

Abstract
Disease-associated SNPs generally do not implicate target genes, as most disease SNPs are regulatory. Many SNP-to-gene (S2G) linking strategies have been developed to link regulatory SNPs to the genes that they regulate in cis. Here, we developed a heritability-based framework for evaluating and combining different S2G strategies to optimize their informativeness for common disease risk. Our optimal combined S2G strategy (cS2G) included 7 constituent S2G strategies and achieved a precision of 0.75 and a recall of 0.33, more than doubling the recall of any individual strategy. We applied cS2G to fine-mapping results for 49 UK Biobank diseases/traits to predict 5,095 causal SNP-gene-disease triplets (with S2G-derived functional interpretation) with high confidence. We further applied cS2G to provide an empirical assessment of disease omnigenicity; we determined that the top 1% of genes explained roughly half of the SNP-heritability linked to all genes, and that gene-level architectures vary with variant allele frequency.
Editor summary:
A heritability-based framework for evaluation of SNP-to-gene linking methods is used to construct an optimal, combined approach and applied to 49 traits. Analysis of trait omnigenicity suggests gene-level architecture varies depending on variant frequency.
Introduction
While genome-wide association studies (GWAS) have successfully identified thousands of loci associated with human diseases and complex traits, they generally do not identify the underlying causal variants, target genes, cell-types and biological functions, thus limiting the translation of GWAS findings into discoveries that will enhance disease treatment1–3. Although recent advances in fine-mapping techniques have improved our ability to nominate causal variants4–7, identifying the underlying target genes remains a critical challenge, as causal variants are predominantly regulatory SNPs8–11 that do not necessarily regulate the closest genes12–17. Large gene expression quantitative trait loci (eQTL) data sets18,19 have proven useful in linking disease variants to their target genes through colocalization analyses20–24 or transcriptome-wide association studies14,17,25, but explain a limited proportion of disease SNP-heritability15,26,27, likely due to limited representation of disease-relevant cell-types/states28. Many other functional assays and computational approaches have recently been developed to link regulatory SNPs to their target genes in cis in a broad set of cell-types26,29–38; for example, EpiMap enhancers37 and ABC enhancers34,38 are linked to their target genes using correlation of enhancer activity with gene expression across cell-types and a quantitative combination of enhancer activity and 3D contact frequencies, respectively. Combining SNP-to-gene (S2G) linking strategies has previously been proposed as an appealing approach to improve SNP-to-gene linking39–42. However, it is currently unclear how S2G strategies should be prioritized in the context of GWAS, limiting our ability to pinpoint disease genes.
Here, we developed a framework for evaluating and combining S2G strategies to optimize their informativeness for human disease risk, leveraging polygenic analyses of disease SNP-heritability. We applied this framework to GWAS summary statistics for 63 diseases and complex traits, evaluating 50 S2G strategies and constructing an optimal combined S2G strategy (cS2G) informed by GWAS data. We applied cS2G to genome-wide fine-mapping results for 49 diseases and complex traits from the UK Biobank7,43 to pinpoint disease genes at thousands of GWAS loci and to enable an empirical assessment of the “omnigenic model”44–46.
Results
Overview of Methods
A SNP-to-gene (S2G) linking strategy is defined as an assignment of raw linking values between each SNP and zero or more candidate target genes, which we converted into linking scores such that each SNP has a sum of linking scores ≤1 (Figure 1a; see Methods). We analyzed 50 S2G strategies that link SNPs to their target genes in cis (Supplementary Table 1 and Supplementary Note), including 13 main strategies (Table 1). Correlations and overlap proportions between S2G strategies (Supplementary Table 2) indicate low concordance between the strategies. In our primary analyses, we restricted each S2G strategy by restricting each SNP to the gene(s) with the highest linking score, as we observed that this led to slightly higher precision (Extended Data Figure 1).
Figure 1: Overview of S2G framework.
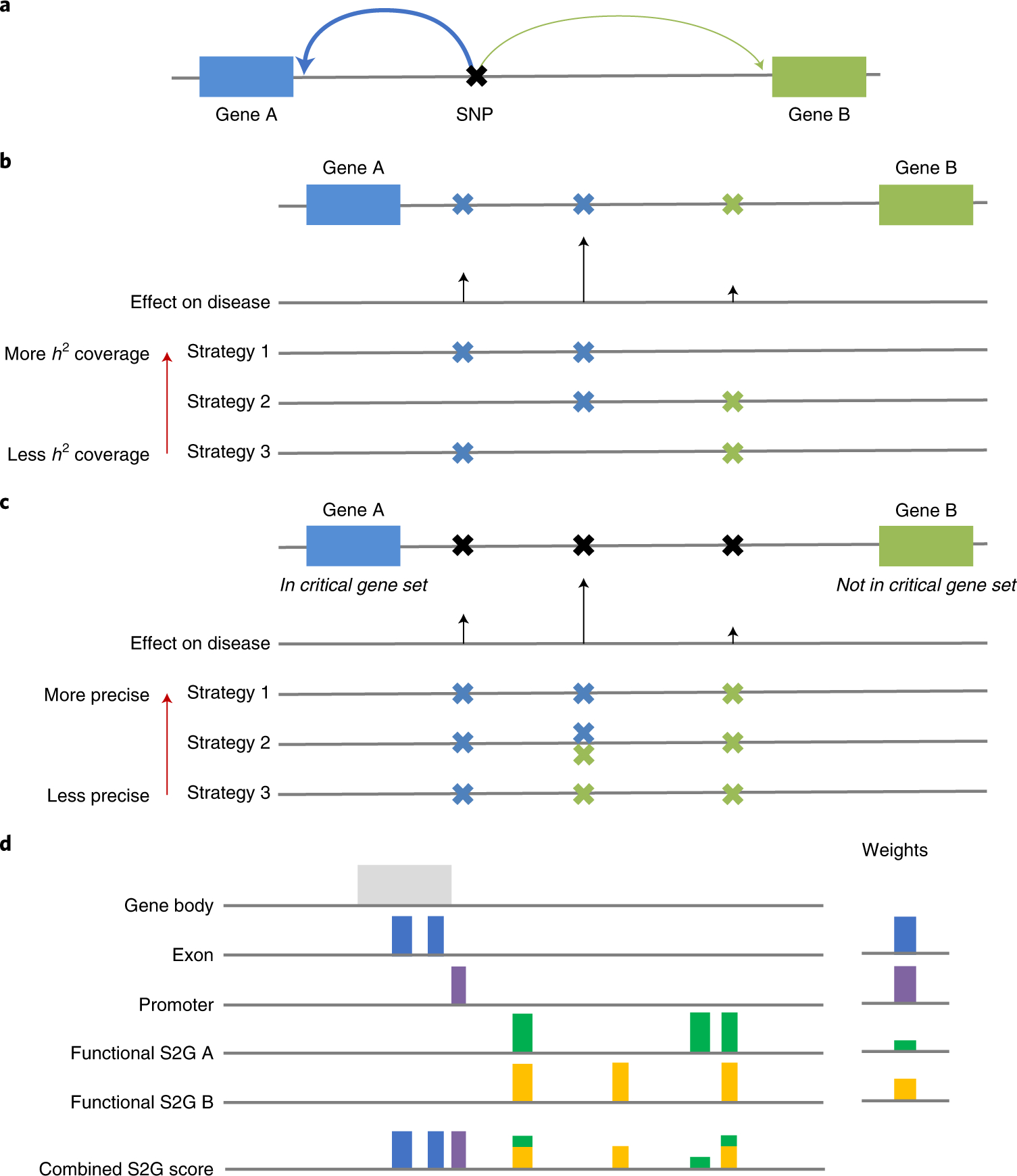
(a) Toy example of SNP linked to two genes (arrow widths denote linking scores). (b) Toy example of h2 coverage. Strategy 1 (which links SNPs with larger effects on disease) has more h2 coverage than strategy 2, which has more h2 coverage than strategy 3 (which links SNPs with smaller effects on disease). (c) Toy example of using critical gene sets to define precision. Strategy 1 (which links the middle SNP with high effect on disease to the gene from the critical gene set) is more precise than strategy 2 (which links the middle SNP to both genes), which is more precise than strategy 3 (which links the middle SNP to the gene that is not from the critical gene set). Recall is defined as the product of the h2 coverage and precision. (d) Toy example of combined S2G strategy. The combined S2G strategy is a linear combination of constituent S2G strategies.
Table 1:
Description of the 13 main SNP-to-gene (S2G) strategies.
| S2G strategy | Description | % SNPs linked | h2 coverage |
|---|---|---|---|
|
| |||
| High confidence (2) | |||
| Exon* | Exons60 +/− 20bp | 2.3% | 9.8% |
| Promoter* | TSSs61 +/− lkb ∩ promoter annotations11,30 | 2.0% | 5.8% |
|
| |||
| Non-functionally informed (3) | |||
| Gene body | Gene body62,60,61 | 43.1% | 55.4% |
| Gene±100kb | Gene body62,60,61 +/− 100kb56 | 73.2% | 85.4% |
| Closest TSS | Gene with closest TSS61 | 100% | 100% |
|
| |||
| Fine-mapped cis-eQTL (2) | |||
| GTEx fine-mapped cis-eQTL* | Fine-mapped GTeX v8 cis-eQTLs18,26 in 54 cell-types | 9.5% | 19.4% |
| eQTLGen fine-mapped blood cis-eQTL* | Fine-mapped eQTLGen cis-eQTLs19,26 in blood | 2.2% | 6.2% |
|
| |||
| Enhancer-gene linking (3) | |||
| Roadmap enhancer-gene linking | Correlation between Roadmap enhancers and gene expression across 127 cell-types29,30,32 | 8.6% | 16.3% |
| EpiMap enhancer-gene linking* | Correlation between EpiMap enhancers and gene expression across 833 cell-types37 | 7.5% | 13.8% |
| Activity-By-Contact (ABC)* | Hi-C linked enhancers in 167 cell-types34,38 | 7.7% | 17.3% |
|
| |||
| PCHi-C (2) | |||
| Jung PCHi-C | Promoter capture Hi-C in 27 cell-types35 | 39.9% | 43.9% |
| Javierre PCHi-C blood | Promoter capture Hi-C in 17 blood cell-types31 | 27.2% | 35.5% |
|
| |||
| scATAC-seq (1) | |||
| Cicero blood/basal* | Correlation between scATAC-seq peaks and gene promoter peaks across 61,806 blood/basal cells33,36 | 1.0% | 2.9% |
included in our combined S2G strategy (cS2G).
For each of 13 main S2G strategies (in 6 categories), we provide a brief description and report the % SNPs linked (proportion of common SNPs that are linked to at least one gene) and h2 coverage (meta-analyzed across 63 independent traits). When combining S2G strategies, we did not include the 3 non-functionally informed strategies because a fundamental goal of cS2G is to provide functional interpretation of GWAS findings. A description of all 50 S2G strategies analyzed is provided in Supplementary Table 1.
To evaluate each S2G strategy’s informativeness for pinpointing disease genes, we leveraged polygenic analyses of disease SNP-heritability (we denote SNP-heritability as h2 throughout the paper). We aimed to define and estimate parameters that correspond to an S2G strategy’s h2 coverage (proportion of total disease SNP-heritability that is linked to genes), precision (proportion of disease SNP-heritability linked to genes that is linked to the correct target gene), and recall (proportion of total disease SNP-heritability that is linked to the correct target gene). First, we defined h2 coverage as the proportion of SNP-heritability explained by all SNPs linked to one or more genes (weighted by their linking scores) (Figure 1b). Second, we defined precision as the relative excess SNP-heritability enrichment of SNPs linked to a critical gene set (see below) vs. SNPs linked to all genes, as compared to the (gold-standard) Exon S2G strategy; this definition is based on the intuition that a precise S2G strategy is more likely (than an imprecise strategy) to link a disease SNP to a critical gene (Figure 1c). Third, we defined recall as the product of the h2 coverage and precision. We estimated these quantities by applying stratified LD score regression (S-LDSC) to 63 independent diseases and complex traits (average N=314K; Supplementary Table 3), meta-analyzing results across traits. We jointly analyzed SNP annotations derived from the 50 S2G strategies for ~10M SNPs with a minor allele count ≥5 in a 1000 Genomes Project European reference panel47.
Our definitions of precision and recall rely on a critical gene set (see above). We used a non-trait-specific training critical gene set to construct an optimal combined S2G strategy (see below), and trait-specific validation critical gene sets to evaluate the optimal combined S2G strategy while avoiding overfitting. We defined the training critical gene set as the top 10% of genes with the most highly constrained exons and conserved promoters, and the validation critical gene set for a given trait as the top 10% of genes ranked by the PoPS method48; the excess overlap between the gene sets was limited (see Supplementary Figure 1 for distribution of overlap across diseases/traits) and did not impact our results (see Supplementary Note). These gene sets attained high SNP-heritability enrichment (Supplementary Table 4), validating their informativeness for disease.
We constructed combined S2G strategies as linear combinations of linking scores from constituent S2G strategies (Figure 1d). We developed an optimization framework maximizing the recall while constraining precision (defined using the training critical gene set) to be ≥0.75; high precision is important for maximizing the utility of functional follow-up studies. The precision and recall of the resulting combined S2G strategies were subsequently evaluated using the validation critical gene sets.
Further details are provided in the Methods section. We have released open-source software implementing our framework (see Code Availability), and have made all S2G strategies, SNP annotations and critical gene sets analyzed publicly available (see Data Availability).
