

Abstract
F1Fo ATP synthase functions as a biological generator and makes a major contribution to cellular energy production. Proton flow generates rotation in the Fo motor that is transferred to the F1 motor to catalyze ATP production, with flexible F1/Fo coupling required for efficient catalysis. F1Fo ATP synthase can also operate in reverse, hydrolyzing ATP and pumping protons, and in bacteria this function can be regulated by an inhibitory ε subunit. Here we present cryo-EM data showing E. coli F1Fo ATP synthase in different rotational and inhibited sub-states, observed following incubation with 10 mM MgATP. Our structures demonstrate how structural transitions within the inhibitory ε subunit induce torsional movement in the central stalk, thereby enabling its rotation within the Fο motor. This highlights the importance of the central rotor for flexible coupling of the F1 and Fo motors and provides further insight into the regulatory mechanism mediated by subunit ε.
Subject terms: Bioenergetics, Cryoelectron microscopy
Cryo-EM structures of E. coli F1Fo ATP synthase highlight the role of the inhibitory ε subunit in regulating the torsional movement of the central stalk within the Fo motor and central stalk flexibility in coupling the F1 and Fo motors.
Introduction
A key component in the generation of cellular energy is the F1Fo ATP synthase, a biological rotary motor that converts proton motive force (pmf) to adenosine tri-phosphate (ATP) in both oxidative phosphorylation and photophosphorylation1–4. The enzyme is based on two rotary motors, termed F1 and Fo, that are coupled together by two stalks: a central rotor stalk and a peripheral stator stalk (Fig. 1a). The central stalk is comprised of subunits ε and γ and rotates during the enzyme’s catalytic cycle. The peripheral stalk is a homodimer comprised of b subunits, and connects the non-rotating components together. During ATP synthesis, the membrane-bound Fo motor converts the pmf into rotation of the Fo rotor ring, this rotation is transferred to the F1 motor via the central rotor which in turn induces conformational changes in the catalytic subunits. These conformational changes alter the chemical environment in the catalytic sites within F1 so that ATP is synthesized from ADP and inorganic phosphate (Pi)5,6. The F1 motor has pseudo three-fold symmetry5,7 with six dwell positions8, but the symmetry of the Fo varies across species with the Escherichia coli enzyme having tenfold rotational symmetry (10 c subunits in the rotor ring9). Because of the symmetry mismatch between the F1 and Fo motors, it has been hypothesized that they need to be flexibly coupled for efficient function10, with the peripheral and/or central stalks flexing to alleviate the symmetry mismatch between them11,12.
Fig. 1. The ε subunit of E. coli F1Fo ATP synthase.

a Schematic of E. coli F1Fo ATP synthase with subunits colored and labeled. The ε subunit is shown in green in the inhibited up conformation. b Crystal structure of the ε subunit in the up conformation (PDB:3oaa16), with α, β, and γ subunits removed for clarity. c Crystal structure of the isolated ε subunit in the down conformation (PDB:1aqt20).
F1Fo ATP synthase can operate in reverse, hydrolyzing ATP and pumping protons. However, cells have evolved inhibitory mechanisms to avoid wasteful hydrolysis of ATP that could occur under certain physiological conditions. Bacterial ATP synthases appear to utilize a range of different mechanisms for inhibition, with nucleotides, ions and conformational changes making contributions13–15. E. coli and other related bacteria exploit an internal subunit, termed ε, that can sense the environment and inhibit the enzyme under certain conditions13. In E. coli, subunit ε can be divided into N-terminal (εNTD) and C-terminal (εCTD) domains. The εNTD is a β-sandwich with two five-stranded sheets whereas the εCTD is formed from two short helices, residues 87–102 and 110–136 (referred to as εCTH1 and εCTH2 respectively), connected by a linker (Fig. 1b). It has been hypothesized that the εCTD is able to inhibit the F1 motor by extending up and blocking rotation under certain conditions16,17. Multiple structural studies examining E. coli F1 or F1Fo ATP synthase either in the absence of nucleotide18 or in the presence of AMPPNP16 or MgADP19, have shown the εCTD oriented in an extended up position (Fig. 1b), whereas the isolated ε subunit has also been crystallized in a condensed down position20 (Fig. 1c). Previously, we reported ~5 Å resolution structure of E. coli F1Fo ATP synthase following incubation with 10 mM MgATP21, which confirmed that the εCTD transitions to a condensed down conformation via a half-up intermediate.
To define the transitions of the εCTD in greater detail and understand how it regulates function, we have used cryo-EM to examine detergent-solubilized E. coli F1Fo ATP synthase22 following a 45 s incubation with 10 mM MgATP. These conditions allow the enzyme to be observed operating in the hydrolysis direction, and are similar to the concentrations found in E. coli undergoing aerobic respiration23 (though with much lower Pi concentration24). Strikingly, the improved detail in this study shows that the transition of the εCTD to a down sub-state is associated with a substantial torsional flexing of the central stalk, as evident by rotation of the εNTD about the γ subunit. This flexing, in combination with bending in the peripheral stalk, mediates rotation in the Fo motor. Our work provides a structural framework for role of the central stalk in flexible coupling within F1Fo ATP synthases and suggests a hypothesis of how pmf could modulate the enzyme’s structure and function.
Results
Cryo-EM analysis of F1Fo ATP synthase following incubation with MgATP
Cryo-EM maps of E. coli F1Fo ATP synthase in the presence of 10 mM MgATP were obtained using methods similar to those in previous studies18,19,21,25 (Supplementary Figs. 1 and 2) and provided superior structural information than was observed previously21. Previous work18 had identified three major conformational states of the enzyme in which the central stalk is rotated by ~120° relative to peripheral stalk. These states are termed State 1, State 2 and State 3, with the order referring to the enzyme operating in ATP hydrolysis direction (Supplementary Fig. 2). An overall resolution of 2.7–3.0 Å was achieved for the three rotational states, which enabled bound nucleotides to be identified and modeled (Fig. 2a). The three catalytic β subunits in the present study were identified as containing β1 (βDP); MgADP, β2 (βE); ADP and β3 (βTP); MgATP (Fig. 2a). Compared to the same enzyme imaged in the presence of 10 mM MgADP19, the central stalk had rotated as a rigid body by ~10° counterclockwise, when viewed from the membrane (Fig. 2b and Supplementary Fig. 3), the εCTH2 has dissociated from the central stalk, and the β1 (βDP) subunit has closed to contact the γ subunit (Fig. 2c) and bind MgADP. Comparison of the relative position of the rotor axel between known structures8,16,19,26–28, highlighted that the F1-ATPase was in a similar rotary position to that of observed of Geobacillus stearothermophilus (also termed Bacillus PS3) in the catalytic dwell8 (Supplementary Fig. 3), indicating that this enzyme was in a similar state. In the maps of these three states, the details of the Fo region remained ambiguous and hence further data processing was performed to verify the location of the εCTD and c-ring, as described in the following section.
