

Abstract
Recent comprehensive analyses of mtDNA and orthogonal RNA‐sequencing data revealed that in numerous human cancers, mtDNA copy numbers and mtRNA amounts are significantly reduced, followed by low respiratory gene expression. Under such conditions (called mt‐Low), cells encounter severe cell proliferation defects; therefore, they must acquire countermeasures against this fatal disadvantage during malignant transformation. This study elucidated a countermeasure against the mt‐Low condition‐induced antiproliferative effects in hepatocellular carcinoma (HCC) cells. The mechanism relied on the architectural transcriptional regulator HMGA2, which was preferably expressed in HCC cells of the mt‐Low type in vitro and in vivo. Detailed in vitro analyses suggest that HMGA2 regulates insulin‐like growth factor binding protein 1 (IGFBP1) expression, leading to AKT activation, which then phosphorylates the cyclin‐dependent kinase inhibitor (CKI), P27KIP1, and facilitates its ubiquitin‐mediated degradation. Accordingly, intervention in the HMGA2 function by RNAi resulted in an increase in P27KIP1 levels and an induction of senescence‐like cell proliferation inhibition in mt‐Low‐type HCC cells. Conclusively, the HMGA2/IGFBP1/AKT axis has emerged as a countermeasure against P27KIP1 CKI upregulation under mt‐Low conditions, thereby circumventing cell proliferation inhibition and supporting the tumorigenic state. Notably, similar to in vitro cell lines, HMGA2 was likely to regulate IGFBP1 expression in HCC in vivo, thereby contributing to poor patient prognosis. Considering the significant number of cases under mt‐Low or the threat of CKI upregulation cancer‐wide, the axis is noteworthy as a vulnerability of cancer cells or target for tumor‐agnostic therapy inducing irreversible cell proliferation inhibition via CKI upregulation in a large population with cancer.
Keywords: hepatocellular carcinoma, HMGA2, IGFBP1, mitochondria deficiency, P27KIP1
mtDNA copy numbers and mtRNA amounts are reduced, followed by low respiratory gene expression in numerous cancer cells. High expression of HMGA2 activates the IGFBP1/AKT pathway, circumventing mitochondria deficiency‐caused cell proliferation defects. Intervention in the HMGA2 function results in an increase in P27KIP protein levels and an induction of senescence‐like cell cycle arrest in cancer cells.

Abbreviations
- CHX
cycloheximide
- CKI
cyclin‐dependent kinase inhibitor
- Dox
doxycycline
- HCC
hepatocellular carcinoma
- HMGA
high mobility group AT‐hook
- IGF
insulin‐like growth factor
- IGFBP
insulin‐like growth factor binding protein
- p27
P27KIP1
- pri.Hep
primary hepatocyte
- SA‐β‐Gal
senescence‐associated β‐galactosidase
- Tet
tetracycline
- TUBE
tandem ubiquitin‐binding entity
1. INTRODUCTION
Cancer cells, while aggressively proliferating and invading into ectopic environments, undergo various inherent stress types collectively referred to as oncogenesis‐associated cellular stresses such as proteotoxic, metabolic, and oxidative stresses. 1 Accordingly, cancer cells need to evolve countermeasures to tolerate the stresses, especially to subvert the stress‐induced proapoptotic and antiproliferative effects by exploiting a molecule or pathway useful for overcoming the stresses. An intervention in such countermeasures deprives cells of their coping ability with the stresses and potentially leads to death or proliferation inhibition, thereby providing an attractive therapeutic target against cancer.
Recent comprehensive analyses of mtDNA and orthogonal RNA‐sequencing data revealed that the mtDNA copy number is reduced at statistically significant levels in seven of the 15 tumor types 2 and many cancer types have a tendency toward lower levels of mtRNAs. 3 Consequently, respiratory activity is suppressed in several cases. 3 According to these analyses, liver cancer was defined as an mtDNA‐depleted type. 2 , 4 These aberrant mitochondrial conditions (called mt‐Low from this point forwards in this study) can cause stress, leading to severe cell proliferation defects due to downregulation of the E2F1 transcriptional network and upregulation of CDKN1A (p21CIP1) and CDKN1B (P27KIP1) CKIs at least in part. 5 Our recent work defined the role of the FOXM1/BMYB transcriptional complex as one of the countermeasures against such antiproliferative effects in mt‐Low‐type HCC cells. 6 In particular, the complex played a role in downregulation of p21CIP1 expression, thereby sustaining cell proliferation.
This study elucidated a countermeasure against P27KIP1 (p27)‐induced antiproliferative effects. The process is directed by a molecular axis driven by the architectural transcriptional regulator, HMGA2, which is aberrantly expressed in nearly all tumor types. 7 , 8 , 9 Several genetic mechanisms, including chromosomal translocation and truncation of 3′UTR, have been potentially associated with HMGA2 re‐expression in mesenchymal tumorigenesis. 10 Particularly, 3′UTR of HMGA2 provides a targeting region for miRNAs, such as let‐7, thereby impacting HMGA2 expression. 7 Although a substantial body of evidence has suggested that HMGA2 has oncogenic roles, 7 , 8 the protein is molecularly versatile, and its roles remain to be fully elucidated. This study reports a stress‐coping role for HMGA2 in mt‐Low‐type cancer, which is a new function of HMGA2 in oncogenesis. Specifically, HMGA2 is preferably expressed in mt‐Low‐type HCC cells and tumor tissue, and has a critical role in counteracting p27 antiproliferative protein upregulation by activating the IGFBP1/AKT pathway in cancer cells.
2. MATERIALS AND METHODS
Full Materials and Methods are available as Appendix S1.
3. RESULTS
3.1. p27 CKI and HMGA2 expression in mt‐Low hepatocytes
Previously, we characterized seven liver cancer cell lines and categorized four of them, HLF, HuH‐7, JHH‐2, and JHH‐4, as mtDNA‐less types based on the mtDNA quantity, TFAM expression, and the D‐loop mutations. 6 These cell lines also exhibited low‐level mtRNA and membrane potential (ΔΨm) compared with pri.Hep (Figure S1A). Accordingly, they are referred to as mt‐Low cells from this point forwards. Similar to these in vitro cell lines, mtRNA levels, which correlate with respiratory chain expression levels, 3 were lower in 61% (17 out of 28) of primary HCC tissues when compared with paired nontumorous peritumor samples. Therefore, more than half in vitro cell lines and in vivo HCC cases are likely to be mt‐Low types, in agreement with large‐scale analyses 2 , 4 (Figure S1B and Table S1; mtRNA [T/N] < 1). These mt‐Low cases are to overcome the deterioration of cell proliferation, particularly the upregulation of two CKIs to maintain a tumorigenic state. 5 , 6
In this study, we elucidated how mt‐Low cells deal with the upregulation of p27 CKI, which occurs following p21CIP1 induction under mt‐Low stress conditions (Figure 1A). Notably, in mt‐Low cell lines, HMGA2, an HMGA protein member previously implicated in mtDNA‐depleted phenotype of epithelial cells, 11 was expressed at higher levels (Figure 1B,C; HMGA2). In in vivo tissues, HMGA2 was expressed in 56% (19 out of 34 cases) of HCC cases (Table S1), but not in nontumorous peritumor tissues except for a single case, and importantly, in the HMGA2‐positive cases, the mtRNA levels were lower than negative cases (Figure 1D). Between the groups categorized with other indices, the mtRNA level differences were apparently insignificant (Figure S1C). These results suggested that HMGA2 was preferably expressed in HCC under mt‐Low conditions, both in vivo and in vitro. Additionally, in vivo HMGA2 expression was detected in 77.8% (seven out of nine) of poorly differentiated cases and 100% (four out of four) of cases with upper‐grade fibrosis (Table S1). Consistent with these results, survival analyses using The Cancer Genome Atlas (TCGA) dataset indicated that high HMGA2 expression correlated with poor prognoses in patients with HCC (Figure 1E).
