

Abstract
Visual inspection of histopathology slides is one of the main methods used by pathologists to assess the stage, types and sub-types of lung cancer tumors. Adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) are the most prevalent sub-types of lung cancer and their distinction requires visual inspection by an experienced pathologist. In this study, we trained a deep convolutional neural network (inception v3) on whole-slide images obtained from The Cancer Genome Atlas to accurately and automatically classify them into LUAD, LUSC or normal lung tissue. The performance of our method is comparable to that of pathologists, with a 0.97 average Area Under the Curve (AUC). Our model was validated on independent datasets of frozen tissues, formalin-fixed paraffin-embedded tissues and biopsies. Furthermore, we trained the network to predict the ten most commonly mutated genes in LUAD. We found that six of them – STK11, EGFR, FAT1, SETBP1, KRAS and TP53 – can be predicted from pathology images with AUCs from 0.733 to 0.856, as measured on a held-out population. These findings suggest that deep learning models can assist pathologists in the detection of cancer sub-types or gene mutations. Our approach can be applied to any cancer type and the code is available at https://github.com/ncoudray/DeepPATH.
Keywords: Computational Biology, Cancer, Precision Medicine, Image Analysis, Computer Vision and Pattern Recognition, Quantitative Methods, Deep-learning
Introduction
According to the American Cancer Society (www.cancer.org) and the Cancer Statistics Center (cancerstatisticscenter.cancer.org), over 150,000 lung cancer patients succumb to their disease each year (154,050 expected for 2018), while another 200,000 new cases are diagnosed on a yearly basis (234,030 expected for 2018). It is one of the most widely spread cancers in the world, due to smoking, but also exposure to toxic chemicals like radon, asbestos and arsenic. Adenocarcinoma and squamous cell carcinoma are the two most prevalent types of non-small cell lung cancer1, and each are associated with discrete treatment guidelines. In the absence of definitive histologic features, this important distinction can be challenging, time-consuming, and will require confirmatory immunohistochemical stains. Lung cancer type classification is a key diagnostic process because the available treatment options, including conventional chemotherapy and more recently targeted therapies, differ for LUAD and LUSC2. Also, a LUAD diagnosis will prompt the search for molecular biomarkers and sensitizing mutations, and thus has a great impact on treatment options3,4. For example, EGFR (epidermal growth factor receptor) mutations, present in about 20% of LUAD, and ALK rearrangements (anaplastic lymphoma receptor tyrosine kinase), present in less than 5% of LUAD5, currently have targeted therapies approved by the Food and Drug Administration (FDA)6,7. Mutations in other genes, such as KRAS and TP53 are very common (about 25% and 50% respectively), but have proven particularly challenging drug targets so far5,8. Lung biopsies are typically used to diagnose lung cancer type and stage. Virtual microscopy of stained images of tissues is typically acquired at magnifications of 20x to 40x, generating very large two-dimensional images (10,000 to over 100,000 pixels in each dimension) that are oftentimes challenging to visually inspect in an exhaustive way. Furthermore, accurate interpretation can be difficult and the distinction between LUAD and LUSC is not always clear, particularly in poorly differentiated tumors, where ancillary studies are recommended for accurate classification9,10. To assist experts, automatic analysis of lung cancer whole-slide images has been recently studied to predict survival outcomes11 and classification12. In the latter, Yu et al. combined conventional thresholding and image processing techniques with machine learning methods, such as random forest classifiers, SVM or Naïve Bayes classifiers, achieving an Area Under the Curve (AUC) of ~0.85 in distinguishing normal from tumor slides, and ~0.75 in distinguishing LUAD from LUSC slides. More recently, the use of Deep Learning was used for the classification of breast, bladder and lung tumors, achieving AUC of 0.83 in TCGA tumor slide classification of lung tumor types13. Analysis of plasma DNA values also shown to be a good predictor of the presence of non-small cell cancer with AUC~0.9414, while the use of immunochemical markers gives AUC of ~0.941 in distinguishing LUAD from LUSC15. Here, we demonstrate how the field can further benefit from Deep Learning, by presenting a strategy based on Convolutional Neural Networks (CNNs) that not only outperforms previously published work, but also achieves accuracies that are comparable to pathologists. Most importantly, our models maintain their performance when tested on independent datasets of both frozen and formalin-fixed paraffin-embedded (FFPE) tissues as well as on images obtained from biopsies. The development of new inexpensive and more powerful technologies (in particular Graphics Processing Units) has made possible the training of larger and more complex neural networks16,17. This resulted in the design of several deep CNNs, capable of accomplishing complex visual recognition tasks. Such algorithms have already been successfully used for segmentation18 or classification of medical images19, and more specifically for whole-slide image applications such as nuclei detection20, renal tissue segmentation21 and glomeruli localization22, breast cancer diagnosis23,24, colon tumor analysis25, glioma grading in brain tumors26, epithelial tissue identification in prostate cancer27 or osteosarcoma diagnosis28. CNNs have also been studied for classifying lung patterns on CT (Computerized Tomography) scans, achieving an f-score of ~85.5%29. To study the automatic classification of lung cancer whole-slide images, we used the inception v3 architecture30 and whole-slide images of hematoxylin and eosin (H&E) stained lung tissue from TCGA obtained by surgical excision followed by frozen section preparation. In 2014, Google won the ImageNet Large-Scale Visual Recognition Challenge by developing the GoogleNet architecture31 which increased the robustness to translation and non-linear learning abilities by using micro-architecture units called inception. Each inception unit includes several non-linear convolution modules at various resolutions. Inception architecture is particularly useful for processing the data in multiple resolutions, a feature that makes this architecture suitable for pathology tasks. This network has already been successfully adapted to other specific types of classifications like skin cancers32 and diabetic retinopathy detection33.
Results
A deep learning framework for the automatic analysis of histopathology images
The purpose of this study was to develop a deep learning model for the automatic analysis of tumor slides using publicly available whole-slide images available in TCGA34 and subsequently test our models on independent cohorts collected at our institution. The TCGA dataset characteristics and our overall computational strategy are summarized in Figure 1 (see Methods for details). We used 1634 whole-slide images from the Genomic Data Commons database: 1176 tumor tissues and 459 normal (Figure 1a). The 1634 whole-slide images were split into three sets: training, validation and testing (Figure 1b). Importantly, this ensures that our model is never trained and tested on tiles (see below) obtained from the same tumor sample. Because the sizes of the whole-slide images are too large to be used as direct input to a neural network (Figure 1c), the network was instead trained, validated and tested using 512×512 pixel tiles, obtained from non-overlapping “patches” of the whole-slide images. This resulted in tens to thousands of tiles per slide depending on the original size (Figure 1d). Based on the computational strategy outlined in Figure 1, we present two main results. First, we develop classification models that classify whole-slide images into normal lung, lung adenocarcinoma (LUAD) or lung squamous cell carcinoma (LUSC) with an accuracy significantly higher than previous work (AUC of 0.97 compared to 0.7512 and 0.8313) and comparable to pathologists. Unlike previous work12,13, the performance of our classification models was tested on several independent datasets: biopsies, and surgical resection specimens either prepared as frozen sections or as formalin-fixed, paraffin-embedded (FFPE) tissue sections. Second, starting with the LUAD regions, as predicted by the LUAD vs LUSC vs normal classification model, we utilize the same computational pipeline (Figure 1) to train a new model in order to predict the mutational status of frequently mutated genes in lung adenocarcinoma using whole-slide images as the only input. The entire workflow of our computational analysis is summarized in Supplementary Figure 1.