Fig. 2. Nucleotide occupancy and conformational changes in the F1 motor following incubation with MgATP.
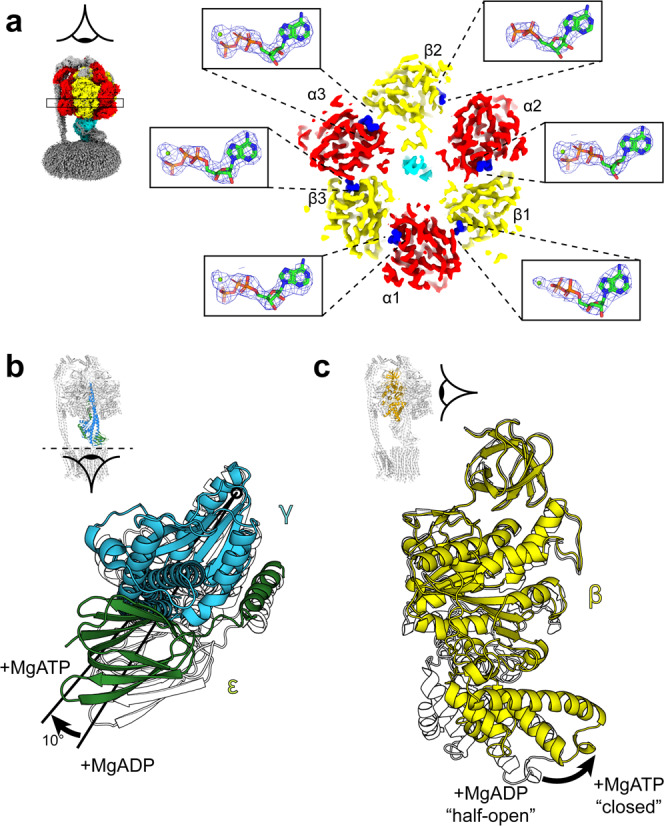
a Horizontal section of the State 2 E. coli F1Fo ATP synthase cryo-EM map and details of the β subunit catalytic site occupancies (with equivalent mitochondrial F1 nomenclature:5 β1 = βDP, β2 = βE, β3 = βTP, as named for the E. coli enzyme16). β1 contains MgADP, β2 contains ADP, and β3 contains MgATP. Section of map contoured to 0.028 in ChimeraX62 and mesh for nucleotides contoured to isolevel 10 in PyMol (Schrödinger). b, c Comparison of the F1 motor after incubation with MgATP (this study; γ in blue, ε in green and β in yellow) or MgADP (PDB:6oqv19; subunits shown as outline). b The central stalk (subunits γ and ε) is rotated ~10° clockwise when viewed from the membrane (structures are aligned to the F1 β barrel crown). c β1 closes inwards from a half-open to closed conformation.
Masked 3D classification focusing on the central rotor highlighted sub-states in which the εCTD adopted either a condensed down conformation or an extended half-up conformation (Fig. 3 and Supplementary Fig. 2). The maps generated for each of these sub-states showed the position of the εCTD, with local resolution estimates of 4–5 Å (Supplementary Fig. 4). However, due to the likely high flexibility of this sample, it was still difficult to unequivocally assign the position of the membrane domain subunits (a, b, and c subunits) in the maps. To obtain clearer information in this region, a refinement that focused on the Fo motor was performed (Supplementary Fig. 2) and produced maps of sufficient detail to enable fitting of the membrane region. These maps were combined (using Phenix29; maps and models provided as Supplementary Data 1–4) with the maps obtained without focused refinement to visualize overall changes between sub-states.
Fig. 3. E. coli F1Fo ATP synthase ε subunit in three conformational sub-states.
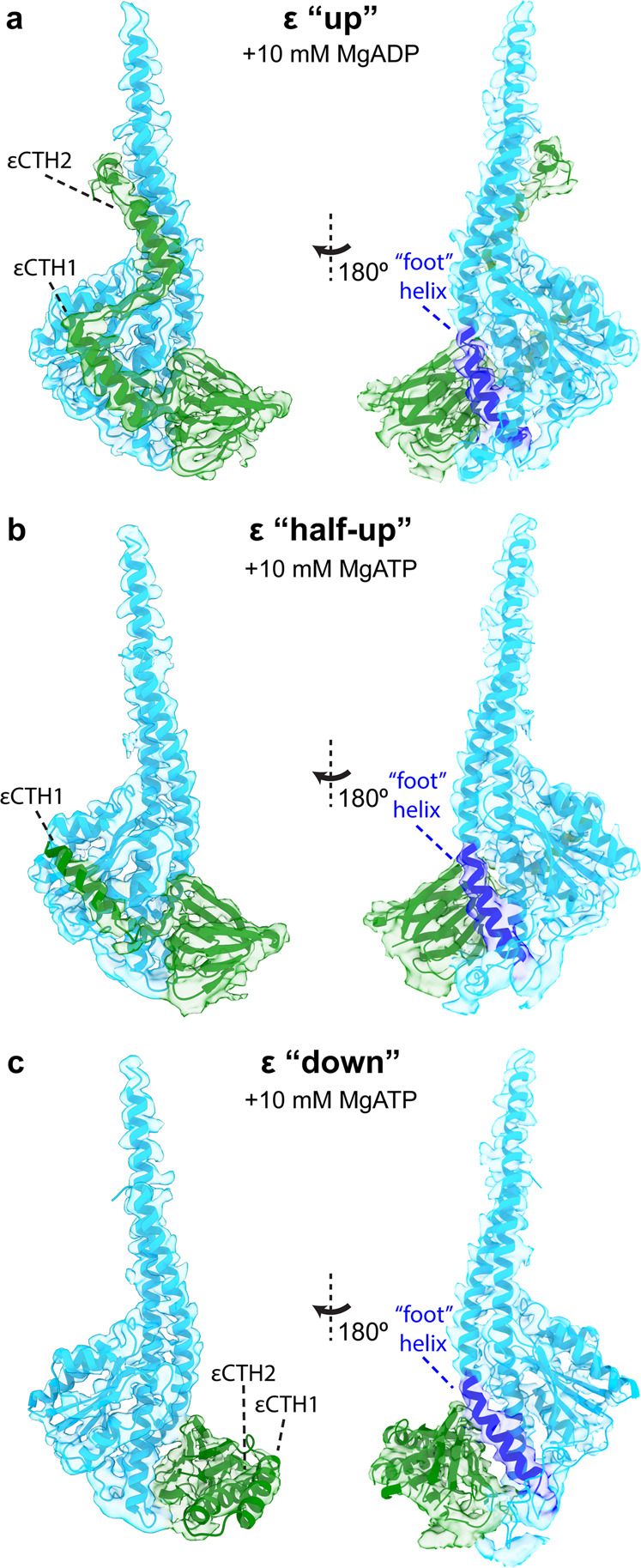
Cryo-EM maps (transparent surface) and molecular models (cartoon representation) of the E. coli F1Fo ATP synthase rotor in three conformation sub-states. Subunit γ in light blue and ε in green, with the foot helix of subunit γ labeled in dark blue. a The εCTD up sub-state observed after addition of 10 mM MgADP (PDB:6oqv; EMDB:2017119). b The εCTD half-up sub-state observed after addition of 10 mM MgATP (State 2 half-up in this study). c The εCTD down sub-state observed after addition of 10 mM MgATP (State 2 down in this study). See Supplementary Fig. 5 for close up views of the cryo-EM maps for the εCTD and γ foot.
