

Abstract
Reprogrammed metabolism is a hallmark of colorectal cancer (CRC). CRC cells are geared toward rapid proliferation, requiring nutrients and the removal of cellular waste in nutrient-poor environments. Intestinal stem cells (ISCs), the primary cell of origin for CRCs, must adapt their metabolism along the adenoma-carcinoma sequence to the unique features of their complex microenvironment that include interactions with intestinal epithelial cells, immune cells, stromal cells, commensal microbes, and dietary components. Emerging evidence implicates modifiable risk factors related to the environment, such as diet, as important in CRC pathogenesis. Here, we focus on describing the metabolism of ISCs, diets that influence CRC initiation, CRC genetics and metabolism, and the tumor microenvironment. The mechanistic links between environmental factors, metabolic adaptations, and the tumor microenvironment in enhancing or supporting CRC tumorigenesis are becoming better understood. Thus, greater knowledge of CRC metabolism holds promise for improved prevention and treatment.
Keywords: colorectal cancer, metabolism, diet, microbiome
1. INTRODUCTION
Colorectal cancer (CRC) is the second leading cause of cancer death worldwide, claiming nearly one million lives in 2020 (1). The majority of CRC cases and deaths in the United States are potentially attributable to modifiable risk factors, including diet (29%), physical inactivity (16%), alcohol intake (13%), smoking (11%), and excess body weight (5%), on the basis of large-scale meta-analyses (2). CRC incidence has been increasing in countries with a rising human development index, especially in those under age 50, correlating with changes in diet and lifestyle (3). Therefore, there is much interest in studying the role of nutrient consumption or metabolism in CRC tumorigenesis.
CRC initiation and progression are driven by the accumulation of somatic mutations in oncogenes and tumor suppressor genes in colon stem cells (4, 5). In the adenoma-carcinoma model, sequential mutations over a period of years in the wingless/integrated (WNT), epidermal growth factor receptor (EGFR), tumor protein 53 (TP53), and transforming growth factor beta (TGF-β) signaling pathways result in CRC formation and progression. A small subset of CRCs, such as those found in Lynch syndrome, are driven by mutations in DNA mismatch repair genes. Further characterization of the molecular events underlying CRC initiation and progression led to the establishment of four CRC consensus molecular subtypes (CMSs): microsatellite unstable (CMS1), canonical WNT and MYC (MYC proto-oncogene, BHLH transcription factor) signaling driven (CMS2), metabolic dysregulation driven by mutant KRAS (KRAS proto-oncogene, GTPase) (CMS3), and mesenchymal-associated (CMS4) (6). A common feature of all CRC CMSs is alterations in metabolic pathways that supply the energy and nutrients necessary to support proliferation. As such, a major focus of research in CRC pathogenesis is to understand the mechanisms of dysregulated cancer metabolism.
The field of cancer metabolism was born in the 1920s when Otto Warburg reported that cancers metabolize substantially more glucose and subsequently produce more lactate relative to healthy tissues (7), an observation that became known as the Warburg effect. Nearly a century later, there has been an explosion of cancer metabolism research to understand how metabolism is reprogrammed in cancer cells and how metabolic changes in cancer can be therapeutically exploited (8). Genetic and pharmacological studies have demonstrated that the Warburg effect is required for cancer growth and is not simply a bystander effect (9). Cancer cells—and rapidly proliferating cells in general—perform aerobic glycolysis, not primarily for energy production but to shuttle glycolytic intermediates and lactate into the cellular building blocks (e.g., amino acids, lipids, and nucleic acids) necessary for increasing biomass (10). Cancer metabolism has also become clinically important: Antimetabolites serve as chemotherapeutics and fluorodeoxyglucose (FDG) positron emission tomography scans are used to stage cancers by monitoring glucose metabolism in cancerous tissue in comparison with the normal surrounding tissue. Cancer metabolism is now recognized as a hallmark of cancer (11).
This review aims to discuss our current understanding of CRC metabolism, with a focus on characterizing the metabolism of intestinal stem cells (ISCs), environmental factors that influence CRC initiation, CRC genetics and metabolism, and the microbiome.
2. INTESTINAL STEM CELLS AND THEIR ROLE IN CANCER
The epithelial lining of the small intestine and colon constitute the largest external surface area of the body, facilitating efficient nutrient transfer while serving as a barrier to microbes (12). Approximately 8 to 10 L of fluid passes through the small intestine daily, where protruding finger-like villi (not found in the colon) aid in the digestion and absorption of nutrients (e.g., amino acids, carbohydrates, lipids, and vitamins) and water (13). Approximately 1.5 to 2 L of ileal effluent passes through the ileocecal valve, and 90% of the effluent is absorbed in the colon each day as water and electrolytes (14). The colon harbors approximately 99% of the approximately 38 trillion microbes composing the human microbiome (15). This nutrient absorption and barrier function is achieved by a one-cell-thick epithelial layer that turns over every 5 to 7 days, maintained by the highly proliferative leucine-rich repeat–containing G protein–coupled receptor 5 positive (LGR5+) ISCs at the base of the intestinal crypts (16).
The ISCs produce transit-amplifying cells that undergo several divisions to differentiate into all epithelial cells lining the intestine, primarily consisting of absorptive (e.g., enterocytes and M cells) and secretory (e.g., goblet, tuft, Paneth, and enteroendocrine) cells. The colonic crypt and associated cell types are illustrated in Figure 1. ISC proliferation depends on ligands of the WNT signaling pathway and their R-spondin cofactors, with a gradient of WNT signaling strongest at the crypt base and weakest at the villus tip (17, 18). A decrease in WNT ligands and an increase in bone morphogenic protein (BMP) signaling higher up the crypt drives differentiation (19). Cells differentiate as they move away from the base of the crypt and up the villi until they eventually undergo anoikis and are sloughed off into the lumen of the intestine. Paneth cells are the exception, as they differentiate toward the base of the crypt. Paneth cells are secretory epithelial cells that specialize in maintaining intestinal homeostasis by secreting antimicrobial compounds and by contributing WNT ligands for the proliferation of ISCs (20). While small intestine ISCs are interspersed with WNT-producing Paneth cells, the colon ISCs are interspersed with Paneth-related, regenerating family member 4 positive (REG4+), and CD24+ deep crypt secretory cells in mice (21, 22). In humans, Paneth cells are also present in the right and transverse colon. Paneth cells primarily maintain ISCs in the small intestine by secreting WNT, whereas GLI family zinc finger 1 positive (GLI1+) mesenchymal cells and family forkhead box 11 positive (FOX11+)/platelet-derived growth factor receptor alpha positive (PDGFRα+) telocytes supply essential WNT signals to maintain the colon ISCs (23, 24). Precise cell specification during differentiation is guided by the amount of WNT ligand, BMP signaling, NOTCH signaling, and other cytokines and growth factors in the microenvironment (25). In addition to maintaining the epithelial lining of the intestine, ISCs are also the primary cell of origin for CRC, and their stemness pathways are hijacked during CRC initiation and progression (16).
Figure 1.
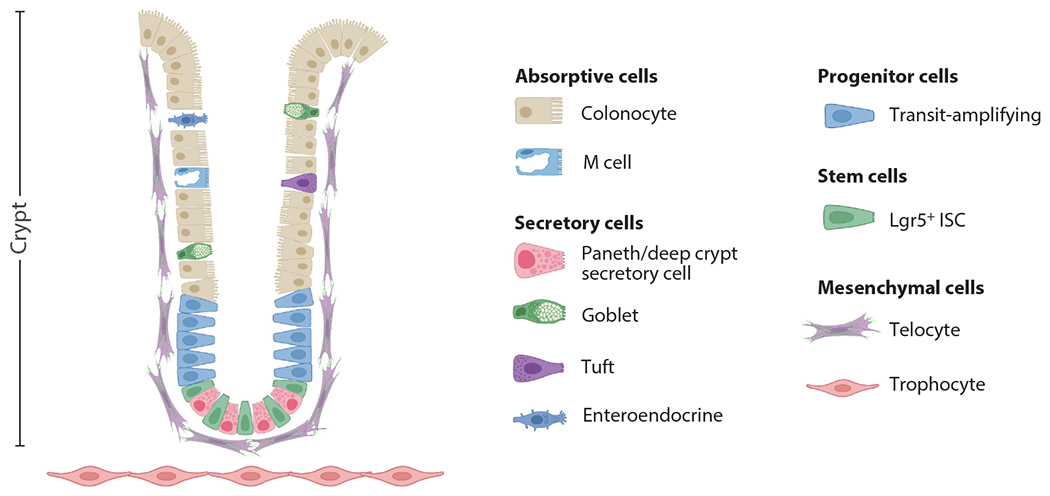
Schematic of the colonic crypt and associated epithelial and mesenchymal cell types. Figure adapted from images created with BioRender.com. Abbreviations: ISC, intestinal stem cell; Lgr5+, leucine-rich repeat–containing G protein–coupled receptor 5 positive.
2.1. Intestinal Stem Cells
ISCs of the small intestine and colon are highly proliferative relative to stem cells from many other tissue compartments, meaning that their metabolic demands are relatively high. ISCs fulfill their metabolic needs, in part, by undergoing aerobic glycolysis, like cancer cells (26). While glycolysis is elevated in ISCs, glycolysis is even higher in adjacent Paneth cells (26). ISCs are spatially associated and metabolically linked with Paneth cells. Paneth cells play an important role in maintaining ISCs through the secretion of EGF, TGF-α, WNT3, and the NOTCH ligands DLL1 and DLL4 (21). However, Paneth cells are dispensable for ISC self-renewal and differentiation (27, 28). A study used organoid models to report that Paneth cells may metabolically support the needs of proliferating ISCs by secreting lactate. High oxidative phosphorylation (OXPHOS) in ISCs, in turn, produces reactive oxygen species (ROS) that induce crypt differentiation through mitogen-activated protein kinase p38 signaling (29). However, another study found that ISCs do not fuel OXPHOS with lactate due to a reduction in the expression of mitochondrial pyruvate carrier (MPC) (30). Thus, while the importance of Paneth cells for ISC function is controversial, they may play an important role in maintaining the metabolic needs of ISCs.
Another metabolic function that differs between ISCs and their differentiated progeny is pyruvate utilization. The expression of MPC, the gatekeeper of pyruvate transfer from the cytosol to the mitochondria (31), is low in ISCs but high in their differentiated progeny (30). ISCs shuttle pyruvate away from energy production through OXPHOS in the mitochondria and toward the production of biosynthetic molecules necessary for increasing cell biomass. Conversely, differentiated cells directly utilize pyruvate for energy production. While ISCs do not convert pyruvate into energy within the mitochondria, ISCs largely generate energy through other mechanisms, such as fatty acid and amino acid oxidation (29, 30). Thus, the downregulation of MPC in ISCs leads to a greater reliance on fatty acids for energy production.
Fatty acids provide energy for ISCs and thus promote their proliferation. As discussed in Section 3.4, high-fat diet (HFD)-induced obesity stimulates components of the WNT/β-catenin pathway and fatty acid oxidation through peroxisome proliferator-activated receptor (PPAR) signaling, thereby increasing stemness (32–34). Moreover, ISC proliferation is enhanced by cholesterol, a major constituent of the cell membrane. Increased intestinal cholesterol availability, through diet or genetic events, promotes tumorigenesis in Apcmin/+ mice (a commonly used model of CRC) (35). Given how diet can directly impact the composition of cells in the intestinal crypt, differing metabolism in ISC progenitors is not simply a byproduct of differentiation but one of several drivers of differentiation (36).
