

Summary
Flavor is essential to consumer preference of foods and is an increasing focus of plant breeding programs. In fruit crops, identifying genes underlying volatile organic compounds has great promise to accelerate flavor improvement, but polyploidy and heterozygosity in many species have slowed progress.
Here we use octoploid cultivated strawberry to demonstrate how genomic heterozygosity, transcriptomic intricacy and fruit metabolomic diversity can be treated as strengths and leveraged to uncover fruit flavor genes and their regulatory elements.
Multi‐omics datasets were generated including an expression quantitative trait loci map with 196 diverse breeding lines, haplotype‐phased genomes of a highly‐flavored breeding selection, a genome‐wide structural variant map using five haplotypes, and volatile genome‐wide association study (GWAS) with > 300 individuals. Overlaying regulatory elements, structural variants and GWAS‐linked allele‐specific expression of numerous genes to variation in volatile compounds important to flavor. In one example, the functional role of anthranilate synthase alpha subunit 1 in methyl anthranilate biosynthesis was supported via fruit transient gene expression assays.
These results demonstrate a framework for flavor gene discovery in fruit crops and a pathway to molecular breeding of cultivars with complex and desirable flavor.
Keywords: eQTL, fruit flavor, GWAS, phased genome assemblies, regulatory elements, strawberry, structural variant map
Introduction
Fruit flavor is an elusive trait, influenced by many factors including genetics, environments and cultural practices (Knee, 2001). Breeders increasingly are focused on meeting the needs of consumers, but genetic improvement of flavor is challenging as a consequence of the chemical and genetic complexities of the flavor phenotype (Klee & Tieman, 2018). These challenges are accentuated in heterozygous, polyploid species. For example, fewer significant single nucleotide polymorphisms (SNPs) were detected in genome‐wide association study (GWAS) of tetraploid blueberry when diploid models were applied (Ferrão et al., 2018); in octoploid strawberry, structural variation underlying a locus affecting volatile production was difficult to resolve using a single reference genome (Oh et al., 2021).
Recent advances have been made via chemical–sensory studies to identified specific volatiles associated with consumer preference (Tieman et al., 2012; Fan et al., 2021). Although important volatile compounds in fruit crops are being identified, too little is known about the metabolomic and genetic diversity within species and breeding populations. Some volatiles have been lost during domestication and breeding as a combined result of negative selection and linkage drag in tomato and watermelon (Zhou et al., 2016; Tieman et al., 2017; Zhu et al., 2018). Likewise, gain and loss of terpene compounds during strawberry domestication and its genetic causes have been investigated (Aharoni et al., 2004).
Recent advances in sequencing technology and analytical approaches have opened new opportunities to understand the chemistry and genetics of fruit flavor. Genome‐wide association studies have revealed loci for flavor in a variety of fruit crops (Cao et al., 2016; Tieman et al., 2017; Ferrão et al., 2020). Meanwhile, genomes‐wide expression quantitative trait loci (eQTL) studies have the capability to bridge the gaps between GWAS signals and their underlying causative genes. Integration of GWAS and eQTL studies has led to discovery of a master metabolite regulator in tomato and a flesh‐color‐determining gene in melon (Galpaz et al., 2018; Zhu et al., 2018). Long‐read sequencing now allows assembly of genomes with high contiguity, and when coupled with parental short‐read data (Koren et al., 2018), the two haplotypes of a heterozygous individual can be fully resolved. Phased assemblies have improved variant discovery, especially for large structural variants (SVs; Ebert et al., 2021). The extent, diversity and impact of SVs increasingly are being studied in horticultural crops (Alonge et al., 2020) and have been shown to alter fruit flavor, fruit shape and sex determination (Alonge et al., 2020; Massonnet et al., 2020; Guan et al., 2021). Great opportunity exists to coherently integrate these multi‐omics resources for the discovery of flavor genes.
Garden strawberry (Fragaria × ananassa) is an allo‐octoploid species (2n = 8x = 56) with highly palatable nonclimacteric fruit (Given et al., 1988; Fan et al., 2021). It increasingly has been utilized as a model for Rosaceae fruit crops genomics and flavor research as a result of its short generation time, wide cultivation and high value. Through exploration of spatiotemporal changes in gene expression and homolog search, several flavor genes have been cloned and validated, including an alcohol dehydrogenase (ADH; Wolyn & Jelenkovic, 1990; Mitchell & Jelenkovic, 1995) and several alcohol acyltransferases (SAAT, FaAAT2; Aharoni et al., 2000; Cumplido‐Laso et al., 2012) for esters, a nerolidol synthase 1 (FaNES1; Aharoni et al., 2004) for terpenes and a quinone oxidoreductase (FaQR; Raab et al., 2006) for furaneol. Recently, QTL studies and transcriptome data analyses for strawberry volatiles using biparental crosses have detected QTL and causative genes for mesifurane and gamma‐decalactone (Zorrilla‐Fontanesi et al., 2012; Chambers et al., 2014; Sánchez‐Sevilla et al., 2014; Cruz‐Rus et al., 2017; Barbey et al., 2021; Oh et al., 2021). Nevertheless, low mapping resolution and a lack of subgenome‐specific markers have hampered further characterization of causal genes underlying other QTL. This problem recently was addressed by the development of 50K Fana SNP array using probe DNA sequences physically anchored to the octoploid ‘Camarosa’ genome (Edger et al., 2019; Hardigan et al., 2020, 2021a). High heterozygosity combined with an allopolyploid genome presents difficulties for resolving causative genes and their haplotypes. To further the goal of discovering causative genes affecting flavor in strawberry, association studies with larger sample sizes and additional genetic resources such as eQTL and additional genomes are required. Furthermore, these resources must span the breadth of natural variation in breeding germplasm.
Here we present multi‐omics resources consisting of an eQTL study representing the genetic diversity of strawberry breeding programs in the US, phased genome assemblies of a highly‐flavored University of Florida breeding selection, a structural variation map in octoploid strawberry and a volatile GWAS of 305 individuals. These are combined to leverage the extensive metabolomic, genomic and regulatory complexity in strawberry for the discovery of natural variation in genes affecting flavor. Ultimately, the functional alleles identified will be selected in breeding to achieve superior flavor.
Materials and Methods
eQTL mapping
The eQTL population consisted of 196 genotypes including 133 newly sequenced accessions (Supporting Information Table S1). The University of Florida genotypes were grown at GCREC and collected in the spring of 2020 and 2021. The University of California‐Davis collection of diverse selections from multiple breeding programs were grown at either Santa Maria CA or Oxnard CA, for day‐neutral and short‐day accessions, respectively, and collected in the spring of 2021. Four UC genotypes were collected at both sites to ensure sequencing and SNP quality. Total RNA was extracted from a bulk of three fully ripe fruits using a Spectrum™ Plant Total RNA Kit (Sigma‐Aldrich), after flash freezing in liquid nitrogen. Illumina (San Diego, CA, USA) 150‐bp pair‐end sequencing was performed on the Illumina NovoSeq platform by Novogene Co. (Sacramento, CA, USA). On average, 6.9 Gb of sequence data were obtained for each sample. Raw RNA‐Seq data of 63 samples from previous published studies were retrieved from the NCBI SRA database (Table S1).
In order to quantify gene expression, short reads were trimmed for adapter sequences and low‐quality reads with Trimmomatic v.0.39 and aligned against the reference genome (Edger et al., 2019) using Star v.2.7.6a in the two‐pass mode (Dobin et al., 2013). Only unique aligned reads were scored by HTSeq v.0.11.2 in the union mode with the ‘‐‐nonunique none’ flag supplied with the latest Fragaria_ananassa_v1.0.a2 annotation (Liu et al., 2021). All count files were compiled in R and normalized with the deseq package (Love et al., 2014). To generate the marker dataset for eQTL mapping, SNPs and InDels were called using the mpileup and call commands. Markers were further hard‐filtered using bcftools with the following steps: (1) individual calls with lower than sequencing depth of three were set to missing using + setGT plugin; (2) marker sites with quality < 30, missing rate > 0.3, heterozygous call rate > 0.98, minor allele frequency < 0.05, or number of alternative alleles > 1 (only retain biallelic sites) were purged; (3) the filtered markers were imported and analyzed in R, and only markers showing more than three matched calls in four duplicated sample pairs were retained. A total of 491 896 markers passed the three stages of filtering. The missing calls were imputed, and all calls were phased using Beagle v.5.2 using the default settings (Browning et al., 2018).
The eQTL mapping was performed for 62 181 fruit expressed genes (average normalized count > 3) using the filtered markers. Linear mixed models (LMM) implemented in GEMMA were used for association analysis (Zhou & Stephens, 2012). The relationship matrix was computed in GEMMA and supplied to explain relationship within populations, and the top five principal components with a total of 25.0% variance explained were imported as covariates to reduce effects from population stratification to signify the genetic variance underlying the target traits. The Bonferroni corrected 5% significance threshold was used, determined the by number of LD‐pruned markers (0.05/168476 = 2.96 × 10−7). The approach to define an eQTL was similar to that used in previous studies (Liu et al., 2017; Li et al., 2020). Briefly, we first clustered all significant markers with distance < 100 kb and purged clusters with fewer than three markers. The lead marker with lowest P‐value was used to identify the eQTL, and boundaries of eQTL were defined as the furthest flanking significant markers. Clusters in LD (r 2 > 0.1) were merged and boundaries were updated. The longest distance between cis‐eQTL boundaries and eGene boundaries was limited to 500 kb. Trans‐eQTL hotspots were searched using the density function in R (Notes S1). Proportion of phenotypic variance explained by lead markers (pve) were computed using the formula , where is the SNP effect size, maf represents minor allele frequency and var(y) is the phenotypic variance. The eQTL also were mapped with the 50K Fana SNP array (Hardigan et al., 2020) for 185 individuals using a similar approach (Notes S1; Dataset S1).
Population genetic analysis
In order to examine structure and admixture in the eQTL population, a series of population genetic analyses were conducted using a pruned marker dataset (r 2 > 0.4 in a window of 100 kb). After pruning, 168 476 SNPs were retained. Population structure modeling was conducted with FastStructure v.20150112 using default parameters (Raj et al., 2014). Values of k between 3 and 10 were tested, and a model with k = 4 had the highest marginal likelihood. A maximum‐likelihood tree was built using snphylo v.20140701 with 1000 bootstrap replicates (Lee et al., 2014). A phylogenetic tree was visualized with R/ggtree v.3.14 (Yu et al., 2017). LD decay was analyzed with PopLDdecay v.3.4 using the unpruned marker set (Zhang et al., 2019). Principal component analysis as well as pairwise measurement was performed in the SNPRelate package v.1.26 in R (Zheng et al., 2012).
Genome assembly
Fragaria × ananassa FL 15.89‐25 carrying multiple favorable alleles for genes affecting flavor was selected for sequencing. High molecular weight DNA was extracted from etiolated leaf tissue. Sequencing was performed by high‐fidelity (HiFi) long‐read sequencing on the Pacbio Sequel II platform. gDNA was sheared to c. 17 kb average and the library prepared with 6 μg of sheared gDNA size‐selected by Blue Pippin to enrich large fragments and remove fragments smaller than 8 kb. The size‐selected library was sequenced with two 8 M SMRT cells using sequencing chemistry v.2 and polymerase v.2.0. Two SMRT cells yielded a total of 31.1 Gb HiFi reads with an average read length of 15.2 kb. The parents ‘Florida Beauty’ (female; Whitaker et al., 2017) and FL 12.115‐10 (male) were sequenced with Illumina NovaSeq 150 bp pair‐end. Total lengths of 35.1 and 33.9 Gb short‐read data were obtained for ‘Florida Beauty’ and FL 12.115.10, respectively.
A de novo trio‐binning assembly was built using Hifiasm v.0.11 coupled with Yak v.0.1 with the parameter ‘‐D10’ (Cheng et al., 2021), provided with parental short reads and HiFi reads from FL 15.89‐25. One incorrectly phased contig was identified in the phasing evaluation and visualized in Bandage v.0.8.1 (Wick et al., 2015). The mis‐phased contig was divided at the break point and reassigned to the correct haplotype assembly. Pseudochromosomes were constructed according to a reference‐based approach using Ragtag v.1.0 with parameters ‘‐C ‐f 10000 ‐‐remove‐small’ (Alonge et al., 2019) based on the ‘Camarosa’ reference genome (Edger et al., 2019). The unscaffolded contigs were concatenated into chr0. Hereafter we use F12 and Bea to refer to the haploid genomes corresponding to FL 12.115.10 and ‘Florida Beauty’, respectively. Details of genome annotation and evaluations of genome assembly and annotation can be found in Notes S2.