Subunit ε stabilizes the conformation of the central stalk
In this study we observed the εCTD in two conformations, a half-up and a down sub-state. Although the resolution of the maps was not atomic (Fig. 3 and Supplementary Fig. 4) they contained clear detail (4-5 Å resolution) that showed the position of the α-helices and enabled the docking and fitting of known crystal20 and cryo-EM19 models (Supplementary Fig. 5), providing a clear picture of the molecular arrangement in this system. In the half up sub-states, no density for εCTH2 was observed, however εCTH1 remained attached to subunit γ (Fig. 3b). In the down sub-state, both the εCTH1 and εCTH2 were folded on to each other as seen in the isolated E. coli ε subunit crystal structure20 (Fig. 3c). The sub-states of State 2 showed the best detail for the two εCTD conformations (Fig. 3 and Supplementary Fig. 4) and allowed the c subunits to be assigned to Fo rotational sub-states based on their interaction with the εNTD (Supplementary Fig. 6), assuming that the c-ring remains bound during rotation, which is likely because the interface between the ring and central rotor has a buried surface of ~1200 Å230,.
When the State 2 εCTD half-up and down structures were compared (Fig. 4) additional clear differences beyond the εCTD up and down conformation were observed. When aligned on the stator a subunit (Fig. 4b), the Fo ring rotates the equivalent of two c subunits in a clockwise direction when viewed from the membrane (akin to the synthesis direction). This rotation of the Fo motor was facilitated mainly by a twisting of the central stalk (~50°), but also by flexing of the peripheral stalk (~15°) (Fig. 5), with the remainder facilitated by small rearrangements within the complex. The twisting of the central stalk involves the εNTD rotating about the γ subunit (Fig. 4c) and this is mediated by a ~10° bending at the foot of the N-terminal γ subunit helix (defined by residues γ39-57) (Figs. 3c and 4d). When the εCTD is half-up, εCTH1 provides an additional link between the εNTD and subunit γ, stabilizing the conformation of the central stalk together (Figs. 3b and 6). When the εCTD is in the down conformation, the central stalk is no longer stabilized by the εCTH1:γ interaction, likely increasing its torsional freedom, and the εNTD rotates relative to the γ subunit. The rotational movement of the εNTD observed between the half-up and down sub-states increases the distance between the εNTD and εCTH1 binding site on subunit γ (Fig. 6), reducing the likelihood of the εCTH1:γ interaction.
Fig. 4. Structural rearrangements of E. coli F1Fo ATP synthase between the half-up and down conformations.

Comparison of State 2 εCTD half-up conformation (top panels) with State 2 εCTD down conformation (bottom panels). a Overall view describing the εCTD (green) transition and rotation of the c-ring (gray with one c subunit colored black). b When superposed on subunit a (orange), the c-ring rotates two c subunits in the Fo motor (one c subunit colored black with black arrow—relative rotation of c subunits identified using the interaction between εNTD and c subunit; Supplementary Fig. 6). c When superposed on the β barrel crown of the α and β subunits, the εNTD rotates ~50° about the γ subunit and the peripheral stalk flexes ~15°. d When superposed on N-terminus of subunit γ, the foot helix (residues γ39-57) bends and twists to accommodate the movement of the εNTD.
Fig. 5. Movements of the central and peripheral stalks.

Side views showing the comparison of State 2 half-up and down sub-states. a Intact F1Fo complexes shown as tube cartoon. b The peripheral stalk (dimer of b subunits) bends counterclockwise and c the central stalk twists clockwise, facilitating a rotation of two c subunits in the Fo motor.
Fig. 6. Rotation of the εNTD increases the distance to the εCTH1 binding site.
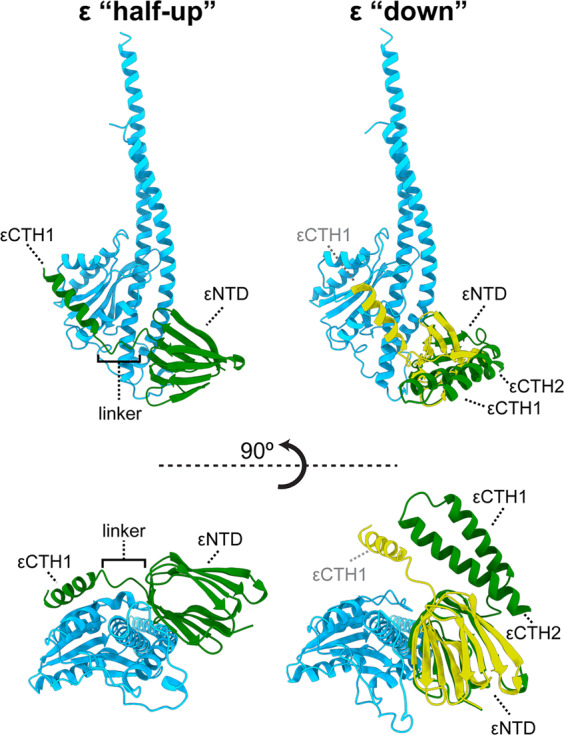
To highlight the relative rotation of the ε subunit between the half-up and down sub-states, the State 2 half-up ε subunit was superposed onto the State 2 down εNTD (shown in yellow). After the εNTD rotates about the γ subunit, the distance between εCTH1 and its binding site on γ is increased in the down sub-state and is less likely to attach to the central stalk.
