

Abstract
DNA and RNA sequencing technologies have revolutionized biology and biomedical sciences, sequencing full genomes and transcriptomes at very high speeds and reasonably low costs. RNA sequencing (RNA-Seq) enables transcript identification and quantification, but once sequencing has concluded researchers can be easily overwhelmed with questions such as how to go from raw data to differential expression (DE), pathway analysis and interpretation. Several pipelines and procedures have been developed to this effect. Even though there is no unique way to perform RNA-Seq analysis, it usually follows these steps: 1) raw reads quality check, 2) alignment of reads to a reference genome, 3) aligned reads’ summarization according to an annotation file, 4) DE analysis and 5) gene set analysis and/or functional enrichment analysis. Each step requires researchers to make decisions, and the wide variety of options and resulting large volumes of data often lead to interpretation challenges. There also seems to be insufficient guidance on how best to obtain relevant information and derive actionable knowledge from transcription experiments. In this paper, we explain RNA-Seq steps in detail and outline differences and similarities of different popular options, as well as advantages and disadvantages. We also discuss non-coding RNA analysis, multi-omics, meta-transcriptomics and the use of artificial intelligence methods complementing the arsenal of tools available to researchers. Lastly, we perform a complete analysis from raw reads to DE and functional enrichment analysis, visually illustrating how results are not absolute truths and how algorithmic decisions can greatly impact results and interpretation.
Keywords: RNA sequencing, differential expression, functional analysis, machine learning, multi-omics
Introduction
RNA sequencing (RNA-Seq) is a technique used to determine the presence and abundance of RNA/transcripts in a biological sample at a specific time, revealing which genes are expressed and more generally what genomic regions are transcribed [1]. RNA-Seq studies, also referred to as massively parallel RNA sequencing or RNA high-throughput sequencing, typically convert RNA into cDNA and subsequently use next generation sequencing (NGS), a cost-effective technology that sequences millions of DNA fragments in parallel, providing high depth sequenced reads at a relatively quick speed [2]. For NGS purposes, high depth implies each fragment is sequenced several times to decrease detection errors, and this resulting increased accuracy has revolutionized fields such as functional genomics, gene expression profiling and personalized medicine [3].
The specific workflows for DNA and RNA sequencing are slightly different (see Supplementary Material S1), and recent methods have notably been developed for direct RNA sequencing, where RNA is not converted into cDNA and there is no reliance on amplification steps [4]. However, the choice of a specific RNA-Seq pipeline also depends on the research objective and may include mRNA sequencing, whole transcriptome sequencing [5], small (non-coding) RNA sequencing including miRNA sequencing [6], targeted RNA sequencing [7], single-cell RNA sequencing [8], ribosome profiling or others [9].
There have been many excellent reviews on RNA-Seq technologies and data analysis [9–12]. However, most reviews stop at differential expression (DE) analysis and do not include functional profiling and pathway analysis, a crucial step for translating biological insights into molecular mechanisms or clinical applications. Moreover, the rapid development of novel tools and technologies calls for updated information. For example, Anders et al. [10] provided a comprehensive RNA-Seq DE analysis protocol with code and parameters. This protocol, however, employed TopHat2 [13], which was then popular but now not as much used for reads mapping. Likewise, with the rapid development of multi-omics technologies and artificial intelligence, RNA-Seq has integrated with these latest technologies, which has not been previously covered.
To address these needs, we explain in detail the common steps followed in current RNA-Seq and outline differences and similarities of different popular options, including traditional and recent approaches utilizing machine and deep learning. We additionally perform a complete analysis from raw reads to DE and functional enrichment analysis, visually illustrating how different methods can lead to different results and conclusions, emphasizing the need to both conduct comprehensive comparative analyses and justify specific study choices.
RNA-Seq steps
NGS-based RNA-Seq analysis usually follows five steps.
Step 1: Read alignment
The first step in any RNA-Seq analysis pipeline is to align the reads to a reference genome (see Supplementary S2 for read format details). Alignment is the process of matching reads to specific regions of the genome or transcriptome, and a read will be considered ‘aligned’ if the algorithm could find its position on the reference genome or it will be ‘unaligned’ otherwise. The percentage of successfully and uniquely aligned genes will serve as a quality measure of both the reads and the alignment algorithm [14]. In ideal circumstances, all reads will be aligned to one and just one position of the reference genome. However, sometimes reads cannot be uniquely aligned due to repetitive sequences within shared domains of paralogous genes [11], and the aligner algorithm can sometimes simply fail to find matching positions for some reads.
Generally, the alignment step is computationally expensive, requiring major CPU usage and temporary disk storage [15]. Popular alignment tools such as BLAST [16] and BLAT [17] were developed for relatively small datasets, and so they are unsuited to align large NGS data. As a consequence, new methods were designed for this purpose, such as Bowtie [18], Subread [19] and STAR [20]. Interested readers can find detailed descriptions in Supplementary S3, and a comprehensive performance evaluation of the most common alignment algorithms in [21].
A separate hurdle and consideration is that RNA reads not only originate from exons, but many actually originate from introns or exon–intron junctions [22]. Transcripts originating from introns can be similar in number to those coming from exons, and it has been noted that large amounts of intronic sequences are not explained by the fact that intronic regions are larger [23]. In turn, when a read overlaps an intron–exon boundary, parts of the read will be mapped to non-contiguous sites in the reference genome, and so the mapping procedure may fail to successfully assign this read. This also means splice junctions, namely sites of former introns in mature mRNA, can be rather problematic to align, and so it is preferable to detect them ab initio [24].
Finally, the raw reads will be aligned to the reference genome as a sequence alignment/map (SAM) file or a binary alignment/map (BAM) file, and the next step in the pipeline is to know which genes or exons they were matched to. It is important to note that read alignments without a reference genome may be performed, but such algorithms usually underperform when compared to reference-guided methods [25]. There could be instances where alignment-free techniques would be advantageous, however, such as for long RNAs, but these algorithms typically fail to accurately quantify expression in low-expressed genes and small RNAs [26, 27].
Step 2: Reads summarization
Once reads have been aligned to a reference genome or transcriptome, the next step is to map those reads to known genes, exons or transcripts (annotation) and quantify them in terms of a count matrix. The process of counting mapped reads is summarization [28], and there are various computational tools to do this such as TopHat [22] and featureCounts [28]. Depending on the biological sample, one will have to choose which features will be used to summarize the aligned reads, where features refer to continuous biological sequences (genes, exons or transcripts). Further, for the summarization step, one needs an annotated reference genome or annotation file of a reference genome to link to, and then count the number of reads that were aligned to each feature in the annotated genome. Typically, a significant number of reads will not be mapped into any known features, even in the case of well annotated organisms like a human or mouse [14], because those reads map to genomic regions outside annotated genes or exons.
The four most common annotation databases are currently RefSeq, UCSC, Ensembl and GENCODE (see Supplementary S4). Generally, it is good practice to choose one annotation source and keep its notation until the end of the analysis. We note that it has also been proposed to utilize a less complex gene annotation, such as RefSeq, when conducting an experiment that focuses on reproducible and robust gene expression estimates, and using more complex genome annotation, such as Ensembl, when conducting exploratory research [29].
There are subtleties that make the summarization a little more complicated than intuited. Firstly, summarization programs must work with both DNA and RNA sequences. These programs are required to work with both single and pair-ended reads. Moreover, the summarization algorithm must accommodate splice variants. Lastly, indels in aligned fragments can significantly increase the computational cost of the read counting step, especially when the number of features is large.
Altogether, there are two common read counting approaches for RNA-Seq data: 1) reads that match annotated exons are counted, and 2) counting at the gene level. The first approach attempts to match reads to exons and count the number of matches for each exon, typically used to test splice variants between groups, while the second counts all reads that align to any exon inside each gene, thus counting reads instead of exons and requires a gene annotation file. Moreover, as previously mentioned, one of the main problems when summarizing RNA-Seq reads is alternative splicing, meaning single genes can express different transcript isoforms, and reads and subsequent counts do not distinguish between them [28]. Therefore, programs and algorithms to specifically summarize RNA-Seq reads are needed. Two popular tools for counting aligned reads are featureCounts [28] and HTSeq-count [30] (see Supplementary S5 for details). The output after summarization is a count matrix indicating the number of aligned reads to each feature in each sample of the experiment and will be the input data for DE analysis.
Step 3: Differential expression analysis
In this step, one needs to define a statistic to measure gene expression levels. The default choice are counts, which entails the number of reads of each feature (i.e. transcript in the context of an RNA-Seq analysis). Counts are the raw measurement of transcript abundance, and other measures such as reads per kilobase million (RPKMs), fragments per kilobase million (FPKMs), counts per million reads mapped (CPM) and transcripts per kilobase million (TPM) normalize according to gene length and/or millions of base pairs.
The goal of DE analysis is to identify genes whose patterns of expression significantly differ across phenotypes or conditions of interest. One simple way to proceed would be to select genes with the highest log-fold difference in expression level when comparing phenotypes or conditions; however, this would not account for biological variation, which is different from gene to gene. Common statistical approaches use parametric tests such as a t-test, or non-parametric tests like the Mann–Whitney test. These tests indeed perform reasonably well when analyzing microarray data, but they perform very poorly with RNA-Seq data [31–33]. There are several reasons for this, for instance: the t-test assumes a continuous distribution, appropriate for microarray data where the raw data are fluorescence intensities on a continuous scale [34], but RNA-Seq counts are intrinsically discrete; and RNA-Seq techniques test gene by gene, without using the information from other genes, thus neglecting variability. Due to these challenges, new methods were specially created to analyze DE in RNA-Seq data. Two of the most popular ones are DESeq2 and edgeR (see Supplementary S6 for details).
