

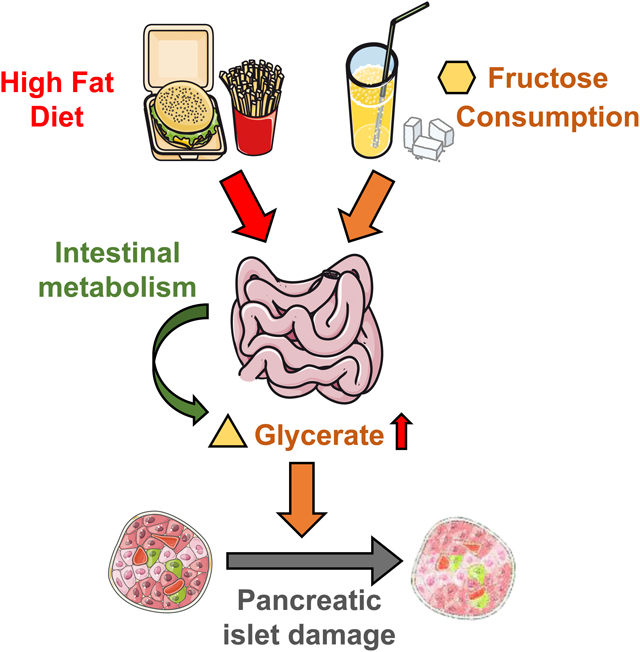
Summary:
Dietary fructose, especially in the context of a high-fat western diet, has been linked to type 2 diabetes. Although the effect of fructose on liver metabolism has been extensively studied, a significant portion of the fructose is first metabolized in the small intestine. Here we report that dietary fat enhances intestinal fructose metabolism, which releases glycerate into the blood. Chronic high systemic glycerate levels induce glucose intolerance by slowly damaging pancreatic islet cells and reducing islet sizes. Our findings provide a link between dietary fructose and diabetes that is modulated by dietary fat.
Graphical Abstract
eTOC blurb
Dietary fructose has been linked to metabolic syndromes such as diabetes. Wu et al. discovered that fructose metabolism in the small intestine releases glycerate into the blood circulation, which damages pancreatic islet cells and induces glucose intolerance. Dietary fat further elevates intestinal fructose metabolism and resulting circulating glycerate levels, supporting the long-term effect of western diet on exacerbating metabolic syndromes.
Introduction
Type II diabetes mellitus (T2DM) has doubled in prevalence over the last two decades, becoming a major threat to global health (Bhupathiraju and Hu, 2016; Unnikrishnan et al., 2017). Although T2DM is associated with genetics, the development of glucose intolerance is also a consequence of lifestyle and dietary patterns (Bhupathiraju and Hu, 2016; Ng et al., 2014; Nguyen and El-Serag, 2010; Wang et al., 2012). Ample evidence associates the consumption of fructose-containing sweetened beverages, as part of the western diet, with the development of T2DM (Cordain et al., 2005; Hannou et al., 2018). Dietary fat, a prominent component of the western diet, has also been implicated in obesity and T2DM (Forouhi et al., 2018; Soltis et al., 2017). Studies that analyzed the combined effects of fat and mono-saccharide consumption found that fructose, but not glucose, exaggerates the deleterious effects of dietary fat (Herman and Samuel, 2016; Koo et al., 2009; Koo et al., 2008; Softic et al., 2017). Reciprocally, pathogenic effects of fructose are more prominent among mice fed a high-fat diet, although the mechanisms remain unknown (Softic et al., 2017).
High fructose feeding is a well-established model for inducing insulin resistance (Liu et al., 2020; Softic et al., 2017). Fructose ingestion does not impact insulin-secreting β-cells directly, as low expression of fructose transporters impedes their metabolism of fructose (Curry, 1989), but synergizes with glucose in signaling to β-cells (Bartley et al., 2019; Kyriazis et al., 2012). Although hyperinsulinemia could result from increased calorie intake and adiposity upon high fructose consumption (Augustin et al., 2015; Rendeiro et al., 2015), fructose metabolism in the liver seems to be the main contributor to metabolic dysfunction (Andres-Hernando et al., 2020; Ishimoto et al., 2012). Compared with glucose catabolism, unchecked fructose catabolism can initiate rapidly (Herman and Samuel, 2016; Laughlin, 2014) and produce a flood of acetyl-CoA and glycerol-3-phosphate (Gro3P) that can fuel de novo synthesis of triglycerides and the development of steatosis (Softic et al., 2017; Softic et al., 2019). Metabolites from both dietary fats and carbohydrates can regulate each other through master transcriptional regulators like carbohydrate response element-binding protein (Chrebp or Mlxipl) (Kim et al., 2016), peroxisome proliferator-activated receptor gamma (Pparg), and sterol regulatory element-binding protein-1c (Srebp-1c or Srebf) (Haas et al., 2012). Hepatic fructose metabolism can also deplete local adenosine triphosphate (ATP) and generate significant amounts of uric acid (Jensen et al., 2018), although their association with clinical hepatic insulin resistance and steatosis is still under debate (Li et al., 2017).
Recent reports demonstrated that the intestine largely shields the liver and other internal organs from the effects of fructose consumption, such as enhanced lipogenesis and ectopic fat deposition, by converting a substantial amount of ingested fructose to various metabolites (Jang et al., 2020; Kim et al., 2017). Only when ingested fructose exceeds the maximum fructose metabolism capacity of the small intestine does unconverted fructose “spillover” to the liver (via the portal vein) and the colon (Jang et al., 2018). These findings indicate that the metabolism of fructose is not localized to the liver and that a systematic evaluation of its fate is warranted. However, little is known about the contribution of fructose-derived metabolites from the small intestine during the development of T2DM, particularly in a liver-independent fashion.
We observed that a high-fat diet (HFD) altered fructose metabolism in the mouse small intestine, resulting in an enrichment of organic acid pools from the fructolysis pathway but not the citric acid cycle. In particular, fructose-derived glycerate was sustained at high levels in systemic circulation, and chronic glycerate treatment caused damage to pancreatic islet cells, especiall β-cells, in vivo. These data suggest that fat consumption enhances intestinal fructose metabolism and circulation of its derived glycerate, which can chronically damage pancreatic islet cells and lead to subsequent glucose intolerance. Our model provides a mechanism linking T2DM pathogenesis to the consumption of a western diet rich in fructose and fat.
Results
High-fat Diet Enhances Fructose Metabolism in the Small Intestine
We examined the effects of dietary fat on intestinal fructose metabolism by measuring the expression of relevant enzymes in the jejunum of mice fed HFD or control diet (CD), with matched sucrose content, and tracing the conversion of an isotopically labeled oral fructose bolus (Figure 1A). Prolonged HFD feeding resulted in increased weight and liver mass compared to prolonged CD feeding (Figures S1A–S1C). Acute HFD feeding for 3 days caused minimal changes in carbohydrate and lipid metabolism genes (Figure 1B). However, the intestines of the HFD-fed mice expressed higher levels of the fructolytic enzymes ketohexokinase (Khk, both Khk-a and Khk-c isoforms) and aldolase B (Aldob) as well as solute carrier family 2 member 5 (Slc2a5 or Glut5) , which mediates ingested fructose absorption, than did those of CD-fed mice (Figure 1C). This suggests that the mice fed HFD had a stronger adaptive response to oral fructose. Chronic HFD feeding led to similar increases in fructose metabolism gene and protein expression (Figures S1D–S1F). We observed changes in gene expression of transcriptional regulators for carbohydrate and lipids after fructose bolus; mice on HFD vs. CD had significant increases in both Pparg and Srebp-1c mRNA levels, but mixed responses for Chrebp isoforms (Figure 1C).
Figure 1. The effects of dietary fat on intestinal fructose metabolism.
