

Abstract
Diseases in the central nervous system (CNS) are often difficult to treat. Antibody- and protein-based therapeutics hold huge promises in CNS disease treatment. However, proteins are restricted from entering the CNS by the blood–brain barrier (BBB). To achieve enhanced BBB crossing, antibody-based carriers have been developed by utilizing the endogenous macromolecule transportation pathway, known as receptor-mediated transcytosis. In this report, we first provided an overall review on key CNS diseases and the most promising antibody- or protein-based therapeutics approved or in clinical trials. We then reviewed the platforms that are being explored to increase the macromolecule brain entry to combat CNS diseases. Finally, we have analyzed the lessons learned from past experiences and have provided a perspective on the future engineering of novel delivery vehicles for antibody- and protein-based therapies for CNS diseases.
Keywords: Antibody, BBB, Bispecific, TfR, InsR
Statement of Significance: Beyond simply reviewing the previous state-of-the-art in antibody delivery crossing the BBB, we summarized a comprehensive list of design tips to generate successful bispecific BBB-crossing antibodies.
INTRODUCTION
Protein- and antibody-based therapeutics delivery into the brain offer huge potential in treating a broad spectrum of central nervous system (CNS) diseases, including cancer (e.g., glioblastoma (GBM) and glioma), neurodegenerative diseases (e.g., Parkinson’s disease (PD) and Alzheimer’s disease (AD)), autoimmune diseases (e.g., multiple sclerosis), nervous system diseases (e.g., amyotrophic lateral sclerosis), and genetic disorders (e.g., lysosomal storage diseases (LSDs)). However, a significant hurdle for protein and antibody therapeutics to enter the brain is the blood–brain barrier (BBB). BBB is formed by a continuous monolayer of brain endothelial cells (BECs), pericytes, and astrocytes. The continuous non-fenestrated capillaries integrity is maintained by tight junctions (such as occludin, claudins, and other junctional adhesion molecules) sealing the BECs together [1]. Key nutrients, such as amino acids, glucose, and iron, are transported across the BBB by specific carrier receptors [2]. The tight regulation of peripheral substances in the CNS maintains the homeostasis of the CNS. Lipophilic molecules <400 Da can diffuse through the BBB [3, 4], with macromolecules showing severely restricted transportation. Beyond BECs, the integrity of BBB is also maintained by the pericytes and astrocytes surrounding the BECs [5–9]. Astrocyte end-feet and pericytes cover up to 99% of the basal capillary membrane [10, 11]. Additional contributions from factors, such as angiopoietin-1 and angiotensin II, also promote the integrity of the BBB [12, 13]. The multi-cellular neurovascular barrier regulates the passage of macromolecules across the BBB, which poses substantial challenges in targeting the CNS with antibodies and proteins. The biology of BBB is thoroughly reviewed elsewhere [14] and therefore is not a focus of this review.
BBB restricts peripheral proteins and antibodies from entering the CNS. Protein passage through the BBB is mostly non-specific and is <0.1% of peripheral circulating proteins enter the CNS [15, 16]. Although neurodegenerative diseases are believed to cause dysfunctions of BBB with higher permeability, it has been demonstrated that the BBB integrity was well maintained, preventing passive BBB permeability of IgG injected peripherally in multiple AD mouse models, including PS2-amyloid precursor protein (APP), Tau transgenics, and APOE4 knock-in mice [17]. It was estimated that only 0.01% of the peripheral antibody molecules may enter the CNS [18]. With the extremely low concentration of antibodies crossing the BBB, it is difficult for therapeutic antibodies to achieve the concentrations required for the desired therapeutic effects. Therefore, increasing the antibody uptake into CNS is imperative for the exploration of a broader spectrum of targets and increased the chance of success in targeting CNS diseases with therapeutic antibodies or proteins.
Since plasma proteins may permeate and maintain physiological concentrations by BBB-specific transporting systems [19], scientists have successfully demonstrated an increased brain delivery of protein and antibody therapeutics by targeting those endogenous transporting receptors, which is the focus of this review. Delivering antibodies to the CNS via invasive strategies, such as ultrasound, microbubbles, and direct injection into the brain (e.g., intracerebroventricular delivery), are reviewed elsewhere and are not included in this review [20, 21].
TARGETING BRAIN DISORDERS BY PROTEIN- AND ANTIBODY-BASED THERAPEUTICS
Protein- and antibody-based therapies are the emerging drug modalities for CNS diseases, with many drug candidates the in preclinical development and clinical trials. We focus here on the CNS disease targets and drug candidates which are approved for clinical use or in clinical studies (Table 1).
Table 1.
Examples of CNS diseases targeted by macromolecules in clinical use or clinical trials
| Target | Ab or protein | Mechanism/outcomes | Status (pre-clinical, clinical stages, and approval) | Ref |
|---|---|---|---|---|
| Disease: glioma/GBM | ||||
| EGFR | Cetuximab (Eli Lilly) | EGFR inhibitor, well tolerated with limited activity in recurrent high-grade glioma | Phase II | [22] |
| VEGF | Bevacizumab (Roche) | VEGP blocker, no difference in OS, a modest increase in progression-free survival when combined with chemotherapy | Approved to treat GBM in adult patients | [23] |
| Disease: AD | ||||
| Aβ | Bapineuzumab (Élan and Wyeth) | Binding Aβ AA 1–5, clears both fibrillar and soluble Aβ, failed phase 2 and 3 clinical trials | Phase 3 trial, terminated 08.2012 | [24–27] |
| Aβ | Solanezumab (Eli Lilly) | Binding Aβ AA 16–26, increases clearance of monomers, failed phase 3 clinical trials | Phase 3 trial, terminated 05.2017 | [28–30] |
| Aβ | Gantenerumab (Hoffmann-La Roche) | Binding Aβ AA 3–12, 18–27, reduced Aβ plaques by recruiting microglia, failed phase 2/3 trial | Phase 2/3 trial terminated in 12.2014, and the ongoing phase 2/3 trial (NCT01760005) for individuals at risk for and with early-stage autosomal-dominant AD | [31–34] |
| Aβ | Crenezumab (Genentech) | Binding Aβ AA 13–24, 10-fold higher affinity for oligomers versus monomers, phase 3 trial terminated | Phase 3 trial terminated 05.2019 due to an interim analysis showing crenezumab was unlikely to meet its primary endpoint | [35–38] |
| Aβ | Aducanumab (Biogen) | Binding Aβ AA 3–6, selectively binds Aβ aggregates, phase 3 trial (NCT02477800) showed a dose-dependent reduction in brain amyloid and some CSF phospho-Tau reduction | FAD approved | [39] |
| Trem2 | AL002 (Alector) | Activates TREM2 | Phase II for participants with early AD (NCT04592874) | [40] |
| Disease: PD | ||||
| α-synuclein | Prasinezumab (Hoffmann-La Roche) | C-terminal of αSyn, targeting aggregated α-synuclein, antibody showed good safety and tolerability with CSF antibody presence | Phase II is ongoing (NCT03100149) | [41,42] |
| α-synuclein | Cinpanemab (Biogen) | binds to α-synuclein residues 1–10, with 800-fold higher affinity for aggregated over monomeric α-synuclein, showed good safety and tolerability with CSF antibody presence | Phase II is ongoing (NCT03318523) | [43] |
| Disease: LSDs | ||||
| α-l-iduronidase | Aldurazyme (Genzyme) | Replace defective IDU, catalyzes the hydrolysis of unsulfated alpha-L-iduronosidic linkages in dermatan sulfate | Approved | [44–46] |
| idursulfase | Elaprase (Takeda Pharmaceutical) | Replace defective IDS, breakdown of heparan and dermatan sulfate | Approved | [47,48] |
| Disease: migraine | ||||
| CGRP | Erenumab (Amgen) | Blocks CGRP | Approved | [49] |
| CGRP | Galcanezumab (Eli Lilly) | Blocks CGRP | Approved | [50] |
| CGRP | Fremanezumab (Teva Pharmaceuticals) | Blocks CGRP | Approved | [51] |
| CGRP | Eptinezumab (Lundbeck Seattle) | Blocks CGRP | Approved | [52–54] |
Glioblastoma
Over 200 000 brain cancers are reported in the USA annually, and GBM is the most common, invasive, and neurologically destructive brain cancer with high morbidity and mortality [55]. With a 6.8% 5-year survival rate, treatment options for GBM are urgently needed. A significant portion of GBM patients (>50%) have epidermal growth factor receptor (EGFR) amplification and mutations, which is associated with poor prognosis [56–58]. EGFR overexpression or mutation affects the cancer proliferation, angiogenesis, and apoptosis. Cetuximab against wild-type EGFR and ΔEGFR has been approved for the treatments of solid tumors but not for GBM. A phase II clinical trial examining cetuximab treatment in patients with recurrent high-grade glioma showed that cetuximab was well tolerated but exhibited limited activity in this patient population [22].
The vascular endothelial growth factor (VEGF) is another heavily studied target. VEGF is highly expressed in glioma cells and is directly associated with the poor prognosis and malignancy of gliomas [59–61]. The irregular vasculature in GBM suggests that angiogenesis plays a role in the GBM development and that VEGF is a potential therapeutic target [62,63]. Bevacizumab is an FDA-approved anti-VEGF antibody for many types of cancer. However, a meta-analysis of four clinical trials, including 607 patients, demonstrated combining chemo-radiotherapy, while bevacizumab showed no overall survival (OS) improvement [23].
Alzheimer’s disease
AD is the most common form of dementia and is characterized by amyloid plaques and neurofibrillary tangles in the brain. In the amyloid hypothesis, the amyloid-β (Aβ) peptide causes synaptic dysfunction and neurodegeneration [64]. Therefore, therapeutically removing Aβ is the most studied approach for AD treatment, with one recently FDA-approved antibody therapy [39]. Other therapeutic approaches have been proposed, which include inhibitors of the synthetic enzyme gamma-secretase and beta-secretase and Aβ aggregation. While we focus on the amyloid targeting antibodies, readers may refer to reviews on other AD therapeutic approaches, such as inhibiting beta-secretase 1 (BACE1), to reduce the Aβ burden [65–69].
Aducanumab (BIIB037), a fully human IgG1 monoclonal antibody (mAb), was the first antibody therapy approved by the FDA for the AD treatment [39]. It binds to AA3–6 present only on aggregated species of Aβ such as oAβ and fibrils. Aducanumab demonstrated reduced brain fibrillar Aβ in a dose- and time-dependent manner in a phase 1b clinical trial [39]. Aducanumab showed a dose-dependent reduction in brain amyloid and some cerebrospinal fluid (CSF) phospho-Tau reduction in a phase 3 study (NCT02477800). Aducanumab was approved by the FDA under the accelerated approval pathway. In addition to aducanumab, several other amyloid antibodies have advanced to clinical trials, which include bapineuzumab, solanezumab, gantenerumab, and crenezumab. These antibodies are summarized in Table 1.
Bapineuzumab (AAB-001) is a humanized IgG1 that binds to AA1–5 of Aβ and targets both fibrillar and soluble Aβ [70]. Bapineuzumab was shown to be safe in a phase I study [24]. Bapineuzumab showed no differences for the primary efficacy endpoints in phase 2 trials for patients with mild to moderate AD [25,26]. However, APOE ε4 noncarriers showed potential treatment differences in later prespecified exploratory analyses. Moreover, bapineuzumab failed to show benefits in the primary outcomes in a large phase 3 study comprising 1121 carriers and 1331 noncarriers with mild to moderate AD [27].
