

Abstract
Accessing fascinating organic and biological significant indolines via dearomatization of indoles represents one of the most efficient approaches. However, it has been difficult for the dearomatization of the electron deficient indoles. Here we report the studies leading to developing a photoredox mediated Giese-type transformation strategy for the dearomatization of the indoles. The reaction has been implemented for chemoselectively breaking indolyl C=C bonds embedded in the aromatic system. The synthetic power of this strategy has been demonstrated by using structurally diverse indoles bearing common electron-withdrawing groups including (thio)ester, amide, ketone, nitrile and even aromatics at either C2 or C3 positions and ubiquitous carboxylic acids as radical coupling partner with high trans-stereoselectivity (>20:1 dr). This manifold can also be applied to other aromatic heterocycles including pyrroles, benzofurans and benzothiophenes. Furthermore, enantioselective dearomatization of indoles has been achieved by a chiral camphorsultam auxiliary with high diastereoselectivity.
Subject terms: Synthetic chemistry methodology, Catalyst synthesis
Dearomatisation of indoles allows efficient access to indolines, but often is incompatible with electron-withdrawing substituents. Here a photoredox Giese-type dearomatisation of indoles yields 2,3-disubstituted indolines bearing electron-withdrawing groups.
Introduction
The indolines have fascinated organic and medicinal chemists for decades1–8. The molecular architecture is a common core featured in numerous natural products, biologically active compounds particularly pharmaceutics and agrochemicals. This biogenically produced privileged structure9 provides highly biologically relevant three-dimensional chemical space for effective interaction with biological targets. Therefore, quickly accessing the framework with the capacity of engineering functional and stereochemical diversity can streamline the target- and diversity-oriented synthesis for biological studies and drug discovery.
The dearomatization of arenes has become a powerful platform for the facile construction of highly valued molecular architectures10–15. The dearomatization of indoles constitutes the most efficient strategy for accessing indolines1–8. Indole is an electron-rich aromatic system containing enamine embedded C2–C3 π bond and strong nucleophilic C3 carbon. The reactivity has dictated indole dearomatization methodology development1–8 since Woodward’s pioneering study using a Pictet-Spengler type reaction to break the aromatic tryptamine in total synthesis of strychnine in 195416. Impressively, this important array of reactivity from the intrinsically nucleophilic indoles upon activation by various tailored electrophiles has become a powerful manifold for the synthesis of structurally diverse indolines as it enables regioselective reactivity, facile ring formation, and efficient skeleton rearrangement1–8. Moreover, this reactivity has been leveraged beyond the 2e transfer pathway. Single-electron transfer (SET) involved oxidation-induced C–H functionalization of the nucleophilic indoles has been elegantly realized as powerful alternatives for indole dearomatization17, particularly mild, green visible light photocatalytic and electrochemical methods (Fig. 1a)3,15,18–29.
Fig. 1. The synthesis of indolines by radical engaged dearomatization of indoles.

a Dearomatization of indoles via oxidative SET of C2=C3 bond (known). b Giese reaction: radical addition to simple α, β-unsaturated systems. c Dearomatization of indoles via a Giese-type transformation (this work).
Despite the great success, it has been challenging for the dearomatization of electron-poor indoles, as evidenced by only a handful of examples30–33, which rely on ionic activation mode. Indoles bearing electron-withdrawing groups (EWGs) at N, C2 or C3 positions tend to make the C2=C3 π bond more difficult to react with electrophilic partners or in an oxidative SET process. The reduced C2=C3 π bond electron density can be reflected by the significant difference of their redox potentials. For example, Eredox of N,3-dimethyl indole is ca. +0.4 V vs SCE34, while N-methyl 3-acetyl indole is ca. +1.0 V vs SCE34. Therefore, an unique activation paradigm is needed to address this unmet synthetic challenge.
Giese reaction involving the reductive conjugation addition of radicals to electron-deficient C=C double bonds servers as a powerful tool for new C–C bond formation35. The original conditions using stoichiometric amounts of trialkyl tin reagents have motivated organic chemists to develop more practical protocols. Recent efforts on the study of photoredox catalysis under mild reaction conditions have made the process greener and more atom economical (Fig. 1b)36–47. In this process, the unsaturated C=C bond is transformed into a saturated C–C bond in a conjugate addition manner. We questioned whether the reaction could be applied for breaking unsaturated C=C bonds embedded in the indole aromatic structure. Specifically, we envisioned the incorporation of an EWG into C2 or C3 position of indoles, which could be viewed as the Michael acceptors for the Giese type transformation. The successful realization of this process could offer a distinct approach for the dearomatization of less developed electron-deficient indoles and would also expand the scope of the Giese reaction.
However, implementing the strategy faces significant roadblocks. Unlike an isolated C=C bond in a typical Giese reaction (Fig. 1b), breaking the unconventional C=C bond in stable indole aromatic systems overcomes a higher energy barrier. The precedent studies of direct addition of an electrophilic radical to the electron-rich C2=C3 bond of indoles in electrophilic aromatic substitution processes provide encouraging possibility48,49. Nonetheless, the reversed reactivity of the addition of a nucleophilic radical to an electron-poor C2=C3 bond of indoles is unknown. Moreover, even though incorporation of EWGs into the C2 or C3 positions of indoles could reverse the polarity from the innate nucleophilic to electrophilic system and serve as a potential radical acceptor, the weakly electron-deficient indoles render the Giese reaction more difficult because more electron deficient, less hindered α, β-unsaturated systems are generally used for effective nucleophilic radical addition36–47. Furthermore, in the photoredox process, possible oxidation of the weakly electron-deficient indole systems could complicate the process.
Herein we wish to disclose the results of the investigation, which leads to a photoorganocatalytic strategy for the dearomatization of electron-deficient indoles. An unconventional Giese-type transformation is successfully implemented for the first time (Fig. 1c). Notably, the protocol uses naturally abundant carboxylic acids as radical precursors50–55 for reacting with various functionalized indoles bearing common EWGs at either C2 or C3 positions including (thio)ester, amide, ketone, nitrile, and even neural H and phenyl moieties. Furthermore, this mild dearomatization method displays a broad substrate scope and a wide array of functional group tolerance and thus enables to deliver a wide array of 2,3-disubstituted indolines with high trans-stereoselectivity (>20:1 dr). This approach can also be applied to other aromatic heterocycles such as pyrroles, benzofurans, and benzothiophenes for the dearomatization. Finally, enantioselective dearomatization of indoles has been achieved by a chiral camphorsultam auxiliary with high ee (up to 98%).
