

Abstract
Background
Mammary carcinogenesis possesses great challenges due to the lack of effectiveness of the multiple therapeutic options available. Gene therapy-based cancer treatment strategy provides more targeting accuracy, fewer side effects, and higher therapeutic efficiency. Downregulation of the oncogene mTOR by mTOR-siRNA is an encouraging approach to reduce cancer progression. However, its employment as means of therapeutic strategy has been restricted due to the unavailability of a suitable delivery system.
Methods
A suitable nanocarrier system made up of 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) has been developed to prevent degradation and for proficient delivery of siRNA. This was followed by in vitro and in vivo anti-breast cancer efficiency analysis of the mTOR siRNA-loaded neutral liposomal formulation (NL-mTOR-siRNA).
Results
In our experiment, a profound reduction in MCF-7 cell growth, proliferation and invasion was ascertained following extensive downregulation of mTOR expression. NL-mTOR-siRNA suppressed tumour growth and restored morphological alterations of DMBA-induced breast cancer. In addition, neutral liposome enhanced accumulation of siRNA in mammary cancer tissues facilitating its deep cytosolic distribution within the tumour, which allows apoptosis thereby facilitating its anti-tumour potential.
Conclusion
Hence, the current study highlighted the augmented ground for therapies aiming toward cancerous cells to diminish mTOR expression by RNAi in managing mammary carcinoma.
Subject terms: Breast cancer, Drug delivery
Overview of anti-cancer effect triggered by mTOR-siRNA-loaded neutral liposomal nano-formulation (NL-mTOR-siRNA) against DMBA-induced mammary carcinogenesis in Sprague Dawley rats.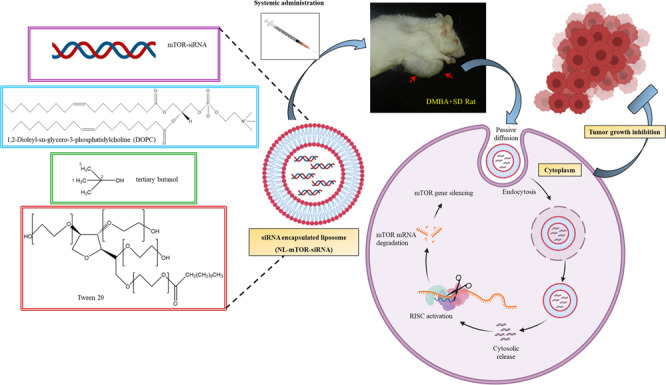
Background
Mammary carcinogenesis with 2.26 million newly diagnosed cases in 2020 [1] has become a serious illness in women and attract the focus of biomedical researchers. The latest report remains worrisome as the number of fresh invasive breast carcinoma touches 276,480 in the USA [2] while in India it stands at 712,758 [3]. Resistance to available breast cancer therapies consisting of chemo-, hormonal- and radiation therapy along with metastasis is the prime reason for patient-related deaths. Henceforth, there is an urgent requirement to design a new therapeutic approach by targeting cell signalling pathways that are entangled in tumourigenesis of the mammary gland [4].
Mammalian target of rapamycin (mTOR), a serine/threonine kinase [5], is a central regulator of mRNA translation, gene transcription, protein synthesis, lipid synthesis, cell proliferation, cell differentiation, glucose metabolism, apoptosis and autophagy [6] and belongs to phosphatidylinositide 3-kinase-related kinase (PIKK) family [7]. Aberrant hyperactivation of the PI3K-AKT-mTOR cascade is reported to contribute to tumourigenesis, especially in breast, liver, prostate, gastric and colorectal cancer cells [8, 9]. Mutation of mTOR signalling in breast cancer is very common with nearly 70% of mammary tumour harbouring genetic alterations that cause mTOR hyperactivation [10]. Being a potent oncogene, its therapeutic targeting by FDA-approved drugs is found to be very beneficial for the management of ER and HER2-positive breast cancer. However, targeting these signalling has many obstacles including identification of a prognostic biomarker for tumour response, rational therapy design to surpass adaptive resistance, along with delivery method optimisation to reduce toxicity while still preventing the target gene [11]. This observation underscores the need for alternative molecular biomarker-guided therapeutic approaches for the management of breast cancer.
In recent times, RNAi technology is appeared to be an important aspect of molecular therapies for life-threatening diseases by silencing targeted genes in a sequence-specific manner [12]. Small interfering RNA (siRNA) is a kind of RNAi tool, the discovery of which in the 1990s offer major advancement in the field of nucleic-acid-based therapeutics [13]. It works on the principle of suppressing the expression of a target gene through DICER and RNA-induced silencing complex (RISC), resulting in mRNA breakdown and averting corresponding protein expression [6, 14]. The easy and economical method of synthesising [15, 16] along with its infrequent administration and self-administrable potential makes it an obvious choice over other nucleic acid therapies [17]. Remarkable evolution in siRNA therapy is apparent amidst approval of the first siRNA drug patisiran in 2018 by US FDA barely a decade after Andrew Z. Fire and Craig C. Mello were awarded with Nobel Prize in Physiology or Medicine that is followed by Givosiran and Lumasiran hitting the market [18]. This gives a strong signal that siRNA drugs are going to stay and opened new avenues with enormous capability for treating human diseases.
The discovery of RNA interference (RNAi) technology paved the path for nucleic acid molecules to be familiarised as next-generation therapeutic drugs. However, its translation into in vivo settings remains challenging due to the absence of a proper delivery technique [19–21]. Main hurdles in this regard include (A) susceptibility of siRNA degradation by serum nucleases present in blood or body fluids resulting in smaller half-life (nearly 7 min), (B) difficulty in crossing the cell membrane to reach the cytoplasm due to high molecular weight and negatively charged backbone limiting cellular internalisation, (C) rapid clearance by kidney through glomerular filtration leading to poor tumour penetration and (D) inadequacy in delivery to targeted cell in vivo [22].
Non-viral vectors comprising nanomaterials are emerging as a prominent carrier system. Liposomes are sphere particles constructed by adding one/more phospholipid bilayers to assist in the successful delivery of oligonucleotides, peptides and siRNA. These nanocarriers shield the biodegradable drug till it reaches its target cells, making it very attractive [23]. It does not directly affect cancer cells, but they passively congregate inside cancer tissues as it provides a greater penetrability and retention effect [16]. Therapeutic use of cationic liposome is constrained by its toxicity in human cells as a consequence of free radical production and lung inflammation [24] and anionic liposome has negligible silencing efficacy due to the absence of complexation enhancing electrostatic interactions of lipids-siRNA along with the limitation of macrophage uptake [25]. However, liposome prepared of neutral lipid was proved to be efficacious with a high rate of in vivo tumour reducing capability [26]. Therefore, in this research, we adopt a neutral nano-liposomal carrier to explore the impact of therapeutic targeting of the mTOR gene in suppressing mammary carcinogenesis in both in vitro and in vivo settings.
Materials and methods
Materials required
siRNA used for in vitro and in vivo studies was purchased in three formulations. A fluorescence (Alexa 555) labelled non-silencing siRNA sequence i.e., Alexa 555 tagged C-siRNA (Qiagen, Valencia, CA), with no equivalence to any recognised mRNA sequence as exhibited by BLAST search was employed to study uptake into MCF-7 cells and tissue distribution. To investigate in vitro and in vivo mTOR downregulation, mTOR-siRNA (ThermoFisher Scientific) targeting mTOR mRNA was purchased. A non-silencing siRNA construct without any tagging (siRNA sequence same as above), i.e. C-siRNA was employed as a control in mTOR-targeting experimentations.
Preparation and characterisation of DOPC-based neutral liposome with siRNA
1,2-Dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) was used to formulate neutral liposomal nanoparticles as NL (empty liposome), NL-C-siRNA (C-siRNA-loaded liposome) and NL-mTOR-siRNA (mTOR-siRNA-loaded liposome) as per previously described protocol [27] and stored as a lyophilised powder. Encapsulation efficiency (EE) reflecting loading of siRNA into nano-liposome was determined with spectrophotometry [27]. Lipid-siRNA powder was dispersed in 0.9% saline followed by 3 min of sonication to produce liposomes for in vivo administration at 15 µg/mL concentration to obtain the required dose for injection [27].
Nano-liposomes were characterised by particle size (PS), zeta potential (ZP) and polydispersity index (PDI) by employing Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25 °C in triplicate [28]. Morphological characterisation gives useful information about the relative structure, alignment of siRNA molecules and assembly of siRNA lipids. NL-C-siRNA was characterised using the field emission scanning electron microscope (FESEM) (Sigma 300, ZEISS, Carl Zeiss) [29], atomic force microscope (AFM) (NT-MDT, Russia; Solver Pro-4) [30] and transmission electron microscope (TEM) [31]. To analyse stability, alteration in PS, ZP and PDI was carried out for 4 weeks following storage at −20 °C, 4 °C and 25 °C [32]. Release of siRNA from NL-C-siRNA at different time intervals was conducted following a dialysis process in phosphate-buffered saline (PBS) of pH 5.5 or 7.4 using UV–vis spectrometry (U.V-1601; Shimadzu, Japan; at 260 nm) [29]. The percentage of siRNA release was calculated as: [(OD of siRNA in PBS/initial total content of siRNA)] × 100. Haemolysis assay based on red blood cell lysis was selected to estimate blood compatibility of sample in vitro. Haemolysis of NL-C-siRNA (50 mg/mL) was measured using a microplate reader (Biotek, USA) at 540 nm following the established method by taking Triton X-100 (0.5%) as positive control and PBS as negative control [33].