The main output of both methods is a table with log-fold changes for each gene in the list (usually base 2 logarithm), a test statistic (Wald statistic for DESeq2, different options for edgeR), and the corresponding P-value. Genes with low P-values will be considered differentially expressed, users typically define a pre-fixed cutoff, and the results below such threshold are considered statistically significant. Individual tests for each gene is straightforward, easy to understand and to a certain extent quite popular, but it has serious drawbacks: first, multiple testing will produce a large number of false discoveries. This can be resolved by using multiple test control techniques to adjust for the false discovery rate (FDR), at the expense of losing sensibility, namely a lower probability of finding significant results [35]. Second, raw data in RNA-Seq are feature counts, namely the number of reads aligned to each genetic feature. A longer genetic feature, for instance a very long gene, will have more aligned reads and a higher number of counts by virtue of being long, not because it has a higher level of expression. Therefore, count variance and gene length will be inversely proportional, and the power of the test and the probability of detecting true DE is a function of gene length [36]. Normalizing raw counts to gene lengths, using measures as RPKM and FPKM, addresses this bias. However, the most serious drawback is that the most differentially expressed genes (DEGs), meaning those with the lowest P-values or highest log-fold changes, may not necessarily be most relevant or explanatory for the phenotypes or conditions of interest in a study. To address this key issue, prior knowledge of the phenomenon of interest is essential, and any such knowledge must be accounted for during analysis. Finally, alternative splicing allows a gene to encode multiple proteins, called isoforms, in eukaryotes. This occurs when exons are joined in different combinations due to inclusion or exclusion of exons, which may affect quantification of gene expression levels during RNA-Seq analysis [37] (see Supplementary S6).
Step 4: Gene set analysis
Identifying DEGs may not on its own yield meaningful biological interpretations. Genes do not act in isolation, gene expression is a very complex coordinated process, and expression levels in different genes can depend on each other [38]. To account for this, one must cluster genes according to functionality, similarity, biological relationships or other relevant classifications. Likewise, one would want to test DE in these groups of genes, usually called gene sets, instead of individual genes. Such a large-scale analysis is typically called gene set analysis (GSA), not to be confused with gene set enrichment analysis (GSEA), which is a particular technique for GSA and we will later cover in detail.
Gene sets are groupings of genes deemed related in some relevant manner, such as simultaneous participation in a signaling pathway, related or dependent expression patterns, similar biological process, etc. [39]. A collection of gene sets can be subsequently formed by grouping based on relationships such as physical proximity, chemical interactions, their contribution to a certain phenotype or medical condition, or else. Therefore, there are many possibilities for constructing gene sets and hierarchies, and the appropriate choice will depend on the study question. Two popular examples are the Gene Ontology (GO) and the KEGG pathways (see Supplementary S7 for details).
Over-representation analysis
Over-representation analysis consists of a hypergeometric test, defined to investigate the over-representation of different GO sets or pathways in a group of DEGs. This test is separated into two analyses, up and down regulated DEGs, and a gene set or pathway will be considered significantly over-represented if the chances of observing at least as many of them in the DE set is too low, assuming random sampling.
For over-representation analysis, one needs to first define a threshold to filter out all genes that are not DE (see an example in Supplementary S8). Other than such threshold being rather arbitrary, it is important to keep in mind that in an RNA-Seq experiment the messenger RNA (mRNA) and non-coding RNA (ncRNA) molecules are cut into millions of small pieces, each one producing a read when successfully aligned and mapped to a feature. Assuming all reads have similar length, longer genes will have more counts than shorter ones, simply because the number of reads is the number of fragments successfully aligned to each genetic feature. With counts being the statistic used to perform DE analysis, the probability of detecting real expression differences is higher in longer genes than in shorter ones, leading to the bias towards longer genes previously discussed. This is one example showing how a statistical procedure originally developed for microarray data is not well suited for RNA-Seq data but is still commonly used. Method adaptations to address this have been proposed, with one of the simplest approaches being weighting the t-statistic in DE analysis by gene length to try to correct for it [40]. GOseq [41] quantifies the likelihood of DE as a function of gene or transcript length and subsequently incorporating this function into the statistical test of each feature’s significance. Yet another method is SeqGSA [42], where gene test statistics are re-standardized according to a special value called the maxmean statistic using a randomization process. In this approach, gene length is considered by weighting each gene’s contribution by its length during the randomized re-standardization process.
Lastly, one of the drawbacks of over-representation analysis is the need to introduce a somewhat artificial cutoff to filter out genes that are not considered DE. One popular method that does not require such a threshold but rather focuses on a summary statistic for all gene expression levels is GSEA.
Gene set enrichment analysis
GSEA is a computational method that determines whether an a priori defined set of genes shows statistically significant consistent differences between two study groups, biological states or phenotypes. It is different to over-representation analysis and a technically more complex manner to investigate differences in the expression patterns of gene sets and pathways [43]. The main goal of GSEA is to create a ranked list of genes according to DE levels and then test for overrepresentation of different gene sets at the top or at the bottom of that ranked gene list. The general idea is that a gene set overrepresented at the top of the ranked list will have a certain degree of up-regulation, while a gene set overrepresented at the bottom of the list will exhibit down-regulation. More specifically, up-regulated at the positive position can be said to happen when most, if not all, of the genes of a given set appear at the beginning of the ranked list, and up-regulated at the negative position when most of those appear at the end of the list. Note that, although GSEA does not support gene sets with up- and down- regulated genes, there are approaches to perform this type of analysis. See for instance [44].
In both versions, pre-ranked and not, GSEA will produce a normalized enrichment score (NES) for every gene set or pathway in the collection. Upon FDR specification, GSEA will output a list of gene sets with positive enrichment scores, meaning over-represented at the beginning of the ranked list, and a list of gene sets with negative enrichment scores, which are correspondingly over-represented at the end of the ranked list. In this case, however, a positive enrichment score indicates a correlation between the gene set and the first phenotype, while a negative enrichment score represents correlation with the second phenotype. We refer readers to Supplementary S9 for details and examples.
Lastly, the GSEA algorithm has been adapted and extended to specifically work with RNA-Seq data [45, 46]. Other additional methodologies have been developed to use it alongside linear regression models [47].
Step 5: Functional enrichment analysis
DAVID functional classification tool
One possible drawback of enrichment analyses such as over-representation and GSEA is that gene sets or pathways may not capture complex relationships between genes and so-called terms, namely elements of gene ontologies that represent gene product properties. Also, relationships between genes and annotated terms are not one-to-one: one term is associated with many different genes, and each gene is included in many different gene sets and pathways. Accordingly, it is assumed genes will have many annotations in common if there is a subset of genes with similar functionalities in a list, namely that they are together in various gene sets and pathways. Conversely, one could say gene sets and pathways have features in common, since they share many common genes. Overall, there is a complex ‘many-to-many-genes-to-terms’ relationship between genes, gene sets and pathways, and GSA methods can fail to capture it in full.
The Database for Annotation, Visualization and Integrated Discovery (DAVID) provides an algorithm to identify overlaps between user-suppled list of genes and curated databases, condensing the list of genes or associated biological terms into organized classes called biological modules (see Supplementary S10 for details). It also catalogs groups of genes sharing common biology and groups of gene sets and pathways sharing common genes [48–50]. This method replaces gene sets and pathways for more complex, broader biological modules that reduce redundant results, aiming for easier understanding while still providing meaningful information about complex biological networks. DAVID uses Fisher’s exact test [49, 50] to processes information in a manner similar to other tools such as GOToolBox and ingenuity pathway analysis (IPA) [51–54]. One key is its clustering algorithm, which allows genes and terms to simultaneously be in different clusters, unlike traditional techniques such as hierarchical or K-means clustering. Nevertheless, the selection of a cutoff for significance values remains arbitrary and, like other methods, modifications will alter enrichment results.
Ingenuity analysis
IPA is a commercial bioinformatics software that allows canonical pathway analysis of RNA-Seq data against a manually curated pathway database [51]. Similar to DAVID, IPA identifies the over-represented pathways by using Fisher’s exact test to measure any significant overlaps of a user-provided gene list and pre-defined gene sets. Additionally, IPA uses the z-score to assess the consistency between the observed gene expression pattern and the expected gene sets [51]. Compared with KEGG, which consisted of 551 pathways at the time this article was written, the number of available pathways in IPA was 734. We also note that although some pathways can be found in both databases, their definition in terms of gene contents can be largely different and contains only a small portion of overlapped genes [55]. It is thus expected nonidentical enriched pathways will be obtained, since different pathway databases (e.g. hallmark gene sets, KEGG and IPA) and/or over-representation analyses are applied. Important biological signatures can still be captured using these different yet well-curated databases and sophisticated approaches, however. For example, researchers demonstrated that IL-2 signaling plays a crucial role in regulating Treg cell homeostasis and function, which was consistent with their GSEA analysis, showing that IL-2-STAT5 signaling was the most enriched hallmark pathway [56]. Using the same RNA-Seq data, IPA also identified IL-2 as one of the most important upstream regulators, albeit IL-2 signaling pathway was only ranked 52 (−log(P-value) = 5.67) out of the 299 statistically significant canonical pathways (P-value < 0.05) (see Supplementary Data – Enriched Pathways).
Sequencing and analysis of non-coding RNAs
Recent advances in high-throughput sequencing technology have provided an impressive platform for profiling ncRNAs in the transcriptome. This has led to the development of a plethora of ncRNA sub-class specific computational tools and databases for their identification and characterization. These techniques and tools have helped uncover the significance of ncRNAs in various physiological mechanisms and regulations during disease pathogenesis.
Long non-coding RNAs
Long non-coding RNAs (lncRNAs) are defined as ncRNAs longer than 200 nucleotides [57]. The functions of lncRNAs include modulating gene expression [58–60] and regulating chromosomal as well as epigenetic modifications [61]. LncRNAs are profiled by sequencing transcriptomes using polyA-selected or stranded ribosomal RNA (rRNA)-depleted libraries from total RNA samples with high sequencing depth (≥30 million) [62–65].
Throughout the years, several machine-learning tools [66], utilizing gold-standard datasets from GENCODE and RefSeq as training sets, have been developed for the characterization of lncRNAs [67] using classifiers including support vector machine (SVM), logistic regression, Random Forest and Deep-Learning. These algorithms have employed both alignment-based features (e.g. sequence conservation, phylogenetic analysis) and alignment-free features (e.g. physiochemical properties, Open reading frame-related and secondary structure related) [66]. Two notable lncRNA detection tools include lncRScan-SVM [68], which uses SVM to distinguish lncRNAs from mRNAs, and LncFinder [69], which accurately identifies lncRNAs by employing 5 different classifiers. Specific tools like iSeeRNA [70] and linc-SF [71] were also developed for the identification of long intergenic non-coding RNAs (lincRNAs).
Increased interest in lncRNA research and innovations allowing their accurate characterization have helped identify thousands of lncRNAs. This has led to the development of several lncRNA databases. Comprehensive databases like LNCipedia [72] and LNCBook [73] have been created using experimentally verified lncRNAs from several pre-existing databases. There are also specialized databases (LncRNADisease 2.0 [74], Lnc2Cancer 3.0 [75], etc.) and predictors (NNLDA [76], IDLDA [77], gGATLDA [78], etc.) available that have been curated for lncRNAs and disease association.
