

Abstract
Studying viral–host protein–protein interactions can facilitate the discovery of therapies for viral infection. We use high-throughput yeast two-hybrid experiments and mass spectrometry to generate a comprehensive SARS-CoV-2–human protein–protein interactome network consisting of 739 high-confidence binary and co-complex interactions, validating 218 known SARS-CoV-2 host factors and revealing 361 novel ones. Our results show the highest overlap of interaction partners between published datasets and of genes differentially expressed in samples from COVID-19 patients. We identify an interaction between the viral protein ORF3a and the human transcription factor ZNF579, illustrating a direct viral impact on host transcription. We perform network-based screens of >2,900 FDA-approved or investigational drugs and identify 23 with significant network proximity to SARS-CoV-2 host factors. One of these drugs, carvedilol, shows clinical benefits for COVID-19 patients in an electronic health records analysis and antiviral properties in a human lung cell line infected with SARS-CoV-2. Our study demonstrates the value of network systems biology to understand human–virus interactions and provides hits for further research on COVID-19 therapeutics.
The severity of the global COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) highlights the need to fill in the gaps in our understanding of the interplay between this virus and its hosts. Once inside a cell, viruses interact with intracellular proteins to hijack host mechanisms that facilitate viral replication and evasion of an immune response1. Studying viral–host protein–protein interactions (PPIs) is therefore crucial for understanding the mechanisms of the viral infection and the host response and to develop new strategies for disease treatment and prevention2–5. Interaction networks are especially important as proteins generally act not in isolation but in concert with their neighborhood of interacting partners. Such interactomes can thus reveal biological pathways and processes impacted by the viral proteome, allowing for the discovery of novel drug targets.
Here we leverage high-throughput yeast two-hybrid (Y2H) and tandem mass tag affinity purification followed by mass spectrometry (TMT-AP–MS) to generate a binary and co-complex SARS-CoV-2–human protein–protein interactome network, which we propose to be a more complete resource for exploration of the viral–host interactome (Fig. 1a). We adopted this approach for several reasons. To date, Y2H and AP–MS are the only two methods available for mapping protein–protein interactome networks on a proteomic scale6,7. Pioneering studies on the earliest SARS-CoV-2–human interactomes utilized label-free AP–MS as their sole method for interaction mapping2–5. While both Y2H and AP–MS alone produce high-quality interactome datasets, they fundamentally capture different yet complementary aspects of the full network; specifically, Y2H interactions often represent key connections between different protein complexes and pathways8. Thus, Y2H and AP–MS together can provide a more comprehensive view of the topological and biological properties of the interactome8. Moreover, labeled (for example, TMT-based) AP–MS has been shown to provide more precise, accurate and reproducible quantification of proteins compared to label-free AP–MS-based approaches, which is an important criterion when trying to identify true protein interactions and generate high-quality interactome networks9–19.
Fig. 1 |. SARS-CoV-2–human protein interactome.
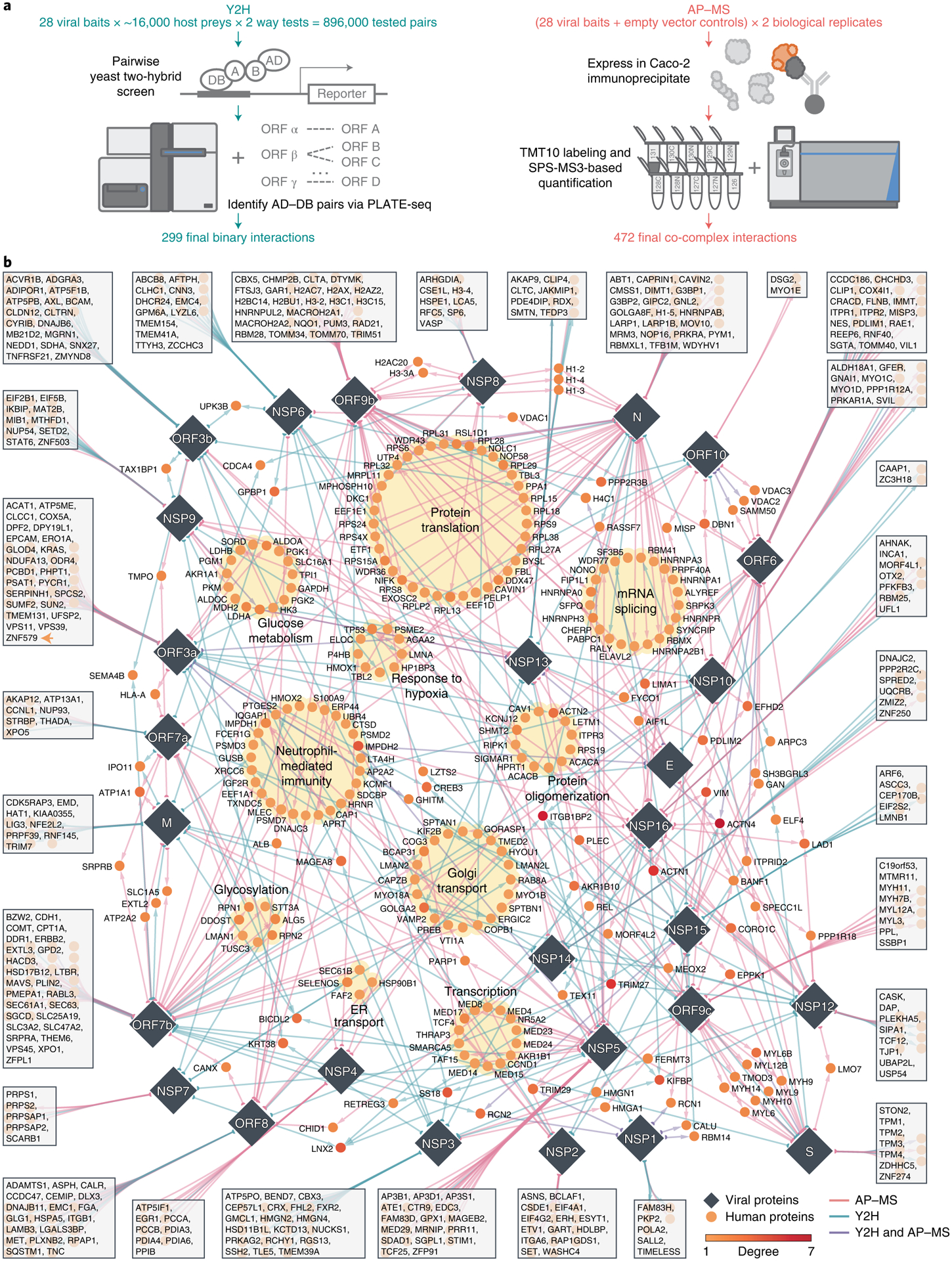
a, Pipelines using Y2H and AP–MS for detecting SARS-CoV-2–human protein–protein interactions. b, Edges between viral proteins (diamonds) and human proteins (circles) represent protein–protein interactions. Edge colors indicate the methods used to detect the protein–protein interaction. Several biological processes that are significantly enriched in these human proteins (Supplementary Fig. 2 and Supplementary Table 2) are highlighted with yellow background. Human proteins that interact with only one SARS-CoV-2 protein are shown in the box connected to that specific protein. The interactome can be found in Supplementary Table 1.
Here we used both Y2H and quantitative TMT-AP–MS to generate a total of 739 high-confidence interactions among 579 human proteins and 28 SARS-CoV-2 proteins. Our interactome had an unprecedented scale and coverage compared with existing ones. Using our interactome, we identified important pathways such as protein translation, mRNA splicing, Golgi transportation, neutrophil-mediated immunity and glucose metabolism. Moreover, we prioritized host-targeting therapies by searching U.S. FDA-approved and investigational drugs for their potential anti-SARS-CoV-2 effect using state-of-the-art network proximity methods. Using two large independent COVID-19 patient databases, we found that usage of one of the top candidates, carvedilol, was associated with a lowered risk (17–20%) of a positive COVID-19 test. Experimental validation shows that carvedilol inhibits SARS-CoV-2 infection with a half-maximal effective concentration (EC50) of 4.1 μM. Altogether, these results suggest that our comprehensive SARS-CoV-2–human protein interactome offers substantial opportunities for understanding the pathobiological process of SARS-CoV-2 in human and identifying host-targeting therapies for COVID-19.
Results
A comprehensive SARS-CoV-2–human protein–protein interactome
To generate a binary SARS-CoV-2–human protein–protein interactome, we systematically tested all pairwise combinations of 28 SARS-CoV-2 proteins (GenBank accession MN908947) against ~16,000 human proteins (hORFeome V8.1)20 using high-throughput Y2H screens8,21–24 (Fig. 1a). We treated each protein as both a bait and a prey, yielding over 896,000 (28 × ~16,000 × 2) total tested pair combinations. Before screening, all autoactivating DNA-binding domain (DB) ORF clones were removed from further tests (see Methods). To increase experimental throughput, viral ORF activating domain (AD) and DB clones were mated against pools of 24 human ORF DB or AD clones, respectively. Following auxotrophic selection, AD–DB pairs were identified via PLATE-seq24 to generate a list of candidate interactions (Methods). Interaction candidates were then subsequently re-tested using Y2H to ascertain high reproducibility. In all, we report a total of 299 high-quality binary SARS-CoV-2–human PPIs via our high-throughput Y2H screen, 267 of which were unique to this assay in this study (Supplementary Table 1).
To complement our binary SARS-CoV-2–human protein–protein interactome, we independently expressed each of the 28 SARS-CoV-2 proteins in the human intestinal epithelial cell line Caco-2 (HTB-37; ATCC) to identify viral–host co-complex interactions using TMT-AP–MS proteomics (Fig. 1a). We used Caco-2 as our cell line model owing to its endogenous expression of angiotensin-converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2) required for SARS-CoV-2 cell entry and S protein priming, respectively25, the extensive use of the line in SARS-CoV and SARS-CoV-2 infection studies26,27, supported by known in vivo replication of SARS-CoV-2 in gastrointestinal cells28,29 and desirable cell culture characteristics including robust transfectability and rapid propagation. All Strep-, Myc-, or FLAG-tagged SARS-CoV-2 baits and their corresponding empty vector controls were transfected in biological duplicates, followed by subsequent affinity purification, TMT labeling and synchronous precursor selection (SPS) MS3-based quantification. We filtered for interactions that met stringent fold change (FC) and P value cutoffs (Methods). In all, we report a total of 472 high-confidence co-complex SARS-CoV-2–human PPIs via AP–MS, 440 of which were unique to this assay in this study (Supplementary Table 1). Altogether, our orthogonal approaches generated a network composed of 739 interactions among 28 viral and 579 host proteins (Supplementary Table 1).
We visualized the SARS-CoV-2–human protein–protein interactome through a network shown in Fig. 1b. The colors of the edges between the viral proteins (represented as diamond nodes) and the host proteins (represented as circle nodes) indicate the methods that detected the interaction. Host proteins that interact with a single viral protein are shown in boxes connected to their interacting partner. Several human proteins interact with multiple SARS-CoV-2 proteins, such as ACTN4, ITGB1BP2, TRIM27 and ACTN1, while the majority of human proteins (469, 81%) interact with only one SARS-CoV-2 protein (Supplementary Fig. 1a). Among the viral proteins, N, ORF7b and ORF9b achieved the highest network degrees, whereas E, NSP7 and NSP1 have the lowest network degrees (Supplementary Fig. 1b). In terms of the shared interacting partners, overall, the viral proteins showed low overlap (Supplementary Fig. 1c), consistent with a previously published SARS-CoV-2 interactome network2,3. We examined the overlap of host factors for Y2H and AP–MS separately and found overall low overlap of host factors as well (Supplementary Fig. 1d,e).
For the entire interactome, functional enrichment analysis revealed significantly overrepresented biological processes (Supplementary Fig. 2a and Supplementary Table 2), including protein translation, transcription and neutrophil-mediated immunity (highlighted with yellow background in Fig. 1b). Semantic analysis shows major biological process categories such as ‘ribosome biogenesis,’ ‘rRNA metabolic process,’ and ‘viral gene expression’ (Supplementary Fig. 2b). Pathway enrichment analysis show top enriched pathways such as ‘protein processing in the endoplasmic reticulum,’ ‘tight junction,’ ‘glycolysis,’ ‘ribosome,’ and ‘protein export’ (Supplementary Fig. 2c and Supplementary Table 2). For individual SARS-CoV-2 proteins, many pathways and biological processes are shared in these viral proteins (Supplementary Fig. 3). For example, NSP12, NSP13 and NSP16 share biological processes such as ‘regulation of cellular component movement,’ ‘negative regulation of cell morphogenesis involved in differentiation’ and ‘negative regulation of substrate adhesion-dependent cell spreading’ (Supplementary Fig. 3a); ORF7a, ORF7b, ORF8 and NSP4 share the pathway ‘protein processing in endoplasmic reticulum’ (Supplementary Fig. 3b).
Given the surge of COVID-19-related studies since 2020, we repeated the enrichment analyses using gene set libraries generated before and after the start of the pandemic to evaluate whether bias was introduced to the gene sets. By comparing Gene Ontology (GO) biological process 2018 versus GO biological process 2021 and Kyoto Encyclopedia of Genes and Genomes (KEGG) human pathway 2021 versus KEGG human pathway 2019, we found that our enrichment analyses were not biased by the addition of the COVID-19 research. In both comparisons, we found that terms such as ‘ribosome biogenesis’, ‘rRNA processing’ and ‘rRNA metabolic process’ and pathways such as ‘Ribosome’ were significantly enriched in the gene set libraries both before and after the pandemic, with similar odds ratio and combined score (Supplementary Fig. 3c).
Overall, our interactome is comprised of abundant information that can be utilized for the identification of COVID-19-relevant pathobiology and host-targeting therapies. We also developed an interactive visualization tool for our interactome which can be accessed from https://github.com/ChengF-Lab/COVID-19_PPI.
Coverage and quality of our interactome
To ensure the authenticity when applying our interactome for downstream studies, we first evaluated the quality through several means. We examined three previously published SARS-CoV-2–human protein–protein interactome networks2,4,5. All three of these interactomes were generated using AP–MS-based methods alone. Overall, we found that the host factors of these interactomes significantly overlap (Fisher’s exact test, false discovery rate (FDR) < 0.05) (Fig. 2a, Extended Data Fig. 1a and Supplementary Table 3), although each interactome still identified a large number of unique factors. This could be explained by differences in the cell line models used (Gordon et al.3 and Li et al.4 used HEK293T/17; Stukalov et al.5 used A549) as well as distinct computational and/or experimental methodologies implemented in their respective studies. Nonetheless, we found that our interactome had the highest overlap of interaction partners among published SARS-CoV-2–human protein–protein interactome networks (Fig. 2a, Extended Data Fig. 1a and Supplementary Table 3), suggesting that our interactome had a high level of coverage. We also found that Y2H confirmed many interactions and resulted in identifying slightly more overlapping host factors with previous studies (Fig. 2a), confirming the complementary nature of our Y2H assay, which was performed in a highly sensitive screen outside of human cells. Overall, AP–MS achieved significantly more overlapping viral–human protein–protein interactions across studies when known human pathways and complexes are taken into consideration (Extended Data Fig. 1b).
Fig. 2 |. Characteristics of the interactome and validation of novel SARS-CoV-2–human interactions.
