

Abstract
Aberrant activation of the hypoxia‐inducible transcription factor HIF‐1 and dysfunction of the tumor suppressor p53 have been reported to induce malignant phenotypes and therapy resistance of cancers. However, their mechanistic and functional relationship remains largely unknown. Here, we reveal a mechanism by which p53 deficiency triggers the activation of HIF‐1‐dependent hypoxia signaling and identify zinc finger and BTB domain‐containing protein 2 (ZBTB2) as an important mediator. ZBTB2 forms homodimers via its N‐terminus region and increases the transactivation activity of HIF‐1 only when functional p53 is absent. The ZBTB2 homodimer facilitates invasion, distant metastasis, and growth of p53‐deficient, but not p53‐proficient, cancers. The intratumoral expression levels of ZBTB2 are associated with poor prognosis in lung cancer patients. ZBTB2 N‐terminus‐mimetic polypeptides competitively inhibit ZBTB2 homodimerization and significantly suppress the ZBTB2–HIF‐1 axis, leading to antitumor effects. Our data reveal an important link between aberrant activation of hypoxia signaling and loss of a tumor suppressor and provide a rationale for targeting a key mediator, ZBTB2, to suppress cancer aggressiveness.
Keywords: cancer, HIF‐1, hypoxia, p53, ZBTB2
Subject Categories: Cancer, Metabolism, Signal Transduction
Aberrant activation of HIF‐1 and p53 deficiency induce malignancy and therapy‐resistance. ZBTB2 links activation of hypoxia signaling and tumor suppressor dysfunction, thereby promoting cancer aggressiveness, thus representing a target to treat p53‐deficient cancers.
Introduction
Aberrant activation of hypoxia‐responsive signaling is one of the typical features of malignant tumors and is associated with angiogenesis, therapy resistance, immune evasion, metabolic reprogramming, invasion, and metastasis of cancers (Hanahan & Weinberg, 2011; Koyasu et al, 2018). Hypoxia‐inducible factor 1 (HIF‐1), a heterodimeric transcription factor composed of HIF‐1α and HIF‐1β (also known as ARNT) subunits, is a master regulator of the biological response to hypoxia (Semenza, 2003, 2010; Koyasu et al, 2018). HIF‐1 activity is mainly regulated through two kinds of oxygen‐dependent hydroxylation of HIF‐1α. The first is at two proline residues, P402 and P564, by prolyl‐4‐hydroxylases (PHDs) for proteolysis via the von Hippel–Lindau protein (pVHL)‐dependent ubiquitin–proteasome pathway. The second is at the asparagine residue, N803, by factor inhibiting HIF‐1 (FIH‐1) for inactivation of the p300/CREB‐binding protein (CBP)‐dependent transactivation activity (Maxwell et al, 1999; Ohh et al, 2000; Ivan et al, 2001; Jaakkola et al, 2001; Mahon et al, 2001; Hirota & Semenza, 2005; Koyasu et al, 2018). These mechanisms of HIF‐1 inactivation are well‐established, and factors suppressing them have been associated with the poor prognosis of cancer patients (Goto et al, 2015; Zeng et al, 2015; Yeom et al, 2016; Kobayashi et al, 2017; Koyasu et al, 2018). However, factors that actively induce HIF‐1 activity remain poorly understood. Another important cancer hallmark is the inactivation of tumor suppressors, such as p53 (Baker et al, 1989; Hanahan & Weinberg, 2011). As p53 functions as a homotetrameric transcription factor, heterozygous dominant‐negative mutations that potentially abrogate the activities of wild‐type p53 are observed in at least 50% of malignant tumors. In addition, because p53 is degraded via the MDM2‐mediated ubiquitin–proteasome pathway, MDM2 overexpression is often seen in various cancers (Whibley et al, 2009; Levine, 2020). Although molecular mechanisms regulating the activity of each HIF‐1 and p53 have been characterized to some extent, as above, functional and mechanistic links between them remain largely unclear. Previously, we found that HIF‐1 was inactivated when p53 became functional in the presence of an inhibitor of MDM2, Nutlin‐3, in a cancer cell line with high levels of endogenous MDM2 expression (Fig EV1), which led us to hypothesize that there is an uncharacterized factor linking HIF‐1 and p53.
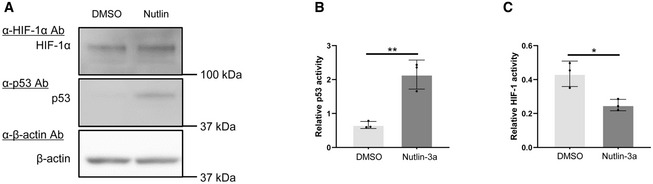
Figure EV1. Relationship between p53 and HIF‐1.
-
A–CU2OS cells transfected with p53‐Luc (B; #219083, Agilent Technologies) or p5HRE‐Luc (C) were treated with DMSO (for control) or 2.5 μM of Nutlin‐3a, cultured under < 0.1% oxygen conditions for 24 h, and subjected to western blotting using the indicated antibodies (A) and luciferase assays (B, C). pRL‐CMV was used as an internal control (B, C).
Data information: Mean ± s.d. The number of technical replicates in all of the experimental groups was 3 (B, C), and reproducibility of the results was confirmed at least three times by biologically independent experiments (A–C) = 3, *P < 0.05, **P < 0.01, Student's t‐test.
Source data are available online for this figure.
Results
ZBTB2 links p53 deficiency to HIF‐1 activation
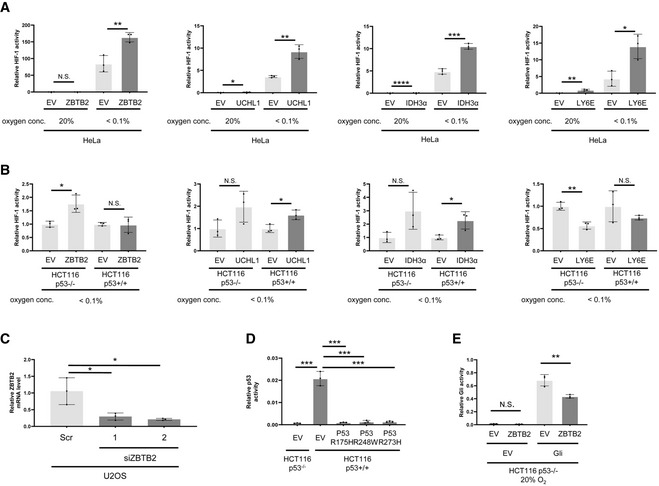
We established a genetic screening method to explore novel genes responsible for the activation of HIF‐1 (Goto et al, 2015; Zeng et al, 2015) and found 10 candidate genes (Goto et al, 2015; Zeng et al, 2015; Yeom et al, 2016). Luciferase assays using the 5HREp‐luc reporter gene, which expresses firefly luciferase under the control of the HIF‐1‐dependent promoter, 5HREp (Harada et al, 2005), demonstrated that overexpression of each of 4 of the 10 candidates significantly upregulated HIF‐1 activity in functional p53‐deficient HeLa cells (Fig EV2A; ZBTB2, UCHL1, IDH3α, and LY6E). Next, we employed the HCT116 cell line and its p53‐null derivative (herein HCT116 p53+/+ and p53−/−, respectively; Bunz et al, 1998) in the same luciferase reporter assay to identify factors that upregulate HIF‐1 only in the absence of functional p53 from the four genes (Fig EV2B). Through this two‐step screening, we eventually identified ZBTB2 as a key factor associating p53 deficiency with HIF‐1 activation; the forced expression of ZBTB2 upregulated 5HREp‐luc reporter activity only in the absence of functional p53 under hypoxic conditions (Figs 1A and EV2B). The upregulation was not observed under normoxic conditions because of the absence of HIF‐1α (Fig 1A). When we performed the same experiment using U2OS cells, in which the p53 function is suppressed by the aberrant overexpression of MDM2 (Florenes et al, 1994) and ZBTB2 expression is detectable, knockdown of ZBTB2 significantly suppressed 5HREp‐luc reporter activity (Figs 1B and EV2C). The positive impact of ZBTB2 on HIF‐1 activity in the p53‐null cells was abrogated by the overexpression of wild‐type p53, depending on its expression levels (Fig 1C). After overexpressing the most common dominant‐negative mutants of p53 in human cancers, R175H, R248W, or R273H (Harris, 1996; Fig EV2D), the positive impact of ZBTB2 on HIF‐1 was recovered in HCT116 p53+/+ cells (Fig 1D). There was no positive effect of ZBTB2 on another transcription factor, Gli, a key transcription factor of Hedgehog signaling, when evaluated with the Gli‐responsive luciferase reporter plasmid (Katagiri et al, 2018), indicating that ZBTB2 is not an activator of global transcription (Fig EV2E). Together, these results clearly show that ZBTB2 specifically activates HIF‐1 in the absence of functional p53.
Figure EV2. Relationships among HIF‐1, p53, and ZBTB2.
-
A, BThe indicated cells transfected with p5HRE‐Luc were transiently co‐transfected with pEF6/ZBTB2 (ZBTB2), pcDNA4/UCHL1 (UCHL1), pcDNA4/IDH3α (IDH3α), pcDNA4/Ly6E (Ly6E), or each of their corresponding control vectors (EV), pEF6/myc‐His B, pcDNA4/myc‐His A, pcDNA4/myc‐His A, or pcDNA4/myc‐His A, respectively. The cells were then cultured under the indicated oxygen conditions for 24 h and subjected to the luciferase assay.
-
CThe indicated cells were treated with scramble‐siRNA (Scr) or ZBTB2‐siRNA (siZBTB2), cultured under < 0.1% oxygen conditions for 24 h, and subjected to qRT–PCR for ZBTB2 mRNA levels.