Lineage tracing experiments demonstrated that LGR5+ ISCs in the colon are the cell of origin of most CRCs (37). LGR5, a seven-transmembrane receptor for R-spondin, strongly enhances WNT signaling by neutralizing the RNF43/ZNRF3 transmembrane E3 ligases that remove WNT receptors from the membrane (38). These LGR5+ ISCs give rise to transit-amplifying cells with a finite number of divisions. While LGR5+ ISCs of the colon have by far the greatest potential to transform into CRC, genetic mouse models of CRC initiation have demonstrated that transit-amplifying cells and/or differentiated cells may also initiate cancer in specific contexts such as inflammation, obesity, or microenvironmental changes (39).
2.2. Mesenchymal Niche
The subepithelial mesenchyme supporting the ISCs has become better characterized and appreciated in recent years. Studies using single-cell RNA sequencing, organoid cocultures, immunofluorescent cell sorting, genetic engineering, and microscopy have provided a more in-depth understanding of how the mesenchyme supports ISCs (40). These experiments have demonstrated that the mesenchymal cell populations provide signals to maintain the ISC niche. GLI1+ mesenchymal cells and FOXL1+/PDGFRα+ telocytes provide WNT ligands to ISCs (23, 24). Additionally, a population of CD81+ and PDGFRαlo fibroblasts, termed trophocytes, is located just below the crypt and secretes the BMP antagonist, Gremlin1 (41). While we now have some understanding of how the supporting mesenchyme guides the differentiation of the epithelium, much less is known about the metabolism of the intestinal mesenchyme.
3. ENVIRONMENTAL FACTORS THAT INFLUENCE COLORECTAL CANCER INITIATION
CRC remains the second-leading cause of cancer death globally and in the United States (1). From 2008 to 2017, CRC death rates have decreased 3% annually in individuals aged 65 years and older and decreased 0.6% annually in individuals who are 50 to 64 years of age in the United States (3). The decrease in CRC deaths in those 50 years of age and older is thought to be due to more widespread screening (predominantly with colonoscopy and stool-based tests) and removal of precancerous adenomas (42). However, during the same 10-year period in the United States, the death rate of CRC increased by 1.3% annually in individuals younger than 50 years of age (3). This increase in the death rate of individuals younger than 50 years of age is also occurring in many other high-income countries, including Australia, Canada, Germany, and the United Kingdom (3). The reason for the increase in CRC cases and deaths in young individuals is unknown but is thought to be due to an increase in lifestyle risk factors.
Over half of all CRC cases and deaths in the United States are potentially attributable to environmental or otherwise modifiable risk factors on the basis of large-scale pooled analyses and meta-analyses (2). Most of the population-attributable fraction of CRC deaths is related to dietary habits: low dietary fiber (10.3%), processed meat consumption (8.2%), red meat consumption (5.4%), and low dietary calcium (4.9%) (2). The connection between diet and CRC may be partially explained by nutrient intake altering metabolite levels in the tumor microenvironment, thereby impacting cancer cell metabolism and growth. Many studies have linked an elevated body-mass index (BMI) with an increased risk of developing CRC (43). The relationship between BMI and early-onset CRC is especially pronounced for individuals experiencing obesity at a young age. In one recent study, individuals with a BMI above 30 at age 20, at age 30, and 10 years before CRC diagnosis/interview were at 2.56-, 2.06-, and 1.88-fold increased risk of developing early-onset CRC, respectively (44).
A summary of the effects of various diets on CRC risk and ISC biology is listed in Table 1.
Table 1.
Impact of diverse diets on CRC risk and ISC biology
| Diet | CRC risk | Effects on ISCs |
|---|---|---|
| Calorie restriction | Decreased in humans and animal models (45–47, 52, 53) | Increased ISC number allows outcompeting of mutant ISCs (59) |
| Fasting | Fasting phase: decreased (54, 62) Refeeding: unknown, potentially increased (66) |
Fasting phase: lower cellular proliferation (61) Refeeding phase: increased proliferation (54, 56–58) |
| Vitamin D | Decreased in humans and animal models (67–72) | Enhances expression of stemness genes and reduces proliferation (74) |
| Vitamin C | Unknown (75–77) | Unknown |
| Fructose | Increased in animal models and some epidemiological associations (79, 81, 82) | Fuels aerobic glycolysis and increases de novo fatty acid synthesis (80) |
| HFD-induced obesity | Increased in animal models (32, 33, 92, 93) Obesity increases CRC risk in humans (44, 86) No effect was observed for HFD, without overeating, on CRC risk in humans (28) |
Increased proliferation and tumorigenicity (32, 33, 92, 93) Decreased MHC-II expression (34) |
| Carbohydrate-rich diet | Unknown | Unknown |
| Ketone bodies | Unknown in humans Decreased in animal models (103, 105) |
Enhanced NOTCH activity, ISC self-renewal, postinjury regeneration, and decreased secretory cell differentiation (104) Decreased proliferation (105) |
| Weight loss | Decreased (106–111) | Unknown |
Abbreviations: CRC, colorectal cancer; HFD, high-fat diet; ISC, intestinal stem cell; MHC-II, major histocompatibility complex class II.
3.1. Calorie Restriction and Fasting
Calorie restriction (CR) without malnutrition has been associated with an increase in overall survival and a decrease in the incidence of cancer in many animal studies (45). Our understanding of the effects of CR on life span and cancer incidence has largely come from animal, epidemiological, and observational studies because of the difficulty of conducting diet-based randomized clinical trials in humans over decades. The most relevant data to humans that support the benefit of CR come from two prospective studies on rhesus monkeys by the University of Wisconsin (UW) and National Institute of Aging (NIA) (46). Adult-onset CR with a 30% reduction of calories compared with controls over the lifetime of the rhesus monkeys resulted in a statistically significant increase in overall survival in the UW study [hazard ratio (HR) = 1.865; 95% confidence interval (CI) of 1.119 to 3.108] and no statistical difference in the NIA study. However, the NIA study was not powered to observe a survival difference. Morbidity and cancer incidence on CR was significantly lower in both the UW (HR = 2.665; 95% CI of 1.527 to 4.653) and NIA (HR = 2.063; 95% CI of 1.169 to 3.641) studies. These positive findings for CR in rhesus monkeys have been corroborated in many animal models. For example, in the Apcmin/+ mouse model, a 40% CR diet reduced the frequency of colorectal polyps by 57% (47). The benefits of CR observed in animal studies and the potential for a drug to provide the same life extension without the inherent challenges of maintaining a CR lifestyle have motivated scientists to investigate mechanisms underlining the effects of CR on survival and protection against cancer.
The mechanisms by which CR extends survival and reduces CRC incidence have been well studied. In animal studies of CR, a decrease in insulin-like growth factor (IGF)-1 signaling is posited as one of the life-extending mechanisms of CR. IGF-1 is a major mediator of growth hormone (GH) signaling and other anabolic processes. Corroborating this hypothesis are genetic studies that identified a higher frequency of heterozygous inactivating mutations in IGFR1, the receptor for IGF-1, in female centenarians compared with controls (48). An Igfr1 knockout mouse model demonstrated an increase in average life expectancy of 33% for females and 14% for males compared with wild-type mice (49). Conversely, patients with enhanced IGF-1 signaling, such as those with acromegaly, have a twofold increased risk of gastrointestinal cancer compared with controls (50). IGF-1 signaling was examined in 218 nonobese young and middle-aged males and females for two years in the longest randomized trial of CR in humans (51). The researchers targeted a CR of 25%, though after six months the average CR was only 9.1%, which may have contributed to no observed decrease in absolute IGF-1 levels in the CR cohort. However, the authors noted a reduction in the activity of IGF-1 and concluded that the absolute levels of IGF-1 may not have decreased due to the higher proportion of calories that came from protein in the participants’ diet (51). Daily CR was shown to decrease the risk of CRC and metastasis greater than fasting-mimicking diets in mice (52). Additional pathways influenced by CR and believed to extend life include reduced mTORC1 (mammalian target of rapamycin complex 1) signaling, activation of GCN2 (eukaryotic translation initiation factor 2 alpha kinase 4), reduced protein synthesis, increased activity of FGF21 (fibroblast growth factor 21), and activation of sirtuins, all of which have been summarized in a recent review (53).
A related dietary intervention to CR studied for its role in preventing cancer and extending life span is fasting. While CR is typically applied chronically with a 20% to 40% reduction of the normal caloric intake and consistent frequency of meals, fasting is defined by periods of abstinence, ranging from hours to weeks, from calorie-containing food and beverages followed by unrestricted caloric consumption. There are many types of fasts, with intermittent fasting (IF) (e.g., alternate day fasting) and periodic fasting (PF) (e.g., fasting lasting three days or longer every two or more weeks) being the most common (54, 55). Fasting may or may not change the overall number of consumed calories. Furthermore, unlike CR, fasting creates unique fasting, refeeding, and postrefeeding phases. The fasting phase is characterized by ketogenesis, a cellular stress response, cellular lipolysis, and autophagy. Through these evolutionarily conserved stress response programs, fasting has been observed to extend life span in Escherichia coli, Saccharomyces cerevisiae, Caenorhabditis elegans, and mice and to decrease IGF-1 in mice and humans (54).
Short-term fasting increases the function of stem cells in many adult tissues during the refeeding phase (54, 56, 57). Our lab studied the effects of a 24-h fast on the metabolism and regenerative capacity of ISCs in young and aged mice. We found that short-term fasting enhances fatty-acid oxidation (FAO) in mouse ISCs, resulting in an ISC proregenerative effect in intestinal crypts grown as organoids in nutrient-rich media from young and aged mice (58). While aging reduces ISC number and function in mice, enhanced FAO through PPARδ agonist treatment restores the regenerative function of aging stem cells. Conversely, inhibiting FAO through genetic disruption of carnitine palmitoyltransferase 1A (CPT1A), the rate-limiting enzyme in FAO, nullifies the regenerative effects of fasting. Thus, ISCs experience a period of elevated proliferation during the refeeding phase, dependent on FAO.
Several preclinical studies have identified mechanisms by which CR and fasting inhibit the progression of cancer. CR may directly reduce CRC risk by increasing the number of ISCs, thereby allowing normal ISCs to outcompete mutant ISCs (59). Additionally, a recent study of CR in an allograft mouse model of pancreatic adenocarcinoma and lung adenocarcinoma found that CR induced a significant reduction in tumor size, with the mechanism linked to an imbalance of the ratio of unsaturated to saturated lipids available to the tumor (60). In a mouse model of CRC, fasting reduced CRC’s proliferation by inhibiting aerobic glycolysis via the upregulation of the cholesterogenic gene, Fdt1 (farnesyl-diphosphate farnesyltransferase 1) (61). Fdt1 acted as a tumor suppressor by inhibiting the AKT/mTOR/HIF1-α pathway (61). Intermittent fasting reduced the size of CRC in another study with a similar mouse model, but the authors focused on the role of tumor-associated macrophages (TAMs) (62). It is generally accepted that M2-polarized TAsMs support tumor growth by blocking tumor immune surveillance and supporting angiogenesis. By suppressing M2 polarization of macrophages and decreasing extracellular adenosine, fasting inhibits CRC growth in vitro and in vivo (62). Thus, studies in preclinical models have identified many mechanisms by which CR and fasting reduce tumorigenesis.
CR and fasting have been explored clinically as adjuvant therapy to enhance the effects of chemotherapy and other cancer treatments (63, 64). However, CR and fasting have not yet proven effective in the treatment of CRC in clinical trials (65). While CR can be tolerated by healthy individuals, patients diagnosed with cancer are at greater risk of weight loss, cachexia, and sarcopenia from tumor-derived factors. Given the nutritional concerns of CR and fasting, CR mimetics, such as metformin, resveratrol, hydroxycitrate, and rapamycin and its analogs, are being assessed in clinical trials (63). The current mismatch between the promise shown in preclinical models and the effect of fasting and CR in the clinic warrants further exploration.
