

Abstract
Cardiovascular disease is the leading contributor to years lost due to disability or premature death among adults. Current efforts focus on risk prediction and risk factor mitigation‚ which have been recognized for the past half-century. However, despite advances, risk prediction remains imprecise with persistently high rates of incident cardiovascular disease. Genetic characterization has been proposed as an approach to enable earlier and potentially tailored prevention. Rare mendelian pathogenic variants predisposing to cardiometabolic conditions have long been known to contribute to disease risk in some families. However, twin and familial aggregation studies imply that diverse cardiovascular conditions are heritable in the general population. Significant technological and methodological advances since the Human Genome Project are facilitating population-based comprehensive genetic profiling at decreasing costs. Genome-wide association studies from such endeavors continue to elucidate causal mechanisms for cardiovascular diseases. Systematic cataloging for cardiovascular risk alleles also enabled the development of polygenic risk scores. Genetic profiling is becoming widespread in large-scale research, including in health care–associated biobanks, randomized controlled trials, and direct-to-consumer profiling in tens of millions of people. Thus, individuals and their physicians are increasingly presented with polygenic risk scores for cardiovascular conditions in clinical encounters. In this scientific statement, we review the contemporary science, clinical considerations, and future challenges for polygenic risk scores for cardiovascular diseases. We selected 5 cardiometabolic diseases (coronary artery disease, hypercholesterolemia, type 2 diabetes, atrial fibrillation, and venous thromboembolic disease) and response to drug therapy and offer provisional guidance to health care professionals, researchers, policymakers, and patients.
Keywords: AHA Scientific Statements; atrial fibrillation; diabetes, type 2; genome-wide association studies; multifactorial inheritance; predictive genetic testing; venous thromboembolism
Cardiovascular disease is the leading cause of morbidity and mortality in the United States and globally.1 In the United States, 121.5 million people have cardiovascular disease1 and 26 million have diabetes (estimates from 2013 to 2016 data), each with continually increasing prevalences.1 Furthermore, cardiovascular diseases and their complications contribute to the substantial and ever-increasing health care costs in the United States.1
In an effort to reduce cardiovascular disease prevalence and its complications, much effort has focused on prevention.2 The cornerstone of prevention is early risk prediction coupled with risk mitigation. The Framingham Risk Score, now nearly 25 years old, represents an early example of synthesizing multiple clinical risk factors into a single estimated risk for coronary artery disease (CAD) and stroke.3,4 With the incorporation of additional cohorts, including individuals of non-European ancestry, the Pooled Cohort Equations (PCE) provide the contemporary 10-year risk estimator for atherosclerotic cardiovascular disease (ASCVD) recommended by cardiovascular professional societies in the United States.5,6 However, the PCE may systematically underperform in some groups and has reduced discrimination among younger adults and older adults.7–11
Incorporation of genetics into risk prediction frameworks offers the opportunity to refine risks, potentially earlier in life, toward the creation of earlier and tailored risk reduction strategies.11a Having a parent with a history of premature CAD is associated with an ≈50% higher odds of developing cardiovascular disease independent of clinical risk factors.12 Furthermore, twin studies (comparing monozygotic twins with dizygotic twins) have shown that variation in the development of CAD,13 atrial fibrillation (AF),14 and diabetes15,16 is attributable to common genetic variations. These observations support the notion that genetics may be additive in risk prediction.13,14,16,17
Monogenic risk variants (defined in Table 1) are typically rare and confer a large risk of disease (such as low-density lipoprotein [LDL] receptor variants causing familial hypercholesterolemia [FH]).These variants have been recognized for decades and represent the current scope of clinical cardiovascular genetics.18 However, monogenic risk variants are present in only a small minority of patients and explain only a small proportion of heritable cardiovascular disease risk in familial aggregation studies (eg, many families do not have monogenic variants and still have cardiovascular disease).19,20 This phenomenon, as well as evidence from twin studies,21 supports the polygenic basis of the development of cardiometabolic diseases: Common genetic variation (ie, present in at least 1% of the population) contributes substantially to risk.
Table 1.
Common Statistical Genetics Terms Used in This Scientific Statement
| Terms | Definition |
|---|---|
| AUC | The probability that the statistical model will correctly classify a participant as having or not having a disease (discrimination) |
| C statistic | The probability that the statistical model will correctly classify a participant who will go on to develop or not go on to develop a disease (discrimination) |
| NRI | A measure of how a new model, typically with the addition of ≥1 more risk factors, reclassifies participants |
| SNV | A variation in nucleotide base pair compared with what is expected at that location in the human genome, ie, an A instead of a G |
| LD | The nonrandom association of alleles that tend to be inherited together more often than chance; a function of allele ages, genomic distance, and local recombination rates |
| Minor allele frequency | The prevalence of the least common allele among the possible allele combinations at a genomic site |
| Monogenic risk variants | Rare and typically disruptive or protein-truncating variants that confer large risks of disease; typically follow classic mendelian patterns of inheritance |
| Polygenic risk variants | Commonly occurring variants in the population that typically confer an individually small risk of developing a disease |
AUC indicates area under the receiver-operating characteristic curve; LD, linkage disequilibrium; NRI, net reclassification index; and SNV, single nucleotide variant.
Indeed, over the past 15 years, increasingly larger genome-wide association studies (GWAS) have confirmed the polygenic basis of cardiometabolic diseases. GWAS has shown that many single nucleotide variants (SNVs) scattered across the genome are associated with many cardiometabolic diseases. Each of these variants is of individually small risk, but collectively, they account for substantial cardiovascular disease risk.20,22,23
GWAS showed the association of many SNVs and cardiometabolic disease. Polygenic risk scores (PRSs; also known as polygenic scores) are the weighted summations of these SNVs. The summation of these SNVs (eg, PRS) has been shown to confer significant risk (Figure 1). For multiple cardiovascular diseases, PRSs are independently associated with respective cardiovascular diseases.20 Thus, PRSs are proposed as tools to improve the prediction of common, complex cardiovascular diseases.24
Figure 1. Development of a PRS.
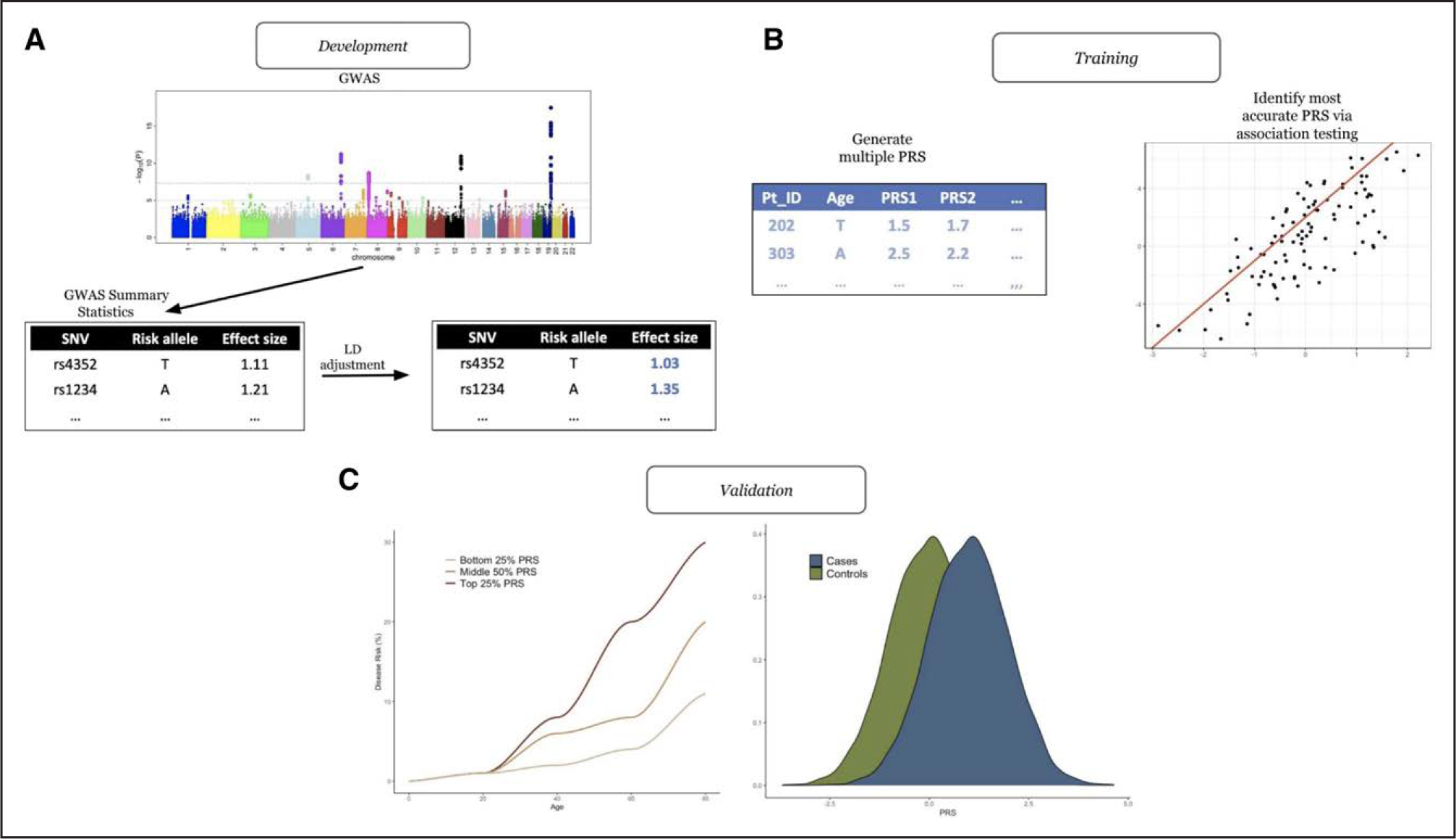
A, Development of polygenic risk scores (PRSs). This typically involves attaining single nucleotide variant (SNV) effect sizes from a genome-wide association study (GWAS) and then adjusting these SNV effect sizes to account for linkage disequilibrium (LD). B, Training of the PRS. Typically, numerous PRSs are created per participant. Each PRS is then assessed through various association testing, and the most accurate PRS is collected. C, Validation of the PRS. The most accurate PRS is then validated in an independent cohort of participants.
Universities, academic medical centers, and direct-to-consumer genetics companies are now able to provide PRSs to research participants, patients, and consumers.25,26 This scientific statement reviews the current science, clinical implementation considerations (eg, efficacy and cost) across stakeholders (health care professionals, patients, and health care administrators), and outstanding questions about the clinical use of PRSs for selected cardiovascular diseases. We focus on 5 cardiometabolic diseases (CAD, hypercholesterolemia, type 2 diabetes [T2D], AF, and venous thromboembolic disease) and offer provisional guidance to health care professionals, researchers, policymakers, and patients on the use of PRSs in cardiovascular disease risk assessment and risk reduction.
In line with the National Heart, Lung, and Blood Institute 2020 guidelines on the use and reporting of race, ethnicity, and ancestry27 and the American Heart Association (AHA) presidential advisory on structural racism,28 throughout this scientific statement, we use the term ethnicity to refer to social categories including both race and ethnicity. We support the AHA presidential advisory definition of race as “a social construct primarily based on phenotype, ethnicity, and other indicators of social differentiation that results in varying access to power and social and economic resources.”28 The term ancestry is used for inferences from genetic data. When we state the ethnicity or ancestry of participants, we attained this information directly from the primary literature. Last, we note that the concepts of ethnicity and ancestry are not synonymous but may be correlated.29
WHAT ARE PRSs?
PRSs (or polygenic scores) are the weighted sum of the risk conferred by multiple disease-associated SNVs across the genome. Constructing a PRS requires a list of SNVs with their accompanying effect sizes (a quantification of the association of the SNV with the disease) from an external data set, typically acquired from a GWAS (Figure 1). Next, methods are used to account for the extensive correlation between SNVs throughout the genome (known as linkage disequilibrium [LD]).18 Common methods include P value thresholding (only including SNVs that are below a predefined P value), LD pruning (randomly removing a SNV from a pair that are in LD, in which LD is typically classified by the correlation between SNVs, quantified by r2), and clumping (similar to LD pruning but the SNVs with the lower P value [of the pair] is selected). All of these earlier methods exclude several SNVs on the basis of an arbitrarily selected P value threshold or at random. Advances in statistical genetics have led to a number of new methods that do not exclude SNVs. These methods include bayesian approaches (such as LDpred,22 Bayesian Sparse Linear Mixed Models,30 AnnoPred,31 LDpred-funct,32 PRS-CS,33 and PleioPred34) and penalized regression (Lassosum35) and include all SNVs but with reweighted effect sizes baselined on at least the P and r2 values.
Current research is focused on ensuring equity of PRS use and predictive ability across all ancestry groups. Transethnic PRSs from meta-analyses of GWAS across multiple ancestries may help with portability.36,37 Large biorepositories such as the UK Biobank, Million Veteran Program, and Electronic Medical Records and Genomics Network have aided in the performance of high-throughput and high-sample-size genome-wide discovery efforts of a wide range of cardiometabolic disease.20,38 In addition, biobanks located in areas with higher levels of ancestry diversity have contributed a critical view of genetic association in understudied populations.39,40
POLYGENIC VERSUS MONOGENIC RISK VARIANTS
Monogenic risk variants are rare (minor allele frequency <1%, defined as the frequency of the second most common allele that occurs within a selected population41) and are typically disruptive or truncating coding sequence variants that confer a large risk of disease. In this scientific statement, rare variants are denoted as minor allele frequency <1%; common polygenic risk variants are denoted as minor allele frequency ≥1%. Monogenic risk variants follow classic mendelian patterns of inheritance. For example, monogenic risk variants in the genes encoding the LDL receptor (LDLR), apolipoprotein B (APOB), and proprotein convertase subtilisin/kexin type 9 (PCSK9) are examples of causes of FH, a monogenic disease that causes severe hypercholesterolemia and is a significant risk factor for early-onset myocardial infarction.42–45 Other examples of monogenic risk variants include variants in GCK for diabetes,46 KCNQ1 for AF,47 and F5 for venous thromboembolic disease.48,49
The risk of developing a disease is influenced by both monogenic and polygenic risk variants. In those with FH monogenic risk variants, LDL cholesterol (LDL-C) concentrations varied, as did their risk of developing CAD. This variation aligned each participant’s LDL-C PRS; that is, those with a low LDL-C PRS and monogenic FH had, on average, lower LDL-C and a lower risk of CAD compared with those with a high LDL-C PRS and monogenic FH.19,50 This concept has also been extended to cardiomyopathies; for example, the risk of developing hypertrophic cardiomyopathy depends on monogenic risk variants (eg, MYH7) and a person’s PRS.51 These observations support the liability threshold model, that is, the notion that multiple factors—monogenic, polygenic, and nongenetic—may each contribute to a threshold necessary for disease development.18
Because genetic testing for monogenic causes of suspected inherited cardiovascular conditions was covered in a prior scientific statement,52 we focus on PRSs in the present scientific statement.
Atrial Fibrillation
AF is the most prevalent cardiac arrhythmia in the United States, with >12 million individuals projected to be diagnosed with AF by 2030.1,53 AF prevalence increases with age and commonly coexists with other cardiovascular diseases.54 It is a well-recognized independent risk factor for stroke55 and can contribute to and worsen heart failure.56
Although many common cardiovascular diseases are risk factors for developing AF,57 genetic variation also has been shown to contribute.20,58 Loss-of-function variants in the Titin (TTN) gene are enriched among individuals with early-onset AF (2.1% prevalence) versus control subjects (1.1% prevalence; odds ratio, 1.76 [95% CI, 1.04–2.97]).59,60
Common genetic variation also contributes to AF risk. The first appreciation of this was in twin studies, which showed that the likelihood of developing AF was higher in monozygotic twins compared with dizygotic twins (hazard ratio [HR], 2.0 [95% CI, 1.3–3.0]).14 More recently, 134 distinct AF-associated loci have been identified through GWAS meta-analyses, all of which are common and of individually small effect.61,62
The increasing number of common genetic variants associated with AF has enabled the development of AF PRSs (Table 2).23,63 PRSs constructed to date have shown a consistent predictive benefit (C statistic, or area under the receiver-operating characteristics curve [AUC], varying from 0.61–0.78).20,23,64–69 Recent advancement in statistical methods, most notably the liberalizing of included SNVs in PRSs, has led to increased predictive accuracy (see the What Are PRSs? section).23,64,65 These improvements contributed to the accuracy of risk prediction in 2 ways. First, they improve the accuracy of the prediction of AF in the absence of clinical risk factors (eg, early-onset AF). Second, they enhance prediction when PRSs and clinical risk factors are combined. Concerning the latter, AF prediction is improved with the addition of PRSs to clinical risk factors (age, height, weight, systolic and diastolic blood pressures, smoking status, blood pressure–lowering medication, diabetes, heart failure, and history of myocardial infarction) through both discrimination (the C statistic changed from 0.725 [95% CI, 0.719–0.732] to 0.734 [95% CI, 0.728–0.741]) and reclassification (net reclassification index [NRI], 10% (95% CI, 4.2%–15.7%]) metrics (Figure 2).23,63 Concerning the former, PRSs are consistently predictive of early-onset AF23,70; 1 study showed that those in the top 10th percentile of PRS risk had an odds ratio of 5.70 (95% CI, 2.60–13.95) for developing early-onset AF compared with those in the bottom 90th percentile of PRS risk.70 Another study showed that those in top 2.5th percentile of PRS risk developed the disease ≈6.64 years before those in the 20th to 80th percentile.23 Similarly, of those who developed AF before 60 years of age, 27.9% were at high PRS risk (>5% 5-year risk of developing AF determined by the PRS model only), whereas only 4.9% of these participants with early AF were deemed high risk by the common clinical risk tool, CHARGE-AF (Cohorts for Heart and Aging Research in Genomic Epidemiology model for AF).23
Table 2.