Circular RNAs
Circular RNAs (circRNAs) are a sub-class of RNAs that are usually grouped under lncRNAs. CircRNAs are characterized as covalently closed and non-polyadenylated circular transcripts that are formed by either exon or protein driven back-splicing mechanisms [79–83]. The lack of 5′ and 3′ terminals in circRNAs promotes their stability in comparison to linear RNAs and makes them more resistant to exonuclease degradation [84]. Hence, circRNA sequencing usually involves the extra step of treating the samples with RNase R for circRNA enrichment by degrading the linear RNAs in the sample, in addition to the rRNA depleted library preparation protocol, but with high sequencing depth, circRNAs can also be identified in samples without RNase R treatment [85, 86]. CircRNAs can also be sequenced utilizing Nanopore long-read sequencing with a modified RNA-Seq sample preparation protocol with rRNA depletion and additional polyA tailing prior to RNase R treatment [87].
With the rediscovery of circRNAs and its several key functions including regulation of miRNA expressions [88–90], interaction with RNA-binding proteins [91, 92], transcription regulation [93] and more, tools were developed for identification, characterization and molecular network interaction analysis of circRNAs. In its initial phase, most circRNA detection software (CIRCExplorer2 [94], CIRI2 [95], KNIFE [96], find_circ [84]) employed alignment-based approaches. The latter was achieved by splitting unmapped junctions/back-spliced junctions (BSJ) and aligning them to the reference genome in reverse, by local alignment, or by constructing a pseudo-reference of potential BSJs for read alignment [97]. In addition, specific tools like CIRIquant [98] and CirComPara [99] offer a comprehensive pipeline for the quantification of circRNAs incorporating above-mentioned identification tools. With the construction of robust circRNA databases (circBase [100], CIRCpedia [94], CircRNADb [101] and circFunBase [102]), recently several machine-learning (PredcircRNA [103], PredicircRNATool [104]) and deep-learning algorithms (DeepCirCode [105], JEDI [106]) were also introduced. Using sequence- and structure-specific features, these tools predict back-splicing events and thus circRNA.
Specialized tools and databases are available for the downstream functions and interaction networks of circRNAs. For instance, CIRCInteractome [107] provides information on potential miRNA targets and RNA-binding proteins of any given circRNAs. Databases like Circ2Disease [108], circRNADisease [109] and Circ2Traits [110] contain experimentally validated information on disease associated circRNAs. CIRI-AS [104] performs alternative splicing analysis for circRNAs.
MicroRNAs
MicroRNAs (miRNAs) are small ncRNAs that are ~20–23 nt long [111] and play crucial regulatory roles in gene silencing [112, 113] and translation repression [112, 114, 115]. Small RNA sequencing technology has advanced significantly in the recent years, gradually gaining traction as a preferred approach for profiling miRNAs and other small ncRNAs (See Supplementary S11). The standard workflow of small RNA-Seq library construction includes reverse transcription of small RNA, which has been isolated using size exclusion gel or size selection magnetic beads, to cDNA, extension of the cDNA fragments by ligation of two adaptors or polyadenylation, and PCR amplification [116] followed by sequencing (depth ≥ 5 million reads) [117]. In efforts to reduce possible PCR amplification biases during sequencing, some small RNA library preparation kits include unique molecular identifiers (UMIs), which enables the distinction of molecules that have been amplified [118].
Many computational tools are available for the detection and characterization of miRNAs. Currently, among over 60 miRNA databases, miRBase [119] is widely used for miRNA read alignment. Also, several web-based and locally available algorithms have been developed for the identification of known miRNAs as well as novel miRNAs (miRMaster 2.0 [120], CAP-miRSeq [121], mirTools2.0 [122], miRNAkey [123], etc.) and isomiRs (isomiR2Function [124], miRge [125], etc.), which are miRNA variations with respect to a reference sequence. All the software and databases related to miRNAs have been listed on a web-based platform, Tools4miR [126]. Among these, miRDeep2 [127] has been most popular algorithm for miRNA identification. miRDeep2 is a deep-sequencing tool based on the miRNA biogenesis that uses RNA-fold [128] to predict the secondary structures and characteristics and determine potential miRNAs.
Recent developments
Metatranscriptomics
The microbiome, which is defined as ‘a characteristic microbial community occupying a reasonable well-defined habitat which has distinct physio-chemical properties’ [142], has drawn great attention over the last decade. Because microbial metabolism can significantly contribute to host health or fluctuations in natural complex ecosystems [143,144], it is crucial to identify the involved microbes and understand how they act and respond under specific conditions. Metatranscriptomics can help this goal by capturing the transcripts and active genes of a whole microbial community, in contrast to transcriptome of a single type of cell/organism with traditional RNA-Seq. Here we focus on the metatranscriptomics bioinformatics workflow, whereas the sequencing workflow can be found in Supplementary S12.
The metatranscriptomics bioinformatics workflow includes quality control of raw reads, assembly-based or read-based analysis, taxonomic and functional annotation, and DE analysis. The first step is to clip sequence adapters, trim low-quality bases, and remove undesired reads (e.g. short fragments after trimming, host mRNA or rRNA due to incomplete removal prior to sequencing). Commonly used QC tools for NGS data include FastQC [145] and Trimmomatic [146]. For rRNA removal after sequencing, SortMeRNA [147] and barrnap [148] can be used. The retained high-quality reads can be subsequently analyzed through either assembly-based or read-based approaches. The former requires more computational resources and only reconstructs transcripts with enough coverage, yet it is suitable for discovering novel expressed genes or when the reference genomes are unavailable or inadequate. Further, there are few assemblers specifically developed for metatranscriptomic data (e.g. IDBA-MT and IDBA-MTP), although some designed for metagenomics or traditional RNA-Seq, such as MEGAHIT, SPAdes, metaSPAdes and Trinity, are still used in metatranscriptomics research [149–154]. The read-based approach, on the other hand, is relatively computationally inexpensive and more sensitive to lowly expressed genes, but largely depends on the accuracy and adequacy of reference databases. Sequence aligners, such as BWA, Bowtie2, DIAMOND and SOAP2, are commonly used to search against reference databases [155–158]. The contigs or mapped reads can thereafter be annotated against databases (e.g. GO, KEGG, Uniprot, COG, Pfam, etc.) for functional profiling. They can also be mapped to the reference databases or, if available, the recovered genomes from paired metagenomics analysis, to understand and quantify microbial transcripts among all transcripts. DE analysis for metatranscriptomics is still in its early stage of development, so methods for traditional RNA-Seq, such as DESeq2 and edgeR, remain used in metatranscriptomics research [159–161]. However, because transcript abundance in a microbial community varies strongly due to gene-copy variation, spurious DE signals can be introduced if this is not accounted for [162]. To that effect, several models have been proposed for metatranscriptomic data normalization, including RNA-level within-taxon total-sum-scaling and, when paired metagenomic data are available, scaling by DNA-level estimated taxon abundance [162,163].
Several packages have integrated most, if not all, of the steps for metatranscriptomic data analysis. Packages incorporating assembly-based approach are SqueezeMeta, IMP, MUFFIN, DiTing, CoMW and MetaPro [164–169], and those applying read-based approach are SAMSA2, MetaTrans, HUMAnN3, COMAN, FMAP and ASaiM-MT [170–175]. For long-read assembly, Canu was wrapped in SqueezeMeta for both PacBio and Oxford Nanopore data processing [176]. Lastly, it is worth noting there is currently no consensus on the best practice for metatranscriptomic data analysis, and the corresponding statistic methods and tools are continuously developing.
Machine learning approaches
In RNA-Seq data, the number of features is generally significantly larger than the number of samples (i.e. ‘large p, small N’ problem). Traditional statistical methods rely on stringent and often hard-to-verify assumptions, making it difficult to control false-positives when dealing with high-dimensional low-sample-size data [186]. In the context of RNA-Seq data, this high-dimensionality will produce many spurious or undetected genes when performing DE analysis with traditional statistical methods, even if the actual false-positive and false-negatives are low. To address this, methods banking on machine learning (ML) and deep learning (DL) approaches have been proposed. A brief introduction to ML/DL can be found in Supplementary S13 and the applications of ML/DL in transcriptomics/metatranscriptomics are covered in the following paragraphs and Table 1.
Table 1.
Applications of ML/DL in (meta-)transcriptomics
| Method | Application | Source code | References |
|---|---|---|---|
| GA/kNN | Identification of differentially expressed genes | https://www.niehs.nih.gov/research/resources/software/biostatistics/gaknn | [129] |
| GA/kNN, gradient boosting |
Identification of differentially expressed genes | NA | [130] |
| E-M algorithm | De novo assembly of meta-transcriptomics data | https://sourceforge.net/projects/dnpipe | [131] |
| Logistic regression w/ L2 regularization | Identification of predictive microbial taxa and KOs from meta-transcriptomics | NA | [132] |
| Random forest, gradient boosting | Construction of predictive models from meta-transcriptomics | https://github.com/armbrustlab/trophic-mode-ml | [133] |
| CNN/Grad-Cam | Identification of marker genes and classification of cancer types | NA | [134] |
| CNN/Grad-Cam | Identification of marker genes and classification of oral cancer types | NA | [135] |
| CNN/saliency maps | Identification of marker genes and classification of cancer types | https://github.com/chenlabgccri/CancerTypePrediction | [136] |
| Deep NN | Alternative splicing analysis | https://github.com/Xinglab/DARTS | [137] |
| CNN and DeepLIFT | Regulatory mechanisms identification | https://github.com/stasaki/DEcode | [138] |
| Mixing observation | Data augmentation | NA | [139] |
| Autoencoder | Cell content inference from bulk RNA-Seq data (which is typically done w/ scRNA-Seq data) | https://github.com/xindd/DCNet | [140] |
| ICA | Identification of novel regulons | https://github.com/avsastry/modulome-workflow | [141] |
CNN: Convolutional neural network; E-M: expectation–maximization; GA: genetic algorithm; ICA: independent component analysis; kNN: k nearest neighbors; NN: neural network.
One key task in RNA-Seq analysis is to identify DEGs. The underlying assumption is here that if genes are differentially expressed, they might contribute to the trait/phenotype/disease being studied. ML, however, instead of making such an assumption, simply finds a combination of features (transcripts) that can largely discriminate two groups. For example, the genetic algorithm k-nearest neighbors method (GA/kNN) has long been proposed to select a subset of discriminative genes (usually also DEGs) from RNA-Seq data between two groups (e.g. control versus experimental group) [129]. This method has been used to identify discriminative genes from RNA-Seq data of 31 tumor types, showing greater than 90% accuracy for classification task [130]. Interestingly, using a different ML approach (gradient boosting machines), the authors observed a comparable classification performance, yet the gene set selected by the two methods slightly overlapped. This highlights the need for experimental confirmation of gene sets identified as biologically relevant, when ML techniques are applied to discover DEGs.