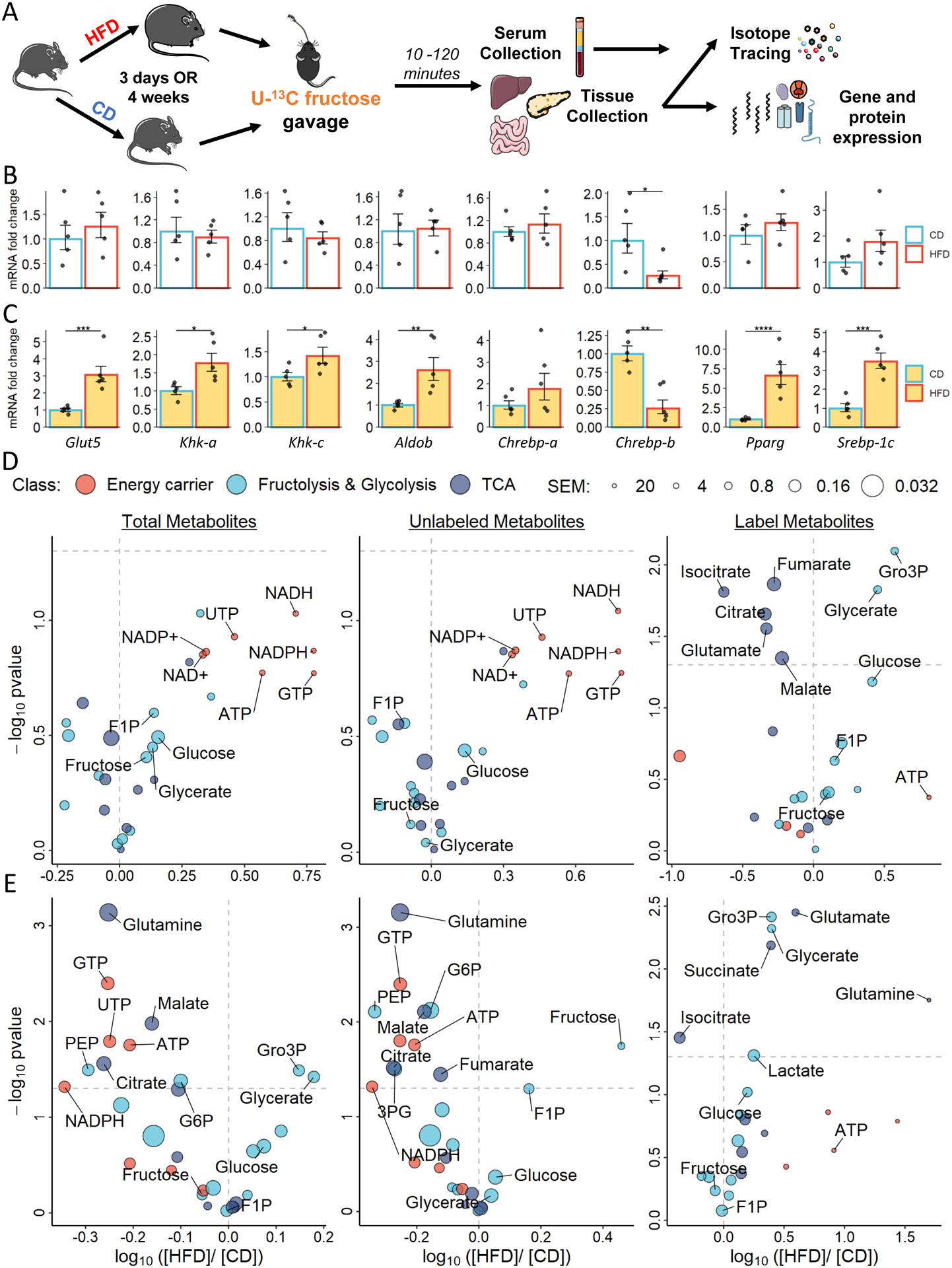
A. Experimental design of the tracing experiment and in the jejunum, pancreas, and liver (N = 3 – 5).
B and C. mRNA expression in jejunum in mice fed on HFD for 3 days; yellow shading depicts jejunum samples that were harvested 10 minutes after the fructose gavage.
D and E. Metabolite log-ratio of the HFD oevr CD mice after fed for 3 days (D) and 4 weeks (E). F1P: fructose-1-phosphate; NAD+: nicotinamide adenine dinucleotide; NADP+: nicotinamide adenine dinucleotide phosphate; GTP: guanosine triphosphate; UTP: uridine triphosphate; SEM: standard error mean. Data are represented as mean ± SEM. T-test was used.*: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001. See also Figures S1.
Lipid Availability Alters Fructolytic Enzyme Expression in Murine and Human Intestinal Organoids
To confirm whether the effect of lipid availability on the intestinal expression of fructose-related metabolic genes is direct, we grew primary 2D intestinal organoids on hydrogel with an exposed apical surface (Wang et al., 2017) (Figure S1G) and treated them with dietary fat components. Apical treatment of murine jejunum organoids with palmitate, the major component of dietary fat, upregulated Glut5, Khk, and AldoB RNA and protein expression (Figures S1H–S1J). Oleate, a more benign lipid species (Palomer et al., 2018), induced less expression of fructolytic enzymes than palmitate treatment. We then derived primary 2D organoids using intestines harvested from human organ donors from the Duke Intestinal Transplant Center. Similar to murine organoids, palmitate and, to a lesser extent, oleate increased the expression of fructolytic enzymes in the human jejunum organoids (Figures S1K–S1M).
High-Fat Diet Alters Enteric Fructose Handling
Next, we sought to examine the impact of HFD on the fructose assimilation in the jejunum after oral administration of a bolus containing universally 13C-labeled fructose (U13C-fructose 1 g/kg). After acute HFD feeding, larger pools of labeled glycerate, a fructose-specific metabolite, and glycerol-3-phosphate (Gro-3P), a lipogenic precursor, were detected in the jejunum of HFD-fed mice compared to CD-fed counterparts (Figure 1D). Similar isotopic tracing in mice that were chronically fed on these diets showed that HFD increased intestinal fructose metabolic flux, resulting in larger total pools of the fructolytic intermediates glycerate, Gro3P, glyceraldehyde-3-phosphate (G3P), and dihydroxyacetone phosphate (DHAP) detected in the jejunum, compared to CD (Figure 1E). Although acute HFD feeding results in down-regulation of labeled citrate, malate, and glutamate, the changes were not sustained upon chronic high-fat feeding (Figures 1D and 1E). Minimal labeling was detected in tricarboxylic acid (TCA) cycle-related metabolites in both groups (Figures S1N and S1O), which suggests that the jejunum of HFD-fed (vs. CD-fed) animals has higher fructose metabolic capacity but does not convert ingested fructose directly into energy.
Fructose-derived Glycerate Persists in Systemic Circulation
The effect of a HFD on hepatic fructose metabolism was less pronounced than in the jejunum. Consistently, we only found subtle changes in fructose metabolism genes in the liver (Figure S2A). The hepatic concentration of labeled fructose was 80% lower than that of the jejunum, with relatively low labeling of fructolysis intermediates (Figures S2B and S2C). Overall, the observation of lower levels of hepatic fructolysis intermediates is consistent with previous reports that fructose is first metabolized in the small intestine (Jang et al., 2018).
Various fructose metabolites can reach the circulation system via the hepatic portal vein (Figure 2A). Mice acutely fed with HFD exhibited reduced TCA incorporation of fructose in the portal vein (Figure 2B). Conversely, HFD-fed mice had larger fructolytic metabolite pools in their portal vein (PV) serum than CD-fed mice, a trend that was also observed in the systemic circulation (SC) (Figure 2B). Among all labeled metabolites with circulating levels that significantly differed between HFD and CD animals, glycerate was the only fructose-specific metabolite and had a distinct labeling kinetic, as revealed by cluster analysis (Figures 2A and 2B). We quantified the absolute concentration of fructose-derived glycerate in the circulation. Circulating glucose and glycerate were significantly up-regulated in HFD mice despite the slightly lower unconverted fructose tracer level in HFD mice (Figures 2C–2E). Although circulating fructose levels peaked at 15 minutes after the oral bolus, sustained labeled glucose was detected in both the PV and SC (Figure 2D). Furthermore, fructose-derived glycerate in the SC was significantly up-regulated in mice fed HFD even after a drastic drop of glycerate after 30 minutes (Figure 2E).
Figure 2. Serum metabolomic profile after U13C-fructose oral gavage.
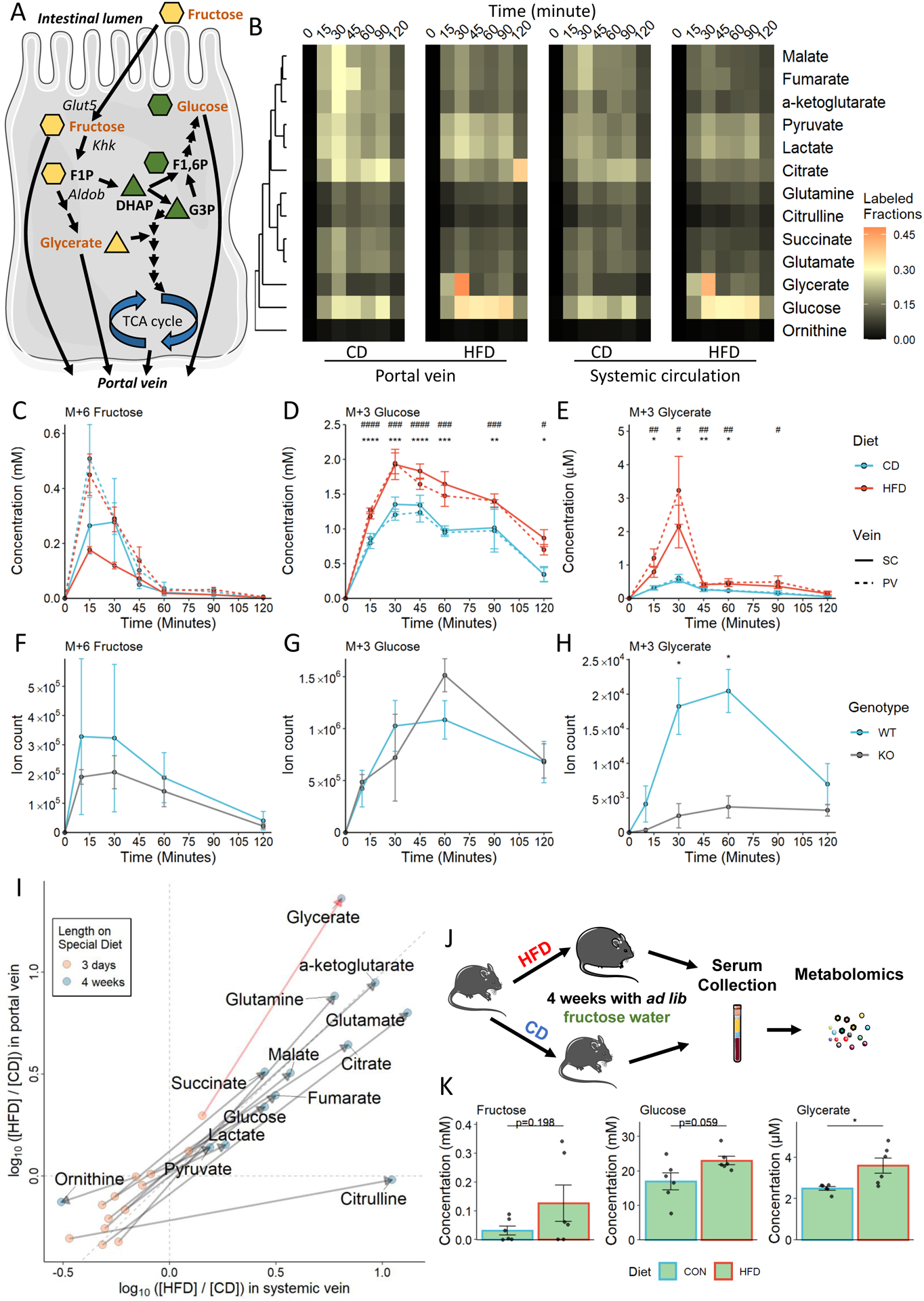
A. Fructose metabolism overview. The shape of each metabolite depicting the number of carbons in the backbone.
B. Heatmap showing circulating labeled (M ≥ +1) metabolite fractions.
C - E. Absolute quantification of 13C -labeled metabolites in each of the circulation pools. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 between HFD and CD groups in PV serum; #: p < 0.05, ##: p < 0.01, ###: p < 0.001, ####: p < 0.0001 between HFD and CD groups in SC serum.