-
DThe indicated cells transiently transfected with p53‐Luc (#219083, Agilent Technologies) were additionally transfected with pcDNA3/p53 R175H, R248W, R273H, or pcDNA3 (EV) as indicated, cultured under < 0.1% oxygen conditions for 24 h, and subjected to the luciferase assay.
-
EThe indicated cells transiently transfected with either pGL3/(3′Gli‐BS)4‐Luc or its empty vector, pGL3‐Luc (EV), were additionally transfected with either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (EV), cultured 20% oxygen conditions for 24 h, and subjected to the luciferase assay.
Data information: pRL‐CMV was used as an internal control in every luciferase assay (A, B, D, E). Mean ± s.d. The number of technical replicates in all of the experimental groups was 3, and the reproducibility of the results was confirmed at least three times by biologically independent experiments (A–E). N.S., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student's t‐test.
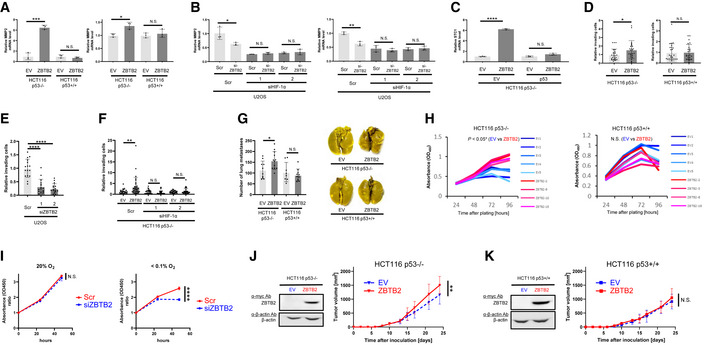
Figure 1. ZBTB2 links p53 deficiency to HIF‐1 activation.
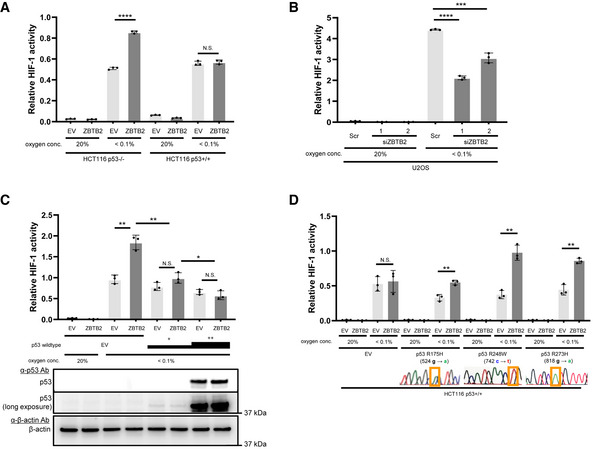
- The indicated cells co‐transfected with p5HRE‐Luc and either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (empty vector: EV) were cultured under the indicated oxygen conditions for 24 h for the luciferase assay.
- U2OS cells co‐transfected with p5HRE‐Luc and the indicated siRNA were cultured under the indicated oxygen conditions for 24 h for the luciferase assay.
- HCT116 p53−/− cells co‐transfected with p5HRE‐Luc, either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (EV), and either pcDNA3/p53 (at two concentrations: + and ++) or pcDNA3 (EV), were cultured under the indicated oxygen conditions for 24 h for the luciferase assay and western blotting using the indicated antibodies.
- HCT116 p53+/+ cells co‐transfected with p5HRE‐Luc, either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (EV), and pcDNA3/p53 R175H, R248W, R273H, or pcDNA3 (EV), were cultured under the indicated oxygen conditions for 24 h for the luciferase assay.
Data information: Mean ± s.d. The number of technical replicates in all of the experimental groups was 3, and the reproducibility of the results was confirmed at least three times by biologically independent experiments (A–D). N.S.: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student's t‐test (A–D).
Source data are available online for this figure.
ZBTB2 promotes invasion, distant metastasis, and growth of xenografted tumors when functional p53 is absent
Then, we investigated the influence of the ZBTB2‐mediated activation of HIF‐1 on tumor malignancy under hypoxia. Quantitative reverse‐transcription PCR (qRT–PCR) analyses demonstrated that the forced expression of ZBTB2 significantly induced the expression of invasion‐related HIF‐1‐downstream genes, such as matrix metalloproteinase (MMP) 2 and MMP9, only in the absence of functional p53 (Figs 2A and EV3A). Consistently, the knockdown of endogenous ZBTB2 in U2OS cells significantly reduced the expression of MMP2 and MMP9 under hypoxia (Fig 2B). The effect of ZBTB2 silencing was not observed in the absence of HIF‐1α, suggesting that ZBTB2 induced MMP expression in a HIF‐1‐dependent manner (Fig 2B). Similarly, ZBTB2 overexpression, in the presence of p53 deficiency, significantly upregulated the expression of stanniocalcin‐1 (STC1), which is a HIF‐1 target gene facilitating growth as well as invasion of cancer cells (Yeung et al, 2005; Chen et al, 2019; Fig 2C).
Figure 2. ZBTB2 promotes invasion, distant metastases, and growth of tumors when functional p53 is deficient in vivo .
-
A–FThe indicated cells transiently transfected with either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (EV) (A, C, D, F), either pcDNA3/p53 (p53) or pcDNA3 (EV) (C), or with the indicated siRNA (B, E, F) were cultured under < 0.1% O2 conditions for 24 h and subjected to qRT–PCR for the indicated mRNA (A–C) or to the transwell invasion assay (D‐F).
-
GThe number of metastatic colonies in lungs in the pulmonary metastasis model using the indicated cells stably transfected with ZBTB2 expression vector (ZBTB2) or its empty vector (EV) (left) and the representative images of lungs (right).
-
H, IThe indicated cells stably transfected with the ZBTB2 expression vector (ZBTB2) or its empty vector (EV) (H) or transiently transfected with siScr or siZBTB2 (I) were cultured under < 0.1% O2 (H) or the indicated oxygen conditions (I) for the indicated periods and subjected to the in vitro cell proliferation assay.
-
J, KGrowth of the indicated tumor xenografts with or without ZBTB2 overexpression was analyzed.
Data information: Mean ± s.d. The number of technical replicates was 3 (A–F, I), at least 8 (G), and 5 and 4 for EV and ZBTB2, respectively (H), and 7 (J, K), and reproducibility of the results was confirmed at least three times by biologically independent experiments (A–K). N.S., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student's t‐test (A–K).
Source data are available online for this figure.
Figure EV3. The ZBTB2–HIF‐1 axis promotes invasion and metastasis of p53‐deficient cancers.
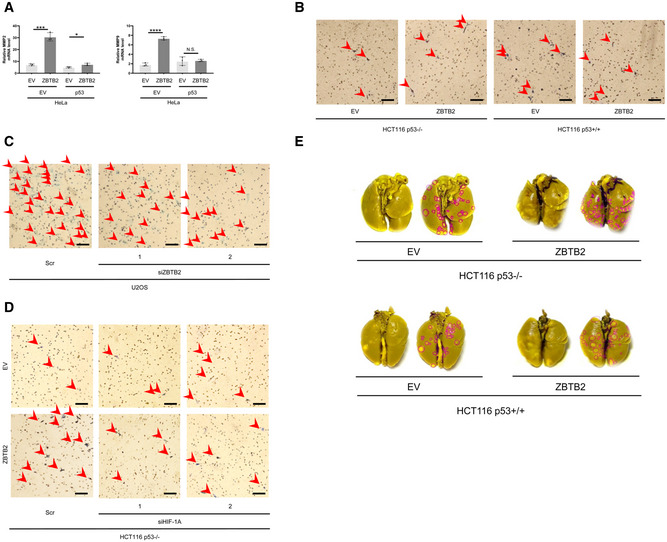
-
AHeLa cells, whose p53 activity is usually suppressed, were transiently transfected with either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (empty vector: EV) and with either pcDNA3/p53 (p53) or pcDNA (EV), cultured under < 0.1% oxygen conditions for 24 h, and subjected to qRT–PCR to quantify the mRNA levels of the indicated genes. Mean ± s.d. The number of technical replicates in all of the experimental groups was 3. N.S., not significant, *P < 0.05, ***P < 0.001, ****P < 0.0001, Student's t‐test.
- B–D
-
ERepresentative images of lungs with metastatic tumors in Fig 2G. Red ROIs represent metastatic colonies in each lung.
Data information: Reproducibility of the result was confirmed at least three times by biologically independent experiments (A–E).
Source data are available online for this figure.
To evaluate the importance of the ZBTB2‐HIF‐1 axis in tumor invasion, we carried out transwell invasion assays using matrigel‐coated inserts. The forced expression of ZBTB2 significantly promoted the invasion of p53−/− but not p53+/+ cells under hypoxia (Figs 2D and EV3B). On the other hand, silencing of endogenous ZBTB2 significantly decreased the number of invading U2OS cells (Figs 2E and EV3C). HIF‐1α silencing completely suppressed the ZBTB2‐mediated invasion of p53−/− cells (Figs 2F and EV3D). These results clearly show that ZBTB2 enhances the invasion of p53‐deficient cells in a HIF‐1‐dependent manner. Next, we analyzed the impact of ZBTB2 on metastatic tumor formation in lungs by transplanting cancer cell suspension through a tail vein. The murine model for pulmonary metastasis demonstrated that ZBTB2 facilitated distant metastasis of HCT116 p53−/− cells, but not that of HCT116 p53+/+ cells (Figs 2G and EV3E).