Figure 1. Data and strategy:
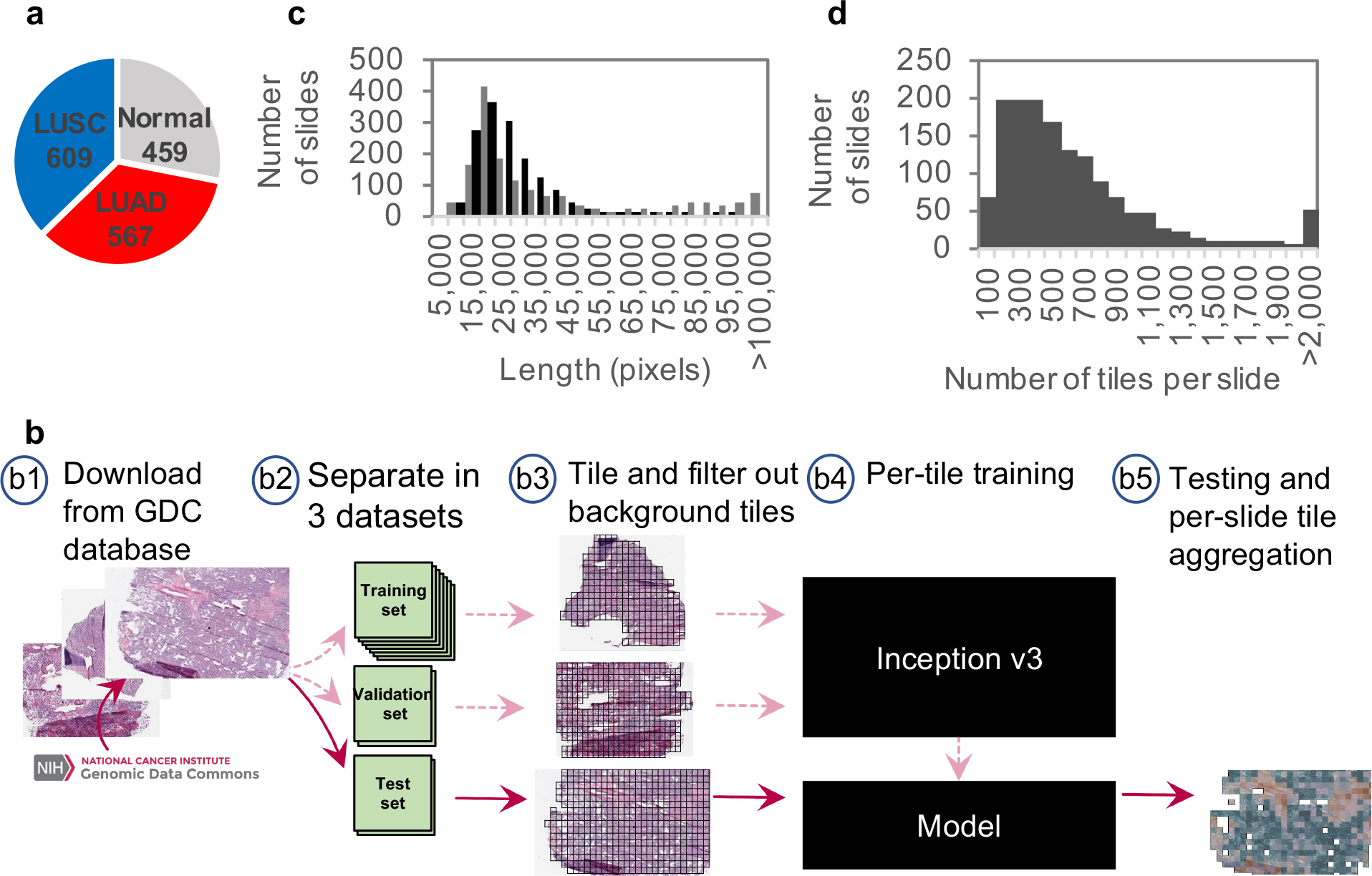
(a) Number of whole-slide images per class. (b) Strategy: (b1) Images of lung cancer tissues were first downloaded from the Genomic Data Common database; (b2) slides were then separated into a training (70%), a validation (15%) and a test set (15%); (b3) slides were tiled by non-overlapping 512×512 pixels windows, omitting those with over 50% background; (b4) the Inception v3 architecture was used and partially or fully re-trained using the training and validation tiles; (b5) classifications were performed on tiles from an independent test set and the results were finally aggregated per slide to extract the heatmaps and the AUC statistics. (c) Size distribution of the images widths (gray) and heights (black). (d) Distribution of the number of tiles per slide.
Deep learning models generate accurate diagnosis of lung histopathology images
Using the computational pipeline of Figure 1, we first trained inception v3 to recognize tumor versus normal. To assess the accuracy on the test set, the per-tile classification results were aggregated on a per-slide basis either by averaging the probabilities obtained on each tile, or by counting the percentage of tiles positively classified, thus generating a per-slide classification (see Methods for details). The two approaches yielded an Area Under the ROC Curve (AUC) of 0.990 and 0.993 (Supplementary Table 1 and Supplementary Figure 2a) respectively for normal-vs-tumor classification, outperforming the AUC of ~0.85 achieved by the feature-based approach of Yu et al.12, of ~0.94 achieved by plasma DNA analysis14 and comparable or better than molecular profiling data (Supplementary Table 2). Next, we tested the performance of our approach on the more challenging task of distinguishing LUAD and LUSC. First, we tested whether convolutional neural networks can outperform the published feature-based approach, even when plain transfer learning is used. For this purpose, the values of the last layer of inception v3 – previously trained on the ImageNet dataset to identify 1,000 different classes – were initialized randomly and then trained for our classification task. After aggregating the statistics on a per slide basis (Supplementary Figure 2b), this process resulted in an Area Under the Curve (AUC) of 0.847 (Supplementary Table 1), i.e. a gain of ~0.1 in AUC compared to the best results obtained by Yu et al12 using image features combined with random forest classifier. The performance can be further improved by fully training inception v3 leading to AUC of 0.950 when the aggregation is done by averaging the per-tile probabilities (Supplementary Figure 2c). These AUC values are improved by another 0.002 when the tiles previously classified as “normal” by the first classifier are not included in the aggregation process (Supplementary Table 1). We further evaluated the performance of the deep learning model by training and testing the network on a direct three-way classification into the three types of images (Normal, LUAD, LUSC). Such an approach resulted in the highest performance with all the AUCs improved to at least 0.968 (Supplementary Figure 2d and Supplementary Table 1). In addition to working with tiles at 20x magnification, we investigated the impact of the magnification and field of view of the tiles on the performance of our models. Since low-resolution features (nests of cells, circular patterns) may also be useful for lung cancer type classification, we trained on slides showing larger field of views by creating 512×512 pixels tiles of images at 5x magnification. The binary and three-way networks trained on such slides led to similar results (Supplementary Figure 2e–f and Supplementary Table 1). Supplementary Figure 2g,h and Supplementary Table 2 summarize and compare the performance of the different approaches explored in this study and in previous work.
Comparison of deep learning model to pathologists
We then asked three pathologists (two thoracic pathologists and one anatomic pathologist) to independently classify the whole-slide H&E images in the test set by visual inspection alone, independently of the classification provided by TCGA. Overall, the performance of our models was comparable to that of each pathologist (Supplementary Figure 2b–f, pink cross). Supplementary Figure 2i shows that 152 slides in our test set have a true positive probability above 0.5 (according to our model), and for 18 slides, this probability is below 0.5. 50% of the slides incorrectly classified by our model were also misclassified by at least one of the pathologists, while 83% of those incorrectly classified by at least one of the pathologists (45 out of 54) were correctly classified by the algorithm. We then measured the agreement between the TCGA classification and that of each pathologist, their consensus and finally our deep learning model (with an optimal threshold leading to sensitivity and specificity of 89% and 93%) using Cohen’s Kappa statistic (Supplementary Table 3). We observed that the agreement of the deep learning model with TCGA was slightly higher (0.82 vs 0.67 for pathologist 1, 0.70 for pathologist 2, 0.70 for pathologist 3, and 0.78 for the consensus), but not reaching statistical significance (p-values 0.035, 0.091, 0.090 and 0.549 respectively, estimated by a two-sample two-tailed z-test score). Regarding time effort, it can take a pathologist one to several minutes to analyze a slide depending on the difficulty to distinguish each case. Furthermore, in the absence of definitive histologic features, confirmatory immunohistochemical stains are required and can delay diagnosis for up to 24 hours. The processing time of a slide by our algorithm depends on its size; currently, it takes ~20 seconds to calculate per-tile classification probabilities on 500 tiles (the median number of tiles per slide is <500) on a single Tesla K20m GPU. Considering the possibility of using multiple GPUs to process tiles in parallel, classification using our model can be executed in a few seconds. The scanning time of each slide using the Aperio scanner (Leica) is currently 2–2.5 minutes for a slide at 20x, but with the 2017 FDA’s approval of the new ultra-fast digital pathology scanner from Philipps35, this step will probably not be a bottleneck anymore in the near future.