FIGURE 1.
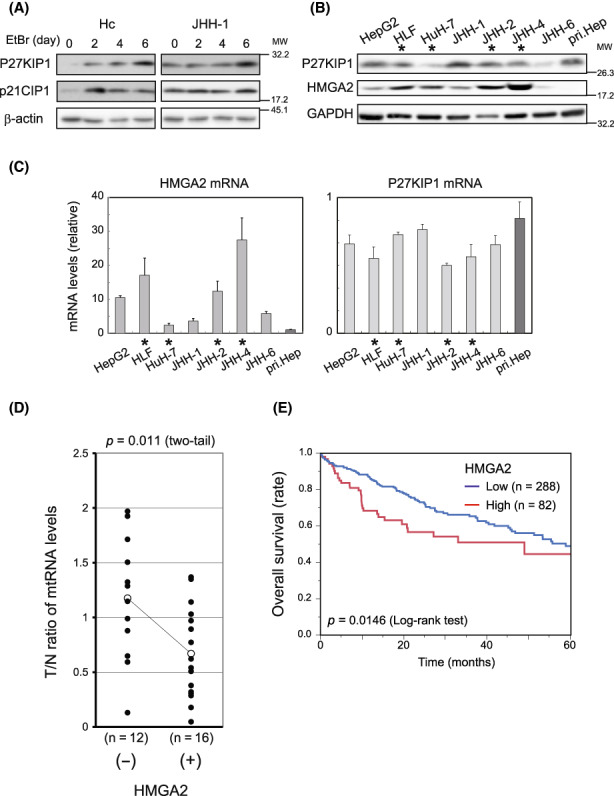
P27KIP1, p21CIP1 And HMGA2 expression in mitochondria‐deficient hepatocytes. (A) Immortalized hepatocytes (Hc) and JHH‐1 cells were cultured in the presence of 250 ng/ml ethidium bromide (EtBr) and 50 μg/ml uridine and western blotting was performed. β‐Actin, loading control. (B, C) Western blotting (B) and qPCR (C) were performed in hepatocellular carcinoma (HCC) cell lines and pri.Hep. Cell lines with asterisks (*), mt‐Low types. GAPDH, the loading control (B). In (C), the values normalized to the corresponding values of GAPDH are shown relative to that of pri.Hep. The values are mean ± SD from triplicates. (D) mtRNA levels were determined (Section 2) in pairs of HCC tumor (T) and adjacent nontumorous (N) tissues obtained from patients (Table S1). The ratio of the value in T relative to that in N in each pair was plotted in the group with amplification of HMGA2 positive (+) and negative (−). The statistically significance was assessed using an unpaired t‐test. (E) A Kaplan–Meier plot, showing patients categorized by HMGA2 expression levels (High [z‐score > 0], and Low [z‐score < 0]), was generated using the liver hepatocellular carcinoma dataset (TCGA, Firehose Legacy) in the cBioPortal (Section 2).
3.2. HMGA2 is essential for sustaining cell proliferation in mt‐Low HCC cells
Interestingly, HMGA2 levels precisely mirrored those of FOXM1/BMYB in the HCC cell lines. 6 Moreover, the levels tended to inversely correlate with those of the p27 protein, but not mRNA, levels (Figure 1B,C; P27KIP1). These observations raised the possibility that HMGA2 has a role in promoting cell proliferation in mt‐Low HCC cells in association with p27 protein levels, similar to FOXM1/BMYB in association with p21CIP1. 6 To test this hypothesis, we first examined how HMGA2 influences cell proliferation in the mt‐Low cells. In the experiments, we used the mt‐Low HCC cells, JHH‐2, JHH‐4, and HLF, exhibiting high‐level HMGA2 expression, and reduced the HMGA2 levels using RNAi. In Figure 2A, the cells were treated with pooled siRNAs for HMGA2 or the scrambled control (Ctr), and in Figure 2B, shRNA (sequences #1 and #2 for HMGA2 [HMGA2#1 and #2] or nontargeting [NT])‐expressing constructs based on the Tet‐Off system (Dox responsive) were virally transduced into cells. Under these HMGA2‐knockdown conditions (Figure S2A), the cell numbers were significantly lower than the control after 5–9 days of culture (Figure 2A,B).
FIGURE 2.
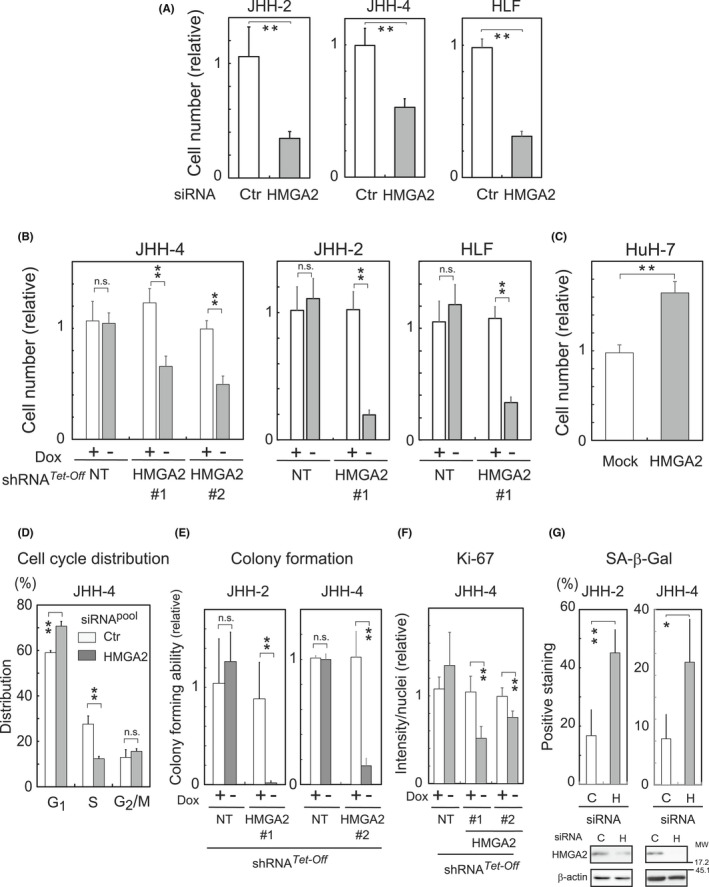
HMGA2 effects on hepatocellular carcinoma (HCC) cell proliferation. (A, B, D–G) HMGA2 was knocked down with pooled siRNA or Dox‐responsive (Tet‐Off) shRNA (sequences #1 and #2) (Figure S2A). Ctr, scrambled siRNA; NT, nontarget shRNA. Cell numbers counted after the treatment with siRNA (A) or incubation with (+) or without (−) Dox (B) for 5–9 days are presented relative to the control. (C) Cell numbers stably expressing HMGA2 or control (Mock) were counted after 4 days of culture. Cell cycle distribution (D), colony‐forming ability (relative to the control; Dox+) (E), Ki‐67 expression levels (n > 50/image) (F), and SA‐β‐Gal activity (percentages of positive cells) (n > 50/sample) (G) were examined in cells incubated with siRNA for 4 days (D), with (+) or without (−) Dox for 2 weeks (E) or 7–9 days (F), and with siRNA for 1 week (G), respectively (Section 2). In (F), Ki‐67 immunostaining intensities on images were normalized with the corresponding number of nuclei, and values are shown relative to those of the control Dox + (+). In (G), HMGA2 knockdown under the conditions was confirmed by western blotting (bottom). C, control; H, HMGA2 pooled siRNA. The values are mean ± SD from triplicates or more. **p < 0.005; *p < 0.05; n.s., not significant.