Subsequently, we analyzed the impact of ZBTB2 on cancer cell proliferation based on the data of Fig 2C. A colorimetric cell proliferation assay using stable transfectants with the ZBTB2 expression vector revealed that forced ZBTB2 expression significantly accelerated the growth of HCT116 p53−/− but not HCT116 p53+/+ cells under hypoxic conditions in vitro (Fig 2H). On the other hand, a loss‐of‐function study demonstrated that the proliferation of a functional p53‐deficient cancer cell line, HeLa, was significantly delayed by silencing ZBTB2 under hypoxia, where HIF‐1α expression was induced (Fig 2I). The positive impact of ZBTB2 on cell proliferation was reflected in the growth of xenografted tumors; the stable overexpression of ZBTB2 significantly promoted the growth of HCT116 p53−/− but not HCT116 p53+/+ xenografted tumors in immunodeficient mice (Fig 2J and K).
ZBTB2 expression is associated with poor prognoses in patients with p53‐deficient cancers
To validate our findings in human tumors, we next performed immunohistochemical analysis for ZBTB2, p53, and one of the most representative HIF‐1‐downstream genes, carbonic anhydrase 9 (CA9), in human lung cancers. Tumor samples were categorized into four groups, “ZBTB2 low” and “ZBTB2 high,” according to the expression levels of ZBTB2 in each wild‐type p53 and mutant p53 patient (Fig 3A). Fisher's exact test for the four groups demonstrated that the proportion of CA9‐expressing tumors was significantly larger in patients with ZBTB2‐high tumors compared with those with ZBTB2‐low tumors when p53 was mutated (Fig 3B). Next, we analyzed the correlation between the ZBTB2 expression levels and prognosis in patients stratified by the p53 status (Fig 3C). Kaplan–Meier analysis revealed that high ZBTB2 expression levels were associated with poor disease‐free survival of patients with mutant p53 tumors, but not with wild‐type p53 tumors (Fig 3C). We subsequently performed TCGA analysis regarding human lung adenocarcinoma. When we detected p53 mutation as low expression levels of p53 downstream genes, such as BAX, ZMAT3, and CEACAM1, overall survival of patients with both ZBTB2 high expression and p53 mutation in their tumor was poor compared with patients with other types of tumors (Fig 3D–F) (Miyashita & Reed, 1995; Israeli et al, 1997; Sappino et al, 2012).
Figure 3. ZBTB2 expression is associated with poor prognoses in patients with p53‐deficient cancers.
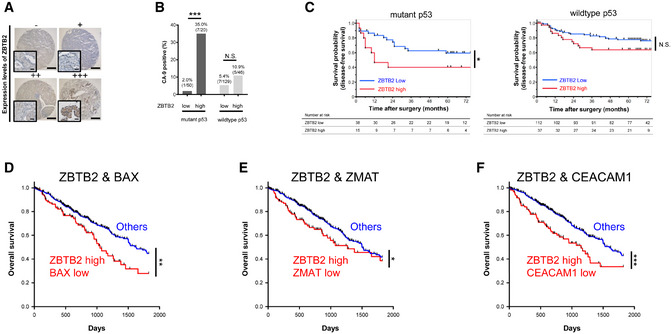
-
ATissue microarrays of human lung adenocarcinoma (243 samples) stained with anti‐ZBTB2 antibodies. High‐magnification images are shown on the left lower side of each image. Scale bar, 500 and 100 μm in the low‐ and high‐magnification images, respectively.
-
BFisher's exact test for the percentage of CA9‐positive tumors in the four indicated groups. Percentages and absolute numbers of each group are indicated in the graph.
-
CKaplan–Meier analysis of the disease‐free survival of the lung cancer patients stratified by the expression levels of ZBTB2 and p53 status.
-
D–FTCGA‐based Kaplan–Meier analysis of overall survival of lung adenocarcinoma patients stratified by the expression levels of ZBTB2 and those of BAX (D), ZMAT3 (E), and CEACAM1 (F).
Data information: N.S., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, Fisher's exact test (B) and log‐rank tests (C–F).
Source data are available online for this figure.
Homodimerization of ZBTB2 via its N‐terminus region increases the transactivation activity of HIF‐1α
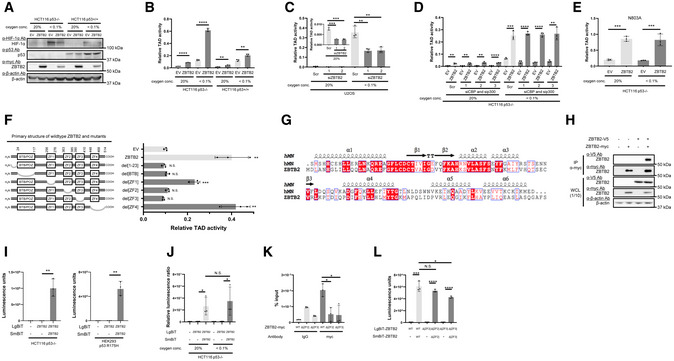
Next, we aimed to elucidate the molecular mechanisms underlying the positive impact of ZBTB2 on HIF‐1 activity. Immunoblotting demonstrated that the expression level of HIF‐1α protein was not increased by ZBTB2 overexpression in either p53‐proficient or p53‐deficient cells (Fig 4A). It was difficult to observe the influence of ZBTB2 on HIF‐1α protein levels under normoxic conditions because its basal expression was below detectable levels in our experimental setting (Fig 4A). We subsequently investigated the influence of ZBTB2 on the transactivation activity of HIF‐1α protein using the luciferase reporter system with a plasmid expressing the Gal4 DNA‐binding domain (Gal4 DBD) fused to the HIF‐1α transactivation domain (TAD: 531–826 a.a.; HIF‐1α TAD) (Mahon et al, 2001). A proline residue corresponding to P564 of HIF‐1α, which is involved in the oxygen‐dependent degradation of the Gal4 DBD‐HIF‐1α TAD fusion protein, was substituted for alanine (Gal4 DBD‐HIF‐1α TAD P564A) to evaluate the transactivation activity of TAD. The luciferase assay revealed that ZBTB2 overexpression markedly stimulated the transactivation activity of TAD P564A in HCT116 p53−/− but not p53+/+ cells (Figs 4B and EV4A). The transactivation activity was completely suppressed when HCT116p53−/− cells were introduced with a large amount of p53 expression vector (Fig EV4B). The knockdown of endogenous ZBTB2 expression significantly reduced the transactivation activity of HIF‐1α TAD P564A in U2OS cells (Fig 4C). Because the transactivation activity of HIF‐1α is known to be enhanced through the association with histone acetyltransferases, p300/CBP, when the N803 residue of HIF‐1α is unhydroxylated (Mahon et al, 2001; Lando et al, 2002), we tested whether p300/CBP is involved in the regulation of the ZBTB2 function. Silencing of p300/CBP was confirmed to have no effect on the ZBTB2‐mediated activation of transactivation activity of TAD P564A in p53−/− cells (Fig 4D). Moreover, the 5HREp‐luc reporter assay showed that forced expression of ZBTB2 enhanced HIF‐1 activity even in the presence of a constitutively active mutant of HIF‐1α, HIF‐1α 3A, which avoids suppression by not only PHDs and VHL but also FIH‐1 due to three point mutations, P402A, P564A, and N803A (Fig EV4C) (Kobayashi et al, 2017; Katagiri et al, 2018) SI‐3. Consistently, the ZBTB2‐dependent activation of TAD activity was still observed even when the asparagine residue, N803, was additionally substituted for alanine (Gal4 DBD‐HIF‐1α TAD P564A and N803A; Fig 4E). Taken together, these results revealed that ZBTB2 activity relies on the enhancement of transactivation activity, but not on that of stability, of HIF‐1α protein, and that it is independent of the known regulatory mechanism by p300/CBP and FIH‐1.
Figure 4. Homodimerization of ZBTB2 via its N‐terminus increases the transactivation activity of HIF‐1α.
-
AThe indicated cells transiently transfected with pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (EV) were cultured under the indicated oxygen conditions for 24 h and subjected to western blotting using the indicated antibodies.
-
B–FThe indicated cells transiently transfected with both pG5H1bLuc and either pcDNA6/Gal4 DBD‐HIF‐1α TAD P564A (B–D, F) or pcDNA6/Gal4 DBD‐HIF‐1α TAD P564A and N803A (E) were additionally co‐transfected with pEF6/myc‐His B (EV), pEF6/ZBTB2 (ZBTB2), pEF6/ZBTB2 del[1–23] (del[1–23]), pEF6/ZBTB2 del[BTB/POZ] (del[BTB]), pEF6/ZBTB2 del[ZF1] (del[ZF1]), pEF6/ZBTB2 del[ZF2] (del[ZF2]), pEF6/ZBTB2 del[ZF3] (del[ZF3]), or pEF6/ZBTB2 del[ZF4]‐ (del[ZF4]), as indicated (B, D–F) or with the indicated siRNA (C, D). The cells were then cultured under the indicated oxygen conditions for 24 h and subjected to the luciferase assay.
-
GProtein sequence alignments of the N‐terminus regions of hMN and ZBTB2.
-
HAfter transient transfection with the expression vectors for ZBTB2‐V5, ZBTB2‐myc, or their empty vector (−), ZBTB2‐myc protein was immunoprecipitated using the anti‐myc antibody and co‐precipitated ZBTB2‐V5 was detected using the anti‐V5 antibody (upper). One‐tenth of the whole‐cell lysate (WCL) was subjected to immunoblotting with the indicated antibodies (lower).
-
I, JThe indicated cells transiently transfected with or without either pcDNA4/ZBTB2‐LgBiT or pcDNA4/LgBiT and either pcDNA4/ZBTB2‐SmBiT or pcDNA4/SmBiT, as indicated, were cultured under < 0.1% oxygen (I) or the indicated oxygen conditions (J) and subjected to the split luciferase complementation assay.
