

Abstract
Sickle cell syndromes, including sickle cell disease (SCD) and sickle cell trait (SCT), are associated with multiple kidney abnormalities. Young patients with SCD have elevated effective renal plasma flow and glomerular filtration rates (GFR), which decrease to normal ranges in young adulthood and subnormal levels with advancing age. The pathophysiology of SCD-related nephropathy is multifactorial — oxidative stress, hyperfiltration and glomerular hypertension are all contributing factors. Albuminuria, which is an early clinical manifestation of glomerular damage, is common in individuals with SCD. Kidney function declines more rapidly in individuals with SCD than in those with sickle cell trait or in healthy individuals. Multiple genetic modifiers, including APOL1, HMOX1, HBA1 and HBA2 variants are also implicated in the development and progression of SCD-related nephropathy. Chronic kidney disease and rapid decline in estimated glomerular filtration rate are associated with increased mortality in adults with SCD. Renin–angiotensin–aldosterone system inhibitors are the standard of care treatment of albuminuria in SCD, despite a lack of controlled studies demonstrating their long-term efficacy. Multiple studies of novel therapeutic agents are ongoing, and patients with SCD and kidney failure should be evaluated for kidney transplantation. Given the high prevalence and severe consequences of kidney disease, additional studies are needed to elucidate the pathophysiology, natural history and treatment of SCD-related nephropathy.
Introduction
Sickle cell syndromes, including sickle cell trait (SCT) and sickle cell disease (SCD), are characterized by the presence of sickle haemoglobin (HbS), which results from a single base missense mutation in the HBB gene on chromosome 11 that leads to a glutamic acid-to-valine substitution in the haemoglobin A β subunit1. SCD is inherited as an autosomal co-dominant trait and affects individuals who are homozygous for the βS allele (HbSS) as well as individuals with various compound heterozygous variants, including sickle haemoglobin C disease (HbSC), sickle β-thalassemia (HbSβ) and other less common genotypes, in which the βS allele is co-inherited with other haemoglobin variants. The β0 thalassemic mutation describes a complete absence of production of β-globin, whereas β+ refers to decreased production of β-globin, resulting in the presence of variable amounts of normal haemoglobin (HbA). Patients with HbSS or HbSβ0 thalassemia typically have a more severe disease phenotype compared to those with HbSC or HbSβ+ thalassemia1.
Globally, sub-Saharan Africa and India have the highest burden of SCD. In the USA, SCD is considered an orphan disease and affects approximately 100,000 individuals2. In contrast to North America, where sickle cell anaemia is detected in around 2,600 births per year, an estimated 230,000 children were born with sickle cell anaemia in sub-Saharan Africa in 20103. Individuals who are heterozygous for the βS allele have SCT, which, in the United States, is estimated to occur in 6–9% of the African-American population4. Worldwide, the number of individuals with SCT exceeds 300 million, with prevalence rates that can exceed 25% in regions where malaria is endemic, such as Nigeria and tribal regions of India5.
The polymerization of HbS following deoxygenation is the primary event in the pathophysiology of SCD6. The presence of intracellular rigid HbS polymers affects the morphology of red blood cells (RBCs), which disrupts their ability to circulate through blood vessels and increases their susceptibility to haemolysis. Sickle RBCs also express high amounts of adhesion molecules, which enhances their binding to other cells. The clinical manifestations of SCD seem to be a consequence of haemolytic anaemia and vaso-occlusive events that lead to ischaemia-reperfusion injury7. Vaso-occlusion results from the adhesive interactions of leukocytes and sickle RBCs with endothelial cells and matrix proteins, which cause microvascular obstruction and subsequent tissue ischaemia7.
Individuals with SCD typically have functional and structural kidney abnormalities. The kidney is particularly sensitive to vaso-occlusion-induced hypoxia owing to its high oxygen consumption and a unique microenvironment that promotes the polymerization of deoxygenated HbS. As more patients with SCD survive to adulthood, the prevalence of end-organ damage, including chronic kidney disease (CKD), is increasing8. In addition to its morbidity burden, kidney disease is associated with increased mortality in patients with SCD.
In this Review, we examine the spectrum of kidney abnormalities in SCD and SCT, and their pathophysiology. We also discuss the epidemiology of kidney disease in individuals with SCD, including challenges in assessment and early detection of kidney dysfunction, and consider the management and treatment of patients with SCD and advanced kidney disease.
Kidney abnormalities in SCD
SCD is characterized by the presence of multiple alterations in kidney function (FIG. 1), including abnormalities in the proximal and distal tubules (for example, increased reabsorption of phosphate and β2-microglobulin, and increased secretion of uric acid and creatinine9), as well as changes in renal haemodynamics that promote hyperfiltration and glomerular damage.
Figure 1: Proposed mechanisms and biomarkers of sickle cell nephropathy.
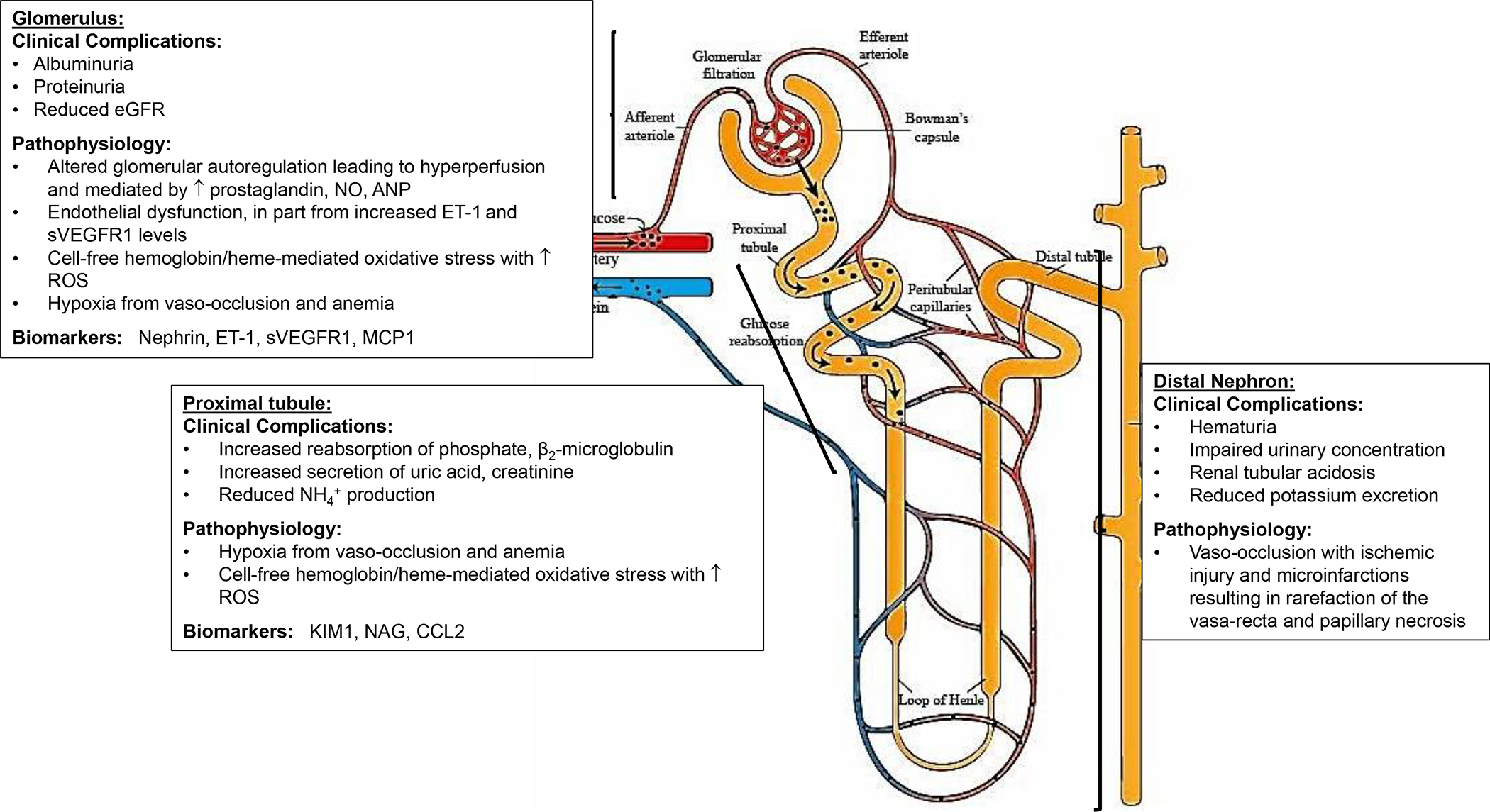
Sickle cell disease is complicated by multiple functional and structural abnormalities that occur along the nephron — clinical complications include hyposthenuria, haematuria, albuminuria and progressive estimated glomerular filtration rate (eGFR) decline. Medullary ischaemia drives localized prostaglandin release and results in marked vasodilation, increasing effective renal blood flow and GFR. In the glomerulus, the pathogenesis of albuminuria seems to be multifactorial — ischaemia-reperfusion injury, haemolysis, oxidative stress, hyperfiltration and glomerular hypertension have all been implicated. ANP, atrial natriuretic peptide; CCL2, CC-chemokine ligand 2; ET1: endothelin 1; KIM1: kidney injury molecule 1; NAG, N-acetyl-β-d-glucosaminidase; NH4+: ammonium ion; NO, nitric oxide; sVEGFR1, soluble vascular endothelial receptor 1; RBC, red blood cell; ROS, reactive oxygen species.
Impaired urinary concentration
Polymerization of deoxygenated HbS increases in the setting of relative hypoxia, acidosis and hyperosmolarity of the inner renal medulla10. Consequent microvascular occlusions in this kidney compartment result in ischaemic injury and microinfarction, and lead to the loss of juxtamedullary nephrons, which are essential for maximal urine concentration. Accordingly, impaired urine concentration is the most commonly recognized kidney abnormality in SCD9. The outer medulla is relatively spared from SCD-related damage and patients can thus concentrate urine under most circumstances, but not following substantial water deprivation or volume loss11. Following overnight water deprivation, urine osmolality in patients with HbSS was substantially lower (414 ± 10 mmol/kg) than that observed in healthy individuals (911 ± 39 mmol/kg)12. Low urine osmolality in SCD did not improve with vasopressin, which confirms that these individuals do not have central diabetes insipidus, in which vasopressin deficiency induces polyuria13. However, urinary endothelin 1 (ET1) levels in patients with SCD were significantly higher than in healthy controls prior to and following overnight water deprivation, and correlated negatively with urine osmolality14. ET1 has been suggested to induce nephrogenic diabetes insipidus in SCD through a combination of kidney fibrosis and the lack of improvement of low urine osmolality with vasopressin. Nocturnal enuresis is common in children and young adults with SCD and might be driven, in part, by an inability to concentrate urine (that is, hyposthenuria)15.
Urinary acidification
Urinary acidification in the distal nephron relies on the maintenance of a high tubule-to-lumen proton gradient, which is an energy-dependent process that is compromised by medullary ischaemia9. Defective urinary acidification in SCD manifests similarly to incomplete distal renal tubular acidosis16, typically resulting in overt metabolic acidosis after the stress of an additional acid load. Abnormal responses to acute acidification are more likely to occur in older patients with SCD, and in those with reduced glomerular filtration rate (GFR), low plasma bicarbonate, low urine ammonium ion excretion and low fasting urine osmolality16.
Potassium excretion
Impaired distal tubule secretion may result in defective potassium excretion in SCD. These defects occur despite intact aldosterone and renin responses16,17 although cases of hyporeninemic hypoaldosteronism have been reported18. Of note, serum potassium does not typically increase, even with potassium loading, until GFR is reduced17, possibly owing to potassium shifts from extracellular to intracellular compartments.
Supranormal renal haemodynamics
Compared with the general population, children and young adults with SCD have supranormal renal haemodynamics that are characterized by elevated effective renal plasma flow (ERPF) and GFR, even when accurately measured12. These patients have a decreased filtration fraction, which indicates that the increase in ERPF substantially exceeds the GFR increase. Notably, both GFR and ERPF decline towards normal levels during adolescence and then continue to decrease with further ageing19. The pathophysiological factors that alter GFR and ERPF seem to result from aberrant glomerular autoregulation that affects both afferent and efferent arteriolar tone. Medullary ischaemia drives localized prostaglandin release and subsequent vasodilation, which increases renal blood flow and GFR20. Use of NSAIDs, which block prostaglandin production, leads to a greater reduction in GFR in patients with SCD that in those without SCD12, which suggests that glomerular filtration is partly maintained by prostaglandin-mediated afferent arteriolar vasodilation.
Although no uniform definition exists, glomerular hyperfiltration in SCD is commonly defined as estimated glomerular filtration rate (eGFR) >130 ml/min/1.73 m2 in women and >140 ml/min/1.73 m2 in men21–23. In a multicentre study of infants (aged 9–24 months) with SCD, GFR ≥ 110 ml/min/1.73 m2 measured directly by plasma clearance of 99mTc-DTPA was observed in > 50% of patients aged between 9–12 months and higher GFR values were observed in older infants24. Other studies have demonstrated that increases in eGFR persist in older children and young adulthood25,26,27 with a subsequent decrease after 30 years of age21–23,28,27. In mice, glomerular hyperfiltration occurred earlier in males than in females, and preceded progressive GFR decline29. In young children, eGFR assessed by cystatin C was not significantly different between sexes, although, by age 13, male patients had significantly lower eGFR than female patients30. A study of adult patients with SCD reported that hyperfiltration was inversely associated with age, male sex, haemoglobin level, weight and angiotensin-converting enzyme (ACE) inhibitor and/or angiotensin receptor blocker (ARB) use, but positively associated with hydroxyurea use27. Normalization of hyperfiltration was less likely with higher baseline eGFR and more likely in male patients. The finding that glomerular permeability to albumin is significantly greater in male sickle mice and delayed in female mice, combined with the prevention of hyperfiltration in sickle mice following treatment with selective endothelin A receptor (ETA) antagonists, regardless of sex, suggests that ET1-dependent ultrastructural changes in the glomerular filtration barrier might partly explain sex differences in hyperfiltration31.
Impaired glomerular filtration
Albuminuria, which is an early marker of glomerular injury, is common in SCD. Up to 27% of children and young adults32–42, and 68% of older patients with SCD43–58 have albuminuria, although persistent albuminuria was not confirmed in most studies (Supplementary Table 1). Albuminuria is more prevalent in patients with HbSS or HbSβ0 than in those with other sickle cell genotypes53,54, and is associated with older age33–38,42,50,53,56, low haemoglobin concentrations35,36,38,39,42,46,50,51,54, high levels of haemolysis markers39,42,50, high levels of kidney injury molecule 1 (KIM1)50 and elevated echocardiography-measured tricuspid regurgitant jet velocity (TRV)52,57. TRV, which provides an estimate of pulmonary artery systolic pressure, is a surrogate measure of pulmonary vasculopathy.
Increasing evidence suggests that SCD-related glomerulopathy is progressive. Initial hyperfiltration progresses to albuminuria, gradual eGFR decline and eventual kidney failure. In a longitudinal study of patients with HbSS or HbSβ0 aged between 5 and 21 years, persistent albuminuria was observed at an earlier age and more frequently in those with hyperfiltration (determined by cystatin C)59. Moderate albuminuria might occur more often in adult patients with SCD and hyperfiltration than in those without hyperfiltration22. A prospective, multicentre, longitudinal study of children and adults with sickle cell anaemia confirmed that albuminuria increases with age50; urinary albumin-to-creatinine ratio (UACR) increased at a linear rate of 3.5 mg/g per year. Other prospective and retrospective cohort studies have reported an increase in CKD development over time in patients with SCD60,61. Baseline severe albuminuria and increasing systolic blood pressure are reportedly associated with CKD development and progression61.
