

Abstract
Inhibitors directed towards PARP1 and PARP2 are approved agents for the treatment of BRCA1 and BRCA2 related cancers. Other members of the PARP family have also been implicated in cancer and are being assessed as therapeutic targets in cancer and other diseases. Recently an inhibitor of PARP7 (RBN-2397) has reached early-stage human clinical trials. Here, we performed a genome-wide CRISPR screen for genes that modify the response of cells to RBN-2397. We identify the polycyclic aromatic hydrocarbon receptor AHR and multiple components of the cohesin complex as determinants of resistance to this agent. Activators and inhibitors of AHR modulate the cellular response to PARP7 inhibition, suggesting potential combination therapy approaches.
Keywords: PARP7, PARP7 inhibitor, AHR, CRISPR screen, resistance
Introduction
ADP-ribosyltransferases, collectively referred to as PARP proteins, are a family of 17 related proteins that post-translationally modify target proteins by the addition of ADP-ribose moieties using NAD+ as a substrate. The best characterized PARP, PARP1, has important roles in the DNA damage response pathway and, particularly, in Base Excision Repair (BER)/single strand break repair (SSBR) (1). Inhibitors of PARP1 have received considerable attention as synthetic lethal therapeutic agents for the treatment of BRCA1 and BRCA2 mutant tumors as well as those with other defects in the repair of double strand breaks by homologous recombination (2). Five different PARP inhibitors are now clinically approved internationally for the treatment of BRCA-related ovarian, breast, prostate and pancreatic cancers.
Some PARP enzymes such as PARP1, PARP2, tankyrase 1 (TNKS1 aka PARP5a) and tankyrase 2 (TNKS2 aka PARP5b), attach multiple ADP-ribose residues to target proteins to form poly (ADP) ribose chains. However, most PARPs are mono-ADP ribosyl (MAR) transferases (MARTs) and catalyze the addition of only a single ADP ribose to target proteins (1). PARP enzymes have been described to have roles in diverse cellular functions including DNA repair, immune function, telomere integrity and cellular metabolism. PARP7, also known as TiPARP (3) is a MART containing a C-terminal catalytic domain which predominantly attaches ADP-ribose to cysteine residues in its target proteins, as well as a CCCH-type Zinc finger and a central PAR binding WWE motif (4). PARP7 expression is induced by treatment with TCDD (2,3,7,8-tetra chlorodibenzo-p-dioxin), polycyclic aromatic hydrocarbons and other ligands of the aryl hydrocarbon receptor (AHR) (5). Subsequently, PARP7 acts to negatively regulate the expression of the AHR-target cytochrome P450 genes, CYP1A1 and CYP1B1 (6).
PARP7 has also been identified as a key component of the innate immune system (7). Using chemical genetics and proteomics approaches, PARP7 was shown to modify the catalytically dead PARP13 which is involved in regulating the antiviral innate immune response. In a number of cell types, PARP7 inactivation enhances DNA/RNA or virus induction of interferon beta levels potentially suppressing viral infection. PARP7 also MARylates TBK1 which is a protein kinase that regulates the type I interferon (IFN) response and antiviral immunity (7). These activities lead to PARP7-mediated suppression of the IFN-response upon viral infection.
PARP7 has also been implicated in cancer pathogenesis; the PARP7 gene is amplified and over-expressed in a subset of cancers, particularly those with squamous histology (8). Moreover, it has recently been proposed that PARP7 is a negative regulator of DNA and RNA sensing in cancer cells and inhibition of PARP7 may promote IFN responses in tumors (8). These observations drove efforts to develop PARP7 inhibitors (PARP7i) as potential cancer therapeutics and the discovery of a potent PARP7i RBN-2397 (8). This agent caused enhanced IFN signaling in tumor models, directly inhibiting cell proliferation and activating the immune system to control tumor growth in animal models. Early-stage clinical trials are currently underway with RBN-2397 (https://clinicaltrials.gov/ct2/show/NCT04053673). To explore genetic determinants of sensitivity and resistance to PARP7i we undertook genome-wide CRISPR knockout screens. We found that inactivation of the AHR gene and sister chromatid cohesion components is associated with PARP7i resistance. Furthermore, pharmacological modulation of AHR signaling can alter cellular responses to RBN-2397.
Materials and methods
Cell culture
Human NCI-H1373 cells (CRL-5866), MDA-MB-468 (HTB-132), SW620 (CCL-227), CAL27 (CRL-2095), and NCI-H2347 (CRL-5942) were obtained from the American Type Culture Collection (ATCC, Manassas VA, USA). HAP1, NCI-H1373 and MDA-MB-468 cells were authenticated by short tandem repeat (STR) profiling and tested for mycoplasma at Genetica. NCI-H1373 and NCI-H2347 cells were grown in Roswell Park Memorial Institute (RPMI) 1640 Medium (ATCC modification) with 10% fetal bovine serum and 1% penicillin-streptomycin. HEK293T, SW620, CAL27 and MDA-MB-468 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum and 1% penicillin-streptomycin. Human HAP1 (C631) cells were purchased from Horizon Discovery and grown in Iscove’s Modified Dulbecco’s Medium (IMDM) with 10% fetal bovine serum and 1% penicillin-streptomycin.
Construction of plasmids
The primers used for the construction of plasmids are listed below are in Supplementary Table 1. All plasmids were verified by Sanger sequencing. To construct the V5-Turbo-ID-PARP7 plasmid, we PCR amplified a 2.1 kb PARP7 fragment from the TIPARP cDNA (Origene, RC230398) and a 1.1 kb V5-TurboID-NES fragment from V5-TurboID-NES_pCDNA3 (Addgene plasmid #107169) and used Gibson cloning kit (NEB E5510) to clone these fragments into a Nhe1/Pme1 digested PB-Tre backbone (Addgene plasmid #63800). To construct the PB-TRE-Myc-DDK-AHR plasmid, a Myc-DDK-tagged-AHR fragment was PCR amplified from AHR (Myc-DDK-tagged) plasmid (Origene, RC209832) using PB-TRE-AHR-F and PB-TRE-Myc-DDK-AHR-R Primers and Gibson cloning was used to clone this fragment into the Nhe1/Pme1 digested PB-TRE backbone (Addgene plasmid #63800). The pLenti-C-mGFP-AHR plasmid was made by ligating an AHR cDNA fragment was removed from the AHR (Myc-DDK-tagged) plasmid using SgfI (AsiSI) and MluI into a SgfI (AsiSI)/MluI-digestsed pLenti-C-mGFP-IFI16-P2A-Puro backbone (Origene, RC202193L4). The lentiCRISPRv2 sgRNA plasmids were constructed using the method previously described by the Zhang lab (9,10) and the sgRNA oligos in Supplementary Table 2. A detailed protocol is available at. V5-TurboID-NES_pCDNA3 was a gift from Alice Ting (Addgene plasmid #107169; http://n2t.net/addgene:107169; RRID: Addgene_107169). PB-TRE-dCas9-VPR was a gift from George Church (Addgene plasmid #63800; http://n2t.net/addgene:63800; RRID: Addgene_63800).
CRISPR screens
A lentiviral preparation of the Human sgRNA library Brunello in lentiCRISPRv2 was a gift from David Root and John Doench (Addgene #73178). 480 million NCI-H1373 cells were infected with this library at a multiplicity of infection (MOI) of ~0.3 to ensure that each cell was infected with <1 sgRNA. After 24 h of infection with Brunello library, the infected NCI-H1373 cells were selected for 3 days with 2 μg/ml puromycin. Thereafter, 200 million cells were harvested as the baseline counts for the T00 control, and another 600 million cells were re-plated in 60 150-mm dishes, the 200 million re-plated cells were treated with 0.001‰ DMSO or 10 nM RBN-2397 or 20 nM RBN-2397 (Medchemexpress, HY-136174) for 7 days, respectively. Genomic DNA from 20~30 million cells (250-fold library coverage) was amplified using the p5 and p7 primers indicated in Supplementary Table 3 and purified using a published protocol (https://media.addgene.org/cms/filer_public/61/16/611619f4-0926-4a07-b5c7-e286a8ecf7f5/broadgpp-sequencing-protocol.pdf). Purified products were subjected to Agilent Bioanalyzer and NGS using an Illumina HiSeq4000 (SE50/SE65 reads). MAGEcK software was used to quantify sgRNAs and identify hits (11). The Gene Ontology pathways enriched of top hits was performed using DAVID (DAVID Bioinformatics Resources 6.8) (12,13).
RNA interference
Knockdown experiments were performed using Lipofectamine RNAiMAX transfection (Thermo Fisher Scientific, 13778075) and 25nM Dharmacon siRNA SMARTpools per the manufacturers protocol. SMARTpools, including the ON-TARGETplus Non-targeting Pool was used as control are listed in Supplementary Table 4. Knockdown cells were analyzed or used for cell viability assay 48 hours after transfection.