-
KHeLa cells overexpressing wild‐type or the indicated mutant of ZBTB2 (ZF2 or ZF3 deletion) were cultured under < 0.1% oxygen conditions and subjected to immunoprecipitation with anti‐myc antibody. Coprecipitated DNA was subjected to the qRT–PCR experiment using primers against HRE regions of the STC1 promoter.
-
LThe same experiment as I was conducted using the indicated combination of pcDNA4/ZBTB2‐LgBiT (WT), pcDNA4/ZBTB2ΔZF2‐LgBiT (ΔZF2), or pcDNA4/ZBTB2ΔZF3‐LgBiT (ΔZF3), and pcDNA4/ZBTB2‐SmBiT (WT), pcDNA4/ZBTB2ΔZF2‐SmBiT (ΔZF2), or pcDNA4/ZBTB2ΔZF3‐SmBiT (ΔZF3).
Data information: Mean ± s.d. The number of technical replicates in all of the experimental groups was 3 (B–F, I–L), and reproducibility of the results was confirmed at least three times by biologically independent experiments (A–L). N.S., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student's t‐test (B–F, I–L).
Source data are available online for this figure.
Figure EV4. Mechanistic relationships among HIF‐1, p53, and ZBTB2.
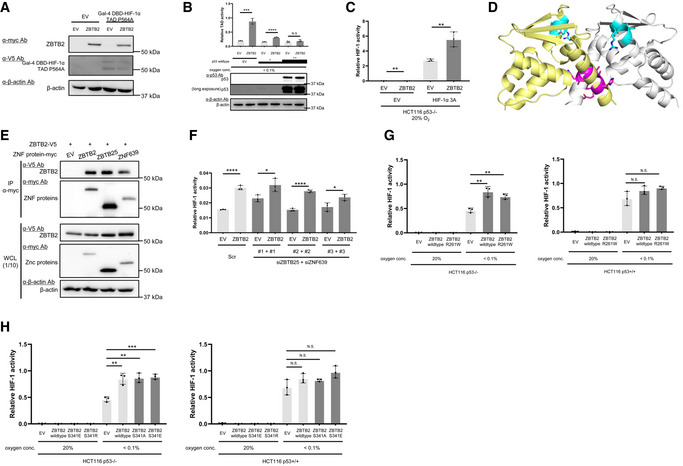
- Western blotting to confirm the expression of the indicated proteins under < 0.1% oxygen conditions in the luciferase assay in order to evaluate the HIF‐1α TAD activity in Fig 4B.
- Luciferase assay for HIF‐1α TAD activity was conducted using HCT116 p53−/− cells after transient transfection of low (+) or high (++) concentrations of the p53 expression vector or its empty vector (EV). Western blotting to confirm the forced expression of p53 was also conducted.
- Cells transiently co‐transfected with p5HRE‐Luc, either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (empty vector: EV), and either pcDNA4A/ HIF‐1α 3A (HIF‐1α 3A) or pcDNA4A (EV) were cultured under 20% oxygen conditions for 24 h and subjected to the luciferase assay.
- Crystal structure of the hMN homodimer (PDB: 2VPK). The 8–11 a.a. residues (EHLL) and 51–54 a.a. residues (AIYR) are highlighted in pink and blue, respectively.
- Twenty‐four hours after co‐transfection of the expression vector for ZBTB2‐V5 (pcDNA4/ZBTB2) with that for either of zinc finger (ZNF) protein, ZBTB2‐myc (pEF6/ZBTB2), ZBTB25‐myc (pEF6/ZBTB25), or ZNF639‐myc (pEF6/ZNF639), or with their empty vector (EV: pEF6/myc‐His B), the myc‐tagged proteins were immunoprecipitated using the anti‐myc‐tag antibody (middle) and co‐precipitated ZBTB2‐V5 was detected using anti‐V5 tag antibody (upper). One‐tenth of the whole‐cell lysate (WCL) was subjected to immunoblotting with the indicated antibodies (lower).
- HCT116 p53−/− cells transfected with either pEF6/ZBTB2 (ZBTB2) or pEF6/myc‐His B (EV) were treated with either scramble‐siRNA (Scr) or both ZBTB25‐siRNA (siZBTB25) and ZNF639‐siRNA (siZNF639) as indicated, cultured under < 0.1% oxygen conditions for 24 h, and subjected to the luciferase assay.
- The indicated cells were transiently co‐transfected with p5HRE‐Luc and pEF6/ZBTB2 (ZBTB2 wild‐type), pEF6/ZBTB2 R261W (ZBTB2 R261W), or pEF6/myc‐His B (empty vector: EV), cultured under the indicated oxygen conditions for 24 h, and subjected to the luciferase assay.
- The indicated cells were transiently co‐transfected with p5HRE‐Luc and pEF6/ZBTB2 (ZBTB2 wild‐type), pEF6/ZBTB2 S341E (ZBTB2 S341E), pEF6/ZBTB2 S341R (ZBTB2 S341R), or pEF6/myc‐His B (empty vector: EV), cultured under the indicated oxygen conditions for 24 h, and subjected to the luciferase assay.
Data information: pRL‐CMV was used as an internal control in every luciferase assay (B, C, F–H). Mean ± s.d. The number of technical replicates in all of the experimental groups was 3 (A–C, F–H), and reproducibility of the results was confirmed at least three times by biologically independent experiments (A–C, E–H). N.S., not significant, *P < 0.05, *P < 0.01, ***P < 0.001, ****P < 0.0001, Student's t‐test.
Source data are available online for this figure.
Next, we constructed a series of plasmids, each of which expressed one of the systematic deletion mutants of ZBTB2 (Fig 4F, left), to identify regions essential for the upregulation of HIF‐1α transactivation activity. Deletion mutants lacking the N‐terminus 23 a.a. region, BTB/POZ domain, zinc finger domain 2 (ZF2), or ZF3 failed to upregulate the transactivation activity of TAD P564A in p53−/− cells (Fig 4F). Peptide sequence alignment analysis revealed that the ZBTB2 N‐terminus 1–113 a.a. region including both the N‐terminus 1–23 a.a. region and the BTB/POZ domain showed 46% similarity with the N‐terminus region of human myoneurin (hMN) protein (Fig 4G). Although the crystal structure of the ZBTB2 protein is not available as yet, that of the N‐terminus fragment of hMN is available in the RCSB Protein Data Bank (https://www.rcsb.org/; PDB ID: 2VPK). It shows that the N‐terminus 1–21 a.a. fragments of hMN cross each other and may be essential to form a head‐to‐head homodimer (Fig EV4D), suggesting that ZBTB2 may also form a homodimer using the corresponding region. To investigate this, we performed co‐immunoprecipitation followed by immunoblotting (co‐IP‐IB) analysis and confirmed that V5‐tagged ZBTB2 proteins were co‐immunoprecipitated with myc epitope‐tagged ZBTB2 proteins (Fig 4H). To further test the ZBTB2 homodimer formation using another experimental setting, we performed a split luciferase complementation assay, in which each of a large and small fragment of split luciferase was fused with ZBTB2; therefore, luciferase fragments are close to each other and form complemented luciferase upon the interaction of two ZBTB2 proteins. The ZBTB2 homodimer formation was actually confirmed as luciferase bioluminescence (Fig 4I), and its efficiency was not influenced by the extracellular oxygen conditions (Fig 4J).
Next, we analyzed the reason why the deletion mutants lacking each ZF2 and ZF3 failed to upregulate the transactivation activity of the HIF‐1α protein. Because zinc‐finger domains were originally reported to function in DNA binding, we hypothesized that ZF2 and ZF3 function in the recruitment of ZTBT2 to the promoter region of genes whose expressions are under the control of the ZBTB2–HIF‐1 axis. Chromatin immunoprecipitation followed by the quantitative PCR (ChIP‐qPCR) experiment confirmed that wild‐type ZBTB2 was recruited to the promoter regions of a downstream gene, STC1, but the deletion mutants of each ZF2 and ZF3 were not (Fig 4K). On the other hand, the split luciferase complementation assay to evaluate ZBTB2 homodimer formation demonstrated that ZBTB2 protein lacking ZF2 or ZF3 still had the ability to form a homodimer (Fig 4L). Taken together, these results clearly indicate that both the homodimer formation mediated by its N‐terminus region and the recruitment to promoter regions of HIF‐1‐downstream genes mediated by ZF2 and ZF3 are essential for ZBTB2 to induce the expression of HIF‐1 downstream genes.
Recently, ChIP‐seq analysis for ZBTB2 revealed that ZBTB2 forms a heterodimer with each of the two other zinc finger proteins, ZBTB25 and zinc finger protein 639 (ZNF639) (Karemaker & Vermeulen, 2018). A co‐IP‐IB analysis showed that V5‐tagged ZBTB2 proteins were co‐immunoprecipitated with each myc epitope‐tagged ZBTB25 and ZNF639 (Fig EV4E). However, silencing either ZBTB25 or ZNF639 did not cancel the effect of ZBTB2 on HIF activity (Fig EV4F), suggesting that ZBTB25 and ZNF639 do not function with ZBTB2 for the activation of HIF‐1.
Since a previous report demonstrated that ZBTB2 harbored hot‐spot mutations at R261 (Gylfe et al, 2013), we generated a plasmid that expresses the ZBTB2 mutant with the most common point mutation, R261W. The 5HRE‐luc reporter assays demonstrated that the ZBTB2 R261W mutant stimulated HIF‐1 activity to the same extent as wild‐type ZBTB2 in HCT116 p53−/− cells, suggesting that R261W mutation does not influence ZBTB2 activity (Fig EV4G). We also generated plasmids that express mutant ZBTB2 with S341A or S341E because the serine residue located at 341 a.a. has been reported to be phosphorylated (UniProtKB, https://www.uniprot.org/uniprot/Q8N680). The reporter assays demonstrated that these mutants also exhibited the same extent of activity as wild‐type ZBTB2, suggesting that the phosphorylation has no impact on ZBTB2 activity (Fig EV4H).