Over 30% of adults with sickle cell anaemia experience rapid eGFR decline23,62 (that is, an annual eGFR loss >3 ml/min/1.73m2), which is higher than the prevalence reported in African American adults (11.5%)63; patients with SCD and proteinuria have a particularly high annual eGFR decline (−3.51 ml/min/1.73 m2) 64. Single-site studies have reported eGFR decline rates of 2.05 and 2.35 ml/min/1.73 m2 per year in patients with HbSS and HbSβ0 thalassemia, respectively23,64. These declines are steeper than those reported for African Americans in the general population (1.27 ± 1.97 ml/min/1.73 m2 per year)63 suggesting that GFR declines more rapidly in patients with sickle cell anaemia than in the general African American population; other studies have confirmed this accelerated rate of GFR decline65,66. Faster decline of measured GFR (mGFR) in SCD was also confirmed in a small longitudinal cohort of patients with sickle cell anaemia from Jamaica (mean follow up of 13.7 years)67. In a multicentre analysis, rapid eGFR decline of >3 ml/min/1.73 m2 per year was independently associated with age, male sex and prior stroke65, whereas eGFR loss >5·0 ml/min/1.73 m2 per year was independently associated with low haemoglobin and prior stroke65.
Haematuria
Haematuria is common in SCD but it is typically mild, painless and self-limited. Often, haematuria occurs owing to vascular occlusion in the renal medulla. In ~80% of cases, the bleeding originates in the left kidney68 and can be attributed to compression of the left renal vein by the aorta and superior mesenteric artery (termed the ‘nutcracker syndrome’)69. In addition, haematuria can result from microinfarction-induced papillary necrosis, which can be diagnosed radiologically.
Management of haematuria is usually conservative and includes bed rest, maintenance of high urinary flow, urine alkalinization and RBC transfusion, if needed70. Vasopressin therapy has had variable success in patients who do not respond to conservative management71. The use of low-dose antifibrinolytics to treat haematuria is reportedly safe and effective72, but should be used cautiously owing to potential for urinary obstructions caused by blood clots. Embolization of involved kidney blood vessels and balloon tamponade have been used for refractory bleeding73. Unilateral nephrectomy has also been performed in extremes cases74 but is not recommended because bleeding might recur in the remaining kidney.
Acute kidney injury
The Kidney Disease Improving Global Outcomes criteria that define acute kidney injury (AKI) include an increase in serum creatinine by ≥0.3 mg/dl within 48 hours or 50% increase in serum creatinine within 7 days from baseline75. The risk of AKI in SCD is increased compared with that of individuals with HbAA and is even more common during hospitalizations for acute chest syndrome76–78 or admission to the intensive care unit (ICU)79. Decline in GFR following AKI is significantly faster in patients with SCD compared with healthy controls80, and a history of AKI predicts an increased long-term risk of CKD81. In adult patients with SCD, hospital admissions complicated by AKI are associated with longer length of hospital stay and an increased risk of inpatient mortality78,81. Potential risk factors for developing AKI in patients with SCD include older age, higher white blood cell count, lower haemoglobin concentration, lower systolic blood pressure at the time of admission and the use of ketorolac or vancomycin during hospitalization78,82.
Mechanisms underlying sickle cell nephropathy
Although the pathophysiology of SCD-related nephropathy is incompletely understood, vaso-occlusion with subsequent ischaemia-reperfusion injury, haemolysis, oxidative stress and hyperfiltration seem to have important roles (FIG. 1). Glomerular hypertension might also have a pathophysiological role in SCD-related nephropathy. ACE inhibitors attenuate glomerular hypertension following partial ablative nephrectomy in non-sickle rodents and, in patients with SCD, these drugs lead to a rapid and reversible decrease in proteinuria55,83.
Haemolysis and oxidative stress
In the kidneys of transgenic sickle mice (that is, mice that express human HbS), lipid peroxidation, which is an indicator of oxidative stress, is enhanced compared with wild-type controls84,85. An increase in oxidative stress might be partly mediated by exposure of the kidney to cell-free haemoglobin and haem that are released into the vasculature following haemolysis. Cell-free haemoglobin filters freely through the glomerulus and haemoglobinuria is observed in 15–42% of patients with SCD at steady-state48,86,87. Magnetic resonance imaging showed that kidney iron deposition correlated positively with the degree of haemolysis but not with the transfusion burden, which was assessed by quantifying iron concentration in the liver, suggesting a role for chronic filtration of cell-free hemoglobin and heme 88,89. Data from independent SCD cohorts showed that haemoglobinuria is positively associated with CKD stage and risk of CKD progression86. Haem oxygenase 1 (HMOX1) is an inducible, rate-limiting enzyme that degrades haem into biliverdin, carbon monoxide and iron, and protects against chronic exposure to cell-free hemoglobin 86. HMOX1 mRNA and protein levels are higher in the kidneys of transgenic sickle mice than in those of control mice 90. Increased HMOX1 staining was detected in the renal tubules of a patient with SCD, whereas kidneys from healthy controls did not express HMOX185. After weekly administration of intravenous cell-free haemoglobin, HMOX1-knockout mice had significantly greater tubulo-interstitial inflammation and fibrosis than wild-type mice91. These findings suggest that HMOX1 might protect the kidney from chronic exposure to cell-free haemoglobin and its anti-inflammatory actions might also be cytoprotective.
Paediatric patients with SCD and AKI have a larger decrease in haemoglobin from baseline levels than patients without AKI77,78. Haemopexin and α−1-microglobulin (A1M) are major scavengers of extracellular haem from the circulation. Haemopexin deficiency in SCD is associated with a compensatory increase in A1M, resulting in a substantially higher A1M-to-haemopexin ratio in individuals with SCD compared than in healthy controls92. A1M-to-haemopexin ratio is associated with markers of hemolysis and AKI in both patients and mice with SCD. Haemopexin deficiency promotes AKI in sickle mice under haemolytic stress, but AKI was prevented with infusions of purified haemopexin prior to the induction of haemolytic stress, highlighting a potential role for haemopexin deficiency as a risk factor for AKI.
Angiotensin receptor signalling seems to be required for urine concentration but promotes glomerular pathologic conditions in SCD. Reactive oxygen species (ROS) increase the conversion of oxidized angiotensinogen to angiotensin II (ATII), and ROS levels are elevated in patients with SCD compared with healthy individuals93,94. Blockade of ATII signalling by ACE inhibitors or ARBs in transgenic sickle mice reduces profibrotic transforming growth factor β1 (TGFβ1)–SMAD 2/3 signalling in the glomerulus and ameliorates albuminuria, glomerulosclerosis and mesangial cell proliferation94. ATII acts via two major receptor subtypes — type 1 (AT1R) and type 2 (AT2R). AT1R is expressed throughout the kidney, including in the vasculature, glomeruli and tubules, and participates in the regulation of renal haemodynamics, sodium reabsorption and glomerular filtration95. By contrast, AT2R is primarily expressed in the vasculature and proximal tubule. AT2R signalling seems to oppose AT1R signalling and promotes vasodilation via nitric oxide and bradykinin, promotes natriuresis and has anti-inflammatory effects96. In transgenic sickle mice, genetic deficiency in AT1R, but not AT2R, prevents the development of albuminuria and focal segmental glomerulosclerosis lesions, and the increase in TGFβ1-SMAD2/3 signalling94. However, the reduced ability to concentrate urine observed in sickle mice worsened following AT1R inhibition with ACE inhibitors (and, to a lesser extent, with ARBs). These data suggest that increased AT1R signalling promotes glomerular pathology, although AT signalling, via both AT1R and AT2R, is also required to maintain the ability to concentrate urine in SCD.
Endothelial dysfunction
Albuminuria in SCD is associated with endothelial dysfunction, as demonstrated by impairment in flow-mediated dilation of the brachial artery97. Soluble vascular endothelial growth factor receptor 1 (sVEGFR1; also known as sFLT1),98 contributes to the pathophysiology of preeclampsia, which is characterized by hypertension, proteinuria and endothelial dysfunction99, and sVEGFR1 levels are elevated in patients with SCD100,101, particularly in those with severe albuminuria52. Serum sVEGFR1 levels also correlate directly with markers of haemolysis102. sVEGFR1 acts as an VEGF antagonist and inhibits Akt phosphorylation, which prevents the activation of endothelial nitric oxide (NO) synthase and reduces the generation of NO103. This sVEGFR1-induced decrease in NO bioavailability leads to endothelial dysfunction. The positive association between soluble vascular cell adhesion molecule 1 (sVCAM1) with sVEGFR1 levels, combined with the positive association between sVCAM1 and albuminuria suggest that sVEGFR1 contributes to albuminuria in SCD by promoting endothelial dysfunction52.
ET1 is a potent vasoconstrictor secreted by endothelial cells in response to injurious stimuli, including shear stress and hypoxaemia, and exposure to haemin, inflammatory cytokines, angiotensin II or thrombin104. Binding of ET1 to ETA leads to vasoconstriction, inflammation, mitogenesis, and nociception105. Plasma and urinary levels of ET1 correlate positively with albuminuria in SCD14,97. In transgenic sickle mice, selective ETA antagonism reduced glomerular ROS production and mRNA expression of oxidative stress markers, and significantly decreased protein and nephrin excretion in urine104. Long-term treatment of sickle mice with ambrisentan, which is a selective ETA antagonist, preserved GFR to levels observed in HbAA-expressing control mice, prevented proteinuria, albuminuria and nephrinuria, and reduced the urinary excretion of KIM1 and N-acetyl-β-d-glucosaminidase (NAG)105. Furthermore, ambrisentan prevented proximal tubular brush border loss, interstitial fibrosis and immune cell infiltration, and diminished tubular iron deposition.
Genetic modifiers of kidney disease risk
Several genetic modifiers that affect the severity of haemolysis, the risk of kidney disease in people of African descent, cell-free haemoglobin processing and inflammatory chemokine clearance have been implicated in SCD-related nephropathy (TABLE 1). The α-globin genes HBA1 and HBA2 are typically duplicated on chromosome 16, which creates four α-globin genes (αα/αα); the α−3.7k and α−4.2k deletions result in decreased production of α-globin chains. Alpha thalassemia caused by inheritance of single or double α-deletions is observed in approximately one-third of patients with SCD and leads to lower intracellular HbS concentration and less haemolytic anaemia than in patients with SCD and intact α-globin genes.106. Co-inheriting α-thalassemia consistently protects against albuminuria in sickle cell anemia40,44,107,108, but this effect is attenuated in patients with HbSC44.
Table 1:
Replicated gene variants implicated in sickle cell nephropathy
| Gene | Gene product | Variants | Effects associated with variant in patients with SCD | Refs |
|---|---|---|---|---|
| HBA1 | Haemoglobin subunit α1 | α-3.7K deletion α-4.2K deletion |
Reduction in albuminuria, higher eGFR and lower risk of CKD progression | 40,44,107,108,117 |
| APOL1 | Apolipoprotein L1, which is a major component of the trypanosome lytic factor complex | G1 (S342G or S342G and I384M substitution) G2 (N388 and Y389 deletion) |
Homozygous or compound heterozygous inheritance of APOL1 G1 & G2 variants: associated with increased risk of urine dipstick-defined proteinuria, higher albuminuria, lower eGFR, higher CKD stage, faster CKD progression and increased risk of kidney failure | 112,113,115–117,226 |
| HMOX1 | Haem oxygenase 1, which is a rate-limiting inducible enzyme that metabolizes haem to biliverdin, carbon monoxide and iron | GT-repeat in promoter region or rs743811 | Long GT-tandem repeats (> 25): lower baseline eGFR and higher AKI risk rs743811: lower eGFR, greater albuminuria, higher CKD stage and increased risk of kidney failure |
82,112,116 |
| ACKR1 | Atypical chemokine receptor 1 (also known as the Duffy antigen receptor 1, which is a receptor for Plasmodium vivax and serves as a chemokine-scavenging receptor | rs2814778 | Higher risk of urine dipstick-defined proteinuria and more severe albuminuria | 114,120 |
AKI, acute kidney injury; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; SCD, sickle cell disease.
Homozygous or compound heterozygous inheritance of APOL1 G1 and G2 risk variants occurs in 10–15% of African Americans and is an important risk factor for CKD in African Americans without diabetes109–111. Trypanosomes are susceptible to apolipoprotein 1 (APOL1) but Trypanosoma brucei rhodesiense neutralizes APOL1 via a serum resistance-associated protein (SRA) and is thus resistant to its toxicity. The APOL1 G1 and G2 variants are not susceptible to the effects of SRA and can therefore protect against T. b. rhodesiense 110. Co-inheritance of APOL1 risk variants is observed in 7–16% of patients with SCD40,112–116, and is associated with increased risk of haemoglobinuria, albuminuria and low eGFR. Patients with SCD and APOL1 risk variants have a 7-fold higher risk of CKD progression117 and a 7–30-fold greater risk of kidney failure112,113.
The HMOX1 rs743811 variant is also associated with lower eGFR, higher UACR, worsening CKD stage and kidney failure in SCD, although the effect of this variant on HMOX1 expression or function is unknown112. Another variant is characterized by the presence of long GT-tandem repeats in the HMOX1 promoter (rs3074372) that lead to reduced HMOX1 expression in cultured cells after H2O2 exposure118. However, data on the association of long GT-tandem repeats (>25) in HMOX1 with compromised kidney function in patients with SCD are inconsistent82,112,40,116.
The atypical chemokine receptor 1 (ACKR1; also known as the Duffy antigen receptor) is expressed on RBCs and provides a chemokine sink that reduces systemic chemokine levels and prevents white blood cell activation119.The ACKR1 rs2814778 polymorphism is located in the promoter region of the gene and decreases GATA binding, which is necessary for the transcription and expression of ACKR1 in RBCs120. In patients with HbSS from the United States, the absence of ACKR1 expression on RBCs was associated with lower white blood cell counts, and increased risk of proteinuria and albuminuria114,120. However, the association between RBC ACKR1 expression and kidney dysfunction was not observed in patients with SCD from Egypt, although the study included patients with and without HbSS121. These discrepancies might be due to differences in the SCD genotype and/or environmental exposures between cohorts.
Detection of kidney disease in SCD
Kidney disease is associated with increased mortality in SCD58,62,122–126. Worsening eGFR is consistently an independent predictor of early mortality58,125. In the Cooperative Study of Sickle Cell Disease (CSSCD), which is a multicentre natural history study, kidney failure (defined as 20% increase in baseline creatinine and creatinine clearance <100 ml/min) was associated with a high risk of early death in patients with HbSS122. Rapid kidney function decline, regardless of the annual eGFR decline threshold (>3·0 mL/min/1·73 m2 or >5·0 mL/min/1·73 m2), is associated with increased mortality in SCD.62,65 Multiple studies also confirm the substantially higher mortality rate among patients with SCD and kidney failure than in patients with kidney failure without SCD.123,124,126 Identifying the patients with SCD at greatest risk of progressive loss of kidney function is therefore crucial to enable optimal disease management and reduce the risk of mortality, but this task might be more complex in SCD than in other CKD aetiologies — persistent albuminuria is a reliable early marker of kidney injury, but estimating GFR remains challenging.
Albuminuria
Owing to its association with eGFR decline, the presence of albuminuria warrants intervention to prevent progressive loss of kidney function127. Given that albuminuria might present in childhood in patients with SCD, current recommendations suggest screening by age 10 and at least annually thereafter; a positive result should be followed up with testing of a subsequent first-morning void sample or a repeat spot sample to confirm albuminuria (FIG. 2)127,128. Screening for albuminuria earlier than age 10 might be beneficial in children with SCD and hyperfiltration, a family history of kidney disease or known APOL1 risk variants, who might be at risk of developing albuminuria earlier in life59,115. However, current data are insufficient to recommend routine screening for APOL1 risk variants.
Figure 2: Approach to screening, evaluation and management of CKD in SCD.