While there are many reported benefits to fasting, there is a potential protumor susceptibility from increased cellular proliferation during the refeeding phase. Unlike the fasting phase, which is associated with lower cellular proliferation, increased cell death, increased atrophy, and decreases in glucose, insulin, and IGF-1, the refeeding phase of fasting is characterized by an increase in growth factors and cellular proliferation. Greater cellular proliferation during the refeeding phase may elevate the potency of carcinogens and accelerate tumorigenesis. A study of rats fasted for four days followed by refeeding and administration of a subnecrogenic dose of the carcinogen diethylnitrosamine (DENA) found significantly more hepatocyte foci/nodules compared with controls on an ad libitum diet given the same dose of DENA (66). Thus, at least one study suggests that the refeeding phase of fasting can increase susceptibility to carcinogens.
3.2. Vitamins D and C
Vitamin D’s role in preventing and reducing CRC has been studied in dozens of observational studies and clinical trials. In an international pooled study of 17 cohorts, higher levels of circulating 25-hydroxyvitamin D3 were associated with a 19% reduction in risk of CRC in women and a 7% reduction in risk for men (67). Vitamin D may also improve CRC-specific survival: Stage III CRC patients in the highest quintile of vitamin D score had significantly improved recurrence-free and overall survival compared with those in the lowest (adjusted HR for death or recurrence 0.62; 95% CI of 0.44 to 0.86) (68). Low postoperative vitamin D levels have been associated with significantly worse survival (adjusted HR for CRC-specific mortality 0.68; 95% CI of 0.50 to 0.90) (69). One meta-analysis of randomized controlled trials found that patients with CRC who took vitamin D supplements manifested a 24% increase in CRC-specific survival (70), and another meta-analysis of five clinical trials reported that vitamin D supplementation increased overall cancer survival by 13% (71). Although promising, the correlation between vitamin D status and survival may not be causal; this association may be due to confounders such as those in poorer health getting less sunlight or the levels of vitamin D being impacted by the severity of the disease. Nonetheless, on the basis of these compelling observational data, it seems reasonable to assess vitamin D levels in patients with newly diagnosed CRC and to replete those with low levels (serum 25-hydroxyvitamin D3 < 20 mg/mL or 50 nmol/L).
Many studies have examined possible antineoplastic mechanisms for vitamin D’s role in preventing and treating CRC. The active form of vitamin D, calcitriol (1α,25-dihydroxyvitamin D3), acts as a steroid hormone, binding to the vitamin D receptor (VDR) and forming a heterodimer with the retinoid X receptor to control gene expression by complexing with vitamin D response elements in multiple regulatory regions of the genome (72). While numerous cell types express VDR, the intestines have the highest transcriptomic expression of VDR after the parathyroid glands (73). In normal colon organoids, calcitriol induces the expression of stemness genes and reduces proliferation (74). Conversely, in CRC organoids, calcitriol induces differentiation and inhibits proliferation. Several of the actions of calcitriol in CRC are to induce differentiation through inhibition of β-catenin transcriptional activity (72). Other antineoplastic mechanisms of calcitriol include inhibiting inflammation, invasion and metastasis, and angiogenesis and inducing apoptosis (72).
In contrast to the relatively strong epidemiological and clinical data to support the antineoplastic effects of vitamin D, the use of vitamin C to treat CRC has a controversial history owing to conflicting results in clinical trials (75). Although previous randomized controlled trials failed to show efficacy of vitamin C to treat cancer, the limitations of those trials and recent promising research on preclinical models have revived interest, and there are now several ongoing randomized clinical trials (75). Vitamin C administration at physiologically nontoxic concentrations in the millimolar range is selectively toxic to KRAS and BRAF mutant human CRC cell lines (76). The mechanism is believed to be the uptake of the oxidized form of vitamin C via glucose transport 1 (GLUT1), which is upregulated in KRAS and BRAF mutant CRC, inhibiting glycolysis (76). Another recent study suggested that vitamin C may inhibit CRC growth through a T cell–dependent mechanism (77). In mouse models of mismatch repair-deficient cancers with a high tumor mutational burden, the combination of vitamin C and immune checkpoint inhibitors could significantly enhance survival and cure a subset of mice (77). While these animal studies are promising, the role of vitamin C in the prevention or treatment of CRC has not been demonstrated in human patients.
3.3. Fructose
Fructose has become a large part of the diet in the United States, with the average daily consumption reaching 50 g in 2004 (78). Dietary fructose is primarily absorbed by the small intestinal cells through the GLUT5 apical transporter and stored in the cell through phosphorylation by ketohexokinase (78). While low doses of fructose are absorbed in the small intestine, high doses of fructose can saturate the absorptive and catabolic enzymes of the small intestine, allowing fructose to reach the colon (78). Thus, the rise in obesity incidence and concomitant rise in CRC incidence may be linked to an increase in consumption of high-fructose corn syrup (HFCS) in sweetened beverages. There was a positive association between sweetened beverage intake in adolescence and conventional adenomas on colonoscopy in the Nurses’ Health Study II population (79).
The association between fructose consumption, obesity, and tumorigenesis may be explained by the effect of fructose on the intestine in animal models (80). HFCS increases the survival of intestinal epithelial cells in several mouse models, thereby extending intestinal villi length by 25% to 40% (80). Longer villi enhance nutrient absorption and result in an increase in adiposity in mice fed an HFD (80). Moreover, fructose-1-phosphate was found to inhibit the M2 isotype of pyruvate kinase and extend survival in hypoxic intestinal epithelial cells, thereby promoting tumor growth. In addition, HFCS at moderate doses increases the number and grade of intestinal tumors formed in Apc-deficient mouse models by fueling aerobic glycolysis and contributing to an increase in de novo fatty acid synthesis (81). Fructose metabolism may also promote CRC metastasis to the liver. CRC liver metastasis upregulates aldolase B expression, the rate-limiting enzyme in the utilization of fructose. Targeting aldolase B, or its upstream regulator, GATA binding protein 6, reduces CRC liver metastasis in mice. Moreover, dietary fructose restriction suppresses CRC liver metastasis and extends survival in a CRC mouse model (82). Other cancers appear to selectively utilize fructose, with an increased number of fructose transporters, Glut5 and Glut9, identified in malignant versus benign prostate tumors (83). Although the role of fructose in CRC development in humans is not well understood, there are compelling preclinical studies showing a mechanistic link between dietary fructose consumption, obesity, and CRC tumorigenesis (79, 81, 82). A summary of the impact of a high-fructose diet on CRC tumorigenesis is shown in Figure 2a.
Figure 2.
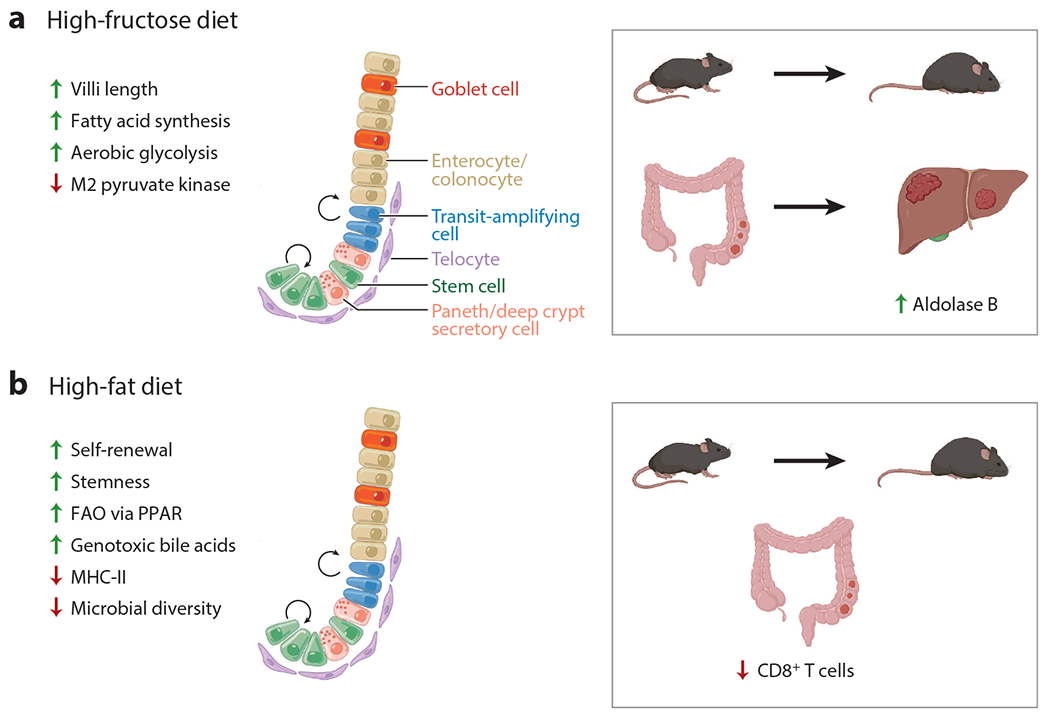
Effect of pro-obesity diets on intestinal epithelium and CRC tumorigenesis. (a) A high-fructose diet increases villi length, fatty acid synthesis, and aerobic glycolysis and reduces M2 pyruvate kinase expression, promoting CRC tumorigenesis and liver metastasis. Arriving CRC cells adapt to the liver by using readily available fructose as a carbon source by upregulating aldolase B. (b) A high-fat diet induces increases in ISC self-renewal, stemness, FAO via PPAR, and genotoxic bile acids, a decrease in MHC-II expression on intestinal epithelial cells, dysbiosis, and a reduction in CD8+ T cells, thereby promoting CRC tumorigenesis. Figure adapted from images created with BioRender.com. Abbreviations: CRC, colorectal cancer; FAO, fatty-acid oxidation; ISC, intestinal stem cell; MHC-II, major histocompatibility complex class II; PPAR, peroxisome proliferator-activated receptor.
3.4. High-Fat Diet and Obesity
There is mixed evidence for an association between fat intake and CRC in humans but strong evidence for an association between obesity and CRC. A recent systemic review and meta-analysis of prospective studies from 18 articles identified no association between total fat, saturated fatty acid, monounsaturated fatty acid, and polyunsaturated fatty acid intake and the risk of CRC (28). The mixed findings in these individual studies may be the result of different fat sources, such as fat from red and processed meats that are consistently associated with CRC risk, contributing to the primary fat content of the diet of the individuals studied. Nonetheless, dietary fat is believed to contribute to the growing obesity epidemic, on the basis of short-term feeding studies in humans and in mice (84, 85). A large body of evidence strongly links obesity (BMI greater than or equal to 30 kg/m2) to CRC; obesity is the second-highest modifiable risk factor for all cancers (86). A recent case-control study found an association between excess body weight and CRC risk, where the adjusted odds ratio for the first to fourth quartile of excess body weight was 1.25 and 2.54, respectively (87). In addition, CRC patients with obesity have a fivefold increased risk of death compared with normal-weight counterparts (BMI < 25) (88). Conversely, weight loss through bariatric surgery is associated with reduced cancer incidence (89, 90).