Results
Exploration and optimization of the Giese-type reaction
In the initial exploratory studies, we chose N-Boc indole methyl ester 1a as a radical acceptor and Boc-alanine 2a as a radical precursor for the proposed Giese-type reaction. It is believed that the carboxylate 2a can be selectively oxidized by the photocatalyst (PC, Ir[dF(CF3)ppy]2(dtbpy))PF6) to give the corresponding radical while the oxidation of the C2=C3 π bond is difficult because the E1/2* is +1.21 V (vs SCE)50 of the PC and 2a salt Eox is around +1.00 V (vs SCE)56 while the Eox of the indole methyl ester 1a is +1.94 V (vs SCE, see Supplementary Methods Section 1.8). Irradiation of a mixture of 1a (0.2 mmol) with N-Boc-Alaine 2a (0.26 mmol) in the presence of (Ir[dF(CF3)ppy]2(dtbpy))PF6 (5 mol%) and Cs2CO3 (0.2 mmol) in DMF (0.1 M) under N2 atmosphere with a 5 W blue LED strip was performed accordingly (Table 1, entry 1). Indeed, the desired indoline product 3a was obtained with yield of 56% after 36 h irradiation without the observed oxidation of 1a (entry 1). It is also noted that the reaction proceeded highly trans selectively. Encouraged by the results, we devoted efforts to optimize reaction conditions. When 4CzIPN was used as a PC57, a nearly quantitative yield was obtained (entry 2). Probing other parameters such as switching solvent to MeCN (entry 3), shortening time (entry 4) and lowering the amount of base (entry 5) revealed that the reaction performed in DMF for 36 h with 1 equiv. of Cs2CO3 and 5 mol% catalyst (entry 2) could give the best reaction yield. As expected, PC (entry 7) and visible light (entry 8) were indispensable for this process. These findings led to establishing the optimal protocol used for probing the scope of an organophotocatalytic dearomatization of indoles.
Table 1.
Optimization of reaction conditions.
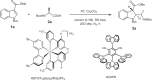 | ||||
|---|---|---|---|---|
| Entry | PC (5 mol%) | Solvent (0.1 M) | Time (h) | Yield (%)a |
| 1 | (Ir[dF(CF3)ppy]2(dtbpy))PF6 | DMF | 36 | 56 |
| 2 | 4CzIPN | DMF | 36 | 99b |
| 3 | 4CzIPN | MeCN | 36 | 81 |
| 4 | 4CzIPN | DMF | 24 | 75 |
| 5 | 4CzIPN | DMF | 24 | 83c |
| 6d | 4CzIPN | DMF | 36 | 91d |
| 7 | None | DMF | 36 | NRe |
| 8 | 4CzIPN | DMF | 36 | NRf |
Unless otherwise specified, to an oven-dried 10 mL-Schlenk tube equipped with a stir bar, was added 1a (55.0 mg, 0.2 mmol), PC (8.0 mg, 5 mol%), 1b (45.5 mg, 0.26 mmol), Cs2CO3 (65.0 mg, 0.2 mmol) and solvent (2.0 mL). The mixture was degassed by freeze-pump-thaw method, then sealed with parafilm. The solution was then stirred at rt under the irradiation of a 5 W blue LED strip for the indicated time.
PC photoredox catalyst, DMF dimethylformamide, NMP N-Methyl-2-pyrrolidone.
a1H NMR yield.
bIsolated yield.
c0.1 mmol Cs2CO3 was used.
d3 mol% PC was used.
eNo reaction.
fNo light.
Scope of indoles and other heteroaromatics
With optimized reaction conditions in hand, we first evaluated the radical-engaged dearomatization reactions utilizing various electron-deficient indoles as substrates. As shown in Fig. 2, this methodology serves as a mild and efficient approach for the synthesis of a wide range of 2,3-disubstituted indoline derivatives in high yields (up to 99%) (Fig. 2) (Supplementary Methods Section 1.5 and Supplementary Data 1). Notably, the protocol works for indoles bearing various EWGs beyond ester. Ketone (3b and 3c), amide (3c and 3g), thioester (3d) and cyanide (3f) can be served to afford broadly functionalized indolines in high yields. Furthermore, commonly used nitrogen protecting groups such as Tos (3 h), Bz (3i), Ac (3j) and Cbz (3k) are tolerated very well. Incorporation of various substituents (e.g., MeO, F, Cl, Br) into the benzene ring in the indole skeleton does not affect dearomatization efficiency (3l, 3m, 3n, 3o, 3q and 3r). Unexpectedly, in addition to electron-deficient indoles, the protocol works smoothly for indoles containing electron neutral H (3o) and phenyl (3p) moieties. Moreover, instead of EWG at C3, indole possessing C2 ester group also works well (3q and 3r). Remarkably, other aromatic structures such as benzofuran, benzothiophene and pyrrole can attend the process and produce 2,3-dihydro-1H-pyrrole (3s), 2,3-dihydrobenzofuran (3t), and 2,3-dihydrobenzo[b]thiophene (3u), respectively in high yields. The obtained trans products were confirmed by the single X-ray analysis of trans-3k (CCDC-1994584, Supplementary Methods Section 1.10: Supplementary Tables 1–3 and Supplementary Data 3: CIF file). The unsuccessful indole substrates listed in Fig. 2 suggest that two EWGs are necessary for this process.
Fig. 2. Scope of indoles and other hetereoaromatics.

Unless specified, see the general procedure in Supplementary Methods Section 1.5 for the experimental protocol.