Targeting ZBTB2 homodimer formation as a rational strategy to inhibit the growth of functional p53‐deficient cancers
We subsequently explored amino acid residues required for ZBTB2 homodimerization. The crystal structure of the N‐terminus fragment of hMN showed that 4 a.a. residues, EHLL, in the 8–11 a.a. region may be located at the interface of the homodimer by the 1–21 a.a. fragments (Fig EV4D). In addition, 4 other a.a residues, AIYR, in the 51–54 a.a. region may be located at another interface (Fig EV4D). To analyze their importance in ZBTB2 homodimerization, we constructed a homology model of the ZBTB2 structure based on the hMN crystal structure and performed molecular dynamics (MD) simulations. The resulting structure shows that the L8‐L11 (LILL) and M51‐V54 (MLFV) regions in ZBTB2 correspond to the E8‐L11 (EHLL) and A51‐R54 (AIYR) regions in hMN, respectively (Fig EV4D; Datasets EV1 and EV2; Fig 5A). Moreover, the L8‐L11 region was found to be located at the homodimer interface (Figs 5A and EV4D). To investigate whether these amino acids play a critical role in ZBTB2 homodimer formation and HIF‐1 activation, we next performed a luciferase assay using plasmids expressing deletion mutants of ZBTB2 lacking LILL (ZBTB2 del[8–11]) or MLFV (ZBTB2 del[51–54]). The luciferase assay revealed that these mutants failed to upregulate the transactivation activity of HIF‐1α (Fig 5B). Consistently, both the co‐IP‐IB experiment and split luciferase complementation assay demonstrated that the ZBTB2 mutant with substitution of LILL to AAAA (herein ZBTB2 4A) completely lost activity for homodimerization (Fig 5C and D). MD simulations indicate that the LILL to AAAA substitution leads to a significant increase in structural fluctuation, not only in the L3‐L23 region (where L8‐L11 is located) but also in A86‐L89 and P106‐L113 regions (Fig 5E and F). Intriguingly, the three regions are located on the same plane, whose normal line is the C 2 axis of the ZBTB2 homodimer (Fig 5A). It is likely that fluctuation in the L3‐L23 region can propagate via the adjacent A86‐L89 region toward the P106‐L113 region. This may explain why the ZBTB2 4A mutant failed to upregulate the transactivation activity of HIF‐1 (Fig 5G), and why stable overexpression of the ZBTB2 4A mutant did not enhance the growth of the HCT116 p53−/− tumor xenograft in immunodeficient mice (Fig 5H). Together, these results indicate that the L8‐L11 region is critical for ZBTB2 homodimer formation and the capacity to activate HIF‐1, and that inhibition of ZBTB2 dimer formation could be a potential strategy to suppress the growth of functional p53‐deficient cancers.
Figure 5. Targeting ZBTB2 homodimer formation as a rational strategy to inhibit the growth of functional p53‐deficient cancers.
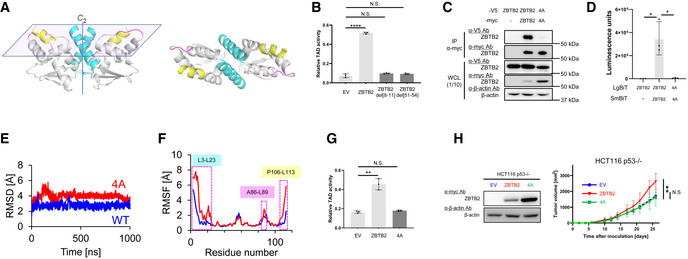
- A homology model of the N‐terminus of ZBTB2 based on the hMN crystal structure. L3‐23L (cyan), A86‐L89 (pink), and P106‐L113 (yellow). (Left) Side view. (Right) Top view.
- HCT116 p53−/− cells co‐transfected with pG5H1bLuc, pcDNA6/Gal4 DBD‐HIF‐1α TAD P564A, and pEF6/ZBTB2 (ZBTB2), pEF6/ZBTB2 del[8–11] (ZBTB2 del[8–11]), pEF6/ZBTB2 del[51–54] (ZBTB2 del[51–54]), or pEF6/myc‐His B (EV), as indicated, were cultured under < 0.1% oxygen conditions for 24 h and subjected to the luciferase assay.
- After transient transfection with or without the expression vectors for ZBTB2‐V5, ZBTB2 4A‐V5, ZBTB2‐myc, ZBTB2 4A‐myc, or their empty vector, as indicated, the myc‐fused proteins were immunoprecipitated using an anti‐myc antibody and co‐precipitated ZBTB2‐V5 or ZBTB2 4A‐V5 was detected (upper). One‐tenth of the whole‐cell lysate (WCL) was subjected to immunoblotting with the indicated antibodies (lower).
- HCT116 p53−/− (left) cells transiently transfected with or without pcDNA4/ZBTB2‐LgBiT (ZBTB2), pcDNA4/ZBTB2 4A‐LgBiT 4A (4A), or pcDNA4/LgBiT (−) and pcDNA4/ZBTB2‐SmBiT (ZBTB2), pcDNA4/ZBTB2 4A‐SmBiT (4A), or pcDNA4/SmBiT (−), as indicated, were cultured under < 0.1% oxygen and subjected to the split luciferase complementation assay.
- Root mean square deviation (RMSD) of the backbone Cα atoms with respect to the MD‐initial structure for the native (blue) and 4A mutant (red) ZBTB2 monomer proteins.
- RMSF of the backbone Cα atoms for the native (blue) and 4A mutant (red) ZBTB2 monomer proteins.
- The same kind of luciferase assay to quantify the transactivation activity as (B) was carried out using pEF6/ZBTB2 (ZBTB2), pEF6/ZBTB2 4A (4A), or pEF6/myc‐His B (EV).
- Growth of the indicated tumor xenografts was analyzed.
Data information: Mean ± s.d. The number of technical replicates was 3 (B, D, G) and 7 (H), and reproducibility of the results was confirmed at least three times by biologically independent experiments (B–D, G, H). N.S., not significant, *P < 0.05, **P < 0.01, ****P < 0.0001, Student's t‐test (B, D, G, H).
Source data are available online for this figure.
ZBTB2 N‐terminus‐mimetic polypeptides competitively inhibit ZBTB2 homodimerization and exhibit therapeutic effects toward p53‐deficient cancers
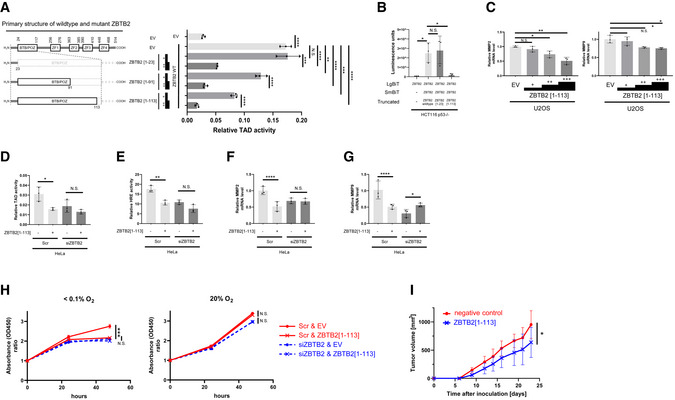
Next, we examined whether a molecule that mimics the conformation of the N‐terminus region of ZBTB2 works as an antagonist to inhibit ZBTB2 homodimer formation. For this proof‐of‐concept study, we employed polypeptides derived from the N‐terminus of ZBTB2 1–23, 1–91, and 1–113 a.a. (herein ZBTB2 [1–23], [1–91], and [1–113], respectively; Fig 6A, left) and found that every peptide significantly inhibited the transactivation activity of HIF‐1α in a concentration‐dependent manner (Fig 6A). The longer polypeptide, ZBTB2 [1–113], particularly showed the highest inhibitory effect on transactivation activity (Fig 6A) and on ZBTB2 homodimer formation evaluated by the split luciferase complementation assay (Fig 6B). Consequently, the inhibitory polypeptide ZBTB2 [1–113] suppressed the expression of both MMP2 and MMP9 in an expression‐level‐dependent manner (Fig 6C). The ZBTB2 [1–113] polypeptide significantly suppressed the transactivation activity of HIF‐1α protein (Fig 6D), HIF activity (Fig 6E), the expression of MMP2 (Fig 6F) and MMP9 (Fig 6G), and proliferation (Fig 6H) of cancer cells only when the expression of endogenous ZBTB2 was not silenced in vitro (Fig 6D–H). A tumor growth assay in vivo demonstrated that the ZBTB2 [1–113] polypeptide significantly delayed the growth of a xenografted tumor with functional p53‐deficient cells (Fig 6I). Through these results, we could validate the proof of concept that a molecule that conformationally mimics the N‐terminus of ZBTB2 antagonizes ZBTB2 homodimer formation and exhibits therapeutic effects against p53‐deficient cancers by inhibiting the ZBTB2–HIF‐1 axis.
Figure 6. ZBTB2 N‐terminus‐mimetic polypeptides competitively inhibit ZBTB2 homodimerization and exhibit therapeutic effects toward p53‐deficient cancers.
-
AThe same kind of luciferase assay as in Fig 5B and G was carried out by transfecting with either pEF6/ZBTB2 (ZBTB2 wt) or its empty vector (EV). To evaluate the inhibitory activity of the three truncated mutants of ZBTB2, ZBTB2 [1–23], ZBTB2 [1–91], and ZBTB2 [1–113], against wt ZBTB2, low (+) and high (++) concentrations of the corresponding expression vectors were additionally used to co‐transfect the cells.