Solanezumab (LY2062430), a humanized IgG1 mAb which binds to AA16–26 of Aβ, targets the soluble pool of Aβ and increases the clearance of Aβ monomers [71–73]. Solanezumab showed mobilization of Aβ42 from plaques in phase 1 and 2 studies [28,29]. However, solanezumab did not meet the co-primary cognitive and functional endpoints in two phase 3 double-blind, placebo-controlled trials. The more prespecified secondary analysis demonstrated that mild subjects benefit more from solanezumab treatment [30].
Gantenerumab, a fully human IgG1 mAb that binds to AA3–12 and AA18–27 of Aβ, is a conformational epitope in Aβ fibrils [31]. Phase 1 trials indicated that gantenerumab is safe and well tolerated [32]. However, a lack of significant treatment effects on brain Aβ levels led to the termination of phase 2/3 trials in December 2014 [33]. Gantenerumab is also being evaluated in a phase 2/3 trial in individuals at risk for and with early-stage autosomal-dominant AD (NCT01760005) [34].
Crenezumab (MABT5102A) is a humanized antibody on an IgG4 backbone to minimize the activation of fragment crystallizable (Fc) gamma receptors [35]. Crenezumab binds to AA13–24 of Aβ and recognizes Aβ monomers, oligomers, and fibrils, with a 10-fold higher affinity for oligomers over monomers [35–37]. Although well tolerated in phase I studies, crenezumab failed to show benefits for the primary or secondary outcomes in a phase 2 trial (NCT01343966) [35]. Later post hoc subgroup analysis of the high-dose cohort showed benefits in the mildest subgroup. Moreover, failure to reach the primary endpoint in participants with prodromal to mild AD led to the termination of a phase 3 study (NCT02670083).
Parkinson’s disease
PD is the most common neurodegenerative disorder after AD. The presence of aggregated and misfolded alpha-synuclein (αSyn) is an important neuropathological marker for PD. Aggregated αSyn forms intraneuronal filamentous inclusions located in perikaryon and/or neurites of degenerating neurons in PD patients [74–76]. Antibodies have been developed and tested for the PD treatment by targeting different regions and forms of αSyn, including the N-terminus, C-terminus, central region, protofibrils, oligomers, and fibrils, which have been reviewed in detail elsewhere [14,77,78]. Prasinezumab, a humanized IgG1 mAb targeting the αSyn C-terminus, has shown good safety and tolerability in ascending-dose trials with intravenous infusions [41,42]. It significantly reduced serum levels of free-to-total αSyn [41,42]. A phase 2, multinational study of PRX002/RO7046015 in newly diagnosed PD patients was initiated in the summer of 2017 (PASADENA Study, ClinicalTrials.gov identifier NCT03100149). Cinpanemab is another αSyn antibody which is in clinical trials for PD [43].
Lysosomal storage diseases
LSDs are inherited metabolic disorders caused by defective lysosomal enzymes which inflict on multiple organs [79]. Deficiencies in the brain result in developmental delay, mild-to-severe mental retardation, ataxia, seizures, and profound neurodegeneration [80–82]. Hurler syndrome is caused by the deficiency of α-l-iduronidase. Alpha-l-iduronidase (IDU) is important in the metabolism of heparan and dermatan sulfate, and its deficiency results in the accumulation of undegradable sulfated glycosaminoglycans (GAG) material throughout the body. Hurler syndrome is manifested as the developmental delay and cognitive decline, and patients usually die within the first year of life [83]. Recombinant human α-l-iduronidase (laronidase) (rhIDU, Aldurazyme) has been studied in clinical trials, showing improved forced vital capacity and reduced symptoms of airway obstruction [44–46]. In addition to rhIDU, recombinant human IDS (idursulfase, Elaprase) improved the forced vital capacity and increased exercise tolerance in clinical trials. IDS treatment demonstrated a 25% reduction in liver and spleen volumes. Significant improvement in elbow joint mobility was also observed [47,48].
Migraine
Migraine is a headache disorder that directly affects more than one in seven people worldwide. Calcitonin gene-related peptide (CGRP) plays a major role in migraine [84]. There has been significant progress in blocking CGRP by antibodies for migraine prevention. At least four CGRP targeting antibodies have been approved for migraine prevention in recent years, which include Erenumab (human IgG2 antibody), Galcanezumab (humanized IgG4 antibody), Fremanezumab (humanized IgG2 antibody), and Eptinezumab (humanized IgG1 antibody) [49–52].
TRANSPORT ROUTES FOR PROTEINS AND ANTIBODIES ACROSS THE BBB
There are two transport routes for proteins and antibodies across the BBB: uncontrolled non-specific protein brain entry and controlled endogenous transport systems on the BBB. The nonspecific nature of the absorptive-mediated transport greatly limits the therapeutic potential of therapeutic proteins and antibodies, as indiscriminate cellular uptake is not only a major disadvantage in terms of off-target effects, but it also may lead to the suboptimal pharmacokinetic properties of the therapies [85]. Exploring the endogenous transport systems on the BBB is more desirable for the CNS delivery of protein and antibody therapies. Receptor-mediated transcytosis (RMT) and carrier-mediated transport (CMT) are the major endogenous transport systems on the BBB [14,21,86]. The substrate-selective CMT is responsible for the delivery of small molecule nutrients that include glucose, amino acids, monocarboxylic acids, hormones, ions, and vitamins [86,87]. RMT delivers larger molecules via the vesicular trafficking of the ligand-receptor complexes [86], such as transferrin [88,89], insulin [90,91], leptin [92,93], tumor necrosis factor-alpha (TNFα) [94], and EGF [95]. Therefore, RMT is the most studied approach for delivering the protein and antibody therapies to the brain [96]. RMT uses endogenous receptors expressed on the luminal side of the BBB, which transport macromolecule nutrients, including iron-bound transferrin, insulin, and leptin, into the brain side via vesicular trafficking of the ligand-receptor complexes [88,90,92]. The RTM transport route requires binding to the extracellular domain of the receptor, subsequent endocytosis, and transcytosis to the luminal (brain) side of the capillary endothelium into the interstitial space [14,96–98].
Receptor-mediated transcytosis
Most of the studies on delivering proteins and antibodies have been on RMT receptors, such as the transferrin receptor (TfR), the insulin receptor (InsR), CD98hc, and FC5 antibody binding receptor, which is the focus of this review (Table 2). Figure 1 illustrates a TfR-mediated transport of a bispecific antibody, which binds to the TfR, through the BBB into the brain. Once inside the brain, the other arm of the bispecific antibody engages the disease target. Other RMT receptors, such as the low-density lipoprotein receptor-related protein 1 and the folate receptor, have shown to enhance the BBB passage of large cargos other than proteins and antibodies, such as virus-like particles and nanoparticles, which have been reviewed by others and are not covered in the current review [99,100].
Table 2.
Summary of previously reported cases to deliver macromolecule therapeutics through the BBB
| Therapeutic target/disease | Ab/protein design/engineering/key characterization | Biological effects (uptake/therapeutic effects) | Status (pre-clinical, clinical stages, and approval) | Ref |
|---|---|---|---|---|
| Transcytosis receptor: InsR | ||||
| IDUA/Hurler’s syndrome | HIR Mab-IDUA fusion (valanafusp alpha), bivalent antibody-enzyme fusion, high-affinity HIR binding, unaltered antibody binding, and enzyme activity upon fusion | In Rhesus monkeys, brain uptake of HIR Mab-IDUA fusion showed >12-fold increase versus IDUA as measured by radioactivity. In a 52-week, open label phase 1–2 trial on the safety and efficacy in pediatric MPSI patients with cognitive impairment, the pediatric subjects had CNS involvement with a mean enrollment Development Quotient (DQ) of 36.1 ± 7.1. The DQ, and the cortical gray matter volume of the brain, were stabilized by valanafusp alpha treatment. Somatic manifestations were stabilized, or improved, based on urinary glycosaminoglycan levels, hepatic and spleen volumes, and shoulder range of motion. | Phase 2 clinical trial | [101–104] |
| IDS/Hunter’s syndrome | HIR Mab-IDS fusion, bivalent antibody-enzyme fusion, high-affinity HIR binding, unaltered antibody binding and enzyme activity (363 ± 37 U/μg) upon fusion, uptake by Hunter fibroblasts | In Rhesus monkeys, brain uptake of HIR Mab-IDS fusion showed >10-fold increase versus GDNF (BBB impermeable marker) as measured by radioactivity. | Preclinical in Rhesus monkey | [105,106] |
| ASA/MLD | HIR Mab-ASA fusion, bivalent antibody-enzyme fusion, high affinity HIR binding EC50 = 0.34 ± 0.11 nM, retains high ASA enzyme activity, 20 ± 1 units/mg. Endocytosis in MLD fibroblasts. | In Rhesus monkeys, brain uptake of HIR Mab-ASA fusion showed >25-fold increase versus control IgG as measured by radioactivity. Enhanced brain distribution was confirmed also by radioautography. | Preclinical in Rhesus monkey | [107] |
| SGSH/Sanfilippo type A syndrome | HIR Mab-SGSH fusion, bivalent antibody-enzyme fusion, high affinity HIR binding EC50 = 0.33 ± 0.05 nM, retains high ASA enzyme activity, 4712 ± 388 units/mg. Endocytosis in MPSIIIA fibroblasts. | In Rhesus monkeys, brain uptake of HIR Mab-SGSH fusion showed >35-fold increase versus control IgG as measured by radioactivity. | Preclinical in Rhesus monkey | [108] |
| NAGLU/MPSIIIB | HIR Mab-NAGLU fusion, bivalent antibody-enzyme fusion, high-affinity HIR binding, unaltered antibody binding and enzyme activity upon fusion, uptake in MPSIIIB fibroblasts normalizes intracellular NAGLU enzyme activity | In Rhesus monkeys, brain uptake of HIR Mab-NAGLU fusion showed >15-fold increase versus control IgG as measured by radioactivity. | Preclinical in Rhesus monkey | [109] |
| PON1/atherosclerosis | HIR Mab-PON1 fusion, bivalent antibody-enzyme fusion, and fusion protein were rapidly removed from the blood, primarily by the liver | In Rhesus monkeys, brain uptake HIR Mab-PON1 fusion showed >25-fold increase versus control IgG as measured by radioactivity. | Preclinical in Rhesus monkey | [110] |
| EPO | HIR Mab-EPO fusion, bivalent antibody-protein fusion, high affinity HIR binding EC50 = 0.21 ± 0.05 nM, retains high EPO activity, ED50 = 0.30 ± 0.01 nM. | In Rhesus monkeys, brain uptake HIR Mab-EPO fusion showed >16-fold increase versus EPO as measured by radioactivity. | Preclinical in Rhesus monkey | [111] |
| GDNF/PD | HIR Mab-GDNF fusion, bivalent antibody-protein fusion, high affinity HIR binding EC50 = 0.87 ± 0.13 nM, retains high GDNF activity in binding GFPα1, ED50 = 1.68 ± 0.17 nM. | In a rat middle, cerebral artery occlusion model stroke volume was reduced by 77%. In the Rhesus monkey, no improvement of Parkinsonian motor symptoms and induced a dose-dependent hypersensitivity reaction | Preclinical in Rhesus monkey (failure) | [112,113] |