Characteristics of Studies of PRSs With the Highest NRI When Comparing a Clinical Risk Tool With a Clinical Risk Tool With PRSs Integrated Within
| Disease, authors (y) | SNVs, n | Participants, n cases/N total* | Biobank | Participants: ancestry/ethnicity, age, y | Outcome 1: HR/OR (95% CI) (included covariates controlled for)* | Outcome 2: AUC/C statistic (95% CI) (included covariates controlled for)* | Outcome 3: AUC/C-statistic (95% CI): clinical risk tool vs PRS+clinical risk tool | Outcome 4: NRI (95% CI) comparison |
|---|---|---|---|---|---|---|---|---|
| AF, Mars et al23 (2020) | 6 171 733 | 12 809/135 300 For model comparing clinical risk model, 229/21 030 |
FinnGenn | European, 59.2±16.6 | HR per SD, 1.62 (1.59–1.65) (collection year, genotyping array/batch and the first 10 principal components of ancestry, and stratified the models by sex) | C statistic, 0.751 (0.744–0.757) (collection year, genotyping array/batch and the first 10 principal components of ancestry, and stratified the models by sex) | CHARGE-AF, 0.725 (0.719–0.732) CHARGE-AF+PRS, 0.734 (0.728–0.741) | Using >5% risk threshold over 5 y, 10.4 (4.1–16.7) CHARGE-AF vs CHARGE-AF+PRS |
| CAD, Riveros-Mckay et al63 (2021) | >3 500 000 | 4247/186 541 | UK Biobank | European, 40–69 | HR per SD, 1.90 (1.86–1.95) (age, sex, principle components of ancestry) | C statistic, 0.633 (0.625–0.641) (age, sex, principle components of ancestry) | AHA/ACC PCE, 0.76 (0.75–0.76) AHA/ACC PCE+PRS, 0.79 (0.78–0.79) | Using >7.5% threshold over 10 y, 5.9% (4.7%–7.0%) AHA/ACC PCE vs AHA/ACC PCE+PRS |
| T2D, Mars et al23 (2020) | 6 437 380 | 17 519/135 300 For model comparing clinical risk model, 1346/10 561 |
FinnGenn | European, 59.2±16.6 | HR per SD, 1.74 (1.72–1.77) (collection year, genotyping array/batch and the first 10 principal components of ancestry, and stratified the models by sex) | C statistic, 0.763 (0.758–0.767) (collection year, genotyping array/batch and the first 10 principal components of ancestry, and stratified the models by sex) | ADA, 0.835 (0.831–0.839) ADA+PRS, 0.845 (0.841–0.849) | NRI using 10-y risk ≥33% risk threshold, 4.5% (3.0%–6.1%) ADA criteria |
ADA indicates American Diabetes Association; AF, atrial fibrillation; AHA/ACC, American Heart Association/American College of Cardiology; AUC, area under the receiver-operating characteristic curve; CAD, coronary artery disease; CHARGE-AF, Cohorts for Heart and Aging Research in Genomic Epidemiology model for atrial fibrillation; HR, hazard ratio; NRI, net reclassification index; OR, odds ratio; PCE, Pooled Cohort Equation; PRS, polygenic risk score; SNV, single nucleotide variant; and T2D, type 2 diabetes.
Data from validation data set/analysis.
Figure 2. Predictive accuracy of polygenic risk scores when combined with clinical risk tools, compared to clinical risk tools alone.
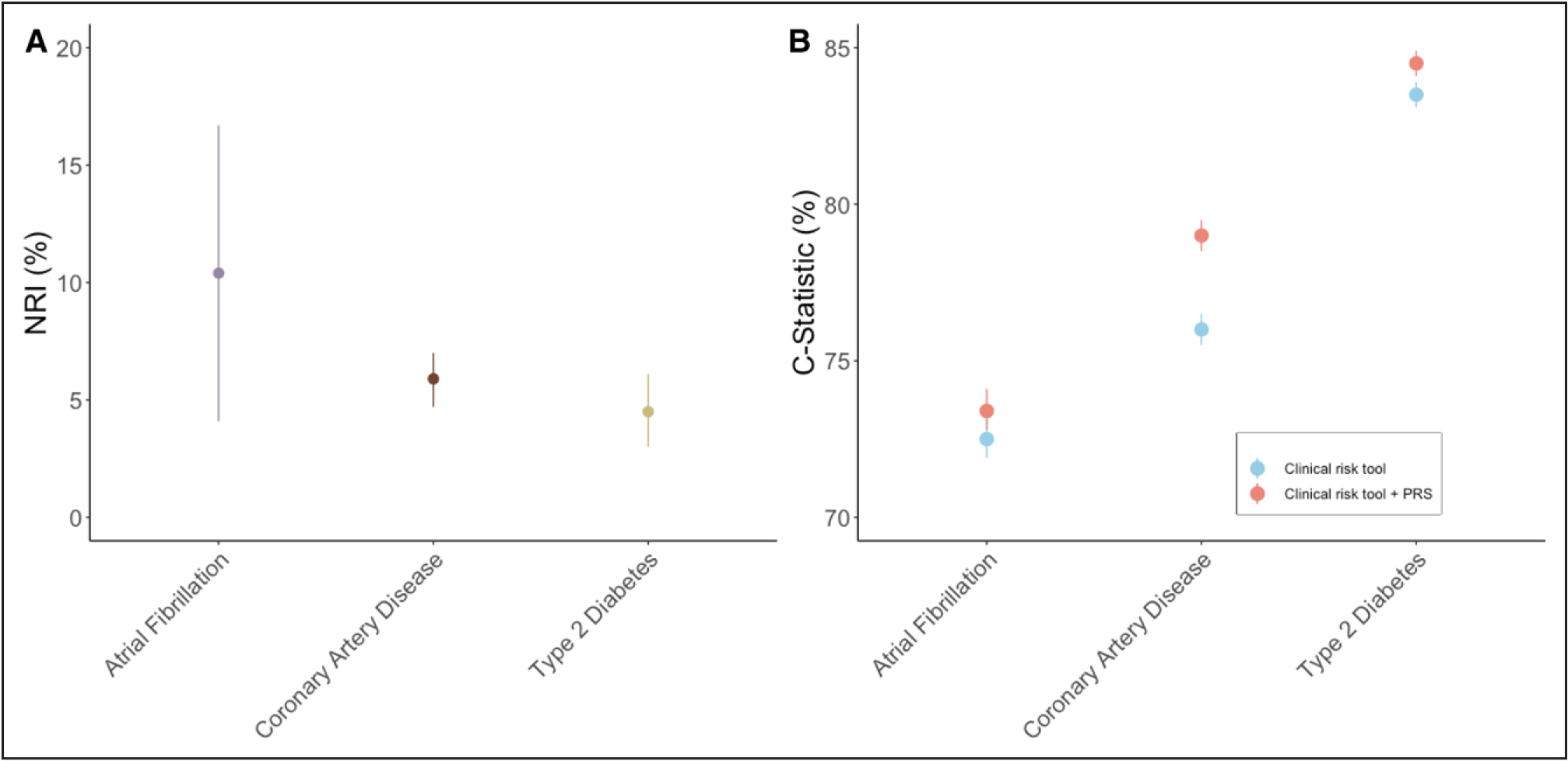
A, Net reclassification index (NRI). Comparison of clinical risk tools with and without the integration of polygenic risk score (PRS).23,63 The clinical risk tool used atrial fibrillation was CHARGE-AF (Cohorts for Heart and Aging Research in Genomic Epidemiology model for atrial fibrillation) with a risk threshold of >5% over 5 years. Variables included in CHARGE-AF were age, height, weight, systolic blood pressure (SBP), diastolic blood pressure (DBP), smoking status, blood pressure–lowering medication, diabetes, heart failure, and history of myocardial infarction. The clinical tool for coronary artery disease was the American Heart Association/American College of Cardiology Pooled Cohort Equation with a 7.5% risk threshold over 10 years and included the following variables: age, diabetes, sex, race, smoking, total cholesterol, high-density lipoprotein (HDL), systolic blood pressure, and treatment for hypertension. The clinical tool for type 2 diabetes was the American Diabetes Association risk score, which had a 33% risk threshold over 10 years and included the following variables: age, sex, body mass index, history of stroke or coronary heart disease, parental history of diabetes, SBP, DBP, HDL, and triglycerides. All differences are statistically significant. B, Comparison of C statistics between clinical risk scores (same as stated in A) and a risk tool with a PRS integrated into the clinical risk tool. All differences are statistically significant.23,63
Diversity of included participants remains an issue; only 1 study of AF PRS focused primarily on non-European participants. This study included Japanese participants and showed results consistent with the studies focused on Europeans (the AUC for the model combining clinical risk factors and PRSs in Japanese participants was 0.84 [95% CI, 0.80–0.86], 6% higher than the model including clinical risk factors alone without PRS).64
In addition to the prediction of AF, AF PRSs may have a role in the prediction of AF-associated complications such as ischemic stroke.71 Evidence from GWAS shows a shared genetic cause between AF and ischemic stroke: Loci at the PITX2 and ZFHX3 genes are associated with risk of AF and ischemic stroke.72,73 Several AF PRSs are predictive of stroke.62,74,75 Similarly, ischemic stroke PRSs appear to be predictive of strokes specifically in patients with AF, even beyond conventional clinical risk factors (eg, CHA2DS2-VASc score).71 Early analyses of the ability of PRSs to predict AF recurrence are inconclusive, with larger studies likely required to investigate whether PRSs play a role.69
For adult patients, established AF risk prediction tools (CHARGE-AF76) are improved with the addition of PRSs (across sexes and age groups [18–85 years]; Figure 2).23 Furthermore, given the lack of clinical risk factors, only PRSs can predict the development of early-onset AF.23,70 Future studies should focus on AF surveillance and risk mitigation strategies for high AF PRSs, as well as cost-effectiveness. The decreasing costs of genetic testing and the ability to calculate PRSs for a large number of diseases from 1 test increase the likelihood of cost-effectiveness, but this is yet to be formally studied for AF.
Coronary Artery Disease
Given the aggregation of CAD in families, particularly when occurring earlier in life, genetic variation has long been expected to influence CAD risk.77 CAD heritability, or the proportion of phenotype explained by the additive sum of genetic factors, is estimated to be 40% to 60%.13,78
FH is a well-recognized monogenic condition.44 Retrospective analyses indicate that those with FH variants have a greater relative and absolute clinical benefit from statins for the prevention of incident CAD.79 Current guidelines and US Food and Drug Administration (FDA) labels support additionally aggressive pharmacological LDL-C lowering among individuals recognized to have FH.80
GWAS of CAD in the general population has shown that common genetic variation also influences the risk for CAD.81 Increasingly large CAD GWAS continues to identify novel genomic loci; 167 separate genomic loci have been identified to be significantly associated with CAD to date.82 Systematic pleiotropy analyses indicate that the majority of these loci do not influence CAD risk through well-recognized risk factors.83
The discovery of common genetic variants associated with CAD has enabled the development of PRSs for the prediction of CAD (Table 2).23,63 Initial PRSs focused on the simple summation of significantly associated independent CAD risk alleles, and subsequent PRSs weighted these alleles by predicted CAD effects. Contemporary scores have focused on liberalizing variant inclusion through varied statistical approaches.20,38 Scores have been shown to be strong predictors of subclinical coronary atherosclerosis and independently prognostic of CAD risk (1.4- to 1.6-fold per SD of the CAD PRS), including self-reported family history and comparable or more powerful predictors of CAD than individual clinical risk factors (smoking, T2D, lipid measures, hypertension; Figure 3).20,23,38,63,84–88 Because the top fifth percentile of CAD PRS is associated with an 8–mg/dL increase in LDL-C, conventional clinical risk scores do not readily detect high CAD PRSs.85,89 Furthermore, the top 95th percentile (1 in 20) of a CAD PRS score is associated with a 3-fold odds for CAD, similar to that associated with FH (1 in 313) without accompanying severe hypercholesterolemia.20,85 Furthermore, in analyses among individuals with prevalent ASCVD, a CAD PRS was independently associated with incident major cardiovascular events (1.1- to 1.2-fold per SD of the CAD PRS).90–92 An initial study in ARIC (Atherosclerosis Risk in Communities) and MESA (Multi-Ethnic Study of Atherosclerosis; n=7237) showed little improvement in the C statistic with the addition of PRSs to PCE to predict incident events.93 More recent studies using more contemporary statistical approaches and validation in larger cohorts (n=352 660 and n=≈250 000) have shown slightly greater performance.63,94 Furthermore, it is likely that different age groups will derive varying benefit from PRSs. For example, NRI reached a peak of 15.4% (95% CI, 11.6%–19.3%) for younger subgroups and an improvement of the C statistic of 5% (0.05 [95% CI, 0.03–0.07]).
Figure 3. Predictive ability of polygenic risk scores for coronary artery disease.
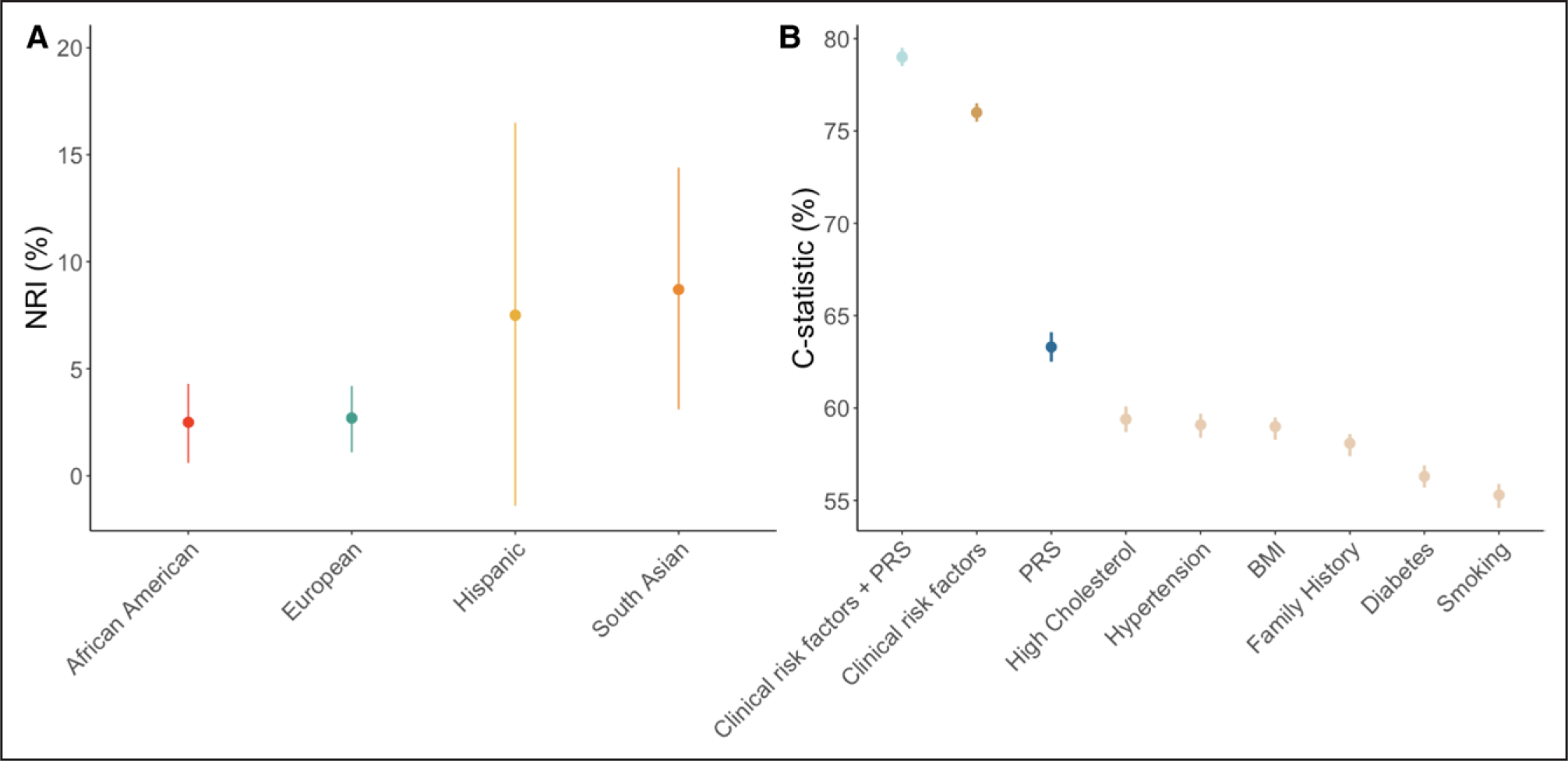
A, Net reclassification index (NRI) comparing clinical risk tools and a risk tool with a polygenic risk score (PRS) integrated into the clinical risk tool for coronary artery disease across multiple ethnicities.84 African American includes Black Caribbean and Black African, and South Asian includes Indian, Bangladeshi, or Pakistani. Copyright © 2021 The Authors. Published by Elsevier Inc. Creative Commons CC-BY license. This is an open access article distributed under the terms of the Creative Commons CC-BY license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. You are not required to obtain permission to reuse this figure. B, NRI for the American Heart Association/American College of Cardiology Pooled Cohort Equations (AHA/ACC PCE) tool+PRS, clinical risk factors collectively as the AHA/ACC PCE tool, PRS, and individual clinical risk factors for coronary artery disease.38,63 BMI indicates body mass index. Copyright © 2018 The Authors. Creative Commons CC-BY license. Published by Elsevier on behalf of the American College of Cardiology Foundation. This is an open access article distributed under the terms of the Creative Commons CC-BY license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. You are not required to obtain permission to reuse this figure.