In contrast, cell numbers were higher than the control in a reverse experiment (Figure 2C), where an HMGA2‐expressing construct or the empty vector (Mock) was introduced into the cells. In this experiment, we used HuH‐7, a mt‐Low cell line exhibiting lower HMGA2 levels than the other mt‐Low cells. These experiments clearly demonstrated that HMGA2 plays a role in sustaining cell proliferation in mt‐Low HCC cells. HMGA2‐silenced cells accumulated at the G1/S boundary in the cell cycle (Figure 2D and Figure S2B) and consequently, the number of colonies that formed after 2 weeks was markedly lower (Figure 2E and Figure S2C) with a concomitant decrease in the levels of Ki‐67, which is a proliferation marker (Figures 2F and Figure S2D,E). In these cells, SA‐β‐Gal activity (Figure 2G and Figure S3A) and one or two of hepatic senescence‐associated secretory phenotype‐related mRNAs 12 were increased (Figure S3B).
3.3. HMGA2 engages in p27 protein‐level regulation
Subsequently, HMGA2 was implicated in the regulation of p27 protein levels using RNAi (Figure 3A–D). In Figure 3A,B, the p27 protein levels increased in the HCC cell lines either transduced with shRNA (JHH‐4 and HLF) or treated with siRNA (JHH‐2 and HLF) for HMGA2. In contrast, the p27 protein levels decreased when the HMGA2 levels were exogenously increased in HepG2 (Figure 3E) and HuH‐7 cells (described below). These results demonstrated that HMGA2 negatively regulated p27 levels. Given that the effect on the mRNA levels was cell line‐dependent or not significant as it was moderate in JHH‐4 (Figure 3C; JHH‐4) and HepG2 (Figure 3F), insignificant in HLF (Figure 3C,D; HLF), or not observed in JHH‐2 (Figure 3D; JHH‐2), HMGA2 was likely to regulate mainly protein levels. Figure 1B,C also supported this notion, indicating that the p27 mRNA levels were neither significantly different among the hepatocyte cell lines nor exactly correlated with those of the protein levels.
FIGURE 3.
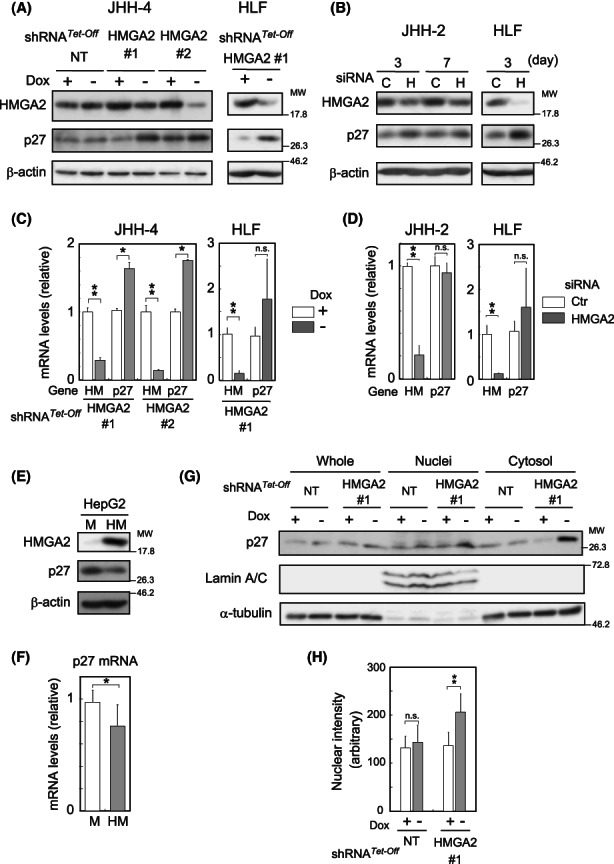
Upregulation and downregulation of p27 levels in HMGA2‐silenced/expressing cells. (A–D) In (A) and (C) Dox‐responsive shRNA (HMGA2 #1 and #2)‐expressing cells were incubated with (+) or without (−) Dox for 4 (A) or 3 days (C). NT, nontarget shRNA. In (B) and (D) cells were treated with siRNA for the indicated days (B) or 3 days (D). C or Ctr, control; H, HMGA2 pooled siRNA. Western blotting (A, B) and qPCR (C, D) were performed. β‐Actin, the loading control. In (C) and (D), the values are shown relative to the control (Dox + or Ctr) after normalization to the corresponding values to TATA‐box binding protein. Gene: Hm; HMGA2, p27; P27KIP1. The values are mean ± SD from triplicates. (E, F) Western blotting (E) and qPCR (F) were performed in HMGA2 (HM)‐expressing or control (M) cells as described above. (G) JHH‐4 cells expressing HMGA2#1 shRNA and the control as in (A) were lysed, fractionated into cytosol and nuclei, and analyzed by western blotting with the indicated antibodies. α‐Tubulin and lamin A/C, the loading controls for whole and cytosol, and nuclei fractions. (H) JHH‐4 cells as in (A) were examined by immunocytochemistry using a p27 antibody (Figure S4A). The nuclear staining intensities measured using the FV10‐ASW Viewer software (Ver.4.2b) are presented as the mean ± SD (n = 19–21). **p < 0.005; *p < 0.05; n.s., not significant.
p27, albeit originally identified as a CKI, has now emerged as a multifunctional protein, functioning in both the cytoplasm and the nucleus while shuttling between the two compartments. 10 Under HMGA2‐knockdown conditions, p27 levels were increased in the nuclei and the cytosol, as indicated by western blot analysis following subcellular fractionation (Figure 3G). Immunocytochemistry further verified the p27 nuclear localization and quantitative analysis showed that it statistically significantly increased under these conditions (Figure 3H and Figure S4A). Therefore, under the HMGA2‐knockdown conditions, p27 is upregulated and probably works as a conventional CKI in the nucleus. To substantiate this possibility, we introduced a p27‐expressing construct into the cells and observed how it affects cell proliferation. As expected, cell proliferation was suppressed with increased SA‐β‐Gal activity in the JHH‐4 and JHH‐2 cells (Figure 4A–D). In contrast, when the p27 upregulation was interrupted by p27 shRNA expression under HMGA2‐knockdown conditions, the SA‐β‐Gal activity increase was markedly attenuated and Ki‐67 expression was reversed, suggesting that cells regained their proliferative potential (Figure 4E). However, cell viability was decreased to ~70% by the interruption of p27 expression, and cell number was not recovered (unpublished data), possibly due to disturbance in p27‐mediated induction of cellular senescence, which can confer resistance to cell death. 13 Taken together, the results revealed that HMGA2 suppressed p27 CKI upregulation inducing senescence‐like cell proliferation inhibition in mt‐Low HCC cells.