CRISPR Cas9 ribonucleotide protein preparation and transfection (CRISPR RNPs)
Knockdown experiments by CRISPR RNPs were performed using Lipofectamine CRISPRMAX Transfection Reagent (Thermo Fisher Scientific, CMAX00008) and Cas9 protein (Berkeley MacroLab) and Dharmacon tracrRNA and crRNA for each target (Supplementary Table 5). Transfection according to the Lipofectamine CRISPRMAX Transfection Reagent manufacturer’s protocol. Briefly, mix 1 μl 160 μM crRNA with 1 μl 160 μM tracrRNA, incubated for 30 minutes at 37°C to complex the gRNAs and then added 2 μl 40 μM Cas9 protein to sgRNA complex and incubated at 37°C for 15 min. Finally, mix the Cas9-crRNA-tracrRNA mixture with 10 μl Cas9 Plus Reagent and 10 μl CRISPRMAX Reagent, and incubate for 10 minutes and then added CRISPR RNPs complex to cells. After 48 hours, we analyzed the transfected cells or proceed cell viability assay.
Lentiviral packaging
Lentivirus was prepared as previously described (14). Briefly, 15 million HEK293T cells were transfected 15 million HEK293T cells were grown overnight on 15 cm poly-L-Lysine coated dishes and then transfected with 6 ug pMD2.G (Addgene plasmid # 12259 ; http://n2t.net/addgene:12259 ; RRID:Addgene_12259), 18 ug dR8.91 (since replaced by second generation compatible pCMV-dR8.2, Addgene plasmid #8455) and 24 ug lentiCRISPR-V2 sgRNA plasmids or pBOB-EF1-FastFUCCI-Puro (Addgene plasmid # 86849 ; http://n2t.net/addgene:86849 ; RRID:Addgene_86849) or pLenti6-H2B-mCherry (Addgene plasmid #89766) using the lipofectamine 3000 transfection reagent per the manufacturer’s protocol (Thermo Fisher Scientific, Cat #L3000001). pMD2.G and dR8.91 were a gift from Didier Trono. pBOB-EF1-FastFUCCI-Puro was a gift from Kevin Brindle & Duncan Jodrell. The following day, media was refreshed with the addition of viral boost reagent at 500x as per the manufacturer’s protocol (Alstem, Cat #VB100). Viral supernatant was collected 48 hours post transfection and spun down at 300 g for 10 minutes, to remove cell debris. To concentrate the lentiviral particles, Alstem precipitation solution (Alstem, Cat #VC100) was added, mixed, and refrigerated at 4°C overnight. The virus was then concentrated by centrifugation at 1500 g for 30 minutes, at 4°C. Finally, each lentiviral pellet was resuspended at 100x of original volume in cold DMEM+10%FBS+1% penicillin-streptomycin and stored until use at −80°C.
Establishment of Individual CRISPR Knockout Cells
To generate knockout clones for individual genes, NCI-H1373 cells were transduced with lentiV2 plasmids containing sgRNAs of hit genes (two guides per gene chosen from the Brunello CRISPR library) or a nontargeting sgRNA (sgNC). Infected cells were selected for 3 days with 2 μg/ml puromycin and single cell cloned by limiting dilution. Individual clones were screened and validated by western.
Cell viability assay
Short-term survival assays were performed as previously described (15). In brief, cells were seeded into 96-well plates at a concentration of 1000 cells per well. The next day, cells were treated with RBN-2397, tapinarof (Selleck Chemicals, S9700), L-kynurenine (Invivogen, tlrl-kyn) and/or CH-223191 (Sigma, C8124) at the indicated concentrations. After 7 days, cell viability was assed Cell Titre-Glo assay (Promega, G7572).
Western Blotting
Cell lysates were prepared in Pierce RIPA Buffer (Thermo Fisher Scientific, 89901) containing protease inhibitor and PhosStop cocktails (Roche, 5892970001 and 4906845001), separated on an SDS-polyacrylamide gel and transferred to a PVDF membrane. The following antibodies were used for immunoblotting: Anti-AHR (CST, 83200S); Anti-MED12 (CST, 14360S); Anti-Beta-Actin (CST, 5125S); Anti-MAU2(SCC4) (Abcam, ab183033;). Anti-V5-Tag (CST, 13202); Anti-Myc-Tag (CST, 2040S); Anti-GAPDH (CST, 3683S); Anti-p21 Waf1/Cip1 (CST, 2947T); Anti-p27 Kip1 (CST, 9313T); Anti-Rb (CST, 3686T); Anti-Phospho-Rb (Ser807/811) (CST, 8516S); Anti-Histone H3 (CST, 3683); Anti-Phospho-Histone H3 (Ser10) (CST, 3377S); Anti- Streptavidin (Thermo Fisher Scientific, S911); Anti-Rabbit HRP antibody (CST, 7074P2) and Anti-Mouse HRP Antibody (CST, 7076S).
Real-time quantitative PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen, 74104), and 500 ng of total RNA was used to prepare cDNA using the PrimeScript™ RT Master Mix (TAKARA, RR036A) according to the manufacturer’s instructions. qRT-PCR was performed in triplicate for each target sequence using iTaq Universal SYBR Green Supermix (BIO-RAD, 1725121) on a Bio-Rad CFX96 using the primers in Supplementary Table 6.
TurboID sample preparation for LC-MS/MS analysis
Cells stably expressing TurboID constructs were incubated in media containing 500 μM biotin to induce biotin labeling of PARP7-interacting proteins. After 10 minutes, plates were placed on ice and washed 5 times with ice-cold PBS to stop labeling reaction. Cells were detached from the flask by gently pipetting a stream of PBS directly onto the cells, harvested and pelleted by centrifuging at 300g for 3 min. Cell pellets were lysed in 1.5 mL of RIPA lysis buffer for 5 min at 4 °C and lysates were clarified by centrifugation at 15,000g for 10 min at 4 °C. To enrich biotinylated proteins, 250 μL of Streptavidin Magnetic Beads (Thermo Fisher Scientific, PI88817) were washed twice with RIPA buffer, incubated with clarified lysates containing ~1 mg protein for each sample with rotation for 1 h at room temperature, and further incubated overnight with rotation at 4°C. The beads were subsequently washed twice with 1 mL of RIPA lysis buffer, once with 1 mL of 1 M KCl, once with 1 mL of 0.1 M Na2CO3, once with 1 mL of 2 M urea in 10 mM Tris-HCl (pH 8.0), and twice with 1 mL of RIPA lysis buffer. The beads were then resuspended in 1 mL of fresh RIPA lysis buffer and transferred to a new Eppendorf tube. Proteins bound to streptavidin beads (~300 μL of slurry) were washed twice with 400 μL of 50 mM Tris HCl buffer (pH 7.5) followed by two washes with 2 M urea/50 mM Tris (pH 7.5) buffer. Beads were incubated with 80 μL of 2 M urea/50 mM Tris containing 1 mM DTT and 0.4 μg trypsin (Promega, PRV5280) for 1 h at 25 °C with shaking. After on-bead trypsin digestion, the supernatant was removed and transferred to a fresh lobind tube (Axygen, MCT-175-L-C). The streptavidin beads were washed twice with 60 μL of 2 M urea/50 mM Tris buffer (pH 7.5) and the washes were combined with the on-bead digested supernatant. The eluate was alkylated with 10 mM iodoacetamide for 45 min in the dark at 25 °C with shaking and subsequently reduced with 4 mM DTT for 30 min at 25 °C with shaking. An additional 0.5 μg of trypsin was added to the sample for further digestion overnight at 25 °C with shaking. After overnight digestion, samples were acidified (to pH<3) by adding trifluoroacetic acid (TFA). Samples were desalted with 50 mg Sep-Pak C18 cartridges (Waters, WAT-WAT054955) and dried by speedvac, as previously described (16,17).
MS data acquisition and analysis
MS data acquisition and analysis was performed as previously described (17). Briefly, samples were resuspended in 20 μL of 1% formic acid (FA) and separated by a reverse-phase gradient on a Nanoflow C18 column. Each sample was directly injected into the Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific) by Easy-nLC 1200 (Thermo Fisher Scientific) and collected and analyzed for 75 minutes. Raw MS data were searched against the uniprot canonical isoforms of the human proteome (downloaded March 21, 2018), and using the default settings in MaxQuant (version 1.6.2.10), with a match-between-runs enabled (18) Peptides and proteins were filtered to 1% false discovery rate in MaxQuant. Protein MS1 intensity as determined by MaxQuant were analyzed by SAINTexpress (version 3.6.1) (19). Parental HAP1 cells treated with doxycycline and biotin were used as background controls for TurboID-PARP7. Proteins with BFDR ≤ 0.01 were regarded as hits. All mass spectrometry raw data files and search parameters/results have been deposited to the ProteomeXchange Consortium via the PRIDE (20) partner repository and can be accessed under the dataset identifier PXD028733.