Cell lines, culture conditions and reagents
MCF-7 cells were acquired from National Centre for Cell Science Pune, India and preserved in RPMI-1640 (Gibco, USA) culture medium added with 10% foetal bovine serum (FBS), streptomycin (100 mg/mL) along with penicillin (100 U/mL) in a dampened environment with 5% CO2 at 37 °C along with 95% of both air and humidity. siRNA was transfected into MCF-7 cells as per earlier established methods where 1 mg of NL-C-siRNA or NL-mTOR-siRNA were transfected into each well [34].
In vitro cellular uptake of siRNA-loaded neutral liposome
Confocal microscopy was adopted for qualitative assessment of in vitro uptake of fluorescence-labelled C-siRNA liposomal formulation. Concisely, MCF-7 cells were cultured in confocal dishes at a density of 1 × 104 cells per well for 24 h. Thereafter, media was exchanged with fresh media comprising of RPMI with 10% foetal bovine serum. After 4 h of incubation with Alexa 555-labelled NL-C-siRNA and naked C-siRNA at 37 °C, cells were bathed thrice with PBS and subjected to 10 min of fixation with paraformaldehyde (4%). Finally, Hoechst was added and incubated for 10 min for staining nuclei before viewing under a confocal microscope (FLOWVIEW, Olympus) to observe blue and red fluorescence reflecting Hoechst and Alexa 555-labelled siRNA, respectively [35].
Quantitative uptake of Alexa 555-labelled NL-C-siRNA was done using flow cytometry (FCM). MCF-7 cells were cultured in 12-well plates (1 × 105 cells/well) for 24 h using 0.5 mL of media. Next, siRNA formulations were added to cells suspended in RPMI containing 10% FBS and incubated for 4 h. Cold PBS was used to cleanse MCF-7 cells thrice followed by trypsinisation and harvesting. Fluorescence (red) intensity of Alexa-555 was measured using BD FACS Calibur flow cytometry and analysed by the FCS Express software (BD Bioscience, USA) [35].
In vitro mTOR silencing effects of NL-mTOR-siRNA
To investigate in vitro mTOR silencing capability of NL-mTOR-siRNA against MCF-7 cells, the qualitative assessment of phosphorylated mTOR (Ser2448) was carried out by mTOR pSer2448 in vitro ELISA (ab168358) kit as per the manufacturer’s protocol using a microplate reader at 450 nm wavelength [36].
In addition, the efficacy of NL-mTOR-siRNA (25 nmol/L) on mTOR protein expression in MCF-7 cells was evaluated via western blot study following the established protocol [37]. MCF-7 cells were treated with NL-C-siRNA and NL-mTOR-siRNA and a total of 1 × 106 cells were collected. After rinsing the cells with phosphate-buffered saline (PBS), it was treated with ice-cold Lysis buffer (500 μL) composed of 0.1 mM of ethylenediaminetetraacetic acid (EDTA), 50 mM of NaCl, 10 mM of HEPES having pH 7.9, 0.5% of triton (X-100), 1 mM of dithiothreitol (DTT), 1 mM of phenylmethylsulfonyl fluoride, 5 μL of phosphatase inhibitor and 5 μL of protease inhibitor cocktails. Then, the lysate was centrifuged (1000 rpm, 4 °C, for 10 min) to isolate the cytoplasmic extract and the supernatant was subsequently centrifuged at 14000 rpm to yield a clear fragment of cytoplasmic protein which was stored in −80 °C. Later, nuclear pellet obtained was blend together with 500 μL of buffer (composed of 0.1 mM of both EGTA and EDTA, NaCl (500 mM), 0.1% of NP40, DTT (1 mM), HEPES of pH 7.9 (10 mM), PMSF (1 mM) along with protease inhibitor (5 mL) cocktail) and vortexed (4 °C, 15 min). The extract was centrifuged once again (14,000 × g, 10 min, 4 °C) and placed in liquid nitrogen. Bradford protein assay was used to estimate the protein content in the sample.
To perform SDS-PAGE analysis, same quantity of protein samples was taken along with successive transfer of proteins obtained electrophoretically into a nitrocellulose membrane (Millipore). Sites of the blots that were not specific, were barred by treating with 5% w/v of non-fat dry milk in Tris-buffer saline Tween-20 (TBST) (25 °C, 60 min). Then, the blots were incubated for 12 h using distinct antibodies on 4 °C and washed thrice by using TBST (5 min every time). HRP-conjugated secondary antibody was employed for re-incubating the blot (in 1:20,000 dilution, 25 °C) for nearly 60 min. The membrane (ThermoFisher Scientific) was developed on film (CLXposure, 8 × 10 in) with the help of chemiluminescent substrate. Primary antibodies of mTOR (Cell Signalling Technology, Danvers, USA) were used in a specific dilution (1:500) in 5% w/v of BSA in TBST-T. Image J software (NIH, Washington, USA) was applied for protein band densitometry assay with β-actin as internal standard.
In vitro anti-cancer effects of NL-mTOR-siRNA
The decrease in the concentration of viable cells was assessed using MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay after NL-mTOR-siRNA treatment as per established protocols [38] and relative inhibition was computed as (Abs control − Abs treatment)/Abs control × 100%.
In vitro metastasis assay including adhesion assay [39], Matrigel invasion assay [38] and scratch assay [38] were performed following methods of previous researchers to assess the anti-metastatic potential of NL-mTOR-siRNA against MCF-7 cells. In brief, the MTT experiment was conducted to determine cell adhesion rate (%) and was calculated as OD treated/OD control × 100%. Likewise, the matrigel invasion test was conducted by employing a transwell (Sigma-Aldrich) with an 8.0 μm pore size with help of haematoxylin and eosin (H&E) stains. In the scratch assay, the decrease in migration of cells into wounded monolayer after treatment with NL-mTOR-siRNA was determined using a phase-contrast microscope (Leica).
In vivo anti-tumour study
Female Sprague Dawley (SD) rats were acquired from the animal house, Birla Institute of Technology, Mesra, Ranchi (1972/PH/BIT/113/20/IAEC) and the entire survey was carried out as per the Institutional Animal Ethics Committee regulations. Rodents were housed in polystyrene cages and 55-day-old rats were employed for in vivo study after 1 week of acclimatisation to the laboratory environment. Animals were given with standard pelleted diet together with water ad libitum.
Twenty-four rats were obtained from the animal house facility and split into four groups (6 animals in each group) to check the effect of NL-mTOR-siRNA in reducing DMBA-induced breast cancer (Fig. S1) as group I (CTRL/SD/DMBA−/−), group II(SD/DMBA+/+), group III (SD/DMBA+/+ NL-C-siRNA) and group IV (SD/DMBA+/+ NL-mTOR-siRNA). Breast carcinoma was developed as per already established methods. 7,12-Dimethylbenz[a]anthracene or DMBA (20 mg) in olive oil (0.5 mL) was utilised to develop breast carcinoma through the air pouch technique [31]. Progression in tumour development was checked by palpation every 7 days. Following 90 days of cancer induction, animals were treated with NL-C-siRNA (group III) and NL-mTOR-siRNA (group IV) twice weekly (150 µg/kg body wt.) by intravenous administration (i.v) for 4 weeks. The dose was selected as per available literature [40]. Alteration in body weight, as well as tumour volume, was assessed during the whole experimentation. Two-dimensional measurement of tumour volume assessed [41] using a Vernier calliper and calculated as V = 4/3π(r1)2r2 (where r1 is the shortest radius and r2 is the longest radius).
Upon completion of the treatment schedule, cervical dislocation was done to sacrifice the animals [40]. Subsequently, tumour was separated and photographed. Tumour growth inhibition rate was calculated as (1 − Tumour volume of treatment group/Tumour volume of the untreated group) × 100% [1].
Lastly, part of the isolated tumour tissue was put in liquid N2 for molecular study. Western blot and RT-PCR analysis were done according to the method described earlier [37] using GAPDH as internal control. Image J software (NIH, Washington, USA) was applied for the protein band densitometry assay. The forward and backward sequence of primers of mTOR and GAPDH gene was given as:
For mTOR: Forward Primer: GCTTTGACGCAGGTGCTAAG Sequence (5′>3′) (16–35 location), Reverse Primer: TGTCCCCATAACCGGAGTAGG Sequence (5′>3′) (118–98 location); for GAPDH: Forward Primer: TGGATTTGGACGCATTGGTC Sequence (5′>3′) (333–352 location), Reverse Primer: TTTGCACTGGTACGTGTTGAT Sequence (5′>3′) (543–523 location).