| TNFR/inflammation | HIR Mab-TNFR fusion, bivalent antibody-protein fusion, high-affinity HIR binding, unaltered antibody binding, and TNFR activity upon fusion | In Rhesus monkeys, brain uptake HIR Mab-TNFR fusion showed >30-fold increase versus TNFR-Fc as measured by radioactivity. | Preclinical in Rhesus monkey | [114] |
| Aβ peptide/AD | HIR Mab-scFv fusion, bivalent antibody-scFv fusion, high affinity HIR binding EC50 = 1.9 ± 0.1 nM, unaltered antibody binding with Aβ1–40 upon fusion with ED50 = 2.0 ± 0.8 nM | Rapidly cleared from plasma and was transported across the primate BBB. In vivo reached 1% ID/100 g tissue, HIR Mab-scFv fusion showed >10-fold increase versus anti-Aβ Mab as measured by radioactivity. | Preclinical in Rhesus monkey | [115,116] |
| Transcytosis receptor: TfR | ||||
| BACE1/AD | Anti-mouse TfR/anti-BACE1, KiH, monovalent binding. Bispecific antibody maintains the activity of BACE1 antibody in inhibiting Aβ production. High-affinity anti-TfR was found to induce TfR lysosomal degradation, causing reduced TfR surface level and curbing brain accumulation. Antibody effector function and complement binding were found to be responsible for acute toxicity and reticulocyte reduction upon injection. | For anti-TfR/BACE1 bispecific using therapeutic dosing, 3–5-fold increased brain uptake (vs. control IgG) 24 hr after injection as measured by sandwich ELISA. Single IV injection at 25 or 50 mg/kg reduced brain Aβ concentration by more than 50% within 24–48 h. | Preclinical in mouse | [117–119] |
| Aβ/AD | IgG-sFab, KiH, monovalent binding to TfR, and bivalent binding to Aβ. Monovalent sFab design showed lower TfR degradation than bivalent dFab design. | sFab design showed 55-fold higher plaque staining versus dFab design after intravenous injection into PS2APP mice. Weekly sFab design injection for 3 months showed a significant decrease of plaque number in both cortex and hippocampus in APPPS2 mice (vs. parent Aβ antibody mAb31). | Preclinical in mouse | [120] |
| BACE1/AD | Anti-human/cyno TfR/anti-BACE1, KiH, monovalent binding. Non-competing with Tf, binding TfR at apical domain. Optimal affinity is required since either high affinity or low affinity showed lower brain uptake. Compared to high-affinity anti-TfR, optimal affinity against TfR showed no degradation of TfR upon injection. Effector-less antibodies with N297G mutation showed no toxicity in monkeys. | TfR/anti-BACE1 with the optimal affinity of the anti-TfR arm showed a 3–5-fold increase in brain uptake in both human TfR KI mice and cynomolgus monkeys. In addition, therapeutic effects were observed as reduced Aβ concentration in both plasma and CNS (brain of mice and CSF of monkeys). | Preclinical in cynomolgus monkey | [121] |
| BACE1/AD | Directed-evolution engineered Fc fragment binding apical domain of human TfR, which did not impede Tf-TfR or Fc-FcRn interactions. ATV:BACE1 has monovalent binding to TfR and bivalent binding to BACE1. The ATV molecules have a native IgG structure and stability. Dose-dependent cellular internalization. | ATV:BACE1 showed >20-fold increase in brain IgG uptake than BACE1 antibody in human TfR KI mice. The increased brain uptake also showed >2-fold decrease in brain Aβ load. ATV:BACE1 showed a 50% reduction of CSF Aβ40 in cynomolgus monkeys and maintained the effects for 14 days after a single IV injection. In comparison, the BACE1 antibody showed no CSF Aβ40 reduction. | Preclinical in cynomolgus monkey | [122] |
| IDS/Hunter’s syndrome | Directed-evolution engineered Fc fragment binding apical domain of human TfR. IDS fusion to the N′ of engineered Fc fragment (ETV:IDS) was chosen due to unimpeded TfR binding. ETV:IDS maintains the biological activity of IDS in MPS II patient fibroblasts. | ETV:IDS showed 3–5-fold higher brain uptake than Fc:IDS in human TfR KI mice. Single or multiple doses of ETV:IDS reduced brain and CSF sGAG levels by 3–5-fold in comparison to IDS. ETV:IDS significantly reduced GAGs in neurons, astrocytes, and microglia and reduced lysosomal lipid accumulation in the brain. | Preclinical in mouse | [123,124] |
| PGRN/frontotemporal dementia | Directed-evolution engineered Fc fragment binding apical domain of human TfR. PGRN fusion to the C′ of engineered Fc fragment (PTV:PGRN) was chosen due to the need for a free PGRN C-terminus to interact with sortilin. Affinity to huTfR is 130 nM, PGRN activity is retained: EC50s sortilin 2 nM, prosaposin 5 nM. | PTV:PGRN rescued various Grn−/− phenotypes in primary murine macrophages and human iPSC-derived microglia, including oxidative stress, lysosomal dysfunction, and endomembrane damage. Peripherally delivered PTV:PGRN corrected levels of BMP, glucosylsphingosine, and disease pathology in Grn−/− CNS, including microgliosis, lipofuscinosis, and neuronal damage. | Preclinical in mouse | [125] |
| TNFR/inflammation | Anti-mouse TfR-TNFR fusion (cTfRMAb-TNFR), bivalent antibody-protein fusion, high-affinity TNFα binding Kd = 96 ± 34 pM | In mice, brain uptake of cTfRMAb-TNFR showed >45-fold increase versus control IgG after IV injection as measured by radioactivity. Neuroprotection in mouse MCAO model with 45%, 48%, 42%, and 54% reduction in hemispheric, cortical, and subcortical stroke volumes, and neural deficit, respectively, upon 1 mg/kg intravenous injection. | Preclinical in mouse | [126,127] |
| GDNF/PD | Anti-mouse TfR-GDNF fusion (cTfRMAb-GDNF), bivalent antibody-protein fusion. No chronic toxicity in mice after 2 mg/kg twice-weekly IV injection for 12 consecutive weeks. Chronic injection induced no anti-drug antibodies. | Chronic cTfRMAb-GDNF treatment showed no impact on brain uptake of cTfRMAb-GDNF. In comparison to the OX26 antibody, a 30-fold increase in brain uptake was observed. In a mouse PD model, chronic cTfRMAb-GDNF injection significantly reduced symptoms of PD. | Preclinical in mouse | [128,129] |
| Aβ/AD | cTfRMAb-scFv fusion, bivalent antibody-scFv fusion targeting both mouse TfR and Aβ. | Daily sc injections of 5 mg/kg of the cTfRMAb-ScFv fusion protein for 12 consecutive weeks reduced amyloid plaque in the cortex and hippocampus 1-year-old PS1/APP AD double transgenic PSAPP mice. | Preclinical in mouse | [130] |
| EPO/MCAO and AD | cTfRMAb-EPO fusion, bivalent antibody-EPO fusion targeting both mouse TfR and EPOR. Retains high EPO activity, ED50 = 0.33 ± 0.04 nM. | The brain uptake of the cTfRMAb-EPO fusion protein was 2.0 ± 0.1% ID/g of the brain following iv administration. cTfRMAb-EPO showed an 81% reduction in hemispheric stroke volume and a 78% reduction of the neural deficit in a permanent mouse MCAO model after a 1 mg/kg IV injection. APP/PS1 mice treated with cTfRMAb-EPO fusion protein showed significantly lower cortical and hippocampal Aβ peptide number and immune-positive area, decreased hippocampal synaptic loss, and cortical microglial activation and improved spatial memory compared with APP/PS1 saline controls. | Preclinical in mouse | [131–133] |
| IDUA/Hurler’s syndrome | cTfRMAb-IDUA fusion, bivalent anti-mouse TfR antibody-IDUA. Unaltered IDUA enzyme activity upon fusion 776 ± 79 units/μg. | Treatment of the MPSI mice with the cTfRMAb-IDUA reduced intracellular lysosomal inclusion bodies by 73% in the brain (versus saline-treated mice) with IV injections twice a week for 8 weeks at a 1 mg/kg dose. | Preclinical in mouse | [134] |
| IL-1/neuropathic pain | Anti-mouse TfR-IL-1RA fusion. Anti-TfR used affinity engineered 8D3 rat anti-mouse TfR. Bivalent anti-mouse TfR 8D3-IL-1RA fusion as chimeric human IgG1 molecules with the S239D/A330L/I332E triple mutation significantly enhances cellular cytotoxicity. | Lower-affinity 8D3 variants showed 30–50-fold increased brain antibody uptake, and the 8D3 variant with 130 nM affinity to mouse TfR showed the maximum brain uptake. IL-1RA-anti-TfR fusions showed dose-dependent inhibition of IL-1β-induced IL6 secretion in NIH-3 T3 cells. 8D3130-IL-1RA fusions showed a significant reversal of partial nerve ligation–induced mechanical hyperalgesia. | Preclinical in mouse | [135] |
| TREM2/AD | A TREM2 antibody in TVD-Ig format with C′ scFv of anti-mouse TfR. Monovalent TfR engagement was confirmed to have broad brain parenchyma distribution. | Weekly treatment of 5XFAD mice (a model of AD) with Ab18 TVD-Ig/αTfR showed a considerable reduction of amyloid burden with increased microglia migration to and phagocytosis of amyloid plaques, improved synaptic and neuronal marker intensity, improved cognitive functions, reduced endogenous tau hyperphosphorylation, and decreased phosphorylated neurofilament H immunostaining. | Preclinical in mouse | [136,137] |
| Transcytosis receptor: CD98hc | ||||
| BACE1/AD | Anti-mouse CD98hc/BACE1 KiH is a bispecific antibody with monovalent binding. CD98hc was identified to be highly expressed in BECs via proteomic study. The ideal CD98hc/BACE1 bispecific antibody has IC50 = 164.4 nM against CD98hc in a competition assay for therapeutic dosing (50 mg/kg). Antibody showed no interference of CD98hc expression, stability, and function. | The lower affinity (CD98hc, IC50 = 164.4 nM) CD98hc/BACE1 bispecific antibody showed a 3-fold increase in brain concentration (measured by ELISA) at therapeutic dosing 50 mg/kg. Both high- and low-affinity CD98hc/BACE1 bispecific antibodies showed a 50% reduction in brain Aβ concentration | Preclinical in mouse | [138] |
| Transcytosis receptor: an unknown receptor that binds FC5 and FC5-derived antibodies | ||||
| Neuropeptides dalargin and neuropeptide Y/Inflammatory pain | Neuropeptides dalargin and neuropeptide Y are chemically conjugated with VHH FC5. FC5 was previously identified to cross both human and mouse BBB by phage-display using llama single-domain library. In the current design, bivalent FC5 fusion to human Fc was used for conjugation. FC5 was previously shown to bind luminal alpha (2,3)-sialoglycoprotein receptor which triggers clathrin-mediated endocytosis. | 30-fold enhanced apparent brain exposure of FC5-conjugates compared with control domain Antibody-Fc fusions after systemic dosing in rats. FC5 conjugates showed dose-dependent inhibition of thermal hyperalgesia in the Hargreaves model after a single systemic injection. | Preclinical in rat | [139–141] |
| mGluR1/thermal hyperalgesia | mGluR1-BBB was the fusion between FC5 and antibody targeting mGluR1 (mGluR1-IgG). Fusion protein showed unaltered mGluR1 antagonist activity in comparison to the parent mGluR1-IgG. mGluR1-BBB showed dose-dependent binding to rat (SV-ARBEC) and mouse (bEnd.3) BECs. | mGluR1-BBB showed up to a 10-fold increase in CSF concentration versus parent mGluR1-IgG. Significant inhibition of thermal hyperalgesia by BBB-mGluR1 treatment in rats with induced chronic inflammatory pain. | Preclinical in rat | [142] |
| IL-1RA/mechanical hyperalgesia | The BBB targeting antibody was screened by phage display in scFv format for competition with FC5 for binding mouse BEC line, bEND.3 (named as BBBt). | The BBBt-IL-1RA showed a 3–4-fold increase in brain concentration versus control IgG. Single IV injection of BBBt-IL-1RA showed long-lasting reversal of mechanical hyperalgesia in the Seltzer model of neuropathic pain versus control IgG-IL-1RA. | Preclinical in mouse | [143] |
EPO, erythropoietin.