Nuclight labeled cell construction
To generate Nuclight labeled cells, 2 million NCI-H1373 cells were seeded in 10 cm dishes and transduced with Incucyte Nuclight Lentivirus (Essen, 4476, MOI = 3) in the presence of 8 μg/mL polybrene (Sigma, TR-1003-G). The media was changed after 24h and, after 48 hours, media containing 1μg/ml puromycin was added. Cells were selected for 72 hours and then expanded without selection. Nuclight fluorescent cells were enriched by flow cytometry on a FACSAria 3 (BD).
AHR induced expression cell construction
NCI-H1373 cells were co-transfected with the PiggyBac Transposase vector (System Bioscience, NC1271867) and PB-TRE-Myc-DDK-AHR. The next day, the medium was changed and cells were cultured for an additional 48 hours before being selected with Hygromycin (20 μg/ml) for 5–7 days. During selection, media containing fresh Hygromycin was replaced every 2 days. After selection cells were expanded and AHR expression was induced with 1μg/ml doxycycline and confirmed by Western blot. To measure the effect of AHR expression on cell proliferation, cells were seeded into 96-well plates. 24 hours later AHR expression was induced by doxycycline (1μg/ml) and cell proliferation (Cell confluence) was measured using an Incucyte® ZOOM Live-Cell Analysis System.
Annexin-V and Caspase3/7 assay
Nuclight-labeled NCI-H1373 cells were seeded in 96-well plate (5000 cells/well) and treated with RBN-2397 and/or tapinarof or CH-223191 at the indicated concentrations. The next day, Annexin V Green Dye or Caspase-3/7 Green Dye was added and an Incucyte® ZOOM Live-Cell Analysis System was used to quantify the number of fluorescent cells.
Quantitative FastFUCCI assay
2 million NCI-H1373 cells were seeded in 10 cm dishes and transduced with pBOB-EF1-FastFUCCI-Puro lentivirus in the presence of 8 μg/mL polybrene. After 24 hours, the media was changed and after 48 hours fresh medium containing 1μg/ml puromycin was added to select infected cells. Cells were selected for 72 hours and then expanded. Red fluorescent cells (i.e. cells in G1) were enriched by flow cytometry using a FACSAria 3 (BD). FastFUCCI expressing cells were seeded into 96-well plates and treated with the indicated concentrations of RBN-2397 and/or tapinarof or CH-223191. After 48 hours, fluorescent cells were scanned using an IN Cell Analyzer 6500 System and then analyzed by IN Cell Developer (Cytiva).
AHR subcellular distribution assay
2 million nuclight labeled NCI-H1373 cells were seeded in 10 cm dishes and transduced with pLenti-C-mGFP-AHR lentivirus in the presence of 8 μg/mL polybrene. After 24 hours, the media was changed, and cells were grown for an additional 48 hours before GFP and Nuclight Red dual fluorescent cells were enriched by flow cytometry (BD FACSAria 3). Dually fluorescent cells were seeded into 96-well plates and treated with the indicated concentrations of RBN-2397, Olaparib, tapinarof and/or CH-223191 the next day. After 2 hours of exposure to drugs, fluorescent cells were scanned by IN Cell Analyzer 6500 System and then analyzed by IN Cell Developer (Cytiva). To assess endogenous AHR localization, NCI-H1373 cells were seeded in 96-well plate (5000 cells/well) and treated the next day with the indicated concentrations of RBN-2397, olaparib and/or tapinarof. After 2 hours of exposure to drugs, treated cells were fixed in pre-cooled methanol at −20°C for 20 min, blocked in 3% bovine serum albumin for 15 min, incubated with Anti-AHR (CST, 83200S) antibodies for 1 h, and then incubated with Goat anti-Rabbit IgG Secondary Antibody, Alexa Fluor 488 (ThermoFisher, A-11008) secondary antibodies for 30 min. Final staining with DAPI for 10 minutes. Fluorescent cells were scanned by IN Cell Analyzer 6500 System and then analyzed by IN Cart (Cytiva).
Mitosis assay
2 million NCI-H1373 cells were seeded in 10 cm dishes and transduced with pLenti6-H2B-mCherry lentivirus (Addgene plasmid. #89766) (21) in the presence of 8 μg/mL polybrene. pLenti6-H2B-mCherry was a gift from Torsten Wittmann. After 24 hours, the media was changed and after 48 hours fresh medium containing 1μg/ml puromycin was added to select infected cells. Cells were selected for 72 hours and then expanded. Red fluorescent cells were enriched by flow cytometry using a FACSAria 3 (BD). H2B-mCherry expressing cells were seeded into micro dish and treated with the indicated concentrations of RBN-2397. After 8 hours, fluorescent cells were scanned using a Spinning Disk Confocal (Yokagawa CSU22) every 10 mins and then analyzed.
IL1α/β ELISA assay
NCI-H1373 cells were seeded in 96-well plate (20000 cells/well) and treated with RBN-2397 and/or tapinarof or CH-223191 at the indicated concentrations. After 48 hours, the media was collected, and ELISA was performed with Human IL-1 alpha/IL-1F1 DuoSet ELISA (R&D Systems, DY-200) or Human IL-1 beta/IL-1F2 DuoSet ELISA (R&D Systems, DY-201) according to the manufacturer’s instructions.
DepMap data analysis
CCLE Expression data and CERES Gene effect scores (Avana) for AHR and TIPARP (PARP7) from 1377 and 902 cell lines, respectively were downloaded from the 21Q3 Public Cancer Dependency Map portal (https://depmap.org/portal/download/) (22–26). A lower CERES score indicates a higher likelihood that the gene is essential for the cell line. A CERES score of 0 indicates that a gene is not essential, while a score of −1 is comparable with the median of all pan-essential genes, i.e., the genes that are essential for every cell line. Expression and dependency scores were plotted pairwise using R studio (27–29) (http://www.rstudio.com/, https://ggplot2.tidyverse.org). Pearson correlations were calculated to assess correlation.
Statistical analyses
All data, if applicable, were presented as mean ± SD. Significant differences were determined by Student’s t-test. p < 0.05 was considered statistically significant.
Data Availability
The mass spectrometry raw data files and search parameters/results generated in this study been deposited to the ProteomeXchange Consortium via the PRIDE (20) partner repository and can be accessed under the dataset identifier PXD028733.
The data from the Broad Institute’s Cancer Dependency Map project analyzed in this study are available on figshare: https://figshare.com/articles/dataset/DepMap_21Q3_Public/15160110.
Other datasets generated and/or analysed during the current study are available in the extended data file or from the corresponding author on reasonable request.
Results
Genome-wide CRISPR screen identifies genes that influence PARP7 inhibitor sensitivity
To identify genes involved in response to PARP7 inhibitors (PARP7i), we performed genome-wide CRISPR knockout screens. We selected the lung adenocarcinoma cell line NCI-H1373, which is sensitive to the selective PARP7i RBN-2397 as our model (8). We tested NCI-H1373 cell viability after 7 days of RBN-2397 treatment and confirmed that this cell line is very sensitive (SF50 = 10nM) to this compound (Fig. 1a). We screened the Brunello Human CRISPR knockout pooled library which contains 76,441 unique sgRNAs, targeting 19,114 genes (30). The vector for this library carries Cas9 and individual sgRNAs targeting single genes. Lentiviral particles encompassing this library were transduced into NCI-H1373 cells with a representation of 1800 cells per gRNA. Transduced cells were then selected with puromycin and, after 3 days, divided into three arms which were treated with the solvent DMSO (0.0001%), 10 nM RBN-2397 or 20 nM RBN-2397. These concentrations were chosen to enable both positive and negative selection screening for sensitization and resistance hits. After 7 days of treatment, surviving cells were grown out and genomic DNA was extracted from each arm. sgRNA fragments were amplified by PCR and subjected to NGS sequencing on a HiSeq 4000. Between 60–100 million reads were sequenced for each experimental arm with an average sequencing depth of ~1000 reads/sgRNA (Fig. 1b and Supplementary Fig. 1a). MAGeCK software (11) was used to determine the guide frequency in each arm and to systematically identify genes that were positively or negatively selected in the cells treated with PARP7i (Fig. 1c and Supplementary Fig. 1a, 1b).
Fig. 1. Whole-genome CRISPR-Cas9 screen in NCI-H1373 cells.

A, Survival curve of NCI-H1373 cells exposed to increasing concentration of RBN-2397. Data shown as mean values ± SD; n = 6. B, Schematic of the workflow for CRISPR screens performed in NCI-H1373. C, Volcano plot comparing sgRNA counts in DMSO vs RBN-2397(10 nM) CRISPR treatment arms. x axis is gene-level LFC (median of LFC for all sgRNAs per gene, scaled). y axis is the −log10(adjp) as calculated by MAGeCK. Genes enriched (LFC > 0.6 and −log10(adjp) > 2,) and depleted (LFC < 0.6 and −log10(adjp) > 2) in the RBN-2397 treated cells are indicated in cyan and red, respectively. D, The top 10 Gene Ontology (GO) terms of significantly enriched hits. E, Top hits ranked by MAGeCK.