εCTD helix mutants alter enzyme function
To further investigate the role of each of the εCTD helices on enzyme function and the impact the half-up sub-state has on ATP hydrolysis, two ε subunit truncation mutants were generated. An εΔCTH2 mutant (lacking residues ε105–124; Fig. 7a) would lack the ability to form the classical autoinhibited up sub-state16 or the uninhibited down sub-state, but would retain the ability to form the half-up sub-state and stabilize the central stalk conformation. An εΔCTH1 + 2 mutant (lacking residues ε82–124; Fig. 7b) lacking both εCTD helices would mimic the uninhibited down sub-state and prevent the up or half-up conformations, freeing the central stalk from the bridge made by the εCTH1.
Fig. 7. Subunit ε attenuates ATPase activity.

a εΔCTH2 truncation retains the εCTH1 which can bind to the γ subunit. b εΔCTH1 + 2 retains only the εNTD akin to the εCTD down conformation. c Cryo-EM maps of the 3 states generated using the εΔCTH2 truncation mutant after exposure to 10 mM MgATP showed E. coli F1Fo ATP synthase in the half-up sub-state, with εCTH1 attached to the subunit γ. The 3 states have been rotated successively by 120° to show the position of subunit ε. d Coomassie stained SDS PAGE (uncropped image provided as Supplementary Data 5) of purified E. coli F1Fo ATP synthase WT, εΔCTH2 and εΔCTH1 + 2. Subunits labeled and minor contamination bands identified as; (i) Ribonuclease E, (ii) GroEL, and (iii) ElaB. e ATP regeneration assays of WT (containing full length subunit ε), εΔCTH2 (ε1–104) and εΔCTH1 + 2 (ε1–81). All data points and mean are shown (raw traces are in Supplementary Fig. 8 and values in Supplementary Data 6). Removal of εCTH2 results in higher ATP turnover than WT. Removal of both εCTH1 and εCTH2 shows higher ATP turnover than removal of only εCTH2.
First, to confirm that the εΔCTH2 truncation was still able to form the half-up sub-state, we obtained cryo-EM maps of the εΔCTH2 truncation in the presence of 10 mM MgATP (Fig. 7c and Supplementary Fig. 7). Although processing and masking methods similar to those used on the wild-type enzyme were performed on these data, only a single conformation of the εΔCTH2 subunit was observed. The maps of this sample showed the εCTH1 was bound to the γ subunit in a manner analogous to that observed in the wild-type half-up sub-state. ATP regeneration assays performed on wild-type and truncation mutants showed different turnover rates (Fig. 7d, e). As expected, εΔCTH1 + 2 showed the highest turnover of ATP because it is unable to form either the up or half-up inhibited sub-states. However, εΔCTH2 showed a higher turnover than wild-type enzyme but lower turnover than εΔCTH1 + 2, indicating that the half-up position of εCTH1and its interaction with γ subunit somehow modulates the enzyme turnover.
Discussion
The cryo-EM and functional studies presented here provide information on how F1Fo ATP synthase changes conformation after addition of 10 mM MgATP. The central stalk not only transitions to a down state, but also twists, allowing rotation in the Fo motor. This twisting, in combination with flexing in the peripheral stalk, may facilitate flexible coupling between the F1 and Fo motors and suggests a hypothesis of how subunit ε can modulate E. coli F1Fo ATP synthase function.
Although the enzyme is likely rotating and hydrolyzing ATP during the freezing process21, we did not observe sub steps (e.g., the binding dwell) in the enzyme beyond the three catalytic dwells and ε/c-ring sub rotation. This is likely due to the limited time the enzyme would spend outside the catalytic dwell under these imagining conditions, with single molecule studies needing external load, in the form of increased medium, to observe sub-states9. Assuming that the F1Fo enzyme has similar turnover to the F1-ATPase, single molecule studies suggest that the enzyme would be in the catalytic dwell ~97% of the time31, and hence too small number of particles for efficient sorting. Further to this, the relative number of particles between each state differed substantially and the proportional differences were different than that observed for the same enzyme incubated with MgADP19. Other work using single molecule methods has suggested that the 3:10 symmetry mismatch between the F1 and Fo motors would cause asymmetry in the c-ring rotation32–34, and the structural data of E. coli F1Fo incubated with either MgADP19 or MgATP certainly corroborates this.
Flexible coupling between the F1 and Fo motors is necessary to facilitate efficient enzyme function, however whether this flexibility originates from the peripheral or central stalk has been controversial35–40. To date, structural studies have only shown flexibility within the peripheral stalk12, with the central stalk remaining rigid in all rotational sub-steps observed19,41–46. Previously we have shown that, in E. coli, the peripheral stalk can flexibly couple the F1 and Fo motors, facilitating a single c subunit step in the Fo motor without flexing of the central stalk19. In the present study we observe that the Fo motor rotates two c subunit steps in the clockwise direction (when viewed from the membrane, i.e. the synthesis direction) when the εCTD transitions from the half-up to the down conformation (Figs. 4b and 8a). Torsional flexing of the central stalk facilitates the majority of this Fo rotation, while the peripheral stalk flexes to accommodate the remainder (Fig. 5). In this experiment MgATP was added prior to grid freezing (similar but not identical conditions to that seen in E. coli undergoing aerobic respiration) and therefore the enzyme would be rotating in the counterclockwise direction (when viewed from the membrane, i.e. the hydrolysis direction). Hence, the rotation we observe in Fo when the εCTD transitions from the half-up to down conformation is in the opposite direction to that in which Fo is being driven during ATP hydrolysis. The counter rotation we observe here could be due to several factors. Single molecule studies33,47 have identified rotation in Fo motor of a similar magnitude and direction, suggesting this could be indicative of sub stepping in the synthesis direction. Another possibility could be that that the drag, that would be experienced at the stator/lipid/detergent interfaces, results in the delay of the Fo ring during rotation that we observe here (Fig. 8a).
Fig. 8. Rotation in the Fo motor in the half-up and down sub-states.

a The Fo rotor of the State 2 down sub-state trails the State 2 half-up sub-state by two c subunit steps in the synthesis direction. b When the Fo motor is driven in the synthesis direction, the central stalk is unclamped and the εNTD is pushed away from the εCTH1 binding site on γ, reducing the likelihood of the up conformation. c When the Fo motor is driven in the reverse direction, the central stalk is closed and the εNTD is pushed towards the εCTH1 binding site on γ, increasing the likelihood of the up conformation.