Scope of carboxylic acids
Next, we probed the structural alternation of carboxylic acids under the optimal reaction conditions (Fig. 3) (Supplementary Methods Section 1.6 and Supplementary Data 1). Again, this strategy provides a preparative power for the synthesis of various trans-selective 2,3-disubstituted indolines on account of their easy availability. Naturally abundant amino acids without requiring protection of side-chain functionalities serve as a good source to install α-amino alkyl groups at C2 position of indoline (3v-3ae). The mild reaction enables incorporation of highly strained structures (3ae and 3ao). In addition, radicals generated from α-oxygen carboxylic acids efficiently engage in the dearomative process of electron-deficient indoles as well (3af-3ai). Next, non-α-heteroatom alkyl radicals bearing four-, five- and six- rings were probed. The corresponding products 3aj, 3ak, 3am, 3ao and 3aq were delivered in high yields. This protocol was also successfully expanded to bridged carboxylic acids (3an and 3ap) as alkyl radical precursors. As for hindered structures and less reactive radicals, stronger light power and a more amount of acid are needed to achieve good yields (3ad, 3ae, 3aj–3aq). It should also be pointed out that under the mild reaction conditions, this radical-based method exhibits broad functional group tolerance, as demonstrated for free hydroxyl (3y), acetal (3ai), thioether (3ab), amide (3aa), and heteroaromatic groups (3x and 3z). Especially, electron-deficient indole was chemoselectively reacted in the presence of electron-rich indole, demonstrated by the case of 3x. Furthermore, a dipeptide can also effectively participate in this process. In the scope study, we found that four unsuccessful carboxylic acids could not partipatite in the process (Fig. 3).
Fig. 3. Scope of carboxylic acids.

Unless specified, see the general procedure in Supplementary Methods Section 1.5 for the experimental protocol.
Late stage dearomatization of indoles and gram-scale synthesis of indolines
To further demonstrate the utility of this mild dearomatization strategy, we performed a series of late-stage modifications on natural products. As shown in Fig. 4a (Supplementary Methods Section 1.5), the standard protocol was successfully applied to natively and selectively modify natural products (+)-menthol, cholesterol and (+)-δ-tocopherol to give trans-indoline-based analogues 3as, 3aw and 3ax in 99, 61 and 80% yield, respectively. Moreover, pentose and hexose derived indoles gave the desired products 3at − 3au. Finally, trans-indoline containing dipeptide 3av was prepared in high yield (83%). This approach can be applied in a gram scale (2 mmol) synthesis of indolines without loss of yields (Figs. 4b, 3k and 3aj) (Supplementary Methods Section 1.5). Moreover, the obtained indolines can go further transformation such as deprotection of N-Boc indoline and reduction of methyl ester to alcohol (4k and 4aj) (Supplementary Methods Section 1.5 and Supplementary Data 1).
Fig. 4. Late-stage dearomatization of indoles and gram-scale synthesis of indolines.

a Late-stage dearomatization of indoles. b Large scale synthesis and synthetic elaboration.
Asymmetric dearomatization of indoles
With the success of efficient radical engaged dearomatization of indoles and other heteroaromatics, we seek to realize the asymmetric version of this synthetically useful approach. The development of radical engaged reactions including asymmetric dearomatization of indoles has been a formidably challenging task in photoredox catalysis and is much less developed, but highly sought area58,59. It is observed that so far, all reported but limited asymmetric examples employ the electron-rich indoles18,22,60–62. In this study, we proposed to realize the asymmetric manner using a chiral auxiliary induced chirality strategy because the carboxylate can be served as a handle for the incorporation of a chiral auxiliary63. However, they are rarely employed in radical processes64,65 probably because of the high reactivity of radial species.
Commonly used Evans chiral oxazolidinone auxiliary66 was explored in our initial attempt. Under the above established optimal reaction conditions, the reaction of chiral oxazolidinone (5a) with N-Boc alanine (2b) gave the product in quantitative yield but only poor distereoselectivity (1.6:1 dr, Table 2, entry 1). It appears that solvent has little effect on the distereoselectivity (entries 1–3), but noticed impact on reaction yield (entries 1, 2 and 5). As Lewis acid (LA) is often used to improve dr by reducing the rotation of the auxiliary through chelating two C = O bonds, we screened several LAs. However, no enhancement of dr value was observed, which probably attributes to the disruption of the chelation interaction by highly polar solvent DMF (entries 6–9). Switching to L-proline derived indole 5b did not produce an encouraging result (entry 10). Gladly, when chiral camphorsultam67 was employed as a chiral auxiliary, excellent dr value (>20:1) was obtained in high yield (99%, entry 11). Shortening reaction time decreased the yield slightly (entry 12). These studies led to a protocol for the asymmetric dearomatization of electron-deficient indoles for the first time and provide an efficient approach to the synthesis of medicinally valued chiral indoline derived amino acids (β- and/or γ-amino acids).
Table 2.
Optimization of asymmetric dearomatization reaction conditions.
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Additive | Solvent (0.1 M) | Yield(%)a | Drb |
| 1 | 5a | None | DMF | 99c | 1.6:1 |
| 2 | 5a | None | MeCN | 78 | 1.3:1 |
| 3 | 5a | None | NMP | 99 | 1.6:1 |
| 4d | 5a | None | DMF | 99 | 1.8:1 |
| 5d | 5a | None | EtOAc | <5 | Not determined |
| 6 | 5a | BF3 (10 mol%) | DMF | 61 | 1.4:1 |
| 7e | 5a | Zn(OAc)2 (1.0 equiv.) | DMF | <5 | Not determined |
| 8e | 5a | Mg(OAc)2 (1.0 equiv.) | DMF | 52 | 1.5:1 |
| 9e | 5a | LiOAc (1.0 equiv.) | DMF | 65 | 1.4:1 |
| 10 | 5b | None | DMF | 77 | 1.1:1 |
| 11 | 5c | None | DMF | 99c | >20:1 |
| 12 | 5c | None | DMF | 91%f | >20:1 |
Unless otherwise specified, to an oven-dried 10 mL-Schlenk tube equipped with a stir bar, was added 5 (0.1 mmol), photoredox catalyst (4CzIPN, 4.0 mg, 5 mol%), 2b (23.0 mg, 0.13 mmol), Cs2CO3 (32.0 mg, 0.1 mmol) and solvent (1.0 mL). The mixture was degassed by freeze-pump-thaw method, then sealed with parafilm. The solution was then stirred at rt under the irradiation of a 5 W blue LED strip for 36 h.
DMF dimethylformamide, NMP N-Methyl-2-pyrrolidone.
a1H NMR yield.
bDetermined by 1H NMR.
cIsolated yield.
dCsOAc (1.0 equiv.) used as base.
eNo base used.
fReaction time: 24 h.