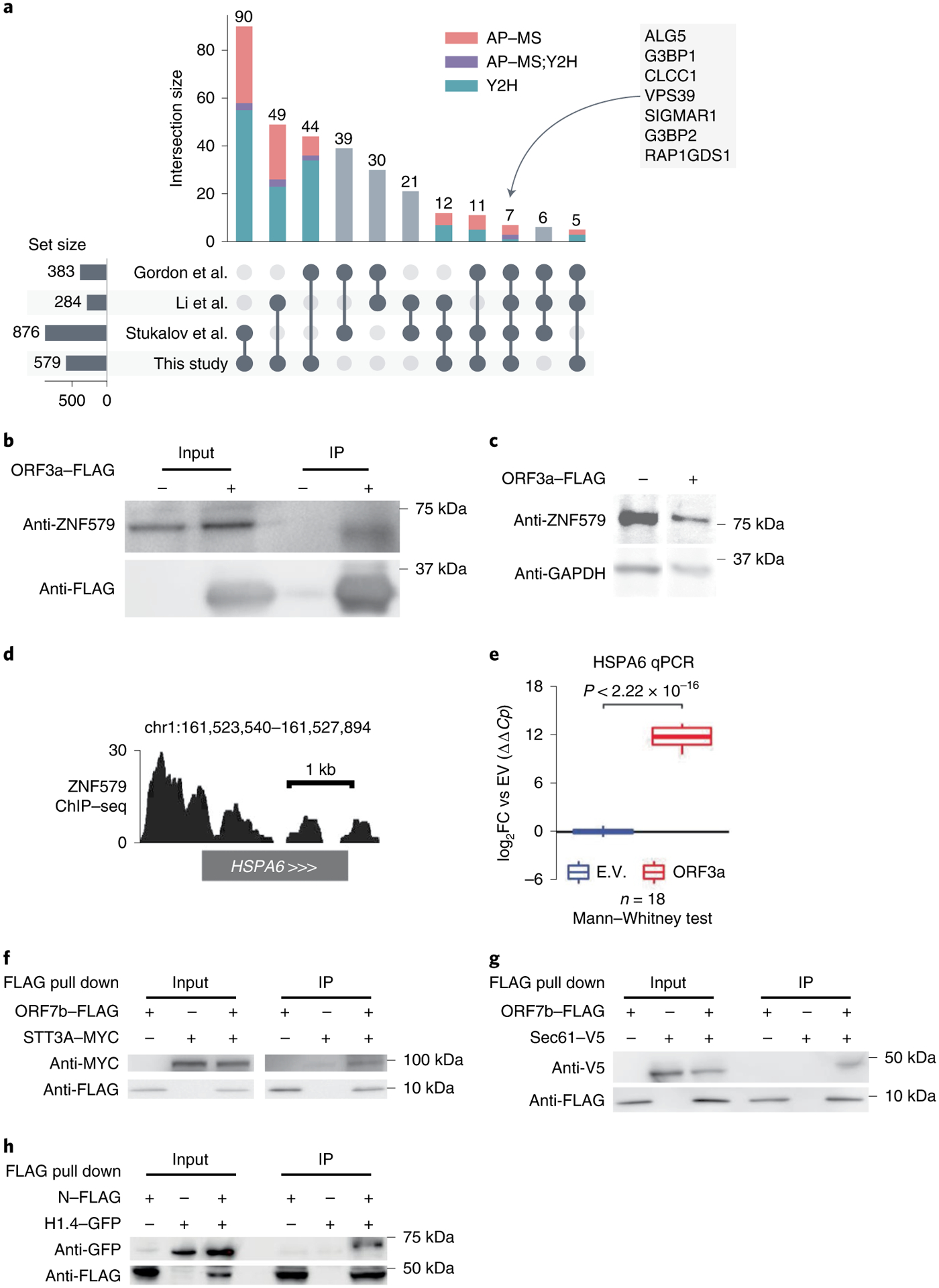
a, UpSet plot showing the overlap of SARS-CoV-2–human protein–protein interactions from four studies (Supplementary Table 3). Each bar shows the interactions shared by only the marked studies at the bottom. Composition of each bar in terms of the source of the interactions are indicated by different colors. b, Co-IP confirming ORF3a–ZNF579 interaction in HEK293T cells following transfection with ORF3a–FLAG or empty vector. The experiment was repeated independently three times with similar results. c, Western blot showing levels of ZNF579 along with GAPDH as a loading control in HEK293T cells following transfection with ORF3a–FLAG or empty vector. Experiment was repeated independently three times with similar results. d, ChIP–seq for ZNF579 in MCF7 cells from the ENCODE consortium at the HSPA6 locus. Signal is log2FC over input. e, Expression of HSPA6 after transfection with ORF3a–FLAG or empty vector (E.V.). Two transfection replicates were probed with two primer pairs to HSPA6 at three different template dilutions in technical triplicate (18 total reactions for each condition). Expression is normalized to GAPDH and then to the empty vector average using the ΔΔCt method. Box plots display the median as the center line, the 1st and 3rd quartiles as hinges and 1.5× interquartile range as whiskers. Significance was assessed using the two-tailed Mann Whitney U test. f,g Co-IP confirming ORF7b–STT3A and ORF7b–Sec61 interactions in HEK293T cells following transfection with ORF7b–FLAG or empty vector and STT3A–MYC or Sec61–V5, respectively. Each experiment was repeated independently three times with similar results. h, Co-IP confirming N-histone H1.4 interaction in HEK293T cells following transfection with N or empty vector and histone H1.4. Experiment was repeated independently three times with similar results.
In comparison to the published SARS-CoV-2–human protein–protein interactome networks2,4,5, our interactome validated 218 (38%) human host factors previously reported, including ALG5, G3BP1, CLCC1, VPS39, SIGMAR1, G3BP2 and RAP1GDS1, which are identified in all four interactomes (Fig. 2a). Our interactome offers 361 (62%) newly discovered human host factors which in total interact with SARS-CoV-2 proteins in 493 interactions. For S protein, which plays a key role in the entry of SARS-CoV-2 into host cells30, we identified 24 interacting partners. Among these interacting partners of S protein, we found that CORO1C31 and STON232, which are known to be present in the cell membrane, suggesting potential cell entry of SARS-CoV-2 through these human proteins in addition to known mechanisms.
We next performed more comparisons between these published interactomes and our own. First, we found that our interactome was enriched in several disease-relevant pathways and biological processes that were not enriched in previous interactomes, for example, ‘Coronavirus disease,’ ‘ribosome biogenesis,’ and ‘rRNA metabolic process’33–36 (Supplementary Table 2). Next, we examined whether these datasets contained interaction partners that coincided with genes that had expression changes in response to SARS-CoV-2 infection. To this end, we performed differential expression analysis for several bulk and single-cell RNA sequencing (RNA-seq) datasets from COVID-19 models or patients (Methods). For the single-cell dataset37, for which we compared the gene expression in SARS-CoV-2+ and SARS-CoV-2− cells, we found that our interactome showed significant overlap (Fisher’s exact test, FDR < 0.05) with the differentially expressed genes (DEGs) in more cell types than that of other interactomes (Extended Data Fig. 1c and Supplementary Table 3). Using four bulk RNA-seq/proteomics datasets that contained samples such as upper airway and bronchial epithelial cells38–41, we found that our interactome had a comparable number of significant overlaps to other datasets and showed the highest overall Jaccard index and overlap coefficient with the bulk RNA-seq datasets (Extended Data Fig. 1d and Supplementary Table 3). These results suggest that our interactome is highly enriched in genes differentially expressed in response to SARS-CoV-2 infection.
We next inspected the evolutionary features of the SARS-CoV-2 human host factors (Extended Data Fig. 1e,f and Supplementary Table 4). Previous studies have shown that virus host factors have more conserved dN/dS rates compared to non-virus host factors42,43. Our SARS-CoV-2–human interactome showed more purifying selection (quantified by lower non-synonymous versus synonymous substitution rate ratio (dN/dS ratio)), as well as a lower evolutionary rate ratio, compared to the random background from the human protein interactome. These bioinformatics observations further indicated high evolutionary conservation of host factors of SARS-CoV-2 identified by our Y2H and TMT-AP–MS proteomics platforms, consistent with previous studies44,45.
Gene expression patterns in disease-related tissues capture important information for revealing the pathogenesis of the disease and identifying potential treatments46–48. We therefore examined the expression of the human host factors in different tissues (Supplementary Fig. 4a,b and Supplementary Table 5) using the GTEx data49. By normalizing the expression of each gene across different tissues (tissue specificity; Methods), we found that lung ranked the 7th out of 33 tissues in terms of the number of host factors with positive tissue specificity (Supplementary Fig. 4a), suggesting that lung is one of the tissues where these host factors have high expression50.
Altogether, these results show the high quality of the SARS-CoV-2–human interactome identified in this study and strongly encouraged us to further look into the pathobiology of COVID-19 and potential treatment using our interactome.
Our interactome identifies potential COVID-19 pathobiology
ORF3a is a SARS-CoV-2 accessory protein that has been reported to induce apoptosis in HEK293T cells51 and to suppress the innate immune response52–54 via unclear molecular mechanisms. Our interactome revealed that ORF3a physically interacts with ZNF579 (Fig. 1b), a previously uncharacterized human protein likely to be a transcription factor. We were able to validate this interaction using co-immunoprecipitation (co-IP) western blotting (Fig. 2b). Furthermore, we found that the level of ZNF579 protein is decreased after overexpression of ORF3a in HEK293T cells (Fig. 2c). As a result, we hypothesized that the presence of ORF3a in cells might trigger changes in the transcriptional state of human genes that are normally regulated by ZNF579. Using ENCODE chromatin immunoprecipitation–sequencing (ChIP–seq) data55,56, we found that there is a significant enrichment of genes known to be dysregulated in SARS-CoV-2 infection among targets bound by ZNF579 (Extended Data Fig. 2a,b). These overlapped genes participate in several disease-relevant pathways such as ‘ribosome’33–36, ‘coronavirus disease’ and various infection-related pathways in several COVID-19 relevant cell types (Extended Data Fig. 2c). Specifically, ZNF579 is bound strongly to the promoter of HSPA6 (Fig. 2d). Using quantitative PCR (qPCR), we found that overexpression of ORF3a in HEK293T cells causes massive induction of HSPA6 (Fig. 2e). These results indicate that the multifunctional SARS-CoV-2 accessory protein ORF3a can induce expression of HSPA6, presumably by disrupting ZNF579, which is likely to normally exert repressive activity at the HSPA6 promoter. This represents an additional previously unknown activity of this multifunctional viral accessory protein.
Notably, the oligosaccharyltransferase (OST) complex subunits STT3A/B, RPN1/2 and DDOST57 were all shown to be present in our Y2H and AP–MS interactome datasets, which we further validated using co-IP (Fig. 2f). The OST complex catalyzes the N-glycosylation of nascent polypeptides in the endoplasmic reticulum58. Glycoproteins are critical for normal cell–cell interactions, RNA replication and pathogenesis59–61. Interestingly, OST inhibition has been shown to have activity against Dengue virus, Zika virus, West Nile virus, yellow fever viruses and HSV162–64 by affecting the viral replication. The OST complex was also found to be crucial for innate immune responses triggered by lipopolysaccharide65. Additionally, we also found Sec61 (Fig. 2g), which is a major component of the ER translocon that facilitates the entry of nascent polypeptides into the ER lumen for protein processing66. Evidence suggests that Sec61 may participate in the replication of several viruses like Ebola virus, Influenza virus, HIV and Dengue virus66–68. Thus, we hypothesize that OST and Sec61 may also participate in SARS-COV-2 replication and/or the host immune response, offering potential targets for host-targeting therapy development.
The SARS-CoV-2 nucleocapsid (N) protein binds to the viral RNA genome and is multifunctional in viral RNA transcription, replication and genome condensation69–71. N protein is conserved and stable with ~90% amino acid homology to the SARS-COV N protein72. From our dataset, we confirmed known interactions, including the stress granule core protein G3BP1/2 also found in three other interactome datasets. In addition to these known interactions, we identified an interaction between histone H1.4 and N protein. To validate this histone H1.4 and N protein interaction, we overexpressed both N protein and histone H1.4 to perform co-IP, confirming their interaction (Fig. 2h). Histone H1, also known as linker histone, mainly functions in chromatin condensation and transcriptional repression73,74. Accumulating evidence suggests that linker histone is essential in the pathogenesis of several diseases, particularly for viral infection74. There is also evidence that histone H1 could influence IFN and inhibit influenza replication75, in addition to playing a role in the regulation of viral gene expression76. Thus, we hypothesize that this viral–host interaction could also be involved in mediation viral replication and/or gene expression.
Discovery of host-targeting therapies for COVID-19
Using our newly discovered SARS-CoV-2–human protein–protein interactome network, we performed network-based drug screening for more than 2,900 FDA-approved or investigational drugs77 (Fig. 3a and Supplementary Table 6). We obtained a list of 189 FDA-approved drugs with significantly closer network proximities to the SARS-CoV-2 host factors, among which 44 had clinical trials for SARS-CoV-2 (Supplementary Table 7). To refine this list, we obtained the antiviral profiles of the top 189 drugs from NCATS (https://opendata.ncats.nih.gov/covid19/assays, National Center for Advancing Translational Sciences) and evaluated each drug for their desired antiviral properties (Methods). From this, we obtained a curated list of 23 drugs with significant network proximities to the SARS-CoV-2 host factors as well as desired anti-SARS-CoV-2 activities in at least two NCATS assays (Fig. 3b, Supplementary Fig. 5, Supplementary Table 8).
Fig. 3 |. Discovery of interactome-based host-targeting therapies for COVID-19.