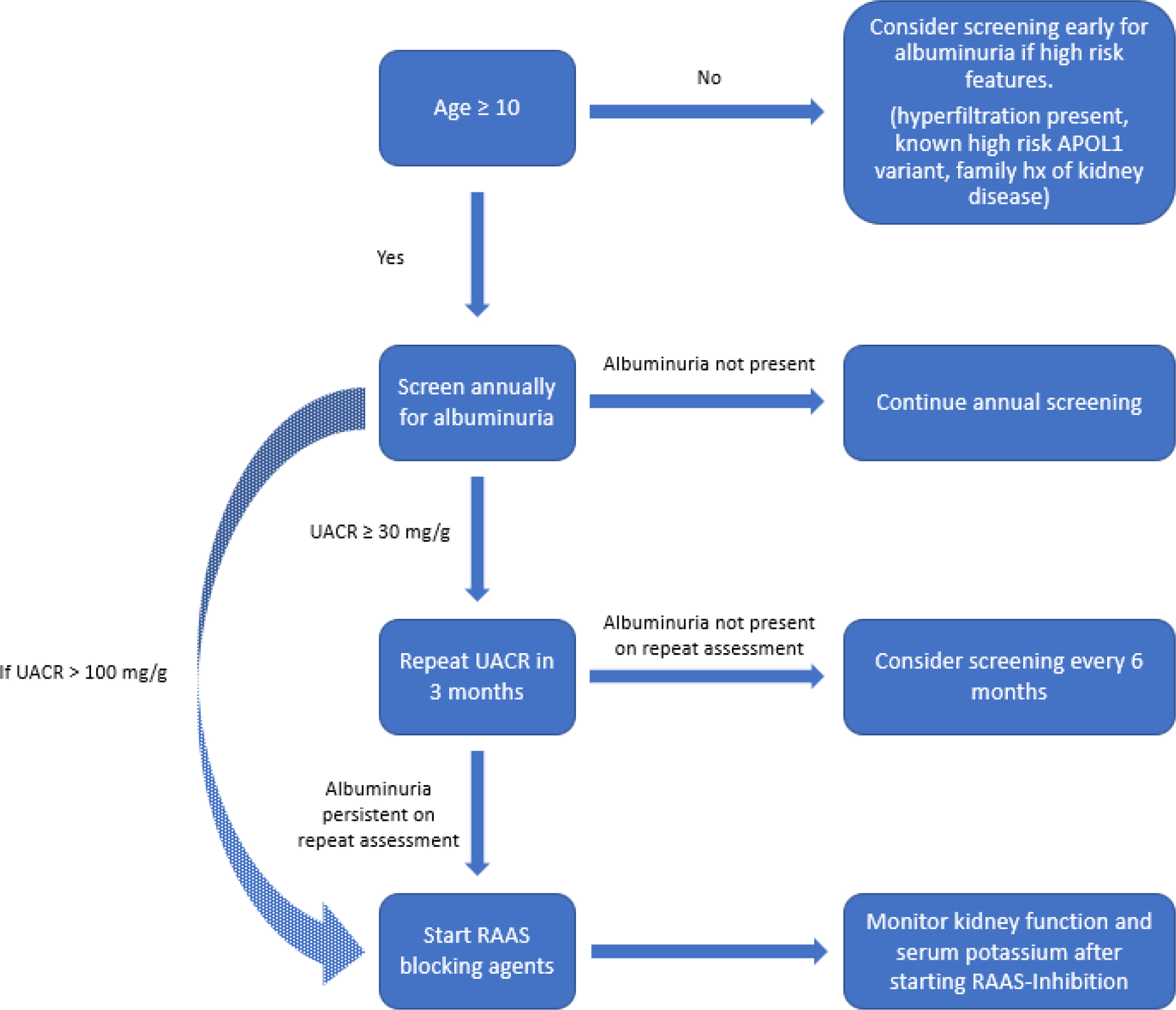
From age 10, patients should be screened for albuminuria at least annually. Repeat evaluation using first-morning void or random urine sample should be obtained to confirm albuminuria, if the initial test is positive. Earlier screening might be considered in the presence of high-risk factors, including glomerular hyperfiltration at an early age, a strong family history of kidney disease or the known presence of APOL1 risk-variants. Renin–angiotensin–aldosterone system (RAAS) blocking agents should be considered in patients with persistent albuminuria or urinary albumin-to-creatinine ratio (UACR) ≥ 100 mg/g. Kidney function (GFR) and serum potassium should be monitored after starting RAAS inhibition. *Although APOL1 testing is available clinically, current data do not support routine screening for APOL1 risk variants.
Persistent albuminuria is more likely to occur in patients with high baseline levels. In a prospective, multicentre study of patients with SCD, among those with baseline UACR ≥100 mg/g, 83% had persistent albuminuria (defined as UACR ≥ 30 mg/g on more than two annual measurements) compared with only 16% of those with baseline UACR <100 mg/g50. If a high UACR is detected on a first assessment, the likelihood that albuminuria might persist warrants immediate intervention.
Measuring and estimating GFR
CKD is currently defined as the presence of kidney damage (e.g. urinary albumin excretion ≥30 mg/day or equivalent) or eGFR <60 ml/min/1.73 m2 for ≥ 3 months, irrespective of the cause129. Enhanced tubular secretion of creatinine in patients with SCD results in an overestimation of GFR when traditional creatinine-based assessments are used — up to a 30% difference when comparing creatinine clearance to the gold standard of inulin clearance12. Consequently, patients with SCD might already have substantial kidney impairment at the time of diagnosis and the use of a higher eGFR cut-off to define CKD in these patients has therefore been suggested64. In 98 Jamaicans with HbSS, various creatinine-based equations were compared with GFR measured by 99mTc-DTPA nuclear renal scan130. The mean mGFR in this group was 94.9 ± 27.4 ml/min/1.73 m2.and the creatinine-based MDRD equation produced the greatest bias, with a mean difference of 70.4 ml/min/1.73 m2 compared with mGFR; the chronic kidney disease epidemiology (CKD-EPI) equation estimates were the closest to mGFR but still had a bias of 41.2 ml/min/1.73 m2. A prior study from France reported a similar CKD-EPI bias (30.2 ml/min/1.73 m2), which improved substantially (10.7 ml/min/1.73 m2) when the adjustment for race was removed131.
Cystatin C is an alternative GFR biomarker that is less influenced by muscle mass or diet than creatinine because it is produced by all nucleated cells, whereas creatinine is released following the breakdown of creatine in muscle. Moreover, in contrast to creatinine, cystatin C is not secreted by proximal tubule cells and its levels are thus less skewed by the enhanced tubular secretion observed in patients with SCD. In a Jamaican cohort of SCD adults, cystatin C correlated better with mGFR than serum creatinine and, when used in the CKD-EPI cystatin C formula, produced better agreement with mGFR than the CKD-EPI creatinine-based formula132. Using the CKD-EPI equation with both creatinine and cystatin C led to greater bias and less precision than using creatinine alone. A subsequent US study of 14 adults with HbSS yielded similar results but reported less bias with CKD-EPI cystatin C than that observed in the Jamaican cohort, although the combined cystatin C–creatinine estimation still had the greatest bias133. Currently, the CKD-EPI equation using cystatin C alone seems to be the optimal choice for estimating GFR in adults (TABLE 2). However, given that cystatin C testing is not widely available, the CKD-EPI creatinine equation without race adjustment should be used as an alternative. In 2021, an updated CKD-EPI equation was developed excluding race as a coefficient; to date, this equation has not been evaluated in patients with SCD134.
Table 2:
Bias of eGFR equations compared with measured GFR
| eGFR equation | Bias (ml/min/1.73 m2) | |||
|---|---|---|---|---|
| Adult Population | Paediatric population | |||
| Arlet 2012 131 |
Asnani 2013
130
Asnani 2015 132 |
Yee 2017 133 | Lebensburger 2020 136 | |
| Cockcroft–Gault | 45.3 | 37.9 | 58.9 | NA |
| MDRD | 48.7 | 70.4 | 82.5 | NA |
| MDRD (without adjustment for race) | 20.7 | NA | NA | NA |
| CKD-EPI | 30.2 | 41.2 | 45.4 | NA |
| CKD-EPI (without adjustment for race) | 10.7 | NA | NA | NA |
| CKD-EPI_CysC | NA | 28.3 | 0.2 | NA |
| CKD-EPI_Cr–CysC | NA | 41.8 | 29.7 | NA |
| JSCCS_Cr | NA | 0.67 | −12.0 | NA |
| JSCCS_CysC-Cr | NA | NA | 17.9 | NA |
| CKiD-_Cr–CysCa | NA | NA | NA | 17.0 |
| Filler_CysC | NA | NA | NA | 12.5 |
| Schwartz_CysC | NA | NA | NA | 47.7 |
| Schwartz_Cr–BUN | NA | NA | NA | 10.7 |
| Schwartz_Cr | NA | NA | NA | −21.4 |
BUN, blood urea nitrogen; CKD-EPI, chronic kidney disease epidemiology collaboration equation; CKiD, chronic kidney disease in children study equation; Cr, creatinine; CysC, cystatin C; eGFR, estimated glomerular filtration rate; JSCCS, Jamaica sickle cell cohort study equation; MDRD, modification of diet in renal disease study equation.
CKiD_Cr–CysC had best correlation with measured GFR.
In the paediatric population, eGFR equations based on creatinine and/or cystatin C have also been examined. The BABY-HUG study evaluated infants with SCD and determined that the Chronic Kidney Disease in Children (CKiD) Schwartz formula, which uses both serum creatinine and cystatin C, had the best agreement with measured99mTc-DTPA-GFR adjusted for body surface area135. In another study of 198 patients with mean age of 8.2 (range 2.1–18) years, the CKiD Schwartz formula also correlated best with mGFR, with reasonably low (but not the lowest) bias compared with the other eGFR equations tested (TABLE 2)136. The CKiD Schwartz formula might therefore be the best current option for estimating GFR in children.
Ascertaining the best approach to estimate GFR in patients with SCD is crucial because CKD definitions, which are used in management decisions regarding medication adjustment, dialysis access planning and transplantation referral, are based on eGFR values. If eGFR is overestimated, patients with undiagnosed CKD might experience delays in care and transplant referral, and receive inappropriately-dosed medications. These concerns parallel the discussions of health disparities related to use of Black race as a variable in eGFR estimation137. When comparing a cohort of individuals with SCD with African Americans in the National Health and Nutrition Examination Survey cohort, manifestations of CKD, including hyperkalaemia, acidosis and elevations in alkaline phosphatase, occurred at higher eGFR values in patients with SCD (80 ml/min/1.73 m2) than in African Americans in the National Health and Nutrition Examination Survey cohort (40 ml/min/1.73 m2)138. These data further support the use of a higher than typical eGFR cutoff, as measured by conventional measures, for identifying CKD in patients with sickle cell disease (that is eGFR <90 ml/min/1.73m2 rather than <60 ml/min/1.73m2)64.
Role of kidney biopsy
Generally, in patients with SCD with a prior history of albuminuria or hyperfiltration, and in the absence of other urinary or systemic findings, a biopsy is not necessary to confirm the diagnosis of SCD-related CKD. However, as with any patient with a new presentation for CKD, a full assessment should be made to assess other potential causes. For example, in the setting of abrupt onset of nephrotic syndrome, dysmorphic haematuria or features of other systemic diseases, a kidney biopsy might be helpful to rule out other causes of kidney disease139. Although no available data suggest an inherently higher risk of kidney biopsy-related adverse events in SCD patients, post-biopsy bleeding events might be more consequential given the degree of anaemia already present in most patients.
Biomarkers of kidney disease
Biomarkers might enable the detection of subclinical damage and highlight mechanisms underlying sickle cell nephropathy. Proteins of interest include biomarkers of endothelial dysfunction (ET1 and sVEGFR1), of glomerular (nephrin) and tubular (KIM1 and NAG) injury, and inflammation (CC-chemokine ligand 2 (CCL2) (FIG. 1).
Nephrin, which is an integral component of the slit diaphragm, maintains the podocyte foot process architecture140. Elevated urinary nephrin precedes the development of albuminuria in animal models of kidney injury and might be a more sensitive biomarker of nephropathy than albuminuria or reduced eGFR in patients with diabetes141. Urinary nephrin levels correlate positively with albuminuria in adults22 and children41 with SCD.
NAG is a lysosomal enzyme found predominantly within proximal tubular cells and is released into the urine after kidney injury. In patients with diabetes, hyperglycaemia is associated with elevated urinary NAG levels and precedes the development of albuminuria142. KIM1 is a transmembrane protein that is overexpressed on the luminal side of proximal tubule cells after acute or chronic tubulointerstitial injury143. Urine concentrations of NAG and KIM1 were positively associated with increased haemoglobinuria, albuminuria and proteinuria in several independent cohorts of patients with SCD112,144,145.
CCL2 is another potentially useful biomarker in SCD. This protein is a chemoattractant for blood monocytes and tissue macrophages, and is released by glomerular and tubular cells in response to pro-inflammatory stimuli146. Glomerular macrophage infiltration increases in SCD mice compared with controls, and blocking macrophage activation of the RON-kinase pathway ameliorated glomerular hypertrophy and endothelial injury in SCD mice147. Urine CCL2 concentration correlates positively with interstitial macrophage accumulation in lupus nephritis and in diabetic kidneys.148,149 In an adult cohort of patients with SCD, urinary CCL2 was positively associated with lipid peroxidation, nitric oxide consumption and albuminuria150; a direct correlation with albuminuria was also observed in children with SCD151.
Metabolomic profiling might identify additional biomarkers and provide insight into pathophysiologic processes that drive kidney damage in SCD. Six metabolites — betaine, proline, dimethylamine, glutamate, leucine and lysine — were elevated in adult SCD patients with versus without albuminuria ≥ 30 mg/g152. In patients with primary focal segmental glomerulosclerosis153, which is a histopathologic lesion that is commonly observed in SCD-related kidney disease, those with proteinuria >3 gm/day had elevated urinary concentrations of proline and dimethylamine compared to those with proteinuria < 3 gm/day55. Dimethylamine is a metabolic product of asymmetric dimethylarginine (ADMA), which is an endogenous inhibitor of nitric oxide synthases that might have deleterious effects on endothelial function154. Plasma ADMA concentrations are positively associated with haemolysis, elevated TRV and early mortality in SCD155. In another cohort of patients with SCD, high plasma ADMA levels and quinolinic acid were associated with rapid eGFR decline23. In cohorts with non-SCD kidney disease, plasma ADMA was associated with increased progression to kidney failure, cardiovascular morbidity and early mortality154.
Other biomarkers and mechanistic pathways in sickle cell nephropathy might be identifiable through the urine proteome. Ceruloplasmin, which is a ferroxidase enzyme that is elevated in acute and chronic inflammatory conditions, acts as a pro-oxidant by donating free copper ions156. A 31-fold higher urine concentration of ceruloplasmin was detected in patients with HbSS and haemoglobinuria, and in this cohort, urinary ceruloplasmin concentrations were progressively higher with increased CKD stage157. Orosomucoid is an acute phase protein expressed in response to tissue injury and is associated with diabetic or systemic lupus erythematosus-related kidney disease158,159. Urinary orosomucoid was also detected in patients with HbSS and haemoglobinuria, and was associated with CKD stage160,161.
Treatment of sickle cell nephropathy
The treatment of SCD-related kidney disease has focused on either therapies that have been demonstrated to improve other SCD-related complications (for example, hydroxyurea or chronic red blood cell transfusion therapy) or adopted from diabetic or hypertensive-related kidney disease (for example, ACE inhibitors or ARBs).