Figure 1.
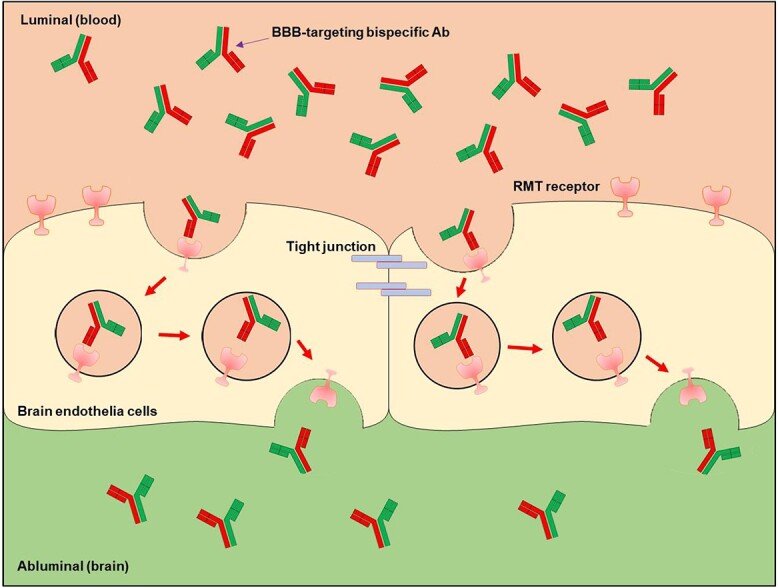
Illustration of the transcytosis of carrier-cargo fusion antibodies through the BBB. Here the illustration uses a bispecific antibody as the example, although other formats also work under the same mechanisms, as shown in Fig. 2. Bispecific antibodies with BBB targeting moiety and cargo bind RMT receptor on the surface of BECs. Then, the antibody-receptor complex undergoes transcytosis to the abluminal side into the brain parenchyma.
The transferrin receptor
There are two known TfRs, TfR1 and TfR2. TfR1 (or TfR) has been extensively studied for the delivery of protein and antibody therapies across the BBB due to its high expression level in BECs [99]. TfR is a type II transmembrane receptor and is a homodimer linked by disulfide bonds at Cys89 and Cys98 [144]. The extracellular domain of TfR consists of three domains, apical domain, helical domain, and protease-like domain. TfR binds iron-bound holo-Tf via the helical and protease-like domains. At neutral pH, iron binds tightly to Tf, and the iron/Tf complex is trafficked intracellularly via TfR. In the low-PH endosomes, iron is released from Tf. The apo-Tf which is complex with TfR is trafficked back to the cell surface. At neutral pH, the low affinity of apo-Tf to TfR results in the release of apo-Tf [144,145]. TfR is ubiquitously expressed, with erythroid cells and proliferating cells expressing high levels due to the metabolic demand for iron [146,147]. BECs were reported to express TfR as well [99]. Transferrin was shown to be transported through the BBB, indicating the TfR pathway can naturally transport macromolecules across the BBB [148].
Using transferrin directly as the carrier is not practical for multiple reasons, including the high circulating endogenous concentration of transferrin at 3 mg/mL. Therefore, using antibodies to target TfR as the carrier for transporting cargo through the BBB is more feasible. To avoid the interference of normal TfR functions of shuttling iron/Tf and apo-Tf across cell membranes and the BBB, the antibody carrier should not compete with Tf binding to TfR [136]. For example, currently discovered carriers targeting TfR often bind at the apical domain of TfR, which is distant from the sites where Tf binds TfR [121–124]. TfR-based BBB-crossing antibody carriers are often used to deliver three types of cargos: enzymes, recombinant proteins, and therapeutic antibodies. In each category, we discuss representative examples of the distinct designs and their effects on efficacy.
An example of anti-TfR/enzyme fusion is the TfRMAb-IDUA for the treatment of LSDs [134]. TfRMAb-IDUA is an anti-mouse TfR antibody-IDUA fusion, with TfR Mab at the N-terminus and IDUA at the C-terminus (Fig. 2, design (1)). The TfR Mab has a high affinity of 1.8 nM. TfRMAb-IDUA fusion was also validated for unaltered IDUA enzyme activity upon fusion at 776 ± 79 units/μg. Treatment of the MPSI mice with TfRMAb-IDUA reduced the intracellular lysosomal inclusion bodies by 73% in the brain (vs. saline-treated mice) with IV injections twice a week for 8 weeks at a 1 mg/kg dose [134].
Figure 2.
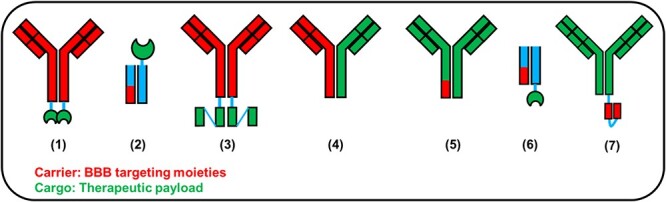
Illustration of common designs of BBB targeting antibody or protein designs. The illustration was based on individual references listed in the articles. (1) TfRMAb-IDUA [134], (2) ETV:IDS [123,124], (3) cTfRMAb-ScFv [130], (4) anti-TfR/BACE1 [121], (5) ATV:BACE1 [122], (6) PTV:PGRN [125], (7) Ab18 TVD-Ig/αTfR [137].
Another example of anti-TfR/enzyme fusion was an enzyme transport vehicle (ETV) fused with the lysosomal enzyme iduronate 2-sulfatase (IDS), known as ETV:IDS, which delivers IDS for treating Hunter’s syndrome [123,124]. IDS is fused to the N-terminus of an engineered Fc fragment that binds to human TfR (called the ETV) (Fig. 2, design (2)). Different from other anti-TfR-based carriers, ETV used directed-evolution engineered Fc fragment binding apical domain of human TfR but without compromising FcRn binding, which preserves the long circulating half-life of the construct. The ETV has low-affinity monovalent binding (130 nM) to TfR. The design possesses multiple advantages such as (1) the native Fc configuration maintains antibody PK profiles, (2) low affinity to TfR allows efficient release of antibodies after entering the brain, and (3) lower affinity to TfR reduces peripheral clearance. ETV:IDS maintains the biological activity of IDS in MPS II patient fibroblasts. ETV:IDS showed 3–5-fold higher brain uptake than Fc:IDS in human TfR KI mice [123,124]. Single or multiple doses of ETV:IDS reduced the brain and CSF sGAG levels by 3–5-fold in comparison to IDS. ETV:IDS significantly reduced GAGs in neurons, astrocytes, and microglia and reduced the lysosomal lipid accumulation in the brain [123,124].
An example of bivalent TfRMAb fusion with an amyloid Ab single-chain variable fragments (scFv) is shown in Fig. 2, design (3). This design has the TfR MAb at the N-terminus and amyloid Ab scFv at the C-terminus [130]. The entire construct is a bivalent fusion with bivalent TfR MAb and bivalent amyloid Ab scFv. Daily subcutaneous (sc) injections of 5 mg/kg of the cTfRMAb-ScFv fusion for 12 consecutive weeks reduced the amyloid plaque in the cortex and hippocampus in 1-year-old presenilin (PS)-1/APP AD double transgenic mice [130].
Another example of an anti-TfR-based carrier is a bispecific antibody that consists of an anti-TfR antibody and an anti-BACE1 antibody (anti-TfR/BACE1) (Fig. 2, design (4)) [121]. The anti-BACE1 inhibits the BACE1 activities and thus reduces soluble amyloid peptides in the brain. Different from the cTfRMAb-ScFv fusion, the anti-TfR and anti-BACE1 are both monovalent. The bispecific antibody was assembled into a heterodimer on the knob-into-hole (KiH) platform. Anti-TfR binds to both human and cynomolgus monkey TfR with low affinities, 270 nM binds to huTfR, and 810 nM binds to cynoTfR. The advantage of low-affinity TfR binding is less degradation of TfR in the BBB and more efficient release of anti-TfR/BACE1 upon entering the brain. Anti-TfR/BACE1 has abolished the Fc effector functions to avoid depletion of the red blood cells (RBCs). Anti-TfR/BACE1 showed a 3–5-fold increase in brain uptake in both human TfR KI mice and cynomolgus monkeys. In addition, therapeutic effects were observed as reduced Aβ concentration in both plasma and CNS (both in mice and monkeys) [121]. Other examples using a similar design are listed in Table 2.
Another distinct design is the antibody transport vehicle (ATV): BACE1, which has the Fc fragment of a bivalent BACE1 Ab replaced with an engineered Fc fragment that binds to huTfR at low affinity (120 nM binds to huTfR and 2200 nM binds to cynoTfR) (Fig. 2, design (5)) [122]. ATV:BACE1 has monovalent binding to TfR and bivalent binding to BACE1. The ATV molecules have a native IgG structure and stability. ATV:BACE1 showed >20-fold increase in brain uptake than the BACE1 antibody in human TfR KI mice. The increased brain uptake also showed an >2-fold decrease in brain Aβ load. ATV:BACE1 showed a 50% reduction of CSF Aβ40 in cynomolgus monkeys and maintained the effect for 14 days after a single IV injection. In comparison, the BACE1 antibody showed no CSF Aβ40 reduction [122].
Zhao et al. isolated a mouse TfR-specific antibody from the phage-displayed antibody libraries and constructed αTfR-containing bispecific antibodies to facilitate the delivery of antibody therapies crossing the BBB. In the bispecific antibody design, monovalent TfR engagement showed a 5-fold higher brain antibody concentration than the bivalent TfR engagement [136]. It was found that the bivalent TfR engagement trapped the antibody in the vasculature, while monovalent TfR engagement showed broad brain parenchyma distribution. Further mechanistic studies found that bivalent TfR engagement significantly increased the lysosomal degradation of TfR, whereas monovalent TfR engagement did not. Using the TfR targeting bispecific antibody, the authors successfully enhanced triggering receptor expressed on myeloid cells 2 (TREM2) antibody delivery into the mouse brain by 10-fold. Compared to the bispecific TREM2 antibody without TfR engagement, weekly treatment of 5XFAD mice (a model of AD) with Ab18 tetra-variable domain (TVD)-immunoglobulin (Ig)/αTfR (Fig. 2, design (7)) showed a considerable reduction of amyloid burden, with an increased microglia migration to and phagocytosis of amyloid plaques, improved synaptic and neuronal marker intensity, improved cognitive functions, reduced endogenous tau hyperphosphorylation, and decreased phosphorylated neurofilament H immunostaining [137].
The insulin receptor
The InsR is a single-pass type I transmembrane receptor with two subunits (α- and β-chains) connected by disulfide bonds. Insulin binds to two distinct sites on each subunit of the receptor, crosslinking the two receptor halves to create a high binding affinity [149,150]. Insulin-receptor binding activates the intracellular tyrosine kinase catalytic domain, which then triggers the downstream signaling cascades and regulates various biological functions, such as glucose uptake. Insulin-bind to InsR can be internalized [151]. InsR is highly expressed by BECs, and it transports insulin across the BBB by an active and energy-dependent process since insulin is indispensable to the brain glucose metabolism. However, insulin is not an appropriate carrier for transportation due to the potential dysfunction of carbohydrate metabolism. Therefore, using InsR antibodies as carriers for transporting cargo across the BBB is more feasible. Anti-InsR-based BBB-crossing carriers have been studied to deliver enzymes or recombinant functional proteins and therapeutic antibodies. In each category, we discuss representative examples of distinct designs and the effects of the different configurations on efficacy.