Although they represent vastly different technologies, comparisons are often made with coronary artery calcium (CAC) scoring and PRSs. These diagnostic approaches have not been compared directly. However, data suggest that both technologies improve the predictive accuracy of clinical risk factors alone (PCE),63,95 and their predictable abilities appear to be at least comparable. For example, in a cohort of 1688 middle-aged adults, CAC scoring improved prediction when combined with the PCE: the C statistic increased from 0.633 with clinical risk factors alone (PCE) to 0.678 with PCE and CAC (ΔC, 0.045 [95% CI, 0.015–0.101]). Furthermore, there was significant improvement in NRI when CAC was added to the PCE: NRI, 6.7% (95% CI, 1.8%–11.6%).95 In comparison, a recent PRS validated in 186 451 participants showed that the C statistic increased from 0.76 with clinical risk factors alone (PCE) to 0.79 (ΔC, 0.03 [95% CI, 0.02–0.04]), and there was significant improvement in NRI when combined with the PCE (C statistic, 0.76 [95% CI, 0.75–0.76]; NRI, 5.8% [95% CI, 4.7%–7.0%]).63 These data were ascertained from different cohorts of varying sample sizes and not directly compared. Evidence also suggests a correlation between PRS and CAC scores.96,97 This indicates that stratified use of the respective technologies may be beneficial, but this topic requires further prospective study.
Observational analyses in epidemiological studies or post hoc analyses within completed randomized controlled trials for CAD prevention have yielded hypotheses for strategies to reduce CAD risk in the setting of a high CAD PRS. Because a CAD PRS is largely additive of nongenetic factors, adherence to a healthful diet mitigates CAD risk regardless of CAD PRS; however, given the worse prognosis, the absolute risk reduction from a healthful diet among those with a high CAD PRS may be greater than for those without a high CAD PRS.88,98 In a single-arm study, disclosure of a higher CAD PRS may influence favorable lifestyle behaviors.99 Beyond lifestyle changes, it appears that PRS risk correlates with medication efficacy. Subgroup analyses in primary prevention statin trials indicated that those with high CAD PRS versus others derived greater relative and absolute clinical benefit from statins versus placebo (46% versus 26%; Pheterogeneity=0.05) without differences in LDL-C reduction.86,91 Similarly, among individuals with established ASCVD on statins, greater benefit from PCSK9 monoclonal antibodies was observed among those with high versus low CAD PRS (37% versus 13%; Pinteraction=0.04).90,92 Data exploring the correlation between PRS and medication efficacy are further explored in the PRSs for Pharmacogenomics section. Similarly, a healthy lifestyle has been shown to reduce disease risk across all deciles of PRS risk, although the greatest reductions are in those with highest PRS risk.100 A randomized controlled trial among patients without CAD showed that disclosure of a high CAD PRS versus low CAD PRS or no PRS disclosure to patients and clinicians led to lower LDL-C concentrations.101
The integration of PRS for CAD into clinical practice will likely rely on its inclusion in current cardiovascular risk prediction tools. Currently, the AHA, American College of Cardiology (ACC), and many other international organizations recommend determining the 10-year cardiovascular risk for all adult patients 40 to 75 years of age with the AHA/ACC ASCVD risk calculator.2 For adult patients, the addition of PRS to the AHA/ACC ASCVD risk calculator significantly improves prediction for cardiovascular events (Figure 2).23,38,63,89,94 This enhanced prediction is independent of conventional clinical risk factors (Figure 3), is detectable before the emergence of clinical risk factors, and is appreciable across a spectrum of ages, sexes, and, increasingly, ancestries and ethnicities (South Asian, Black/African American/Black Caribbean/Black African, Chinese, and Japanese).23,36,38,63,84,94,102,103 The inclusion of PRS in the AHA/ACC ASCVD risk calculator significantly improves prediction, and early evidence suggests that targeted screening may be cost-effective (further discussed in the Considerations for Payers section),104 particularly because evidence suggests that conventional tools are missing participants at high PRS risk.89
Among asymptomatic middle-aged adults at borderline/intermediate risk by conventional clinical risk factors, a high CAD PRS has been shown to aid in reclassifying statin prescriptions.89 Among asymptomatic younger adults, a high CAD PRS may prompt more intensive earlier efforts for lifestyle modification and potentially earlier statin initiation akin to severe hypercholesterolemia to mitigate high lifetime risk for CAD.105
Hypercholesterolemia
Lipids, or cholesterol and triglycerides, are carried in the blood by lipoprotein macromolecules such as LDL and high-density lipoprotein. The properties of circulating lipoproteins are dictated by the relative concentrations of cholesterol, triglycerides, phospholipids, and proteins and biochemical alterations thereof. A reduction of apolipoprotein B–containing lipoproteins such as LDL-C is associated with a reduction in major adverse cardiovascular disease events through multiple pharmacological classes in randomized controlled clinical trials.37,106–109
Common genetic variants, summed as a PRS, can also predict lipid concentrations (Table 3). For example, the top 95th percentile of an LDL-C PRS carries an LDL-C effect (≈30 mg/dl) similar to that in individuals with an FH variant among those of European ancestry. Among those with severe hypercholesterolemia, 2% have an FH variant, whereas 23% are in the top fifth percentile of an LDL-C PRS.111 Furthermore, an LDL-C PRS explains some variation in LDL-C concentrations among individuals with FH variants.50
Table 3.
Characteristics of Published Studies That Compared Risk Estimates for Carriers of Monogenic-Risk Variants Versus Individuals With a High Polygenic Score
| Disease, author (y) | SNVs, n | Participants: n cases/N total | Biobank | Participants: ancestry/ethnicity, age, y, other | HR/OR PRS (95% CI) (included covariates controlled for) | HR/OR monogenic risk (95% CI) (included covariates controlled for) | HR/OR PRS+monogenic risk (95% CI) (included covariates controlled for) |
|---|---|---|---|---|---|---|---|
| Hypercho-lestolemia, Fahed et al19 (2020) | 6 630 150 | 6432/12 852 | UK Biobank | European, 40–69 | OR for top 20% PRSs, 2.28 (2.10–2.48) (age, sex, and the first 4 principal components of ancestry) vs 20%–80% PRS risk | OR for FH carriers (LDLR, APOB, or PCSK9 mutations), 3.21 (1.72–5.99) (age, sex, and the first 4 principal components of ancestry) compared with noncarriers | OR for FH carriers (LDLR, APOB, or PCSK9 mutations) and top 20% PRS risk, 12.61 (2.96–53.62) (age, sex, and the first 4 principal components of ancestry) OR for FH carriers (LDLR, APOB, or PCSK9 mutations) and bottom 20% PRS risk, 1.30 (0.39–4.32). Both compared noncarriers and 20% to 80% PRS risk. |
| VTE, Klarin et al110 (2019) | 297 | WHI: 690/10 975 MVP: 2100/55 965 | MVP 2.1, UK Biobank: development, MVP 3.0, WHI: validation | European, 19–100 | HR for top 5% risk in WHI, 2.51 (1.97–3.19) (age, 10 principal components of ancestry, and hormone therapy intervention status during the active phase of the WHI-HT) OR for top 5% risk in MVP, 2.89 (2.52–3.30) (age, sex, and 5 principal components of ancestry) compared with the bottom 95% risk |
HR for factor V Leiden in WHI (F5 p.R506Q), 2.34 (1.86–3.35) HR for factor V Leiden in WHI (F2 G20210A), 3.35 (1.10–10.23) (age, 10 principal components of ancestry, and hormone therapy intervention status during the active phase of the WHI-HT) OR for factor V Leiden in MVP (F5 p.R506Q), 2.97 (2.63–3.36) OR for factor V Leiden in MVP (F2 G20210A), 2.61 (2.19–3.12) (age, sex, and 5 principal components of ancestry) |
“In addition, we observed that this risk was further compounded for individuals among the top 5% with increased polygenic VTE risk who were also F5 Leiden or F2 G20210A carriers.”110 |
FH indicates familial hypercholesterolemia; HR, hazard ratio; MVP, Million Veteran Program; OR, odds ratio; PRS, polygenic risk score; SNV, single nucleotide variant; VTE, venous thromboembolism; WHI, Women’s Health Initiative; and WHI-HT, Women’s Health Initiative hormone therapy.
Because monogenic carriers meet current clinical guidelines for intervention, a logical extension would be considering individuals with similar risk levels attributable to polygenic risk status.
LDL-C PRSs, in addition to predictive LDL-C concentrations, are predictive for ASCVD events.19,112 Recent work has indicated that an LDL-C PRS may predict ASCVD event risk in addition to cross-section LDL-C measures. Among nearly 49 000 participants of the UK Biobank, ASCVD risks were compared for those with FH variants, the top 95th percentile of an LDL-C PRS, and those with hypercholesterolemia to a similar degree without the aforementioned genetic factors.113 Compared with nongenetic hypercholesterolemia, FH and polygenic hypercholesterolemia were associated with a 1.93- and 1.26-fold risk, respectively, for ASCVD events (coronary and carotid revascularization, myocardial infarction, ischemic stroke, and all-cause mortality).
LDL-C PRSs have also been correlated with risks for other cardiovascular disease, including aortic stenosis,114 abdominal aortic aneurysm, peripheral arterial disease, and venous thromboembolic disease.110 However, the prognostic capabilities of LDL-C PRSs that are independent of LDL-C measurement for these conditions have not been well studied to date.
PRSs for other lipid fractions also have been evaluated for ASCVD risk prediction. High-density lipoprotein cholesterol PRSs have limited prognostic utility for ASCVD prediction. Although triglyceride PRSs generally support an association with CAD events, the clinical utility of a triglyceride PRS is not well established. Lipoprotein(a) is a highly heritable LDL-like lipoprotein, additionally harboring apolipoprotein a, which is independently predictive of ASCVD events.115 An LPA PRS is strongly predictive of both lipoprotein(a) and ASCVD events.116 In fact, an LPA genetic risk score performs similarly to lipoprotein(a) measurement for the prediction of CAD events.117
For individuals undergoing genetic testing for severe hypercholesterolemia, comprehensive evaluation for monogenic and polygenic determinants will increase genetic testing diagnostic yield compared with FH gene panel testing alone and may refine CAD risk prediction beyond conventional lipid measures. Lipid and CAD PRSs will likely have distinct yield, according to the presence and extent of hypercholesterolemia. Both may have roles in identifying individuals for whom aggressive and early lipid-lowering therapies are indicated; this hypothesis requires prospective evaluation. Future work focusing on lipid and lipoprotein PRSs in the context of therapeutic interventions may better clarify clinical relevance.
Type 2 Diabetes
Epidemiological research has long appreciated the clustering of T2D within families.16 The scientific pursuit of monogenic causes of T2D has identified some causal genes (eg, peroxisome proliferator-activated receptor γ [PPARG])118; however, recent GWAS has revealed that a polygenic, rather than monogenic, predisposition accounts for substantially more T2D heritability.119
The advent of GWAS revealed the polygenic architecture of T2D,119,120 which, in turn, facilitated the development of PRSs for T2D.20,121 Early T2D PRSs showed the predictive ability of summated genetic loci.122 With increasing GWAS sample sizes and improvements in PRS methodology, recent T2D PRSs have become more accurate (Table 2).20,23,63,121 For instance, a 2018 meta-analysis of 32 GWAS including participants of exclusively European ancestry included 171 249 variants and showed a 9-fold increase in risk between the lowest 2.5% and the highest 2.5% PRSs.121 This same study showed that T2D prediction with PRS alone was similar to the prediction from body mass index, age, and sex (AUC reported as 66% for both, no 95% CI reported). Similarly, a 2018 study of participants of European ancestry in the UK Biobank included 6 917 436 variants and showed a linear increase in risk with higher PRS20; participants in the highest 10% PRS had a 2.5-fold risk, and participants in the highest 1% PRS were at a 3.3-fold risk (compared with the remaining 90% and 99%, respectively). This same study showed an AUC of 0.72 (95% CI, 0.72– 0.73) for PRS (inclusive of age, sex, genotyping array, and the first 4 principal components of ancestry as covariates).20 Last, a 2020 study showed those in the top 2.5th percentile risk were at 3.5-fold increased risk compared with those in the middle 20th to 80th percentile risk, and those in the bottom 2.5th percentile risk had an ≈80% reduction in lifetime risk compared with the middle 20th to 80th percentile risk.23 This same study showed a modest improvement in risk prediction when PRS was added to clinical risk factors: The C statistic modestly improved from 0.84 (95% CI, 0.83–0.84) with clinical risk factors only to 0.85 (95% CI, 0.84–0.85) with the inclusion of PRS.23 This same study showed more convincing improvement in prediction with the NRI metric: The addition of PRS to the above American Diabetes Association clinical risk factors criteria showed an NRI of 4.5% (95% CI, 3.0%–6.1%; Figure 2).23 Although older studies previously showed little improvement in discrimination compared with cumulative clinical risk factor models, more recent studies, substantially improved with the inclusion of a larger number of SNVs and trained on a larger sample size, showed improvement in prediction with the inclusion of PRS.122–124 For example, a 2010 study that used a 40-SNV PRS123 reported an AUC of 0.54 (95% CI, 0.50–0.58) compared with a 2020 study that used a 6 437 380–SNV PRS (0.763 [95% CI, 0.758–0.767]).23
Nevertheless, the early identification of those at high risk of T2D is just one potential application of PRS and one that remains of unclear therapeutic value. Whether determined by clinical or genetic risk (or both), the primary benefits of early identification of T2D risk are targeted prevention through lifestyle modification and more rigorous surveillance. Recent evidence suggests that stratifying risk by PRS may help identify high-risk subgroups for whom successful lifestyle modification is associated with greater absolute reduction in the risk of incident diabetes.125 However, older evidence suggests more modest effects across T2D PRS strata.98,126 Therefore, it may be argued that those who are identified as high risk through PRSs should be screened at a younger age or at more frequent intervals, but data supporting the use of PRSs for this application are needed.
A T2D PRS may also predict treatment responsiveness. This is further described in the PRSs for Pharmacogenomics section. In brief, a 2020 study showed that participants with a higher T2D PRS had greater reductions in hemoglobin A1c (HbA1c) in response to sulfonylurea therapy (P=0.02).127 The authors also investigated the relationship between PRS and glycemic surrogates of metformin response but found no association.127
In regard to glycemic control, a 2016 study showed that a PRS modified the effect of intensive glycemic control on cardiovascular mortality in the ACCORD randomized trial (Action to Control Cardiovascular Risk in Diabetes)128: Participants with a high T2D PRS were found to have a 3-fold risk of cardiovascular mortality with intense glycemic control (HR, 3.08 [95% CI, 1.82–5.21]), whereas those with a low T2D PRS had a substantial mortality benefit from intensive glycemic control (HR, 0.24 [95% CI, 0.07–0.86]).128
This application of genetic data to guide risk stratification of T2D complications and treatment response mirrors efforts to cluster patients with T2D with the use of clinical factors, which has demonstrated some early promise for guiding clinical care.129,130 Five genetically defined subtypes of diabetes (severe autoimmune diabetes, severe insulin-deficient diabetes, severe insulin-resistant diabetes, mild obesity-related diabetes, and mild age-related diabetes)131 have distinct underlying pathophysiologies with varying diabetes-associated complications.130,132
The predictive accuracy of the American Diabetes Association risk tool is increased with the addition of a PRS.23 The cost-effectiveness of screening remains less clear. Although the cost of genetic testing continues to fall, the effect of therapies that may be offered to those at high risk remains an active area of research. Given that an advantage of PRS is the possibility to calculate numerous PRSs from 1 test, cost-effectiveness analyses that consider risk prediction of both CAD and T2D may be advantageous (because the cost of a PRS to calculate a number of diseases will be the same as the cost to calculate 1 disease). Furthermore, a scientific opportunity remains for PRSs to help personalize T2D pharmacological management in terms of medication responsiveness and earlier prevention of microvascular and macrovascular complications.
Venous Thromboembolic Disease
Acute venous thromboembolism (VTE), which comprises deep venous thrombosis and pulmonary embolism, occurs in around 1000 000 individuals yearly in the United States1 and is the among the leading causes of acquired harm and preventable death in hospitalized patients.133 Although inherited monogenic thrombophilias (eg, factor V Leiden and prothrombin G20210A) increase the relative risk of VTE by ≈3- to 5-fold, the role of genetic testing in informing therapy has remained limited134,135 because of uncertainties about the effects of inherited thrombophilias on recurrent VTE risk,136–138 the lack of data demonstrating that thrombophilia testing improves outcomes,139 and the risks of prolonged anticoagulation.
Recent advances in the genetics of VTE have produced increasingly robust GWAS summary statistics enabling the development of PRS predictive of VTE. With the use of summary statistics from a GWAS comprising 30 234 VTE cases, a 37-variant VTE PRS was recently constructed (Table 3).140 Among 6573 cases and 20 515 controls from the UK Biobank, individuals in the lowest fifth percentile of genetic risk had a 50% lower odds of having had VTE (odds ratio, 0.51 [95% CI, 0.42–0.6]), and those in the upper 95th percentile had a 3.2-fold odds of having had VTE (odds ratio, 3.2 [95% CI, 2.9–3.5]) compared with those in the intermediate range. Similarly, another recent analysis of 26 066 separate VTE cases ultimately yielded a 297-variant PRS for VTE, inclusive of many of the SNVs or correlated SNVs in the 37-variant score.110 After exclusion of the factor V Leiden and prothrombin G20210A variants, the 297-variant PRS was applied to an independent set of 2100 cases with VTE and 53 865 VTE controls.110 In the same study, across 10 975 women in the Women’s Health Initiative with 690 incident VTE events over 25 years, the women in the top 95th percentile of polygenic risk had a 2.5-fold risk of incident VTE (HR, 2.5 [95% CI, 2.0–3.2]); this was comparable in effect size to the risk conferred by factor V Leiden (HR, 2.4 [95% CI, 1.9–3.4]) and prothrombin G20210A (HR, 3.3 [95% CI, 1.1–10.2]).110
Current guidelines recommend prophylactic anticoagulation for patients with factor V Leiden in certain clinical situations (surgery, hospitalization, peripartum, and postpartum).141 Given the comparable risk of factor V Leiden and those at highest PRS risk, future research could examine whether there are groups who would benefit from VTE PRS–aided use of prophylactic anticoagulation in high-risk scenarios. Furthermore, future studies can examine the predictive performance of the current risk scores (the Padua score for medical inpatients142 and the Caprini score for surgical inpatients143) with the integration of PRS. Because prophylaxis for VTE is generally anticoagulation, the potential risks should also be studied in prospective studies.