Structural variant analysis
Five high‐quality, long‐read‐based haploid assemblies including the F12 and Bea haplotypes of FL 15.89‐25, PHASE1&2 haplotypes of University of California, Davis cultivar ‘Royal Royce’ (Hardigan et al., 2021a) and the WONG haplotype of Korean inbred line ‘Wongyo 3115’ (Lee et al., 2021) were used to build a cumulative SV map. Individual haploid assemblies (F12 and Bea contig sets were used to avoid false positives introduced from scaffolding) were first mapped to the PHASE1 haplotype using Minimap2 v.2.17. The SVs then were called for individual haplotypes with SVIM‐asm v.1.02 (Heller & Vingron, 2020). Jasmine v.1.14 was used to merge SVs across haplotypes (Alonge et al., 2020). The merged SV map was annotated with genomic features using Vcfanno v.0.3.3 (Pedersen et al., 2016). Sampling and analysis for fruit allele‐specific expression were similar to the protocol given in eQTL mapping. Details are given in Notes S3.
Genome‐wide association study for strawberry volatiles
The GWAS population consisted of 305 genotypes, including progenies from 11 biparental crosses and elite breeding lines from the strawberry breeding program at the University of Florida. Among the 305 individuals, 164 were developed in a previous study (Barbey et al., 2021). We added 141 individuals, mostly from three experimental crosses. These were ‘Mara de Bois’ בFlorida Elyana’ (family 16.63, n = 20), ‘Florida Beauty’ × FL 15.89‐25 (family 18.50, n = 66), and FL 15.34‐82 × FL 15.89‐25 (family 18.51, n = 53). The fruits were harvested from experimental fields at the Gulf Coast Research and Education Center (GCREC) in Wimauma, Florida. Three harvests were conducted on 3 Dec, 17 Dec and 17 Jan during the 2018–2019 season. Individuals from family 16.63 were harvested two times on 16 Feb and 2 Mar 2017. Following harvests, fruits were immediately frozen in liquid nitrogen, then ground into fine powder and mixed with an equal volume of saturated NaCl solution and an internal standard of 3‐hexanone (final concentration of 1 ppm). Volatile quantification of three biological replicates (harvests) was performed with GC–MS, similar to the previous study (Barbey et al., 2021) with some modifications to the volatile quantification process (Notes S4).
Genotyping of the new individuals was performed using the FanaSNP 50K array (Hardigan et al., 2020) including 5809 markers previously included in the iStraw 90K and iStraw 35K SNP arrays (Bassil et al., 2015; Verma et al., 2016). The 164 individuals genotyped with the 35K array were imputed to the FanaSNP array using AlphaImpute2 v.0.02 (Whalen & Hickey, 2020). An additional 161 elite lines representing the UF breeding pool also were genotyped with the FanaSNP array to improve imputation accuracy. After filtering using minor allele frequency (MAF) > 0.05, a total of 34 056 SNPs were retained for the GWAS (Dataset S2). Because the GWAS population mostly constituted biparental crosses, strong relatedness and stratification existed in the population. To account for population structure, we used a mixed linear model implemented in fastGWA (GCTA v.1.93.2beta) including the top 10 principal components with a total of 48.2% variance explained and a relationship matrix derived from SNPs with all of the small off‐diagonal elements (< 0.05) set to 0, which approximated the pedigree‐based relationship matrix (Jiang et al., 2019; Notes S5).
Transient fruit gene expression assay
Transient expression in strawberry fruits by agroinfiltration was performed according to Hoffmann et al. (2006). Briefly, the full‐length cDNA of FaASa1 and 208‐bp partial cDNA flanked by attB1 and attB2 sequences were synthesized by Integrated DNA Technologies Inc. (Coralville, IA, USA) (Table S2). Gateway vectors pK7GWIWG2(II) and pMDC32 were used for RNAi and overexpression assays, respectively. The positive clones were confirmed by PCR and Sanger sequencing. The FaASa1‐OX, FaASa1‐RNAi and two empty vectors, pK7GWIWG2(II) and pMDC32, were transformed into Agrobacterium tumefaciens strain EHA105. In each experiment, 10 attached white fruits from glasshouse‐grown FL 15.89‐25 plants were agro‐infiltrated for each construct. Injection was stopped when the whole fruit was saturated with the Agro‐suspension using a 5‐ml syringe. Fruits were harvested 5–7 d after infiltration depending on ripening status. Three to four fruits were mixed and immediately frozen in liquid nitrogen before volatile quantification and RNA extraction. The whole experiment was repeated three times. For FaOMT overexpression (sequences of synthesized gene fragment and primers can be found in Table S2), detached white fruits of ‘Chandler’ and ‘Camarosa’ were used for agroinfiltration.
Additional miscellaneous methods including Mendelian randomization, sensory data, RT‐PCR, haplotyping analysis and high‐resolution melting marker testing can be found in Notes S6.
Results
High‐resolution eQTL mapping
In order to gain a holistic view of allelic diversity and genetic regulatory networks for fruit‐expressed genes in cultivated strawberry, we analyzed the transcriptomes of 196 individuals, mainly from North America, including 133 newly sequenced genotypes. After pruning (r 2 < 0.4 in a 100‐kb window), 168 476 marker sites remained for population genetic analysis. Based on a maximum‐likelihood phylogenetic tree, principal component analysis (PCA) and population structure modeling (k = 3), the UC and UF populations formed distinct clusters as expected (Fig. 1a–c). The UC population had high uniformity with small contribution from outside sources (Fig. 1a). Zero to thirty percent of UC contribution was admixed in the UF population. The UF population could be further classified into two subgroups, supported by principal component 3 (Fig. 1b) and a population structure model using k = 4 (Fig. S1). Despite a clear divergence between UF and UC populations, a smaller (0.057) was found than in previous estimations (Hardigan et al., 2021b). This smaller could be explained in part by recent concordant efforts to introgress exotic resources to expand the flavor profiles. All ‘cosmopolitan’ cultivars from various sources showed an admixture structure (Fig. 1a). In addition to high‐density markers, low LD among markers is essential for high‐resolution eQTL mapping. We observed rapid LD decay across populations (Fig. S1). Short‐range LD (r 2 ≈ 0.2) decayed at a distance < 250 bp in both UF and UC populations (Fig. S1).
Fig. 1.
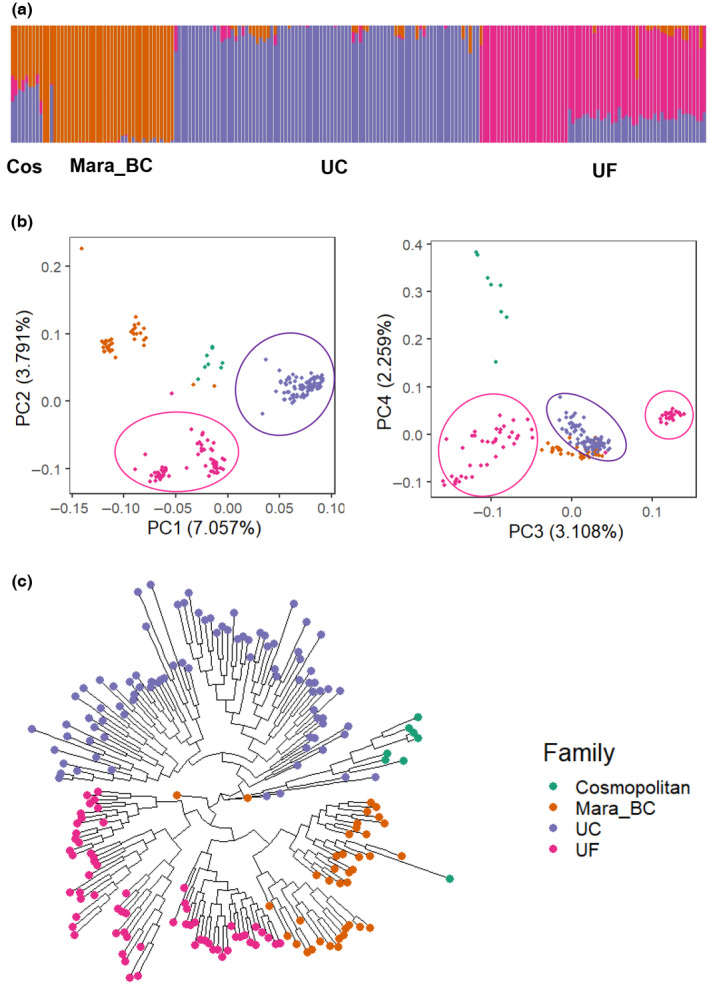
Population genetic analysis of the strawberry (Fragaria × ananassa) expression quantitative trait loci (eQTL) population. (a) Model‐based clustering of 196 accessions using a pruned SNP dataset including 168 476 SNP loci. The k (number of clusters) was set to 3. Populations are grouped by geographical origin. Cos, ‘cosmopolitan’, varieties were not developed from UF and UC breeding programs; Mara_BC, the first‐generation backcross of UF elite germplasm to ‘Mara des Bois’; UF, University of Florida germplasm; UC, University of California germplasm. (b) Principal component analysis (PCA) plots of the first two principal components (PCs, left) and the third and fourth PCs (right). Both plots support classification based on breeding programs. Purple and pink eclipses highlight the clusters of the UC and the UF materials. (c) Maximum‐likelihood phylogenetic tree with six cosmopolitan cultivars rooted in the same cluster as UC accessions and showing clear divergence between breeding programs.
The eQTL mapping using 491 896 markers revealed 68 535 eQTL for 33 397 fruit expressed genes (eGenes; Fig. 2a; Dataset S3). About 53.7% of eGenes (n = 62 181) had at least one eQTL. Among eGenes, the mean number of eQTL was 2.05, with a maximum of 65 (FxaC_9g21450). Many eGenes exhibited complex genetic controls, containing both distant and local regulatory elements. In total, 45 083 trans‐eQTL were identified for 20 650 eGenes, compared to 23 452 cis‐eQTLs for 22 731 eGenes. Although only 873 lead SNPs (the most significant SNP) located within eGenes, 37.0% of cis‐regulated eGenes had at least one significant marker inside the gene, and the median shortest distance between a significant marker within a cis‐eQTL and its associated eGene was only 3787 bp (Fig. 2b). Cis‐eQTL generally had larger effects than trans‐eQTL (Student's t‐test, P < 2.2e‐16), supported by both higher pve and −logP values (Fig. 2c). Interestingly, there was a significant enrichment ( test, P‐value < 2.2e‐16) of homoeologous trans‐eQTL and intrachromosomal trans‐eQTL (Fig. 2a,d), in line with eQTL mapping results in allotetraploid cotton and maize (Liu et al., 2017; Li et al., 2020). About 35.4% of trans‐eQTL were located between homoeologous chromosomes (Fig. 2d). Furthermore, the homoeologous trans‐eQTL exhibited significantly larger effects on gene expression than other trans‐eQTL (Tukey's honestly significant difference test, P < 0.05) including intrachromosomal trans‐eQTL (Fig. S2). The dominant Fagaria vesca‐like subgenome (Edger et al., 2019) harbored the largest number of nonhomoeologous trans‐eQTL (Fig. 2d, ‐test, P = 0.0004), but a similar number of homoeologous trans‐eQTL as the F. iinumae‐like subgenome.
Fig. 2.