-
BThe same kind of split luciferase complementation assay as in Fig 5D was carried out by co‐transfecting with pcDNA4/ZBTB2‐LgBiT (ZBTB2) and either pcDNA4/ZBTB2‐SmBiT (ZBTB2) or pcDNA4/SmBiT (−). To evaluate the inhibitory activity of the two truncated mutants of ZBTB2, ZBTB2 [1–23] and ZBTB2 [1–113], against wt ZBTB2, the corresponding expression vectors were additionally used to co‐transfect the cells.
-
CU2OS cells transiently transfected with three concentrations (+, ++, +++) of pEF6/ZBTB2 [1–113] or pEF6/myc‐His B (EV) were cultured under < 0.1% oxygen conditions and subjected to qRT–PCR for indicated mRNA.
-
D–GThe indicated cells were co‐transfected with either siScr or siZBTB2 and the expression vector for ZBTB2 or its empty vector (EV), cultured under < 0.1% oxygen conditions, and subjected to the luciferase assay for transactivation activity of HIF‐1α (D), that for HIF‐1 activity (E), and the q‐RT–PCR experiments for the indicated mRNA (F, G).
-
HHeLa cells transiently transfected with either siScr or siZBTB2 and with either ZBTB2[1–113] expression vector or its empty vector were cultured under 20% (left) or < 0.1% O2 (right) oxygen conditions for the indicated periods and subjected to the in vitro cell proliferation assay.
-
ITumor growth assay using the indicated cells with s.c. transplantation.
Data information: Mean ± s.d. The number of technical replicates was 3 (A–H) and 10 (I), and reproducibility of the results was confirmed at least three times by biologically independent experiments (A–I). N.S., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student's t‐test (A–I).
Discussion
The relationship between HIF‐1‐dependent hypoxia signaling and p53‐deficiency remains an important missing link in cancer research, and factors linking the two have drawn marked attention to better understand the nature of cancer and develop novel anticancer strategies. Here, we identified ZBTB2 and advanced our limited understanding. The present study demonstrates that ZBTB2 upregulates HIF‐1 transactivation activity and enhances the invasiveness, distant tumor metastasis, and growth of p53‐deficient cancers. Moreover, this study provides a rationale for targeting ZBTB2 homodimerization by a mimetic of the ZBTB2 N‐terminus to inhibit the ZBTB2–HIF‐1 axis for treating p53‐deficient cancers.
Whether p53 works as a negative regulator for HIF‐1 remains controversial; several lines of evidence suggest that p53 triggers HIF‐1α proteolysis in MDM2‐dependent and independent manners (Wang et al, 2019). Meanwhile, it is also true that p53 has been reported to cooperate with HIF‐1 (Amelio et al, 2018). The present study might provide a clue to solve this conundrum from the viewpoint of basal ZBTB2 expression levels. We found that p53 suppressed HIF‐1 activity without decreasing HIF‐1α protein levels only when we exogenously introduced the ZBTB2 expression vector into HeLa cells, in which endogenous ZBTB2 expression was moderate (Fig 1A). Likewise, in U2OS cells, in which endogenous ZBTB2 expression was extremely high, HIF‐1 activity was suppressed when p53 activity was induced by treatment with an MDM2 inhibitor, Nutlin‐3a (Fig EV1). These results suggest that p53 efficiently suppresses HIF‐1 activity when cells express high levels of ZBTB2, although it is critical to further test this hypothesis.
We identified critical amino acid residues for ZBTB2 homodimer formation. The polypeptides derived from the ZBTB2 N‐terminus significantly inhibited ZBTB2 dimerization and transactivation activity of HIF‐1; particularly, the ZBTB2 [1–113] polypeptide exhibited the highest inhibitory activity compared with shorter polypeptides, such as ZBTB2 [1–91], and [1–23] polypeptides. The difference in the potency probably resulted from the 1–113 polypeptide physically antagonizing the ZBTB2 homodimerization more efficiently than the others due to its bulkiness and strong affinity for the target region. However, these results were obtained only by overexpression of the polypeptides in cells, and it is not clear whether the ZBTB2 [1–113] polypeptide can exhibit the same potency when it is externally supplied to cells because the permeability of high‐molecular‐weight materials across the plasma membrane is low in general. To develop an antagonist/inhibitor for ZBTB2 homodimerization, it is critical to pay attention to the balance between its bulkiness/molecular weight and permeability across the plasma membrane.
Jeon et al (2009) reported that ZBTB2 suppressed p53 function through multiple mechanisms; however, we could not confirm this, at least in our experimental setting. However, there are possibilities that ZBTB2 might be responsible for multiple functions, and which of them functions might be dependent on the cell context. If ZBTB2 and p53 suppress each other's functions, ZBTB2 might be a modulator of p53 in a feedback loop.
While our findings reveal both the function and mechanism of action of ZBTB2 and provide a rationale for targeting ZBTB2 homodimerization by conducting a series of in vitro and in vivo proof‐of‐concept studies, they also raise further questions. First, detailed mechanisms underlying how ZBTB2 upregulates HIF‐1 transactivation activity, how the ZBTB2–HIF‐1 axis facilitates growth and distant metastasis of cancers, and how it is suppressed by p53 remain to be elucidated. Also, since it is true that ZBTB2 does not necessarily activate HIF‐1 in every p53−/−deficient cell, it is important to elucidate what causes the cell context dependency. Moreover, it is unclear whether there are ZBTB2 mutations in human tumors, and if there are, whether they are associated with the prognosis of cancer patients. Furthermore, although we confirmed the proof of concept to target the ZBTB2 homodimerization in vitro and in vivo, we did not examine whether a ZBTB2 N‐terminus‐mimetic polypeptide actually exhibits a therapeutic effect when it is externally provided. Approaching these areas will lead to a better understanding of the nature of cancer and will help establish a novel therapeutic strategy targeting the ZBTB2–HIF‐1 axis.
Materials and Methods
Cell culture and reagents
HeLa, U2OS, HEK293, and HEK293TN were purchased from the American Type Culture Collection. HCT116 p53+/+ and HCT116 p53−/− were gifts from Prof. Bert Vogelstein. Cells were maintained in 10% FBS‐Dulbecco's modified Eagle's medium and incubated at 37°C in a well‐humidified incubator with 5% CO2 and 95% air for normoxic incubation or in the RUSKINN INVIVO2 500 (Ruskinn) for hypoxic incubation at < 0.1%. Double‐stranded RNAs for the transient silencing of ZBTB2 (Stealth siRNAs, HSS126563 and HSS183975, Thermo Fisher Scientific), HIF‐1α (Stealth siRNAs, HSS104775 and HSS179231, Thermo Fisher Scientific), and HIF‐2α (Stealth siRNAs, HSS103261 and HSS176569, Thermo Fisher Scientific), and for the negative control (Stealth RNAi siRNA negative control, Thermo Fisher Scientific) were purchased from Life Technologies.
Plasmid constructs
To construct pEF6/ZBTB2 and pcDNA4/ZBTB2, cDNA encoding the human zbtb2 gene was amplified from cDNA of HeLa cells using the primers listed in Table EV1 and inserted between EcoRI and EcoRV sites of pEF6/myc‐His B and pcDNA4/V5 His B (Invitrogen), respectively. To construct pEF6/ZBTB2 R261W, S341A, S341E, del[1–23], del[BTB/POZ], del[ZF1], del[ZF2], del[ZF3], del[ZF4]‐, del[8–11], del[51–54], 4A, [1–23], [1–91], and [1–113], cDNAs encoding the corresponding mutants were introduced between EcoRI and EcoRV sites of pEF6/myc‐His B, as summarized in Table EV1. pcDNA6/Gal4 DBD‐HIF‐1α TAD P564A and pcDNA6/Gal4 DBD‐HIF‐1α TAD P564A and N803A were constructed as described previously (Zeng et al, 2015). pG5H1bLuc was described previously (Jiang et al, 1996; Mahon et al, 2001). To construct pcDNA3/p53, pcDNA3/p53 R175H, pcDNA3/p53 R248W, and pcDNA3/p53 R273H, coding sequences of the wild‐type human tp53 gene, R175H, R248W, and R273H mutants were inserted between BamHI and EcoRI recognition sites of pcDNA3, respectively. To construct pEF6/ZBTB25 and pEF6/ZNF639, cDNA encoding human zbtb25 and human znf639 genes was amplified from cDNA of HeLa cells using the primers listed in Table EV1 and inserted between the EcoRI and EcoRV sites and between SpeI and EcoRV of pEF6/myc‐His B, respectively. To construct pcDNA4/ZBTB2‐SmBiT and pcDNA4/ZBTB2 4A‐SmBiT, cDNA encoding the wild‐type human zbtb2 gene and human zbtb2 gene, in which LILL of the 8–11 a.a. region is substituted with 4 alanine residues, was inserted between EcoRI and EcoRV sites of pcDNA4B (Invitrogen), respectively, and the smaller sequence (small BiT; SmBiT, Promega) of the gene encoding the NanoLuc luciferase was additionally inserted between XhoI and XbaI sites of the same vector. Similarly, to construct pcDNA4/ZBTB2‐LgBiT and pcDNA4/ZBTB2 4A‐LgBiT, the larger sequence (large BiT; LgBiT Promega) of the gene encoding the NanoLuc luciferase was inserted between XhoI and XbaI sites of the vector.