Several mouse studies have shown a mechanistic association between an ad libitum HFD and CRC. These studies typically use a dietary fat content composed of 60% of calories resulting in the mice consuming up to 40% more daily calories, thereby inducing obesity (91). As such, an ad libitum HFD that promotes overeating does not distinguish the effects of the higher fat content in the diet, increased calorie intake that causes obesity, and obesity itself. Obesity is a key confounder to note when discussing HFD-induced tumorgenicity because obesity is an established risk factor for CRC (44). We and others have found that a pro-obesity HFD enhances intestinal tumorigenesis by increasing the number, proliferation, and function of ISCs (32, 33, 92, 93). ISCs increase their proliferation and tumorigenicity through PPAR-mediated activation of an FAO metabolic program; the increase in tumorigenesis is sensitive to FAO inhibition by pharmacologic inhibitors and genetic disruption (33). Another distinct path by which an HFD promotes CRC tumorigenesis is through increasing the production of specific bile acids (94). A recently described mechanism for bile-acid-induced CRC tumorigenesis came from a study of Apc-deficient mice on an HFD. The authors found that Apc mutation and HFD increase levels of the bile acids tauro-β-muricholic acid and deoxycholic acid, antagonizing the intestinal farnesoid X receptor (FXR) and inducing proliferation and DNA damage in Lgr5+ ISCs (95). Affirming these findings, the protumorigenic effect of these bile acids could be blocked by agonists of intestinal FXR (95). Thus, there are clear protumorigenic effects of an ad libitum HFD in mice with multiple identified mechanisms, which are summarized in Figure 2b.
The immune system and microbiome play important roles in the protumorigenic effects of an ad libitum HFD in mice. Using immunocompetent C57BL/6 background mouse models, our group showed that an HFD contributes to CRC tumorigenesis through a microbiome–ISC–immune cell interaction (34). Major histocompatibility complex class II (MHC-II) is expressed on the surface of antigen-presenting cells for recognition by CD4+ T cells. While antigen-presenting cells are most commonly macrophages and dendritic cells, intestinal epithelial cells can also monitor and present local antigens on MHC-II, presumably for microbial and tumor immune surveillance (96). An HFD causes intestinal epithelial and Lgr5+ ISCs to lose expression of MHC-II, which is not seen in leptin-deficient mice (db/db) that develop obesity on normal chow, suggesting that the decrease in MHC-II expression is specific to the higher fat content of the diet. HFD lowers MHC-II expression via a reduction in the microbial diversity of the intestines, specifically the loss of Helicobacter sp. and Odoribacter sp. Germ-free mice and mice given broad-spectrum antibiotics produce a similar decrease in microbial diversity and MHC-II expression (34). Thus, decreased MHC-II expression on intestinal epithelial cells from an HFD may promote tumorigenesis through decreasing tumor immune surveillance (34).
Using a less physiological mouse model of a subcutaneously transplanted CRC cell line (i.e., MC38), another group identified an immune-related role of an HFD contributing to tumorigenesis through reducing CD8+ T cells in the tumor microenvironment (97). An HFD metabolically reprograms the CRC tumor to increase fat uptake, thereby depleting the availability of lipids in the tumor microenvironment for CD8+ T cells. Finally, a recent report concluded that an HFD promotes CRC by inducing intestinal microbial dysbiosis (i.e., increased Alistipes sp. and decreased Parabaceroides distasonis), metabolomic dysregulation (i.e., increased protumorigenic lysophosphatidic acid), and gut barrier dysfunction using the azoxymethane (AOM) and Apcmin/+ CRC mouse models (98). Interestingly, the protumorigenic effect of the HFD were abrogated with the administration of antibiotics, reaffirming previous research, suggesting that microbial dysbiosis contributes to HFD-mediated CRC tumorigenesis (99). Thus, there is clear evidence that a pro-obesity HFD impacts ISC proliferation and tumorigenicity through multiple mechanisms. A summary of immune- and microbiome-mediated effects of an HFD on CRC tumorigenesis is shown inFigure 3.
Figure 3.
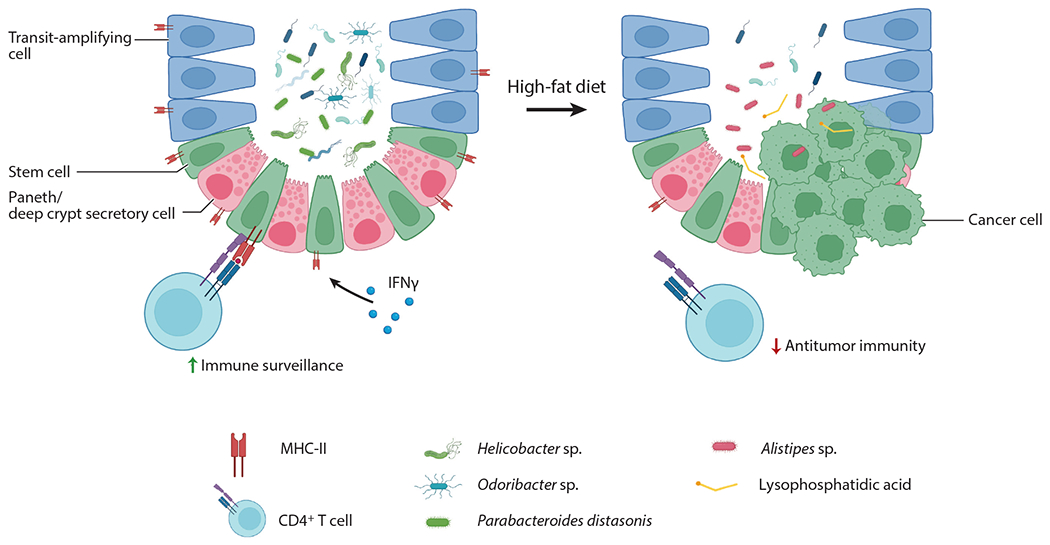
An HFD promotes CRC tumorigenesis through intestinal dysbiosis and decreased tumor immune surveillance. Beyaz et al. (32) found that an HFD reduces microbial diversity, particularly Helicobacter sp. and Odoribacter sp., leading to a decrease in MHC-II expression on intestinal epithelial cells and a reduction in antitumor immunity. Yang et al. (98) reported that an HFD promotes tumorigenesis through a shift in protective Parabacteroides distasonis to Alistipes sp. and an increase in the concentration of the protumorigenic fatty acid, lysophosphatidic acid. Figure adapted from images created with BioRender.com. Abbreviations: CRC, colorectal cancer; HFD, high-fat diet; IFN, interferon; MHC-II, major histocompatibility complex class II.
3.5. Carbohydrate-Rich Diet
There is limited epidemiological and animal study evidence that a high-carbohydrate diet increases the risk of CRC (100). Most of the research on the effects of a high-carbohydrate diet in animal models coadminister an HFD (pro-obesity, Western diet), recapitulating many of the effects of an HFD (101). The importance of specific carbohydrates on CRC tumorigenesis is poorly understood and is an area of needed attention.
3.6. Ketone Bodies
Ketone bodies, consisting of β-hydroxybutyrate, acetoacetate, and acetone, are an alternative fuel source to glucose produced primarily by the liver from fatty acids during periods of fasting, prolonged physical activity, or a ketogenic diet (KD). While ketone bodies were first discovered as toxic byproducts in the urine of patients succumbing to diabetic ketoacidosis, the mild levels of ketones produced in nondiabetic and otherwise healthy individuals under nonstarvation conditions are not known to be toxic. The KD generally consists of 75% of calories from fat, 20% of calories from protein, and 5% of calories from carbohydrates and is used clinically to help control drug-resistant epilepsy (102). In theory, a KD could deprive cancer cells of the glucose consumed during aerobic glycolysis, thereby slowing the growth of tumors. However, there is currently no randomized controlled trial evidence for any effect of a KD on CRC initiation or treatment. Nonetheless, a recent meta-analysis in preclinical cancer models found that KD supplementation significantly prolonged survival by an average of 76% and reduced tumor volume (103).
Our research group recently studied the effects of ketone bodies on intestinal epithelial cells in mice (104). We found that ketone bodies produced from LGR5+ ISCs enhanced NOTCH activity, ISC self-renewal, and postinjury regeneration, while decreasing secretory cell differentiation (104). This effect was achieved, at least in part, through β-hydroxybutyrate inhibiting histone deacetylase. A recent study found that a KD suppressed intestinal tumor growth in an AOM/dextran sodium sulfate mouse model of CRC and that these effects were reproduced by administering β-hydroxybutyrate (105). The mechanism of β-hydroxybutyrate suppressing intestinal tumorigenesis in this study was believed to be downstream of β-hydroxybutyrate binding to the surface receptor HCAR2 (hydroxycarboxylic acid receptor 2) and altering transcriptional regulation through HOPX (homeodomain-only protein). Nonetheless, the mechanisms of how a KD may impair tumor growth are not well understood and may depend on the fatty acid composition of the KD and fatty acid desaturation activity of the tumor. In an allograft mouse model of pancreatic adenocarcinoma and lung adenocarcinoma, CR, but not a KD, decreased tumor volume (60). While CR and a KD lowered blood glucose, CR reduced while a KD increased circulating and intratumoral lipid levels. The authors found that stearyl coreductase (SCD), which synthesizes monounsaturated fatty acids, is required for cancer cells to proliferate in a lipid-depleted environment. CR reduces circulating and intratumoral lipid levels and SCD expression, thereby creating an imbalance in the ratio of unsaturated to saturated lipids to tumors, slowing cancer growth. A KD reduces SCD expression, but the monounsaturated fatty acids in the KD maintain the ratio of unsaturated to saturated lipids that are important for tumor growth. However, substituting monounsaturated fatty acids in the KD with saturated fatty acids by switching from a lard-based to palm-oil-based KD slows tumor growth. In summary, a KD, and specifically the ketone body β-hydroxybutyrate, enhances ISC self-renewal and function while also potentially inhibiting CRC growth through distinct mechanisms in animal models.
3.7. Weight Loss
Obesity increases the risk of type 2 diabetes, cardiovascular disease, most types of cancer, early-onset CRC, and other comorbidities (44, 86). Weight loss after bariatric surgery appears to reduce the risk of these comorbidities, though there have been mixed findings in retrospective studies for CRC risk reduction after gastric bypass (106). There are several variations of bariatric surgery, with gastric bypass but not gastric banding or sleeve gastrectomy, associated with an increase in CRC risk in one national study in the United Kingdom (107). However, a large, multicenter cohort of severely obese patients who received bariatric surgery—mostly gastric bypass—demonstrated a 33% and 41% reduced risk of developing any cancer and CRC, respectively (108). Additionally, a nationwide study in a French population found that the risk of CRC in obese patients after bariatric surgery (with gastric bypass as the second-most common procedure) went down 34%, matching the risk of the general population (109). A recent meta-analysis reported that patients who underwent any bariatric surgery had more than a 35% reduction in their risk of developing CRC (110).
How bariatric surgery reduces the risk of CRC is not well understood. Whether the reduction in CRC risk is due to a decrease in precancerous adenomas was explored in a retrospective study at a large academic medical center. Patients who received their index colonoscopy at least one year after bariatric surgery were compared with patients who received their bariatric surgery after index colonoscopy (111). Adenomatous polyps were found in 16.8% of patients who received their bariatric surgery before index colonoscopy, compared with 35.5% of patients who received bariatric surgery after index colonoscopy (111). Thus, there is evidence bariatric surgery reduces CRC initiation and risk, but more research is needed to find a biological mechanism linking bariatric surgery and CRC risk.
4. COLORECTAL CANCER GENETICS AND METABOLISM
The metabolism of ISCs shifts toward a protumorigenic program beginning with the earliest initiating genetic events in the adenoma-carcinoma sequence (112). In this section, we discuss the effects of the activation of oncogenes and inactivation of tumor suppressor genes on the metabolism of CRC.