FIGURE 4.
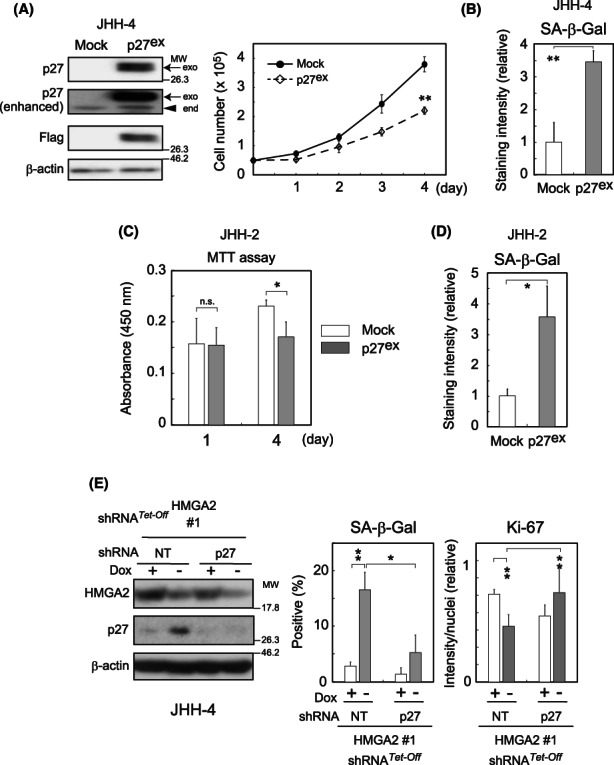
The role of p27 in inhibiting cell proliferation. (A–D) Cells were infected with p27 (Flag‐tagged)‐expressing (p27ex) or control (Mock) lentivirus constructs. After 3 days of selection with blasticidin, the cells were plated and proliferation was assessed by counting the cell number (A) or MTT assay (C). Western blotting confirmed the expression of Flag‐tagged p27 (A). Arrows and arrowhead indicate exogenous and endogenous p27 proteins, respectively. β‐Actin, the loading control. In (C), the values represent the means of absorbance at 450 nm ± SD from triplicates. The SA‐β‐Gal activity was determined on days 6 (B) or 2 (D) after the selection (Section 2). The values obtained from triplicate samples are shown as relative to the control (Mock) (n > 100). (E) The lentiviral constructs stably expressing shRNA for p27 or nontargeted controls (NT) were transduced into Dox‐responsive HMGA2‐silenced JHH‐4 cells. The western blot analysis, SA‐β‐Gal staining (n > 50/sample), and Ki‐67 immunostaining (n > 50/image) were performed after incubation under Dox (+) or (−) conditions for 3, 6, and 9 days as in Figure 2F,G, respectively. **p < 0.005; *p < 0.05; n.s., not significant.
3.4. HMGA2 regulates p27 ubiquitination and turnover via AKT activity
Regarding the mechanisms underlying HMGA2‐mediated p27 level suppression, we first examined how HMGA2 expression could affect p27 protein turnover using CHX, considering the above‐described possibility that HMGA2 targeted post‐transcriptional mechanisms (Figures 1B,C and 3A–F). Figure 5A shows that p27 was markedly stabilized under HMGA2‐knockdown conditions, suggesting that HMGA2 is engaged in the p27 protein turnover regulation. As the p27 protein turnover is mostly regulated via a ubiquitin‐mediated pathway, 14 we subsequently assessed the p27 protein ubiquitination levels. The results obtained by the TUBE method 15 suggested that more p27 was ubiquitinated under the control than HMGA2‐knockdown conditions (Figure 5B). Therefore, HMGA2 was suggested to regulate p27 protein turnover by promoting ubiquitination.
FIGURE 5.
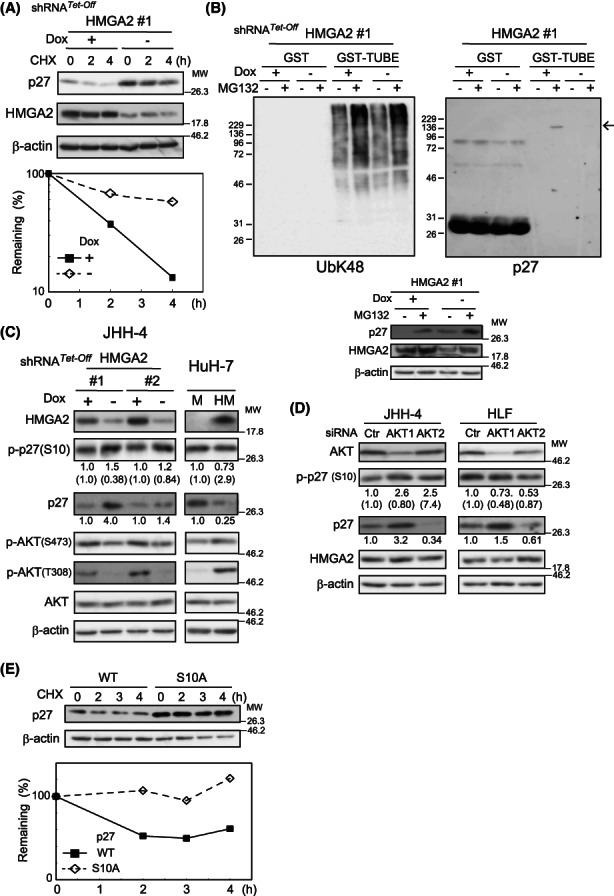
AKT activity‐mediated HMGA2 regulation of p27 protein turnover. (A) Dox‐responsive HMGA2 #1 shRNA‐expressing HLF cells incubated for 4 days under Dox (+) or (−) were treated with CHX (100 μg/ml), and the p27 amount was quantified by western blotting using ImageJ software. After normalization to β‐actin values, the remaining p27 amounts were plotted. (B) Dox‐responsive HMGA2#1 shRNA‐expressing JHH‐4 cells incubated under Dox (+) or (−) conditions for 4 days were treated with (+) or without (−) 10 μM MG132 for 6.5 h and lysed. The lysates were pulled down with GST or GST‐TUBE (Section 2) and analyzed with the antibodies against p27 and polyubiquitin K‐48‐linkage (UbK48). An arrow indicates ubiquitinated p27 proteins. Aliquots of the lysates were analyzed by western blotting with the indicated antibodies (bottom). β‐Actin, the loading control. (C) Dox‐responsive HMGA2#1 and #2 shRNA‐expressing JHH‐4 cells incubated under Dox (+) or (−) conditions for 4 days and HuH‐7 cells stably expressing HMGA2 (HM) or the control (M) were analyzed by western blotting. The p27 and p‐p27(S10) band intensities quantified with ImageJ software are shown relative to the controls (Dox + or M) after normalization with the corresponding intensities of the loading control of β‐actin. The p‐p27(S10) intensities were also normalized with the p27 values and are shown in parentheses. (D) Cells were treated with pooled AKT1 and 2 or control (Ctr) siRNA for 3 days and analyzed as in (C). (E) JHH‐4 cells expressing the wild‐type (WT) and mutant (S10A)‐p27 were treated with CHX (100 μg/ml), and the p27 amount was quantified by western blotting using ImageJ software and plotted as in (A).