A human InsR (HIR) antibody 83–14 has been extensively studied as a HIR-mediated brain delivery vehicle [101–104]. It is noted that antibody 83–14 is non-competing with insulin for binding InsR [99]. In this design, the anti-InsR 83–14 is typically configured at the N-terminus and the enzyme or protein is typically configured at the C-terminus with the entire construct as bivalent antibody-enzyme/protein fusion (Fig. 2, design (1)). A specific example is the HIR MAb-IDUA fusion (valanafusp alpha), which is under evaluation in phase 2 clinical trials for the treatment of Hurler’s syndrome. Valanafusp alpha is a bivalent HIR MAb fused to IDUA, with HIR MAb at the N-terminus and IDUA at the C-terminus (Fig. 2, design (1)). HIR antibody was verified to maintain high-affinity InsR binding, and the IDUA maintains its enzymatic activities. Preclinical studies in Rhesus monkeys showed a >12-fold brain distribution increase versus IDUA as measured by radioactivity [105,106]. In a 52-week open-label phase 1–2 trial on the safety and efficacy in pediatric MPSI patients with cognitive impairment, valanafusp alpha demonstrated the effects in stabilizing the cortical gray matter volume of the brain, urinary glycosaminoglycan levels, spleen volumes, and shoulder range of motion [101–104]. Other HIR MAb-enzyme/protein fusion examples that are being studied in preclinical nonhuman primates are listed in Table 2. All the HIR MAb-enzyme/protein fusions demonstrated a 10–40-fold increase in the Rhesus monkey protein brain distribution versus the enzymes/proteins themselves with HIR MAb measured by radioactivity.
Fusion of anti-InsR with therapeutics antibodies is less studied, and no molecules in this category have reached human clinical trials. One representative example is the HIR MAb-anti-amyloid fusion in which the HIR MAb was fused with amyloid Ab as a bivalent fusion protein with HIR MAb at the N-terminus and the amyloid Ab scFv at the C-terminus (Fig. 2, design (3)). High affinity HIR binding, EC50 = 1.9 ± 0.1 nM, and high Aβ-binding activity, ED50 = 2.0 ± 0.8 nM, were validated. In Rhesus monkeys, the HIR MAb-anti-amyloid antibody fusion showed a >10-fold increase in brain uptake versus anti-amyloid antibody alone. The amount of HIR MAb-anti-amyloid antibody fusion entering BBB reached 1% injected dose (ID)/100 g tissue as measured by radioactivity [115,116].
Beyond the well-studied TfR and InsR, efforts are being made to identify new receptors that may bring higher efficiency for transporting protein/antibody cargos across the BBB, with a focus on new RMT receptors highly expressed on the BECs. Some RMT receptors are listed in Table 2.
CONSIDERATIONS IN ENGINEERING CARRIER-CARGO FUSIONS FOR BRAIN DELIVERY OF PROTEIN/ANTIBODY THERAPIES
Highly expressed at BBB
The ideal RMT receptor candidates are often highly expressed in the BECs such as TfR. Proteomics study of the species of interest will aid the identification of other highly expressed RMT receptors in the brain capillary or BECs for brain delivery [138]. Species-specific RMT receptor expression profiles are often different from species to species, and the conclusion learned from rodents needs to be validated in nonhuman primates or humans. For example, InsR is highly expressed in human BBB but much less in mouse BBB, resulting in less transportation to mouse brains which limits the use of mice as preclinical animal models [99]. Detailed case studies on the optimized design of carrier-cargo fusions for brain delivery are summarized in Table 3 and are depicted in Fig. 3.
Table 3.
Summary of engineering tips learned from previous cases studying macromolecule BBB crossing
| Factor to consider | Description/details | Ref |
|---|---|---|
| Category: choice of receptors for the carrier | ||
| Highly expressed at BBB | Highly expressed in the BECs Identify novel RMTs by proteomics |
[138,152–154] |
| Antibody cross-reactivity | Validate expression in species-of-interest Preclinical studies require cross-reactivity to nonhuman primates’ RMT |
[103,121,155] [122,139–143,156] |
| Category: antibody format engineering | ||
| Fusion to naturally existing transcytosis ligands should be avoided | Being blocked by natural ligands present at high concentration Perturbing normal biological functions |
[144,155,157] |
| Antibody and fusion format | Valency of the antibody targeting RMT is important Bivalency may not be preferred for the carrier, but may be preferred for the cargo Protein fusion order or configuration matters, depending on the optimal functions Fusion protein produced using genetic engineering is preferred over conjugation due to more homogeneity Overall structure stability Optimal activities of the cargo should be retained |
[102,108,109,117–120,122] |
| Antibody affinity to RMT targets | High-affinity binding may induce receptor degradation and compromise further transcytosis thus limiting brain uptake High-affinity binding promotes peripheral receptor-mediated clearance, causes significant shorter circulation and lower BBB crossing efficiency High-affinity binding prevents the release of transported antibodies, instead, being trapped in the vasculature |
[102,106,115–118,120,121,135,138] |
| Antibody effector function | Abolish Fc effector functions if causing toxicities | [119] |
| Antibody binding RMT receptor epitope | Avoid competing with natural ligands | [120,121] |
| Antibody endocytosis | Verify endocytosis by in vitro cell models | [107–109,120,122,142,158] |
| Category: in vivo characterization | ||
| Blood circulation time | Longer circulation time is preferred to increase exposure to brain Introducing Fc mutations that increase FcRn binding at pH = 6.0 |
[117,121,135,159] |
| Verify brain distribution | Quantifying antibody concentration from brain lysate is not sufficient Broad brain distribution should be confirmed using imaging |
[102,107,117,121] |
| Quantification of brain uptake | Quantification using ELISA or radioactivity Brain capillary depletion or perfusion with PBS should be performed before quantification in order to exclude possible interferences from blood |
[102,107,117,121] |
| Confirm biological effects or therapeutic efficacy | Using appropriate animal models to confirm improved therapeutic efficacy is required as the final verification of true brain entry | [120–123,126–129,131–133,135,138,141–143] |
Figure 3.
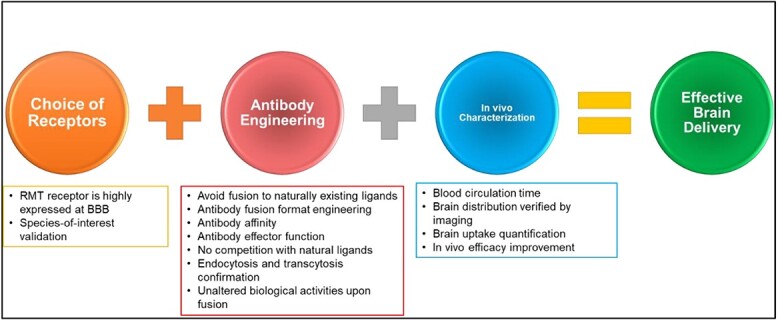
Illustration showing the summarized considerations for obtaining effective brain antibody and protein delivery. Authors should consider first the choice of receptors, which include the RMT receptor is highly expressed at BBB and species-of-interest validation. In antibody engineering, authors should consider several factors, including avoiding fusion to naturally existing ligands, antibody fusion format engineering, antibody affinity, antibody effector function, no competition with natural ligands, endocytosis, and transcytosis confirmation, and unaltered biological activities upon fusion. At last, the in vivo evaluation is needed to validate the therapeutic efficacy and delivery efficiency, including blood circulation time, brain distribution verified by imaging, brain uptake quantification, and in vivo efficacy improvement.
Antibody cross-reactivity
Antibody cross-reactivity is an important consideration. Antibodies often target specific epitopes in the RMT receptors, which may not be conserved between species. For example, the majority of the reported TfR antibodies do not cross-react between humans and mice and even between mice and rats or human and nonhuman primates [117–119,121]. This implies that it is often necessary to generate transgenic mouse models with the desired receptor knock-in to evaluate the biological and physical properties of the drug molecule preclinically. Because it is not readily feasible to generate transgenic nonhuman primate models with the desired receptor knock-in, it is essential to have antibodies that are cross-reactive to nonhuman primate and human RMT receptors.
Avoid natural ligands as carriers
Although RMT receptors transport natural ligands, it is often not feasible to use natural ligands directly as the carrier due to multiple concerns. First, natural ligands present in high concentrations with strong binding, which could block the receptor engagement of the carrier-cargo fusions. Second, the natural ligand triggers signaling upon binding to the receptor. If using a natural ligand as the carrier, dysfunctional physiological effects may result in adverse side effects. An example is insulin, which triggers carbohydrate metabolism after binding to InsR. Third, the natural ligand fusion may perturb normal biological functions by competing with natural ligands. Therefore, most of the known successful anti-RMT platforms use antibodies that do not compete with natural ligands binding to the receptor.
Optimal fusion design
Specific optimization may be needed for the optimal efficiency of the carrier-cargo constructs. For the optimal functions of the carrier, for example, bivalent targeting TfR was found to cause lysosomal degradation of TfR, significantly reducing the overall transportation efficiency [120,136]. Also, the avidity effects from bivalent anti-TfR binding resulted in >100-fold stronger binding to TfR [117–119]. Without a proper level of dissociation from TfR, even if the anti-TfR-cargo is transported inside the brain, they cannot diffuse into the brain parenchyma, resulting in being trapped inside the vasculature [117–119,121,136]. The overall structural stability of the carrier-cargo fusion is also critical. For example, compared to the traditional bispecific antibody anti-TfR/BACE1, the ATV:BACE1 is more preferred due to better overall stability, which can improve the PK profiles and subsequently increase delivery to the brain [122].
For the optimal activities of the cargos, both fusion configurations and valency need to be determined case by case for individual cargos. For example, the ATV:BACE1 was preferred over the anti-TfR/BACE1 because the bivalent anti-BACE1 from ATV:BACE1 neutralizes BACE1 significantly better [122]. Moreover, the ETV enzyme delivery platform was optimized for different enzymes. While IDS tolerates being fused at the C-terminus, progranulin (PGRN) requires a free C-terminus to be functional [123–125] (Fig. 2, design (6)). In the anti-InsR-enzyme fusions, fusion proteins are often validated for similar InsR binding and enzyme activity in comparison to the parent antibodies and enzymes [101–106].
Optimal affinity to RMT receptors
High affinity to RMT receptors may not be always optimal for the carrier antibody. As learned from TfR, high-affinity TfR binding induces receptor degradation by promoting trafficking to the lysosome [117–119]. The reduction of TfR will compromise subsequent transcytosis, thus limiting brain uptake. Additionally, due to the broad peripheral TfR expression, high-affinity anti-TfR showed significantly shorter circulation time due to peripheral clearance [117–119]. Shorter circulation time limits the exposure to the brain since transcytosis at the BBB interface is concentration dependent. More importantly, the high-affinity TfR antibody will remain bound to TfR after transcytosis into the brain side, trapping the antibody near vasculature and limiting broad brain distribution [120]. Endocytosis and transcytosis of candidate carrier antibodies should be verified. For example, a TfR targeting antibody was verified for the cellular uptake and transcytosis using BEC lines, such as hCMEC/D3, bEnd3 cells, and SV-ARBEC [120]. The anti-HIR antibody 83–14 was also validated for endocytosis in vitro [99].
Avoid antibody effector functions
Antibody Fc engineering may be required to eliminate the Fc-effector function–mediated toxicities. For example, TfR is known to be expressed during RBC development; therefore, designs with uncompromised Fc effector functions were shown to deplete RBC and cause acute toxicity upon injection [117–119]. The binding to BECs may cause cell killing and lead to significant side effects if the RMT receptors have broad peripheral distribution.