Next, we evaluated the scope of the asymmetric process under the optimized reaction conditions (Fig. 5) (Supplementary Methods Section 1.6 and Supplementary Data 1 and 2). To get more accurate dr value of dearomatization indoline products, the camphorsultam auxiliary was removed by either hydrolysis (Condition A) or reduction (Condition B) to analyze the ee value of derived compounds such as ester or alcohol using chiral HPLC. The results reveal that this radical engaged asymmetric strategy serves as a general approach to enantioenriched trans 2,3-disubstituted indolines 7 with high enantioselectivity (up to 98% ee). α-Amino alkyl groups were successfully introduced to deliver pharmaceutically valued chiral β,γ-diamino acids with both high ee values and yields by using natural amino acids (7a–d). Besides, radicals produced from α-oxygen carboxylic acids reacted with indole substrates smoothly (7e–m). Notable, various functional groups were tolerated well under the reaction conditions such as allylic (7e), propargyl (7f), benzyl (7h) and halogens (7j and 7k). Moreover, indolines bearing heteroaromatics were successfully obtained (7m–o). Again, the radical approach enables the incorporation of highly sterically demanding structures (7p–v), which are particularly challenging using catalytic ionic methods with high level of enantioselectivity. We also probed indoles with different protecting groups (PGs) on nitrogen. It was found that Boc and Cbz (7w) were untouched while other PGs such as Ac, Bz, Tos, and pivaloyl were sensitive to the reaction conditions. It seems that chiral moieties do not affect the newly formed stereogentic centers, as seen in the synthesis of chiral indoline saccharide derivatives with both good ee value and yields (7x and 7y). The gram scale (1 mmol) synthesis of 8c was also realized in 72% yield and 94% ee and the camphorsultam auxiliary was recovered at the same time (81% yield) (Supplementary Methods Section 1.7). The absolute conformation of the products 7 and 8 were determined to 2R and 3S by converting a known chiral compound (Supplementary Methods Section 1.7)68. In all cases except products 8a–c as alcohols, chiral methyl esters 7 were used for chiral HPLC analysis. Finally, unsuccessful substrates provided in Fig. 5 reveal the limitations of this methodology.
Fig. 5. Scope of asymmetric dearomatization of indoles.

Unless specified, see the general procedure in Supplementary Methods Section 1.6 for the experimental protocol.
Discussion
A Giese-type process has been successfully implemented for the dearomatization of electron-poor indole systems in this study. The method can also be viewed as a decarboxylative Michael addition process, which has been intensively studied in recent years35,50–55. However, indoles are not the same as simple alkenes and to the best of our knowledge, breaking the stable C2=C3 bond embedded in the aromatic indoles with this strategy has not been reported. Furthermore, in a typical decarboxylative Michael addition process35,50–55, less hindered electron-deficient vinyls are generally used for effective transformation, whereas this process accomplishes with more complicated α, β-unsaturated systems. Therefore, the process significantly expands the scope of the synthetic strategy. Moreover, the process offers a distinct approach to highly vauled indolines. The dearomative structures are complementary to those of well studied electron-rich systems. The tethered EWGs such as ester, ketone, amide, nitrile, etc are versatile handles for further synthetic elaboration.
It is noted that although intermolecular radical addition to the indole C2=C3 double bond has been documented48,49, these processes also use electron-rich structures. Mechanistically, our dearomatization activation strategy is completely different from that of these radical engaged Friedel-Crafts type methods48,49. In our approach, an oxidative process is implemented to generate a nucleophilic radical for an addition to an electron-deficient C=C bond (Fig. 6a). The process delivers a dearomative product. In contrast, an opposite photoredoxcatalytic reductive activation produces an electrophilic radical for reacting with the electron-rich indole C2=C3 double bond. Therefore, an aromatic product is obtained instead via subsequent oxidation to accomplish the catalytic cycle48,49.
Fig. 6. Proposed reaction mechanism and mechanistic studies.

See Supplementary Methods Section 1.9 for the experimental protocol.
What we learned from this study is that the successful realization of the distinct indole dearomatization method lies in the rationalization of the reactivities of the radical species in organophotoredox catalytic cycle and enables achieving the chemoselectivity. In the intensively studied photoredox mediated indole dearomatization processes, direct oxidation of electron-rich indoles can be achieved because they have relatively low redox potentials. For example, Eredox of N,3-dimethyl indole is ca. +0.4 V vs SCE34. However, indoles bearing EWGs at N, C2 or C3 positions have much higher Eredox. For instance, N-methyl 3-acetyl indole is ca. +1.0 V vs SCE34 while methyl N-Boc-3-indole carboxylate (1a, Eredox + 1.94 vs SCE, see Supplementary Methods Section 1.8) is even bigger. The significant difference suggests oxidative dearomatization of electrophilic indoles is difficult. The overlooked, reversed reactivity offers a unique opportunity for developing distinct dearomative methods, which have been successfully carried out in this study. Critically, the selective controlling reactivity makes the process possible (Fig. 6a). The excited 4CzIPN* (E*redox + 1.35 vs SCE)57 can selectively oxidize N-Boc-alanine Cs salt I (as a representative example, Eredox + 1.00 vs SCE)56 without crossover oxidation of methyl N-Boc-3-indole carboxylate (1a, Eredox + 1.94 vs SCE). The resulting nucleophilic radical II then undergoes a Giese-type addition process with 1a to produce radical III. The radical III is facial selectively reduced by 4CzIPN•− to give the trans- anion IV and 4CzIPN to complete the redox cycle. Finally, protonation of anion IV delivers observed trans-dearomatization product 3a. The pathway is consistent with the photoredox Giese reaction. Furthermore, our control experiments support the proposed pathway (Supplementary Methods Section 1.9). The radical engaged process is verified by a radical scavenger TEMPO suppressed reaction (Fig. 6b). The anion intermediate IV is validated by a D2O quenching experiment. To get more insights of the reactivity of 1a compared with the classic Michael acceptor in Giese reaction, a competition experiment using 1a and cyclopent-2-en-1-one reacting with N-Boc-glycine was performed. Interestingly, we didn’t observe trans-3v, but only product 12 coming from cyclopent-2-en-1-one with a yield of 87%. This result shows the much less reactivity of 1a than commonly used electron-deficient olefins.