PRSs FOR PHARMACOGENOMICS
Evidence supporting PRSs for cardiovascular pharmacogenomics is evolving. Until recently, candidate gene studies including only 1 or a few genes have been the focus of most pharmacogenomic research studies144–146 and clinical implementation programs.147 The development of pharmacogenomic PRSs has been hindered by unique challenges for pharmacogenomic GWAS such as securing adequate sample sizes of patients treated with the same drug, along with the necessary drug and phenotypic data (eg, dose, frequency, adherence, drug response metrics).148 Large consortia are working toward overcoming this barrier (eg, International Clopidogrel Pharmacogenomics Consortium149 and International Warfarin Pharmacogenetics Consortium150). Researchers are also using creative approaches to calculate pharmacogenomic PRSs such as scanning multiple candidate genes151,152 or testing disease-associated PRSs for associations with cardiovascular drug responses.86,91 These efforts have led to several currently published studies applying PRSs to cardiovascular pharmacogenomics.
These pharmacogenomic PRSs have been applied with 4 different goals: (1) predicting drug efficacy, (2) predicting drug toxicity, (3) reviving drugs that failed in clinical trials (ie, by finding a genetic subgroup of responders, although successful examples have not been demonstrated to date), and (4) predicting adverse cardiovascular reactions for noncardiovascular drugs. For the prediction of drug efficacy, the strongest evidence currently supports PRSs predicting statin response. A PRS predictive of CAD risk also predicted the absolute and relative benefit from statins for the primary and secondary prevention of CAD.86,91 Similarly, a 2019 study showed that patients with a high PRS for CAD had a larger risk reduction (absolute and relative) in major adverse cardiovascular events and death when treated with alirocumab (a PCSK9 inhibitor): Participants at high PRS risk (>90th percentile) had a 6% absolute reduction (95% CI not reported) compared with a 1.5% absolute reduction in those at low PRS risk (≤90th percentile).90 Participants at high PRS risk had a relative risk reduction by alirocumab of 37% (HR, 0.63 [95% CI, 0.46–0.86]) compared with a 13% reduction in the low-PRS group (HR, 0.87 [95% CI, 0.78–0.98]).90 A 2020 study of another PCSK9 inhibitor, evolocumab, found a similar relationship in which patients with higher PRS risk benefited more from evolocumab therapy.92 The absolute risk reduction for major vascular events improved significantly across an increase in the genetic risk category: 0.7%, 0.9%, and 4.0% absolute risk reduction in low-, intermediate-, and high-genetic-risk groups, respectively (Ptrend=0.04). Participants at high PRS risk had a relative risk reduction by evolocumab of 31% (HR, 0.69 [95% CI, 0.55–0.86]) compared with 9% (HR, 0.91 [95% CI, 0.79–1.03]) in the intermediate-PRS-risk group and 8% in the low-PRS-risk group (HR, 0.92 [95% CI, 0.72–1.18]; Ptrend=0.07).
Beyond CAD, there is also a building body of evidence to support the correlation between T2D PRS and treatment responses. A 2020 study showed that a T2D PRS was predictive of a reduction in HbA1c in patients taking a sulfonylurea127; for every 1-SD increase in T2D PRS, there was a 0.06% (0.07 mmol/mol) decrease in HbA1c level in response to sulfonylurea therapy (P=0.02).127 Similarly, participants in the highest decile of the T2D PRS had a 0.27±0.12% greater HbA1c reduction compared with those in the lowest decile (P=0.03).127 The authors also investigated the relationship between PRS and glycemic surrogates of metformin response but found no association.127
Evidence also supports PRS predicting clinical benefit from β-blockers (predicting survival benefit in patients with heart failure with reduced ejection fraction),153 angiotensin-converting enzyme inhibitors (predicting cardiovascular mortality, nonfatal myocardial infarction, or resuscitated cardiac arrest),154 calcium channel blockers (predicting all-cause death, nonfatal myocardial infarction, or nonfatal stroke),155 and clopidogrel (predicting ischemic events and cardiovascular mortality).152 These PRSs for clinical responses to β-blockers, angiotensin-converting enzyme inhibitors, and calcium channel blockers were not based on disease PRS; rather, they were derived from drug×SNV interaction tests for clinical outcomes in GWAS or multiple candidate genes. The PRS for clopidogrel was also not derived from disease PRS but rather candidate SNVs independently associated with high on-clopidogrel platelet reactivity.
A further study assessed the ability of a PRS derived from a GWAS of the QT interval to predict the outcome of drug-induced QT interval prolongation and torsades de pointes,156 demonstrating the potential for PRSs derived from intermediate phenotypes to predict cardiovascular drug toxicities. PRSs have also been used in an attempt to revive a drug that failed in clinical trials. However, a CAD PRS did not successfully identify a genetic subgroup of patients who benefited from evacetrapib, a cholesterol ester transfer protein inhibitor.157
CONSIDERATIONS FOR HEALTH CARE SYSTEMS
Criteria for Implementing PRSs in Cardiovascular Clinical Practice
We suggest 3 broad criteria to be considered by health care systems before implementation of PRSs for cardiovascular care: (1) efficacy, (2) harm, and (3) logistics. First, estimations of benefit overall and across subgroups from observational data sets are likely to be key initial driving forces. Although issuing definitive criteria for the clinical implementation is beyond the scope of this scientific statement, we suggest in broad terms that the clinical efficacy of PRS is likely appropriate when either of the following is achieved: (1) The integration of PRS into clinical risk tools substantially improves their accuracy, or (2) PRS risk tools can identify participants at a risk at least equivalent to that of individuals with monogenic risk variants (such as LDLR for FH; Table 4). In regard to the first point, for most of the disease examples in this scientific statement (AF, CAD, and T2D), the predictive accuracy of established clinical risk factor models is improved with the addition of PRS (ie, PRS improves prediction when incorporated into the PCE for ASCVD, the CHARGE-AF risk model [AF], and the American Diabetes Association risk model [T2D]).23,63
Table 4.
Potential Clinical Utility of PRSs
| Disease/risk factor | Potential clinical utility of PRS |
|---|---|
| CAD | Earlier identification for lifestyle therapies and statins, potentially for those with very high CAD PRSs Earlier screening for subclinical atherosclerosis to time the initiation of pharmacotherapies Use as a risk-enhancing factor for primary prevention in middle-aged patients at borderline-intermediate 10-y ASCVD risk |
| AF | Earlier AF detection and resultant prophylactic anticoagulation, potentially with monitoring devices Rigorous control of additive clinical risk factors for AF |
| T2D | Earlier lifestyle modification Potential consideration of prophylactic hypoglycemic medications with concomitant additional T2D clinical risk factors Genomic stratification may optimize hypoglycemic choice |
| VTE | Rigorous VTE risk-reducing strategies in the context of high-risk scenarios (prolonged travel, major surgery, etc) |
| Hypercholesterolemia | Earlier institution and earlier uptitration of lipid-lowering pharmacotherapies analogous to FH |
| Pharmacogenomics | Personalized drug therapy regimens that increase drug efficacy and decrease toxicities, eg, personalized β-blocker target dose in patients with HFrEF or the prevention of drug-induced QT prolongation |
AF indicates atrial fibrillation; ASCVD, atherosclerotic cardiovascular disease; CAD, coronary artery disease; FH, familial hypercholesterolemia; HFrEF, heart failure with reduced ejection fraction; PRS, polygenic risk score; T2D, type 2 diabetes; and VTE, venous thromboembolism.
Lone AF refers to AF in the absence of other cardiovascular risk factors (typically in young adults).
Furthermore, statin allocation, particularly in scenarios of clinical equipoise for primary CAD prevention among middle-aged adults, is harmonized with the 2019 ACC/AHA cholesterol guidelines.2 Therefore, recent analyses in 3 ethnically and geographically distinct hospital biobanks have shown the capability of using a CAD PRS to allocate statins for primary prevention among middle-aged adults when 10-year estimated risk by clinical risk factors is borderline to intermediate.89 Post hoc pharmacogenomic analyses in randomized controlled trials may also help with refining treatment allocation.90 Whether screening earlier in life identifies individuals at sufficiently high lifetime risk to treat with statins on the basis of a CAD PRS alone, akin to the risk conferred by severe FH, requires further study. Preliminary analyses indicate that disclosure alone of a CAD PRS may improve health-related behaviors, but results are inconsistent to date.99,101,158 Limited experience with T2D PRS has not shown behavioral modification after disclosure.159 Improved risk prediction and allocation of established effective and safe therapies may be sufficient to motivate clinical use.
Second, with increasingly widespread availability, it is important for any guidance on responsible clinical use to address the potential harms. Although most current studies may estimate prognosis or treatment effects with retrospective analyses, estimations of harms remain challenging. There have been concerns about exacerbating existing racial disparities in health care with the use of existing PRSs. Initial and large GWAS still largely comprise individuals of European ancestry,40 which poses recalibration challenges, although ongoing efforts to genetically profile non-Europeans and novel methods are continuing to bridge this gap and have shown comparable predictive accuracy between multiple ethnicities (Figure 3).18,84 In addition, in some scenarios such as a high AF PRS or high VTE PRS, higher-risk prevention strategies such as initiation or extension of the duration of anticoagulation prophylaxis may be considered. For these riskier protocols, prospective randomized controlled trials addressing both safety and efficacy are necessary.
Third, several logistical and educational considerations exist. To date, genomic data for PRS calculation are largely external to the health care system (ie, research study or direct-to-consumer testing product) with few exceptions.160 Although germline genetic profiling is a static biomarker, associations are also functions of age and other potential nongenetic factors, as well evolving evidence refining interpretation of the human genome. Information technology systems should be robust to new knowledge to use new algorithms and to revise clinical decision support on the basis of static genotypes. Furthermore, population genetics literacy among both clinicians and patients remains limited, potentially hindering putative benefits and potentially exacerbating harms.1–3,5 The extent to which ancillary support such as specialized clinicians, including genetic counselors, is needed will likely vary across health systems. Last, health care systems should consider the regulatory guidance for use of PRSs (described in the Considerations for Commercial Genetics Organizations section)
Calibrating PRSs to the Population of a Health Care System
Similar to prediction equations based on clinical risk factors, performance of PRSs for disease prediction is sensitive to characteristics of the population in whom they are applied.39,40,161–163 Systemic underperformance will be strongly correlated with the extent of dissimilarity between the derivation and applied data sets. For example, ASCVD risk estimation from the PCE varies when applied to different cohorts.161,164,165 Cohort-specific calibration approaches have been demonstrated to improve performance for the PCE for ASCVD.161,164,165 Similarly, recalibration of PRS models for distinct genetic ancestries in target populations has been shown to improve performance. For example, a T2D PRS derived from individuals of European ancestry was reweighted according to Latino haplotypes with resulting improvement in T2D prediction among people of Hispanic ancestry.166 However, simple variant filtration based on predicted functional impact may also improve transancestry transferability.167 Similarly, prediction approaches tailored separately for men and women may improve on current approaches. For admixed individuals, partial PRSs corresponding to deconvoluted ancestries recombined168 and linear combinations of ancestry-specific PRSs166 have shown recent promise.
Current PRS studies are presented in percentiles according to the cohort studied. Therefore, varying genotyping platforms limit the ability to generate universal raw scores. Because percentiles are therefore a function of cohort ethnicity, admixture, and genotyping platform, internal calibration procedures are likely also necessary. Nevertheless, universal harmonization efforts would improve generalizations of PRS applicability beyond single health systems. The ultimate goal for application of PRSs should be their representation in absolute risks, not percentiles.
CONSIDERATIONS FOR COMMERCIAL GENETICS ORGANIZATIONS
Genetic testing in the United States falls under the College of American Pathologists (CAP) and the Clinical Laboratory Improvement Amendments (CLIA; part of the Centers for Medicare & Medicaid Services).169 The FDA has traditionally asserted its authority to oversee all diagnostic tests, including laboratory developed tests (LDTs). However, the FDA has for decades practiced “enforcement discretion,” leaving regulation of LDTs almost entirely to the CLIA/CAP process. The FDA clarified direct authority in 2010, intervening with several direct-to-consumer genetic testing companies,170 and in 2019 to 2020 with several pharmacogenomics testing companies.171,172 In 2020, however, in a ruling that derived direction from a Presidential Executive Order, the Department of Health and Human Services made FDA regulation of LDTs harder, requiring the agency to implement notice-and-comment rulemaking in any regulation of LDTs.173 Although the high-level regulatory authority for LDTs remains unclear and unstable, in practical terms, the FDA is not resourced to regulate the thousands of LDTs. This means that implementation of tests such as PRSs currently continues to fall under CLIA/CAP regulation.173 There are no specific guidelines for CLIA/CAP certification of PRS tests, but CLIA/CAP certification of any new test has relied on peer-reviewed publications.
Furthermore, commercial genetics companies need to consider how they report an individual’s risk and those responsible for medical follow-up when deemed necessary. The most easily understood and meaningful way of presenting one’s PRS remains somewhat unclear. Future research addressing effective PRS risk communication is needed.
Commercial direct-to-consumer genetics companies also need to consider medical follow-up workflows for individuals with a high PRS. For example, if an individual is determined to be in the top 1% of PRSs for T2D, should the commercial genetics company alert the patient’s primary care physician, recommend visiting with their primary care physician, recommend increased frequency of HbA1c screening tests, or provide some other guidance? These difficult-to-resolve questions highlight the advantage of commercial genetics companies collaborating with established health care systems.
Last, it would be advantageous if genetics companies welcomed open science practices. Both the advancement and integrity of genomic science have been greatly aided by collaborations, sharing of data (although maintaining privacy and confidentiality of the individuals who contributed their DNA), and open examination of methods. A continuation of these practices will help ensure the integrity and accuracy of both commercial and academic PRSs.
CONSIDERATIONS FOR PAYERS
How to Consider the Financial Integration of PRSs Into Clinical Practice
The goal for any health care system is to maximize population health, and payers are tasked with considering interventions that will achieve this in a financially responsible manner.174 Once the scientific accuracy of PRS is confirmed in its intended population (described in the Considerations for Health Care Systems section) and the regulatory standards are met (described in the Considerations for Commercial Genetics Organizations section), policymakers can begin to consider the financial implications.
For cost-effectiveness studies for PRS, the following costs should be considered: the one-off costs of genotyping (and the associated infrastructure) and algorithm development and ongoing costs such as laboratory and bioinformatics staff.175 Assessing these direct costs is particularly challenging, given the rapidly decreasing costs of genetic profiling; sequencing technology costs have dropped substantially over the past 10 years.176,177
Conversely, there are unique potential savings with the use of PRSs. With 1-time broad genetic profiling, PRSs for numerous conditions can be generated with trivial incremental costs. In addition, PRSs may provide health care savings through earlier targeted prevention and mitigation of future costly medicines or procedures, for example, through deimplementing screening for PRS-determined lower-risk groups. Such estimates are likely to vary per condition according to condition heritability, prognostic performance of the PRS, the intervention invoked, including efficacy by PRS, and several others.
There is a paucity of cost-effectiveness studies for cardiometabolic PRS. Those that are available focus largely on CAD.178–180 The existing literature uses different methodological approaches, includes a variety of populations, and shows mixed results. A 2018 simulation study of people 45 to 65 years of age examined the cost-effectiveness of a 27-SNV PRS guiding statin treatment in the primary prevention for ASCVD for those at low borderline 10-year ASCVD risk (2.5%–7.5%).178 Although the authors conclude that this strategy is not more cost-effective than treating all at low borderline risk, secondary analyses indicated that the use of a 27-SNV PRS might be cost-effective in some scenarios, including when 10-year ASCVD risk was closer to 7.5%. Because the majority of patients at low borderline risk are not currently recommended for statins, this scenario may not suitably reflect the anticipated use of a CAD PRS. Furthermore, PRSs have become more sophisticated since this study; among many other advances, the inclusion of more SNVs has significantly enhanced predictive accuracy (contemporary CAD scores include millions of SNVs).35,181 These methodological advances are reflected in a 2019 study that assessed the cost-effectiveness of a 49 310–SNV CAD PRS.180 This study used a bayesian decision-tree approach based on adults >45 years of age living in Finland and found that the addition of a PRS to clinical risk factors may be cost saving compared with clinical risk factors alone for certain patient segments.180 More recent data suggest that targeted screening with PRSs in addition to clinical risk factors is likely to be cost-effective through the prevention of “7% more cardiovascular disease events than conventional risk prediction alone.”104 As PRSs continue to improve, this benefit is expected to increase. It is notable that a PRS alone has a predictive accuracy that comparable to or greater than that of many individual clinical risk factors (including T2D and hypercholesterolemia).38,63 Further research would likely be beneficial, but the decreasing costs of genetic testing, the comparable cost to current biomarker tests (eg, the cost of an HbA1c test is comparable to the cost of a PRS),182 and the ability to calculate PRSs for a large number of diseases from 1 test increase the likelihood of cost-effectiveness. These data support cost-effectiveness among middle-aged adults at intermediate risk. Last, there are encouraging data on the cost-effectiveness of PRSs for noncardio-metabolic diseases, which has led to the inclusion of PRSs in current clinical risk tools such as in the Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm/CanRisk for breast cancer.183,184
Interpretation and Relevance of PRSs in the Context of Insurance Policies
In the United States, the Genetic Information Nondiscrimination Act prohibits the use of genetic information in the provision of health insurance or employment hiring, firing, pay, or promotion.185 However, this protection does not extend to life and disability insurance. Numerous other countries have government policies (on a spectrum of government intervention) that protect patients from insurance exclusion while maintaining market sustainability.186
CHALLENGES AND FUTURE DIRECTIONS
The overarching goals of a cardiovascular PRS include disease prevention by identifying at-risk individuals for improved disease surveillance and better-informed treatment plans. Although there are promising applications for PRS, several limitations should be acknowledged that would benefit from future work.