We next investigated how HMGA2 promotes p27 ubiquitination. Given that HMGA2 is a transcriptional regulator, it is likely to regulate gene expression, thereby modulating a signaling pathway that affects ubiquitination rather than regulates protein ubiquitination directly. As ubiquitin‐dependent p27 proteolysis is tightly coupled with the phosphorylation of several amino acids on the protein, 14 , 16 we observed the impact of HMGA2 expression on p27 phosphorylation at position S10, which is the major phosphorylation site of this protein. 17 , 18 To compare the net change in phosphorylation levels per protein, we normalized the levels of gross phosphorylation to those of the corresponding protein to offset the protein‐level increase or decrease. Of note, we observed a decrease and increase in the net p27 protein phosphorylation levels at S10 [p‐p27(S10)], which are shown in parentheses under the panel Figure 5C, concomitantly with the stabilization and destabilization of the protein in HMGA2‐silenced (JHH‐4) and HMGA2‐expressing (HuH‐7) cells, respectively (Figure 5C).
More notably, AKT activity was altered in parallel with p27 S10 phosphorylation. AKT has been identified as one of the kinases responsible for the phosphorylation at S10. 19 Figure 5C shows that AKT activation, detected by the phosphorylation at T308 and S473, declined in HMGA2‐silenced JHH‐4 cells. AKT activity reduction was similarly observed in JHH2 and HLF cells under HMGA2‐knockdown conditions (Figure S4B). In contrast, AKT activity was augmented in HMGA2‐expessing HuH‐7 cells (Figure 5C; HuH‐7). Therefore, AKT has emerged as a possible mediator of HMGA2 function, regulating p27 protein turnover, potentially via S10 phosphorylation. To prove AKT involvement in p27 protein turnover regulation more directly, we interfered with its activity and observed the effects. In cells treated with siRNAs for AKT1 and AKT2, AKT was effectively silenced with AKT1 siRNA and under these conditions, p27 protein levels increased concurrently with decreased net phosphorylation at S10 (Figure 5D). Similar results were obtained by the pharmacological inhibition of AKT activity using the PI3K inhibitor, LY294002, and the AKT1 inhibitor, A‐674563 (Figure S4C,D). Collectively, these data suggested that HMGA2 regulated p27 protein turnover via S10 phosphorylation by AKT.
In this scenario, S10 phosphorylation is a key event, coupling the HMGA2/AKT pathway to p27 protein degradation. This notion is compatible with the role of S10 phosphorylation in stimulating nuclear export of p27. 19 Given that the subcellular localization of a protein determines its accessibility to the ubiquitination machinery, S10 phosphorylation indirectly but inevitably influences the ubiquitination process followed by protein degradation. Moreover, S10 phosphorylation is reportedly involved in the ubiquitination process directly in the cytoplasm. 20 In fact, p‐p27(S10) was exclusively detected in the cytoplasm (Figure S4E). These findings strongly suggest that S10 phosphorylation plays a role in promoting p27 degradation in the cytoplasm. However, results to the contrary have also been reported. 17 Therefore, we have directly addressed the significance of the phosphorylation in the protein degradation by examining the stability of a mutant p27 form (S10A), in which the serine residue at position 10 was converted to alanine. Figure 5E shows that S10A p27 was stabilized more than the wild‐type. The CHX chase experiment confirmed that amino acid conversion increased protein stability and supported the degradation‐promoting role of S10 phosphorylation. The values observed in the long incubation periods in Figure 5E exceeded owing to the effects of CHX on the loading control of β‐actin.
3.5. HMGA2 regulates AKT activity via IGFBP1 expression
So far, our data have defined a critical role for AKT in the HMGA2‐directed regulation of p27 protein turnover. However, it remained unclear how HMGA2 regulates AKT activity. AKT is activated by various growth factors in a PI3K‐dependent manner. Among the growth factors, insulin‐like growth factor (IGF) is one of the most relevant in hepatocarcinogenesis. 21 Therefore, we focused on the IGF system and searched for an HMGA2 mediator of AKT activation. A survey of genes whose expression levels were affected by HMGA2 identified the candidate, IGFBP1. Specifically, among IGFBP family members (Figure 6A,B) and others related to the IGF system examined (Figure S5A), IGFBP1 was the only one whose expression was significantly and commonly affected by HMGA2 knockdown in both JHH‐2 and JHH‐4 cells: in Figure 6A, HMGA2 knockdown with shRNA reduced IGFBP1 mRNA and protein levels in JHH‐4 cells. Similarly, HMGA2 siRNA treatment reduced IGFBP1 mRNA and protein levels in JHH‐2 cells. IGFBP3 expression was also affected. However, between the two cell lines, the responses were different: IGFBP3 expression was increased in JHH‐4 and decreased in JHH‐2 cells (Figure 6A,B). Therefore, IGFBP1 mRNA and protein levels, but not those of other family members, were unambiguously reduced in response to HMGA2 knockdown in both cell lines, suggesting that HMGA2 contributes to the upregulation of IGFBP1 expression. Similar to these cell lines, IGFBP1 expression was upregulated in 5 of 16 HMGA2‐positive HCC tissues (Table S1; IGFBP1 mRNA). Importantly, IGFBP1 was involved in the activation of AKT. As shown (Figures 6D; JHH‐4 and S5B), IGFBP1 knockdown decreased AKT activity in JHH‐4 cells.
FIGURE 6.
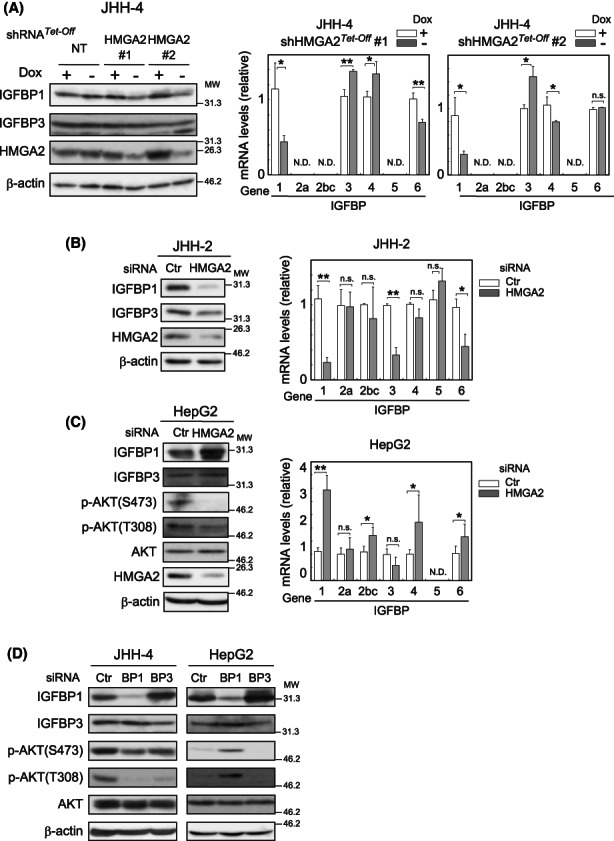
HMGA2 Regulation of AKT activity via IGFBP1 expression. (A–C) Dox‐responsive shRNA HMGA2 #1 and #2‐expressing and the control (NT) cells incubated under Dox (+) or (−) conditions (A), or JHH‐2 (B) and HepG2 (C) cells treated with HMGA2 and the control (Ctr) pooled siRNA were examined by western blotting after 4 days and by qPCR after 3 days. β‐Actin, the loading control. The values represent the mean ± SD from triplicates and are shown relative to the control (Dox + or Ctr) after normalizing the corresponding values to TATA‐box binding protein. **p < 0.005; *p < 0.05; n.s., not significant. (D) Cells were treated with IGFBP1 (BP1), IGFBP3 (BP3), and control (Ctr) pooled siRNA for 3 days and analyzed by western blotting.