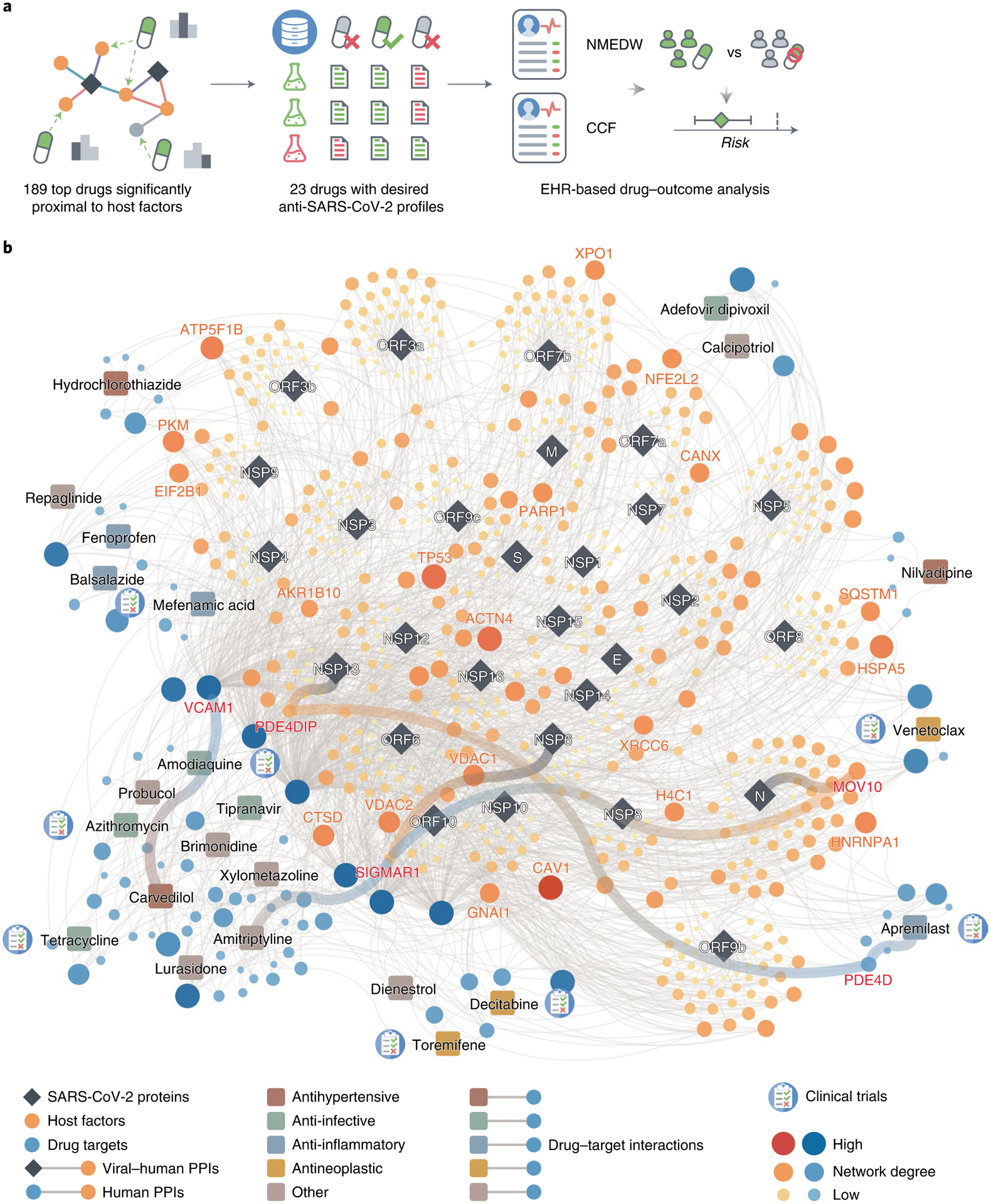
a, Work flow of drug repurposing for COVID-19 using our interactome. We ranked the drugs by their proximity to the SARS-CoV-2 host factors (Supplementary Table 7), filtered the top drugs by their NCATS anti-SARS-CoV-2 profiles (Supplementary Table 8) and finally analyzed their drug–outcome relationship using EHR data (Table 1 and Supplementary Table 9,10). b, The top 23 drugs can target the SARS-CoV-2 host factors directly or through protein–protein interactions with their targets.
Overall, these top drugs fall into several major categories, including anti-infective (amodiaquine, azithromycin, tetracycline, adefovir dipivoxil, tipranavir), anti-inflammatory (apremilast, mefenamic acid, balsalazide, fenoprofen), antihypertensive (carvedilol, hydrochlorothiazide, nilvadipine) and antineoplastic (toremifene, decitabine, venetoclax). Among these drugs, apremilast, toremifene, decitabine, amodiaquine, azithromycin are currently being or have been tested in clinical trials for SARS-CoV-2. These top 23 drugs offer candidate treatments for SARS-CoV-2 infections across diverse mechanism-of-actions identified from our human–SARS-CoV-2 interactome. For example, balsalazide, toremifene, tetracycline, venetoclax, tipranavir and brimonidine may inhibit viral replication by inhibiting papain-like protease 3CL (Supplementary Fig. 5 and Supplementary Table 8). Other drugs, such as carvedilol and hydrochlorothiazide, may directly inhibit viral entry by disrupting the spike–ACE2 PPI (Supplementary Fig. 5 and Supplementary Table 8). We also found some literature evidence that may provide mechanistic insights for these drugs against SARS-CoV-2 (Fig. 3b). For example, apremilast is a phosphodiesterase 4D (PDE4D) inhibitor78, which interacts with PDE4DIP79, a direct target of NSP13. Amitriptyline activates SIGMAR180, while NSP6 interact with SIGMAR1 to inhibit host autophagosome formation to facility coronavirus replication81. SIGMAR1 also interact with MOV10 (an RNA helicase, also a host factor targeted by the N protein), which exhibits antiviral activity against RNA viruses82.
To test whether our interactome identified drugs that could not be predicted by previously published datasets, we compared the screening results using different interactomes and their combinations. We found that 16 drugs identified by our interactome could not be predicted by any of the other three interactomes or their combinations (Extended Data Fig. 3a). Of the top 23 drugs with desired anti-SARS-CoV-2 profiles, 6 were among the 16 drugs that can only be identified by our interactome (Extended Data Fig. 3a). We also found that among the seven drugs identified by combining all four interactomes that could not be identified by any interactome individually, three drugs (Extended Data Fig. 3b) were found to have desired anti-SARS-CoV-2 profiles (Supplementary Fig. 6).
Among the top 23 drug candidates, toremifene achieved significantly closer network proximity (Z = −2.19, FDR = 0.037) and has a desired anti-SARS-CoV-2 profile. Previous studies show that toremifene blocks various viral infections efficiently, including SARS-CoV-283 (half-maximal inhibitory concentration (IC50) = 3.58 μM), SARS-CoV-184 (EC50 = 11.97 μM), MERS-CoV85 (EC50 = 12.9 μM) and Ebola virus86 (IC50 = ~1 μM). Indeed, NCATS data show that toremifene is active across four assays: spike–ACE2 protein–protein interaction (half-maximal activity concentration (AC50) = 11.92 μM), SARS-CoV pseudotyped particle entry (AC50 = 15.85 μM), MERS-CoV pseudotyped particle entry (AC50 = 31.62 μM) and 3CL enzymatic activity (AC50 = 5.01 μM) (Supplementary Fig. 5). Mechanistically, a previous study showed that toremifene may inhibit SARS-CoV-2 cell entry by blocking the S and NSP14 proteins87. These comprehensive validations show potential implications of SARS-CoV-2 interactome-predicted drugs (for example, toremifene) offer candidates to be tested further in COVID-19 patients.
Validation of interactome-predicted drugs
Further, we used subject matter expertize to select candidate drugs for patient-level data validation and experimental validation on the basis of a combination of factors: (1) strength of the interactome network-based prediction associations (a stronger network proximity score in Supplementary Table 7); (2) novelty of predicted drugs; (3) availability of sufficient patient data for meaningful evaluation (exclusion of infrequently used medications); and (4) ideal pharmacokinetics properties in the lung of interactome-predicted drugs. Applying these criteria resulted in two top candidate drugs, carvedilol (Z = −2.195, FDR = 0.03) and hydrochlorothiazide (Z = −2.428, FDR = 0.005), which are originally approved for treatment of hypertension.
To identify the drug–outcome relationships of these drugs, we used a state-of-the-art active user-design approach47,88, which is based on large-scale electronic health record (EHR) data. Using the Northwestern Medicine Enterprise Data Warehouse (NMEDW) COVID-19 dataset (481,526 total patients, 66,541 COVID-19 positive cases, Table 1), we found that both carvedilol (odds ratio = 0.8, 95% confidence interval 0.68–0.94, P = 0.008) and hydrochlorothiazide (odds ratio = 0.62, 95% confidence interval 0.47–0.82, P < 0.001) were associated with a significantly lowered risk of positive COVID-19 test after confounding adjustment (age, sex, race and comorbidities) using a propensity-score matching approach47,88 (Fig. 4a,b and Supplementary Table 9). The effect of carvedilol was consistent for different race and sex subgroups (Fig. 4a and Supplementary Table 9). To validate these observations, we used a second EHR database from the Cleveland Clinic COVID-19 registry as an external validation set (168,712 total individuals, 83,340 SARS-CoV-2 positive cases; Supplementary Table 10). We found that carvedilol had a sufficient number of usage cases for the drug–outcome evaluation. By comparing individuals with and without carvedilol usages (propensity-score-matched by age, sex, race and/or comorbidities), we found that carvedilol usage was associated with a 17% (odds ratio = 0.83, 95% confidence interval 0.78–0.88, P < 0.001) significantly lowered risk of COVID-19 positive test (Fig. 4c). This protective effect was also consistent when we examined subgroups from the registry in terms of race and sex (Fig. 4c).
Table 1 |.
Patient Characteristics of NMEDW dataset
| All patients | SARS-CoV-2 positive patients | |||
|---|---|---|---|---|
| Carvedilol− | Carvedilol+ | Carvedilol− | Carvedilol+ | |
| Total | 478,536 | 2,990 | 66,289 | 252 |
| Age | 44.67±21.74 | 67.37±15.08 | 41.86±21.09 | 63.52±17.20 |
| Sex, male | 202,994 (42.4) | 1,674 (56.0) | 30,550 (46.1) | 146 (57.9) |
| Race | ||||
| Black | 41,858 (8.7) | 725 (24.2) | 6,136 (9.3) | 73 (29.0) |
| White | 343,549 (71.8) | 1,927 (64.4) | 46,493 (70.1) | 143 (56.7) |
| Other | 60,273 (12.6) | 274 (9.2) | 9,407 (14.2) | 27 (10.7) |
| Comorbidity | ||||
| AIDS HIV | 1,843 (0.4) | 36 (1.2) | 248 (0.4) | 5 (2.0) |
| CD | 32,555 (6.8) | 1,336 (44.7) | 3,498 (5.3) | 103 (40.9) |
| CPD | 87,868 (18.4) | 1,249 (41.8) | 12,078 (18.2) | 123 (48.8) |
| CHF | 26,973 (5.6) | 2,042 (68.3) | 3,331 (5.0) | 172 (68.3) |
| Dementia | 8,567 (1.8) | 358 (12.0) | 1,297 (2.0) | 42 (16.7) |
| Diabetes with cc | 20,670 (4.3) | 1,359 (45.5) | 3,031 (4.6) | 139 (55.2) |
| Diabetes without cc | 54,322 (11.4) | 1,629 (54.5) | 8,301 (12.5) | 150 (59.5) |
| HP | 5,380 (1.1) | 298 (10.0) | 568 (0.9) | 27 (10.7) |
| Malignancy | 47,660 (10.0) | 805 (26.9) | 4,606 (6.9) | 67 (26.6) |
| MST | 23,690 (5.0) | 413 (13.8) | 2,385 (3.6) | 36 (14.3) |
| MLD | 29,730 (6.2) | 583 (19.5) | 3,711 (5.6) | 53 (21.0) |
| MSLD | 2,906 (0.6) | 119 (4.0) | 307 (0.5) | 11 (4.4) |
| MI | 7,913 (1.7) | 630 (21.1) | 1,017 (1.5) | 54 (21.4) |
| PUD | 11,785 (2.5) | 309 (10.3) | 1,289 (1.9) | 32 (12.7) |
| PVD | 23,925 (5.0) | 1,158 (38.7) | 2,626 (4.0) | 99 (39.3) |
| RD | 26,068 (5.4) | 1,820 (60.9) | 3,479 (5.2) | 181 (71.8) |
| RHD | 13,887 (2.9) | 232 (7.8) | 1,607 (2.4) | 18 (7.1) |
Age is shown as mean ± standard deviation. All other characteristics are shown as number of cases (percentage). P values were calculated by two-sided t-test for age and Fisher’s exact test for other variables. AIDS HIV, acquired immunodeficiency syndrome and human immunodeficiency virus; CD, cerebrovascular disease; CPD, chronic pulmonary disease; CHF, congestive heart failure; diabetes with/without cc, diabetes with/without chronic complications; HP, hemiplegia or paraplegia; MST, metastatic solid tumor; MLD, mild liver disease; MSLD, moderate or severe liver disease; MI, myocardial infarction; PUD, peptic ulcer disease; PVD, peripheral vascular disease; RD, renal disease; RHD, rheumatic disease.
Fig. 4 |. Population-based and experimental validation of interactome-predicted drugs.
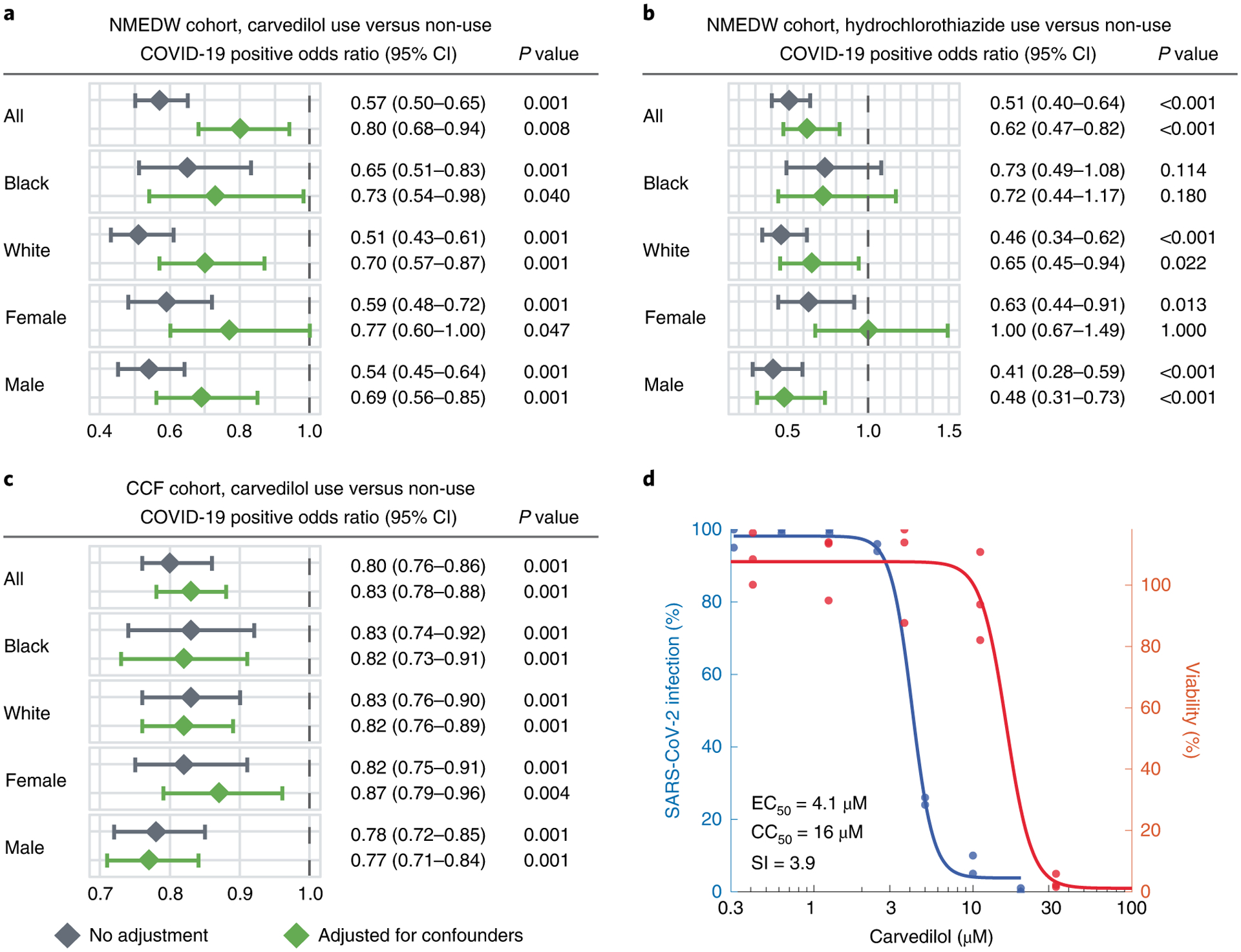
a–c, Drug–outcome evaluation using the NMEDW and CCF COVID-19 databases. Odds ratio was used to evaluate the carvedilol effect to the positive laboratory test result of COVID-19. Statistically significant at P < 0.05 and odds ratio < 1 indicate that the carvedilol user with lower odds of COVID-19 positive testing. The central diamond box denotes the odds ratio and the error bar denotes the 95% confidence interval. Patients were matched with propensity score using age, sex, race and other comorbidities (Table 1) to reduce various confounding factors. Statistics derived from cohort details shown in Table 1. d, Experimental validation of the anti-SARS-CoV-2 activity of carvedilol showed an EC50 value of 4.1 μM and low cell toxicity. CC50, half-maximal cytotoxic concentration; SI, selectivity index (SI = CC50/EC50).