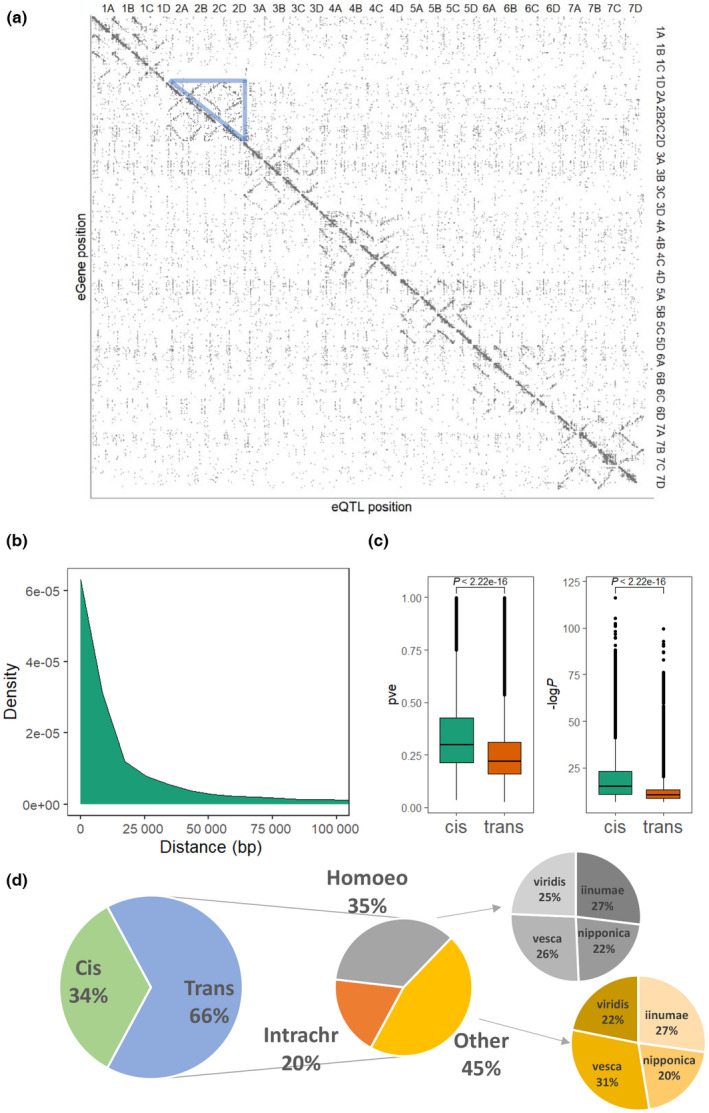
Genome‐wide characterization of expression quantitative trait loci (eQTL) in strawberry (Fragaria × ananassa). (a) The lead marker positions of eQTL are plotted against the middle positions of mapped eGenes. Each dot represents a significant eQTL. The grayscale of the dot reflects the −logP value. The blue triangle highlights homoeologous trans‐eQTL on chromosomes 2A to 2D. (b) Density plot of minimum distance between significant cis‐eQTL markers and their associated eGenes. (c) Box plots comparing proportions of phenotypic variance explained by lead markers (pve, left) and −logP (right) between cis‐ and trans‐eQTL. Boxes are delimited by upper and lower quantiles. Two whiskers represent highest/lowest values; dots represent outliers; and horizontal lines represent medians. (d) Proportions of cis‐eQTL and trans‐eQTL. Trans‐eQTL are categorized into homoeologous trans‐eQTL (homoeo), intrachromosomal trans‐eQTL (intrachr) and other trans‐eQTL. Homoeologous trans‐eQTL/other trans‐eQTL are further divided into four groups according to their subgenome ancestry.
The eQTL mapping using the 50K Fana SNP array captured 17 562 cis‐eQTL for 17 441 eGenes (Fig. S3a; Datasets [Link], [Link]), which made up 74.9% of cis‐eQTL identified through RNAseq variants. Moreover, the median distance of cis‐eQTL to eGene was only 9029 bp, only about two‐fold larger than using RNAseq variants (Fig. S3b). However, trans‐eQTL were poorly detected by the Fana SNP array, with only 5936 trans‐eQTL passing the significant threshold (p < 0.05/49331). The possible reason for lower power in detecting tran‐eQTL was low marker density.
Similar to other eQTL studies (Liu et al., 2017; Albert et al., 2018), we found that trans‐eQTL were not evenly distributed, but instead congregated at scattered hotspots. We identified a total of 2141 hotspots (Dataset S6). The largest eQTL hotspot was located at 8788 309–8795 572 bp on Chr 5A, associated with 928 eGenes. A total of 384 master regulators (Dataset S6) were putatively identified leveraging Mendelian Randomization (MR) tests (Notes S6), including an E3 ubiquitin ligase BIG BROTHER‐like gene (FxaC_20g13890, P_MR = 3.18e‐12) for the largest trans‐eQTL hotspot on Chr 5D (Notes S7; Fig. S4).
The structural variant landscape in strawberry
Because a substantial number of regulatory elements were found for fruit‐expressed genes, a structural variant (SV) map would greatly facilitate the identification of potential causative SVs underlying the regulatory elements. To construct an SV map, we first assembled a phased genome of an UF accession. The genome of FL 15.89‐25 was assembled into 1480 and 672 phased contigs with N50 of 12.8 and 12.4 Mb, respectively (Fig. S5a), with similar contiguity to other recent high‐quality octoploid strawberry genomes (Table 1). A Kmer‐based approach revealed 97.1% and 99.2% completeness for the haploid assemblies based on parental Illumina short reads, which were corroborated by 98.1% and 98% completeness of the BUSCO eudicots odb10 genes (Table 1). Phasing quality was evaluated by parent‐specific Kmers; the average switching error and hamming error were 0.19% and 0.18% for the F12 haploid assembly (Fig. S5b–d), comparable to phased genomes in other species (Cheng et al., 2021). The phased contigs were scaffolded into pseudochromosomes based on alignment to the ‘Camarosa’ reference genome, with 96.0% (795.1 Mb) and 92.8% (778.6 Mb) of phased contigs placed on 28 pseudochromosomes for the respective F12 and Bea haploid assemblies (Fig. S6), consistent with previous flow cytometry estimations (720/813 Mb) (Akiyama et al., 2001; Edger et al., 2019). There were only 88 and 79 gaps in the final scaffolds, averaging 3.14 and 2.82 per chromosome for the respective F12 and Bea assemblies (Fig. S6). Scaffolding quality was evaluated by a linkage map and public Hi‐C data (Lee et al., 2021; Fig. S7). High collinearity was observed between haplotypes (Fig. S8).
Table 1.
Assembly quality evaluations of the FL15.89‐25 genome (Fragaria × ananassa).
| Assembly matrices | F12 haplotype | Bea haplotype | ‘Royal Royce’ PHASE1 a | ‘Wongyo 3115’ |
|---|---|---|---|---|
| Contig Size (Mb) | 827.3 | 839.4 | 784.7 | 805.7 |
| Number of contigs | 1480 | 672 | 136 | 323 |
| Scaffolded size (Mb) | 795.1 | 778.6 | 784.5 | 805.7 |
| Number of scaffolded contigs | 116 | 107 | 81 | 323 |
| Largest contig (Mb) | 23.2 | 26.7 | 27.6 | 22.9 |
| N50 (Mb) | 12.8 | 12.4 | 11.0 | 9.8 |
| N90 (Mb) | 3.4 | 2.5 | ||
| Completeness (%) | 97.1 | 99.2 | ||
| BUSCO (%) | 98.1 | 98.0 | 98.1 | 94.1 b |
| BUSCO frag (%) | 0.1 | 0.1 | 0.8 | |
| QV | 60 | 58 | ||
| Switching error (%) | 0.19 | 0.24 | ||
| Hamming error (%) | 0.18 | 0.24 |
Completeness was evaluated by Kmer based completeness and BUSCO score of complete and fragmented genes. Assembly accuracy was measured by QV and phasing quality was evaluated by switching error and hamming error. Switch error rate is the percentage of adjacent SNP pairs that are wrongly phased. Phasing hamming error rate is the percentage of SNP sites that are wrongly phased.
Assembly matrices of ‘Royal Royce’ and ‘Wongyo 3115’ were retrieved from published papers (Lee et al., 2021; Hardigan et al., 2021a).
Embryophyta gene set was used for BUSCO evaluation, different from Eudicot gene set used for other assemblies.
The FL 15.89‐25 assemblies and three additional haploid assemblies (Lee et al., 2021; Hardigan et al., 2021a) were utilized to explore SV diversity in garden strawberry. These geographically and genetically diverse accessions empowered the discovery of SVs across all chromosomes except for a large portion of Chr 4B which may be under strong purifying selection (Fig. 3c). Individual haplotypes had between 31 574 and 60 453 SVs relative to the PHASE1 assembly of ‘Royal Royce’ (Fig. 3a; Table S3), with the WONG haplotype harboring the most SVs, consistent with the larger genetic distance of Asian populations to North American populations (Fig. 3c). Insertions and deletions were the most common SV types, together consisting of 88.3–94.1% (Fig. 3a; Table S3) of SVs. All SVs across haplotypes were then merged into a nonredundant set of SVs (Dataset S7). In total, 56 342 deletions, 60 983 insertions, 12 016 translocations, 166 interspersed duplications, 236 tandem duplications and 137 inversions were identified. Unlike the SV composition of a tomato population in which the majority of SVs were singletons (Alonge et al., 2020), an average of 62.6% strawberry SVs were shared by at least two haplotypes (Fig. 3b). We observed a gradually reduced number of new SVs every time a new haplotype was merged (Fig. 3d), suggesting this SV map surveys a substantial portion of SV diversity in cultivated strawberry. The majority of SVs were < 1 kb (73.0%), whereas only 3.3% were > 10 kb (Fig. 3e). Structural variations were present extensively in exons (8675 deletions and 3782 insertions; Fig. 3f), introns (6715 deletions and 9922 insertions) and promoter regions (15 495 deletions and 18 566 insertions). Transposable elements (TEs) were rich resources of SVs. We identified 34 379 deletions overlapped with TEs, especially inverted tandem repeats (TIRs) and long terminal repeats (LTRs), consisting of 61.0% of total deletions, significantly higher than the genome‐wide TE percentage of 38.42% (χ 2 test, P < 2.2e‐16).
Fig. 3.
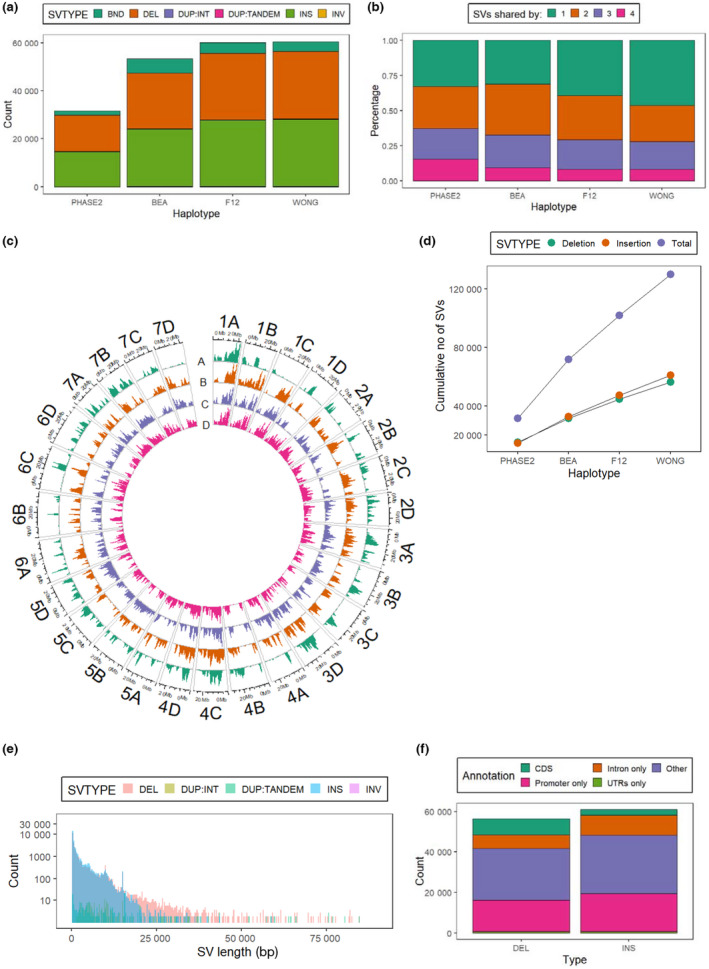
The structural variant landscape in garden strawberry (Fragaria × ananassa). (a) Counts of different types of structural variations (SVs) relative to the PHASE1 haplotype of FaRR1. BND, translocation; DEL, deletion; DUP : TANDEM, tandem duplication; DUP : INT, interspersed duplication; INS, insertion; INV, inversion. (b) Percentage of SVs that are singleton or shared by two, three or four other haplotypes in each haplotype. (c) Genome‐wide distribution of SV density in PHASE2(A), BEA(B), F12(C) and WONG(D) haplotypes relative to the PHASE1 haplotype. (d) Cumulative number of SVs when new haplotypes are sequentially merged. Colors of the dots represent SV types. (e) Size distribution of SVs. Colors represent different SV types. (f) Counts of deletions and insertions overlapped with different genome annotations.