The genome-wide screen resulted in a significant enrichment of 1150 genes (P < 0.05) in the 10 nM RBN-2397 treatment arm and 1295 genes (P < 0.05) in the 20 nM RBN-2397 treatment arm compared with the DMSO control group. Amongst these, 542 genes were enriched at both concentrations of RBN-2397 (Supplementary Fig. 1c). In contrast, 1101 and 1138 genes were significantly depleted (P < 0.05) in the cells treated with 10 nM RBN-2397 and 20 nM RBN-2397, respectively, compared with the DMSO treated cells. Amongst these, 346 genes were depleted at both concentrations of RBN-2397 (Supplementary Fig. 1d). Pathway analysis of the 50 genes with the most significant P values in cells treated with 10 nM RBN-2397 compared to the DMSO treated control (DAVID Bioinformatics Resources 6.8) showed an enrichment of genes involved in a variety of cellular pathways (Fig. 1d). In particular, we noted that multiple components of the sister chromatin cohesion pathway (NIPBL, MAU2, SMC1A, SMC3 and RAD21) were amongst the top hits. In addition, an enrichment of sgRNAs directed against MYD88, IL1R1 and IRAK1 genes in RBN-2397-treated cells suggests that IL1 signaling contributes to PARP7i response (Fig. 1e). Intriguing, AHR, a known substrate and regulator of PARP7 appeared in the list of top enriched genes (Fig. 1c, 1e and Supplementary Fig. 1b). Finally, PARP7 (TIPARP) itself scored as a hit just outside the top 50 most significant hits but still with high significance (p <0.0005) (Fig. 1e). Pathway analysis of 346 depleted genes common to both treatment arms did not show a strong pathway enrichment compared with DMSO treated cells but six depleted gene hits (MTMR2, PTPRA, MTMR14, TPTE, PTPRK, ACP1) are included from the protein tyrosine phosphatase activity pathway (Supplementary Fig. 1e).
Identification of PARP7 associated proteins via proximity labelling analysis
A physical interaction between proteins is an important indicator of a shared function. We therefore sought to identify PARP7 interacting proteins. To this end, we used a proximity labeling technique that allows tagging of proteins located adjacent to a target protein (31). We fused a PARP7 open reading frame with a V5-TurboID engineered biotin ligase domain in a PB-TRE-VPR vector backbone and stably integrated this doxycycline-inducible expression construct (TurboID-PARP7) into HAP-1 cells (Fig. 2a). TurboID-PARP7 cells were then treated with doxycycline (1μg/ml) for 48 h to induce the expression of TurboID-PARP7. After treatment with 500 μM biotin for 10 min to allow biotinylation of PARP7-interacting proteins, cells were collected, lysed, and subjected to streptavidin pull down. Western blots for biotinylated proteins confirmed that this TurboID-PARP7 system was functional (Fig. 2b). The proteins pulled down with Streptavidin-agarose were then digested with trypsin and subject to tandem liquid chromatography mass spectrometry (LC-MS/MS). This analysis resulted in the identification of 1538 (BFDR ≤ 0.01) TurboID-PARP7 associated proteins (Fig. 2c). Upon inspection, we noted that among the 103 hits displaying both genetic and physical interactions with PARP7 were multiple sister chromatid cohesion genes including NIPBL, SMC1A, SMC3, RAD21 and MYD88.
Fig. 2. AHR and NIPBL interact with PARP7.
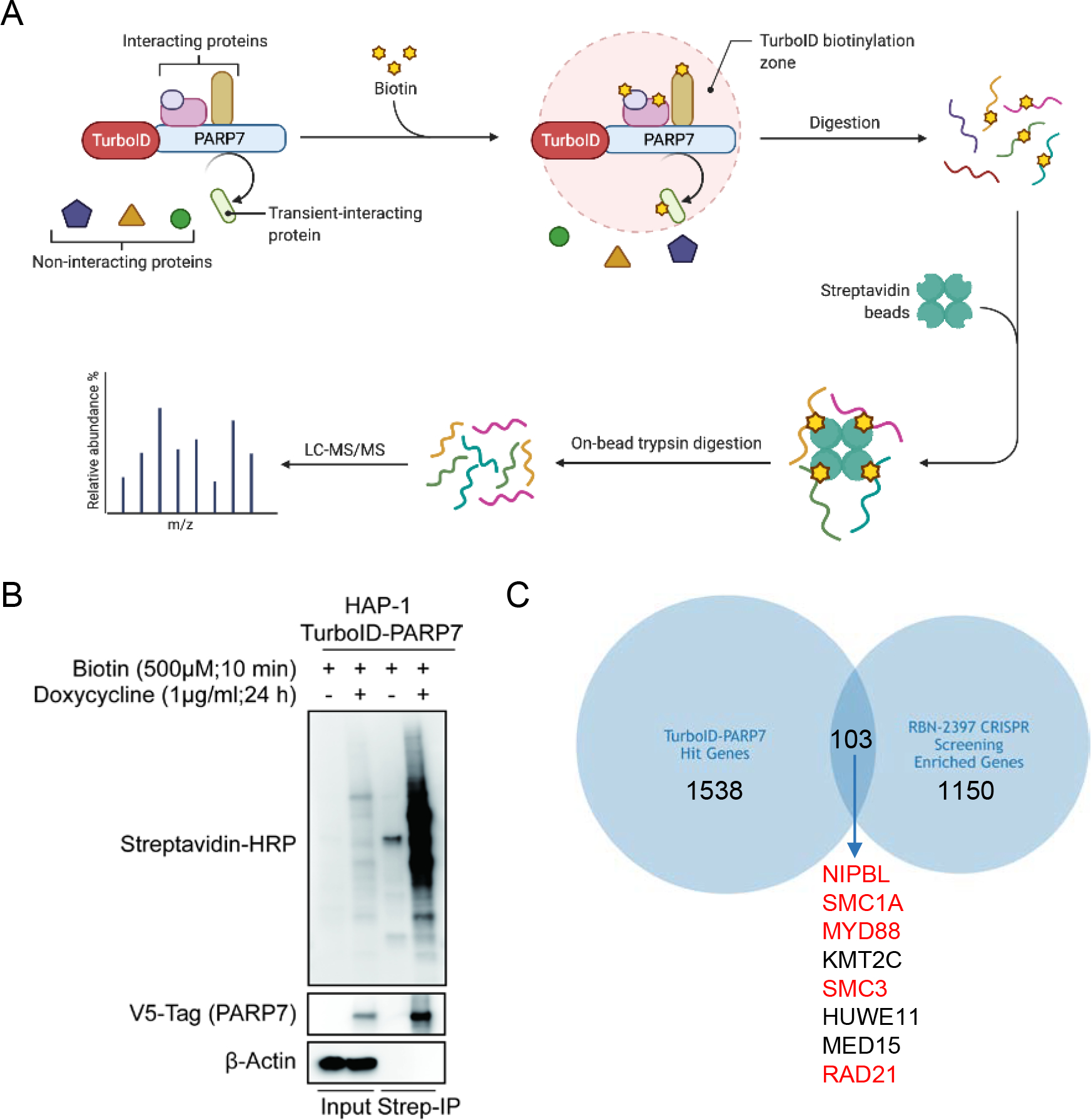
A, Schematic diagram of proximity dependent biotin labeling strategy to identify PARP7- interacting proteins. B, Western blot analysis of total cell extracts and streptavidin binding samples prepared from cells with/without doxycycline-indued PARP7 expression. Lysates were run in duplicate and probed separately for Streptavidin/V5 and β-actin. C, Venn diagram of LC-MS/MS hits (BFDR ≤ 0.01) and CRISPR screen hits (P < 0.05). Hits ranked in the top 20 by MAGeCK are highlighted in red.
AHR, NIPBL-MAU2 and MED12 mediate sensitivity to PARP7i
To orthogonally validate candidate targets from our pooled genome-wide CRISPR screen, we used siRNA to knockdown top hits, including AHR, MAU2, NIPBL and MED12 in NCI-H1373 cells and measured changes in sensitivity to the PARP7i RBN-2397 (Fig. 3a and Supplementary Fig. 2a). To varying degrees, siRNA knockdown of each target was associated with resistance to RBN-2397 (4.8 to 32.8 fold) (Fig. 3b). We also used the all-in-one lentiCRISPR V2 plasmid and CRISPR RNPs to confirm that knocking out these targets also conferred PARP7i resistance to NCI-H1373 cells (Fig. 3c and Supplementary Fig. 2b). Further supporting a role for these genes in PARP7i response, knockdown of these targets in four additional cell lines MDA-MB-468, SW620, CAL27 and NCI-H2347 (Fig. 3d, 3e and Supplementary Fig. 2c, Supplementary Fig. 3) was also associated with PARP7i resistance. Together these results provide strong evidence that loss or silencing of AHR, MAU2, NIPBL or MED12 can cause resistance to RBN-2397 in multiple genetic contexts.