In HepG2, HMGA2 knockdown also significantly affected IGFBP1 mRNA and protein levels (Figure 6C). However, in striking contrast with JHH‐2 and JHH‐4 cells, the HMGA2 knockdown resulted in an IGFBP1 mRNA increase, opposite to the cases in JHH‐2 and 4. Therefore, in this case, HMGA2 affected IGFBP1 expression negatively. Notably, IGFBP1 expression was downregulated in 11 of 16 HMGA2‐positive HCC tissues (Table S1; IGFBP1 mRNA). Interestingly, the IGFBP1‐silencing effect on AKT activity was also opposite to that of JHH‐4 in HepG2 cells. In HepG2 cells, IGFBP1 siRNA treatment activated AKT in contrast with inactivation in JHH‐4 cells (Figure 6D; HepG2). In short, HMGA2 regulates IGFBP1 expression positively or negatively, depending on the cell lines or the impact of IGFBP1 on AKT activity. It is important to note that, in either case, HMGA2 activates AKT via IGFBP1 regulation. In previous studies, the insulin‐like growth factor 2 mRNA binding protein (IGF2BP2) was identified as an HMGA2 target during embryogenesis and in mesenchymal cells 22 , 23 ; however, its expression was unaffected by HMGA2 in HCC cell lines (unpublished data).
Finally, we analyzed HCC tissue data to validate the in vitro HMGA2 regulation of IGFBP1 expression. Given that IGFBP1 expression was affected positively or negatively by HMGA2, we plotted fold changes of IGFBP1 mRNA level in HCC tumors and compared between HMGA2‐positive and HMGA2‐negative HCCs. Figure 7A shows that in HMGA2‐negative (−) group, most values were clustered at lower levels, indicating that IGFBP1 expression was minimally affected in the HMGA2‐negative HCCs. In contrast, values varied considerably over a wide range in HMGA2‐positive (+) group, suggesting that HMGA2 expression had a significant impact on IGFBP1 expression in vivo HCCs. Of note, survival analyses using TCGA dataset underscored the significance of HMGA2 regulation of IGFBP1 expression for patient prognoses. Given the bifurcate regulation of IGFBP1 by HMGA2 as described above, we compared overall survival between two patient groups, one with high HMGA2 expression combined with high or low IGFBP1 expression, and the other with low HMGA2 and median IGFBP1 expression. In the former group, high HMGA2 expression regulated IGFBP1 expression positively or negatively, and presumably activated the axis, thereby supporting tumor growth. As expected, patients in the former group [HMGA2 (H)/IGFBP1 (H + L)] showed poor prognoses when compared with the latter group [HMGA2 (L)/IGFBP1 (M)] (Figure 7B). Importantly, the prognoses were poorer than those of all the cases with high HMGA2 presented in Figure 1E.
FIGURE 7.
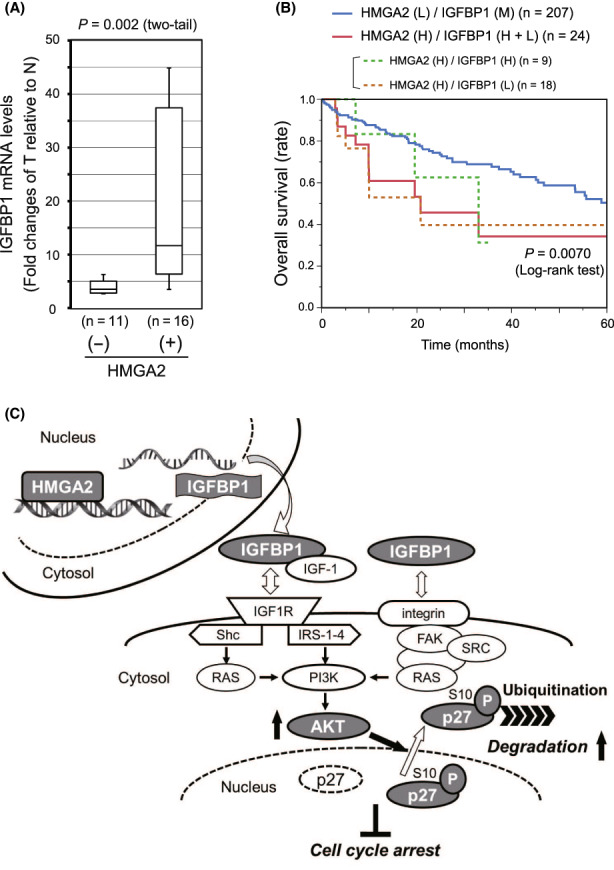
HMGA2 and IGFBP1 expression, and patient prognosis in the hepatocellular carcinoma (HCC) cases. (A) IGFBP1 mRNA amounts were determined by qPCR in pairs of HCC tumor (T) and adjacent nontumorous (N) tissues obtained from patients (Table S1). After normalization with the nuclear‐encoded control mRNAs (Section 2), the fold change of the value in T relative to that in N in each pair was plotted in the group with HMGA2 amplification positive (+) and negative (−). The statistical significance of the differences was assessed by Mann–Whitney U‐test. (B) A Kaplan–Meier plot showing patients categorized by combined HMGA2 and IGFBP1 expression levels, was generated using the liver hepatocellular carcinoma dataset (TCGA, Firehose Legacy) in the cBioPortal (Section 2). HMGA2 (H), high (z‐score > 0); (L), low (z‐score < 0); IGFBP1 (H + L), high (z‐score > 1) + low (z‐score < −1); (M), median (−1 < z‐score < 1). (C) Schematic representation of the degradative regulation of p27 protein levels by the HMGA2/IGFBP1/AKT axis as a countermeasure against antiproliferative cyclin‐dependent kinase inhibitor (CKI) effects.
