. Similarly, mutations in RPGRIP1L (NPHP8/MKS5) cause both NPHP, MKS and JBTS, consistent with its role in scaffolding both the NPHP and MKS modules in C. elegans and mammals.
Aside from the scaffolding proteins CEP290 and RPGRIP1L, a small degree of overlap in ciliopathy phenotypes between NPHP and MKS modules does appear to exist; TMEM67/MKS3 is linked to MKS, and curiously, NPHP as well, whereas NPHP1 is associated with JBTS. At least one TZ‐localized protein not conserved outside of Choanoflagellates and metazoans, namely the CEP290‐interacting IQCB1 (NPHP5/SLSN5) protein, is linked to NPHP as well as MKS or SLSN, respectively (Sang et al, 2011; Barbelanne et al, 2015). It should also be noted that the above‐mentioned NPHP and MKS module proteins also have shared links to one or more additional disorders, including Leber congenital amaurosis (LCA), oral‐facial‐digital syndrome (OFD), and COACH syndrome (Fig 2). Some TZ proteins are not presently associated with either MKS or NPHP. For example, mutations in TCTN1 and AHI1 are linked to JBTS, TMEM138 is associated with JBTS and OFD, and finally, disruption of RPGRIP1 or LCA5 results in retinal phenotypes alone (cone‐rod dystrophy and LCA, or LCA; Reiter & Leroux, 2017).
Interestingly, a complex of several ciliary proteins localized just distally to the TZ is linked to either NPHP or MKS and has physical and functional connections to the TZ (Shiba et al, 2010; Sang et al, 2011; Warburton‐Pitt et al, 2012; Hoff et al, 2013). One component, INVS (Inversin; also known as NPHP2) defines a ciliary subdomain called the “inversin compartment” that is associated with cilium assembly/disassembly, renal function, establishing left‐right asymmetry as well as Wnt and Hedgehog signaling (Shiba et al, 2009; Lienkamp et al, 2012; Warburton‐Pitt et al, 2012; Zhang et al, 2019). Other proteins in the inversin compartment include NPHP3 (MKS7/SLSN3), NEK8 (NPHP9) and ANKS6 (NPHP16), and likely also ANKS3, whose disruption in zebrafish results in an NPHP‐like phenotype (Shiba et al, 2010; Hoff et al, 2013; Yakulov et al, 2015).
In brief, the links between TZ‐localized MKS and NPHP module proteins and their MKS and NPHP namesake ciliopathies are mostly consistent, suggesting a genotype‐phenotype relationship. However, several exceptions exist, and how the loss of functionality for any given TZ‐localized or inversin compartment‐localized protein specially results in NPHP, MKS, or any other recurring ciliopathies, including SLSN, JBTS, and OFD, represents one of the most challenging questions in ciliary biology.
Functional modules at the TZ with potentially missing pieces
Altogether, the multispecies experimental and human genetic data accumulated thus far argues for the existence of two modules, NPHP and MKS, that require two major scaffolding proteins (“assembly factors”), namely CEP290 and RPGRIP1L, for their correct assembly at the TZ (Fig 2). These collectively participate in creating a gate at the base of cilia and have functional connections with its bordering inversin compartment.
There are likely to be additional, undiscovered TZ components. It is unclear, for instance, what protein(s) make up the typically Y‐shaped axoneme‐to‐membrane connectors. CEP290 and RPGRIP1L represent excellent candidates. However, they are not unambiguously structural features of the TZ; for example, as described below, CEP290 localization does not necessarily span the entire region containing Y‐links. Additionally, some TZ proteins are specific to an organism, or cilium type. For example, at least two TZ proteins from C. elegans, DYF‐17 and TZA‐3 lack detectable human homologues (Phirke et al, 2011; Jensen et al, 2016). Trypanosomes have a full complement of evolutionarily conserved TZ proteins, but several TZ‐localized proteins seem unique and are not conserved across different species (Dean et al, 2016).
In the following sections, we focus on how individual TZ components and their organization within a ciliary subdomain containing Y‐links relate to their roles in maintaining a selective diffusion barrier.
Spatial organization of transition zone proteins
Unlike the protein‐rich doublet microtubule axoneme and accessory domains of the motile cilium (Ishikawa, 2013; Lacey et al, 2019; Ma et al, 2019; Khalifa et al, 2020; Poghosyan et al, 2020), atomic or high‐resolution details of the TZ are not available. A cryo‐electron tomography reconstruction of a photoreceptor connecting cilium hints at potential, albeit poorly resolved axoneme‐to‐membrane Y‐shaped connectors (Robichaux et al, 2019). A similar approach used on motile cilia from bovine trachea did not reveal connectors, potentially because the sample was detergent‐extracted (Greenan et al, 2020). It did, however, uncover connections between the microtubule doublets unique to the TZ. A separate deep‐etch study of guinea pig tracheal cilia shows an interesting association between Y‐links and circular ridge structures on the inner TZ membrane surface, which the authors suggested may anchor the V‐shaped ends and connect the necklace particles (Arima et al, 1984).
Placement of transition zone proteins by microscopy
By standard confocal microscopy, TZ proteins tagged with fluorescent reporters, or detected with antibodies, usually appear as roughly circular puncta or pill‐shaped structures immediately distal of the basal body (Winkelbauer et al, 2005; Craige et al, 2010; Garcia‐Gonzalo et al, 2011; Sang et al, 2011; Williams et al, 2011). The size of the TZ varies in different species, but is typically ~270 nm in diameter and less than ~750 nm in length (the photoreceptor connecting cilium is notably longer). The use of TEM, and the advent of super‐resolution microscopy in particular, has been extremely useful for mapping proteins both axially and longitudinally within these small TZ regions. It is difficult to unite all of the localization patterns observed in different TZ structures across divergent species, and even different cell types. For example, there are significant differences in TZ composition/structures within Drosophila olfactory, auditory and spermatocyte TZ regions (Jana et al, 2018). Nevertheless, we discuss findings below that suggest a tentative, simplified 3D‐like view of TZ protein organization (Fig 3).
Figure 3. Spatial organization of evolutionarily conserved transition zone proteins.

Tentative spatial organization of evolutionarily conserved transition zone (TZ) proteins based on their ciliary subcellular localization in mammalian cells and other species (see text for details). The longitudinal cutout view shows one slice of the ninefold symmetrical microtubule (MT) basal body triplets and TZ doublets (brown layer), together with Y‐link structures that connect the axoneme to the ciliary membrane (blue layer). Since the exact relationship between the TZ proteins and the Y‐link is unclear, the Y‐shaped structure is simply drawn as a projection. In this model, CEP290 and RPGRIP1L (shaped as cuboids; green layer) are interacting, core scaffolding proteins found close to the axoneme MT doublets, with CEP290 being more proximal (closer to the basal body) compared with other TZ proteins in some cilia. RPGRIP1L is required for the localization of all TZ proteins, including NPHP module (triangular prisms) and MKS module (cylinders) components. CEP290 is most closely affiliated with the MKS module. MKS and NPHP module proteins are situated either at or within the membrane (blue layer) or in an intermediate position between the membrane and axoneme (red layer). TF, transition fiber.
Super‐resolution microscopes of the SIM, STED, and STORM varieties show that all TZ proteins form rings just distal to the basal body‐associated transition fibers, whose components (e.g., CEP164) also form rings (Tanos et al, 2013; Lee et al, 2014; Lambacher et al, 2015; Yang et al, 2015; Mojarad et al, 2017; Shi et al, 2017; Jana et al, 2018; Yang et al, 2018; Gogendeau et al, 2020). Averaging ring diameters for different TZ proteins reveals an onion peel‐type of arrangement that can be divided roughly into three layers (Fig 3).
The smallest rings, indicative of close proximity to the axoneme (shown as brown), comprise the two known scaffolding proteins, CEP290 and RPGRIP1L (green). Both proteins contain expansive coiled coils, which potentially make direct contact with the axoneme (Drivas et al, 2013; Jensen et al, 2018; Gogendeau et al, 2020; Wu et al, 2020). CC2D2A, another protein with coiled coils (as well as a C2 domain), interacts with CEP290 (Gorden et al, 2008), but is positioned closer to the TZ membrane. Transmembrane proteins such as TMEM67, TMEM216, TMEM231 and members of the tectonic protein family (TCTN2 and TCTN3) form the largest rings (blue). In between the two layers are MKS1, B9D1, and B9D2 (red). Although these latter three proteins possess a B9 domain that is structurally related to the lipid/calcium‐binding C2 domains, the motif may enable protein‐protein interactions between them (and likely other proteins) rather than direct associations with the membrane (Remans et al, 2014; Okazaki et al, 2020).
Lastly, the “core” NPHP4 protein may be hierarchically positioned at the level of B9 domain‐containing proteins, potentially extending closer to the axoneme and membrane (Takao et al, 2017), while the “peripheral” NPHP1 protein appears proximal to the membrane (Fig 3). Both are predicted to harbor C2 domains (Zhang & Aravind, 2012).
Analysis and modeling of the highest‐resolution images suggest a more refined view of TZ positioning with respect to Y‐links. By STORM microscopy, TMEM231, NPHP1, and RPGRIP1L appear to form nine discontinuous, paired puncta (Shi et al, 2017). This arrangement suggests a specific association with the bifurcated ends of each Y‐link that associates with the membrane.
Other approaches for positioning the TZ proteins relative to the Y‐links will prove to be helpful. Given that visualizing proteins typically requires labeling with large primary and secondary antibodies, using the smaller self‐labeling Halotag protein and bright fluorophores, coupled to super‐resolution microscopy, would be an improvement. Expansion microscopy, combined with super‐resolution approaches, might be particularly well suited to study the 3D organization of proteins within the TZ (Sahabandu et al, 2019). Comprehensively tagging TZ proteins at their N and C terminus would help define their span and spatial orientation within the context of the electron‐dense Y‐link structures. This latter approach was applied to a subset of proteins analyzed (NPHP4, AHI1 and NPHP5) by Takao et al (2017), who used bimolecular fluorescence complementation (BIFC) to help define pairs of stably, or transiently, interacting proteins within the axial plane of the TZ (Takao et al, 2017). In Drosophila olfactory neurons, CEP290 tagged with GFP at the C or N terminus appears as small “contiguous” rings, or larger‐diameter rings consisting of nine distinct puncta, respectively (Jana et al, 2018). This suggests that the coiled coil‐containing CEP290 protein is elongated, and spans from very close to the microtubule axoneme (C terminus) to the membrane (or close to the membrane) at each Y‐link (N terminus).
Are there two functionally distinct regions in the TZ?
Longitudinally, an intriguing arrangement of TZ proteins is observed. In Chlamydomonas and mammalian cells, CEP290 occupies a proximal region of the TZ that is closest to the transition fibers (Craige et al, 2010; Yang et al, 2015). By contrast, the distal region is occupied by NPHP4 in Chlamydomonas, as well as RPGRIP1L and MKS module proteins in mammalian cells (Awata et al, 2014; Yang et al, 2015; Fig 3). In Drosophila, a bipartite TZ can also be observed in olfactory and auditory neurons (Jana et al, 2018). Several TZ proteins, including CEP290, localize to a proximal region, but only CEP290 can also be found (albeit less prominently) within a distal region.
In Chlamydomonas, there are some ultrastructural differences between the proximal and distal regions of the TZ (Craige et al, 2010). This is also observed in other motile cilia, for example, in Trypanosomes (Dean et al, 2016; Trépout et al, 2018). But in metazoan primary cilia, including the photoreceptor connecting cilium, or C. elegans, there is no clear structural demarcation between the proximal and distal regions readily observed by electron microscopy. Yet, in multiple C. elegans TZ mutants, both fluorescently tagged CEP‐290 and MKS‐5 (RPGRIP1L) change their appearance from one continuous signal to a slightly longer, bipartite signal (Jensen et al, 2015; Schouteden et al, 2015; Li et al, 2016a), hinting at two separable TZ sections. In mouse photoreceptors, disruption of the connecting cilium‐localized Spata7 protein selectively dislodges several conserved TZ proteins (AHI1, NPHP1, NPHP4, CEP290, RPGRIP1, and RPGR) from a distal—but not proximal—region of the connecting cilium (Dharmat et al, 2018). SPATA7, only found in vertebrates, appears to have a photoreceptor‐specific function and is associated with LCA and retinitis pigmentosa (Wang et al, 2009).
Why the TZ can be subdivided into proximal and distal regions is uncertain. One can only speculate at this point, but the answer could help uncover the mechanism of the TZ as a whole. For example, one region could organize the membrane diffusion barrier while the second may harbor a soluble gate functionality, both of which are covered in greater detail in later sections. Alternatively, the two regions could incorporate both barriers, but each may have different gating properties.
Formation and maintenance of the transition zone
As touched on earlier, there is evidence of a hierarchical assembly of TZ proteins, with “core” scaffolding (CEP290 and RPGRIP1/RPGRIP1L) proteins setting the stage for the incorporation of the MKS and NPHP modules. While the steps involved in the assembly of individual TZ proteins are not well understood, the process likely occurs early in ciliogenesis and requires IFT proteins for maintenance.
Assembly of the transition zone during early ciliogenesis
Initiation of ciliogenesis requires two principal events (Avidor‐Reiss & Leroux, 2015; Baehr et al, 2019; Chen et al, 2020). The first is the maturation of the centrosomal mother centriole into a basal body, which acts as the foundation for building the doublet microtubule axoneme. The second involves the physical association of the basal body with the incipient ciliary membrane. This membrane interaction requires the basal body transition fibers (Schmidt et al, 2012). Depending on the cell type, it may also involve the prior migration of the basal body to the plasma membrane, with or without prior association with a ciliary vesicle that is transported to, and fuses with, the plasma membrane (Garcia‐Gonzalo & Reiter, 2012; Reiter et al, 2012; Wang & Dynlacht, 2018). The trigger for TZ assembly is not well understood, but likely involves multiple proteins. At least in most metazoans/vertebrates, two basal body proteins (CBY1 and FSD1), localized at or near the distal end of the basal body, are required for the recruitment of AHI1 and the scaffolding protein RPGRIP1L to the TZ, respectively (Lee et al, 2014; Tu et al, 2018). CBY1 itself interacts with other basal body proteins, DZIP1 and its paralog DZIP1L, which help initiate ciliary bud and TZ formation (Wang et al, 2018a). A CBY1‐binding protein, FAM92, is further implicated in this early stage of ciliogenesis in both mammalian and Drosophila cells (Li et al, 2016b; Lapart et al, 2019).