Testing on independent cohorts demonstrates generalizability of the neural network model
The model was then evaluated on independent datasets of lung cancer whole-slide images obtained from frozen sections (98 slides), formalin-fixed paraffin-embedded (FFPE) sections (140 slides), as well as lung biopsies (102 slides) obtained at the NYU Langone Medical Center (Figure 2a–c). In this case, the diagnosis made by the pathologists based on morphology and supplemented by immunohistochemical stains (TTF-1 and p40 for LUAD and LUSC respectively) when necessary was used as the gold standard. Each TCGA image is almost exclusively composed of either LUAD cells, LUSC cells, or normal lung tissue. As a result, several images in the two new datasets contain features the network has not been trained to recognize, making the classification task more challenging. We observed that features including blood clot, blood vessels, inflammation, necrotic regions, and regions of collapsed lung are sometimes labelled as LUAD, bronchial cartilage is sometimes labelled as LUSC, and fibrotic scars can be misclassified as normal or LUAD. As demonstrated in Supplementary Figure 3a, TCGA images have significantly higher tumor content compared to the independent datasets, and tumor content correlates with the ability of the algorithm to generalize on these new unseen samples. To reduce the bias generated by some of these particular features that are found outside the tumor areas and only test the ability of our network to dissociate LUAD / LUSC / Normal tissues regions, the AUCs in Figure 2 were computed on regions of high tumor content, manually selected by a pathologist. Considering that new types of artifacts were also observed on some older slides (dull staining, uneven staining, air bubbles under the slide cover leading to possible distortion), the results obtained on these independent cohorts are very encouraging. At 20x magnification, more tiles are fully covered by some of these “unknown” features, whereas at 5x magnification, the field of view is larger and contains features known by the classified (tumor or normal cells) in many more tiles, allowing a more accurate per-tile classification. This, in turn, leads to a more accurate per-slide classification. Taken together, these observations may explain why the AUC of the classifier on 5x magnified tiles is mostly higher than the one from 20x magnified tiles. Interestingly, even though the slides from FFPE and biopsy sections were preserved using a different technique from those in the TCGA database, the performance remains satisfactory (Figure 2b). For the biopsies, we noticed that poor performance was associated with regions where fibrosis, inflammation or blood was also present, but also in very poorly differentiated tumors. Sections obtained from biopsies are usually much smaller, which reduces the number of tiles per slide, but the performance of our model remains consistent for the 102 samples tested (AUC~0.834–0.861 using x20 magnification and 0.871–0.928 using the 5x magnification; Figure 2c) and the accuracy of the classification does not correlate with the sample size or the size of the area selected by our pathologist (Supplementary Figure 4; R2 = 9.5e-5). In one third of the cases collected, the original diagnosing pathologist was not able to visually determine the tumor type; TTF-1 and p40 stains were therefore used to identify LUAD and LUSC cases respectively. Interestingly, when splitting the dataset, we noticed that our model is able to classify those difficult cases as well: at 20x, the LUAD/LUSC’s AUCs for those difficult cases are 0.809/0.822 (CIs=[0.639–0.940 / 0.658–0.951]), which is only slightly lower than the slides considered obvious for the pathologists (AUC of 0.869/0.883 with CIs=[0.753–0.961 / 0.777–0.962]. Finally, we tested whether it is possible to replace the manual tumor selection process by an automatic computational selection. To this end, we trained inception v3 to recognize tumor areas using the pathologist’s manual selections. Training and validation was done on two out of the three datasets and testing was performed on the third one. For example, to test the performance of the tumor selection model on the biopsies, we trained the model to recognize the tumor area on the frozen and FFPE samples, then applied this model to the biopsies and finally applied the TCGA-trained 3-way classifier on the tumor area selected by the automatic tumor selection model. The per tile AUC of the automatic tumor selection model (using the pathologist’s tumor selection as reference) was 0.886 [CIs=0.880–0.891] for the biopsies, 0.797 [CIs=0.795–0.800] for the frozen samples, and 0.852 [CIs=0.808–0.895] for the FFPE samples. As demonstrated in Supplementary Figure 3a (right-most bar of each graph), we observed that the automatic selection resulted in a performance that is comparable to the manual selection (slightly better AUC in Frozen, no difference in FFPE and slighly worse in biopsies; see also Supplementary Figure 3b).
Figure 2. Classification of presence and type of tumor on alternative cohorts:
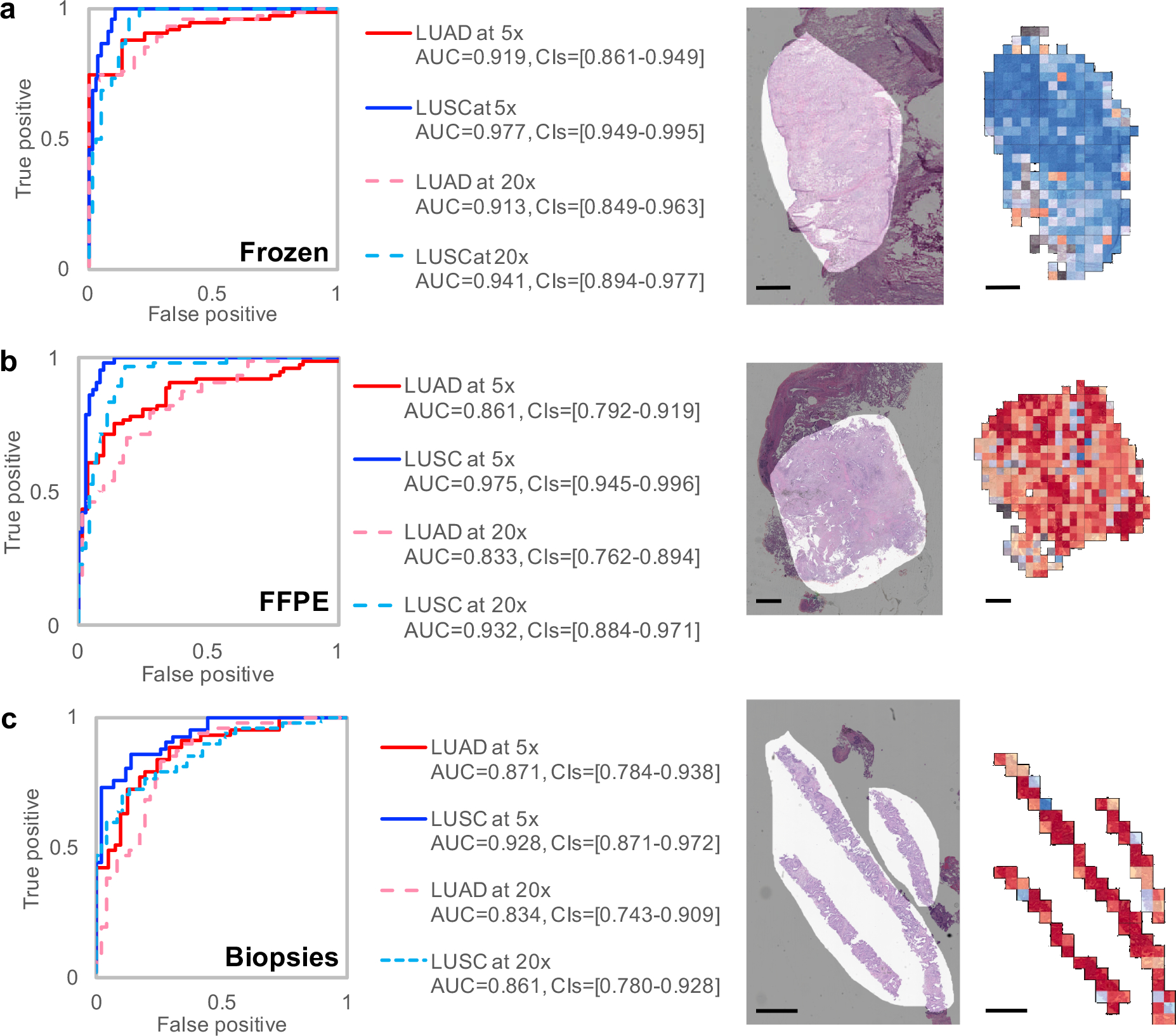
Receiver Operating Characteristic (ROC) curves (left) from tests on (a) frozen sections (n=98 biologically independent slides), (b) formalin-fixed paraffin-embedded (FFPE) sections (n=140 biologically independent slides) and (c) biopsies (n=102 biologically independent slides) from NYU Langone Medical Center. On the right of each plot, we show examples of raw images with an overlap in light grey of the mask generated by a pathologist and the corresponding heatmaps obtained with the three-way classifier. Scale bars are 1 mm.