Leveraging the power of ML in metatranscriptomics is still an under-researched topic, but it has shown to improve the quality of de novo assembly of metatranscriptomic data by learning the abundance information of transcript contigs [131]. Another application, in parallel with traditional RNA-Seq, is to identify informative microbial taxa and KOs from metatranscriptomic data [132]. Yet another desirable application is to directly build a predictive model based on metatranscriptomic data. As one example, a random forest-based feature selection method was used to obtain a subset of expressed genes from protist metatranscriptomes to predict the in situ trophic status of marine protists [133].
Beyond traditional ML approaches, DL has also been used for DEGs discovery. For example, researchers [134–136] applied convolutional neural network (CNN) to identify key transcripts from RNA-Seq data of different phenotypes. The RNA-Seq data were normalized and embedded into two-dimensional images, followed by representation learning using a CNN model. Finally, a heatmap based on Guided-Grad CAM [187] was generated to visualize and extract the important transcripts from the model output. Additional applications of ML/DL, beyond DEGs discovery, include differential alternative splicing inference [137], regulatory mechanisms identification [138] and RNA-Seq data augmentation [139]. Other DL applications include the identification of cell landscapes based on RNA-Seq data (rather than scRNA-Seq) using autoencoder models [140]. Using unsupervised learning methods, one can build a transcriptional regulatory network to identify key regulons and elucidate the function of undercharacterized regulons [141,188].
Though being powerful and promising methods, ML and DL are no panacea. In some cases, classical statistical approaches may still be useful. For example, ML approaches including RF and SVM–RFE have been used to select relevant genes for GSA with quantitative trait loci, but the performance of the selected gene sets based on biologically relevant criteria did not outperform those obtained using simple univariate gene selection methods [189]. Aside, one criticism of using ML/DL in biological systems is the resulting inscrutable black box model, which, even if highly accurate, cannot facilitate our understanding of biological processes or molecular mechanisms. Methods have been proposed to explain the prediction/classification from ML/DL models, such as TreeExplainer [190] and class activation maps [191], and have recently been applied to interpret the performance of black-box ML models for tissue types’ prediction from RNA-seq data [192]. However, deciphering underlying biological principles could highly benefit from constructing inherently interpretable ML models [193]. Overall, more sophisticated algorithms and methods have yet to be developed for extracting meaningful information from high-dimensional expression data, DE analysis and GSA, as well as integrating with other ‘omics’ data.
Multi-omics integration
Recent advances in high-throughput technologies enable us to study complex biological systems through various ‘omics’ methods beyond RNA-Seq, including genomics, proteomics, and metabolomics, to name a few. Integrating omics data can provide novel insights, broadening our understanding of complex molecular mechanisms. For instance, researchers have combined transcriptomic, epigenomic and proteomic data to identify potential epigenetic drivers involved in Alzheimer’s disease [194].
A typical multi-omics workflow includes data collection, data pre-processing, data integration and data analysis. For data collection, researchers can generate different types of omics data from the same samples, and then analyze by individual omics and collectively as multi-omics data. Alternatively, the data set can be integrated with multi-omics databases to study single-nucleotide polymorphisms (SNPs), insertions and deletions (indels), structure variations (SVs) and other molecular interactions and mechanisms. There are publicly available multi-omics databases for various research topics including cancer [195], cancer driver genes [196], tissues [197], aging biology [198], maize [199,200], microbiome [201], and early embryos [202], among many others. Typically, each omics data set can be raw counts or abundances matrices containing sample IDs and feature identifiers (e.g. transcripts, proteins or metabolites) as the first row or column. Data pre-processing may include data filtering, quality checking, normalization, missing value imputation and batch effects correction. These steps help reduce false discoveries and spurious results. Tools incorporating pre-processing step as part of their workflow include miodin [183], BIOMEX [182], OmicsAnalyst [181], Mergeomics 2.0 [180] and TIMEOR [179]. After the pre-processing step, integrative analysis is usually performed to systemically discover underlying, complex biological mechanisms. Depending on the principle applied, integrative methods can be categorized into multivariate, similarity, correlation, network, fusion and Bayesian approaches [203]. The widely used R package mixOmics [204] provides a number of multivariate methods for multi-omics data integration, including principal component analysis (PCA), canonical correlation analysis (CCA), partial least squares (PLS), MINT [205] and DIABLO [206]. Besides data integration, network analysis can be used to reveal the association, interaction, and even causation between pairs of omics features. One common and simple approach is correlation network analysis, which calculates the association between biological features in a pairwise manner. Another popular network analysis approach is based on dimension reduction techniques, such as PCA and PLS, to compress the information into a limited number of feature combinations. Web-based platforms such as 3Omics [184] and OmicsAnalyst [181] offer correlation network analysis, whereas xMWAS [177] and PaintOmics 3 [178] provide dimension reduction-based network analysis. Recently, a novel tool TIMEOR [179] was developed to uncover the causal regulatory mechanism networks by analyzing time-series multi-omics data. For gene set or pathway enrichment analysis for multi-omics data, the R package multiGSEA [185] and many other aforementioned platforms can complement the need of such analysis. Table 2 summarizes the functionality of a non-exhaustive list of workflows for different types of multi-omics data.
Table 2.
Examples of publicly available workflows for multi-omics data integration, analysis, and/or visualization
| xMWAS | PaintOmics 3 | TIMEOR | Mergeomics 2 | OmicsAnalyst | BIOMEX | miodin | 3Omics | multiGSEA | |
|---|---|---|---|---|---|---|---|---|---|
| Implementation | R, online | Online | Online, command line | Online | Online | R | R | Online | R |
| Functionality | |||||||||
| Pre-processing | x | x | x | x | x | ||||
| Data integration | x | x | x | x | x | x | |||
| Network analysis | x | x | x | x | x | x | |||
| Enrichment analysis | x | x | x | x | x | x | x | ||
| Pathway analysis | x | x | x | x | x | x | |||
| Time series analysis | x | x | |||||||
| Visualization | x | x | x | x | x | x | x | x | |
| Accepted-omics data (besides transcriptomics) | |||||||||
| Metabolomics | x | x | x | x | x | x | x | ||
| Proteomics | x | x | x | x | x | x | x | x | |
| Genomics | x | x | x | x | |||||
| Epigenomics | x | x | |||||||
| Region-based omicsa | x | x | |||||||
| Regulatory omicsb | x | x | x | x | |||||
| Reference | [177] | [178] | [179] | [180] | [181] | [182] | [183] | [184] | [185] |
aChIP-seq, ATAC-seq or Methyl-seq.
bmiRNAs or other transcription factors.
Lastly, ML techniques have also been applied to multi-omics integration, to gain insights into molecular mechanisms. For example, with the combination of multi-omics data and ML-based network models, researchers identified chemical compounds modes of action, which cannot be found using single-omics data [207]. The integration of RNA-Seq data and information on copy number alterations with the application of unsupervised ML algorithms (autoencoder and K-means clustering) can also generate new features, and this has been illustrated by research associated with ultra-high-risk neuroblastoma [208]. Overall, the application of ML on multi-omics analysis is still in its inchoate stage and we refer interested readers to [209] for a comprehensive review on this topic.
Results
To demonstrate the typical RNA-Seq analysis process and evaluate the effect of reference genome databases, DE methods, and enrichment analysis methods, we re-analyzed murine data previously published in the literature [56]. This dataset contains three replicates from Foxp3+CD4+ T regulatory (Treg) cells and T follicular regulatory (TFR) cells, respectively. The reads quality was checked with FastQC prior to downstream analysis. The overall flowchart of the RNA-Seq analysis is presented in Figure 1 and additional detailed steps can be found in Supplementary S14.
Figure 1.
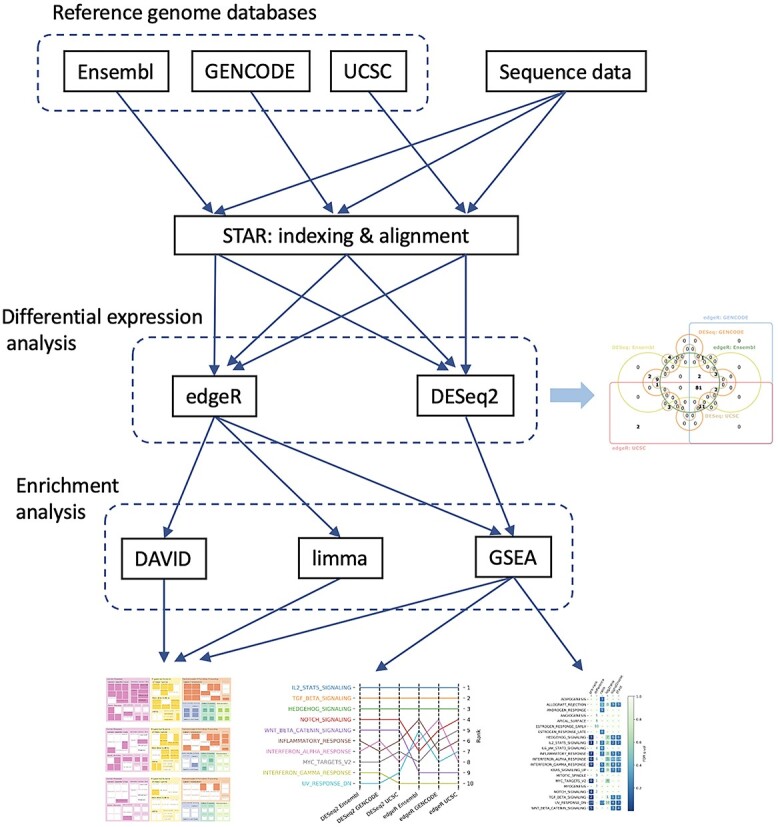
Flowchart describing the major steps of bioinformatics analyses in this study.