Stable cell lines and colorimetric cell proliferation assay in vitro
pCDH/EV, pCDH/ZBTB2, and pCDH/ZBTB2 4A were applied for the pPACKH1 HIV Lentivector Packaging Kit (System Biosciences) to prepare the corresponding lentiviruses, according to the manufacturer's instructions. HCT116 p53−/− and HCT116 p53+/+ cells infected with each lentivirus were selected with puromycin to prepare HCT116p53−/−/EV, HCT116p53−/−/ZBTB2, HCT116p53−/−/ZBTB2 4A, HCT116p53+/+/EV, and HCT116p53+/+/ZBTB2 cells for the in vivo tumor growth assay. From the puromycin‐resistant bulk cells, HCT116 p53−/−/EV clone #1, 2, 3, 4, and 5 cell lines, HCT116 p53−/−/ZBTB2 clone #2, 9, 10, and 15 cell lines, HCT116 p53+/+/EV clone #1, 2, 3, 4, and 5 cell lines, and HCT116 p53+/+/ZBTB2 clone #2, 3, 8, and 16 cell lines were isolated for the in vitro colorimetric cell proliferation assay using Cell Count Reagent SF (Nacalai tesque).
Luciferase assay, western blotting, and qRT–PCR
Cells (1 × 104 cells per well in a collagen‐coated 24‐well plate for the luciferase assay and 2 × 105 cells per well in a collagen‐coated 6‐well plate for western blotting and qRT–PCR) were transfected with the indicated plasmids using the Polyfection Transfection Reagent (QIAGEN). Twenty‐four hours after the transfection, the cells were treated under normoxic (20% O2) or hypoxic (0.1% O2) conditions for the indicated periods. The cell lysate was harvested in 100 μl Passive Lysis Buffer (Promega) or 100 μl Cell Lytic Buffer (Sigma‐Aldrich) for the luciferase assay or western blotting, respectively; total RNA was harvested in Sepasol‐RNA I Super G (Nacalai tesque) according to the manufacturer's instructions. The luciferase assay was performed using the Dual Luciferase Assay kit according to the manufacturer's instructions (Promega). The plasmid pRL‐CMV (Promega) was used in every luciferase assay as an internal control to calculate relative luciferase activity. Western blotting was performed using anti‐HIF‐1α antibody (500‐fold dilution, BD Bioscience), anti‐ZBTB2 antibody (ab235604, 1,000‐fold dilution, Abcam), anti‐myc epitope tag antibody (1,000‐fold dilution, Cell Signaling), anti‐p53 antibody (1,000‐fold dilution, Santa Cruz Cat# sc‐126), and anti‐β‐actin antibody (500‐fold dilution, Santa Cruz Cat# sc‐69879) as primary antibodies, and anti‐mouse and anti‐rabbit IgG horseradish peroxidase linked whole antibodies (5,000‐fold dilution; GE Healthcare) as secondary antibodies. ECL Plus Western Blotting Detection System (GE Healthcare) was used for detection according to the manufacturer's instructions. Images of bands were acquired using the digital photo scanner Amersham Imager 600 (GE Healthcare). Images have been cropped for presentation. Uncropped images are presented in Source Data. For qRT–PCR, total RNA (1 μg) was then subjected to reverse transcription using RNA LA PCR kit (AMV) version 1.1 (Takara Bio), according to the manufacturer's instructions. The mRNA levels of the indicated genes were quantified using primers (Takara Primer Set ID HA173965 for MMP2 mRNA, HA129244 for MMP9 mRNA, HA067803 for human β‐actin mRNA, HA074624 for human HIF‐1α, HA159619 for human HIF‐2α, and HA193533 for ZBTB2 mRNA), as described previously. ACTB mRNA levels were used as an internal control.
Co‐immunoprecipitation followed by immunoblotting analysis
Twenty‐four hours after cells were transfected with the indicated plasmids, they were harvested in 250 μl Cell Lytic Buffer (Sigma‐Aldrich). The ZBTB2‐myc or other mutated ZBTB2‐myc protein was immunoprecipitated with anti‐myc antibody (#2276, Cell Signaling) using Immunoprecipitation Kit Dynabeads Protein G (Life Technologies) according to the manufacturer's instructions. Western blotting was performed as above using anti‐V5 antibody (1000‐fold dilution, Invitrogen).
ChIP‐qPCR assay
The experiment was conducted as described previously (Kobayashi et al, 2017). In detail, 48 h after cells (1.6 × 106 cells/dish in φ100‐mm dish) were transfected with the indicated plasmids, they were treated with 1% paraformaldehyde in PBS (−) for cross linkage between DNA and protein, added with glycine at the final conc. of 136 mM for quenching, washed with PBS (−), and then harvested with 1 ml of Lysis buffer (50 mM Tris–HCl pH 8.0, 1% SDS, and 10 mM EDTA). The lysates were sonicated for 24 min (30 s ON–30 s OFF; 24 cycles), and centrifuged at 16,100 g. The resultant supernatants were diluted with IP buffer (16.7 mM Tris–HCl pH 8.0, 1.2 mM EDTA, 167 mM NaCl, 1.1% Triton X‐100, and 0.01% SDS). IP was carried out with the anti‐myc antibody (Cell Signaling) or purified mouse IgG1 κ isotype control (BD Pharmingen) and using Dynabeads Protein G Immunoprecipitation Kit (Thermo Fisher). Precipitates were sequentially washed with low salt buffer, high salt buffer, LiCl wash buffer, and TE buffer. To purify DNA, samples were first incubated in 100‐times‐diluted TE buffer at 65°C overnight for the de‐crosslinking reaction, and DNA was extracted with the QIAquick PCR purification kit (Qiagen). The precipitated DNA levels were quantified using the qRT–PCR technique, as described above, using the following primers: Forward: 5′‐CTAGCAAAATAGGCTGTCCC‐3″, Reverse: 5′‐CAAGCCTTTCCTCCCCTCTC‐3′.
Split luciferase complementation assay
LgBiT‐ZBTB2 and SmBit‐ZBTB2 with or without the indicated mutations were co‐expressed in the indicated cells plated in 96‐well plates. Twenty‐four hours after they were transfected with the indicated plasmids, cells were treated under hypoxic (0.1% O2) conditions for 24 h and protein–protein interaction was evaluated as luciferase bioluminescence according to the manufacturer's instructions with a microplate reader (GloMax, Promega).
Transwell invasion assay
Cells were seeded onto the matrigel‐coated insert of the transwell chamber (5 × 104 cells per insert well; Corning BioCoat 24 well #354480), pre‐treated with serum‐reduced DMEM medium (1% FBS) for 24 h, and cultured under hypoxic (0.1% O2) conditions for 24 h in DMEM medium supplemented with 10% FBS in bottom wells, according to the manufacturer's instructions. Invaded cells were stained with Giemsa solution.
In vivo studies using mice
Animal studies using mice were performed at a temperature ranging 24°C ± 2°C and humidity ranging 50% ± 10% in a specific‐pathogen‐free facility of the Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University. For in vivo tumor growth assay, cancer cell suspensions (1 × 106 cells per mouse) were subcutaneously transplanted into the right hind leg of 8‐week‐old female specific‐pathogen‐free athymic nude mice (BALB/c nu/nu; Japan SLC Inc.). The tumor volume was calculated as (length × width2 × 1/2) mm3. All measurements were done in random order, with the investigator being blinded to the groups.
For pulmonary metastasis assay, cancer cell suspensions (4 × 106 cell/mouse) were transplanted into the tail veins of athymic nude mice (BALB/c nu/nu; Japan SLC Inc.), and lungs with metastatic tumors were surgically excised 36 days later and fixed with Bouin's solution. Number of metastatic colonies were counted externally.
Immunohistochemical analysis
Formalin‐fixed and paraffin‐embedded tissue microarrays of human lung adenocarcinoma were subjected to immunohistochemical staining with anti‐ZBTB2 rabbit polyclonal antibody (Thermo Fisher Scientific) and the anti‐p53 antibody through a standard technique, as described previously (Goto et al, 2015). ZBTB2 expression levels were evaluated by two independent investigators in a blind fashion without being informed of the therapeutic outcome. According to the intensity and extensity of ZBTB2 signals, samples were classified into four groups, −, +, ++, and +++, and categorized as low (−, +, and ++) or high (+++).
TCGA analysis
An open‐access RNA‐seq dataset of The Cancer Genome Atlas Lung Adenocarcinoma (TCGA‐LUAD) was obtained from GDC Data Portal (https://portal.gdc.cancer.gov). Samples (N = 506) were stratified by the expression levels (FPKM) of ZBTB2 or p53‐regulated genes (BAX, CEACAM1, and ZMAT3); the top 50% of samples with the highest expression were categorized as the “high” group, while the others were the “low” group. In the case of the combinatorial stratification, samples with “high” expression of ZBTB2 and “low” expression of BAX, CEACAM1, or ZMAT3 were categorized as “ZBTB2‐high/BAX‐low,” “ZBTB2‐high/CEACAM1‐low,” or “ZBTB2‐high/ZMAT3‐low” group, respectively, while all the rest were “others” group. Five‐year overall survival in each group was estimated by the Kaplan–Meier method and compared by log‐rank test.
Homology modeling and MD simulations
The ZBTB2 monomer structure was modeled with the SWISS‐MODEL web interface (Kiefer et al, 2009), using the hMN crystal structure (PDB code, 2VPK). The ZBTB2 4A mutant structure was modeled from the ZBTB2 structure. The ZBTB2 assembly was soaked in a water box. After structural optimization, the system was heated from 0.001 to 300 K for 5.0 ps with a time step of 0.05 fs. A production run was conducted over 1,000 ns with a time step of 2 fs and the SHAKE algorithm for hydrogen constraint. All simulations were performed using NAMD version 2.11 (Phillips et al, 2005) with the AMBER‐ff14SB force field (Maier et al, 2015). To calculate RMSD, the initial structure was used as a reference structure.
Ethics
All animal experiments were approved by the Animal Research Committee of Kyoto University. We performed all experiments according to the guidelines governing animal care in Japan. The study protocols using the samples of lung cancer patients were approved by the Ethics Committee of Kyoto University Hospital. Written informed consent was obtained from every patient. The study was performed in accordance with the Helsinki Declaration. Preparations of tissue microarrays mounted with human lung tumors were described previously (Fujimoto et al, 2013).