Besides, we were interested in how the chiral auxilary induces diasteroselectivity. Based on Curran’s work69,70, a model for the asymmetric dearomatization was proposed (Fig. 6c). The sulfonyl group, which is spatially located close to C2, is believed to play a major role in the inducement of asymmetric selectivity. Radical attacking C2 from the Re face(bottom) is favored due to less steric interaction between radical and equatorial β oxygen of the sulfonyl group. While axial α oxygen blocks the radical addition from the Si (top) face because of their strong steric interaction. The resulting configuration of the product from this model is conistent with our experimental results.
Conclusion
In summary, we have developed an unprecedented versatile organophotoredox process for the dearomatization of electron-poor indoles. A distinct strategy that a photoredox mediated Giese-type transformation is introduced to break the electron-deficient aromatic C=C bonds for the first time. The preparative power of the dearomatization strategy has been demonstrated by the use of naturally abundant carboxylic acids and readily available structurally diverse indoles bearing common EWGs including (thio)ester, amide, ketone, nitrile, and even aromatics at either C2 or C3 positions. A wide array of 2,3-disubstituted indolines with high anti-stereoselectivity (>20:1 dr) are prepared. This powerful manifold can also be applied to other aromatic heterocycles such as pyrroles, benzofurans, and benzothiophenes for dearomatization. Furthermore, enantioselective dearomatization of indoles has been achieved by a chiral camphorsultam auxiliary with high enantioselectivity (up to 98% ee). The simplicity, efficiency, and board scope of this distinct synthetic strategy will be appreciated by organic and medicinal chemists to rapid access a library of synthetically and biologically important indolines.
Methods
Typical procedure for the synthesis of racemic products 3 (Figs. 2–4)
To an oven-dried 10 mL-Schlenk tube equipped with a stir bar, was added indole derivatives (0.2 mmol, 1.0 equiv.), 4CzIPN (8.0 mg, 5 mol%), acid (0.26 mmol, 1.3 equiv.), Cs2CO3 (65.0 mg, 0.2 mmol, 1.0 equiv.) and DMF (2.0 mL). The mixture was degassed by freeze-pump-thaw method, then sealed with parafilm. The solution was then stirred at room temperature under the irradiation of a 5w blue LED strip or two 40 w blue LED lamps for 36 h. After completion of the reaction, the mixture was diluted with 20 mL of water and extracted by EtOAc (3 × 10 mL). The organic layer was collected, dried by Na2SO4 and concentrated under vacuum. The residue was purified by flash column chromatography to afford the product.
Typical procedure for the synthesis of products 7 and 8 (Fig. 5)
To an oven-dried 10 mL-Schlenk tube equipped with a stir bar, was added chiral auxiliary attached indole derivatives 5 (0.1 mmol, 1.0 equiv.), photocatalyst 4CzIPN (4.0 mg, 5 mol%), acid (0.13–0.2 mmol, 1.3–2.0 equiv.), Cs2CO3 (32.0 mg, 0.1 mmol, 1.0 equiv.) and DMF (1.0 mL). The mixture was degassed by freeze-pump-thaw method, then sealed with parafilm. The solution was then stirred at room temperature under the irradiation of a 5w blue LED strip or two 40 w blue LED lamps for the indicated time. After completion of the reaction, the mixture was diluted with 10 mL of water and extracted by EtOAc (3 × 5 mL). The combined organic layers were washed by 10 mL of brine, dried by Na2SO4, and concentrated under vacuum. The residue was used in the next step without purification.
Condition A
the residue was dissolved in 0.8 mL of THF and 0.2 mL of water. To the solution was added LiOH (12 mg, 0.5 mmol) and H2O2 (113 μL, 30% (w/w) in water) at rt. The reaction was stirred at rt for 10 h, after which 10 mL of EtOAc was added. The mixture was washed by 0.5 M NaOH (3 × 5 mL). The combined aqueous layers were collected, washed once with 5 mL of Et2O and acidified with 2 M HCl to pH = 2–3. The aqueous solution was extracted by EtOAc (3 × 5 mL). The combined organic layers were dried by Na2SO4 and concentrated under a vacuum. The residue was used in next step without purification. The residue was dissolved in 0.75 mL of Et2O and 0.25 mL of MeOH. To the solution was added (trimethylsilyl)diazomethane solution (2 M, 110 μL) at 0 °C under nitrogen atmosphere, the mixture was stirred at room temperature for 20 min. The reaction mixture was concentrated in vacuo and purified by flash column chromatography or preparative TLC plate to afford the product.
Condition B
The residue was dissolved in 0.75 mL of EtOH and 0.25 mL of Et2O. To the solution was added LiCl (21 mg, 0.5 mmol) and NaBH4 (31.5 mg, 0.5 mmol) at rt. The reaction was stirred at rt for 3 h, after which the solution was concentrated and purified by column chromatography or preparative TLC plate to afford the product.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
Financial support of this research from the University of Arizona is gratefully acknowledged. The Foundation for Basic and Applied Research of Guangdong Province (2019A1515110489) for Dr. Ziyuan Zhou; This work supported by the high-performance computing platform of Peking University.
Author contributions
Y.Z., P.J., Y.D., H.H., C.W., Z.Z. and F.G. planned, conducted, and analyzed the experiments. W.W. planned, designed, and directed the project, and Y.Z. and W.W. wrote the manuscript.
Data availability
1H and 13C NMR spectra for products 3, 7, and 8: see Supplementary Figs. 3–192 in Suppmentary Data 1.
Chiral HPLC analysis for products 7, 8, and 11: see Supplementary Figs. 193–254 in Suppmentary Data 2.
The authors declare that all the other data including Supplementary Methods and compound structural characterization data, supporting the findings of this study are available within this paper, its Supplementary Information file. The X-ray crystallographic coordinates (Supplementary Methods: Supplementary Tables 1–3 and Supplementary Data 3: CIF file) for structures trans-3k reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC-1994584. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-021-00460-y.