Evaluation of S2G strategies
We estimated the h2 coverage, precision and recall for the 50 S2G strategies (Supplementary Table 5); we used the (trait-specific) validation critical gene sets to perform these evaluations. Results for the 13 main S2G strategies are reported in Figure 2 and Supplementary Figure 2. The Exon and Promoter strategies attained high precision (1.00 for Exon (by definition) and 0.80 for promoters), but low h2 coverage (0.06–0.10) and thus low recall (0.05–0.10). On the other hand, the Closest TSS strategy attained low precision (0.34), but the highest h2 coverage (1.00) and recall (0.34). In addition to Exon and Promoter, 5 other main S2G strategies attained high precision (>0.5) but low recall (0.02–0.13): the 2 fine-mapped cis-eQTL strategies, the 2 enhancer-gene linking strategies informed by gene expression, and the scATAC-seq strategy. Interestingly, S2G strategies using fine-mapped cis-eQTLs26 attained significantly higher precision than S2G strategies using all cis-eQTL (0.68±0.07 vs. 0.40±0.04 in GTeX, 0.81±0.11 vs. 0.29±0.03 in eQTLGen), consistent with previous reports of low precision for strategies using all cis-eQTL17,49 and emphasizing the advantage of fine-mapped cis-eQTL for more precise analyses of GWAS data.
Figure 2: Accuracy of individual S2G strategies and combined S2G (cS2G) strategy.
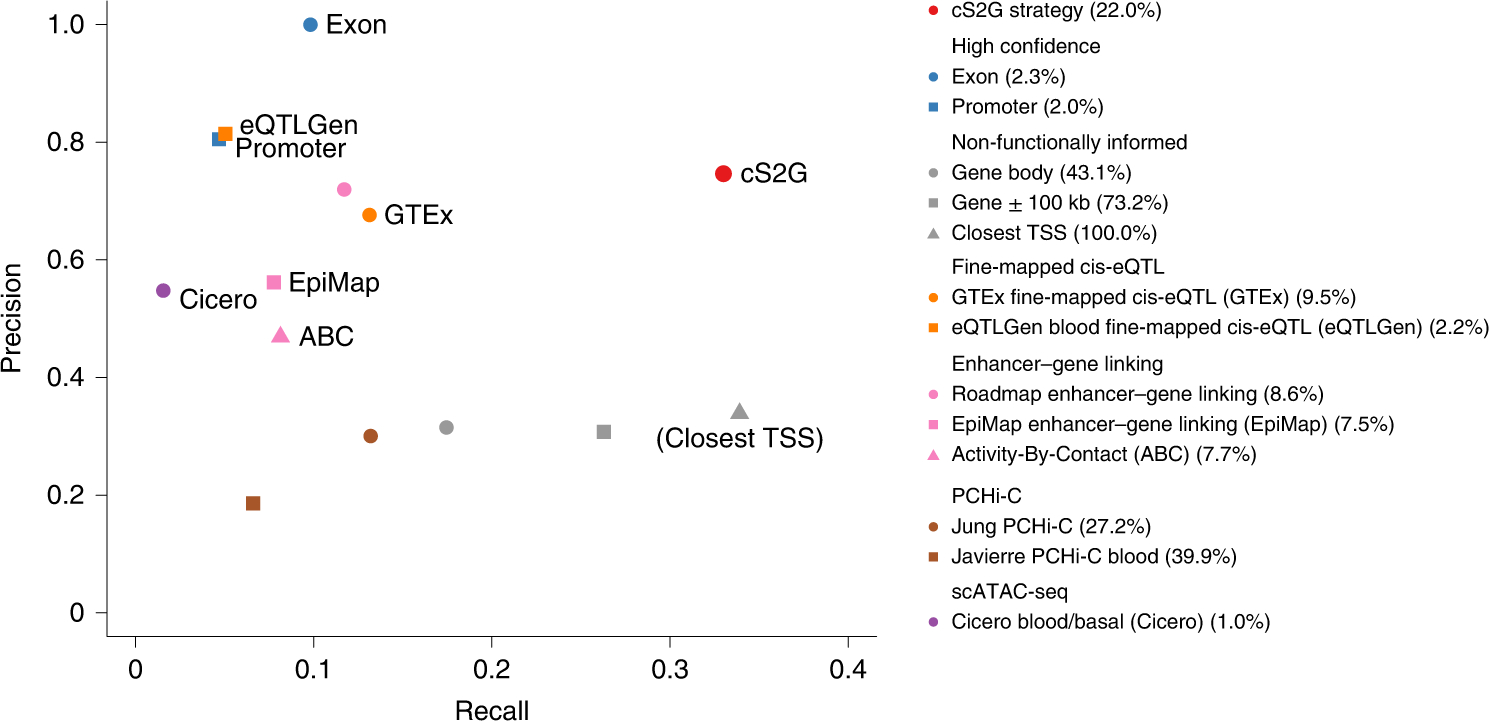
We report the precision and recall of the 13 main S2G strategies from Table 1 and the cS2G strategy (estimated using trait-specific validation critical gene sets and meta-analyzed across 63 independent traits). Colored font denotes the cS2G strategy and its 7 constituent S2G strategies (gray font in parentheses denotes the Closest TSS strategy). Numbers in parentheses in legend denotes the proportion of common SNPs that are linked to at least one gene (as in Table 1). We note that our evaluation of these S2G strategies is impacted by their widely varying underlying biosample sizes (see Methods), in addition to differences in functional assays and SNP-to-gene linking methods. Standard errors are reported in Supplementary Figure 2, and numerical results are reported in Supplementary Table 5; standard errors for all S2G strategies linking >2.5% of common SNPs were ≤0.12 for precision and ≤0.03 for recall, with smaller standard errors for S2G strategies linking larger proportions of common SNPs.
The 50 S2G strategies included 27 S2G strategies based on physical distance to TSS. As expected, we observed that proximal closest TSS are likely to implicate target genes (e.g. precision of 0.78 for closest TSS range <1kb), whereas distal closest TSS are much less likely to implicate target genes (e.g. precision of 0.15 for closest TSS range 100–500kb) (Extended Data Figure 2 and Supplementary Table 5). We further determined that closest TSS are moderately likely to systematically implicate target genes: precision of 0.34, decreasing to 0.17 for 2nd closest TSS and 0.062 for 5th closest TSS. The mean value of 0.043 for 6th-20th closest TSS suggests that genes located in the same regions as causal disease genes have a slightly elevated probability of being causal. Further validation of our precision metric is provided in the Supplementary Note and Supplementary Figures 3–4.
We next investigated whether functionally informed S2G strategies restricted to trait-specific tissues and cell-types were more precise for the corresponding traits. We determined that S2G strategies defined using all available tissues and cell-types achieved higher precision than S2G strategies restricted to blood and immune cell-types (Extended Data Figure 3) in analyses restricted to 11 autoimmune diseases and blood cell traits (average N=257K; Supplementary Table 3), perhaps due to limited biosample size; these results support including all available cell-types in current efforts to pinpoint disease genes17,27 (see Discussion).
In summary, we developed and validated a polygenic framework for evaluating S2G strategies, inferring high precision but low recall for many functionally informed S2G strategies, and low precision but relatively high recall for non-functionally informed strategies (such as Closest TSS).
Combining S2G strategies
We constructed an optimal combined S2G strategy (cS2G) by considering linear combinations of linking scores from 10 functionally informed main S2G strategies (Table 1), maximizing the recall while constraining precision to be ≥0.75 using the (non-trait-specific) training critical gene set. The resulting cS2G strategy included 7 constituent S2G strategies: Exon, Promoter, eQTLGen blood fine-mapped cis-eQTL, GTEx fine-mapped cis-eQTL, EpiMap enhancer-gene linking, ABC, and Cicero blood/basal (ordered from highest to lowest weight; Supplementary Table 6). The cS2G strategy linked 22% of common SNPs (minor allele frequency (MAF) ≥5%) to at least one gene and 99.6% of genes to at least one common SNP (average of 1.20 genes per linked common SNP and 79 common SNPs per linked gene). Despite the high weights for Exon and Promoter, 43% of linked common SNPs were not linked to the gene with closest TSS; the mean distance to the gene TSS for all cS2G links involving common SNPs was 96kb (Supplementary Figure 5). The number of common SNPs linked per gene was correlated to gene-body length (r2=0.13), but uncorrelated to gene-body length after correcting for exon and promoter length (r2=0.01). Strikingly, only 18% (resp. 3.3%) of the cS2G links were supported by at least 2 (resp. 3) of 7 constituent S2G strategies (Supplementary Table 7), consistent with the low correlations between the constituent S2G strategies (Supplementary Table 2). This provides a strong motivation for combining multiple S2G strategies.
We evaluated the cS2G strategy using the (trait-specific) validation critical gene sets and meta-analyzed the results across the 63 independent diseases and complex traits. The cS2G strategy attained h2 coverage of 0.44 (s.e. 0.01), precision of 0.75 (s.e. 0.06), and recall of 0.33 (s.e. 0.03) (Figure 2 and Supplementary Table 5), more than doubling the precision and/or recall of any individual strategy; this implies that 33% of SNP-heritability can be linked to causal genes with 75% confidence using cS2G. Notably, cS2G attained much higher precision than two previously proposed combined strategies, GeneHancer39 (0.14) and Open Targets42 (0.33) (Supplementary Table 5). Additional experiments comparing cS2G strategy to alternative approaches for combining S2G strategies are described in the Supplementary Note and Supplementary Tables 8–11.
In summary, we constructed an optimal combined S2G strategy (cS2G), incorporating 7 constituent S2G strategies, which more than doubles the precision and/or recall of any individual strategy; our evaluation of cS2G was based on validation critical gene sets that were distinct from the training critical gene set used to optimize cS2G.
Validating the combined S2G strategy using curated examples
We sought to validate the combined S2G strategy using a manually curated list of 17 disease-associated loci (including 12 loci from ref.50) containing 25 experimentally validated causal SNP-gene pairs (Table 2), and reflecting the very limited set of experimentally validated disease-associated SNP-gene pairs50. We restricted these analyses to SNPs that had a linked gene with cS2G linking score >0.5, consistent with our goal of attaining high precision for each individual SNP analyzed to maximize the utility of functional follow-up studies. 16 of the 25 pairs had a linked gene with cS2G linking score >0.5. The cS2G prediction of the target gene matched the experimentally validated gene for 11 of these 16 loci, yielding a precision of 11/16=0.69 (s.e.= 0.12) and a recall of 11/25= 0.44 (s.e.=0.10) (Table 2 and Supplementary Table 12). The precision was lower than our estimate based on validation critical gene sets (0.75) (and lower than the precision of one constituent strategy; 0.78 (s.e.=0.14) for EpiMap), whereas the recall was higher than our estimate based on validation critical gene sets (0.33) (and higher than the recall of any constituent strategy); however, these differences were not statistically significant due to the small number of experimentally validated SNP-gene pairs (Supplementary Table 13). Interestingly, of the 11 pairs that were correctly linked to the experimentally validated gene, 8 pairs were linked by at least two cS2G constituent strategies. However, we failed to identify the well-studied rs1421085-IRX5/IRX3 link12, as none of the constituent S2G strategies linked rs1421085 to either IRX5 or IRX3; we also failed to identify the well-studied rs12740374-SORT1 link51, as rs12740374 is an exonic SNP for CELSR2, outweighing the link to SORT1 by the GTEx fine-mapped cis-eQTL strategy.
Table 2:
Validation of combined S2G (cS2G) strategy using experimentally validated causal SNP-gene pairs associated to disease.
| Position (hg19) | SNP | Gene | Disease/trait | cS2G prediction | ||
|---|---|---|---|---|---|---|
| Gene | Score | Annotations | ||||
|
| ||||||
| 1:109,817,590 | rs12740374 | SORT1 | LDL50,51 | CELSR2 * | 0.91 | Exon |
|
| ||||||
| 1:162,020,969 | rs7539120 | NOS1AP | QT interval63 | - | - | - |
|
| ||||||
| 2:60,718,043 | rs1427407 | BCL11A | Fetal hemoglobin level50,64 | - | - | - |
| 2:60,725,451 | rs7606173 | BCL11A | 1.00 | EpiMap, ABC, Cicero | ||
|
| ||||||
| 3:38,767,315 | rs6801957 | SCN5A | QRS prolongation65 | SCN5A | 1.00 | EpiMap, ABC |
|
| ||||||
| 4:90,674,431 | rs356168 | SNCA | Parkinson’s disease50,66 | SNCA | 1.00 | EpiMap, ABC |
|
| ||||||
| 5:56,031,822 | rs17432750 | MAP3K1 | Breast cancer50,67 | MAP3K1 | 0.66 | EpiMap |
| 5:56,052,695 | rs62355900 | - | - | - | ||
| 5:56,053,479 | rs74345699 | - | - | - | ||
| 5:56,134,276 | rs16886397 | MAP3K1 | 1.00 | ABC | ||
|
| ||||||
| 6:12,903,957 | rs9349379 | EDN1 | Vascular diseases68 | PHACTR1 | 1.00 | GTEx |
|
| ||||||
| 6:105,706,878 | rs1743292 | BVES | Cardiac QT interval and QRS duration50,69 | - | - | - |
| 6:105,720,538 | rs1772203 | POPDC3 | 1.00 | ABC | ||
|
| ||||||
| 6:117,210,052 | rs339331 | RFX6 | Prostate cancer50,70,71 | RFX6 | 1.00 | EpiMap, ABC |
|
| ||||||
| 6:135,418,635 | rs7775698 | MYB | Fetal hemoglobin level50,72 | MYB | 1.00 | EpiMap |
| 6:135,418,637 | rs66650371 | - | - | - | ||
| 6:135,431,640 | rs9494142 | HBS1L | 1.00 | ABC | ||
|
| ||||||
| 7:12,284,008 | rs1990620 | TMEM106B | Fronto-temporal dementia50,73 | - | - | - |
|
| ||||||
| 8:11,351,220 | rs1382568 | BLK | Systemic lupus50,74 | - | - | - |
| 8:11,351,912 | rs922483 | BLK | 0.76 | Exon, ABC, Cicero | ||
|
| ||||||
| 8:81,290,387 | rs2370615 | PAG1 | Allergy50,75 | ZBTB10 | 0.51 | ABC, Cicero |
|
| ||||||
| 10:12,307,894 | rs11257655 | CAMK1D | Type 2 diabetes50,76 | CAMK1D | 0.90 | GTEx, EpiMap |
|
| ||||||
| 10:6,094,697 | rs61839660 | IL2RA | Inflammatory bowel disease77 | IL2RA | 1.00 | ABC, Cicero |
|
| ||||||
| 16:53,800,954 | rs1421085 | IRX5/IRX3 | Obesity12,50 | - | - | - |
|
| ||||||
| 20:55,990,405 | rs737092 | RBM38 | Red blood cell count78 | RBM38 | 0.96 | eQTLGen, ABC |
: rs12740374 was linked to SORT1 using GTEx with cS2G linking score = 0.07 (which is less than 0.5). Predicted genes that match the experimentally validated gene are denoted in bold font.