In order to investigate whether SVs were related to allele‐specific expression (ASE) in fruit, we performed total RNA sequencing for four biological replicates of FL 15.89‐25 ripe fruit (Notes S3, S8). Consistent density distributions of allelic expression ratios were observed across replicates (Fig. S9a). A total of 12 503 genes (Fig. S10; Dataset S8) (P adjusted < 0.05, 48.8% of expressed heterozygous genes) exhibited significant ASE. Extreme expression ratios (0 and 1) were inflated, with 3415 genes showing extremely imbalanced expression in which the dominant allele contributed to > 90% of gene expression (Fig. S9a). After integrating with SV results, we identified 2457 of 12 503 ASE genes overlapping SVs at the intragenic region, compared to significantly fewer (1860 of 13 097) non‐ASE genes overlapping with SVs (Fig. S10; χ 2 test, P < 2.2e‐16). However, we did not observe a higher rate of SVs at the 2 kb promoter region of ASE genes (3443 vs 3496). KEGG enrichment analysis identified several secondary metabolic pathways enriched in ASE genes including terpenoid backbone biosynthesis (P adjusted = 1.07e‐6, ko00900), phenylpropanoid biosynthesis (P adjusted = 3.36e‐4, ko00940), alpha‐linolenic acid metabolism (P adjusted = 4.46e‐3, ko00592, Fig. S9b), fatty acid biosynthesis (P adjusted = 5.74e‐3, ko00061) and flavonoid biosynthesis (P adjusted = 7.94e‐3, ko00941).
Volatile GWAS
In order to investigate the genetic control of fruit volatiles, we performed volatile phenotyping and SNP array genotyping with 49 330 markers on a panel of 305 accessions from the UF strawberry breeding program, with 59 individuals overlapped with the eQTL panel (Table S1). A total of 97 volatiles including esters, terpenes, aldehydes, alcohols, acids, ketones and lactones were quantified (Fig. S11). Based on relationships among volatiles, we identified at least five clusters belonging to the same chemical class or biosynthetic pathway, including clusters of eight aldehydes, three ethyl esters, three hexanoic acid derivatives, seven medium‐chain esters and three terpenes (Fig. S11). Generally high narrow‐sense heritability () was observed across volatiles (Table S4), ranging from 0.212 to 0.916, with a mean of 0.660. The highest value of was found for mesifurane (0.916) and the lowest for octanoic acid, ethyl ester (0.212).
Genome‐wide association study identified 62 signals (P < 1.85 × 10−5) for 35 volatiles (Fig. 4a; Table S4). The lead SNP effects varied from 0.27 to 2.44 (1.2‐ to 5.4‐fold change), with the largest effect for methyl anthranilate (Fig. 4d; Table S5). Two hotspots which contained multiple signals of volatiles belonging to the same class or pathway were found for medium‐chain esters (Fig. 4b) and for terpenes (Fig. 4c), which also were detected to in previous studies (Barbey et al., 2021; Fan et al., 2021) and reflected in chemical relationships (Fig. S11). Our GWAS results confirmed previous homoeologous group assignments for these volatile QTL and clarified their subgenome and physical positions. The SNP AX‐166515537 was the lead SNP for three esters, and a 14 Mb region on Chr 6A shared signals for six medium‐chain esters. An LD analysis revealed three linkage blocks (Fig. S12a). The distal region of Chr 3C was associated with six volatiles including five terpenes (Fig. 4c). This 3.1‐Mb region did not display clear LD block separation (Fig. S12b). Two significant markers for medium‐chain ester hotspot and methyl thiolacetate were tested for their predictability of flavor characteristics (Notes S9; Fig. S13).
Fig. 4.
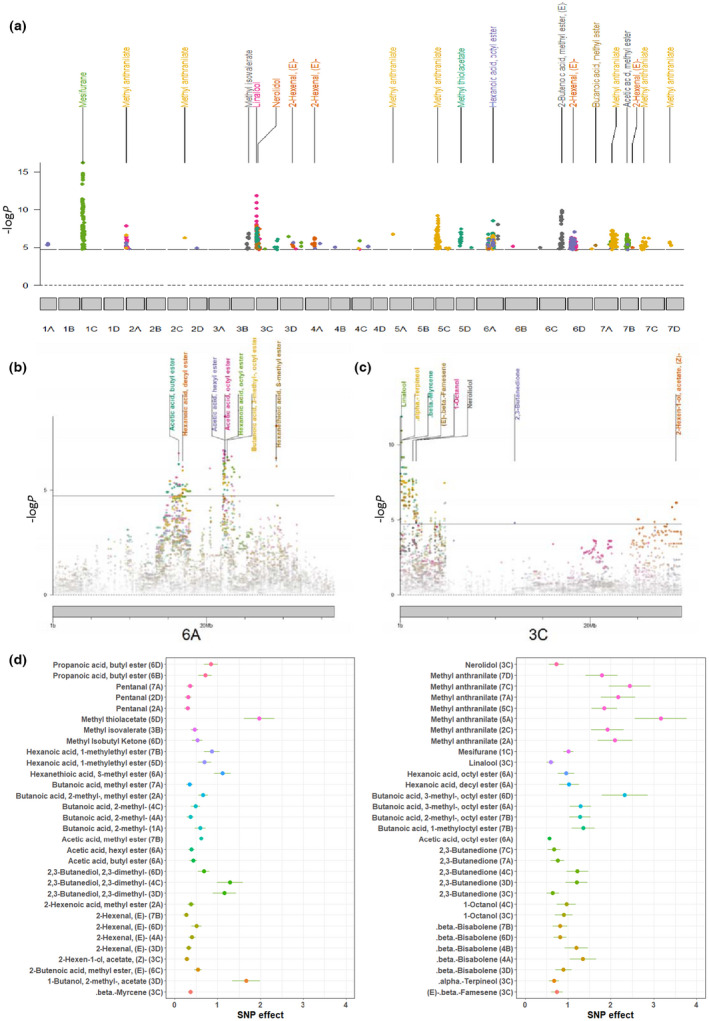
Genome‐wide association study (GWAS) analysis of strawberry (Fragaria × ananassa) fruit volatiles. (a) Manhattan plot of all significant GWAS signals for 35 volatiles. A fraction of significant GWAS peaks are labeled by their associated chemical names. Nonsignificant markers were removed. (b, c) Chromosome‐views of GWAS hotspots for medium‐chain esters (b) on chromosome 6A and terpenes on chromosome 3C. (d) Absolute single nucleotide polymorphism (SNP) effect (beta from linear mixed model) of all 62 GWAS signals, with whiskers the length of one SE. Chromosome labeled next to chemical name. Chemical abundances are log‐transformed.
Some abundant volatiles including: 2‐hexenal, (E)‐; butanoic acid, 2‐methyl‐; and pentanal (Figs 4d, S14) were associated with multiple DNA variants (number of signals ≥ 3), suggesting polygenic inheritance. Pentanal (Fig. S14a) was associated with three loci, together explaining 30.7% of phenotypic variation in a GLM model. Significantly higher pentanal content was observed in genotypes with three doses of the alternative allele at two loci (Fig. S15a). Epistatic interactions emerged among pentanal loci as significant interactions were detected between lead SNPs AX‐184916449 and AX‐184236819 (F‐test, P = 0.002), as well as AX‐184916449 and AX‐184422639 (F‐test, P = 4.07e‐06). Similar dosage effects and polygenic patterns were observed for 2,3‐butanedione and 2‐hexenal, (E)‐ (Figs S14b,c, S15b,c).
Known flavor genes and their regulatory elements
In order to investigate natural variation of known genes underlying volatile‐GWAS peaks, a list of 13 previously curated and functionally validated genes (Aharoni et al., 2000, 2004; Raab et al., 2006; Landmann et al., 2007; Cumplido‐Laso et al., 2012; Zorrilla‐Fontanesi et al., 2012; Chambers et al., 2014; Molina‐Hidalgo et al., 2017; Pillet et al., 2017; Dai et al., 2020; Lu et al., 2021) affecting flavor in Fragaria × ananassa was compiled (Table S6). Their best corresponding annotations in the ‘Camarosa’ reference genome were identified via homolog search and fruit expression, but both FaFAD1 and FaNES1 (Aharoni et al., 2004; Oh et al., 2021) had no match. Our eQTL results revealed 31 eQTLs for seven genes affecting flavor including three cis‐eQTL for FaAAT2 (FxaC_11g40760), FaGT2 (FxaC_6g04890) and FaOMT (FxaC_28g01250; Table S6). Our GWAS also revealed a weak signal on Chr 7D for mesifurane, overlapping with the cis‐eQTL of FaOMT (Fig. 5a; Zorrilla‐Fontanesi et al., 2012). Using four unlinked significant SNPs (r 2 < 0.5) from the cis‐eQTL, haplotype analysis revealed nine haplotypes (freq > 2/392) including haplotypes 4, 7 and 8 with reduced expression (Figs 5b, S16a). The functional haplotype2 of FaOMT for mesifurane production was confirmed through transient overexpression (Fig. 5c). Previously, a natural InDel (InDel1) at the promoter region was proposed to contribute to the complete silencing of FaOMT (Zorrilla‐Fontanesi et al., 2012). A novel SV (InDel2) of 28 bp at the first exon (Fig. 5d) was revealed through inspection of multiple haplotypes of FL 15.89‐25 (hap2/hap6) and ‘Royal Royce’ (hap2/hap8). InDel2 caused a frameshift, leading to a premature stop at the fourth codon following the SV (Fig. 5d). This InDel2 also was captured by resequencing data from ‘Florida127’ (Fig. S16b). A functional HRM marker was designed based on the causal InDel2 and tested in a panel of breeding selections (n = 38, two harvests) and a new test cross (n = 19, two field replicates, three harvests). This co‐dominant HRM marker predicted mesifurane content, with an additive effect of allele dosage (Figs 5e, S16c), consistent with transient overexpression results (Fig. 5c).
Fig. 5.
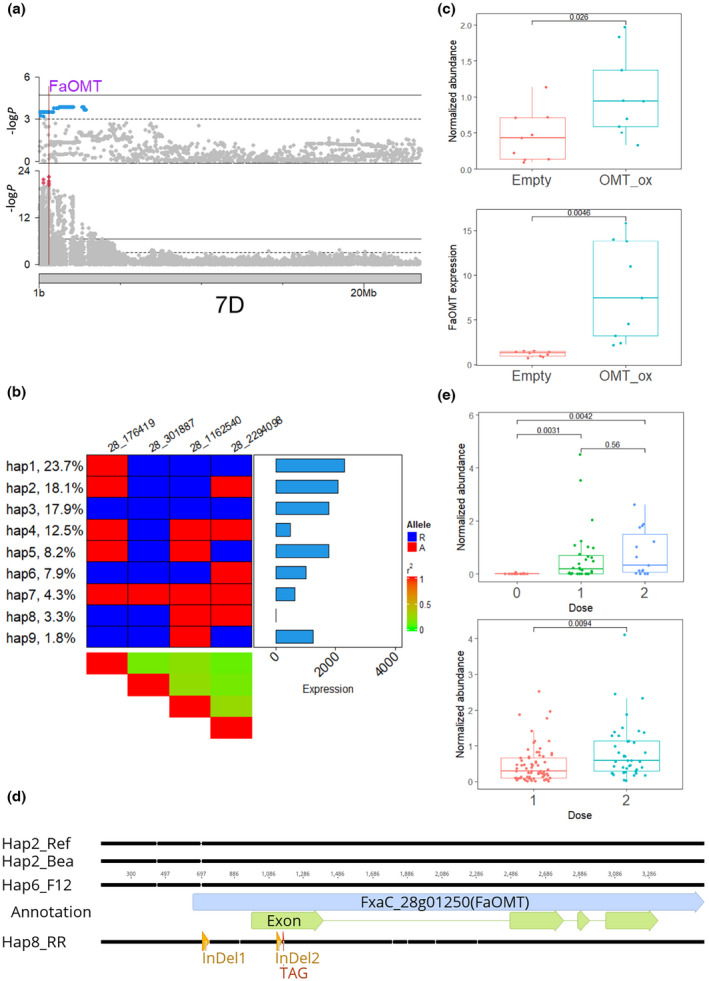
Genetic analysis of mesifurane in strawberry (Fragaria × ananassa). (a) Manhattan plots of genome‐wide association study (GWAS) results for mesifurane (upper panel) and expression quantitative trait loci (eQTL) results for FaOMT (lower panel) on choromosome 7D. The physical position of FaOMT is marked with a vertical line. Blue dots represent all significant markers in mesifurane GWAS results and red dots represent top five makers of FaOMT eQTL. (b) Nine haplotypes of FaOMT identified with four unlinked significant markers (r 2 < 0.5). Markers were named according to chrID_position. Left annotation shows haplotype frequency. Right annotation shows haplotype effect in unit of normalized count. The central heatmap shows marker genotype. Blue represents the reference allele; red represents the alternative allele. (c) Transient overexpression of FaOMT in strawberry fruit. Upper panel shows normalized abundance of mesifurane in fruits agroinfiltrated with either FaOMT overexpression construct (blue) or pMDC32 control (red). Lower panel shows FaOMT expression quantified with quantitative (q)PCR. Boxes are delimited by upper and lower quantiles. Two whiskers represent highest/lowest values; dots represent individual values; and horizontal lines represent medians. (d) Alignment among four haplotypes of FaOMT. Two haplotype 2s are from the reference genome and the Bea haploid assembly. Haplotype 6 is derived from the F12 haploid assembly. The nonfunctional haplotype 8 is from the ‘Royal Royce’ assembly. (e) Functional high‐resolution melting marker tested in selected breeding lines (upper) and a test cross (lower). The normalized abundance of mesifurane is plotted with the dosage of functional allele of FaOMT. A significant gene dosage effect was observed in the test cross. Box plot annotations are the same as for plot (c).