4. DISCUSSION
High mobility group AT‐hook is one of the high mobility group (HMG) families, the largest and best characterized nonhistone chromosomal protein group. 24 Among the HMG families, HMGA is characterized by a unique DNA‐binding AT‐hook motif and further divided into two subfamilies, HMGA1 and HMGA2. 7 The members are so‐called chromatin modifiers, which modify various high‐ordered DNA architectures by the AT‐hook, thereby regulating transcription. 25 Interestingly, the HMGA proteins can regulate the transcription of various genes either positively or negatively, depending on the cell type and cellular context, thereby coordinating complex biological processes. 24 , 25 , 26 , 27 , 28 , 29 , 30 Regarding HMGA2, which is highly expressed during development and re‐expressed in neoplasia, a body of evidence suggests its roles in cell cycle regulation 31 , 32 , 33 , 34 and/or neoplastic transformation. 7 , 8 Mechanistically, HMGA2 regulated cell proliferation by mostly targeting cell cycle regulators, such as E2F1, cyclin A, and the activating protein‐1 (AP‐1) complex. 32 , 33 , 34 Alternatively, HMGA2 was involved in AKT activation 35 , 36 , 37 or p27 expression regulation. 37 , 38 However, the precise mechanisms underlying HMGA2 functions were largely unexplored in these studies.
In hepatocytes, HMGA2 reportedly promoted epithelial‐to‐mesenchymal transition (EMT). 39 However, HMGA2 effects on EMT marker expression were inconsistent among the cell lines in our experiments (Figure S6). Therefore, HMGA2 appeared to induce EMT but only partially in HCC cells. Instead, our study shed light on the stress‐coping role of HMGA2 in oncogenesis, which is different from the oncogenic roles suggested by previous studies. 7 , 8 , 9 In particular, HMGA2 drives the HMGA2/IGFBP1/AKT axis and, in combination with the FOXM1/BMYB complex, 6 HMGA2 counteracts stresses, such as mt‐Low stress, which can induce cell proliferation inhibition via p21CIP1 and p27 CKI upregulation. In the emerging axis (Figure 7C), HMGA2 regulates IGFBP1 expression upstream, leading to AKT activation, and AKT post‐transcriptionally suppresses p27 levels via S10 phosphorylation, followed by ubiquitination and degradation in the cytoplasm. 14 , 19 , 20 AKT also suppresses p27 expression at the transcriptional level, 40 , 41 although p27 transcriptional regulation is cellular context dependent or not significant in HCC cells.
IGFBP1 is one of six IGFBPs, binding IGF1 and 2, and mainly controlling locally available IGF amounts as their carrier. 42 Depending on the species with different modifications, IGFBP1 inhibits or potentiates IGF actions: it potentiates the IGF actions, when the IGF binding affinity declines. 42 In this study, IGFBP1 activated AKT in JHH‐4 cells for which IGFBP1 is likely to act as a positive IGF signaling regulator or alternatively, activates AKT in an IGF‐independent manner. 42 In contrast, IGFBP1 negatively affected AKT activation in HepG2 cells. In this regard, HepG2 cells were reported to secrete primarily phosphorylated IGFBP1 forms 43 with an IGF binding affinity higher than that of the nonphosphorylated form and inhibitory effects on IGF actions. 44 In in vivo HCCs, IGFBP1 potentially activates IGF signaling in one‐third of HMGA2‐positive cases as in JHH‐4 cells, while suppresses it in two third of HMGA2‐positive cases as in HepG2 cells.
Recent large‐scale sequencing has failed to newly identify genes mutated at high frequency in cancers. 45 Given such limitations, one of the future directions of cancer therapy is targeting cancer vulnerability or addiction to a molecule/pathway supporting the oncogenic state rather than cancer‐driving pathways. 1 The stress‐coping system provided by HMGA2 can be listed as a pertinent target for such a strategy. From this study, without the HMGA2/IGFBP1/AKT axis, mt‐Low‐type cancer cells succumb to senescence‐like cell proliferation inhibition. Although the axis was related to mt‐Low cases in this study, other oncogenesis‐associated stresses may also impose similar CKI upregulation‐related threats to cancer cells. Therefore, intervening in the activation of HMGA2/IGFBP1/AKT axis could be a tumor‐agnostic therapy based on mtDNA copy number, mtRNA amounts, and/or HMGA2 expression levels. Additionally, the effects of intervention in the axis are predicted to be almost exclusive to cancer cells because HMGA2 expression is virtually absent in adult tissue. 7 , 8 We believe that this study will contribute to next‐generation therapies with few adverse effects and may help a large population of patients with broad‐spectrum cancer types.
AUTHOR CONTRIBUTIONS
Tsuyoshi Maruyama: Investigation, Validation, Formal analysis, Writing (original draft). Koji Saito: Clinical sample collection and data analysis. Masato Higurashi: Investigation, Validation, Clinical data analysis. Fumihiro Ishikawa: Methodology, Writing (review and editing). Yohko Kohno: Clinical sample collection and data analysis. Kazunori Mori: Validation, Writing (review & editing). Motoko Shibanuma: Conceptualization, Supervision, Project administration. All authors have read the journal's authorship agreement and approved the submitted manuscript.
FUNDING INFORMATION
This work was supported by JSPS KAKENHI Grant Number JP26460399.
DISCLOSURE
The authors have no conflicts of interest to declare.
APPROVAL OF THE RESEARCH PROTOCOL
The research has been approved by the Institutional Review Board of Showa University and performed in accordance with the Declaration of Helsinki with patient consent.
INFORMED CONSENT
Written informed consent was obtained from the patients.
Supporting information
Appendix S1
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
We are deeply grateful to Dr. Takeshi Aoki and Dr. Masahiko Murakami from Showa Univ., Sch. Med., Dep. Surgery, Div. General and Gastroenterological Surgery, and Dr. Masafumi Takimoto from Dep. Pathology for their contribution to collection of clinical samples. We also thank Dr. Hidetoshi Tahara (Hiroshima University) for his kind gift of immortalized human hepatocytes, Hc3716‐hTERT. We thank Mr. Yoshihiro Harada, Mr. Yuya Sato, and Ms. Aya Kawashima for contributing to this work as part of their bachelor's degrees. The authors would also like to thank Enago (www.enago.jp) for English language review.