SCD-specific therapy
Hydroxyurea (also known as hydroxycarbamide) is a ribonucleotide reductase inhibitor that increases fetal haemoglobin (HbF) and reduces rates of vaso-occlusive crises, acute chest syndrome and RBC transfusion requirements in patients with SCD162. The BABY HUG study was a prospective, randomized, placebo-controlled, double-blind study that evaluated the effects of hydroxyurea on organ damage in infants (mean age 14 months) with HbSS or HbSβ0 thalassemia135. Infants treated with hydroxyurea for 24 months had higher urine osmolality and lower renal volumes, but no effect was observed on mGFR. An initial analysis of the Hydroxyurea Study of Long-Term Effects study, which was a prospective, observational study of children with SCD treated with hydroxyurea for accepted clinical indications25, showed that escalation of hydroxyurea to maximum tolerated dose for three years increased HbF levels, reduced haemolysis and led to a decrease in mGFR from a hyperfiltration range (mean 167 ± 46 ml/min/1.73 m2) to values closer to the normal range (mean 145 ± 27 ml/min/1.73 m2). Moreover, analysis from Hydroxyurea Study of Long-Term Effects and the Sickle Cell Clinical Research and Intervention Program demonstrated greater risk of persistent albuminuria in children starting hydroxyurea after 10 years of age (HR 2.3; 95% CI 1.2–4.6)163. The effect of hydroxyurea on albuminuria in adults with HbSS was prospectively evaluated in an open-label, single-centre study164. Treatment with hydroxyurea at a mean dose of 15 mg/kg for six months improved UACR (from median of 27 mg/g to 15 mg/g; P < 0.01) and the UACR improvement was primarily observed in patients with moderate albuminuria. These studies suggest that hydroxyurea might protect against sickle cell nephropathy, particularly in the early stages of disease, but confirmatory prospective studies are required. The Siklos on Kidney Function and Albuminuria Clinical Trial (NCT03806452) is an ongoing phase IIb, multicentre, double-blind, randomized, placebo-controlled study focused on adults (age ≥18 years) with HbSS or HbSβ0 thalassemia, and albuminuria (UACR 27–885 mg/g). Participants are being randomly assigned to receive hydroxyurea (15 mg/kg per day) or placebo, and the primary outcome is a 30% improvement in albuminuria after 6–12 months of therapy. Hydroxyurea can lead to myelosuppression and the optimal dose that preserves kidney function while maintaining safe blood counts still needs to be determined.
Chronic RBC transfusion increases haemoglobin concentration, reduces HbS and improves oxygen delivery165. Data from paediatric SCD cohorts suggest that RBC transfusion might protect against development of hyposthenuria and moderate albuminuria, particularly if started before 10 years of age166. In a paediatric study of patients with HbSS or HbSβ0 thalassemia, albuminuria was less prevalent in those randomly assigned to receive chronic RBC transfusion in the Transcranial Doppler with Transfusions Changing to Hydroxyurea study (10%) than in two age-matched cohorts of patients who did not receive chronic transfusions (14–22%)167. The benefits of using chronic transfusion to stabilize or improve kidney function in adults with SCD are less clear. Sickle Cell Disease and Cardiovascular Risk-Red Cell Exchange Trial is an ongoing prospective, randomized, multi-centre study (NCT04084080) comparing the effect of chronic RBC exchange transfusion to that of standard of care on cardiovascular and kidney complications, including albuminuria, eGFR and CKD progression.
Kidney-specific therapy
Several small prospective cohorts have evaluated the efficacy of ACE inhibitors and ARBs in sickle cell nephropathy. Urine protein excretion was reduced by 57% in 10 adults with HbSS and proteinuria after two weeks of enalapril (5–10 mg/day)55. In another study, 22 normotensive adults with HbSS and persistent moderate albuminuria were randomly assigned to receive captopril (6.25 mg/day for month 1, 12.5 mg/day for months 2–3 and 25 mg/day for months 4–6) or placebo168. Captopril was well tolerated and led to a 37% reduction in albuminuria compared with a 17% increase in the placebo group. Other studies have investigated losartan, an ARB that selectively inhibits AT1R while preserving the potentially renoprotective signalling through AT2R169. A single-centre study of patients aged ≥ 10 years with HbSS or HbSβ0 thalassemia and persistent albuminuria were treated with hydroxyurea and losartan and reported a reduction in albuminuria of (−134 mg/min median decrease in albumin excretion rate) in 12 patients who received a short-term course of losartan (4–10 weeks); a reduction in albuminuria persisted in 8 patients who received losartan for ≥12 months170. In a multicentre study of 36 children and adults with HbSS or HbSβ0 thalassemia, 58% of those with moderate albuminuria and 83% of those with severe albuminuria treated with losartan met the primary endpoint of ≥25% reduction in albuminuria171. These improvements in albuminuria after treatment with ACE inhibitors or ARBs are consistent with observations in transgenic sickle mice94. No prospective studies have evaluated the effects of ACE inhibitors or ARBs on eGFR decline, but retrospective data suggest that patients with SCD receiving these agents have a slower eGFR decline (−2.8 ml/min/1.73 m2 per year) than those not receiving such therapy (−4.7 ml/min/1.73 m2 per year)125. Given the predisposition of patients with SCD to develop hyperkalaemia, potassium levels should be monitored in patients treated with renin-angiotensin-aldosterone blockade.
Novel therapeutic options
Several studies have focused on the renoprotective effects of therapies that target endothelial dysfunction. Ambrisentan was evaluated in a Phase I, double-blind, placebo-controlled study of 26 patients with SCD172. UACR declined in patients randomly assigned to receive 12 weeks of ambrisentan (−37 mg/g), whereas it increased in those receiving placebo (+92 mg/g). The cholesterol-lowering agent atorvastatin improves endothelial function by upregulating nitric oxide and reducing oxidative stress mediators173. A 6-week exploratory, randomized, double-blind, crossover pilot study of 13 patients reported trends of reduced plasma ET1 and soluble P-selectin with atorvastatin therapy compared with placebo174. In transgenic sickle mice, 8 weeks of atorvastatin lowered levels of sVCAM1, attenuated urine concentration defects, reduced albuminuria and KIM1 levels, and improved GFR175.
In 2019, the FDA approved the use of voxelotor and crizanlizumab to treat SCD. Voxelotor reversibly binds and stabilizes oxygenated haemoglobin, which prevents HbS polymerization. In a phase III, double-blind, randomized, placebo-controlled trial, a 24-week course of voxelotor significantly improved haemoglobin concentrations and reduced haemolysis in patients with SCD176. A pilot study (NCT04335721) is recruiting patients with HbSS or HbSβ0 thalassemia with a combination of albuminuria and haemoglobinuria to determine the effects of 48 weeks of voxelotor on the kidney. Crizanlizumab, which is a P-selectin inhibitor, reduced the rate and time-to-first vaso-occlusive pain episodes in patients with SCD in a double-blind, randomized, placebo-controlled, phase II study177. Another phase II, multicentre, randomized study, is evaluating the kidney effects of 52 weeks of crizanlizumab compared with standard of care alone in patients with SCD (NCT04053764).
Management of advanced CKD in SCD
Given the myriad complications associated with SCD, when these patients develop advanced CKD, several important management issues must be considered.
Anti-hypertensive therapy
Historically, hypertension was reported to be uncommon in SCD patients as blood pressure has been thought to be typically lower than in otherwise healthy individuals. The Cooperative Study of Sickle Cell Disease reported lower systolic and diastolic blood pressure over most age ranges for patients with SCD compared with control individuals matched for age, race and sex178; hypertension was diagnosed in < 10% of adults with SCD179. Investigations have evaluated what was previously termed relative systemic hypertension (that is, systolic blood pressure 120–139 mmHg or diastolic blood pressure 70–89 mmHg)178,180 and found associations with an elevated serum creatinine as well as high risks of stroke and early mortality 178,180. Notably, these ranges are now considered elevated blood pressure or stage 1 hypertension in the general population181. Subsequent data suggest hypertension is more common than previously thought in patients with SCD179,182. Although most studies have not reported its prevalence with current definitions, one study from adults with SCD in Ghana noted elevated systemic hypertension in 45% of patients and hypertension (defined as >140/90 mmHg) in 19%182. By modern definitions, all of these patients would have elevated blood pressure or hypertension. Recommendations for blood pressure targets in patients with SCD are not currently available and thus the use of targets similar to those established for the general population have been suggested127. Of note, although thiazide diuretics are often suggested as the most appropriate first-line agent for treating hypertension 181,183, in patients with SCD diuretics are often avoided owing to the risk of volume depletion (discussed below). Therefore, an ACE inhibitor or ARB (particularly in the setting of albuminuria or CKD), or a calcium-channel blocker might be reasonable first-line agents to treat hypertension181.
Diuretic use
Diuretics are generally avoided in patients with SCD owing to the risk that volume depletion and subsequent dehydration might precipitate vaso-occlusive crises184. However, with worsening CKD, volume retention might become clinically relevant, particularly as ageing patients accumulate additional comorbidities, including right ventricular failure and congestive heart failure. In these settings, diuretics are warranted and can be used cautiously. Patients should be educated to stop any prescribed diuretics during intercurrent acute illnesses to avoid excess dehydration.
Erythropoiesis-stimulating agents
Erythropoiesis-stimulating agents (ESAs) have been used in patients with SCD who are intolerant to optimal hydroxyurea doses due to reticulocytopenia and can allow an increase in hydroxyurea dosage185. As CKD progresses and endogenous erythropoiesis wanes, anaemia worsens and the use of ESAs might be necessary to maintain acceptable haemoglobin levels. A retrospective study of 32 patients treated with an ESA for at least one year demonstrated no increase in vaso-occlusive crises after ESA initiation186, despite earlier anecdotal reports; data from an insurance claims repository also showed that crisis rates after ESA initiation were similar in patients with SCD receiving ESAs (n=24) and those SCD patients not receiving ESAs186. Patients with the best haemoglobin responses had received both an ESA and hydroxyurea.
Based on current recommendations, patients with SCD and CKD who experience a drop in absolute reticulocyte count might benefit from concomitant ESA and hydroxyurea therapy127. Although higher ESA doses might be required than in patients with CKD not due to SCD, the target haemoglobin should be individually tailored to maintain quality of life and reduce the need for transfusions, and should not exceed 10–11 g/dl187.
Iron chelation therapy
Iron overload is common in SCD owing to frequent RBC transfusions. When indicated, management requires chelation therapy with one of three available agents — deferoxamine administered parenterally, or the oral agents deferasirox and deferiprone187,188. Deferasirox is associated with a (usually reversible) acute rise in creatinine and should thus be used cautiously and with close monitoring in patients with CKD187. Of note, in a long-term study of 62 adult and paediatric patients treated with deferasirox for 5 years, only two patients had AKI events and neither event was considered to be drug-related189. Deferasirox excretion is primarily faecal, whereas deferiprone is primarily excreted via urine187. A pilot study in eight patients without SCD who were receiving haemodialysis evaluated two doses of deferasirox (10 or 15 mg/kg daily). The optimal dosing remains uncertain — the lower dose did not achieve therapeutic levels and the higher dose achieved greater than expected levels, but no adverse events occurred190. Recurrent hypocalcaemia in a patient with SCD receiving haemodialysis while on deferasirox has been reported191, and although the risk is probably low, patients treated with haemodialysis should be monitored closely when treated with deferasirox. Deferoxamine can be used in patients receiving dialysis but must be administered during dialysis to allow removal, especially given the risk of Yersinia sepsis and mucormycosis associated with this agent187,192. Deferoxamine has been administered intraperitoneally in patients receiving peritoneal dialysis193.
Kidney failure
In two US studies (from 1992–1997 and 2005–2009), patients with SCD comprised only 0.1% of the entire population with kidney failure123,194; information to guide optimal management of these patients remains minimal. Several studies have demonstrated that, among patients with kidney failure, those with SCD have higher mortality and lower likelihood of transplantation than those without SCD123,194,195. Of note, the mortality risk of SCD in patients with kidney failure is attenuated by kidney transplant194. Moreover, compared with patients with SCD and kidney failure who are awaiting transplantation, kidney recipients with SCD trended to lower mortality, even at 90 days following transplant (relative risk 0.14; P=0.056)196. Mortality among patients with SCD and kidney failure is also associated with lower haemoglobin levels, higher ESA dose, higher ultrafiltration rates, presence of a dialysis catheter, hypoalbuminaemia and the use of high sodium dialysate (>145meq/L)195.
Dialysis
Patient with SCD and kidney failure who establish nephrology care in the pre-dialysis period have a reduction in mortality in the first year of dialysis compared with patients without pre-dialysis nephrology care123. In one French study, dialysis initiation did not affect the incidence of vaso-occlusive crises in patients with SCD124. Of note, patients with SCD are at a higher risk of vascular access failure than patients with kidney failure without SCD195, perhaps owing to activated coagulation and endothelial injury in SCD. Peritoneal dialysis might be beneficial in SCD to avoid overly aggressive volume removal, bleeding complications and vascular access complications, and should perhaps be considered as a first option in the able patient. Indeed, initial data from a US Renal Data System-based study suggested a survival benefit for patients with SCD and kidney failure receiving peritoneal dialysis compared with hemodialysis.197 However, similar to most individuals with kidney failure, the use of peritoneal dialysis among SCD patients with kidney failure is low (5%). For patients receiving haemodialysis, high sodium concentrates, high ultrafiltration rates and low dialysis temperatures (owing to vasoconstriction) should be avoided, as these conditions could promote vaso-occlusive crises.
Kidney transplantation
Kidney transplantation remains a viable option for patients with SCD and advanced kidney disease or kidney failure, with acceptable graft survival and probable survival advantage over dialysis. However, patients with SCD who develop kidney failure might have additional disease-related comorbidities and medical barriers to transplantation45,52, which might partly explain why they are less likely to receive a kidney transplant123,124,194.
Several studies have evaluated kidney transplant outcomes in SCD. The rates of delayed graft function and one-year graft survival from the period 1984–1996 in the US Renal Data System were similar in kidney transplant recipients with SCD (78%) and those without SCD (77%)196. However, patients with SCD had lower graft survival at three years than those without SCD (48% versus 60%), and lower one-year (78 versus 90%) and three-year patient survival (59% versus 81%). Of note, although mortality was also greater in transplant recipients with SCD than in those without SCD according to data collected from 1988–1999 and 2000–2011, the overall survival at 6 years had improved for SCD patients in the more recent time period (68.8% versus 55.7%)198. Compared with transplant recipients with kidney failure due to diabetes, 6-year mortality was similar (73.1% vs. 74.1%) in the more recent time period198. Moreover, a 2019 study of kidney transplant recipients with SCD in France did not report a difference in death-censored graft survival compared with recipients without SCD (median follow up of 17.4 months)199. A somewhat contrasting US Organ Procurement and Transplantation Network/United Network for Organ Sharing study evaluated patients with SCD transplanted between 2010 and 2019 reported lower patient and graft survival in recipients with SCD compared with recipients without SCD, including in recipients with diabetes200. Outcomes in patients with SCD during this period were similar to those seen in an earlier period (2000–2009); however, unlike in prior eras, patients with diabetes fared as well as non-diabetic patients, suggesting that improved outcomes in diabetes may have accounted for the observed difference between the studies. Another Organ Procurement and Transplantation Network/United Network for Organ Sharing study of data from the period 1998–2017 compared 189 kidney transplant recipients with SCD with patients with SCD who remained on the waiting list, and noted that transplantation was associated with a 20% reduction in the 10-year absolute risk of death, which is similar to the transplantation benefit observed in patients without SCD201. Overall, these studies demonstrating favourable transplant outcomes in SCD patients suggest that SCD alone should not be a contraindication to kidney transplantation. Further, the improved mortality with transplant highlights that early referral for transplantation, if appropriate, is paramount.
Specific interventions might improve transplant outcomes in patients with SCD. Pre-transplant (in particular for planned living donation), RBC exchange transfusion of leuko-reduced blood, which minimizes HLA alloimmunization, can be considered to reduce the proportion of HbS and thereby the likelihood of sickling 187,202; warming of the allograft and intraoperative hyperoxygenation of the recipient might also be beneficial202. Although prior recommendations have suggested corticosteroids should be minimized as they are thought to potentially trigger SCD-related pain crises, steroid-containing immunosuppression regimens have been used successfully202. Post-transplantation, hydroxyurea might reduce ongoing SCD-related allograft injury, but should be used carefully when co-administered with other myelosuppressive medications (such as sulfamethoxazole, valganciclovir or mycophenolate)203. A 2020 study of kidney transplant recipients with SCD reported that post-transplant exchange transfusion was associated with better patient and graft survival, and preserved eGFR without a greater risk of rejection or development of donor-specific antibodies compared with historical SCD transplant recipients who had not received exchange transfusion post-transplant 204.