F - H. Quantification of C13-labeled metabolites in SC serum in wild-type (WT) and intestine-specific Khk-c knockout (KO) mice.
I. Log fold change of the labeled (M ≥ +1) metabolites in the portal vein and systemic vein after 3-day or 4-week HFD feeding; serums were harvested 10 minutes after the fructose gavage. J. Experimental design of ad lib fructose drinking water experiment.
K. Absolute quantification of fructose-derived metabolites in systemic circulation.
N = 3 – 6 mice. Data are represented as mean ± SEM. T-test was used.*: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001. See also Figure S2.
HFD increased glycerate levels in PV serum more than in SC serum, consistent with intestinal fructolysis. To identify the major production site of glucose and glycerate, we used mice lacking Khk-c expression specifically in the Villin-expressing enteric epithelium (Khk-ciKO) in an independent fructose tracing experiment with a focus on the labeled metabolites in the SC. The amounts of unconverted fructose and labeled glucose in Khk-ciKO mice were comparable to those in wild-type controls (Figures 2F and 2G), consistent with intact hepatic fructose catabolism. In contrast, the labeled glycerate was markedly reduced in Khk-ciKO mice, indicating that the intestine is the major site of glycerate production (Figure 2H).
Elevated Circulating Glycerate Upon Chronic High Fat Diet with Fructose-supplemented Water
Given the acute effects of HFD on altered fructose metabolism and serum metabolites, we asked whether a chronic western diet amplifies these phenotypes. We observed that chronic HFD further enhanced the fructose conversion and metabolite production in the small intestine, which resulted in elevated fructose-derived metabolites, including glycerate, in the circulation (Figure 2I). The cooperative effect of HFD and fructose was determined by providing fructose-supplemented water to mice fed on either HFD or CD (Figure 2J). Metabolomics on SC serum revealed a significant up-regulation of glycerate in HFD-fed mice (Figure 2K). Taken together, these results suggest that in the context of a chronic western diet, dietary fat modifies fructose metabolism in the small intestine and promotes the production of glycerate.
Glycerate kinase mutation is Associated with Diabetes
High blood levels of glycerate are present in patients with D-glycerate aciduria, a rare disease diagnosed by abnormally high circulating glycerate levels and a loss-of-function mutation to glycerate kinase (Antony et al., 1969; Duran et al., 1987; Van Schaftingen, 1989). To explore the potential impact of circulating glycerate on diabetes, we analyzed the customized SNP-array of 5 million individuals from the UK Biobank database (Bomba et al., 2017). Analysis showed that glycerate kinase mutant carriers were significantly predisposed to diabetes (Table S1). This association is independent of common diabetes risk factors, including age, sex, obesity, cholesterol level (both high- and low-density), and triglyceride level. This suggests that the chronically elevated circulating glycerate levels may be a risk factor for diabetes via a yet unknown mechanism.
Glycerate Treatment Prompts Glucose Intolerance
We compared the phenotypic effects of elevated circulating glycerate and fructose by assessing the glucose metabolic phenotype of animals receiving daily intraperitoneal (IP) injections of these compounds (Figure 3A). Glycerate injection resulted in a surge of serum glycerate that had fast clearance with an estimated half-life of 24.8 minutes (Figure S3A). Consistently, we found no bioaccumulation of glycerate over daily injection as the glycerate level were the same among the groups 24-hour after the nutrient injection (Figure S3B). The glycerate-treated mice were not associated with obesity, and glycerate treatment did not significantly alter the liver mass (Figures S3C and S3D). Mice receiving a daily glycerate injection showed signs of glucose handling disruption. The impairments among mice receiving glycerate injection were exacerbated after prolonged treatment as revealed by the larger area under the curve (AUC) and incremental area under the curve (iAUC) when compared to the control treatment group during an IP glucose tolerance test (IPGTT) (Figures 3B and 3C). Conversely, the fructose injection group did not show significant alterations in glucose handling. IP insulin releasing test (IPIRT) demonstrated that glycerate-injected animals were deficient in insulin even after an injected bolus of glucose, as shown by the reduction in AUC of circulating insulin levels measured following an IP glucose bolus (Figures 3D and 3E). A group of animals injected with 0.1 g/kg glycerate daily had milder reductions in glucose tolerance and overall circulating insulin levels when compared to the animals injected with 1 g/kg glycerate daily (Figures 3B–3E). In an experiment to determine the kinetics of the changes in insulin release upon glucose challenge (Figure 3F), the reduction in overall circulating insulin began as early as day 10 of glycerate treatment (Figure 3G).
Figure 3. Metabolic profiling of mice upon injection of nutrients.
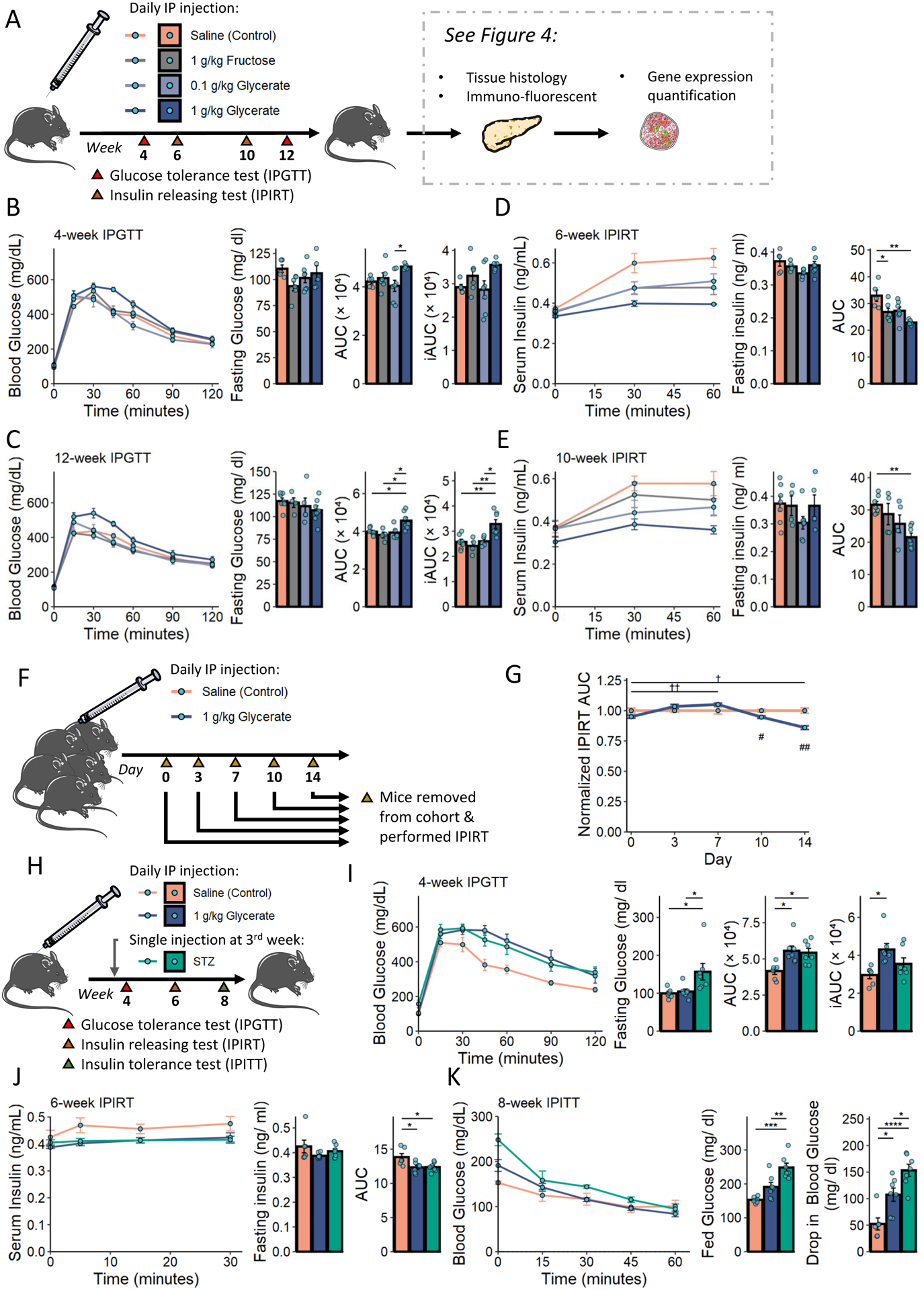
A. Experimental design of the nutrient administration studies.
B - E. When fed on control diet, IPGTT was performed at 4th week (N = 5 – 7 mice) and 12th week (N = 4 – 7 mice), and IPIRT was performed at 6th week (N = 5 – 7 mice) and 10th week (N = 4 – 7 mice).
F - G. A separate cohort of mice was treated with short-term saline or glycerate. AUC of serum insulin levels upon an IPIRT (N = 3 – 5). Results were normalized to the control-treated group on the day of the experiment. †: p < 0.05, ††: p < 0.01 after Bonferroni correction of the test comparing each time point to day 0 in the glycerate treated group; #: p < 0.05, ##: p < 0.01 when comparing control and glycerate treated groups.
H. Experimental design for phenotypic comparison of insulin resistance and insulin deficiency.
I - K. IPGTT was performed at 4th week, IPIRT was performed at 6th week, and IPITT was performed at 8th week (N = 6 – 7 mice).
Data are represented as mean ± SEM. T-test was used for comparison between 2 groups and the post-hoc Tukey correction was performed after ANOVA was used for comparison more than 2 groups.*: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001. See also Figure S3 and Table S1.