It is notable that the TZ appears to be built concurrently with the extension of doublet microtubules from the basal body. Studies in both protists and vertebrate cells show the presence of Y‐link connections, and the appearance of ciliary necklace at the earliest moment of ciliogenesis (Menco, 1980; Hufnagel, 1983; Chailley & Boisvieux‐Ulrich, 1985; Reiter et al, 2012). In developing retinal photoreceptors, Y‐link structures can be observed in rod‐like cilia before the outer segment has developed disks (Besharse et al, 1985). In this immature connecting cilium, Y‐links appear fully formed in the proximal region, but are absent from more distal regions, suggesting that, perhaps not surprisingly, doublet microtubules are extended just prior to Y‐link assembly.
Why is the TZ assembled so early during ciliogenesis? We know that a ciliary axoneme can be made in the absence of TZ ultrastructure in nonmotile (C. elegans) and motile (Chlamydomonas) cilia (Craige et al, 2010; Jensen et al, 2015). In principle, the ciliary gate could be built independently, following the complete elongation of the axoneme. One possibility is that compartmentalization is necessary to rapidly form an emerging ciliary structure that is preloaded with signal transduction proteins, such as smoothened of the Hedgehog pathway in vertebrates (Lu et al, 2015). Such a signaling compartment may begin to function before the completion of, or may be required to regulate, ciliogenesis.
Establishing the correct lipid composition in the growing ciliary compartment is likely important as well. The proper formation and function of the TZ itself depend on at least one lipid species, PIP2, and proteins such as INPP5E that regulate PIP2 levels within the cilium (Dyson et al, 2017; Gupta et al, 2018).
Role of IFT in TZ assembly or maintenance and other IFT‐TZ connections
In most eukaryotes, IFT is required for building the microtubule‐based axoneme (Ishikawa & Marshall, 2011; Avidor‐Reiss & Leroux, 2015). However, studies in different model systems indicate that TZ formation is independent of IFT. For example, in C. elegans, abrogating the “core” IFT modules (IFT‐A and IFT‐B), or the IFT‐associated BBS adapter subunits involved in trafficking signaling proteins, does not impair the localization of TZ proteins and formation of Y‐links at the base of cilia (Perkins et al, 1986; Williams et al, 2011). In Chlamydomonas, the TZ stubs are visible in an IFT mutant where the rest of the axoneme is missing (Pazour et al, 2000).
Yet, four studies since 2018 have revealed a role for IFT in the complete assembly and/or maintenance and function of the TZ in C. elegans and mammalian cells (Jensen et al, 2018; Scheidel & Blacque, 2018; Vuolo et al, 2018; De‐Castro et al, 2022). Collectively, the findings reveal that retrograde IFT may be involved in recovering TZ proteins which diffuse inside the cilium, supporting the correct assembly—and function—of the TZ. This is important, as it suggests that some IFT‐associated ciliopathy phenotypes may be partially caused by a disruption of the ciliary gate. In this regard, it is notable that mutations in established TZ proteins (e.g., MKS1 and CEP290) can result in Bardet–Biedl syndrome, which is normally caused by defects in IFT‐associated cargo transport (Leitch et al, 2008). Genetic interactions between TZ and BBS genes are also reported in mice and C. elegans (Yee et al, 2015; Goetz et al, 2017). How, and if, the loss of TZ proteins ostensibly influences the BBS protein complex is unclear. IFT particles transit the TZ continuously, and one TZ protein (B9D2) interacts with the IFT protein TTC30B/IFT70B (Zhao & Malicki, 2011). Hence, it is interesting that all ciliated protists with a TZ have an IFT system; those which lack IFT proteins and undergo “cytosolic ciliogenesis” are devoid of a TZ (Avidor‐Reiss & Leroux, 2015). There are likely to be more functional connections between the IFT system and the TZ left to discover (see Park & Leroux (2022) and below).
Functional mechanism of the transition zone diffusion barrier
Cilium homeostasis depends on the TZ to maintain a distinct composition of ciliary proteins. For instance, disruption of the TZ in vertebrates leads to the mislocalization of ciliary proteins such as rhodopsin or Hedgehog signaling‐associated Smoothened, which collectively engenders many if not most of the cellular and ciliopathy phenotypes (Reiter & Leroux, 2017). But coincidently, loss of TZ function also leads to a redistribution of PIP2 and probably other lipid species. In several species, PIP2 concentration is lower inside the cilium than outside, and in C. elegans, loss of TZ function eliminates this partitioning of the phosphoinositide (Jensen et al, 2015). This latter finding suggests that the TZ might specifically act as a diffusion barrier for lipids, which would help maintain the unique lipid compositions of cilia, as previously discussed. Interestingly, the TZ is necessary for the ciliary enrichment of phosphatases (INPP5E, INPP5B and OCRL in vertebrates) that convert PIP2 to PI4P (Bielas et al, 2009; DiTirro et al, 2019). Hence, the ciliary gate likely also indirectly contributes to the partitioning of at least some lipid species by compartmentalizing enzymes that can modify lipid species in situ. In either case, the TZ influences ciliary lipid composition, and such a function is also important for maintaining ciliary homeostasis.
At the cellular level, the consequence of an altered PIP2‐PI4P lipid gradient may be a failure of the IFT system to release trafficked GPCR proteins into cilia (Mukhopadhyay et al, 2010; Luo et al, 2012; Sun et al, 2012; Luo et al, 2013; Brear et al, 2014; Garcia‐Gonzalo et al, 2015). Such a defect might explain why mutations in INPP5E cause JBTS (Bielas et al, 2009; Jacoby et al, 2009), a ciliopathy linked to several TZ‐associated proteins (Fig 2). More generally, these observations suggest fascinating connections between the ciliary abundance of a phosphoinositide and over 35 proteins associated with JBTS—all of which have ciliary roles, but not all of which are restricted to the TZ (Parisi, 2019).
Understanding how the TZ may function as a selective diffusion barrier for proteins and likely also for lipids remains largely unresolved (Williams et al, 2011; Reiter et al, 2012; Jensen et al, 2015; Trimble & Grinstein, 2015; Garcia‐Gonzalo & Reiter, 2017; Gonçalves & Pelletier, 2017; Nachury, 2018; Nechipurenko, 2020). Below, we explore four potential mechanisms. First, the TZ may harbor one or more “protein picket fence” barriers that physically block the movement of membrane‐associated proteins. Second, the TZ may promote the formation of a unique lipid microdomain that simultaneously acts as a lipid and membrane protein diffusion barrier. Third, the TZ membrane may have unique physical properties that can also hinder the free lateral diffusion of lipids and membrane‐associated proteins. Lastly, the TZ appears to incorporate a meshwork of proteins that behaves like a sieve and limits the diffusion of soluble (and perhaps also membrane) proteins. These potential functions, which are not mutually exclusive, are summarized in Fig 4.
Figure 4. Proposed mechanisms for transition zone function as a diffusion barrier for membrane and soluble proteins.
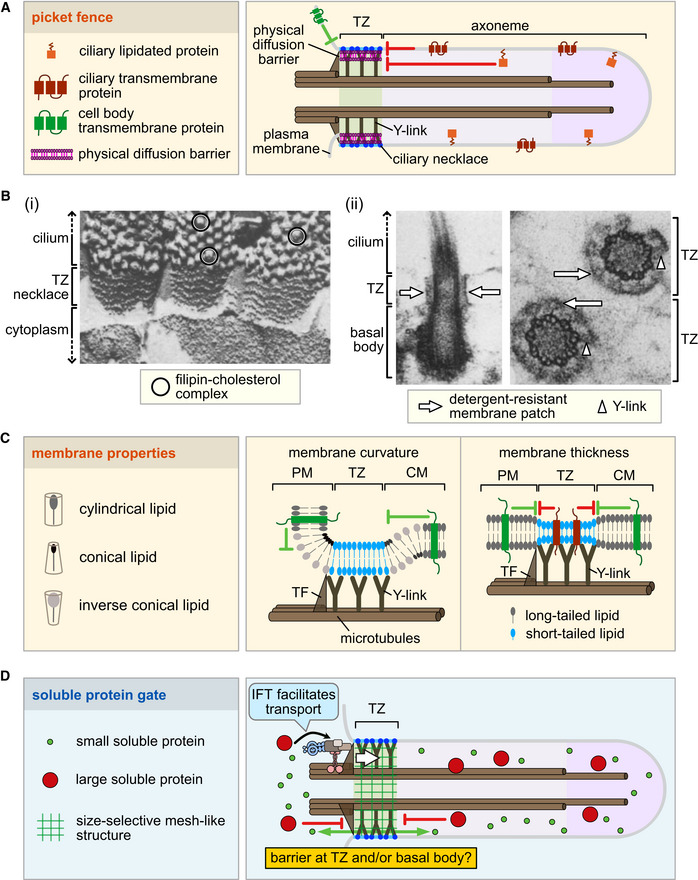
At least four potential, nonmutually exclusive properties of the transition zone (TZ) may explain its role as a ciliary gate. (A) A protein picket fence or “physical diffusion barrier,” consisting of septins and/or additional proteins, may be present within the TZ region, potentially in association with Y‐links and the ciliary necklace. This barrier may prevent membrane‐associated proteins and lipids from outside or inside of the cilium from freely diffusing across the barrier. (B) A condensed, lipid‐ordered microdomain at the TZ may restrict the diffusion of lipids and membrane‐associated proteins. (i) The TZ, which harbors TZ necklaces, is devoid of filipin‐sterol complexes (protrusions) relative to more distal regions of the cilium (where some are circled). These freeze‐fracture views of tracheal cells incubated with filipin suggest the lack of or inaccessibility of free cholesterol in the TZ. Modified from ref. (Montesano, 1979). (ii) The TZ has a detergent‐resistant membrane. Detergent extraction of photoreceptors removes plasma and ciliary membranes, but leaves continuous or interrupted membrane patches at the TZ (white arrows), visible in longitudinal (left) and transverse (right) electron micrographs. The positions of some Y‐links are shown with arrowheads. Modified from ref. (Anderson, 1974). (C) Distinct lipid bilayer properties of the TZ, such as curvature and thickness, may be conferred by different lipid species with different aliphatic and head groups (cylindrical, conical, inverse conical, long‐tailed, short‐tailed), the Y‐links, as well as specific lipid‐binding and membrane‐shaping proteins (red). These properties may influence the diffusion of lipids and membrane‐associated proteins across the TZ. PM, plasma membrane; CM, ciliary membrane; TF, transition fiber. (D) A mesh‐like protein gate, consisting of nucleoporins and potentially other proteins, restricts the movement of larger soluble proteins into and out of the ciliary compartment. Although this sieve‐type gate (green mesh) depends on the TZ, several studies suggest that nucleoporins are localized at the basal body, making its exact site of action unclear (see text for details). The transport of certain soluble proteins (including tubulins) across the gate may benefit from trafficking systems such as IFT (shown at the basal body before entering the cilium).
Protein picket fence as a physical barrier to diffusion
The most conspicuous aspect of the TZ is the presence of connecting structures, typically Y‐shaped, that span from the axoneme to the ciliary membrane (Fisch & Dupuis‐Williams, 2011; Reiter et al, 2012; Barker et al, 2014; Figs 1A and 4Bii). As a barrier, these structures seem imperfect: they are discontinuous, leaving large evenly spaced gaps at the TZ membrane and within the TZ internal space. Septins, which are cytoskeletal proteins that can form ring‐shaped, membrane‐associated physical barriers, represent excellent candidates for filling in such gaps at the membrane (Oh & Bi, 2011; Fung et al, 2014; Fig 4A). Septins form scaffolds that help, for example, partition the emerging bud from the mother yeast cell.
Various motile and nonmotile cilia are reported to contain septins (Palander et al, 2017). In IMCD3 kidney cells, SEPT2 forms a ring‐shaped structure just distal to the transition fibers, as expected for a TZ protein (Hu et al, 2010). Consistent with having a gating function, knockdown of SEPT2 leads to altered localization of ciliary proteins and an increase in the free‐diffusion rate of several ciliary membrane proteins from the cytoplasm into the cilium. In Xenopus, SEPT7 similarly forms a ring at the ciliary base, and loss of SEPT2 and SEPT7 impairs ciliogenesis (Kim et al, 2010). Another study further showed that loss of SEPT2 disrupts the assembly of some TZ proteins, providing evidence for a functional connection between septins and the TZ (Chih et al, 2011). The lone septin from Chlamydomonas was similarly found to concentrate at the base of cilia, potentially at the TZ (Kim et al, 2010).
Interestingly, some reports also position septins, not at the TZ, but more distally and widespread within the ciliary compartment (Ghossoub et al, 2013; Kim et al, 2016; Palander et al, 2017). Septins are capable of forming gauze‐like structures that could conceivably act as barriers (Garcia et al, 2011; Oh & Bi, 2011). However, how septins, not forming distinct rings, might function in the cilium will require additional experimental efforts.
There is evidence that the TZ may be dynamic and that TZ‐like structures can form outside of their canonical positions. In both insect and vertebrate sperm, the annulus, which separates a “cytosolic” axoneme and a membrane‐ensheathed or “compartmentalized” axoneme. The vertebrate sperm annulus is known to harbor multiple septins (Toure et al, 2011). Interestingly, during the formation of these motile cilia, the annulus is initially just distal to the basal body and then migrates away, keeping a tight association with its ciliary membrane (Avidor‐Reiss & Leroux, 2015). Although not a canonical TZ, the annulus—a well‐established diffusion barrier—contains a full assortment of TZ proteins in Drosophila, and CEP290 and MKS1 in mice. It may also harbor axoneme‐membrane connectors (Basiri et al, 2014).
Some TZ proteins are also present at cell–cell junctions in mammalian cells (NPHP1, NPHP4, and AHI1) and at least one ciliated cell type in C. elegans (MKS‐6/CC2D2A; Eley et al, 2008; Delous et al, 2009; Nguyen et al, 2014). These extra‐ciliary functions within structures that act as diffusion barriers (Kong et al, 2018) could hint, upon further investigation, at new functional interaction partners and gating mechanisms for TZ proteins.
Other yet‐to‐be‐discovered proteins may be involved in forming a picket fence‐like membrane diffusion barrier at the base of cilia. For example, proteins involved at the axon initial segment may offer clues (Box 2). This neuronal domain contains a tethered lattice or network of proteins and separates the cell body from the axon (Huang & Rasband, 2018) in a manner resembling the TZ.
Box 2. In need of answers.
The transition zone (TZ), with its distinctive Y‐link microtubule‐to‐membrane connectors, represents one of the more structurally interesting and complex cellular domains of eukaryotes. But despite our knowledge of at least 15 widely conserved components localizing to this region, ascribing specific molecular roles for any particular protein is challenging. Even collectively, how the known TZ proteins function to create a dynamic gate remains unclear.
