

Abstract
BACKGROUND:
The sinoatrial node (SAN) functions as the pacemaker of the heart, initiating rhythmic heartbeats. Despite its importance, the SAN is one of the most poorly understood cardiac entities because of its small size and complex composition and function. The Hippo signaling pathway is a molecular signaling pathway fundamental to heart development and regeneration. Although abnormalities of the Hippo pathway are associated with cardiac arrhythmias in human patients, this pathway’s role in the SAN is unknown.
METHODS:
We investigated key regulators of the Hippo pathway in SAN pacemaker cells by conditionally inactivating the Hippo signaling kinases Lats1 and Lats2 using the tamoxifen-inducible, cardiac conduction system (CCS)-specific Cre driver Hcn4CreERT2 with Lats1 and Lats2 conditional knockout (CKO) alleles. In addition, the Hippo signaling effectors Yap and Taz were conditionally inactivated in the SAN. To determine the function of Hippo signaling in the SAN and other CCS components, we conducted a series of physiologic and molecular experiments including telemetry electrocardiogram recording, echocardiography, Masson’s Trichrome staining, calcium imaging, immunostaining, RNAscope, Cleavage Under Targets and Tagmentation (CUT&Tag) sequencing using antibodies against Yap1 or H3K4me3, quantitative real-time PCR, and Western blotting. We also performed comprehensive bioinformatics analyses of various datasets.
RESULTS:
We found that Lats1/2 inactivation caused severe sinus node dysfunction. Compared with the controls, Lats1/2 CKO mutants exhibited dysregulated calcium handling and increased fibrosis in the SAN, indicating that Lats1/2 function through both cell-autonomous and non–cell-autonomous mechanisms. It is notable that the Lats1/2 conditional knockout phenotype was rescued by genetic deletion of Yap and Taz in the cardiac conduction system. These rescued mice had normal sinus rhythm and reduced fibrosis of the SAN, indicating that Lats1/2 function through Yap and Taz. Cleavage Under Targets and Tagmentation sequencing data showed that Yap potentially regulates genes critical for calcium homeostasis such as Ryr2 and genes encoding paracrine factors important in intercellular communication and fibrosis induction such as Tgfb1 and Tgfb3. Consistent with this, Lats1/2 CKO mutants had decreased Ryr2 expression and increased Tgfb1 and Tgfb3 expression compared with control mice.
CONCLUSIONS:
We reveal, for the first time to our knowledge, that the canonical Hippo-Yap pathway plays a pivotal role in maintaining SAN homeostasis.
Keywords: Hippo signaling, sinus node dysfunction, fibrosis, TGF-β, calcium homeostasis
INTRODUCTION
The cardiac conduction system (CCS) initiates and propagates electrical depolarization, which coordinates the synchronized contraction of the cardiac muscle. Disturbances in the CCS give rise to cardiac arrhythmias, a major cause of morbidity and mortality worldwide.1–3 The CCS consists of the sinoatrial node (SAN, or sinus node), atrioventricular node (AVN), bundle of His, bundle branches, and Purkinje fibers.4, 5 The SAN is the primary pacemaker structure of the heart, and is a heterogeneous tissue characterized by clusters of pacemaker cells (PCs) encompassed within fibrotic components including collagens, elastin, and cardiac fibroblasts (CFs).6 PCs are characterized as highly specialized cardiomyocytes, which can generate electrical impulses to spontaneously and directly control the heart rate.7, 8 SAN intranodal fibrotic tissue provides important structural and functional integrity for PCs, along with electrically insulating PCs from the surrounding atrial myocardium to efficiently regulate normal sinus rhythm.9, 10 However, increased SAN fibrosis under pathologic conditions may cause cardiac arrhythmias such as tachycardia-bradycardia arrhythmias, cardiac arrest, atrial fibrillation, and ventricular arrhythmias. The function of the SAN is highly sensitive to changes in ionic currents, such as those regulated by intracellular calcium signaling11 and energy metabolism12 in the adult heart. In addition, the risk for sinus node dysfunction (SND, also known as sick sinus syndrome) increases with other factors including aging, hypertension, and concomitant conduction disease.13 The incidence of SND is predicted to double over the next 50 years.14 Therefore, understanding the molecular machinery controlling SAN homeostasis in adulthood has considerable clinical significance.
Hippo signaling is a highly conserved kinase cascade that plays critical roles in cardiac development, regeneration, and homeostasis. When Hippo signaling is active, the downstream Hippo effectors Yap/Taz are phosphorylated by the Hippo core kinases Lats1/2. Phosphorylated Yap/Taz are retained in the cytoplasm and are eventually degraded. When Hippo signaling is off, Yap/Taz act as transcriptional cofactors that can translocate into the nucleus and interact with other transcription factors such as TEA domain transcription factor family members (TEADs) to regulate gene expression.15, 16 Hippo signaling plays critical roles during both embryonic and postnatal stages, including the regulation of cardiomyocyte proliferation and renewal.17–23 According to the DECIPHER (DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources) database, patients with sequence variants in Hippo pathway components had cardiac defects and arrhythmias. Altered Hippo pathway activity has been found in patients with arrhythmogenic and hypertrophic cardiomyopathy heart failure.20, 24, 25 Compared with normal human heart samples, myocardial samples from patients with arrhythmogenic cardiomyopathy had greater Hippo signaling activity and increased phosphorylated YAP level.24 Analysis of RNA-seq data indicated that expression of Mst1, a core kinase of Hippo signaling and an upstream regulator of Lats1/2, was significantly less in the SAN of failing human hearts than in SAN of donor hearts without heart failure or a history of arrhythmia.26 Recent bioinformatics analysis of gene expression profiles predicted that Hippo signaling was the most significantly enriched pathway in the left atrium of atrial fibrillation patients.27 These studies suggest the potential role of Hippo signaling in the CCS. Hippo signaling has been studied in different cardiac contexts, yet its function in the CCS has not been investigated.
To uncover the function of Hippo signaling in the SAN, we conditionally deleted Lats1 and Lats2 in the CCS in the Hcn4CreERT2; Lats1/2flox/flox (Lats1/2 CKO) mouse model. Hcn4CreERT2 is a tamoxifen-inducible CCS-specific Cre,28 which is specifically expressed in the developing and adult SAN and in the other components of the conduction system from fetal stages onward. We found that Lats1/2 CKO mice developed cardiac conduction disorders such as sinus pauses, atrioventricular (AV) blocks, irregular RR intervals, and abnormal P-waves. Furthermore, Lats1/2 deletion disrupted the intrinsic calcium homeostasis of PCs, making their spontaneous firing rate irregular. We also found that the absence of Lats1/2 increased SAN fibrosis by non–cell-autonomous fibroblast proliferation; however, PCs in both control and Lats1/2 CKO mice were not proliferative. We further determined that Lats1/2 function through Yap and Taz and performed CUT&Tag sequencing to uncover Yap target genes in the SAN. We found that Hippo-Yap signaling regulates the key calcium homeostasis genes such as sarcoplasmic reticulum (SR) calcium channel Ryr2 (encoding ryanodine receptor type 2, RyR2) and important genes for cell communication and fibrosis induction such as Tgfb1 and Tgfb3. For the first time to our knowledge, we show that Hippo-Yap signaling is required for maintaining SAN homeostasis through both cell-autonomous and non–cell-autonomous mechanisms.
METHODS
All animal studies and procedures were conducted in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and were approved by The University of Texas Health Science Center at Houston Institutional Animal Care and Use Committee. Detailed methods and supporting data are available in the Supplementary Material. Study materials are available from the corresponding author upon reasonable request.
Mice
Hcn4CreERT2,28 Lats1flox/flox,17 Lats2flox/flox,17 Yap flox/flox,19 and Taz flox/flox,19 mouse lines were previously described. Six- to eight-week-old mice were intraperitoneally injected with 150 mg tamoxifen (10 mg/ml) per day for 2 days to induce Hcn4CreERT2 activity.
Telemetry Electrocardiography
Telemetry electrocardiograms (ECGs) were recorded over 24 hours in conscious and ambulatory mice using telemetry transmitters ETA-F10 (Data Sciences International) as previously described.29
Echocardiography
Echocardiography was performed on anesthetized mice as previously described30 by using a VisualSonics Vevo 2100 system with a 550-s probe.
Confocal Ca2+ imaging of PCs
SAN tissues were isolated as previously described.12 Confocal Ca2+ imaging studies were performed in isolated PCs from SAN tissues as previously described.31, 32
CUT&Tag and sequencing data analysis
CUT&Tag assays in nuclei from SAN tissues were performed as previously described33 by separately using anti-H3K4me3 and anti-Yap1 antibodies. The CUT&Tag sequencing (GSE202641) raw reads were aligned by using Bowtie234 and were further processed with Picard and Samtools.35 Cut&Tag signals in the genome were visualized by using the bamCoverage module of deepTools.36 SEACR37 was used to call peaks, and peaks were annotated with Homer.38 Metascape was used for gene ontology analysis.39 For conserved sequence analysis, the phastCons60way score40 file of mm10 was downloaded from the UCSC Genome Browser.
ATAC-seq data processing and analysis
Raw ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) reads of PCd and right atrial cardiomyocytes ATAC-seq data (GSE14851541) were mapped to the mm10 build of the mouse genome using BWA with default settings. For SAN-like pacemaker cells and ventricle-like cardiomyocytes ATAC-seq data (GSE14604442), raw ATAC-seq reads were mapped to the hg19 build of the mouse genome using BWA with default settings.
Statistical Analysis
Quantitative data are presented as the mean±SEM. All statistical tests, error bars, P-values, and n numbers of sample sizes and replicates are reported in the corresponding figure legends.
RESULTS
Lats1 and Lats2 deletion in the CCS results in cardiac conduction disorders
To determine whether Hippo signaling functionally affects the CCS, we generated inducible CCS-specific knockout models of the Hippo signaling kinases Lats1 and Lats2 by intercrossing the floxed Lats1/2 (Lats1 flox/flox; Lats2 flox/flox) mice with the inducible CCS-specific Cre Hcn4CreERT2 mice.28 Mice were treated with tamoxifen at 6–8 weeks of age (Figure 1A). Both Hcn4CreERT2 and Lats1/2flox/flox mice were used as controls. Yap is an effector of Hippo signaling and is phosphorylated by Lats1 and Lats2 when Hippo signaling is active.43–45 To detect Hippo signaling activity in the SAN, we co-stained phosphorylated (inactive) Yap-Ser127 (pYap) and the PC marker Hcn4 in sections of SAN tissue from both control and Lats1/2 CKO mice.46–48 Compared with control SANs (Figure 1B), SANs of Lats1/2 CKO mice had decreased pYap levels in Hcn4-positive PCs (Figure 1C). Consistent with this, active Yap (aYap) protein level was greater in the PC nuclei of Lats1/2 CKO mice (Figure 1E) than in those of controls (Figure 1D). These data indicated efficient CCS-specific inactivation of Lats1/2 in Lats1/2 CKO mice.
Figure 1. Inducible cardiac conduction system (CCS)-specific Lats1/2 deletion induces cardiac arrhythmias.