One limitation is that the current state-of-the-art polygenic risk models include only common variants.20,22,187–189 The advantages of including rare variants in PRS models have yet to be investigated. Recent studies have shown how rare and low-frequency variants can explain a substantial fraction of heritability.190–193 The number of discoveries of this type of variants is limited by lack of power; for cardiovascular diseases, only a few studies have identified rare and low-frequency variants that confer a high risk of disease (similar to variants that cause monogenic disease).194–197 To be able to capture information across the allele frequency and effect size spectrum, PRS models built from common variants can be improved by incorporating well-characterized, rare, high-risk variants. This is likely to be achieved as sequencing moves to whole-genome sequencing. Future studies should prioritize data from whole-genome sequencing to facilitate PRS models that incorporate common and rare variants, that is, a full allelic spectrum polygenic score. In the absence of whole-genome data on patients for whom a PRS is to be calculated, genotyping array data can be imputed to whole-genome sequencing data. However, imputation accuracy is typically low for rare variants, and poorly imputed variants can affect the quality of the PRS. Other options include high-coverage whole-genome sequencing,85 whole-exome sequencing and genotyping array, and high-coverage sequencing of individual genes and low-coverage sequencing across the genome.66 Technology developments that improve cost and efficiency will improve the accessibility for this type of test in the clinical practice.
A second limitation is the reduced transferability of PRSs across many different populations. Most training data are derived from a single population, typically of European ancestry. This limits the utility of current data for use in non-European populations,198 although there are promising data that current PRSs show similar predictive accuracy across ethnicity groups (Figure 3).36,84,103 Despite the increase in the proportion of GWAS and PRSs that include non-Europeans, having more non-Europeans in future biobanks and studies remains an urgent priority.166,199–203 To some degree, this has already begun. The Million Veteran Program has nearly 30% non-European individuals204; Biobank Japan has recruited exclusively from Japan205; and East London Genes and Health was established to recruit British South Asians.206 Increasingly large health care–associated biobanks represent the genomic diversity of their sociodemographically diverse patients.207 Newer biobanks such as All of Us Research Program and East London Genes and Health have focused on the recruitment of individuals traditionally underrepresented in biomedical research through parallel recruiting strategies206: recruiting from community settings aided by local organizing groups and recruiting from health care settings. Community engagement approaches that seek to understand more prevalent conditions among diverse ancestral groups may serve as a research engagement strategy. In the short term, computational strategies to improve PRS transferability to non-European populations will complementarily improve PRS generalizability.84,203,208 Similarly, future PRS studies may consider tailoring risk models by sex. Because there is substantial variation in the incidence of and risk factors for cardiovascular disease between sexes, enhanced prediction may be achieved by future cardiovascular risk models that include sex-specific PRSs and sex-specific clinical risk factors. This should be a focus of future research.
Future PRS research should be encouraged in existing and planned randomized controlled trials to conduct post hoc analyses to further assess clinical utility. Specifically, further work should examine the incremental value of PRSs over clinical scores, as well as treatment changes based on PRSs and resultant clinical outcomes. Although this strategy has already been effective in identifying subgroups who may benefit from PRS-directed treatment,86,91 there is a paucity of data for non-CAD diseases and for nondrug interventions. Moreover, given the cumulative lifetime effect of genetic risk, it would be of particular value to recruit younger adults (eg, <40 years of age) or even children and adolescents (eg, <18 years of age) into PRS research studies. PRSs have the unique advantage of assessing risk before the emergence of clinical risk factors and therefore can act as a modulator of screening practices; the intensification or deintensification of screening of children and young adults on the basis of a PRS would be a valuable focus of future research.
The apparent correlation in PRSs among family members has implications for cascade screening, particularly if a PRS is not already available for family members of a proband with a high PRS.209 Furthermore, earlier identification of at-risk individuals and earlier treatment that reduces causal risk factors such as LDL-C will likely provide stronger mitigation of risk for atherosclerotic diseases such as CAD. However, the clinical efficacy and cost-effectiveness of cascade screening with PRSs are yet to be fully explored.
Beyond the technical and analytical limitations, potential negative consequences of PRS adoption into cardiovascular clinical practice should be carefully considered. These potential considerations include, but are not limited to, widening care disparities related to access to PRSs,40 unequal benefit of PRSs across race and ethnicity groups, misinterpretation or misapplication of PRS information attributable to clinician knowledge or patient understanding, threats to patient well-being related to genetic data security and to coverage practices of insurers, and escalating health care costs.
The use of PRSs for CAD also merits further discussion. As the most studied phenotype to date, the efficacy of PRSs for CAD has been examined in numerous studies and produced mixed results. These mixed results may be attributable to varying sample sizes, varying statistical approaches, reliance on single accuracy metrics, and aggregated results across various subgroups. Studies in smaller biobanks (MESA and ARIC) have generally produced nonsignificant results compared with studies of larger biobanks (UK Biobank,63,94 Malmö Diet and Cancer Study,87 Women’s Genome Health Study88), and statistical approaches continue to advance and produce more accurate PRSs.36,63
The transparency and reproducibility of PRSs are essential as clinical integration is considered. In an effort to improve transparency, the PRS reporting standards writing committee recently published a reporting guideline.169 The guideline acts as a checklist for researchers performing PRS studies, outlining the minimum information that should be stated in a research article to ensure that the work is transparent and able to be reproduced. In broad terms, the reporting guideline includes information on study design and recruitment, participant demographics (including ancestry), genetic data, nongenetic variables, risk model development, and evaluation, including discrimination and calibration. This reporting guideline can act as a checklist not only for researchers but also in the appraisal of PRS studies by payers, health care systems, and policymakers considering implementation and reimbursement of certain PRSs. Furthermore, the polygenic score catalog is a freely available, open-access resource of published PRSs.210
Although we review 5 cardiometabolic diseases in this scientific statement, the polygenic bases for other cardiometabolic diseases are increasingly being characterized. Examples include stroke (hemorrhagic and ischemic), left ventricular end-systolic volume,211 hypertrophic cardiomyopathy,51 and heart failure.212 Similar encouraging data exist for channelopathies and related traits, including QT interval prolongation213,214 and Brugada syndrome.215
CONCLUSIONS
The identification of monogenic risk variants predisposing to cardiovascular conditions has been used clinically to inform surveillance and management plans. Relatively recent advances in population genetics have uncovered the polygenic basis of these and other cardiovascular conditions in most patients. These observations point to the possibility of using genetic profiling to inform clinical practice in significantly larger groups of individuals than for whom monogenic cardiovascular variants are considered. As a result of exponential increases in the proportion of individuals with broad genetic profiling, cardiovascular PRSs are beginning to enter clinical practice. Such PRSs may be appropriately considered in select scenarios, given the current evidence base. The evolving literature aims to continue to narrow the current knowledge gaps and to improve the performance and communication of PRSs.
Below, we recap the pertinent points covered in previous sections:
What Are PRSs? PRSs are single scores reflecting the cumulative weighted risk of individual genetic variation for a set of traits. These individual genetic variants confer an incrementally small disease risk, but summated, they have been shown to be predictive of many cardiovascular diseases.
Polygenic Versus Monogenic Risk Variants: Monogenic risk variants are typically single, protein-truncating variants conferring a relatively large risk of disease. Examples of monogenic risk variants for cardiovascular disease include LDLR for FH, GCK for diabetes,46 KCNQ1 for AF,47 and F5 for venous thromboembolic disease.48 PRSs independently associate with disease risk and, at particularly high scores, may yield similar estimated disease risk as monogenic risk variants.20
Atrial Fibrillation: PRSs for AF have consistently shown incremental predictive capabilities in addition to clinical risk factors.20,23,64–69 Proposed utility has been to refine the identification of individuals meriting close surveillance for AF development.
Coronary Artery Disease: CAD is perhaps the most studied cardiovascular phenotype for PRSs.20,23,38,85–88,90–94 Among middle-aged adults, a CAD PRS performs similarly to conventional risk factors and provides additional prognostic information for CAD,23,38,63,94 but the clinical significance of this improvement is contentious.24,94 Observational evidence suggests that CAD PRSs may have utility in guiding pharmacological management (particularly for LDL-C lowering) attributable to increased estimated disease risk and greater estimated treatment benefit.88,98
Hypercholesterolemia: LDL-C PRSs have been shown to be predictive of LDL-C concentrations,111 including severe hypercholesterolemia.111 Because an LDL-C PRS is predictive of ASCVD events independently of LDL-C,112 whether it should be used to help allocate novel LDL-C–lowering medicines (as FH variants currently allow) requires further study.
Type 2 Diabetes: Early research suggested that PRSs for T2D had a predictive ability similar to that of clinical risk factors.122–124 More recent evidence suggests that PRSs may be additive to clinical risk factors.23 However, the identification of those at high risk of T2D currently has unclear value; lifestyle modification and metformin treatment for T2D prevention did not appear to have different effects across genetic risk strata.98,126 Nevertheless, T2D PRSs may help guide T2D management through both sulfonylurea responsiveness127 and intensity of glucose management.128
Venous Thromboembolic Disease: A VTE PRS is associated with incident venous thromboembolic disease (VTE) risk.110,140 Because the clinical utility of identifying inherited thrombophilias is unknown, the clinical utility of a VTE PRS also remains unknown. The benefits and risk of prolonged anticoagulation in those at high risk (determined using both clinical and genetic factors) require further study.
PRSs for Pharmacogenomics: Pharmacogenetic PRSs have addressed the following: In regard to PRSs for drug efficacy, most research has focused on statins with CAD PRSs,86,91 sulfonylureas with T2D PRSs,127 and PCSK9 inhibitors with CAD PRSs,90 whereas drug toxicity PRSs have focused on QTc prolongation with QTc PRSs.156 Ongoing disease-associated PRS analyses in completed clinical trials will continue to inform pharmacogenetics. Discovery genetic association analyses within clinical trials may enable novel pharmacogenomic PRSs.
Criteria for Implementing PRSs in Cardiovascular Clinical Practice: Estimates of incremental efficacy and harm and logistical challenges are key aspects for health care systems to consider when evaluating PRSs. Clinician and patient education on their interpretation and limitations may require additional infrastructure and personnel.
Calibrating PRSs to the Population of a Health Care System: Recalibration of PRSs to target populations may improve performance but may lead to challenges in transferability.166
Considerations for Commercial Genetics Organizations: Commercial genetics organizations should be aware of the current, but likely changing, regulatory approval process for LDTs. Currently, LDT regulation falls largely under CLIA/CAP regulation,173 but a series of decisions by the FDA over the past couple years seem to indicate that this may change in the future.
How to Consider the Financial Integration of PRSs Into Clinical Practice: There are a paucity of cost-effectiveness studies addressing cardiovascular PRSs. Decreasing genotyping costs and the negligible incremental direct costs of virtually limitless PRSs lead to modeling challenges.
Interpretation and Relevance of PRSs in the Context of Insurance Policies: The Genetic Information Nondiscrimination Act protects people in the United States from being discriminated against on the basis of their genetic information, specifically the provision of health insurance or employment hiring, firing, pay, or promotion.185 However, this protection does not extend to life and disability insurance.
Challenges and Future Directions: The addition of PRSs to clinical risk tools consistently enhances the predictive ability. However, significant but surmountable challenges exist. The lack of diversity of participant inclusion in biobanks, GWAS, and consequently PRSs is a major current limitation. The lack of inclusion of non-Europeans in PRS construction makes the use of PRS in non-European populations suboptimal. Recognizing this major limitation, large samples of non-Europeans are being enrolled in newer biobanks. For some populations, study of large numbers of non-European participants has already begun (ie, Japan Biobank, Million Veteran Program), but much work is still required. Further future work should address the inclusion of rare variants in scores, improvement in phenotyping in biobanks, and inclusion of PRSs within randomized controlled trials; future work on other cardiometabolic diseases also is needed.
Disclosures
Writing Group Disclosures.
| Writing group member | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| Jack W. O’Sullivan | Stanford Cardiology | None | None | None | None | None | None | None |
| Pradeep Natarajan | Massachusetts General Hospital Cardiovascular Research Center | Apple†; Amgen*; Astra-Zeneca*; Boston Scientific*; Fondation Leducq†; Novartis*; NIH† (all investigator-initiated grants) | None | Allelica* | None | Named as inventors on a government-owned US patent application related to the use of genetic risk prediction for venous thromboembolic disease filed by the US Department of Veterans Affairs in accordance with federal regulatory requirements*; TenSixteen Bio†; geneXwell*; Vertex (immediate family members)† | Apple†; AstraZeneca*; Genentech/Roche†; Novartis†; Blackstone Life Sciences*; Foresite Labs†; TenSixteen Bio†; Invitae* | Massachusetts General Hospital (Paul and Phyllis Fireman Chair in Vascular Medicine)†; Vertex (medical director; immediate family members)† |
| Euan A. Ashley | Stanford University School of Medicine Center for Inherited Cardiovascular Disease | Bristol Myers Squibb (academic grant)*; Takeda (academic grant)*; Google*; Verily* | None | None | None | Personalis*; DeepCell*; Silicon Valley Exercise Analytics* | Nuevocor*; Cathay Capital (unpaid)*; Third Rock Ventures (unpaid)*; Medical Excellence*; Foresite Capital*; Novartis (unpaid)*; Samsung (unpaid)*; Analog Devices (unpaid)*; Apple†; Illumina (unpaid)*; Nanopore (unpaid)*; SequenceBio (unpaid)*; Genome Medical (unpaid)*; Disney (unpaid)* | AstraZeneca (non-executive director)* |
| Scott M. Damrauer | University of Pennsylvania School of Medicine | US Department of Veterans Affairs (genetic research on cardiovascular disease)†; Renalytix AI (precision medicine approaches to CKD progression)† | Pending patent (named as a coinventor on a government-owned US patent application related to the use of genetic risk prediction for venous thromboembolic disease filed by the US Department of Veterans Affairs in accordance with federal regulatory requirements)* | None | None | Abbott*; United Health*; Gilead*; Daiichi Sankyo*; Abbvie*; Teva* | None | None |
| Jasmine A. Luzum | University of Michigan Clinical Pharmacy College of Pharmacy | NHLBI (K08 HL146990)* | None | None | None | None | None | None |
| Carla Marquez-Luna | Icahn School of Medicine at Mount Sinai | None | None | None | None | None | None | None |
| Christopher J. O’Donnell | VA Boston Healthcare System | None | None | None | None | None | None | Novartis Institute for Biomedical Research (global head, translational medicine cardiovascular and metabolic disease)† |
| Sridharan Raghavan | Denver Veterans Affairs Medical Center | Department of Veterans Affairs (active research grant funding, including partial salary support)†; US National Institutes of Health (active research grant funding)*; Boettcher Foundation (active research grant funding, including partial salary support)† | None | None | None | None | None | None |
| Cristen J. Willer | University of Michigan Medical School | None | None | None | None | None | None | None |
This table represents the relationships of writing group members that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all members of the writing group are required to complete and submit. A relationship is considered to be “significant” if (a) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (b) the person owns 5% or more of the voting stock or share of the entity, or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
Reviewer Disclosures.
| Reviewer | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| Andrew P. Landstrom | Duke University School of Medicine | None | None | None | None | None | None | None |
| Naveen L. Pereira | Mayo Clinic Rochester | None | None | None | None | None | None | None |
| Samuli Ripatti | Institute for Molecular Medicine, Finland, FIMM (Finland) | None | None | None | None | None | None | None |
| Jerome I. Rotter | Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center | NIH*; Sanford Health System* | None | None | None | None | None | None |
This table represents the relationships of reviewers that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all reviewers are required to complete and submit. A relationship is considered to be “significant” if (a) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (b) the person owns 5% or more of the voting stock or share of the entity, or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Significant.
Footnotes
The American Heart Association makes every effort to avoid any actual or potential conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the writing panel. Specifically, all members of the writing group are required to complete and submit a Disclosure Questionnaire showing all such relationships that might be perceived as real or potential conflicts of interest.
This statement was approved by the American Heart Association Science Advisory and Coordinating Committee on April 14, 2022, and the American Heart Association Executive Committee on May 16, 2022. A copy of the document is available at https://professional.heart.org/statements by using either “Search for Guidelines & Statements” or the “Browse by Topic” area. To purchase additional reprints, call 215–356-2721 or email Meredith.Edelman@wolterskluwer.com.
The American Heart Association requests that this document be cited as follows: O’Sullivan JW, Raghavan S, Marquez-Luna C, Luzum JA, Damrauer SM, Ashley EA, O’Donnell CJ, Willer CJ, Natarajan P; on behalf of the American Heart Association Council on Genomic and Precision Medicine; Council on Clinical Cardiology; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Lifestyle and Cardiometabolic Health; and Council on Peripheral Vascular Disease. Polygenic risk scores for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2022;146:e93–e118. doi: 10.1161/CIR.0000000000001077
The expert peer review of AHA-commissioned documents (eg, scientific statements, clinical practice guidelines, systematic reviews) is conducted by the AHA Office of Science Operations. For more on AHA statements and guidelines development, visit https://professional.heart.org/statements. Select the “Guidelines & Statements” drop-down menu, then click “Publication Development.”