Alteration in breast tissue morphology was detected through histopathology and Field Emission Scanning Electron Microscopy (FESEM). The isolated tumour and breast tissues preserved in buffered formalin were embedded in paraffin wax and fine sections (6–8 μm) of these paraffin blocks were cut using a rotary microtome. Then, the H&E dye staining technique was employed according to the standard experimental protocol [31]. FESEM of breast tissue was done after formalin fixation (10%v/v), dehydration with increasing strengths (70%, 80%, 90%, 100% v/v) of ethanol and subsequent drying. A FESEM (Sigma 300, ZEISS, Carl Zeiss) was employed to detect the difference in the architecture of mammary tissues of various groups [31].
Next, to study the immunohistochemistry of mTOR and Ki-67 (proliferative marker) breast tissues (5–6 µm) preserved in buffered formalin were immunolabelled with Monoclonal rabbit anti-p-mTOR (Ser2448) (49F9) antibody (Cell Signalling, Danvers, MA, USA) and Ki-67 mouse monoclonal antibody (Thermo Scientific, 50-5698-82 (eBioscience), MA5-11358, USA), respectively, after peroxide quenching, following a 3-stage protocol of immune-peroxidase. Alteration in immunohistochemistry was detected using a high-resolution microscope (Leica DME microscope; Merck, India) [42].
In vivo uptake into tumour tissue
To estimate in vivo uptake into tumour tissue, a single-dose fluorescently labelled siRNA was injected after stabilisation of tumour which was assessed by palpation (90 days after tumour induction). To compare the distribution of siRNA within tumour, we injected Alexa-555-tagged NL-C-siRNA (in liposomal formulation) and Alexa-555-tagged naked C-siRNA (without liposomal formulation) by intravenous administration. 5 µg of Alexa-555-tagged siRNA as a 200 µL dose was administered intravenously and breast tissues were harvested at 1-h, 6-h, 48-h, 4-day, 7-day or 10-day intervals.
For immunofluorescence analysis, breast tumour tissues were collected after sacrificing the animal and quickly frozen in an OCT medium. A thin slice (30 µm) was dissected out and acetone fixed. Then, slides containing tissues were rinsed with PBS followed by staining with 1.0 µg/mL DAPI (4′,6-diamidino-2-phenylindole) for 10 min (for staining the nuclei), cleansed, added with propyl gallate and a coverslip was placed and observed under a confocal microscope (FLOWVIEW, Olympus) [27].
Tissue distribution study
To examine the distribution of siRNA into different organs, animals were treated with Alexa-555-tagged NL-C-siRNA (in liposomal formulation) and Alexa-555-tagged naked C-siRNA (without liposomal formulation) and euthanised after 4 h. Tissues obtained were subjected to homogenisation, centrifugation and fluorescence intensity of supernatants collected were measured through a plate reader (Bioscan, Washington, DC) at a wavelength of 555/565 nm. The quantity of siRNA present in each sample was determined using a standard curve [43].
Pharmacokinetic experiment
Animals were administered with a single dose of 5 µg Alexa-555 tagged NL-C-siRNA (in liposomal formulation) and Alexa-555 tagged naked C-siRNA (without liposomal formulation) as a 200 µL dose by intravenous administration via tail vein. Blood was withdrawn from the retro-orbital sinus of experimental animals under anaesthesia at different time points (2, 12, 24, and 48 h), centrifuged and fluorescence intensity was measured through a fluorescence plate reader (Bioscan, Washington, DC). Alexa 555 excitation/emission wavelength was 555/565 nm [22].
In vivo apoptosis study through TUNEL assay
The paraffin-embedded breast tissue sections (30 mm) of each experimental group were subjected to TUNEL assay to determine their intra-tumoural late-stage apoptosis. The tissues were stained using ClickiT plus TUNEL assay kit (Thermo-Fisher) containing Alexa Fluor 594 for in situ detection of apoptosis according to the product manual and viewed under a confocal microscope (FLOWVIEW, Olympus) [31].
Preliminary biotoxicity assessment
Healthy animals were employed to carry out the experimentation by distributing them into three groups with six rats in each group. An identical treatment protocol to that of the anti-tumour efficacy study was followed to assess preliminary biotoxicity of siRNA-loaded nano-formulation.
Alteration in body weight was examined by comparing body weights of animals at the end of the experimental period to determine any toxic effect caused by liposomal formulations.
Animals were sacrificed on completion of their treatment period and major organs including kidney, lungs, liver, heart, spleen and brain were removed and alteration in histopathology of tissues (6–8 mm) was observed after H&E staining using a microscope (Leica) [22].
Further, biochemical alteration in glucose (Glu), alkaline phosphatase (ALP), aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatine kinase (CK), high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL), albumin (ALB), uric acid (UA), triglyceride (TG), total cholesterol (TC) and total protein (TP) [38] was assessed.
Statistical analysis
One-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison tests was employed to establish statistical significance within data obtained from this investigation by taking a uniform sample size. The significance level was assessed at p < 0.05. Data obtained were denoted as mean ± standard error of mean (SEM).
Results
Characterisation of siRNA-loaded neutral liposomal nanoparticle
In this study, we synthesised neutral nano-liposome using DOPC and characterised it (Fig. 1). DOPC-based empty liposome (NL) were found to have PS of 65.34 ± 0.58 nm (Fig. 1a) with PDI 0.319 ± 0.04; after siRNA encapsulation (NL-C-siRNA), it displayed small enhancement in size, i.e. 74.12 ± 0.58 nm (Fig. 1b). Liposomes having charges between +10 and −10 mV are considered neutral [44]. Zeta potential was detected as −1.04 and −1.53 mV for NL and NL-C-siRNA, respectively (Fig. 1c, d). Loading efficiency obtained was 70% as analysed with UV-vis spectrophotometer, implying efficient loading of siRNA, which was beneficial for delivery. Surface morphology of NL-C-siRNA assessed by TEM (Fig. 1e), FESEM (Fig. 1f) and AFM (Fig. 1g) confirmed a multifaceted, smooth surface with an almost spheric shape.
Fig. 1. Morphological characterisation of DOPC-based neutral nanoliposome.

a Particle size distribution of NL and b NL-C-siRNA. c Zeta potential of NL and d NL-C-siRNA. e Transmission electron microscopy (TEM). f Field emission scanning electron microscopy (FESEM). g Atomic force microscopy (AFM) of NL-C-siRNA. h siRNA release curve of NL-C-siRNA in PBS with pH 5.5 and 7.4, where data are represented as mean ± SEM and n = 3. (DOPC 1,2-dioleoyl-sn-glycero-3-phosphocholine, NL empty neutral liposome, NL-C-siRNA control siRNA-loaded neutral nanoliposome).
Release of siRNA is demonstrated with two different pH (Fig. 1h) depicting more release at pH 5.5 than at pH 7.4, i.e. gradual in both pH over a period of 48 h. The release was continued up to 72 h before reaching the stationary phase. Further, the cumulative release reached nearly 80% after 96 h without an initial burst release effect.
Stability, being an essential criterion, was examined in RT, 4 °C or −20 °C (Fig. S2). As per pictographs, PS and PDI of NL and NL-C-siRNA liposomes exhibited minor diversion up to a tolerable limit in 28 days of incubation at −20 °C temperature, indicating the absence of particle aggregation. However, powder stored at 4 °C and RT showcases diversified particle size and varied PDI on rehydration, probably the reason being the fluctuating humidity levels. Further, DOPC, a zwitterionic lipid in physiological pH is having a fluid phase temperature of −22 °C [45], maybe the reason for instability at 4 °C and RT.
The haemolytic nature of liposomes was checked in blood collected from experimental animals (Fig. 2a, b). No visual haemolysis was observed in the negative control group (PBS) and low haemolysis (3.8%) in the experimental group (NL-C-siRNA) with obviously broken erythrocytes visible in the positive control group (treated with Triton X-100), indicating good haemocompatibility of formulation.
Fig. 2. Haemolysis assay and cellular uptake analysis of neutral nanoliposome.

a Images of haemolysis on red blood cells. b The percentage of haemolysis (%) in each group to determine the serum stability of the formulation (n = 3). c Intracellular uptake study showing CLSM images of MCF-7 cells subjected to treatment with naked C-siRNA and NL-C-siRNA, the blue fluorescence (Hoechst: dark blue) indicates cell nuclei location and the red fluorescence (Alexa 555: Red) denotes intracellular uptake and internalisation of NL-C-siRNA. d Flow cytometry analysis of MCF-7 cells incubated with NL-C-siRNA for quantification of cellular uptake. (NL-C-siRNA control siRNA-loaded neutral nanoliposome).