A variety of terpenes were associated with markers at the distal region of Chr 3C (Fig. 1c). Alignments of both new haplotype assemblies to the reference genome revealed that the whole 1.61‐Mb distal arm of Chr 3C was missing in the reference genome (Fig. S17a). A homolog search identified two FaNES1 genes (Aharoni et al., 2004) between 1520 037 and 1530 908 bp on Chr 3C_F (Fig. 6a), whereas the FaNES1t had very low transcript accumulation among all eQTL individuals (mean normalized count = 0.25). The two tandem genes only differed by several nonsynonymous mutations including a mutation I266V located directly upstream of the conserved substrate‐binding DDxxD motif (Fig. S17b). Among all genes within the 1.61 Mb region, Pearson correlation analysis indicated that FaNES1 expression has the highest correlation with linalool production (r = 0.65) and high correlations with beta‐myrcene, alpha‐terpineol, (E)‐beta‐famesene and nerolidol also (mean r = 0.61). eQTL mapping using makers derived from aligning RNAseq data to the F12 haploid assembly revealed a significant peak spanning the whole 1.61‐Mb region (Fig. 6b), with the lead SNPs at 93 4298 and 1166 597 bp on Chr 3C_F. Only one haplotype (hap3) showed reduced expression (Fig. S17c). Short read mapping of ‘Florida Beauty’ (hap3/hap4) to Chr 3C_F revealed a 24‐bp potential causal deletion (1520 457–1520 480 bp) in the first exon of FaNES1 (Figs 6a, S18). Evidence suggests that the 1.61‐Mb distal region was under strong selection: compared to the 1.61–5‐Mb region on Chr 3C_F, a significantly lower frequency of the AA genotype (homozygous for minor allele, mean AA%: 0.61% vs 5.7%; Fig. 6c), nucleotide diversity (mean : 1.87e‐4 vs 4.81e‐4; Fig. 6d) and minor allele frequency (mean MAF: 0.068 vs 0.158; Fig. 6d) were detected. Although there was another terpene synthase gene (germacrene d synthase‐like, maker‐Fvb3‐3_F‐augustus‐gene‐8.13) in this region, no positive correlation (r = −0.07) was found between terpene abundances and expression of this gene. In addition to the aforementioned two putative causal variants, we also detected four SVs overlapping with either genic or promoter regions of genes affecting flavor (Table S6; Figs S19, S20), including the deletion spanning the entire FaFAD1 gene, with an accurate re‐delineated the boundaries of the SV (Notes S10). By examining its flanking regions, we determined that this SV probably was mediated by illegitimate recombination (Fig. S19).
Fig. 6.
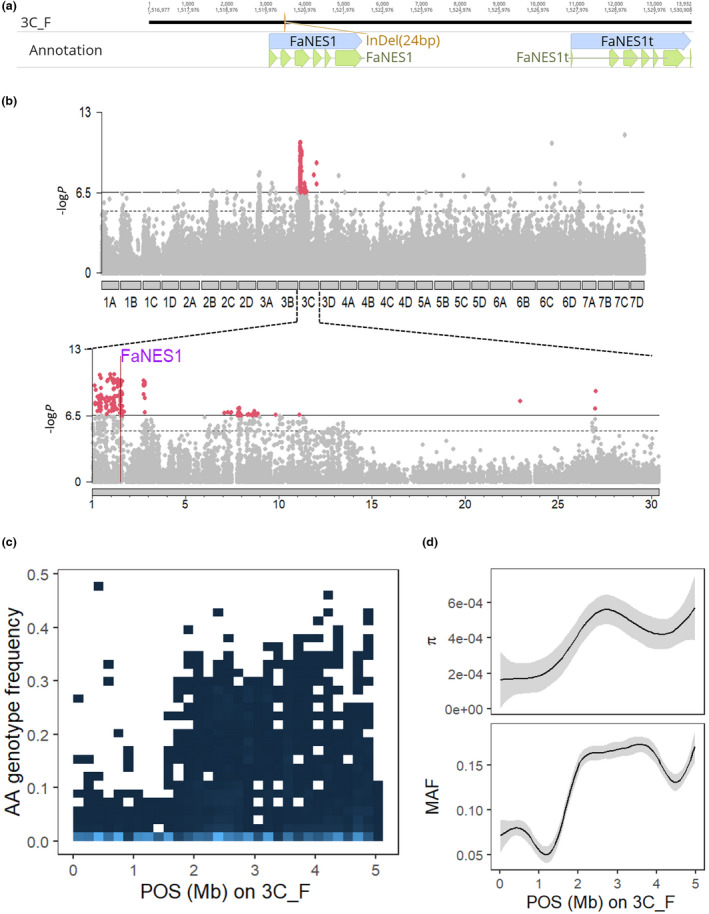
Genetic analysis of terpenes in strawberry (Fragaria × ananassa). (a) Gene models and physical positions of FaNES1 and its tandem duplicate FaNES1t. The putative causal 24‐bp deletion detected using ‘Florida Beauty’ resequencing data is highlighted. (b) Genome‐wide and chromosome view of expression quantitative trait loci (eQTL) results for FaNES1. The F12 haploid assembly was used as the reference. The physical position of FaNES1 was marked on 3C_F. Significant markers are colored in red. (c) 2D‐density plot shows AA genotype frequencies of markers derived from aligning RNAseq data to the F12 haplotype assembly. Each square represents marker count in the embedded range of physical position and genotypes frequency. Lighter color represents higher frequency. (d) Smoothed functions of nucleotide diversity (pi) and minor allele frequency (MAF) for markers ranging from 0 b to 5 Mb on chromosome 3C_F. The shaded area covers a 95% confidence interval.
Discovery of novel genes affecting flavor
Novel candidate genes for 14 volatile GWAS signals were identified based on MR scores, expression correlations and evidence of biological function (Table 2). A summary of these are as follows: a cis‐eQTL for a putative esterase gene (FxaC_20g11130) co‐localized with a GWAS peak for methyl thioacetate on Chr 5D, explaining 41% of expression variation; a putative alpha/beta‐hydrolase (FxaC_5g03020) was assigned as a candidate for butanoic acid, 2‐methyl‐, methyl ester on Chr 2A; a putative triglyceride lipases (FxaC_15g35150) had a cis‐eQTL co‐segregating with butanoic acid, 2‐methyl‐ GWAS peak on Chr 4C; two transcription factors including a putative WRKY3 on Chr 6C and zinc finger protein on Chr 7B were linked to biosynthesis of 2‐butenoic acid, methyl ester, (E)‐ and acetic acid, methyl ester, respectively; and a putative cinnamoyl CoA reductase 1 (CCR, FxaC_21g33200) was localized at the GWAS hotspot for medium‐chain esters on Chr 6A. High correlations (mean r = 0.42) were detected between expression of FxaC_21g33200 and abundances of: acetic acid, butyl ester; acetic acid, hexyl ester; acetic acid, octyl ester; hexanoic acid, octyl ester; and hexanoic acid, decyl ester. This highly expressed CCR in fruit potentially could catalyze substrates including precursor CoAs of medium‐chain esters, because a homolog of CCR was validated to convert benzoyl‐CoA to benzaldehyde (Liu et al., 2019). A putative anthranilate phosphoribosyltransferase (FxaC_19g06300) catalyzing the reaction of free anthranilate to N‐(5‐phospho‐d‐ribosyl)‐anthranilate was associated with the Chr 5C methyl anthranilate (MA) GWAS peak. An opposite marker effect (AX‐184023191) was detected for FxaC_19g06300 expression and MA production, suggesting that two reactions compete for free anthranilate. The best candidate for the Chr 7C MA GWAS peak was a putative beta‐hydroxyisobutyryl‐CoA hydrolase 1. Loss‐of‐function mutants in Arabidopsis were defective in beta‐oxidization of fatty acids and insensitive to auxin stimulus (indole‐3‐butyric acid; Zolman et al., 2001). It is possible that this gene regulates MA biosynthesis because it co‐localized with two trans‐eQTL for a putative anthranilate synthase beta subunit 1 (FxaC_16g27660) and a putative beta glucosidase 41 (FxaC_12g27490), both potentially involved in MA biosynthesis.
Table 2.
Candidate genes underlying genome‐wide association study (GWAS) peaks for strawberry (Fragaria × ananassa).
| CHR | Top SNP | POS | AF1 | BETA | SE | −logP | Volatile | Gene_CHR | Gene | P_MR | Prediction |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5D | AX184285180 | 5666 122 | 0.14 | −1.76 | 0.37 | 5.81 | Methyl thiolacetate | 5D | FxaC_20g11130 | 3.65E‐11 | Esterase |
| 4C | AX184169435 | 22 136 266 | 0.24 | 0.49 | 0.11 | 5.17 | Butanoic acid, 2‐methyl‐ | 4C | FxaC_15g35150 | 1.62E‐02 | Triglyceride lipases |
| 2A | AX184291444 | 1661 505 | 0.40 | 0.42 | 0.09 | 4.96 | Butanoic acid, 2‐methyl‐, methyl ester | 2A | FxaC_5g03020 | 2.10E‐02 | alpha/beta‐Hydrolases |
| 6C | AX123362701 | 31 363 992 | 0.56 | 0.55 | 0.09 | 8.74 | 2‐Butenoic acid, methyl ester, (E)‐ | 6C | FxaC_23g48290 | 2.40E‐05 | WRKY3 |
| 6A | AX123358920 | 16 899 973 | 0.41 | −1.03 | 0.24 | 4.86 | Hexanoic acid, decyl ester | 6A | FxaC_21g33200 | 2.98E‐02 | Cinnamoyl CoA reductase 1 |
| 6A | AX123358920 | 16 899 973 | 0.41 | −0.92 | 0.19 | 5.94 | Hexanoic acid, octyl ester | 6A | FxaC_21g33200 | 4.45E‐03 | Cinnamoyl CoA reductase 1 |
| 6A | AX123358920 | 16 899 973 | 0.41 | −0.47 | 0.09 | 6.12 | Acetic acid, octyl ester | 6A | FxaC_21g33200 | 7.96E‐03 | Cinnamoyl CoA reductase 1 |
| 6A | AX184534745 | 16 383 672 | 0.33 | 0.44 | 0.09 | 6.24 | Acetic acid, butyl ester | 6A | FxaC_21g33200 | 8.21E‐02 | Cinnamoyl CoA reductase 1 |
| 6A | AX184534745 | 16 383 672 | 0.33 | 0.31 | 0.07 | 4.77 | Acetic acid, hexyl ester | 6A | FxaC_21g33200 | 5.98E‐02 | Cinnamoyl CoA reductase 1 |
| 7B | AX184266735 | 9032 057 | 0.12 | −0.61 | 0.14 | 4.90 | Acetic acid, methyl ester | 7B | FxaC_26g17010 | 2.30E‐01 | Zinc finger type family protein |
| 5C | AX184023191 | 1596 661 | 0.65 | 1.48 | 0.34 | 4.88 | Methyl anthranilate | 5C | FxaC_19g06300 | 5.38E‐03 | Anthranilate phosphoribosyltransferase |
| 7C | AX184857983 | 4634 089 | 0.11 | 2.44 | 0.49 | 6.29 | Methyl anthranilate | 7C | FxaC_27g10310 | 3.85E‐10 | beta‐Hydroxyisobutyryl‐CoA hydrolase1 |
| 7A | AX184379101 | 24 047 189 | 0.52 | 1.51 | 0.33 | 5.31 | Methyl anthranilate | 7D | FxaC_28g10190 | 3.76E‐03 | Anthranilate synthase alpha subunit 1 |
| 7D | AX166516935 | 4666 935 | 0.24 | 1.88 | 0.40 | 5.68 | Methyl anthranilate | 7A | FxaC_25g43700 | 4.93E‐01 | Anthranilate synthase alpha subunit 1 |
Two eQTL of homoeologous anthranilate synthase alpha subunit 1 genes (FxaC_25g43700 & FxaC_28g10190) were identified co‐localizing with two GWAS peaks for MA on Chr 7A and Chr 7D (Table 2; Fig. S21b). Based on previous RNAseq data from different tissue samples (Sánchez‐Sevilla et al., 2017), FaASa1 (FxaC_28g10190) had the highest fruit expression among four homoeologs, and qPCR confirmed its dramatic increase of expression during ripening (Fig. S21a). A total of eight haplotypes were detected using three unlinked significant markers at cis‐eQTL (Fig. 7a), and variability in expression of FaASa1 was observed across haplotypes (Fig. 7b). The alignment among multiple assemblies and short read mapping of four additional genotypes revealed a deletion of 1904 bp adjacent to the 3′‐UTR region, and multiple SNPs in haplotypes with reduced expression (Fig. S22). The function of FaASa1 was validated through transient overexpression and RNAi of FaASa1 in strawberry fruit (Fig. 7c). A significant increase in MA production was detected in fruit transiently overexpressing FaASa1 via agroinfiltration (Student's t‐test, P = 0.022). Conversely, a significant elimination of MA was found in the RNAi‐mediated gene silenced fruit (Student's t‐test, P = 0.00039). Taken together, we identified four putative causal genes underlying MA GWAS peaks contributing to natural variation of MA content and functionally confirmed the involvement of FaASa1 (Fig. S21b). These genes may function at different steps in the pathway (Fig. 7d) to quantitatively influenced anthranilate availability or modulate FaAAMT (Pillet et al., 2017).