REFERENCES
- 1.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, et al. ; on behalf of the American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052 [DOI] [PubMed] [Google Scholar]
- 2.Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, Himmelfarb CD, Khera A, Lloyd-Jones D, McEvoy JW, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines [published corrections appear in Circulation. 2019;140:e649–e650, Circulation. 2020;141:e60, and Circulation. 2020;141:e774]. Circulation. 2019;140:e596–e646. doi: 10.1161/CIR.0000000000000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet. 2014;383:999–1008. doi: 10.1016/S0140-6736(13)61752-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837 [DOI] [PubMed] [Google Scholar]
- 5.Yadlowsky S, Hayward RA, Sussman JB, McClelland RL, Min YI, Basu S. Clinical implications of revised Pooled Cohort Equations for estimating atherosclerotic cardiovascular disease risk. Ann Intern Med. 2018;169:20–29. doi: 10.7326/M17-3011 [DOI] [PubMed] [Google Scholar]
- 6.Goff DC Jr, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, Greenland P, Lackland DT, Levy D, O’Donnell CJ, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(suppl 2):S49–S73. doi: 10.1161/01.cir.0000437741.48606.98 [DOI] [PubMed] [Google Scholar]
- 7.Ridker PM, Cook NR. Statins: new American guidelines for prevention of cardiovascular disease. Lancet. 2013;382:1762–1765. doi: 10.1016/S0140-6736(13)62388-0 [DOI] [PubMed] [Google Scholar]
- 8.Mora S, Wenger NK, Cook NR, Liu J, Howard BV, Limacher MC, Liu S, Margolis KL, Martin LW, Paynter NP, et al. Evaluation of the Pooled Cohort Risk Equations for cardiovascular risk prediction in a multiethnic cohort from the Women’s Health Initiative. JAMA Intern Med. 2018;178:1231–1240. doi: 10.1001/jamainternmed.2018.2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khera R, Pandey A, Ayers CR, Carnethon MR, Greenland P, Ndumele CE, Nambi V, Seliger SL, Chaves PHM, Safford MM, et al. Performance of the Pooled Cohort Equations to estimate atherosclerotic cardiovascular disease risk by body mass index. JAMA Netw Open. 2020;3:e2023242. doi: 10.1001/jamanetworkopen.2020.23242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emdin CA, Khera AV, Natarajan P, Klarin D, Baber U, Mehran R, Rader DJ, Fuster V, Kathiresan S. Evaluation of the Pooled Cohort Equations for prediction of cardiovascular risk in a contemporary prospective cohort. Am J Cardiol. 2017;119:881–885. doi: 10.1016/j.amjcard.2016.11.042 [DOI] [PubMed] [Google Scholar]
- 11.Karmali KN, Goff DC Jr, Ning H, Lloyd-Jones DM. A systematic examination of the 2013 ACC/AHA Pooled Cohort risk assessment tool for atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64:959–968. doi: 10.1016/j.jacc.2014.06.1186 [DOI] [PubMed] [Google Scholar]; 11a. Ashley EA, Hershberger RE, Caleshu C, Ellinor PT, Garcia JGN, Her-rington DM, Ho CY, Johnson JA, Kittner SJ, MacRae CA, et al. ; on behalf of the American Heart Association Advocacy Coordinating Committee. Genetics and cardiovascular disease: a policy statementf from the American Heart Association. Circulation. 2012;126:142–157. doi: 10.1161/CIR.0b013e31825b07f8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lloyd-Jones DM, Nam BH, D’Agostino RB Sr, Levy D, Murabito JM, Wang TJ, Wilson PW, O’Donnell CJ. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults: a prospective study of parents and offspring. JAMA. 2004;291:2204–2211. doi: 10.1001/jama.291.18.2204 [DOI] [PubMed] [Google Scholar]
- 13.Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, De Faire U. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med. 2002;252:247–254. doi: 10.1046/j.1365-2796.2002.01029.x [DOI] [PubMed] [Google Scholar]
- 14.Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, Christensen K. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–383. doi: 10.1161/CIRCEP.108.786665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance: a population-based twin study. Diabetologia. 1999;42:139–145. doi: 10.1007/s001250051131 [DOI] [PubMed] [Google Scholar]
- 16.Newman B, Selby JV, King MC, Slemenda C, Fabsitz R, Friedman GD. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia. 1987;30:763–768. doi: 10.1007/BF00275741 [DOI] [PubMed] [Google Scholar]
- 17.Crous-Bou M, Harrington LB, Kabrhel C. Environmental and genetic risk factors associated with venous thromboembolism. Semin Thromb Hemost. 2016;42:808–820. doi: 10.1055/s-0036-1592333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aragam KG, Natarajan P. Polygenic scores to assess atherosclerotic cardiovascular disease risk. Circ Res. 2020;126:1159–1177. doi: 10.1161/CIRCRESAHA.120.315928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL, Lai C, Brockman D, Philippakis A, Ellinor PT, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun. 2020;11:3635. doi: 10.1038/s41467-020-17374-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, Posthuma D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–709. doi: 10.1038/ng.3285 [DOI] [PubMed] [Google Scholar]
- 22.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, Genovese G, Loh PR, Bhatia G, Do R, et al. ; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) study. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet. 2015;97:576–592. doi: 10.1016/j.ajhg.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mars N, Koskela JT, Ripatti P, Kiiskinen TTJ, Havulinna AS, Lindbohm JV, Ahola-Olli A, Kurki M, Karjalainen J, Palta P, et al. ; FinnGen. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med. 2020;26:549–557. doi: 10.1038/s41591-020-0800-0 [DOI] [PubMed] [Google Scholar]
- 24.Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet. 2019;28:R133–R142. doi: 10.1093/hmg/ddz187 [DOI] [PubMed] [Google Scholar]
- 25.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufman DJ, Baker R, Milner LC, Devaney S, Hudson KL. A survey of U.S adults’ opinions about conduct of a nationwide Precision Medicine Initiative® cohort study of genes and environment. PLoS One. 2016;11:e0160461. doi: 10.1371/journal.pone.0160461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guidelines on the use and reporting of race, ethnicity, and ancestry in the NHLBI Trans-Omics for Precision Medicine (TOPMed) program. Accessed March 11, 2021. https://www.nhlbiwgs.org/guidelines-use-and-reporting-race-ethnicity-and-ancestry-topmed
- 28.Churchwell K, Elkind MSV, Benjamin RM, Carson AP, Chang EK, Lawrence W, Mills A, Odom TM, Rodriguez CJ, Rodriguez F, et al. ; on behalf of the American Heart Association. Call to action: structural racism as a fundamental driver of health disparities: a presidential advisory from the American Heart Association. Circulation. 2020;142:e454–e468. doi: 10.1161/CIR.0000000000000936 [DOI] [PubMed] [Google Scholar]
- 29.Race, Ethnicity, and Genetics Working Group. The use of racial, ethnic, and ancestral categories in human genetics research. Am J Hum Genet. 2005;77:519–532. doi: 10.1086/491747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X, Carbonetto P, Stephens M. Polygenic modeling with bayesian sparse linear mixed models. PLoS Genet. 2013;9:e1003264. doi: 10.1371/journal.pgen.1003264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y, Lu Q, Powles R, Yao X, Yang C, Fang F, Xu X, Zhao H. Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput Biol. 2017;13:e1005589. doi: 10.1371/journal.pcbi.1005589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Márquez-Luna C, Gazal S, Loh P-R, Kim SS, Furlotte N, Auton A, 23andMe Research Team, Price AL. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat Commun. 2021;12:6052. doi: 10.1038/s41467-021-25171-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge T, Chen CY, Ni Y, Feng YA, Smoller JW. Polygenic prediction via bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1776. doi: 10.1038/s41467-019-09718-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, Lu Q, Liu W, Zhang Y, Li M, Zhao H. Joint modeling of genetically correlated diseases and functional annotations increases accuracy of polygenic risk prediction. PLoS Genet. 2017;13:e1006836. doi: 10.1371/journal.pgen.1006836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mak TSH, Porsch RM, Choi SW, Zhou X, Sham PC. Polygenic scores via penalized regression on summary statistics. Genet Epidemiol. 2017;41:469–480. doi: 10.1002/gepi.22050 [DOI] [PubMed] [Google Scholar]
- 36.Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, Momozawa Y, Matsunaga H, Ieki H, Ozaki K, Onouchi Y, et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet. 2020;52:1169–1177. doi: 10.1038/s41588-020-0705-3 [DOI] [PubMed] [Google Scholar]
- 37.Graham SE, Clarke SL, Wu KH, Kanoni S, Zajac GJM, Ramdas S, Surakka I, Ntalla I, Vedantam S, Winkler TW, et al. ; VA Million Veteran Program; Global Lipids Genetics Consortium. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600:675–679. doi: 10.1038/s41586-021-04064-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, Lai FY, Kaptoge S, Brozynska M, Wang T, et al. ; UK Biobank CardioMetabolic Consortium CHD Working Group. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol. 2018;72:1883–1893. doi: 10.1016/j.jacc.2018.07.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD, Kenny EE. Human demographic history impacts genetic risk prediction across diverse populations. Am J Hum Genet. 2017;100:635–649. doi: 10.1016/j.ajhg.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. doi: 10.1016/j.jacc.2016.03.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benn M, Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37:1384–1394. doi: 10.1093/eurheartj/ehw028 [DOI] [PubMed] [Google Scholar]
- 44.Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, et al. ; on behalf of the American Heart Association Atherosclerosis, Hypertension, and Obesity in Young Committee of Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and Council on Lifestyle and Cardiometabolic Health. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association [published correction appears in Circulation. 2015;132:e397]. Circulation. 2015;132:2167–2192. doi: 10.1161/CIR.0000000000000297 [DOI] [PubMed] [Google Scholar]
- 45.Gidding SS, Sheldon A, Neben CL, Williams HE, Law S, Zhou AY, Wilemon K, Ahmed CD, Kindt I. Patient acceptance of genetic testing for familial hypercholesterolemia in the CASCADE FH Registry. J Clin Lipidol. 2020;14:218–223.e2. doi: 10.1016/j.jacl.2020.02.001 [DOI] [PubMed] [Google Scholar]
- 46.Anık A, Çatlı G, Abacı A, Böber E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 2015;28:251–263. doi: 10.1515/jpem-2014-0384 [DOI] [PubMed] [Google Scholar]
- 47.Mahida S, Lubitz SA, Rienstra M, Milan DJ, Ellinor PT. Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc Res. 2011;89:692–700. doi: 10.1093/cvr/cvq381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Stefano V, Leone G. Resistance to activated protein C due to mutated factor V as a novel cause of inherited thrombophilia. Haematologica. 1995;80:344–356. [PubMed] [Google Scholar]
- 49.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae [DOI] [PubMed] [Google Scholar]
- 50.Oetjens MT, Kelly MA, Sturm AC, Martin CL, Ledbetter DH. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun. 2019;10:4897. doi: 10.1038/s41467-019-12869-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, Waring A, Ormondroyd E, Kramer CM, Ho CY, et al. ; HCMR Investigators. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. 2021;53:135–142. doi: 10.1038/s41588-020-00764-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC; on behalf of the American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020;13:e000067. doi: 10.1161/HCG.0000000000000067 [DOI] [PubMed] [Google Scholar]
- 53.Colilla S, Crow A, Petkun W, Singer DE, Simon T, Liu X. Estimates of current and future incidence and prevalence of atrial fibrillation in the U.S. adult population. Am J Cardiol. 2013;112:1142–1147. doi: 10.1016/j.amjcard.2013.05.063 [DOI] [PubMed] [Google Scholar]
- 54.Chugh SS, Blackshear JL, Shen WK, Hammill SC, Gersh BJ. Epidemiology and natural history of atrial fibrillation: clinical implications. J Am Coll Cardiol. 2001;37:371–378. doi: 10.1016/s0735-1097(00)01107-4 [DOI] [PubMed] [Google Scholar]
- 55.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke. 1991;22:983–988. doi: 10.1161/01.str.22.8.983 [DOI] [PubMed] [Google Scholar]
- 56.Odutayo A, Wong CX, Hsiao AJ, Hopewell S, Altman DG, Emdin CA. Atrial fibrillation and risks of cardiovascular disease, renal disease, and death: systematic review and meta-analysis. BMJ. 2016;354:i4482. doi: 10.1136/bmj.i4482 [DOI] [PubMed] [Google Scholar]
- 57.Verdecchia P, Angeli F, Reboldi G. Hypertension and atrial fibrillation: doubts and certainties from basic and clinical studies. Circ Res. 2018;122:352–368. doi: 10.1161/CIRCRESAHA.117.311402 [DOI] [PubMed] [Google Scholar]
- 58.Campbell CM, Campbell JD, Thompson CH, Galimberti ES, Darbar D, Vanoye CG, George AL. Selective targeting of gain-of-function KCNQ1 mutations predisposing to atrial fibrillation. Circ Arrhythm Electrophysiol. 2013;6:960–966. doi: 10.1161/CIRCEP.113.000439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi SH, Weng LC, Roselli C, Lin H, Haggerty CM, Shoemaker MB, Barnard J, Arking DE, Chasman DI, Albert CM, et al. ; DiscovEHR Study and the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. 2018;320:2354–2364. doi: 10.1001/jama.2018.18179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoneda ZT, Anderson KC, Quintana JA, O’Neill MJ, Sims RA, Glazer AM, Shaffer CM, Crawford DM, Stricker T, Ye F, et al. Early-onset atrial fibrillation and the prevalence of rare variants in cardiomyopathy and arrhythmia genes. JAMA Cardiol. 2021;6:1371–1379. doi: 10.1001/jamacardio.2021.3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239. doi: 10.1038/s41588-018-0171-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riveros-Mckay F, Weale ME, Moore R, Selzam S, Krapohl E, Sivley RM, Tarran WA, Sørensen P, Lachapelle AS, Griffiths JA, et al. An integrated polygenic tool substantially enhances coronary artery disease prediction. Circ Genom Precis Med. 2021;14:e003304. doi: 10.1161/CIRCGEN.120.003304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okubo Y, Nakano Y, Ochi H, Onohara Y, Tokuyama T, Motoda C, Amioka M, Hironobe N, Okamura S, Ikeuchi Y, et al. Predicting atrial fibrillation using a combination of genetic risk score and clinical risk factors. Heart Rhythm. 2020;17(pt A):699–705. doi: 10.1016/j.hrthm.2020.01.006 [DOI] [PubMed] [Google Scholar]
- 65.Kloosterman M, Santema BT, Roselli C, Nelson CP, Koekemoer A, Romaine SPR, Van Gelder IC, Lam CSP, Artola VA, Lang CC, et al. Genetic risk and atrial fibrillation in patients with heart failure. Eur J Heart Fail. 2020;22:519–527. doi: 10.1002/ejhf.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Homburger JR, Neben CL, Mishne G, Zhou AY, Kathiresan S, Khera AV. Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome-wide polygenic scores. Genome Med. 2019;11:74. doi: 10.1186/s13073-019-0682-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weng LC, Preis SR, Hulme OL, Larson MG, Choi SH, Wang B, Trinquart L, McManus DD, Staerk L, Lin H, et al. Genetic predisposition, clinical risk factor burden, and lifetime risk of atrial fibrillation. Circulation. 2018;137:1027–1038. doi: 10.1161/CIRCULATIONAHA.117.031431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi Seung H, Jurgens SJ, Weng L-C, Pirruccello JP, Roselli C, Chaffin M, Lee CJ-Y, Hall AW, Khera AV, Lunetta KL, et al. Monogenic and polygenic contributions to atrial fibrillation risk. Circ Res. 2020;126:200–209. doi: 10.1161/CIRCRESAHA.119.315686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shoemaker MB, Husser D, Roselli C, Al Jazairi M, Chrispin J, Kühne M, Neumann B, Knight S, Sun H, Mohanty S, et al. Genetic susceptibility for atrial fibrillation in patients undergoing atrial fibrillation ablation. Circ Arrhythm Electrophysiol. 2020;13:e007676. doi: 10.1161/CIRCEP.119.007676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lazarte J, Dron JS, McIntyre AD, Skanes AC, Gula LJ, Tang AS, Tadros R, Laksman ZW, Hegele RA, Roberts JD. Role of common genetic variation in lone atrial fibrillation. Circ Genom Precis Med. 2021;14:e003179. doi: 10.1161/CIRCGEN.120.003179 [DOI] [PubMed] [Google Scholar]
- 71.O’Sullivan JW, Shcherbina A, Justesen JM, Turakhia M, Perez M, Wand H, Tcheandjieu C, Clarke SL, Rivas MA, Ashley EA. Combining clinical and polygenic risk improves stroke prediction among individuals with atrial fibrillation. Circ Genom Precis Med. 2021;14:e003168. doi: 10.1161/CIRCGEN.120.003168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, et al. ; AFGen Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium; International Genomics of Blood Pressure (iGEN-BP) Consortium; INVENT Consortium; STARNET; BioBank Japan Cooperative Hospital Group; COMPASS Consortium; EPIC-CVD Consortium; EPIC-InterAct Consortium; International Stroke Genetics Consortium (ISGC); METASTROKE Consortium; Neurology Working Group of the CHARGE Consortium; NINDS Stroke Genetics Network (SiGN); UK Young Lacunar DNA Study; MEGASTROKE Consortium. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, Gulcher J, Mathiesen EB, Njølstad I, Nyrnes A, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876–878. doi: 10.1038/ng.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tada H, Shiffman D, Smith JG, Sjögren M, Lubitz SA, Ellinor PT, Louie JZ, Catanese JJ, Engström G, Devlin JJ, et al. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke. 2014;45:2856–2862. doi: 10.1161/STROKEAHA.114.006072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lubitz SA, Yin X, Lin HJ, Kolek M, Smith JG, Trompet S, Rienstra M, Rost NS, Teixeira PL, Almgren P, et al. ; AFGen Consortium. Genetic risk prediction of atrial fibrillation. Circulation. 2017;135:1311–1320. doi: 10.1161/CIRCULATIONAHA.116.024143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alonso A, Krijthe BP, Aspelund T, Stepas KA, Pencina MJ, Moser CB, Sinner MF, Sotoodehnia N, Fontes JD, Janssens AC, et al. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: the CHARGE-AF Consortium. J Am Heart Assoc. 2013;2:e000102. doi: 10.1161/JAHA.112.000102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.White PD. Genes, the heart and destiny. N Engl J Med. 1957;256:965–969. doi: 10.1056/NEJM195705232562101 [DOI] [PubMed] [Google Scholar]