Fig. 3. Loss of AHR and NIPBL-MAU2 mediates resistance to PARP7 inhibitor.

A, Western blots of NCI-H1373 cell lysates collected 48h after transfection with siNC, siAHR, siNIPBL, siMAU2 or siMED12. B, Dose response survival curves NCI-H1373 cells treated with RBN-2397 for 7 days showing the effect of AHR, MAU2, MED12 and NIPBL knockdown. Data shown as mean values ± SD; n = 3. C, Dose response curves of NCI-H1373 cells treated with RBN-2397 for 7 days showing the effect of knocking out AHR, MAU2, MED12 and NIBPL. Data shown as mean values ± SD; n = 3. D, Western blots of MDA-MB-468 cells transfected with siNC, siAHR, siMAU2, siMED12 or siNIPBL for 48 h. E, Dose response curves of MDA-MB-468 cells treated with RBN-2397 for 7 days showing the effect of AHR, MAU2, MED12 and NIPBL knockdown. Data shown as mean values ± SD; n = 3.
Modulating AHR activity influences sensitivity to RBN-2397
Of our lead hits, AHR was of particular interest because it is already linked to PARP7. PARP7 transcription is regulated by AHR activity and PARP7 is recruited to AHR bound promoters to repress transcription of AHR target genes (5,7). To better understand how AHR activity mediates the cytotoxicity of RBN-2397, we first generated AHR knockout cells using an AHR lenti V2 plasmid (Fig. 4a) and confirmed resistance to RBN-2397 (Fig. 4b). We next explored how AHR activity altered sensitivity to RBN-2397 using the AHR inhibitor CH-223191 and the AHR agonists L-kynurenine and tapinarof to modulate AHR activity in both WT and AHR knockout NCI-H1373 cell lines. We empirically determined a concentration of the AHR inhibitor CH-223191 which as a single agent did not affect NCI-H1373 cell growth (Supplementary Fig. 4a) and found that combination of CH-223191 and RBN-2397 induced resistance of NCI-H1373 to RBN-2397 (Fig. 4c). Notably, the degree of resistance conferred by CH-223191 is similar to that seen in the AHR knockout cell lines (Fig. 4b, 4c) further supporting the conclusion that AHR is necessary for RBN-2397 cytotoxicity. We next tested the effect of combining the AHR agonist L-kynurenine with RBN-2397. Again, we evaluated a range of L-kynurenine doses to identify a concentration that did not affect viability in NCI-H1373 (Supplementary Fig. 4b) and found that treating cells with L-kynurenine made them more sensitive to PARP7i (Fig. 4c and Supplementary Fig. 4b). Another AHR agonist, tapinarof, had a similar effect on RBN-2397 sensitivity (Supplementary Fig. 4c). These changes in sensitivity were abrogated in AHR knockout cell lines, showing that the effects of these agents in this system were AHR dependent (Fig. 4c and Supplementary Fig. 4d). Tapinarof also considerably increased the sensitivity of MDA-MB-468 cells to RBN-2397 (Supplementary Fig. 4e). We also tested the effect of the induced overexpression of an AHR transgene and found AHR expression slowed the growth of NCI-H1373 cells and increased sensitivity to RBN-2397 (Fig. 4d, 4e and Supplementary Fig. 4f). Together, these studies suggest that AHR plays an important role in mediating the cellular response to PARP7 inhibitors.
Fig. 4. AHR function mediates sensitivity to PARP7 inhibitor.
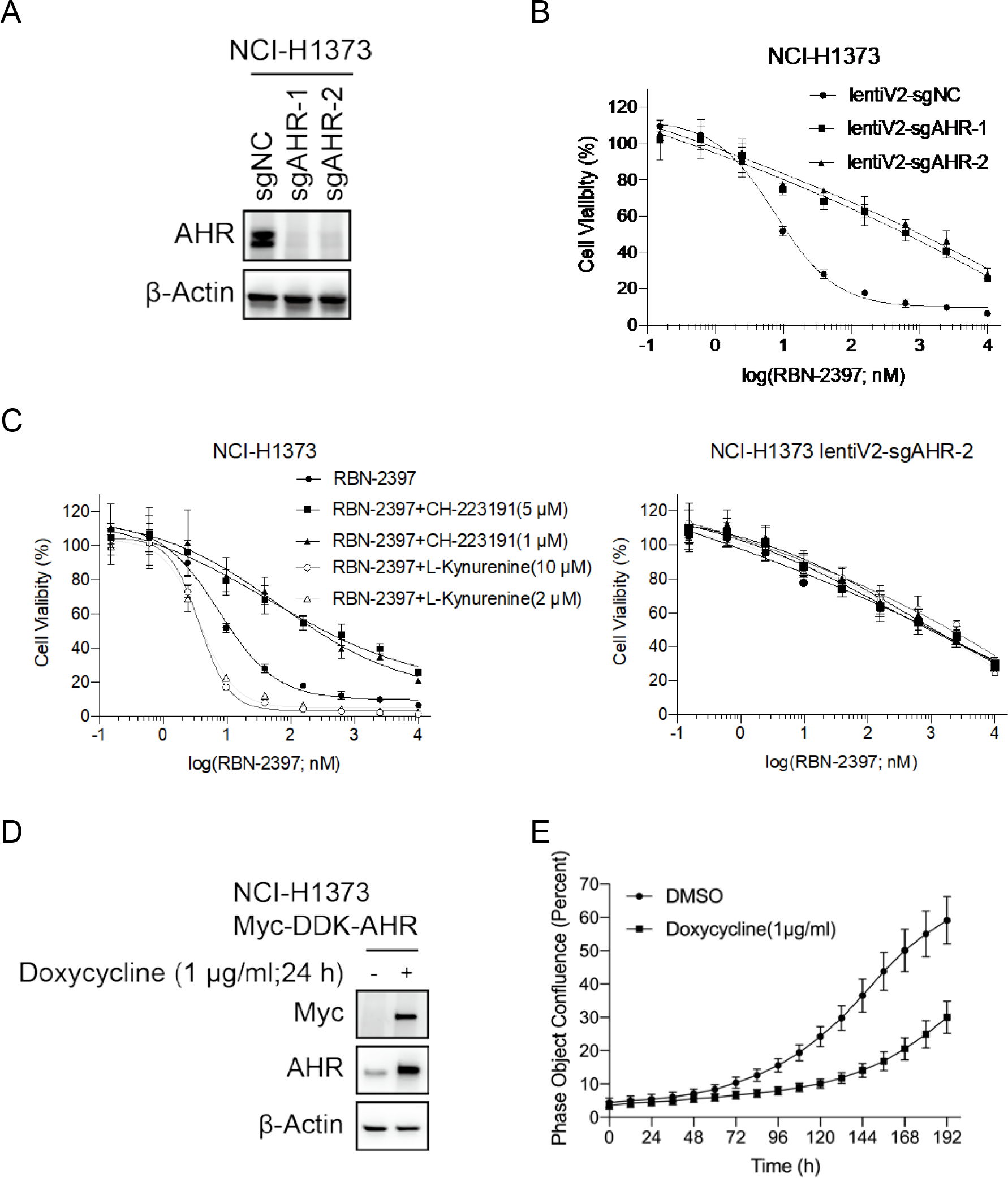
A, Western blots of cell lysates from NCI-H1373 cells and AHR knockout pools. B, Dose response curves of parental and AHR-knockout NCI-H1373 cells treated with RBN-2397 for 7 days. Data shown as mean values ± SD; n = 3. C, Dose response curves of parental (left) and AHR-knockout NCI-H1373 cells (right) treated with RBN-2397 alone or combined with indicated concentrations of CH-223191/L-kynurenine. Data shown as mean values ± SD; n = 3. D, Western blots of cell lysates from NCI-H1373 parental cells and cells induced to express a myc-tagged AHR cDNA transgene. E, Growth curves showing the effect of doxycycline-induced AHR overexpression. Data shown as mean values ± SD; n = 15.
RBN-2397 activates AHR-mediated transcription and promotes AHR nuclear localization
Because PARP7 is reported to influence the expression of AHR target genes, we next examined how treatment with RBN-2397 affected AHR effector genes. Using real-time quantitative PCR, we found increased mRNA levels for the AHR target genes PARP7, CYP1A1, CYP1B1, AHRR, PAI2, IL6, IL1A and IL1B in parental NCI-H1373 cells treated with RBN-2397, but not in isogenic AHR knockout cells (Fig. 5a and Supplementary Fig. 4g and 5a). Addition of both RBN-2397 and the AHR inhibitor CH-223191 decreased mRNA levels of AHR target genes compared to RBN-2397 alone while treatment with both RBN-2397 and the AHR agonist tapinarof further increased mRNA levels above either single agent (Fig. 5a and Supplementary Fig. 5a). ELISA assays showed that IL1α and IL1β expression changes that mirrored the mRNA levels (Fig. 5b). RBN-2397 and AHR inhibitors or agonists did not significantly increase AHR and ARNT transcription levels (Supplementary Fig. 5b).