In vitro cellular uptake of siRNA-loaded liposome
Owing to their significance in course of gene transfection, the expanse of cellular uptake of siRNA preparations into MCF-7 cells was tested and evaluated both qualitatively by CLSM (Fig. 2c) and quantitatively by flow cytometry (Fig. 2d). Internalisation of naked C-siRNA into MCF-7 cells were very low marked by a faint band of red fluorescence. On the contrary, encapsulating siRNA cargo into the liposomal carrier (NL-C-siRNA) resulted in a visible increase in fluorescence intensity depicting enhanced cellular internalisation. Further, flow cytometry analysis demonstrated tremendous internalisation (78%) of NL-C-siRNA, which might be associated with higher loading efficiency.
In vitro anti-cancer effects of NL-mTOR-siRNA
mTOR inhibitory potential of NL-mTOR-siRNA estimated through in vitro ELISA was shown in Fig. S3. No significant alteration in mTOR level was observed on treatment with NL-C-siRNA compared to untreated cells; however, the evident reduction was observed on NL-mTOR-siRNA (50 nmol/L) treatment (p < 0.001) implying its mTOR silencing potential. Likewise, 25 nmol/L of NL-mTOR-siRNA is capable of reducing the mTOR expression (p < 0.001) as verified through western blot analysis (Fig. 3a).
Fig. 3. In vitro activity of mTOR siRNA-loaded neutral liposome.

a In vitro mTOR inhibitory activity of NL-C-siRNA (50 nmol/L) and NL-mTOR-siRNA (25 nmol/L) using western blot analysis. b Pictorial representation and c graphical representation of MTT assay for cell viability analysis following treatment of MCF-7 cells with NL-C-siRNA (50 nmol/L) and NL-mTOR-siRNA (10, 25 and 50 nmol/L). d, f Inhibitory effects of NL-C-siRNA and NL-mTOR-siRNA on the invasion of MCF-7 cells as represented by photomicrographs and graph showing in vitro invasion and measured using transwells coated with Matrigel. e, g Representative photomicrographs of wounded monolayers of MCF-7 cells taken at 24 h to determine the effect of NL-C-siRNA and NL-mTOR-siRNA on the migration of MCF-7 breast cancer cells. The migration was estimated by measuring the cells within the wounded region after 24 h. h Cell adhesion rate of MCF-7 cells at 30, 60 and 90 min after treatment with NL-C-siRNA and NL-mTOR-siRNA respectively. The data are represented as mean ± SEM, n = 3 and significance of difference is indicated as ***p < 0.001 and nsp > 0.05. (NL-C-siRNA control siRNA-loaded neutral liposome, NL-mTOR-siRNA mTOR-siRNA-loaded neutral liposome, MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]).
Elevated mTOR expression is coupled with a lower survival rate and aggressive tumour growth [35]. Efficacy of mTOR knockdown by siRNA in inhibiting MCF-7 cell growth was asserted through MTT assay (Fig. 3b, c), illustrating the degradation of cancer cells in a concentration-dependent manner on NL-mTOR-siRNA treatment (IC50 = 25.36 nmol/L).
Prevention of tumour metastasis is a vital aspect of carcinogenesis management [28]. The prominent role of mTOR in mammary carcinogenesis prompted us to investigate the potency of NL-mTOR-siRNA to influence in vitro breast cancer metastasis, which signified remarkable suppression in MCF-7 cell invasion (Fig. 3d, f) and migration (Fig. 3e, g) along with less cell adhesion at every time point (Fig. 3h) after depletion of mTOR by NL-mTOR-siRNA. This result advocates the involvement of mTOR in breast cancer metastasis and validates the capability of NL-mTOR-siRNA to restrict breast cancer metastasis successfully.
In vivo siRNA uptake and pharmacokinetic study
The specific distribution of siRNA at tumour sites at the optimum therapeutic amount is required for in vivo anti-tumour therapy. In vivo uptake into mammary tissues of SD/DMBA+/+ assessed through confocal microscopy (Fig. 4) images established a greater intracellular fluorescence signal in tissue section of animals administered with Alexa-555 tagged NL-C-siRNA; however, the fluorescence intensity in tissues of animals given with Alexa-555 tagged naked C-siRNA was very less. It confirms the deep cytosolic distribution of siRNA within the tumour. The above data suggest that encapsulation of siRNA into liposomal formulation enhanced uptake of siRNA in breast cancer tissues. Additionally, diffusion of Alexa-555 labelled siRNA into major organs (heart, liver, lung, kidney and spleen) was determined, indicating significant deposition and retention of siRNA nanoliposomes in the liver and spleen (Fig. S4A). The reason behind this deposition may be the presence of specialised cells in both liver (hepatocyte) and spleen (lymphocytes and erythrocytes). Being functional organs of the mononuclear phagocytic system (MPS) or reticuloendothelial system (RES), both these organs have a leaky vasculature [46, 47] that allows plasma protein to cross the endothelial membrane and promote lymphatic drainage [48]. Likewise, siRNA liposomal nanoparticles due to their nanosized characteristic extravasate through this fenestration and are then, engulfed by RES resulting in its accumulation in these organs [46].
Fig. 4. In vivo uptake of siRNA-loaded neutral liposome into the tumour tissue.

CLSM images of tumour tissues subjected to treatment with Alexa-555-tagged naked C-siRNA and Alexa-555-tagged NL-C-siRNA. Blue fluorescence (DAPI: blue) indicates cell nuclei location and the red fluorescence (Alexa-555: red) denotes intra-tumoural uptake of Alexa 555-tagged siRNA. (NL-C-siRNA control siRNA-loaded neutral liposome, CLSM confocal laser scanning microscopy, DAPI 4′,6-diamidino-2-phenylindole).
The pharmacokinetic study was carried out to quantitatively determine the circulation time of siRNA cargo in blood by administering a single dose of fluorescent siRNA (Fig. S4B). It is noted that the plasma concentration of siRNA without a carrier (naked C-siRNA) was very less due to rapid clearance from systemic circulation within 15 min following intravenous injection. In contrast, liposomal nano-carrier (NL-C-siRNA) provides good stability and prolonged circulation time since encapsulation into DOPC shields siRNA cargo by forming lipid bilayers and prevents degradation by enzymes and rapid elimination through kidney upon intravenous administration, leading to boosted efficacy [1].
mTOR silencing induced in vivo anti-cancer activity
Changes in anatomical parameters are directly linked to physiological irregularities [49]. From Fig. S5A, we can observe that a single dose of carcinogen caused marked regression (p < 0.001) in body weight (group II, III and IV) opposite to non-cancerous rats (group I). However, mTOR depletion by NL-mTOR-siRNA treatment enhanced (p < 0.001) the body weight of animals (group IV).
The mammary tumour was noticeable 14 days after subcutaneous injection of 20 mg of DMBA and attain promotional phase after three months [50]. Anti-tumour therapy with NL-mTOR-siRNA (group IV) done at this phase resulted in marked tumour regression (p < 0.001) as compared to other groups (SD/DMBA+/+ and SD/DMBA+/+ NL-C-siRNA) (Fig. 5a, b). Further, at the end of the treatment schedule (28 days), tumour growth inhibition (Fig. S5B) in group IV (SD/DMBA+/+ NL-mTOR-siRNA) was significantly higher (p < 0.001) validating the therapeutic potential of NL-mTOR-siRNA.
Fig. 5. In vivo anti-tumour efficacy of mTOR siRNA-loaded neutral liposome in DMBA-induced breast tumour model in SD rats.

a Alteration in tumour volume of cancer-bearing mice following treatment with NL-C-siRNA and NL-mTOR-siRNA. b Images of tumours of each group excised after the end of the treatment protocol. c Photomicrographic depiction of breast tissues isolated from different experimental groups as analysed by FESEM to determine the structural and morphological alterations. d Histopathological evaluation (×400) of breast tissues obtained from different groups of experimental animals, where scale bar represent 50 µm section. Normal control: CTRL/SD/DMBA−/−, induced control: SD/DMBA+/+, cancerous animals subjected to treatment with NL-C-siRNA: SD/DMBA+/+ NL-C-siRNA, cancerous animals subjected to treatment with NL-mTOR-siRNA: SD/DMBA+/+ NL-mTOR-siRNA. The data are represented as mean ± SEM values and n = 6, ***p < 0.001 and nsp > 0.05. (NL-C-siRNA control siRNA-loaded neutral liposome, NL-mTOR-siRNA mTOR-siRNA-loaded neutral liposome, DMBA 7,12-dimethylbenz[a]anthracene, SD Sprague Dawley, FESEM field emission scanning electron microscope).
Morphological alterations in breast cancer tissues evaluated by FESEM (Fig. 5c) indicated membrane ruffles (MR) along with compact microvilli (MV) covering, depicting the invasiveness of breast cancer. Also, collagen fibres deriving from adjacent matrices were found crosslinking with each other to produce a network. However, treatment with NL-mTOR-siRNA markedly reduced MR and MV along with the prominent reduction in collagen fibre network that almost disappeared, advocating the efficacy of NL-mTOR-siRNA.