We found that carvedilol not only showed favorable results in the EHR-based validation, but also has a promising antiviral profile from NCATS, showing high potencies for multiple desired activities (Supplementary Fig. 5). The NCATS profile of carvedilol is comparable to that of remdesivir, whose profile was deemed highly desirable89. We then investigated the anti-SARS-CoV-2 activity of carvedilol experimentally. We treated A549-ACE2 cells with 0.3–20 μM of carvedilol for 2 h followed by infection with SARS-CoV-2 at a multiplicity of infection of 0.5 and incubation for 2 days. Carvedilol showed a low cell toxicity (Fig. 4d). Cells were subsequently fixed and immunostained to detect for S protein, which was used as a marker for infection. We found that carvedilol inhibited SARS-CoV-2 infection with an EC50 value of 4.1 μM (Fig. 4d), mechanistically supporting our SARS-CoV-2–human interactome-based prediction and EHR-based findings. Lastly, we conducted drug–target network analysis of the targets of carvedilol and SARS-CoV-2 host factors (Extended Data Fig. 4 and Supplementary Table 11). We found that carvedilol could potentially affect the SARS-CoV-2 host factors (that is, VCAM1 and KCNH2) through PPIs with its targets (Extended Data Fig. 6).
Discussion
In this study, we used high-throughput Y2H and quantitative TMT-AP–MS to generate a binary and co-complex SARS-CoV-2–human protein–protein interactome network, expanding the known map produced solely by label-free AP–MS2–5. This interactome validated 218 previously published SARS-CoV-2 host factors and revealed 361 novel ones. In comparison with previous interactomes2–5, this interactome has higher overlaps among the interactomes and differentially expressed genes captured by bulk and single-cell RNA-seq of SARS-CoV-2 infection. The host factors we identified, particularly those altered in response to SARS-CoV-2 infection, will contribute to understanding the disease pathobiology of COVID-19 and prioritizing potential drug targets.
Among the novel interacting partners for S protein, we identified several human proteins that may play important roles in SARS-CoV-2 infection. CORO1C31 and STON232 are expressed on the cell membrane. CORO1C is highly expressed in lung (Supplementary Table 5). STON2 is ubiquitously expressed and involved in endocytic machinery32. It is possible that SARS-CoV-2 can enter host cells through binding of S protein not only to ACE2, NRP190,91 and BSG92, but also other (unknown) factors such as CORO1C and STON2. We also noticed two proteins, EPPK193 and SPECC1L94, that both express on the cell junctions. It has been suggested that SARS-CoV-2 could spread through cell-to-cell transmission95. These cell junction proteins that can be targeted by SARS-CoV-2 S protein may facilitate its cell-to-cell transmission.
We identified a previously uncharacterized human transcription factor, ZNF579, that interacts with SARS-CoV-2 accessory protein ORF3a, and report that this interaction leads to the de-repression of HSPA6. Notably, HSPA6 is significantly upregulated after SARS-CoV-2 infection in cell culture models38, indicating that the disruption of ZNF579 by ORF3a may be relevant in the context of infection. HSPA6 is a HSP70 family molecular chaperone, which are known to be involved in the entry, replication, assembly and release of various viral pathogens96. We speculate that SARS-CoV-2 has evolved this activity to ensure sufficient levels of molecular chaperones are available to assist with the production of viral proteins in cells.
Next, using this newly discovered SARS-CoV-2–human protein–protein interactome, we studied drug repurposing and identified 23 candidate drugs with desired anti-SARS-CoV-2 profiles. In this study, we used undirected human protein interactome network and degree preserved node shuffling technique. We also tested different variations of the proximity analysis and found that using directed human protein interactome network and using degree preserved link shuffling resulted in overall highly consistent Z-scores compared to the original results in this study (Extended Data Fig. 5). Although some of these drugs can directly target the host factors, most of them indirectly affect the host factors through PPIs with their targets (Fig. 3b). For example, our predicted drug candidates are validated by well-established NCATS assays (Supplementary Fig. 5). In addition, among the drugs which did not have NCATS assay results, alprazolam (Iranian Registry of Clinical Trials: IRCT20211015052773N1), l-citrulline (ClinicalTrials.gov IDs: NCT04404426 and NCT04570384), nadroparin (European Union Clinical Trials Register: EUCTR2020-001709-21-FR, EUCTR2020-001739-28-BE, EUCTR2020-005884-29-IT), vortioxetine (ClinicalTrials.gov ID: NCT05047952) and myrrh (Clinical Trials Registry—India: CTRI/2020/07/026669, CTRI/2020/12/029575, CTRI/2021/01/030825, Australian New Zealand Clinical Trials Registry: ACTRN12622000215729) are in clinical trials for COVID-19. Nevertheless, future experimental and clinical studies for our novel predicted drug candidates are warranted.
Further, we have identified carvedilol and hydrochlorothiazide as potential host-targeting treatments for COVID-19 supported by multiple lines of evidence (strong network proximities to SARS-CoV-2 host factors, significantly reduced SARS-CoV-2 positive test risks in patients using these drugs on the basis of large-scale EHR data and experimental validation of anti-SARS-CoV-2 activity). As drug repurposing focuses on drugs that are already in existing patient databases, we are able to test hypotheses using EHR data as we88,97 and other teams98,99 demonstrated. The unique strengths of EHRs include their provision of large patient populations useful for detecting small differences and the availability of a large number of patient factors recorded without risk of recall bias, allowing for high-dimensional covariate adjustment to minimize confounding47,98,100. Our findings are consistent with previous reports that hydrochlorothiazide98 and carvedilol99 have potential beneficial effects for COVID-19 patients. Another beta-blocker metoprolol (Z = −2.327, FDR = 0.003) was also among the top 189 drugs (Supplementary Table 7), which has been tested in a small clinical trial with positive effects101. These results confirm that the unique integration of SARS-CoV-2–human interactome findings and patient analysis approaches using two large-scale EHR databases from two independent health care systems, along with in vitro antiviral observations, offer a powerful strategy for discovery of COVID-19 therapeutics. This kind of systems biology strategy can be applied to future pandemics as well.
To understand the potential mechanisms of the anti-SARS-CoV-2 activity of carvedilol, we examined the mechanism-of-action of carvedilol using network analysis (Extended Data Fig. 4 and Supplementary Table 11). Among the 579 unique host factors, 237 (41%) have PPIs with carvedilol targets. A large portion of the human proteins in the enriched pathways (protein translation (26/37, 70%), mRNA splicing (14/21, 67%), glucose metabolism (9/15, 60%) and neutrophil-mediated immunity (14/27, 52%)) have PPIs with carvedilol targets, suggesting a potential mechanism-of-action in which carvedilol inhibits SARS-CoV-2 replication through multiple important pathways such as protein translation and mRNA splicing. We found several carvedilol targets that exhibited closer network distance to the virus host factors and COVID-19 pathways, such as GJA1, KCNH2, NDUFC2, VCAM1 and VEGFA (Extended Data Fig. 4). For example, VCAM1 plays important roles and has elevated levels in COVID-19102,103, and carvedilol can inhibit expression of VCAM1104. These results offer hypotheses that can be tested for the anti-SARS-CoV-2 effect of carvedilol. Yet, future experimental validation to decipher the anti-SARS-CoV-2 mechanism-of-action of carvedilol is highly warranted as well104.
We acknowledge several limitations. For Y2H, expression in yeast cells or translocation into the nuclei may be inefficient for some viral or human proteins, which can lead to false negatives. While fragment libraries can improve the coverage of Y2H screens, they can also lead to self-activating false positives105. For AP–MS, although overexpression of bait proteins can lead to false positives, it is not a limitation in the context of viral–host interaction studies. Viral proteins are inherently exogenous to host cells and expressed at a high level upon natural infection. Many of the overexpression promoters used for AP–MS are, in fact, viral-derived. Another factor is the reported limitation of ratio compression in TMT, which has been alleviated by the development of the SPS MS3-based acquisition work flow106,107. Moreover, as confirmed by previous studies on direct comparisons between TMT-based and label-free quantification108,109, TMT-based quantification has better precision (that is, consistency of measurements across replicates), while label-free quantification has large numbers of missing values, even with strategies like match between runs108. Thus, TMT approaches have better statistical power to detect abundance changes for more proteins, leading to better coverage108,109. The network-based SARS-CoV-2 treatment discovery may be affected by the incompleteness of the human protein–protein interactome and drug–target network. Therefore, we relied not only on the network discoveries, but also incorporated other types of evidence, such as EHR-based validation and experimental validation. Our EHR-based validation is retrospective and can only be applied to commonly used drugs owing to data availability. Although we adjusted for several confounding factors, other unknown factors may still have effect on the results of EHR-based validation. Therefore, the drugs identified in this study must be validated using randomized clinical trials before they can be used in patients with COVID-19. Lastly, we focused on the SARS-CoV-2–human protein interactome in this study. Combining multiple data resources (such as clustered regularly interspaced short palindromic repeats (CRISPR)110, genome-wide association studies111, rare variants112, synthetic-lethality-based genetics interactions113 and metabolomics and proteomics114) may help build up comprehensive knowledge of COVID-19 using various advanced computational (for example, genome-scale metabolic modeling115) and multi-omics data integration approaches.
Methods
SARS-CoV-2 ORF clones
ORF3b (plasmid no. 141384; Addgene), NSP4 (plasmid no. 141369; Addgene), NSP12 (plasmid no. 141378; Addgene), NSP13 (plasmid no. 141379; Addgene) and NSP14 (plasmid no. 141380; Addgene) were a gift from N. Krogan, University of California, San Francisco. NSP6 (plasmid no. 149309; Addgene) and NSP16 (plasmid no. 141269; Addgene) were a gift from F. Roth, University of Toronto, which we cloned into our pHAGE-CMV-GAW-3xMyc-IRES-PURO construct using Gateway. E, M, N, NSP1, NSP2, NSP3, NSP5, NSP7, NSP8, NSP9, NSP10, NSP15, ORF3a, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, ORF10 and S, cloned into pCAG-FLAG and pcDNA6B-FLAG constructs, were a gift from P.-H. Wang, Shandong University. All SARS-CoV-2 ORFs were codon-optimized and expressed in either pLVX-EF1alpha-2xStrep-IRES-Puro (plasmid no. 141395; Addgene), pHAGE-CMV-3xMyc-IRES-PURO, pCAG-FLAG, or pcDNA6B-FLAG mammalian expression vectors.
Y2H
Y2H screens were carried out as previously described8,21–24. In brief, viral ORFs were cloned into pDEST-AD and pDEST-DB vectors using Gateway LR to generate N-terminal ORF fusions. Similarly, human ORFeome 8.1 (ref.20) was cloned into pDEST-AD and pDEST-DB vectors. All AD and DB expression clones were transformed into Y2H Saccharomyces cerevisiae strains MATa Y8800 and MATα Y8930 (genotype: leu2-3, 112 trp1-901 his3Δ200 ura3-52 gal4Δ gal80Δ GAL2∷ADE2 GAL1∷HIS3@LYS2 GAL7∷lacZ@MET2 cyh2R), respectively. To screen out autoactivating DB-ORFs, all DB-ORF MATα Y8930 transformants were mated pairwise against empty pDEST-AD MATa Y8800 transformants and scored for growth on SC-Leu-Trp+3AT and SC-Leu-Trp-Ade plates, where DB-ORFs that triggered reporter activity were removed from further experiments. To increase screening throughput, 24 human ORF AD or DB clones were pooled into single human ORF AD or DB wells, respectively. Viral ORF AD and DB clones were then mated pairwise against pools of human ORF DB and AD clones, respectively, for the first round of Y2H testing (called phenotyping I). Mated transformants were incubated overnight at 30 °C before being plated onto SC-Leu-Trp to select for mated diploid yeast. After another overnight incubation at 30 °C, diploid yeast was plated onto SC-Leu-Trp-His+3AT and SC-Leu-Trp-Ade selection plates. After another overnight incubation at 30 °C, plates were replica cleaned and incubated again for 3 days at 30 °C for colony picking. All yeast colonies picked from phenotyping I were individually subjected to another round of Y2H testing called phenotyping II. Positive colonies from phenotyping II were processed for PLATE-seq to identify each AD-X and DB-Y pair. Finally, for every AD-Y and DB-X identified by PLATE-seq, we performed pairwise Y2H testing of each identified pair to ensure high reproducibility.
PLATE-seq
Each colony was picked into 96-well plates containing 15 μl 2.5 mg ml−1 Zymolyase (catalog no. E1004; Zymo Research) and incubated for 45 min at 37 °C followed by 10 min at 95 °C to prepare yeast cell lysate used as PLATE-seq DNA template. PLATE-seq was carried out as previously described24. In brief, plasmid(s) from individual wells of 96-well plates were PCR amplified using a plasmid-specific forward primer and a reverse primer consisting of a well-position-specific barcode and TruSeq 3′ sequencing adapter. Amplicons derived from the same 96-well plate were pooled and purified using QIAquick PCR Purification Kit (catalog no. 28104; Qiagen). Each amplicon pool was subject to Tn5 tagmentation to fragment the amplicons and append adapters consisting of a plate-specific barcode and TruSeq 5′ sequencing adapter. Tagmented DNA was purified using QIAquick PCR Purification Kit (catalog no. 28104; Qiagen) and pooled across all 96-well plates. These pools were then subjected to low-cycle PCR both to extend the TruSeq end adapters with sequences compatible for binding to the Illumina flow cell and to enrich for only DNA fragments consisting of TruSeq adapter sequences on both ends of the plate-specific and well-position-specific barcodes. PLATE-seq libraries were paired-end sequenced on an Illumina MiSeq.
Affinity purification
Caco-2 (HTB-37; ATCC) cells were cultured in EMEM (catalog no. 30–2003; ATCC) with 15% fetal bovine serum (FBS) (catalog no. 30–2020; ATCC) at 37 °C with 5% CO2. All 28 SARS-CoV-2 ORFs were codon-optimized and cloned into mammalian expression vectors that contained Strep, Myc, or FLAG affinity tags. SARS-CoV-2 ORF plasmids and corresponding empty vectors were individually transfected in biological duplicates into Caco-2 cells using Lipofectamine 3000 Transfection reagent (catalog no. L3000001; Invitrogen) following manufacturer’s instructions. Cells were harvested 72 h after transfection and lysed using RIPA lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% (v/v) Nonidet P 40 Substitute, 5 mM EDTA, phosphatase inhibitor (catalog no. 4906845001; Roche) and protease inhibitor cocktail (catalog no. 11873580001; Roche)). Samples were incubated for 30 min at 4 °C and then centrifuged at 13,000g for 15 min at 4 °C. Supernatants were collected and incubate with either MagStrep ‘type3’ XT beads (catalog no. 2-4090-002; IBA Lifesciences), Myc-Trap Agarose (catalog no. yta-10; ChromoTek) or Anti-FLAG M2 Affinity Gel (catalog no. A2220; Millipore) overnight at 4 °C. Strep-tagged samples were washed with 10x Buffer W (catalog no. 2-1003-100; IBA Lifesciences) three times at 4 °C. Myc- and FLAG-tagged samples were washed with RIPA buffer. Strep-tagged samples were eluted using 10x Buffer BXT (catalog no. 2-1042-025; IBA Lifesciences). Myc- and FLAG-tagged samples were eluted using IP elution buffer (100 mM Tris-HCl [pH 7.5], 1% (v/v) SDS) and incubated for 15 min at 65 °C. Other primary antibodies used in this study include c-Myc monoclonal antibody (catalog no. 13-2500; Invitrogen) and monoclonal anti-FLAG M2 antibody (catalog no. F3165; Sigma-Aldrich).