A long time in blood circulation is preferred
A longer circulation time of the carrier-cargo molecules is preferred to increase exposure to the brain. Due to the broad expression of RMT receptors, such as TfR and InsR in the periphery, the BBB-crossing antibodies often exhibit rapid clearance from the blood. For example, the HIR MAb-IDUA fusion showed an even quicker clearance than the IDUA enzyme in circulation [101–104]. Anti-TfR antibodies show a significantly shorter circulation time than the control IgGs [117–119].
Verify brain distribution
Quantifying antibody concentration in the brain lysate is not sufficient to confirm the actual brain entry and broad distribution into the brain parenchyma. Even if a high concentration is observed in the brain lysate, the antibodies could just be trapped inside the vasculature [117–120]. Therefore, brain distribution should be confirmed using immunostaining or radioautography. For example, low-affinity anti-TfR was found to co-localize with neuronal marker NeuN using the immunofluorescent imaging of brain sections [122]. Also, monovalent sFab was found to engage plaques, while the bivalent dFab was found to remain mostly inside microvessels [120]. Radioautography was used to confirm the brain tissue distribution of injected radioisotope-labeled HIR MAb-IDUA fusion protein [101–104].
Depending on the labeling method and sensitivity of the assay, quantification using enzyme-linked immunosorbent assay (ELISA) or radioactivity has been explored. For injection with trace dosing (0.1–0.5 mg/kg), radioisotope labeling is suitable due to its high sensitivity [101–104]. For injection at therapeutic dosing (>20 mg/kg), quantification by ELISA has been successful [117–119]. For example, when quantifying anti-TfR brain distribution, the brain antibody concentration was quantified using sandwich ELISA. It should be noted that brain capillary depletion or perfusion with PBS should be performed before quantification to exclude possible interferences from blood, which has a much higher antibody concentration than that in the brain tissues.
Verify therapeutic efficacy
The most important proof of drug brain delivery is therapeutic efficacy. Using appropriate animal models to confirm the improved therapeutic efficacy is required as the final verification of true brain entry. A major issue is that most of the disease models are only available in mice and rats, rendering NHP and human correlation more difficult. If a biomarker is available in the nonhuman primate, it is preferred to predict human therapeutic outcomes. For example, the anti-TfR-BACE1 bispecific antibody was shown to reduce the brain and plasma Aβ concentration in NHP in addition to mice [121,122].
PERSPECTIVE
RMT requires endocytosis of the protein therapeutics, which implies that antibody drug conjugates (ADCs) are not likely to be deliverable through RMT due to toxicities to the endothelial cells. In addition, the RMT receptors may not be specific to cancer, and thus bispecific ADC will cause strong side effects, depending on the distribution of the RMT receptors in normal organs. Hypothetically, delivery of ADCs via TfR will likely result in lethal toxicities considering the broad distribution of TfR in the body. Similarly, any therapeutic modalities that rely on the effector functions of antibodies are not suitable for delivery using RMT, which was also illustrated in the case of TfR-mediated antibody delivery. Anti-TfR with bispecific antibodies with full effector functions also causes lethal side effects, such as RBC depletion. Therefore, discovery of novel RMT targets with CNS-restricted expression is necessary to avoid systemic toxicities. The advancement of RNA-seq and proteomics will accelerate the identification of novel CNS-specific RMT targets.
The widely used BBB crossing platforms, such as TfR and InsR, are not ideal RTM receptor systems, as they can lead to fast clearance from the blood due to their broad peripheral distribution, which limits brain entry. While strategies, such as low-affinity TfR binding antibodies and monovalent carrier design, can minimize the fast clearance in blood, high and more frequent dosing may also be needed to compensate for the clearance and achieve the therapeutic concentration inside the brain. Therefore, there is a need to identify new RMT receptors that are more specific to the brain. Future engineering approaches will focus on further boosting the BBB crossing efficiency, thus allowing a much lower dose to be used. As more protein- and antibodies-based therapies target CNS diseases with enhanced BBB crossing properties entering human clinical trials, we will gain new insights on how to further improve brain exposure to macromolecule therapeutics.
ACKNOWLEDGEMENTS
We thank Dr. Georgina To’a Salazar for discussions during the writing process.
Contributor Information
Peng Zhao, Texas Therapeutics Institute, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center at Houston, 1825 Pressler Street, Houston, Texas, USA.
Ningyan Zhang, Texas Therapeutics Institute, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center at Houston, 1825 Pressler Street, Houston, Texas, USA.
Zhiqiang An, Texas Therapeutics Institute, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center at Houston, 1825 Pressler Street, Houston, Texas, USA.
FUNDING
This study was funded in part by The Cancer Prevention and Research Institute of Texas (CPRIT) (RP190561) and Welch Foundation (AU-0042-20030616).
CONFLICT OF INTEREST STATEMENT
The University of Texas System has filed patent applications (TREM2 ANTIGEN BINDING PROTEINS AND USES THEREOF, 63/243431 and TfR ANTIGEN BINDING PROTEINS AND USES THEREOF, 63/243453) on the TfR and TREM2 targeting antibodies. P.Z., N.Z., and Z.A. are named as the inventors of the patent applications. Z.A. holds the position of deputy editor-in-chief for Antibody Therapeutics and is blinded from reviewing or making decisions for the manuscript.
DATA AVAILABILITY
No data were involved in the writing of the manuscript. For the information on the references and figures, the corresponding authors should be contacted.
ETHICS AND CONSENT STATEMENT
The current paper does not involve any animal or human experiments; therefore, consent was not required.
ANIMAL RESEARCH STATEMENT
No animal experiments were conducted.
LIST OF ABBREVIATIONS
AD, Alzheimer’s disease; ADCs, antibody drug conjugates; Aβ, amyloid-β; APP, amyloid precursor protein; ATV, antibody transport vehicle; BACE1, beta-secretase 1; BBB, blood–brain barrier; BECs, brain endothelial cells; CGRP, calcitonin gene-related peptide; CMT, carrier-mediated transport; CNS, central nervous system; CSF, cerebrospinal fluid; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ELISA, enzyme-linked immunosorbent assay; EPO, erythropoietin; ETV, enzyme transport vehicle; Fc, fragment crystallizable; GAGs, sulfated glycosaminoglycans; GBM, glioblastoma; HIR, human insulin receptor; IDS, iduronate 2-sulfatase; IDU, alpha-l-iduronidase; InsR, insulin receptor, Ig, immunoglobulin; KiH, knob-into-hole; LSDs, lysosomal storage diseases; mAb, monoclonal antibody; PD, Parkinson’s disease; PGRN, progranulin; PS, presenilin; rhIDU, recombinant human α-l-iduronidase (laronidase); RMT, receptor-mediated transcytosis; sc, subcutaneous; scFv, single-chain variable fragment; Syn, synuclein; Tf, transferrin; TfR, transferrin receptor; TNFα, tumor necrosis factor-alpha; TREM2, triggering receptor expressed on myeloid cells 2; TVD, tetra-variable domain; VEGF, vascular endothelial growth factor.
References
- 1. Tietz, S, Engelhardt, B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol 2015; 209: 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee, G, Dallas, S, Hong, Met al. . Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev 2001; 53: 569–96. [PubMed] [Google Scholar]
- 3. Brightman, MW, Reese, TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol 1969; 40: 648–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reese, TS, Karnovsky, MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 1967; 34: 207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ek, CJ, Dziegielewska, KM, Stolp, Het al. . Functional effectiveness of the blood-brain barrier to small water-soluble molecules in developing and adult opossum (Monodelphis domestica). J Comp Neurol 2006; 496: 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Janzer, RC, Raff, MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987; 325: 253–7. [DOI] [PubMed] [Google Scholar]
- 7. Stewart, PA, Wiley, MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail--chick transplantation chimeras. Dev Biol 1981; 84: 183–92. [DOI] [PubMed] [Google Scholar]
- 8. Lai, C-H, Kuo, K-H. The critical component to establish in vitro BBB model: pericyte. Brain Res Rev 2005; 50: 258–65. [DOI] [PubMed] [Google Scholar]
- 9. Gaillard, PJ, van derSandt, ICJ, Voorwinden, LHet al. . Astrocytes increase the functional expression of P-glycoprotein in an in vitro model of the blood-brain barrier. Pharm Res 2000; 17: 1198–205. [DOI] [PubMed] [Google Scholar]
- 10. Persidsky, Y, Ramirez, SH, Haorah, Jet al. . Blood–brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 2006; 1: 223–36. [DOI] [PubMed] [Google Scholar]
- 11. Chow, BW, Gu, C. The molecular constituents of the blood–brain barrier. Trends Neurosci 2015; 38: 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hori, S, Ohtsuki, S, Hosoya, KIet al. . A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem 2004; 89: 503–13. [DOI] [PubMed] [Google Scholar]
- 13. Wosik, K, Cayrol, R, Dodelet-Devillers, Aet al. . Angiotensin II controls occludin function and is required for blood–brain barrier maintenance: relevance to multiple sclerosis. J Neurosci 2007; 27: 9032–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Freskgård, P-O, Urich, E. Antibody therapies in CNS diseases. Neuropharmacology 2017; 120: 38–55. [DOI] [PubMed] [Google Scholar]
- 15. Poduslo, JF, Curran, GL, Berg, CT. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A 1994; 91: 5705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felgenhauer, K. Protein size and cerebrospinal fluid composition. Klin Wochenschr 1974; 52: 1158–64. [DOI] [PubMed] [Google Scholar]
- 17. Bien-Ly, N, Boswell, CA, Jeet, Set al. . Lack of widespread BBB disruption in Alzheimer's disease models: focus on therapeutic antibodies. Neuron 2015; 88: 289–97. [DOI] [PubMed] [Google Scholar]
- 18. Yu, YJ, Watts, RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013; 10: 459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang, AC, Stevens, MY, Chen, MBet al. . Physiological blood–brain transport is impaired with age by a shift in transcytosis. Nature 2020; 583: 425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He, Q, Liu, J, Liang, Jet al. . Towards improvements for penetrating the blood-brain barrier-recent progress from a material and pharmaceutical perspective. Cell 2018; 7: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Banks, WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 2016; 15: 275–92. [DOI] [PubMed] [Google Scholar]
- 22. Neyns, B, Sadones, J, Joosens, Eet al. . Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol 2009; 20: 1596–603. [DOI] [PubMed] [Google Scholar]
- 23. Yang, S-B, Gao, K-D, Jiang, Tet al. . Bevacizumab combined with chemotherapy for glioblastoma: a meta-analysis of randomized controlled trials. Oncotarget 2017; 8: 57337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Black, RS, Sperling, RA, Safirstein, Bet al. . A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord 2010; 24: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salloway, S, Sperling, R, Gilman, Set al. . A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009; 73: 2061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rinne, JO, Brooks, DJ, Rossor, MNet al. . 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 2010; 9: 363–72. [DOI] [PubMed] [Google Scholar]
- 27. Salloway, S, Sperling, R, Fox, NCet al. . Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med 2014; 370: 322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siemers, ER, Friedrich, S, Dean, RAet al. . Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease. Clin Neuropharmacol 2010; 33: 67–73. [DOI] [PubMed] [Google Scholar]