4.1. Metabolic Pathways Altered in Colorectal Cancer
The Warburg effect, or aerobic glycolysis, is one of many metabolic programs shared between healthy proliferating cells and cancer to meet the energy and biosynthesis demands of proliferation (10). ISCs are highly glycolytic, like CRC cells, compared with their differentiated counterparts higher up the crypt/villus axis (26). ISCs require the same building blocks for cell divisions and therefore share many other similarities with CRC. For example, the regenerative response after injury of ISCs is enhanced by TIGAR (TP53-inducible glycolysis and apoptosis regulator), increasing the flux of glucose carbons toward the pentose phosphate pathway (113). TIGAR also promotes CRC tumorigenesis and is more highly expressed after APC loss (114). Many of the biological pathways required for ISC proliferation and regeneration are upregulated by perturbations in oncogenes and tumor suppressor genes accrued during the adenoma-carcinoma sequence.
Genetic perturbations in oncogenes and tumor suppressor genes alter metabolism to support CRC tumorigenesis, starting at the adenoma stage of the adenoma-carcinoma sequence (112). The most common CRC oncogenic drivers include KRAS (30%), PIK3CA (19%), and BRAF (14%); common tumor suppressors include APC (56%), TP53 (53%), FAT4 (22%), LRP1B (21%), KMT2D (14%), ACVR2A (14%), FBXW7 (13%), SMAD4 (13%), and PTEN (6.7%) (115). Activation of the WNT signaling pathway is present in more than 90% of CRCs, achieved through the loss of function of APC, activating mutations in β-catenin, or overexpression of frizzled receptors (116). Somatic mutations in KRAS (117), TP53 (118), WNT pathway genes (114, 119), and PTEN (120) promote tumorigenesis, in part, by driving metabolic changes. KRAS mutations trigger a major shift in metabolism to promote cellular proliferation, activating MYC and downstream glucose uptake (e.g., upregulating GLUT1 receptors), glutamine uptake, micropinocytosis, and autophagy and mitophagy (117, 121). Loss of TP53 also contributes myriad effects on the cancer cell’s metabolism, including increased lipid synthesis and glucose influx into the cell (118). Likewise, loss of APC can lead to downstream activation of TIGAR, MYC target genes, glycolysis, and angiogenesis (114, 119). PTEN mutations and the PI3K signaling pathway promote CRC proliferation through the activation of glycolysis and other metabolic pathways (120). Thus, the proliferation of CRC cells is fueled by metabolic changes that occur from genetic perturbations in common oncogenes and tumor suppressors.
Mutations found in noncanonical tumor suppressors and oncogenes may also alter metabolism to promote CRC tumorigenesis. Somatic mutations in mitochondrial DNA acquired from aging may cause OXPHOS defects, leading to protumorigenic metabolic remodeling through the upregulation of the de novo serine synthesis pathway (122). Additional serine fuels CRC proliferation through serine’s conversion to pyruvate via the enzyme serine racemase (123). Inhibition of serine racemase decreases the size and proliferation of CRC cells in vitro and in vivo, indicating that serine racemase may be a novel and promising therapeutic target (123). Other defects of mitochondria function, such as the inactivation of MPC, which transfers pyruvate into the mitochondria, also promote CRC tumorigenesis (124). Inactivation of MPC in mice and flies is sufficient to promote intestinal tumorigenesis, while overexpression of MPC within the fly is sufficient to suppress tumorigenesis. CRCs also metabolically adapt under nutrient stress. One adaptation co-opted by CRCs in the context of glucose withdrawal is the inactivation of protein kinase C zeta (PKCϛ). PKCϛ deficiency promotes a shift in metabolism toward the utilization of glutamine through the serine biosynthetic pathway, thereby enhancing intestinal tumorigenesis in Apcmin/+ mice (125). Other lesser-known pathways similarly contribute to CRC tumorigenesis, including the transsulfuration enzyme cystathionine-beta-synthase (CBS) that produces hydrogen sulfide. Upregulation of CBS in precancerous lesions and CRC was found to contribute to tumorigenesis through increased glycolysis, nucleotide synthesis, pentose-phosphate pathway, and lipogenesis (126). Thus, there are many noncanonical metabolic alterations, potentially susceptible to therapeutic interventions, that promote the progression of CRC.
4.2. Colorectal Cancer Metastasis
Almost all CRC mortality is caused by metastases, with the most common sites being the liver and lungs. Metastasizing tumor cells must adapt to new metabolic conditions throughout invasion, intravasation, circulation, extravasation, and growth within different microenvironments. A subpopulation of CRC cells, called cancer stem cells (CSCs), are believed to have the greatest metastatic potential. Unlike other CSCs, CRC CSCs appear to retain their epithelial characteristics, rather than adopting a quasi-mesenchymal phenotype through the epithelial-to-mesenchymal transition program (127). These CSCs have been characterized by several markers, including the thrombopoietin (TPO) receptor, CD110, which functionally boosts CRC metastasis (128). TPO promoted CRC liver metastasis by CD110+ CSC through activating lysine degradation, inducing a shift in redox status and activation of WNT signaling (129). Given the dynamic metabolic dependencies of CRC throughout the metastatic cascade, metabolism has been a major focus of ongoing research to understand mechanisms of CRC metastasis (130).
Increased FAO, glutathione production, and prostaglandin E2 (PGE2) boost the ability of CRC cells to metastasize. Increased FAO by an HFD was found to expand CSCs and promote liver metastases through the expression of NANOG, a transcription factor important for stem cell self-renewal (131). A recent study found that FAO upregulation can also help CRC cells become resistant to anoikis, a specialized form of apoptosis caused by detachment from the extracellular matrix (132). Small-molecule inhibition of CPT1A, the rate-limiting step of FAO, leads to fewer liver and lung metastases in mouse models of CRC. Glutathione production appears to similarly increase the metastatic ability of CRC cells. Increased glutaminase 1 (GLS1), an enzyme that hydrolyzes glutamine to glutamate, is associated with worse clinical outcomes, and GLS1 deficiency suppresses CRC growth and metastasis in a mouse model (133). The mechanism underlying increased CRC metastasis from GLS1 upregulation is not fully understood but may relate to an increase in glutathione production from the greater availability of glutamate, one of the three amino acid building blocks of glutathione (134). Increased glutathione may promote a CSC phenotype through upregulation of NANOG (135). Others have found that the induction of glutathione is critical for CRC liver metastasis, though downstream of liver and red blood cell pyruvate kinase L/R (136). Additionally, PGE2 treatment of immunocompromised mice transplanted with a human CRC cell line with intact TP53 increases CRC cell invasiveness and ability to form liver and lung metastases (137). The mechanism of increased CRC metastasis by PGE2 occurred downstream of TP53 inhibition and is believed to also be due to an expansion of CSCs (137, 138). Thus, metabolic alterations in FAO, glutathione production, and PGE2 production have distinct roles in driving CRC metastasis.
The liver poses a harsh, hypoxic environment for arriving CRC cells, requiring complex metabolic adaptations for survival. To provide energy for proliferation in a low-oxygen environment, CRC cells release creatine kinase, brain-type (CKB). CKB converts extracellular creatine into phosphocreatine that could be transferred back into the CRC cells through the phosphocreatine transporter, SLC6A8, to generate ATP (139). Importantly, CKB inhibition is effective at reducing CRC liver metastases in mouse models and is upregulated along with SLC6A8 in human liver metastases (139). Blocking phosphocreatine import by CRC cells using a small-molecule SLC6A8 transport inhibitor reduces CRC progression and metastasis in vitro and in mouse models, especially in combination with other anticancer drugs (140). Treating metastatic CRC patients in a phase 1 clinical trial with this small-molecule inhibitor of SLC6A8 resulted in an increase in serum and urine creatinine, mirroring the pharmacodynamics observed in mouse models. An additional source of energy and biomass comes from the utilization of high levels of fructose found in the liver. CRC liver metastases take advantage of the high concentration of fructose through the upregulation of aldolase B, the rate-limiting enzyme of fructose metabolism (82). Notably, reducing dietary fructose diminishes liver metastatic growth (82). Another metabolic adaptation that may help CRC cells to survive in the liver is the ability to synthesize nucleotides under hypoxia, via the upregulation of the gluconeogenic enzyme, phosphoenolpyruvate carboxykinase 1 (PCK1) (141). Inhibiting nucleotide synthesis with leflunomide, an inhibitor of dihydroorotate dehydrogenase (DHODH), decreases CRC liver metastases in a CRC mouse model. As such, PCK1 and DHODH have been proposed as potential therapeutic targets of CRC metastatic progression. There are currently more than a dozen inhibitors of metabolic targets that have shown promise for cancer metastasis prevention and treatment in preclinical models (130). Thus, CRCs establish metastases using unique metabolic adaptations that may be therapeutically targeted. A summary of the metabolic pathways adapted for CRC initiation, progression, and metastasis is detailed in Figure 4.
Figure 4.
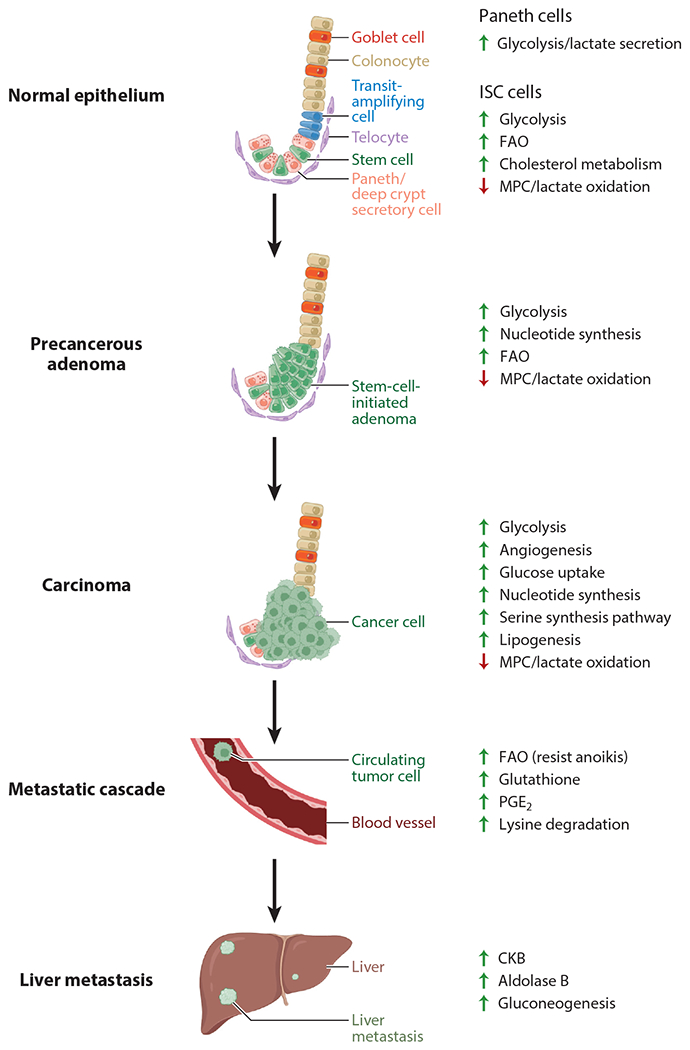
Metabolic pathways exploited in CRC initiation and progression. At homeostasis, ISCs sustain high proliferation through upregulated glycolysis, FAO, cholesterol metabolism, shuttling lactate toward biosynthetic pathways through downregulation of MPC, and lactate secretion from adjacent Paneth cells. Adenomas exploit these pathways and sustain high proliferation through perturbations in oncogenes and tumor suppressors, enhancing glycolysis, nucleotide synthesis, and FAO. CRC further manipulates these same pathways and others, including the serine synthesis pathway, angiogenesis, and lipogenesis. Additional metabolic adaptations are required for metastasis, including increased FAO to resist anoikis, glutathione production, PGE2, and lysine degradation. CRC cells arriving at the liver adapt to the harsh microenvironment through upregulating CKB for extracellular sources of energy, aldolase B for fructose metabolism, and gluconeogenesis. Figure adapted from images created with BioRender.com. Abbreviations: CKB, creatine kinase, brain-type; CRC, colorectal cancer; FAO, fatty-acid oxidation; ISC, intestinal stem cell; MPC, mitochondrial pyruvate carrier; PGE2, prostaglandin E2.