Statistical analysis
The reproducibility of results was confirmed by repeating the same experiments at least three times. The significance of differences was determined using Student's t‐test, Fisher's exact test, and the log‐rank test. A P‐value < 0.05 was considered significant.
Author contributions
Sho Koyasu: Conceptualization; data curation; formal analysis; funding acquisition; validation; investigation; visualization; writing—original draft; writing—review and editing. Shoichiro Horita: Data curation; software; validation; investigation; visualization; writing—original draft. Keisuke Saito: Data curation; software; investigation; visualization. Minoru Kobayashi: Funding acquisition; investigation. Hiroshi Ishikita: Data curation; software; investigation; writing—original draft. Christalle CT Chow: Investigation. Gouki Kambe: Investigation. Shigeto Nishikawa: Resources; data curation; investigation; visualization; writing—original draft. Toshi Menju: Resources; data curation; investigation; visualization. Akiyo Morinibu: Investigation. Yasushi Okochi: Investigation. Yoshiaki Tabuchi: Investigation. Yasuhito Onodera: Data curation; software; investigation; visualization; writing—original draft. Norihiko Takeda: Investigation; writing—review and editing. Hiroshi Date: Resources; investigation. Gregg L Semenza: Resources; methodology; writing—review and editing. Ester M Hammond: Resources; supervision; methodology; writing—review and editing. Hiroshi Harada: Conceptualization; resources; data curation; software; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing—original draft; project administration; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We would like to thank Prof. Hiroyuki Kouji, Oita University, for his critical suggestions and helpful discussion about the protein structure of ZBTB2, Prof. Masahiro Sonoshita, Hokkaido University, for technical advice, and Ms. Kumi Johchi and Ms. Sawako Hayami for technical assistance. This study was supported by the Research Project on Development of New Drugs (17933766), the Translational Research Grant Seeds A (A101), the AMED‐CREST (21gm1110010s0203), and the Promotion of Cancer Research and Therapeutic Evolution (P‐PROMOTE; 22ama221417h0001) to H.H. from the Japan Agency for Medical Research and development (AMED), by the Core‐to‐Core Program (JPJSCCA20200009) to H.H. and KAKENHI (18KK0241, 19K22595, 20H03621, 20H05033, 21KK0144, and 21K19445 to H.H., 21K07727 to M.K., and 16H06891, 17J07699, and 19K17164 to S.K.) from the Japan Society for the Promotion of Science (JSPS), and by the research grant programs of the Princess Takamatsu Cancer Research Fund, Uehara Memorial Foundation, Takeda Science Foundation, Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical Care, Kobayashi Foundation for Cancer Research, Yasuda Medical Foundation, and the Foundation for Promotion of Cancer Research to H.H. This study was conducted through the Joint Usage Programs of the Radiation Biology Center, Kyoto University, and of the Institute for Integrated Radiation and Nuclear Science, Kyoto University. S.K. was a JSPS postdoctoral research fellow, SPD.
EMBO Reports (2023) 24: e54042.
Data availability
This study includes no data deposited in external repositories.
References
- Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, Marini A, Antonov AA, Le Quesne J, Baena Acevedo JD et al (2018) p53 mutants cooperate with HIF‐1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci USA 115: E10869–E10878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y et al (1989) Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244: 217–221 [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Chen F, Zhang Z, Pu F (2019) Role of stanniocalcin‐1 in breast cancer. Oncol Lett 18: 3946–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florenes VA, Maelandsmo GM, Forus A, Andreassen A, Myklebost O, Fodstad O (1994) MDM2 gene amplification and transcript levels in human sarcomas: relationship to TP53 gene status. J Natl Cancer Inst 86: 1297–1302 [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Yoshizawa A, Sumiyoshi S, Sonobe M, Kobayashi M, Koyanagi I, Aini W, Tsuruyama T, Date H, Haga H (2013) Stromal plasma cells expressing immunoglobulin G4 subclass in non‐small cell lung cancer. Hum Pathol 44: 1569–1576 [DOI] [PubMed] [Google Scholar]
- Goto Y, Zeng L, Yeom CJ, Zhu Y, Morinibu A, Shinomiya K, Kobayashi M, Hirota K, Itasaka S, Yoshimura M et al (2015) UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF‐1alpha. Nat Commun 6: 6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylfe AE, Kondelin J, Turunen M, Ristolainen H, Katainen R, Pitkanen E, Kaasinen E, Rantanen V, Tanskanen T, Varjosalo M et al (2013) Identification of candidate oncogenes in human colorectal cancers with microsatellite instability. Gastroenterology 145: 540–543.e22 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Harada H, Kizaka‐Kondoh S, Hiraoka M (2005) Optical imaging of tumor hypoxia and evaluation of efficacy of a hypoxia‐targeting drug in living animals. Mol Imaging 4: 182–193 [DOI] [PubMed] [Google Scholar]
- Harris CC (1996) p53 tumor suppressor gene: from the basic research laboratory to the clinic‐‐an abridged historical perspective. Carcinogenesis 17: 1187–1198 [DOI] [PubMed] [Google Scholar]
- Hirota K, Semenza GL (2005) Regulation of hypoxia‐inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys Res Commun 338: 610–616 [DOI] [PubMed] [Google Scholar]
- Israeli D, Tessler E, Haupt Y, Elkeles A, Wilder S, Amson R, Telerman A, Oren M (1997) A novel p53‐inducible gene, PAG608, encodes a nuclear zinc finger protein whose overexpression promotes apoptosis. EMBO J 16: 4384–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFalpha targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468 [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ et al (2001) Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 292: 468–472 [DOI] [PubMed] [Google Scholar]
- Jeon BN, Choi WI, Yu MY, Yoon AR, Kim MH, Yun CO, Hur MW (2009) ZBTB2, a novel master regulator of the p53 pathway. J Biol Chem 284: 17935–17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Semenza GL, Bauer C, Marti HH (1996) Hypoxia‐inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 271: C1172–C1180 [DOI] [PubMed] [Google Scholar]
- Karemaker ID, Vermeulen M (2018) ZBTB2 reads unmethylated CpG Island promoters and regulates embryonic stem cell differentiation. EMBO Rep 19: e44993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T, Kobayashi M, Yoshimura M, Morinibu A, Itasaka S, Hiraoka M, Harada H (2018) HIF‐1 maintains a functional relationship between pancreatic cancer cells and stromal fibroblasts by upregulating expression and secretion of sonic hedgehog. Oncotarget 9: 10525–10535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T (2009) The SWISS‐MODEL repository and associated resources. Nucleic Acids Res 37: D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Morinibu A, Koyasu S, Goto Y, Hiraoka M, Harada H (2017) A circadian clock gene, PER2, activates HIF‐1 as an effector molecule for recruitment of HIF‐1alpha to promoter regions of its downstream genes. FEBS J 284: 3804–3816 [DOI] [PubMed] [Google Scholar]
- Koyasu S, Kobayashi M, Goto Y, Hiraoka M, Harada H (2018) Regulatory mechanisms of hypoxia‐inducible factor 1 activity: two decades of knowledge. Cancer Sci 109: 560–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML (2002) Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295: 858–861 [DOI] [PubMed] [Google Scholar]
- Levine AJ (2020) p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer 20: 471–480 [DOI] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL (2001) FIH‐1: a novel protein that interacts with HIF‐1alpha and VHL to mediate repression of HIF‐1 transcriptional activity. Genes Dev 15: 2675–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11: 3696–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 399: 271–275 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299 [DOI] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG (2000) Ubiquitination of hypoxia‐inducible factor requires direct binding to the beta‐domain of the von Hippel‐Lindau protein. Nat Cell Biol 2: 423–427 [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappino AP, Buser R, Seguin Q, Fernet M, Lesne L, Gumy‐Pause F, Reith W, Favaudon V, Mandriota SJ (2012) The CEACAM1 tumor suppressor is an ATM and p53‐regulated gene required for the induction of cellular senescence by DNA damage. Oncogenesis 1: e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2003) Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 3: 721–732 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2010) Defining the role of hypoxia‐inducible factor 1 in cancer biology and therapeutics. Oncogene 29: 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Guan D, Zhang XP, Liu F, Wang W (2019) Modeling the regulation of p53 activation by HIF‐1 upon hypoxia. FEBS Lett 593: 2596–2611 [DOI] [PubMed] [Google Scholar]
- Whibley C, Pharoah PD, Hollstein M (2009) p53 polymorphisms: cancer implications. Nat Rev Cancer 9: 95–107 [DOI] [PubMed] [Google Scholar]
- Yeom CJ, Zeng L, Goto Y, Morinibu A, Zhu Y, Shinomiya K, Kobayashi M, Itasaka S, Yoshimura M, Hur CG et al (2016) LY6E: a conductor of malignant tumor growth through modulation of the PTEN/PI3K/Akt/HIF‐1 axis. Oncotarget 7: 65837–65848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung HY, Lai KP, Chan HY, Mak NK, Wagner GF, Wong CK (2005) Hypoxia‐inducible factor‐1‐mediated activation of stanniocalcin‐1 in human cancer cells. Endocrinology 146: 4951–4960 [DOI] [PubMed] [Google Scholar]
- Zeng L, Morinibu A, Kobayashi M, Zhu Y, Wang X, Goto Y, Yeom CJ, Zhao T, Hirota K, Shinomiya K et al (2015) Aberrant IDH3alpha expression promotes malignant tumor growth by inducing HIF‐1‐mediated metabolic reprogramming and angiogenesis. Oncogene 34: 4758–4766 [DOI] [PubMed] [Google Scholar]