Evaluation
The DEGs can be selected based on a somewhat arbitrary cutoff for the P-value, adjusted P-value, or log-fold change, or even selected from the list of the top ranked genes. To assess different choices, we first considered the genes with an adjusted P-value < 0.05. As shown in Figure 2a, a total of 6247, 6268 and 6202 DEGs were identified by edgeR using Ensembl, GENCODE and UCSC database, respectively; likewise, 6255, 6289 and 6286 DEGs were identified by DESeq2, and 5612 genes were identified regardless of databases and DE methods. When the significant DEGs generated by different methods with the same reference genome were compared, approximately 5% of genes identified by edgeR (4.32–5.46%) did not show statistical significance when DESeq2 was used, and vice versa (5.56–5.72%). When the gene lists generated by the same DE method with different databases were compared, a total of 6572 and 6626 distinct DEGs were identified by edgeR and DESeq2, respectively, with 5879 and 5924 of which were common among databases. In addition, the results from Ensembl and GENCODE were more consistent than that of UCSC, as the former two gene lists shared additional 368 and 328 genes using edgeR and DESeq2, respectively. Next, instead of setting a cutoff for DEGs, we selected the top 100 ranked DEGs based on their P-values (Figure 2b). A total of 121 distinct genes were included and 82 of them were ranked among the top 100 DEGs regardless of databases and DE methods. There were 11 and 9 genes exclusively presented in the top ranked lists using edgeR and DESeq2, respectively. Using the latest mm39 reference genome from GENCODE with DESeq2, 6353 genes passed the cutoff of p-value of 0.05, of which 6261 genes were also identified in mm10 reference genome (data not shown), suggesting that the impact of different versions of reference genome database on DEGs discovery might be minor.
Figure 2.
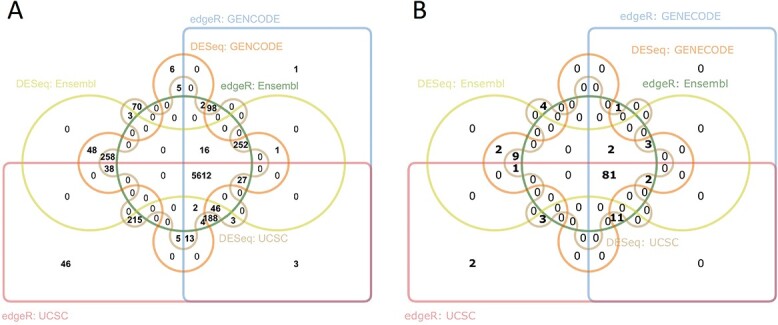
Venn diagram of (A) DE genes with adjusted P value < 0.05 and (B) top 100 DE genes based on three reference genome databases (Ensembl, GENCODE and UCSC) and two DE analysis methods (edgeR and DESeq2).
Using the same gene list for GSEA, we showed that the enriched gene sets varied according to different ranking functions (Figure 3). When the results were evaluated based on the presence/absence of the top 10 enriched gene sets, identical results were observed when signal-to-noise and t-test statistic were applied to rank the input gene list. These results were also comparable to that using log-to-ratio with Jaccard similarity J = 0.82. The top 10 gene sets derived from the pre-rank list were similar to those from signal-to-noise, t-test and log-to-ratio functions with J = 0.67. The difference and ratio functions generated relatively distinct patterns though, with J = 0.15 ± 0.06 (mean ± SD) and J = 0.33 ± 0.14, respectively. When considering the FDR of the five ranking functions, 9 out of the top 10 enriched gene sets passed the recommended FDR cutoff of 25% using ratio, signal-to-noise and t-test functions, whereas no single gene set was below the FDR cutoff using difference and log-to-ratio functions. These results clearly demonstrate that, even when the input gene list was the same, the enriched gene sets’ output might still be distinct when different ranking functions are applied.
Figure 3.
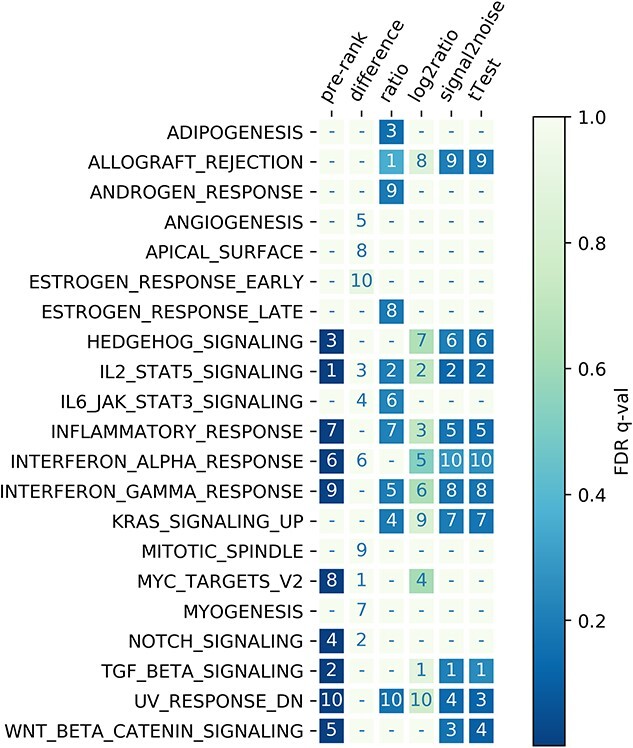
Top 10 up-regulated hallmark gene sets obtained from pre-ranked gene list (sgn(logFC)*log(P-value)) and DESeq2-normalized counts data with GSEA built-in ranking functions including difference statistic, ratio statistic, log-ratio statistic, signal-to-noise statistic and t-test statistic. The number indicates the rank of the gene set based on the NES and the color represents the FDR q-value of the gene set. The dash symbol (–) indicates the gene set is not listed among the top 10 based on the NES.
Next, we compared the outputs of the same enrichment analysis method (here, pre-ranked GSEA) with six DE gene lists generated from three reference genome databases and two DE methods. Ranked according to the NES, the top 10 enriched gene sets were identical (Figure 4), albeit the rank for each gene set was slightly different between datasets, accounting for up to 5% of DE gene difference between each dataset. Notably, the top three enriched gene sets remained the same regardless of the reference genome databases or DE methods. In addition, the top 10 enriched gene sets identified by pre-ranked GSEA with gene list from mm39 were also found in the analysis using mm10, despite the slight discrepancy of ranking order between each other. This result suggests that, despite reference genome databases and DE methods, with the same enrichment analysis method and parameters, consistent output despite different ranking order can be obtained. Note, however, that we only tested two DE methods that rely on the same assumptions.
Figure 4.
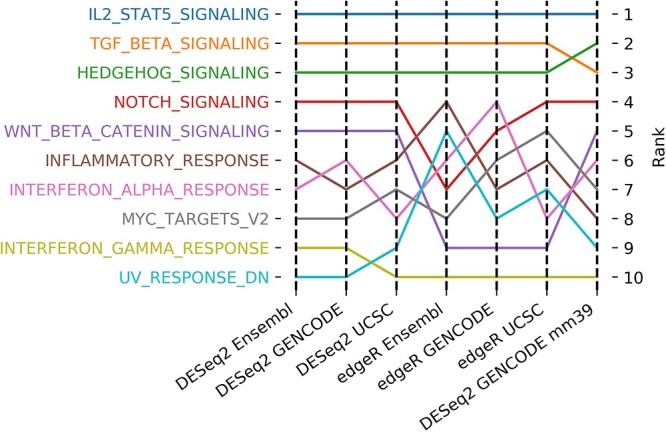
Comparison of top 10 up-regulated hallmark gene sets using GSEA with input pre-ranked gene lists from the combinations of different reference genome databases (Ensembl, GENCODE and UCSC) and DE analysis methods (edgeR and DESeq2), along with the latest reference genome (mm39). The solid line indicates a hallmark gene set and the rank is based on the NES.
Using multiple pathway enrichment analysis methods, a total of 62, 32 and 5 enriched pathways were identified by DAVID, limma and GSEA, respectively. When pairwise comparing the enriched pathways, we found 18, 2 and 1 shared pathway(s) in common between DAVID and limma, DAVID and GSEA, and limma and GSEA (Figure 5), respectively. Only one pathway (phosphatidylinositol signaling) was found by all the three methods. Overall, the results show the distinct over-representation patterns among the enrichment analysis methods using the same dataset.
Figure 5.

Treemap showing the KEGG pathway enrichment analysis using (A) DAVID, (B) limma’s kegga function and (C) GSEA. Each rectangle represents a single KEGG pathway, and the pathways are clustered and colored based on BRITE hierarchies. The uncolored (white) rectangle indicates the pathway is not significantly enriched; namely, that it does not pass the FDR q-value threshold of 0.05 for DAVID and limma, and 0.25 as suggested by GSEA.
Conclusions
RNA-Seq has become ubiquitous for studying the gene expression and transcript identification of cells or organisms. Transcriptomics methods are still under active development and computational theories, analysis pipelines and integrated databases are continuously proposed. In this review paper, we first provided a detailed introduction to RNA-Seq analysis steps from raw reads to functional enrichment analysis, for both coding and non-coding RNAs. Next, we summarized the recent advances related to ML in RNA-Seq, metatranscriptomics, integrative multi-omics and ncRNA-Seq technologies. Lastly, by analyzing a real-world data with different analysis options, we demonstrated that the results can be unintentionally impacted by the choice of methods.
Although the reference genome annotations were slightly different among databases, and different DE methods were used, similar DEGs were identified in our analysis. This slight difference affected the statistical significance of the enriched pathways but, overall, the same enriched pathways were captured by using GSEA regardless of the source of the reference annotation. The consistent results can be partly attributed to the use of similar assumptions and normalization approaches in edgeR and DESeq2. Other RNA-Seq normalization approaches with different assumptions are available, and they can largely affect the downstream analysis and engender inflated false positives if chosen unwisely [210]. That said, reporting the version information of reference database and software is still important for transparency and reproducibility [211]. In addition, we showed that GSEA, one of the most popular enrichment analysis methods, can yield completely different gene sets from the same input gene list due to the choice of ranking functions, which is based on different statistics and assumptions. For example, the default ranking function of GSEA (i.e. signal-to-noise) assumes normal distribution of data without outliers, and the statistical power diminishes when the assumptions are not met [212].