Our understanding of TZ mechanisms is almost entirely limited to observing the terminal phenotypes of having lost TZ protein function (e.g., in a mutant) prior to ciliogenesis. Typically, with this approach, researchers find that other TZ components are mislocalized, and various membrane or soluble proteins have inappropriately entered or exited the ciliary compartment (Chih et al, 2011; Garcia‐Gonzalo et al, 2011; Williams et al, 2011; Reiter et al, 2012; Gonçalves & Pelletier, 2017; Nachury, 2018). In brief, ciliary homeostasis is compromised. Few studies assess the loss of TZ function following cilium formation, although conditional knockout mice can help address this shortcoming. This was illustrated nicely by Datta et al (2019), who showed that disruption of CEP290 postciliogenesis results in the mislocalization of several membrane‐associated proteins, including rhodopsin (Datta et al, 2019).
Several gaps in knowledge must be filled to understand the molecular characteristics of the TZ ciliary region more precisely. This includes uncovering the actual physical/chemical properties of the TZ membrane; ascertaining the structures of TZ proteins, and their three‐dimensional organization with respect to the doublet microtubules, Y‐links and overlying membrane; and finally, how individual TZ proteins contribute separately and as a whole to the different selective barriers for membrane proteins, soluble proteins, and potentially, trafficking system.
What is the lipid composition at the TZ, and how does it differ from that of the cell body and more distal ciliary membrane? To what extent does this contribute to making the TZ a membrane diffusion barrier? The TZ has been isolated from Chlamydomonas, which excises this ciliary region after the axoneme has retracted and basal bodies are re‐purposed for cell division (Diener et al, 2015). Presuming that the overlying membrane of the isolated TZs is still relatively intact, probing its lipid composition could be addressed experimentally, and compared with the flanking cell body and ciliary membranes. Assessing lipid composition in various TZ mutants would clarify how the TZ proteins might contribute to forming a unique lipid microdomain. In parallel, fluorescently tagged membrane‐associated proteins and reporters for different lipid species (e.g., phospholipids, phosphoinositides, and sterols) could be used to assess the lipid properties, and the diffusion characteristics, within the TZ in both wild‐type cells and different TZ mutants. Here, one must appreciate the complexity and limitations of these approaches: disruption of certain TZ proteins often causes wholesale mislocalization of other TZ proteins, and so more specific mutations or truncations of specific TZ proteins will almost certainly be required to maintain the overall integrity of the TZ while assessing the functions of individual TZ proteins.
There may also be additional proteins in the TZ, whose identification would help clarify some of the TZ structural properties and functional mechanisms. Detergent‐extracted TZ fragments from Trypanosomes were subjected to mass spectrometry analyses, uncovering both known and interesting TZ protein candidates (Dean et al, 2016). Proteomic analyzes of isolated Chlamydomonas TZs similarly revealed many of the known components (CEP290, NPHP, and MKS module proteins), as well as additional candidates (Diener et al, 2015). The latter include, for example, ANK2, which is not yet linked to cilia but is known to regulate the diameter of axons by linking microtubules to the neuronal membrane (Stephan et al, 2015). Incidentally, the related ANK3 (ankyrin‐G) protein plays a crucial role in forming a diffusion barrier at the axon initial segment, which separates the cell soma from the axon (Huang & Rasband, 2018). In photoreceptors, ankyrin‐G is found distal to the TZ (outer segment) and influences the targeting of a CNG channel required for vision (Kizhatil et al, 2009).
Yet‐to‐be‐identified TZ proteins include those that make up Y‐links, whose composition remains unknown. The ninefold array of Y‐links spans from the axoneme to the membrane, but leaves—at least by electron microscopy—prominent gaps at the membrane and within the TZ lumen. Are there protein(s) at the membrane that “fill” the discontinuous regions between Y‐links and membrane? Septins, discussed in this review, represent an intriguing possibility. Gaps are present between Y‐links in the TZ lumen; is this where nucleoporins, which could form a size‐selective mesh/matrix, reside? Are they anchored via the NPHP module, as the evidence seems to suggest? (Park & Leroux, 2022) Cryo‐EM reconstructions of a TZ, and positioning known components within this framework—as it has been done for understanding the complex and intricate organization of proteins making up the axoneme of motile cilia—would be enormously useful (Ma et al, 2019).
Finally, a complex interplay between different transport machineries, including IFT and LIFT, and the TZ they must cross, seems apparent. IFT generally operates in the absence of a TZ, so what is the nature, and purpose of the observed physical/functional interactions? Does the TZ play a role in cargo selectivity? Loading and/or unloading? Much as trying to understand how the TZ acts as a diffusion barrier, this area of research is ripe for additional discoveries.
Specialized membrane domain as a diffusion barrier
Another distinct possibility for how the TZ functions as a gate is that its membrane sports a lipid composition and properties that are substantively different from that of the rest of the ciliary membrane—which is itself different from the cell body membrane. Such a discrete specialized lipid microdomain would likely influence the area‐partitioning of lipids and membrane protein diffusion across the TZ (Trimble & Grinstein, 2015).
A unique composition of cholesterol and other lipids at the TZ?
Evidence for a lipid microdomain first came from the staining of both motile and nonmotile cilia freeze‐fracture preparations with filipin (Montesano, 1979; Chailley et al, 1983; Cuevas & Gutierrez Diaz, 1985). In these experiments, only membrane regions containing free cholesterol deform in a predictable manner, creating a blanket of small (~30 nm) protuberances. Remarkably, the necklace region at the TZ is almost entirely devoid of filipin staining, in sharp contrast to the ciliary membrane immediately distal to this region (Fig 4Bi). This demarcation in filipin staining is even apparent during the earliest stages of ciliogenesis, when ciliary necklaces are first visible (Chailley & Boisvieux‐Ulrich, 1985).
Intriguingly, a protein known to bind cholesterol—caveolin 1 (CAV1)—is enriched within the TZ in different mammalian cell types, including photoreceptors (Kersten et al, 2009; Schou et al, 2017). This suggests that any cholesterol present at the TZ may be sequestered, unavailable to bind filipin. Caveolins form hairpins that insert into the membrane (Martinez‐Outschoorn et al, 2015). They are generally associated with invaginations of the plasma membrane (caveolae), but also help organize more planar, ordered membrane domains of 5‐100 nm in size (Lajoie et al, 2009). Confinement of CAV1 to the TZ requires NPHP4, as well as KIF13B, a protein with a C2 domain related to RPGRIP1L that itself interacts with NPHP4 (Schou et al, 2017). Loss of CAV1 from the TZ appears to disrupt ciliary gating, since Sonic Hedgehog‐induced accumulation of SMO in the cilium is impaired when it (or KIF13B) is disrupted. It is tempting to suggest that CAV1 plays an essential role in organizing a more highly ordered lipid microdomain at the TZ, supporting its role as a membrane diffusion barrier. Such a possibility is consistent with Laurdan staining experiments, which indicate that the lipid bilayer at the ciliary base is more condensed relative to its surrounding membrane (Vieira et al, 2006).
Some lipids may occupy specific domains within, or immediately next to, the TZ. Super‐resolution microscopy in mammalian cells uncovered localized populations of PIP2 at the distal tip of the basal body transition fibers and PIP3 just distal to this within the TZ (Conduit et al, 2021). Much more work is needed to define the lipid compositions of membranes within and surrounding the TZ.
A detergent‐resistant membrane patch
A remarkable property of the TZ membrane, first observed by Richard Anderson in 1974 (Anderson, 1974), is revealed when oviduct cells containing motile cilia are subjected to detergent. After this treatment, both cell body and ciliary membranes are extracted (removed), but the TZ membrane remains associated with Y‐links (Fig 4Bii). This membrane patch remnant is also observed in Chlamydomonas (Goodenough, 1983; Craige et al, 2010), and in mammalian photoreceptors, where it retains wheat germ agglutinin‐binding glycoproteins (Horst et al, 1987). In Trypanosomes, a pull‐down with a TZ‐specific protein from detergent‐treated cells uncovered most known TZ proteins, and notably, many additional membrane‐associated proteins (Dean et al, 2016). Thus, the TZ membrane and associated proteins have detergent‐resistant properties that seem conserved in evolution.
This detergent‐resistant TZ membrane patch is lost in the cep290 Chlamydomonas mutant, which implies that intact Y‐link structures are required for its formation (Craige et al, 2010). While Chlamydomonas CEP290 is reported to be dynamically associated with the TZ, in that it can relocalize to another cell during gamete mating, several membrane‐associated TZ proteins in C. elegans (TMEM‐107/TMEM107, MKS‐2/TMEM216, and MKS‐6/CC2D2A) appear to be substantially immobile compared with other membrane proteins (Lambacher et al, 2015). The latter result is consistent with a stable membrane patch, containing membrane proteins, that is stably anchored to this ciliary domain.
In some detergent preparations, the leftover membrane patch appears to be concentrated at the ends of Y‐shaped structures, resulting in discontinuous rings of ninefold symmetry visible by electron microscopy (Horst et al, 1987). However, the membrane appears uninterrupted (continuous) in other micrographs (Fig 4Bii). It is unclear how a discontinuous arrangement of Y‐links would help organize a continuous lipid microdomain encircling the entire ciliary base. One possibility is that the membrane‐associated TZ proteins might help nucleate a long‐range lipid composition and organization that propagates throughout (encircles) the region (Sonnino & Prinetti, 2010). If true, there may be transient breaks in continuity in the barrier that makes it “leaky,” for example, during the passage of the large multiprotein IFT particles that just fit between the Y‐links. Such a leaky barrier may create the need for maintenance/replenishment of at least some ciliary proteins/lipids to preserve ciliary homeostasis.
Unique physical properties at or near the TZ
Membrane curvature
The TZ is, positionally, at a unique junction with respect to membranes. At the level of transition fibers, immediately proximal to the TZ, the positively curved cell body membrane undergoes a dramatic change to one of negative curvature, to project outward. Then, to create a tube‐shaped TZ compartment of ~270 nm in diameter, the membrane within the TZ region rapidly reverses its curvature, becoming again more positively curved.
Changes in curvature are usually accommodated by the presence of lipids with varied headgroups and tails that exhibit differential packing propensities in the inner and outer leaflets (McMahon & Boucrot, 2015; Bozelli & Epand, 2020; Fig 4C). These, in turn, are often enabled by membrane‐shaping proteins. Such curved membrane properties can restrict the diffusion of lipids and membrane‐associated proteins and could in principle contribute to some of the gating functionality at the ciliary base (Trimble & Grinstein, 2015; Garcia et al, 2018). In addition to CAV1 (Schou et al, 2017), other proteins within or in proximity to the TZ may influence its unique membrane properties. Candidates include the membrane‐remodeling ESCRT proteins, which were identified in the Chlamydomonas TZ proteome (Diener et al, 2015), and are known to bind and stabilize membranes with positive curvature. Indeed, an ESCRT subunit, CHMP4B, was recently shown to localize to primary cilia and influence both ciliogenesis and the integrity of existing cilia (Jung et al, 2020).
It is perplexing how the loss of TZ Y‐link structures, positioned next to an energetically unfavorable membrane structure, does not appear to prevent the formation of the ciliary cylinder in C. elegans, Chlamydomonas or vertebrate photoreceptors (Craige et al, 2010; Jensen et al, 2015; Schouteden et al, 2015; Li et al, 2016a; Wiegering et al, 2018). One possibility is that, as mentioned earlier, the transition fiber connections themselves may provide a tight, stabilizing connection at the ciliary base. We also cannot exclude the possibility the integrity of the ciliary structure is determined by additional axoneme‐to‐membrane connectors, present within the TZ and/or throughout the cilium, that remain to be identified.
Bilayer thickness
Certain regions of a lipid bilayer can have an inhomogeneous thickness, creating hydrophobic mismatches at the interface (Andersen & Koeppe, 2007). This can form a diffusion barrier between certain types of lipids and membrane proteins at the boundary (Trimble & Grinstein, 2015; Fig 4C). For example, short‐lipid bilayers may accommodate proteins with small transmembrane domains and exclude those with longer hydrophobic segments. With this in mind, the tight connection between Y‐links and the membrane, and the presence of multiple transmembrane proteins, may alter membrane thickness at the TZ (Fig 4C). However, as far as we are aware, this has not been reported but could be investigated by TEM analysis of wild‐type and mutants either lacking Y‐links or for example one or more mostly membrane‐associated MKS module protein(s).
Evidence for a ciliary soluble protein gate
A distinct function of the ciliary base is to regulate the diffusion of soluble proteins into, and likely out of, the organelle (Awata et al, 2014; Takao & Verhey, 2016; Verhey & Yang, 2016; Garcia‐Gonzalo & Reiter, 2017; Jensen & Leroux, 2017). A size‐selective, mesh‐like structure may be responsible for such gating (Fig 4D).
In 2012, the Verhey group reported on a size‐dependent barrier at the entrance of primary cilia, based on ciliary accessibility of microinjected fluorescent dextran beads in an hTERT‐RPE cell line (Kee et al, 2012). The following year, Lin et al (2013) used a mammalian cell line (NIH 3T3) and an assortment of differently sized fluorescent protein reporters and a ciliary protein trap system to observe an exponentially decreasing diffusion coefficient for proteins of increasing size (MW) and Stokes radius (Lin et al, 2013). A small protein of ~40 kDa had ready access to the interior of primary cilia; by contrast, a larger protein of >600 kDa had significantly more limited access. Their findings suggested an average mesh radius of ~8 nm. Another study published the same year by the Nachury laboratory made use of permeabilized mammalian cells to administer a panel of protein reporters with different sizes (Breslow et al, 2013). Those with a MW of >70 kDa (4.3 nm) were found to exhibit limited entry into cilia.
Collectively, these and other studies provide evidence for a diffusion barrier that restricts or slows the entry of larger proteins and protein complexes. But what is the nature of the proteins associated with this soluble protein gate? Studies across two evolutionarily distant species now point to TZ‐localized proteins. In Chlamydomonas, loss of the core scaffolding proteins CEP290 and RPGRIP1L, or NPHP4, results in changes to the ciliary abundance of generally larger soluble proteins (Craige et al, 2010; Awata et al, 2014; Lin et al, 2018). Similarly, RPGR mutant mouse photoreceptors exhibit altered levels of proteins within their outer segments (Rao et al, 2015).