5. MICROBIOME
The healthy human colon harbors hundreds to thousands of different microbial species that collectively make up the approximately 38 trillion microbes in the microbiome (15, 142). These microbes largely reside within the outer mucus layer coating the crypts, though certain strains may access the inner mucus layer and epithelium (142). A few dominant bacterial phyla—Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria—compose most of the microbiome. The functions of the gut microbiome include digesting nutrients from food, synthesizing vitamins and other nutrients, detoxifying metabolites, and secreting antimicrobial products, among an expanding repertoire of appreciated functions (143). Evidence for the microbiome’s functional role in health and disease has rapidly accumulated, with studies now showing that the composition and diversity of the microbiome regulate ISC biology and influence CRC tumorigenesis (143–145). Numerous factors, from diet to medications, impact the microbiome. Interestingly, several risk factors for CRC development impact the composition of the gut microbiome: low dietary fiber, obesity, physical inactivity, and red and processed meats (143). Whether these CRC risk factors induce changes in the microbiome that then mediate CRC tumorigenesis is currently unknown. Intestinal microbes secrete an estimated three million metabolites and compounds, yet little is known about how these metabolites and compounds impact human health (146). A recent study in mice identified a mechanistic link between lactate-producing bacteria and enhanced ISC proliferation (144). While ISCs directly use lactate to fuel proliferation, the microbe-secreted lactate activates GPR81 on the surface of Paneth and stromal cells, thereby increasing WNT3 secretion, which promotes ISC stemness and proliferation. Thus, the gut microbiome likely has a major impact on ISC function and CRC tumorigenesis, although mechanisms by which specific microbial metabolites regulate intestinal homeostasis and cancer initiation remain poorly understood.
Microbes commonly found in the human microbiome can promote or protect against CRC. Genotoxic pks+ E. coli, Fusobacterium nucleatum, enterotoxigenic Bacteroides fragilis, and other microbes are associated with CRC (145). For example, pks+ E. coli are found in the colon of 55% of patients with CRC versus 19% of control patients (147). The pks operon enables the production of the colibactin genotoxin. Colibactin directly causes DNA damage, promoting CRC tumorigenesis, through alkylating adenosines (148). Interestingly, a shift in the microbiome toward harboring pathogenic bacteria—Alistipes sp. Marseille-P5997 and Alistipes sp. 5CPEGH6—is associated with enhanced CRC tumorigenesis on an HFD (98). While pks+ E. coli and other microbes contribute to CRC tumorigenesis, certain microbes can also protect against CRC development. The absence of gut microbes (i.e., germ-free mice) is associated with enhanced CRC tumorigenesis—potentially from decreased MHC-II expression—which supports the concept that commensal microbes protect against CRC (34, 149, 150). Microbes can also protect against CRC tumorigenesis by the effects of several diverse microbially secreted compounds. Many microbial species metabolize tryptophan into metabolites that serve as ligands of the AhR receptor transcription factor, thereby enhancing DNA repair and slowing cellular proliferation by inhibiting the WNT pathway (146). Additionally, reuterin, a microbial metabolite from Lactobacillus reuteri, was found to protect against CRC through increasing protein oxidation, decreasing ribosome biogenesis, and decreasing protein translation in nascent CRC tumors (150). A growing body of knowledge suggests an important role for commensal microbes and their metabolic products in CRC pathogenesis, with the potential for future clinical therapeutic applications.
6. THERAPY
Targeting metabolic processes to treat CRC requires the identification of an adequate therapeutic index to avoid or limit toxicity in normal tissues. CRC treatment is currently driven by disease stage; stage I cancer (i.e., limited to the colonic mucosa) can be cured with surgical or endoscopic resection, while stage IIIA to stage IV disease (i.e., invasion into the local lymphatics or distant metastasis) is typically treated with chemotherapy (151). 5-Fluorouracil (5-FU) is the most common therapeutic agent, typically as part of FOLFOX (5-FU, leucovorin, and oxaliplatin) or FOLFIRI (5-FU, leucovorin, and irinotecan) regimens. While 5-FU acts on cancer cells through many mechanisms, including activation of cancer cell apoptosis and autophagy, its main mechanism of action is to inhibit cellular thymidylate synthase, which prevents DNA replication (152). Therefore, although 5-FU was developed in the 1950s before dysregulated metabolism was accepted as a hallmark of cancer, it should be seen as a therapeutic that targets an essential metabolic pathway.
Several novel approaches have been developed to target metabolic vulnerabilities in KRAS mutant CRC. Alternatively, other small-molecule compounds have been found to also target KRAS-dependent metabolic processes. For instance, the dual RAS/MEK inhibitor RO5126766 decreases FDG uptake and cell proliferation in KRAS and BRAF mutant CRC cell line xenograft models (153). Glutamine metabolism has been studied as a strategy to treat KRAS mutant CRC. KRAS mutant CRC cells adapt to glutamine deprivation by upregulating asparagine synthetase expression and producing more asparagine; l-asparaginase plus rapamycin inhibits the growth of KRAS mutant tumors in vivo (154). As discussed in prior sections, vitamin C has been studied in preclinical models as a treatment for highly glycolytic KRAS and BRAF mutant CRC by inducing oxidative stress from depleted glutathione. Increased ROS levels lead to inactivation of the glycolytic enzyme GAPDH and subsequent cell death (76).
Several studies have examined KRAS-independent metabolic targets in CRC. 2-Deoxy-d-glucose (2-DG) is a glucose molecule in which the 2-hydroxyl group is replaced with hydrogen, such that it cannot undergo further glycolysis, and thus competitively inhibits the production of glucose-6-phosphate from glucose. 2-DG preferentially kills CRC cells both by inhibiting glycolysis and by upregulating death receptor 5, which increases sensitivity to TRAIL-induced apoptosis (155). The use of the antidiabetic drug metformin is associated with reduced risk for CRC and has therefore been proposed as a therapeutic agent for CRC (156). Metformin reduces adenoma formation and aberrant crypt foci formation in mouse models by inhibiting the mitochondrial electron transport chain complex 1 and by activating AMP-activated protein kinase signaling (157, 158). Another study reported that metformin inhibits CRC formation in a rat model by suppressing PKM2-dependent glycolysis (159). Finally, metformin inhibits the proliferation of chemotherapy-resistant breast cancer stem cells, suggesting a possible approach for treating CRC patients who have failed standard chemotherapy (160). Potential future approaches for targeting metabolism in CRC include identifying metabolic vulnerabilities in immune cells in the tumor microenvironment, tailoring therapies to patients on the basis of genetically defined metabolic targets, targeting metabolic reprogramming during cancer progression and/or metastasis, and administering specific metabolites such as β-hydroxybutyrate.
7. CONCLUSIONS AND FUTURE DIRECTION
Dysregulated nutrient consumption and cellular metabolism are key mechanisms of colorectal carcinogenesis. The cell of origin for most CRCs, the LGR5+ ISC, is highly proliferative and shares similar metabolic features with CRC. Perturbations in canonical oncogenes and tumor suppressor genes drive metabolic adaptations to maintain the biomass and energy needs of proliferating CRC cells without homeostatic growth factor signaling. The metabolic adaptations of CRC also extend into perturbations of noncanonical genes, providing additional avenues to acquire a selective advantage for nutrient challenges encountered throughout CRC initiation and progression. These recent basic science discoveries are beginning to translate into potential therapies for patients. New clinical targets of CRC metabolism, such as the phosphocreatine transporter, SLC6A8, are already in clinical trials. Other metabolic adaptations of CRC are prime targets for chemoprevention and treatment, with potential for combination therapies. Additionally, novel classes of therapies may arise from the rapidly maturing microbiome field, where associations of specific microbes with CRC are becoming reinforced with mechanistic data to support causation by microbe-produced metabolites. Further research on the microbiome holds promise for CRC and disease prevention by finding strategies to achieve and maintain an optimal microbiome—replacing pathogenic microbial strains with those known to be protective or administering microbiome-derived metabolites. A pro-obesity HFD significantly contributes to CRC tumorigenesis in animal models, originating from effects on the intestinal stem and progenitor cells and microbiome; further research is required to translate these discoveries into specific therapies for patients with obesity-associated CRC. More research is also needed to understand the impact of specific modifiable risk factors, such as consumption of alcohol and red meat, on CRC development. In addition, metabolic responses to modifiable risk factors may underlie the growing incidence of CRC in younger individuals under the age of 50. As such, a growing body of preclinical and clinical research suggests that metabolic adaptations are important drivers of ISC function, CRC initiation, and CRC metastasis.
ACKNOWLEDGMENTS
We thank Negin Amouei for assisting with the literature review.
DISCLOSURE STATEMENT
J.C.S. is a board member and scientific advisor to Mercy BioAnalytics Inc. J.C.S. is supported by National Institutes of Health (NIH) grant F30CA260789. Ö.H.Y. is a science advisory board member for AVA LifeScience and receives research funding from and is a consultant for Microbial Machines. Ö.H.Y. is supported by NIH grants P30CA014051, R01CA211184, R01CA245314, R01CA034992, R01DK126545, R01CA257523, and U54CA224068; a Pew-Stewart Trust scholar award; the Kathy and Curt Marble Cancer Research Award; a Koch Institute–Dana-Farber/Harvard Cancer Center Bridge Project grant; and the Massachusetts Institute of Technology (MIT) Stem Cell Initiative via Foundation MIT. J.R. is supported by NIH grants R37CA259363, R21CA256414, R21DK125911, R41EB032693, R01CA254108, R01CA256530, and R01CA244359; US Department of Defense grant W81XWH-20-1-0203; and a Duke–NC State Translational Research Grant.