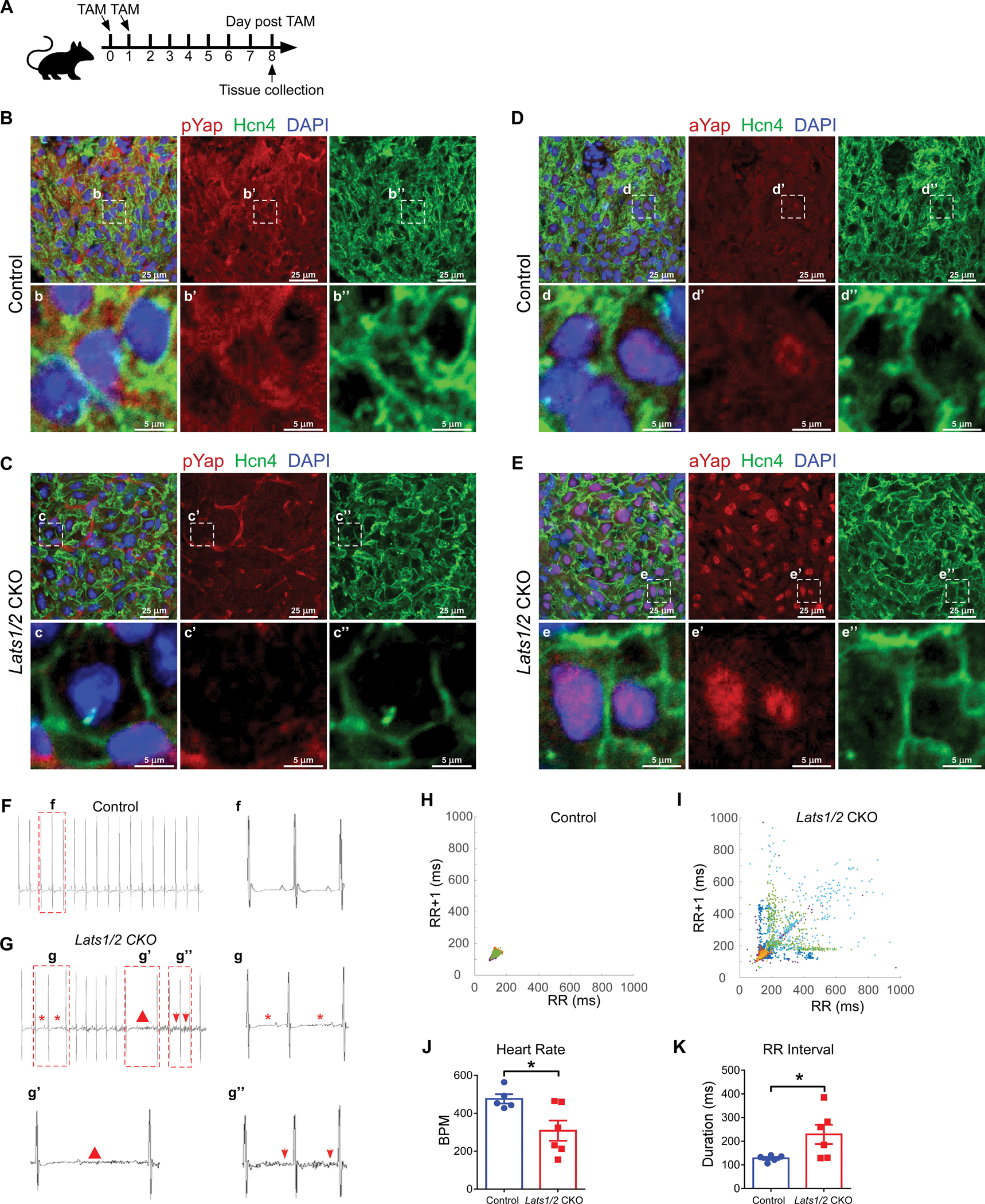
A. Experimental strategy for CCS-specific Cre activation and tissue collection. Cre activity was induced with 2 consecutive intraperitoneal injections of tamoxifen. B-C. Representative images of immunofluorescence staining of pYap in the SAN after tamoxifen induction in control (B) and Lats1/2 CKO (C) mice. Hcn4 (green) was used to label pacemaker cells (PCs) in the SAN. pYap (red) is an indicator of Hippo signaling activity. Nuclei were labeled with DAPI (blue). Lower panels show higher-magnification views of the boxed areas in the upper panels. Scale bar, 25 μm and 5 μm. D-E. Representative images of immunofluorescence staining of aYap in the SAN after tamoxifen induction in control (D) and Lats1/2 CKO (E) mice. aYap was stained with red; Hcn4 (green) was used to label PCs in SAN. Nuclei were labeled with DAPI (blue). Lower panels show higher-magnification views of the boxed areas in the upper panels Scale bar, 25 μm and 5 μm. F-G. Representative electrocardiogram (ECG) recordings in control and Lats1/2 CKO mice. Star, irregular RR intervals (g); triangle, AV blocks (g’); arrows, abnormal P waves (g”). H-I. Representative Poincaré plots showing beat-to-beat RR interval variability from control (n=5) and Lats1/2 CKO (n=6) mice. Each color represents individual RR/RR+1 intervals from a different mouse. J-K. Heart rates and RR intervals of control and Lats1/2 CKO mice. Control, n=5; Lats1/2 CKO, n=6. Data are shown as means ± s.e.m. Statistical significance was determined by t-test, *p<0.05.
To further elucidate the functional role of Hippo signaling in the CCS, we obtained telemetry ECG recordings in both control and Lats1/2 CKO mice with implanted telemetry transmitters for over 24 hours. Before tamoxifen treatment, we recorded ECGs from controls (including Hcn4CreERT2 and Lats1/2flox/flox mice) and Lats1/2 CKO mice and found no obvious ECG differences between these mice. At 8 days after tamoxifen injection, the most frequently observed phenotypes in Lats1/2 CKO mice were cardiac conduction disorders including AV block and SND, which manifested as lower heart rates and longer RR intervals than in control mice (Figure 1F–1G). Poincaré plot analysis of the RR intervals demonstrated tremendous beat-to-beat variation in Lats1/2 CKO mice (Figure 1H–1I). The heart rate was lower (Figure 1J), and the average RR interval was significantly longer in Lats1/2 CKO mice (Figure 1K). In contrast, average P duration, PR interval, QRS complex duration, and QTc duration were not significantly different between control and Lats1/2 CKO mice (Figure S1J–S1M), suggesting that that atrial and ventricular conduction and the other components of the CCS were less affected by Lats1/2 deficiency. Together, these findings indicate that the CCS-specific deletion of Lats1/2 disrupts the function of the SAN.
Lats1 and Lats2 deficiency in the CCS does not cause altered heart contractile function
Hippo signaling is known to play an important role in regulating cardiomyocyte proliferation and heart size in mouse embryos, as well as cardiomyocyte size in adult mice.21, 49–51 Therefore, we further determined whether Lats1/2 deletion in the CCS alters heart size, SAN PC size, and general cardiac function. No obvious difference was observed in the size of Lats1/2 CKO hearts and control hearts (Figure S1A–S1B). Co-staining of wheat germ agglutinin (WGA, plasma membrane marker) and Hcn4 (Figure S1C–S1D) indicated no significant difference in PC size between Lats1/2 CKO and control mice (Figure S1E). Likewise, echocardiography data showed no obvious between-group differences in cardiac contractile function and dimensions (Figure S1F–S1I). Further analysis of echocardiographic measurements indicated no significant differences between control and Lats1/2 CKO in parameters such as left ventricle internal diameter, left ventricle posterior wall thickness, left ventricle volume, stroke volume, cardiac output, ejection fraction, and fractional shortening (Table S1). Together, these results indicated that the CCS-specific deletion of Lats1/2 did not affect overall heart size, pacemaker cell size, or cardiac function, suggesting that phenotypes caused by Lats1/2 deletion were intrinsic to the CCS rather than secondary effects.
Loss of Lats1/2 in PCs promotes fibrosis in the SAN
To investigate whether SND in Lats1/2 CKO mice was associated with structural abnormalities of the SAN region, we evaluated collagen deposition and fibrosis in the SAN region. Masson’s Trichrome staining revealed that compared with control SANs (Figure 2A), Lats1/2 CKO SANs (Figure 2B) exhibited an increase in blue-stained collagen content, although no obvious differences were observed between control and Lats1/2 CKO mice in other cardiac components such as the right atria, cardiac valves, and ventricular septal regions (Figure S2A–S2B). We further performed immunohistochemistry (IHC) experiments to label the extracellular matrix protein collagen 1 (Col1a1) in tissue sections of control and Lats1/2 CKO mice and used the co-staining of Hcn4 to indicate the SAN region. Col1a1 levels were significantly greater in SANs of Lats1/2 CKO samples than in those of controls (Figure 2C–2E), indicating that Lats1/2 deficiency increased collagen deposition. Expression level of the fibroblast marker vimentin (Vim) was also higher in the SAN region of Lats1/2 CKO hearts than in that of control hearts (Figure 2F–2G). These data revealed fibrotic remodeling and increased fibrosis in the SAN caused by the deletion of Lats1/2 in PCs.
Figure 2. Lats1/2 deletion in CCS results in SAN fibrosis and fibroblast proliferation.
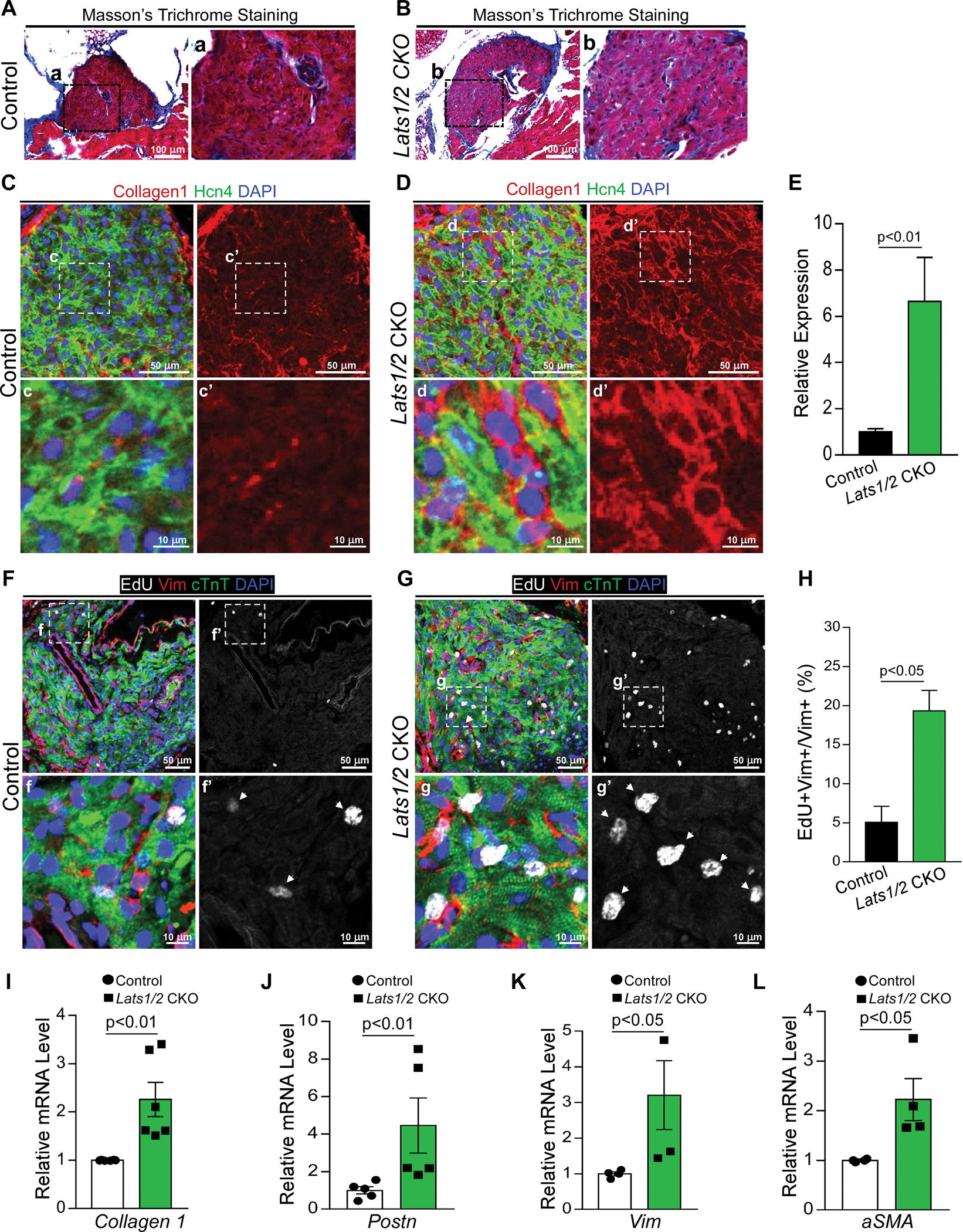
A-B. Masson’s Trichrome staining in control and Lats1/2 CKO SANs. Panels on the right show higher magnification of the boxed area from the panels on the left. Scale bar, 100 μm. C-E. Representative immunofluorescence confocal images from control (C) and Lats1/2 CKO (D) SANs after Hcn4-Cre activation showing collagens (Col1a1, red), PCs (Hcn4, green), and nuclei labeling (DAPI, blue). Panels on the right show higher magnification of the boxed area in panels on the left. Collagen 1 quantification (E), measured by pixel intensity. Data in (E) represent means ± s.e.m; statistical significance was determined by Mann–Whitney test. Scale bar, 25 μm and 5 μm. F-H. Representative images of EdU-labeled control (F) and Lats1/2 CKO (G) SANs. Samples were pulse-chased with EdU (white); fibroblasts and PCs were labeled with vimentin (red) and cTnT (green) respectively. Nuclei were stained with DAPI (blue). Arrows point to EdU-positive cells. Panels on the right show higher magnification of the boxed area in panels on the left. (H) Quantification of fibroblast proliferation. Representative image of proliferating fibroblast cells shown in F and G. Statistical significance was determined using the Mann–Whitney test (P<0.05). Scale bar, 25 μm and 5 μm. I-L. Quantitative real-time polymerase chain reaction validation of Collagen 1 (I), Periostin (Postn) (J), Vimentin (Vim) (K), and Acta2 (α-SMA) (L) in control and Lats1/2 CKO SANs. Each dot represents an independent biological replicate (n≥4). Data are shown as means ± s.e.m. Statistical significance was determined by Mann–Whitney test.