For each of 25 experimentally validated causal SNP-gene pairs at 17 disease- associated loci, we report the cS2G predictions: predicted gene with cS2G linking score >0.5 (if applicable), corresponding cS2G linking score, and constituent S2G annotation(s). Further details are provided in Supplementary Table 12. GTEx: GTEx fine-mapped cis-eQTL; eQTLGen: eQTLGen blood fine-mapped cis-eQTL; EpiMap: EpiMap enhancer-gene linking; ABC: Activity-By-Contact; Cicero: Cicero blood/basal.
We obtained similar results when analyzing a larger curated list consisting of disease genes validated with high confidence without strictly requiring experimental validation42 (see Supplementary Note and Supplementary Tables 14–16).
In summary, these analyses provide a promising validation of the potential of cS2G to pinpoint causal disease genes.
Leveraging the cS2G strategy to pinpoint disease genes
In previous work, we showed that functionally informed fine-mapping under the PolyFun framework robustly identifies a large number of fine-mapped SNPs for 49 UK Biobank diseases/traits (using N=337K unrelated British samples7,43) with well-calibrated posterior inclusion probabilities (PIP)7. Here, we leveraged the high precision and recall of our combined S2G (cS2G) strategy to predict target genes of 9,670 predicted causal SNP-disease pairs with PIP >0.5 from PolyFun analyses (7,675 unique SNPs) (Supplementary Table 17).
Restricting to SNPs that had a linked gene with cS2G linking score >0.5, we predicted 7,111 causal SNP-gene-disease triplets (5,384 unique SNPs; 3,401 unique genes) (Figure 3a and Supplementary Table 17). The mean PIP of the 7,111 SNP-gene-disease triplets was equal to 0.80. We further assigned a confidence score to each of the triplets by multiplying the PIP of the constituent SNP and its precision (see Methods and Supplementary Tables 17–18). The average confidence score was 0.64, implying that 64% of the 7,111 SNP-gene-disease triplets (4,554 triplets) predict the correct causal SNP and target gene (Figure 3a and Supplementary Table 18). Distributions of confidence scores are reported in Figure 3b. Using cS2G, we detected 5,095 triplets with confidence score >0.5 (72% of its detected triplets), which is 1.84 times larger than what could be attained using any individual functionally informed constituent S2G strategy (only 2,763 for Exon, even though the Exon strategy had the highest confidence scores, consistent with its high precision), and 1.58 times higher than what could be attained using the Closest TSS strategy (Figure 3b and Supplementary Table 18). In many instances, multiple causal SNPs were linked to the same gene (e.g. 119 genes were each linked to at least 5 different fine-mapped SNPs for 5 different diseases/traits; Supplementary Table 19), implying that a single gene can be causal for different diseases/traits using different causal SNP-gene links; we note that SNPs linked to the same gene are generally not in LD with each other (mean r2=0.09 across 3,900 pairs).
Figure 3: SNP-gene-disease triplets identified by cS2G and other S2G strategies.
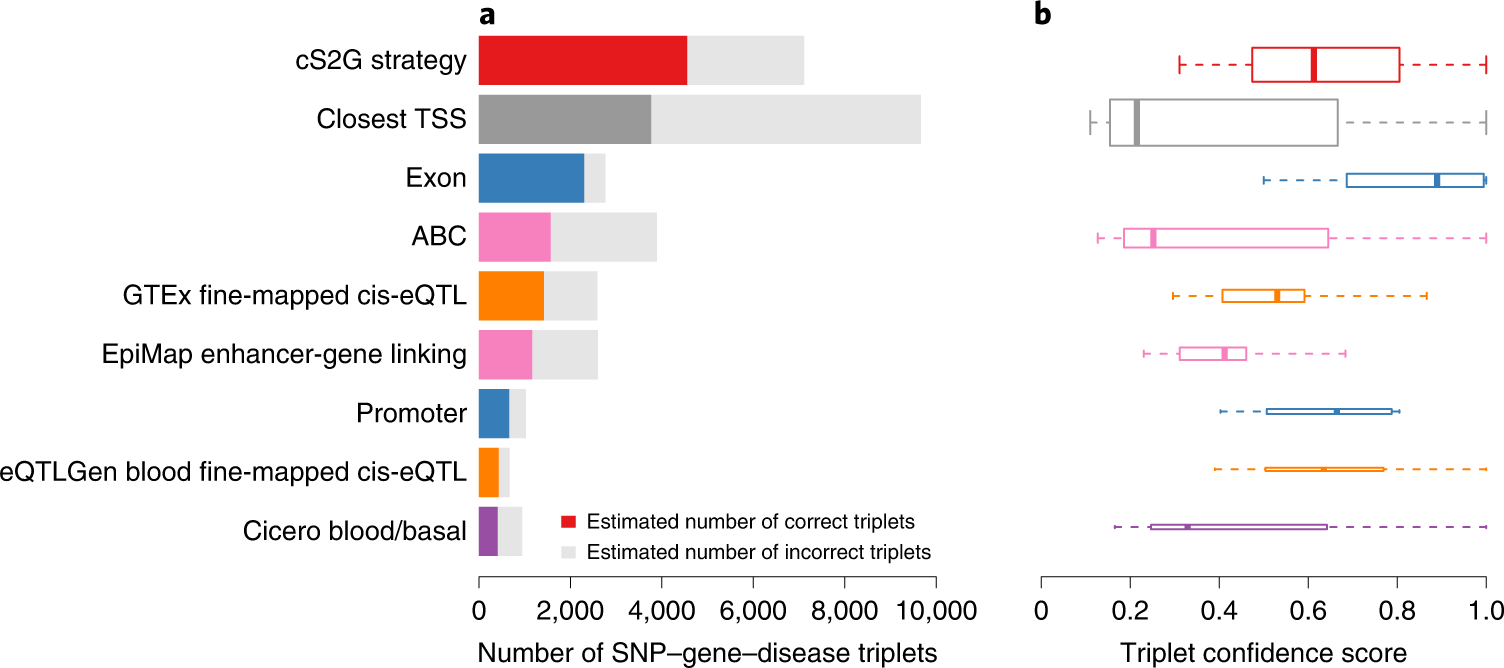
(a) We report the number of SNP-gene-disease triplets identified by cS2G, its 7 constituent strategies, and the Closest TSS S2G strategy. For each strategy, we estimated the number of correct triplets based on the mean confidence score across triplets; the estimated number of correct triplets is denoted as a colored bar, and the estimated number of incorrect triplets is denoted as a grey bar. (b) We report the distribution of confidence scores of SNP-gene-disease triplets for each S2G strategy. The median value of confident scores is displayed as a band inside each box; boxes denote values in the second and third quartiles; the length of each whisker is 1.5 times the interquartile range, defined as the width of each box; the height of each box is proportional to the total number of triplets linked by each strategy (7,111, 9,664, 2,763, 3,889, 2,589, 2,604, 1,029, 674 and 943 for the 9 plotted S2G strategies). The list of SNP-gene-disease triplets predicted by cS2G is reported in Supplementary Table 17. Numerical results are reported in Supplementary Table 18.
The SNP-gene-disease triplets predicted by cS2G included 2,163 triplets involving distal regulatory fine-mapped SNPs that were not in the gene body (or promoter) of the target gene, of which 532 were supported by at least 2 of the functionally informed constituent S2G strategies used by cS2G (Supplementary Table 17). We highlight 4 examples (Figure 4). Briefly, these examples included instances where two independent fine-mapped SNPs were linked to the same gene (Figure 4a and Figure 4b) and instances where the functionally informed constituent S2G strategies either implicated (Figure 4c) or failed (Figure 4d) to implicate a plausible critical tissue/cell-type (Supplementary Table 20), highlighting both the benefit of aggregating S2G links across multiple cell-types to infer SNP-gene pairs, and the challenge of identifying the causal cell-type of action. Further details of each example are provided in the Supplementary Note.
In summary, our cS2G strategy predicted 5,095 causal SNP-gene-disease triplets with high confidence, far more than any other S2G strategy; these triplets represent, to our knowledge, the largest high-confidence SNP-gene-disease resource with S2G-derived functional interpretation to date.
Leveraging the cS2G strategy to assess disease omnigenicity
Previous work has proposed an “omnigenic model” in which all genes expressed in disease-critical cell-types impact the function of core disease genes and thus impact disease SNP-heritability44–46. This work raises intense interest in estimating components of disease SNP-heritability contributed by each gene, but this has yet to be empirically assessed, due to the challenges of linking SNPs to genes. We leveraged our combined S2G (cS2G) strategy to estimate the SNP-heritability causally explained by SNPs linked to each gene for 49 UK Biobank diseases/traits (Supplementary Table 21) with functionally informed genome-wide fine-mapping results (not restricted to GWAS loci) available for all MAF ≥0.1% SNPs7. We re-estimated the SNP-heritability linked to ranked gene sets by running S-LDSC11,52,53 on summary statistics computed from N=122K European-ancestry UK Biobank validation samples that were distinct from the training samples used for fine-mapping (to avoid winner’s curse; see Methods and ref.7). We also estimated the effective number of causal genes (Ge) for each trait (using fourth moments of per-gene effects, analogous to Me for SNPs54), and estimated this quantity separately for per-gene h2 linked to common (MAF≥5%) and low-frequency (0.1%≤MAF<5%) SNPs.
The top 200 (resp. top 2,000) genes explained 52±6% (resp. 96±8%) of the disease SNP-heritability linked to genes in cis using the cS2G strategy (h2gene, which captures 53±3% of h2), meta-analyzed across a subset of 16 independent diseases/traits (Figure 5a and Supplementary Tables 22–23). Estimates directly based on the N=337K training samples used for fine-mapping were very similar, implying minimal effects of winner’s curse (Figure 5a). Results were similar when restricting to genes expressed in disease-critical cell-types55 (as proposed in ref.44) (Supplementary Figure 6) and when using a restricted set of N=49K validation samples that were unrelated to the N=337K training samples (Supplementary Figure 7). Interestingly, repeating the analysis using the Closest TSS S2G strategy implicated a far more polygenic gene-level architecture that required the top 1,000 (resp. top 10,000) genes to explain 48±2% (resp. 85±2%) of h2gene (Figure 5a and Supplementary Table 22); these results demonstrate the benefits of using more precise S2G strategies to infer more accurately infer gene-level architectures. We caution that the primary analysis using cS2G may still slightly overestimate gene-level polygenicity, because even the cS2G strategy is not perfectly precise. We further caution that all our findings pertain to effects of SNPs in cis (see Discussion), potentially leading to underestimation of gene-level polygenicity.
Figure 5: Empirical assessment of disease omnigenicity using cS2G.