Fig. 5. RBN-2397 activates AHR-mediated transcription and increases AHR nuclear/cytoplasmic ratio.

A, RT-qPCR data showing relative mRNA levels of AHR transcriptional targets in NCI-H1373 cells treated with the indicated combinations of RBN-2397, CH-223191 and/or tapinarof for 24 h. Data shown as mean values ± SD; n = 2. B, protein levels of IL1-α and IL1-β detected by ELISA in supernatants of NCI-H1373 cells treated with the indicated combinations of RBN-2397, CH-223191 and/or tapinarof for 48 h. Data shown as mean values ± SD; n = 3. C, Representative images of Nuclight Red labeled NCI-H1373 cells expressing an GFP-AHR transgene treated with RBN-2397, tapinarof or a combination of both drugs for 2 h. DMSO treated cells are shown as a vehicle control. Scale bar is 60 μm. D, Relative nuclear/cytoplasmic GFP intensity ratio of cells from (C). Data shown as mean values ± SD; At least 3000 cells were analyzed each group, from triplicate wells.
The cellular distribution of AHR is the result of a dynamic balance between nuclear import and export. In the absence of ligand, AHR can shuttle between the nucleus and the cytosol. Upon ligand binding, AHR localizes to the nucleus where it binds with ARNT and activates transcription of target genes by binding to Dioxin Responsive Elements (DREs) in their promoters (3,32). Loss of PARP7 causes nuclear accumulation of AHR suggesting PARP7 may regulate AHR by promoting nuclear export and proteasomal degradation (33). To investigate the effects of RBN-2397 and AHR agonists on the cellular localization of AHR, we stably expressed a GFP-AHR transgene in NCI-H1373 cells and treated cells with each compound for 2 hours. While neither RBN-2397 or the clinical PARP1 inhibitor olaparib alone changed the cellular distribution of AHR, there was an increase in the ratio of nuclear:cytoplasmic AHR after 2 hours of treatment with tapinarof. The combination of RBN-2397 and tapinarof further increased the proportion of AHR in the nucleus (Fig. 5c, d). Tapinarof also caused an increase in the ratio of nuclear:cytoplasmic endogenous AHR with RBN-2397 further shifting the distribution in favor of nuclear localization (Supplementary Fig 5c, 5d). Taken together with the qPCR data, these observations provide evidence that RBN-2397 activates the transcriptional expression of AHR target genes, possibly by increasing the proportion of active AHR in the nucleus.
PARP7 regulates cell cycle arrest and apoptosis through AHR activity
To understand how AHR activity mediates the cytotoxicity of RBN-2397, we used the FastFUCCI system (34) (Fig. 6a) to examine the cell cycle of parental and AHR knockout NCI-H1373 cells treated with RBN-2397 alone or RBN-2397 combined with AHR inhibitor CH-223191 or AHR agonist tapinarof. FUCCI transgenes were stably expressed in NCI-H1373 parental cells and AHR knockout cells, and fluorescent cells were imaged and analyzed using an IN Cell Analyzer 6500 System (Cytiva). We found that 10 nM, 20 nM and 100 nM of RBN-2397 induced a G1 arrest in NCI-H1373 parental cells in a concentration-dependent manner, but there was no corresponding change in AHR knockout cells (Fig. 6b). In parental cells, treatment with AHR inhibitor CH-223191 and RBN-2397 partially reversed the G1 arrest induced by RBN-2397, and the combination of AHR agonist tapinarof and RBN-2397 enhanced the arrest. These changes were not detected in AHR knockout cells (Fig. 6b). Previous studies have showed that AHR can induce G1 arrest by activating the transcription of p27 and p21 (35) which bind to cyclin-CDK complexes to inhibit their activity. To assess the impact of RBN-2397 on these genes we performed real-time quantitative PCR and found a slight increase in p27 and p21 mRNA levels in cells treated with RBN-2397 (Fig. 6c and Supplementary Fig. 6a). Simultaneous treatment with the AHR agonist tapinarof further increased transcript levels. In contrast, addition of the AHR inhibitor CH-223919 reversed the increase in p27 and p21 mRNA levels on RBN-237 treatment (Fig. 6c and Supplementary Fig. 6a). Western blot analysis showed that RBN-2397 treatment also caused an increase in p27 protein levels as well as a decrease in the levels of Rb and phosphorylated H3 in NCI-H1373 parental cells without affecting isogenic AHR knockout cells (Fig. 6d).
Fig. 6. RBN-2397 induces G1 arrest and apoptosis through AHR activity.

A, Schematic of the FastFUCCI cell cycle reporter system14. Red, yellow and green fluorescent cells correspond to the cells in G1, G1/S and S/G2-M, respectively. B, Cell cycle analysis of FastFUCCI labeled NCI-H1373 treated with the indicated drugs for 48 h. C, RT-qPCR data showing the relative mRNA levels of p27/CDKN1B in parental and AHR knockout cells treated with the indicated drugs for 24 h. D, Western blots of cell lysates from parental and AHR knockout NCI-H1373 cells treated with the indicated concentrations of RBN-2397 for 48 h. E, Fraction of parental (top) and AHR knockout (bottom) NCI-H1373 cells staining positive for Annexin V treatment with the indicated drugs for 24 h. Data shown as mean values ± SD; n = 3.
In addition to regulating the cell cycle, increased p27 and p21 are both associated with apoptosis. To explore the possibility that RBN-2397 induced apoptosis in NCI-H1373 cells, we assessed apoptosis in real time using Annexin V Green Dye and Caspase-3/7 Green Dye. Parental cells treated with RBN-2397 showed a concentration-dependent increase in the proportion of Annexin V and Caspase-3/7 positive cells while no difference was detected in AHR knockout cell lines (Fig. 6e and Supplementary Fig. 6b). Consistent with the growth inhibition results (Fig. 4a-c), the combination of the AHR agonist CH-223191 and RBN-2397 partially reversed the increase in RBN-2397-induced apoptosis-positive cells whereas the AHR agonist tapinarof further increased the proportion of RBN-2397-induced apoptosis-positive cells (Fig. 6e and Supplementary Fig. 6c, 6d). Again, consistent with the growth inhibition assay, few apoptotic cells were detected in the AHR knockout lines and the levels of apoptosis were consistent across all treatments (Fig. 6e and Supplementary Fig. 6c, 6d).
To further investigate how RBN-2397 disrupts the cell cycle, we stably expressed a H2B:mCherry transgene in NCI-H1373 cells and monitored mitotic progression in the presence and absence of PARP7i. While untreated cells completed mitosis normally (Supplementary Fig. 7a), approximately half of the cells treated with 1 μM RBN-2397 failed to divide within 80 minutes and, among those that did undergo mitosis, almost all of the resulting cells displayed nuclear blebbing or micronuclei consistent with abnormal chromosome segregation (Supplementary Fig. 7b). Together, these results argue that RBN-2397 exerts a cytotoxic effect in part by activating AHR to induce cell cycle arrest and apoptosis. At high doses, RBN-2397 prevents cells from completing mitosis and, for cells that do progress, chromosome segregation errors promote a G1 arrest.
Discussion
Much attention has been paid to the role of PARP1 and PARP2 in DNA repair especially in single strand break repair/base excision repair. Inhibitors of PARP1 and PARP2 can elicit synthetic lethality in cells defective in BRCA1, BRCA2 and other components of double strand break repair by homologous recombination. This observation has led to five PARP inhibitors becoming standard of care therapies world-wide for the treatment of four different BRCA-related cancers: breast, ovarian, prostate and pancreatic. The clinical success of PARP1/2 inhibitors has fueled interest in exploring the therapeutic value in inhibiting the catalytic activity of other members of the PARP family. The lesser studied PARP proteins have been implicated in multiple cellular processes including regulation of innate immunity. Amongst these, PARP7 can act with PARP13 to induce interferon beta in response to viral infection (7). Moreover, in cancer cells, PARP7 inhibition has been shown to increase IFN responses (8). Interestingly, inhibitors of PARP7 act in both a cell autonomous and non-cell autonomous fashion to inhibit cancer cell growth (8).