Although single molecule36,47,48 and molecular dynamics studies35 have indicated that the central rotor can be flexible, the present work provides a better understanding of how this flexibility is both conferred and modulated by the εCTD. When the εCTD is in the up or half-up conformation, the εCTH1 binds to the γ subunit, clamping the central rotor together (Figs. 8 and 9). This clamping would stabilize the central stalk in a closed position, increasing the stiffness of the rotor which, in turn, has the potential to impede enzyme turnover by decreasing the flexibility of the intact enzyme. When the εCTD subunit is in the down conformation, εCTH1 can no longer bridge between the εNTD and the γ subunit, thereby enabling the complex to open up to an unclamped conformation and allow a greater degree of flexible coupling between the F1 and Fo motors (Figs. 6, 8 and 9). The ATPase assays presented here suggest that, when the enzyme is in half-up conformation (εΔCTH2), although the turnover is reduced compared to a fully active enzyme (εΔCTH1 + 2), it remains greater than for an enzyme that is able to be fully autoinhibited (WT) (Fig. 7). Together these data indicate that the εCTD is able to modulate central rotor flexibility in addition to inserting into the F1-ATPase to physically block rotation. An in-depth study on yeast F1Fo ATP synthase46, using similar methods to those in this study and which presented the enzyme after incubation with 10 mM ATP, revealed the enzyme in many rotary states. In all structures the rotor was in the same conformation and showed none of the torsional flexing that we observed in the present study, suggesting that the torsional flexing we observe may be limited to the bacterial or E. coli enzyme, and may not always be seen with other species.
Fig. 9. Schematic of clamping by εCTH1.
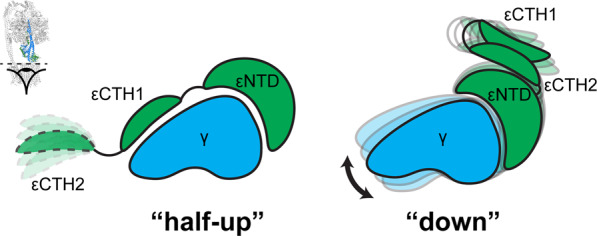
When the εCTH1 is in the half-up state and bound to subunit γ, the central stalk is clamped and increases the stiffness of the rotor. When εCTH1 is in the down state, the central stalk is no longer clamped and has increased flexibility.
The precise function of ATP in regulating the ε subunit of F1Fo ATP synthase appears to vary between bacterial species. For example, the εCTD of Bacillus PS3 F1Fo ATP synthase forms a single helix49 that does not bridge the γ subunit in the same manner as the E. coli εCTD (Supplementary Fig. 9). Furthermore, the isolated Bacillus PS3 ε subunit has been shown to bind ATP50–52 with micromolar binding affinity (Kd = 4.3 μM53) and a FRET sensor based on this subunit has been used to visualize ATP levels inside living cells54. Hence, in Bacillus PS3, cellular ATP concentrations are likely to regulate the εCTD conformation and the single εCTD helix is unable to clamp the central stalk in the manner we observe in E. coli ATP synthase. The ATP binding affinity for the isolated E. coli subunit ε has been shown to be much weaker than Bacillus PS3, with little discrimination between ATP and ADP (Kd of ~20 mM for both ATP and ADP13,52). The crystal structure of the isolated ε subunit from E. coli also demonstrates that the εCTD can form the down conformation in the absence of ATP20. Further to this, studies on Caldalaklibacillus thermarum F1-ATPase have also shown that removal of the ε subunit ATP binding site has little effect on activity or conformation of the εCTD55. Hence, other mechanisms in addition to ATP binding could potentially control the conformation of the εCTD in E. coli and other related bacteria.
The cryo-EM maps generated in this study suggest another mechanism whereby the conformation of subunit ε could be controlled by the pmf. When F1Fo ATP synthase is operating in synthesis mode with high pmf, torque generated in the Fo motor would drive the Fo ring clockwise (Fig. 8b). This torque would pull the εNTD clockwise and increase the distance between it and the εCTH1 binding site on subunit γ, reducing the likelihood of εCTH1 binding and favoring the εCTD down conformation (Figs. 6 and 8b). If the pmf were to be reversed, the torque in Fo would rotate the εNTD anticlockwise and decrease the distance to the εCTH1 binding site on subunit γ, increasing the likelihood of εCTH1 binding and consequently the εCTD up conformation (Figs. 6 and 8c). In this way, the function of E. coli F1Fo ATP synthase could be modulated by a combination of the cellular ATP concentration and the pmf (i.e., the torque in the Fo motor). Hence, the εCTD would act as a ratchet, inhibiting F1Fo ATP synthase when the pmf is insufficient to drive ATP synthesis, allowing the bacterium to grow and quickly adapt to unfavorable conditions. A recent in vivo crosslinking study investigating the physiological relevance of subunit ε demonstrated that the εCTD down state would be most prevalent in conditions that produce high pmf56, suggesting that the ε subunit does change conformation in response to pmf in addition to ATP concentration. Hence, the data presented here illustrates both flexible coupling in the central stalk and a potential mechanism whereby this torsional flexing could impact the regulation of F1Fo ATP synthase.
Methods
Cloning and protein purification
Mutant constructs were made by overlap extension PCR and using the following primers:
εΔCTH2: Forward primer 5’-aagcgaaacgtaaggctgaagagcactaacaccggcttgaaaagcacaaa-3’
Reverse primer 5’-tggcttttgtgcttttcaagccggtgttagtgctcttcagccttacgttt-3’
εΔCTH1 + 2: Forward primer 5’-aacgtgaccgttctggccgactaacaccggcttgaaaagcacaaa-3’
Reverse primer 5’-ggcttttgtgcttttcaagccggtgttagtcggccagaacggtcacgtt-3’
E. coli F1Fo ATP synthase protein was prepared as described in Sobti et al.21,25. Cysteine-free E. coli ATP synthase (all cysteine residues substituted with alanine and a His-tag introduced on the β subunit) was expressed in E. coli DK8 strain22. Cells were grown at 37 °C in LB medium supplemented with 100 μg/ml ampicillin for 5 h. The cells were harvested by centrifugation at 5000 × g, providing ~1.25 g cells per liter of culture. Cells were resuspended in lysis buffer containing 50 mM Tris/Cl pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 2.5% glycerol and 1 μg/ml DNase I, and processed with three freeze thaw cycles followed by one pass through a continuous flow cell disruptor at 20 kPSI. Cellular debris was removed by centrifuging at 7700 × g for 15 min, and the membranes collected by ultracentrifugation at 100,000 × g for 1 h. The ATP synthase complex was extracted from membranes at 4 °C for 1 h by resuspending the pellet in extraction buffer consisting of 20 mM Tris/Cl, pH 8.0, 300 mM NaCl, 2 mM MgCl2, 100 mM sucrose, 20 mM imidazole, 10% glycerol, 4 mM digitonin and EDTA-free protease inhibitor tablets (Roche). Insoluble material was removed by ultracentrifugation at 100,000 × g for 30 min. The complex was then purified by binding on Talon resin (Clontech) and eluted in 150 mM imidazole, and further purified with size exclusion chromatography on a 16/60 Superose 6 column equilibrated in a buffer containing 20 mM Tris/Cl pH 8.0, 100 mM NaCl, 1 mM digitonin, and 2 mM MgCl2. The purified WT protein was then concentrated to 11 μM (6 mg/ml), and snap frozen and stored for grid preparation while the εΔCTH2 and εΔCTH1 + 2 mutants were concentrated and frozen at 9 μM (5 mg/ml)
Cryo-EM grid preparation
One μl of 100 mM ATP/100 mM MgCl2 (pH 8.0) was added to an aliquot of 9 μl of purified cysteine-free E. coli F1Fo ATP synthase (WT at 11 μM and εΔCTH2 mutant at 9 μM) and the sample was incubated at 22 °C for 30 s, before 3.5 μl was placed on glow-discharged holey gold grid (UltrAufoils R1.2/1.3, 200 Mesh). Grids were blotted for 4 s at 22 °C, 100% humidity and flash-frozen in liquid ethane using a FEI Vitrobot Mark IV (total time for sample application, blotting, and freezing was 45 s).