We also addressed differences depending on the choice of the enrichment analysis approach. We have demonstrated that, by using the same gene list with different analysis methods, almost non-overlapping pathways can be obtained. This is expected as competitive methods (which consider all genes in the list, such as GSEA) are generally more conservative compared to self-contained methods (which consider only genes in the gene set of interest, such as the method implemented by DAVID) [213]. However, there is no consensus about the best practice of enrichment analysis for a given RNA-Seq experiment [214]. Mathur et al. [213] evaluated the run time and statistical power of GSEA and three other enrichment analysis methods by using simulated gene sets with different proportions of DEGs, and GSEA was recommended as one of the most powerful methods. Yet another study [215] found that, compared with self-contained methods, competitive methods tend to have low reproducibility in terms of gene sets they found; on the other hand, that self-contained methods, especially those applying multivariate statistics, have a better performance in terms of false positive controls, statistical power, robustness to sample size and reproducibility. Geistlinger et al. [216] has provided guidelines for the choice of gene set enrichment methods based on the input gene lists and question of interest.
Overall, we described in detail the most popular RNA-Seq analysis options and, instead of providing a gold standard or best practice for RNA-Seq analysis, we pinpointed the differences that may raise due to selected methods. Researchers thus need to cautiously interpret the clinical or biological relevance of the statistically significant features derived from choice of analysis methods and, wherever possible, conduct experimental validation after RNA-Seq analyses, which also holds true for scRNA-Seq.
Key Points
This manuscript explains RNA-Seq analysis from start to end including current popular software options and their similarities, differences, advantages and disadvantages.
We present a comprehensive summary of RNA-Seq technologies and applications, including non-coding RNA analysis, multi-omics, meta-transcriptomics and use of artificial intelligence-aided methods.
We show how different RNA-Seq results may be obtained according to selected computational methods. Overall, results need to be cautiously interpreted and validated.
Supplementary Material
Author Biographies
Jiung-Wen Chen is a Biology PhD student at the University of Alabama at Birmingham. His main research interests are host–microbiome interactions, microbiome-associated data analysis and machine learning applications in microbiome studies.
Lisa Shrestha is a Genetics and Genomics PhD student in the Graduate Biomedical Sciences Program, University of Alabama at Birmingham. Her main research interests include non-coding RNA biology, neurodegeneration and bioinformatics.
George Green is a Biology PhD student at the University of Alabama at Birmingham. His main research interests are bioinformatics and the effects of nutrition and the microbiome.
André Leier is assistant professor in the Department of Genetics and the Department of Cell, Developmental and Integrative Biology, Heersink School of Medicine, University of Alabama at Birmingham, USA. He is also an associate scientist in the UAB Comprehensive Cancer Center. His research interests are in biochemical engineering, gene therapies, bioinformatics, machine learning, computational and systems biomedicine.
Tatiana T. Marquez-Lago is associate professor in the Departments of Genetics, Microbiology, and Cell, Developmental and Integrative Biology, Heersink School of Medicine, University of Alabama at Birmingham, USA. Her research interests include multiscale modelling and simulations, artificial intelligence, bioengineering and systems biomedicine. Her interdisciplinary lab studies stochastic gene expression, antibiotic resistance and host–microbiota interactions.
Contributor Information
Jiung-Wen Chen, Department of Biology, University of Alabama at Birmingham, Birmingham, AL, USA.
Lisa Shrestha, Department of Genetics, University of Alabama at Birmingham, School of Medicine, Birmingham, AL, USA.
George Green, Department of Biology, University of Alabama at Birmingham, Birmingham, AL, USA.
André Leier, Department of Genetics, University of Alabama at Birmingham, School of Medicine, Birmingham, AL, USA; Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, School of Medicine, Birmingham, AL, USA.
Tatiana T Marquez-Lago, Department of Genetics, University of Alabama at Birmingham, School of Medicine, Birmingham, AL, USA; Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, School of Medicine, Birmingham, AL, USA; Department of Microbiology, University of Alabama at Birmingham, School of Medicine, Birmingham, AL, USA.
Author contributions
T.T.M.L. conceptualized the study and supervised the project. J-W.C. performed bioinformatics analyses. All authors contributed manuscript content and assisted with reviews and edits.
Data availability
All data is available as supplementary material (4 files), or readily available in the literature as noted by the corresponding references.
Funding
This work was supported by NIH 1R01HD102690.
References
- 1. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Childhood - Educ 2013;98(6):236–8(Practice edition). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee C-Y, Chiu Y-C, Wang L-B, et al. Common applications of next-generation sequencing technologies in genomic research. Transl Cancer Res 2013;2(1):33–45. [Google Scholar]
- 4. Furlan M, Tanaka I, Leonardi T, et al. Direct RNA sequencing for the study of synthesis, processing, and degradation of modified transcripts. Front Genet 2020;11:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang IS, Kim S. Analysis of whole transcriptome sequencing data: workflow and software. Genomics Inform 2015;13(4):119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seashols-Williams S, Lewis C, Calloway C, et al. High-throughput miRNA sequencing and identification of biomarkers for forensically relevant biological fluids. Electrophoresis 2016;37(21):2780–8. [DOI] [PubMed] [Google Scholar]
- 7. Mercer TR, Gerhardt DJ, Dinger ME, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol 2011;30(1):99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolodziejczyk AA, Kim JK, Svensson V, et al. The technology and biology of single-cell RNA sequencing. Mol Cell 2015;58(4):610–20. [DOI] [PubMed] [Google Scholar]
- 9. Kukurba KR, Montgomery SB. RNA sequencing and analysis. Cold Spring Harb Protoc 2015;2015(11):951–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anders S, McCarthy DJ, Chen Y, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 2013;8(9):1765–86. [DOI] [PubMed] [Google Scholar]
- 11. Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biol 2016;17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berge KV, Hembach KM, Soneson C, et al. RNA sequencing data: Hitchhiker's guide to expression analysis. Ann Rev Biomed Data Sci 2019;2(1):139–73. [Google Scholar]
- 13. Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol 2010;11(12):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuznetsova I, Karpievitch YV, Filipovska A, et al. Review of machine learning algorithms in differential expression analysis. arXiv preprint arXiv:1707.09837. 2017. [Google Scholar]
- 16. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215(3):403–10. [DOI] [PubMed] [Google Scholar]
- 17. Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao Y, Smyth GK, Shi W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 2013;41(10):e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baruzzo G, Hayer KE, Kim EJ, et al. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Methods 2017;14(2):135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25(9):1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kapranov P, St Laurent G, Raz T, et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is 'dark matter' un-annotated RNA. BMC Biol 2010;8:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia X. RNA-Seq approach for accurate characterization of splicing efficiency of yeast introns. Methods 2020;176:25–33. [DOI] [PubMed] [Google Scholar]
- 25. Hayer KE, Pizarro A, Lahens NF, et al. Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data. Bioinformatics 2015;31(24):3938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zielezinski A, Vinga S, Almeida J, et al. Alignment-free sequence comparison: benefits, applications, and tools. Genome Biol 2017;18(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu DC, Yao J, Ho KS, et al. Limitations of alignment-free tools in total RNA-seq quantification. BMC Genomics 2018;19(1):510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 29. Wu PY, Phan JH, Wang MD. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics 2013;14(Suppl 11):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 2013;14:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schurch NJ, Schofield P, Gierlinski M, et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016;22(6):839–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeanmougin M, Reynies A, Marisa L, et al. Should we abandon the t-test in the analysis of gene expression microarray data: a comparison of variance modeling strategies. PLoS One 2010;5(9):e12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Planet PJ, DeSalle R, Siddall M, et al. Systematic analysis of DNA microarray data: ordering and interpreting patterns of gene expression. Genome Res 2001;11(7):1149–55. [DOI] [PubMed] [Google Scholar]
- 35. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 1995;57(1):289–300. [Google Scholar]
- 36. Zheng W, Chung LM, Zhao H. Bias detection and correction in RNA-Sequencing data. BMC Bioinformatics 2011;12:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang W, Chen L. Alternative splicing: human disease and quantitative analysis from high-throughput sequencing. Comput Struct Biotechnol J 2021;19:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emmert-Streib F, Glazko GV. Pathway analysis of expression data: deciphering functional building blocks of complex diseases. PLoS Comput Biol 2011;7(5):e1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li W, Fontanelli O, Miramontes P. Size distribution of function-based human gene sets and the split-merge model. R Soc Open Sci 2016;3(8):160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bullard JH, Purdom E, Hansen KD, et al. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 2010;11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Young MD, Wakefield MJ, Smyth GK, et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 2010;11(2):R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ren X, Hu Q, Liu S, et al. Gene set analysis controlling for length bias in RNA-seq experiments. BioData Min 2017;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313(5795):1929–35. [DOI] [PubMed] [Google Scholar]
- 45. Wang X, Cairns MJ. Gene set enrichment analysis of RNA-Seq data: integrating differential expression and splicing. BMC Bioinformatics 2013;14(Suppl 5):S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X, Cairns MJ. SeqGSEA: a Bioconductor package for gene set enrichment analysis of RNA-Seq data integrating differential expression and splicing. Bioinformatics 2014;30(12):1777–9. [DOI] [PubMed] [Google Scholar]
- 47. Oron A, Gentleman R. GSEAlm: linear model toolset for gene set enrichment analysis. Bioconductor package version 1.0. 2008. https://www.bioconductor.org/packages/release/bioc/html/GSEAlm.html.