Kidney abnormalities in SCT
SCT was traditionally thought to be a benign state. Although less likely to occur than in patients with SCD, HbS can still polymerize in individuals with SCT205. SCT is associated with multiple kidney complications, although they are often less severe than those observed in SCD. Rhabdomyolysis and sudden death are the most recognized complications of SCT, but kidney complications, including impaired urinary concentration, haematuria and papillary necrosis, occur commonly206. The prevalence of supranormal renal haemodynamics in individuals with SCT is unclear but a small study of Congolese children suggested that hyperfiltration was more common in children with SCT than in HbAA controls207. Impaired urinary concentration in patients with SCT is attenuated by co-inheritance of α-thalassemia208. High intracellular HbS concentration is a crucial risk factor for HbS polymerization. In a Nigerian study, individuals with SCT who had papillary necrosis had a higher mean HbS concentration than those without papillary necrosis (34% versus 25%, respectively; P<0.05)209. Another important consideration in patients with SCT with gross haematuria is the evaluation for renal medullary carcinoma (BOX 1)206.
Box 1 – Renal medullary carcinoma.
Renal medullary carcinoma (RMC) is a rare, aggressive malignancy most commonly described in young individuals with sickle cell syndromes, predominantly SCT227,228. The reason for the high prevalence in sickle cell syndromes is not fully known but may be related to chronic medullary hypoxia. Data obtained from patients suggest that high-intensity exercise may be a risk factor for RMC in individuals with SCT, with additional studies in SCT mice showing higher renal medullary hypoxia compared to wild-type controls following high-intensity exercise229. These data suggest that high-intensity exercise may be a modifiable risk factor for RMC in individuals with SCT. Loss of expression of the chromatin remodeling factor and tumour suppressor SMARCB1, strong expression of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor, and positivity for cellular tumour antigen p53 have all been implicated in RMC development230,231. Patients commonly present with haematuria, flank pain, weight loss and abdominal masses, and the disease is often metastatic at presentation. In a systematic review of 217 cases, 88% of patients had SCT, 8% had SCD, 50% were children and the risk of RMC was more than two-fold higher in males228. Interestingly, RMC has been noted to have a predilection for the right kidney, seen in 70% of patients227,228. Isolated haematuria, or in combination with abdominal or flank pain, was present at diagnosis in 66% of cases and tumour-related mortality was 95%228. Although screening is not currently recommended, the poor prognosis of RMC demands a thorough evaluation of individuals with sickle cell syndromes who present with haematuria.
Multiple studies have confirmed a relationship between SCT and CKD. In a large meta-analysis of 15,975 African Americans, participants with SCT had an increased risk of baseline CKD and incident CKD, as well as a higher rate of eGFR decline, compared with individuals without SCT210. Another population-based cohort of 9,909 African Americans in the Reasons for Geographic and Racial Differences in Stroke study, confirmed an association between SCT and incident CKD; patients with SCT had a nearly two-fold higher risk of kidney failure than those without hemoglobinopathies211. Of note, co-inheritance of high-risk APOL1 variants conferred no additional risk. In another large patient data registry that included self-identified Black patients who had haemoglobin electrophoresis, patients with SCT had a faster rate of eGFR decline (0.45 ml/min/1.73 m2 greater) than individuals with normal haemoglobin electrophoresis66. Additional studies in Hispanic and Latinx populations noted a similar association between SCT and kidney disease risk212. Interestingly, data from the African American Study of Kidney Disease and Hypertension found no association between SCT and CKD progression or kidney failure risk in patients with established CKD213. These findings suggest that SCT is a risk factor for development of CKD but might not influence disease progression. Collectively, these data suggest that SCT is a modest risk factor for the development of CKD, although further studies assessing whether SCT affects the risk of CKD progression are needed. At present, no data suggest that screening for CKD in patients with SCT is beneficial. However, in those with other CKD risk factors (for example, hypertension, diabetes or family history of CKD), awareness of this heightened risk might enable patients to adopt lifestyle modifications that reduce the risk of disease development and inform clinicians to be particularly observant for the development of CKD. These considerations are particularly important as SCT status is determined at birth in many countries due to universal newborn SCD screening programs.
The association between SCT and AKI is less well established. Several case reports have described AKI in individuals with SCT, often with associated rhabdomyolysis. One study using diagnostic codes found no association between SCT and AKI214, but another that evaluated African Americans in the US Military reported that the risk of AKI was higher in individuals with SCT detected by hemoglobin electrophoresis than in those without SCT (odds ratio (OR) 1.74; 95% CI 1.17–2.59)215. A 2021 study that used the same previously noted multicentre registry of Black patients with haemoglobin electrophoresis found that individuals with SCT had a 1.6-fold higher risk of incident severe AKI (defined as creatinine ≥1.5 times above baseline for ≥ 72 hours) than those without haemoglobin variants80. Following AKI, individuals with SCT also had a more rapid eGFR decline than those without SCT80.
Among patients with kidney failure receiving dialysis, SCT was associated with the use of higher doses of ESAs than those used in patients without detectable hemoglobin variants to achieve similar haemoglobin levels216,217. Data on outcomes for individuals with SCT who undergo kidney transplantation are scarce202, although immediate post-transplant complications have been reported218. With regard to organ donation, some centres screen living donors for SCT during assessment of eligibility. In a 2008 US survey, only 17.5% of responding transplant centres had an established policy for donors with SCT but 37.2% reported excluding these donors most or all of the time219. In the UK, 20% of respondent centres reported a formal policy although all would consider SCT donors202. In the absence of data to suggest adverse outcomes after kidney donation, evaluation of SCT donors should be performed on a case-by-case basis.
CONCLUSIONS
Kidney disease is highly prevalent in SCD but many questions remain about its pathophysiology, natural history and optimal treatment, and additional studies are required. Given the limitations of current eGFR equations, the CKD-EPI equation using cystatin C alone presently seems to be the optimal choice. However, as cystatin C tests are not widely available, the CKD-EPI creatinine equation without race adjustment should be used in adults, whereas the combined cystatin C and creatinine CKiD Schwartz formula might be preferable in children. This is consistent with recent recommendations by the NKF-ASN taskforce that the CKD-EPI creatinine equation without the race variable be used in all US laboratories to estimate GFR in adults, as well as increased efforts to facilitate routine and timely use of cystatin C to confirm eGFR, especially in adults at risk of or have CKD220. The most recent 2021 CKD-EPI equation that has eliminated inclusion of race has not yet been evaluated in the SCD population134. Current recommendations suggest screening children with SCD for albuminuria by the age of 10 and at least annually thereafter. The use of genetic, proteomic and metabolomic tools, which could perhaps be integrated to calculate risk scores, might facilitate the early identification of individuals at risk of kidney disease.
Early identification of kidney damage and subsequent initiation of disease-modifying therapies, adequate blood pressure control and avoidance of nephrotoxic agents (BOX 2) could slow progression of CKD and decrease risk of death in SCD. Albuminuria associated with CKD development and progression as well as rapid eGFR decline in SCD, consistent with the relationship of albuminuria as an independent risk predictor of progressive CKD and ESKD in diabetic and non-diabetic individuals221,222. Although ACE inhibitors, ARBs and hydroxyurea decrease albuminuria in short-term studies, no long-term studies have yet been performed to show that reduction of albuminuria will slow kidney disease progression. Sodium-glucose co-transporter 2 (SGLT2) inhibitors have demonstrated benefits in diabetic kidney disease and other forms of kidney disease with albuminuria223–225. Combined with these data and with the roles of hyperfiltration and glomerular hypertension in SCD nephropathy, these drugs should be evaluated in SCD patients at risk of progressive kidney disease. At present, however, no data exist to support their use in SCD, in which adverse events such as volume depletion may be a particular concern. Adequately designed studies that determine the long-term effects of disease-modifying therapies on progressive CKD are highly warranted. Several studies have shown that the survival of patients with SCD and kidney failure improves after kidney transplantation and these patients should therefore be considered for this treatment modality. More data are needed to define optimal immunosuppressive treatments after kidney transplant, and the role of chronic RBC transfusion and other disease-modifying therapies to minimize the recurrence of kidney disease. Although SCT is less severe than SCD, it is also associated with kidney abnormalities and CKD. Because SCT is identified via universal newborn screening programs in many resource-rich countries, clinicians should consider its potential contribution to CKD risk. Answers to these and other questions will require carefully designed clinical and laboratory studies and collaboration among multiple centres.
Box 2 – Pain management and nephrotoxicity.
Avoidance of nephrotoxic agents might prevent additional kidney damage in SCD. Administration of NSAIDs, which are important for treating vaso-occlusive pain, causes a more pronounced decline in GFR and renal plasma flow in patients with SCD compared with healthy individuals12. In a paediatric SCD cohort, the odds ratio of AKI increased 1.8-fold for each additional day of ketorolac therapy during a vaso-occlusive crisis78. In adult patients with SCD, vancomycin use during a vaso-occlusive crisis was associated with 4.5-fold greater risk of AKI82.
Morphine is an opioid that is commonly used to manage SCD-related pain but might have a pathophysiologic role in SCD-related nephropathy. Chronic treatment of sickle mice with morphine increased glomerular volume, mesangial cell proliferation, parietal cell metaplasia, podocyte effacement and microvillus transformation; HMOX1 activity and albuminuria also increased232. Morphine-related kidney injury was ameliorated by the non-selective opioid antagonist, naloxone. In a single-centre preliminary study, we found that the use of opioid analgesics was associated with albuminuria in adult patients with SCD233 but further studies are required confirm this finding and further evaluate the association of opioid analgesics with albuminuria in SCD.
Supplementary Material
Key Points.
Albuminuria is common in patients with sickle cell disease (SCD) and predicts the progression of chronic kidney disease (CKD).
Haematuria is usually benign in individuals with sickle cell trait (SCT) and SCD, but might be a presenting symptom of renal medullary carcinoma.
The pathophysiology of SCD-related nephropathy is likely driven by hyperfiltration, increased oxidant stress and glomerular hypertension.
Genetic modifiers, including APOL1, HMOX1, HBA1 and HBA2 variants, are implicated in the development and/or progression of CKD.
Kidney function declines more rapidly in individuals with sickle cell trait and SCD than in the general African American population; baseline CKD and rapid decline in estimated glomerular filtration rate are associated with increased mortality in SCD.
Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and hydroxyurea decrease albuminuria in short-term studies; adequately controlled studies are required to evaluate the long-term effects of these agents on progressive kidney disease.
Acknowledgements
K.I.A. and V.K.D. are supported by FDA grant FD006030 and NIH grant HL159376. S.L.S is supported by NIH grants HL146788, HL153161 and HL159376.
Glossary terms
- Compound heterozygous variants
refers to the presence of two different mutated alleles at a particular gene locus, e.g. inheritance of βS allele and βC allele.
- Nocturnal enuresis
persistence of night-time urination in bed two or more times per week for at least 3 months after the age of 5.
- Hyporeninemic hypoaldosteronism
is characterized by both diminished renin release and an intra-adrenal defect, which result in decreased systemic and intra-adrenal angiotensin II production and a decline in aldosterone secretion
- Dysmorphic haematuria
abnormally shaped red blood cells in the urine whose presence suggest glomerular injury
- Relative hypertension
defined as systolic blood pressure 120–139 mmHg or diastolic blood pressure 70–89 mmHg
- Exchange transfusion
a procedure in which a patient’s blood or components (e.g. red blood cells) of it are replaced by other blood or blood products
Footnotes
Competing interests
K.I.A. has received research funding from Novartis and Global Blood Therapeutics, served on advisory boards for Novartis, Global Blood Therapeutics, Novo Nordisk, Editas Medicine, Forma Therapeutics and Agios Pharmaceuticals, and as a consultant for Roche. S.L.S. receives research funding support from Novartis, Pfizer and Global Blood Therapeutics, and served on advisory boards for Novartis and Global Blood Therapeutics. V.K.D. has served on advisory boards for Novartis, Bayer and Travere.