Glucose intolerance associated with insulin deficiency is a common phenotype of type 1 diabetes, characterized by the loss of insulin-producing pancreatic β-cells (Chen et al., 2017; Prentki and Nolan, 2006). To compare the effects of glycerate to a β-cell damage model on glucose homeostasis, we examined a new cohort of CD-fed mice that received daily injections of saline or glycerate with a subset of mice that only received a single dose of the toxin streptozotocin (STZ) after 3 weeks of CD feeding (Parilla et al., 2018) (Figure 3H). Although mice treated with STZ had higher fasting glucose levels than those in saline- and glycerate-injected groups, they showed similar glucose clearance upon glucose challenge (Figure 3I). The aberrant glucose handling in the STZ-injected group was associated with impaired fasting glucose while the glycerate-injected group was associated with impaired glucose tolerance. Notably, animals in both STZ-treated and glycerate-injected groups had reduced overall circulating insulin levels compared to saline controls during an IPIRT performed after 6 weeks of treatment (Figure 3J). To directly test the insulin sensitivity of these mice fed on CD, we employed the IP insulin tolerance test (IPITT) at 8 weeks and found that all treated animals remained insulin sensitive (Figure 3K). Specifically, although STZ-treated groups had elevated blood glucose levels compared to the control group, all animals reached similar blood glucose levels upon receiving insulin injection. The elevated non-fast blood glucose levels were normalized in both glycerate- and STZ-treated groups as those mice experienced a more drastic drop in blood glucose levels upon insulin injection.
Mice receiving the same treatment regimes described in Figures 3A and 3H but fed on HFD were used to examine the compounded effects of diet and nutrient injections. We observed similar glucose clearance between the glycerate-treated group and the control group in two independent cohorts (Figures S3E–S3G), although the AUC and iAUC of IPGTT for HFD-fed mice were larger than that of mice fed with CD. Conversely, the STZ- and glycerate-treated HFD-fed mice demonstrated a reduction in total serum insulin levels during an IPIRT compared with control mice (Figure S3H). Compared to the observed significant drop in blood glucose level after insulin injection among mice fed on CD, mice fed on HFD showed signs of insulin resistance while the drop in blood glucose level was not statistically significant between the control and the glycerate treated groups (Figure S3I). Taken together, our data suggest that glycerate-injected animals are glucose intolerant due to a reduced overall circulating insulin rather than insulin resistance. The reduced overall circulating insulin upon daily glycerate injection was also observed in mice fed with HFD, which suggests that insulin resistance and reduced circulating insulin may fuel the development of diabetes independently.
Elevated Glycerate Induces Islet Dysfunction
Given the dampened insulin release in mice treated with glycerate, we then performed imaging mass spectrometry on pancreatic tissue to trace the deposition of U13C-fructose-derived metabolites on mice fed on CD vs. HFD (Figure 1A). We first validated the usage of M+3 glycerate as a tracer of fructose-derived glycerate in jejunum tissue where the glycerate is likely produced (Figure S4A). Using the same scheme, we observed increased deposition of fructose-derived glycerate into the pancreas in acute HFD-fed animals compared to CD (Figures S4B and S4C).
We examined the physiological effects of daily glycerate injections on the pancreas 12 weeks after treatment was initiated. Histological analysis of the pancreas showed reductions in the cross-section islet diameter of CD-fed animals receiving daily glycerate and fructose injections (Figures 4A and 4B); HFD-fed mice showed comparable islet diameter among the groups (Figures S4D and S4E). Immunofluorescence studies revealed elevated apoptotic signaling in the pancreatic islets of mice treated with glycerate compared to those treated with fructose or control (Figures 4C–4F). The proapoptotic (cleaved) form of Caspase-3, though rare, was found almost exclusively in islets of mice treated with glycerate (Figures 4C and 4E). Apoptosis of pancreatic acinar cells, which do not produce insulin, was comparable among the three groups of animals (Figure 4D). Conversely, glycerate treatment up-regulated the activated caspase-3 signals within the pancreatic islet β-cells (Figure 4F). Co-staining of activated caspase-3 with glucagon or proinsulin, marking pancreatic α-cell and β-cell respectively, showed that 62 – 88% of glycerate treatment-induced apoptotic cells expressed either insulin/ proinsulin while 32% of apoptotic cells expressed glucagon (Figures 4G–4I). Moreover, glycerate treatment augmented the expression of the apoptotic genes Caspase-8 (Casp8) and apoptotic peptidase activating factor 1 (Apaf1) in islets of mice treated with glycerate compared with control (Figure 4J). Cell counting assays demonstrated the growth inhibitory effect of glycerate in immortalized mouse pancreatic β-cell lines (Figures S4F and S4G). Treating these β-cell lines with glycerate also enhanced the mRNA expression of Casp8 and Apaf1 (Figure S4H).
Figure 4. The effect of fructose-derived glycerate on pancreatic islet cells.
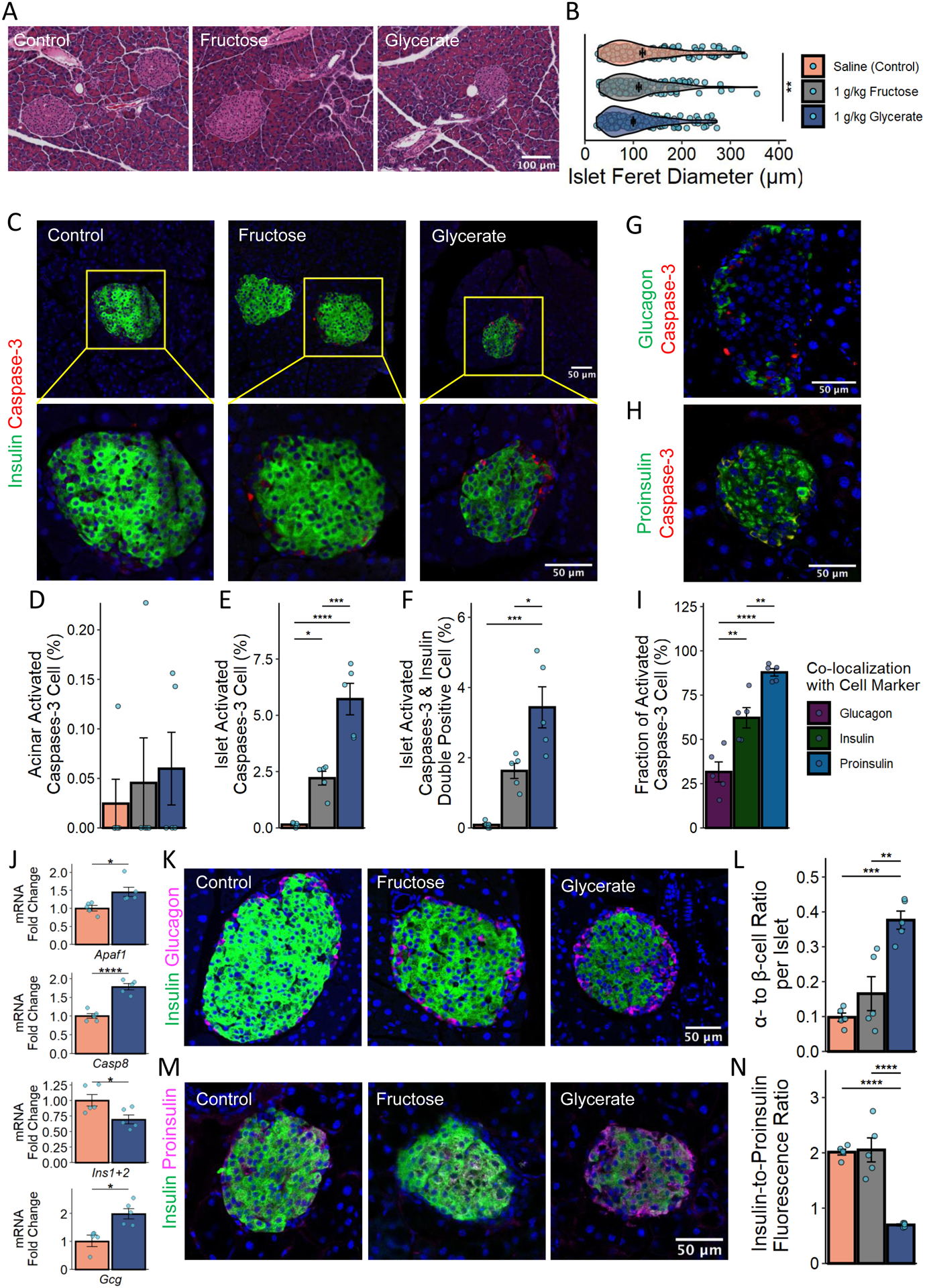
A & B. H&E staining of pancreas and quantification after 12 weeks of control (197 islets), fructose (159 islets), or glycerate (213 islets) treatment of mice fed on CD.
C - F. Insulin (green) and activated Caspase-3 (red) staining and the quantification of active Caspase-3 positive cells within the islet and other pancreatic regions.
G. Glucagon (green) and activated Caspase-3 (red) staining of the pancreas among mice receiving glycerate treatment.
H. Proinsulin (gree) and activated Caspase-3 (red) staining of the pancreas among mice receiving glycerate treatment.
I. Quantification of the proportion of activated caspase-3 positive cells that were also co-expressing the indicated pancreatic islet cell marker.
J. mRNA expression in isolated mouse islets after 12 weeks of treatment.
K & L. Insulin-Glucagon staining and the quantification of the α-cell to β-cell ratio.
M & N. Insulin-Proinsulin staining and the quantification of the insulin to proinsulin ratio. N = 5 mice for C-N. Data are represented as mean ± SEM. T-test was used for comparison between 2 groups and the post-hoc Tukey correction was performed after ANOVA was used for comparison more than 2 groups.*: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001. See also Figure S4.