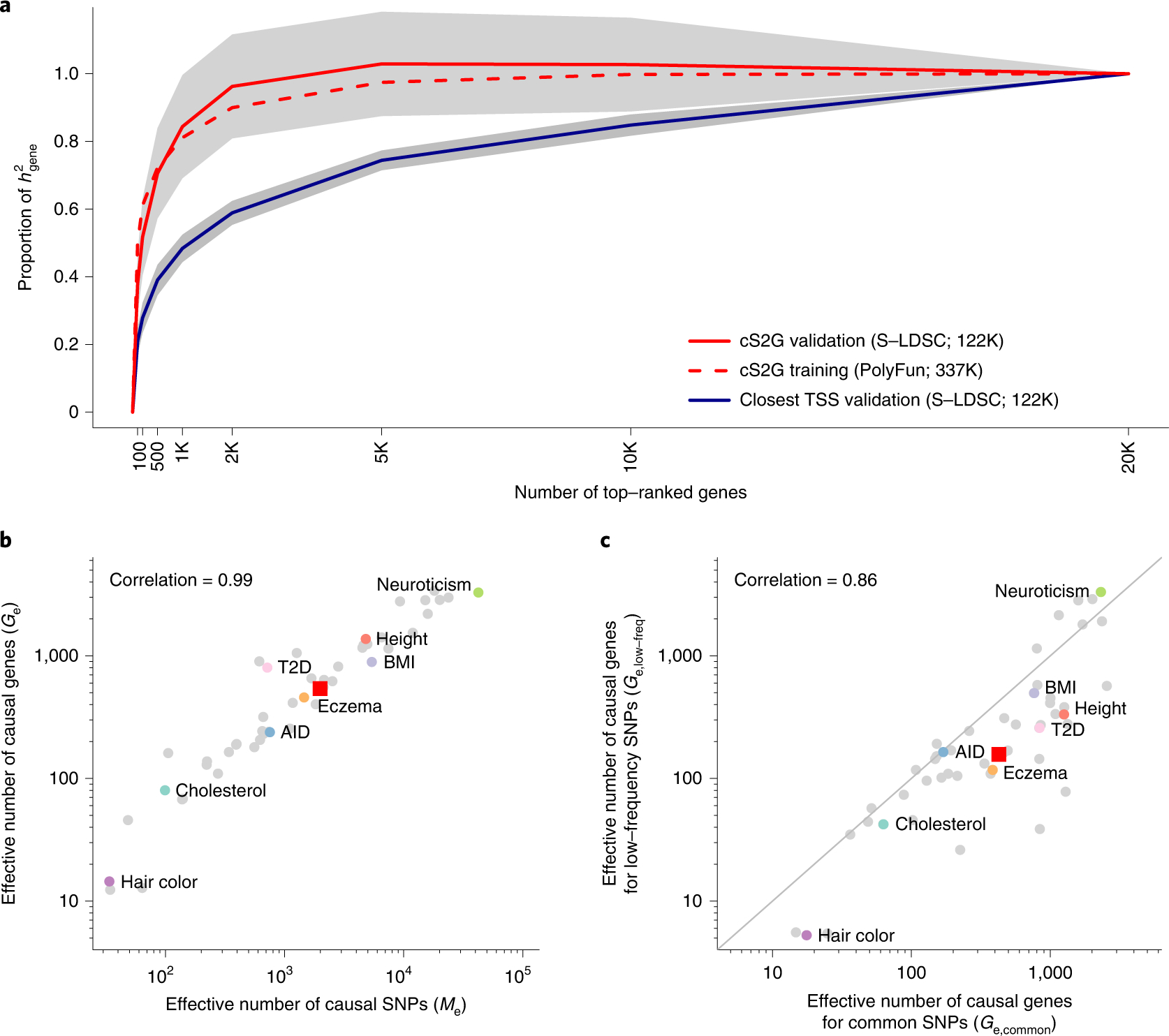
(a) We report the proportion of SNP-heritability linked to genes (h2gene) explained by genes ranked by top per-gene h2, as inferred using three approaches (see text). Grey shading denotes 95% confidence intervals for cS2G-validation and Closest TSS-validation around meta-analyzed values. We forced the s.e. of the proportion of h2gene explained by all genes to be 0 (see Methods). We note that values greater than 1 are outside the biologically plausible 0–1 range, but allowing point estimates outside the biologically plausible 0–1 range is necessary to ensure unbiasedness. Results were meta-analyzed across 16 independent UK Biobank traits. (b) We report the effective number of causal SNPs54 (Me) and the effective number of causal genes (Ge) for 49 UK Biobank diseases/traits, with representative traits in colored font. (c) We report the effective number of causal genes for per-gene h2 linked to common SNPs (Ge,common) and the effective number of causal genes for per-gene h2 linked to low-frequency SNPs (Ge,low-frea) for 49 UK Biobank diseases/traits, with representative traits in colored font. In (b) and (c), red squares denote median values across 16 independent traits and correlations are computed on log-scale values. Numerical results are reported in Supplementary Table 22 and Supplementary Table 24. AID: Autoimmune disease; BMI: Body mass index; Cholesterol: Total cholesterol; T2D: Type 2 diabetes.
We estimated the effective number of causal genes (Ge) for each trait. Estimates of Ge varied widely, from 3,289 (neuroticism) to 1,375 (height) to 80 (total cholesterol) with a median of 540 (across 16 independent traits), and were strongly correlated (log-scale r=0.99) to estimates of the effective number of independently associated SNPs (Me; median=1,991), a SNP-based measure of disease/trait polygenicity54 (Figure 5b and Supplementary Table 24; the strong correlation provides a validation of both Ge and Me).
We further estimated Ge,common (resp. Ge,low-frea) by restricting per-gene h2 explained by causal SNPs to the SNP-heritability causally explained by common (resp. low-frequency) SNPs linked to each gene. Gene-level architectures were more polygenic for common vs. low-frequency SNPs, with median Ge,common of 427 vs. median Ge,low-freq of 157 (median ratio of 2.8) across the 16 independent traits (Figure 5c and Supplementary Table 21), consistent with more polygenic SNP architectures for common vs. low-frequency SNPs due to the action of negative selection (Me,common / Me,low-freq=3.9 in ref.54). Surprisingly, there was low concordance between genes underlying gene-level architectures for common vs. low-frequency SNPs (Extended Data Figure 4 and Supplementary Table 24–25). However, we observed consistent excess overlap between the top 200 genes contributing to h2gene,common (resp. h2gene,low-freq) and two disease-specific gene sets56,57 (Extended Data Figures 5–6), suggesting that common and low-frequency variant gene-level architectures are driven by different genes pertaining to similar biological processes.
In summary, our cS2G strategy provided a quantitative assessment of the “omnigenic model”44–46, implicating a far less polygenic gene-level architecture than the Closest TSS S2G strategy. We inferred a more polygenic gene-level architecture for common variants as compared to low-frequency variants, with little overlap between genes underlying these gene-level architectures despite shared biological processes.
Discussion
We developed a polygenic framework to evaluate and combine S2G strategies; in particular, our framework is a substantial advance over previous approaches for evaluating S2G strategies using curated lists of disease-associated SNP-gene pairs42,48 (see Supplementary Note for further discussion). We applied our framework to construct a combined S2G (cS2G) strategy that achieved a precision of 0.75 and a recall of 0.33, more than doubling the precision and/or recall of any individual strategy. We applied cS2G to fine-mapping results for 49 UK Biobank diseases/traits to predict 5,095 causal SNP-gene-disease triplets (with S2G-derived functional interpretation) with high confidence, including 2,163 triplets involving distal regulatory fine-mapped SNPs that were not in the gene body (or promoter) of the target gene; notable examples included CDKN1C in type 2 diabetes, BCL6 in asthma, PDCD1 in eczema, and LAMP1 in HDL, all of which were supported by multiple S2G strategies. We further applied cS2G to provide a quantitative assessment of the “omnigenic hypothesis”44–46, concluding that the top 200 (1%) of ranked genes explained roughly half of the SNP-heritability linked to all genes; this implies that gene-level architectures in cis are largely driven by a relatively modest number of top genes.
Our findings have several implications for downstream analyses. First, we recommend that GWAS fine-mapping studies employ cS2G to powerfully link fine-mapped SNPs to their target genes; we note that, as with previous S2G approaches, cS2G can be combined with similarity-based approaches leveraging genome-wide patterns of associated genes (as proposed in ref.48). Second, our framework can be used to optimize (and combine) S2G strategies that may be developed in the future; the development of new S2G strategies remains a key priority, as our cS2G strategy—despite its large improvement over other S2G strategies—attained a modest recall of 33%, implying that only 1/3 of disease SNP-heritability can be explained by causal disease SNPs linked to their correct target genes. Third, our results highlight the advantages of enhancer-gene linking strategies such as EpiMap and ABC in future efforts to improve S2G strategies; their advantages include cost effective experiments targeting multiple cell-types (EpiMap and ABC provide links for 833 and 167 cell-types, respectively), and high potential for linking rare variants to genes. Fourth, our findings support the hypothesis that rare variant association studies58,59 will provide biological insights complementary to those of GWAS—both because we observed little overlap between genes underlying common variant and low-frequency variant gene-level architectures, and because we determined that low-frequency variant gene-level architectures were less polygenic (median ratio of 2.8); we expect these differences to be even more pronounced for rare variant architectures. Finally, cS2G can improve identification of gene sets that are enriched for disease SNP-heritability (although it has not been optimized for this specific purpose; Supplementary Figure 8, also see ref.55); we further note the importance of including appropriate SNP annotations in the model used by S-LDSC in analyses of enriched gene sets, in order to avoid biased enrichment estimates (see Methods and Supplementary Figure 9). Investigating the relative performance of different combined S2G strategies in analyses of gene sets that are enriched for disease SNP-heritability is a direction for future research.
We note several limitations of our work. We included all available tissues and cell types in the constituent S2G strategies of cS2G, as we observed that this led to higher precision (Extended Data Figure 3), perhaps due to limited biosample size. However, S2G links involving disease-critical tissues/cell-types are central to understanding biological mechanisms (Figure 4, Supplementary Table 20, Supplementary Note). As larger data sets become available, it may become practical to define disease-specific combined S2G strategies that restrict to disease-critical tissues and cell types, furthering the goal of pinpointing the causal cell-types of action of SNP-gene-disease triplets. Additional limitations are discussed in the Supplementary Note. Despite these limitations, our results convincingly demonstrate both the advantages of using our polygenic framework to evaluate and combine S2G strategies, and the effectiveness of using our cS2G strategy to pinpoint disease genes.
Figure 4: Examples of high-confidence SNP-gene-disease triplets identified by cS2G.
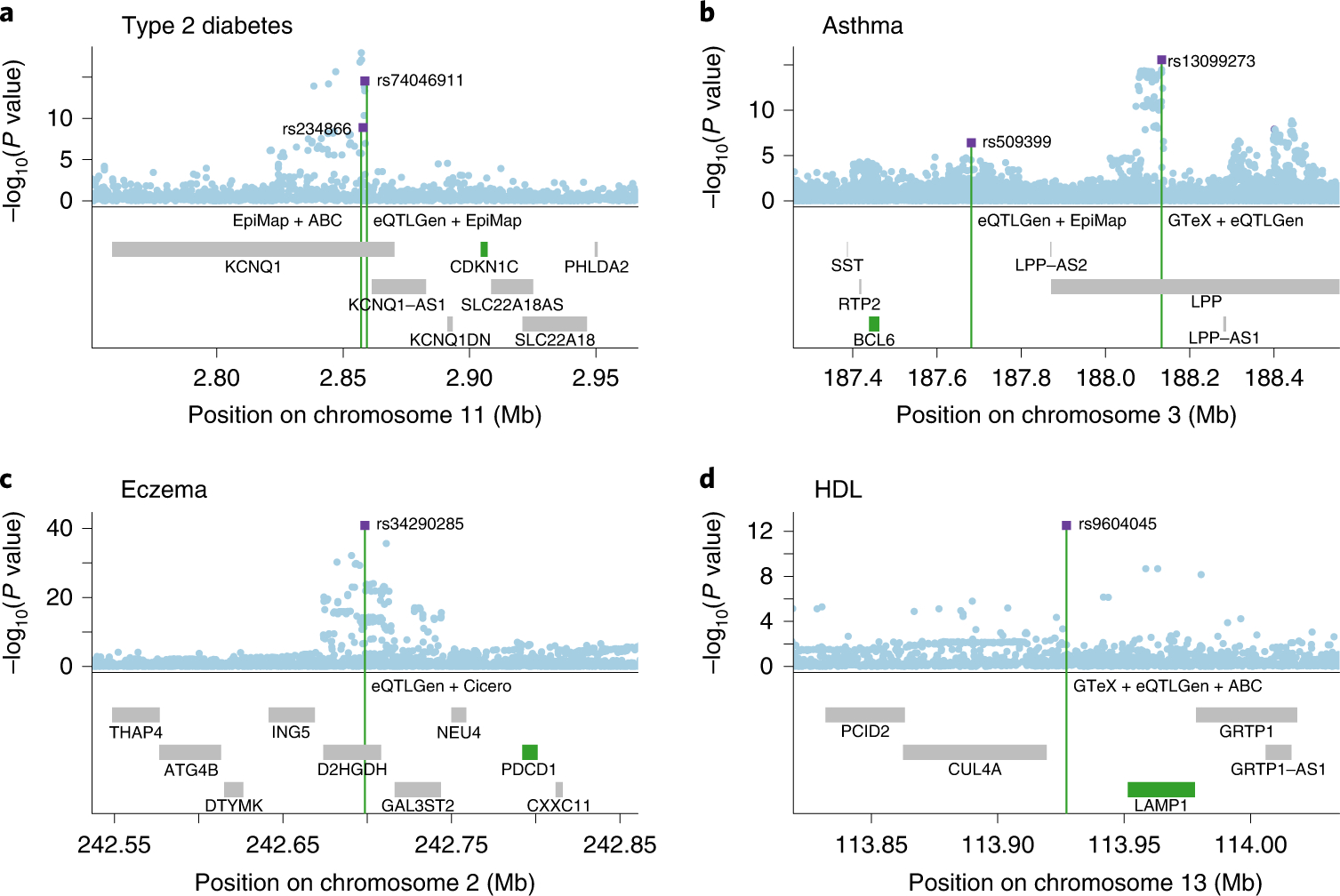
We report four examples where cS2G predicts target genes for distal regulatory fine-mapped SNPs (i.e. not in promoter or gene body) for (a) type 2 diabetes, (b) asthma, (c) eczema, and (d) high-density lipoprotein (HDL) cholesterol. We plot the −log10 GWAS P values of each SNP (top) and the gene body of the genes in the locus (bottom). Fine-mapped SNPs are denoted as purple squares, target genes are denoted in green, and constituent S2G strategies implicating the target gene are denoted in purple. All fine-mapped SNPs in these examples have posterior inclusion probability (PIP) >0.9 for the corresponding disease/trait, except rs13099273 for asthma (PIP=0.58). S2G links for all 13 main S2G strategies are reported in Supplementary Table 17, and tissues/cell-types for constituent strategies of cS2G are reported in Supplementary Table 20. GTEx: GTEx fine-mapped cis-eQTL; eQTLGen: eQTLGen blood fine-mapped cis-eQTL; EpiMap: EpiMap enhancer-gene linking; ABC: Activity-By-Contact; Cicero: Cicero blood/basal.
Methods
Ethics statement
This study relied on analyses of publicly available genetic and genomic datasets and so did not require ethical approval.