Proteomic sample preparation
IP eluates were reduced using 200 mM TCEP for 1 h at 55 °C. Samples were then alkylated using 375 mM iodoacetamide for 30 min at room temperature in the absence of light. Samples were digested using Trypsin Gold, mass spectrometry grade (catalog no. V5280; Promega) at an enzyme-to-substrate ratio of 1:100 and incubated overnight with nutation at 37 °C. Peptide concentrations were measured using Pierce Quantitative Colorimetric Peptide Assay (catalog no. 23275; Thermo Scientific). Samples were normalized and resuspended using 1 M triethylammonium bicarbonate for TMT experiments (catalog no. 90114; Thermo Scientific). Samples were labeled using TMT10plex Isobaric Mass Tagging Kit (catalog no. 90113; Thermo Scientific) or TMTsixplex Isobaric Label Reagent Set (catalog no. 90061; Thermo Scientific) at a (w/w) label-to-peptide ratio of 10:1 for 1 h at room temperature. Labeling reactions were quenched by the addition of 5% hydroxylamine and immediately pooled and dried using a SpeedVac. Labeled peptides were enriched and fractionated using Pierce High pH Reversed-Phase Peptide Fractionation Kit according to the manufacturer’s protocol (catalog no. 84868; Thermo Scientific).
Liquid chromatography–tandem mass spectrometry
Fractions were analyzed using an EASY-nLC 1200 System (catalog no. LC140; Thermo Scientific) equipped with an in-house 3 μm C18 resin-(Michrom BioResources) packed capillary column (75 μm × 25 cm) coupled to an Orbitrap Fusion Lumos Tribrid Mass Spectrometer (catalog no. IQLAAEGAAPFADBMBHQ; Thermo Scientific). The mobile phase and elution gradient used for peptide separation were as follows: 0.1% formic acid in water as buffer A and 0.1% formic acid in 80% acetonitrile as buffer B; 0–5 min, 5%-10% B; 5–65 min, 10–55% B; 66–67 min, 55%-95% B; 67–68 min, 2% B; 68–72 min, 95% B; 72–80 min, 5% B; with a flow rate set to 200 nl min−1. MS1 precursors were detected at m/z = 375–1500 and resolution = 120,000. A CID-MS2-HCD-MS3 method was used for MSn data acquisition. Precursor ions with charge of 2+ to 7+ were selected for MS2 analysis at resolution = 30,000, isolation width = 0.4 m/z, maximum injection time = 50 ms and CID collision energy at 35%. 6 SPS precursors were selected for MS3 analysis and ions were fragmented using HCD collision energy at 65%. Spectra were recorded using Thermo Xcalibur Software v.4.1 (catalog no. OPTON-30965; Thermo Scientific) and Tune application v.3.0 (Thermo Scientific). Raw data were searched using Proteome Discoverer Software 2.3 (Thermo Scientific) against an UniProtKB human database containing all SARS-COV-2 proteins. Search parameters specified precursor mass and fragment mass tolerance of 15 p.p.m. Peptide-spectrum matches (PSMs) were searched with SEQUEST HT and Percolator and filtered at FDR < 1%.
See Supplementary Table 12a for viral bait vector and tag information, Supplementary Table 12b,c for TMT10/TMT6 channel label information, including bait and control tags; and Supplementary Table 12d for isotope impurity correction information. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD035805.
Downstream proteomic analysis
We applied the MSstatsTMT116 linear-model-based pipeline (optimized for our experimental setup and purpose) to identify high-confidence viral–host interactions from TMT-AP–MS datasets. In brief, PSMs filtered at 1% FDR were selected for quantification by (1) the number of reporter intensity values per fraction, (2) percent isolation interference and (3) precursor intensity values to select for one instance of a peptide peak. If more than one PSM passed these criteria, then the average of the reporter ion intensities per channel of these PSMs were taken to represent the quantification of the peptide peak. The reporter intensity values of selected PSMs were log transformed, weighed with their respective precursor intensities and averaged to obtain protein level quantification values. Our pipeline’s novelty lies in its ability to retain useful information separated across fractions at the PSM level while ensuring no violation of the assumption of independence, such that our linear fixed-effects model with conditions (for example, sample versus control), as a fixed effect, can be utilized. An improved P value calculation was used through empirical Bayes estimation of prior variance as implemented using limma package v.3.46.0 (ref.117) in R platform v.4.0.3.
The FC and P values obtained from this linear-model-based approach are used to generate volcano plots for each viral bait protein compared to control. A baseline cutoff was set at a FC of greater than 2 and P value of less than 5%, on top of which a hyperbolic curve is optimized using the distribution of the log-transformed FCs of all identified proteins to identify high-confidence interactors. As a result, the actual cutoffs used for each AP–MS experiment are often significantly more stringent than the baseline values. A PSM cutoff, along with a peptide-coverage percent cutoff (that is, the percentage of all possible trypsin-digested peptides, accounting for up to two missed cleavages that can be found), on the basis of the number of the PSMs of the viral protein’s and peptide-coverage percentage, is also implemented before the optimization of this hyperbolic curve.
Co-immunoprecipitation
HEK293T (CRL-3216; ATCC) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (catalog no. 30-2002; ATCC) supplemented with 10% FBS (catalog no. 30-2020; ATCC) and incubated at 37 °C with 5% CO2. Cells were seeded onto six-well plates and grown until reaching 70–80% confluency. SARS-CoV-2 N, ORF3a, ORF7b, histone H1.4 or N + histone H1.4, Sec61 or ORF7b + Sec61, STT3A or ORF7b + STT3A and empty vector controls were transfected into cells by combining 2 μg of DNA with 10 μl of 1 mg ml−1 PEI (catalog no. 23966; Polysciences) and 150 μl Opti-MEM (catalog no. 31985062; Gibco). After 24 h incubation, cells were gently washed three times with Dulbecco’s PBS (1×) (catalog no. 14040117; Gibco), resuspended with 200 μl cell lysis buffer (10 mM Tris-HCl [pH 8.0], 137 mM NaCl, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 2 mM EDTA and protease inhibitor cocktail (catalog no. 11873580001; Roche)) and incubated on ice for 30 min. Extracts were then cleared by centrifugation at 16,000g for 10 min at 4 °C. To perform co-immunoprecipitation (co-IP), 100 μl cell lysate was incubated with 5 μl Red Anti-FLAG M2 Affinity Gel (catalog no. F2426; Millipore) overnight at 4 °C under gentle rotation. Bound proteins were then washed three times with cell lysis buffer, eluted with 50 μl elution buffer (10 mM Tris-HCl [pH 8.0], 1% (v/v) SDS) and incubated for 10 min at 65 °C. Cell lysates and co-IP samples were then treated with 6× SDS protein loading buffer (1 M Tris-HCl pH 6.8, 10% (v/v) SDS, 50% (v/v) glycerol, 0.03% (v/v) bromophenol blue and 10% (v/v) β-mercaptoethanol), subjected to SDS-PAGE, and transferred onto PVDF membranes (catalog no. GE10600023; Amersham). For immunoblotting analysis, V5 Tag monoclonal antibody (catalog no. R960-25; Invitrogen), c-Myc monoclonal antibody (catalog no. 13-2500; Invitrogen), monoclonal anti-FLAG M2 antibody (catalog no. F1804; Sigma-Aldrich), or ZNF579 polyclonal antibody (catalog no. A303-275A; Bethyl Laboratories) were used at 1:1,000 dilutions.
Quantitative PCR
HEK293T cells were cultured as above, and ORFa–FLAG or empty vector were introduced with Lipofectamine 2000 transfection reagent (catalog no. 11668030; Invitrogen) according to the manufacturer’s instructions. Transfection experiments were performed in duplicate. Medium was replaced 6 h after transfection, and RNA was harvested using TRIzol Reagent (catalog no. 15596018; Invitrogen). Reverse transcription was performed with the Maxima First Strand cDNA Synthesis Kit for qPCR with reverse transcription (RT–qPCR), with dsDNase (catalog no. K1671; Thermo Scientific). qPCR was performed on a LightCycler 480 System using LightCycler FastStart DNA Master SYBR Green I (catalog no. 03003230001; Roche Diagnostics). We used two primer sets for HSPA6 (FWD1: CAAGGTGCGCGTATGCTAC, REV1: GCTCATTGATGATCCGCAACAC, FWD2: CATCGCCTATGGGCTGGAC, REV2: GGAGAGAACCGACACATCGAA), and performed three technical replicates of three concentrations of cDNA (1:10, 1:100, 1:1,000) for each replicate, and then compared expression levels normalized to GAPDH using the ΔΔCp method.
Interactome comparative analysis
We compared our SARS-CoV-2–human interactome to a collection of three previously reported interactomes3–5, and compared with ours in terms of the overlap (Fisher’s exact test) with the differentially expressed genes in SARS-CoV-2 from several SARS-CoV-2 RNA-seq/proteomics datasets. These datasets include: (1) a single-cell dataset that contains CD8, epithelial (epi)-ciliated, epi-secretory, epi-squamous, macro, mono and natural killer cells from BALF37. We performed comparisons of virus+ versus virus− cells for each cell type; (2) bulk RNA-seq of human bronchial epithelial cells infected with SARS-CoV-238 (GSE147507), denoted as SARS2-DEG; (3) proteomic dataset of human Caco-2 cells infected with SARS-CoV-239, denoted as SARS2-DEP; (4) bulk RNA-seq of upper airway from COVID-19 patients versus non-COVID-19 patients (GSE156063)40, denoted as DE-NS; (5) bulk RNA-seq of peripheral blood mononuclear cell (PBMC) isolated from COVID-19 patients versus non-COVID-19 patients (GSE157103)41, denoted as DE-PBMC. For differential expression analysis, a cutoff of |log2FC| > 0.5 and FDR < 0.05 was considered significant. We calculated the Jaccard index (J) and overlap coefficient (C)118 for two gene sets A and B as below:
| (1) |
| (2) |
Functional enrichment analysis
Functional enrichment of our SARS-CoV-2 host factors were analyzed using Enrichr119 against the KEGG and GO biological process datasets. Pathways a GO terms with FDR < 0.05 were considered significantly enriched. GO terms were summarized using Revigo120.
Selective pressure and evolutionary rates
The non-synonymous and synonymous substitution rate ratio (dN/dS ratio)121 and the evolutionary rate ratio122 of our SARS-CoV-2 host factors were evaluated as described in a previous study123. For dN/dS ratio, dN/dS < 1 was considered purifying selection; dN/dS = 1 was considered neutral evolution; and dN/dS > 1 was considered positive Darwinian selection. The evolutionary rate ratio >1 was regarded as a fast rate and <1 as a slow rate122.
Tissue gene expression specificity
We evaluated the gene expression specificity of the SARS-CoV-2 host factors in 33 tissues using the RNA-Seq data from GTEx V8 (ref.49) (https://www.gtexportal.org/home/). The expression specificity of gene i in tissue t was defined as
| (3) |
where Ei was the mean and σi was the standard deviation of the expression of gene i across all considered tissues, and Eit was the mean expression of gene i in tissue t.
Construction of human protein–protein interactome and drug–target network
The human protein–protein interactome and the drug–target network were used to screen for drugs against the SARS-CoV-2 host factors. The human protein–protein interactome, composed of 17,706 protein nodes and 351,444 unique PPI edges was constructed in our previous studies46,47,124,125. In brief, several types of high-quality PPI evidence gathered from public databases and datasets were considered, including: binary PPIs identified by high-throughput yeast two-hybrid in three datasets47,126,127; low- or high-throughput experimentally discovered kinase–substrate interactions from KinomeNetworkX128, PhosphoNetworks129, Human Protein Resource Database130, DbPTM 3.0 (ref.131), Phospho.ELM132 and PhosphositePlus133; signaling networks identified using low-throughput experiments in SignaLink2.0 (ref.134); protein complexes revealed by robust affinity purification-mass spectrometry in BioPlex v.2.016135; and curated PPIs from Instruct136, IntAct137, BioGRID138, MINT139, PINA140 and InnateDB141 that were identified by yeast two-hybrid studies, affinity purification-mass spectrometry, protein three-dimensional structures, or low-throughput experiments. For comparison, we also built a directed version of the human protein interactome using the PPI direction information (including kinase–substrate and signaling networks) from PhosphositePlus133 and SignaLink2.0 (ref.134).
The drug–target network was constructed using several data sources as described in our recent studies46,47,124: DrugBank database (v.4.3)142, BindingDB143, ChEMBL (v.20)144, Therapeutic Target Database145, PharmGKB database146 and IUPHAR/BPS Guide to PHARMACOLOGY147. Binding affinities Ki, Kd, IC50 or EC50 ≤ 10 μM were used as cutoff for the drug–target interactions. All networks were visualized using Cytoscape 3.8.0148. Clinical trial information was retrieved from the International Clinical Trials Registry Platform (assessed in May 2022).
Network proximity-based drug and drug combination screening
The ‘closest’ network proximity measure was used to screen for 2,938 FDA-approved or investigational drugs. The ‘closest’ distance dAB for two gene/protein sets A (for example, drug targets) and B (for example, SARS-CoV-2 host factors) was calculated as:
| (4) |
where d(a, b) is the shortest path length of a and b in the human protein–protein interactome. Network proximity dAB was further normalized to obtain a Z-score using a permutation test with randomly selected proteins from the interactome with similar degree distributions to A and B (degree preserved node shuffling). Permutation tests were repeated 1,000 times. We prioritized drugs by Z < −2 and FDR < 0.05. For comparison, we also conducted degree preserved link shuffling using the double edge swap method to swap the links ten times the size of the human protein interactome times. For individual drug–target level network proximity to the disease modules, we used the ‘shortest’ measure that measures the average shortest distances of a target to the disease proteins (host factors):
| (5) |
The antiviral profiles of the prioritized drugs were retrieved from NCATS (https://opendata.ncats.nih.gov/covid19/assays). NCATS contains experimental high-throughput screening results for drugs from a series of screenings (some accompanied by counter screens) to evaluate their anti-SARS-CoV-2 potential. We included the following screening results: SARS-CoV-2 cytopathic effect and its counter screen SARS-CoV-2 cytopathic effect (host tox counter)/cytotoxicity; human fibroblast toxicity; spike–ACE2 protein–protein interaction (AlphaLISA) and its counter screen spike–ACE2 protein–protein interaction (TruHit Counter); ACE2 enzymatic activity; SARS-CoV pseudotyped particle entry (CoV-PPE) and its counter screen SARS-CoV pseudotyped particle entry counter screen (CoV-PPE_cs); MERS-CoV pseudotyped particle entry (MERS-PPE) and its counter screen MERS-CoV pseudotyped particle entry counter screen (MERS-PPE_cs); and 3CL enzymatic activity. on the basis of the NCATS SARS-CoV-2 data, we further selected a list of top drugs from the network proximity-based prioritization that show ideal activities in at least two of these screenings.