References
- 1.Roche SP, Tendoung J-JY, Tréguier B. Advances in dearomatization strategies of indoles. Tetrahedron. 2015;71:3549–3591. doi: 10.1016/j.tet.2014.06.054. [DOI] [Google Scholar]
- 2.Zheng C, You SL. Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 2019;36:1589–1605. doi: 10.1039/C8NP00098K. [DOI] [PubMed] [Google Scholar]
- 3.Chen J-B, Jia Y-X. Recent progress in transition-metal-catalyzed enantioselective indole functionalizations. Org. Biomol. Chem. 2017;15:3550–3567. doi: 10.1039/C7OB00413C. [DOI] [PubMed] [Google Scholar]
- 4.Steven A, Overman LE. Total synthesis of complex cyclotryptamine alkaloids: Stereocontrolled construction of quaternary carbon stereocenters. Angew. Chem. Int. Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612. [DOI] [PubMed] [Google Scholar]
- 5.Festa AA, Voskressensky LG, Van der Eycken EV. Visible light-mediated chemistry of indoles and related heterocycles. Chem. Soc. Rev. 2109;48:4401–4423. doi: 10.1039/C8CS00790J. [DOI] [PubMed] [Google Scholar]
- 6.Bandini M, Eichholzer A. Catalytic functionalization of indoles in a new dimension. Angew. Chem. Int. Ed. 2009;48:9608–9644. doi: 10.1002/anie.200901843. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D, Song H, Qin Y. Total synthesis of indoline alkaloids: a cyclopropanation strategy. Acc. Chem. Res. 2011;44:447–457. doi: 10.1021/ar200004w. [DOI] [PubMed] [Google Scholar]
- 8.Hua T-B, Xiao C, Yang Q-Q, Chen J-R. Recent advances in asymmetric synthesis of 2-substituted indoline derivatives. Chin. Chem. Lett. 2019;31:331–323. [Google Scholar]
- 9.Xu W, Gaviab DJ, Tang Y. Biosynthesis of fungal indole alkaloids. Nat. Prod. Rep. 2014;31:1474–1787. doi: 10.1039/C4NP00073K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roche SP, Porco JA., Jr. Dearomatization strategies in the synthesis of complex natural products. Angew. Chem. Int. Ed. 2011;50:4068–4093. doi: 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng C, You S-L. Catalytic asymmetric dearomatization by transition-metal catalysis: a method for transformations of aromatic compounds. Chem. 2016;1:830–857. doi: 10.1016/j.chempr.2016.11.005. [DOI] [Google Scholar]
- 12.Wertjes WC, Southgate EH, Sarlah D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 2018;47:7996–8017. doi: 10.1039/C8CS00389K. [DOI] [PubMed] [Google Scholar]
- 13.Fan L, Liu J, Bai L, Wang Y, Luan X. Rapid assembly of diversely functionalized spiroindenes by a three-component palladium-catalyzed C−H amination/phenol dearomatization domino reaction. Angew. Chem. Int. Ed. 2017;56:14257–14261. doi: 10.1002/anie.201708310. [DOI] [PubMed] [Google Scholar]
- 14.Bartoli G, Bencivenni G, Dalpozzo R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010;39:4449–4465. doi: 10.1039/b923063g. [DOI] [PubMed] [Google Scholar]
- 15.Okumura M, Sarlah D. Visible-light-induced dearomatizations. Eur. J. Org. Chem. 2020;2020:1259–1273. doi: 10.1002/ejoc.201901229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cannon JS, Overman LE. Is there no end to the total syntheses of strychnine? Lessons learned in strategy and tactics in total synthesis. Angew. Chem. Int. Ed. 2012;51:4288–4311. doi: 10.1002/anie.201107385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaraju K, Ma D. Oxidative coupling strategies for the synthesis of indole alkaloids. Chem. Soc. Rev. 2018;47:8018–8029. doi: 10.1039/C8CS00305J. [DOI] [PubMed] [Google Scholar]
- 18.Gentry EC, Rono LJ, Hale ME, Matsuura R, Knowles RR. Enantioselective synthesis of pyrroloindolines via noncovalent stabilization of indole radical cations and applications to the synthesis of alkaloid natural products. J. Am. Chem. Soc. 2018;140:3394–3402. doi: 10.1021/jacs.7b13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An J, Zou Y-Q, Yang Q-Q, Wang Q, Xiao W-J. Visible light-induced aerobic oxyamidation of indoles: a photocatalytic strategy for the preparation of tetrahydro-5h-indolo[2,3-b]quinolinols. Adv. Synth. Catal. 2013;355:1483–1489. doi: 10.1002/adsc.201300175. [DOI] [Google Scholar]
- 20.Wu K, Du Y, Wang T. Visible-light-mediated construction of pyrroloindolines via an amidyl radical cyclization/carbon radical addition cascade: rapid synthesis of (+/−)-flustramide B. Org. Lett. 2017;19:5669–5672. doi: 10.1021/acs.orglett.7b02837. [DOI] [PubMed] [Google Scholar]
- 21.Muhmel S, Alpers D, Hoffmann F, Brasholz M. Iridium(III) photocatalysis: a visible-light-induced dearomatizative tandem [4+2] cyclization to furnish benzindolizidines. Chem. –Eur. J. 2015;21:12308–12312. doi: 10.1002/chem.201502572. [DOI] [PubMed] [Google Scholar]
- 22.Liang K, et al. Enantioselective radical cyclization of tryptamines by visible light-excited nitroxides. J. Org. Chem. 2018;83:10948–10958. doi: 10.1021/acs.joc.8b01597. [DOI] [PubMed] [Google Scholar]
- 23.Zhu M, Zheng C, Zhang X, You S-L. Synthesis of cyclobutane-fused angular tetracyclic spiroindolines via visible-light-promoted intramolecular dearomatization of indole derivatives. J. Am. Chem. Soc. 2019;141:2636–2644. doi: 10.1021/jacs.8b12965. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M, Duan Y, Li W, Cheng Y, Zhu C. Visible-light-induced aerobic dearomative reaction of indole derivatives: access to heterocycle fused or spirocyclo indolones. Chem. Commun. 2016;52:4761–4763. doi: 10.1039/C6CC00818F. [DOI] [PubMed] [Google Scholar]