According to the results of the histopathological study (Fig. 5d), normal mammary tissue (CTRL/SD/DMBA−/−) was characterised by normal parenchyma, acini and ductules. Further, interstitial tissue made up of fatty tissues was seen along with thin layers of connective tissue lining lobular units. A thick layer of basement membrane surrounding the ductal epithelial cells can be seen along with a single layer of dark cells surrounding acini. However, a distorted breast tissue architecture was observed in group II (SD/DMBA+/+) portrayed by ductal hyperplasia indicated as irregular breast ducts near the proliferating lumen. Abnormally large alveolar cells and cribriform pattern of the cells signified proliferative lesions. The widespread proliferation of epithelial cells results in fused glandular structure representing ductal carcinoma (adenocarcinoma). Neoplastic transformation of the epithelial ductal cells is also evident and characterised by nuclear polymorphism (abnormal increase in the nucleus), hyper‑chromatinization and chromatid clumping. Administration of NL-C-siRNA in group III (SD/DMBA+/+ NL-C-siRNA) shows no evidence of restoration of cellular morphology. On the other hand, NL-mTOR-siRNA intervention (SD/DMBA+/+ NL-mTOR-siRNA) causes amelioration in the cellular anomalies of the breast tissue depicted by alleviated cell proliferation and inversion of ductal hyperplasia resulting in a certain degree of restoration in the breast ducts and fatty tissues. This observation characterises return of normalcy in NL-mTOR-siRNA treated group.
Western blot (Fig. 6a) and RT-PCR (Fig. 6b) analysis carried out on mammary tissues demonstrated a highly significant (p < 0.001) increase in expressions of mTOR protein and mRNA in SD/DMBA+/+ animals as compared to CTRL/SD/DMBA−/− animals. NL-mTOR-siRNA proficiently reimposed the alteration towards normalcy, signifying gene silencing efficacy of NL-mTOR-siRNA. No significant change in NL-C-siRNA-treated group was observed. Immunohistochemical analysis (Fig. 6c) was performed to support the results obtained in western blot and RT-PCR analysis, which revealed a significant reduction in mTOR expression in tumour tissue following target gene silencing by NL-mTOR-siRNA.
Fig. 6. In vivo mTOR downregulation following NL-mTOR-siRNA administration.

a Western blot analysis, b RT-PCR study, c immunohistochemical analysis for evaluation of mTOR expression in breast tissue depicting therapeutic silencing of mTOR restores altered mTOR expression following target gene silencing by NL-mTOR-siRNA. d Immunohistochemical observations of proliferation markers revealed expression of Ki-67 (brown colouration) in mammary tissues of experimental rats. Normal control: CTRL/SD/DMBA−/−, induced control: SD/DMBA+/+, cancerous animals subjected to treatment with NL-C-siRNA: SD/DMBA+/+ NL-C-siRNA, cancerous animals subjected to treatment with NL-mTOR-siRNA: SD/DMBA+/+ NL-mTOR-siRNA. The data are represented as mean ± SEM values and n = 6, ***p < 0.001 and nsp > 0.05. (NL-C-siRNA control siRNA-loaded neutral liposome, NL-mTOR-siRNA mTOR-siRNA-loaded neutral liposome, DMBA 7,12-dimethylbenz[a]anthracene, SD Sprague Dawley).
To examine the effect of NL-mTOR-siRNA on the proliferation kinetics of tumour cells, Ki-67 expression was estimated through immunohistochemistry of cancer tissues (Fig. 6d). An elevated level of Ki-67 positive (brown colouration) was detected in SD/DMBA+/+ animals. This proliferation rate was found to be decreased highly in SD/DMBA+/+ NL-mTOR-siRNA group due to the protection provided by NL-mTOR-siRNA treatment.
mTOR silencing invoked tumour growth inhibition by inducing apoptosis
A TUNEL assay was performed on the tumours excised from experimental animals and the rates of apoptosis were enumerated for further exploiting the mechanism associated with the anti-tumour activity (Fig. 7a, b). Minimal purple fluorescence signals denoting Alexa Flour 594 were visible in the SD/DMBA+/+ rats. While cancer-induced animals treated with NL-mTOR-siRNA (SD/DMBA+/+ NL-mTOR-siRNA) revealed significantly advanced apoptosis. Thus, the extent of intra-tumoural apoptosis and TUNEL positive cells, respectively, clearly suggested the successful tumour growth inhibition by mTOR downregulation.
Fig. 7. Induction of apoptosis in DMBA-induced mammary tumours by mTOR siRNA-loaded neutral liposome.

a Representative TUNEL fluorescent images of section of tumour excised from rats of individual groups. Blue fluorescence (Hoechst: dark blue) indicates cell nuclei location and the purple fluorescence (Alexa Flour 594: purple) denotes apoptotic cell location. The scale bars are represented as 100 µm. b Estimation of rate of tumour apoptosis in rats belonging to each group in accordance to images of TUNEL assay in which each value shows mean ± SEM (n = 6). Induced control: SD/DMBA+/+, cancerous animals subjected to treatment with NL-C-siRNA: SD/DMBA+/+ NL-C-siRNA, cancerous animals subjected to treatment with NL-mTOR-siRNA: SD/DMBA+/+ NL-mTOR-siRNA. ***p < 0.001, **p < 0.01, *p < 0.05 and nsp > 0.05. (NL-C-siRNA control siRNA-loaded neutral liposome, NL-mTOR-siRNA mTOR-siRNA-loaded neutral liposome, DMBA 7,12-dimethylbenz[a]anthracene, SD Sprague Dawley).
Preliminary biotoxicity assessment of siRNA-loaded liposomal particle
Toxicity caused by systemic administration of siRNA is widely known. Thus, the safety of siRNA-loaded liposomes was preliminarily assessed in healthy rats. Intravenous injection of NL-mTOR-siRNA for 28 days does not invoke any kind of toxic effect on animals. We did not observe any sign of behavioural alteration as compared to normal animals. Alteration in body weight, histopathology and blood biochemistry parameters were studied for bio-toxicity investigation. Animal weight was not varied substantially amongst various groups (Fig. S5C) indicating no significant effect on the eating and drinking habit of animals. No signs of pathological alterations and biotoxicity (such as; oedema, lesion, bleeding, inflammation, hyperaemia, or any other reactions) [38] were observed in histological analysis (Fig. 8). Moreover, non-significant differences were found in biochemical indicators of all groups (Fig. S6), with all these parameters neither aberrantly higher nor lower suggesting non-toxic nature of NL-mTOR-siRNA in experimental animals.
Fig. 8. Histopathological analysis of rats treated with or without mTOR siRNA-loaded neutral liposome.

H&E staining of vital organs (liver, heart, kidney, lungs, spleen and brain) tissues to determine biotoxicity isolated from animals belonging to different experimental groups (×400): Normal control: CTRL/SD/DMBA−/−, animals subjected to treatment with NL-C-siRNA: CTRL/SD/DMBA−/+ NL-C-siRNA, animals subjected to treatment with NL-mTOR-siRNA: CTRL/SD/DMBA−/+ NL-mTOR-siRNA. Scale bar represent 50 µm section. (H&E haematoxylin and eosin, NL-C-siRNA control siRNA-loaded neutral liposome, NL-mTOR-siRNA mTOR-siRNA-loaded neutral liposome, DMBA 7,12-dimethylbenz[a]anthracene, SD Sprague Dawley).
Discussion
Statistics from the International Agency for Research on Cancer of the World Health Organisation (WHO) reported that breast cancer has become the most regularly spotted cancer throughout the world and has been continuously increasing in the past decade because of treatment failure and disease recurrence [1]. The emergence of RNAi technology involving siRNA as therapeutic drugs has been a game-changer in addressing these issues but its translation into the clinical setting faces obstacles of shorter half-life, inadequate uptake and toxicity [15]. In accordance with our findings, a neutral nano-liposomal carrier system entrapping a high amount of mTOR-siRNA can be an efficient platform in providing significant silencing efficacy against mTOR that further inhibits the growth and progression of mammary carcinogenesis.
In the present study, neutral liposome has been synthesised using DOPC. Enhancement in size of particles has been noticed following siRNA complexation, suggesting multilayer formation, beneficial for siRNA encapsulation [51]. PDI values smaller than 0.3 indicates homogeneous dispersion of particles [52]. Limited variations in particle size and PDI confirmed NL-C-siRNA to be stable in storage allowing its use for experimental purposes. Further, once packaged, siRNAs are stable, resistant to degradation by serum nucleases and show sustained release from liposomes that characterised their haemocompatibility behaviour in bloodstream [53]. Synthesised nano-formulation showed sustained release over a prolonged period in physiological conditions favouring tumour accumulation [1]. Moreover, siRNA release was better at pH 5.5 which resembles tumour micro-environment as compared to physiological pH, 7.4 [29]. The pharmacokinetic study displayed a higher percentage of siRNA remaining in blood up to 48 h following encapsulation, owing to the employment of neutral lipids that provide long blood circulation by shielding siRNA from degradation.