A potent and specific PARP7i, RBN-2397, is now being explored in clinical trials (8) and it is important to understand determinants of sensitivity and resistance to this agent and to identify optimal combinatorial treatments. To address these needs, we performed genome-wide CRISPR knockout screens in the presence of RBN-2397 and discovered multiple genes that when inactivated conferred resistance to this agent. Amongst the top hits were NIPBL and MAU2, two components of the Cohesin complex. Using an unbiased proteomics analysis we showed close physical proximity between PARP7 and Cohesin components in cells (Fig. 2). Cohesin is a multi-subunit protein complex that is essential for cohesion between sister chromatids after replication. Loss of Cohesin function leads to chromosome missegregation and genome instability (36). In addition to this canonical role, multiple other functions for Cohesin have been proposed including transcription and DNA repair (37). Loss of the Cohesin components STAG2 or STAG3 has been previously associated with drug resistance, in particular to BRAF inhibitors through MAP kinase reactivation (38). Loss of MED12 has been previously linked to resistance to cancer drugs including BRAF inhibitors also through MAPK inactivation (39) and our data show that loss of the Mediator component MED12 is also associated with PARP7i resistance. NIPBL colocalizes with MED12 extensively on chromatin to regulate gene expression (40) linking together our Cohesin and MED12 observations and raising the possibility that MAPK signaling may also play a role in PARP7i response. Future work to explore the association between Cohesin components and cellular response to PARP7i may allow a mechanistically understanding and define patient populations that may not benefit clinically from PARP7i treatment.
Another major observation arising from our genome-wide CRISPR knockout screens was that inactivation of AHR rendered cells resistant to PARP7i. We find that that PARP7i caused a dose-dependent G1 arrest and apoptosis in NCI-H1373, but not in AHR knockout cells. Cells treated with both RBN-2397 and an AHR inhibitor did not exhibit a G1 arrest suggesting that PARP7i toxicity is mediated through AHR in PARP7i-sensitive cells. AHR is a receptor for endogenous and exogenous ligands and activation of AHR can promote cell proliferation. Cytoplasmic AHR is found bound to multiple proteins including HSP90, AHR- interacting protein, and AHR-activated 9 protein (32). Upon ligand binding, AHR translocates to the nucleus where it forms a heterodimer with ARNT and induces transcription through binding to specific sequences (Dioxin Responsive Elements - DREs) in the promoters of target genes. AHR/DRE-regulated genes include the cytochrome P450 encoding genes CYP1A1 and CYP1B1, the aryl hydrocarbon receptor repressor AHRR as well as PARP7. Upon activation, AHR is exported from the nucleus and degraded, an event triggered in part by PARP7- mediated mono-ADP ribosylation acting in concert with the E3 ubiquitin ligase CUL4B (33).
Silencing of PARP7 causes an increase in AHR activity in some cells (4,5) and liver specific knock-in of a mutation (H532A) that abrogates PARP7 catalytic activity induces increased sensitivity to TCDD-induced liver toxicity in mice likely through enhanced Ahr activity (41). Using AHR agonists and antagonists, we showed that pharmacological modulation of AHR activity altered sensitivity to RBN-2397 in cells that were wild-type for AHR but not in AHR knockout cells. We also provide evidence that that treatment with RBN-2397 increases mRNA levels of AHR target genes in parental NCI-H1373 cells but not in isogenic AHR knockout cells. Finally, we find that PARP7 inhibition results in an increased proportion of AHR in the nucleus. Together, these data demonstrate that AHR plays a central role the cellular response to RBN-2397 and suggest that the cytotoxicity of PARP7i may be due to activation of AHR. We note that PARP7 itself was a statistically significant resistance hit in our screen (Fig. 1e). While it may seem paradoxical that loss of PARP7 renders cells resistant to a PARP7i, we note that PARP7 is required for viability in NCI-H1373 cells (8) and this result may reflect a latency in the death of PARP7 deficient cells during screening. It is also possible that PARP7 protein itself is required for the inhibitory effects of RBN-2397, as has been shown for PARP1 inhibitors (42–44).
Extending previous observations (4,41), we propose a model (Fig. 7a) whereby PARP7 inhibition leads to AHR activation and the enhanced expression of downstream effector genes. These downstream targets include CYP1A1 and CYP1B1 which have been linked to AHR-dependent apoptosis (45,46), and the cell cycle inhibitors, p21 and p27(47). In the absence of AHR or when AHR activity is attenuated pharmacologically, cells become less sensitive to PARP7 inhibition. Conversely, AHR agonists enhance the effects of PARP7 inhibition. To substantiate this model, we analyzed publicly available expression and CRISPR dependency data from more than 900 cancer cell lines in the Cancer Dependency Map (DepMap) database (www.depmap.org) (22–26) and found the following correlative relationships between AHR and PARP7: (i) cells that have higher PARP7 expression are more sensitive to loss of PARP7 as has been previously noted (8) (Fig. 7b); (ii) in cells with higher AHR expression, loss of PARP7 is more toxic (Fig. 7c); and (iii) sensitivity to PARP7 loss is correlated with a growth advantage upon AHR knockout, consistent with the enhanced proliferation we observed in NIH-H1373 cells upon AHR knockout (Fig. 4e, Fig. 7d). Each of these correlations is very highly statistically significant (P < 10−9-10−16) and together they argue for AHR acting as a brake on cell growth in PARP7-sensitive cells. Although these associations do not establish direct causation, they provide additional support for our model that sensitivity to PARP7 inhibition requires active AHR and that AHR inactivation may induce resistance.
Fig. 7. Proposed model for PARP7i cytotoxicity.
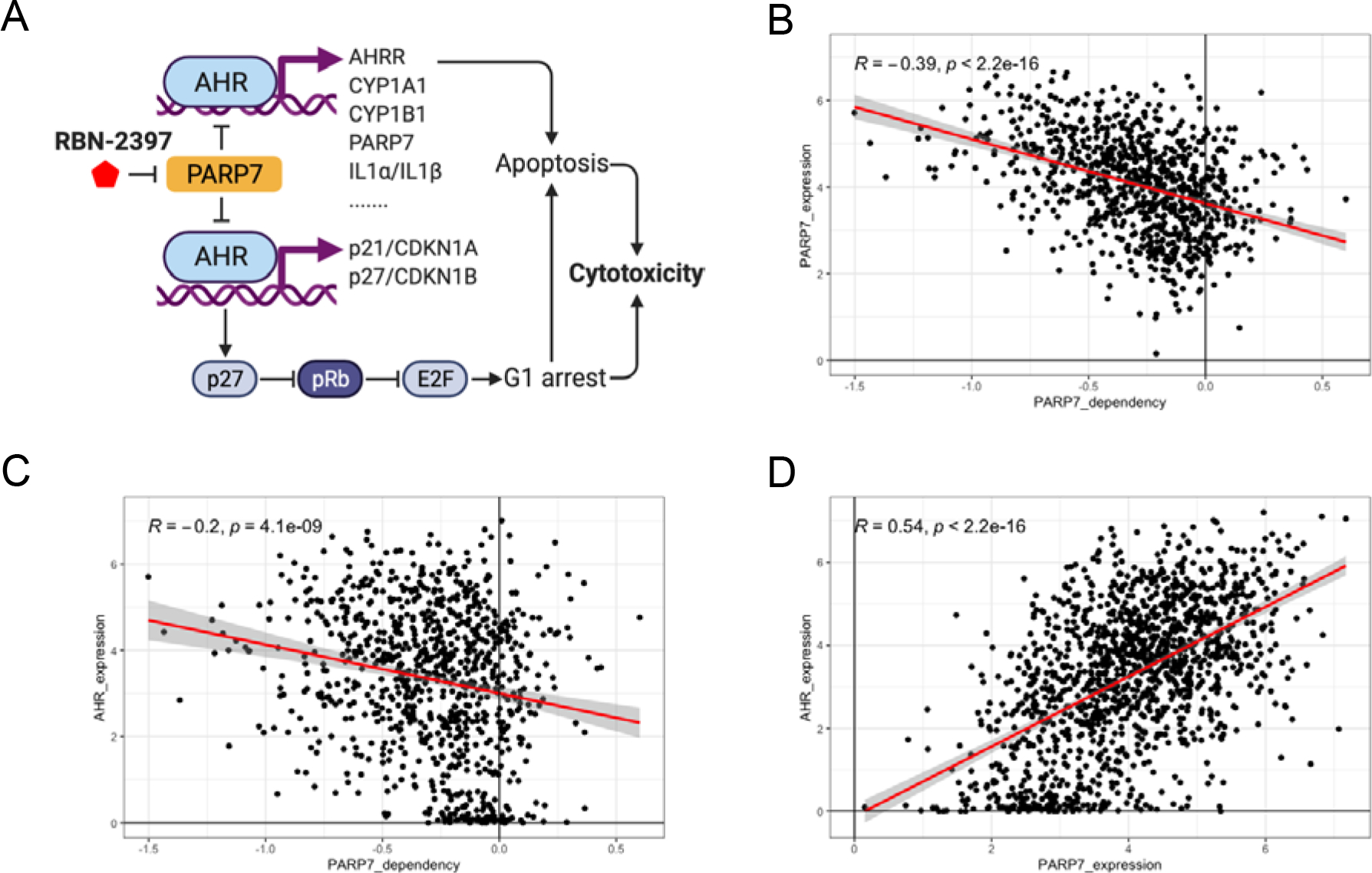
A, Model showing how PARP7i may exert cytotoxicity by regulating the transcription/expression of some AHR target genes that have known roles in cell cycle regulation and apoptosis. B-D, Scatter plots comparing DepMap expression and dependency scores across approximately 900 cell lines show a negative association between PARP dependency scores vs PARP expression (B) and PARP7 dependency scores vs AHR expression (E) and a positive correlation between PARP7 expression vs AHR expression (D). A lower dependency score indicates a higher likelihood that the gene is essential in a given cell line. A score of 0 indicates that a gene is not essential, while a score <−0.5 suggest essentiality and a score of 1 is comparable with the median of pan-essential genes. Regression lines are in red with a 95% confidence interval shown in gray. Pearson correlation coefficients (R) and p-values are indicated on each plot.