WT cryo-EM data collection and data processing
Grids were transferred to a Thermo Fisher Talos Arctica transmission electron microscope (TEM) operating at 200 kV and screened for ice thickness and particle density. Suitable grids were subsequently transferred to a Thermo Fisher Titan Krios TEM operating at 300 kV equipped with a Gatan BioQuantum energy filter and K3 Camera at the Pacific Northwest Centre for Cryo-EM at OHSU. Images were recorded automatically using SerialEM v3.7 at ×81,000 magnification yielding a pixel size of 0.54 Å (K3 operating in super resolution mode). A total dose of 48 electrons per Å2 was used spread over 77 frames, with a total exposure time of 3.5 s. In all, 8620 movie micrographs were collected (Supplementary Fig. 1). MotionCorr257 was used to correct local beam-induced motion and to align resulting frames, with 9 × 9 patches and binning by a factor of two. Defocus and astigmatism values were estimated using Gctf37 and 8215 micrographs were selected after exclusion based on ice contamination, drift, astigmatism. Approximately 1000 particles were manually picked and subjected to 2D classification to generate templates for template picking in cryoSPARC58, yielding 869,147 particles. These particles were binned by a factor of four and subjected to 2D classification generating a final dataset of 429,638 particles. The locations of these particles were then imported into Relion59, re-extracted at full resolution, and further classified into 3D classes using a low pass filtered cryo-EM model generated from a previous study18, yielding the three main states related by a rotation of the central stalk (State 1, State 2, and State 3 with 100,831, 215,003, and 113,804 particles, respectively). Focused classification, using a mask comprising the lower half of the central rotor, was implemented without performing image alignment in Relion 3.0, yielding the half-up and the down sub-classes in each of the three main states. A further Fo focused classification without image shifts was performed on each of the half-up and down sub-classes to elucidate the position of Fo subunits in the respective sub-states. See Supplementary Fig. 2 for a flowchart describing this classification.
εΔCTH2 mutant cryo-EM data collection and data processing
Grids were transferred to a Thermo Fisher Talos Arctica transmission electron microscope (TEM) operating at 200 kV and screened for ice thickness and particle density. Suitable grids were subsequently transferred to a Thermo Fisher Titan Krios TEM operating at 300 kV equipped with a Gatan BioQuantum energy filter and K2 Camera at the Molecular Horizons, University of Wollongong. Images were recorded automatically using EPUv2.7 at ×120,000 magnification yielding a pixel size of 1.13 Å. A total dose of 55 electrons per Å2 was used spread over 50 frames, 3680 movie micrographs were collected. All the processing was subsequently performed in cryoSPARC58. Initial particles were picked using the blob protocol which were 2D classified to create templates to the pick the entire dataset. Extracted particles were subjected to multiple rounds of 2D classification, ab initio reconstruction, heterogenous refinement to sort the particles into discreet structures. See Supplementary Fig. 7 for a flowchart describing this classification. Additional processing was performed using masks and focused refinement but did not yield any maps showing alternate conformations, so are not included in the flow chart. Supplementary Table 1 contains a summary of data collection/processing statistics and Supplementary Fig. 10 for FSC curves.
Model building
Models were built and refined in Coot60, PHENIX29, and ISOLDE61 using pdbs 6OQT, 6OQV, 6OQW19 (E. coli ATP synthase incubated with MgADP), and 1AQT20 (isolated E. coli ATP synthase subunit ε) as guides. See Supplementary Table 1 for a summary of refinement and validation statistics.
ATP regeneration assays
ATP regeneration assays were performed as in Sobti et al.25. Five μg of detergent solubilized protein was added to 100 mM KCl, 50 mM MOPS pH 7.4, 1 mM MgCl2, 1 mM ATP, 2 mM PEP, 2.5 units/ml pyruvate kinase, 2.5 units/ml lactate dehydrogenase and 0.2 mM NADH and monitored for OD at 340 nm at for up to 20 min (Supplementary Fig. 8).
Statistics and reproducibility
The cryo-EM analyses were performed on single protein preparations. We have performed similar experiments on similar protein preparations (more than ten times using a range of microscopes) with similar outcomes18,19,21,25. Detailed statistics for the sample size, data collection and analysis of cryo-EM data are provided in Supplementary Table 1, and Fourier shell correlation curves are provided in Supplementary Fig. 10. The activity assays were performed in technical triplicate, with only means provided as the raw data (Supplementary Fig. 8 and Data 6) could be plotted and interpreted without error bars (Fig. 7e).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We wish to thank and acknowledge Dr. Craig Yoshioka and Dr. Claudia López (Oregon Health & Sciences University (OHSU)) for data collection and processing expertise. We would also like to thank Dr Emily Furlong (The Victor Chang Cardiac Research Institute) for critically reviewing the manuscript. A.G.S. was supported by a National Health and Medical Research Council Fellowship APP1159347 and Grant APP1146403. We acknowledge the use of the Victor Chang Innovation Centre, funded by the NSW Government, and the Electron Microscope Unit at UNSW Sydney, funded in part by the NSW Government. We also acknowledge he use of the University of Wollongong Cryogenic Electron Microscopy Facility at Molecular Horizons. A portion of this research was supported by NIH grant U24GM129547 and performed at the Pacific Northwest Centre for Cryo-EM at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research.