- 78.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. doi: 10.1056/NEJM199404143301503 [DOI] [PubMed] [Google Scholar]
- 79.Besseling J, Hovingh GK, Huijgen R, Kastelein JJP, Hutten BA. Statins in familial hypercholesterolemia: consequences for coronary artery disease and all-cause mortality. J Am Coll Cardiol. 2016;68:252–260. doi: 10.1016/j.jacc.2016.04.054 [DOI] [PubMed] [Google Scholar]
- 80.Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines [published correction appears in Circulation. 2019;139:e1182–e1186]. Circulation. 2019;139:e1082–e1143. doi: 10.1161/CIR.0000000000000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, Leed A, Weale ME, Spencer CCA, Aguet F, et al. ; CARDIoGRAMplusC4D Consortium. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397. doi: 10.1038/ng.3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114:1241–1257. doi: 10.1093/cvr/cvy084 [DOI] [PubMed] [Google Scholar]
- 83.Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NG, Jansen H, Kanoni S, Nelson CP, Ferrario PG, König IR, et al. ; Wellcome Trust Case Control Consortium; MORGAM Investigators; Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69:823–836. doi: 10.1016/j.jacc.2016.11.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weale ME, Riveros-Mckay F, Selzam S, Seth P, Moore R, Tarran WA, Gradovich E, Giner-Delgado C, Palmer D, Wells D, et al. Validation of an integrated risk tool, including polygenic risk score, for atherosclerotic cardiovascular disease in multiple ethnicities and ancestries. Am J Cardiol. 2021;148:157–164. doi: 10.1016/j.amjcard.2021.02.032 [DOI] [PubMed] [Google Scholar]
- 85.Khera AV, Chaffin M, Zekavat SM, Collins RL, Roselli C, Natarajan P, Lichtman JH, D’Onofrio G, Mattera J, Dreyer R, et al. Whole-genome sequencing to characterize monogenic and polygenic contributions in patients hospitalized with early-onset myocardial infarction. Circulation. 2019;139:1593–1602. doi: 10.1161/CIRCULATIONAHA.118.035658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R, Sartori S, Fuster V, Reilly DF, Butterworth A, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135:2091–2101. doi: 10.1161/CIRCULATIONAHA.116.024436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, Devlin JJ, Kathiresan S, Shiffman D. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J. 2016;37:561–567. doi: 10.1093/eurheartj/ehv462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, Chasman DI, Baber U, Mehran R, Rader DJ, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–2358. doi: 10.1056/NEJMoa1605086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aragam KG, Dobbyn A, Judy R, Chaffin M, Chaudhary K, Hindy G, Cagan A, Finneran P, Weng LC, Loos RJF, et al. Limitations of contemporary guidelines for managing patients at high genetic risk of coronary artery disease. J Am Coll Cardiol. 2020;75:2769–2780. doi: 10.1016/j.jacc.2020.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Damask A, Steg PG, Schwartz GG, Szarek M, Hagström E, Badimon L, Chapman MJ, Boileau C, Tsimikas S, Ginsberg HN, et al. ; Regeneron Genetics Center and the ODYSSEY OUTCOMES Investigators. Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the ODYSSEY OUTCOMES trial. Circulation. 2020;141:624–636. doi: 10.1161/CIRCULATIONAHA.119.044434 [DOI] [PubMed] [Google Scholar]
- 91.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, Nordio F, Hyde C, Cannon CP, Sacks F, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271. doi: 10.1016/S0140-6736(14)61730-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marston NA, Kamanu FK, Nordio F, Gurmu Y, Roselli C, Sever PS, Pedersen TR, Keech AC, Wang H, Lira Pineda A, et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the FOURIER trial. Circulation. 2020;141:616–623. doi: 10.1161/CIRCULATIONAHA.119.043805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, Shaffer CM, Kundu S, Robinson-Cohen C, Psaty BM, Rich SS, et al. Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA. 2020;323:627–635. doi: 10.1001/jama.2019.21782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elliott J, Bodinier B, Bond TA, Chadeau-Hyam M, Evangelou E, Moons KGM, Dehghan A, Muller DC, Elliott P, Tzoulaki I. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA. 2020;323:636–645. doi: 10.1001/jama.2019.22241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel J, Pallazola VA, Dudum R, Greenland P, McEvoy JW, Blumenthal RS, Virani SS, Miedema MD, Shea S, Yeboah J, et al. Assessment of coronary artery calcium scoring to guide statin therapy allocation according to risk-enhancing factors: the Multi-Ethnic Study of Atherosclerosis. JAMA Cardiol. 2021;6:1161–1170. doi: 10.1001/jamacardio.2021.2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saad Shaukat MH, Stys P, Sjovold A, Hajek C, Petrasko P, Pham M, Stys V, Petrasko M, Rynders B, Singh K, et al. Concordance of high polygenic CAD Risk score with high coronary artery calcium score & low polygenic CAD risk score with low coronary artery calcium score: a real world experience. Circulation. 2021;144(suppl):A12277–A12277. Abstract 12277. [Google Scholar]
- 97.Natarajan P, Bis JC, Bielak LF, Cox AJ, Dörr M, Feitosa MF, Franceschini N, Guo X, Hwang SJ, Isaacs A, et al. ; CHARGE Consortium. Multiethnic exome-wide association study of subclinical atherosclerosis. Circ Cardiovasc Genet. 2016;9:511–520. doi: 10.1161/CIRCGENETICS.116.001572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Said MA, Verweij N, van der Harst P. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK Biobank Study. JAMA Cardiol. 2018;3:693–702. doi: 10.1001/jamacardio.2018.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Widen E, Junna N, Ruotsalainen S, Surakka I, Mars N, Ripatti P, Partanen JJ, Aro J, Mustonen P, Tuomi T, et al. How communicating polygenic and clinical risk for atherosclerotic cardiovascular disease impacts health behavior: an observational follow-up study. Circ Genom Precis Med. 2022;15:e003459. doi: 10.1161/CIRCGEN.121.003459 [DOI] [PubMed] [Google Scholar]
- 100.Ye Y, Chen X, Han J, Jiang W, Natarajan P, Zhao H. Interactions between enhanced polygenic risk scores and lifestyle for cardiovascular disease, diabetes, and lipid levels. Circ Genom Precis Med. 2021;14:e003128. doi: 10.1161/CIRCGEN.120.003128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kullo IJ, Jouni H, Austin EE, Brown SA, Kruisselbrink TM, Isseh IN, Haddad RA, Marroush TS, Shameer K, Olson JE, et al. Incorporating a genetic risk score into coronary heart disease risk estimates: effect on low-density lipoprotein cholesterol levels (the MI-GENES clinical trial). Circulation. 2016;133:1181–1188. doi: 10.1161/CIRCULATIONAHA.115.020109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang QQ, Sallah N, Dunca D, Trivedi B, Hunt KA, Hodgson S, Lambert SA, Arciero E, Genes & Health Research Team, Wright J, Griffiths C, et al. Transferability of genetic loci and polygenic scores for cardiometabolic traits in British Pakistanis and Bangladeshis. medRxiv. Preprint posted online June 24, 2021. doi: 10.1101/2021.06.22.21259323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu X, Liu Z, Cui Q, Liu F, Li J, Niu X, Shen C, Hu D, Huang K, Chen J, et al. A polygenic risk score improves risk stratification of coronary artery disease: a large-scale prospective Chinese cohort study. Eur Heart J. 2022;43:1702–1711. doi: 10.1093/eurheartj/ehac093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sun L, Pennells L, Kaptoge S, Nelson CP, Ritchie SC, Abraham G, Arnold M, Bell S, Bolton T, Burgess S, et al. Polygenic risk scores in cardiovascular risk prediction: a cohort study and modelling analyses. PLoS Med. 2021;18:e1003498. doi: 10.1371/journal.pmed.1003498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Natarajan P Polygenic risk scoring for coronary heart disease: the first risk factor. J Am Coll Cardiol. 2018;72:1894–1897. doi: 10.1016/j.jacc.2018.08.1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cholesterol Treatment Trialists’ (CTT) Collaboration, Fulcher J, O’Connell R, Voysey M, Emberson J, Blackwell L, Mihaylova B, Simes J, Collins R, Kirby A, Colhoun H, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385:1397–1405. doi: 10.1016/S0140-6736(14)61368-4 [DOI] [PubMed] [Google Scholar]
- 107.Schwartz GG, Steg GP, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–2107. doi: 10.1056/nejmoa1801174 [DOI] [PubMed] [Google Scholar]
- 108.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, et al. ; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664 [DOI] [PubMed] [Google Scholar]
- 109.Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–2206. doi: 10.1056/NEJMoa1400284 [DOI] [PubMed] [Google Scholar]
- 110.Klarin D, Busenkell E, Judy R, Lynch J, Levin M, Haessler J, Aragam K, Chaffin M, Haas M, Lindström S, et al. ; INVENT Consortium; Veterans Affairs’ Million Veteran Program. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat Genet. 2019;51:1574–1579. doi: 10.1038/s41588-019-0519-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Natarajan P, NHLBI TOPMed Lipids Working Group, Peloso GM, Zekavat SM, Montasser M, Ganna A, Chaffin M, Khera AV, Zhou W, Bloom JM, et al. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nat Commun. 2018;9:3391. doi: 10.1038/s41467-018-05747-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728 [DOI] [PubMed] [Google Scholar]
- 113.Trinder M, Francis GA, Brunham LR. Association of monogenic vs polygenic hypercholesterolemia with risk of atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;5:390–399. doi: 10.1001/jamacardio.2019.5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smith JG, Luk K, Schulz CA, Engert JC, Do R, Hindy G, Rukh G, Dufresne L, Almgren P, Owens DS, et al. ; Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE) Extracoronary Calcium Working Group. Association of low-density lipoprotein cholesterol-related genetic variants with aortic valve calcium and incident aortic stenosis. JAMA. 2014;312:1764–1771. doi: 10.1001/jama.2014.13959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, Willeit P, Young R, Surendran P, Karthikeyan S, et al. ; European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC-CVD) Consortium. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a mendelian randomization analysis. JAMA Cardiol. 2018;3:619–627. doi: 10.1001/jamacardio.2018.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Trinder M, Uddin MM, Finneran P, Aragam KG, Natarajan P. Clinical utility of lipoprotein(a) and LPA genetic risk score in risk prediction of incident atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;6:1–9. doi: 10.1001/jamacardio.2020.5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang Y, Chan L. Monogenic diabetes: what it teaches us on the common forms of type 1 and type 2 diabetes. Endocr Rev. 2016;37:190–222. doi: 10.1210/er.2015-1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, Ma C, Fontanillas P, Moutsianas L, McCarthy DJ, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–47. doi: 10.1038/nature18642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, Huffman JE, Assimes TL, Lorenz K, Zhu X, et al. ; HPAP Consortium; Regeneron Genetics Center; VA Million Veteran Program. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52:680–691. doi: 10.1038/s41588-020-0637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, Florez JC, Wilson PW, D’Agostino RB Sr, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–2219. doi: 10.1056/NEJMoa0804742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Talmud PJ, Hingorani AD, Cooper JA, Marmot MG, Brunner EJ, Kumari M, Kivimäki M, Humphries SE. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ. 2010;340:b4838. doi: 10.1136/bmj.b4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869 [DOI] [PubMed] [Google Scholar]
- 125.Raghavan S, Jablonski K, Delahanty LM, Maruthur NM, Leong A, Franks PW, Knowler WC, Florez JC, Dabelea D; Diabetes Prevention Program Research Group. Interaction of diabetes genetic risk and successful lifestyle modification in the Diabetes Prevention Programme. Diabetes Obes Metab. 2021;23:1030–1040. doi: 10.1111/dom.14309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hivert MF, Jablonski KA, Perreault L, Saxena R, McAteer JB, Franks PW, Hamman RF, Kahn SE, Haffner S, Meigs JB, et al. ; DIAGRAM Consortium; Diabetes Prevention Program Research Group. Updated genetic score based on 34 confirmed type 2 diabetes loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes. 2011;60:1340–1348. doi: 10.2337/db10-1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li JH, Szczerbinski L, Dawed AY, Kaur V, Todd JN, Pearson ER, Florez JC. A polygenic score for type 2 diabetes risk is associated with both the acute and sustained response to sulfonylureas. Diabetes. 2021;70:293–300. doi: 10.2337/db20-0530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shah HS, Gao H, Morieri ML, Skupien J, Marvel S, Paré G, Mannino GC, Buranasupkajorn P, Mendonca C, Hastings T, et al. Genetic predictors of cardiovascular mortality during intensive glycemic control in type 2 diabetes: findings from the ACCORD clinical trial. Diabetes Care. 2016;39:1915–1924. doi: 10.2337/dc16-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dennis JM, Shields BM, Henley WE, Jones AG, Hattersley AT. Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol. 2019;7:442–451. doi: 10.1016/S2213-8587(19)30087-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, Vikman P, Prasad RB, Aly DM, Almgren P, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6:361–369. doi: 10.1016/S2213-8587(18)30051-2 [DOI] [PubMed] [Google Scholar]
- 131.Udler MS, Kim J, von Grotthuss M, Bonàs-Guarch S, Cole JB, Chiou J, Anderson CD, METASTROKE and the ISGC, Boehnke M, Laakso M, Atzmon G, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 2018;15:e1002654. doi: 10.1371/journal.pmed.1002654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aly DM, Dwivedi OP, Prasad RB, Käräjämäki A, Hjort R, Åkerlund M, Mahajan A, Udler MS, Florez JC, McCarthy MI, et al. , Regeneron Genetics Center. Aetiological differences between novel subtypes of diabetes derived from genetic associations. medRxiv. Preprint posted online September 30, 2020. 10.1101/2020.09.29.20203935 [DOI] [Google Scholar]
- 133.Henke PK, Kahn SR, Pannucci CJ, Secemksy EA, Evans NS, Khorana AA, Creager MA, Pradhan AD; on behalf of the American Heart Association Advocacy Coordinating Committee. Call to action to prevent venous thromboembolism in hospitalized patients: a policy statement from the American Heart Association [published corrections appear in Circulation. 2020;141:e932 and Circulation. 2021;143:e249]. Circulation. 2020;141:e914–e931. doi: 10.1161/CIR.0000000000000769 [DOI] [PubMed] [Google Scholar]
- 134.Connors JM. Thrombophilia testing and venous thrombosis. N Engl J Med. 2017;377:1177–1187. doi: 10.1056/NEJMra1700365 [DOI] [PubMed] [Google Scholar]
- 135.Nicholson M, Chan N, Bhagirath V, Ginsberg J. Prevention of venous thromboembolism in 2020 and beyond. J Clin Med Res. 2020;9:2467. doi: 10.3390/jcm9082467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Federici EH, Al-Mondhiry H. High risk of thrombosis recurrence in patients with homozygous and compound heterozygous factor V R506Q (factor V Leiden) and prothrombin G20210A. Thromb Res. 2019;182:75–78. doi: 10.1016/j.thromres.2019.07.030 [DOI] [PubMed] [Google Scholar]
- 137.Segal JB, Brotman DJ, Necochea AJ, Emadi A, Samal L, Wilson LM, Crim MT, Bass EB. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. 2009;301:2472–2485. doi: 10.1001/jama.2009.853 [DOI] [PubMed] [Google Scholar]
- 138.Lijfering WM, Middeldorp S, Veeger NJ, Hamulyák K, Prins MH, Büller HR, van der Meer J. Risk of recurrent venous thrombosis in homozygous carriers and double heterozygous carriers of factor V Leiden and prothrombin G20210A. Circulation. 2010;121:1706–1712. doi: 10.1161/CIRCULATIONAHA.109.906347 [DOI] [PubMed] [Google Scholar]
- 139.Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost. 2008;6:1474–1477. doi: 10.1111/j.1538-7836.2008.03055.x [DOI] [PubMed] [Google Scholar]
- 140.Lindström S, Wang L, Smith EN, Gordon W, van Hylckama Vlieg A, de Andrade M, Brody JA, Pattee JW, Haessler J, Brumpton BM, et al. ; Million Veteran Program; CHARGE Hemostasis Working Group. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood. 2019;134:1645–1657. doi: 10.1182/blood.2019000435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, Huisman M, King CS, Morris TA, Sood N, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149:315–352. doi: 10.1016/j.chest.2015.11.026 [DOI] [PubMed] [Google Scholar]
- 142.Barbar S, Noventa F, Rossetto V, Ferrari A, Brandolin B, Perlati M, De Bon E, Tormene D, Pagnan A, Prandoni P. A risk assessment model for the identification of hospitalized medical patients at risk for venous thromboembolism: the Padua Prediction Score. J Thromb Haemost. 2010;8:2450–2457. doi: 10.1111/j.1538-7836.2010.04044.x [DOI] [PubMed] [Google Scholar]
- 143.Caprini JA, Arcelus JI, Hasty JH, Tamhane AC, Fabrega F. Clinical assessment of venous thromboembolic risk in surgical patients. Semin Thromb Hemost. 1991;17(suppl 3):304–312. [PubMed] [Google Scholar]
- 144.Center for Drug Evaluation, Research. Table of pharmacogenomic biomarkers. 2020. Accessed November 11, 2020. https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling
- 145.Center for Devices, Radiological Health. Table of pharmacogenetic associations. 2020. Accessed November 11, 2020. https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations
- 146.CPIC. Guidelines. Accessed November 11, 2020. https://cpicpgx.org/guidelines/