Congruent with the model of glycerate-induced β-cell damage, daily injections reduced the number of insulin-positive cells within the pancreatic islet (Figures S4I and S4J) and the amount of mature insulin (Figures S4K and S4L). Moreover, in mice injected with glycerate (vs. fructose or control), a larger fraction of islet cells expressed glucagon (Figures S4I and S4J), which led to an increase in the α-cell to β-cell ratio that is commonly observed in diabetic pancreatic islets (Mezza et al., 2019) (Figures 4K and 4L). These results were further confirmed by decreased expression of Insulin I and Insulin II (Ins1+2) and increased expression of Glucagon (Gcg) mRNA in isolated islets from mice treated with glycerate (Figure 4J). Although islets from glycerate-treated mice maintained their expression of chromogranin A (Chga) in islets and displayed normal levels of proinsulin staining (Figures S4K–S4N), the decrease in the insulin-to-proinsulin ratio indicated insufficient post-translational processing of proinsulin or β-cell degranulation (Figures 4M and 4N). Taken together, these data suggest that high glycerate levels dysregulate the endocrine function of the pancreas via β-cell damage.
Discussion
The western diet, composed of large percentages of fat and fructose, has been associated with the development of T2DM. The combination of these two nutrients exacerbated metabolic syndromes in animal models (Softic et al., 2017). Fructose is known to promote lipid synthesis, but less is known about the reciprocal interaction (Bouwman et al., 2020). Our data suggest that HFD enhances adaptive intestinal fructose metabolism via sustained high mRNA levels of Glut5, Khk, and Aldob. The rapid kenetics of the observed adaptive response may have resulted from the suppression of mRNA degradation rather than transcriptional induction and warrant further study (Morita et al., 2019). Although fructose may remain unabsorbed by the CD-fed mouse intestine and instead be taken up by the colonic microbes (Jang et al., 2018), a large percentage of fructose is converted to metabolites (including glucose and glycerate) in the small intestine before entering the portal vein in HFD-fed mice. With both acute and chronic HFD feeding, absorbed fructose is released into the systemic circulation as glycerate that exerts negative effects on β-cells and insulin regulation. We also found that an HFD coupled with fructose water enhanced systemic levels of fructose-derived glycerate, the effect of which may accumulate over the long term to increase the risk of pancreas dysfunction.
Khk knockout studies have shown that hepatic fructose metabolism is a major contributor to steatosis (Andres-Hernando et al., 2020). Although detrimental effects to the liver are likely to be the primary consequences associated with fructose consumption and subsequent liver assimilation (Jang et al., 2018), glycerate from intestinal fructose metabolism may add a new piece to the puzzle of the disrupted whole-body metabolism. Therefore, a whole-body inhibition (vs. liver alone) of fructose metabolism may further protect mice from the development of diabetes. Studies have shown that T2DM is characterized by β-cell loss preceded by hyperinsulinemia (Prentki and Nolan, 2006), and our data suggests that glycerate promotes the gradual loss of functional β-cells in the pancreatic islet via apoptosis. Although the effects of upregulated glycerate on human whole-body metabolism are likely masked by insulin resistance in situations of hypercaloric intake (as also recapitulated by the HFD feeding in our in vivo experiments), we found the β-cell damage from glycerate is independent of insulin resistance. Thus, our model suggests that prolonged exposure of β-cells to glycerate, such as when following a western diet, may contribute to the eventual transition from the compensatory phase of β-cell expansion to the β-cell failure indicative of T2DM (Prentki and Nolan, 2006).
Fructose was first introduced to the western diet to control diabetes based on its low glycemic index (Hawkins et al., 2002). This study provides further evidence of a suppressive effect on innate insulin secretion by fructose via fructose-derived glycerate. A recent report that describes glycerate supplementation eliciting insulin secretion in vitro supports the idea of active uptake of glycerate by β-cells (Davis et al., 2020). We similarly observed acute enhancement of insulin secretion after 10 days of glycerate treatment (Figure 3G). However, the observed apoptosis of β-cells upon chronic exposure to glycerate might outweigh the acute insulin stimulatory effect of glycerate. The potential negative effect of glycerate is also consistent with the clinical association between missense mutations of glycerate kinase and diabetes.
Limitations of Study
Other direct and indirect effects of glycerate on other metabolically active organs merit further investigation to establish the systemic metabolic effects of glycerate (Zehavi et al., 2019). The effects of glycerate on other pancreatic islets cell populations in addition to the β-cell warrant further studies as we see up to one-third of apoptotic signals were from other islet cells (Rodriguez-Diaz et al., 2019; Unger Roger and Orci, 2010). Given the daily treatment regime rather than regular meal intakes by the mice, the 1 g/kg glycerate dose used in this study is at a supra-physiological level. The animals in our study cleared the supra-physiological glycerate load quickly and the effects of glycerate appear to be dose-dependent as a lower dose of glycerate injection resulted in milder effects on glucose and insulin homeostasis. However, the slow accumulation of these milder effects may resemble the slow and progressive development of diabetes in humans. As humans have lower metabolism rates when compared to mice, glycerate appearance and clearance from circulation after fructose consumption is likely slower (Agoston, 2017). Although the current study provides mounting evidence of glycerate-associated pancreatic islet damage via apoptosis, the detailed molecular cascades remain elusive and the effect of glycerate on islet cell proliferation needs further elucidation (Dai et al., 2017; Teta et al., 2005; Tschen et al., 2009). The glycerate kinase was reported to be responsible for glycerate clearance, but the aldehyde dehydrogenase upstream of glycerate is ill-defined. Additional investigation into the function of glycerate will require the identification of its receptor or transporter and enzymes. Future drug discovery targeting glycerate metabolism will rely on the elucidation of these components of glycerate metabolism.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiling Shen (xiling.shen@duke.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Metabolomics data have been deposited at NIH Common Fund’s National Metabolomics Data Repository (NMDR) and are publicly available as of the date of publication (Sud et al., 2016). Accession number is listed in the key resources table. Original western blot images and analyzed data are available as Data S1. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse Glut5 | NSJ Bioreagents | Cat# R30827 |
| Anti-Human GLUT5 | NSJ Bioreagents | Cat# R31175 |
| Anti-Khk | Thermo Fisher Scientific | Cat# PA5-29004, RRID: AB_2546480 |
| Anti-Aldob | Thermo Fisher Scientific | Cat# PA5-30218, RRID: AB_2547692 |
| Anti-β-Actin | Cell Signaling Technology | Cat# 12262, RRID: AB_2566811 |
| Anti-Insulin (mouse) | Thermo Fisher Scientific | Cat# 53-9769-82, RRID: AB_2574469 |
| Anti-Insulin (rabbit) | Cell Signaling Technology | Cat# 9016, RRID: AB_2797688 |
| Anti-Cleaved Caspase-3 | Cell Signaling Technology | Cat# 9664, RRID: AB_2070042 |
| Anti-Glucagon | Cell Signaling Technology | Cat# 2760, RRID: AB_659831 |
| Anti-Proinsulin | R and D Systems | Cat# MAB13361, RRID: AB_2126534 |
| Anti-Chromogranin A | Proteintech | Cat# 10529-1-AP, RRID: AB_2081122 |
| Anti-Glucagon (mouse) | Novus Biologicals | Cat# NBP2-21803AF647 |
| Biological samples | ||
| Human jejunum tissue | Duke Transplant Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Glyceric Acid | TCI | D0602 |
| D-(−)-Fructose | Sigma | F0127 |
| D-(+)-Glucose | Sigma | G8270 |
| Streptozocin | Sigma | S0130 |
| U-13C fructose | Sigma | 587621 |
| DL-Glyceric-2,3,3-d3 acid calcium salt dihydrate | Sigma | 616672 |
| D-Glucose-6,6-d2 | Sigma | 488720 |
| D-Fructose-1,1,3,4,5,6,6-d7 | Sigma | 729051 |
| PowerUp™ SYBR™ Green Master Mix | Thermo Fisher Scientific | A25742 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | 89900 |
| Halt™ Protease Inhibitor Cocktail | Thermo Fisher Scientific | 87786 |
| GlutaMAX™ | Thermo Fisher Scientific | 35050061 |
| HEPES | Thermo Fisher Scientific | 15630080 |
| Primocin® | InvivoGen | ant-pm-1 |
| B-27™ | Thermo Fisher Scientific | 12587010 |
| N-Acetyl-L-cysteine | Fisher | ICN19460305 |
| Murine EGF | PeproTech | AF-315-09 |
| Nicotinamide | Sigma | N0636 |
| Gastrin | AnaSpec | AS-64149 |
| Prostaglandin E2 (PGE2) | Cayman Chemicals | 14010 |
| A 83-01 | Sigma | SML0788 |
| SB202190 | LC Laboratories | S-1700 |
| Y27632 | ApexBio | A3008-200 |
| JAG-1 peptide | AnaSpec | AS-61298 |
| Sucrose | Fisher | BP220-1 |
| Sorbitol | Fisher | BP439-500 |
| EDTA | Thermo Fisher Scientific | 15575-038 |
| 1,4-Dithiothreitol (DTT) | Sigma | 10197777001 |
| Matrigel® Growth Factor Reduced Basement Membrane Matrix | Corning | 354230 |
| Rat Tail Collagen I | Corning | 354236 |
| Sodium palmitate | Sigma | P9767 |
| Sodium oleate | Sigma | O7501 |
| Bovine Albumin Fraction V (7.5% solution) | Thermo Fisher Scientific | 15260037 |
| Insulin human | Sigma | 1342106 |
| Collagenase P | Millipore Sigma | 11213865001 |
| Fluoro-Gel II Mounting Medium | EMS | 17985-50 |
| Critical Commercial assays | ||
| Quick-RNA Miniprep Kit | ZYMO | R1055 |
| RNAqueous™-Micro Total RNA Isolation Kit | Thermo Fisher Scientific | AM1931 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368814 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 |
| STELLUX® Chemi Rodent Insulin ELISA kit | ALPCO | 80-INSMR-CH01 |
| Cell Counting Kit 8 | Abcam | ab228554 |
| Deposited data | ||
| Metabolomics data | This paper | http://dx.doi.org/10.21228/M84127 |
| Raw and unprocessed data | This paper | Data S1 |
| Experimental models: Cell lines | ||
| L-WRN | ATCC | CRL-3276™ |
| NIT1 | ATCC | CRL-2055™ |
| βTC-6 | ATCC | CRL-11506™ |
| Experimental models: Organisms/strains | ||
| C57BL6/J | Jackson lab | 000664 |
| Khk-ciKO | Cholsoon Jang lab | N/A |
| Oligonucleotides | ||
| qPCR primers | This paper | Table S2 |
| Software and algorithms | ||
| Fiji 2.0.0 | Schindelin et al., 2012 | https://imagej.net |
| R version 4.0.3 (2020-10-10) | R Foundation | https://www.r-project.org |
| EI-MAVEN v0.11.0 | Agrawal et al., 2019 | https://www.elucidata.io/el-maven |
| MSiReader v1.03c | Bokhart et al., 2018 | https://msireader.wordpress.ncsu.edu/ |
| Other | ||
| High fat diet (HFD) | Research diet | D12492 |
| Control diet (CD) | Research diet | D12450J |
EXPERIMENT MODEL AND SUBJECT DETAILS
Cell lines
Two female mouse pancreatic β-cell lines, β-TC-6 and NIT-1, were used. β-TC-6 and NIT-1 cells were maintained and expanded in DMEM supplemented with 15% FBS and Ham’s F12K medium supplemented with 10% FBS respectively. The cells were cultured at 37°C with 5% CO2. The same conditions were also used during the experiments. Cell authentication was done by ATCC and experiments were performed with cells that were within 15 passages from the original ATCC stock.