COVID-19 patient data observations
Two independent datasets revealed corroborating evidence for the drug carvedilol, which was identified by our interactome prioritization framework. The first dataset (discovery dataset) was from the NMEDW. We first identified 512,198 patients who had SARS-CoV-2 reverse transcription-polymerase chain reaction (RT–qPCR) test results recorded in NMEDW. Patients with a positive RT–qPCR test were considered COVID-19 positive, where the earliest time of the test was recorded as the effective time. Patients that did not have any positive or presumptive positive RT–qPCR tests and the latest PCR test was negative (excluding pending and undetermined results) were considered COVID-19 negative, where the latest time of the test was recorded as the effective time. By these metrics, 29,224 patients with pending or undetermined results were removed, yielding 482,974 patients of interest. We excluded patients without age or sex information yielding a cohort of 481,526 patients, 66,541 of which were COVID-19 positive. We then extracted the carvedilol (and other drugs) administration information for all patients in the final cohort. If a patient had a carvedilol administration record with an administration date in the 6-month time window leading to the effective RT–qPCR result date and an administered dose > 0, the patient was considered carvedilol+. We also extracted comorbidity information of the cohort for propensity-score matching, for which we used the Charlson comorbidity index. All comorbidities and corresponding patient numbers are listed in Table 1.
The second dataset (external validation dataset) was an institutional review board-approved COVID-19 registry dataset that included 168,712 individuals tested for SARS-CoV-2 infection (83,340 of which were positive cases) from March 8th to May 26th 2021, at the Cleveland Clinic in Ohio and Florida, United States (Supplementary Table 10). Pooled oropharyngeal and nasopharyngeal swab specimens were used to test for SARS-CoV-2 by RT–qPCR assay in the Cleveland Clinic Pathology and Laboratory Medicine Institute. All SARS-CoV-2 testing followed the guidelines established by the Centers for Disease Control and Prevention of United States. The dataset included baseline demographic information, medications and COVID-19 test results. We used REDCap149 electronic data capture tools to extract the patient data from the electronic health records (EPIC Systems), and the data were manually checked by a professional team trained on uniform sources for the study variables. A carvedilol exposure group (carvedilol+) included patients that were actively taking carvedilol at the time of SARS-CoV-2 testing. Positive laboratory test results for COVID-19 were used as the primary outcome. Propensity score was used to match age, sex and race to reduce various confounding factors. Odds ratio was used to evaluate the carvedilol benefit to primary outcome. All analyses were conducted by MatchIt package v.4.0.0 in the R v.4.1.0 platform.
Anti-SARS-CoV-2 activity assay for carvedilol
A549 (CCL-185; ATCC) cells exogenously expressing ACE2 (A549-ACE2) were a gift from B. R. Tenoever, Icahn School of Medicine at Mount Sinai. A549-ACE2 cells were cultured in DMEM (catalog no. 11965092; ThermoFisher) with 10% FBS (catalog no. 100–106; GeminiBio) and used for SARS-CoV-2 infection. SARS-CoV-2 virus (nCoV/Washington/1/2020) was provided by the Biocontainment Laboratory University of Texas Medical Branch Galveston National Laboratory, Texas, United States. Vero E6 (CRL-1586; ATCC) cells were used to propagate and titer SARS-CoV-2. SARS-CoV-2 infections were performed under biosafety level 3 conditions at the Biocontainment Laboratory University of Chicago Howard T. Ricketts Laboratory, Illinois, United States. A549-ACE2 cells cultured in DMEM with 2% FBS were treated with carvedilol for 2 h at the indicated concentrations. Cells were infected with a multiplicity of infection of 0.5 in medium containing the appropriate concentration of drug. Forty-eight hours after infection, cells were fixed with 10% formalin (catalog no. 305–510; Fisherbrand), blocked and probed with mouse anti-SARS-CoV-2-spike antibody (catalog no. GTX632604; GeneTex) diluted 1:1,000 for 4 h, rinsed and probed with anti-mouse-HRP (catalog no. MP7401; Vector Labratories) for 1 h, washed and then developed with DAB substrate (catalog no. 34065; ThermoScientifc) for 10 min. Spike positive cells (n > 40) were quantified by light microscopy as blinded samples. A sigmoid fit was used to extract EC50 values using MATLAB (2020b).
To measure the effect of carvedilol on cell viability, cells were treated with various concentrations of carvedilol diluted in DMEM with 2% FBS for 48 h. The drug solution was then removed and cells were fixed with 10% formalin solution. The cells were stained with Crystal Violet 0.25% for 30 min. The plate was spun dried in a tabletop centrifuge and absorbance of each well was measured using a TECAN Infinite 200 Pro at 595 nm. The percent survival was calculated relative to cells treated with dimethyl sulfoxide.
Extended Data
Extended Data Fig. 1 |. Characteristics of the interactome.
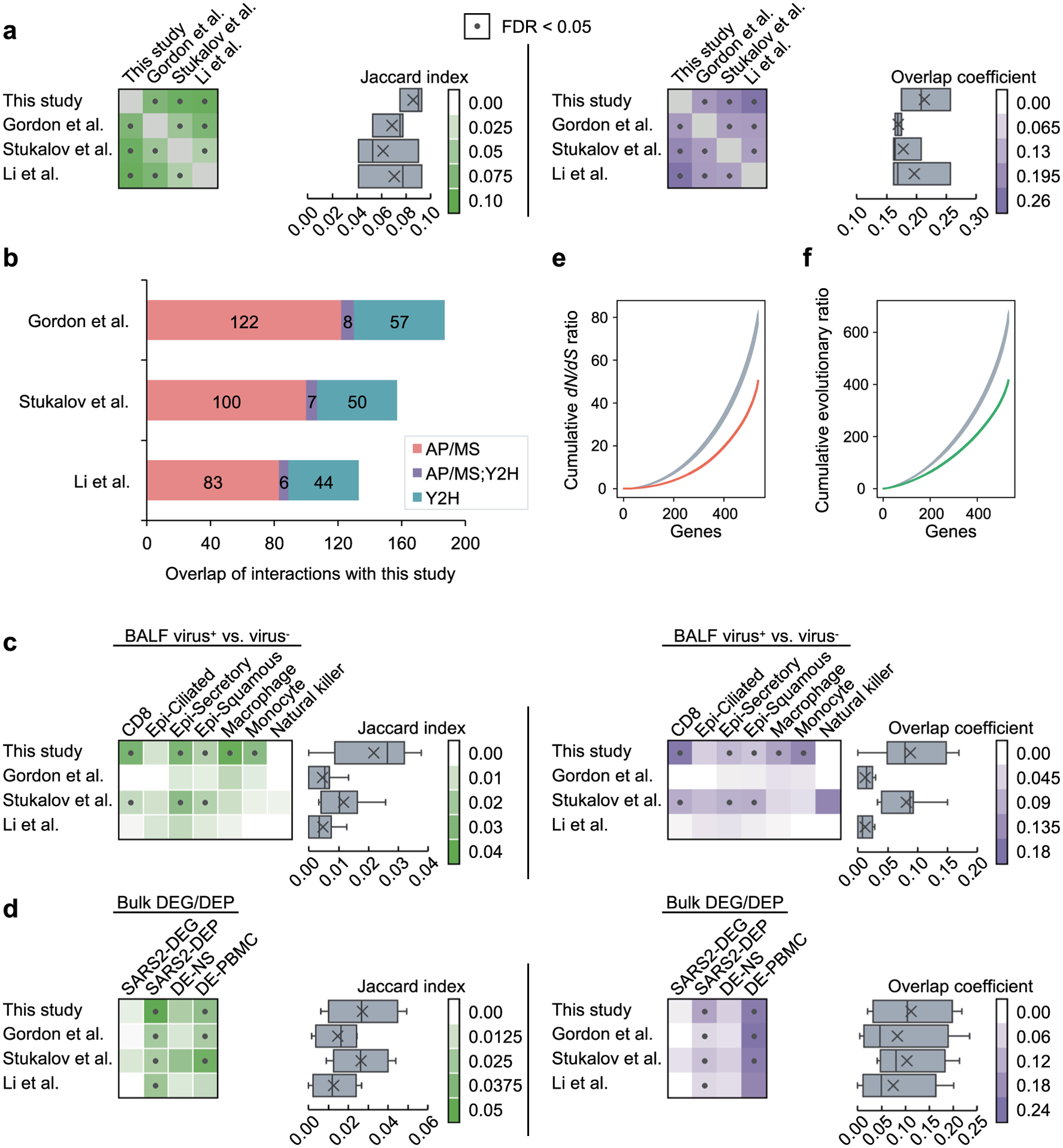
(a) Overlap of the host factors among the four interactomes compared in this study. Heatmaps show the Jaccard indexes (green) and overlap coefficients (purple) of the host factors against other gene sets. Dots indicate FDR < 0.05 by Fisher’s exact test. In the box plots, boxes range from lower to upper quartiles, center lines indicate medians, whiskers show 1.5 × interquartile ranges and crosses show mean values. (b) The overlap of the interactions in our interactome with the other three interactomes by considering the protein complexes and pathways. If two host factors interacting with the same viral protein are known to interact with each other in the literature, we consider the two viral-host interactions as overlapping. (c) Overlap of the host factors with the differentially expressed genes in SARS-CoV-2+ vs. SARS-CoV-2− cells in seven cell types from COVID-19 patient samples. Epi - epithelial. (d) Overlap of the host factors with the differentially expressed genes from four bulk RNA-seq/proteomics datasets. (e, f) Biological characteristics of the SARS-CoV-2 host factors. The host factors have lower non-synonymous to synonymous substitutions (dN/dS) ratios (e) and lower evolutionary ratios (f) compared to random background (gray, mean ± standard deviation of 100 repeats using genes randomly selected by degree preserved node shuffling). Genes were sorted in ascending order in terms of dN/dS ratio or evolutionary ratio.
Extended Data Fig. 2 |. ZNF579 targets significantly overlap with the differentially expressed genes in SARS-CoV-2 infected patient samples.
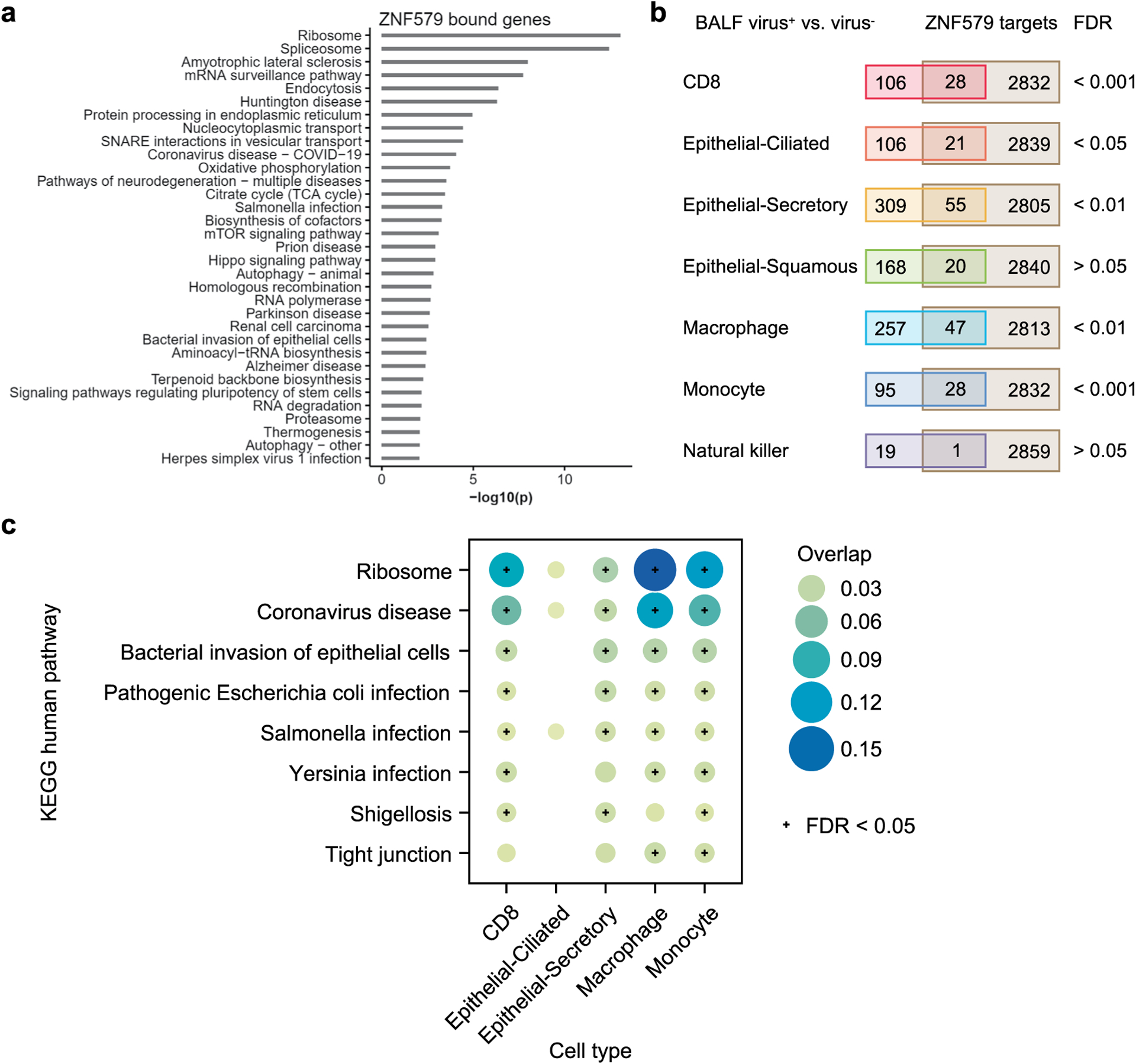
(a) Enriched KEGG pathways of genes associated with ZNF579 binding by ChIP-seq (ENCODE:ENCSR018MQH). Genes were considered to be bound by ZNF579 if a ChIP-seq peak overlapped with the promoter region (-1000 to transcription start site). (b) Overlap of ZNF579 targets and differentially expressed genes (DEGs) in bronchoalveolar lavage fluid (BALF) SARS-CoV-2+ vs. SARS-CoV-2− samples. See Methods for the source of the single-cell dataset. Fisher’s exact tests show that the overlaps are significant (FDR < 0.05) for five cell types, including CD8, epithelial-ciliated, epithelial-secretory, macrophage and monocyte. (c) The enriched pathways of the overlapping ZNF579 targets and DEGs in the five cell types. Pathways that are significantly enriched in at least two cell types are shown.
Extended Data Fig. 3 |. Comparison of the drug screening results using different interactomes and their combinations.