Predicting gene mutational status from whole-slide images
We then focused on the LUAD slides and tested whether CNNs can be trained to predict gene mutations using images as the only input. For this purpose, gene mutation data, for matched patient samples, were downloaded from TCGA. To make sure the training and test sets contain enough images from the mutated genes, we only selected those which were mutated in at least 10% of the available tumors. From each LUAD slide, only tiles classified as LUAD by our classification model were utilized for this task in order to avoid biasing the network to learn LUAD-specific vs LUSC-specific mutations and focus instead on distinguishing mutations relying exclusively on LUAD tiles. Inception v3 was modified to allow multi-output classification (see Methods for details): training and validation was conducted on ~212,000 tiles from ~320 slides, while testing was performed on ~44,000 tiles from 62 slides. Box plot and ROC curves analysis (Figure 3a–b and Supplementary Figure 5) show that six frequently mutated genes seem predictable using our deep learning approach: AUC values for STK11, EGFR, FAT1, SETBP1, KRAS and TP53 were found between 0.733 and 0.856 (Table 1). Availability of more data for training is expected to improve the performance significantly. As mentioned earlier, EGFR already has targeted therapies. STK11 (Serine/Threonine protein Kinase 11), also known as Liver Kinase 1 (LKB1), is a tumor suppressor inactivated in 15–30% of non-small cell lung cancers36 and is also a potential therapeutic target: it has been reported that phenformin, a mitochondrial inhibitor, increases survival in mice37. Also, it has been shown that STK11 mutations may play a role in KRAS mutations which, combined, result in more aggressive tumors38. FAT1 is an ortholog of the Drosophila fat gene involved in many types of cancers and its inactivation is suspected to increase cancer cell growth39. Mutation of the tumor suppressor gene TP53 is thought to be more resistant to chemotherapy leading to lower survival rates in small-cell lung cancers40. As for SETBP1 (SET 1 binding protein), like KEAP1 and STK11, has been identified as one of the signature mutations of LUAD41. Finally, for each gene, we compared the classification achieved by our deep learning model with the allele frequency (Figure 3c). Among the gene mutations predicted with a high AUC, in four of them, classification probabilities (as reported by our model) are associated with allele frequency: FAT1, KRAS, SETBP1 and STK11, demonstrating that these probabilities may reflect the percentage of cells effectively affected by the mutation. Looking, for example, at the predictions performed on the whole-slide image from Figure 4a, our process successfully identifies TP53 (allele frequency of 0.33) and STK11 (allele frequency of 0.25) as two genes most likely mutated (Figure 4a). The heatmap shows that almost all the LUAD tiles are highly predicted as showing TP53-mutant-like features (Figure 4b), and two major regions with STK11-mutant-like features (Figure 4c). Interestingly, when the classification is applied on all tiles, it shows that even tiles classified as LUSC present TP53 mutations (Figure 4d) while the STK11 mutant is confined to the LUAD tiles (Figure 4e). These results are realistic since, as mentioned earlier, STK11 is a signature mutations of LUAD41 while TP53 is more common in all human cancers. Future work on deep leaning models visualization tools42 would help identify and characterize the features used by the neural network. To visualize how the mutations and tiles are organized in the multi-dimensional space of the network, we used as before a t-SNE representation43 with the values of the last fully connected layer used an inputs. On the resulting plots (Supplementary Figure 6a), each dot represents a tile and its color is proportional to the probability of the gene to be mutated, as estimated by our model. The tile-embedded representation (Supplementary Figure 6b) allows the visual comparison of tiles sharing similar predicted mutations. Clusters of specific mutations can be seen at the surroundings of the plot. The top left group for example shows tiles were the aggressive double mutants KRAS and STK11 are both present, while the small one at the top shows tiles with KEAP1/SETBP1 and the cluster on the top right has been associated with the triple mutation of FAT1/LRP1B/TP53. Future analysis with laser capture microdissection could provide some additional spatial information and study the limits and precision of such a method44. Although our current analysis does not define yet the specific features used by the network to identify mutations, our results suggest that such genotype-phenotype correlations are detectable. Determining mutation status from a histological image and bypassing additional testing is important in lung cancer in particular as these mutations often carry prognostic as well as predictive information. Previous work has shown associations between clinically important mutations and specific patterns of lung adenocarcinoma45,46, as well as the histologic changes that correspond with the evolution of resistance47. More recently, Chiang and colleagues empirically demonstrated the relationship between a defining mutation and the unique morphology of a breast cancer subtype48. Some of the mutations with high AUCs highlighted in our study (like STK11 and TP53) have been shown to affect cell polarity and cell shape49,50, two features that are not routinely assessed during the pathologic diagnosis. We note that our model was not able to detect ALK mutations although such tumors have been associated with specific histologic features, such as a solid pattern with signet ring cells or a mucinous cribriform pattern51,52. Although the prevalence of ALK mutations are very low (reportedly ranging from 1.8%–6.4%53), their presence is routinely determined via immunohistochemistry as these tumors may respond to ALK inhibitors6,7. To confirm that our models can be applied to independent cohorts, we tested the prediction of the EGFR mutant using 63 whole-slide images of lung resection specimens with known EGFR mutational status: 29 EGFR-mutant and 34 EGFR wild-type samples. This independent dataset has some important differences from the TCGA dataset which may negatively impact the evaluation of the TCGA-based model: (1) the samples were not frozen but were instead preserved using FFPE, and (2) only 22 samples had sequencing data to support the EGFR mutational status with high specificity and sensitivity; the rest of the samples (i.e. 65% of the test set) have been analyzed by immunohistochemical (IHC) stains54, a technique known for its high specificity but low sensitivity55,56 and which solely identifies the two most common mutations54 (L858R and E746_A750del). On the other hand, data from the TCGA dataset used for training were identified with NGS (Next-Generation Sequencing tools Illumina HiSeq 2000 or Genome Analyzer II). Our TCGA model has therefore been trained to detect not only L858R and E746_A75-del but many other EGFR mutants and deletions such as G719A, L861Q or E709_T710delinsD for example. Despite these caveats, we believed that it would still be important to demonstrate that our TCGA-derived models can at least perform significantly better than random in the independent NYU cohort. Indeed, the results show an AUC of 0.687 (confidence intervals = 0.554–0.811) with higher AUC (0.750, CIs=[0.500–0.966]) in samples validated by sequencing compared to those tested by IHC (AUC=0.659, CIs=[0.485–0.826]). Although the sequencing-based AUC of 0.75 is lower than the one estimated on the TCGA test set (0.83), we believe that most of this difference can be attributed to the difference in the sample preparation (frozen versus FFPE). We noticed that the discrepancy (~0.08) is similar to the difference observed in the AUCs of LUAD from the TCGA dataset (0.97) and the FFPE dataset (0.83). In the classification task, this issue was solved by lowering the magnification to 5x. However, this is not useful for the mutation prediction task, because it appears that 20x is necessary to capture predictive image features (the TCGA EGFR mutation prediction model at 5x has a random performance). Still, we believe that the 0.75 AUC we obtained on the sequencing-validated subset of EGFR-mutant cases demonstrates that the model can generalize on independent datasets.
Figure 3. Gene mutation prediction from histopathology slides give promising results for at least 6 genes:
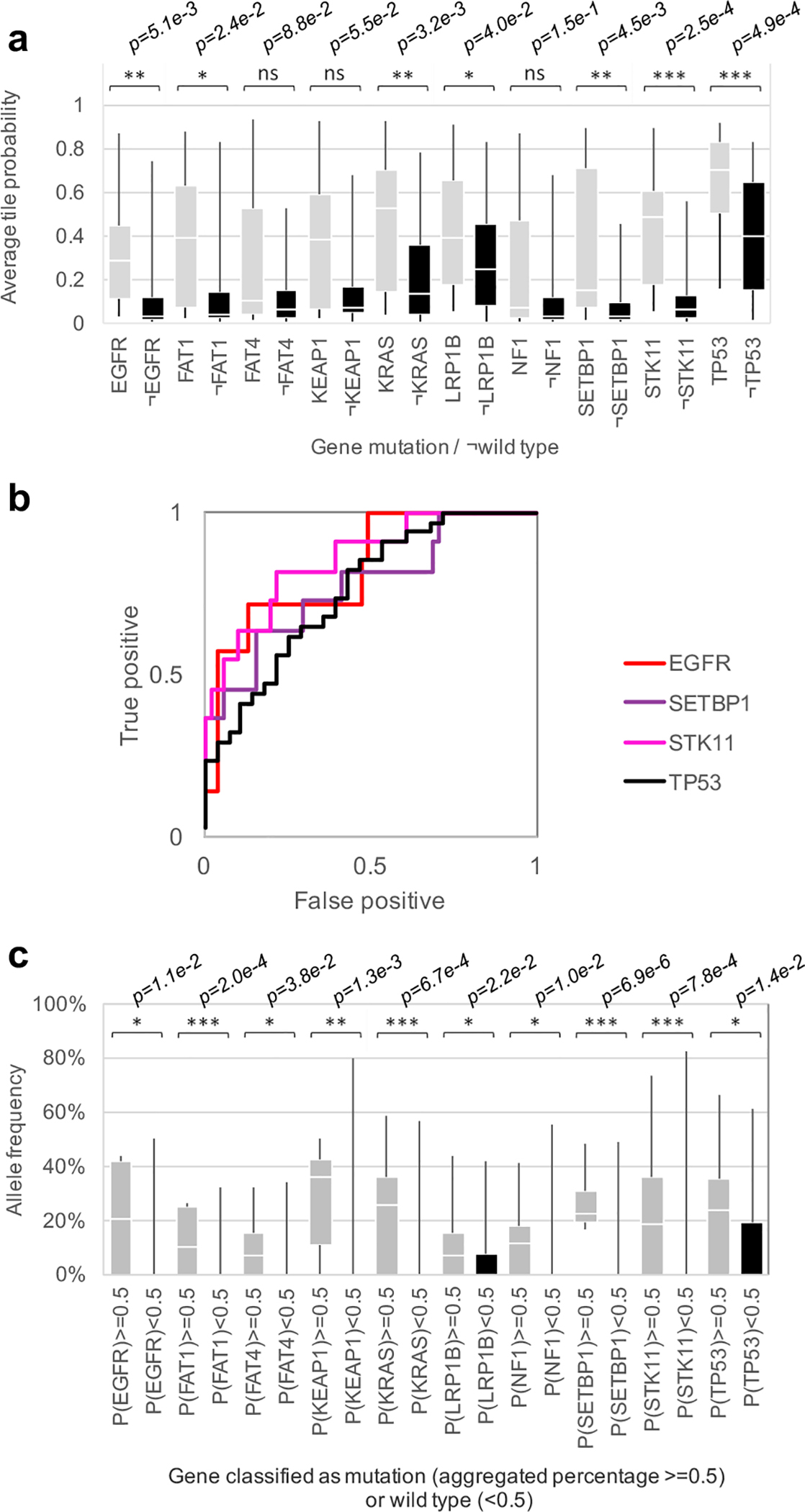
(a) Mutation probability distribution for slides where each mutation is present or absent (tile aggregation by averaging output probability). (b) ROC curves associated with the top four predictions (a). (c) Allele frequency as a function of slides classified by the deep learning network as having a certain gene mutation (P≥0.5), or the wild-type (P<0.5). p-values estimated with two-tailed Mann-Whitney U-test are shown as ns (p>0.05), * (p≤0.05), ** (p≤0.01) or *** (p≤0.001). For a, b and c, n=62 slides from 59 patients. For the two box plots, whiskers represent the minima and maxima. The middle line within the box represents the median.