SNP-to-gene (S2G) strategies
A SNP-to-gene (S2G) linking strategy k is defined as an assignment of a raw linking value Ak,j,g, between each SNP j and zero or more candidate target genes g, which we converted into a linking score ψk,j,g such that each SNP has a sum of linking scores ≤1 (we allowed Σg ψk,j,g < 1 to allow for incomplete SNP-to-gene linking; see below). We considered only links related to a list of 19,995 genes, including 17,871 protein-coding genes, that pass our quality control procedure (see Data Availability). Specifically, we selected genes in Ensembl61 (grch37, accessed on 2019-04-24), GENCODE60 (release 19), and RefSeq62 (refGene, version 2017-03-08) databases that have a unique identifier in all the datasets, overlapping starting and ending gene positions, and similar strand information. We verified that restricting our analysis to 17,871 protein-coding genes (instead of 19,995 protein-coding and non-protein-coding genes) had little impact on our results (Supplementary Figure 10).
We considered 50 S2G strategies (Table 1, Supplementary Table 1, and Supplementary Note); in each case we first considered raw linking values Ak,j,g, which we next converted into linking scores ψk,j,g. For all but one strategy (Hi-C distance, see Supplementary Note), we converted raw linking values Ak,j,g (as defined above) into linking scores ψk,j,g such that each SNP has a sum of linking scores over genes being 0 or 1 (linking score ψk,j,g should not be interpreted as probabilities). We note that some S2G strategies include instances of SNP-gene links with low linking scores, based on the definitions of raw linking values for each strategy. When an S2G strategy linked a SNP to multiple genes, we restricted each S2G strategy such that each SNP was restricted to the gene(s) with the highest linking score (regardless of whether this linking score was high or low in absolute terms; no specific threshold), as we observed that this led to slightly higher precision (Extended Data Figure 1).
Correlations and overlap proportions between the S2G strategies were computed on all SNP-gene links observed by at least one of 34 S2G strategies (we omitted 6th closest TSS to 20th closest strategy and Hi-C due to computational constraints) (Supplementary Table 2). Overlap proportion between a strategy k and a strategy k’ is defined as the proportion of SNP-gene links reported by strategy k that are also reported by strategy k’ (these values are not symmetric). We defined a subset of 13 independent S2G strategies (different from the 13 main strategies) with r2 < 0.1 when comparing h2 coverage, precision or recall estimates.
We note that the functionally informed S2G strategies are derived from functional assays with widely varying biosample sizes. For example, PCHi-C datasets used 17/27 cell-types, enhancer maps such as EpiMap or ABC used multiple functional assays for 127 and 833 cell-types, and cis-eQTLs such as GTeX and eQTLGen used 17,382 and 31,684 RNA-seq samples. Thus, our evaluation of S2G strategies should not be viewed as an evaluation of the underlying functional assays.
Evaluation of S2G strategies
To evaluate each S2G strategy’s informativeness for pinpointing disease genes, we aimed to define and estimate parameters that correspond to an S2G strategy’s h2 coverage (proportion of total disease SNP-heritability that is linked to genes), precision (proportion of linked disease SNP-heritability that is linked to the correct target gene), and recall (proportion of total disease SNP-heritability that is linked to the correct target gene).
First, we defined h2 coverage as the proportion of SNP-heritability explained by all SNPs linked to one or more genes (weighted by their linking scores):
| (1) |
where is the SNP-heritability explained by common SNPs linked all genes using k, and h2 is the SNP-heritability explained by common SNPs.
Second, we defined precision as the relative excess SNP-heritability enrichment of SNPs linked to a critical gene set (see below) vs. SNPs linked to all genes, as compared to the (gold-standard) Exon S2G strategy. More precisely, the precision of an S2G strategy k was defined as
| (2) |
with
| (3) |
where is the SNP-heritability explained by common SNPs linked to the critical gene set using k, M(genes:critical,S2G:k) is the number of common SNPs linked to the critical gene set using k, and M(genes:all,S2G:k) is the number of common SNPs linked to all genes using k. We note that this definition relies on the hypothesis that genes in the critical gene set are enriched for causal disease genes (as observed empirically, see below), and the hypothesis that the Exon S2G strategy is a perfectly precise strategy (even though it suffers from low h2 coverage). Third, we defined recall as the product of the h2 coverage and precision.
We estimated these quantities using polygenic analyses of disease SNP-heritability by applying stratified LD score regression (S-LDSC; v1.0.1) with the baseline-LD model (v2.2)11,52,53 to 63 independent diseases and complex traits (average N = 314K; Supplementary Table 3), meta-analyzing results across traits. All traits had z-score>6 for non-zero SNP-heritability, following previous recommendations52. We removed the major histocompatibility complex (MHC) region during the regression step because of its unusual LD patterns and genetic architecture11. We analyzed SNP annotations for ~10M SNPs with a minor allele count ≥5 in a 1000 Genomes Project European reference panel47. We jointly considered the 97 SNP annotations of the baseline-LD model v2.2 (refs.52,53), 50 S2G-derived SNP annotations constructed by restricting SNPs linked to genes of the critical gene set, and 30 S2G-derived SNP annotations constructed by restricting SNPs linked to all 19,995 genes (we did not include SNP annotations constructed using all genes for the 20 closest TSS S2G strategies, as these would include all SNPs), for a total of 177 SNP annotations. Jointly considering all these S2G-derived SNP annotations was crucial to maximize the accuracy of (Supplementary Figure 9). This strongly demonstrates the importance of including appropriate SNP annotations in the model used by S-LDSC in analyses estimating the proportion of SNP-heritability explained by enriched gene sets55, in order to avoid biased enrichment estimates. We note that precisions from preliminary analyses of Extended Data Figure 1 were estimated using the baseline-LD model and S2G-derived SNP annotations constructed from the Exon, Promoter, Gene body, Gene body +/− 100kb, and Closest TSS S2G strategies; using this restricted set of S2G strategies attenuated the bias of .
We estimated values of h2 coverage and gene-enrichment for each disease/trait, estimated their standard errors using a genomic block-jackknife with 200 blocks, meta-analyzed results across the 63 independent traits using a fixed-effect meta-analysis, and used these values to estimate precision and recall. Precision and recall were estimated from meta-analyzed h2 coverage and gene-enrichment values (instead of meta-analyzing precision and recall across traits) to guarantee robust estimates. Precision and recall standard errors were estimated by using the 200 h2 coverage and gene-enrichment estimates from the block-jackknife procedure, but meta-analyzed using the same h2 coverage and gene-enrichment standard errors. We note that performing fixed effect meta-analyses when computing overall estimates of precision/recall assigns low weights to traits with large standard errors. For example, including 5 traits with low SNP-heritability (< 0.02, despite z-score>6 for non-zero SNP-heritability) had little impact on our results: when removing these traits, estimates of precision changed from 0.747 (s.e. 0.061) to 0.753 (s.e. 0.062) and estimates of recall changed from 0.330 (s.e. 0.027) to 0.332 (s.e. 0.027).
Training and validation critical gene sets
Our definitions of precision and recall rely on a critical gene set. We used a non-trait-specific training critical gene set to construct an optimal combined S2G strategy, and trait-specific validation critical gene sets to evaluate the optimal combined S2G strategy while avoiding overfitting (for comparison purposes, we also used the validation critical gene sets to evaluate individual S2G strategies). Training and validation critical gene sets rely on information from exons and promoters to guarantee high-confidence SNP-gene links.
We defined a non-trait-specific training critical gene set as the top 10% of genes with the most highly constrained exons and conserved promoters. Specifically, for each gene we multiply its pLI score79 (estimating gene probability to be intolerant to loss-of-function mutations) by the fraction of bases of its promoter (defined using the Promoter S2G strategy) that is conserved (defined using 4 baseline-LD conserved SNP annotations80,81) (note that 17,554 out of our 19,995 genes had a pLI score). As we observed a correlation between this score and gene body length (r = 0.18), we created 10 bins of genes based on their gene body length, and selected the genes with the top 10% of this score (1,760 genes in total).
We defined the validation critical gene set for a given trait as the top 10% of genes ranked by the PoPS method48 (we note that PoPS gene scores are based on a leave-one-chromosome-out approach, implying that gene scores should be independent of their surrounding SNPs). By default, the initial step of PoPS is to apply MAGMA82 to compute gene-level association statistics, which relies on linking SNPs to genes using a gene body S2G strategy. To limit the impact of the gene body S2G strategy in our analyses, we modified PoPS to only link SNPs that are in exons or promoters (note that this led to nearly similar SNP-heritability enrichment and gene-enrichment values; Supplementary Table 4). 16,728 out of our 19,995 genes had a PoPS score, leading to validation critical gene sets with 1,673 genes.
Across the 63 diseases/traits analyzed, the overlap between the training gene set (which does not vary across disease/traits) and the validation gene sets (which does vary across diseases/traits) had a mean of 20% (vs. 10% expected by chance), mean of 20%, standard deviation of 4.1%, and range from 13%−28% (Supplementary Figure 1). We also observed substantial excess overlap of housekeeping genes (1,997 genes ) in the training critical gene set (357 genes; excess overlap = 2.0) and validation critical gene sets (median of 281 genes across 63 traits; median excess overlap = 1.7).
Combining S2G strategies
We constructed combined S2G strategies as linear combinations of linking scores from K S2G strategies. Specifically, for each SNP j and gene g we computed a combined S2G linking score
| (4) |
where ψk,j,g is the linking score between SNP j and gene g for S2G strategy k, and wk is the weight associated to strategy k. We used Ψj,g = Σk wk × ψk,j,g when ΣgΣk wk × ψk,j,g < 1, and Ψj,g = Σk wk × ψk,j,g / ΣgΣk wk × ψk,j,g otherwise; allowing Σg Ψj,g to be < 1 allows to give small combined S2G linking scores to SNPs with linking score available only for imprecise S2G strategies. Here, we allowed weights wk to have a maximum value of 100, to prioritize S2G strategies with higher precision in the case where two S2G strategies link the same SNP to different genes. For example, if we have an S2G strategy 1 with high precision and low recall and an S2G strategy 2 with reasonable precision and high recall, then assigning weights of 100 and 1 allows to create a combined S2G strategy that will leverage the high precision of S2G strategy 1 when a SNP is linked to different genes using S2G strategies 1 and 2, while maximizing recall using S2G strategy 2.
To estimate the optimal weights wk, we developed an optimization framework to identify the weights maximizing the recall while constraining precision (defined using the training critical gene set) to be ≥0.75. Indeed, providing high precision maximizes the utility of functional follow-up studies. First, we computed for each SNP its expected per-SNP heritability by meta-analyzing across the 63 independent traits the S-LDSC regression coefficients estimated with the baseline-LD model and the 80 S2G-derived SNP annotations of the training gene set and all genes. Second, we defined a function taking as input a vector of weights w, computed for each SNP the expected per-SNP heritability linked to the critical gene set and to all genes using the 80 S2G-derived SNP annotations, and outputting precision and recall. Finally, we found the vector w maximizing recall while constraining precision to be ≥0.75. (We note that the precision of 0.75 for the cS2G strategy computed using the validation critical gene sets is independent of the threshold of precision ≥0.75 in the training critical gene set used to optimize the cS2G strategy (estimated precision using the training critical gene set was equal to 0.81 during the optimization process; Supplementary Table 8).) Specifically, we considered a grid of values for w, going from 0 to 2.5 with a 0.1 step, and the values 5, 7.5, 10, 25, 50, 75, and 100 (33 total values). We created a custom optimization framework that (a) starts by giving a weight of 1 to the Exon strategy and 0 to all other S2G strategies investigated, (b) computes precision and recall by increasing a single value of w at a time (the higher weight on the grid), and keeps for next step the weight vector maximizing recall (constraining precision to be ≥0.75), (c) computes precision and recall by decreasing a single value of w at a time (the lower weight on the grid), and keeps for next step the weight vector maximizing recall (constraining precision to be ≥0.75), (d) computes precision and recall by randomly modifying a single value of w at a time, and keeps for next step the weight vector maximizing recall (constraining precision to be ≥0.75), and (e) restarts from (b) till the recall does not improve. We repeated this algorithm 5 times, and kept the weight vector providing the maximum recall. We note that we investigated 5 different optimization algorithms from the R software (methods Nelder-Mead, BFGS, CG, L-BFGS-B and SANN from the optim function; R version 3.6.1 was used in our analyses), but none of them reached higher recall than our algorithm. When giving as a starting point the outputs of our custom algorithm, these 5 algorithms did not converge to significantly different weight vectors and recalls.
We note that we allowed weights to have a maximum value of 100, to prioritize S2G strategies with higher precision in the case where two S2G strategies link the same SNP to different genes. For example, in the case of our cS2G strategy (Supplementary Table 6), if a SNP is linked to gene A through the Exon S2G strategy (weight = 100), and to gene B through the Cicero S2G strategy (weight = 1), then the cS2G linking score is 100/101 for gene A (stronger evidence from Exon), and 1/101 for gene B. We note that weights of 10 and 0.1 for Exon and Cicero (rather than 100 and 1), would have assigned the same linking scores in the case of the SNP described above, but would have assigned lower linking scores to SNPs that are linked to genes only through Cicero. However, we note that fixing the weights of the 7 constituent S2G strategies of the cS2G strategy to the same value only slightly underperformed cS2G in both precision and recall (0.71 vs. 0.75 and 0.31 vs. 0.33, respectively; Supplementary Table 8), as expected given the low overlap of SNPs annotated by these 7 strategies (Supplementary Table 7).