2D Murine and Human Organoids
Crypts were isolated from the Jejunum of adult (8–12 week) C57BL6/J male mice as previously described. In brief, the jejunum was resected from the middle of the small intestine, flushed with cold shake buffer consisting of 10 uM Y27632 (ApexBio, with 5.6 mM Na2HPO4, 8 mM KH2PO4 96.2 mM NaCl, 1.6 mM KCl, 43.4 mM Sucrose, and 54.9 mM Sorbitol), opened longitudinally, and then chelated in murine isolation buffer (shake buffer plus 3 mM EDTA). After 15 minutes, the tissue was transferred to shake buffer and gently agitated for 2 minutes. The tissue fragments were transferred back to murine isolation buffer and chelated for an additional 30 minutes; crypt/villi units were then released from the tissue by rigorous shaking for 4 minutes. Crypts were separated from the solution using a 100 μm cell strainer. Crypts were plated on a collagen hydrogel (Wang et al., 2017) prepared from rat tail collagen (Corning) at 1 crypt/μm2 in an expansion medium (EM) consisting of 50% Advanced DMEM/F-12 (Thermo Scientific, with HEPES, GlutaMax, and Primocin) plus 50% L-WRN conditioned medium (home-made from L-WRN cells), B27 (ThermoFisher), 1 mM N-Acetyl-L-cysteine (Fisher), 50 ng/mL EGF (Peprotech), 10 mM Nicotinamide (Sigma), 10 nM Gastrin (AnaSpec), 10 nM PGE2 (Cayman Chemicals), 3 uM SB202190 (LC Laboratories), and 10 uM Y27632 (ApexBio). Organoids were grown into exposed organoids and passaged every 5–7 days. Passaged organoids were expanded to maturity (5–7 days) and treated for 24 hours in a growth factor-depleted differentiation medium (DM) consisting of 100% Advanced DMEM/F-12 (Thermo Scientific, with HEPES, GlutaMax, and Primocin) plus B27 (ThermoFisher), 1mM N-Acetyl-L-cysteine (Fisher), 50 ng/mL EGF (Peprotech), 10 mM Nicotinamide (Sigma), 10 nM Gastrin (AnaSpec), 10 nM PGE2 (Cayman Chemicals), and 500 nM A 83-01 (Sigma) supplemented with either BSA, oleic-BSA, or palmitic-BSA.
Human organoids were grown using crypts isolated from a segment of fresh transplant grade jejunum tissue from male donors (Duke Transplant Center) as previously described (Wang et al., 2017) Mucosa (5.1 g) was resected from a 4-cm segment of the small bowel, washed in shake buffer, minced, and chelated in human isolation buffer (shake buffer plus 2 mM EDTA and 20mM DTT) for 90 minutes. Crypt/villi units were released from the tissue fragments by rigorous shaking and isolated by filtration. Filtered crypt units were plated on collagen hydrogel (Wang et al., 2017) at 1 crypt/μm2 and grown in EM with passaging every 7–10 days. The resulting human organoids were converted from exposed to embedded (Matrigel, Corning) and back to exposed platforms, demonstrating renewal capacity. Late passage exposed organoids were expanded to maturity (7 days) and differentiated in DM for 24 hours supplemented with either BSA, oleic-BSA, or palmitic-BSA.
Animals
All animal procedures were reviewed and approved by the Duke University Institutional Care and Use Committee (Protocol Registry Number A235-18-10). Procedures were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals and all relevant regulations for animal testing and research.
All experiments were conducted on 8–12-week-old C57BL6/J male mice (The Jackson Laboratory). Mice were kept in the institution’s conventional animal housing with barriers and a controlled light cycle. In the feeding studies, animals were randomly assigned to high-fat diet (HFD, D12492, 60% Kcal) or control diet with matched sucrose content (CD, D12450J, 10% Kcal) groups upon arrival. Separate cohorts of 8-week-old C57BL6/J mice were fed CD or HFD and received a daily intraperitoneal (IP) injection of glycerate (adjusted to pH 7.4 with sodium hydroxide), fructose, or saline to mimic the everyday fructose ingestion and resultant spike in circulating glycerate and fructose. A single IP injection of the β-cell toxin streptozotocin (STZ) (100 mg/kg, Sigma) was used as a positive control for induction of diabetes. For ad lib fructose feeding experiments, mice were fed fructose in the drinking water (15% w/v) for 4 weeks.
METHOD DETAILS
Real-time PCR
Tissue from jejunum and liver were ground to homogenous powder, and total RNA was purified using a Quick-RNA Miniprep Kit (ZYMO). Total RNA was isolated from replicate murine and human organoids using an RNAqueous™-Micro Total RNA Isolation Kit (ThermoFisher). RNA samples were converted into cDNA with a High-Capacity cDNA Reverse Transcription Kit (ThermoFisher), and Real-time PCR was then performed on a StepOne™ Real-Time PCR System using PowerUp™ SYBR™ Green Master Mix (ThermoFisher). The primers used are listed in Table S2.
Western Blot
Ground tissues and organoid samples were lysed in ice-cold RIPA Lysis and Extraction Buffer (ThermoFisher) containing a protease inhibitor and phosphatase inhibitor cocktail (ThermoFisher). Protein concentrations were determined using a BCA Protein Assay Kit (ThermoFisher), and approximately 30 μg of each sample protein was loaded into NuPAGE™ 4 to 12%, Bis-Tris, Mini Protein Gel. The separated proteins were transferred to 0.45 μm PVDF Membrane at 30V for 2 hours. After transfer, the membranes were blocked with 5% nonfat milk and then incubated with primary antibodies overnight at 4 °C. After three washes with TBST, the blots were incubated for 1h with horseradish peroxidase-conjugated secondary antibodies (Abcam) for 1h at room temperature. After washing, the peroxidase signal of each blot was captured by ECL chemiluminescence (Bio-Rad).
Isotopically Tracing Ingested Fructose
After 3 days or 4 weeks of either HFD or CD feeding, each mouse was administrated a 1g/kg U-13C fructose oral gavage. Mice were anesthetized with 2% isoflurane, and their abdominal cavities were opened at the specified time. Abdominal tissues were displaced to identify the portal vein, and a 27G needle was used to immediately collect portal vein blood samples (~100 μL). Immediately after, cardiac blood was collected from the right ventricle using a 25G needle (~1 mL). Jejunum and liver tissues were quickly resected and snap-frozen in liquid nitrogen. Blood samples were placed on ice in the absence of anticoagulant for 20 minutes and centrifuged at 16,000 × g for 10 minutes at 4 °C to separate the serum and plasma fractions of each sample. Serum and tissue samples were kept at −80 °C until further analysis.
Intestine-specific Khk-c knockout mice and control littermates were gavaged with 1 g/kg U-13C fructose, and tail blood was collected at 0, 10, 30, 60, and 120 minutes after the gavage.
Metabolite Extraction
Metabolites were extracted using a protocol optimized for water-soluble polar metabolite analysis using liquid chromatography coupled with mass spectrometry. All extraction buffers were stored at −20°C prior to usage and immediately preceding the metabolite extraction.
For serum samples, a 10-μL serum aliquot was used for fructose tracing. Metabolites were extracted with 40 μL of ice-cold methanol and incubated at −20 °C for 20 minutes. The clean supernatant was collected after centrifugation for 10 minutes at the highest speed, and the leftover pellet was further treated with 200 μL cold extraction buffer (40:40:20 v/v/v methanol:acetonitrile:water solution) and left to incubate on crushed ice for an additional 10 minutes. Following an additional 10 minutes of centrifugation at the highest speed, the clean supernatant was collected and pooled with the supernatant from the first collection.
For tissue samples, the extraction buffer used was a (v/v/v) solution of 40:40:20 (methanol:acetonitrile:water) + 0.1 M formic acid. An aliquot volume equivalent to 20x the sample weight was added to the Eppendorf tube with the homogenized sample, vortexed for 10 seconds, and left to chill on crushed ice for 10 minutes. The samples were then centrifuged for 10 minutes at the highest speed at 4°C. The supernatant was transferred to a correspondingly labeled and chilled Eppendorf tube, and the process was repeated once more. The total volume of supernatant was then centrifuged for an additional 10 minutes. After centrifugation, a final 500μL aliquot of the homogenate was then pipetted to a second clean Eppendorf tube, to which 44μL of 15% (m/v) NH4HCO3 was added to neutralize the acid in the buffer and precipitate the protein. This is the final sample extract to be vialed and loaded to the instrument for analysis. Metabolite extracts were stored at −80 °C until analysis.