Loss of Lats1/2 promotes fibroblast but not pacemaker cell proliferation in the SAN
Hippo signaling has a well-known and critical role in cell proliferation. Therefore, to determine the impact on proliferation caused by loss of Lats1/2, we injected control and Lats1/2 CKO mice with 5-ethynyl-2’-deoxyuridine (EdU) for four consecutive days before the heart was harvested at 8 days after tamoxifen injection. EdU signal was barely detectable in cardiac troponin-T (cTnT)-positive PCs of both control and Lats1/2 CKO hearts, indicating that PCs were not proliferative after Lats1/2 deletion (Figure 2F–2H). However, EdU labeling indicated that vimentin-positive fibroblast cells were significantly more proliferative in the SAN region of Lats1/2 CKO hearts than in control hearts (Figure 2F–2H). To examine expression of fibrosis-regulated genes, we isolated RNA from dissected SAN tissues of control and Lats1/2 CKO mouse hearts. qRT-PCR analysis revealed that mRNA expression of the fibrosis-associated genes Col1a1, Vimentin, Acta2 (encoding alpha-smooth muscle actin, aSMA) and Periostin (Postn) were upregulated in Lats1/2 CKO SANs compared to control SANs (Figure 2I–2L). These data suggested that Lats1/2 deletion in the CCS increased SAN fibrosis and non–cell-autonomous fibroblast proliferation.
Lats1/2 deficiency disrupts calcium homeostasis in SAN PCs
Spontaneous beating and rhythmic electrical impulses of the SAN are regulated by the If-dependent voltage membrane clock and the intracellular Ca2+ clock, which is controlled by the localized subsarcolemmal Ca2+ release via RyR2 on the SR.52, 53 Therefore to study potential mechanisms responsible for SND at the cellular level, we evaluated cellular Ca2+ activity by performing confocal Ca2+ imaging in isolated PCs of control (Figure 3A) and Lats1/2 CKO mice (Figure 3B). We found that PCs isolated from Lats1/2 CKO mice exhibited greater variability in the spontaneous Ca2+ transient rates and presented with an overall reduced frequency of spontaneous firing (Figure 3C–3F). No obvious difference was observed in the amplitude of the caffeine-induced Ca2+ transients between control and Lats1/2 CKO PCs (Figure 3C–3D), indicating that the control and Lats1/2 CKO PCs have similar SR Ca2+ content. However, compared with controls, PCs isolated from Lats1/2 CKO hearts had weaker activity and decreased response to caffeine challenge (Figure 3C–3D). Quantification of spontaneous Ca2+ transient rates (Figure 3E) and distribution of spontaneous Ca2+ transient rates (Figure 3F) in isolated PCs of control and Lats1/2 CKO mice further indicated the irregular and overall reduced frequency of spontaneous firing. These data revealed that Lats1/2 deficiency disrupted PC Ca2+ activity, suggesting a critical role of Lats1/2 in maintaining PC Ca2+ homeostasis.
Figure 3. Lats1/2 deficiency disrupts the calcium homeostasis of pacemaker cells.
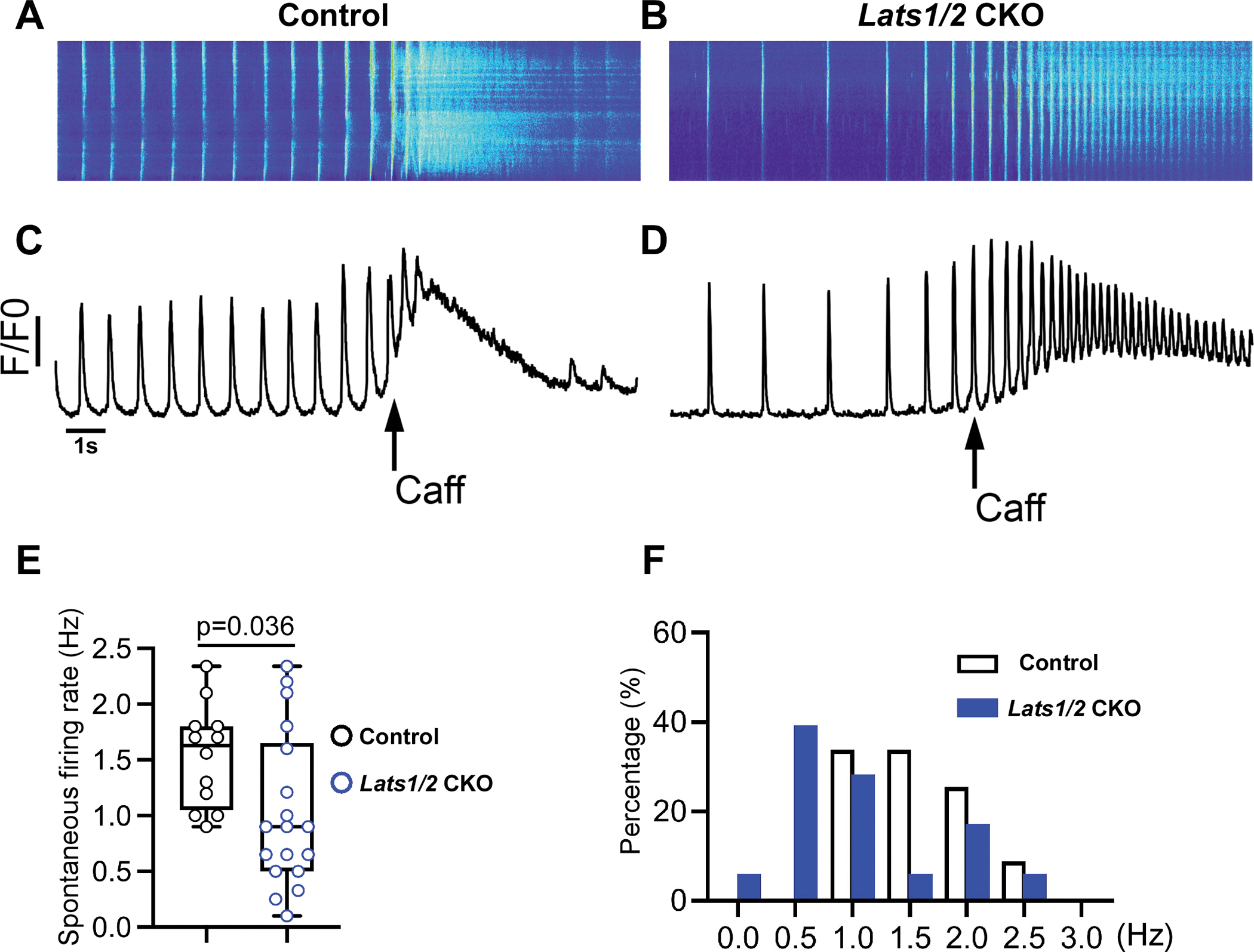
A-D. Ca2+ transient recordings obtained from control (A, C) and Lats1/2 CKO (B, D) PCs in the presence of caffeine (Caff). A-B. Representative confocal line-scan images. C-D Spatial average of fluorescence signal normalized to baseline (F/F0) over the entire field of observation of isolated PCs from control and Lats1/2 CKO. PCs from Lats1/2 mutant showed reduced and irregular frequency of spontaneous firing rate compared to controls. During caffeine challenge, Lats1/2 CKO PCs also had a weaker response and reduced activity of the caffeine-induced Ca2+ signal compared to controls. Caff, caffeine. E-F. Summary data for spontaneous firing rate. (E) Quantification of spontaneous Ca2+ transient rates in SAN cells of control and Lats1/2 CKO mice (n=12 cells/3 mice in control, n=18 cells/6 mice in Lats1/2 CKO, P=0.036 by Student t-test). (F) Distribution of spontaneous Ca2+ transient rates in isolated PCs from control and Lats1/2 CKO.
Lats1/2 maintains SAN homeostasis through the canonical Hippo-Yap/Taz pathway
To determine whether Yap/Taz, the downstream effectors of Lats1/2, were critical for SAN homeostasis, we specifically deleted Yap/Taz in the CCS of Lats1/2 CKO mice by generating Hcn4CreERT2; Lats1/2f/f; Yap/Tazf/f mice. After tamoxifen injection, we confirmed ablation of Yap/Taz in Lats1/2 CKO mice by the reduction in pYap immunofluorescence (IF) staining (Figure 4A–4C). Just as Lats1/2 deficiency caused no obvious cardiac structure and function changes in the CCS, Yap and Taz deletion in the CCS of Lats1/2 CKO did not alter cardiac structure, contractile function and SAN PC size (Figure S3). Notably, ablation of Yap/Taz in Lats1/2 CKO mice rescued cardiac arrhythmias observed in Lats1/2 CKO mice to an extent comparable to that of control mice (Figure 4D–4K). These results suggested that restriction of Yap/Taz activity by Lats1/2 kinases-mediated phosphorylation was essential for SAN homeostasis. Masson’s Trichrome staining showed that SAN fibrosis was suppressed in Hcn4CreERT2; Lats1/2f/f; Yap/Tazf/f mice compared to Lats1/2 CKO mice (Figure 5A–5C). We also observed reduced levels of Col1a1 and vimentin immunostaining in SANs of Hcn4CreERT2; Lats1/2f/f; Yap/Tazf/f mice compared with Lats1/2 CKO mice (Figure 5D–5I).
Figure 4. Yap/Taz deletion rescues arrhythmic phenotype in Lats1/2 mutants.