- 147.Luzum JA, Pakyz RE, Elsey AR, Haidar CE, Peterson JF, Whirl-Carrillo M, Handelman SK, Palmer K, Pulley JM, Beller M, et al. ; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102:502–510. doi: 10.1002/cpt.630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Giacomini KM, Yee SW, Mushiroda T, Weinshilboum RM, Ratain MJ, Kubo M. Genome-wide association studies of drug response and toxicity: an opportunity for genome medicine. Nat Rev Drug Discov. 2017;16:1. doi: 10.1038/nrd.2016.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bergmeijer TO, Reny JL, Pakyz RE, Gong L, Lewis JP, Kim EY, Aradi D, Fernandez-Cadenas I, Horenstein RB, Lee MTM, et al. ; ICPC Investigators. Genome-wide and candidate gene approaches of clopidogrel efficacy using pharmacodynamic and clinical end points: rationale and design of the International Clopidogrel Pharmacogenomics Consortium (ICPC). Am Heart J. 2018;198:152–159. doi: 10.1016/j.ahj.2017.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.International Warfarin Pharmacogenetics Consortium, Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, Limdi NA, Page D, Roden DM, Wagner MJ, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009;360:753–764. doi: 10.1056/NEJMoa0809329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Brugts JJ, Isaacs A, Boersma E, van Duijn CM, Uitterlinden AG, Remme W, Bertrand M, Ninomiya T, Ceconi C, Chalmers J, et al. Genetic determinants of treatment benefit of the angiotensin-converting enzyme-inhibitor perindopril in patients with stable coronary artery disease. Eur Heart J. 2010;31:1854–1864. doi: 10.1093/eurheartj/ehq169 [DOI] [PubMed] [Google Scholar]
- 152.Lewis JP, Backman JD, Reny JL, Bergmeijer TO, Mitchell BD, Ritchie MD, Déry JP, Pakyz RE, Gong L, Ryan K, et al. ; ICPC Investigators. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel-treated patients. Eur Heart J Cardiovasc Pharmacother. 2020;6:203–210. doi: 10.1093/ehjcvp/pvz045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lanfear DE, Luzum JA, She R, Gui H, Donahue MP, O’Connor CM, Adams KF, Sanders-van Wijk S, Zeld N, Maeder MT, et al. Polygenic score for β-blocker survival benefit in European ancestry patients with reduced ejection fraction heart failure. Circ Heart Fail. 2020;13:e007012. doi: 10.1161/CIRCHEARTFAILURE.119.007012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Oemrawsingh RM, Akkerhuis KM, Van Vark LC, Redekop WK, Rudez G, Remme WJ, Bertrand ME, Fox KM, Ferrari R, Danser AH, et al. ; PERGENE Investigators. Individualized angiotensin-converting enzyme (ACE)-inhibitor therapy in stable coronary artery disease based on clinical and pharmacogenetic determinants: the PERindopril GENEtic (PERGENE) risk model. J Am Heart Assoc. 2016;5:e002688. doi: 10.1161/JAHA.115.002688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McDonough CW, Gong Y, Padmanabhan S, Burkley B, Langaee TY, Melander O, Pepine CJ, Dominiczak AF, Cooper-Dehoff RM, Johnson JA. Pharmacogenomic association of nonsynonymous SNPs in SIGLEC12, A1BG, and the selectin region and cardiovascular outcomes. Hypertension. 2013;62:48–54. doi: 10.1161/HYPERTENSIONAHA.111.00823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Strauss DG, Vicente J, Johannesen L, Blinova K, Mason JW, Weeke P, Behr ER, Roden DM, Woosley R, Kosova G, et al. Common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk: a pilot study. Circulation. 2017;135:1300–1310. doi: 10.1161/CIRCULATIONAHA.116.023980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Emdin CA, Bhatnagar P, Wang M, Pillai SG, Li L, Qian H-R, Riesmeyer JS, Michael Lincoff A, Nicholls SJ, Nissen SE, et al. Genome-wide polygenic score and cardiovascular outcomes with evacetrapib in patients with high-risk vascular disease. Circ Genom Precis Med. 2020;13:e002767. doi: 10.1161/circgen.119.002767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Goldstein BA, Knowles JW, Salfati E, Ioannidis JP, Assimes TL. Simple, standardized incorporation of genetic risk into non-genetic risk prediction tools for complex traits: coronary heart disease as an example. Front Genet. 2014;5:254. doi: 10.3389/fgene.2014.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Grant RW, O’Brien KE, Waxler JL, Vassy JL, Delahanty LM, Bissett LG, Green RC, Stember KG, Guiducci C, Park ER, et al. Personalized genetic risk counseling to motivate diabetes prevention: a randomized trial. Diabetes Care. 2013;36:13–19. doi: 10.2337/dc12-0884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Aquilante CL, Kao DP, Trinkley KE, Lin CT, Crooks KR, Hearst EC, Hess SJ, Kudron EL, Lee YM, Liko I, et al. Clinical implementation of pharmacogenomics via a health system-wide research biobank: the University of Colorado experience. Pharmacogenomics. 2020;21:375–386. doi: 10.2217/pgs-2020-0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cook NR, Ridker PM. Calibration of the Pooled Cohort Equations for atherosclerotic cardiovascular disease: an update. Ann Intern Med. 2016;165:786–794. doi: 10.7326/M16-1739 [DOI] [PubMed] [Google Scholar]
- 162.Damen JA, Hooft L, Schuit E, Debray TP, Collins GS, Tzoulaki I, Lassale CM, Siontis GC, Chiocchia V, Roberts C, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416. doi: 10.1136/bmj.i2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kerminen S, Martin AR, Koskela J, Ruotsalainen SE, Havulinna AS, Surakka I, Palotie A, Perola M, Salomaa V, Daly MJ, et al. Geographic variation and bias in the polygenic scores of complex diseases and traits in Finland. Am J Hum Genet. 2019;104:1169–1181. doi: 10.1016/j.ajhg.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pennells L, Kaptoge S, Wood A, Sweeting M, Zhao X, White I, Burgess S, Willeit P, Bolton T, Moons KGM, et al. ; Emerging Risk Factors Collaboration. Equalization of four cardiovascular risk algorithms after systematic recalibration: individual-participant meta-analysis of 86 prospective studies. Eur Heart J. 2019;40:621–631. doi: 10.1093/eurheartj/ehy653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sussman JB, Wiitala WL, Zawistowski M, Hofer TP, Bentley D, Hayward RA. The Veterans Affairs Cardiac Risk Score: recalibrating the atherosclerotic cardiovascular disease score for applied use. Med Care. 2017;55:864–870. doi: 10.1097/MLR.0000000000000781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Márquez-Luna C, Loh PR, Price AL; South Asian Type 2 Diabetes (SAT2D) Consortium; SIGMA Type 2 Diabetes Consortium. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet Epidemiol. 2017;41:811–823. doi: 10.1002/gepi.22083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Amariuta T, Ishigaki K, Sugishita H, Ohta T, Koido M, Dey KK, Matsuda K, Murakami Y, Price AL, Kawakami E, et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat Genet. 2020;52:1346–1354. doi: 10.1038/s41588-020-00740-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marnetto D, Pärna K, Läll K, Molinaro L, Montinaro F, Haller T, Metspalu M, Mägi R, Fischer K, Pagani L. Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat Commun. 2020;11:1628. doi: 10.1038/s41467-020-15464-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wand H, Lambert SA, Tamburro C, Iacocca MA, O’Sullivan JW, Sillari C, Kullo IJ, Rowley R, Dron JS, Brockman D, et al. Improving reporting standards for polygenic scores in risk prediction studies. Genet Genom Med. 2021;591:211–219. doi: 10.1038/s41586-021-03243-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Allyse MA, Robinson DH, Ferber MJ, Sharp RR. Direct-to-consumer testing 2.0: emerging models of direct-to-consumer genetic testing. Mayo Clin Proc. 2018;93:113–120. doi: 10.1016/j.mayocp.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 171.Center for Drug Evaluation, Research. Direct-to-consumer tests. 2019. Accessed November 11, 2020. https://www.fda.gov/medical-devices/vitro-diagnostics/direct-consumer-tests
- 172.Center for Devices, Radiological Health. Genetic tests with unapproved claims to predict patient response. 2020. Accessed November 11, 2020. https://www.fda.gov/medical-devices/safety-communications/fda-warns-against-use-many-genetic-tests-unapproved-claims-predict-patient-response-specific
- 173.Assistant Secretary for Public Affairs (ASPA). Rescission of guidances and other informal issuances. 2020. Accessed December 1, 2020. https://www.hhs.gov/coronavirus/testing/recission-guidances-informal-issuanc-es-premarket-review-lab-tests/index.html
- 174.Gavan SP, Thompson AJ, Payne K. The economic case for precision medicine. Expert Rev Precis Med Drug Dev. 2018;3:1–9. doi: 10.1080/23808993.2018.1421858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Phillips KA, Deverka PA, Hooker GW, Douglas MP. Genetic test availability and spending: where are we now? Where are we going? Health Aff (Millwood). 2018;37:710–716. doi: 10.1377/hlthaff.2017.1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.National Human Genome Research Institute. The cost of sequencing a human genome. Accessed November 9, 2020. https://www.genome.gov/about-genomics/fact-sheets/sequencing-human-genome-cost
- 177.Biesecker LG, Burke W, Kohane I, Plon SE, Zimmern R. Next-generation sequencing in the clinic: are we ready? Nat Rev Genet. 2012;13:818–824. doi: 10.1038/nrg3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jarmul J, Pletcher MJ, Hassmiller Lich K, Wheeler SB, Weinberger M, Avery CL, Jonas DE, Earnshaw S, Pignone M. Cardiovascular genetic risk testing for targeting statin therapy in the primary prevention of atherosclerotic cardiovascular disease. Circ Cardiovasc Qual Outcomes. 2018;11:e004171. doi: 10.1161/CIRCOUTCOMES.117.004171 [DOI] [PubMed] [Google Scholar]
- 179.Sussman J, Marrero W, Burke J, Lavieri M, Hayward RA. Cost-effectiveness and decision analysis of polygenic risk scores in statin use for primary prevention. Circ Cardiovasc Qual Outcomes. 2018;11(suppl):A101–A101. Abstract 101. [Google Scholar]
- 180.Hynninen Y, Linna M, Vilkkumaa E. Value of genetic testing in the prevention of coronary heart disease events. PLoS One. 2019;14:e0210010. doi: 10.1371/journal.pone.0210010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Privé F, Arbel J, Vilhjálmsson BJ. LDpred2: better, faster, stronger. Bioinformatics. 2020;36:5424–5431. doi: 10.1093/bioinformatics/btaa1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shao H, Lin J, Zhuo X, Rolka DB, Gregg EW, Zhang P. Influence of diabetes complications on HbA1c treatment goals among older U.S. adults: a cost-effectiveness analysis. Diabetes Care. 2019;42:2136–2142. doi: 10.2337/dc19-0381 [DOI] [PubMed] [Google Scholar]
- 183.Pashayan N, Morris S, Gilbert FJ, Pharoah PDP. Cost-effectiveness and benefit-to-harm ratio of risk-stratified screening for breast cancer: a life-table model. JAMA Oncol. 2018;4:1504–1510. doi: 10.1001/jamaoncol.2018.1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee A, Mavaddat N, Wilcox AN, Cunningham AP, Carver T, Hartley S, Babb de Villiers C, Izquierdo A, Simard J, Schmidt MK, et al. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med. 2019;21:1708–1718. doi: 10.1038/s41436-018-0406-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Genetic Information Discrimination. Accessed November 9, 2020. https://www.eeoc.gov/genetic-information-discrimination
- 186.Bélisle-Pipon JC, Vayena E, Green RC, Cohen IG. Genetic testing, insurance discrimination and medical research: what the United States can learn from peer countries. Nat Med. 2019;25:1198–1204. doi: 10.1038/s41591-019-0534-z [DOI] [PubMed] [Google Scholar]
- 187.International Schizophrenia Consortium Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Stahl EA, Wegmann D, Trynka G, Gutierrez-Achury J, Do R, Voight BF, Kraft P, Chen R, Kallberg HJ, Kurreeman FA, et al. ; Diabetes Genetics Replication and Meta-analysis Consortium; Myocardial Infarction Genetics Consortium. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet. 2012;44:483–489. doi: 10.1038/ng.2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lloyd-Jones LR, Zeng J, Sidorenko J, Yengo L, Moser G, Kemper KE, Wang H, Zheng Z, Magi R, Esko T, et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat Commun. 2019;10:5086. doi: 10.1038/s41467-019-12653-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mancuso N, Rohland N, Rand KA, Tandon A, Allen A, Quinque D, Mallick S, Li H, Stram A, Sheng X, et al. ; PRACTICAL Consortium. The contribution of rare variation to prostate cancer heritability. Nat Genet. 2016;48:30–35. doi: 10.1038/ng.3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, et al. Efficient bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47:284–290. doi: 10.1038/ng.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zeng J, de Vlaming R, Wu Y, Robinson MR, Lloyd-Jones LR, Yengo L, Yap CX, Xue A, Sidorenko J, McRae AF, et al. Signatures of negative selection in the genetic architecture of human complex traits. Nat Genet. 2018;50:746–753. doi: 10.1038/s41588-018-0101-4 [DOI] [PubMed] [Google Scholar]
- 193.Schoech AP, Jordan DM, Loh PR, Gazal S, O’Connor LJ, Balick DJ, Palamara PF, Finucane HK, Sunyaev SR, Price AL. Quantification of frequency-dependent genetic architectures in 25 UK Biobank traits reveals action of negative selection. Nat Commun. 2019;10:790. doi: 10.1038/s41467-019-08424-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Do R, Stitziel NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, et al. ; NHLBI Exome Sequencing Project. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–106. doi: 10.1038/nature13917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.UK10K Consortium, Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JRB, Xu C, Futema M, Lawson D, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, Teslovich TM, Caulkins L, Koesterer R, Barajas-Olmos F, et al. ; Broad Genomics Platform; DiscovEHR Collaboration; CHARGE; LuCamp; ProDiGY; GoT2D; ESP; SIGMA-T2D; T2D-GENES; AMP-T2D-GENES. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. 2019;570:71–76. doi: 10.1038/s41586-019-1231-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sevim Bayrak C, Zhang P, Tristani-Firouzi M, Gelb BD, Itan Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020;12:9. doi: 10.1186/s13073-019-0709-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Durvasula A, Lohmueller KE. Negative selection on complex traits limits phenotype prediction accuracy between populations. Am J Hum Genet. 2021;108:620–631. doi: 10.1016/j.ajhg.2021.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Coram MA, Candille SI, Duan Q, Chan KH, Li Y, Kooperberg C, Reiner AP, Tang H. Leveraging multi-ethnic evidence for mapping complex traits in minority populations: an empirical Bayes approach. Am J Hum Genet. 2015;96:740–752. doi: 10.1016/j.ajhg.2015.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bitarello BD, Mathieson I. Polygenic scores for height in admixed populations. G3 (Bethesda). 2020;10:4027–4036. doi: 10.1534/g3.120.401658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, Feldman M, Peterson R, Domingue B. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun. 2019;10:3328. doi: 10.1038/s41467-019-11112-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gurdasani D, Barroso I, Zeggini E, Sandhu MS. Genomics of disease risk in globally diverse populations. Nat Rev Genet. 2019;20:520–535. doi: 10.1038/s41576-019-0144-0 [DOI] [PubMed] [Google Scholar]
- 203.Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, Highland HM, Patel YM, Sorokin EP, Avery CL, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hunter-Zinck H, Shi Y, Li M, Gorman BR, Ji S-G, Sun N, Webster T, Liem A, Hsieh P, Devineni P, et al. ; VA Million Veteran Program. Measuring genetic variation in the multi-ethnic Million Veteran Program (MVP). bioRxiv. Preprint posted online January 7, 2020. doi: 10.1101/2020.01.06.896613v1.full [DOI] [Google Scholar]
- 205.Nagai A, Hirata M, Kamatani Y, Muto K, Matsuda K, Kiyohara Y, Ninomiya T, Tamakoshi A, Yamagata Z, Mushiroda T, et al. ; BioBank Japan Cooperative Hospital Group. Overview of the BioBank Japan project: study design and profile. J Epidemiol. 2017;27(suppl):S2–S8. doi: 10.1016/j.je.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Finer S, Martin HC, Khan A, Hunt KA, MacLaughlin B, Ahmed Z, Ashcroft R, Durham C, MacArthur DG, McCarthy MI, et al. Cohort profile: East London Genes & Health (ELGH), a community-based population genomics and health study in British Bangladeshi and British Pakistani people. Int J Epidemiol. 2020;49:20–21i. doi: 10.1093/ije/dyz174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, Sanderson SC, Kannry J, Zinberg R, Basford MA, et al. ; eMERGE Network. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013;15:761–771. doi: 10.1038/gim.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–366. doi: 10.1038/nrg2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Reid NJ, Brockman DG, Leonard CE, Pelletier R, Khera AV. Concordance of a high polygenic score among relatives: implications for genetic counseling and cascade screening. Circ Genom Precis Med. 2021;14:e003262. doi: 10.1161/CIRCGEN.120.003262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.The polygenic score (PGS) catalog. Accessed March 24, 2021. https://www.pgscatalog.org/
- 211.Pirruccello JP, Bick A, Wang M, Chaffin M, Friedman S, Yao J, Guo X, Venkatesh BA, Taylor KD, Post WS, et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat Commun. 2020;11:2254. doi: 10.1038/s41467-020-15823-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, Venkataraman GR, Wainberg M, Ollila HM, Kiiskinen T, et al. ; FinnGen. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53:185–194. doi: 10.1038/s41588-020-00757-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Turkowski KL, Dotzler SM, Tester DJ, Giudicessi JR, Bos JM, Speziale AD, Vollenweider JM, Ackerman MJ. Corrected QT interval–polygenic risk score and its contribution to type 1, type 2, and type 3 long-QT syndrome in probands and genotype-positive family members. Circ Genom Precis Med. 2020;13:e002922. doi: 10.1161/CIRCGEN.120.002922 [DOI] [PubMed] [Google Scholar]
- 214.Noseworthy PA, Havulinna AS, Porthan K, Lahtinen AM, Jula A, Karhunen PJ, Perola M, Oikarinen L, Kontula KK, Salomaa V, et al. Common genetic variants, QT interval, and sudden cardiac death in a Finnish population-based study. Circ Cardiovasc Genet. 2011;4:305–311. doi: 10.1161/CIRCGENETICS.110.959049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wijeyeratne YD, Tanck MW, Mizusawa Y, Batchvarov V, Barc J, Crotti L, Bos JM, Tester DJ, Muir A, Veltmann C, et al. SCN5A mutation type and a genetic risk score associate variably with Brugada syndrome phenotype in SCN5A families. Circ Genom Precis Med. 2020;13:e002911. doi: 10.1161/CIRCGEN.120.002911 [DOI] [PMC free article] [PubMed] [Google Scholar]