While the presence of many transmembrane proteins at the TZ might point to a mechanism for creating a membrane diffusion barrier, it is unclear how the known TZ proteins might act molecularly to create a barrier for soluble proteins. A promising clue regarding the soluble protein gate was first suggested by the unexpected discovery by the Verhey laboratory in 2010 that ciliary import of the IFT‐kinesin motor protein, KIF17, depends on three factors shared with nuclear import (Dishinger et al, 2010). One is a motif on KIF17 with similarities to a nuclear localization signal (NLS). The second is a functional interaction with the nuclear import protein importin‐ß2. Finally, KIF17 import depends on a concentration gradient of the small GTPase Ran that parallels that of nuclear import, with cilia (or the nucleus) containing higher levels of the GTP‐bound form (RanGTP).
Further evidence for a cilium‐nuclear import connection came from several studies implicating nucleoporins, which are best known for their size‐dependent gating functions at nuclear pores (Takao & Verhey, 2016; Johnson & Malicki, 2019). Disruption of several nucleoporins, including the central Phe‐Gly (FG) NUP62 and NUP98 proteins, influenced the entry of KIF17 or various soluble proteins (Kee et al, 2012; Takao et al, 2014; Endicott & Brueckner, 2018). A study in Xenopus found that knockdown of NUP188 or its binding partner NUP93 interferes with ciliogenesis during embryonic development, while not affecting the function of the nuclear pore complex (Del Viso et al, 2016). An additional component of the inner‐ring nucleoporin complex, NUP205, is now similarly implicated in cilium formation (Marquez et al, 2020).
These findings suggest seemingly analogous functions for nucleoporins at the entrance of the nuclear and ciliary compartments. However, the exact localization of nucleoporins, and hence where the soluble protein gate might be operating, remains unclear. One study could not find nucleoporins in proximity to cilia (Breslow et al, 2013). Others report the position of several nucleoporins at or near the basal body (Kee et al, 2012; Endicott et al, 2015; Takao et al, 2017; Marquez et al, 2020). In one case, super‐resolution microscopy shows a barrel‐shaped distribution around the basal body centrioles (Del Viso et al, 2016). How nucleoporins might function within this enclave rather than at the TZ, is perplexing. Intriguingly, interactions between nucleoporins and at least two TZ components (NPHP4 and RPGRIP1L) were observed by Yeast‐2‐Hybrid (Blasius et al, 2019). It remains to be seen whether there is a clear‐cut functional relationship between the TZ and nucleoporins.
Altogether, it appears as if the TZ has a number of different attributes that create a unique subcellular domain, which acts as diffusion barriers for membrane and soluble proteins. These TZ properties likely contribute, collectively, to the maintenance of ciliary homeostasis, including a unique lipid and membrane protein composition. However, the ciliary compartment is not static, and we need to consider the transport systems that move into and out of the cilium, passing through the TZ along the way, to make it dynamic and adaptable to different cellular requirements.
Transport across the ciliary gate
If the TZ functions as a gate, it must by necessity be permissive for the entry and exit of certain proteins or protein complexes (Jensen & Leroux, 2017). Some soluble and membrane proteins may simply diffuse into—and out of—the ciliary compartment without being impeded or requiring assistance (Nachury et al, 2010; Ye et al, 2013; Nachury & Mick, 2019; Craft Van De Weghe et al, 2020). In this case, ciliary enrichment may be achieved via a capture mechanism. Retention could be, for example, through association with the axoneme or particular ciliary membrane subcompartment with a specific lipid composition. But a subset of proteins relies on large, multisubunit transport systems that are able to somehow enter and/or exit cilia. Below, we discuss the two major systems, IFT and LIFT, and another specific for a class of signaling proteins, which must cross the TZ barrier to transport ciliary cargo.
Intraflagellar transport (IFT)
The IFT system docks at or near the transition fibers (Box 1) and shuttles between the ciliary base and the ciliary tip (Figs 1 and 5; Reiter et al, 2012; Sung & Leroux, 2013). There is evidence that IFT complexes in C. elegans move more slowly within the TZ compared with more distal regions of the axoneme, and removal of TZ Y‐links partially restores its speed (Jensen et al, 2015; Mangeol et al, 2015; Oswald et al, 2018; Yang et al, 2019; De‐Castro et al, 2022). Single‐particle tracking of IFT proteins reveals a similar reduction in IFT velocities at the TZ of mammalian cilia (Yang et al, 2019). A small proportion of IFT/BBS‐associated ciliary proteins can also be observed to stall or accumulate within the mammalian TZ (Shi et al, 2017; Yang et al, 2019). Interestingly, Ye et al (2018) report the stalling of IFT/BBS‐bound ciliary cargo (such as GPR161) within a compartment or “airlock,” situated between the TZ and a periciliary barrier (Nachury & Mick, 2019). Because this barrier is in close proximity to the transition fibers, it is conceivable that it represents prolonged docking events (physical interactions) between IFT particles and a transition fiber component such as FBF1 (Wei et al, 2013).
Figure 5. Trafficking systems that cross the transition zone ciliary gate create a dynamic signaling compartment.

Intraflagellar transport (IFT) and other transport systems such as lipidated protein intraflagellar targeting (LIFT) cross the TZ and therefore make the cilium a dynamic compartment. The LIFT system, which includes the lipidated protein‐binding chaperones UNC119 and PDE6D, facilitates the transport of lipid‐modified proteins such as INPP5E and NPHP3 into cilia. Cargo release is mediated by effectors, including ARL3 and RP2. At least for some ciliary cargos, including GPCR proteins, the IFT system (including IFT‐A complex‐associated TULP3) makes use of a PIP2‐PI4P lipid gradient established by INPP5E to bind cargo outside the cilium (high PIP2) and release it inside the cilium (low PIP2). Functional interactions between IFT and LIFT (shown as question mark) have been documented but require further investigation (see text for details).
The reason for the slow‐down behavior of IFT at the TZ is unclear. The movement of IFT trains, which are intimately associated with the membrane (Wingfield et al, 2018; Wang et al, 2018b), could decrease in a particular lipid environment. Motility could also be impeded by the size‐selective mesh discussed above, given that the IFT particles are large, spanning from the axoneme to the ciliary membrane. Indeed, the IFT‐kinesin motor, KIF17, interacts with and depends on nucleoporin(s) for translocation into cilia (Kee et al, 2012; Takao et al, 2014). Moreover, a recent study by De‐Castro et al (2022) observed that C. elegans IFT particles/trains with reduced levels of IFT‐dynein motor activity are unable to cross or “power through” the TZ during their retrograde transport back to the base (De‐Castro et al, 2022; Park & Leroux, 2022). Most interestingly, the authors ascribe this functionality to the NPHP module: disruption of NPHP4 restores the ability of the underpowered IFT particles to reach the base, whereas loss of CEP290 or the MKS module did not. These findings suggest that the NPHP module might specifically anchor nuclear pore components at the TZ, in line with the discovery that NPHP4 interacts with the nucleoporin NUP62 (Takao et al, 2017).
Lastly, IFT proteins may directly, albeit transiently, associate with TZ proteins themselves. Physical interactions between NPHP4 and KIF17, and between B9D2 and an IFT subunit (TTC30B/IFT70B), have been reported (Zhao & Malicki, 2011; Takao et al, 2017). This may help explain why some IFT proteins, as well as IFT cargo, notably the Hedgehog signaling protein Smoothened, are observed to stall within the TZ region (Shi et al, 2017). Consistent with a functional interaction between TZ components and the IFT/BBSome complexes, genetic interactions between the two were uncovered in both C. elegans and mice (Yee et al, 2015; Goetz et al, 2017; Bentley‐Ford et al, 2021). However, it is unclear whether these interactions represent a conserved functional feature of the TZ, suggested by Shi et al (2017) to be a waypoint (or hub) for ciliary protein trafficking (Shi et al, 2017).
Lipidated protein intraflagellar targeting (LIFT)
Another trafficking system, which we term lipidated protein intraflagellar targeting (LIFT), must also cross the TZ to transport lipid‐modified proteins such as INPP5E and NPHP3 into mammalian cilia (Stephen & Ismail, 2016; Jean & Pilgrim, 2017; Jensen & Leroux, 2017; Roy & Marin, 2019; Frederick et al, 2020; Fig 5). The proteins implicated in this process include UNC119, PDE6D, RP2, ARL3, and ARL13B. Both UNC119 and PDE6D function as chaperones, shielding the lipidated cargo protein to facilitate transport, and perhaps more specifically, to traverse the TZ. Once within the cilium, cargo release involves the GTPase activating protein (GAP) RP2 and guanine nucleotide exchange factor (GEF) ARL3. Hence, lipid modifications play a critical role in trafficking some proteins to cilia (Roy & Marin, 2019). As demonstrated in C. elegans, once inside the ciliary compartment, the lipid modifications and presence of a functional TZ function together help ensure proper retention of the cargo protein (Cevik et al, 2010, 2013).
The large size of the chaperone‐cargo complex may be restrictive and require a specific entry mechanism (Jensen & Leroux, 2017). Various tantalizing connections between LIFT components and the IFT machinery are known, for example, as found in C. elegans and mammalian cells; ARL13B is directly associated with IFT, impairment of IFT affects RP2 localization, PDE6D interacts with RAB28, an IFT‐cargo protein, and the LIFT‐cargo protein INPP5E also requires IFT for ciliary enrichment (Humbert et al, 2012; Cevik et al, 2013; Jensen et al, 2018; Han et al, 2019).
Intriguingly, a bona fide component of the photoreceptor connecting cilium (TZ)—discussed above in the context of rhodopsin compartmentalization—is lipidated and represents another well‐established LIFT‐associated protein in mammalian cilia (Watzlich et al, 2013; Rao et al, 2016). RPGR itself is needed for the correct targeting of the LIFT cargo INPP5E. Hence, RPGR may participate in a handover (capture and release)‐type of mechanism to assist trafficking across the TZ (Jensen & Leroux, 2017). Collectively, the above findings point to an interplay between LIFT, IFT, and the TZ that collectively ensures cargo transit across the ciliary gate and into the organelle.
The transport systems and the TZ also work to regulate ciliary lipid composition, which itself influences the trafficking of ciliary cargo (Fig 5). Once correctly localized to the cilium, INPP5E converts PIP2 to PI4P, helping to maintain ciliary PIP2 levels low. At high PIP2 levels outside the ciliary compartment, the interaction of ciliary proteins, including GPCRs with IFT, via the IFT‐associated tubby protein TULP3 is strengthened; at lower PIP2 levels inside the mammalian cilium, cargo is released (Mukhopadhyay et al, 2010; Garcia‐Gonzalo et al, 2015). INPP5E is not only targeted by LIFT/IFT to the cilium, but also requires functional TZ—both the NPHP and MKS modules—for enrichment in this compartment and regulation of Hedgehog signaling (Roberson et al, 2015; Slaats et al, 2016; Ning et al, 2021). As such, the TZ plays essential roles in ciliary lipid homeostasis, trafficking, and signal transduction.
A transport system for cyclic nucleotide and GPCR signaling
Yet other means of regulated trafficking across the TZ are likely. One such transport system is required for the ciliary enrichment of adenylate and guanylate cyclases, enzymes that have prokaryotic origins and may represent perhaps the earliest signaling molecules to function within eukaryotic cilia (Johnson & Leroux, 2010). At least in C. elegans, all studied cilium‐associated cGMP‐producing guanylate cyclases depend on the DAF‐25 protein, the mammalian orthologue of ANKMY2, for their ciliary localization (Fujiwara et al, 2010; Jensen et al, 2010; Nguyen et al, 2014; van der Burght et al, 2020). In the absence of DAF‐25, the membrane‐bound cyclases are prevented from crossing the TZ barrier and entering cilia. Notably, this transport function appears linked, in a manner not yet understood, to IFT (Jensen et al, 2010; van der Burght et al, 2020).
Interestingly, mammalian Ankmy2 was shown to be required for the correct localization of cAMP‐producing adenylate cyclases to cilia (Somatilaka et al, 2020), including ADCY3, which itself depends on TZ proteins for correct ciliary localization (Garcia‐Gonzalo et al, 2011). Hence, just as shown for the TZ, ANKMY2 regulates the Hedgehog signaling pathway (Saita et al, 2014; Somatilaka et al, 2020). In C. elegans, DAF‐25 also participates in the ciliary targeting of a subset of G protein‐coupled receptors (GPCRs), which themselves depend on IFT‐mediated transport (Mukhopadhyay et al, 2010; Brear et al, 2014). Whether ANKMY2 plays a similar role in GPCR trafficking in mammalian cells, and is functionally associated with the IFT system and the TZ, remains to be seen.
In summary, it is increasingly apparent that shedding additional light on the mechanisms by which ciliary proteins traverse the TZ, aided or unaided, will be necessary to fully understand how the TZ functions as a dynamic ciliary gate.
Conclusions and future prospects
In the span of about a dozen years, tremendous knowledge has been gained from studying the composition, overall organization, and role of the transition zone as a selective diffusion barrier for membrane and soluble proteins. Many TZ components are evolutionarily conserved, having emerged in the primordial eukaryote to function within an essential ciliary subdomain. This has allowed researchers to use powerful and complementary model systems, including Trypanosoma, Tetrahymena, Chlamydomonas, C. elegans, Drosophila, zebrafish, mouse, and mammalian tissue culture cells, to dissect how the TZ operates as a gate at the base of both motile and nonmotile cilia.
Many questions regarding the molecular mechanisms employed by the TZ remain, however (Box 2). Until we determine the structures of individual TZ proteins, position them into well‐defined complexes as is being done for the different IFT modules (Wingfield et al, 2018; Webb et al, 2020; Jordan & Pigino, 2021), and begin ascribing specific roles to each of the proteins, how the TZ operates at the molecular level to compartmentalize cilia in a dynamic manner will remain mostly enigmatic.
While the primary function of the TZ may be as a ciliary gate, its position at the ciliary base influences other important aspects of ciliary function that also deserve attention. Ciliary (flagellar) autotomy, discussed only briefly, is not well studied but is, for example, of interest in respiratory airway motile cilia. These can deciliate in response to stressors and viral infections—perhaps including COVID‐19, which is notably known to affect smell and hence probably olfactory cilia (Li et al, 2020) and whose previously uncharacterized protein (ORF10) has now been directly implicated in the rapid loss of cilia (Wang et al, 2022).