Metabolomics Analysis
UHPLC chromatography conditions. The HILIC separation was performed on a Vanquish Horizon UHPLC system (Thermo Fisher Scientific, Waltham, MA) with XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μm particle size, Waters, Milford, MA) using a gradient of solvent A (95%:5% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4), and solvent B (20%:80% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4). The gradient was 0 min, 100% B; 3 min, 100% B; 3.2 min, 90% B; 6.2 min, 90% B; 6.5 min, 80% B; 10.5 min, 80% B; 10.7 min, 70% B; 13.5 min, 70% B; 13.7 min, 45% B; 16 min, 45% B; 16.5 min, 100% B and 22 min, 100% B (Su et al., 2020). The flow rate was 300 μl/min. The injection volume was 5 μL and the column temperature was 25 °C. The autosampler temperature was set to 4°C and the injection volume was 5 μL.
Fullscan mass spectrometry. The full scan mass spectrometry analysis was performed on a Thermo Q Exactive PLUS with a HESI source which was set to a spray voltage of −2.7kV under negative mode and 3.5kV under positive mode. The sheath, auxiliary, and sweep gas flow rates of 40, 10, and 2 (arbitrary unit), respectively. The capillary temperature was set to 300°C, and the aux gas heater was 360°C. The S-lens RF level was 45. The m/z range was set to 72 to 1000 m/z under both positive and negative ionization mode. The AGC target was set to 3e6, and the maximum IT was 200 ms. The mass resolution (full-width half maximum) was set to 70,000 @ m/z = 200.
Mass Spectrometry Imaging by IR-MALDESI-MSI
Pancreas tissues were first equilibrated to −15°C then sectioned to 20 μm thickness using a Leica CM1950 cryostat (Buffalo Grove, IL, USA). Cut sections were then thaw-mounted on clean microscope slides (1 mm height, plain, Fisher Scientific, Pittsburgh, PA) and stored at −80 °C until IR-MALDESI-MSI analysis. Tissues were first blocked into four groups, each containing one replicate of each condition. Tissues were both cut and imaged in randomized order within these blocks to minimize sampling bias.
Mass spectrometry imaging (MSI) was performed using the infrared matrix-assisted laser desorption electrospray ionization (IR-MALDESI) source (Caleb Bagley et al., 2021; Robichaud et al., 2013) coupled to an Exploris 240 Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) to achieve high resolution and accurate mass measurements. To facilitate desorption of targeted analytes, a thin layer of ice was applied to the surface each tissue section by first purging the source enclosure with nitrogen gas to 10% humidity, stabilizing the sample stage at −8 °C, and exposing the stage to ambient humidity until the ice matrix formed (Robichaud et al., 2014). The enclosure was then resealed and purged again until humidity returned to 10%, where it was maintained throughout analysis.
To ablate targeted tissue regions a 2970-nm wavelength laser was used with a single burst of ten pulses to produce 1 mJ of energy at a rate of 10 kHz. X and Y stage movements of 100 μm were used to achieve oversampling (Nazari and Muddiman, 2015). Ablated analytes were post-ionized in the orthogonal electrospray plume, established by applying a voltage of approximately 3 kV to the electrospray solvent (1 mM acetic acid in 50:50 water/acetonitrile.) Mass spectrometry analysis of ionized molecules was performed in negative mode with internal calibrant used to achieve high mass accuracy (<2.5 ppm) within the 85–225 m/z range. Automatic gain control (AGC) was disabled. A mass resolution power of 240,000FWHM at 200 m/z was used with a fixed injection time (15 ms) to synchronize timing of the ablation plume with ion collection in the C-trap of the mass spectrometer.
Collected raw data files were converted from the XCalibur.RAW format to mzML files via the ProteoWizard tool, MSConvert (Chambers et al., 2012), and then to imzML format using imzMLConverter (Race et al., 2012). Images were generated from the imzML files in MSiReader v1.03c (Bokhart et al., 2018).
Intraperitoneal Glucose Tolerance Test
Intraperitoneal glucose tolerance tests (IPGTTs) were performed on mice after 4 and 12 weeks of daily IP injections. Animals were fasted overnight before a 1-μL sample of tail vein blood was taken and measured for baseline glucose levels (AlphaTRAK 2). Each animal then received a 2g/kg body weight IP bolus of glucose, with additional glucose measurements taken from tail vein blood 15, 30, 45, 60, 90, and 120 minutes after the injection.
Intraperitoneal Insulin Releasing Test
Intraperitoneal insulin releasing tests (IPIRTs) were performed on mice after 6 and 10 weeks of daily IP injections. Animals were fasted overnight before a ~20-μL blood sample was taken from the tail vein. Animals then received a 2g/kg body weight IP bolus of glucose. Additional blood samples were collected 5, 15, 30, and 60 minutes following the bolus. Insulin concentration was quantified using the STELLUX® Chemi Rodent Insulin ELISA kit (ALPCO).
Intraperitoneal Insulin Tolerance Test
Intraperitoneal insulin tolerance tests (IPITTs) were performed on mice after 8 weeks of daily IP injections. A 1-μL sample of tail vein blood was taken and measured for baseline glucose levels (AlphaTRAK 2). Each animal received a 1 U/kg body weight IP bolus of human insulin (Sigma), and additional 1-μL samples of tail vein blood was collected 15, 30, 45, and 60minutes after the bolus.
Isolation of Mouse Pancreatic Islets
The isolation of pancreatic islets from mice treated with daily IP injections of glycerate or saline for 12 weeks was performed as described previously (Li et al., 2009). Briefly, the mice were fully anesthetized, sterilized with 70% alcohol, and killed by cervical dislocation. An incision around the upper abdomen was made, and the ampulla was clamped. A 30G needle was inserted into the common bile duct, and 3 mL of 1mg/mL Collagenase P was slowly injected. The pancreas was removed, and the isolated islets were hand-picked following pancreas digestion. Total RNA was isolated from individual mice islets using a RNAqueous™-Micro Total RNA Isolation Kit (ThermoFisher). Real-time PCR was performed with the primers listed in Table S2.
Histological and Immunohistochemistry
The animals were sacrificed at the end of 12 weeks. Pancreas samples were cut and fixed with 4% paraformaldehyde for 24 hours, dehydrated in ethanol, and embedded in paraffin. Longitudinal pancreatic sections were cut at a 5-μm thickness, collected at 250-μm intervals, and mounted on polylysine-coated slides.
For immunofluorescence staining, N > 2 sections per treatment group (N = 5) were incubated with Anti-Insulin antibody (ThermoFisher), Anti-Cleaved Caspase-3 Antibody (CST), Anti-Glucagon Antibody (CST), Anti-Proinsulin Antibody (R and D Systems), and Anti-Chromogranin A Antibody (Proteintech) overnight at 4 °C. Bound primary antibodies were detected using secondary antibodies following a 1-hour incubation. The sections were then incubated with Fluoro-Gel II Mounting Medium (EMS), and images were taken using the Zeiss 780up confocal microscope. Islets were imaged with the grouping information blinded to the investigator, and individual islets (N > 50 per group) were used for quantitative analyses.
Cell proliferation assays
The dose-dependent response and time-series response of β-cell lines to glycerate treatment in vitro were performed in 96-well plates. Cell numbers were counted by CCK-8 cell counting kits (Abcam) 48 hours after treatment at specified doses (N = 3 – 4). PBS was used as the zero dose control. To trace the action of glycerate on cell growth over time, cells (N= 3 – 5) were treated with 5 mM glycerate, and cell counting was performed at 0 hours, 24 hours, and 48 hours. To quantify the mRNA responses upon glycerate treatments, cells were seeded on 24-well plates. Total mRNA extraction was performed 24 hours after treating the cells with 5 mM glycerate or PBS control using a Quick-RNA Miniprep Kit (ZYMO). Real-time PCR was performed with the primers listed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
qPCR and western blot quantification
The mRNA fold-change was deduced by the 2−ΔΔCt method, where ΔCt was normalized to the expression of actin. The intensity of detected protein bands was quantified by using Image J and normalized to the intensity of the actin band.
Metabolomic quantification
For LC-MS, the data is presented as total ion count unless otherwise specified. Peak picking was performed in EI-MAVEN with default parameters (Agrawal et al., 2019). Comparisons between the HFD and CD groups were done using the student’s t-test and adjusted by the Benjamini-Hochberg procedure. Due to the small sample size, we set the false discovery rate (FDR) to 0.2. Hierarchical clustering of metabolites was performed using the complete linkage method on the Euclidean distance.
For MSI data, abundances of stable isotope-labeled peaks (M+3 glycerate, 108.0293 m/z) were corrected for potential signal overlap from the naturally abundant species of each molecule using the percent isotope enrichment (PIE) tool in MSiReader (Mellinger et al., 2021). Isotopic distributions were computed using the R-based IsoSpec package (Lacki et al., 2020). Tissue regions of interest were selected to include pancreas tissue only and exclude peripheral fat tissue found in adjacent tissue staining. Wilcoxon rank-sum test was used to compare the abundance quantification between CD and HFD groups.
Genetic Association Analysis
Diabetes was defined according to the ICD code 250.x (ICD-9) or E10, E11, and E13 (ICD-10). Glycerate kinase allele statuses were inferred by the single nucleotide polymorphism (SNP) at any of rs144605797, rs150662353, rs9813489 loci. These are more common missense mutations with minor allele frequency between 0.02% – 1% in the BioVU cohort and have been predicted to have at least a moderate impact on protein function (Bastarache et al., 2018). All available imputed SNP array genotyping data were downloaded from the UK BioBank. A logistic regression model was fitted using a diabetes event as the dependent variable and glycerate kinase mutation carrier (dosage > 0.5 of any alternative alleles), age, sex, overweight status (BMI > 25), ethnicity, trunk fat, total cholesterol, high-density cholesterol, low-density cholesterol, and triglyceride level as independent variables.
Metabolic profiling quantification
For IPGTTs and IPIRT, the area under the curve (AUC) was deduced by the trapezoidal rule on raw glucose or insulin readings over time. The incremental area under the curve (iAUC) was deduced using similar methods but the glucose levels were first normalized by subtracting the baseline glucose level.