Leveraging the combined S2G strategy to pinpoint disease genes
We analyzed 9,670 predicted causal SNP-disease pairs with posterior inclusion probability (PIP) >0.50 from functionally informed fine-mapping of 49 UK Biobank diseases/traits using PolyFun + SuSiE using N=337K unrelated British UK Biobank samples7,43. SNPs in the MHC region were removed from the fine-mapping analyses7. We selected a PIP threshold of 0.5 as this threshold was carefully validated using simulations in ref.7, and a threshold of 0.5 was also used for cS2G linking score and triplet confidence score. For this purpose, we created S2G and cS2G strategies for 19M imputed UK Biobank SNPs with MAF ≥ 0.1% (we note that these analyses include both SNPs and indels, but we use the term SNP throughout the manuscript for simplicity). We predicted causal SNP-gene-disease triplets by restricting to SNPs that had a linked gene with cS2G linking score >0.5 (98% of the cS2G linked SNPs). We note that the proportion of fine-mapped SNPs linked by the cS2G strategy (5,384/7,675 = 0.70) was higher than its h2 coverage (0.44) due to the excess of exon and promoter SNPs (2,629/5,384 SNPs are in exons or promoters, consistent with the use of functional priors in fine-mapping analyses).
We further assigned a confidence score to each SNP-gene-disease triplet with a cS2G linking score >0.5 by multiplying their corresponding PIP and precision. To account for the excess of exon and promoter SNPs, we assigned the precision of Exon (i.e. 1.00) if the link was validated through the Exon S2G strategy, the precision of Promoter (i.e. 0.80) if the link was validated through the Promoter S2G strategy, and an estimated precision for SNPs that are not in exons or promoters (precisionOther) otherwise. We estimated precisionOther for an S2G strategy k using the following formula
| (5) |
where coverageExon, coveragePron, and coverageOther are the proportion of h2 coverage explained by SNPs in exons, promoters, and SNPs that are not in exons or promoters, respectively.
To predict the candidate cell-type of action for these triplets, we (i) focused on SNP-gene pairs provided by GTEx fine-mapped cis-eQTL, EpiMap enhancer-gene linking, and/or ABC, (ii) restricted to cell-types where the SNP-gene pair has been observed (out of 54, 833 and 167, respectively), and (iii) reported the cell-type with the most significant regression coefficient in an S-LDSC analysis conditioned to the baseline model (as performed in refs.11,56).
Leveraging the combined S2G strategy to empirically assess disease omnigenicity
To estimate the SNP-heritability explained by SNPs linked to each gene g for 49 UK Biobank diseases/traits (per-gene SNP-heritability h2gene,g), we used PolyFun + SuSiE estimates of posterior mean squared causal effect sizes for 19M imputed SNPs with MAF ≥0.1% estimated on N=337K unrelated British UK Biobank samples7,43 (SNPs in the MHC region were removed from these analyses7). First, we computed unadjusted per-gene SNP-heritability , where Ψj,g is the cS2G linking score between SNP j and gene g. Then, we computed (adjusted) per-gene SNP-heritability , with being a trait specific cS2G linking score. The motivation of this additional step is to improve the cS2G linking scores of SNPs linked to multiple genes by integrating evidence from linked to a single gene; we note that this step changed per-gene SNP-heritability for only a small number of genes, as most SNPs linked using cS2G have large cS2G linking score (87% of the linked SNPs have a maximum cS2G linking score >0.95) (see Supplementary Figure 11). We also estimated per-gene SNP-heritabilities linked to common SNPs (MAF≥5%) as , and per-gene SNP-heritabilities linked to low-frequency SNPs (0.1%≤MAF<5%) as . We note that these estimates of per-gene SNP-heritabilities do not account for LD between probabilistically fine-mapped SNPs, leading to genome-wide underestimation of variance explained by SNPs; this underestimation could in principle vary across genes. However, this limitation does not impact our main conclusions, because (i) it impacts only estimates of per-gene SNP-heritabilities derived from PolyFun + SuSiE, and not S-LDSC estimates of proportions of h2gene explained by the resulting 8 gene sets (see next paragraph) as reported in Figure 5a; and (ii) estimates of proportions of h2gene derived from PolyFun + SuSiE (based on N=337K training samples) and S-LDSC estimates of proportions of h2gene (based on N=122K new validation samples) were strongly concordant (Figure 5a).
To estimate the proportion of SNP-heritability linked to genes (h2gene) explained by genes with the top per-gene SNP-heritability, we partitioned genes into 8 gene sets ranked by per-gene SNP-heritability explained (top 100, 200, 500, 1,000, 2,000, 5,000, 10,000 and 19,995), and re-estimated the SNP-heritability linked to each ranked gene set by running S-LDSC with the baseline-LD model on summary statistics computed from N=122K European-ancestry UK Biobank samples that were distinct from the N=337K British UK Biobank samples used to estimate and (in order to avoid winner’s curse, analogous to what was performed in ref.7). We defined h2gene as the SNP-heritability linked to all 19,995 genes. The proportion of h2gene explained by the X top genes was computed as the proportion of SNP-heritability explained by the X top genes divided by h2gene. The standard error of the proportion of h2gene explained by the X top genes was computed as the standard error of the proportion of h2 explained by the X top genes divided by h2gene (viewing the denominator h2gene as a constant); we believe this to be a reasonable approximation, as the numerator has greater uncertainty than the denominator (except when including all genes), and the errors are correlated such that this approximation is conservative. We note that a ratio of meta-analyzed values (meta-analyzed proportion of SNP-heritability explained by X top genes divided by meta-analyzed proportion of SNP-heritability explained by all genes) is more robust than a meta-analyzed value of ratios (meta-analyzing the proportion of SNP-heritability explained by X top genes divided by the proportion of SNP-heritability explained by all genes). In Figure 5a, we forced the s.e. of the proportion of h2gene explained by all genes (a quantity that must equal 1) to be 0.
We estimated the effective number of causal genes (Ge) for each trait using per-gene SNP-heritability and the formula of ref.54. Specifically, we defined Ge = 3G/κ, with G the total number of genes and . These estimates relied on per-gene SNP-heritability explained by causal SNPs, and were thus directly based on the N=337K samples used for fine-mapping; as noted above, the impact of winner’s curse on our analyses was minimal. We extended this formula to per-gene h2 linked to common and low-frequency SNPs to estimate Ge,common and Ge,low-frea, respectively. Per-gene h2 were directly estimated on the N=337K samples used for fine-mapping as we observed that the impact of winner’s curse on our analyses was minimal.
Statistics and reproducibility
No statistical method was used to predetermine sample size. We restricted our analyses to well-powered (z-score>6 for non-zero SNP-heritability52) GWAS datasets of European ancestry. We removed the MHC region during the S-LDSC regression step11. We did not use any study design that required randomization or blinding.
Extended Data
Extended Data Figure 1: S2G strategy linking each SNP to best gene leads to higher precision than linking SNPs to multiple target genes.
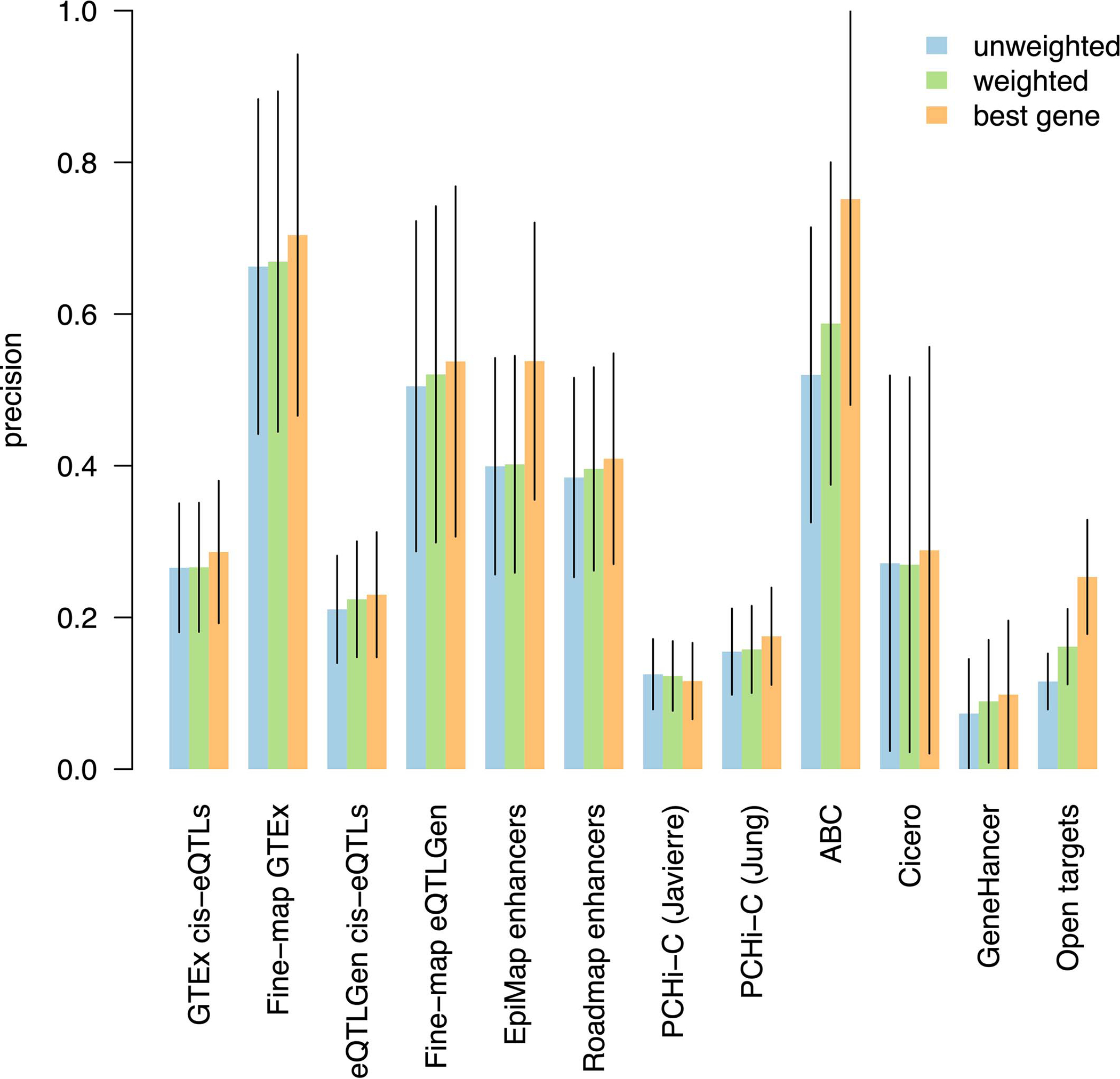
We report the precision of S2G strategies linking SNPs to target genes using three difference approaches for converting raw linking values into linking scores: by assigning to each gene with non-zero raw linking value the same linking score (unweighted), by assigning to each gene a linking score proportional to its raw linking value (weighted), and by retaining only the gene(s) with the highest linking score (best gene). Values were estimated using non-trait-specific training critical gene set and meta-analyzed across 63 independent traits. Error bars represent 95% confidence intervals around meta-analyzed values. For most of the S2G strategies the precision was very similar (except for EpiMap, ABC and Open Targets), but the precision was generally highest for the “best gene” strategy. However, we note that this choice does not reflect biological reality, in which a regulatory element may target more than one gene, and that refinements to this choice are a direction for future research.
Extended Data Figure 2: Precision of 27 S2G strategies based on physical distance to TSS.
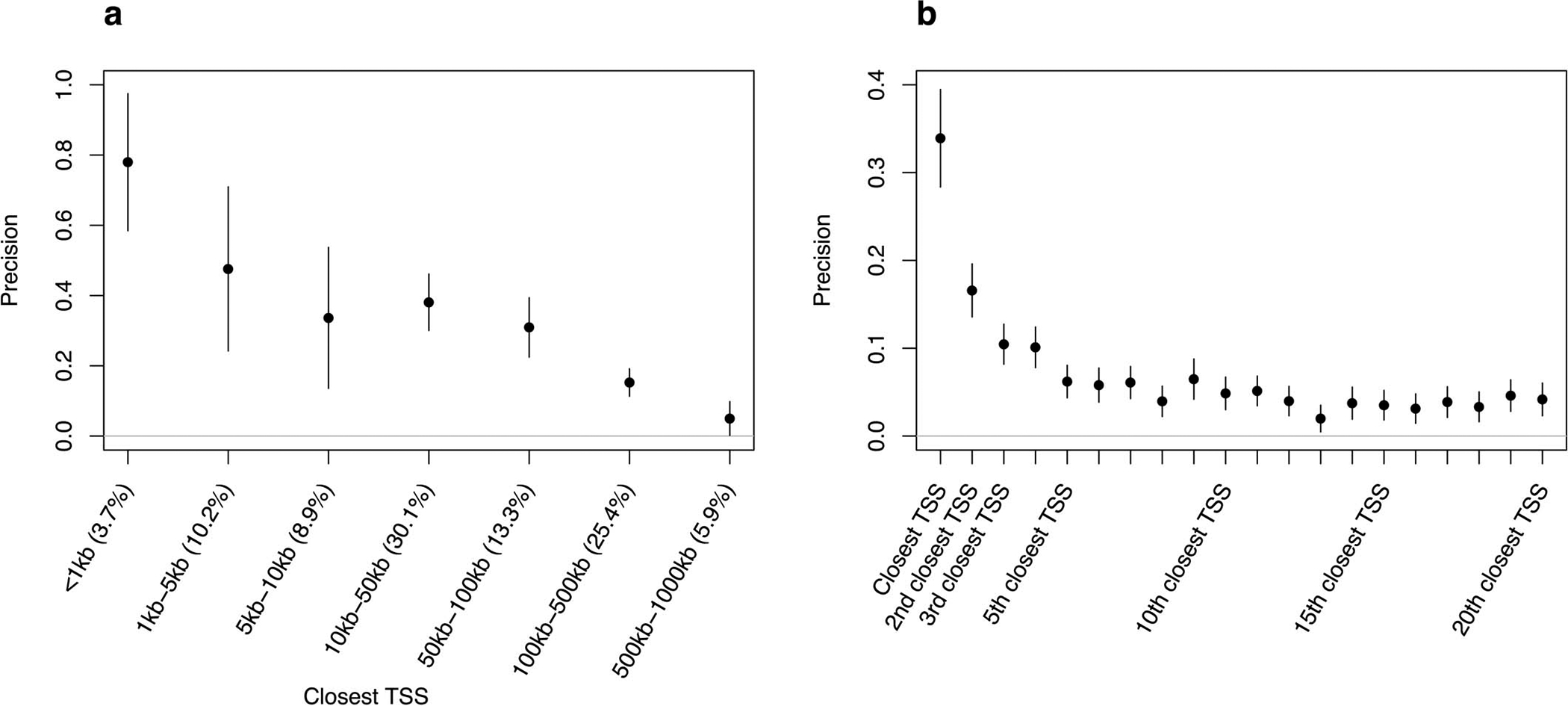
We report precision of the closest TSS strategy as a function of the distance between a SNP and its closest TSS (a) (numbers between parentheses represent the fraction of common SNPs linked by the strategy), and the precision of the ith closest TSS (each strategy links 100% of the SNPs) (b). Values were estimated using trait-specific validation critical gene sets and meta-analyzed across 63 independent traits. Error bars represent 95% confidence intervals around meta-analyzed values. The mean value of 0.043 for 6th-20th closest TSS suggests that genes located relatively close to causal disease genes have a slightly elevated probability of being causal. Numerical results including values of recall and corresponding standard errors are reported in Supplementary Table 5.