- 25.Wu K, Du Y, Wei Z, Wang T. Synthesis of functionalized pyrroloindolines via a visible-light-induced radical Cascade reaction: rapid synthesis of (+/−)-flustraminol B. Chem. Commun. 2018;54:7443–7446. doi: 10.1039/C8CC03575J. [DOI] [PubMed] [Google Scholar]
- 26.Alpers D, Gallhof M, Witt J, Hoffmann F, Brasholz M. A photoredox-induced stereoselective dearomative radical (4+2)-cyclization/1,4-addition cascade for the synthesis of highly functionalized hexahydro-1H-carbazoles. Angew. Chem., Int. Ed. 2017;56:1402–1406. doi: 10.1002/anie.201610974. [DOI] [PubMed] [Google Scholar]
- 27.Ma J, et al. Gadolinium photocatalysis: dearomative [2+2] cycloaddition/ring-expansion sequence with indoles. Angew. Chem. Int. Ed. 2020;59:9639–9645. doi: 10.1002/anie.202001200. [DOI] [PubMed] [Google Scholar]
- 28.Liu K, et al. Electrooxidation enables highly regioselectivedearomative annulation of indole and benzofuran derivatives. Nat. Commun. 2020;11:3. doi: 10.1038/s41467-019-13829-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Dou Y, Guillot R, Cyrille Kouklovsky C, Vincent G. Electrochemical dearomative 2,3-difunctionalization of indoles. J. Am. Chem. Soc. 2019;141:2832–2837. doi: 10.1021/jacs.8b13371. [DOI] [PubMed] [Google Scholar]
- 30.Morimoto N, Morioku K, Suzuki H, Takeuchi Y, Nishina Y. Lewis acid and fluoroalcohol mediated nucleophilic addition to the C2 position of indoles. Org. Lett. 2016;18:2020–2023. doi: 10.1021/acs.orglett.6b00629. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Shao Y, Liu Y. Nucleophilic addition of Grignard reagents to 3-acylindoles: Stereoselective synthesis of highly substituted indoline scaffolds. Org. Lett. 2012;14:3978–3981. doi: 10.1021/ol301750b. [DOI] [PubMed] [Google Scholar]
- 32.Nandi RK, et al. Triflic acid as an efficient Brønsted acid promoter for the umpolung of N-Ac indoles in hydroarylation reactions. Adv. Synth. Catal. 2018;360:161–172. doi: 10.1002/adsc.201701074. [DOI] [Google Scholar]
- 33.Hill JE, Lefebvre Q, Fraser LA, Clayden J. Polycyclic indoline derivatives by dearomatizing anionic cyclization of indole and tryptamine-derived ureas. Org. Lett. 2018;20:5770–5773. doi: 10.1021/acs.orglett.8b02468. [DOI] [PubMed] [Google Scholar]
- 34.Colonna, M. et al. A correlation between half-wave and ionization potentials for indoles and indolizines. J. Chem. Soc. Perkin Trans. II 1229-1231 (1986).
- 35.Giese B. Formation of C-C bonds by addition of free radicals to alkenes. Angew. Chem. Int. Ed. Engl. 1983;22:753–764. doi: 10.1002/anie.198307531. [DOI] [Google Scholar]
- 36.Jasperse CP, Curran DP, Fevig TL. Radical reactions in natural product synthesis. Chem. Rev. 1991;91:1237–1286. doi: 10.1021/cr00006a006. [DOI] [Google Scholar]
- 37.Protti S, Dondi D, Fagnoni M, Albini A. Assessing photochemistry as a green synthetic method. Carbon–carbon bond forming reactions. Green. Chem. 2009;11:239–249. doi: 10.1039/B810594D. [DOI] [Google Scholar]
- 38.Ryu I, Uehara S, Hirao H, Fukuyama T. Tin-free Giese reaction and the related radical arbonylation using alkyl iodides and cyanoborohydrides. Org. Lett. 2008;10:1005–1008. doi: 10.1021/ol7031043. [DOI] [PubMed] [Google Scholar]
- 39.Chu L, Ohta C, Zuo Z, MacMillan DWC. Carboxylic acids as a traceless activation group for conjugate additions: a three-step synthesis of (±)-pregabalin. J. Am. Chem. Soc. 2014;136:10886–10889. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qin T, et al. Nickel-catalyzed Barton decarboxylation and Giese reactions: a practical take on classic transforms. Angew. Chem. Int. Ed. 2017;56:260–265. doi: 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Millet A, Lefebvre Q, Rueping M. Visible-light photoredox-catalyzed Giese reaction: decarboxylative addition of amino acid derived α-amino radicals to electron-deficient olefins. Chem. Eur. J. 2016;22:13464–13468. doi: 10.1002/chem.201602257. [DOI] [PubMed] [Google Scholar]
- 42.Ravelli D, Montanaro S, Zema M, Fagnoni M, Angelo Albini AA. Tin-free, radical photocatalyzed addition to vinyl sulfones. Adv. Synth. Catal. 2011;533:3295–3300. doi: 10.1002/adsc.201100591. [DOI] [Google Scholar]
- 43.Liu H, et al. One-pot photomediated Giese reaction/Friedel–Crafts hydroxyalkylation/oxidative aromatization to access naphthalene derivatives from toluenes and enones. ACS Catal. 2018;8:6224–6229. doi: 10.1021/acscatal.8b00481. [DOI] [Google Scholar]
- 44.ElMarrouni A, Ritts CB, Balsells J. Silyl-mediated photoredox-catalyzed Giese reaction: Addition of non-activated alkyl bromides. Chem. Sci. 2018;9:6639–6646. doi: 10.1039/C8SC02253D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanegusuku ALG, Castanheiro T, Ayer SK, Roizen JL. Sulfamyl radicals direct photoredox-mediated Giese reactions at unactivated C(3)-H bonds. Org. Lett. 2019;21:6089–6095. doi: 10.1021/acs.orglett.9b02234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee GS, Hong SH. Formal Giese addition of C(sp3)–H nucleophiles enabled by visible light mediated Ni catalysis of triplet enone diradicals. Chem. Sci. 2018;9:5810–5815. doi: 10.1039/C8SC01827H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramirez NP, Jose C, Gonzalez-Gomez JC. Decarboxylative Giese-type reaction of carboxylic acids promoted by visible light: a sustainable and photoredox-neutral protocol. Eur. J. Org. Chem. 2017;2017:2154–2163. doi: 10.1002/ejoc.201601478. [DOI] [Google Scholar]