Table 1.
Area Under the Curve (AUC) achieved by the network trained on mutations (with 95% CIs).
| Mutations | Per tile AUC | Per slide AUC after aggregation by… | |
|---|---|---|---|
| … average predicted probability | … percentage of positively classified tiles | ||
| STK11 | 0.845 [0.838–0.852] | 0.856 [0.709–0.964] | 0.842 [0.683–0.967] |
| EGFR | 0.754 [0.746–0.761] | 0.826 [0.628–0.979] | 0.782 [0.516–0.979] |
| SETBP1 | 0.785 [0.776–0.794] | 0.775 [0.595–0.931] | 0.752 [0.550–0.927] |
| TP53 | 0.674 [0.666–0.681] | 0.760 [0.626–0.872] | 0.754 [0.627–0.870] |
| FAT1 | 0.739 [0.732–0.746] | 0.750 [0.512–0.940] | 0.750 [0.491–0.946] |
| KRAS | 0.814 [0.807–0.829] | 0.733 [0.580–0.857] | 0.716 [0.552–0.854] |
| KEAP1 | 0.684 [0.670–0.694] | 0.675 [0.466–0.865] | 0.659 [0.440–0.856] |
| LRP1B | 0.640 [0.633–0.647] | 0.656 [0.513–0.797] | 0.657 [0.512–0.799] |
| FAT4 | 0.768 [0.760–0.775] | 0.642 [0.470–0.799] | 0.640 [0.440–0.856] |
| NF1 | 0.714 [0.704–0.723] | 0.640 [0.419–0.845] | 0.632 [0.405–0.845] |
n = 62 slides from 59 patients
Figure 4. Spatial heterogeneity of predicted mutations.
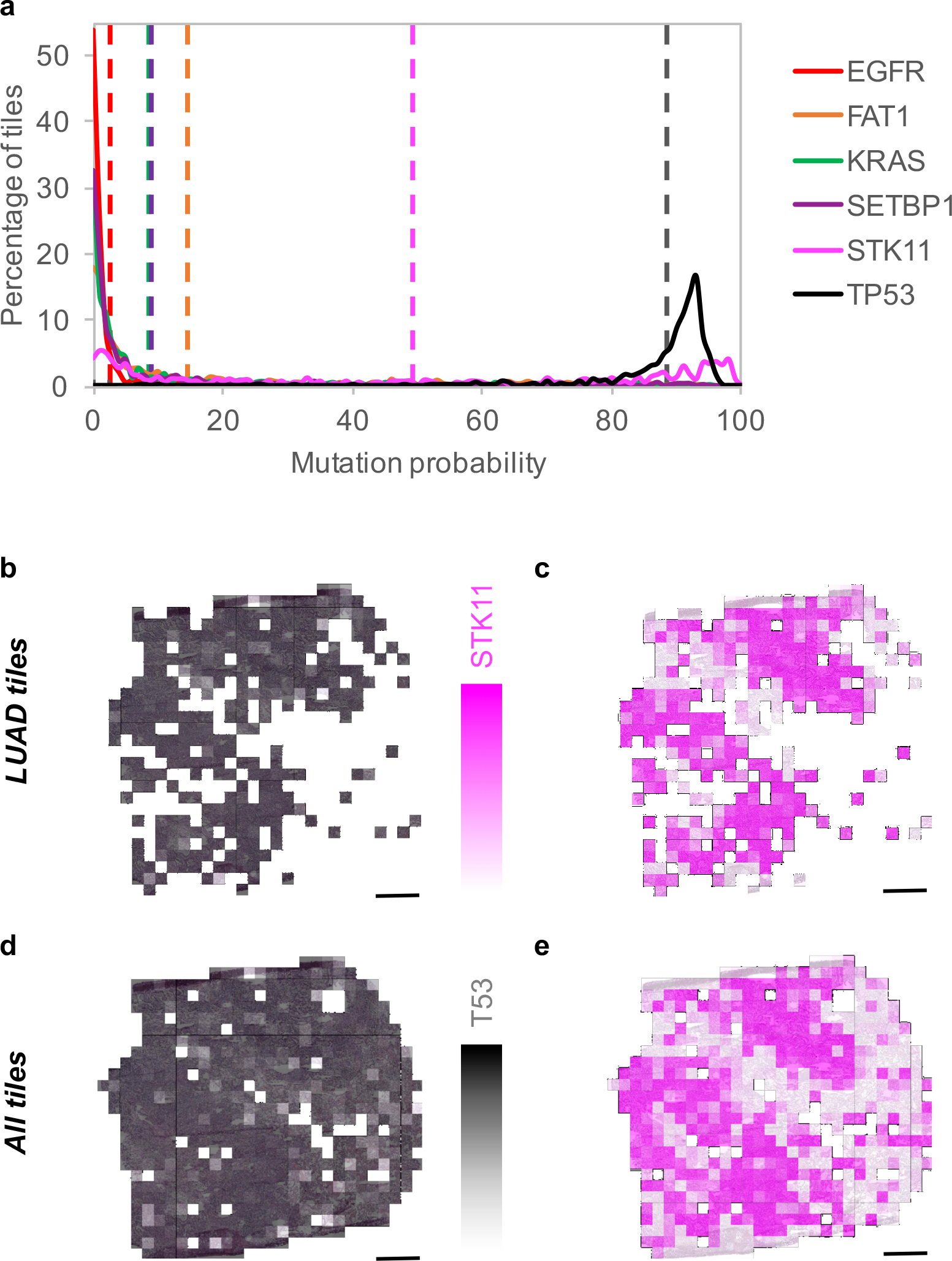
(a) Probability distribution on LUAD tiles for the 6 predictable mutations with average values in dotted lines (n=327 non-overlapping tiles). The allele frequency is 0.33 for TP53, 0.25 for STK11 and 0 for the 4 other mutations. (b) heatmap of TP53 and (c) STK11 when only tiles classified as LUAD are selected, and in (d) and (e) when all the tiles are considered. Scale bars are 1 mm.