Extended Data Figure 3: Precision of functional S2G strategies using all available cell-types and tissues or restricted to blood and immune cell-types and tissues.
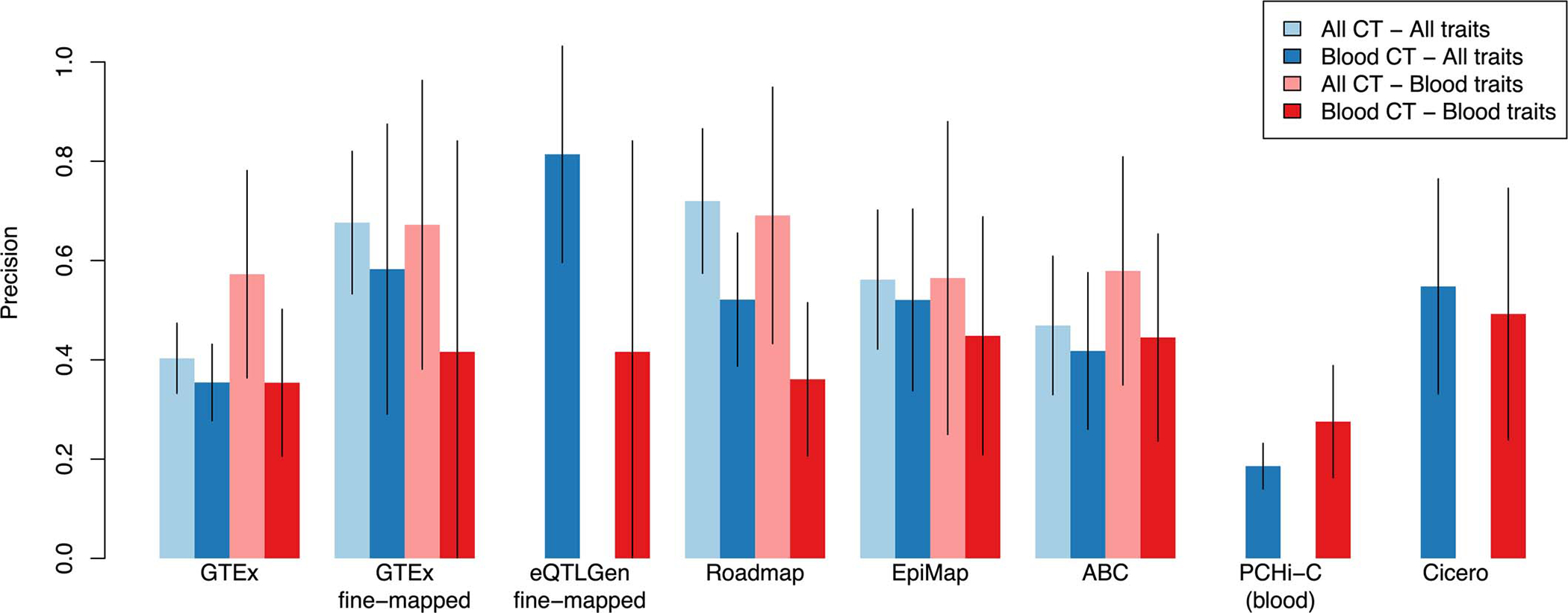
We report the precisions of functional S2G strategies built using either all available cell-types and tissues (All CT; in light color) and/or blood and immune cell-types and tissues (Blood CT; in dark color) meta-analyzed across 63 independent traits (All traits; in blue) and 11 blood cell traits and autoimmune diseases (Blood traits; in red) (UK Biobank all auto-immune diseases, Crohn’s Disease, Rheumatoid Arthritis, Ulcerative Colitis, Lupus, Celiac, Platelet Count, Red Blood Cell Count, Red Blood Cell Distribution Width, Eosinophil Count, White Blood Cell Count; see Supplementary Table 3). Error bars represent 95% confidence intervals around meta-analyzed values. We considered 5 S2G strategies with data available for cell-types and tissues: GTEx cis-eQTLs (GTEx), GTEx fine-mapped cis-eQTL (GTEx fine-mapped), Roadmap enhancer-gene linking (Roadmap), EpiMap enhancer-gene linking (EpiMap), and Activity-By-Contact (ABC). We considered 3 S2G strategies with data available only for blood and immune cell-types and tissues: eQTLGen fine-mapped blood cis-eQTL (eQTLGen fine-mapped), PCHi-C (blood), and Cicero blood/basal (Cicero). We observed 1) that S2G strategies using data from all cell-types and tissues were more precise than S2G strategies restricted to blood and immune cell-types and tissues in both analyses of all traits (light blue vs. dark blue) and blood cell traits and autoimmune diseases (light red vs. dark red), and 2) that S2G strategies using data from blood and immune cell-types and tissues are more precise in all traits than in blood cell traits and autoimmune diseases (dark blue vs. dark red).
Extended Data Figure 4: Proportion of common and low-frequency variant heritability linked to genes explained by each individual gene.
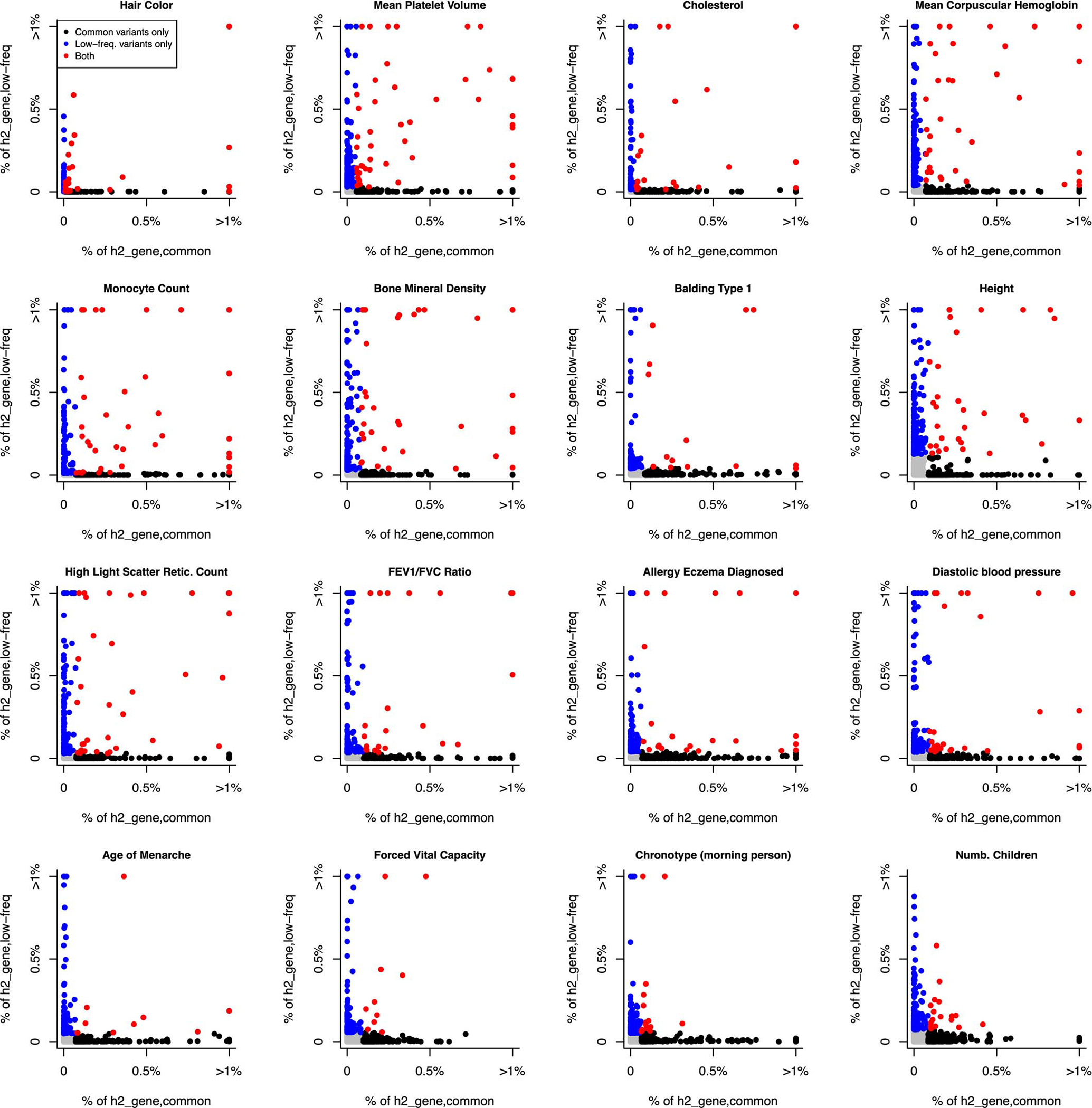
We report the proportion of common and low-frequency variant heritability linked to genes (h2gene,common and h2gene,low-freq, respectively) explained by each individual gene in 16 independent UK Biobank traits. Genes in the top 200 genes (top 1% of all genes) contributing to both h2gene,common and h2gene,low-freq are denoted in red (median of 26 genes across the 16 traits), genes in the top 200 genes contributing to only h2gene,common (resp. h2gene,low-freq) are colored in black (resp. blue) (median of 174 genes each), and remaining genes are colored in grey (median of 19,621 genes, with values close to 0 on both axes). We observe low concordance between per-gene contributions to gene architectures for common vs. low-frequency SNPs.
Extended Data Figure 5: Excess overlap between top genes contributing to common and low-frequency variant heritability linked to genes and disease-specific Mendelian disorder genes.
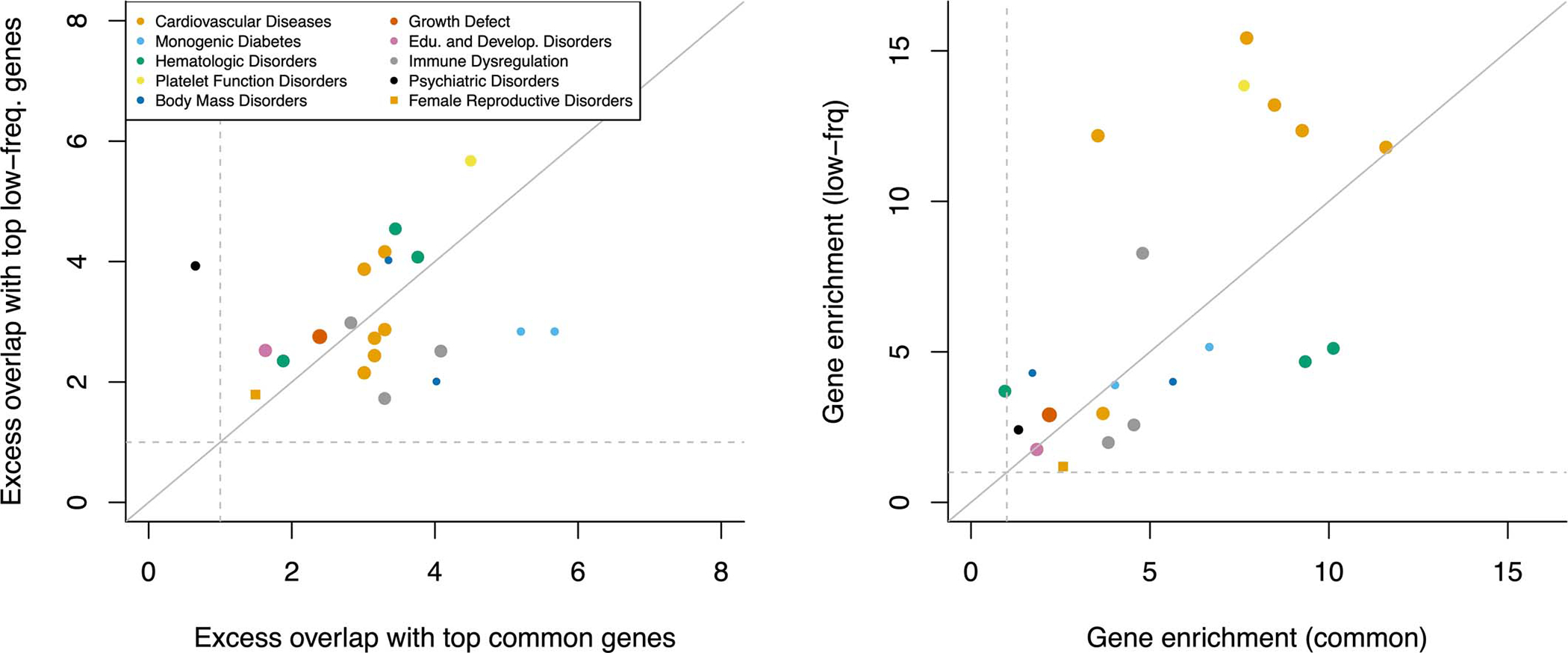
We report the excess overlap between phenotype-specific Mendelian disorder genes57 and the top 200 genes contributing to common and low-frequency variant heritability linked to genes (left), and the gene enrichment of disease-specific Mendelian disorder genes (i.e. [SNP-heritability linked to Mendelian disorder genes / SNP-heritability linked to all genes] / [number of Mendelian disorder genes / total number of genes]) across common and low-frequency variants (right). Each dot represents a disease/trait - Mendelian disorder gene set pair, and is colored by the Mendelian disorder gene set. These two results suggest that both the set of top 200 genes and the per-gene heritability estimates are unlikely to be driven by noisy estimates arising from finite sample size. We restricted analyses to 21 traits analyzed in ref. 57.