Generally, efficient cancer therapeutics needs a higher accumulation of drug at tumour site [1]. Encapsulated siRNA exhibited significant uptake into breast cancer cells in flow cytometry and CLSM because nanoparticles with an average particle size of 100 nm diameter express better cellular uptake [54]. Further, due to the hyperproliferative nature of cancer tissue, it requires extra energy from its neighbouring tissues making tumour environment more acidic [55]. The unorganised and perforated endothelial membrane containing pores of around 100-780 nm size permit entry of nanoparticles by passive diffusion, known as the enhanced permeability and retention (EPR) effect [56]. Hence, nanoparticles with particle sizes smaller than 100 nm provide ideal silencing of targeted genes and allow better tumour penetration and accumulation [57]. Positive/negative/neutral charge on formulation affects their uptake into various cancer tissues [58]. Neutral liposomal particles move faster than cationic (>10 mV) and anionic (<−20 mV) liposomes in a hydrogel mimicking tumour environment as observed by Lieleg et al. [59], indicating high tumour penetrability. Likewise, Nomura et al. [60] reported a greater accumulation of neutral liposomes in highly perfused areas (184 times more than cationic liposomes). The non-specific micropinocytosis mechanism facilitates the uptake of neutral liposomes into cells, facilitating endosomal escape because of leaky macropinosomes [61]. Hence, using neutral lipid-like DOPC, provide a balance between proficient siRNA uptake by tumour cells and discharge of siRNA contents from liposome to the cytoplasm following cellular internalisations [27]. Together these data, supported neutral liposomes as a potential nanocarrier with an adequate tumour accumulation and efficient endosomal escape, endorsing its employment for in vivo experimentations.
mTOR, a prime driver of cell survival and drug resistance, is overexpressed in nearly all types of cancers. In cancer conditions, PI3K/ AKT overexpression stimulate phosphorylation and activation of the mTOR complex by inactivating the p70S6k1/mTOR complex through negative feedback regulation. mTOR activations promote unchecked mitochondrial process along with ribosomal biosynthesis and angiogenesis that results in enhanced protein synthesis for cell growth and proliferation [62–64]. This evidence suggests mTOR deregulation in mammary carcinogenesis [65]. Ma et al. [66] pointed out the increased levels of p-mTOR in mammary carcinoma tissues to that of normal tissues. Further, aberrant expression of mTOR incites cancer cells to metastasise and invade adjacent healthy tissues [67], bloodstream and lymphatic system to form metastasis in distant tissues [68]. mTOR, its upstream PI3K and downstream S6K play a role in regulating tumour cell motility, invasion, angiogenesis and metastasis [69–71]. Since metastasis has a prominent role in cancer-related death, its prevention can be an important tool for its management [72, 73]. In course of our experiment, an evident reduction in mTOR expression following NL-mTOR-siRNA treatment corroborates with mTOR silencing potential of NL-mTOR-siRNA. Observations of the MTT experiment provide evidence of cytotoxic effects of NL-mTOR-siRNA against MCF-7 cells. Furthermore, observations of cell metastasis experiments endorse its efficacy in preventing the way towards tumour cell adhesion, migration and invasion.
Following the successful establishment of anti-breast cancer efficacy of NL-mTOR-siRNA in cells, it was evaluated for in vivo anti-breast cancer potential. The mammary gland of rat is extra sensitive toward the development of tumour and replicates human mammary carcinogenesis, for which rats are generally used to determine the anti-tumoural efficacy of various therapies [74]. Our investigation is the first in producing proof of breast tumour inhibition by in vivo knockdown of mTOR using NL-mTOR-siRNA (i.v.) as a therapeutic drug. Anatomical alterations like changes in body weight indicate abnormalities and physiological anomalies. Reduced body mass index reflects cancer cachexia. Deduction in body weight, anorexia, muscle fatigue, early satiety, malabsorption, altered body metabolism and unspecific anaemia are prominent features of cachexia [49]. We found rats administered with NL-mTOR-siRNA displayed significant improvement in body weight analogues to findings of past researchers for anti-tumour drugs [37, 42, 75, 76]. Additionally, tumour volume of rats was alleviated after treatment with NL-mTOR-siRNA. Photomicrographic images of histopathology, FESEM, immunohistochemical analysis along with relative mTOR mRNA and protein expression studies revealed the therapeutic potential of NL-mTOR-siRNA. Moreover, studies done in past by employing neutral liposome nanocarrier showed satisfactory in vivo gene silencing results. Landen et al. [27] formulated siRNA/DOPC neutral liposome to ascertain therapeutic delivery of EphA2 gene-specific siRNA into tumour, resulting in a subsequent decrease in protein expression and reduced tumour growth. Likewise, Halder et al. [26] examined in vivo therapeutic potential of FAK siRNA in a neutral liposomal (DOPC) construct and observed a significant downregulation of in vivo FAK expression resulting in up to 72% reduction in tumour weight.
Ki-67 is a nuclear protein associated with cellular proliferation which represents the growth factor expressed in dividing cells. The elevated Ki-67 level denoting abnormal proliferation in DMBA-induced mammary cancer rats has been well-established [42]. As per our findings, downregulation of this proliferation marker following mTOR silencing can propose the chemotherapeutic potential of NL-mTOR-siRNA.
To clarify the mechanism of NL-mTOR-siRNA invoked tumour growth reduction, the TUNEL assay was carried out since enough evidence are available to support the role of mTOR as an apoptosis inhibitor [7, 77]. Apoptosis, a naturally occurring cell-death mechanism, is helpful in removing infected, impaired and undesirable cells from a living being [78] and thereby regulates cancer oncogenesis. In relation to the existing data, the result obtained from our experimentation is very encouraging as depicted by significantly high apoptotic cell death in breast cancer tissues following NL-mTOR-siRNA treatment. It indicates that NL-mTOR-siRNA exerts anti-breast cancer activity through the apoptotic cell-death mechanism.
Lastly, the need for biotoxicity assessment of NL-mTOR-siRNA arises before its future employment in clinical trials. During the experiments, mTOR-siRNA was well tolerated in Sprague Dawley rats. Histopathological examinations and blood biochemical parameters indicated no substantial alterations among various groups, suggesting no evident toxicity of NL-mTOR-siRNA which is in accordance with previous studies [34] reasoned by limited toxicologic effects of nano-liposomes on the biological system [27]. The composition of formulation affects cellular toxicity too. Liposomes consisting of a higher percentage of neutral lipid exhibited lesser toxicity [79]. Research done in past showed that treatment with one/two dose of DOPC-liposome (75–225 µg/kg) does not evoke any signs of adverse effects [80]. Biodistribution experiments by Landen et al. revealed considerable deposition of EphA2-siRNA-DOPC in the liver, kidney, and lung tissues, however, it is not associated with cytotoxicity [25]. It is analogous to the findings of Wagner et al. which demonstrated multiple doses of EphA2-siRNA-DOPC administered to experimental subjects (non-human) did not exhibit any substantial hepatotoxicity [80]. Further, we have provided potent inhibition of mTOR by injecting NL-mTOR siRNA (0.15 mg/kg) which is 60-120 times smaller in contrast to another research where 10 mg/kg of cationic liposomes-siRNA was being administered [81]. These biotoxicity data indicated that NL-mTOR-siRNA does not contain toxicity for further clinical development.
In summary, according to our knowledge, this experiment is the first in describing the effectual delivery of mTOR-siRNA to DMBA-induced breast cancerous rats via DOPC-based neutral nanoliposomes. Consequently, a profound reduction in tumour growth, proliferation and invasion was ascertained following extensive downregulation of mTOR expression, thereby providing a proof of concept of an RNAi-based therapeutic strategy of NL-mTOR-siRNA as a superior choice for the management of breast carcinoma. Due to the prominent role of mTOR in apoptosis and autophagy, it can be noted that NL-mTOR-siRNA may exert anti-carcinogenic action by upregulation of pro-apoptotic protein, which needs further study and validation. Subsequently, it provides definite ground for use of NL-mTOR-siRNA in humans after the successful establishment of clinical trials.
Supplementary information
Acknowledgements
We acknowledge the support provided by the Central Instrumentation facility (CIF), Birla Institute of Technology, Mesra, Ranchi and HR-TEM facility of Vellore Institute of Technology in the characterisation of liposomal preparation.
Author contributions
RS and SPP conducted the experiments and wrote the manuscript with equal contribution. SPP and RS was responsible for confocal microscopy, Western blot, immunohistochemistry and flow cytometry. RS and SJ took part in the in vivo experiments. SJ analysed the data and revised the manuscript. SPP designed the research plan, analysed the data and revised the manuscript. All authors have approved the manuscript.
Funding
This study was supported by Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Ranchi, India. This work has been funded by UGC (201819-NFO-2018-19-OBC-ORI-80495).