- 48. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 49. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007;8(9):R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kramer A, Green J, PollardJ, Jr, et al. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014;30(4):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ben-Ari Fuchs S, Lieder I, Stelzer G, et al. GeneAnalytics: an integrative gene set analysis tool for next generation sequencing. RNAseq Microarray Data OMICS 2016;20(3):139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin D, Brun C, Remy E, et al. GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol 2004;5(12):R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arend RC, Londono AI, Montgomery AM, et al. Molecular response to neoadjuvant chemotherapy in high-grade serous ovarian carcinoma. Mol Cancer Res 2018;16(5):813–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soh D, Dong D, Guo Y, et al. Consistency, comprehensiveness, and compatibility of pathway databases. BMC Bioinformatics 2010;11(1):449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Botta D, Fuller MJ, Marquez-Lago TT, et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat Immunol 2017;18(11):1249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet 2015;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Atianand MK, Fitzgerald KA. Long non-coding RNAs and control of gene expression in the immune system. Trends Mol Med 2014;20(11):623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fernandes JCR, Acuna SM, Aoki JI, et al. Long non-coding RNAs in the regulation of gene expression: physiology and disease. Noncoding RNA 2019;5(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goff LA, Groff AF, Sauvageau M, et al. Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A 2015;112(22):6855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang X, Wang W, Zhu W, et al. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int J Mol Sci 2019;20(22):5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li J, Hao M, Yang B, et al. Long non-coding RNAs expression profile and functional analysis of acute ischemic stroke. Medicine (Baltimore) 2020;99(50):e22964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu X, Ma Y, Yin K, et al. Long non-coding and coding RNA profiling using strand-specific RNA-seq in human hypertrophic cardiomyopathy. Sci Data 2019;6(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cui P, Lin Q, Ding F, et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 2010;96(5):259–65. [DOI] [PubMed] [Google Scholar]
- 65. Dahlgren AR, Scott EY, Mansour T, et al. Comparison of poly-A(+) selection and rRNA depletion in detection of lncRNA in two equine tissues using RNA-seq. Noncoding RNA 2020;6(3):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Duan Y, Zhang W, Cheng Y, et al. A systematic evaluation of bioinformatics tools for identification of long noncoding RNAs. RNA 2021;27(1):80–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li J, Zhang X, Liu C. The computational approaches of lncRNA identification based on coding potential: status quo and challenges. Comput Struct Biotechnol J 2020;18:3666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sun L, Liu H, Zhang L, et al. lncRScan-SVM: a tool for predicting long non-coding RNAs using support vector machine. PLoS One 2015;10(10):e0139654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han S, Liang Y, Ma Q, et al. LncFinder: an integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief Bioinform 2019;20(6):2009–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sun K, Chen X, Jiang P, et al. iSeeRNA: identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genomics 2013;14(Suppl 2):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang Y, Li Y, Wang Q, et al. Computational identification of human long intergenic non-coding RNAs using a GA-SVM algorithm. Gene 2014;533(1):94–9. [DOI] [PubMed] [Google Scholar]
- 72. Volders PJ, Anckaert J, Verheggen K, et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res 2019;47(D1):D135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ma L, Cao J, Liu L, et al. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res 2019;47(5):2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bao Z, Yang Z, Huang Z, et al. LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res 2019;47(D1):D1034–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gao Y, Shang S, Guo S, et al. Lnc2Cancer 3.0: an updated resource for experimentally supported lncRNA/circRNA cancer associations and web tools based on RNA-seq and scRNA-seq data. Nucleic Acids Res 2021;49(D1):D1251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hu J, Gao Y, Li J, et al. Deep learning enables accurate prediction of interplay between lncRNA and disease. Front Genet 2019;10:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang Q, Yan G. IDLDA: an improved diffusion model for predicting lncRNA-disease associations. Front Genet 2019;10:1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang L, Zhong C. gGATLDA: lncRNA-disease association prediction based on graph-level graph attention network. BMC Bioinformatics 2022;23(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guo JU, Agarwal V, Guo H, et al. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014;15(7):409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013;19(2):141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 2014;28(20):2233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sanger HL, Klotz G, Riesner D, et al. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A 1976;73(11):3852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Teplova M, Hafner M, Teplov D, et al. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev 2013;27(8):928–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495(7441):333–8. [DOI] [PubMed] [Google Scholar]
- 85. Suzuki H, Zuo Y, Wang J, et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 2006;34(8):e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hanan M, Simchovitz A, Yayon N, et al. A Parkinson's disease circRNAs resource reveals a link between circSLC8A1 and oxidative stress. EMBO Mol Med 2020;12(9):e11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang J, Hou L, Zuo Z, et al. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat Biotechnol 2021;39(7):836–45. [DOI] [PubMed] [Google Scholar]
- 88. Du WW, Yang W, Liu E, et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 2016;44(6):2846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Piwecka M, Glazar P, Hernandez-Miranda LR, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017;357(6357):eaam8526. [DOI] [PubMed] [Google Scholar]
- 90. Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 2016;7:11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Abdelmohsen K, Panda AC, Munk R, et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 2017;14(3):361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Holdt LM, Stahringer A, Sass K, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun 2016;7:12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ashwal-Fluss R, Meyer M, Pamudurti NR, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014;56(1):55–66. [DOI] [PubMed] [Google Scholar]
- 94. Zhang XO, Dong R, Zhang Y, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 2016;26(9):1277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform 2018;19(5):803–10. [DOI] [PubMed] [Google Scholar]
- 96. Szabo L, Morey R, Palpant NJ, et al. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol 2015;16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gao Y, Zhao F. Computational strategies for exploring circular RNAs. Trends Genet 2018;34(5):389–400. [DOI] [PubMed] [Google Scholar]
- 98. Zhang J, Chen S, Yang J, et al. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat Commun 2020;11(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gaffo E, Bonizzato A, Kronnie GT, et al. CirComPara: a multi-method comparative bioinformatics pipeline to detect and study circRNAs from RNA-seq data. Noncoding RNA 2017;3(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Glazar PPP, Rajewsky N. circBase: a database for circular RNAs. RNA 2014;20(11):1666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chen X, Han P, Zhou T, et al. circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci Rep 2016;6:34985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Meng X, Hu D, Zhang P, et al. CircFunBase: a database for functional circular RNAs. Database (Oxford) 2019;2019:baz003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pan X, Xiong K. PredcircRNA: computational classification of circular RNA from other long non-coding RNA using hybrid features. Mol Biosyst 2015;11(8):2219–26. [DOI] [PubMed] [Google Scholar]
- 104. Gao Y, Wang J, Zheng Y, et al. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 2016;7:12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang J, Wang L. Deep learning of the back-splicing code for circular RNA formation. Bioinformatics 2019;35(24):5235–42. [DOI] [PubMed] [Google Scholar]
- 106. Jiang JY, Ju CJ, Hao J, et al. JEDI: circular RNA prediction based on junction encoders and deep interaction among splice sites. Bioinformatics 2021;37(Suppl_1):i289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Dudekula DB, Panda AC, Grammatikakis I, et al. CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol 2016;13(1):34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yao D, Zhang L, Zheng M, et al. Circ2Disease: a manually curated database of experimentally validated circRNAs in human disease. Sci Rep 2018;8(1):11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhao Z, Wang K, Wu F, et al. circRNA disease: a manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis 2018;9(5):475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ghosal S, Das S, Sen R, et al. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet 2013;4:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Annese T, Tamma R, De Giorgis M, et al. microRNAs biogenesis, functions and role in tumor angiogenesis. Front Oncol 2020;10:581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 2019;20(1):21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011;12(2):99–110. [DOI] [PubMed] [Google Scholar]
- 114. Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010;79:351–79. [DOI] [PubMed] [Google Scholar]
- 115. Wilczynska A, Bushell M. The complexity of miRNA-mediated repression. Cell Death Differ 2015;22(1):22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Benesova S, Kubista M, Valihrach L. Small RNA-sequencing: approaches and considerations for miRNA analysis. Diagnostics 2021;11(6):964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Campbell JD, Liu G, Luo L, et al. Assessment of microRNA differential expression and detection in multiplexed small RNA sequencing data. RNA 2015;21(2):164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kivioja T, Vaharautio A, Karlsson K, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 2011;9(1):72–4. [DOI] [PubMed] [Google Scholar]
- 119. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014;42(Database issue):D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fehlmann T, Kern F, Laham O, et al. miRMaster 2.0: multi-species non-coding RNA sequencing analyses at scale. Nucleic Acids Res 2021;49(W1):W397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sun Z, Evans J, Bhagwate A, et al. CAP-miRSeq: a comprehensive analysis pipeline for microRNA sequencing data. BMC Genomics 2014;15:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wu J, Liu Q, Wang X, et al. mirTools 2.0 for non-coding RNA discovery, profiling, and functional annotation based on high-throughput sequencing. RNA Biol 2013;10(7):1087–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ronen R, Gan I, Modai S, et al. miRNAkey: a software for microRNA deep sequencing analysis. Bioinformatics 2010;26(20):2615–6. [DOI] [PubMed] [Google Scholar]
- 124. Yang K, Sablok G, Qiao G, et al. isomiR2Function: an integrated workflow for identifying microRNA variants in plants. Front Plant Sci 2017;8:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Baras AS, Mitchell CJ, Myers JR, et al. miRge - a multiplexed method of processing small RNA-seq data to determine microRNA entropy. PLoS One 2015;10(11):e0143066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lukasik A, Wojcikowski M, Zielenkiewicz P. Tools4miRs - one place to gather all the tools for miRNA analysis. Bioinformatics 2016;32(17):2722–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Friedlander MR, Chen W, Adamidi C, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 2008;26(4):407–15. [DOI] [PubMed] [Google Scholar]
- 128. Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res 2003;31(13):3429–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Li L, Weinberg CR, Darden TA, et al. Gene selection for sample classification based on gene expression data: study of sensitivity to choice of parameters of the GA/KNN method. Bioinformatics 2001;17(12):1131–42. [DOI] [PubMed] [Google Scholar]
- 130. Li Y, Kang K, Krahn JM, et al. A comprehensive genomic pan-cancer classification using The Cancer Genome Atlas gene expression data. BMC Genomics 2017;18(1):508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mohsen H, Tang H, Ye Y. Improving de novo metatranscriptome assembly via machine learning algorithms. Int J Comput Biol Drug Des 2017;10(2):91–107. [Google Scholar]
- 132. Banavar G, Ogundijo O, Toma R, et al. The salivary metatranscriptome as an accurate diagnostic indicator of oral cancer. NPJ Genom Med 2021;6(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lambert BS, Groussman RD, Schatz MJ, et al. The dynamic trophic architecture of open-ocean protist communities revealed through machine-guided metatranscriptomics. Proc Natl Acad Sci U S A 2022;119(7):e2100916119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lyu B, Haque A (eds). Deep learning based tumor type classification using gene expression data. Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. Association for Computing Machinery, 2018.
- 135. Pratama R, Hwang JJ, Lee JH, et al. Authentication of differential gene expression in oral squamous cell carcinoma using machine learning applications. BMC Oral Health 2021;21(1):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mostavi M, Chiu YC, Huang Y, et al. Convolutional neural network models for cancer type prediction based on gene expression. BMC Med Genomics 2020;13(Suppl 5):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhang Z, Pan Z, Ying Y, et al. Deep-learning augmented RNA-seq analysis of transcript splicing. Nat Methods Institute of Electrical and Electronics Engineers, 2019;16(4):307–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tasaki S, Gaiteri C, Mostafavi S, et al. Deep learning decodes the principles of differential gene expression. Nat Mach Intell 2020;2(7):376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Saremi B, Gusmag F, Distl O, et al. A comparison of strategies for generating artificial replicates in RNA-seq experiments. Sci Rep 2022;12(1):7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wang X, Wang H, Liu D, et al. Deep learning using bulk RNA-seq data expands cell landscape identification in tumor microenvironment. Onco Targets Ther 2022;11(1):2043662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sastry AV, Gao Y, Szubin R, et al. The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat Commun 2019;10(1):5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Berg G, Rybakova D, Fischer D, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome 2020;8(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Visconti A, Le Roy CI, Rosa F, et al. Interplay between the human gut microbiome and host metabolism. Nat Commun 2019;10(1):4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cullen CM, Aneja KK, Beyhan S, et al. Emerging priorities for microbiome research. Front Microbiol 2020;11:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 146. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012;28(24):3211–7. [DOI] [PubMed] [Google Scholar]
- 148. Seemann T. Barrnap: bacterial ribosomal RNA predictor 2013. Available from: https://github.com/tseemann/barrnap.