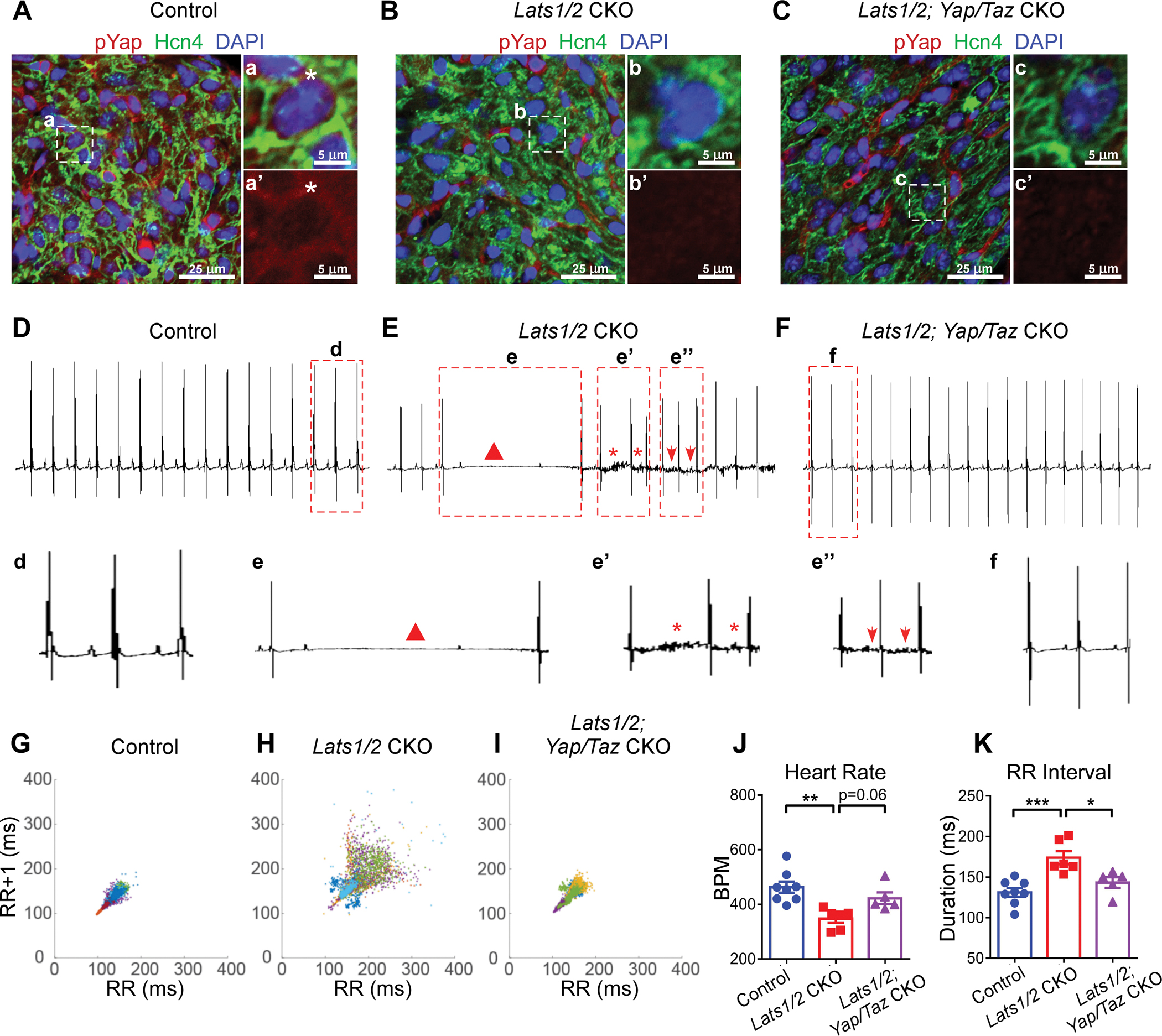
A-C. Representative images of immunofluorescent staining of pYap in SAN after tamoxifen induction in control (A), Lats1/2 CKO (B), and Lats1/2; Yap/Taz CKO (C) mice. Hcn4 (green) was used to label PCs in SAN. pYap (red) is an indicator of Hippo signaling activity. Nuclei were labeled with DAPI (blue). Panels on the right show higher magnification of the boxed area in the panels on the left. Stars, pYap staining. Scale bar, 25 μm and 5 μm. D-F. Representative ECG recordings in control (D), Lats1/2 CKO (E), and Lats1/2; Yap/Taz CKO (F) mice. Triangle, AV blocks (e); star, irregular RR intervals (e’); arrows, abnormal P waves (e”). G-I. Representative Poincaré plots, showing beat-to-beat RR interval variability, from control (n=8), Lats1/2 CKO (n=7) and Lats1/2; Yap/Taz CKO (n=5). Each color represents individual RR/RR+1 intervals from a different mouse. J-K. Heart rate and RR interval of control, Lats1/2 CKO, and Lats1/2; Yap/Taz CKO mice. Data are shown as means ± s.e.m. Statistical significance was determined by one-way ANOVA analysis. *p<0.05, **p<0.01, ***p<0.001.
Figure 5. Yap/Taz deletion in Lats1/2 CKO rescues SAN fibrosis and fibroblast proliferation.
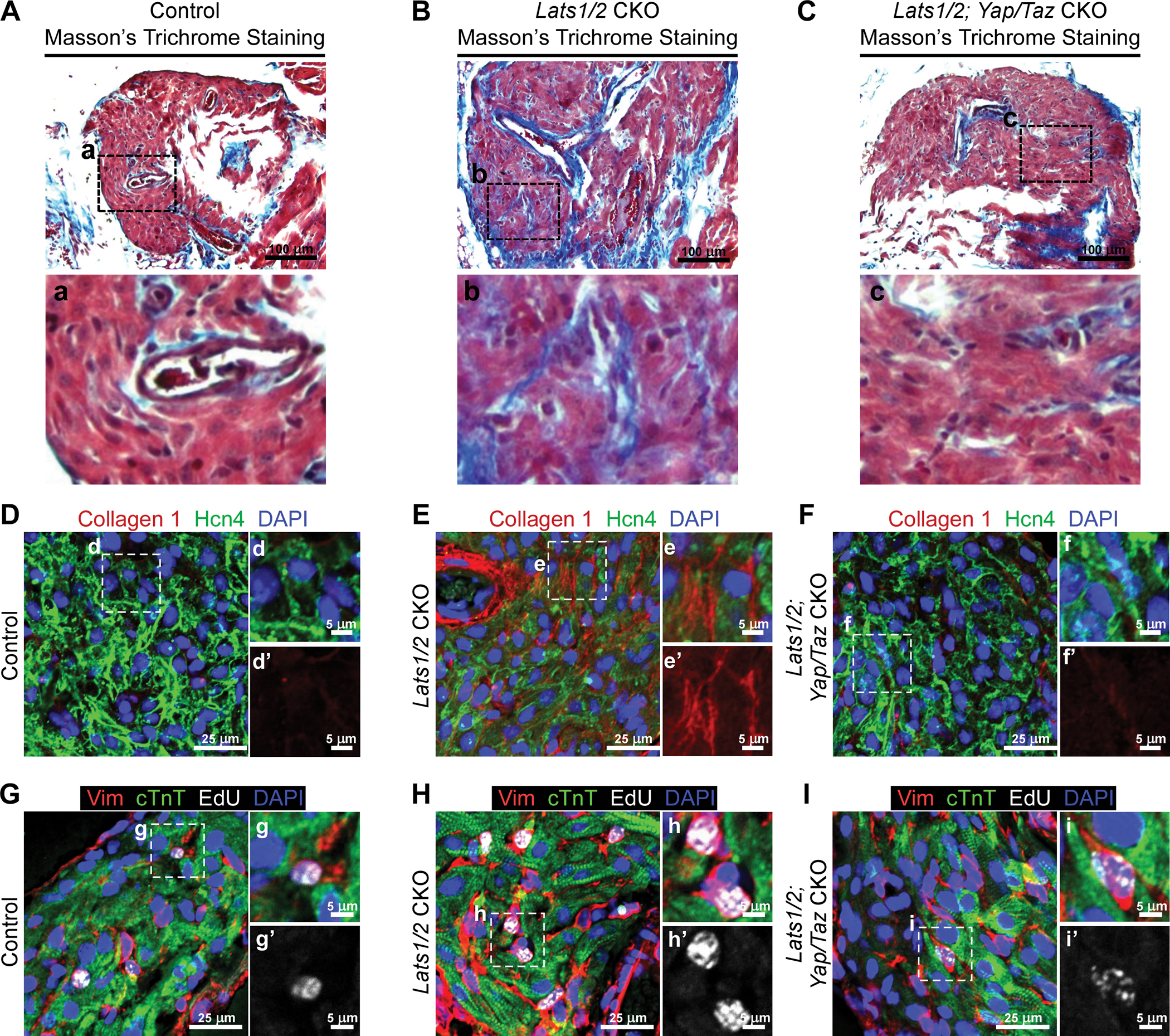
A-C. Masson’s Trichrome staining in control (A), Lats1/2 CKO (B), and Lats1/2; Yap/Taz CKO (C) SANs. Lower panels show higher magnification of the boxed areas from the upper panels. Scale bar, 100 μm. D-F. Representative immunofluorescence confocal images of Collagen 1 (red) from control (D), Lats1/2 CKO (E), and Lats1/2; Yap/Taz CKO (F) SANs. Hcn4 are stained with green, and nuclei are stained with blue. The right-hand panels show higher magnification of the boxed areas in the left-hand panels. Scale bar, 25 μm and 5 μm. G-I. Representative images of EdU-labeled control (G), Lats1/2 CKO (H), and Lats1/2; Yap/Taz CKO (I) SANs. Samples were pulse-chased with EdU (white); fibroblasts and PCs were labeled with vimentin (red) and cTnT (green), respectively. Nuclei were stained with DAPI (blue). Right-hand panels show higher magnification of boxed areas in left-hand panels. Scale bar, 25 μm and 5 μm.
To test whether the ablation of Yap/Taz inhibited proliferation of fibroblasts in the SAN, we injected EdU in control, Lats1/2 CKO, and Hcn4CreERT2; Lats1/2f/f; Yap/Tazf/f mice. In SANs of Hcn4CreERT2; Lats1/2f/f; Yap/Tazf/f mice, vimentin-positive fibroblasts showed less EdU incorporation than those in SANs of Lats1/2 CKOs, while Yap/Taz deletion rescued EdU-positive fibroblasts in SANs to a level comparable with that in the control mice (Figure 5G–5I). Taken together, these data indicated that Lats1/2 in the CCS function through the canonical Hippo signaling pathway mediated by Yap/Taz.
Yap regulates genes essential for intercellular communication, cell-cell adhesion, and calcium homeostasis
To decipher molecular mechanisms underlying cardiac arrhythmias and to identify the downstream targets of the Hippo-Yap pathway involved in SAN homeostasis, we performed CUT&Tag sequencing33 in SANs of control and Lats1/2 CKO mice using antibodies against Yap1, H3K4me3 (as positive control), and IgG (as negative control) (Figure 6A). The heatmaps showed significantly enriched Yap binding sites in Lats1/2 CKOs when compared to controls (Figure 6A). H3K4me3 marks, indicative of active gene expression,54 were significantly greater in Yap binding sites from Lats1/2 CKO than in those from control (Figure S4A), suggesting that Yap in Lats1/2 CKO SANs interacts with promoters/enhancers that are more transcriptionally active. We further examined genomic occupancy by Yap, and found that in control SANs, Yap binding peaks were preferentially located in intergenic and intronic regions, whereas a small fraction of Yap binding peaks were located in promoter, untranslated region (UTR), and exonic regions (Figure 6B). Yap binding peaks in Lats1/2 CKOs were still preferentially located in intergenic and intronic regions, but a higher percentage of peaks were detected in promoter and exonic regions of Lats1/2 CKOs than in those of controls (Figure 6B). When we overlapped and compared Yap binding peaks in controls and Lats1/2 CKOs, we found that Yap binding peaks that exhibited significant differences still had the majority of their genomic occupancy in intergenic and intronic regions, but further increased their genomic occupancy in promoter, exonic, and UTR regions (Figure 6B). Motif analysis of YAP CUT&Tag peaks showed 35 known motifs that were specifically enriched in Lats1/2 CKO (Figure 6C). Notably, TEAD4 (TEA Domain Transcription Factor 4) binding motif is shown among them in Lats1/2 CKO (Figure 6C). TEAD4 is a transcription factor which contains a C-terminal YAP-binding domain55 to interact with Yap and form a transcriptional complex for the regulation of gene expression. We further performed gene ontology (GO) term analysis of genes associated with Yap binding peaks that were significantly changed in Lats1/2 CKOs compared with controls. The top GO terms revealed that Yap-bound genes were involved in cell-cell communication, cell-cell adhesion, calcium ion activity, cell proliferation, action potential and rhythmic process (Figure 6D).
Figure 6. Analysis of CUT&Tag sequencing data.