LITERATURE CITED
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, et al. 2021. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 71:209–49 [DOI] [PubMed] [Google Scholar]
- 2.Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, et al. 2018. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin 68:31–54 [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, et al. 2020. Colorectal cancer statistics, 2020. CA Cancer J. Clin 70:145–64 [DOI] [PubMed] [Google Scholar]
- 4.Fearon ER, Vogelstein B. 1990. A genetic model for colorectal tumorigenesis. Cell 61:759–67 [DOI] [PubMed] [Google Scholar]
- 5.Paterson C, Clevers H, Bozic I. 2020. Mathematical model of colorectal cancer initiation. PNAS 117:20681–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, et al. 2015. The consensus molecular subtypes of colorectal cancer. Nat. Med 21:1350–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warburg O, Wind F, Negelein E. 1927. The metabolism of tumors in the body. J. Gen. Physiol 8:519–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeBerardinis RJ, Chandel NS. 2016. Fundamentals of cancer metabolism. Sci. Adv 2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fantin VR, St-Pierre J, Leder P 2006. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9:425–34 [DOI] [PubMed] [Google Scholar]
- 10.Lunt SY, Vander Heiden MG. 2011. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol 27:441–64 [DOI] [PubMed] [Google Scholar]
- 11.Pavlova NN, Zhu J, Thompson CB. 2022. The hallmarks of cancer metabolism: still emerging. Cell Metab. 34:355–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helander HF, Fandriks L. 2014. Surface area of the digestive tract—revisited. Scand. J. Gastroenterol 49:681–89 [DOI] [PubMed] [Google Scholar]
- 13.Kiela PR, Ghishan FK. 2016. Physiology of intestinal absorption and secretion. Best Pract. Res. Clin. Gastroenterol 30:145–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandle GI. 1998. Salt and water absorption in the human colon: a modern appraisal. Gut 43:294–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 14:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, et al. 2007. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449:1003–7 [DOI] [PubMed] [Google Scholar]
- 17.Pinto D, Gregorieff A, Begthel H, Clevers H. 2003. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 17:1709–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KA, Kakitani M, Zhao J, Oshima T, Tang T, et al. 2005. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 309:1256–59 [DOI] [PubMed] [Google Scholar]
- 19.Kosinski C, Li VS, Chan AS, Zhang J, Ho C, et al. 2007. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. PNAS 104:15418–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lueschow SR, McElroy SJ. 2020. The Paneth cell: the curator and defender of the immature small intestine. Front. Immunol 11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, et al. 2011. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469:415–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasaki N, Sachs N, Wiebrands K, Ellenbroek SI, Fumagalli A, et al. 2016. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. PNAS 113:E5399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degirmenci B, Valenta T, Dimitrieva S, Hausmann G, Basler K. 2018. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 558:449–53 [DOI] [PubMed] [Google Scholar]
- 24.Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, Toth B, Kondo A, et al. 2018. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 557:242–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gehart H, Clevers H. 2019. Tales from the crypt: new insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol 16:19–34 [DOI] [PubMed] [Google Scholar]
- 26.Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ, Gratton E. 2012. Metabolic trajectory of cellular differentiation in small intestine by phasor fluorescence lifetime microscopy of NADH. Sci. Rep 2:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durand A, Donahue B, Peignon G, Letourneur F, Cagnard N, et al.2012. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). PNAS 109:8965–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M, Park K. 2018. Dietary fat intake and risk of colorectal cancer: a systematic review and meta-analysis of prospective studies. Nutrients 10:1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, et al. 2017. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 543:424–27 [DOI] [PubMed] [Google Scholar]
- 30.Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, et al. 2017. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol 19:1027–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, et al. 2012. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, et al. 2016. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531:53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mana MD, Hussey AM, Tzouanas CN, Imada S, Barrera Millan Y, et al. 2021. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 35:109212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beyaz S, Chung C, Mou H, Bauer-Rowe KE, Xifaras ME, et al. 2021. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 28:1922–35.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang B, Rong X, Palladino END, Wang JF, Fogelman AM, et al. 2018. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22:206–220.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei P, Dove KK, Bensard C, Schell JC, Rutter J. 2018. The force is strong with this one: metabolism (over)powers stem cell fate. Trends Cell Biol. 28:551–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, et al. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457:608–11 [DOI] [PubMed] [Google Scholar]
- 38.de Lau W, Peng WC, Gros P, Clevers H. 2014. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 28:305–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huels DJ, Sansom OJ. 2015. Stem versus non-stem cell origin of colorectal cancer. Br. J. Cancer 113:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarthy N, Kraiczy J, Shivdasani RA. 2020. Cellular and molecular architecture of the intestinal stem cell niche. Nat. Cell Biol 22:1033–41 [DOI] [PubMed] [Google Scholar]
- 41.McCarthy N, Manieri E, Storm EE, Saadatpour A, Luoma AM, et al. 2020. Distinct mesenchymal cell populations generate the essential intestinal BMP signaling gradient. Cell Stem Cell 26:391–402.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan BA, Noujaim M, Roper J. 2022. Cause, epidemiology, and histology of polyps and pathways to colorectal cancer. Gastrointest. Endosc. Clin. N. Am 32:177–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karahalios A, English DR, Simpson JA. 2015. Weight change and risk of colorectal cancer: a systematic review and meta-analysis. Am. J. Epidemiol 181:832–45 [DOI] [PubMed] [Google Scholar]
- 44.Li H-J, Boakye D, Chen X-C, Jansen L, Chang-Claude J, et al. 2022. Associations of body mass index at different ages with early-onset colorectal cancer. Gastroenterology 162:1088–97.e3 [DOI] [PubMed] [Google Scholar]
- 45.Fontana L, Partridge L, Longo VD. 2010. Extending healthy life span—from yeast to humans. Science 328:321–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, et al. 2017. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun 8:14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mai V, Colbert LH, Berrigan D, Perkins SN, Pfeiffer R, et al. 2003. Calorie restriction and diet composition modulate spontaneous intestinal tumorigenesis in ApcMin mice through different mechanisms. Cancer Res. 63:1752–55 [PubMed] [Google Scholar]
- 48.Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, et al. 2008. Functionally significant insulin-like growth factor I receptor mutations in centenarians. PNAS 105:3438–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, et al. 2003. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421:182–87 [DOI] [PubMed] [Google Scholar]
- 50.Renehan AG, O’Connell J, O’Halloran D, Shanahan F, Potten CS, et al. 2003. Acromegaly and colorectal cancer: a comprehensive review of epidemiology, biological mechanisms, and clinical implications. Horm. Metab. Res 35:712–25 [DOI] [PubMed] [Google Scholar]
- 51.Fontana L, Villareal DT, Das SK, Smith SR, Meydani SN, et al. 2016. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: a randomized clinical trial. Aging Cell 15:22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pomatto-Watson LCD, Bodogai M, Bosompra O, Kato J, Wong S, et al. 2021. Daily caloric restriction limits tumor growth more effectively than caloric cycling regardless of dietary composition. Nat. Commun 12:6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Green CL, Lamming DW, Fontana L. 2022. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol 23:56–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Longo VD, Mattson MP 2014. Fasting: molecular mechanisms and clinical applications. Cell Metab. 19:181–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longo VD, Di Tano M, Mattson MP, Guidi N. 2021. Intermittent and periodic fasting, longevity and disease. Nat. Aging 1:47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng CW, Adams GB, Perin L, Wei M, Zhou X, et al. 2014. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 14:810–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mihaylova MM, Sabatini DM, Yilmaz OH. 2014. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell 14:292–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, et al. 2018. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell 22:769–78.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruens L, Ellenbroek SIJ, Suijkerbuijk SJE, Azkanaz M, Hale AJ, et al. 2020. Calorie restriction increases the number of competing stem cells and decreases mutation retention in the intestine. Cell Rep. 32:107937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lien EC, Westermark AM, Zhang Y, Yuan C, Li Z, et al. 2021. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 599:302–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weng ML, Chen WK, Chen XY, Lu H, Sun ZR, et al. 2020. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1α pathway suppression. Nat. Commun 11:1869. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Sun P, Wang H, He Z, Chen X, Wu Q, et al. 2017. Fasting inhibits colorectal cancer growth by reducing M2 polarization of tumor-associated macrophages. Oncotarget 8:74649–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Flanagan CH, Smith LA, McDonell SB, Hursting SD. 2017. When less may be more: calorie restriction and response to cancer therapy. BMC Med. 15:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nencioni A, Caffa I, Cortellino S, Longo VD. 2018. Fasting and cancer: molecular mechanisms and clinical application. Nat. Rev. Cancer 18:707–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castejon M, Plaza A, Martinez-Romero J, Fernandez-Marcos PJ, Cabo R, Diaz-Ruiz A. 2020. Energy restriction and colorectal cancer: a call for additional research. Nutrients 12:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tessitore L, Tomasi C, Greco M, Sesca E, Laconi E, et al. 1996. A subnecrogenic dose of diethylnitrosamine is able to initiate hepatocarcinogenesis in the rat when coupled with fasting/refeeding. Carcinogenesis 17:289–92 [DOI] [PubMed] [Google Scholar]
- 67.McCullough ML, Zoltick ES, Weinstein SJ, Fedirko V, Wang M, et al. 2019. Circulating vitamin D and colorectal cancer risk: an international pooling project of 17 cohorts. J. Natl. Cancer Inst 111:158–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuchs MA, Yuan C, Sato K, Niedzwiecki D, Ye X, et al. 2017. Predicted vitamin D status and colon cancer recurrence and mortality in CALGB 89803 (Alliance). Ann. Oncol 28:1359–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zgaga L, Theodoratou E, Farrington SM, Din FV, Ooi LY, et al. 2014. Plasma vitamin D concentration influences survival outcome after a diagnosis of colorectal cancer. J. Clin. Oncol 32:2430–39 [DOI] [PubMed] [Google Scholar]
- 70.Vaughan-Shaw PG, Buijs LF, Blackmur JP, Theodoratou E, Zgaga L, et al. 2020. The effect of vitamin D supplementation on survival in patients with colorectal cancer: systematic review and meta-analysis of randomised controlled trials. Br. J. Cancer 123:1705–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keum N, Lee DH, Greenwood DC, Manson JE, Giovannucci E. 2019. Vitamin D supplementation and total cancer incidence and mortality: a meta-analysis of randomized controlled trials. Ann. Oncol 30:733–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feldman D, Krishnan AV, Swami S, Giovannucci E, Feldman BJ. 2014. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 14:342–57 [DOI] [PubMed] [Google Scholar]
- 73.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347:1260419. [DOI] [PubMed] [Google Scholar]
- 74.Fernandez-Barral A, Costales-Carrera A, Buira SP, Jung P, Ferrer-Mayorga G, et al. 2020. Vitamin D differentially regulates colon stem cells in patient-derived normal and tumor organoids. FEBS J. 287:53–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ngo B, Van Riper JM, Cantley LC, Yun J. 2019. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 19:271–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, et al. 2015. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350:1391–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Magri A, Germano G, Lorenzato A, Lamba S, Chila R, et al. 2020. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med 12:eaay8707. [DOI] [PubMed] [Google Scholar]
- 78.Ferraris RP, Choe JY, Patel CR. 2018. Intestinal absorption of fructose. Annu. Rev. Nutr 38:41–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joh HK, Lee DH, Hur J, Nimptsch K, Chang Y, et al. 2021. Simple sugar and sugar-sweetened beverage intake during adolescence and risk of colorectal cancer precursors. Gastroenterology 161:128–42.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor SR, Ramsamooj S, Liang RJ, Katti A, Pozovskiy R, et al. 2021. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 597:263–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, et al. 2019. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363:1345–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bu P, Chen KY, Xiang K, Johnson C, Crown SB, et al. 2018. Aldolase B-mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell Metab. 27:1249–62.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carreno DV, Corro NB, Cerda-Infante JF, Echeverria CE, Asencio-Barria CA, et al. 2021. Dietary fructose promotes prostate cancer growth. Cancer Res. 81:2824–32 [DOI] [PubMed] [Google Scholar]