Author contributions
M.S., R.I., and A.G.S. conceived the study and wrote the manuscript. M.S. performed the formal analysis of the study. Y.C.Z. performed the reconstitution for ATP regeneration assays and edited the manuscript. J.L.W. and S.H.J.B. aided in cryo-EM data acquisition and analysis. A.G.S. supervised the study.
Peer review
Peer review information
Communications Biology thanks Joachim Weber and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary handling editors: Joanna Timmins and Gene Chong. Peer reviewer reports are available.
Data availability
The models generated and analyzed during the current study are available from the RCSB PDB: 8DBP, 8DBQ, 8DBR, 8DBS, 8DBT, 8DBU, 8DBV, 8DBW. The cryo-EM maps used to generate models are available from the EMDB: 27296, 27297, 27298, 27299, 27300, 27301, 27302, 27303, 27304, 27305, 27306, 27307, 27308, 27309, 27310, 27311, 27312, 27313, 27314, 27315. Phenix combined maps and models for State 2 up and down are provided as Supplementary Data 1–4 with this manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-023-04414-z.
References
- 1.Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013;41:1–16. doi: 10.1042/BST20110773. [DOI] [PubMed] [Google Scholar]
- 2.Kuhlbrandt W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019 doi: 10.1146/annurev-biochem-013118-110903. [DOI] [PubMed] [Google Scholar]
- 3.Stewart AG, Laming EM, Sobti M, Stock D. Rotary ATPases - dynamic molecular machines. Curr. Opin. Struct. Biol. 2014;25:40–48. doi: 10.1016/j.sbi.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Courbon, G. M. & Rubinstein, J. L. CryoEM reveals the complexity and diversity of ATP synthases. Front. Microbiol.10.3389/fmicb.2022.864006 (2022). [DOI] [PMC free article] [PubMed]
- 5.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 6.Boyer PD. The ATP synthase - a splendid molecular machine. Annu. Rev. Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 7.Bianchet M, Ysern X, Hullihen J, Pedersen PL, Amzel LM. Mitochondrial ATP synthase. Quaternary structure of the F1 moiety at 3.6 A determined by x-ray diffraction analysis. J. Biol. Chem. 1991;266:21197–21201. doi: 10.1016/S0021-9258(18)54840-7. [DOI] [PubMed] [Google Scholar]
- 8.Sobti M, Ueno H, Noji H, Stewart AG. The six steps of the complete F1-ATPase rotary catalytic cycle. Nat. Commun. 2021;12:4690. doi: 10.1038/s41467-021-25029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishmukhametov R, Hornung T, Spetzler D, Frasch WD. Direct observation of stepped proteolipid ring rotation in E. coli F0F1-ATP synthase. EMBO J. 2010;29:3911–3923. doi: 10.1038/emboj.2010.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 11.Kinosita K, Jr., Yasuda R, Noji H, Adachi K. A rotary molecular motor that can work at near 100% efficiency. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000;355:473–489. doi: 10.1098/rstb.2000.0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart AG, Lee LK, Donohoe M, Chaston JJ, Stock D. The dynamic stator stalk of rotary ATPases. Nat. Commun. 2012;3:687. doi: 10.1038/ncomms1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sielaff H, Duncan TM, Borsch M. The regulatory subunit epsilon in Escherichia coli FOF1-ATP synthase. Biochim. Biophys. Acta Bioenerg. 2018;1859:775–788. doi: 10.1016/j.bbabio.2018.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, et al. Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature. 2021;589:143–147. doi: 10.1038/s41586-020-3004-3. [DOI] [PubMed] [Google Scholar]
- 15.Milgrom YM, Duncan TM. F-ATP-ase of Escherichia coli membranes: The ubiquitous MgADP-inhibited state and the inhibited state induced by the epsilon-subunit’s C-terminal domain are mutually exclusive. Biochim. Biophys. Acta Bioenerg. 2020;1861:148189. doi: 10.1016/j.bbabio.2020.148189. [DOI] [PubMed] [Google Scholar]
- 16.Cingolani G, Duncan TM. Structure of the ATP synthase catalytic complex F1 from Escherichia coli in an autoinhibited conformation. Nat. Struct. Mol. Biol. 2011;18:701–707. doi: 10.1038/nsmb.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsunoda SP, et al. Large conformational changes of the epsilon subunit in the bacterial F1F0 ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc. Natl Acad. Sci. USA. 2001;98:6560–6564. doi: 10.1073/pnas.111128098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobti, M. et al. Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states. Elife10.7554/eLife.21598 (2016). [DOI] [PMC free article] [PubMed]
- 19.Sobti M, et al. Cryo-EM structures provide insight into how E. coli F1Fo ATP synthase accommodates symmetry mismatch. Nat. Commun. 2020;11:2615. doi: 10.1038/s41467-020-16387-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uhlin U, Cox GB, Guss JM. Crystal structure of the epsilon subunit of the proton-translocating ATP synthase from Escherichia coli. Structure. 1997;5:1219–1230. doi: 10.1016/S0969-2126(97)00272-4. [DOI] [PubMed] [Google Scholar]
- 21.Sobti, M. et al. Cryo-EM reveals distinct conformations of E. coli ATP synthase on exposure to ATP. Elife10.7554/eLife.43864 (2019). [DOI] [PMC free article] [PubMed]
- 22.Ishmukhametov R, Galkin MA, Vik SB. Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1Fo ATP synthase. Biochim. Biophys. Acta. 2005;1706:110–116. doi: 10.1016/j.bbabio.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Bennett BD, et al. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashket ER. Stoichiometry of the H+-ATPase of growing and resting, aerobic Escherichia coli. Biochemistry. 1982;21:5534–5538. doi: 10.1021/bi00265a024. [DOI] [PubMed] [Google Scholar]
- 25.Sobti M, Ishmukhametov R, Stewart AG. ATP synthase: expression, purification, and function. Methods Mol. Biol. 2020;2073:73–84. doi: 10.1007/978-1-4939-9869-2_5. [DOI] [PubMed] [Google Scholar]
- 26.Bowler MW, Montgomery MG, Leslie AG, Walker JE. Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 Å resolution. J. Biol. Chem. 2007;282:14238–14242. doi: 10.1074/jbc.M700203200. [DOI] [PubMed] [Google Scholar]