Cilium length control represents another notable role for TZ‐associated proteins. Disruption of mammalian RPGRIP1L/NPHP8, MKS3, TCTN1, and TCTN2 influences cilium formation and length regulation (Tammachote et al, 2009; Patzke et al, 2010; Garcia‐Gonzalo et al, 2011). In C. elegans, mutations in MKS‐5 (RPGRIP1L) and the NPHP module (NPHP1 and NPHP4) also cause cilium length phenotypes (Williams et al, 2011). It is unclear, however, whether such an effect is direct or indirect—for example, one of the many factors influencing cilium length, including the IFT system, may be mislocalized/deregulated in TZ mutant cilia. More recently, a C. elegans TZ‐localized kinase that appears to not play a role in gating, CDKL‐1, was shown to negatively modulate cilium length, likely by regulating the IFT system (Canning et al, 2018; Park et al, 2021). It joins another CDKL family member from Chlamydomonas, which localizes just distally to the TZ and modulates cilium length (Tam et al, 2013).
Without a doubt, continued studies on the TZ promise to yield important insights into the homeostasis of diverse ciliary signaling compartments that are critically important for human physiology and development, and relevant to a wide array of human diseases.
Author contributions
Michel R Leroux: Conceptualization; writing—original draft; writing—review and editing. Kwangjin Park: Conceptualization; writing—original draft; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Table EV1
Acknowledgments
We apologize to our colleagues whose findings were not covered in this otherwise expansive review of the ciliary transition zone. This work was funded by the Canadian Institutes of Health Research (CIHR; grants MOP‐142243 and PJT‐156042 to M.R.L.). M.R.L. holds a Michael Smith Foundation for Health Research (MSFHR) senior scholar award. K.P. is a recipient of Michael Smith Health Research BC's Research Trainee Awards, and Vanier Canada Graduate Scholarships.
EMBO reports (2022) 23: e55420
References
- AFZELIUS BA (1961) The fine structure of the cilia from ctenophore swimming‐plates. J Biophys Biochem Cytol 9: 383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE (2007) Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 36: 107–130 [DOI] [PubMed] [Google Scholar]
- Anderson RG (1974) Isolation of ciliated or unciliated basal bodies from the rabbit oviduct. J Cell Biol 60: 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST (2019) Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol 15: 199–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima T, Shibata Y, Yamamoto T (1984) A deep‐etching study of the guinea pig tracheal cilium with special reference to the ciliary transitional region. J Ultrastruct Res 89: 34–41 [DOI] [PubMed] [Google Scholar]
- Arslanhan MD, Gulensoy D, Firat‐Karalar EN (2020) A proximity mapping journey into the biology of the mammalian centrosome/cilium complex. Cell 9: 1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashique AM, Choe Y, Karlen M, May SR, Phamluong K, Solloway MJ, Ericson J, Peterson AS (2009) The Rfx4 transcription factor modulates Shh signaling by regional control of ciliogenesis. Sci Signal 2: ra70 [DOI] [PubMed] [Google Scholar]
- Avidor‐Reiss T, Leroux MR (2015) Shared and distinct mechanisms of compartmentalized and cytosolic ciliogenesis. Curr Biol 25: R1143–R1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor‐Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS (2004) Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117: 527–539 [DOI] [PubMed] [Google Scholar]
- Awata J, Takada S, Standley C, Lechtreck KF, Bellvé KD, Pazour GJ, Fogarty KE, Witman GB (2014) NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J Cell Sci 127: 4714–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann‐Gagescu R, Neuhauss SC (2019) The photoreceptor cilium and its diseases. Curr Opin Genet Dev 56: 22–33 [DOI] [PubMed] [Google Scholar]
- Baehr W, Hanke‐Gogokhia C, Sharif A, Reed M, Dahl T, Frederick JM, Ying G (2019) Insights into photoreceptor ciliogenesis revealed by animal models. Prog Retin Eye Res 71: 26–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbelanne M, Hossain D, Chan DP, Peränen J, Tsang WY (2015) Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet 24: 2185–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker AR, Renzaglia KS, Fry K, Dawe HR (2014) Bioinformatic analysis of ciliary transition zone proteins reveals insights into the evolution of ciliopathy networks. BMC Genomics 15: 531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basiri ML, Ha A, Chadha A, Clark NM, Polyanovsky A, Cook B, Avidor‐Reiss T (2014) A migrating ciliary gate compartmentalizes the site of axoneme assembly in Drosophila spermatids. Curr Biol 24: 2622–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18: 73–89 [DOI] [PubMed] [Google Scholar]
- Bentley‐Ford MR, LaBonty M, Thomas HR, Haycraft CJ, Scott M, LaFayette C, Croyle MJ, Andersen RS, Parant JM, Yoder BK (2021) Evolutionarily conserved genetic interactions between nphp‐4 and bbs‐5 mutations exacerbate ciliopathy phenotypes. Genetics 220: iyab209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besharse JC, Forestner DM, Defoe DM (1985) Membrane assembly in retinal photoreceptors. III. Distinct membrane domains of the connecting cilium of developing rods. J Neurosci 5: 1035–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas NJ, Inglis PN, Li C, Robinson JF, Parker JD, Healey MP, Davis EE, Inglis CD, Toivonen T, Cottell DC et al (2009) Functional interactions between the ciliopathy‐associated Meckel syndrome 1 (MKS1) protein and two novel MKS1‐related (MKSR) proteins. J Cell Sci 122: 611–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al‐Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel‐Aleem A, Rosti RO et al (2009) Mutations in INPP5E, encoding inositol polyphosphate‐5‐phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41: 1032–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque OE, Sanders AA (2014) Compartments within a compartment: what C. elegans can tell us about ciliary subdomain composition, biogenesis, function, and disease. Organogenesis 10: 126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque OE, Perens EA, Boroevich KA, Inglis PN, Li C, Warner A, Khattra J, Holt RA, Ou G, Mah AK et al (2005) Functional genomics of the cilium, a sensory organelle. Curr Biol 15: 935–941 [DOI] [PubMed] [Google Scholar]
- Blasius TL, Takao D, Verhey KJ (2019) NPHP proteins are binding partners of nucleoporins at the base of the primary cilium. PLoS One 14: e0222924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozelli JC, Epand RM (2020) Membrane shape and the regulation of biological processes. J Mol Biol 432: 5124–5136 [DOI] [PubMed] [Google Scholar]
- Braun DA, Hildebrandt F (2017) Ciliopathies. Cold Spring Harb Perspect Biol 9: a028191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brear AG, Yoon J, Wojtyniak M, Sengupta P (2014) Diverse cell type‐specific mechanisms localize G protein‐coupled receptors to Caenorhabditis elegans sensory cilia. Genetics 197: 667–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Koslover EF, Seydel F, Spakowitz AJ, Nachury MV (2013) An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. J Cell Biol 203: 129–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujakowska KM, Liu Q, Pierce EA (2017) Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb Perspect Biol 9: a028274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burght SN, Rademakers S, Johnson JL, Li C, Kremers GJ, Houtsmuller AB, Leroux MR, Jansen G (2020) Ciliary tip signaling compartment is formed and maintained by intraflagellar transport. Curr Biol 30: 4299–4306 [DOI] [PubMed] [Google Scholar]
- Canning P, Park K, Gonçalves J, Li C, Howard CJ, Sharpe TD, Holt LJ, Pelletier L, Bullock AN, Leroux MR (2018) CDKL family kinases have evolved distinct structural features and ciliary function. Cell Rep 22: 885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho‐Santos Z, Azimzadeh J, Pereira‐Leal JB, Bettencourt‐Dias M (2011) Evolution: tracing the origins of centrioles, cilia, and flagella. J Cell Biol 194: 165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevik S, Hori Y, Kaplan OI, Kida K, Toivenon T, Foley‐Fisher C, Cottell D, Katada T, Kontani K, Blacque OE (2010) Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J Cell Biol 188: 953–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevik S, Sanders AA, Van Wijk E, Boldt K, Clarke L, van Reeuwijk J, Hori Y, Horn N, Hetterschijt L, Wdowicz A et al (2013) Active transport and diffusion barriers restrict Joubert Syndrome‐associated ARL13B/ARL‐13 to an Inv‐like ciliary membrane subdomain. PLoS Genet 9: e1003977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailley B, Boisvieux‐Ulrich E (1985) Detection of plasma membrane cholesterol by filipin during microvillogenesis and ciliogenesis in quail oviduct. J Histochem Cytochem 33: 1–10 [DOI] [PubMed] [Google Scholar]
- Chailley B, Boisvieux‐Ulrich E, Sandoz D (1983) Evolution of filipin‐sterol complexes and intramembrane particle distribution during ciliogenesis. J Submicrosc Cytol 15: 275–280 [PubMed] [Google Scholar]
- Chen HY, Kelley RA, Li T, Swaroop A (2020) Primary cilia biogenesis and associated retinal ciliopathies. Semin Cell Dev Biol 110: 70–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS (2011) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 14: 61–72 [DOI] [PubMed] [Google Scholar]
- Chu JS, Baillie DL, Chen N (2010) Convergent evolution of RFX transcription factors and ciliary genes predated the origin of metazoans. BMC Evol Biol 10: 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL (1998) Chlamydomonas kinesin‐II‐dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol 141: 993–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit SE, Vanhaesebroeck B (2020) Phosphoinositide lipids in primary cilia biology. Biochem J 477: 3541–3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit SE, Davies EM, Fulcher AJ, Oorschot V, Mitchell CA (2021) Superresolution microscopy reveals distinct phosphoinositide subdomains within the cilia transition zone. Front Cell Dev Biol 9: 634649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft Van De Weghe J, Harris JA, Kubo T, Witman GB, Lechtreck KF (2020) Diffusion rather than intraflagellar transport likely provides most of the tubulin required for axonemal assembly in Chlamydomonas. J Cell Sci 133: jcs249805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB (2010) CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol 190: 927–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas P, Gutierrez Diaz JA (1985) Absence of filipin‐sterol complexes from the ciliary necklace of ependymal cells. Anat Embryol 172: 97–99 [DOI] [PubMed] [Google Scholar]
- van Dam TJ, Townsend MJ, Turk M, Schlessinger A, Sali A, Field MC, Huynen MA (2013) Evolution of modular intraflagellar transport from a coatomer‐like progenitor. Proc Natl Acad Sci U S A 110: 6943–6948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam TJP, Kennedy J, van der Lee R, de Vrieze E, Wunderlich KA, Rix S, Dougherty GW, Lambacher NJ, Li C, Jensen VL et al (2019) CiliaCarta: an integrated and validated compendium of ciliary genes. PLoS One 14: e0216705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta P, Hendrickson B, Brendalen S, Ruffcorn A, Seo S (2019) The myosin‐tail homology domain of centrosomal protein 290 is essential for protein confinement between the inner and outer segments in photoreceptors. J Biol Chem 294: 19119–19136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis E (1956) Electron microscope observations on the submicroscopic organization of the retinal rods. J Biophys Biochem Cytol 2: 319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean S, Moreira‐Leite F, Varga V, Gull K (2016) Cilium transition zone proteome reveals compartmentalization and differential dynamics of ciliopathy complexes. Proc Natl Acad Sci U S A 113: E5135–E5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De‐Castro ARG, Rodrigues DRM, De‐Castro MJG, Vieira N, Vieira C, Carvalho AX, Gassmann R, Abreu CMC, Dantas TJ (2022) WDR60‐mediated dynein‐2 loading into cilia powers retrograde IFT and transition zone crossing. J Cell Biol 221: e202010178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Viso F, Huang F, Myers J, Chalfant M, Zhang Y, Reza N, Bewersdorf J, Lusk CP, Khokha MK (2016) Congenital heart disease genetics uncovers context‐dependent organization and function of nucleoporins at cilia. Dev Cell 38: 478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Hellman NE, Gaude HM, Silbermann F, Le Bivic A, Salomon R, Antignac C, Saunier S (2009) Nephrocystin‐1 and nephrocystin‐4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet 18: 4711–4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentler W (2005) Intraflagellar transport (IFT) during assembly and disassembly of Chlamydomonas flagella. J Cell Biol 170: 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmat R, Eblimit A, Robichaux MA, Zhang Z, Nguyen TT, Jung SY, He F, Jain A, Li Y, Qin J et al (2018) SPATA7 maintains a novel photoreceptor‐specific zone in the distal connecting cilium. J Cell Biol 217: 2851–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener DR, Lupetti P, Rosenbaum JL (2015) Proteomic analysis of isolated ciliary transition zones reveals the presence of ESCRT proteins. Curr Biol 25: 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle AD, Fulton C (1966) Development of the flagellar apparatus of Naegleria. J Cell Biol 31: 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishinger JF, Kee HL, Jenkins PM, Fan S, Hurd TW, Hammond JW, Truong YN, Margolis B, Martens JR, Verhey KJ (2010) Ciliary entry of the kinesin‐2 motor KIF17 is regulated by importin‐beta2 and RanGTP. Nat Cell Biol 12: 703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTirro D, Philbrook A, Rubino K, Sengupta P (2019) The Caenorhabditis elegans Tubby homolog dynamically modulates olfactory cilia membrane morphogenesis and phospholipid composition. Elife 8: e48789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drivas TG, Holzbaur EL, Bennett J (2013) Disruption of CEP290 microtubule/membrane‐binding domains causes retinal degeneration. J Clin Invest 123: 4525–4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK (1995) Flagellar assembly in two hundred and fifty easy‐to‐follow steps. Trends Genet 11: 398–404 [DOI] [PubMed] [Google Scholar]
- Dyson JM, Conduit SE, Feeney SJ, Hakim S, DiTommaso T, Fulcher AJ, Sriratana A, Ramm G, Horan KA, Gurung R et al (2017) INPP5E regulates phosphoinositide‐dependent cilia transition zone function. J Cell Biol 216: 247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley L, Gabrielides C, Adams M, Johnson CA, Hildebrandt F, Sayer JA (2008) Jouberin localizes to collecting ducts and interacts with nephrocystin‐1. Kidney Int 74: 1139–1149 [DOI] [PubMed] [Google Scholar]
- Endicott SJ, Brueckner M (2018) NUP98 sets the size‐exclusion diffusion limit through the ciliary base. Curr Biol 28: 1643–1650.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott SJ, Basu B, Khokha M, Brueckner M (2015) The NIMA‐like kinase Nek2 is a key switch balancing cilia biogenesis and resorption in the development of left‐right asymmetry. Development 142: 4068–4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Bansal R, Antonellis PJ, Berbari NF (2020) Cilia signaling and obesity. Semin Cell Dev Biol 110: 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk N, Lösl M, Schröder N, Gießl A (2015) Specialized cilia in mammalian sensory systems. Cell 4: 500–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett DW, Porter KR (1954) A study of the fine structure of ciliated epithelia. J Morphol 94: 221–281 [Google Scholar]