Extended Data Figure 6: Excess overlap between top genes contributing to common and low-frequency variant heritability linked to genes and differentially expressed gene sets.
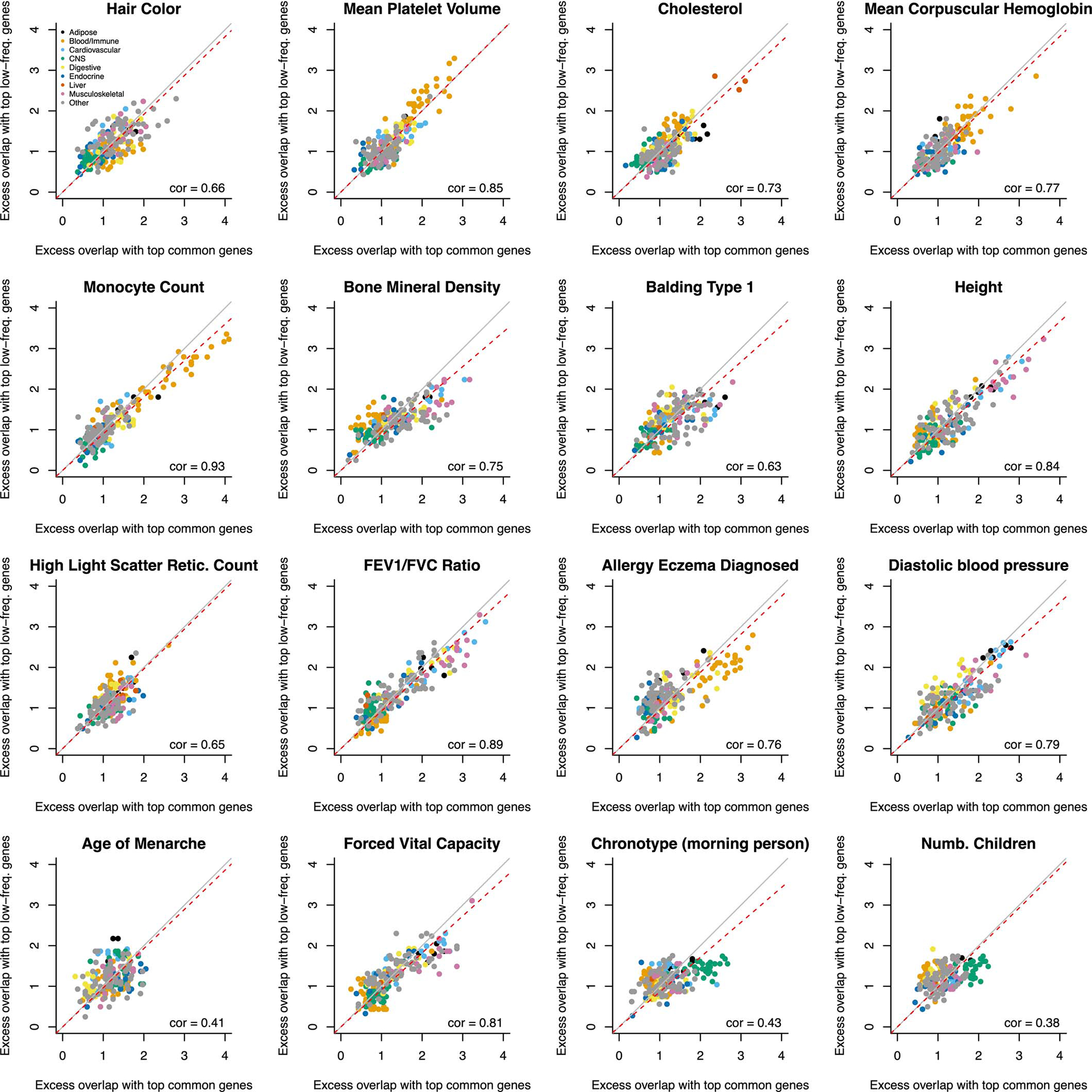
We report the excess overlap between 205 differentially expressed gene sets56 and the top 200 genes contributing to common and low-frequency variants heritability linked to genes across 16 independent UK Biobank traits. Each dot represents a differentially expressed gene set, and is colored by the tissue category. We generally observed excess overlap for disease-critical tissues/cell types. We observed high correlations between excess overlaps for common vs. low-frequency variant architectures, suggesting that common and low-frequency variants architectures are driven by different genes pertaining to similar biological processes.
Supplementary Material
Acknowledgments
We thank X. Jiang, C. Boix and M. Kellis for helpful discussion. S.G. is funded by NIH grant R00 HG010160. A.L.P. is funded by NIH grants U01 HG009379, R01 MH101244, R37 MH107649, R01 MH115676, R01 MH109978 and U01 HG012009. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. This research was conducted using the UK Biobank Resource under Application #16549.
Footnotes
Competing Interests Statement
C.P.F. is now an employee of Bristol Myers Squibb. The remaining authors declare no competing interests.
Code Availability
Code to estimate precision and recall of S2G strategies, and code to create combined S2G strategies have been made publicly available at https://alkesgroup.broadinstitute.org/cS2G/code and https://doi.org/10.5281/zenodo.6415925.
Peer Review Information:
Nature Genetics thanks Guillaume Lettre and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data Availability
The List of 19,995 genes, summary statistics of the 63 independent traits, training and validation critical gene sets, S2G and cS2G strategies, SNP annotations, predicted causal SNP-disease pairs from UK Biobank fine-mapping analyses and from the NHGRI-EBI GWAS catalog, and SNP-heritability causally explained by SNPs linked to each gene have been made publicly available at https://alkesgroup.broadinstitute.org/cS2G and https://doi.org/10.5281/zenodo.6354007. Links for all datasets used to create S2G strategies are provided in Supplementary Table 26.
Access to the UK Biobank resource is available via application at http://www.ukbiobank.ac.uk/.
GWAS catalog https://www.ebi.ac.uk/gwas/api/search/downloads/full
Open Targets SNP-gene pairs https://raw.githubusercontent.com/opentargets/genetics-gold-standards/master/gold_standards/processed/gwas_gold_standards.191108.tsv
SNP-gene pairs from ref.48 https://www.dropbox.com/s/kz2c49rpm2yanf5/all_byCS_rev1.txt?dl=0
References
- 1.Visscher PM et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. The American Journal of Human Genetics 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Claussnitzer M et al. A brief history of human disease genetics. Nature 577, 179–189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaid DJ, Chen W & Larson NB From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, Sarkar A, Carbonetto P & Stephens M A simple new approach to variable selection in regression, with application to genetic fine mapping. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 82, 1273–1300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weissbrod O et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nature Genetics 52, 1355–1363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hindorff LA et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. PNAS 106, 9362–9367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurano MT et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trynka G et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet 45, 124–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claussnitzer M et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. New England Journal of Medicine 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farh KK-H et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature Genetics 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gamazon ER et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nature Genetics 50, 956–967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porcu E et al. Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nature Communications 10, 3300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wainberg M et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet 51, 592–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Võsa U et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet 53, 1300–1310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee D et al. JEPEG: a summary statistics based tool for gene-level joint testing of functional variants. Bioinformatics 31, 1176–1182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hormozdiari F et al. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet 99, 1245–1260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun S et al. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat Genet 49, 600–605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu B, Gloudemans MJ, Rao AS, Ingelsson E & Montgomery SB Abundant associations with gene expression complicate GWAS follow-up. Nat Genet 51, 768–769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics 47, 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hormozdiari F et al. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat. Genet. 50, 1041–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao DW, O’Connor LJ, Price AL & Gusev A Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet. 52, 626–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Umans BD, Battle A & Gilad Y Where Are the Disease-Associated eQTLs? Trends Genet 37, 109–124 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ernst J et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Javierre BM et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Sarkar A, Kheradpour P, Ernst J & Kellis M Evidence of reduced recombination rate in human regulatory domains. Genome Biology 18, 193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pliner HA et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fulco CP et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat Genet 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung I et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat Genet 51, 1442–1449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boix CA, James BT, Park YP, Meuleman W & Kellis M Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nasser J et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fishilevich S et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michailidou K et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GEMO Study Collaborators et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet 52, 56–73 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mountjoy E et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat Genet 53, 1527–1533 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wray NR, Wijmenga C, Sullivan PF, Yang J & Visscher PM Common Disease Is More Complex Than Implied by the Core Gene Omnigenic Model. Cell 173, 1573–1580 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Li YI & Pritchard JK Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 177, 1022–1034.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weeks EM et al. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. medRxiv 2020.09.08.20190561 (2020) doi: 10.1101/2020.09.08.20190561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X & Goldstein DB Enhancer Domains Predict Gene Pathogenicity and Inform Gene Discovery in Complex Disease. The American Journal of Human Genetics 106, 215–233 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallagher MD & Chen-Plotkin AS The Post-GWAS Era: From Association to Function. Am J Hum Genet 102, 717–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Musunuru K et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466, 714–719 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gazal S et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gazal S, Marquez-Luna C, Finucane HK & Price AL Reconciling S-LDSC and LDAK functional enrichment estimates. Nat. Genet. 51, 1202–1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Connor LJ et al. Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. Am. J. Hum. Genet. 105, 456–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jagadeesh KA et al. Identifying disease-critical cell types and cellular processes across the human body by integration of single-cell profiles and human genetics. bioRxiv 2021.03.19.436212 (2021) doi: 10.1101/2021.03.19.436212. [DOI] [Google Scholar]
- 56.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freund MK et al. Phenotype-Specific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits. The American Journal of Human Genetics 103, 535–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Povysil G et al. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet 20, 747–759 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Van Hout CV et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yates AD et al. Ensembl 2020. Nucleic Acids Research 48, D682–D688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapoor A et al. An Enhancer Polymorphism at the Cardiomyocyte Intercalated Disc Protein NOS1AP Locus Is a Major Regulator of the QT Interval. The American Journal of Human Genetics 94, 854–869 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bauer DE et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 342, 253–257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van den Boogaard M et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest 124, 1844–1852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soldner F et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Glubb DM et al. Fine-Scale Mapping of the 5q11.2 Breast Cancer Locus Reveals at Least Three Independent Risk Variants Regulating MAP3K1. The American Journal of Human Genetics 96, 5–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gupta RM et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 170, 522–533.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X et al. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. eLife 5, e10557 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang Q et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat Genet 46, 126–135 (2014). [DOI] [PubMed] [Google Scholar]
- 71.The GAME-ON/ELLIPSE Consortium et al. CAUSEL: an epigenome- and genome-editing pipeline for establishing function of noncoding GWAS variants. Nat Med 21, 1357–1363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stadhouders R et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Invest. 124, 1699–1710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gallagher MD et al. A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. The American Journal of Human Genetics 101, 643–663 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guthridge JM et al. Two Functional Lupus-Associated BLK Promoter Variants Control Cell-Type- and Developmental-Stage-Specific Transcription. The American Journal of Human Genetics 94, 586–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vicente CT et al. Long-Range Modulation of PAG1 Expression by 8q21 Allergy Risk Variants. The American Journal of Human Genetics 97, 329–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fogarty MP, Cannon ME, Vadlamudi S, Gaulton KJ & Mohlke KL Identification of a Regulatory Variant That Binds FOXA1 and FOXA2 at the CDC123/CAMK1D Type 2 Diabetes GWAS Locus. PLOS Genetics 10, e1004633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ulirsch JC et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome research 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lindblad-Toh K et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leeuw C. A. de, Mooij JM, Heskes T & Posthuma D MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Computational Biology 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hounkpe BW, Chenou F, de Lima F & De Paula EV HRT Atlas v1.0 database: redefining human and mouse housekeeping genes and candidate reference transcripts by mining massive RNA-seq datasets. Nucleic Acids Research 49, D947–D955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The List of 19,995 genes, summary statistics of the 63 independent traits, training and validation critical gene sets, S2G and cS2G strategies, SNP annotations, predicted causal SNP-disease pairs from UK Biobank fine-mapping analyses and from the NHGRI-EBI GWAS catalog, and SNP-heritability causally explained by SNPs linked to each gene have been made publicly available at https://alkesgroup.broadinstitute.org/cS2G and https://doi.org/10.5281/zenodo.6354007. Links for all datasets used to create S2G strategies are provided in Supplementary Table 26.
Access to the UK Biobank resource is available via application at http://www.ukbiobank.ac.uk/.
GWAS catalog https://www.ebi.ac.uk/gwas/api/search/downloads/full
Open Targets SNP-gene pairs https://raw.githubusercontent.com/opentargets/genetics-gold-standards/master/gold_standards/processed/gwas_gold_standards.191108.tsv
SNP-gene pairs from ref.48 https://www.dropbox.com/s/kz2c49rpm2yanf5/all_byCS_rev1.txt?dl=0