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This animal study was approved by the Institutional Animal Ethical Committee, Birla Institute of Technology, Mesra, Ranchi (approval no. 1972/PH/BIT/113/20/IAEC). All animal experiments were conducted in accordance with the Institutional Animal Ethical Committee (IAEC) regulation. This study did not include patient participation or analysis of patient data.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Roja Sahu, Shakti Prasad Pattanayak.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-022-02011-1.
References
- 1.Zhou B, Li M, Xu X, Yang L, Ye M, Chen Y, et al. Integrin α2β1 targeting DGEA-modified liposomal doxorubicin enhances antitumor efficacy against breast cancer. Mol Pharm. 2021;18:2634–46. doi: 10.1021/acs.molpharmaceut.1c00132. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Mathur P, Sathishkumar K, Chaturvedi M, Das P, Sudarshan KL, Santhappan S, et al. Cancer statistics, 2020: report from National Cancer Registry Programme, India. JCO Glob Oncol. 2020;6:1063–75. doi: 10.1200/GO.20.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Early Breast Cancer Trialists’ Collaborative Group. Polychemotherapy for early breast cancer: an overview of the randomised trials. Lancet. 1998;352:930–42. doi: 10.1016/S0140-6736(98)03301-7. [DOI] [PubMed] [Google Scholar]
- 5.Gentzler RD, Altman JK, Platanias LC. An overview of the mTOR pathway as a target in cancer therapy. Expert Opin Ther Targets. 2012;16:481–9. doi: 10.1517/14728222.2012.677439. [DOI] [PubMed] [Google Scholar]
- 6.Sahu R, Pattanayak SP. Strategic developments & future perspective on gene therapy for breast cancer: role of mTOR and Brk/PTK6 as molecular targets. Curr Gene Ther. 2020;20:237–58. doi: 10.2174/1566523220999200731002408. [DOI] [PubMed] [Google Scholar]
- 7.Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:1–1. doi: 10.1186/s13578-020-00396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tapia O, Riquelme I, Leal P, Sandoval A, Aedo S, Weber H, et al. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 2014;465:25–33. doi: 10.1007/s00428-014-1588-4. [DOI] [PubMed] [Google Scholar]
- 9.Singh BN, Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem Pharm. 2012;84:1154–63. doi: 10.1016/j.bcp.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Li HQ, Zhou FX, Yu JW, Sun L, Han ZH. Targeting the mTOR pathway in breast cancer. Tumor Biol. 2017;39:1010428317710825. doi: 10.1177/1010428317710825. [DOI] [PubMed] [Google Scholar]
- 11.Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35:515–24. doi: 10.1007/s10555-016-9637-x. [DOI] [PubMed] [Google Scholar]
- 12.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 13.Kapoor M, Burgess DJ, Patil SD. Physicochemical characterization techniques for lipid based delivery systems for siRNA. Int J Pharm. 2012;427:35–57. doi: 10.1016/j.ijpharm.2011.09.032. [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Zhang W, Qi R, Mao ZW, Shen H. Engineering functional inorganic–organic hybrid systems: advances in siRNA therapeutics. Chem Soc Rev. 2018;47:1969–95. doi: 10.1039/C7CS00479F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–27. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabarti S, Finnes HD, Mahipal A. Fibroblast growth factor receptor (FGFR) inhibitors in cholangiocarcinoma: current status, insight on resistance mechanisms and toxicity management. Expert Opin Drug Metab Toxicol. 2022;14:1–4. doi: 10.1080/17425255.2022.2039118. [DOI] [PubMed] [Google Scholar]
- 18.Rossi JJ, Rossi DJ. siRNA drugs: here to stay. Mol Ther. 2021;29:431–2. doi: 10.1016/j.ymthe.2021.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–38. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merritt WM, Bar-Eli M, Sood AK. The dicey role of Dicer: implications for RNAi therapy. Cancer Res. 2010;70:2571–4. doi: 10.1158/0008-5472.CAN-09-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70:3687–96. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Paolo D, Brignole C, Pastorino F, Carosio R, Zorzoli A, Rossi M, et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol Ther. 2011;19:1131–40. doi: 10.1038/mt.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liyanage PY, Hettiarachchi SD, Zhou Y, Ouhtit A, Seven ES, Oztan CY, et al. Nanoparticle-mediated targeted drug delivery for breast cancer treatment. Biochim Biophys Acta Rev Cancer. 2019;1871:419–33. doi: 10.1016/j.bbcan.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filion MC, Phillips NC. Major limitations in the use of cationic liposomes for DNA delivery. Int J Pharm. 1998;162:159–70. doi: 10.1016/S0378-5173(97)00423-7. [DOI] [Google Scholar]
- 25.Foged C, Nielsen HM, Frokjaer S. Liposomes for phospholipase A2 triggered siRNA release: preparation and in vitro test. Int J Pharm. 2007;331:160–6. doi: 10.1016/j.ijpharm.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Halder J, Kamat AA, Landen CN, Han LY, Lutgendorf SK, Lin YG, et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res. 2006;12:4916–24. doi: 10.1158/1078-0432.CCR-06-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landen CN, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–8. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 28.Song Y, Zhou B, Du X, Wang Y, Zhang J, Ai Y, et al. Folic acid (FA)-conjugated mesoporous silica nanoparticles combined with MRP-1 siRNA improves the suppressive effects of myricetin on non-small cell lung cancer (NSCLC) Biomed Pharmacother. 2020;125:109561. doi: 10.1016/j.biopha.2019.109561. [DOI] [PubMed] [Google Scholar]
- 29.Alinejad V, Somi MH, Baradaran B, Akbarzadeh P, Atyabi F, Kazerooni H, et al. Co-delivery of IL17RB siRNA and doxorubicin by chitosan-based nanoparticles for enhanced anticancer efficacy in breast cancer cells. Biomed Pharmacother. 2016;83:229–40. doi: 10.1016/j.biopha.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 30.Kumar K, Maiti B, Kondaiah P, Bhattacharya S. Efficacious gene silencing in serum and significant apoptotic activity induction by survivin downregulation mediated by new cationic gemini tocopheryl lipids. Mol Pharm. 2015;12:351–61. doi: 10.1021/mp500620e. [DOI] [PubMed] [Google Scholar]
- 31.Bose P, Priyam A, Kar R, Pattanayak SP. Quercetin loaded folate targeted plasmonic silver nanoparticles for light activated chemo-photothermal therapy of DMBA induced breast cancer in Sprague Dawley rats. RSC Adv. 2020;10:31961–78(a). doi: 10.1039/D0RA05793B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tekedereli I, Alpay SN, Akar U, Yuca E, Ayugo-Rodriguez C, Han HD, et al. Therapeutic silencing of Bcl-2 by systemically administered siRNA nanotherapeutics inhibits tumor growth by autophagy and apoptosis and enhances the efficacy of chemotherapy in orthotopic xenograft models of ER (−) and ER (+) breast cancer. Mol Ther Nucleic Acids. 2013;2:e121. doi: 10.1038/mtna.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin Y, Liang X, An Y, Dai Z. Microwave-triggered smart drug release from liposomes co-encapsulating doxorubicin and salt for local combined hyperthermia and chemotherapy of cancer. Bioconjug Chem. 2016;27:2931–42. doi: 10.1021/acs.bioconjchem.6b00603. [DOI] [PubMed] [Google Scholar]
- 34.Tekedereli I, Alpay SN, Tavares CD, Cobanoglu ZE, Kaoud TS, Sahin I, et al. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS ONE. 2012;7:e41171. doi: 10.1371/journal.pone.0041171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Cheng Q, Jiang Q, Huang Y, Liu H, Zhao Y, et al. Enhanced endosomal/lysosomal escape by distearoyl phosphoethanolamine-polycarboxybetaine lipid for systemic delivery of siRNA. J Control Release. 2014;176:104–14. doi: 10.1016/j.jconrel.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Acharya R, Chacko S, Bose P, Lapenna A, Pattanayak SP. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci Rep. 2019;9:1–3. doi: 10.1038/s41598-019-52162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haque MW, Bose P, Siddique MU, Sunita P, Lapenna A, Pattanayak SP. Taxifolin binds with LXR (α & β) to attenuate DMBA-induced mammary carcinogenesis through mTOR/Maf-1/PTEN pathway. Biomed Pharmacother. 2018;105:27–36. doi: 10.1016/j.biopha.2018.05.114. [DOI] [PubMed] [Google Scholar]
- 38.Kumar A, Sunita P, Jha S, Pattanayak SP. Daphnetin inhibits TNF‐α and VEGF‐induced angiogenesis through inhibition of the IKK s/IκBα/NF‐κB, Src/FAK/ERK 1/2 and Akt signalling pathways. Clin Exp Pharm Physiol. 2016;43:939–50. doi: 10.1111/1440-1681.12608. [DOI] [PubMed] [Google Scholar]
- 39.Xiao W, Zhang W, Huang H, Xie Y, Zhang Y, Guo X, et al. Cancer targeted gene therapy for inhibition of melanoma lung metastasis with eiF3i shRNA loaded liposomes. Mol Pharm. 2019;17:229–38. doi: 10.1021/acs.molpharmaceut.9b00943. [DOI] [PubMed] [Google Scholar]