Peer review information
TBC
REFERENCES:
- 1.Saraf SL et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatric respiratory reviews 15, 4–12, doi: 10.1016/j.prrv.2013.11.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hassell KL Population estimates of sickle cell disease in the U.S. Am J Prev Med 38, S512–521, doi: 10.1016/j.amepre.2009.12.022 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Piel FB, Hay SI, Gupta S, Weatherall DJ & Williams TN Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 10, e1001484, doi: 10.1371/journal.pmed.1001484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heller P, Best WR, Nelson RB & Becktel J Clinical implications of sickle-cell trait and glucose-6-phosphate dehydrogenase deficiency in hospitalized black male patients. The New England journal of medicine 300, 1001–1005, doi: 10.1056/NEJM197905033001801 (1979). [DOI] [PubMed] [Google Scholar]
- 5.Pecker LH & Naik RP The current state of sickle cell trait: implications for reproductive and genetic counseling. Blood 132, 2331–2338, doi: 10.1182/blood-2018-06-848705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunn HF Pathogenesis and treatment of sickle cell disease. N Engl J Med 337, 762–769, doi: 10.1056/NEJM199709113371107 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Rees DC, Williams TN & Gladwin MT Sickle-cell disease. Lancet 376, 2018–2031, doi: 10.1016/S0140-6736(10)61029-X (2010). [DOI] [PubMed] [Google Scholar]
- 8.Thein SL & Howard J How I treat the older adult with sickle cell disease. Blood 132, 1750–1760, doi: 10.1182/blood-2018-03-818161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allon M Renal abnormalities in sickle cell disease. Archives of internal medicine 150, 501–504 (1990). [PubMed] [Google Scholar]
- 10.Nath KA & Hebbel RP Sickle cell disease: renal manifestations and mechanisms. Nature reviews. Nephrology 11, 161–171, doi: 10.1038/nrneph.2015.8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Statius van Eps LW, Pinedo-Veels C, de Vries GH & de Koning J Nature of concentrating defect in sickle-cell nephropathy. Microradioangiographic studies. Lancet 1, 450–452 (1970). [DOI] [PubMed] [Google Scholar]
- 12.Allon M, Lawson L, Eckman JR, Delaney V & Bourke E Effects of nonsteroidal antiinflammatory drugs on renal function in sickle cell anemia. Kidney international 34, 500–506 (1988). [DOI] [PubMed] [Google Scholar]
- 13.De Jong PE et al. The influence of indomethacin on renal concentrating and diluting capacity in sickle cell nephropathy. Clin Sci (Lond) 63, 53–58, doi: 10.1042/cs0630053 (1982). [DOI] [PubMed] [Google Scholar]
- 14.Tharaux PL et al. Urinary endothelin-1 as a marker of renal damage in sickle cell disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 20, 2408–2413, doi: 10.1093/ndt/gfi111 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Wolf RB, Kassim AA, Goodpaster RL & DeBaun MR Nocturnal enuresis in sickle cell disease. Expert review of hematology 7, 245–254, doi: 10.1586/17474086.2014.892412 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Cazenave M et al. Tubular Acidification Defect in Adults with Sickle Cell Disease. Clinical journal of the American Society of Nephrology : CJASN 15, 16–24, doi: 10.2215/CJN.07830719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeFronzo RA, Taufield PA, Black H, McPhedran P & Cooke CR Impaired renal tubular potassium secretion in sickle cell disease. Annals of internal medicine 90, 310–316, doi: 10.7326/0003-4819-90-3-310 (1979). [DOI] [PubMed] [Google Scholar]
- 18.DeFronzo RA Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney international 17, 118–134, doi: 10.1038/ki.1980.14 (1980). [DOI] [PubMed] [Google Scholar]
- 19.Hatch FE Jr., Azar SH, Ainsworth TE, Nardo JM & Culbertson JW Renal circulatory studies in young adults with sickle cell anemia. J Lab Clin Med 76, 632–640 (1970). [PubMed] [Google Scholar]
- 20.de Jong PE & Statius van Eps LW Sickle cell nephropathy: new insights into its pathophysiology. Kidney international 27, 711–717 (1985). [DOI] [PubMed] [Google Scholar]
- 21.Haymann JP et al. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clinical journal of the American Society of Nephrology : CJASN 5, 756–761, doi: 10.2215/CJN.08511109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vazquez B et al. Hyperfiltration is associated with the development of microalbuminuria in patients with sickle cell anemia. American journal of hematology 89, 1156–1157, doi: 10.1002/ajh.23817 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu JZ et al. Clinical and metabolomic risk factors associated with rapid renal function decline in sickle cell disease. American journal of hematology, doi: 10.1002/ajh.25263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ware RE et al. Renal function in infants with sickle cell anemia: baseline data from the BABY HUG trial. The Journal of pediatrics 156, 66–70 e61, doi: 10.1016/j.jpeds.2009.06.060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aygun B et al. Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. American journal of hematology 88, 116–119, doi: 10.1002/ajh.23365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belisario AR et al. Prevalence and risk factors for albuminuria and glomerular hyperfiltration in a large cohort of children with sickle cell anemia. American journal of hematology 95, E125–E128, doi: 10.1002/ajh.25763 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Derebail VK, Zhou Q, Ciccone EJ, Cai J & Ataga KI Longitudinal study of glomerular hyperfiltration and normalization of estimated glomerular filtration in adults with sickle cell disease. Br J Haematol 195, 123–132, doi: 10.1111/bjh.17723 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asnani MR & Reid ME Renal function in adult Jamaicans with homozygous sickle cell disease. Hematology 20, 422–428, doi: 10.1179/1607845414Y.0000000213 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Kasztan M et al. Hyperfiltration predicts long-term renal outcomes in humanized sickle cell mice. Blood Adv 3, 1460–1475, doi: 10.1182/bloodadvances.2018028878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasztan M, Aban I, Hande SP, Pollock DM & Lebensburger JD Sex differences in the trajectory of glomerular filtration rate in pediatric and murine sickle cell anemia. Blood Adv 4, 263–265, doi: 10.1182/bloodadvances.2019001237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasztan M & Pollock DM Impact of ET-1 and sex in glomerular hyperfiltration in humanized sickle cell mice. Clinical science 133, 1475–1486, doi: 10.1042/CS20190215 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Alvarez O, Montane B, Lopez G, Wilkinson J & Miller T Early blood transfusions protect against microalbuminuria in children with sickle cell disease. Pediatr Blood Cancer 47, 71–76, doi: 10.1002/pbc.20645 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Dharnidharka VR, Dabbagh S, Atiyeh B, Simpson P & Sarnaik S Prevalence of microalbuminuria in children with sickle cell disease. Pediatric nephrology 12, 475–478 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Wigfall DR, Ware RE, Burchinal MR, Kinney TR & Foreman JW Prevalence and clinical correlates of glomerulopathy in children with sickle cell disease. J Pediatr 136, 749–753 (2000). [PubMed] [Google Scholar]
- 35.McBurney PG, Hanevold CD, Hernandez CM, Waller JL & McKie KM Risk factors for microalbuminuria in children with sickle cell anemia. Journal of pediatric hematology/oncology 24, 473–477 (2002). [DOI] [PubMed] [Google Scholar]
- 36.McKie KT et al. Prevalence, prevention, and treatment of microalbuminuria and proteinuria in children with sickle cell disease. Journal of pediatric hematology/oncology 29, 140–144, doi: 10.1097/MPH.0b013e3180335081 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Iwalokun BA, Iwalokun SO, Hodonu SO, Aina OA & Agomo PU Evaluation of microalbuminuria in relation to asymptomatic bacteruria in Nigerian patients with sickle cell anemia. Saudi J Kidney Dis Transpl 23, 1320–1330, doi: 10.4103/1319-2442.103589 (2012). [DOI] [PubMed] [Google Scholar]
- 38.McPherson Yee M et al. Chronic kidney disease and albuminuria in children with sickle cell disease. Clinical journal of the American Society of Nephrology : CJASN 6, 2628–2633, doi: 10.2215/CJN.01600211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubert M et al. Degree of anemia, indirect markers of hemolysis, and vascular complications of sickle cell disease in Africa. Blood 130, 2215–2223, doi: 10.1182/blood-2016-12-755777 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Geard A et al. Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br J Haematol 178, 629–639, doi: 10.1111/bjh.14724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heimlich JB et al. Nephrin as a biomarker of sickle cell glomerulopathy in Malawi. Pediatric blood & cancer 65, doi:ARTNe26993 10.1016/S2352-3026(14)00007-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ranque B et al. Early renal damage in patients with sickle cell disease in sub-Saharan Africa: a multinational, prospective, cross-sectional study. Lancet Haematol 1, e64–73, doi: 10.1016/S2352-3026(14)00007-6 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Ataga KI, Derebail VK & Archer DR The glomerulopathy of sickle cell disease. American journal of hematology 89, 907–914, doi: 10.1002/ajh.23762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Day TG, Drasar ER, Fulford T, Sharpe CC & Thein SL Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica 97, 201–205, doi: 10.3324/haematol.2011.050336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drawz P et al. Kidney Disease among Patients with Sickle Cell Disease, Hemoglobin SS and SC. Clinical journal of the American Society of Nephrology : CJASN 11, 207–215, doi: 10.2215/CJN.03940415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson J, Reid M, Hambleton I & Serjeant GR Albuminuria and renal function in homozygous sickle cell disease: observations from a cohort study. Archives of internal medicine 167, 701–708, doi: 10.1001/archinte.167.7.701 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Ephraim RK et al. Chronic kidney disease is common in sickle cell disease: a cross-sectional study in the Tema Metropolis, Ghana. BMC nephrology 16, 75, doi: 10.1186/s12882-015-0072-y (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolarinwa RA, Akinlade KS, Kuti MA, Olawale OO & Akinola NO Renal disease in adult Nigerians with sickle cell anemia: a report of prevalence, clinical features and risk factors. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia 23, 171–175 (2012). [PubMed] [Google Scholar]
- 49.Arogundade FA et al. An appraisal of kidney dysfunction and its risk factors in patients with sickle cell disease. Nephron. Clinical practice 118, c225–231, doi: 10.1159/000321138 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Niss O et al. Progression of albuminuria in patients with sickle cell anemia: a multicenter, longitudinal study. Blood Adv 4, 1501–1511, doi: 10.1182/bloodadvances.2019001378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laurin LP, Nachman PH, Desai PC, Ataga KI & Derebail VK Hydroxyurea is associated with lower prevalence of albuminuria in adults with sickle cell disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 29, 1211–1218, doi: 10.1093/ndt/gft295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ataga KI et al. Urinary albumin excretion is associated with pulmonary hypertension in sickle cell disease: potential role of soluble fms-like tyrosine kinase-1. European journal of haematology 85, 257–263, doi: 10.1111/j.1600-0609.2010.01471.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guasch A, Navarrete J, Nass K & Zayas CF Glomerular involvement in adults with sickle cell hemoglobinopathies: Prevalence and clinical correlates of progressive renal failure. Journal of the American Society of Nephrology : JASN 17, 2228–2235, doi: 10.1681/ASN.2002010084 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Asnani MR, Fraser RA & Reid ME Higher rates of hemolysis are not associated with albuminuria in Jamaicans with sickle cell disease. PloS one 6, e18863, doi: 10.1371/journal.pone.0018863 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Falk RJ et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med 326, 910–915, doi: 10.1056/NEJM199204023261402 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Aleem A Proteinuria in adult Saudi patients with sickle cell disease is not associated with identifiable risk factors. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia 21, 903–908 (2010). [PubMed] [Google Scholar]
- 57.De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A & Telen MJ Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. American journal of hematology 83, 19–25, doi: 10.1002/ajh.21058 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Elmariah H et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. American journal of hematology 89, 530–535, doi: 10.1002/ajh.23683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lebensburger JD et al. Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. American journal of hematology 94, 417–423, doi: 10.1002/ajh.25390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Powars DR et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Annals of internal medicine 115, 614–620 (1991). [DOI] [PubMed] [Google Scholar]
- 61.Gosmanova EO, Zaidi S, Wan JY & Adams-Graves PE Prevalence and progression of chronic kidney disease in adult patients with sickle cell disease. Journal of investigative medicine : the official publication of the American Federation for Clinical Research 62, 804–807, doi: 10.1097/01.JIM.0000446836.75352.72 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Derebail VK, Zhou Q, Ciccone EJ, Cai J & Ataga KI Rapid decline in estimated glomerular filtration rate is common in adults with sickle cell disease and associated with increased mortality. Br J Haematol 186, 900–907, doi: 10.1111/bjh.16003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Young BA et al. Risk Factors for Rapid Kidney Function Decline Among African Americans: The Jackson Heart Study (JHS ). American journal of kidney diseases : the official journal of the National Kidney Foundation 68, 229–239, doi: 10.1053/j.ajkd.2016.02.046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Derebail VK et al. Progressive Decline in Estimated GFR in Patients With Sickle Cell Disease: An Observational Cohort Study. Am J Kidney Dis 74, 47–55, doi: 10.1053/j.ajkd.2018.12.027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ataga KI et al. Rapid decline in estimated glomerular filtration rate in sickle cell anemia: results of a multicenter pooled analysis. Haematologica Online ahead of print, doi: 10.3324/haematol.2020.267419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olaniran KO et al. Kidney Function Decline among Black Patients with Sickle Cell Trait and Sickle Cell Disease: An Observational Cohort Study. Journal of the American Society of Nephrology : JASN 31, 393–404, doi: 10.1681/ASN.2019050502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asnani M, Serjeant G, Royal-Thomas T & Reid M Predictors of renal function progression in adults with homozygous sickle cell disease. British journal of haematology 173, 461–468, doi: 10.1111/bjh.13967 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25–1985. A 27-year-old man with recurrent bleeding from the left kidney for 13 years. N Engl J Med 312, 1623–1631, doi: 10.1056/NEJM198506203122508 (1985). [DOI] [PubMed] [Google Scholar]
- 69.Ter Maaten JC, G. R, De Jong PE. in Comprehensive Clinical Nephrology (ed Feehally J, Johnson RJ) 665 (2003).
- 70.Oksenhendler E et al. Recurrent hematuria in 4 white patients with sickle cell trait. J Urol 132, 1201–1203, doi: 10.1016/s0022-5347(17)50097-x (1984). [DOI] [PubMed] [Google Scholar]
- 71.John EG, Schade SG, Spigos DG, Cort JH & Rosenthal IM Effectiveness of triglycyl vasopressin in persistent hematuria associated with sickle cell hemoglobin. Arch Intern Med 140, 1589–1593 (1980). [PubMed] [Google Scholar]
- 72.Gabrovsky A, Aderinto A, Spevak M, Vichinsky E & Resar LM Low dose, oral epsilon aminocaproic acid for renal papillary necrosis and massive hemorrhage in hemoglobin SC disease. Pediatr Blood Cancer 54, 148–150, doi: 10.1002/pbc.22295 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Herard A, Colin J, Youinou Y, Drancourt E & Brandt B Massive gross hematuria in a sickle cell trait patient with renal papillary necrosis. Conservative approach using a balloon ureteral catheter to tamponade the papilla bleeding. Eur Urol 34, 161–162, doi: 10.1159/000019703 (1998). [DOI] [PubMed] [Google Scholar]
- 74.Flaster S, Lome LG & Presman D Urologic complications of renal papillary necrosis. Urology 05, 331–336, doi: 10.1016/0090-4295(75)90148-x (1975). [DOI] [PubMed] [Google Scholar]
- 75.Section 2: AKI Definition. Kidney Int Suppl (2011) 2, 19–36, doi: 10.1038/kisup.2011.32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Audard V et al. Acute kidney injury in sickle patients with painful crisis or acute chest syndrome and its relation to pulmonary hypertension. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 25, 2524–2529, doi: 10.1093/ndt/gfq083 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Lebensburger JD et al. Prevalence of acute kidney injury during pediatric admissions for acute chest syndrome. Pediatric nephrology 31, 1363–1368, doi: 10.1007/s00467-016-3370-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baddam S et al. Acute kidney injury during a pediatric sickle cell vaso-occlusive pain crisis. Pediatric nephrology 32, 1451–1456, doi: 10.1007/s00467-017-3623-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCormick M, Richardson T, Warady BA, Novelli EM & Kalpatthi R Acute kidney injury in paediatric patients with sickle cell disease is associated with increased morbidity and resource utilization. Br J Haematol 189, 559–565, doi: 10.1111/bjh.16384 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Olaniran KO, Allegretti AS, Zhao SH, Nigwekar SU & Kalim S Acute Kidney Injury among Black Patients with Sickle Cell Trait and Sickle Cell Disease. Clinical journal of the American Society of Nephrology : CJASN 16, 348–355, doi: 10.2215/CJN.06960520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yeruva SL, Paul Y, Oneal P & Nouraie M Renal Failure in Sickle Cell Disease: Prevalence, Predictors of Disease, Mortality and Effect on Length of Hospital Stay. Hemoglobin 40, 295–299, doi: 10.1080/03630269.2016.1224766 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saraf SL et al. HMOX1 and Acute Kidney Injury in Sickle Cell Anemia. Blood, doi: 10.1182/blood-2018-05-853929 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Olson JL, Hostetter TH, Rennke HG, Brenner BM & Venkatachalam MA Altered glomerular permselectivity and progressive sclerosis following extreme ablation of renal mass. Kidney international 22, 112–126, doi: 10.1038/ki.1982.143 (1982). [DOI] [PubMed] [Google Scholar]