- Fisch C, Dupuis‐Williams P (2011) Ultrastructure of cilia and flagella ‐ back to the future! Biol Cell 103: 249–270 [DOI] [PubMed] [Google Scholar]
- Flower NE (1971) Particles within membranes: a freeze‐etch view. J Cell Sci 9: 435–441 [DOI] [PubMed] [Google Scholar]
- Fraire‐Zamora JJ, Cardullo RA (2010) The physiological acquisition of amoeboid motility in nematode sperm: is the tail the only thing the sperm lost. Mol Reprod Dev 77: 739–750 [DOI] [PubMed] [Google Scholar]
- Frederick JM, Hanke‐Gogokhia C, Ying G, Baehr W (2020) Diffuse or hitch a ride: how photoreceptor lipidated proteins get from here to there. Biol Chem 401: 573–584 [DOI] [PubMed] [Google Scholar]
- Fritz‐Laylin LK, Cande WZ (2010) Ancestral centriole and flagella proteins identified by analysis of Naegleria differentiation. J Cell Sci 123: 4024–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Teramoto T, Ishihara T, Ohshima Y, McIntire SL (2010) A novel zf‐MYND protein, CHB‐3, mediates guanylyl cyclase localization to sensory cilia and controls body size of Caenorhabditis elegans . PLoS Genet 6: e1001211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung KY, Dai L, Trimble WS (2014) Cell and molecular biology of septins. Int Rev Cell Mol Biol 310: 289–339 [DOI] [PubMed] [Google Scholar]
- Gabriel GC, Young CB, Lo CW (2020) Role of cilia in the pathogenesis of congenital heart disease. Semin Cell Dev Biol 110: 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E (2011) Subunit‐dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol 195: 993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, Raleigh DR, Reiter JF (2018) How the ciliary membrane is organized inside‐out to communicate outside‐in. Curr Biol 28: R421–R434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo FR, Reiter JF (2012) Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol 197: 697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo FR, Reiter JF (2017) Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect Biol 9: a028134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo FR, Corbit KC, Sirerol‐Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ et al (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 43: 776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo FR, Phua SC, Roberson EC, Garcia G, Abedin M, Schurmans S, Inoue T, Reiter JF (2015) Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev Cell 34: 400–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghossoub R, Hu Q, Failler M, Rouyez MC, Spitzbarth B, Mostowy S, Wolfrum U, Saunier S, Cossart P, Jamesnelson W et al (2013) Septins 2, 7 and 9 and MAP4 colocalize along the axoneme in the primary cilium and control ciliary length. J Cell Sci 126: 2583–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilula NB, Satir P (1972) The ciliary necklace. A ciliary membrane specialization. J Cell Biol 53: 494–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Bangs F, Barrington CL, Katsanis N, Anderson KV (2017) The Meckel syndrome‐ associated protein MKS1 functionally interacts with components of the BBSome and IFT complexes to mediate ciliary trafficking and hedgehog signaling. PLoS One 12: e0173399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogendeau D, Lemullois M, Le Borgne P, Castelli M, Aubusson‐Fleury A, Arnaiz O, Cohen J, Vesque C, Schneider‐Maunoury S, Bouhouche K et al (2020) MKS‐NPHP module proteins control ciliary shedding at the transition zone. PLoS Biol 18: e3000640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves J, Pelletier L (2017) The ciliary transition zone: finding the pieces and assembling the gate. Mol Cells 40: 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough UW (1983) Motile detergent‐extracted cells of Tetrahymena and Chlamydomonas. J Cell Biol 96: 1610–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D et al (2008) CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy‐associated basal body protein CEP290. Am J Hum Genet 83: 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenan GA, Vale RD, Agard DA (2020) Electron cryotomography of intact motile cilia defines the basal body to axoneme transition. J Cell Biol 219: e201907060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guemez‐Gamboa A, Coufal NG, Gleeson JG (2014) Primary cilia in the developing and mature brain. Neuron 82: 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GD, Coyaud É, Gonçalves J, Mojarad BA, Liu Y, Wu Q, Gheiratmand L, Comartin D, Tkach JM, Cheung SW et al (2015) A dynamic protein interaction landscape of the human centrosome‐cilium interface. Cell 163: 1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Fabian L, Brill JA (2018) Phosphatidylinositol 4,5‐bisphosphate regulates cilium transition zone maturation in Drosophila melanogaster . J Cell Sci 131: jcs218297 [DOI] [PubMed] [Google Scholar]
- Han S, Miyoshi K, Shikada S, Amano G, Wang Y, Yoshimura T, Katayama T (2019) TULP3 is required for localization of membrane‐associated proteins ARL13B and INPP5E to primary cilia. Biochem Biophys Res Commun 509: 227–234 [DOI] [PubMed] [Google Scholar]
- Heller RF, Gordon RE (1986) Chronic effects of nitrogen dioxide on cilia in hamster bronchioles. Exp Lung Res 10: 137–152 [DOI] [PubMed] [Google Scholar]
- Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R et al (2007) A higher‐level phylogenetic classification of the Fungi. Mycol Res 111: 509–547 [DOI] [PubMed] [Google Scholar]
- Hodges ME, Scheumann N, Wickstead B, Langdale JA, Gull K (2010) Reconstructing the evolutionary history of the centriole from protein components. J Cell Sci 123: 1407–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, van Reeuwijk J, Boehlke C, Schell C, Yasunaga T, Helmstädter M et al (2013) ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet 45: 951–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh RA, Stowe TR, Turk E, Stearns T (2012) Transcriptional program of ciliated epithelial cells reveals new cilium and centrosome components and links to human disease. PLoS One 7: e52166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DH, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T (2000) A retinitis pigmentosa GTPase regulator (RPGR)‐deficient mouse model for X‐linked retinitis pigmentosa (RP3). Proc Natl Acad Sci U S A 97: 3649–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst CJ, Forestner DM, Besharse JC (1987) Cytoskeletal‐membrane interactions: a stable interaction between cell surface glycoconjugates and doublet microtubules of the photoreceptor connecting cilium. J Cell Biol 105: 2973–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ (2010) A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329: 436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Rasband MN (2018) Axon initial segments: structure, function, and disease. Ann N Y Acad Sci 1420: 46–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN et al (2011) TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet 89: 713–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel LA (1983) Freeze‐fracture analysis of membrane events during early neogenesis of cilia in Tetrahymena: changes in fairy‐ring morphology and membrane topography. J Cell Sci 60: 137–156 [DOI] [PubMed] [Google Scholar]
- Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, Seo S (2012) ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A 109: 19691–19696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T (2013) 3D structure of eukaryotic flagella/cilia by cryo‐electron tomography. Biophysics 9: 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF (2011) Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol 12: 222–234 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Thompson J, Yates JR, Marshall WF (2012) Proteomic analysis of mammalian primary cilia. Curr Biol: 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PK, Attardi LD (2016) p73 and FoxJ1: programming multiciliated epithelia. Trends Cell Biol 26: 239–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN et al (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41: 1027–1031 [DOI] [PubMed] [Google Scholar]
- Jana SC, Mendonça S, Machado P, Werner S, Rocha J, Pereira A, Maiato H, Bettencourt‐Dias M (2018) Differential regulation of transition zone and centriole proteins contributes to ciliary base diversity. Nat Cell Biol 20: 928–941 [DOI] [PubMed] [Google Scholar]
- Jauregui AR, Barr MM (2005) Functional characterization of the C. elegans nephrocystins NPHP‐1 and NPHP‐4 and their role in cilia and male sensory behaviors. Exp Cell Res 305: 333–342 [DOI] [PubMed] [Google Scholar]
- Jean F, Pilgrim D (2017) Coordinating the uncoordinated: UNC119 trafficking in cilia. Eur J Cell Biol 96: 643–652 [DOI] [PubMed] [Google Scholar]
- Jenal U, Reinders A, Lori C (2017) Cyclic di‐GMP: second messenger extraordinaire. Nat Rev Microbiol 15: 271–284 [DOI] [PubMed] [Google Scholar]
- Jensen VL, Leroux MR (2017) Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr Opin Cell Biol 47: 83–91 [DOI] [PubMed] [Google Scholar]
- Jensen VL, Bialas NJ, Bishop‐Hurley SL, Molday LL, Kida K, Nguyen PA, Blacque OE, Molday RS, Leroux MR, Riddle DL (2010) Localization of a guanylyl cyclase to chemosensory cilia requires the novel ciliary MYND domain protein DAF‐25. PLoS Genet 6: e1001199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen VL, Li C, Bowie RV, Clarke L, Mohan S, Blacque OE, Leroux MR (2015) Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J 34: 2537–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen VL, Carter S, Sanders AA, Li C, Kennedy J, Timbers TA, Cai J, Scheidel N, Kennedy BN, Morin RD et al (2016) Whole‐organism developmental expression profiling identifies RAB‐28 as a novel ciliary GTPase associated with the BBSome and intraflagellar transport. PLoS Genet 12: e1006469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen VL, Lambacher NJ, Li C, Mohan S, Williams CL, Inglis PN, Yoder BK, Blacque OE, Leroux MR (2018) Role for intraflagellar transport in building a functional transition zone. EMBO Rep 19: e45862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Leroux MR (2010) cAMP and cGMP signaling: sensory systems with prokaryotic roots adopted by eukaryotic cilia. Trends Cell Biol 20: 435–444 [DOI] [PubMed] [Google Scholar]
- Johnson CA, Malicki JJ (2019) The nuclear arsenal of cilia. Dev Cell 49: 161–170 [DOI] [PubMed] [Google Scholar]
- Jordan MA, Pigino G (2021) The structural basis of intraflagellar transport at a glance. J Cell Sci 134: jcs247163 [DOI] [PubMed] [Google Scholar]
- Jung E, Choi TI, Lee JE, Kim CH, Kim J (2020) ESCRT subunit CHMP4B localizes to primary cilia and is required for the structural integrity of the ciliary membrane. FASEB J 34: 1331–1344 [DOI] [PubMed] [Google Scholar]
- Karunakaran KB, Chaparala S, Lo CW, Ganapathiraju MK (2020) Cilia interactome with predicted protein‐protein interactions reveals connections to Alzheimer's disease, aging and other neuropsychiatric processes. Sci Rep 10: 15629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee HL, Dishinger JF, Lynne Blasius T, Liu CJ, Margolis B, Verhey KJ (2012) A size‐exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol 14: 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten F, van Wijk E, van Reeuwijk J, van der Zwaag B, Märker T, Peters TA, Katsanis N, Wolfrum U, Keunen JE, Roepman R et al (2009) Whirlin associates with the Cav1.3 ({alpha}1D) channels in photoreceptors, defining a novel member of the Usher protein network. Invest Ophthalmol Vis Sci 51: 2338–2346 [DOI] [PubMed] [Google Scholar]
- Khalifa AAZ, Ichikawa M, Dai D, Kubo S, Black CS, Peri K, McAlear TS, Veyron S, Yang SK, Vargas J et al (2020) The inner junction complex of the cilia is an interaction hub that involves tubulin post‐translational modifications. Elife 9: e52760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Scholey JM (2018) Assembly, functions and evolution of archaella, flagella and cilia. Curr Biol 28: R278–R292 [DOI] [PubMed] [Google Scholar]
- Khanna H (2015) Photoreceptor sensory cilium: traversing the ciliary gate. Cell 4: 674–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie‐Bittach T, Katsanis N et al (2010) Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 329: 1337–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Froese CD, Xie H, Trimble WS (2016) Immunofluorescent staining of septins in primary cilia. Methods Cell Biol 136: 269–283 [DOI] [PubMed] [Google Scholar]
- Kizhatil K, Baker SA, Arshavsky VY, Bennett V (2009) Ankyrin‐G promotes cyclic nucleotide‐gated channel transport to rod photoreceptor sensory cilia. Science 323: 1614–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Naggert JK, Nishina PM (2018) The impact of adherens and tight junctions on physiological function and pathological changes in the retina. Adv Exp Med Biol 1074: 545–551 [DOI] [PubMed] [Google Scholar]
- Kopinke D, Norris AM, Mukhopadhyay S (2020) Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin Cell Dev Biol 110: 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey SE, He S, Scheres SH, Carter AP (2019) Cryo‐EM of dynein microtubule‐binding domains shows how an axonemal dynein distorts the microtubule. Elife 8: e47145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Goetz JG, Dennis JW, Nabi IR (2009) Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol 185: 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambacher NJ, Bruel AL, van Dam TJ, Szymańska K, Slaats GG, Kuhns S, McManus GJ, Kennedy JE, Gaff K, Wu KM et al (2015) TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol 18: 122–131B137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapart JA, Gottardo M, Cortier E, Duteyrat JL, Augière C, Mangé A, Jerber J, Solassol J, Gopalakrishnan J, Thomas J et al (2019) Dzip1 and Fam92 form a ciliary transition zone complex with cell type specific roles in Drosophila . Elife 8: e49307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurencon A, Dubruille R, Efimenko E, Grenier G, Bissett R, Cortier E, Rolland V, Swoboda P, Durand B (2007) Identification of novel regulatory factor X (RFX) target genes by comparative genomics in Drosophila species. Genome Biol 8: R195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L, Ostrowski LE (2020) Motile cilia genetics and cell biology: big results from little mice. Cell Mol Life Sci 78: 769–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YL, Santé J, Comerci CJ, Cyge B, Menezes LF, Li FQ, Germino GG, Moerner WE, Takemaru K, Stearns T (2014) Cby1 promotes Ahi1 recruitment to a ring‐shaped domain at the centriole‐cilium interface and facilitates proper cilium formation and function. Mol Biol Cell 25: 2919–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz‐Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E et al (2008) Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet‐Biedl syndrome. Nat Genet 40: 443–448 [DOI] [PubMed] [Google Scholar]
- Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May‐Simera H, Li H, Blacque OE, Li L, Leitch CC et al (2004) Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 117: 541–552 [DOI] [PubMed] [Google Scholar]