Fig. 7.
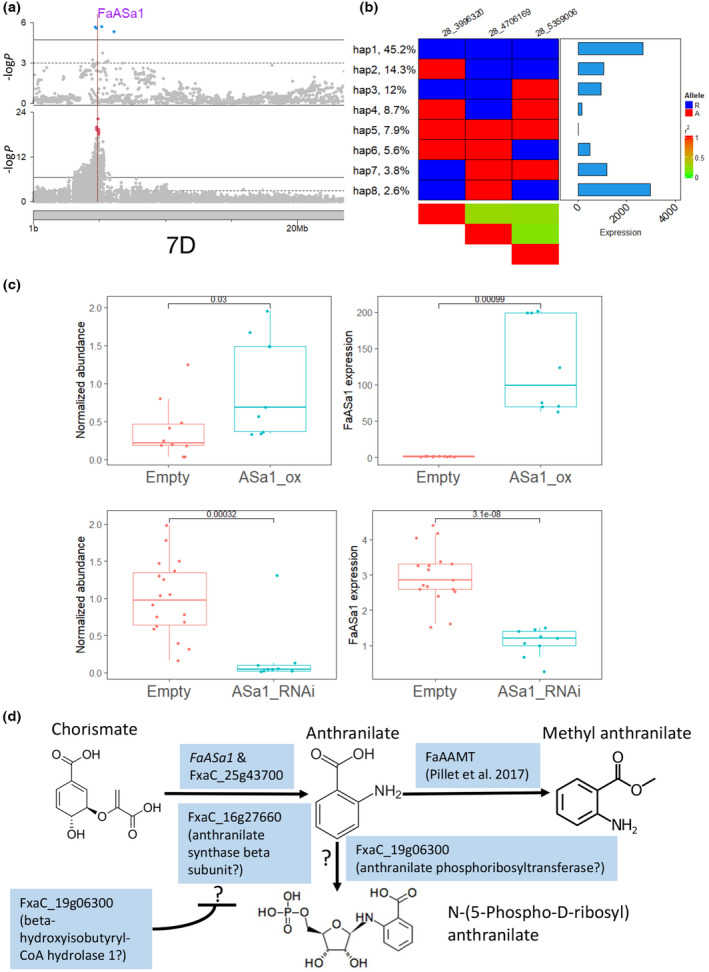
Genetic analysis of methyl anthranilate in strawberry (Fragaria × ananassa). (a) Manhattan plots of genome‐wide association study (GWAS) results for methyl anthranilate (upper panel) and expression quantitative trait loci (eQTL) results for FaASa1 (lower panel) on choromosome 7D. The physical position of FaASa1 is marked with a vertical line. (b) Eight haplotypes of FaASa1 identified using three unlinked significant markers (r 2 < 0.5). Markers are named by chrID_position. Left annotation shows haplotype frequency. Right annotation shows haplotype effect in unit of normalized count. The central heatmap shows marker genotype. Blue represents the reference allele; red represents the alternative allele. (c) Transient fruit assay for FaASa1. Two upper plots show methyl anthranilate abundance (left) in fruits agroinfiltrated with either empty construct (red) or FaASa1 overexpression construct (blue) and FaASa1 expression for the two treatments (right). Two lower plots show the same plots for the empty and FaASa1 RNAi constructs. Boxes are delimited by upper and lower quantiles. Two whiskers represent highest/lowest values; dots represent individual values; and horizontal lines represent medians. (d) Genes putatively required for or regulating methyl anthranilate biosynthesis. FaASa1 and FxaC_19g06300 were found within GWAS peaks and co‐localized with their cis‐eQTL. FxaC_16g27660 was remotely regulated by the 7C GWAS region. Predicted gene functions were enclosed by parenthesis. FxaC_19g06300 has a putative regulatory role in MA biosynthesis. Question marks represent relationships or functions not validated. Blunt‐ended arrow indicates potential regulator.
Discussion
In this study we leveraged eQTL, GWAS and haplotype‐resolved genome assemblies of a heterozygous octoploid to identify allelic variation in flavor genes and their regulatory elements. Fine‐tuning of metabolomic traits such as amylose content in rice (Xu et al., 2021) and sugar content in wild strawberry (Xing et al., 2020) recently were made possible via CRISPR‐Cas9 gene‐editing technology. Similar approaches can be taken in cultivated strawberry for flavor improvement, but not before the biosynthetic genes responsible for metabolites production and their regulatory elements are identified. Our pipeline has proven to be effective in identification of novel causal mutations for flavor genes responsible for natural variation in volatile content and can be further applied to various metabolomic and morphological aspects of strawberry fruit such as anthocyanin biosynthesis (Notes S11; Fig. S23), sugar content and fruit firmness.
These findings also will help breeders to select for genomic variants underlying volatiles important to flavor. New markers can be designed from regulatory regions of key aroma volatiles, including multiple medium‐chain volatiles shown to improve strawberry flavor and consumer liking (Fan et al., 2021), methyl thioacetate contributing to overripe flavor (Du et al., 2011) and methyl anthranilate imparting grape flavor (Pillet et al., 2017). In the present study, a new functional HRM marker for mesifurane was developed and tested in multiple populations (Fig. S16). These favorable alleles of volatiles can be pyramided to improve overall fruit flavor via marker assisted selection. Strawberry also shares common volatiles with a variety of fruit crops. Specific esters are shared with apple (Young et al., 1996), certain lactones are shared with peach (Predieri et al., 2006) and various terpenes are shared with citrus (Feng et al., 2021). Syntenic regions and orthologous genes could be exploited for flavor improvement in those species.
Additional insights were gained for the strawberry gene regulatory landscape, SV diversity, complex interplays among cis‐ and trans‐ regulatory elements, and subgenome dominance. Previously, Hardigan et al. (2021b) and Pincot et al. (2021) showed a large genetic diversity existing in breeding populations of Fragaria × ananassa, challenging previous assumptions that cultivated strawberry lacked nucleotide variation owing to the nature of its interspecific origin and short history of domestication (Gaston et al., 2020). Our work corroborated their findings and showed that even highly domesticated populations harbor substantial expression regulatory elements and structural variants. Over half of the expressed genes in fruit harbored at least one eQTL, and 22 731 eGenes had impactful cis‐eQTL. The distribution of trans‐eQTL is not random, but rather is concentrated at a few hotspots controlled by putative master regulators (Fig. S4; Dataset S6). The aggregation of trans‐eQTL also was observed in plant species such as Lactuca sativa (Zhang et al., 2017) and Zea mays (Liu et al., 2017). Furthermore, we observed a substantial number of trans‐eQTL among homoeologous chromosomes, similar to observations in other allopolyploid plant species (Li et al., 2020; He et al., 2022). In cotton, physical interactions among chromatins from different subgenomes have been identified via Hi‐C sequencing (Wang et al., 2018), supporting a potential regulatory mechanism among homoeologous chromosomes. However, owing to the high similarity among four subgenomes and limited length of Illumina reads, false alignment to incorrect homoeologous chromosomes could arise, leading to ‘ghost’ trans‐eQTL signals. Future studies are needed to scrutinize the homoeologous trans‐eQTL and investigate the mechanism behind this genome‐wide phenomenon. Higher numbers of trans‐eQTL in the Fragaria vesca‐like subgenome are consistent with its dominance in octoploid strawberry (Edger et al., 2019). By contrast, the highly mixed Fragaria viridis‐ and Fragaria nipponica‐ like subgenomes contained much smaller numbers of trans‐eQTL.
The characterization of naturally‐occurring allelic variants underlying volatile abundance has direct breeding applications. First, this will facilitate the selection of desirable alleles via DNA markers. Second, understanding the causal mutations in alleles can guide precision breeding approaches such as gene editing to modify the alleles themselves and/or their level of expression. From a broader perspective, multi‐omics resources such as this one will have value for breeding a wide array of fruit traits. Enhancing consumer satisfaction in fruit ultimately will depend on the improvement of the many traits that together enhance the overall eating experience.
Author contributions
ZF, VMW, PZ, SJK, CRB, KMF and SL designed the study; ZF performed bioinformatic analyses and collected field data; RRA provided advice on the data analyses; DMT and CRB conducted chemical analyses; RF, ZF and CRB collected samples and extracted RNA; ZF and ML performed fruit transient assays; YO and ZF designed and tested HRM markers; and ZF and VMW composed the manuscript. All authors read and approved the final manuscript.
Supporting information
Dataset S1 Single nucleotide polymorphism array genotyping file of expression quantitative trait loci panel.
Dataset S2 Single nucleotide polymorphism array genotyping file of volatile‐genome‐wide association study panel.
Dataset S3 List of lead markers for all expression quantitative trait loci using RNAseq‐called markers.
Dataset S4 Full list of significant markers of expression quantitative trait loci using single nucleotide polymorphism array.
Dataset S5 List of lead markers for all expression quantitative trait loci using single nucleotide polymorphism array.
Dataset S6 List of trans‐expression quantitative trait loci hot spots and the putative master regulators.
Dataset S7 Nonredundant list of structural variants.
Dataset S8 List of allele‐specific expressed genes.
Fig. S1 Model based clustering with k = 4 and LD decay.
Fig. S2 Comparisons of marker r 2 and −logP among different types of expression quantitative trait loci.
Fig. S3 Dot plot maps the lead single nucleotide polymorphisms on Fana array for all expression quantitative trait loci.
Fig. S4 Characterization of trans‐expression quantitative trait loci and their master regulators on chromosome 5D.
Fig. S5 Additional quality evaluations of the assembly.
Fig. S6 Karyoplots of F12 assembly and Bea assembly.
Fig. S7 Hi‐C contact map of Bea haploid assembly.
Fig. S8 Synteny plot between two haplotypes.
Fig. S9 Density distribution of expression ratios of genes.
Fig. S10 Circos plot of allele‐specific expressed genes in ‘FL15.89‐25’.
Fig. S11 Cluster analysis and chemical relationships among volatiles.
Fig. S12 Linkage view of the two hotspots for median‐chain esters and terpenes.
Fig. S13 Marker prediction of overripe and sweetness scores.
Fig. S14 Manhattan plots of pentanal; butanoic acid, 2‐methyl‐; 2‐hexenal, (E)‐.
Fig. S15 Box‐plots of relative abundance of pentanal, 2‐hexenal, (E)‐ and 2,3‐butanedione with different dosage of the alternative allele.
Fig. S16 Dosage effect on mesifurane abundance.
Fig. S17 Chromosomal alignment of 3C to the ‘Camarosa’ reference genome.
Fig. S18 IGV view of short reads alignment to FaNES1.
Fig. S19 Schematics of three haplotypes at the FaFAD1 region.
Fig. S20 Gene models, structural variation locations, genome alignments between haplotypes.
Fig. S21 Tissue‐specific expression of FaASa1.
Fig. S22 Long‐range alignment to the FaASa1 region.
Fig. S23 Genetic variations of genes involved in the anthocyanin pathway.
Notes S1 Expression quantitative trait loci mapping using single nucleotide polymorphism array data.
Notes S2 Genome annotation and evaluation.
Notes S3 Fruit allele‐specific expression within ‘FL15.89‐25’.
Notes S4 Volatile quantification method.
Notes S5 Volatile‐genome‐wide association study method.
Notes S6 Miscellaneous methods.
Notes S7 Trans‐expression quantitative trait loci hotspots.