Maruyama T, Saito K, Higurashi M, et al. HMGA2 drives the IGFBP1/AKT pathway to counteract the increase in P27KIP1 protein levels in mtDNA/RNA‐less cancer cells. Cancer Sci. 2023;114:152‐163. doi: 10.1111/cas.15582
REFERENCES
- 1. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non‐oncogene addiction. Cell. 2009;136:823‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reznik E, Miller ML, Senbabaoglu Y, et al. Mitochondrial DNA copy number variation across human cancers. Elife. 2016;5:e10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reznik E, Wang Q, La K, Schultz N, Sander C. Mitochondrial respiratory gene expression is suppressed in many cancers. Elife. 2017;6:e21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuan Y, Ju YS, Kim Y, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet. 2020;52:342‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mori K, Uchida T, Fukumura M, et al. Linkage of E2F1 transcriptional network and cell proliferation with respiratory chain activity in breast cancer cells. Cancer Sci. 2016;107:963‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Higurashi M, Maruyama T, Nogami Y, et al. High expression of FOXM1 critical for sustaining cell proliferation in mitochondrial DNA‐less liver cancer cells. Exp Cell Res. 2020;389:111889. [DOI] [PubMed] [Google Scholar]
- 7. Mansoori B, Mohammadi A, Ditzel HJ, et al. HMGA2 as a critical regulator in cancer development. Genes. 2021;12:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Unachukwu U, Chada K, D'Armiento J. High mobility group AT‐hook 2 (HMGA2) oncogenicity in mesenchymal and epithelial neoplasia. Int J Mol Sci. 2020;21:3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang S, Mo Q, Wang X. Oncological role of HMGA2 (review). Int J Oncol. 2019;55:775‐788. [DOI] [PubMed] [Google Scholar]
- 10. Abbastabar M, Kheyrollah M, Azizian K, et al. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: a double‐edged sword protein. DNA Repair. 2018;69:63‐72. [DOI] [PubMed] [Google Scholar]
- 11. Shibanuma M, Ishikawa F, Kobayashi M, et al. Critical roles of the cAMP‐responsive element‐binding protein‐mediated pathway in disorganized epithelial phenotypes caused by mitochondrial dysfunction. Cancer Sci. 2012;103:1803‐1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Irvine KM, Skoien R, Bokil NJ, et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J Gastroenterol. 2014;20:17851‐17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hnit SS, Xie C, Yao M, et al. p27(Kip1) signaling: transcriptional and post‐translational regulation. Int J Biochem Cell Biol. 2015;68:9‐14. [DOI] [PubMed] [Google Scholar]
- 15. Yoshida Y, Saeki Y, Murakami A, et al. A comprehensive method for detecting ubiquitinated substrates using TR‐TUBE. Proc Natl Acad Sci U S A. 2015;112:4630‐4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253‐267. [DOI] [PubMed] [Google Scholar]
- 17. Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem. 2000;275:25146‐25154. [DOI] [PubMed] [Google Scholar]
- 18. Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355‐14358. [DOI] [PubMed] [Google Scholar]
- 19. Fujita N, Sato S, Katayama K, Tsuruo T. Akt‐dependent phosphorylation of p27Kip1 promotes binding to 14‐3‐3 and cytoplasmic localization. J Biol Chem. 2002;277:28706‐28713. [DOI] [PubMed] [Google Scholar]
- 20. Lee JG, Kay EP. Involvement of two distinct ubiquitin E3 ligase systems for p27 degradation in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2008;49:189‐196. [DOI] [PubMed] [Google Scholar]
- 21. Adamek A, Kasprzak A. Insulin‐like growth factor (IGF) system in liver diseases. Int J Mol Sci. 2018;19:1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brants JR, Ayoubi TA, Chada K, Marchal K, Van de Ven WJ, Petit MM. Differential regulation of the insulin‐like growth factor II mRNA‐binding protein genes by architectural transcription factor HMGA2. FEBS Lett. 2004;569:277‐283. [DOI] [PubMed] [Google Scholar]
- 23. Li Z, Gilbert JA, Zhang Y, et al. An HMGA2‐IGF2BP2 axis regulates myoblast proliferation and myogenesis. Dev Cell. 2012;23:1176‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reeves R. High mobility group (HMG) proteins: modulators of chromatin structure and DNA repair in mammalian cells. DNA Repair. 2015;36:122‐136. [DOI] [PubMed] [Google Scholar]
- 25. Reeves R, Beckerbauer L. HMGI/Y proteins: flexible regulators of transcription and chromatin structure. Biochim Biophys Acta. 2001;1519:13‐29. [DOI] [PubMed] [Google Scholar]
- 26. Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091‐1100. [DOI] [PubMed] [Google Scholar]
- 27. Zha L, Wang Z, Tang W, Zhang N, Liao G, Huang Z. Genome‐wide analysis of HMGA2 transcription factor binding sites by ChIP on chip in gastric carcinoma cells. Mol Cell Biochem. 2012;364:243‐251. [DOI] [PubMed] [Google Scholar]
- 28. Henriksen J, Stabell M, Meza‐Zepeda LA, Lauvrak SA, Kassem M, Myklebost O. Identification of target genes for wild type and truncated HMGA2 in mesenchymal stem‐like cells. BMC Cancer. 2010;10:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Disney JE, Johnson KR, Magnuson NS, Sylvester SR, Reeves R. High‐mobility group protein HMG‐I localizes to G/Q‐ and C‐bands of human and mouse chromosomes. J Cell Biol. 1989;109:1975‐1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bagga R, Michalowski S, Sabnis R, Griffith JD, Emerson BM. HMG I/Y regulates long‐range enhancer‐dependent transcription on DNA and chromatin by changes in DNA topology. Nucleic Acids Res. 2000;28:2541‐2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou X, Benson KF, Ashar HR, Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI‐C. Nature. 1995;376:771‐774. [DOI] [PubMed] [Google Scholar]
- 32. Fedele M, Visone R, De Martino I, et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell. 2006;9:459‐471. [DOI] [PubMed] [Google Scholar]
- 33. Tessari MA, Gostissa M, Altamura S, et al. Transcriptional activation of the cyclin a gene by the architectural transcription factor HMGA2. Mol Cell Biol. 2003;23:9104‐9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vallone D, Battista S, Pierantoni GM, et al. Neoplastic transformation of rat thyroid cells requires the junB and fra‐1 gene induction which is dependent on the HMGI‐C gene product. EMBO J. 1997;16:5310‐5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu KR, Park SB, Jung JW, et al. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood‐derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res. 2013;10:156‐165. [DOI] [PubMed] [Google Scholar]
- 36. Liu H, Wang X, Liu S, et al. Effects and mechanism of miR‐23b on glucose‐mediated epithelial‐to‐mesenchymal transition in diabetic nephropathy. Int J Biochem Cell Biol. 2016;70:149‐160. [DOI] [PubMed] [Google Scholar]
- 37. Wei CH, Wei LX, Lai MY, Chen JZ, Mo XJ. Effect of silencing of high mobility group A2 gene on gastric cancer MKN‐45 cells. World J Gastroenterol. 2013;19:1239‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Federico A, Forzati F, Esposito F, et al. Hmga1/Hmga2 double knock‐out mice display a "superpygmy" phenotype. Biol Open. 2014;3:372‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luo Y, Li W, Liao H. HMGA2 induces epithelial‐to‐mesenchymal transition in human hepatocellular carcinoma cells. Oncol Lett. 2013;5:1353‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Medema RH, Kops GJ, Bos JL, Burgering BM. AFX‐like Forkhead transcription factors mediate cell‐cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782‐787. [DOI] [PubMed] [Google Scholar]
- 41. Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969‐8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bach LA. IGF‐binding proteins. J Mol Endocrinol. 2018;61:T11‐T28. [DOI] [PubMed] [Google Scholar]
- 43. Jones JI, D'Ercole AJ, Camacho‐Hubner C, Clemmons DR. Phosphorylation of insulin‐like growth factor (IGF)‐binding protein 1 in cell culture and in vivo: effects on affinity for IGF‐I. Proc Natl Acad Sci U S A. 1991;88:7481‐7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin YW, Weng XF, Huang BL, Guo HP, Xu YW, Peng YH. IGFBP‐1 in cancer: expression, molecular mechanisms, and potential clinical implications. Am J Transl Res. 2021;13:813‐832. [PMC free article] [PubMed] [Google Scholar]
- 45. Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11:340‐349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Table S1
Table S2
Table S3