- 149. Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011;29(7):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Leung HC, Yiu SM, Parkinson J, et al. IDBA-MT: de novo assembler for metatranscriptomic data generated from next-generation sequencing technology. J Comput Biol 2013;20(7):540–50. [DOI] [PubMed] [Google Scholar]
- 151. Leung HC, Yiu SM, Chin FY. IDBA-MTP: a hybrid metatranscriptomic assembler based on protein information. J Comput Biol 2015;22(5):367–76. [DOI] [PubMed] [Google Scholar]
- 152. Li D, Liu CM, Luo R, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31(10):1674–6. [DOI] [PubMed] [Google Scholar]
- 153. Nurk S, Meleshko D, Korobeynikov A, et al. metaSPAdes: a new versatile metagenomic assembler. Genome Res 2017;27(5):824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19(5):455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009;25(15):1966–7. [DOI] [PubMed] [Google Scholar]
- 156. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12(1):59–60. [DOI] [PubMed] [Google Scholar]
- 158. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Salazar G, Paoli L, Alberti A, et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 2019;179(5):1068–83 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Yergeau E, Tremblay J, Joly S, et al. Soil contamination alters the willow root and rhizosphere metatranscriptome and the root-rhizosphere interactome. ISME J 2018;12(3):869–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Nowicki EM, Shroff R, Singleton JA, et al. Microbiota and metatranscriptome changes accompanying the onset of gingivitis. MBio 2018;9(2):e00575–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zhang Y, Thompson KN, Huttenhower C, et al. Statistical approaches for differential expression analysis in metatranscriptomics. Bioinformatics 2021;37(Suppl_1):i34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Klingenberg H, Meinicke P. How to normalize metatranscriptomic count data for differential expression analysis. PeerJ 2017;5:e3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Xue CX, Lin H, Zhu XY, et al. DiTing: a pipeline to infer and compare biogeochemical pathways from metagenomic and metatranscriptomic data. Front Microbiol 2021;12:698286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Narayanasamy S, Jarosz Y, Muller EE, et al. IMP: a pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol 2016;17(1):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Van Damme R, Holzer M, Viehweger A, et al. Metagenomics workflow for hybrid assembly, differential coverage binning, metatranscriptomics and pathway analysis (MUFFIN). PLoS Comput Biol 2021;17(2):e1008716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tamames J, Puente-Sanchez F. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front Microbiol 2018;9:3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Anwar MZ, Lanzen A, Bang-Andreasen T, et al. To assemble or not to resemble-a validated comparative metatranscriptomics workflow (CoMW). Gigascience 2019;8(8):giz096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Taj B, Adeolu M, Xiong X, et al. MetaPro: a scalable and reproducible data processing and analysis pipeline for metatranscriptomic investigation of microbial communities. bioRxiv 2021;2021:02.23.432558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Mehta S, Crane M, Leith E, et al. ASaiM-MT: a validated and optimized ASaiM workflow for metatranscriptomics analysis within Galaxy framework. F1000Res 2021;10:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ni Y, Li J, Panagiotou G. COMAN: a web server for comprehensive metatranscriptomics analysis. BMC Genomics 2016;17(1):622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kim J, Kim MS, Koh AY, et al. FMAP: functional mapping and analysis pipeline for metagenomics and metatranscriptomics studies. BMC Bioinformatics 2016;17(1):420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Beghini F, McIver LJ, Blanco-Miguez A, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 2021;10:e65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Martinez X, Pozuelo M, Pascal V, et al. MetaTrans: an open-source pipeline for metatranscriptomics. Sci Rep 2016;6:26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Westreich ST, Treiber ML, Mills DA, et al. SAMSA2: a standalone metatranscriptome analysis pipeline. BMC Bioinformatics 2018;19(1):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27(5):722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Uppal K, Ma C, Go YM, et al. xMWAS: a data-driven integration and differential network analysis tool. Bioinformatics 2018;34(4):701–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Hernandez-de-Diego R, Tarazona S, Martinez-Mira C, et al. PaintOmics 3: a web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res 2018;46(W1):W503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Conard AM, Goodman N, Hu Y, et al. TIMEOR: a web-based tool to uncover temporal regulatory mechanisms from multi-omics data. Nucleic Acids Res 2021;49(W1):W641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Ding J, Blencowe M, Nghiem T, et al. Mergeomics 2.0: a web server for multi-omics data integration to elucidate disease networks and predict therapeutics. Nucleic Acids Res 2021;49(W1):W375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Zhou G, Ewald J, Xia J. OmicsAnalyst: a comprehensive web-based platform for visual analytics of multi-omics data. Nucleic Acids Res 2021;49(W1):W476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Taverna F, Goveia J, Karakach TK, et al. BIOMEX: an interactive workflow for (single cell) omics data interpretation and visualization. Nucleic Acids Res 2020;48(W1):W385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ulfenborg B. Vertical and horizontal integration of multi-omics data with miodin. BMC Bioinformatics 2019;20(1):649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Kuo TC, Tian TF, Tseng YJ. 3Omics: a web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst Biol 2013;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Canzler S, Hackermuller J. multiGSEA: a GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinformatics 2020;21(1):561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Konietschke F, Schwab K, Pauly M. Small sample sizes: a big data problem in high-dimensional data analysis. Stat Methods Med Res 2021;30(3):687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Selvaraju RR, Cogswell M, Das A, et al. (eds) Grad-cam: visual explanations from deep networks via gradient-based localization. Proceedings of the IEEE International Conference on Computer Vision, Institute of Electrical and Electronics Engineers, 2017.
- 188. Lim HG, Rychel K, Sastry AV, et al. Machine-learning from Pseudomonas putida KT2440 transcriptomes reveals its transcriptional regulatory network. Metab Eng 2022;72:297–310. [DOI] [PubMed] [Google Scholar]
- 189. Das S, Rai A, Mishra DC, et al. Statistical approach for gene set analysis with trait specific quantitative trait loci. Sci Rep 2018;8(1):2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Lundberg SM, Erion G, Chen H, et al. From local explanations to global understanding with explainable AI for trees. Nat Mach Intell 2020;2(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Zhou B, Khosla A, Lapedriza A, et al. (eds) Learning deep features for discriminative localization. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. Institute of Electrical and Electronics Engineers, 2016.
- 192. Zhao Y, Shao J, Asmann YW. Assessment and optimization of explainable machine learning models applied to transcriptomic data. Genomics Proteomics Bioinformatics 2022. 10.1016/j.gpb.2022.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Rudin C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat Mach Intell 2019;1(5):206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Nativio R, Lan Y, Donahue G, et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease. Nat Genet 2020;52(10):1024–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19(1A):A68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Liu SH, Shen PC, Chen CY, et al. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res 2020;48(D1):D863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Palmieri V, Backes C, Ludwig N, et al. IMOTA: an interactive multi-omics tissue atlas for the analysis of human miRNA-target interactions. Nucleic Acids Res 2018;46(D1):D770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Aging AC. Aging Atlas: a multi-omics database for aging biology. Nucleic Acids Res 2021;49(D1):D825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Liu H, Wang F, Xiao Y, et al. MODEM: multi-omics data envelopment and mining in maize. Database (Oxford) 2016;2016:baw117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gui S, Yang L, Li J, et al. ZEAMAP, a comprehensive database adapted to the maize multi-omics era. iScience 2020;23(6):101241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Creasy HH, Felix V, Aluvathingal J, et al. HMPDACC: a Human Microbiome Project Multi-omic data resource. Nucleic Acids Res 2021;49(D1):D734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Yan Z, An J, Peng Y, et al. DevOmics: an integrated multi-omics database of human and mouse early embryo. Brief Bioinform 2021;22(6):bbab208. [DOI] [PubMed] [Google Scholar]
- 203. Subramanian I, Verma S, Kumar S, et al. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights 2020;14:1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Rohart F, Gautier B, Singh A, et al. mixOmics: an R package for 'omics feature selection and multiple data integration. PLoS Comput Biol 2017;13(11):e1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Rohart F, Eslami A, Matigian N, et al. MINT: a multivariate integrative method to identify reproducible molecular signatures across independent experiments and platforms. BMC Bioinformatics 2017;18(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Singh A, Shannon CP, Gautier B, et al. DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019;35(17):3055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Patel-Murray NL, Adam M, Huynh N, et al. A multi-omics interpretable machine learning model reveals modes of action of small molecules. Sci Rep 2020;10(1):954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Zhang L, Lv C, Jin Y, et al. Deep learning-based multi-omics data integration reveals two prognostic subtypes in high-risk neuroblastoma. Front Genet 2018;9:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Reel PS, Reel S, Pearson E, et al. Using machine learning approaches for multi-omics data analysis: a review. Biotechnol Adv 2021;49:107739. [DOI] [PubMed] [Google Scholar]
- 210. Evans C, Hardin J, Stoebel DM. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief Bioinform 2018;19(5):776–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Simoneau J, Dumontier S, Gosselin R, et al. Current RNA-seq methodology reporting limits reproducibility. Brief Bioinform 2021;22(1):140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zyla J, Marczyk M, Weiner J, et al. Ranking metrics in gene set enrichment analysis: do they matter? BMC Bioinformatics 2017;18(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Mathur R, Rotroff D, Ma J, et al. Gene set analysis methods: a systematic comparison. BioData Min 2018;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Maleki F, Ovens K, Hogan DJ, et al. Gene set analysis: challenges, opportunities, and future research. Front Genet 2020;11:654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Rahmatallah Y, Emmert-Streib F, Glazko G. Gene set analysis approaches for RNA-seq data: performance evaluation and application guideline. Brief Bioinform 2016;17(3):393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Geistlinger L, Csaba G, Santarelli M, et al. Toward a gold standard for benchmarking gene set enrichment analysis. Brief Bioinform 2021;22(1):545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available as supplementary material (4 files), or readily available in the literature as noted by the corresponding references.