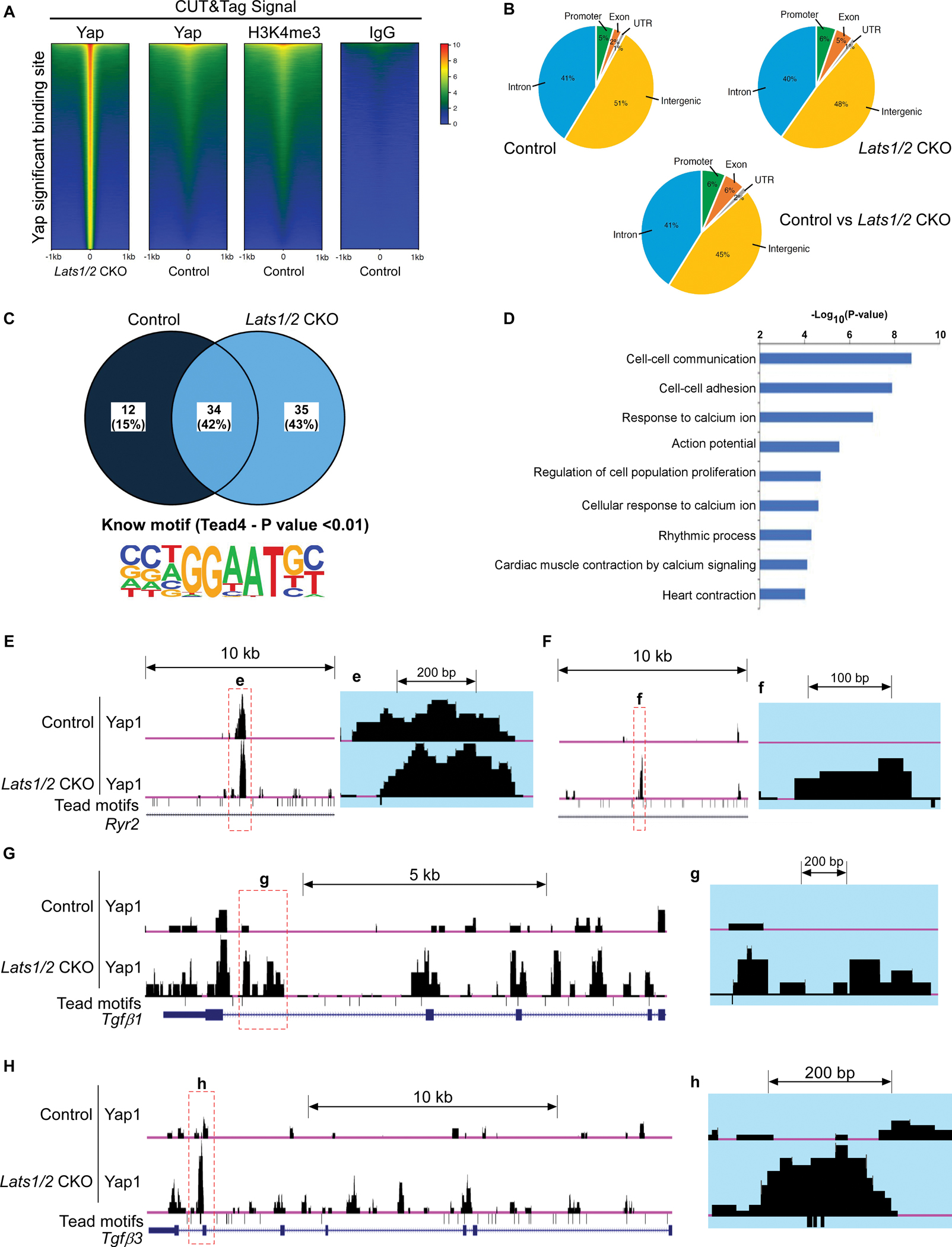
A. Heatmap showing DNA binding peaks determined by CUT&Tag sequencing with Yap, IgG, and H3K4me3 in control and Lats1/2 CKO SANs. Quantification of CUT&Tag enrichment signal by ±1 kb to the center of Yap-associated regions. B. Distribution of Yap-associated regions. C. Motif analysis. The upper panel shows the percentage of identified binding sites containing the consensus binding motif(s). HOMER software was used to identify known motifs underneath Yap1 CUT&Tag peaks. The Tead4 consensus motif (the lower panel) was highly enriched and was represented as a sequence logo position weight matrix. D. Gene ontology (GO) analysis of direct targets of Yap. E-H. UCSC genome browser view of Yap CUT&Tag enriched peaks for the labeled genes. Larger peaks were seen in Lats1/2 CKO. Tead (TEA domain) motifs were also aligned.
Yap regulates the expression of Ryr2 and TGF-β family members
Because of the molecular phenotypes observed in Lats1/2 CKO mice, including disruption of Ca2+ homeostasis in PCs and enhanced fibroblast proliferation in the SAN, we visualized enriched Yap binding peaks of genes reported to regulate these events. CUT&Tag seq data suggested that Yap potentially regulates many genes that function in cell-cell communication and response to calcium ion regulation. For instance, Yap binding peaks were identified in different genomic regions of the key calcium regulator Ryr2 (encoding RyR2, the major component of the SR Ca2+ channel) (Figure 6E–6F) and the paracrine fibrosis inducers Tgfb1 and Tgfb3 (encoding the profibrotic signaling factors TGF-beta ligands TGF-β1 and TGF-β3) (Figure 6G–6H), suggesting their potential regulation by Yap. Importantly, multiple alignment sequence analysis indicated that overall, the Yap binding peaks at Ryr2, Tgfb1, and Tgfb3 genes were evolutionarily conserved among vertebrate species including humans (Figure S4B–S4D).
We further evaluated whether expression of these genes is regulated by Yap. We performed fluorescence in situ hybridization by using RNAscope for Tgfb1 and found that Tgfb1 transcripts were significantly increased in the SAN in Lats1/2 CKOs compared with controls (Figure 7A–7C). Consistently, IF staining showed that the phosphorylated Smad3 (pSmad3, an indicator of TGF-β pathway activation) signal was increased in Lats1/2 CKO SANs compared with control SANs (Figure 7D–7E, S5A–S5B), and Yap/Taz deletion in Lats1/2 CKO SANs rescued pSmad3 to a level comparable with that in control SANs (Figure S5C). qRT-PCR analysis indicated that expression of Tgf-β1 (Figure 7F) and Tgf-β3 (Figure 7G) was significantly upregulated, whereas expression of Ryr2 was significantly downregulated (Figure 7K) in Lats1/2 CKO SANs compared with control SANs. Western blot (WB) analyses confirmed greater pSmad3 levels in Lats1/2 CKO SANs (Figure 7J), further confirming that Lats1/2 deletion enhanced TGF-β signaling in the SAN. Consistently, the results of WB and IHC showed less RyR2 in Lats1/2 CKO SANs (Figure 7H–7J). In summary, Yap in the SAN PCs likely functions as an activator of Tgf-β and a repressor of Ryr2.
Figure 7. Lats1/2 deficiency activated TGF-β signaling and inhibited Ryr2 expression in the SAN.
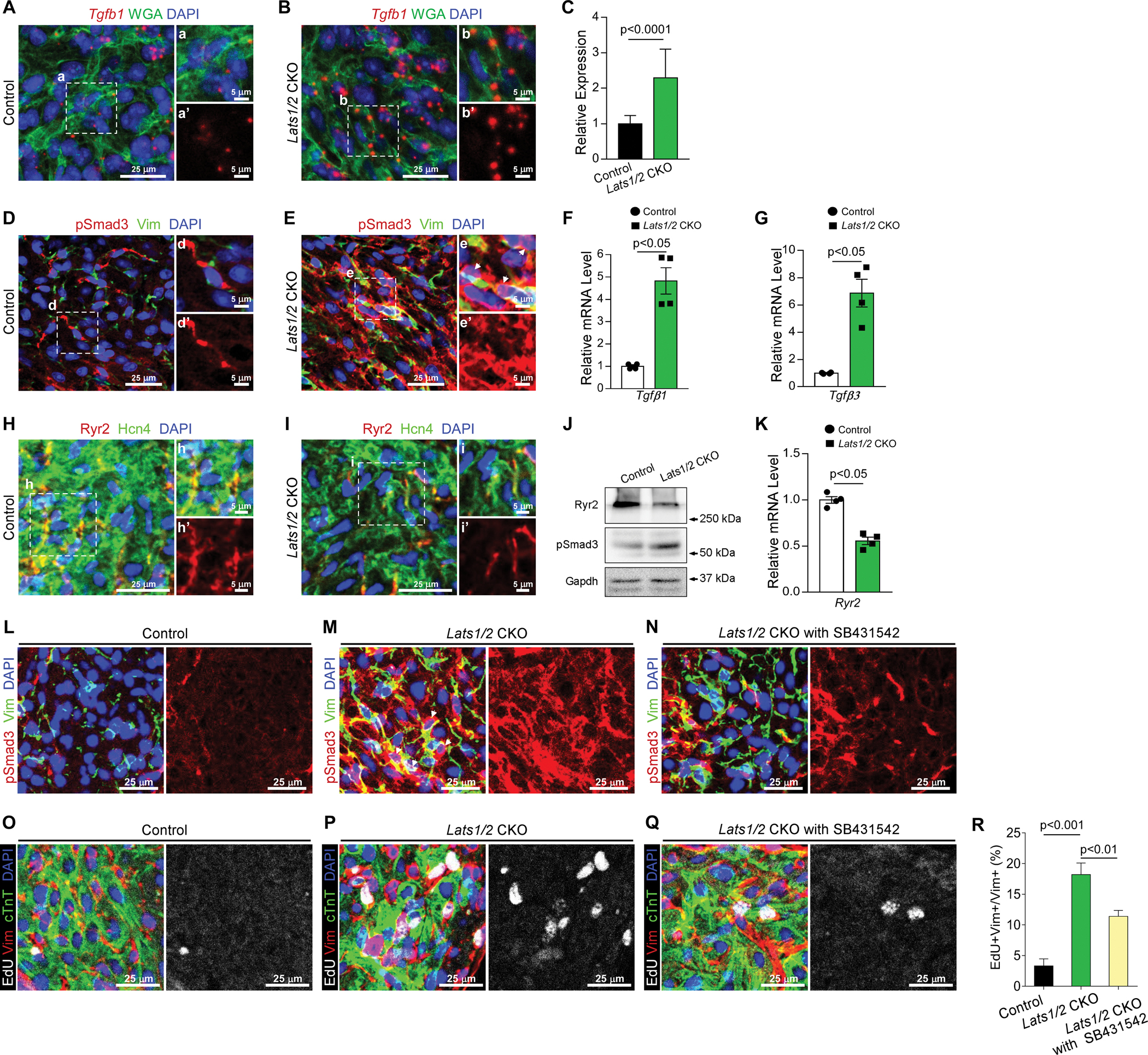
A-C. Representative RNAscope confocal images of Tgfb1 from control (A) and Lats1/2 CKO (B) SANs. Tgfb1 mRNA was stained red; the cell membrane was stained green (by using wheat germ agglutinin [WGA]), and nuclei were stained blue. Tgfb1 mRNA quantification (C), measured by pixel intensity. Data in (C) represent means ± s.e.m; statistical significance was determined using the Mann–Whitney test. Scale bar, 25 μm, 5 μm. D-E. Representative images of immunofluorescence staining of pSmad3 in SAN after tamoxifen induction in control (D) and Lats1/2 CKO (E) mice. Vimentin (green) was used to label fibroblasts in SAN. pSmad3 (red) is an indicator of TGF-β signaling activity. Nuclei were labeled with DAPI (blue). Right-hand panels show higher magnification of the boxed area in the left-hand panels. Arrows, pSmad3 staining in nuclei. Scale bar, 25 μm and 5 μm. F-G. Quantitative real-time PCR (qRT-PCR) data indicated that expression of Tgf-β1 (F) and Tgf-β3 (G) was greater in Lats1/2 CKO SAN than in control SAN. Each dot represents an independent biological replicate (n=4). Data are shown as means ± s.e.m; statistical significance was determined using the Mann–Whitney test. H-I. Representative images of immunofluorescence staining of Ryr2 in SAN after tamoxifen induction in control (H) and Lats1/2 CKO (I) mice. Ryr2 was stained with red; Hcn4 (green) was used to label PCs in SAN. Nuclei were labeled with DAPI (blue). Scale bar, 25 μm and 5 μm. J. Representative western blot images of Ryr2, pSmad3, and corresponding GAPDH loading control. K. qRT-PCR data indicated that expression of Ryr2 was decreased in Lats1/2 CKO compared with control SAN. Each dot represents an independent biological replicate (n=4). Data are shown as means ± s.e.m. Statistical significance was determined using the Mann–Whitney test, p< 0.05. L-N. Representative immunofluorescence confocal images of pSmad3 from SANs of control mice (L), Lats1/2 CKO mice (M), and Lats1/2 CKO mice treated with SB431542 (N). SB431542 is a selective inhibitor of TGF-β signaling. Vimentin (Vim) is stained in green, pSmad3 is stained in red, and nuclei are stained in blue. Arrows, pSmad3 staining in nuclei. Scale bar, 25 μm. O-R. Representative images of EdU-labeling of SANs of control mice (O), Lats1/2 CKO mice (P), and Lats1/2 CKO mice treated with SB431542 (Q). Samples were pulse-chased with EdU (white). Fibroblasts and PCs were labeled with vimentin (red) and cTnT (green), respectively. Nuclei were stained with DAPI (blue). (R) Quantification of fibroblast proliferation. Representative images of the experiment shown in Figure 7O–7Q. Statistical significance was determined by the Kruskal–Wallis test. Scale bar, 25 μm.