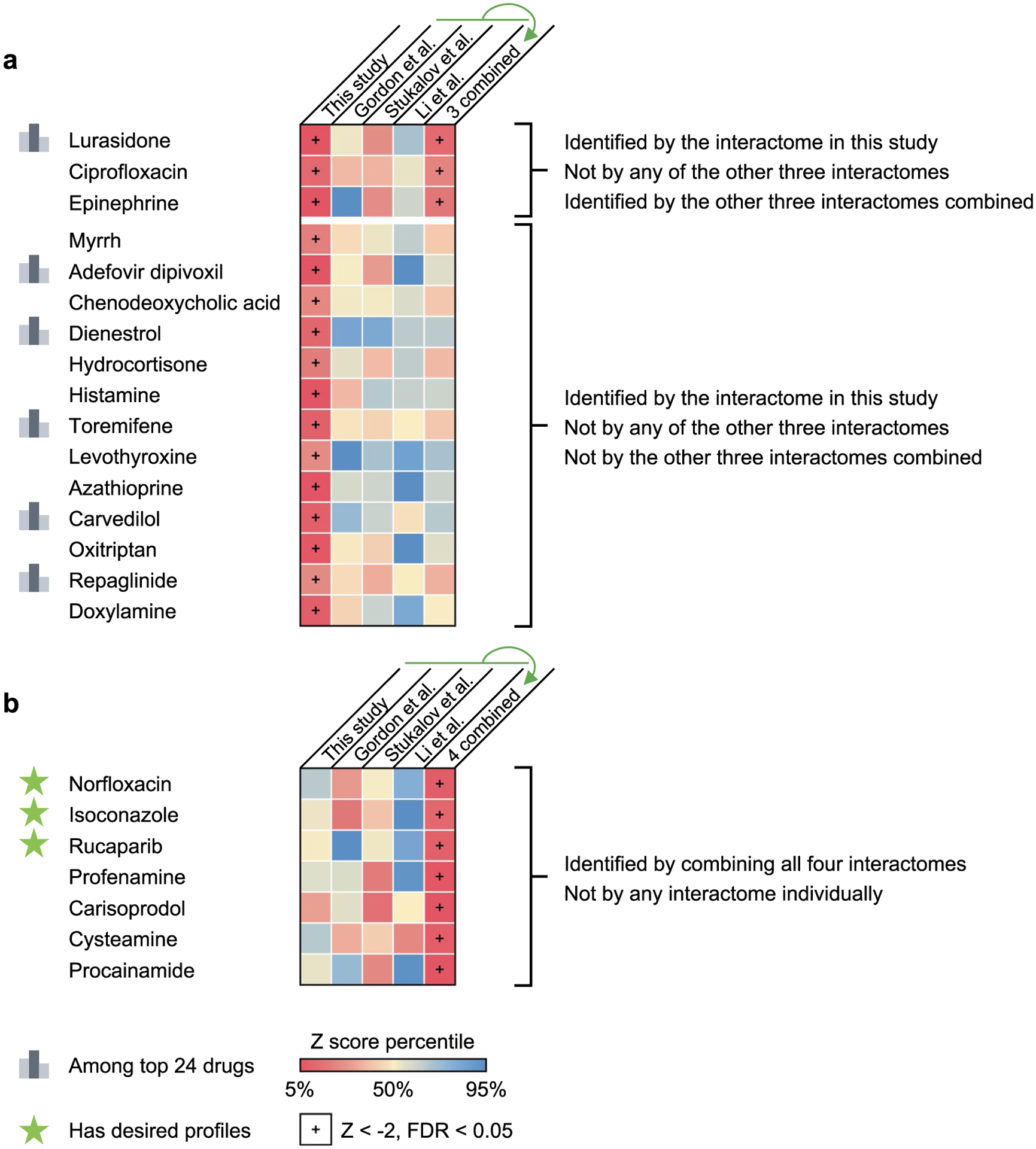
(a) 16 drugs identified by our interactome cannot be identified by any of the other three interactomes (and the interactome combined from them for 13 drugs) compared in this study. 6 of the top 23 drugs with desired anti-SARS-CoV-2 profiles are among these drugs. (b) Drugs identified by combining all four interactomes that could not be identified by any interactome individually. Three drugs (highlighted with a star) were found to have desired anti-SARS-CoV-2 profiles.
Extended Data Fig. 4 |. Carvedilol indirectly targets the SARS-CoV-2 host factors through protein-protein interactions with its targets.
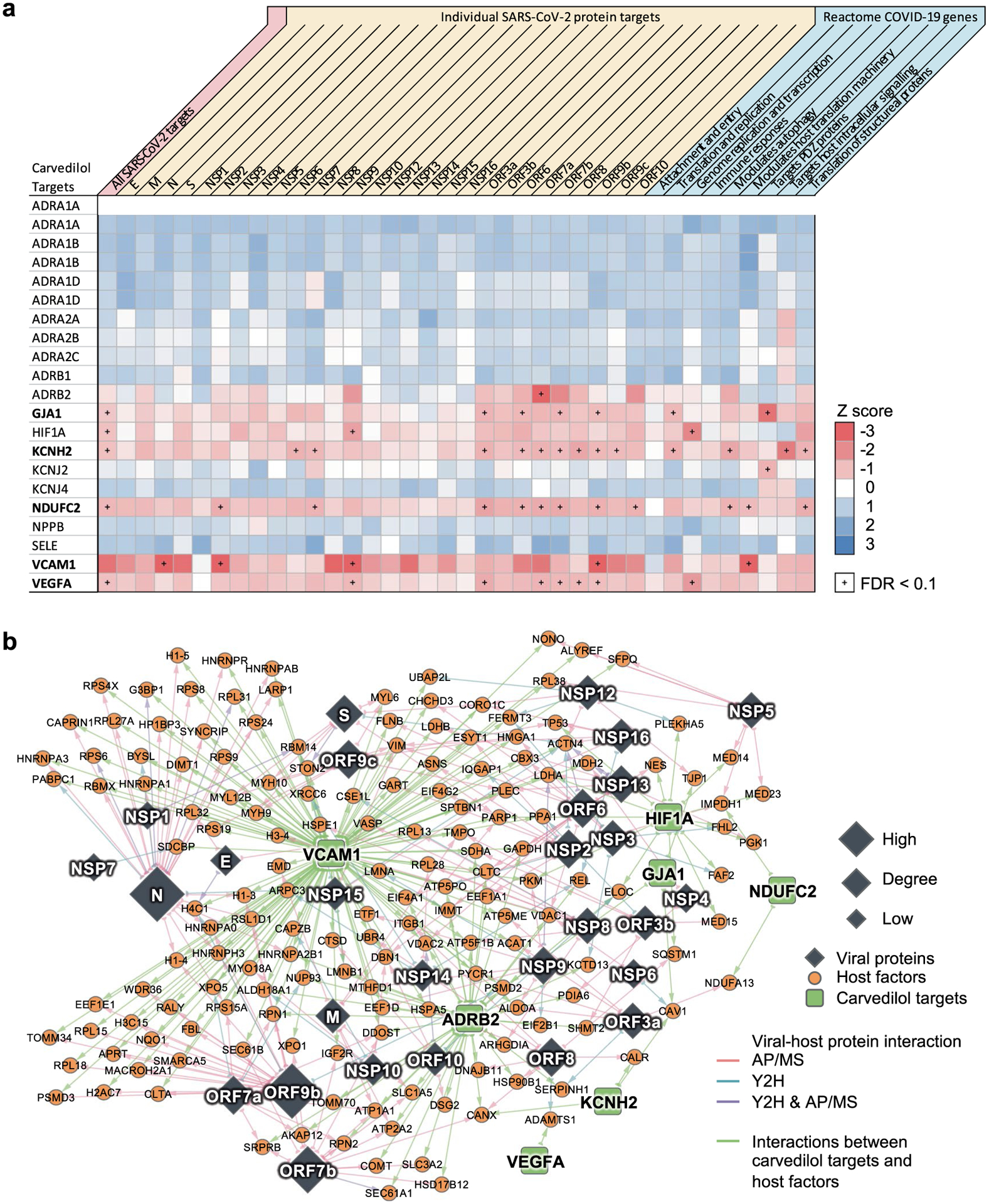
(a) Individual target-level network proximities to the SARS-CoV-2 gene sets (all host factors, host factors for each viral protein and gene sets by different functions from Reactome). Network proximities were computed using the ‘shortest’ method (See Methods). (b) Potential mechanisms-of-action of carvedilol by exploring the protein-protein interactions of its targets and the SARS-CoV-2 host factors.
Extended Data Fig. 5 |. Comparison of the drug screening results using different variations of the network proximity-based screening methods.
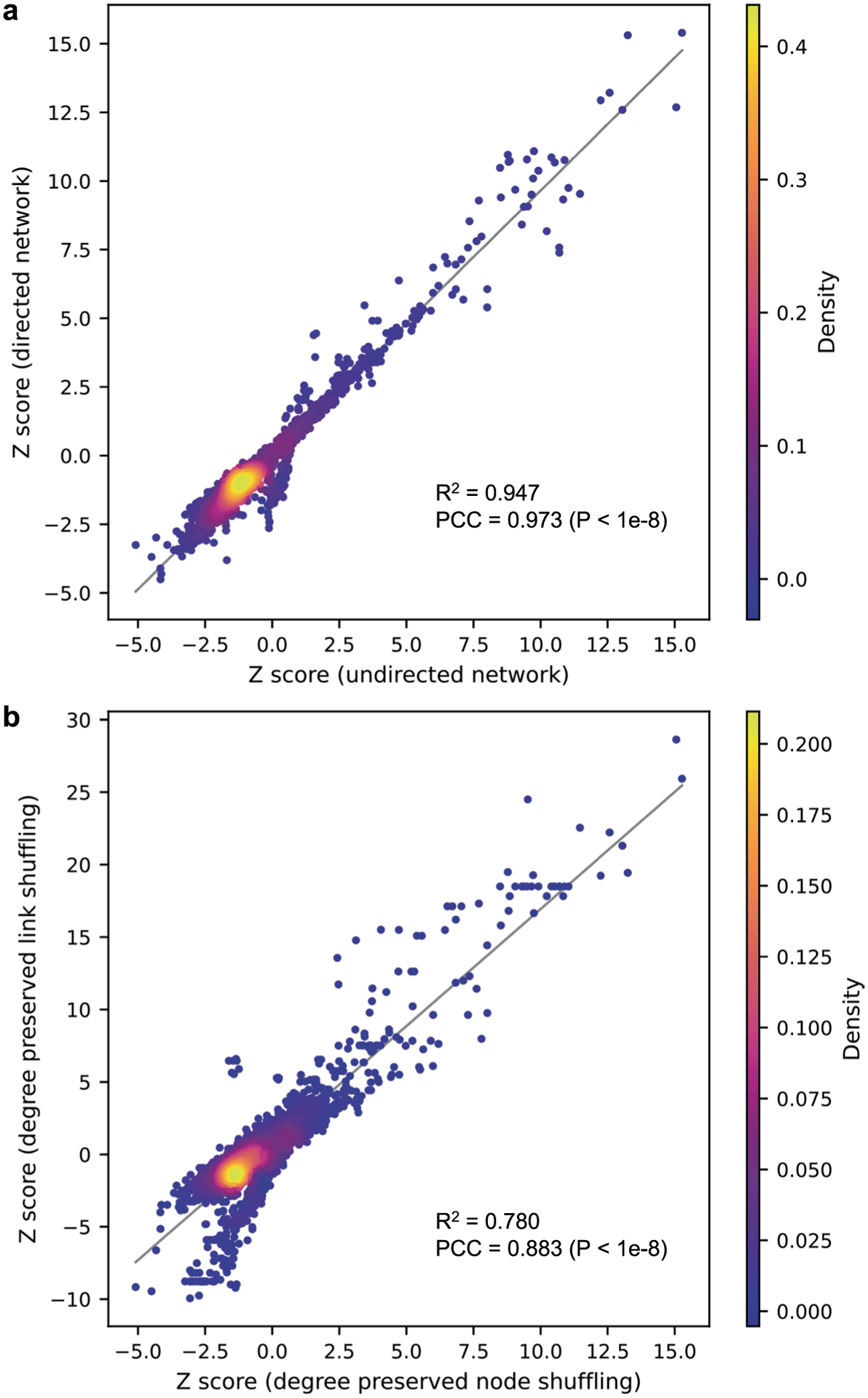
(a) Network proximity-based drug screening using directed human protein-protein interactome vs. undirected human protein-protein interactome. (b) Network proximity-based drug screening using degree preserved edge shuffling vs. degree preserved node shuffling. PCC, Pearson correlation coefficient.
Supplementary Material
Acknowledgements
This work was primarily supported by National Institute on Aging (NIA) grants U01AG073323, R01AG076448 and R01AG066707 to F.C., and NIGMS grant R01GM124559 and Cornell Rapid Research Response to SARS-CoV-2 Seed Grant to H.Y. This work was supported in part by NIA grants 3R01AG066707-01S1, 3R01AG066707-02S1 and R56AG074001 to F.C., National Institute of General Medical Sciences (NIGMS) grants R01GM125639, R01GM130885 and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01DK115398 to H.Y. and NIGMS grant RM1GM139738 and National Human Genome Research Institute (NHGRI) grant UM1HG009393 to H.Y. and J.T.L. C.F. was supported by R35GM122550 and U01HG009391. Y. Luo. was supported in part by R01LM013337. J.J. was supported by F31HG010820. M.I.P. was supported in part by a Cornell University Center for Vertebrate Genomics Scholarship.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-022-01474-0.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The network proximity framework can be found in https://github.com/ChengF-Lab/COVID-19_Map.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41587-022-01474-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-022-01474-0.
Data availability
GTEx v849 data was downloaded from https://gtexportal.org/home/. ENCODE ZNF579 ChIP–seq is available under accession no. ENCSR018MQH55,56. RNA-seq of human bronchial epithelial cells infected with SARS-CoV-2 is available from the Gene Expression Omnibus (GEO) under accession no. GSE147507. RNA-seq of upper airway from COVID-19 patients versus non-COVID-19 patients is available from GEO under accession no. GSE156063. RNA-seq of peripheral blood mononuclear cells isolated from COVID-19 patients versus non-COVID-19 patients is available from GEO under accession no. GSE157103. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD035805. Human protein–protein interactome and drug–target network are available at https://github.com/ChengF-Lab/COVID-19_Map. An interactive version of Fig. 1b is available at https://github.com/ChengF-Lab/COVID-19_PPI. Unaltered scans used to generate Fig. 2b,c,f–h are available in the Source Data file. All other data are available in the supplementary tables. Source data are provided with this paper.