Discussion
Our study demonstrates that convolutional neural networks, such as Google’s inception v3, can be used to assist in the diagnosis of lung cancer from histopathology slides: it almost unambiguously classifies normal vs tumor tissues (~0.99 AUC), distinguishes lung cancer types with high accuracy (0.97 AUC), reaching sensitivity and specificity comparable to that of a pathologist. Interestingly, around half of the TCGA whole-slide images misclassified by the algorithms have also been misclassified by the pathologists, highlighting the intrinsic difficulty in distinguishing LUAD from LUSC in some cases. However, 45 out of 54 of the TCGA images misclassified by at least one of the pathologists were assigned to the correct cancer type by the algorithm, suggesting that our model could be beneficial in assisting the pathologists in their diagnosis. The confusions matrices in Supplementary Table 4 details the discrepancies between the different classifications, while Supplementary Figure 7 shows a few examples where our model correctly classified whole-slide images misclassified by at least one of our pathologists. These images show poorly differentiated tumors that lack the classic histological features of either type (keratinization for LUSC and gland formation/recognizable histological pattern for LUAD). The high accuracy of our model was achieved despite the presence of various artefacts in the TCGA images, related to sample preparation and preservation procedures. However, the TCGA images used to train the deep neural network may not fully represent the diversity and heterogeneity of tissues that pathologists typically inspect, which may include additional features such as necrosis, blood vessels and inflammation. More slides containing such features would be needed to re-train the network in order to further improve its performance. Despite this and the fact that the process was trained on frozen images, tests show very promising results on tumor classification from FFPE sections as well. While it has been suggested that mutations could be predicted from H&E images (AUC of ~0.71 for the prediction of SPOP mutations from prostate cancer H&E images57), before this study, it was unclear whether gene mutations would affect the pattern of tumor cells on a lung cancer whole-slide image, but training the network using presence/absence of mutated genes as a label revealed that there are certain genes whose mutational status can be predicted from image data alone: EGFR, STK11, FAT1, SETBP1, KRAS and TP53. Notably, the presence of STK11 mutations can be predicted with the highest accuracy (~0.85 AUC). A limiting factor in obtaining higher accuracies lies in the small number of slides that contain positive instances (i.e. the gene mutations) available for training, therefore our models can greatly benefit from larger datasets that may become available in the near future. The ability to quickly and inexpensively predict both the type of cancer and the gene mutations from histopathology images could be beneficial to the treatment of cancer patients given the importance and impact of these mutations6,36–41. Overall, this study demonstrates that the use of deep learning convolutional neural networks could be a very promising tool to assist pathologists in their classification of whole-slide images of lung tissues. This information can be crucial in applying the appropriate and tailored targeted therapy to lung cancer patients, increasing thereby the scope and performance of precision medicine that aims at developing a multiplex approach with patient-tailored therapies58. The diagnosis and therapy differ significantly between LUSC to LUAD and may depend on the mutational status of specific genes. In particular, when inspecting frozen section biopsies, pathologists only rely on morphology, and may need immunostaining for the most difficult cases; our algorithm, which still achieves an AUC above 0.8 on biopsies that usually require immune-staining, can be used as an adjunct to telepathology to speed up diagnosis and classification during intraoperative consultation. As a result of advances in our understanding of lung cancer and a concomitant rise in the number and types of treatment options, the role of the pathologist in the diagnosis and management of this disease is significantly more complex than cancer type distinction and even determination of mutational status. While our computational analyses may play a role in the initial diagnosis with the benefit of providing important prognostic information based on an H&E image alone, the pathologist has additional tasks such as staging the tumor and, in an increasing number of cases, estimating response to treatment. In the future, we would ideally extend the classification to other types of less common lung cancers (large cell carcinoma, small cell lung cancer) histological subtypes of LUAD (acinar, lepidic, papillary, micropapillary and solid), as well as non-neoplastic features including necrosis, fibrosis, and other reactive changes in the tumor microenvironment, though the amount of data currently available is insufficient. We hope that by extending our algorithm to recognize a wider range of histologic features, followed by providing a quantitative and spatial assessment as in our heatmaps, we will be able to aid aspects of the pathologist’s evaluation that are well-suited to automated analyses. We hope that this computational approach could play a role in both routine tasks and difficult cases (for example, distinguishing intrapulmonary metastases from multiple synchronous primary lung cancers) in order to allow the pathologist to concentrate on higher-level decisions, such as integrating histologic, molecular, and clinical information in order to guide treatment decisions for individual patients.
Online Methods
TCGA lung cancer whole-slide image dataset
Our dataset comes from the NCI Genomic Data Commons34 which provides the research community with an online platform for uploading, searching, viewing and downloading cancer-related data. All freely available slide images of Lung cancer were uploaded from this source. We studied the automatic classification of “solid tissue normal” and “primary tumor” slides using a set of respectively 459 and 1175 hematoxylin and eosin stained histopathology whole-slide images59,60. Then, the “primary tumor” were classified between LUAD and LUSC types using a set of respectively 567 and 608 of those whole-slide images. The labels provided by the TCGA database were used as our gold standard. Those labels were the result of a consensus as explained by the GDC data curator (personal communication): first, the submitting institutions were asked to review each sample prior sending it to confirm the diagnosis. Then, a slide from the sample was reviewed by a TCGA contracted expert thoracic pathologist. In the event of a disagreement, the slide would be reviewed by one or more other expert thoracic pathologists. Out of the 170 slides in our test set, only 1 image was tagged as leading to inconsistent labels (and about 30 had not information about it).
Image pre-processing generates 987,931 tiles
The slides were tiled in non-overlapping 512×512 pixel windows at a magnification of x20 using the openslide library67 (533 of the 2167 slides initially uploaded were removed because of compatibility and readability issues at this stage). The slides with a low amount of information were removed, that is all the tiles where more than 50% of the surface is covered by background (where all the values are below 220 in the RGB color space). This process generated nearly 1,000,000 tiles.
Deep learning with Convolutional Neural Networks
We used 70% of those tiles for training, 15% for validation, and 15% for final testing (Table 2 and Table 3). The tiles associated with a given slide were not separated but associated as a whole to one of these sets to prevent overlaps between the three sets. Typical CNN consist of several levels of convolution filters, pooling layers and fully connected layers. We based our model on inception v3 architecture36. This architecture makes use of inception modules which are made of a variety of convolutions having different kernel sizes and a max pooling layer. The initial 5 convolution nodes are combined with 2 max pooling operations and followed by 11 stacks of inception modules. The architecture ends with a fully connected and then a softmax output layer. For “normal” vs “tumor” tiles classification, we fully trained the entire network. For the classification of type of cancer, we followed and compared different approaches to achieve the classification: transfer learning, which includes training only the last fully-connected layer, and training the whole network. Tests were implemented using the Tensorflow library (tensorflow.org).
Table 2.
Dataset information for normal vs tumor classification (number of tiles / slides in each category).
| Training | Validation | Testing | |
|---|---|---|---|
| Normal | 132,185 / 332 | 28,403 / 53 | 28,741 / 74 |
| Primary tumor | 556,449 / 825 | 121,094 / 181 | 121,059 / 170 |
Table 3.
Dataset information for LUAD vs LUSC classification (number of tiles / slides in each category).
| Training | Validation | Testing | |
|---|---|---|---|
| LUAD | 255,975 / 403 | 55,721 / 85 | 55,210 / 79 |
| LUSC | 300,474 / 422 | 65,373 / 96 | 65,849 / 91 |
Transfer learning on inception v3
We initialized our network parameters to the best parameter set that was achieved on ImageNet competition. We then fine-tuned the parameters of the last layer of the network on our data via back propagation. The loss function was defined as the cross entropy between predicted probability and the true class labels, and we used RMSProp69 optimization, with learning rate of 0.1, weight decay of 0.9, momentum of 0.9, and epsilon of 1.0 method for training the weights. This strategy was tested for the binary classification of LUAD vs LUSC.
Training the entire inception v3 network
The inception v3 architecture was fully trained using our training datasets and following the procedure described in 70. Similar to transfer learning, we used back-propagation, cross entropy loss, and RMSProp optimization method, and we used the same hyperparameters as the transfer learning case, for the training. In this approach, instead of only optimizing the weights of the fully connected layer, we also optimized the parameters of previous layers, including all the convolution filters of all layers. This strategy was tested on three classifications: normal vs tumor, LUAD vs LUSC and Normal vs LUAD vs LUSC. The training jobs were run for 500,000 iterations. We computed the cross-entropy loss function on train and validation dataset, and used the model with best validation score as our final model. We did not tune the number of layers or hyper-parameters of the inception network such as size of filters. As this training gave the best results, we also investigated the importance of training the network on a larger field of view at the expense of a lower resolution. Whole slide images were tiled at a magnification of 5x (keeping the tile size at 512×512 pixels) and the network was again fully trained.
Identification of gene mutations
To study the prediction of gene mutations from histopathology images, we modified the inception v3 to perform multi-task classification rather than a single task classification. Each mutation classification was treated as a binary classification, and our formulation allowed multiple mutations to be assigned to a single tile. We optimized the average of the cross entropy of each individual classifier. To implement this method, we replaced the final softmax layer of the network with a sigmoid layer, to allow each sample to be associated with several binary labels 71. We used RMSProp algorithm for the optimization, and fully trained this network for 500k iterations using only LUAD whole-slide images, each one associated with a 10-cell vector, each cell associated to a mutation and set to 1 or 0 depending on the presence or absence of the mutation. Only the most commonly mutated genes were used (Table 4), leading to a training set of 223,185 tiles. Training and validation were done over 500,000 iterations (Supplementary Figure 8). The test was then achieved on the tiles, and aggregation on the n=62 test-slides where at least one of these mutations is present was done only if the tile was previously classified as “LUAD” by the Normal/LUAD/LUSC 3-classes classifier.
Table 4.