- Li Y, Calvo SE, Gutman R, Liu JS, Mootha VK (2014) Expansion of biological pathways based on evolutionary inference. Cell 158: 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Jensen VL, Park K, Kennedy J, Garcia‐Gonzalo FR, Romani M, De Mori R, Bruel AL, Gaillard D, Doray B et al (2016a) MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLoS Biol 14: e1002416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Chen X, Fisher C, Siller SS, Zelikman K, Kuriyama R, Takemaru KI (2016b) BAR domain‐containing FAM92 proteins interact with Chibby1 to facilitate ciliogenesis. Mol Cell Biol 36: 2668–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li M, Ou G (2020) COVID‐19, cilia, and smell. FEBS J 287: 3672–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienkamp S, Ganner A, Walz G (2012) Inversin, Wnt signaling and primary cilia. Differentiation 83: S49–S55 [DOI] [PubMed] [Google Scholar]
- Lin YC, Niewiadomski P, Lin B, Nakamura H, Phua SC, Jiao J, Levchenko A, Inoue T, Rohatgi R, Inoue T (2013) Chemically inducible diffusion trap at cilia reveals molecular sieve‐like barrier. Nat Chem Biol 9: 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Guo S, Dutcher SK (2018) RPGRIP1L helps to establish the ciliary gate for entry of proteins. J Cell Sci 131: jcs220905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS (1999) Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci 19: 6267–6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Tan G, Levenkova N, Li T, Pugh ENJ, Rux JJ, Speicher DW, Pierce EA (2007) The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics 6: 1299–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobasso S, Lopalco P, Angelini R, Baronio M, Fanizzi FP, Babudri F, Corcelli A (2010) Lipidomic analysis of porcine olfactory epithelial membranes and cilia. Lipids 45: 593–602 [DOI] [PubMed] [Google Scholar]
- Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS et al (2015) Early steps in primary cilium assembly require EHD1/EHD3‐dependent ciliary vesicle formation. Nat Cell Biol 17: 228–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N, West C, Murga‐Zamalloa C, Sun L, Anderson RM, Wells C, Weinreb RN, Travers JB, Khanna H, Sun Y (2012) OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet 21: 3333–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N, Kumar A, Conwell M, Weinreb RN, Anderson R, Sun Y (2013) Compensatory role of inositol 5‐phosphatase INPP5B to OCRL in primary cilia formation in oculocerebrorenal syndrome of lowe. PLoS One 8: e66727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Stoyanova M, Rademacher G, Dutcher SK, Brown A, Zhang R (2019) Structure of the decorated ciliary doublet microtubule. Cell 179: 909–922.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicki JJ, Johnson CA (2017) The Cilium: cellular antenna and central processing unit. Trends Cell Biol 27: 126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manojlovic Z, Earwood R, Kato A, Stefanovic B, Kato Y (2014) RFX7 is required for the formation of cilia in the neural tube. Mech Dev 132: 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez J, Bhattacharya D, Lusk CP, Khokha MK (2020) Nucleoporin NUP205 plays a critical role in cilia and congenital disease. Dev Biol 469: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Qin H, Rodrigo Brenni M, Rosenbaum JL (2005) Flagellar length control system: testing a simple model based on intraflagellar transport and turnover. Mol Biol Cell 16: 270–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Outschoorn UE, Sotgia F, Lisanti MP (2015) Caveolae and signalling in cancer. Nat Rev Cancer 15: 225–237 [DOI] [PubMed] [Google Scholar]
- Mayer U, Kuller A, Daiber PC, Neudorf I, Warnken U, Schnolzer M, Frings S, Mohrlen F (2009) The proteome of rat olfactory sensory cilia. Proteomics 9: 322–334 [DOI] [PubMed] [Google Scholar]
- McInally SG, Kondev J, Dawson SC (2019) Length‐dependent disassembly maintains four different flagellar lengths in Giardia. Elife 8: e48694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E (2015) Membrane curvature at a glance. J Cell Sci 128: 1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menco M (1980) Qualitative and quantitative freeze‐fracture studies on olfactory and respiratory epithelial surfaces of frog, ox, rat, and dog. IV. Ciliogenesis and ciliary necklaces (including high‐voltage observations). Cell Tissue Res 212: 1–16 [DOI] [PubMed] [Google Scholar]
- Mitchell DR (2017) Evolution of Cilia. Cold Spring Harb Perspect Biol 9: a028290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison HM, Valente EM (2017) Motile and non‐motile cilia in human pathology: from function to phenotypes. J Pathol 241: 294–309 [DOI] [PubMed] [Google Scholar]
- Mojarad BA, Gupta GD, Hasegan M, Goudiam O, Basto R, Gingras AC, Pelletier L (2017) CEP19 cooperates with FOP and CEP350 to drive early steps in the ciliogenesis programme. Open Biol 7: 170114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R (1979) Inhomogeneous distribution of filipin‐sterol complexes in the ciliary membrane of rat tracheal epithelium. Am J Anat 156: 139–145 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK (2010) TULP3 bridges the IFT‐A complex and membrane phosphoinositides to promote trafficking of G protein‐coupled receptors into primary cilia. Genes Dev 24: 2180–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Badgandi HB, Hwang SH, Somatilaka B, Shimada IS, Pal K (2017) Trafficking to the primary cilium membrane. Mol Biol Cell 28: 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrave A, de Wildt P, van Etten I, Pijst H, Scholma C, Kooyman R, Homan W, van den Ende H (1986) Evidence for a functional membrane barrier in the transition zone between the flagellum and cell body of Chlamydomonas eugametos gametes. Planta 167: 544–553 [DOI] [PubMed] [Google Scholar]
- Nachury MV (2018) The molecular machines that traffic signaling receptors into and out of cilia. Curr Opin Cell Biol 51: 124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Mick DU (2019) Establishing and regulating the composition of cilia for signal transduction. Nat Rev Mol Cell Biol 20: 389–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Seeley ES, Jin H (2010) Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol 26: 59–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K, Kozuka‐Hata H, Nonami Y, Ao‐Kondo H, Suzuki T, Nakamura H, Yamakawa K, Oyama M, Inoue T, Takeda S (2012) Proteomic analysis of multiple primary cilia reveals a novel mode of ciliary development in mammals. Biol Open 1: 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechipurenko IV (2020) The enigmatic role of lipids in cilia signaling. Front Cell Dev Biol 8: 777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PA, Liou W, Hall DH, Leroux MR (2014) Ciliopathy proteins establish a bipartite signaling compartment in a C. elegans thermosensory neuron. J Cell Sci 127: 5317–5330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning K, Song E, Sendayen BE, Prosseda PP, Chang KC, Ghaffarieh A, Alvarado JA, Wang B, Haider KM, Berbari NF et al (2021) Defective INPP5E distribution in NPHP1‐related Senior‐Loken syndrome. Mol Genet Genomic Med 9: e1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir I, Papermaster DS (1983) Differential distribution of opsin in the plasma membrane of frog photoreceptors: an immunocytochemical study. Invest Ophthalmol Vis Sci 24: 868–878 [PubMed] [Google Scholar]
- Oh Y, Bi E (2011) Septin structure and function in yeast and beyond. Trends Cell Biol 21: 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki M, Kobayashi T, Chiba S, Takei R, Liang L, Nakayama K, Katoh Y (2020) Formation of the B9‐domain protein complex MKS1‐B9D2‐B9D1 is essential as a diffusion barrier for ciliary membrane proteins. Mol Biol Cell 31: 2259–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski LE, Blackburn K, Radde KM, Moyer MB, Schlatzer DM, Moseley A, Boucher RC (2002) A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics 1: 451–465 [DOI] [PubMed] [Google Scholar]
- Oswald F, Prevo B, Acar S, Peterman EJG (2018) Interplay between ciliary ultrastructure and IFT‐train dynamics revealed by single‐molecule super‐resolution imaging. Cell Rep 25: 224–235 [DOI] [PubMed] [Google Scholar]
- Ounjai P, Kim KD, Liu H, Dong M, Tauscher AN, Witkowska HE, Downing KH (2013) Architectural insights into a ciliary partition. Curr Biol 23: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palander O, El‐Zeiry M, Trimble WS (2017) Uncovering the roles of septins in cilia. Front Cell Dev Biol 5: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA (2019) The molecular genetics of Joubert syndrome and related ciliopathies: the challenges of genetic and phenotypic heterogeneity. Transl Sci Rare Dis 4: 25–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Leroux MR (2022) IFT trains overcome an NPHP module barrier at the transition zone. J Cell Biol 221: e202112015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Li C, Tsiropoulou S, Gonçalves J, Kondratev C, Pelletier L, Blacque OE, Leroux MR (2021) CDKL kinase regulates the length of the ciliary proximal segment. Curr Biol 31: 2359–2373.e7 [DOI] [PubMed] [Google Scholar]
- Parker JD, Hilton LK, Diener DR, Rasi MQ, Mahjoub MR, Rosenbaum JL, Quarmby LM (2010) Centrioles are freed from cilia by severing prior to mitosis. Cytoskeleton 67: 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil H, Tserentsoodol N, Saha A, Hao Y, Webb M, Ferreira PA (2012) Selective loss of RPGRIP1‐dependent ciliary targeting of NPHP4, RPGR and SDCCAG8 underlies the degeneration of photoreceptor neurons. Cell Death Dis 3: e355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzke S, Redick S, Warsame A, Murga‐Zamalloa CA, Khanna H, Doxsey S, Stokke T (2010) CSPP is a ciliary protein interacting with Nephrocystin 8 and required for cilia formation. Mol Biol Cell 21: 2555–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 151: 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB (2005) Proteomic analysis of a eukaryotic cilium. J Cell Biol 170: 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins LA, Hedgecock EM, Thomson JN, Culotti JG (1986) Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev Biol 117: 456–487 [DOI] [PubMed] [Google Scholar]
- Phirke P, Efimenko E, Mohan S, Burghoorn J, Crona F, Bakhoum MW, Trieb M, Schuske K, Jorgensen EM, Piasecki BP et al (2011) Transcriptional profiling of C. elegans DAF‐19 uncovers a ciliary base‐associated protein and a CDK/CCRK/LF2p‐related kinase required for intraflagellar transport. Dev Biol 357: 235–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Huang B, Luck DJ (1977) Two‐dimensional analysis of flagellar proteins from wild‐type and paralyzed mutants of Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A 74: 1600–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poghosyan E, Iacovache I, Faltova L, Leitner A, Yang P, Diener DR, Aebersold R, Zuber B, Ishikawa T (2020) The structure and symmetry of the radial spoke protein complex in Chlamydomonas flagella. J Cell Sci 133: jcs245233 [DOI] [PubMed] [Google Scholar]
- Pratt MB, Titlow JS, Davis I, Barker AR, Dawe HR, Raff JW, Roque H (2016) Drosophila sensory cilia lacking MKS‐proteins exhibit striking defects in development but only subtle defects in adults. J Cell Sci 129: 3732–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevo B, Mangeol P, Oswald F, Scholey JM, Peterman EJ (2015) Functional differentiation of cooperating kinesin‐2 motors orchestrates cargo import and transport in C. elegans cilia. Nat Cell Biol 17: 1536–1545 [DOI] [PubMed] [Google Scholar]
- Quarmby LM (2004) Cellular deflagellation. Int Rev Cytol 233: 47–91 [DOI] [PubMed] [Google Scholar]
- Quax TEF, Albers SV, Pfeiffer F (2018) Taxis in archaea. Emerg Top Life Sci 2: 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KN, Li L, Anand M, Khanna H (2015) Ablation of retinal ciliopathy protein RPGR results in altered photoreceptor ciliary composition. Sci Rep 5: 11137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KN, Zhang W, Li L, Anand M, Khanna H (2016) Prenylated retinal ciliopathy protein RPGR interacts with PDE6δ and regulates ciliary localization of Joubert syndrome‐associated protein INPP5E. Hum Mol Genet 25: 4533–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TS (1965) Olfactory cilia in the frog. J Cell Biol 25: 209–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Leroux MR (2017) Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol 18: 533–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Blacque OE, Leroux MR (2012) The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep 13: 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remans K, Bürger M, Vetter IR, Wittinghofer A (2014) C2 domains as protein‐protein interaction modules in the ciliary transition zone. Cell Rep 8: 1–9 [DOI] [PubMed] [Google Scholar]
- Renaud FL, Swift H (1964) The development of basal bodies and flagella in Allomyces arbusculus . J Cell Biol 23: 339–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringo DL (1967) Flagellar motion and fine structure of the flagellar apparatus in Chlamydomonas. J Cell Biol 33: 543–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson EC, Dowdle WE, Ozanturk A, Garcia‐Gonzalo FR, Li C, Halbritter J, Elkhartoufi N, Porath JD, Cope H, Ashley‐Koch A et al (2015) TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol 209: 129–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux MA, Potter VL, Zhang Z, He F, Liu J, Schmid MF, Wensel TG (2019) Defining the layers of a sensory cilium with STORM and cryoelectron nanoscopy. Proc Natl Acad Sci U S A 116: 23562–23572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman R, Wolfrum U (2007) Protein networks and complexes in photoreceptor cilia. Subcell Biochem 43: 209–235 [DOI] [PubMed] [Google Scholar]
- Roepman R, Letteboer SJ, Arts HH, van Beersum SE, Lu X, Krieger E, Ferreira PA, Cremers FP (2005) Interaction of nephrocystin‐4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis‐associated mutations. Proc Natl Acad Sci U S A 102: 18520–18525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Snell WJ (2010) The ciliary membrane. Curr Opin Cell Biol 22: 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlich P (1975) The sensory cilium of retinal rods is analogous to the transitional zone of motile cilia. Cell Tissue Res 161: 421–430 [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3: 813–825 [DOI] [PubMed] [Google Scholar]
- Roy K, Marin EP (2019) Lipid modifications in cilia biology. J Clin Med 8: 921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahabandu N, Kong D, Magidson V, Nanjundappa R, Sullenberger C, Mahjoub MR, Loncarek J (2019) Expansion microscopy for the analysis of centrioles and cilia. J Microsc 276: 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saita S, Shirane M, Ishitani T, Shimizu N, Nakayama KI (2014) Role of the ANKMY2‐FKBP38 axis in regulation of the Sonic hedgehog (Shh) signaling pathway. J Biol Chem 289: 25639–25654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M et al (2011) Mapping the NPHP‐JBTS‐MKS protein network reveals ciliopathy disease genes and pathways. Cell 145: 513–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P, Christensen ST (2007) Overview of structure and function of mammalian cilia. Annu Rev Physiol 69: 377–400 [DOI] [PubMed] [Google Scholar]
- Scheidel N, Blacque OE (2018) Intraflagellar transport complex A genes differentially regulate cilium formation and transition zone gating. Curr Biol 28: 3279–3287.e2 [DOI] [PubMed] [Google Scholar]
- Schmidt HB, Görlich D (2016) Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem Sci 41: 46–61 [DOI] [PubMed] [Google Scholar]