- 29. Farlow, M, Arnold, SE, vanDyck, CHet al. . Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimers Dement 2012; 8: 261–71. [DOI] [PubMed] [Google Scholar]
- 30. Doody, RS, Thomas, RG, Farlow, Met al. . Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med 2014; 370: 311–21. [DOI] [PubMed] [Google Scholar]
- 31. Bohrmann, B, Baumann, K, Benz, Jet al. . Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers Dis 2012; 28: 49–69. [DOI] [PubMed] [Google Scholar]
- 32. Ostrowitzki, S, Deptula, D, Thurfjell, Let al. . Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol 2012; 69: 198–207. [DOI] [PubMed] [Google Scholar]
- 33. Lasser, R, Ostrowitzki, S, Scheltens, Pet al. . DT-01-03: efficacy and safety of gantenerumab in prodromal Alzheimer's disease: results from scarlet road—a global, multicenter trial. Alzheimers Dement 2015; 11: P331–2. [Google Scholar]
- 34. Bateman, RJ, Benzinger, TL, Berry, Set al. . The DIAN-TU next generation Alzheimer's prevention trial: adaptive design and disease progression model. Alzheimers Dement 2017; 13: 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adolfsson, O, Pihlgren, M, Toni, Net al. . An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci 2012; 32: 9677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ultsch, M, Li, B, Maurer, Tet al. . Structure of crenezumab complex with Aβ shows loss of β-Hairpin. Sci Rep 2016; 6: 39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cummings, JL, Cohen, S, vanDyck, CHet al. . ABBY: a phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018; 90: e1889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salloway, S, Honigberg, LA, Cho, Wet al. . Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer's disease (BLAZE). Alzheimers Res Ther 2018; 10: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sevigny, J, Chiao, P, Bussière, Tet al. . The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 2016; 537: 50–6. [DOI] [PubMed] [Google Scholar]
- 40. Wang, S, Mustafa, M, Yuede, CMet al. . Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med 2020; 217: e20200785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schenk, DB, Koller, M, Ness, DKet al. . First-in-human assessment of PRX002, an anti–α-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 2017; 32: 211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jankovic, J, Goodman, I, Safirstein, Bet al. . Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol 2018; 75: 1206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brys, M., Ellenbogen, A., Fanning, L.et al. . Randomized phase I clinical trial of anti-α-synuclein antibody BIIB054. Movement disorders : official journal of the Movement Disorder Society 2019; 34: 1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kakkis, ED, Muenzer, J, Tiller, GEet al. . Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001; 344: 182–8. [DOI] [PubMed] [Google Scholar]
- 45. Wraith, JE, Clarke, LA, Beck, Met al. . Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004; 144: 581–8. [DOI] [PubMed] [Google Scholar]
- 46. Wraith, JE, Beck, M, Lane, Ret al. . Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics 2007; 120: e37–46. [DOI] [PubMed] [Google Scholar]
- 47. Muenzer, J, Wraith, JE, Beck, Met al. . A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006; 8: 465–73. [DOI] [PubMed] [Google Scholar]
- 48. Muenzer, J, Gucsavas-Calikoglu, M, McCandless, SEet al. . A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab 2007; 90: 329–37. [DOI] [PubMed] [Google Scholar]
- 49. Goadsby, PJ, Reuter, U, Hallström, Yet al. . A controlled trial of erenumab for episodic migraine. N Engl J Med 2017; 377: 2123–32. [DOI] [PubMed] [Google Scholar]
- 50. Goadsby, PJ, Dodick, DW, Leone, Met al. . Trial of Galcanezumab in prevention of episodic cluster headache. N Engl J Med 2019; 381: 132–41. [DOI] [PubMed] [Google Scholar]
- 51. Silberstein, SD, Dodick, DW, Bigal, MEet al. . Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med 2017; 377: 2113–22. [DOI] [PubMed] [Google Scholar]
- 52. Ashina, M, Saper, J, Cady, Ret al. . Eptinezumab in episodic migraine: a randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia 2020; 40: 241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vécsei, L, Majláth, Z, Szok, Det al. . Drug safety and tolerability in prophylactic migraine treatment. Expert Opin Drug Saf 2015; 14: 667–81. [DOI] [PubMed] [Google Scholar]
- 54. Silberstein, S, Diamond, M, Hindiyeh, NAet al. . Eptinezumab for the prevention of chronic migraine: efficacy and safety through 24 weeks of treatment in the phase 3 PROMISE-2 (prevention of migraine via intravenous ALD403 safety and efficacy-2) study. J Headache Pain 2020; 21: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Razpotnik, R, Novak, N, Čurin Šerbec, Vet al. . Targeting malignant brain tumors with antibodies. Front Immunol 2017; 8: 1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brennan, CW, Verhaak, RGW, McKenna, Aet al. . The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li, J, Di, C, Mattox, AKet al. . The future role of personalized medicine in the treatment of glioblastoma multiforme. Pharmacogenomics Pers Med 2010; 3: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li, J, Liang, R, Song, Cet al. . Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco Targets Ther 2018; 11: 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Das, S, Marsden, PA. Angiogenesis in glioblastoma. N Engl J Med 2013; 369: 1561–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Plate, KH, Breier, G, Millauer, Bet al. . Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res 1993; 53: 5822–7. [PubMed] [Google Scholar]
- 61. Ma, C, Li, Y, Zhang, Xet al. . Levels of vascular endothelial growth factor and matrix metalloproteinase-9 proteins in patients with glioma. J Int Med Res 2014; 42: 198–204. [DOI] [PubMed] [Google Scholar]
- 62. Li, Q, Qiao, G, Ma, Jet al. . Downregulation of VEGF expression attenuates malignant biological behavior of C6 glioma stem cells. Int J Oncol 2014; 44: 1581–8. [DOI] [PubMed] [Google Scholar]
- 63. Shibuya, M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct 2001; 26: 25–35. [DOI] [PubMed] [Google Scholar]
- 64. Selkoe, DJ, Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016; 8: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Prins, ND, Scheltens, P. Treating Alzheimer's disease with monoclonal antibodies: current status and outlook for the future. Alzheimers Res Ther 2013; 5: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Graham, WV, Bonito-Oliva, A, Sakmar, TP. Update on Alzheimer's disease therapy and prevention strategies. Annu Rev Med 2017; 68: 413–30. [DOI] [PubMed] [Google Scholar]
- 67. van Dyck, CH. Anti-amyloid-β monoclonal antibodies for Alzheimer's disease: pitfalls and promise. Biol Psychiatry 2018; 83: 311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sumner, IL, Edwards, RA, Asuni, AAet al. . Antibody engineering for optimized immunotherapy in Alzheimer's disease. Front Neurosci 2018; 12: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Katsinelos, T, Tuck, BJ, Mukadam, ASet al. . The role of antibodies and their receptors in protection against ordered protein assembly in neurodegeneration. Front Immunol 2019; 10: 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bard, F, Cannon, C, Barbour, Ret al. . Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000; 6: 916–9. [DOI] [PubMed] [Google Scholar]
- 71. Siemers, ER, Sundell, KL, Carlson, Cet al. . Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement 2016; 12: 110–20. [DOI] [PubMed] [Google Scholar]
- 72. DeMattos, RB, Bales, KR, Cummins, DJet al. . Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2001; 98: 8850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. DeMattos, RB, Bales, KR, Cummins, DJet al. . Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 2002; 295: 2264–7. [DOI] [PubMed] [Google Scholar]
- 74. Braak, H, Del Tredici, K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the Chaff. J Parkinsons Dis 2017; 7: S71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dauer, W, Przedborski, S. Parkinson's disease: mechanisms and models. Neuron 2003; 39: 889–909. [DOI] [PubMed] [Google Scholar]
- 76. Baba, M, Nakajo, S, Tu, PHet al. . Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 1998; 152: 879–84. [PMC free article] [PubMed] [Google Scholar]
- 77. Zella, SMA, Metzdorf, J, Ciftci, Eet al. . Emerging immunotherapies for Parkinson disease. Neurol Ther 2019; 8: 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang, Z, Gao, G, Duan, Cet al. . Progress of immunotherapy of anti-α-synuclein in Parkinson's disease. Biomed Pharmacother 2019; 115: 108843. [DOI] [PubMed] [Google Scholar]
- 79. Neufeld, EF. Lysosomal storage diseases. Annu Rev Biochem 1991; 60: 257–80. [DOI] [PubMed] [Google Scholar]
- 80. Sands, MS, Haskins, ME. CNS-directed gene therapy for lysosomal storage diseases. Acta Paediatr 2008; 97: 22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Marques, ARA, Saftig, P. Lysosomal storage disorders—challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci 2019; 132. [DOI] [PubMed] [Google Scholar]
- 82. Wang, RY, Bodamer, OA, Watson, MSet al. . Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med 2011; 13: 457–84. [DOI] [PubMed] [Google Scholar]
- 83. Braunlin, E, Steinberger, J, DeFor, Tet al. . Metabolic syndrome and cardiovascular risk factors after hematopoietic cell transplantation in severe mucopolysaccharidosis type I (Hurler syndrome). Biol Blood Marrow Transplant 2018; 24: 1289–93. [DOI] [PubMed] [Google Scholar]
- 84. Edvinsson, L, Haanes, KA, Warfvinge, Ket al. . CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat Rev Neurol 2018; 14: 338–50. [DOI] [PubMed] [Google Scholar]
- 85. Hervé, F, Ghinea, N, Scherrmann, J-M. CNS delivery via adsorptive transcytosis. AAPS J 2008; 10: 455–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Abbott, NJ, Patabendige, AA, Dolman, DEet al. . Structure and function of the blood-brain barrier. Neurobiol Dis 2010; 37: 13–25. [DOI] [PubMed] [Google Scholar]
- 87. Ohtsuki, S, Terasaki, T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res 2007; 24: 1745–58. [DOI] [PubMed] [Google Scholar]
- 88. Jefferies, WA, Brandon, MR, Hunt, SVet al. . Transferrin receptor on endothelium of brain capillaries. Nature 1984; 312: 162–3. [DOI] [PubMed] [Google Scholar]
- 89. Visser, CC, Voorwinden, LH, Crommelin, DJAet al. . Characterization and modulation of the transferrin receptor on brain capillary endothelial cells. Pharm Res 2004; 21: 761–9. [DOI] [PubMed] [Google Scholar]
- 90. Duffy, KR, Pardridge, WM. Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res 1987; 420: 32–8. [DOI] [PubMed] [Google Scholar]
- 91. Banks, WA. The source of cerebral insulin. Eur J Pharmacol 2004; 490: 5–12. [DOI] [PubMed] [Google Scholar]
- 92. Golden, PL, Maccagnan, TJ, Pardridge, WM. Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels. J Clin Invest 1997; 99: 14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Banks, WA, Niehoff, ML, Martin, Det al. . Leptin transport across the blood-brain barrier of the Koletsky rat is not mediated by a product of the leptin receptor gene. Brain Res 2002; 950: 130–6. [DOI] [PubMed] [Google Scholar]
- 94. Pan, W, Kastin, AJ. TNFalpha transport across the blood-brain barrier is abolished in receptor knockout mice. Exp Neurol 2002; 174: 193–200. [DOI] [PubMed] [Google Scholar]
- 95. Pan, W, Kastin, AJ. Entry of EGF into brain is rapid and saturable. Peptides 1999; 20: 1091–8. [DOI] [PubMed] [Google Scholar]
- 96. Georgieva, JV, Hoekstra, D, Zuhorn, IS. Smuggling drugs into the brain: an overview of ligands targeting transcytosis for drug delivery across the blood-brain barrier. Pharmaceutics 2014; 6: 557–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pardridge, WM. Drug transport across the blood–brain barrier. J Cereb Blood Flow Metab 2012; 32: 1959–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xiao, G, Gan, L-S. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int J Cell Biol 2013; 2013: 703545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Freskgård, PO, Urich, E. Antibody therapies in CNS diseases. Neuropharmacology 2017; 120: 38–55. [DOI] [PubMed] [Google Scholar]