- 27.Rees DM, Montgomery MG, Leslie AG, Walker JE. Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria. Proc. Natl Acad. Sci. USA. 2012;109:11139–11143. doi: 10.1073/pnas.1207587109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frasch WD, Bukhari ZA, Yanagisawa S. F1FO ATP synthase molecular motor mechanisms. Front. Microbiol. 2022;13:965620. doi: 10.3389/fmicb.2022.965620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Afonine PV, et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Spetzler D, et al. Microsecond time scale rotation measurements of single F1-ATPase molecules. Biochemistry. 2006;45:3117–3124. doi: 10.1021/bi052363n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sielaff, H., Yanagisawa, S., Frasch, W. D., Junge, W. & Borsch, M. Structural asymmetry and kinetic limping of single rotary F-ATP synthases. Molecules10.3390/molecules24030504 (2019). [DOI] [PMC free article] [PubMed]
- 33.Yanagisawa, S. & Frasch, W. D. pH-dependent 11° F(1)F(O) ATP synthase sub-steps reveal insight into the F(O) torque generating mechanism. Elife10.7554/eLife.70016 (2021). [DOI] [PMC free article] [PubMed]
- 34.Yanagisawa S, Frasch WD. Protonation-dependent stepped rotation of the F-type ATP synthase c-ring observed by single-molecule measurements. J. Biol. Chem. 2017;292:17093–17100. doi: 10.1074/jbc.M117.799940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okazaki K, Hummer G. Elasticity, friction, and pathway of gamma-subunit rotation in FoF1-ATP synthase. Proc. Natl Acad. Sci. USA. 2015;112:10720–10725. doi: 10.1073/pnas.1500691112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okuno D, Iino R, Noji H. Stiffness of gamma subunit of F(1)-ATPase. Eur. Biophys. J. 2010;39:1589–1596. doi: 10.1007/s00249-010-0616-9. [DOI] [PubMed] [Google Scholar]
- 37.Zhou M, et al. Ion mobility-mass spectrometry of a rotary ATPase reveals ATP-induced reduction in conformational flexibility. Nat. Chem. 2014;6:208–215. doi: 10.1038/nchem.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muench SP, et al. Subunit positioning and stator filament stiffness in regulation and power transmission in the V1 motor of the Manduca sexta V-ATPase. J. Mol. Biol. 2014;426:286–300. doi: 10.1016/j.jmb.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song CF, et al. Flexibility within the rotor and stators of the vacuolar H+-ATPase. PLoS ONE. 2013;8:e82207. doi: 10.1371/journal.pone.0082207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stewart AG. The molecular V brake. J. Mol. Biol. 2014;426:273–274. doi: 10.1016/j.jmb.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Murphy, B. J. et al. Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1-Fo coupling. Science10.1126/science.aaw9128 (2019). [DOI] [PubMed]
- 42.Spikes, T. E., Montgomery, M. G. & Walker, J. E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl Acad. Sci. USA10.1073/pnas.2013998117 (2020). [DOI] [PMC free article] [PubMed]
- 43.Hahn, A., Vonck, J., Mills, D. J., Meier, T. & Kuhlbrandt, W. Structure, mechanism, and regulation of the chloroplast ATP synthase. Science10.1126/science.aat4318 (2018). [DOI] [PMC free article] [PubMed]
- 44.Guo, H., Suzuki, T. & Rubinstein, J. L. Structure of a bacterial ATP synthase. Elife10.7554/eLife.43128 (2019). [DOI] [PMC free article] [PubMed]
- 45.Srivastava, A. P. et al. High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science10.1126/science.aas9699 (2018). [DOI] [PMC free article] [PubMed]
- 46.Guo H, Rubinstein JL. Structure of ATP synthase under strain during catalysis. Nat. Commun. 2022;13:2232. doi: 10.1038/s41467-022-29893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin JL, Ishmukhametov R, Spetzler D, Hornung T, Frasch WD. Elastic coupling power stroke mechanism of the F(1)-ATPase molecular motor. Proc. Natl Acad. Sci. USA. 2018;115:5750–5755. doi: 10.1073/pnas.1803147115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sielaff H, et al. Domain compliance and elastic power transmission in rotary F(O)F(1)-ATPase. Proc. Natl Acad. Sci. USA. 2008;105:17760–17765. doi: 10.1073/pnas.0807683105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shirakihara Y, et al. Structure of a thermophilic F1-ATPase inhibited by an epsilon-subunit: deeper insight into the epsilon-inhibition mechanism. FEBS J. 2015;282:2895–2913. doi: 10.1111/febs.13329. [DOI] [PubMed] [Google Scholar]
- 50.Krah A, Kato-Yamada Y, Takada S. The structural basis of a high affinity ATP binding epsilon subunit from a bacterial ATP synthase. PLoS ONE. 2017;12:e0177907. doi: 10.1371/journal.pone.0177907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krah A, Huber RG, McMillan DGG, Bond PJ. The molecular basis for purine binding selectivity in the bacterial ATP synthase subunit. Chembiochem. 2020 doi: 10.1002/cbic.202000291. [DOI] [PubMed] [Google Scholar]
- 52.Yagi H, et al. Structures of the thermophilic F1-ATPase epsilon subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1. Proc. Natl Acad. Sci. USA. 2007;104:11233–11238. doi: 10.1073/pnas.0701045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato S, Yoshida M, Kato-Yamada Y. Role of the epsilon subunit of thermophilic F1-ATPase as a sensor for ATP. J. Biol. Chem. 2007;282:37618–37623. doi: 10.1074/jbc.M707509200. [DOI] [PubMed] [Google Scholar]
- 54.Imamura H, et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl Acad. Sci. USA. 2009;106:15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferguson SA, Cook GM, Montgomery MG, Leslie AG, Walker JE. Regulation of the thermoalkaliphilic F1-ATPase from Caldalkalibacillus thermarum. Proc. Natl Acad. Sci. USA. 2016;113:10860–10865. doi: 10.1073/pnas.1612035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, et al. A high-throughput genetically directed protein crosslinking analysis reveals the physiological relevance of the ATP synthase ‘inserted’ state. FEBS J. 2021;288:2989–3009. doi: 10.1111/febs.15616. [DOI] [PubMed] [Google Scholar]
- 57.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 59.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Croll TI. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goddard TD, et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The models generated and analyzed during the current study are available from the RCSB PDB: 8DBP, 8DBQ, 8DBR, 8DBS, 8DBT, 8DBU, 8DBV, 8DBW. The cryo-EM maps used to generate models are available from the EMDB: 27296, 27297, 27298, 27299, 27300, 27301, 27302, 27303, 27304, 27305, 27306, 27307, 27308, 27309, 27310, 27311, 27312, 27313, 27314, 27315. Phenix combined maps and models for State 2 up and down are provided as Supplementary Data 1–4 with this manuscript.