- 84.Nath KA et al. Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. The American journal of pathology 166, 963–972, doi: 10.1016/s0002-9440(10)62318-8 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nath KA et al. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. The American journal of pathology 158, 893–903, doi: 10.1016/S0002-9440(10)64037-0 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saraf SL et al. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. British journal of haematology 164, 729–739, doi: 10.1111/bjh.12690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aleem A Renal abnormalities in patients with sickle cell disease: a single center report from Saudi Arabia. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia 19, 194–199 (2008). [PubMed] [Google Scholar]
- 88.Vasavda N et al. Renal iron load in sickle cell disease is influenced by severity of haemolysis. British journal of haematology 157, 599–605, doi: 10.1111/j.1365-2141.2012.09093.x (2012). [DOI] [PubMed] [Google Scholar]
- 89.Schein A, Enriquez C, Coates TD & Wood JC Magnetic resonance detection of kidney iron deposition in sickle cell disease: a marker of chronic hemolysis. J Magn Reson Imaging 28, 698–704, doi: 10.1002/jmri.21490 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saraf SL et al. Progressive glomerular and tubular damage in sickle cell trait and sickle cell anemia mouse models. Translational research : the journal of laboratory and clinical medicine 197, 1–11, doi: 10.1016/j.trsl.2018.01.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nath KA et al. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney international 59, 106–117, doi: 10.1046/j.1523-1755.2001.00471.x (2001). [DOI] [PubMed] [Google Scholar]
- 92.Ofori-Acquah SF et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood 135, 1044–1048, doi: 10.1182/blood.2019002653 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roy S et al. Increased Oxidative Stress In Sickle Cell Disease Activates The Renin-Angiotensin-TGF-β Pathway To Mediate Sickle Nephropathy. Blood 122, 2211–2211, doi: 10.1182/blood.V122.21.2211.2211 (2013). [DOI] [Google Scholar]
- 94.Roy S et al. Angiotensin receptor signaling in sickle cell anemia has a reno-protective effect on urine concentrating ability but results in sickle glomerulopathy. American journal of hematology 93, E177–E181, doi: 10.1002/ajh.25118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Siragy HM & Carey RM Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am J Nephrol 31, 541–550, doi: 10.1159/000313363 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Matavelli LC & Siragy HM AT2 receptor activities and pathophysiological implications. J Cardiovasc Pharmacol 65, 226–232, doi: 10.1097/FJC.0000000000000208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ataga KI et al. Albuminuria Is Associated with Endothelial Dysfunction and Elevated Plasma Endothelin-1 in Sickle Cell Anemia. PloS one 11, e0162652, doi: 10.1371/journal.pone.0162652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fischer C, Mazzone M, Jonckx B & Carmeliet P FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer 8, 942–956, doi: 10.1038/nrc2524 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Ahmed A New insights into the etiology of preeclampsia: identification of key elusive factors for the vascular complications. Thromb Res 127 Suppl 3, S72–75, doi: 10.1016/S0049-3848(11)70020-2 (2011). [DOI] [PubMed] [Google Scholar]
- 100.Landburg PP, Elsenga H, Schnog JB, Duits AJ & Group CS Increased serum levels of anti-angiogenic factors soluble fms-like tyrosine kinase and soluble endoglin in sickle cell disease. Acta Haematol 120, 130–133, doi: 10.1159/000178143 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Ataga KI et al. Association of soluble fms-like tyrosine kinase-1 with pulmonary hypertension and haemolysis in sickle cell disease. Br J Haematol 152, 485–491, doi: 10.1111/j.1365-2141.2010.08410.x (2011). [DOI] [PubMed] [Google Scholar]
- 102.Youssry I et al. Novel marker for the detection of sickle cell nephropathy: soluble FMS-like tyrosine kinase-1 (sFLT-1). Pediatric nephrology 30, 2163–2168, doi: 10.1007/s00467-015-3172-9 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Dimmeler S, Dernbach E & Zeiher AM Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett 477, 258–262, doi: 10.1016/s0014-5793(00)01657-4 (2000). [DOI] [PubMed] [Google Scholar]
- 104.Heimlich JB et al. Endothelin-1 contributes to the progression of renal injury in sickle cell disease via reactive oxygen species. British journal of pharmacology 173, 386–395, doi: 10.1111/bph.13380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kasztan M et al. Long-Term Endothelin-A Receptor Antagonism Provides Robust Renal Protection in Humanized Sickle Cell Disease Mice. Journal of the American Society of Nephrology : JASN 28, 2443–2458, doi: 10.1681/asn.2016070711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Higgs DR et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. The New England journal of medicine 306, 1441–1446, doi: 10.1056/NEJM198206173062402 (1982). [DOI] [PubMed] [Google Scholar]
- 107.Guasch A et al. Evidence that microdeletions in the alpha globin gene protect against the development of sickle cell glomerulopathy in humans. Journal of the American Society of Nephrology : JASN 10, 1014–1019 (1999). [DOI] [PubMed] [Google Scholar]
- 108.Nebor D et al. Alpha-thalassemia is associated with a decreased occurrence and a delayed age-at-onset of albuminuria in sickle cell anemia patients. Blood cells, molecules & diseases 45, 154–158, doi: 10.1016/j.bcmd.2010.06.003 (2010). [DOI] [PubMed] [Google Scholar]
- 109.Parsa A et al. APOL1 risk variants, race, and progression of chronic kidney disease. The New England journal of medicine 369, 2183–2196, doi: 10.1056/NEJMoa1310345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Genovese G et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845, doi: 10.1126/science.1193032 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Freedman BI et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. Journal of the American Society of Nephrology : JASN 21, 1422–1426, doi: 10.1681/ASN.2010070730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saraf SL et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica 100, 1275–1284, doi: 10.3324/haematol.2015.124875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kormann R et al. Roles of APOL1 G1 and G2 variants in sickle cell disease patients: kidney is the main target. British journal of haematology 179, 323–335, doi: 10.1111/bjh.14842 (2017). [DOI] [PubMed] [Google Scholar]
- 114.Schaefer BA et al. Genetic Modifiers of White Blood Cell Count, Albuminuria and Glomerular Filtration Rate in Children with Sickle Cell Anemia. PloS one 11, e0164364, doi: 10.1371/journal.pone.0164364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zahr RS et al. Children with sickle cell anemia and APOL1 genetic variants develop albuminuria early in life. Haematologica 104, e385–e387, doi: 10.3324/haematol.2018.212779 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Adebayo OC et al. Clinical and genetic factors are associated with kidney complications in African children with sickle cell anaemia. Br J Haematol, doi: 10.1111/bjh.17832 (2021). [DOI] [PubMed] [Google Scholar]
- 117.Saraf SL et al. APOL1, alpha-thalassemia, and BCL11A variants as a genetic risk profile for progression of chronic kidney disease in sickle cell anemia. Haematologica 102, e1–e6, doi: 10.3324/haematol.2016.154153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamada N et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. American journal of human genetics 66, 187–195, doi: 10.1086/302729 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Novitzky-Basso I & Rot A Duffy antigen receptor for chemokines and its involvement in patterning and control of inflammatory chemokines. Front Immunol 3, 266, doi: 10.3389/fimmu.2012.00266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Afenyi-Annan A et al. Lack of Duffy antigen expression is associated with organ damage in patients with sickle cell disease. Transfusion 48, 917–924, doi: 10.1111/j.1537-2995.2007.01622.x (2008). [DOI] [PubMed] [Google Scholar]
- 121.Farawela HM et al. Association between Duffy antigen receptor expression and disease severity in sickle cell disease patients. Hematology 21, 474–479, doi: 10.1080/10245332.2015.1111643 (2016). [DOI] [PubMed] [Google Scholar]
- 122.Platt OS et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 330, 1639–1644, doi: 10.1056/NEJM199406093302303 (1994). [DOI] [PubMed] [Google Scholar]
- 123.McClellan AC et al. High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol 159, 360–367, doi: 10.1111/bjh.12024 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nielsen L et al. Morbidity and mortality of sickle cell disease patients starting intermittent haemodialysis: a comparative cohort study with non- Sickle dialysis patients. British journal of haematology 174, 148–152, doi: 10.1111/bjh.14040 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Thrower A et al. Effect of renin-angiotensin-aldosterone system blocking agents on progression of glomerulopathy in sickle cell disease. British journal of haematology 184, 246–252, doi: 10.1111/bjh.15651 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Viner M et al. The morbidity and mortality of end stage renal disease in sickle cell disease. American journal of hematology 94, E138–E141, doi: 10.1002/ajh.25439 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liem RI et al. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv 3, 3867–3897, doi: 10.1182/bloodadvances.2019000916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shatat IF, Qanungo S, Hudson S, Laken MA & Hailpern SM Changes in Urine Microalbumin-to-Creatinine Ratio in Children with Sickle Cell Disease over Time. Front Pediatr 4, 106, doi: 10.3389/fped.2016.00106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chapter 1: Definition and classification of CKD. Kidney Int Suppl (2011) 3, 19–62, doi: 10.1038/kisup.2012.64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Asnani MR, Lynch O & Reid ME Determining glomerular filtration rate in homozygous sickle cell disease: utility of serum creatinine based estimating equations. PloS one 8, e69922, doi: 10.1371/journal.pone.0069922 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arlet JB et al. Determination of the best method to estimate glomerular filtration rate from serum creatinine in adult patients with sickle cell disease: a prospective observational cohort study. BMC nephrology 13, 83, doi: 10.1186/1471-2369-13-83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Asnani M & Reid M Cystatin C: a useful marker of glomerulopathy in sickle cell disease? Blood cells, molecules & diseases 54, 65–70, doi: 10.1016/j.bcmd.2014.07.018 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Yee MEM et al. Estimation of glomerular filtration rate using serum cystatin C and creatinine in adults with sickle cell anemia. American journal of hematology 92, E598–E599, doi: 10.1002/ajh.24840 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Inker LA et al. New Creatinine- and Cystatin C-Based Equations to Estimate GFR without Race. The New England journal of medicine 385, 1737–1749, doi: 10.1056/NEJMoa2102953 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alvarez O et al. Effect of hydroxyurea treatment on renal function parameters: results from the multi-center placebo-controlled BABY HUG clinical trial for infants with sickle cell anemia. Pediatric blood & cancer 59, 668–674, doi: 10.1002/pbc.24100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lebensburger JD et al. High bias and low precision for estimated versus measured glomerular filtration rate in pediatric sickle cell anemia. Haematologica, doi: 10.3324/haematol.2019.242156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eneanya ND et al. Health inequities and the inappropriate use of race in nephrology. Nat Rev Nephrol, doi: 10.1038/s41581-021-00501-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saraf SL et al. Manifestations of Reduced Kidney Function Occur at a Higher Estimated Glomerular Filtration Rate in Sickle Cell Anemia. Blood 134, 2268–2268, doi: 10.1182/blood-2019-124737 (2019). [DOI] [Google Scholar]
- 139.Zahr RS et al. Kidney biopsy findings in children with sickle cell disease: a Midwest Pediatric Nephrology Consortium study. Pediatric nephrology 34, 1435–1445, doi: 10.1007/s00467-019-04237-3 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Sekulic M & Pichler Sekulic S A Compendium of Urinary Biomarkers Indicative of Glomerular Podocytopathy. Patholog Res Int 2013, 782395, doi: 10.1155/2013/782395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ng DP et al. Nephrinuria associates with multiple renal traits in type 2 diabetes. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 26, 2508–2514, doi: 10.1093/ndt/gfq738 (2011). [DOI] [PubMed] [Google Scholar]
- 142.Kern EFO, Erhard P, Sun WJ, Genuth S & Weiss MF Early Urinary Markers of Diabetic Kidney Disease: A Nested Case-Control Study From the Diabetes Control and Complications Trial (DCCT). American Journal of Kidney Diseases 55, 824–834, doi: 10.1053/j.ajkd.2009.11.009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Han WK, Bailly V, Abichandani R, Thadhani R & Bonventre JV Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney international 62, 237–244, doi: 10.1046/j.1523-1755.2002.00433.x (2002). [DOI] [PubMed] [Google Scholar]
- 144.Voskaridou E et al. Early markers of renal dysfunction in patients with sickle cell/beta-thalassemia. Kidney international 69, 2037–2042, doi: 10.1038/sj.ki.5000248 (2006). [DOI] [PubMed] [Google Scholar]
- 145.Hamideh D et al. Albuminuria correlates with hemolysis and NAG and KIM-1 in patients with sickle cell anemia. Pediatric nephrology, doi: 10.1007/s00467-014-2821-8 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Rovin BH, Yoshiumura T & Tan L Cytokine-induced production of monocyte chemoattractant protein-1 by cultured human mesangial cells. J Immunol 148, 2148–2153 (1992). [PubMed] [Google Scholar]
- 147.Khaibullina A et al. RON kinase inhibition reduces renal endothelial injury in sickle cell disease mice. Haematologica 103, 787–798, doi: 10.3324/haematol.2017.180992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chow FY et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney international 69, 73–80, doi: 10.1038/sj.ki.5000014 (2006). [DOI] [PubMed] [Google Scholar]
- 149.Rovin BH et al. Urine chemokines as biomarkers of human systemic lupus erythematosus activity. Journal of the American Society of Nephrology : JASN 16, 467–473, doi: 10.1681/ASN.2004080658 (2005). [DOI] [PubMed] [Google Scholar]
- 150.dos Santos TE, Goncalves RP, Barbosa MC, da Silva GB Jr. & Daher Ede F Monocyte chemoatractant protein-1: a potential biomarker of renal lesion and its relation with oxidative status in sickle cell disease. Blood cells, molecules & diseases 54, 297–301, doi: 10.1016/j.bcmd.2014.11.019 (2015). [DOI] [PubMed] [Google Scholar]
- 151.Belisario AR et al. Evidence for interactions between inflammatory markers and renin-angiotensin system molecules in the occurrence of albuminuria in children with sickle cell anemia. Cytokine 125, 154800, doi: 10.1016/j.cyto.2019.154800 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Elsherif L, Pathmasiri W, McRitchie S, Archer DR & Ataga KI Plasma metabolomics analysis in sickle cell disease patients with albuminuria - an exploratory study. British journal of haematology 185, 620–623, doi: 10.1111/bjh.15592 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kalantari S et al. (1) H NMR-based metabolomics exploring urinary biomarkers correlated with proteinuria in focal segmental glomerulosclerosis: a pilot study. Magn Reson Chem 54, 821–826, doi: 10.1002/mrc.4460 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Vallance P & Leiper J Asymmetric dimethylarginine and kidney disease--marker or mediator? Journal of the American Society of Nephrology : JASN 16, 2254–2256, doi: 10.1681/ASN.2005050539 (2005). [DOI] [PubMed] [Google Scholar]
- 155.Schnog JB et al. Plasma levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell disease. Annals of hematology 84, 282–286, doi: 10.1007/s00277-004-0983-3 (2005). [DOI] [PubMed] [Google Scholar]
- 156.Lee MJ et al. Serum Ceruloplasmin Level as a Predictor for the Progression of Diabetic Nephropathy in Korean Men with Type 2 Diabetes Mellitus. Diabetes Metab J 39, 230–239, doi: 10.4093/dmj.2015.39.3.230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jerebtsova M et al. Identification of ceruloplasmin as a biomarker of chronic kidney disease in urine of sickle cell disease patients by proteomic analysis. American journal of hematology 93, E45–E47, doi: 10.1002/ajh.24965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jiang H et al. Increased urinary excretion of orosomucoid is a risk predictor of diabetic nephropathy. Nephrology 14, 332–337, doi: 10.1111/j.1440-1797.2008.01053.x (2009). [DOI] [PubMed] [Google Scholar]
- 159.Watson L et al. Urinary monocyte chemoattractant protein 1 and alpha 1 acid glycoprotein as biomarkers of renal disease activity in juvenile-onset systemic lupus erythematosus. Lupus 21, 496–501, doi: 10.1177/0961203311431249 (2012). [DOI] [PubMed] [Google Scholar]