TGF-β inhibitor treatment partially rescues the fibroblast phenotype in Lats1/2 CKO SANs
Because the increased fibrosis and fibroblast proliferation phenotypes observed in Lats1/2 CKO mice may be mediated partially through increased TGF-β signaling, we further evaluated this possibility by treating Lats1/2 CKO mice with SB431542, a TGF-β1 receptor inhibitor. Lats1/2 CKO mice were injected with SB431542 and compared with DMSO-treated Lats1/2 CKO mice and control mice. Although Lats1/2 CKO mice treated with SB431542 (Figure 7N) still showed increased pSmad3 signals compared with control mice (Figure 7L), those pSmad3 signals were greatly reduced compared with Lats1/2 CKO mice treated with DMSO (Figure 7M), indicating an efficient inhibition of TGF-β signaling by SB431542. To test whether TGF-β inhibitor treatment hindered the proliferation of fibroblasts in the SAN, we injected EdU in control mice, Lats1/2 CKO mice treated with DMSO, and Lats1/2 CKO mice treated with SB431542. Significantly more vimentin+EdU+ proliferating fibroblasts were observed in SANs of Lats1/2 CKO mice (Figure 7P) than in SANs of control mice (Figure 7O), while treatment with TGF-β inhibitor rescued this fibroblast phenotype (Figure 7Q–7R). Together, these data further suggested that blocking TGF-β signaling in Lats1/2 CKO mice inhibits Lats1/2 deficiency induced fibroblast proliferation in the SAN.
Yap regulation varies in different cardiac contexts and at different ages
To gain more insight into Yap regulation, we also performed a comprehensive comparative analysis of our Yap CUT&Tag seq data from adult mouse SANs, with published ATAC-seq datasets including PCs and right atrial cardiomyocytes (RACM) from neonatal mouse hearts,41 as well as human embryonic stem cell–derived SAN-like pacemaker cells (SANLPCs) and ventricle-like cardiomyocytes (VLCM).42 We checked all Yap peaks identified from CUT&Tag sequencing and mapped them to ATAC-seq at a genome-wide level. We found that overall, Yap peaks frequently coincide with the ATAC-seq peaks in all ATAC-seq datasets, suggesting that Yap binding is associated with increased chromatin accessibility at a genome-wide level (Figure S6A–S6B). However, chromatin accessibility and Yap binding activity varied among different ATAC-seq datasets (Figure S6A–S6B). As shown in the alignment of adult mouse SAN Yap CUT&Tag data with neonatal mouse PC ATAC-seq datasets (Figure S7A–S7C), Yap interaction with chromatin varies at different ages. Although some overlap occurred between Yap binding sites and accessible chromatin regions (examples are shown in red-boxed regions) in Ryr2 (Figure S7A), Tgfb1(Figure S7B), and Tgfb3 (Figure S7C), variability and differential binding events also occurred (examples are shown in green-boxed regions in Figure S7A–S7C). We also converted human SANLPC and VLCM ATAC-seq datasets by using CrossMap (0.6.1) and aligned them with mouse Yap CUT&Tag data. For examples, Yap binding peaks at Ryr2 (Figure S8A), Tgfb1 (Figure S8B), and Tgfb3 (Figure S8C) regulatory elements varied between PCs and ventricular myocytes (green-boxed regions), with some overlapping peaks (red-boxed regions). Together, these findings suggested that Yap regulation is complicated and varies in different cardiac contexts and at different ages.
DISCUSSION
In this study, we discovered that the CCS-specific deletion of Lats1/2 causes SND and elucidated the mechanisms associated with SND due to Lats1/2 deficiency (Figure 8). Our results indicated that altered Ca2+ homeostasis in SAN PCs and fibrotic remodeling in the SAN are likely causes of SND in Lats1/2 CKO mice. Using CUT&Tag sequencing, we found that Yap potentially regulates genes involved in cell communication and adhesion, as well as in calcium homeostasis. We further found that the reduced expression of the calcium-handling protein RyR2 in the SAN and increased expression of the secreted proteins TGF-β1 and TGF-β3 were likely causes of SND in the context of CCS-specific Lats1/2 deficiency. Moreover, using a double-knockout strategy, we showed that the CCS-specific deletion of Yap/Taz rescued SND and SND-associated fibrotic remodeling caused by Lats1/2 deficiency. Altogether, our study provided the first evidence of an essential role for canonical Hippo signaling in the homeostatic maintenance of the SAN.
Figure 8. Schematic representation of the Hippo signaling–mediated mechanisms underlying the development of SND.
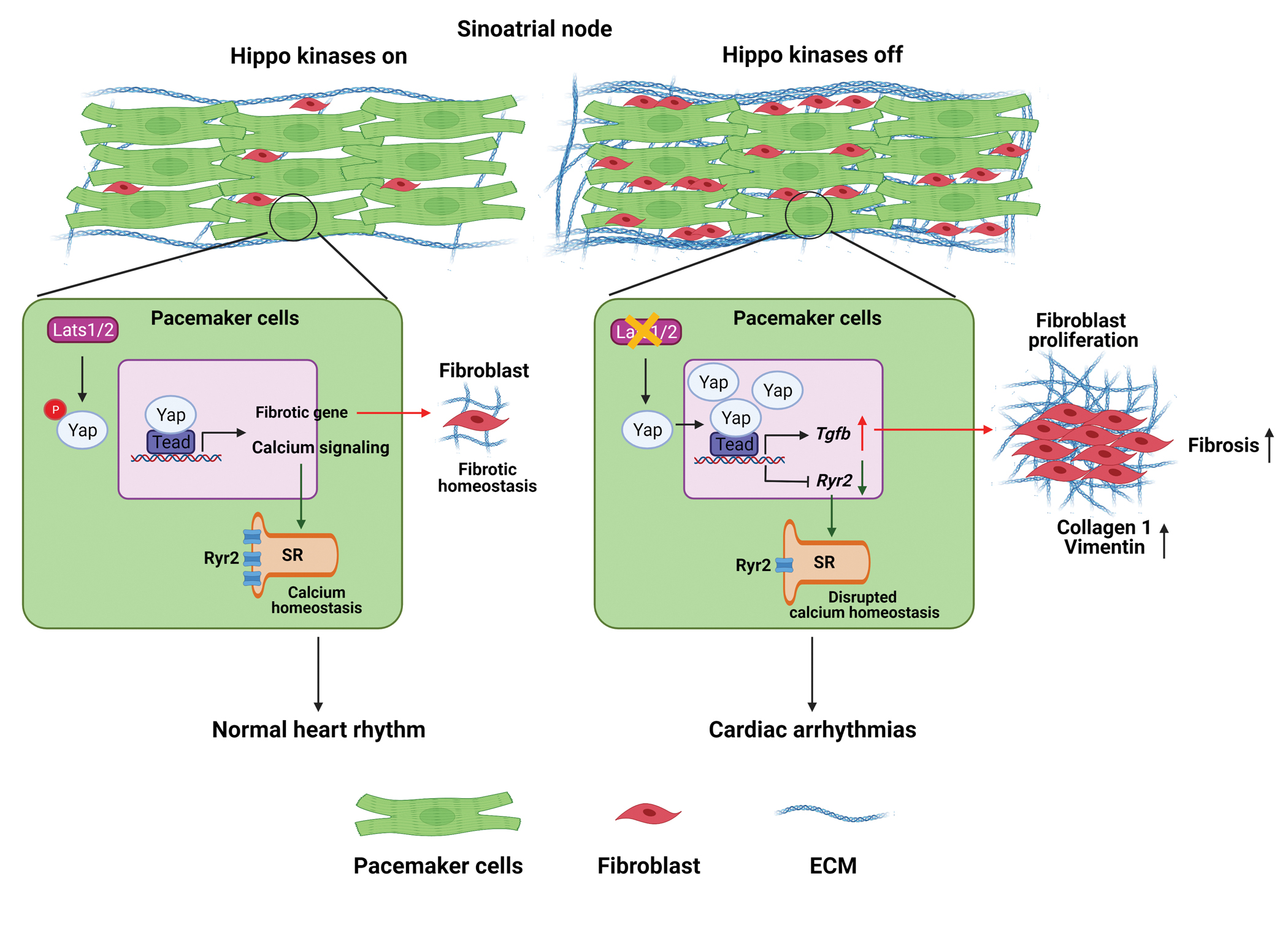
On one hand, the CCS-specific loss of Lats1/2 (‘Hippo kinases off’) can enhance the transcription of calcium-handling protein genes such as Ryr2 and cause the dysfunctional ‘Ca2+ clock’ in PCs, thereby impairing their pacemaking function. On the other hand, the upregulation of genes in the profibrotic pathway, such as Tgf-β, due to the CCS-specific loss of Lats1/2 can promote fibroblast proliferation and increase fibrosis within and near the SAN, which also impairs the propagation of electrical activation from the SAN to the atrial myocardium. Figure was created with Biorender (https://biorender.com/ ).
Although accumulating evidence suggests that Hippo signaling plays pivotal roles in heart development, disease, and regeneration, no other study has provided insight into Hippo signaling in the SAN. The SAN is composed of clusters of PCs controlling heart rhythm. PCs are encompassed by the extracellular matrix, which is required for electrically insulating PCs to maintain regular heartbeats.9 However, excessive fibrosis can block the propagation of pacemaking activity initiated from the PCs.9, 56 Our study reveals that Lats1/2 deficiency in the SAN led to increased fibrosis in the SAN, which could impair electrical conduction both within the SAN and from the SAN to the surrounding atrium. Hippo signaling was previously reported to play an essential role in the transition of epicardial cells to CFs during CF development.57 Deletion of Lats1/2 in adult CFs resulted in CF proliferation and spontaneous self-sustaining fibrosis.30 Previous work also showed that paracrine signaling may be one of the mechanisms of fibroblast-induced PC dysfunction58, 59 and that Tgfb2/3 are transcriptional targets of Yap/Taz.60 Loss of Lats1/2 in primary hepatoblasts increases biliary epithelial cell and fibroblast proliferation by upregulating TGF-β signaling during and after liver development.61 In our study, because Lats1/2 were ablated only in SAN PCs in Lats1/2 CKO mice, it is likely that the upregulation of Tgfb1 and Tgfb3 in PCs led to an increased release of the fibrosis inducers TGF-β1 and TGF-β3. Evidence of TGF-β pathway activation was demonstrated by increased levels of pSmad, ultimately promoting fibrotic remodeling and fibroblast proliferation. In addition to increased pSmad in PCs, increased pSmad was detected in fibroblasts in the SAN, which is an interesting non–cell-autonomous effect caused by Hippo signaling deletion. Importantly, treatment with TGF-β1 receptor inhibitor efficiently inhibited fibroblast proliferation in the SAN of Lats1/2 CKOs, suggesting that Lats1/2 deficiency caused fibroblast proliferation in the SAN through the activation of TGF-β signaling. Here, we have dissected the mechanistic regulation of SAN homeostasis by Lats1/2; however, given the complexity of the CCS, potential roles of Lats1/2 in other components of the CCS should be further investigated in future studies.
We also showed that Lats1/2 function through the canonical Yap/Taz-mediated Hippo pathway in the SAN. Blocking Hippo signaling in cardiomyocytes can activate Yap to reduce fibrosis and promote heart regeneration after injury or under pathologic conditions such as myocardial infarction18 or heart failure in mice.20 In contrast, under physiologic conditions, induction of YAP5SA (an active version of YAP) in cardiomyocytes did not induce interstitial fibrosis in adult mice.62 Notably, one month of YAP overexpression in cardiomyocytes did not induce measurable fibrosis; however, four months of overexpression caused a slight but statistically significant increase in myocardial fibrosis.63 These findings suggested the complexity of fibrosis in response to Hippo signaling under different pathologic and physiologic conditions. Further, the Yap/Taz/Tead complex commonly interacts with other transcription factors to regulate gene expression in different contexts. For instance, in breast cancer cells, Yap/Taz/Tead bind to chromatin with activator protein-1 (ap-1) at composite cis-regulatory elements (CREs) containing Tead and ap-1 motifs.64 In two recent studies, ATAC-seq data showing the mouse and human chromatin accessibility landscape of PCs and cardiomyocytes revealed SAN-specific CREs containing Tbx3, Isl1, and Shox2 motifs.41, 42 The possibility remains that the Yap/Taz/Teads transcription complex can interact with SAN-specific transcription factors to regulate gene expression in the SAN. Our comprehensive comparative analysis of these published ATAC-seq datasets and our Yap CUT&Tag seq data (Figure S6–S8) suggested that Yap regulation varies in PCs and cardiomyocytes, as well as in PCs from mice of different ages. Yap regulation is an interesting yet complicated area of study worthy of more in-depth investigation in the future.