- 48.Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Visible light-mediated intermolecular C-H functionalization of electron-rich heterocycles with malonates. Org. Lett. 2010;12:3104–3107. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]
- 49.O’Brien CJ, et al. Photoredox cyanomethylation of indoles: catalyst modification and mechanism. J. Org. Chem. 2018;83:8926–8935. doi: 10.1021/acs.joc.8b01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tellis JC, et al. Single-electron transmetalation via photoredox/nickel dual catalysis: unlocking a new paradigm for sp3-sp2 cross-coupling. Acc. Chem. Res. 2016;49:1429–1439. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Romero NA, Nicewicz DA. Organic photoredox catalysis. Chem. Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 53.Xuan J, Zhang Z-G, Xiao W-J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed. 2015;54:15632–15641. doi: 10.1002/anie.201505731. [DOI] [PubMed] [Google Scholar]
- 54.Huang H, Jia K, Chen Y. Radical decarboxylative functionalizations enabled by dual photoredox catalysis. ACS Catal. 2016;6:4983–4988. doi: 10.1021/acscatal.6b01379. [DOI] [Google Scholar]
- 55.Jin Y, Fu H. Visible-light photoredox decarboxylative couplings. Asian J. Org. Chem. 2017;6:368–385. doi: 10.1002/ajoc.201600513. [DOI] [PubMed] [Google Scholar]
- 56.Zuo Z, MacMillan DWC. Decarboxylative arylation of α-amino acids via photoredox catalysis: a one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc. 2014;136:5257–5260. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang H, et al. Visible-light-promoted nickel- and organic-dye-cocatalyzed formylation reaction of aryl halides and triflates and vinyl bromides with diethoxyacetic acid as a formyl equivalent. Angew. Chem. Int. Ed. 2017;56:1500–1505. doi: 10.1002/anie.201610108. [DOI] [PubMed] [Google Scholar]
- 58.Meggers E. Asymmetric catalysis activated by visible light. Chem. Commun. 2015;51:3290–3301. doi: 10.1039/C4CC09268F. [DOI] [PubMed] [Google Scholar]
- 59.Brimioulle R, Lenhart D, Maturi MM, Bach T. Enantioselective catalysis of photochemical reactions. Angew. Chem. Int. Ed. 2015;54:3872–3890. doi: 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- 60.Ding W, et al. Photocatalytic aerobic oxidation/semipinacol rearrangement sequence: a concise route to the core of pseudoindoxyl alkaloids. Tetrahedron Lett. 2014;55:4648–4652. doi: 10.1016/j.tetlet.2014.06.102. [DOI] [Google Scholar]
- 61.Bu L, et al. Organocatalytic asymmetric cascade aerobic oxidation and semipinacol rearrangement reaction: a visible light-induced approach to access chiral 2,2-disubstituted indolin-3-ones. Chem. Asian J. 2018;13:2382–2387. doi: 10.1002/asia.201800446. [DOI] [PubMed] [Google Scholar]
- 62.Cheng YZ, Zhao QR, Zhang X, You SL. Asymmetric dearomatization of indole derivatives with N-hydroxycarbamates enabled by photoredox catalysis. Angew. Chem. Int. Ed. 2019;58:18069–18074. doi: 10.1002/anie.201911144. [DOI] [PubMed] [Google Scholar]
- 63.Diaz-Munoz, G., Miranda, I. L., Sartori, S. K., de Rezende, D. C. & Alves Nogueira Diaz, M. Use of chiral auxiliaries in the asymmetric synthesis of biologically active compounds: a review. Chirality31, 776–812 (2019). [DOI] [PubMed]
- 64.Garner P, et al. Development of an effective chiral auxiliary for hydroxyalkyl radicals. J. Org. Chem. 2002;67:6195–6209. doi: 10.1021/jo010880f. [DOI] [PubMed] [Google Scholar]
- 65.Sibi MP, Shankar Manyem S, Zimmerman J. Enantioselective radical processes. Chem. Rev. 2003;103:3263–3269. doi: 10.1021/cr020044l. [DOI] [PubMed] [Google Scholar]
- 66.Evans DA, Bartroli J, Shih TL. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. J. Am. Chem. Soc. 1981;103:2127–2129. doi: 10.1021/ja00398a058. [DOI] [Google Scholar]
- 67.Heravi MM, Vahideh Zadsirjan V. Recent advances in the application of the Oppolzer camphorsultam as a chiral auxiliary. Tetrahedron.: Asymmetry. 2014;25:1061–1090. doi: 10.1016/j.tetasy.2014.07.001. [DOI] [Google Scholar]
- 68.Guo T, Yuan BH, Liu WJ. Highly efficient asymmetric construction of novel indolines and tetrahydroquinoline derivatives via aza-Barbier/C-N coupling reaction. Org. Biomol. Chem. 2017;16:57–61. doi: 10.1039/C7OB02891A. [DOI] [PubMed] [Google Scholar]
- 69.Curran DP, Kim BH, Daugherty J, Heffner TA. The preparation of optically active Δ2-isoxazolines. A model for asymmetric induction in the non lewis acid catalyzed reactions of Oppolzer’s Chiral Sultam. Tetrahedron Lett. 1988;29:3555–3558. doi: 10.1016/0040-4039(88)85291-2. [DOI] [Google Scholar]
- 70.Curran DP, Shen W, Zhang J, Heffner TA. Asymmetric radical addition, cyclization, and annulation reactions with Oppolzer’s Camphor Sultam. J. Am. Chem. Soc. 1990;112:6738–6740. doi: 10.1021/ja00174a059. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
1H and 13C NMR spectra for products 3, 7, and 8: see Supplementary Figs. 3–192 in Suppmentary Data 1.
Chiral HPLC analysis for products 7, 8, and 11: see Supplementary Figs. 193–254 in Suppmentary Data 2.
The authors declare that all the other data including Supplementary Methods and compound structural characterization data, supporting the findings of this study are available within this paper, its Supplementary Information file. The X-ray crystallographic coordinates (Supplementary Methods: Supplementary Tables 1–3 and Supplementary Data 3: CIF file) for structures trans-3k reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC-1994584. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.