References
- 1.Durmus Tekir SD & Ulgen KO Systems biology of pathogen–host interaction: networks of protein–protein interaction within pathogens and pathogen–human interactions in the post-genomic era. Biotechnol. J 8, 85–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon DE et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon DE et al. Comparative host–coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370, eabe9403 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J et al. Virus–host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med. 2, 99–112 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stukalov A et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 594, 246–252 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Rajagopala SV Mapping the protein–protein interactome networks using yeast two-hybrid screens. Adv. Exp. Med. Biol 883, 187–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Causier B Studying the interactome with the yeast two-hybrid system and mass spectrometry. Mass Spectrom. Rev 23, 350–367 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Yu H et al. High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy JP et al. Multiplexed relative quantitation with isobaric tagging mass spectrometry reveals class I major histocompatibility complex ligand dynamics in response to doxorubicin. Anal. Chem 91, 5106–5115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santin YG Uncovering the in vivo proxisome using proximity-tagging methods. Bioessays 41, e1900131 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Berggard T, Linse S & James P Methods for the detection and analysis of protein–protein interactions. Proteomics 7, 2833–2842 (2007). [DOI] [PubMed] [Google Scholar]
- 12.ten Have S, Boulon S, Ahmad Y & Lamond AI Mass spectrometry-based immuno-precipitation proteomics—the user’s guide. Proteomics 11, 1153–1159 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang G, Annan RS, Carr SA & Neubert TA Overview of peptide and protein analysis by mass spectrometry. Curr. Protoc. Protein Sci 62, 16.1.1–16.1.30 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Gingras AC & Raught B Beyond hairballs: the use of quantitative mass spectrometry data to understand protein–protein interactions. FEBS Lett. 586, 2723–2731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asara JM, Christofk HR, Freimark LM & Cantley LC A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics 8, 994–999 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Collier TS et al. Direct comparison of stable isotope labeling by amino acids in cell culture and spectral counting for quantitative proteomics. Anal. Chem 82, 8696–8702 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Megger DA et al. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta 1844, 967–976 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Li Z et al. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res 11, 1582–1590 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Stepath M et al. Systematic comparison of label-free, SILAC, and TMT techniques to study early adaption toward inhibition of EGFR signaling in the colorectal cancer cell line DiFi. J. Proteome Res 19, 926–937 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Team, M. G. C. P. et al. The completion of the mammalian gene collection (MGC). Genome Res. 19, 2324–2333 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das J et al. Cross-species protein interactome mapping reveals species-specific wiring of stress response pathways. Sci. Signal 6, ra38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vo TV et al. A proteome-wide fission yeast interactome reveals network evolution principles from yeasts to human. Cell 164, 310–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fragoza R et al. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun 10, 4141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wierbowski SD et al. A massively parallel barcoded sequencing pipeline enables generation of the first ORFeome and interactome map for rice. Proc. Natl Acad. Sci. USA 117, 11836–11842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann M et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cinatl J et al. Treatment of SARS with human interferons. Lancet 362, 293–294 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoehl S et al. Evidence of SARS-CoV-2 infection in returning travelers from Wuhan, China. N. Engl. J. Med 382, 1278–1280 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamers MM et al. SARS-CoV-2 productively infects human gut enterocytes. Science 369, 50–54 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao F et al. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology 158, 1831–1833 e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Yang C, Xu XF, Xu W & Liu SW Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin 41, 1141–1149 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xavier CP et al. Structural and functional diversity of novel coronin 1C (CRN2) isoforms in muscle. J. Mol. Biol 393, 287–299 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Martina JA, Bonangelino CJ, Aguilar RC & Bonifacino JS Stonin 2: an adaptor-like protein that interacts with components of the endocytic machinery. J. Cell Biol 153, 1111–1120 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J & Hui A Review of Ribosome Interactions with SARS-CoV-2 and COVID-19 mRNA Vaccine. Life 12, 57 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerjee AK et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339 e21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou M et al. The molecular mechanism of multiple organ dysfunction and targeted Intervention of COVID-19 based on time-order transcriptomic analysis. Front Immunol. 12, 729776 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt N et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat. Microbiol 6, 339–353 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren X et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 184, 1895–1913 e19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanco-Melo D et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045 e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bojkova D et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583, 469–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mick E et al. Upper airway gene expression reveals suppressed immune responses to SARS-CoV-2 compared with other respiratory viruses. Nat. Commun 11, 5854 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Overmyer KA et al. Large-scale multi-omic analysis of COVID-19 severity. Cell Syst. 12, 23–40 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sironi M, Cagliani R, Forni D & Clerici M Evolutionary insights into host–pathogen interactions from mammalian sequence data. Nat. Rev. Genet 16, 224–236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng F et al. Systems Biology-Based investigation of cellular antiviral drug targets identified by gene-trap insertional mutagenesis. PLoS Comput. Biol 12, e1005074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rozenblatt-Rosen O et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong Q et al. An inter-species protein–protein interaction network across vast evolutionary distance. Mol. Syst. Biol 12, 865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng F et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun 10, 3476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng F et al. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun 9, 2691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo Y et al. A multidimensional precision medicine approach identifies an autism subtype characterized by dyslipidemia. Nat. Med 26, 1375–1379 (2020). [DOI] [PubMed] [Google Scholar]
- 49.GTEx Consortium. The genotype–tissue expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jing Y et al. Potential influence of COVID-19/ACE2 on the female reproductive system. Mol. Hum. Reprod 26, 367–373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren Y et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol 17, 881–883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia H et al. Evasion of type I Interferon by SARS-CoV-2. Cell Rep. 33, 108234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rui Y et al. Unique and complementary suppression of cGAS-STING and RNA sensing-triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct. Target. Ther 6, 123 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang R et al. ORF3a protein of severe acute respiratory syndrome coronavirus 2 inhibits interferon-activated janus kinase/signal transducer and activator of transcription signaling via elevating suppressor of cytokine signaling 1. Front Microbiol. 12, 752597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis CA et al. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohd Ropidi MI, Khazali AS, Nor Rashid N & Yusof R Endoplasmic reticulum: a focal point of Zika virus infection. J. Biomed. Sci 27, 27 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelleher DJ & Gilmore R An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R (2006). [DOI] [PubMed] [Google Scholar]
- 59.Ruiz-Canada C, Kelleher DJ & Gilmore R Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindenbach BD & Rice CM trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol 71, 9608–9617 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beatty PR et al. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med 7, 304ra141 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Lu H, Cherepanova NA, Gilmore R, Contessa JN & Lehrman MA Targeting STT3A-oligosaccharyltransferase with NGI-1 causes herpes simplex virus 1 dysfunction. FASEB J. 33, 6801–6812 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marceau CD et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puschnik AS et al. A small-molecule oligosaccharyltransferase inhibitor with pan-flaviviral activity. Cell Rep. 21, 3032–3039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parnas O et al. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Linxweiler M, Schick B & Zimmermann R Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct Target. Ther 2, 17002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heaton NS et al. Targeting viral proteostasis limits influenza virus, HIV, and Dengue virus infection. Immunity 44, 46–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iwasa A et al. Contribution of Sec61α to the life cycle of Ebola virus. J. Infect. Dis 204, S919–S926 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cascarina SM & Ross ED A proposed role for the SARS-CoV-2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. 34, 9832–9842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dutta NK, Mazumdar K & Gordy JT The nucleocapsid protein of SARS-CoV-2: a target for vaccine development. J. Virol 94, e00647–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zeng W et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun 527, 618–623 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grifoni A et al. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 27, 671–680 e2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buttinelli M, Panetta G, Rhodes D & Travers A The role of histone H1 in chromatin condensation and transcriptional repression. Genetica 106, 117–124 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Ye X et al. Linker histone in diseases. Int. J. Biol. Sci 13, 1008–1018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu X et al. HIST1H1C regulates interferon-β and inhibits influenza virus replication by interacting with IRF3. Front. Immunol 8, 350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conn KL, Hendzel MJ & Schang LM Linker histones are mobilized during infection with herpes simplex virus type 1. J. Virol 82, 8629–8646 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou Y et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 6, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schafer PH et al. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal 26, 2016–2029 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Perino A, Ghigo A, Scott JD & Hirsch E Anchoring proteins as regulators of signaling pathways. Circ. Res 111, 482–492 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Werling LL, Keller A, Frank JG & Nuwayhid SJ A comparison of the binding profiles of dextromethorphan, memantine, fluoxetine and amitriptyline: treatment of involuntary emotional expression disorder. Exp. Neurol 207, 248–257 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Kumar S et al. Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell 184, 5950–5969 e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuevas RA et al. MOV10 provides antiviral activity against RNA viruses by enhancing RIG-I-MAVS-independent IFN induction. J. Immunol 196, 3877–3886 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeon S et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother 64, e00819–e00820 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dyall J et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother 58, 4885–4893 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cong Y et al. MERS-CoV pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS ONE 13, e0194868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johansen LM et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med 5, 190ra79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martin WR & Cheng F Repurposing of FDA-approved toremifene to treat COVID-19 by blocking the spike glycoprotein and NSP14 of SARS-CoV-2. J. Proteome Res 19, 4670–4677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou Y et al. A network medicine approach to investigation and population-based validation of disease manifestations and drug repurposing for COVID-19. PLoS Biol. 18, e3000970 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kc GB et al. A machine learning platform to estimate anti-SARS-CoV-2 activities. Nat. Mach. Intell 3, 527–535 (2021). [Google Scholar]
- 90.Daly JL et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 370, 861–865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cantuti-Castelvetri L et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang K et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther 5, 283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shimada H et al. Epiplakin modifies the motility of the HeLa cells and accumulates at the outer surfaces of 3-D cell clusters. J. Dermatol 40, 249–258 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Saadi I et al. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am. J. Hum. Genet 89, 44–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zeng C et al. SARS-CoV-2 spreads through cell-to-cell transmission. Proc. Natl Acad. Sci. USA 119, e2111400119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wan Q, Song D, Li H & He ML Stress proteins: the biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct. Target. Ther 5, 125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fang J et al. Endophenotype-based in silico network medicine discovery combined with insurance record data mining identifies sildenafil as a candidate drug for Alzheimer’s disease. Nat. Aging 1, 1175–1188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Israel A et al. Identification of drugs associated with reduced severity of COVID-19—a case-control study in a large population. Elife 10, e68165 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Skayem C & Ayoub N Carvedilol and COVID-19: a potential role in reducing infectivity and infection severity of SARS-CoV-2. Am. J. Med. Sci 360, 300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou Y, Wang F, Tang J, Nussinov R & Cheng F Artificial intelligence in COVID-19 drug repurposing. Lancet Digital Health 2, e667–e676 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clemente-Moragon A et al. Metoprolol in critically ill patients with COVID-19. J. Am. Coll. Cardiol 78, 1001–1011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shen J et al. The epidemiological and mechanistic understanding of the neurological manifestations of COVID-19: a comprehensive meta-analysis and a network medicine observation. Front. Neurosci 15, 606926 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kumar N et al. SARS-CoV-2 spike protein S1-mediated endothelial injury and pro-inflammatory state Is amplified by dihydrotestosterone and prevented by mineralocorticoid antagonism. Viruses 13, 2209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen JW et al. Carvedilol inhibits tumor necrosis factor-α-induced endothelial transcription factor activation, adhesion molecule expression, and adhesiveness to human mononuclear cells. Arterioscler. Thromb. Vasc. Biol 24, 2075–2081 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Walhout AJ & Vidal M A genetic strategy to eliminate self-activator baits prior to high-throughput yeast two-hybrid screens. Genome Res. 9, 1128–1134 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McAlister GC et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ting L, Rad R, Gygi SP & Haas W MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hogrebe A et al. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun 9, 1045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Connell JD, Paulo JA, O’Brien JJ & Gygi SP Proteome-wide evaluation of two common protein quantification methods. J. Proteome Res 17, 1934–1942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Samelson AJ et al. BRD2 inhibition blocks SARS-CoV-2 infection by reducing transcription of the host cell receptor ACE2. Nat. Cell Biol 24, 24–34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roberts GHL et al. Expanded COVID-19 phenotype definitions reveal distinct patterns of genetic association and protective effects. Nat. Genet 54, 374–381 (2022). [DOI] [PubMed] [Google Scholar]
- 112.Mantovani S et al. Rare variants in Toll-like receptor 7 results in functional impairment and downregulation of cytokine-mediated signaling in COVID-19 patients. Genes Immun. 23, 51–56 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pal LR et al. Synthetic lethality-based prediction of anti-SARS-CoV-2 targets. iScience 25, 104311 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shen B et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72 e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng K et al. Genome-scale metabolic modeling reveals SARS-CoV-2-induced metabolic changes and antiviral targets. Mol. Syst. Biol 17, e10260 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang T et al. MSstatsTMT: statistical detection of differentially abundant proteins in experiments with isobaric labeling and multiple mixtures. Mol. Cell Proteom 19, 1706–1723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Menche J et al. Disease networks. uncovering disease–disease relationships through the incomplete interactome. Science 347, 1257601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Supek F, Bosnjak M, Skunca N & Smuc T REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hirsh AE, Fraser HB & Wall DP Adjusting for selection on synonymous sites in estimates of evolutionary distance. Mol. Biol. Evol 22, 174–177 (2005). [DOI] [PubMed] [Google Scholar]
- 122.Bezginov A, Clark GW, Charlebois RL, Dar VU & Tillier ER Coevolution reveals a network of human proteins originating with multicellularity. Mol. Biol. Evol 30, 332–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cheng F et al. Studying tumorigenesis through network evolution and somatic mutational perturbations in the cancer interactome. Mol. Biol. Evol 31, 2156–2169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cheng F, Kovacs IA & Barabasi AL Network-based prediction of drug combinations. Nat. Commun 10, 1197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smith IN, Thacker S, Seyfi M, Cheng F & Eng C Conformational dynamics and allosteric regulation landscapes of germline pten mutations associated with autism compared to those associated with cancer. Am. J. Hum. Genet 104, 861–878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rolland T et al. A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rual JF et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 (2005). [DOI] [PubMed] [Google Scholar]
- 128.Cheng F, Jia P, Wang Q & Zhao Z Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget 5, 3697–3710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hu J et al. PhosphoNetworks: a database for human phosphorylation networks. Bioinformatics 30, 141–142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Keshava Prasad TS et al. Human protein reference database—2009 update. Nucleic Acids Res. 37, D767–D772 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lu CT et al. DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 41, D295–D305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dinkel H et al. Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 39, D261–D267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hornbeck PV et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Csabai L, Olbei M, Budd A, Korcsmaros T & Fazekas D SignaLink: multilayered regulatory networks. Methods Mol. Biol 1819, 53–73 (2018). [DOI] [PubMed] [Google Scholar]
- 135.Huttlin EL et al. The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Meyer MJ, Das J, Wang X & Yu H INstruct: a database of high-quality 3D structurally resolved protein interactome networks. Bioinformatics 29, 1577–1579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Orchard S et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oughtred R et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 47, D529–D541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Licata L et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 40, D857–D861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cowley MJ et al. PINA v2.0: mining interactome modules. Nucleic Acids Res. 40, D862–D865 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Breuer K et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 41, D1228–D1233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Law V et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–D1097 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu T, Lin Y, Wen X, Jorissen RN & Gilson MK BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 35, D198–D201 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gaulton A et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang H et al. Therapeutic target database update 2016: enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 44, D1069–D1074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Whirl-Carrillo M et al. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther 92, 414–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pawson AJ et al. The IUPHAR/BPS guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 42, D1098–D1106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harris PA et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform 42, 377–381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GTEx v849 data was downloaded from https://gtexportal.org/home/. ENCODE ZNF579 ChIP–seq is available under accession no. ENCSR018MQH55,56. RNA-seq of human bronchial epithelial cells infected with SARS-CoV-2 is available from the Gene Expression Omnibus (GEO) under accession no. GSE147507. RNA-seq of upper airway from COVID-19 patients versus non-COVID-19 patients is available from GEO under accession no. GSE156063. RNA-seq of peripheral blood mononuclear cells isolated from COVID-19 patients versus non-COVID-19 patients is available from GEO under accession no. GSE157103. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD035805. Human protein–protein interactome and drug–target network are available at https://github.com/ChengF-Lab/COVID-19_Map. An interactive version of Fig. 1b is available at https://github.com/ChengF-Lab/COVID-19_PPI. Unaltered scans used to generate Fig. 2b,c,f–h are available in the Source Data file. All other data are available in the supplementary tables. Source data are provided with this paper.