- Schmidt KN, Kuhns S, Neuner A, Hub B, Zentgraf H, Pereira G (2012) Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J Cell Biol 199: 1083–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schou KB, Mogensen JB, Morthorst SK, Nielsen BS, Aleliunaite A, Serra‐Marques A, Fürstenberg N, Saunier S, Bizet AA, Veland IR et al (2017) KIF13B establishes a CAV1‐enriched microdomain at the ciliary transition zone to promote Sonic hedgehog signalling. Nat Commun 8: 14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouteden C, Serwas D, Palfy M, Dammermann A (2015) The ciliary transition zone functions in cell adhesion but is dispensable for axoneme assembly in C. elegans . J Cell Biol 210: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serricchio M, Schmid AW, Steinmann ME, Sigel E, Rauch M, Julkowska D, Bonnefoy S, Fort C, Bastin P, Bütikofer P (2015) Flagellar membranes are rich in raft‐forming phospholipids. Biol Open 4: 1143–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Garcia G, Van De Weghe JC, McGorty R, Pazour GJ, Doherty D, Huang B, Reiter JF (2017) Super‐resolution microscopy reveals that disruption of ciliary transition‐zone architecture causes Joubert syndrome. Nat Cell Biol 19: 1178–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba D, Yamaoka Y, Hagiwara H, Takamatsu T, Hamada H, Yokoyama T (2009) Localization of Inv in a distinctive intraciliary compartment requires the C‐terminal ninein‐homolog‐containing region. J Cell Sci 122: 44–54 [DOI] [PubMed] [Google Scholar]
- Shiba D, Manning DK, Koga H, Beier DR, Yokoyama T (2010) Inv acts as a molecular anchor for Nphp3 and Nek8 in the proximal segment of primary cilia. Cytoskeleton 67: 112–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigg MA, Menchen T, Lee C, Johnson J, Jungnickel MK, Choksi SP, Garcia G 3rd, Busengdal H, Dougherty GW, Pennekamp P et al (2017) Evolutionary proteomics uncovers ancient associations of cilia with signaling pathways. Dev Cell 43: 744–762.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman MA, Leroux MR (2009) Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol 19: 306–316 [DOI] [PubMed] [Google Scholar]
- Sim HJ, Yun S, Kim HE, Kwon KY, Kim GH, Yun S, Kim BG, Myung K, Park TJ, Kwon T (2020) Simple method to characterize the ciliary proteome of multiciliated cells. J Proteome Res 19: 391–400 [DOI] [PubMed] [Google Scholar]
- SJOSTRAND FS (1953) The ultrastructure of the innersegments of the retinal rods of the guinea pig eye as revealed by electron microscopy. J Cell Comp Physiol 42: 45–70 [DOI] [PubMed] [Google Scholar]
- Slaats GG, Isabella CR, Kroes HY, Dempsey JC, Gremmels H, Monroe GR, Phelps IG, Duran KJ, Adkins J, Kumar SA et al (2016) MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J Med Genet 53: 62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Northey JG, Garg J, Pearlman RE, Siu KW (2005) Robust method for proteome analysis by MS/MS using an entire translated genome: demonstration on the ciliome of Tetrahymena thermophila. J Proteome Res 4: 909–919 [DOI] [PubMed] [Google Scholar]
- Somatilaka BN, Hwang SH, Palicharla VR, White KA, Badgandi H, Shelton JM, Mukhopadhyay S (2020) Ankmy2 prevents smoothened‐independent hyperactivation of the hedgehog pathway via cilia‐regulated adenylyl cyclase signaling. Dev Cell 54: 710–726.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnino S, Prinetti A (2010) Lipids and membrane lateral organization. Front Physiol 1: 153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin S (1962) Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol 15: 363–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer M, Detwiler PB, Bunt‐Milam AH (1988) Distribution of membrane proteins in mechanically dissociated retinal rods. Invest Ophthalmol Vis Sci 29: 1012–1020 [PubMed] [Google Scholar]
- Sreekumar V, Norris DP (2019) Cilia and development. Curr Opin Genet Dev 56: 15–21 [DOI] [PubMed] [Google Scholar]
- Stephan R, Goellner B, Moreno E, Frank CA, Hugenschmidt T, Genoud C, Aberle H, Pielage J (2015) Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev Cell 33: 5–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen LA, Ismail S (2016) Shuttling and sorting lipid‐modified cargo into the cilia. Biochem Soc Trans 44: 1273–1280 [DOI] [PubMed] [Google Scholar]
- Stolc V, Samanta MP, Tongprasit W, Marshall WF (2005) Genome‐wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc Natl Acad Sci U S A 102: 3703–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subota I, Julkowska D, Vincensini L, Reeg N, Buisson J, Blisnick T, Huet D, Perrot S, Santi‐Rocca J, Duchateau M et al (2014) Proteomic analysis of intact flagella of procyclic Trypanosoma brucei cells identifies novel flagellar proteins with unique sub‐localization and dynamics. Mol Cell Proteomics 13: 1769–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Haley J, Bulgakov OV, Cai X, McGinnis J, Li T (2012) Tubby is required for trafficking G protein‐coupled receptors to neuronal cilia. Cilia 1: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Leroux MR (2013) The roles of evolutionarily conserved functional modules in cilia‐related trafficking. Nat Cell Biol 15: 1387–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda P, Adler HT, Thomas JH (2000) The RFX‐type transcription factor DAF‐19 regulates sensory neuron cilium formation in C. elegans. Mol Cell 5: 411–421 [DOI] [PubMed] [Google Scholar]
- Takao D, Verhey KJ (2016) Gated entry into the ciliary compartment. Cell Mol Life Sci 73: 119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao D, Dishinger JF, Kee HL, Pinskey JM, Allen BL, Verhey KJ (2014) An assay for clogging the ciliary pore complex distinguishes mechanisms of cytosolic and membrane protein entry. Curr Biol 24: 2288–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao D, Wang L, Boss A, Verhey KJ (2017) Protein interaction analysis provides a map of the spatial and temporal organization of the ciliary gating zone. Curr Biol 27: 2296–2306.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam LW, Ranum PT, Lefebvre PA (2013) CDKL5 regulates flagellar length and localizes to the base of the flagella in Chlamydomonas. Mol Biol Cell 24: 588–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammachote R, Hommerding CJ, Sinders RM, Miller CA, Czarnecki PG, Leightner AC, Salisbury JL, Ward CJ, Torres VE, Gattone VH 2nd et al (2009) Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum Mol Genet 18: 3311–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MF (2013) Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev 27: 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J, Morle L, Soulavie F, Laurencon A, Sagnol S, Durand B (2010) Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell 102: 499–513 [DOI] [PubMed] [Google Scholar]
- Tokuyasu K, Yamada E (1959) The fine structure of the retina studied with the electron microscope. IV. Morphogenesis of outer segments of retinal rods. J Biophys Biochem Cytol 6: 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure A, Rode B, Hunnicutt GR, Escalier D, Gacon G (2011) Septins at the annulus of mammalian sperm. Biol Chem 392: 799–803 [DOI] [PubMed] [Google Scholar]
- Trépout S, Tassin AM, Marco S, Bastin P (2018) STEM tomography analysis of the trypanosome transition zone. J Struct Biol 202: 51–60 [DOI] [PubMed] [Google Scholar]
- Trimble WS, Grinstein S (2015) Barriers to the free diffusion of proteins and lipids in the plasma membrane. J Cell Biol 208: 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu HQ, Qin XH, Liu ZB, Song ZQ, Hu HB, Zhang YC, Chang Y, Wu M, Huang Y, Bai YF et al (2018) Microtubule asters anchored by FSD1 control axoneme assembly and ciliogenesis. Nat Commun 9: 5277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Rosti RO, Gibbs E, Gleeson JG (2014) Primary cilia in neurodevelopmental disorders. Nat Rev Neurol 10: 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Yang W (2016) Permeability barriers for generating a unique ciliary protein and lipid composition. Curr Opin Cell Biol 41: 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WL, Simons K (2006) FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin‐Darby canine kidney (MDCK) cells. Proc Natl Acad Sci U S A 103: 18556–18561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuolo L, Stevenson NL, Heesom KJ, Stephens DJ (2018) Dynein‐2 intermediate chains play crucial but distinct roles in primary cilia formation and function. Elife 7: e39655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallmeier J, Nielsen KG, Kuehni CE, Lucas JS, Leigh MW, Zariwala MA, Omran H (2020) Motile ciliopathies. Nat Rev Dis Primers 6: 77 [DOI] [PubMed] [Google Scholar]
- Wang L, Dynlacht BD (2018) The regulation of cilium assembly and disassembly in development and disease. Development 145: dev151407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, den Hollander AI, Moayedi Y, Abulimiti A, Li Y, Collin RW, Hoyng CB, Lopez I, Abboud EB, Al‐Rajhi AA et al (2009) Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet 84: 380–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li J, Takemaru KI, Jiang X, Xu G, Wang B (2018a) Centrosomal protein Dzip1l binds Cby, promotes ciliary bud formation, and acts redundantly with Bromi to regulate ciliogenesis in the mouse. Development 145: dev164236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Taschner M, Ganzinger KA, Kelley C, Villasenor A, Heymann M, Schwille P, Lorentzen E, Mizuno N (2018b) Membrane association and remodeling by intraflagellar transport protein IFT172. Nat Commun 9: 4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu C, Yang B, Zhang H, Jiao J, Zhang R, Liu S, Xiao S, Chen Y, Liu B et al (2022) SARS‐CoV‐2 ORF10 impairs cilia by enhancing CUL2ZYG11B activity. J Cell Biol 221: e202108015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton‐Pitt SR, Jauregui AR, Li C, Wang J, Leroux MR, Barr MM (2012) Ciliogenesis in Caenorhabditis elegans requires genetic interactions between ciliary middle segment localized NPHP‐2 (inversin) and transition zone‐associated proteins. J Cell Sci 125: 2592–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts KJ, Vaknin A, Fuqua C, Kazmierczak BI (2019) New twists and turns in bacterial locomotion and signal transduction. J Bacteriol 201: e00439‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watzlich D, Vetter I, Gotthardt K, Miertzschke M, Chen YX, Wittinghofer A, Ismail S (2013) The interplay between RPGR, PDEdelta and Arl2/3 regulate the ciliary targeting of farnesylated cargo. EMBO Rep 14: 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S, Mukhopadhyay AG, Roberts AJ (2020) Intraflagellar transport trains and motors: insights from structure. Semin Cell Dev Biol 107: 82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Xu Q, Zhang Y, Li Y, Zhang Q, Hu Z, Harris PC, Torres VE, Ling K, Hu J (2013) Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nat Commun 4: 2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Ling K, Hu J (2015) The essential roles of transition fibers in the context of cilia. Curr Opin Cell Biol 35: 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RL, Goodenough DA, Goodenough UW (1977) Membrane particle arrays associated with the basal body and with contractile vacuole secretion in Chlamydomonas. J Cell Biol 72: 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G, Nazlamova L, Hancock JT (2018) Signaling through the primary cilium. Front Cell Dev Biol 6: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Gull K (2011) The evolution of the cytoskeleton. J Cell Biol 194: 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegering A, Dildrop R, Kalfhues L, Spychala A, Kuschel S, Lier JM, Zobel T, Dahmen S, Leu T, Struchtrup A et al (2018) Cell type‐specific regulation of ciliary transition zone assembly in vertebrates. EMBO J 37: e97791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegering A, Dildrop R, Vesque C, Khanna H, Schneider‐Maunoury S, Gerhardt C (2021) Rpgrip1l controls ciliary gating by ensuring the proper amount of Cep290 at the vertebrate transition zone. Mol Biol Cell 32: 675–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Winkelbauer ME, Schafer JC, Michaud EJ, Yoder BK (2008) Functional redundancy of the B9 proteins and nephrocystins in C. elegans ciliogenesis. Mol Biol Cell 19: 2154–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE et al (2011) MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol 192: 1023–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield JL, Lechtreck KF, Lorentzen E (2018) Trafficking of ciliary membrane proteins by the intraflagellar transport/BBSome machinery. Essays Biochem 62: 753–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelbauer ME, Schafer JC, Haycraft CJ, Swoboda P, Yoder BK (2005) The C. elegans homologs of nephrocystin‐1 and nephrocystin‐4 are cilia transition zone proteins involved in chemosensory perception. J Cell Sci 118: 5575–5587 [DOI] [PubMed] [Google Scholar]
- Wu Z, Pang N, Zhang Y, Chen H, Peng Y, Fu J, Wei Q (2020) CEP290 is essential for the initiation of ciliary transition zone assembly. PLoS Biol 18: e3001034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakulov TA, Yasunaga T, Ramachandran H, Engel C, Müller B, Hoff S, Dengjel J, Lienkamp SS, Walz G (2015) Anks3 interacts with nephronophthisis proteins and is required for normal renal development. Kidney Int 87: 1191–1200 [DOI] [PubMed] [Google Scholar]
- Yang TT, Su J, Wang WJ, Craige B, Witman GB, Tsou MF, Liao JC (2015) Superresolution pattern recognition reveals the architectural map of the ciliary transition zone. Sci Rep 5: 14096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TT, Chong WM, Wang WJ, Mazo G, Tanos B, Chen Z, Tran TMN, Chen YD, Weng RR, Huang CE et al (2018) Super‐resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat Commun 9: 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TT, Tran MNT, Chong WM, Huang CE, Liao JC (2019) Single‐particle tracking localization microscopy reveals nonaxonemal dynamics of intraflagellar transport proteins at the base of mammalian primary cilia. Mol Biol Cell 30: 828–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Rajendran A, Valentine MS, Saha M, Ballif BA, Van Houten JL (2013) Proteomic analysis of the cilia membrane of Paramecium tetraurelia . J Proteomics 78: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Breslow DK, Koslover EF, Spakowitz AJ, Nelson WJ, Nachury MV (2013) Single molecule imaging reveals a major role for diffusion in the exploration of ciliary space by signaling receptors. Elife 2: e00654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Nager AR, Nachury MV (2018) BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J Cell Biol 217: 1847–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee LE, Garcia‐Gonzalo FR, Bowie RV, Li C, Kennedy JK, Ashrafi K, Blacque OE, Leroux MR, Reiter JF (2015) Conserved genetic interactions between ciliopathy complexes cooperatively support ciliogenesis and ciliary signaling. PLoS Genet 11: e1005627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder BK (2008) Ciliary function in mammalian development. Preface. Curr Top Dev Biol 85: xv–xix [DOI] [PubMed] [Google Scholar]
- Zhang D, Aravind L (2012) Novel transglutaminase‐like peptidase and C2 domains elucidate the structure, biogenesis and evolution of the ciliary compartment. Cell Cycle 11: 3861–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhuang T, Lin Q, Yang B, Xu X, Xin G, Zhu S, Wang G, Yu B, Zhang T et al (2019) Patched1‐ArhGAP36‐PKA‐Inversin axis determines the ciliary translocation of Smoothened for Sonic Hedgehog pathway activation. Proc Natl Acad Sci U S A 116: 874–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Malicki J (2011) Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J 30: 2532–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table EV1