- 100. Pulgar, VM. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front Neurosci 2019; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Boado, RJ, Zhang, Y, Zhang, Yet al. . Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol Bioeng 2008; 99: 475–84. [DOI] [PubMed] [Google Scholar]
- 102. Boado, RJ, Pardridge, WM. Brain and organ uptake in the Rhesus monkey in vivo of recombinant iduronidase compared to an insulin receptor antibody-iduronidase fusion protein. Mol Pharm 2017; 14: 1271–7. [DOI] [PubMed] [Google Scholar]
- 103. Giugliani, R, Giugliani, L, deOliveira Poswar, Fet al. . Neurocognitive and somatic stabilization in pediatric patients with severe mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): an open label phase 1–2 trial. Orphanet J Rare Dis 2018; 13: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Pardridge, WM, Boado, RJ, Giugliani, Ret al. . Plasma pharmacokinetics of valanafusp alpha, a human insulin receptor antibody-iduronidase fusion protein, in patients with mucopolysaccharidosis type I. BioDrugs 2018; 32: 169–76. [DOI] [PubMed] [Google Scholar]
- 105. Lu, JZ, Boado, RJ, Hui, EKet al. . Expression in CHO cells and pharmacokinetics and brain uptake in the Rhesus monkey of an IgG-iduronate-2-sulfatase fusion protein. Biotechnol Bioeng 2011; 108: 1954–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Boado, RJ, Ka-Wai Hui, E, Zhiqiang, Let al. . Insulin receptor antibody-iduronate 2-sulfatase fusion protein: pharmacokinetics, anti-drug antibody, and safety pharmacology in Rhesus monkeys. Biotechnol Bioeng 2014; 111: 2317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boado, RJ, Lu, JZ, Hui, EKet al. . Pharmacokinetics and brain uptake in the Rhesus monkey of a fusion protein of arylsulfatase a and a monoclonal antibody against the human insulin receptor. Biotechnol Bioeng 2013; 110: 1456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Boado, RJ, Lu, JZ, Hui, EKet al. . Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells. Mol Pharm 2014; 11: 2928–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Boado, RJ, Lu, JZ, Hui, EKet al. . Insulin receptor antibody-α-N-acetylglucosaminidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in sanfilippo type B fibroblasts. Mol Pharm 2016; 13: 1385–92. [DOI] [PubMed] [Google Scholar]
- 110. Boado, RJ, Hui, EK, Lu, JZet al. . CHO cell expression, long-term stability, and primate pharmacokinetics and brain uptake of an IgG-paroxonase-1 fusion protein. Biotechnol Bioeng 2011; 108: 186–96. [DOI] [PubMed] [Google Scholar]
- 111. Boado, RJ, Hui, EK-W, Lu, JZet al. . Drug targeting of erythropoietin across the primate blood-brain barrier with an IgG molecular trojan horse. J Pharmacol Exp Ther 2010; 333: 961. [DOI] [PubMed] [Google Scholar]
- 112. Boado, RJ, Zhang, Y, Zhang, Yet al. . GDNF fusion protein for targeted-drug delivery across the human blood-brain barrier. Biotechnol Bioeng 2008; 100: 387–96. [DOI] [PubMed] [Google Scholar]
- 113. Ohshima-Hosoyama, S, Simmons, HA, Goecks, Net al. . A monoclonal antibody-GDNF fusion protein is not neuroprotective and is associated with proliferative pancreatic lesions in Parkinsonian monkeys. Plos One 2012; 7: e39036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Boado, RJ, Hui, EK, Lu, JZet al. . Selective targeting of a TNFR decoy receptor pharmaceutical to the primate brain as a receptor-specific IgG fusion protein. J Biotechnol 2010; 146: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Boado, RJ, Lu, JZ, Hui, EKet al. . IgG-single chain Fv fusion protein therapeutic for Alzheimer's disease: expression in CHO cells and pharmacokinetics and brain delivery in the Rhesus monkey. Biotechnol Bioeng 2010; 105: 627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Boado, RJ, Zhang, Y, Zhang, Yet al. . Fusion antibody for Alzheimer's disease with bidirectional transport across the blood-brain barrier and abeta fibril disaggregation. Bioconjug Chem 2007; 18: 447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yu, YJ, Zhang, Y, Kenrick, Met al. . Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med 2011; 3: 84ra44. [DOI] [PubMed] [Google Scholar]
- 118. Bien-Ly, N, Yu, YJ, Bumbaca, Det al. . Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med 2014; 211: 233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Couch, JA, Yu, YJ, Zhang, Yet al. . Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med 2013; 5: 181–12183ra157. [DOI] [PubMed] [Google Scholar]
- 120. Niewoehner, J, Bohrmann, B, Collin, Let al. . Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014; 81: 49–60. [DOI] [PubMed] [Google Scholar]
- 121. Yu, YJ, Atwal, JK, Zhang, Yet al. . Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med 2014; 6: 261ra154. [DOI] [PubMed] [Google Scholar]
- 122. Kariolis, MS, Wells, RC, Getz, JAet al. . Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med 2020; 12: eaay1359. [DOI] [PubMed] [Google Scholar]
- 123. Ullman, JC, Arguello, A, Getz, JAet al. . Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci Transl Med 2020; 12. [DOI] [PubMed] [Google Scholar]
- 124. Sonoda, H, Morimoto, H, Yoden, Eet al. . A blood-brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Mol Ther 2018; 26: 1366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Logan, T, Simon, MJ, Rana, Aet al. . Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 2021; 184: 4651–4668.e4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhou, Q-H, Boado, RJ, Hui, EK-Wet al. . Brain-penetrating tumor necrosis factor decoy receptor in the mouse. Drug Metab Dispos 2011; 39: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sumbria, RK, Boado, RJ, Pardridge, WM. Brain protection from stroke with intravenous TNFα decoy receptor-trojan horse fusion protein. J Cereb Blood Flow Metab 2012; 32: 1933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhou, Q-H, Boado, RJ, Hui, EK-Wet al. . Chronic dosing of mice with a transferrin receptor monoclonal antibody-glial-derived neurotrophic factor fusion protein. Drug Metab Dispos 2011; 39: 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Fu, A, Zhou, Q-H, Hui, EK-Wet al. . Intravenous treatment of experimental Parkinson's disease in the mouse with an IgG-GDNF fusion protein that penetrates the blood–brain barrier. Brain Res 2010; 1352: 208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sumbria, RK, Hui, EK-W, Lu, JZet al. . Disaggregation of amyloid plaque in brain of Alzheimer’s disease transgenic mice with daily subcutaneous administration of a tetravalent bispecific antibody that targets the transferrin receptor and the abeta amyloid peptide. Mol Pharm 2013; 10: 3507–13. [DOI] [PubMed] [Google Scholar]
- 131. Zhou, QH, Boado, RJ, Lu, JZet al. . Re-engineering erythropoietin as an IgG fusion protein that penetrates the blood-brain barrier in the mouse. Mol Pharm 2010; 7: 2148–55. [DOI] [PubMed] [Google Scholar]
- 132. Fu, A, Hui, EK-W, Lu, JZet al. . Neuroprotection in stroke in the mouse with intravenous erythropoietin—trojan horse fusion protein. Brain Res 2011; 1369: 203–7. [DOI] [PubMed] [Google Scholar]
- 133. Chang, R, Al Maghribi, A, Vanderpoel, Vet al. . Brain penetrating bifunctional erythropoietin-transferrin receptor antibody fusion protein for Alzheimer's disease. Mol Pharm 2018; 15: 4963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Boado, RJ, Hui, EK-W, Lu, JZet al. . Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous trojan horse-iduronidase fusion protein. Mol Pharm 2011; 8: 1342–50. [DOI] [PubMed] [Google Scholar]
- 135. Webster, CI, Hatcher, J, Burrell, Met al. . Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti–transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. Pain 2017; 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhao, P, Anami, Y, Gao, Pet al. . Enhanced anti-angiogenetic effect of transferrin receptor-mediated delivery of VEGF-trap in a glioblastoma mouse model. MAbs 2022; 14: 2057269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhao, P, Xu, Y, Jiang, Let al. . A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer’s disease. Sci Transl Med 2022; 14: eabq0095. [DOI] [PubMed] [Google Scholar]
- 138. Zuchero, YJ, Chen, X, Bien-Ly, Net al. . Discovery of novel blood-brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron 2016; 89: 70–82. [DOI] [PubMed] [Google Scholar]
- 139. Muruganandam, A, Tanha, J, Narang, Set al. . Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J 2002; 16: 1–22. [DOI] [PubMed] [Google Scholar]
- 140. Abulrob, A, Sprong, H, En Henegouwen, PVBet al. . The blood–brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem 2005; 95: 1201–14. [DOI] [PubMed] [Google Scholar]
- 141. Farrington, GK, Caram-Salas, N, Haqqani, ASet al. . A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J 2014; 28: 4764–78. [DOI] [PubMed] [Google Scholar]
- 142. Webster, CI, Caram-Salas, N, Haqqani, ASet al. . Brain penetration, target engagement, and disposition of the blood-brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J 2016; 30: 1927–40. [DOI] [PubMed] [Google Scholar]
- 143. Thom, G, Hatcher, J, Hearn, Aet al. . Isolation of blood-brain barrier-crossing antibodies from a phage display library by competitive elution and their ability to penetrate the central nervous system. MAbs 2018; 10: 304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wessling-Resnick, M. Crossing the iron gate: why and how transferrin receptors mediate viral entry. Annu Rev Nutr 2018; 38: 431–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lawrence, CM, Ray, S, Babyonyshev, Met al. . Crystal structure of the ectodomain of human transferrin receptor. Science 1999; 286: 779–82. [DOI] [PubMed] [Google Scholar]
- 146. Levy, JE, Jin, O, Fujiwara, Yet al. . Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 1999; 21: 396–9. [DOI] [PubMed] [Google Scholar]
- 147. Marsee, DK, Pinkus, GS, Yu, H. CD71 (transferrin receptor): an effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol 2010; 134: 429–35. [DOI] [PubMed] [Google Scholar]
- 148. Moos, T, Morgan, EH. Transferrin and transferrin receptor function in brain barrier systems. Cell Mol Neurobiol 2000; 20: 77–95. [DOI] [PubMed] [Google Scholar]
- 149. Haeusler, RA, McGraw, TE, Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 2018; 19: 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Ye, L, Maji, S, Sanghera, Net al. . Structure and dynamics of the insulin receptor: implications for receptor activation and drug discovery. Drug Discov Today 2017; 22: 1092–102. [DOI] [PubMed] [Google Scholar]
- 151. Hall, C, Yu, H, Choi, E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp Mol Med 2020; 52: 911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hoshi, Y, Uchida, Y, Tachikawa, Met al. . Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci 2013; 102: 3343–55. [DOI] [PubMed] [Google Scholar]
- 153. Uchida, Y, Ohtsuki, S, Katsukura, Yet al. . Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem 2011; 117: 333–45. [DOI] [PubMed] [Google Scholar]
- 154. Uchida, Y, Tachikawa, M, Obuchi, Wet al. . A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood–brain barrier in ddY, FVB, and C57BL/6J mice. Fluids and Barriers of the CNS 2013; 10: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Pardridge, WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 2012; 32: 1959–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Haqqani, AS, Caram-Salas, N, Ding, Wet al. . Multiplexed evaluation of serum and CSF pharmacokinetics of brain-targeting single-domain antibodies using a NanoLC–SRM-ILIS method. Mol Pharm 2013; 10: 1542–56. [DOI] [PubMed] [Google Scholar]
- 157. Pardridge, WM. Delivery of biologics across the blood–brain barrier with molecular trojan horse technology. BioDrugs 2017; 31: 503–19. [DOI] [PubMed] [Google Scholar]
- 158. Weksler, B, Romero, IA, Couraud, P-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013; 10: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Wang, X, Mathieu, M, Brezski, RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018; 9: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were involved in the writing of the manuscript. For the information on the references and figures, the corresponding authors should be contacted.