- 84.Hu S, Wang L, Yang D, Li L, Togo J, et al. 2018. Dietary fat, but not protein or carbohydrate, regulates energy intake and causes adiposity in mice. Cell Metab. 28:415–31.e4 [DOI] [PubMed] [Google Scholar]
- 85.Astrup A, Buemann B, Western P, Toubro S, Raben A, Christensen NJ. 1994. Obesity as an adaptation to a high-fat diet: evidence from a cross-sectional study. Am. J. Clin. Nutr 59:350–55 [DOI] [PubMed] [Google Scholar]
- 86.Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. 2016. Obesity, inflammation, and cancer. Annu. Rev. Pathol. Mech. Dis 11:421–49 [DOI] [PubMed] [Google Scholar]
- 87.Li X, Jansen L, Chang-Claude J, Hoffmeister M, Brenner H. 2022. Risk of colorectal cancer associated with lifetime excess weight. JAMA Oncol. 8:730–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Daniel CR, Shu X, Ye Y, Gu J, Raju GS, et al. 2016. Severe obesity prior to diagnosis limits survival in colorectal cancer patients evaluated at a large cancer centre. Br. J. Cancer 114:103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wiggins T, Antonowicz SS, Markar SR. 2019. Cancer risk following bariatric surgery—systematic review and meta-analysis of national population-based cohort studies. Obes. Surg 29:1031–39 [DOI] [PubMed] [Google Scholar]
- 90.Stroud AM, Dewey EN, Husain FA, Fischer JM, Courcoulas AP, et al. 2020. Association between weight loss and serum biomarkers with risk of incident cancer in the Longitudinal Assessment of Bariatric Surgery cohort. Surg. Obes. Relat. Dis 16:1086–94 [DOI] [PubMed] [Google Scholar]
- 91.Licholai JA, Nguyen KP, Fobbs WC, Schuster CJ, Ali MA, Kravitz AV 2018. Why do mice overeat high-fat diets? How high-fat diet alters the regulation of daily caloric intake in mice. Obesity 26:1026–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mah AT, Van Landeghem L, Gavin HE, Magness ST, Lund PK. 2014. Impact of diet-induced obesity on intestinal stem cells: hyperproliferation but impaired intrinsic function that requires insulin/IGF1. Endocrinology 155:3302–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.DeClercq V, McMurray DN, Chapkin RS. 2015. Obesity promotes colonic stem cell expansion during cancer initiation. Cancer Lett. 369:336–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Degirolamo C, Modica S, Palasciano G, Moschetta A. 2011. Bile acids and colon cancer: solving the puzzle with nuclear receptors. Trends Mol. Med 17:564–72 [DOI] [PubMed] [Google Scholar]
- 95.Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, et al. 2019. FXR regulates intestinal cancer stem cell proliferation. Cell 176:1098–112.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, et al. 2018. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175:1307–20.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, et al. 2020. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell 183:1848–66.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang J, Wei H, Zhou Y, Szeto CH, Li C, et al. 2022. High-fat diet promotes colorectal tumorigenesis through modulating gut microbiota and metabolites. Gastroenterology 162:135–49.e2 [DOI] [PubMed] [Google Scholar]
- 99.Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, et al. 2014. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514:508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu YS, Wu QJ, Lv JL, Jiang YT, Sun H, et al.2021. Dietary carbohydrate and diverse health outcomes: umbrella review of 30 systematic reviews and meta-analyses of 281 observational studies. Front. Nutr 8:670411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aliluev A, Tritschler S, Sterr M, Oppenlander L, Hinterdobler J, et al. 2021. Diet-induced alteration of intestinal stem cell function underlies obesity and prediabetes in mice. Nat. Metab 3:1202–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Payne NE, Cross JH, Sander JW, Sisodiya SM. 2011. The ketogenic and related diets in adolescents and adults—a review. Epilepsia 52:1941–48 [DOI] [PubMed] [Google Scholar]
- 103.Li J, Zhang H, Dai Z. 2021. Cancer treatment with the ketogenic diet: a systematic review and meta-analysis of animal studies. Front. Nutr 8:594408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheng CW, Biton M, Haber AL, Gunduz N, Eng G, et al. 2019. Ketone body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell 178:1115–31.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dmitrieva-Posocco O, Wong AC, Lundgren P, Golos AM, Descamps HC, et al. 2022. β-Hydroxybutyrate suppresses colorectal cancer. Nature 605:160–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bruno DS, Berger NA. 2020. Impact of bariatric surgery on cancer risk reduction. Ann. Transl. Med 8:S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mackenzie H, Markar SR, Askari A, Faiz O, Hull M, et al. 2018. Obesity surgery and risk of cancer. Br. J. Surg 105:1650–57 [DOI] [PubMed] [Google Scholar]
- 108.Schauer DP, Feigelson HS, Koebnick C, Caan B, Weinmann S, et al. 2019. Bariatric surgery and the risk of cancer in a large multisite cohort. Ann. Surg 269:95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bailly L, Fabre R, Pradier C, Iannelli A. 2020. Colorectal cancer risk following bariatric surgery in a nationwide study of French individuals with obesity. JAMA Surg. 155:395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Almazeedi S, El-Abd R, Al-Khamis A, Albatineh AN, Al-Sabah S. 2020. Role of bariatric surgery in reducing the risk of colorectal cancer: a meta-analysis. Br. J. Surg 107:348–54 [DOI] [PubMed] [Google Scholar]
- 111.Kedrin D, Gandhi SC, Wolf M, Roper J, Yilmaz O, et al. 2017. Bariatric surgery prior to index screening colonoscopy is associated with a decreased rate of colorectal adenomas in obese individuals. Clin. Transl. Gastroenterol 8:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, et al.2017. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. PNAS 114:E7697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, et al. 2013. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev. Cell 25:463–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheung EC, Lee P, Ceteci F, Nixon C, Blyth K, et al. 2016. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 30:52–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, et al. 2019. COSMIC: the catalogue of somatic mutations in cancer. Nucleic. Acids. Res 47:D941–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nie X, Liu H, Liu L, Wang YD, Chen WD. 2020. Emerging roles of Wnt ligands in human colorectal cancer. Front. Oncol 10:1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kimmelman AC. 2015. Metabolic dependencies in RAS-driven cancers. Clin. Cancer Res 21:1828–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Labuschagne CF, Zani F, Vousden KH. 2018. Control of metabolism by p53—cancer and beyond. Biochim. Biophys. Acta Rev. Cancer 1870:32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, et al. 2014. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 33:1454–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Worby CA, Dixon JE. 2014. PTEN. Annu. Rev. Biochem 83:641–69 [DOI] [PubMed] [Google Scholar]
- 121.Mukhopadhyay S, Vander Heiden MG, McCormick F. 2021. The Metabolic landscape of RAS-driven cancers from biology to therapy. Nat. Cancer 2:271–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith AL, Whitehall JC, Bradshaw C, Gay D, Robertson F, et al. 2020. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 1:976–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ohshima K, Nojima S, Tahara S, Kurashige M, Kawasaki K, et al. 2020. Serine racemase enhances growth of colorectal cancer by producing pyruvate from serine. Nat. Metab 2:81–96 [DOI] [PubMed] [Google Scholar]
- 124.Bensard CL, Wisidagama DR, Olson KA, Berg JA, Krah NM, et al. 2020. Regulation of tumor initiation by the mitochondrial pyruvate carrier. Cell Metab. 31:284–300.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ma L, Tao Y, Duran A, Llado V, Galvez A, et al. 2013. Control of nutrient stress-induced metabolic reprogramming by PKCζ in tumorigenesis. Cell 152:599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Phillips CM, Zatarain JR, Nicholls ME, Porter C, Widen SG, et al. 2017. Upregulation of cystathionine-β-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res. 77:5741–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lambert AW, Weinberg RA. 2021. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 21:325–38 [DOI] [PubMed] [Google Scholar]
- 128.Gao W, Chen L, Ma Z, Du Z, Zhao Z, et al. 2013. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology 145:636–46.e5 [DOI] [PubMed] [Google Scholar]
- 129.Wu Z, Wei D, Gao W, Xu Y, Hu Z, et al. 2015. TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+ tumor-initiating cells. Cell Stem Cell 17:47–59 [DOI] [PubMed] [Google Scholar]
- 130.Bergers G, Fendt SM. 2021. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 21:162–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang D, Fu L, Wei J, Xiong Y, DuBois RN. 2019. PPARδ mediates the effect of dietary fat in promoting colorectal cancer metastasis. Cancer Res. 79:4480–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, et al. 2018. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 37:6025–40 [DOI] [PubMed] [Google Scholar]
- 133.Xiang L, Mou J, Shao B, Wei Y, Liang H, et al.2019. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiang L, Xie G, Liu C, Zhou J, Chen J, et al. 2013. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim. Biophys. Acta Mol. Cell Res 1833:2996–3005 [DOI] [PubMed] [Google Scholar]
- 135.Lu H, Samanta D, Xiang L, Zhang H, Hu H, et al. 2015. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. PNAS 112:E4600–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nguyen A, Loo JM, Mital R, Weinberg EM, Man FY, et al. 2016. PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J. Clin. Investig 126:681–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cen B, Lang JD, Du Y, Wei J, Xiong Y, et al. 2020. Prostaglandin E2 induces miR675-5p to promote colorectal tumor metastasis via modulation of p53 expression. Gastroenterology 158:971–84.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang D, Fu L, Sun H, Guo L, DuBois RN. 2015. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 149:1884–95.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Loo JM, Scherl A, Nguyen A, Man FY, Weinberg E, et al. 2015. Extracellular metabolic energetics can promote cancer progression. Cell 160:393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kurth I, Yamaguchi N, Andreu-Agullo C, Tian HS, Sridhar S, et al. 2021. Therapeutic targeting of SLC6A8 creatine transporter suppresses colon cancer progression and modulates human creatine levels. Sci. Adv 7:eabi7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, et al. 2019. PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife 8:e52135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Donaldson GP, Lee SM, Mazmanian SK. 2016. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol 14:20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Song M, Chan AT, Sun J. 2020. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology 158:322–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lee YS, Kim TY, Kim Y, Lee SH, Kim S, et al. 2018. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 24:833–46.e6 [DOI] [PubMed] [Google Scholar]
- 145.Pleguezuelos-Manzano C, Puschhof J, Clevers H. 2022. Gut microbiota in colorectal cancer: associations, mechanisms, and clinical approaches. Annu. Rev. Cancer Biol 6:65–84 [Google Scholar]
- 146.Han H, Safe S, Jayaraman A, Chapkin RS. 2021. Diet-host-microbiota interactions shape aryl hydrocarbon receptor ligand production to modulate intestinal homeostasis. Annu. Rev. Nutr 41:455–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, et al. 2013. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLOS ONE 8:e56964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, et al. 2019. The human gut bacterial genotoxin colibactin alkylates DNA. Science 363:eaar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhan Y, Chen PJ, Sadler WD, Wang F, Poe S, et al. 2013. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 73:7199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hannah B Bell RJR, Goyert J, Gygi SP, Mancias JD, Shah YM. 2022. Reuterin in the healthy gut microbiome suppresses colorectal cancer growth through altering redox balance. Cancer Cell 40:185–200.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Biller LH, Schrag D. 2021. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA 325:669–85 [DOI] [PubMed] [Google Scholar]
- 152.Longley DB, Harkin DP, Johnston PG. 2003. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3:330–38 [DOI] [PubMed] [Google Scholar]
- 153.Tegnebratt T, Lu L, Lee L, Meresse V, Tessier J, et al. 2013. [18 F]FDG-PET imaging is an early non-invasive pharmacodynamic biomarker for a first-in-class dual MEK/Raf inhibitor, RO5126766 (CH5126766), in preclinical xenograft models. EJNMMI Res. 3:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Toda K, Kawada K, Iwamoto M, Inamoto S, Sasazuki T, et al. 2016. Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 18:654–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Carr RM, Qiao G, Qin J, Jayaraman S, Prabhakar BS, Maker AV. 2016. Targeting the metabolic pathway of human colon cancer overcomes resistance to TRAIL-induced apoptosis. Cell Death Discov. 2:16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W, et al. 2011. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: a meta-analysis. Diabetes Care 34:2323–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, et al. 2008. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 99:2136–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hosono K, Endo H, Takahashi H, Sugiyama M, Uchiyama T, et al. 2010. Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol. Carcinog 49:662–71 [DOI] [PubMed] [Google Scholar]
- 159.Jia Y, Ma Z, Liu X, Zhou W, He S, et al. 2015. Metformin prevents DMH-induced colorectal cancer in diabetic rats by reversing the Warburg effect. Cancer Med. 4:1730–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Janzer A, German NJ, Gonzalez-Herrera KN, Asara JM, Haigis MC, Struhl K. 2014. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. PNAS 111:10574–79 [DOI] [PMC free article] [PubMed] [Google Scholar]