Intracellular calcium concentration is a critical regulator of cardiomyocyte function.65, 66 For the spontaneous firing of PCs, automaticity essentially relies on the integrated activity of SR Ca2+ release, voltage-gated ionic currents, and Ca2+ pumps/transporters.52, 53, 67, 68 Our CUT&Tag data suggested potential Yap target genes regulating calcium ion activity and the If-dependent voltage membrane clock, such as Camk2 genes (encoding CaMKII, calcium/calmodulin-dependent protein kinase II). CaMKII is a serine/threonine-specific protein kinase,69, 70 which also regulates Ca2+ homeostasis in SAN cells.71 CaMKII can catalyze the phosphorylation of RYR2,72 increasing Ca2+ release from phosphorylated RYR2. Increased oxidized CaMKII in the SAN induced by angiotensin II led to SAN cell apoptosis and SND.73 Recently, Hippo-Yap signaling has been implicated in calcium homeostasis. In cardiomyocytes, several studies indicated that Yap/Tead can function as direct transcriptional activators of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase-2a (SERCA2a), which is required for SR calcium cycling.74–76 An in vitro study identified the calcium channel PIEZO1 as a transcriptional target of YAP in oral squamous cell carcinoma cells.77 However, the role of Hippo signaling in calcium activity regulation remains largely unknown. In this study, we discovered that Hippo signaling maintains the spontaneous firing rate and calcium activity of PCs. PCs in the SAN of Lats1/2 CKO mice showed abnormal calcium activity compared with controls. Here, we determined that Ryr2 was decreased in the SAN consequent to Lats1/2 deletion. Consistent with our findings, the Ryr-specific inhibitor ryanodine inhibited the beating rate of PCs.78 In addition, Ryr2 was found to be downregulated in cardiomyocytes overexpressing YAP5SA (a constitutively active Yap) compared with expression in control adult mice.62 Although deregulation of Ryr2 expression may partially lead to the abnormal calcium activity in Lats1/2 CKO SANs, the altered calcium homeostasis is probably a concerted change that includes other genes, given that there are other Yap potential target genes implicated in calcium regulation. Therefore, it will be important to further study and understand how Yap regulates calcium homeostasis in the SAN.
Cell-cell adhesions, including tight junction and gap junction, are required for normal cardiac conduction development and function.79–83 Disruption of cell-cell adhesion molecules results in abnormal cardiac conduction, arrhythmias, and cardiomyopathies.5, 84, 85 Zebrafish hearts lacking TAZ (WWTR1) were shown to exhibit abnormal cell-cell junctions.86 Yap CUT&Tag seq data indicated that many genes that regulate cell-cell communication and adhesion are potentially regulated by Yap. For instance, TJP1 (tight junction protein 1, also known as zona occludens 1 [ZO-1]) is an important intercellular tight junctions protein, and its CCS-specific deletion caused AV blocks.87 Cdh2, which encodes CDH2 (also known as N-cadherin) is a direct Yap target88, 89 and is important for cell-cell junction formation in the myocardium.90 Mice with the cardiac-specific deletion of Cdh2 had diminished QRS complex amplitudes80 and sudden death due to ventricular tachyarrhythmias.91 Hippo signaling may also regulate electrical and physical coupling between SAN cells and other cell types through cell-cell adhesion molecules, which we believe could be an important and interesting area of future investigation.
In conclusion, our study reveals a novel and essential role of the canonical Hippo signaling pathway in maintaining CCS homeostasis. Our findings provide a better understanding of the molecular and genetic regulation of calcium homeostasis and fibrosis homeostasis in the SAN, which could benefit preclinical research and lead to novel approaches for the prevention and treatment of cardiac arrhythmias in human patients.
Supplementary Material
Clinical Perspective.
What Is New?
We identified Hippo signaling as an important regulator of SAN homeostasis.
Deletion of the essential Hippo kinases Lats1/2 causes increased fibroblast proliferation and fibrosis in the SAN involving non–cell-autonomous TGF-β signaling.
Hippo signaling regulates calcium homeostasis in pacemaker cells that may be partially mediated by the regulation of genes encoding key calcium handling proteins such as Ryr2.
Deletion of the Hippo effectors Yap and Taz in the SAN can rescue defects caused by Lats1/2 deficiency, revealing that the canonical Hippo signaling pathway plays vital function in SAN homeostasis.
What Are the Clinical Implications?
Our comprehensive studies of various mouse models provide novel insight into the molecular genetic regulation of SAN homeostasis and reveal new potential therapeutic targets for recovery from cardiac arrhythmias.
Dysregulation and genetic variants of the genes encoding Hippo signaling pathway components are associated with human arrhythmias, and here we present a preclinical model for sinus node dysfunction, which may provide mechanistic insights applicable to the treatment of patients with bradycardia disorders.
Acknowledgements
The authors thank the sequencing support from Cancer Genomics Center supported by CPRIT RP180734. We also thank Drs. Eric N. Olson, James F. Martin, and Randy L. Johnson for mouse lines; Dr. Idaliz M. Martínez Traverso for technical support of RNAscope; Drs. Stephen N. Palmer and Nicole Stancel of Scientific Publications at the Texas Heart Institute for editorial assistance, and Kaleigh L. Riggs for statistical analysis assistance.
Sources of Funding
This study was supported by the funding sources from the National Institutes of Health (R56HL142704 and R01HL142704 to J.W.; T32HL07676 to R.G.L.; R01HL163277, R01HL136389 and R01HL147108 to N.L.) and American Heart Association (SDG19840000 and TPA970606 to J.W.; 902940 to M.Z.; 903251 to R.G.L.; EIA936111 to N.L.).
Non-standard Abbreviations and Acronyms
- CCS
cardiac conduction system
- SAN
sinoatrial node/sinus node
- AVN
atrioventricular node
- CUT&Tag
cleavage under targets and tagmentation
- PCs
pacemaker cells
- CFs
cardiac fibroblasts
- SND
sinus node dysfunction
- TEADs
TEA domain transcription factor family members
- SR
sarcoplasmic reticulum
- Ryr2
ryanodine receptor type 2
- Tgfb
transforming growth factor beta
- WGA
wheat germ agglutinin
- EdU
5-ethynyl-2’-deoxyuridine
- cTnT
cardiac troponin-T
- Vim
vimentin
- Col1a1
collagen 1
- Acta2/aSMA
alpha-smooth muscle actin
- Postn
Periostin
- GO
gene ontology
Footnotes
Disclosures
None.
Contributor Information
Mingjie Zheng, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston.
Rich G. Li, Texas Heart Institute, Houston, Texas, USA.
Jia Song, Department of Medicine (Section of Cardiovascular Research), Cardiovascular Research Institute, Baylor College of Medicine, Houston, TX, USA.
Xiaolei Zhao, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston.
Li Tang, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan, China.
Shannon Erhardt, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston; MD Anderson Cancer Center and UTHealth Graduate School of Biomedical Sciences, The University of Texas, Houston, Texas, USA.
Wen Chen, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston.
Bao H. Nguyen, Department of Molecular Physiology and Biophysics, Cardiovascular Research Institute, Baylor College of Medicine, Houston, TX, USA.
Xiao Li, Texas Heart Institute, Houston, Texas, USA.
Min Li, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan, China.
Jianxin Wang, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan, China.
Sylvia M. Evans, Skaggs School of Pharmacy and Pharmaceutical Sciences, Departments of Pharmacology and Medicine, University of California at San Diego, La Jolla, CA 92093, USA.
Vincent M. Christoffels, Medical Biology, Amsterdam Cardiovascular Sciences, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands.
Na Li, Department of Medicine (Section of Cardiovascular Research), Cardiovascular Research Institute, Baylor College of Medicine, Houston, TX, USA.
Jun Wang, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston; MD Anderson Cancer Center and UTHealth Graduate School of Biomedical Sciences, The University of Texas, Houston, Texas, USA.
References
- 1.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. The Journal of clinical investigation. 2005;115:2305–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. The New England journal of medicine. 2001;345:1473–1482 [DOI] [PubMed] [Google Scholar]
- 3.Mandla R, Jung C, Vedantham V. Transcriptional and epigenetic landscape of cardiac pacemaker cells: Insights into cellular specialization in the sinoatrial node. Frontiers in physiology. 2021;12:712666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christoffels VM, Smits GJ, Kispert A, Moorman AF. Development of the pacemaker tissues of the heart. Circulation research. 2010;106:240–254 [DOI] [PubMed] [Google Scholar]
- 5.van Eif VWW, Devalla HD, Boink GJJ, Christoffels VM. Transcriptional regulation of the cardiac conduction system. Nature reviews. Cardiology. 2018;15:617–630 [DOI] [PubMed] [Google Scholar]
- 6.Linscheid N, Logantha S, Poulsen PC, Zhang S, Schrolkamp M, Egerod KL, Thompson JJ, Kitmitto A, Galli G, Humphries MJ, Zhang H, Pers TH, Olsen JV, Boyett M, Lundby A. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nature communications. 2019;10:2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashou AH, Basit H, Chhabra L. Physiology, sinoatrial node. Statpearls. Treasure Island (FL); 2021. [PubMed] [Google Scholar]
- 8.Liang X, Evans SM, Sun Y. Development of the cardiac pacemaker. Cellular and molecular life sciences : CMLS. 2017;74:1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csepe TA, Kalyanasundaram A, Hansen BJ, Zhao J, Fedorov VV. Fibrosis: A structural modulator of sinoatrial node physiology and dysfunction. Frontiers in physiology. 2015;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li N, Hansen BJ, Csepe TA, Zhao J, Ignozzi AJ, Sul LV, Zakharkin SO, Kalyanasundaram A, Davis JP, Biesiadecki BJ, Kilic A, Janssen PML, Mohler PJ, Weiss R, Hummel JD, Fedorov VV. Redundant and diverse intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. Science translational medicine. 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Sun AY, Kim JJ, Graham V, Finch EA, Nepliouev I, Zhao G, Li T, Lederer WJ, Stiber JA, Pitt GS, Bursac N, Rosenberg PB. Stim1-ca2+ signaling modulates automaticity of the mouse sinoatrial node. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E5618–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yavari A, Bellahcene M, Bucchi A, Sirenko S, Pinter K, Herring N, Jung JJ, Tarasov KV, Sharpe EJ, Wolfien M, Czibik G, Steeples V, Ghaffari S, Nguyen C, Stockenhuber A, Clair JRS, Rimmbach C, Okamoto Y, Yang D, Wang M, Ziman BD, Moen JM, Riordon DR, Ramirez C, Paina M, Lee J, Zhang J, Ahmet I, Matt MG, Tarasova YS, Baban D, Sahgal N, Lockstone H, Puliyadi R, de Bono J, Siggs OM, Gomes J, Muskett H, Maguire ML, Beglov Y, Kelly M, Dos Santos PPN, Bright NJ, Woods A, Gehmlich K, Isackson H, Douglas G, Ferguson DJP, Schneider JE, Tinker A, Wolkenhauer O, Channon KM, Cornall RJ, Sternick EB, Paterson DJ, Redwood CS, Carling D, Proenza C, David R, Baruscotti M, DiFrancesco D, Lakatta EG, Watkins H, Ashrafian H. Mammalian gamma2 ampk regulates intrinsic heart rate. Nat Commun. 2017;8:1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easterling M, Rossi S, Mazzella AJ, Bressan M. Assembly of the cardiac pacemaking complex: Electrogenic principles of sinoatrial node morphogenesis. J Cardiovasc Dev Dis. 2021;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen PN, Gronroos NN, Chen LY, Folsom AR, deFilippi C, Heckbert SR, Alonso A. Incidence of and risk factors for sick sinus syndrome in the general population. J Am Coll Cardiol. 2014;64:531–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. Tead mediates yap-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, Guan KL. Tead transcription factors mediate the function of taz in cell growth and epithelial-mesenchymal transition. J Biol Chem. 2009;284:13355–13362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN. Hippo pathway effector yap promotes cardiac regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13839–13844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT, Martin JF. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. 2017;550:260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morikawa Y, Zhang M, Heallen T, Leach J, Tao G, Xiao Y, Bai Y, Li W, Willerson JT, Martin JF. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in hippo-deficient mice. Science signaling. 2015;8:ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Liu S, Heallen T, Martin JF. The hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat Rev Cardiol. 2018 [DOI] [PubMed] [Google Scholar]
- 23.Zheng M, Jacob J, Hung SH, Wang J. The hippo pathway in cardiac regeneration and homeostasis: New perspectives for cell-free therapy in the injured heart. Biomolecules. 2020;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circulation research. 2014;114:454–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang P, Mao B, Luo W, Wei B, Jiang W, Liu D, Song L, Ji G, Yang Z, Lai YQ, Yuan Z. The alteration of hippo/yap signaling in the development of hypertrophic cardiomyopathy. Basic Res Cardiol. 2014;109:435. [DOI] [PubMed] [Google Scholar]