Notes S8 Allele‐specific expression.
Notes S9 Prediction of sensory characteristics using single nucleotide polymorphism markers.
Notes S10 Deletion in FaFAD1.
Notes S11 Regulatory network of anthocyanin biosynthesis.
Table S1 List of individuals used in expression quantitative trait loci study and their origins.
Table S2 List of commercially synthesized DNA and primers.
Table S3 Counts of different types of structural variations relative to the PHASE1 haplotype of FaRR1.
Table S4 Narrow‐sense heritability estimates and single nucleotide polymorphism‐based heritability estimates.
Table S5 Significant genome‐wide association study signals for 36 volatiles.
Table S6 Curated flavor genes identified previously in Fragaria × ananassa and novel structural variations and expression quantitative trait loci identified in our study.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We gratefully thank Gina Fernandez and Jose Guillermo Chacon Jimenez for providing fruit for transient assays. We thank the personnel of the University of Florida strawberry breeding program and the University of California‐Davis strawberry breeding program for fruit collections and other supporting activities. We also thank Marcio F. R. Resende for reviewing and providing feedback on the manuscript. This research was supported by grants to SJK, VMW and SL from the United States Department of Agriculture (doi: 10.13039/100000199), National Institute of Food and Agriculture (NIFA), Specialty Crops Research Initiative (no. 2017‐51181‐26833) and SJK from the California Strawberry Commission (doi: 10.13039/100006760) and University of California, Davis (doi: 10.13039/100007707).
Data availability
The genome sequencing data of FL15.89‐15 are available on NCBI under BioProject accession no. PRJNA804219. Raw RNAseq data iares available on NCBI under BioProject accession no. PRJNA787565. The phased genome assemblies of FL15.89‐15 (https://www.rosaceae.org/Analysis/13738092) and eQTL mapping results including the full list of significant markers using RNAseq‐called SNPs are available on GDR (https://www.rosaceae.org/Publication/13876299).
References
- Aharoni A, Giri AP, Verstappen FWA, Bertea CM, Sevenier R, Sun Z, Jongsma MA, Schwab W, Bouwmeester HJ. 2004. Gain and loss of fruit flavor compounds produced by wild and cultivated strawberry species. Plant Cell 16: 3110–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni A, Keizer LC, Bouwmeester HJ, Sun Z, Alvarez‐Huerta M, Verhoeven HA, Blaas J, van Houwelingen AM, de Vos RC, van der Voet H et al. 2000. Identification of the SAAT gene involved in strawberry flavor biogenesis by use of DNA microarrays. Plant Cell 12: 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama Y, Yamamoto Y, Ohmido N, Ohshima M, Fukui K. 2001. Estimation of the nuclear DNA content of strawberries (Fragaria spp.) compared with Arabidopsis thaliana by using dual‐step flow cytometry. Cytologia 66: 431–436. [Google Scholar]
- Albert FW, Bloom JS, Siegel J, Day L, Kruglyak L. 2018. Genetics of trans‐regulatory variation in gene expression. eLife 7: e35471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge M, Soyk S, Ramakrishnan S, Wang X, Goodwin S, Sedlazeck FJ, Lippman ZB, Schatz MC. 2019. RaGOO: fast and accurate reference‐guided scaffolding of draft genomes. Genome Biology 20: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge M, Wang X, Benoit M, Soyk S, Pereira L, Zhang L, Suresh H, Ramakrishnan S, Maumus F, Ciren D et al. 2020. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182: 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbey CR, Hogshead MH, Harrison B, Schwartz AE, Verma S, Oh Y, Lee S, Folta KM, Whitaker VM. 2021. Genetic analysis of methyl anthranilate, mesifurane, linalool, and other flavor compounds in cultivated strawberry (Fragaria × ananassa). Frontiers in Plant Science 12: 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassil NV, Davis TM, Zhang H, Ficklin S, Mittmann M, Webster T, Mahoney L, Wood D, Alperin ES, Rosyara UR et al. 2015. Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo‐octoploid cultivated strawberry Fragaria × ananassa . BMC Genomics 16: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Zhou Y, Browning SR. 2018. A one‐penny imputed genome from next‐generation reference panels. The American Journal of Human Genetics 103: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Zhou Z, Wang Q, Guo J, Zhao P, Zhu G, Fang W, Chen C, Wang X, Wang X et al. 2016. Genome‐wide association study of 12 agronomic traits in peach. Nature Communications 7: 13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AH, Pillet J, Plotto A, Bai J, Whitaker VM, Folta KM. 2014. Identification of a strawberry flavor gene candidate using an integrated genetic‐genomic‐analytical chemistry approach. BMC Genomics 15: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype‐resolved de novo assembly using phased assembly graphs with hifiasm . Nature Methods 18: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Rus E, Sesmero R, Ángel‐Pérez JA, Sánchez‐Sevilla JF, Ulrich D, Amaya I. 2017. Validation of a PCR test to predict the presence of flavor volatiles mesifurane and γ‐decalactone in fruits of cultivated strawberry (Fragaria × ananassa). Molecular Breeding 37: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumplido‐Laso G, Medina‐Puche L, Moyano E, Hoffmann T, Sinz Q, Ring L, Studart‐Wittkowski C, Caballero JL, Schwab W, Muñoz‐Blanco J et al. 2012. The fruit ripening‐related gene FaAAT2 encodes an acyl transferase involved in strawberry aroma biogenesis. Journal of Experimental Botany 63: 4275–4290. [DOI] [PubMed] [Google Scholar]
- Dai X, Liu Y, Zhuang J, Yao S, Liu L, Jiang X, Zhou K, Wang Y, Xie D, Bennetzen JL et al. 2020. Discovery and characterization of tannase genes in plants: roles in hydrolysis of tannins. New Phytologist 226: 1104–1116. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Song M, Rouseff R. 2011. Identification of new strawberry sulfur volatiles and changes during maturation. Journal of Agricultural and Food Chemistry 59: 1293–1300. [DOI] [PubMed] [Google Scholar]
- Ebert P, Audano PA, Zhu Q, Rodriguez‐Martin B, Porubsky D, Bonder MJ, Sulovari A, Ebler J, Zhou W, Mari RS et al. 2021. Haplotype‐resolved diverse human genomes and integrated analysis of structural variation. Science 372: eabf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Poorten TJ, VanBuren R, Hardigan MA, Colle M, McKain MR, Smith RD, Teresi SJ, Nelson ADL, Wai CM et al. 2019. Origin and evolution of the octoploid strawberry genome. Nature Genetics 51: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Hasing T, Johnson TS, Garner DM, Barbey CR, Colquhoun TA, Sims CA, Resende MFR, Whitaker VM. 2021. Strawberry sweetness and consumer preference are enhanced by specific volatile compounds. Horticulture Research 8: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Gmitter FG Jr, Grosser JW, Wang Y. 2021. Identification of key flavor compounds in citrus fruits: a flavoromics approach. ACS Food Science & Technology 1: 2076–2085. [Google Scholar]
- Ferrão LFV, Johnson TS, Benevenuto J, Edger PP, Colquhoun TA, Munoz PR. 2020. Genome‐wide association of volatiles reveals candidate loci for blueberry flavor. New Phytologist 226: 1725–1737. [DOI] [PubMed] [Google Scholar]
- Ferrão LFV, Benevenuto J, Oliveira IB, Cellon C, Olmstead J, Kirst M, MFR R, Munoz P. 2018. Insights into the genetic basis of blueberry fruit‐related traits using diploid and polyploid models in a GWAS context. Frontiers in Ecology and Evolution 6: 107. [Google Scholar]
- Galpaz N, Gonda I, Shem‐Tov D, Barad O, Tzuri G, Lev S, Fei Z, Xu Y, Mao L, Jiao C et al. 2018. Deciphering genetic factors that determine melon fruit‐quality traits using RNA‐Seq‐based high‐resolution QTL and eQTL mapping. The Plant Journal 94: 169–191. [DOI] [PubMed] [Google Scholar]
- Gaston A, Osorio S, Denoyes B, Rothan C. 2020. Applying the Solanaceae strategies to strawberry crop improvement. Trends in Plant Science 25: 130–140. [DOI] [PubMed] [Google Scholar]
- Given NK, Venis MA, Gierson D. 1988. Hormonal regulation of ripening in the strawberry, a non‐climacteric fruit. Planta 174: 402–406. [DOI] [PubMed] [Google Scholar]
- Guan J, Xu Y, Yu Y, Fu J, Ren F, Guo J, Zhao J, Jiang Q, Wei J, Xie H. 2021. Genome structure variation analyses of peach reveal population dynamics and a 1.67 Mb causal inversion for fruit shape. Genome Biology 22: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardigan MA, Feldmann MJ, Lorant A, Bird KA, Famula R, Acharya C, Cole G, Edger PP, Knapp SJ. 2020. Genome synteny has been conserved among the octoploid progenitors of cultivated strawberry over millions of years of evolution. Frontiers in Plant Science 10: 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardigan MA, Feldmann MJ, Pincot DD, Famula RA, Vachev MV, Madera MA, Zerbe PJ, Mars K, Peluso P, Rank D. 2021a. Blueprint for phasing and assembling the genomes of heterozygous polyploids: application to the octoploid genome of strawberry. BioRxiv . doi: 10.1101/2021.11.03.467115. [DOI]
- Hardigan MA, Lorant A, Pincot DDA, Feldmann MJ, Famula RA, Acharya CB, Lee S, Verma S, Whitaker VM, Bassil N et al. 2021b. Unraveling the complex hybrid ancestry and domestication history of cultivated strawberry. Molecular Biology and Evolution 38: 2285–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Wang W, Rutter WB, Jordan KW, Ren J, Taagen E, DeWitt N, Sehgal D, Sukumaran S, Dreisigacker S et al. 2022. Genomic variants affecting homoeologous gene expression dosage contribute to agronomic trait variation in allopolyploid wheat. Nature Communications 13: 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller D, Vingron M. 2020. SVIM‐asm: structural variant detection from haploid and diploid genome assemblies. Bioinformatics 36: 5519–5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T, Kalinowski G, Schwab W. 2006. RNAi‐induced silencing of gene expression in strawberry fruit (Fragaria × ananassa) by agroinfiltration: a rapid assay for gene function analysis. The Plant Journal 48: 818–826. [DOI] [PubMed] [Google Scholar]
- Jiang L, Zheng Z, Qi T, Kemper KE, Wray NR, Visscher PM, Yang J. 2019. A resource‐efficient tool for mixed model association analysis of large‐scale data. Nature Genetics 51: 1749–1755. [DOI] [PubMed] [Google Scholar]
- Klee HJ, Tieman DM. 2018. The genetics of fruit flavour preferences. Nature Reviews Genetics 19: 347–356. [DOI] [PubMed] [Google Scholar]
- Knee M. 2001. Fruit quality and its biological basis (sheffield biological siences). Sheffield, UK: Blackwell. [Google Scholar]
- Koren S, Rhie A, Walenz BP, Dilthey AT, Bickhart DM, Kingan SB, Hiendleder S, Williams JL, Smith TPL, Phillippy AM. 2018. De novo assembly of haplotype‐resolved genomes with trio binning. Nature Biotechnology 36: 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmann C, Fink B, Schwab W. 2007. FaGT2: A multifunctional enzyme from strawberry (Fragaria × ananassa) fruits involved in the metabolism of natural and xenobiotic compounds. Planta 226: 417–428. [DOI] [PubMed] [Google Scholar]
- Lee H‐E, Manivannan A, Lee SY, Han K, Yeum J‐G, Jo J, Kim J, Rho IR, Lee Y‐R, Lee ES et al. 2021. Chromosome level assembly of homozygous inbred line ‘Wongyo 3115’ facilitates the construction of a high‐density linkage map and identification of QTLs associated with fruit firmness in octoploid strawberry (Fragaria × ananassa). Frontiers in Plant Science 12: 696229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T‐H, Guo H, Wang X, Kim C, Paterson AH. 2014. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang P, You C, Yu J, Zhang X, Yan F, Ye Z, Shen C, Li B, Guo K et al. 2020. Combined GWAS and eQTL analysis uncovers a genetic regulatory network orchestrating the initiation of secondary cell wall development in cotton. New Phytologist 226: 1738–1752. [DOI] [PubMed] [Google Scholar]
- Liu B, Wei G, Hu Z, Wang G. 2019. Benzaldehyde synthases are encoded by cinnamoyl‐CoA reductase genes in cucumber (Cucumis sativus L.). BioRxiv . doi: 10.1101/2019.12.26.889071. [DOI]