- 40.Bertrand JR, Pottier M, Vekris A, Opolon P, Maksimenko A, Malvy C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem Biophys Res Commun. 2002;296:1000–4. doi: 10.1016/S0006-291X(02)02013-2. [DOI] [PubMed] [Google Scholar]
- 41.Sahu R, Kar RK, Sunita P, Bose P, Kumari P, Bharti S, et al. LC-MS characterized methanolic extract of Zanthoxylum armatum possess anti-breast cancer activity through nrf2-keap1 pathway: an in-silico, in-vitro and in-vivo evaluation. J Ethnopharmacol. 2021;269:113758. doi: 10.1016/j.jep.2020.113758. [DOI] [PubMed] [Google Scholar]
- 42.Kumar A, Sunita P, Jha S, Pattanayak SP. 7, 8-Dihydroxycoumarin exerts antitumor potential on DMBA-induced mammary carcinogenesis by inhibiting ERα, PR, EGFR, and IGF1R: involvement of MAPK1/2-JNK1/2-Akt pathway. J Physiol Biochem. 2018;74:223–34. doi: 10.1007/s13105-018-0608-2. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010;18:1650–6. doi: 10.1038/mt.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clogston JD, Patri AK. Zeta potential measurement. Methods Mol Biol. 2011;697:63–70. doi: 10.1007/978-1-60327-198-1_6. [DOI] [PubMed] [Google Scholar]
- 45.Plumb JA. Cell sensitivity assays: clonogenic assay. In: Langdon SP, editor. Cancer cell culture, methods in molecular medicine. Totowa, NJ: Humana Press Inc.; 2004. pp. 159–64. [DOI] [PubMed]
- 46.Teymouri M, Badiee A, Golmohammadzadeh S, Sadri K, Akhtari J, Mellat M, et al. Tat peptide and hexadecylphosphocholine introduction into pegylated liposomal doxorubicin: an in vitro and in vivo study on drug cellular delivery, release, biodistribution and antitumor activity. Int J Pharm. 2016;511:236–44. doi: 10.1016/j.ijpharm.2016.06.117. [DOI] [PubMed] [Google Scholar]
- 47.Barenholz YC. Doxil®—the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–34. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 48.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134:2139–47. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 49.Argilés JM, Azcón-Bieto J. The metabolic environment of cancer. Mol Cell Biochem. 1988;81:3–17. doi: 10.1007/BF00225648. [DOI] [PubMed] [Google Scholar]
- 50.Pattanayak SP, Mazumder PM. Therapeutic potential of Dendrophthoe falcata (Lf) Ettingsh on 7, 12-dimethylbenz (a) anthracene-induced mammary tumorigenesis in female rats: effect on antioxidant system, lipid peroxidation, and hepatic marker enzymes. Comp Clin Pathol. 2011;20:381–92. doi: 10.1007/s00580-010-1008-3. [DOI] [Google Scholar]
- 51.Song F, Sakurai N, Okamoto A, Koide H, Oku N, Dewa T, et al. Design of a novel PEGylated liposomal vector for systemic delivery of siRNA to solid tumors. Biol Pharm Bull. 2019;42:996–1003. doi: 10.1248/bpb.b19-00032. [DOI] [PubMed] [Google Scholar]
- 52.Badran M, Shalaby K, Al-Omrani A. Influence of the flexible liposomes on the skin deposition of a hydrophilic model drug, carboxyfluorescein: dependency on their composition. Sci World J. 2012;2012:134876. doi: 10.1100/2012/134876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang C, Attia AB, Tan JP, Ke X, Gao S, Hedrick JL, et al. The role of non-covalent interactions in anticancer drug loading and kinetic stability of polymeric micelles. Biomaterials. 2012;33:2971–9. doi: 10.1016/j.biomaterials.2011.11.035. [DOI] [PubMed] [Google Scholar]
- 54.Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13:1838–45. doi: 10.1023/A:1016085108889. [DOI] [PubMed] [Google Scholar]
- 55.Prakash S, Malhotra M, Shao W, Tomaro-Duchesneau C, Abbasi S. Polymeric nanohybrids and functionalized carbon nanotubes as drug delivery carriers for cancer therapy. Adv Drug Deliv Rev. 2011;63:1340–51. doi: 10.1016/j.addr.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 56.Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 57.Arranja AG, Pathak V, Lammers T, Shi Y. Tumor-targeted nanomedicines for cancer theranostics. Pharm Res. 2017;115:87–95. doi: 10.1016/j.phrs.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF. Liposome− cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998;37:12875–83. doi: 10.1021/bi980096y. [DOI] [PubMed] [Google Scholar]
- 59.Lieleg O, Baumgärtel RM, Bausch AR. Selective filtering of particles by the extracellular matrix: an electrostatic bandpass. Biophys J. 2009;97:1569–77. doi: 10.1016/j.bpj.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nomura T, Koreeda N, Yamashita F, Takakura Y, Hashida M. Effect of particle size and charge on the disposition of lipid carriers after intratumoral injection into tissue-isolated tumors. Pharm Res. 1998;15:128–32. doi: 10.1023/A:1011921324952. [DOI] [PubMed] [Google Scholar]
- 61.Xia Y, Tian J, Chen X. Effect of surface properties on liposomal siRNA delivery. Biomaterials. 2016;79:56–68. doi: 10.1016/j.biomaterials.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–46. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 63.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 64.Vara JÁ, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 65.Butt G, Shahwar D, Qureshi MZ, Attar R, Akram M, Birinci Y, et al. Role of mTORC1 and mTORC2 in breast cancer: therapeutic targeting of mTOR and its partners to overcome metastasis and drug resistance. Adv Exp Med Biol. 2019;1152:283–92. [DOI] [PubMed]
- 66.Ma BL, Shan MH, Sun G, Ren GH, Dong C, Yao X, et al. Immunohistochemical analysis of phosphorylated mammalian target of rapamycin and its downstream signaling components in invasive breast cancer. Mol Med Rep. 2015;12:5246–54. doi: 10.3892/mmr.2015.4037. [DOI] [PubMed] [Google Scholar]
- 67.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–18. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berven LA, Willard FS, Crouch MF. Role of the p70S6K pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res. 2004;296:183–95. doi: 10.1016/j.yexcr.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 70.Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP‐9. Hepatol Res. 2009;39:177–86. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 71.Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–40. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- 72.Langer EM, Kendsersky ND, Daniel CJ, Kuziel GM, Pelz C, Murphy KM, et al. ZEB1-repressed microRNAs inhibit autocrine signaling that promotes vascular mimicry of breast cancer cells. Oncogene. 2018;37:1005–19. doi: 10.1038/onc.2017.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verrax J, Defresne F, Lair F, Vandermeulen G, Rath G, Dessy C, et al. Delivery of soluble VEGF receptor 1 (sFlt1) by gene electrotransfer as a new antiangiogenic cancer therapy. Mol Pharm. 2011;8:701–8. doi: 10.1021/mp100268t. [DOI] [PubMed] [Google Scholar]
- 74.Mollard S, Mousseau Y, Baaj Y, Richard L, Cook-Moreau J, Monteil J, et al. How can grafted breast cancer models be optimized? Cancer Biol Ther. 2011;12:855–64. doi: 10.4161/cbt.12.10.18139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haque MW, Pattanayak SP. Taxifolin inhibits 7, 12-dimethylbenz (a) anthracene-induced breast carcinogenesis by regulating AhR/CYP1A1 signaling pathway. Pharmacogn Mag. 2017;13:S749–55. doi: 10.4103/pm.pm_315_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bose P, Pattanayak SP, Priyam A. Herniarin, a natural coumarin loaded novel targeted plasmonic silver nanoparticles for light activated chemo-photothermal therapy in preclinical model of breast cancer. Pharmacogn Mag. 2020;16:474–85 (b). doi: 10.4103/pm.pm_223_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang HW, Hu JJ, Fu RQ, Liu X, Zhang YH, Li J, et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kγ mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci Rep. 2018;8:1–3. doi: 10.1038/s41598-018-29308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wideranging implications in tissue kinetics. Br J cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spagnou S, Miller AD, Keller M. Lipidic carriers of siRNA: differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry. 2004;43:13348–56. doi: 10.1021/bi048950a. [DOI] [PubMed] [Google Scholar]
- 80.Wagner MJ, Mitra R, McArthur MJ, Baze W, Barnhart K, Wu SY, et al. Preclinical mammalian safety studies of EPHARNA (DOPC nanoliposomal EphA2-targeted siRNA) Mol Cancer Ther. 2017;16:1114–23. doi: 10.1158/1535-7163.MCT-16-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sonoke S, Ueda T, Fujiwara K, Sato Y, Takagaki K, Hirabayashi K, et al. Tumor regression in mice by delivery of Bcl-2 small interfering RNA with pegylated cationic liposomes. Cancer Res. 2008;68:8843–51. doi: 10.1158/0008-5472.CAN-08-0127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.