In conclusion, our work has uncovered genetic determinants of sensitivity to a PARP7 inhibitor which may have important implications for how these compounds are used clinically. Mutations that disrupt AHR or components of the Cohesin complex represent potential resistance mechanisms to PARP7i and this prediction can be studied in the ongoing clinical trials now in progress. While genetic studies have previously demonstrated that PARP7 plays a key role in regulating AHR signaling and transcriptional activity, our finding extends these observations to small molecule inhibitors of PARP7. Furthermore, we show that AHR is essential for PARP7i toxicity and demonstrate that pharmacological modulation of AHR activity can affect sensitivity to PARP7i suggesting potential combination therapies that may enhance clinical benefit.
Supplementary Material
Acknowledgments
This research was supported by the UCSF Benioff Initiative for Prostate Cancer Research (AA and MK) and NIH U54 CA209891 (NJK and AA). Additional funding was received from the Breast Cancer Research Foundation (AA), The Susan G. Komen Breast Cancer Foundation (AA), the V foundation for Cancer Research (AA), the Gray Foundation (AA), Emerson Collective (AA), the Martha and Bruce Atwater Breast Cancer Research Program via UCSF Helen Diller Family Comprehensive Cancer Center (MK) and UCSF Prostate Cancer Program Research Pilot Funding (MK). The Cancer Dependency Map (DepMap) project is partially funded by CTD2, the Achilles consortium, and The Carlos Slim Foundation in Mexico through the Slim Initiative for Genomic Medicine. Fig. 1b, 2a, 6a and 7a were created using BioRender.com
Footnotes
Conflict of interest: AA is a co-founder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation, Kytarro; a consultant for SPARC, Bluestar, ProLynx, Earli, Cura, Phoenix MD and GSK; a member of the SAB of Genentech and GLAdiator; a member of the boards of Cambridge Science Corporation and Cytomyx; receives grant/research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca which he has benefitted financially (and may do so in the future). NJK is a shareholder of Tenaya Therapeutics and has received stocks from Maze Therapeutics and Interline Therapeutics; has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics and Interline Therapeutics. The laboratory of NJK has received research support from Vir Biotechnology and F. Hoffmann-La Roche.
References
- 1.Luscher B, Ahel I, Altmeyer M, Ashworth A, Bai P, Chang P, et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma Q, Baldwin KT, Renzelli AJ, McDaniel A, Dong L. TCDD-inducible poly(ADP-ribose) polymerase: a novel response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Biophys Res Commun 2001;289:499–506 [DOI] [PubMed] [Google Scholar]
- 4.MacPherson L, Tamblyn L, Rajendra S, Bralha F, McPherson JP, Matthews J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acids Res 2013;41:1604–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthews J. AHR toxicity and signaling: Role of TIPARP and ADP-ribosylation. Current Opinion in Toxicology 2017;2:50–7 [Google Scholar]
- 6.Gomez A, Bindesboll C, Satheesh SV, Grimaldi G, Hutin D, MacPherson L, et al. Characterization of TCDD-inducible poly-ADP-ribose polymerase (TIPARP/ARTD14) catalytic activity. Biochem J 2018;475:3827–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada T, Horimoto H, Kameyama T, Hayakawa S, Yamato H, Dazai M, et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon–mediated antiviral innate defense. Nature Immunology 2016;17:687–94 [DOI] [PubMed] [Google Scholar]
- 8.Gozgit JM, Vasbinder MM, Abo RP, Kunii K, Kuplast-Barr KG, Gui B, et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 2021 [DOI] [PubMed] [Google Scholar]
- 9.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014;343:84–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 13.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research 2009;37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 2018;175:1958–71 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller RE, Brough R, Bajrami I, Williamson CT, McDade S, Campbell J, et al. Synthetic Lethal Targeting of ARID1A-Mutant Ovarian Clear Cell Tumors with Dasatinib. Mol Cancer Ther 2016;15:1472–84 [DOI] [PubMed] [Google Scholar]
- 16.Cho KF, Branon TC, Udeshi ND, Myers SA, Carr SA, Ting AY. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat Protoc 2020;15:3971–99 [DOI] [PubMed] [Google Scholar]
- 17.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583:459–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72 [DOI] [PubMed] [Google Scholar]
- 19.Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, Choi H. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics 2014;100:37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019;47:D442–D50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pemble H, Kumar P, van Haren J, Wittmann T. GSK3-mediated CLASP2 phosphorylation modulates kinetochore dynamics. J Cell Sci 2017;130:1404–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 2017;49:1779–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dempster JM, Boyle I, Vazquez F, Root DE, Boehm JS, Hahn WC, et al. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol 2021;22:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pacini C, Dempster JM, Boyle I, Goncalves E, Najgebauer H, Karakoc E, et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat Commun 2021;12:1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, et al. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019:720243 [Google Scholar]
- 26.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.RStudio T. 2020. RStudio: Integrated Development for R. RStudio, PBC, Boston, MA. [Google Scholar]
- 28.Wickham H. 2016. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York. [Google Scholar]
- 29.Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, et al. Welcome to the Tidyverse. Journal of Open Source Software 2019;4 [Google Scholar]
- 30.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34:184–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 2018;36:880–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr 2008;18:207–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rijo MP, Diani-Moore S, Yang C, Zhou P, Rifkind AB. Roles of the ubiquitin ligase CUL4B and ADP-ribosyltransferase TiPARP in TCDD-induced nuclear export and proteasomal degradation of the transcription factor AHR. J Biol Chem 2021;297:100886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh SB, Mascalchi P, Rodriguez E, Lin Y, Jodrell DI, Richards FM, et al. A quantitative FastFUCCI assay defines cell cycle dynamics at a single-cell level. J Cell Sci 2017;130:512–20 [DOI] [PubMed] [Google Scholar]
- 35.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer 2014;14:801–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waldman T Emerging themes in cohesin cancer biology. Nat Rev Cancer 2020;20:504–15 [DOI] [PubMed] [Google Scholar]
- 37.Mehta GD, Kumar R, Srivastava S, Ghosh SK. Cohesin: functions beyond sister chromatid cohesion. FEBS Lett 2013;587:2299–312 [DOI] [PubMed] [Google Scholar]
- 38.Shen CH, Kim SH, Trousil S, Frederick DT, Piris A, Yuan P, et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat Med 2016;22:1056–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang S, Holzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012;151:937–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010;467:430–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutin D, Long AS, Sugamori K, Shao P, Singh SK, Rasmussen M, et al. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD)-Inducible Poly-ADP-Ribose Polymerase (TIPARP/PARP7) Catalytic Mutant Mice (TiparpH532A) Exhibit Increased Sensitivity to TCDD-Induced Hepatotoxicity and Lethality. Toxicol Sci 2021;183:154–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 2018;9:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen HD, Chen CH, Wang YT, Guo N, Tian YN, Huan XJ, et al. Increased PARP1-DNA binding due to autoPARylation inhibition of PARP1 on DNA rather than PARP1-DNA trapping is correlated with PARP1 inhibitor’s cytotoxicity. Int J Cancer 2019;145:714–27 [DOI] [PubMed] [Google Scholar]
- 44.Murai J, Pommier Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu Rev Canc Biol 2019;3:131–50 [Google Scholar]
- 45.Coelho NR, Pimpao AB, Correia MJ, Rodrigues TC, Monteiro EC, Morello J, et al. Pharmacological blockage of the AHR-CYP1A1 axis: a call for in vivo evidence. J Mol Med (Berl) 2022;100:215–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuo HH, Ahmad R, Lee GQ, Gao C, Chen HR, Ouyang Z, et al. Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4(+) T Cells. Immunity 2018;48:1183–94 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coqueret O New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 2003;13:65–70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry raw data files and search parameters/results generated in this study been deposited to the ProteomeXchange Consortium via the PRIDE (20) partner repository and can be accessed under the dataset identifier PXD028733.
The data from the Broad Institute’s Cancer Dependency Map project analyzed in this study are available on figshare: https://figshare.com/articles/dataset/DepMap_21Q3_Public/15160110.
Other datasets generated and/or analysed during the current study are available in the extended data file or from the corresponding author on reasonable request.