Gene included in the multi-output classification and the percentage of patients with LUAD in the database where the genes are mutated.
| Gene mutated | TP53 | LRP1B | KRAS | KEAP1 | FAT4 | STK11 | EGFR | FAT1 | NF1 | SETBP1 |
|---|---|---|---|---|---|---|---|---|---|---|
| %Patients | 50 | 34 | 28 | 18 | 16 | 15 | 12 | 11 | 11 | 11 |
Statistical Analysis
Once the training phase was finished, the performance was evaluated using the testing dataset which is composed of tiles from slides not used during the training. We then aggregated the probabilities for each slide using two methods: either average of the probabilities of the corresponding tiles, or percentage of tiles positively classified. For the binary LUAD/CLUSC classifiers, n=170 slides from 137 patients, and for the Normal/Tuimor and for the three-way classifiers, n=244 slides from 137 patients. The ROC (Receiver Operating Characteristic) curves and the corresponding AUC (Area Under the Curve) were computed in each case61 using the python library sklearn62. Confidence Intervals (CIs) at 95% were estimated by 1,000 iterations of the bootstrap method63. Tumor slides could contain a certain amount of “normal” tiles. Therefore, we also checked how the ROC & AUC were affected when tiles classified as “normal” were removed from the aggregation. We asked three pathologists to manually label the TCGA test LUAD and LUSC images and compared the agreements between the ratings using the Cohen’s Kappa statistic64,65, comparing it to the binary LUAD/LUSC deep-learning classifier using the optimal threshold of 0.4/0.6 (optimal threshold is here defined as the point of the ROC curve which is closest to the perfect (1,0) coordinate). Heatmaps were also generated for some tested slide to visualize the differences between the two approaches and identify the regions associated with a certain cancer type. To analyze more thoroughly the network trained on gene mutations, we used the Barnes-Hit implementation of the t-Distributed Stochastic Neighbor Embedding (t-SNE) technique43 to reduce the dimensionality and facilitate the visualization of the classes. The values associated with the last fully connected layer were used as an input, and setting theta to 0.5, perplexity to 50, and 10,000 iterations. For the LUAD/LUSC classifier, the t-SNE plot was generated using n=149,790 tiles of 244 slides from 137 patients. For the gene mutation prediction task, the t-SNE plot was generated using n=24,144 tiles of 62 slides from 59 patients. Mutation probability distributions and relationship to allele frequency were analyzed with the two-tailed Mann Whitney U-tests and computed using the same dataset (62 slides from 59 patients).
Visualization of features identified by the three-way classifier in high-confidence tiles
In Supplementary Figure 9, we present examples of LUSC and LUAD slides, together with heatmaps generated by our algorithm, where the color of each tile corresponds to the class assigned by our algorithm (LUAD, LUSC or Normal), while the color shade is proportional to the classification probability. The LUSC image shows most of its tiles with a strong true positive probability for LUSC classification, while in the LUAD image the largest regions indeed have strong LUAD features, with normal cells on the side (as confirmed by our pathologist), and some light blue tiles indicating the existence of LUSC-like features in this tumor. In Supplementary Figure 10, the values of the last fully connected layer are visualized using a t-SNE representation which generates two-dimensional scatterplots of high-dimensional features43. For tiles associated with LUSC, we note a predominance of areas of keratinization, dyskeratotic cells, as well as rare foci of cells with prominent intracellular bridging. Among the tiles denoted LUAD, the predominant feature noted is the presence of distinct gland forming histological patterns such as lepidic and acinar (well differentiated) and micropapillary (poorly differentiated). These include well-differentiated patterns (lepidic and acinar) as well as poorly differentiated types (micropapillary). At the center of the t-SNE, regions that cannot be clearly associated with either LUAD or LUSC are composed of tiles with conspicuous preservation artifact, minute foci of tumor, or areas of interstitial/septal fibrosis. Then, the area designated as normal is composed of tiles showing benign lung parenchyma, focal fibrosis or inflammation, as well as rare LUAD with preservation artifacts. Interestingly, the area with tiles which could not be designated normal/LUAD/LUSC with high confidence shows both benign and malignant lung tissue in a background of dense fibrosis and/or inflammation.
Tests on independent cohorts
To challenge the trained algorithm and identify its limitations, we tested the three-way classifier with different cohorts. Images of lung cancers were obtained from the New York University Langone Medical Center from both frozen (75 of LUAD and 23 of LUSC), FFPE sections (74 LUAD, 66 LUSC) and biopsies (51 LUAD and 51 LUSC). The diagnosis used as true positive for these cases are based on morphology (gland formation for adenocarcinoma and keratinization and intracellular bridges for squamous cells), with the cases classified according to the World Health Organization, and for the more challenging cases, immunostaining was performed. Because biopsies can be much narrower, during the tiling process at 5x magnification, tiles were kept if at least 20% of it was covered by the tissue instead of 50%. As those external slides also contained a lot of elements the network was not trained to identify (blood clot, cartilage), we ran the final tests on regions of interests (ROI) selected by a pathologist. Those regions were selected manually using Aperio ImageScope (Leica Biosystems), and tiles were kept only if it was covered by at least 50% of the ROI for 20x mag tiles, and 10% for the 5x mag tiles. Additionally, we trained several networks to automatically select those ROIs for the NYU dataset (tumor / non-tumor): the first network was trained with the FFPE+Biopsies slides and tested on the Frozen ones, the second trained with the FFPE+Frozen slides and tested on the Biopsy ones, and the third trained with the Frozen+Biopsy slides and tested on the FFPE ones. For each test, we therefore applied this automatic ROI selection followed by the three-way classifier trained on the TCGA dataset, allowing us to compare the performance of the independent cohorts at different levels: using the whole slide image, using ROIs selected by a pathologist, and using ROIs selected by a trained deep-learning architecture. For the mutations, we identified 63 FFPE sections which were tested for EGFR mutations; 34 were identified as wild-type and 29 as mutant. Most of them (41) were analyzed using markers used as immunochemical stains to detect the mutations L858R and E746_A750del. The others (17 and 5 respectively), were analyzed by PCR (Polymerase Chain Reaction) or NGS (Next Generation Sequencing). The tests were run using tumor regions manually selected by a pathologist. Further information on experimental design is available in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We would like to thank the Applied Bioinformatics Laboratories (ABL) at the NYU School of Medicine for providing bioinformatics support and helping with the analysis and interpretation of the data. The Applied Bioinformatics Laboratories are a Shared Resource, partially supported by the Cancer Center Support Grant, P30CA016087 (A.T.), at the Laura and Isaac Perlmutter Cancer Center (A.T.). This work has utilized computing resources at the High-Performance Computing Facility (HPC) at NYU Langone Medical Center. The slide images and the corresponding cancer information were uploaded from the Genomic Data Commons portal (https://gdc-portal.nci.nih.gov) and are in whole or part based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/). These data were publicly available without restriction, authentication or authorization necessary. We thank the GDC help desk for providing assistance and information regarding the TCGA dataset. For the independent cohorts, we only used images information; the NYU dataset we used consists of slide images without identifiable information and therefore does not require approval according to both federal regulations and the NYU School of Medicine Institutional Review Board. For this same reason, written informed consent was not necessary. We thank C. Dickerson, from the Center for Biospecimen Research and Development (CBRD) for scanning the whole-slide images from the NYU Langone Medical Center. We also thank Dr. T. Papagiannakopoulos, Dr. H. Pass and Dr. K.-K. Wong or their valuable and constructive suggestions.
Footnotes
Competing Financial Interests Statement
The authors declare no competing interests.
Accession Code
N/A
Code Availability
The source code can be accessed at https://github.com/ncoudray/DeepPATH.