- 160.Jerebtsova M et al. Urinary orosomucoid is associated with progressive chronic kidney disease stage in patients with sickle cell anemia. American journal of hematology 93, E107–E109, doi: 10.1002/ajh.25036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jerebtsova M et al. Association between plasma and urinary orosomucoid and chronic kidney disease in adults with sickle cell disease. British journal of haematology 190, e45–e48, doi: 10.1111/bjh.16702 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Charache S et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. The New England journal of medicine 332, 1317–1322, doi: 10.1056/NEJM199505183322001 (1995). [DOI] [PubMed] [Google Scholar]
- 163.Zahr RS et al. Hydroxyurea prevents onset and progression of albuminuria in children with sickle cell anemia. American journal of hematology 94, E27–E29, doi: 10.1002/ajh.25329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bartolucci P et al. Six Months of Hydroxyurea Reduces Albuminuria in Patients with Sickle Cell Disease. Journal of the American Society of Nephrology : JASN 27, 1847–1853, doi: 10.1681/ASN.2014111126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rees DC, Robinson S & Howard J How I manage red cell transfusions in patients with sickle cell disease. British journal of haematology 180, 607–617, doi: 10.1111/bjh.15115 (2018). [DOI] [PubMed] [Google Scholar]
- 166.Itano HA, Keitel HG & Thompson D Hyposthenuria in sickle cell anemia: a reversible renal defect. The Journal of clinical investigation 35, 998–1007, doi: 10.1172/JCI103360 (1956). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Alvarez O et al. Kidney function of transfused children with sickle cell anemia: Baseline data from the TWiTCH study with comparison to non-transfused cohorts. American journal of hematology 92, E637–E639, doi: 10.1002/ajh.24871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Foucan L et al. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. The American journal of medicine 104, 339–342 (1998). [DOI] [PubMed] [Google Scholar]
- 169.Kaschina E, Namsolleck P & Unger T AT2 receptors in cardiovascular and renal diseases. Pharmacol Res 125, 39–47, doi: 10.1016/j.phrs.2017.07.008 (2017). [DOI] [PubMed] [Google Scholar]
- 170.Yee ME et al. Losartan therapy decreases albuminuria with stable glomerular filtration and permselectivity in sickle cell anemia. Blood cells, molecules & diseases 69, 65–70, doi: 10.1016/j.bcmd.2017.09.006 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Quinn CT et al. Losartan for the nephropathy of sickle cell anemia: A phase-2, multicenter trial. American journal of hematology 92, E520–E528, doi: 10.1002/ajh.24810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kutlar A et al. Phase-I Study of ETA Receptor Antagonist Ambrisentan in Sickle Cell Disease. Blood 134, doi: 10.1182/blood-2019-130036 (2019). [DOI] [Google Scholar]
- 173.Margaritis M, Channon KM & Antoniades C Statins as regulators of redox state in the vascular endothelium: beyond lipid lowering. Antioxidants & redox signaling 20, 1198–1215, doi: 10.1089/ars.2013.5430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ataga KI et al. A pilot study of the effect of atorvastatin on endothelial function and albuminuria in sickle cell disease. American journal of hematology 94, E299–E301, doi: 10.1002/ajh.25614 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Zahr RS et al. Renal protection by atorvastatin in a murine model of sickle cell nephropathy. British journal of haematology 181, 111–121, doi: 10.1111/bjh.15157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vichinsky E et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. The New England journal of medicine 381, 509–519, doi: 10.1056/NEJMoa1903212 (2019). [DOI] [PubMed] [Google Scholar]
- 177.Ataga KI et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. The New England journal of medicine 376, 429–439, doi: 10.1056/NEJMoa1611770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pegelow CH et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. The American journal of medicine 102, 171–177 (1997). [DOI] [PubMed] [Google Scholar]
- 179.Desai PC et al. Decades after the cooperative study: a re-examination of systemic blood pressure in sickle cell disease. American journal of hematology 87, E65–68, doi: 10.1002/ajh.23278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gordeuk VR et al. Relative systemic hypertension in patients with sickle cell disease is associated with risk of pulmonary hypertension and renal insufficiency. American journal of hematology 83, 15–18, doi: 10.1002/ajh.21016 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Whelton PK et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71, e13–e115, doi: 10.1161/HYP.0000000000000065 (2018). [DOI] [PubMed] [Google Scholar]
- 182.Benneh-Akwasi Kuma A et al. Prevalence of relative systemic hypertension in adults with sickle cell disease in Ghana. PloS one 13, e0190347, doi: 10.1371/journal.pone.0190347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.James PA et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311, 507–520, doi: 10.1001/jama.2013.284427 (2014). [DOI] [PubMed] [Google Scholar]
- 184.Jang T et al. Vaso-occlusive crisis in sickle cell disease: a vicious cycle of secondary events. J Transl Med 19, 397, doi: 10.1186/s12967-021-03074-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Little JA et al. Combination erythropoietin-hydroxyurea therapy in sickle cell disease: experience from the National Institutes of Health and a literature review. Haematologica 91, 1076–1083 (2006). [PMC free article] [PubMed] [Google Scholar]
- 186.Han J et al. Erythropoiesis-stimulating agents in sickle cell anaemia. British journal of haematology 182, 602–605, doi: 10.1111/bjh.14846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Boyle SM, Jacobs B, Sayani FA & Hoffman B Management of the Dialysis Patient with Sickle Cell Disease. Seminars in dialysis 29, 62–70, doi: 10.1111/sdi.12403 (2016). [DOI] [PubMed] [Google Scholar]
- 188.Porter J & Garbowski M Consequences and management of iron overload in sickle cell disease. Hematology Am Soc Hematol Educ Program 2013, 447–456, doi: 10.1182/asheducation-2013.1.447 (2013). [DOI] [PubMed] [Google Scholar]
- 189.Vichinsky E et al. Long-term safety and efficacy of deferasirox (Exjade) for up to 5 years in transfusional iron-overloaded patients with sickle cell disease. Br J Haematol 154, 387–397, doi: 10.1111/j.1365-2141.2011.08720.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Maker GL et al. Pharmacokinetics and safety of deferasirox in subjects with chronic kidney disease undergoing haemodialysis. Nephrology 18, 188–193, doi: 10.1111/nep.12035 (2013). [DOI] [PubMed] [Google Scholar]
- 191.Yusuf B, McPhedran P & Brewster UC Hypocalcemia in a dialysis patient treated with deferasirox for iron overload. American journal of kidney diseases : the official journal of the National Kidney Foundation 52, 587–590, doi: 10.1053/j.ajkd.2008.03.034 (2008). [DOI] [PubMed] [Google Scholar]
- 192.Kontoghiorghes GJ, Kolnagou A, Skiada A & Petrikkos G The role of iron and chelators on infections in iron overload and non iron loaded conditions: prospects for the design of new antimicrobial therapies. Hemoglobin 34, 227–239, doi: 10.3109/03630269.2010.483662 (2010). [DOI] [PubMed] [Google Scholar]
- 193.Falk RJ et al. Iron removal during continuous ambulatory peritoneal dialysis using deferoxamine. Kidney international 24, 110–112, doi: 10.1038/ki.1983.132 (1983). [DOI] [PubMed] [Google Scholar]
- 194.Abbott KC, Hypolite IO & Agodoa LY Sickle cell nephropathy at end-stage renal disease in the United States: patient characteristics and survival. Clinical nephrology 58, 9–15 (2002). [DOI] [PubMed] [Google Scholar]
- 195.Olaniran KO, N. S, Eneanya ND, Zhao S, Ofsthun NJ, Dalrymple LS, Maddux FW, Thadhani RI. Mortality and morbidity among African American patients with sickle cell disease and ESRD initiating dialysis. [Abstract]. J Am Soc Nephrol 30, 1037 (2019).31015255 [Google Scholar]
- 196.Ojo AO et al. Renal transplantation in end-stage sickle cell nephropathy. Transplantation 67, 291–295 (1999). [DOI] [PubMed] [Google Scholar]
- 197.Kwarteng-Siaw M, Heydarpour M, Baker O, Tucker K & Achebe M Morbidity and Mortality Associated with Hemodialysis Versus Peritoneal Dialysis in Patients with End Stage Renal Disease Caused By Sickle Cell Disease. Blood 138, 488–488, doi: 10.1182/blood-2021-144872 (2021). [DOI] [Google Scholar]
- 198.Huang E et al. Improved survival among sickle cell kidney transplant recipients in the recent era. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 28, 1039–1046, doi: 10.1093/ndt/gfs585 (2013). [DOI] [PubMed] [Google Scholar]
- 199.Gerardin C et al. Survival and specific outcome of sickle cell disease patients after renal transplantation. Br J Haematol 187, 676–680, doi: 10.1111/bjh.16113 (2019). [DOI] [PubMed] [Google Scholar]
- 200.Leeaphorn N et al. Outcomes of Kidney Transplant Recipients with Sickle Cell Disease: An Analysis of the 2000–2019 UNOS/OPTN Database. J Clin Med 10, doi: 10.3390/jcm10143063 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bae S et al. Mortality and Access to Kidney Transplantation in Patients with Sickle Cell Disease-Associated Kidney Failure. Clinical journal of the American Society of Nephrology : CJASN 16, 407–414, doi: 10.2215/CJN.02720320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nath J et al. Sickle cell and renal transplant: a national survey and literature review. Exp Clin Transplant 10, 1–7, doi: 10.6002/ect.2011.0098 (2012). [DOI] [PubMed] [Google Scholar]
- 203.Allen A, Scoble J, Snowden S, Hambley H & Bellingham A Hydroxyurea, sickle cell disease and renal transplantation. Nephron 75, 106–107, doi: 10.1159/000189511 (1997). [DOI] [PubMed] [Google Scholar]
- 204.Willis JC et al. Outcomes following kidney transplantation in patients with sickle cell disease: The impact of automated exchange blood transfusion. PloS one 15, e0236998, doi: 10.1371/journal.pone.0236998 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kato GJ et al. Sickle cell disease. Nat Rev Dis Primers 4, 18010, doi: 10.1038/nrdp.2018.10 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Key NS, Connes P & Derebail VK Negative health implications of sickle cell trait in high income countries: from the football field to the laboratory. Br J Haematol 170, 5–14, doi: 10.1111/bjh.13363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Aloni MN et al. Congolese children with sickle cell trait may exhibit glomerular hyperfiltration: A case control study. J Clin Lab Anal 31, doi: 10.1002/jcla.22143 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gupta AK et al. Effects of alpha-thalassemia and sickle polymerization tendency on the urine-concentrating defect of individuals with sickle cell trait. The Journal of clinical investigation 88, 1963–1968, doi: 10.1172/JCI115521 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ahmed SG & Ibrahim UA Haemoglobin-S in sickle cell trait with papillary necrosis. Br J Haematol 135, 415–416, doi: 10.1111/j.1365-2141.2006.06318.x (2006). [DOI] [PubMed] [Google Scholar]
- 210.Naik RP et al. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. Jama 312, 2115–2125, doi: 10.1001/jama.2014.15063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Naik RP et al. Sickle Cell Trait and the Risk of ESRD in Blacks. J Am Soc Nephrol 28, 2180–2187, doi: 10.1681/ASN.2016101086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kramer HJ et al. African Ancestry-Specific Alleles and Kidney Disease Risk in Hispanics/Latinos. J Am Soc Nephrol 28, 915–922, doi: 10.1681/ASN.2016030357 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sood R et al. Sickle cell trait, estimated glomerular filtration rate, and risk of adverse outcomes in chronic kidney disease. American journal of hematology 94, E275–E278, doi: 10.1002/ajh.25588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bucknor MD, Goo JS & Coppolino ML The risk of potential thromboembolic, renal and cardiac complications of sickle cell trait. Hemoglobin 38, 28–32, doi: 10.3109/03630269.2013.832689 (2014). [DOI] [PubMed] [Google Scholar]
- 215.Hu J et al. Sickle cell trait and renal disease among African American U.S. Army soldiers. Br J Haematol 185, 532–540, doi: 10.1111/bjh.15820 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Derebail VK et al. Variant hemoglobin phenotypes may account for differential erythropoiesis-stimulating agent dosing in African-American hemodialysis patients. Kidney international 80, 992–999, doi: 10.1038/ki.2011.247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Derebail VK et al. Sickle trait in African-American hemodialysis patients and higher erythropoiesis-stimulating agent dose. J Am Soc Nephrol 25, 819–826, doi: 10.1681/ASN.2013060575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim L, Garfinkel MR, Chang A, Kadambi PV & Meehan SM Intragraft vascular occlusive sickle crisis with early renal allograft loss in occult sickle cell trait. Hum Pathol 42, 1027–1033, doi: 10.1016/j.humpath.2010.09.013 (2011). [DOI] [PubMed] [Google Scholar]
- 219.Reese PP, Hoo AC & Magee CC Screening for sickle trait among potential live kidney donors: policies and practices in US transplant centers. Transpl Int 21, 328–331, doi: 10.1111/j.1432-2277.2007.00611.x (2008). [DOI] [PubMed] [Google Scholar]
- 220.Delgado C et al. A Unifying Approach for GFR Estimation: Recommendations of the NKF-ASN Task Force on Reassessing the Inclusion of Race in Diagnosing Kidney Disease. J Am Soc Nephrol, doi: 10.1681/ASN.2021070988 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Astor BC et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney international 79, 1331–1340, doi: 10.1038/ki.2010.550 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gansevoort RT et al. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney international 80, 93–104, doi: 10.1038/ki.2010.531 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Heerspink HJL et al. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 383, 1436–1446, doi: 10.1056/NEJMoa2024816 (2020). [DOI] [PubMed] [Google Scholar]
- 224.Perkovic V et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 380, 2295–2306, doi: 10.1056/NEJMoa1811744 (2019). [DOI] [PubMed] [Google Scholar]
- 225.Wanner C et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med 375, 323–334, doi: 10.1056/NEJMoa1515920 (2016). [DOI] [PubMed] [Google Scholar]
- 226.Kopp JB et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. Journal of the American Society of Nephrology : JASN 22, 2129–2137, doi: 10.1681/ASN.2011040388 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Davis CJ Jr., Mostofi FK & Sesterhenn IA Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol 19, 1–11, doi: 10.1097/00000478-199501000-00001 (1995). [DOI] [PubMed] [Google Scholar]
- 228.Alvarez O, Rodriguez MM, Jordan L & Sarnaik S Renal medullary carcinoma and sickle cell trait: A systematic review. Pediatric blood & cancer 62, 1694–1699, doi: 10.1002/pbc.25592 (2015). [DOI] [PubMed] [Google Scholar]
- 229.Shapiro DD et al. Association of High-Intensity Exercise with Renal Medullary Carcinoma in Individuals with Sickle Cell Trait: Clinical Observations and Experimental Animal Studies. Cancers (Basel) 13, doi: 10.3390/cancers13236022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Msaouel P, Tannir NM & Walker CL A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 2044–2049, doi: 10.1158/1078-0432.ccr-17-3296 (2018). [DOI] [PubMed] [Google Scholar]
- 231.Swartz MA et al. Renal medullary carcinoma: clinical, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology 60, 1083–1089, doi: 10.1016/s0090-4295(02)02154-4 (2002). [DOI] [PubMed] [Google Scholar]
- 232.Weber ML et al. Morphine promotes renal pathology in sickle mice. Int J Nephrol Renovasc Dis 5, 109–118, doi: 10.2147/IJNRD.S33813 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Thrower A, Z. L, Derebail VK, Wichlan D, Smith A, Cai J, Ataga KI Opioid analgesics are associated with albuminuria in adult patients with sickle cell anemia [Abstract]. Blood 134, 2308 (2019). [Google Scholar]
- 234.Marsenic O, Couloures KG & Wiley JM Proteinuria in children with sickle cell disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 23, 715–720, doi: 10.1093/ndt/gfm858 (2008). [DOI] [PubMed] [Google Scholar]
- 235.Becton LJ et al. Prevalence and clinical correlates of microalbuminuria in children with sickle cell disease. Pediatric nephrology 25, 1505–1511, doi: 10.1007/s00467-010-1536-8 (2010). [DOI] [PubMed] [Google Scholar]
- 236.Aygun B, Mortier NA, Smeltzer MP, Hankins JS & Ware RE Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatric nephrology 26, 1285–1290, doi: 10.1007/s00467-011-1857-2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.King L, MooSang M, Miller M & Reid M Prevalence and predictors of microalbuminuria in Jamaican children with sickle cell disease. Archives of disease in childhood 96, 1135–1139, doi: 10.1136/archdischild-2011-300628 (2011). [DOI] [PubMed] [Google Scholar]
- 238.Aloni MN et al. Prevalence and determinants of microalbuminuria in children suffering from sickle cell anemia in steady state. Clinical kidney journal 10, 479–486, doi: 10.1093/ckj/sfx058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ekpenyong EE, Ikpeme EE, Bassey EG & Dixon-Umo OT Early Detection of Renal Injury in Children with Sickle Cell Anaemia using Microalbuminuria in a Tertiary Health Institution in Southern Nigeria. West Afr J Med 37, 412–417 (2020). [PubMed] [Google Scholar]
- 240.Ocheke IE, Mohamed S, Okpe ES, Bode-Thomas F & McCullouch MI Microalbuminuria risks and glomerular filtration in children with sickle cell anaemia in Nigeria. Ital J Pediatr 45, 143, doi: 10.1186/s13052-019-0720-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Al-Musawa FE & Al-Saqladi AM Prevalence and correlates of microalbuminuria in Yemeni children with sickle cell disease. Saudi J Kidney Dis Transpl 30, 832–842, doi: 10.4103/1319-2442.265459 (2019). [DOI] [PubMed] [Google Scholar]
- 242.Forrest S, Kim A, Carbonella J & Pashankar F Proteinuria is associated with elevated tricuspid regurgitant jet velocity in children with sickle cell disease. Pediatric blood & cancer 58, 937–940, doi: 10.1002/pbc.23338 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.