- Liu H, Luo X, Niu L, Xiao Y, Chen L, Liu J, Wang X, Jin M, Li W, Zhang Q et al. 2017. Distant eQTLs and non‐coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize. Molecular Plant 10: 414–426. [DOI] [PubMed] [Google Scholar]
- Liu T, Li M, Liu Z, Ai X, Li Y. 2021. Reannotation of the cultivated strawberry genome and establishment of a strawberry genome database. Horticulture Research 8: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA‐seq data with deseq2. Genome Biology 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Luo Z, Li D, Jiang Y, Li L. 2021. FaMYB11 promotes the accumulation of volatile esters by regulating FaLOX5 during strawberry (Fragaria × ananassa) ripening. Postharvest Biology and Technology 178: 111560. [Google Scholar]
- Massonnet M, Cochetel N, Minio A, Vondras AM, Lin J, Muyle A, Garcia JF, Zhou Y, Delledonne M, Riaz S et al. 2020. The genetic basis of sex determination in grapes. Nature Communications 11: 2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell WC, Jelenkovic G. 1995. Characterizing NAD‐and NADP‐dependent alcohol dehydrogenase enzymes of strawberries. Journal of the American Society for Horticultural Science 120: 798–801. [Google Scholar]
- Molina‐Hidalgo FJ, Medina‐Puche L, Cañete‐Gómez C, Franco‐Zorrilla JM, López‐Vidriero I, Solano R, Caballero JL, Rodríguez‐Franco A, Blanco‐Portales R, Muñoz‐Blanco J et al. 2017. The fruit‐specific transcription factor FaDOF2 regulates the production of eugenol in ripe fruit receptacles. Journal of Experimental Botany 68: 4529–4543. [DOI] [PubMed] [Google Scholar]
- Oh Y, Barbey CR, Chandra S, Bai J, Fan Z, Plotto A, Pillet J, Folta KM, Whitaker VM, Lee S. 2021. Genomic characterization of the fruity aroma gene, FaFAD1, reveals a gene dosage effect on γ‐decalactone production in strawberry (Fragaria × ananassa). Frontiers in Plant Science 12: 639345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Layer RM, Quinlan AR. 2016. vcfanno: fast, flexible annotation of genetic variants. Genome Biology 17: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillet J, Chambers AH, Barbey C, Bao Z, Plotto A, Bai J, Schwieterman M, Johnson T, Harrison B, Whitaker VM. 2017. Identification of a methyltransferase catalyzing the final step of methyl anthranilate synthesis in cultivated strawberry. BMC Plant Biology 17: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincot DDA, Ledda M, Feldmann MJ, Hardigan MA, Poorten TJ, Runcie DE, Heffelfinger C, Dellaporta SL, Cole GS, Knapp SJ. 2021. Social network analysis of the genealogy of strawberry: retracing the wild roots of heirloom and modern cultivars. G3 Genes |Genomes |Genetics 11: jkab015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predieri S, Ragazzini P, Rondelli R. 2006. Sensory evaluation and peach fruit quality. Acta Horticulturae 713: 429–434. [Google Scholar]
- Raab T, López‐Ráez JA, Klein D, Caballero JL, Moyano E, Schwab W, Muñoz‐Blanco J. 2006. FaQR, required for the biosynthesis of the strawberry flavor compound 4‐hydroxy‐2,5‐dimethyl‐3(2H)‐furanone, encodes an enone oxidoreductase. Plant Cell 18: 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Stephens M, Pritchard JK. 2014. FastStructure: variational inference of population structure in large SNP data sets. Genetics 197: 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Sevilla JF, Cruz‐Rus E, Valpuesta V, Botella MA, Amaya I. 2014. Deciphering gamma‐decalactone biosynthesis in strawberry fruit using a combination of genetic mapping, RNA‐Seq and eQTL analyses. BMC Genomics 15: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Sevilla JF, Vallarino JG, Osorio S, Bombarely A, Posé D, Merchante C, Botella MA, Amaya I, Valpuesta V. 2017. Gene expression atlas of fruit ripening and transcriptome assembly from RNA‐seq data in octoploid strawberry (Fragaria × ananassa). Scientific Reports 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieman D, Bliss P, McIntyre LM, Blandon‐Ubeda A, Bies D, Odabasi AZ, Rodríguez GR, van der Knaap E, Taylor MG, Goulet C. 2012. The chemical interactions underlying tomato flavor preferences. Current Biology 22: 1035–1039. [DOI] [PubMed] [Google Scholar]
- Tieman D, Zhu G, Resende MFR, Lin T, Nguyen C, Bies D, Rambla JL, Beltran KSO, Taylor M, Zhang B et al. 2017. A chemical genetic roadmap to improved tomato flavor. Science 355: 391–394. [DOI] [PubMed] [Google Scholar]
- Verma S, Bassil NV, van de Weg E, Harrison RJ, Monfort A, Hidalgo JM, Amaya I, Denoyes B, Mahoney L, Davis TM. 2016. Development and evaluation of the Axiom® IStraw35 384HT array for the allo‐octoploid cultivated strawberry Fragaria × ananassa . Acta Horticulturae 1156: 75–82. [Google Scholar]
- Wang M, Wang P, Lin M, Ye Z, Li G, Tu L, Shen C, Li J, Yang Q, Zhang X. 2018. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nature Plants 4: 90–97. [DOI] [PubMed] [Google Scholar]
- Whalen A, Hickey JM. 2020. AlphaImpute2: Fast and accurate pedigree and population based imputation for hundreds of thousands of individuals in livestock populations. BioRxiv . doi: 10.1101/2020.09.16.299677. [DOI]
- Whitaker VM, Osorio LF, Peres NA, Fan Z, Herrington M, Nunes MCN, Plotto A, Sims CA. 2017. ‘Florida Beauty’ strawberry. HortScience 52: 1443–1447. [Google Scholar]
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31: 3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolyn DJ, Jelenkovic G. 1990. Nucleotide sequence of an alcohol dehydrogenase gene in octoploid strawberry (Fragaria × ananassa Duch). Plant Molecular Biology 14: 855–857. [DOI] [PubMed] [Google Scholar]
- Xing S, Chen K, Zhu H, Zhang R, Zhang H, Li B, Gao C. 2020. Fine‐tuning sugar content in strawberry. Genome Biology 21: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lin Q, Li X, Wang F, Chen Z, Wang J, Li W, Fan F, Tao Y, Jiang Y et al. 2021. Fine‐tuning the amylose content of rice by precise base editing of the Wx gene. Plant Biotechnology Journal 19: 11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young H, Gilbert JM, Murray SH, Ball RD. 1996. Causal effects of aroma compounds on royal gala apple flavours. Journal of the Science of Food and Agriculture 71: 329–336. [Google Scholar]
- Yu G, Smith DK, Zhu H, Guan Y, Lam TT‐Y. 2017. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 8: 28–36. [Google Scholar]
- Zhang C, Dong S‐S, Xu J‐Y, He W‐M, Yang T‐L. 2019. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35: 1786–1788. [DOI] [PubMed] [Google Scholar]
- Zhang L, Su W, Tao R, Zhang W, Chen J, Wu P, Yan C, Jia Y, Larkin RM, Lavelle D et al. 2017. RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis. Nature Communications 8: 2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. 2012. A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28: 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M. 2012. Genome‐wide efficient mixed‐model analysis for association studies. Nature Genetics 44: 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Ma Y, Zeng J, Duan L, Xue X, Wang H, Lin T, Liu Z, Zeng K, Zhong Y et al. 2016. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae. Nature Plants 2: 16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Wang S, Huang Z, Zhang S, Liao Q, Zhang C, Lin T, Qin M, Peng M, Yang C et al. 2018. Rewiring of the fruit metabolome in tomato breeding. Cell 172: 249–261. [DOI] [PubMed] [Google Scholar]
- Zolman BK, Monroe‐Augustus M, Thompson B, Hawes JW, Krukenberg KA, Matsuda SPT, Bartel B. 2001. chy1, an Arabidopsis mutant with impaired β‐oxidation, is defective in a peroxisomal β‐hydroxyisobutyryl‐CoA hydrolase. Journal of Biological Chemistry 276: 31037–31046. [DOI] [PubMed] [Google Scholar]
- Zorrilla‐Fontanesi Y, Rambla J‐L, Cabeza A, Medina JJ, Sánchez‐Sevilla JF, Valpuesta V, Botella MA, Granell A, Amaya I. 2012. Genetic analysis of strawberry fruit aroma and identification of O‐methyltransferase FaOMT as the locus controlling natural variation in mesifurane content(C)(W)(OA). Plant Physiology 159: 851–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1 Single nucleotide polymorphism array genotyping file of expression quantitative trait loci panel.
Dataset S2 Single nucleotide polymorphism array genotyping file of volatile‐genome‐wide association study panel.
Dataset S3 List of lead markers for all expression quantitative trait loci using RNAseq‐called markers.
Dataset S4 Full list of significant markers of expression quantitative trait loci using single nucleotide polymorphism array.
Dataset S5 List of lead markers for all expression quantitative trait loci using single nucleotide polymorphism array.
Dataset S6 List of trans‐expression quantitative trait loci hot spots and the putative master regulators.
Dataset S7 Nonredundant list of structural variants.
Dataset S8 List of allele‐specific expressed genes.
Fig. S1 Model based clustering with k = 4 and LD decay.
Fig. S2 Comparisons of marker r 2 and −logP among different types of expression quantitative trait loci.
Fig. S3 Dot plot maps the lead single nucleotide polymorphisms on Fana array for all expression quantitative trait loci.
Fig. S4 Characterization of trans‐expression quantitative trait loci and their master regulators on chromosome 5D.
Fig. S5 Additional quality evaluations of the assembly.
Fig. S6 Karyoplots of F12 assembly and Bea assembly.
Fig. S7 Hi‐C contact map of Bea haploid assembly.
Fig. S8 Synteny plot between two haplotypes.
Fig. S9 Density distribution of expression ratios of genes.
Fig. S10 Circos plot of allele‐specific expressed genes in ‘FL15.89‐25’.
Fig. S11 Cluster analysis and chemical relationships among volatiles.
Fig. S12 Linkage view of the two hotspots for median‐chain esters and terpenes.
Fig. S13 Marker prediction of overripe and sweetness scores.
Fig. S14 Manhattan plots of pentanal; butanoic acid, 2‐methyl‐; 2‐hexenal, (E)‐.
Fig. S15 Box‐plots of relative abundance of pentanal, 2‐hexenal, (E)‐ and 2,3‐butanedione with different dosage of the alternative allele.
Fig. S16 Dosage effect on mesifurane abundance.
Fig. S17 Chromosomal alignment of 3C to the ‘Camarosa’ reference genome.
Fig. S18 IGV view of short reads alignment to FaNES1.
Fig. S19 Schematics of three haplotypes at the FaFAD1 region.
Fig. S20 Gene models, structural variation locations, genome alignments between haplotypes.
Fig. S21 Tissue‐specific expression of FaASa1.
Fig. S22 Long‐range alignment to the FaASa1 region.
Fig. S23 Genetic variations of genes involved in the anthocyanin pathway.
Notes S1 Expression quantitative trait loci mapping using single nucleotide polymorphism array data.
Notes S2 Genome annotation and evaluation.
Notes S3 Fruit allele‐specific expression within ‘FL15.89‐25’.
Notes S4 Volatile quantification method.
Notes S5 Volatile‐genome‐wide association study method.
Notes S6 Miscellaneous methods.
Notes S7 Trans‐expression quantitative trait loci hotspots.
Notes S8 Allele‐specific expression.
Notes S9 Prediction of sensory characteristics using single nucleotide polymorphism markers.
Notes S10 Deletion in FaFAD1.
Notes S11 Regulatory network of anthocyanin biosynthesis.
Table S1 List of individuals used in expression quantitative trait loci study and their origins.
Table S2 List of commercially synthesized DNA and primers.
Table S3 Counts of different types of structural variations relative to the PHASE1 haplotype of FaRR1.
Table S4 Narrow‐sense heritability estimates and single nucleotide polymorphism‐based heritability estimates.
Table S5 Significant genome‐wide association study signals for 36 volatiles.
Table S6 Curated flavor genes identified previously in Fragaria × ananassa and novel structural variations and expression quantitative trait loci identified in our study.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The genome sequencing data of FL15.89‐15 are available on NCBI under BioProject accession no. PRJNA804219. Raw RNAseq data iares available on NCBI under BioProject accession no. PRJNA787565. The phased genome assemblies of FL15.89‐15 (https://www.rosaceae.org/Analysis/13738092) and eQTL mapping results including the full list of significant markers using RNAseq‐called SNPs are available on GDR (https://www.rosaceae.org/Publication/13876299).