Data availability
All relevant data used for training during the current study are available through the Genomic Data Commons portal (https://gdc-portal.nci.nih.gov). These datasets were generated by TCGA Research Network (http://cancergenome.nih.gov/) and they have made them publicly available. Other datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Travis WD et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma. Journal of Thoracic Oncology 6, 244–285 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanna N et al. Systemic therapy for stage IV non–small-cell lung cancer: American Society of Clinical Oncology clinical practice guideline update. Journal of Clinical Oncology 35, 3484–3515 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Chan BA & Hughes BG Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Translational Lung Cancer Research 4, 36–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parums DV Current status of targeted therapy in non-small cell lung cancer. Drugs Today (Barc). 50, 503–525 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Terra SB et al. Molecular characterization of pulmonary sarcomatoid carcinoma: analysis of 33 cases. Modern Pathology 29, 824–831 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Blumenthal GM et al. Oncology Drug Approvals: Evaluating Endpoints and Evidence in an Era of Breakthrough Therapie. The Oncologist 22, 762–767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez-Soler R et al. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. Journal of Clinical Oncology 22, 3238–3247 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Jänne PA et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. The Lancet Oncology 14, 38–47 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Thunnissen E, van der Oord K & Den Bakker M Prognostic and predictive biomarkers in lung cancer. A review. Virchows Archiv 464, 347–358 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Zachara-Szczakowski S, Verdun T & Churg A Accuracy of classifying poorly differentiated non–small cell lung carcinoma biopsies with commonly used lung carcinoma markers. Human pathology 46, 776–782 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Luo X et al. Comprehensive Computational Pathological Image Analysis Predicts Lung Cancer Prognosis. Journal of Thoracic Oncology 12, 501–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu K-H et al. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nature Communications 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khosravi P, Kazemi E, Imielinski M, Elemento O & Hajirasouliha I Deep Convolutional Neural Networks Enable Discrimination of Heterogeneous Digital Pathology Images. EBioMedicine, doi: 10.1016/j.ebiom.2017.12.026. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sozzi G et al. Quantification of Free Circulating DNA As a Diagnostic Marker in Lung Cancer. Journal of Clinical Oncology 21, 3902–3908 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Terry J et al. Optimal immunohistochemical markers for distinguishing lung adenocarcinomas from squamous cell carcinomas in small tumor samples. The American journal of surgical pathology 34, 1805–1811 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Schmidhuber J Deep learning in neural networks: An overview. Neural Networks 61, 85–117 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Greenspan H, Ginneken B. v. & Summers RM Guest Editorial Deep Learning in Medical Imaging: Overview and Future Promise of an Exciting New Technique. IEEE TRANSACTIONS ON MEDICAL IMAGING 35, 1153–1159 (2016). [Google Scholar]
- 18.Qaiser T, Tsang Y-W, Epstein D & RajpootEma N in Medical Image Understanding and Analysis: 21st Annual Conference on Medical Image Understanding and Analysis. (ed Springer International Publishing; ). [Google Scholar]
- 19.Shen D, Wu G & Suk H-I Deep Learning in Medical Image Analysis. Annual Review of Biomedical Engineering 19, 221–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing F, Xie Y & Yang L An automatic learning-based framework for robust nucleus segmentation. IEEE transactions on medical imaging 35, 550–566 (2016). [DOI] [PubMed] [Google Scholar]
- 21.de Bel T et al. in Medical Imaging 2018: Digital Pathology Vol. 10581 (ed International Society for Optics and Photonics.) 1058112 (2018). [Google Scholar]
- 22.Simon O, Yacoub R, Jain S, Tomaszewski JE & Sarder P Simon Olivier, et al. “Multi-radial LBP Features as a Tool for Rapid Glomerular Detection and Assessment in Whole Slide Histopathology Images. Scientific Reports 8, 2032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng J-Z et al. Computer-Aided Diagnosis with Deep Learning Architecture: Applications to Breast Lesions in US Images and Pulmonary Nodules in CT Scans. Scientific Reports 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruz-Roa A et al. Accurate and reproducible invasive breast cancer detection in whole-slide images: A Deep Learning approach for quantifying tumor extent. Scientific Reports 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sirinukunwattana K et al. Locality sensitive deep learning for detection and classification of nuclei in routine colon cancer histology images. IEEE TRANSACTIONS ON MEDICAL IMAGING 35, 1196–1206 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Ertosun MG & Rubin DL in AMIA Annual Symposium Proceedings. (ed American Medical Informatics Association). [Google Scholar]
- 27.Bulten W, Kaa C. A. H.-v. d., Laak J. v. d. & Litjens GJ Automated segmentation of epithelial tissue in prostatectomy slides using deep learning. International Society for Optics and Photonics 10581, 105810S (2018). [Google Scholar]
- 28.Mishra R, Daescu O, Leavey P, Rakheja D & Sengupta A in International Symposium on Bioinformatics Research and Applications. (ed Springer; ) 12–23. [Google Scholar]
- 29.Anthimopoulos M, Christodoulidis S, Ebner L, Christe A & Mougiakakou S Lung Pattern Classification for Interstitial Lung Diseases Using a Deep Convolutional Neural Network. IEEE TRANSACTIONS ON MEDICAL IMAGING 35, 1207–1216 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Szegedy C, Vanhoucke V, Ioffe S, Shlens J & Wojna Z Rethinking the Inception Architecture for Computer Vision. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, 2818–2826 (2015). [Google Scholar]
- 31.Szegedy C et al. Going Deeper With Convolutions. The IEEE Conference on Computer Vision and Pattern Recognition, 1–9 (2015). [Google Scholar]
- 32.Esteva A et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature 542, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gulshan V et al. Development and Validation of a Deep Learning Algorithm for Detection of Diabetic Retinopathy in Retinal Fundus Photographs. JAMA 316, 2402–2410 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Grossman RL et al. Toward a Shared Vision for Cancer Genomic Data. New England Journal of Medicine 375, 1109–1112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abels E & Pantanowitz L Current State of the Regulatory Trajectory for Whole Slide Imaging Devices in the USA. Journal of Pathology Informatics, 8–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Cespedes M et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Research 62, 3659–3662 (2002). [PubMed] [Google Scholar]
- 37.Shackelford DB et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 23, 143–158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makowski L & Hayes DN Role of LKB1 in lung cancer development. British Journal of Cancer 99, 683–688 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris LG et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nature genetics 45, 253–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mogi A & Kuwano H TP53 Mutations in Nonsmall Cell Lung Cancer. Journal of Biomedicine and Biotechnology 2011, 9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kandoth C et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeiler MD & Fergus R in European Conference on Computer Vision. 818–833. [Google Scholar]
- 43.Maaten L. J. P. v. d. Accelerating t-SNE using Tree-Based Algorithms. Journal of Machine Learning Research 15, 3221–3245 (2014). [Google Scholar]
- 44.Bonner RF et al. Laser Capture Microdissection: Molecular Analysis of Tissue. Science 278, 1481–1483 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Ninomiya H et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas. Lung cancer 63, 235–240 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Warth A et al. EGFR, KRAS, BRAF and ALK Gene alterations in lung adenocarcinomas: patient outcome, interplay with morphology and immunophenotype. European Respiratory Journal erj00180–2013 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Sequist LV et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR InhibitorsGenotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Science translational medicine 3, 75ra26–75ra26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiang S et al. IDH2 Mutations Define a Unique Subtype of Breast Cancer with Altered Nuclear Polarity. Cancer research 76, 7118–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baas AF, Smit L & Clevers H LKB1 tumor suppressor protein: PARtaker in cell polarity. Trends in Cell Biology 14, 312–319 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Gloushankova N, Ossovskaya V, Vasiliev J, Chumakov P & Kopnin B Changes in p53 expression can modify cell shape of ras-transformed fibroblasts and epitheliocytes. Oncogene 15, 2985 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Yoshida A et al. Comprehensive histologic analysis of ALK-rearranged lung carcinomas. The American journal of surgical pathology 35, 1226–1234 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Rodig SJ et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clinical cancer research 15, 5216–5223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dearden S, Stevens J, Wu Y-L & Blowers D Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Annals of Oncology 24, 2371–2376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu J et al. Mutation-specific antibodies for the detection of EGFR mutations in non–small-cell lung cancer. Clinical Cancer Research 15, 3023–3028 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Houang M et al. EGFR mutation specific immunohistochemistry is a useful adjunct which helps to identify false negative mutation testing in lung cancer. Pathology-Journal of the RCPA 46, 501–508 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Dimou A et al. Standardization of Epidermal Growth Factor Receptor (EGFR) Measurement by Quantitative Immunofluorescence and Impact on Antibody-Based Mutation Detection in Non–Small Cell Lung Cancer. The American journal of pathology 179, 580–589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaumberg AJ, Rubin MA & Fuchs TJ H&E-stained Whole Slide Deep Learning Predicts SPOP Mutation State in Prostate Cancer. bioRxiv, 064279 (2016). [Google Scholar]
- 58.Donovan MJ et al. A systems pathology model for predicting overall survival in patients with refractory, advanced non-small-cell lung cancer treated with gefitinib. European Journal of Cancer 45, 1518–1526 (2009). [DOI] [PubMed] [Google Scholar]
Methods-Only References
- 59.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanley JA & McNeil BJ The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982). [DOI] [PubMed] [Google Scholar]
- 62.Pedregosa F et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830 (2011). [Google Scholar]
- 63.Efron B & Tibshirani RJ An introduction to the bootstrap. Vol. 56 (1994). [Google Scholar]
- 64.Cohen J A coefficient of agreement for nominal scales. Educational and Psychological Measurement 20, 37–46 (1960). [Google Scholar]
- 65.McHugh ML Interrater reliability: the kappa statistic. Biochemia medica: Biochemia medica 22, 276–282 (2012). [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data used for training during the current study are available through the Genomic Data Commons portal (https://gdc-portal.nci.nih.gov). These datasets were generated by TCGA Research Network (http://cancergenome.nih.gov/) and they have made them publicly available. Other datasets analyzed during the current study are available from the corresponding author on reasonable request.