- 26.Li N, Artiga E, Kalyanasundaram A, Hansen BJ, Webb A, Pietrzak M, Biesiadecki B, Whitson B, Mokadam NA, Janssen PML, Hummel JD, Mohler PJ, Dobrzynski H, Fedorov VV. Altered microrna and mrna profiles during heart failure in the human sinoatrial node. Scientific reports. 2021;11:19328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu SD, Yu JY, Guo Y, Liu XY, Liang T, Chen LZ, Chu YP, Zhang HP. Bioinformatic analysis for the identification of potential gene interactions and therapeutic targets in atrial fibrillation. European review for medical and pharmacological sciences. 2021;25:2281–2290 [DOI] [PubMed] [Google Scholar]
- 28.Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, Chen Y, Chen J, Sun Y, Evans SM. Hcn4 dynamically marks the first heart field and conduction system precursors. Circulation research. 2013;113:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Bai Y, Li N, Ye W, Zhang M, Greene SB, Tao Y, Chen Y, Wehrens XH, Martin JF. Pitx2-microrna pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:9181–9186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, Martin JF. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33:1491–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe EJ, St Clair JR, Proenza C. Methods for the isolation, culture, and functional characterization of sinoatrial node myocytes from adult mice. Journal of visualized experiments : JoVE. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N. Loss of microrna-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circulation. Arrhythmia and electrophysiology. 2014;7:1214–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaya-Okur HS, Janssens DH, Henikoff JG, Ahmad K, Henikoff S. Efficient low-cost chromatin profiling with cut&tag. Nature protocols. 2020;15:3264–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nature methods. 2012;9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. Deeptools2: A next generation web server for deep-sequencing data analysis. Nucleic acids research. 2016;44:W160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meers MP, Tenenbaum D, Henikoff S. Peak calling by sparse enrichment analysis for cut&run chromatin profiling. Epigenetics & chromatin. 2019;12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Molecular cell. 2010;38:576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature communications. 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margulies EH, Blanchette M, Program NCS, Haussler D, Green ED. Identification and characterization of multi-species conserved sequences. Genome research. 2003;13:2507–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galang G, Mandla R, Ruan H, Jung C, Sinha T, Stone NR, Wu RS, Mannion BJ, Allu PKR, Chang K, Rammohan A, Shi MB, Pennacchio LA, Black BL, Vedantham V. Atac-seq reveals an isl1 enhancer that regulates sinoatrial node development and function. Circulation research. 2020;127:1502–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Eif VWW, Protze SI, Bosada FM, Yuan X, Sinha T, van Duijvenboden K, Ernault AC, Mohan RA, Wakker V, de Gier-de Vries C, Hooijkaas IB, Wilson MD, Verkerk AO, Bakkers J, Boukens BJ, Black BL, Scott IC, Christoffels VM. Genome-wide analysis identifies an essential human tbx3 pacemaker enhancer. Circulation research. 2020;127:1522–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. Inactivation of yap oncoprotein by the hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang J, Wu S, Barrera J, Matthews K, Pan D. The hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating yorkie, the drosophila homolog of yap. Cell. 2005;122:421–434 [DOI] [PubMed] [Google Scholar]
- 45.Halder G, Johnson RL. Hippo signaling: Growth control and beyond. Development. 2011;138:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591 [DOI] [PubMed] [Google Scholar]
- 47.Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, Hofmann F, Ludwig A. The hyperpolarization-activated channel hcn4 is required for the generation of pacemaker action potentials in the embryonic heart. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15235–15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moorman AF, Christoffels VM. Cardiac chamber formation: Development, genes, and evolution. Physiological reviews. 2003;83:1223–1267 [DOI] [PubMed] [Google Scholar]
- 49.Matsui Y, Nakano N, Shao D, Gao S, Luo W, Hong C, Zhai P, Holle E, Yu X, Yabuta N, Tao W, Wagner T, Nojima H, Sadoshima J. Lats2 is a negative regulator of myocyte size in the heart. Circulation research. 2008;103:1309–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. Regulation of insulin-like growth factor signaling by yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, Sadoshima J. Proapoptotic rassf1a/mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. The Journal of clinical investigation. 2010;120:3555–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled system of intracellular ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circulation research. 2010;106:659–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiological reviews. 2008;88:919–982 [DOI] [PubMed] [Google Scholar]
- 54.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837 [DOI] [PubMed] [Google Scholar]
- 55.Chen L, Chan SW, Zhang X, Walsh M, Lim CJ, Hong W, Song H. Structural basis of yap recognition by tead4 in the hippo pathway. Genes Dev. 2010;24:290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glukhov AV, Hage LT, Hansen BJ, Pedraza-Toscano A, Vargas-Pinto P, Hamlin RL, Weiss R, Carnes CA, Billman GE, Fedorov VV. Sinoatrial node reentry in a canine chronic left ventricular infarct model: Role of intranodal fibrosis and heterogeneity of refractoriness. Circulation. Arrhythmia and electrophysiology. 2013;6:984–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Wang S, Moise AR, Wythe JD, Martin JF. Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev Cell. 2018;45:153–169 e156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munoz V, Campbell K, Shibayama J. Fibroblasts: Modulating the rhythm of the heart. The Journal of physiology. 2008;586:2423–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fahrenbach JP, Mejia-Alvarez R, Banach K. The relevance of non-excitable cells for cardiac pacemaker function. The Journal of physiology. 2007;585:565–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, Takano Y, Hikasa H, Itoh T, Suzuki SO, Kurihara H, Aishima S, Leask A, Sasaki T, Nakano T, Nishina H, Nishikawa Y, Sekido Y, Nakao K, Shin-Ya K, Mimori K, Suzuki A. Dysregulated yap1/taz and tgf-beta signaling mediate hepatocarcinogenesis in mob1a/1b-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee DH, Park JO, Kim TS, Kim SK, Kim TH, Kim MC, Park GS, Kim JH, Kuninaka S, Olson EN, Saya H, Kim SY, Lee H, Lim DS. Lats-yap/taz controls lineage specification by regulating tgfbeta signaling and hnf4alpha expression during liver development. Nature communications. 2016;7:11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, Krijger PHL, de Laat W, Wehrens XHT, Rodney GG, Martin JF. Yap partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev Cell. 2019;48:765–779 e767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PR, Zhou B, Chen J, Seidman JG, Wang DZ, Pu WT. Cardiac-specific yap activation improves cardiac function and survival in an experimental murine mi model. Circulation research. 2014;115:354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. Genome-wide association between yap/taz/tead and ap-1 at enhancers drives oncogenic growth. Nature cell biology. 2015;17:1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harbor perspectives in biology. 2011;3:a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circulation research. 2017;121:181–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiFrancesco D The role of the funny current in pacemaker activity. Circulation research. 2010;106:434–446 [DOI] [PubMed] [Google Scholar]
- 68.Sah R, Mesirca P, Van den Boogert M, Rosen J, Mably J, Mangoni ME, Clapham DE. Ion channel-kinase trpm7 is required for maintaining cardiac automaticity. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E3037–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulman H, Greengard P. Stimulation of brain membrane protein phosphorylation by calcium and an endogenous heat-stable protein. Nature. 1978;271:478–479 [DOI] [PubMed] [Google Scholar]
- 70.Lisman J, Schulman H, Cline H. The molecular basis of camkii function in synaptic and behavioural memory. Nature reviews. Neuroscience. 2002;3:175–190 [DOI] [PubMed] [Google Scholar]
- 71.Vinogradova TM, Zhou YY, Bogdanov KY, Yang D, Kuschel M, Cheng H, Xiao RP. Sinoatrial node pacemaker activity requires ca(2+)/calmodulin-dependent protein kinase ii activation. Circulation research. 2000;87:760–767 [DOI] [PubMed] [Google Scholar]
- 72.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase ii phosphorylation regulates the cardiac ryanodine receptor. Circulation research. 2004;94:e61–70 [DOI] [PubMed] [Google Scholar]
- 73.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized camkii causes cardiac sinus node dysfunction in mice. The Journal of clinical investigation. 2011;121:3277–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu R, Lee J, Kim BS, Wang Q, Buxton SK, Balasubramanyam N, Kim JJ, Dong J, Zhang A, Li S, Gupte AA, Hamilton DJ, Martin JF, Rodney GG, Coarfa C, Wehrens XH, Yechoor VK, Moulik M. Tead1 is required for maintaining adult cardiomyocyte function, and its loss results in lethal dilated cardiomyopathy. JCI insight. 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhong J, Ouyang H, Zheng S, Guo Z, Chen Y, Zhong Y, Zhong W, Zuo L, Lu J. The yap/serca2a signaling pathway protects cardiomyocytes against reperfusion-induced apoptosis. Aging. 2020;12:13618–13632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xie J, Wang Y, Ai D, Yao L, Jiang H. The role of the hippo pathway in heart disease. The FEBS journal. 2021 [DOI] [PubMed] [Google Scholar]
- 77.Hasegawa K, Fujii S, Matsumoto S, Tajiri Y, Kikuchi A, Kiyoshima T. Yap signaling induces piezo1 to promote oral squamous cell carcinoma cell proliferation. The Journal of pathology. 2021;253:80–93 [DOI] [PubMed] [Google Scholar]
- 78.Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and na(+)-ca(2+) exchanger: Molecular partners in pacemaker regulation. Circulation research. 2001;88:1254–1258 [DOI] [PubMed] [Google Scholar]
- 79.Delgado C, Bu L, Zhang J, Liu FY, Sall J, Liang FX, Furley AJ, Fishman GI. Neural cell adhesion molecule is required for ventricular conduction system development. Development. 2021;148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD, Radice GL. Cardiac-specific loss of n-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circulation research. 2005;97:474–481 [DOI] [PubMed] [Google Scholar]
- 81.Li J, Patel VV, Radice GL. Dysregulation of cell adhesion proteins and cardiac arrhythmogenesis. Clinical medicine & research. 2006;4:42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Rijen HVM, van Veen TAB, Gros D, Wilders R, de Bakker JMT. Connexins and cardiac arrhythmias. Advances in cardiology. 2006;42:150–160 [DOI] [PubMed] [Google Scholar]
- 83.Kanno S, Saffitz JE. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology. 2001;10:169–177 [DOI] [PubMed] [Google Scholar]
- 84.Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovascular research. 2004;62:368–377 [DOI] [PubMed] [Google Scholar]
- 85.Arnolds DE, Chu A, McNally EM, Nobrega MA, Moskowitz IP. The emerging genetic landscape underlying cardiac conduction system function. Birth defects research. Part A, Clinical and molecular teratology. 2011;91:578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lai JKH, Collins MM, Uribe V, Jimenez-Amilburu V, Gunther S, Maischein HM, Stainier DYR. The hippo pathway effector wwtr1 regulates cardiac wall maturation in zebrafish. Development. 2018;145 [DOI] [PubMed] [Google Scholar]
- 87.Dai W, Nadadur RD, Brennan JA, Smith HL, Shen KM, Gadek M, Laforest B, Wang M, Gemel J, Li Y, Zhang J, Ziman BD, Yan J, Ai X, Beyer EC, Lakata EG, Kasthuri N, Efimov IR, Broman MT, Moskowitz IP, Shen L, Weber CR. Zo-1 regulates intercalated disc composition and atrioventricular node conduction. Circulation research. 2020;127:e28–e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park JS, Davis RJ, Mao J. Tead and ap1 coordinate transcription and motility. Cell reports. 2016;14:1169–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skladal P, Pesl M, Caluori G, Pagliari S, Martino F, Maceckova Z, Hajduch M, Sanz-Garcia A, Pugno NM, Stokin GB, Forte G. Yap regulates cell mechanics by controlling focal adhesion assembly. Nature communications. 2017;8:15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aharonov A, Shakked A, Umansky KB, Savidor A, Genzelinakh A, Kain D, Lendengolts D, Revach OY, Morikawa Y, Dong J, Levin Y, Geiger B, Martin JF, Tzahor E. Erbb2 drives yap activation and emt-like processes during cardiac regeneration. Nature cell biology. 2020;22:1346–1356 [DOI] [PubMed] [Google Scholar]
- 91.Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the n-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circulation research. 2005;96:346–354 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.