Pharmacokinetic and dose-response estimation
For estimating the half-life of glycerate upon IP injection, first-order kinetic was assumed as the plot of the natural logarithm of the concentration of glycerate versus time was a straight line. For median effective dose (ED50) estimation, the three-parameter log-logistic function within the R package drc was used with default parameters.
Immunofluorescence quantification
For the number of cells counted for activated caspase 3-, insulin-, proinsulin-, and glucagon-positive cells per islet, we first estimate the cell boundaries using the DAPI staining by Voronoi tessellation, then the expression of the individual marker within the cell was quantified. We defined positive signals as intensities that were 3-fold higher than the average background intensities. We defined a cell that was stained positively with insulin, proinsulin, glucagon, and activated caspase-3 if the positive signals filled >25%, >25%, >25%, and >15% of the cell area respectively. These parameters were chosen based on a preliminary experiment that is not included in the reported quantification analysis. The fractions of insulin- and glucagon-positive cells to DAPI-stained nuclei in islets were represented as β cells/islet and α cells/islet, respectively. Similar quantifications were performed on activated caspase-3 stained tissues. The ratio of α cells/islet to β cells/islet was represented as the α cell/β cell ratio per islet. Insulin and proinsulin fluorescence intensity was expressed as the mean value of fluorescence intensity within the islets. ChgA expression was also quantified by average fluorescence intensity. All quantitative measurements were conducted using Fuji software in a blinded fashion by two independent investigators.
Statistical Analysis
All data were presented as mean ± SEM. Statistical analyses were performed in R. Comparisons between two groups were performed by the student t-test unless otherwise specified. When more than two groups were compared, the post-hoc Tukey correction was performed after analysis of variance (ANOVA). Unless specified otherwise, asterisks in the figures indicate statistical significance *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
Supplementary Material
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figure 1 – 4 and S1 – S4.
Highlights.
High-fat diet increases fructose metabolism in the small intestine
Intestinal fructose metabolism releases glycerate into circulation
Circulating glycerate induces pancreatic islet cell damage
Circulating glycerate induces glucose intolerance
Acknowledgments
This study was supported by the National Institutes of Health grant (NIH) grants R35GM122465, R01DK119795, T32DK007568-30S1, R01GM087964, and R01AA029124, a Department of Defense (DOD) grant W81XWH1910676, National Research Foundation of Korea grants 2021R1A6A3A-14039681 and 2021R1A6A3A-14039132, an AASLD Foundation Pinnacle Research Award in Liver Disease, and an Edward Mallinckrodt, Jr. Foundation Award.
Inclusion and diversity
We worked to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Agoston DV (2017). How to Translate Time? The Temporal Aspect of Human and Rodent Biology. Front Neurol 8, 92–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S, Kumar S, Sehgal R, George S, Gupta R, Poddar S, Jha A, and Pathak S (2019). El-MAVEN: A Fast, Robust, and User-Friendly Mass Spectrometry Data Processing Engine for Metabolomics. Methods Mol Biol 1978, 301–321. [DOI] [PubMed] [Google Scholar]
- Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, and Lanaspa MA (2020). Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab 32, 117–127 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony G, White LW, and Landau BR (1969). Metabolism of D- and L-glyceraldehyde in adipose tissue: a stereochemical probe for glycerokinase activity. J Lipid Res 10, 521–527. [PubMed] [Google Scholar]
- Augustin LSA, Kendall CWC, Jenkins DJA, Willett WC, Astrup A, Barclay AW, Björck I, Brand-Miller JC, Brighenti F, Buyken AE, et al. (2015). Glycemic index, glycemic load and glycemic response: An International Scientific Consensus Summit from the International Carbohydrate Quality Consortium (ICQC). Nutrition, Metabolism and Cardiovascular Diseases 25, 795–815. [DOI] [PubMed] [Google Scholar]
- Bartley C, Brun T, Oberhauser L, Grimaldi M, Molica F, Kwak BR, Bosco D, Chanson M, and Maechler P (2019). Chronic fructose renders pancreatic β-cells hyper-responsive to glucose-stimulated insulin secretion through extracellular ATP signaling. American Journal of Physiology-Endocrinology and Metabolism 317, E25–E41. [DOI] [PubMed] [Google Scholar]
- Bastarache L, Hughey JJ, Hebbring S, Marlo J, Zhao W, Ho WT, Van Driest SL, McGregor TL, Mosley JD, Wells QS, et al. (2018). Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 359, 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhupathiraju SN, and Hu FB (2016). Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ Res 118, 1723–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhart MT, Nazari M, Garrard KP, and Muddiman DC (2018). MSiReader v1.0: Evolving Open-Source Mass Spectrometry Imaging Software for Targeted and Untargeted Analyses. J Am Soc Mass Spectrom 29, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomba L, Walter K, and Soranzo N (2017). The impact of rare and low-frequency genetic variants in common disease. Genome Biol 18, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman LMS, Nieuwenhuizen AG, Swarts HJM, Piga R, van Schothorst EM, and Keijer J (2020). Metabolic effects of the dietary monosaccharides fructose, fructose-glucose, or glucose in mice fed a starch-containing moderate high-fat diet. Physiol Rep 8, e14350–e14350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleb Bagley M, Garrard KP, and Muddiman DC (2021). The development and application of matrix assisted laser desorption electrospray ionization: The teenage years. Mass Spectrom Rev. [DOI] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Cohrs CM, Stertmann J, Bozsak R, and Speier S (2017). Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab 6, 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O’Keefe JH, and Brand-Miller J (2005). Origins and evolution of the Western diet: health implications for the 21st century. The American Journal of Clinical Nutrition 81, 341–354. [DOI] [PubMed] [Google Scholar]
- Curry DL (1989). Effects of Mannose and Fructose on the Synthesis and Secretion of Insulin. Pancreas 4. [DOI] [PubMed] [Google Scholar]
- Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, Phillips N, Levy SE, Greiner DL, Shultz LD, et al. (2017). Age-dependent human β cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. The Journal of Clinical Investigation 127, 3835–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JC, Alves TC, Helman A, Chen JC, Kenty JH, Cardone RL, Liu DR, Kibbey RG, and Melton DA (2020). Glucose Response by Stem Cell-Derived beta Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. Cell Rep 31, 107623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran M, Beemer FA, Bruinvis L, Ketting D, and Wadman SK (1987). D-glyceric acidemia: an inborn error associated with fructose metabolism. Pediatr Res 21, 502–506. [DOI] [PubMed] [Google Scholar]
- Forouhi NG, Krauss RM, Taubes G, and Willett W (2018). Dietary fat and cardiometabolic health: evidence, controversies, and consensus for guidance. BMJ 361, k2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas Joel T., Miao J, Chanda D, Wang Y, Zhao E, Haas Mary E., Hirschey M, Vaitheesvaran B, Farese Robert V., Kurland Irwin J., et al. (2012). Hepatic Insulin Signaling Is Required for Obesity-Dependent Expression of SREBP-1c mRNA but Not for Feeding-Dependent Expression. Cell Metabolism 15, 873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannou SA, Haslam DE, McKeown NM, and Herman MA (2018). Fructose metabolism and metabolic disease. J Clin Invest 128, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins M, Gabriely I, Wozniak R, Vilcu C, Shamoon H, and Rossetti L (2002). Fructose improves the ability of hyperglycemia per se to regulate glucose production in type 2 diabetes. Diabetes 51, 606–614. [DOI] [PubMed] [Google Scholar]
- Herman MA, and Samuel VT (2016). The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol Metab 27, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, et al. (2012). Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A 109, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, and Rabinowitz JD (2018). The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab 27, 351–361 e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Wada S, Yang S, Gosis B, Zeng X, Zhang Z, Shen Y, Lee G, Arany Z, and Rabinowitz JD (2020). The small intestine shields the liver from fructose-induced steatosis. Nat Metab 2, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang DH, et al. (2018). Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol 68, 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, Sargsyan A, Kou HH, Fowler AJ, Liang G, and Herman MA (2017). Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh HL, Kang HJ, Meissen JK, Blatnik M, et al. (2016). ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest 126, 4372–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo H-Y, Miyashita M, Simon Cho BH, and Nakamura MT (2009). Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochemical and Biophysical Research Communications 390, 285–289. [DOI] [PubMed] [Google Scholar]
- Koo HY, Wallig MA, Chung BH, Nara TY, Cho BH, and Nakamura MT (2008). Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim Biophys Acta 1782, 341–348. [DOI] [PubMed] [Google Scholar]
- Kyriazis GA, Soundarapandian MM, and Tyrberg B (2012). Sweet taste receptor signaling in beta cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proceedings of the National Academy of Sciences of the United States of America 109, E524–E532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacki MK, Valkenborg D, and Startek MP (2020). IsoSpec2: Ultrafast Fine Structure Calculator. Anal Chem 92, 9472–9475. [DOI] [PubMed] [Google Scholar]
- Laughlin MR (2014). Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 6, 3117–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DS, Yuan YH, Tu HJ, Liang QL, and Dai LJ (2009). A protocol for islet isolation from mouse pancreas. Nat Protoc 4, 1649–1652. [DOI] [PubMed] [Google Scholar]
- Li X, Meng X, Timofeeva M, Tzoulaki I, Tsilidis KK, Ioannidis JP, Campbell H, and Theodoratou E (2017). Serum uric acid levels and multiple health outcomes: umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. Bmj 357, j2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li T, Liao Y, Wang Y, Gao Y, Hu H, Huang H, Wu F, Chen YG, Xu S, et al. (2020). Triose Kinase Controls the Lipogenic Potential of Fructose and Dietary Tolerance. Cell Metab 32, 605–618 e607. [DOI] [PubMed] [Google Scholar]
- Mellinger AL, Garrard KP, Khodjaniyazova S, Rabbani ZN, Gamcsik MP, and Muddiman DC (2021). Multiple Infusion Start Time Mass Spectrometry Imaging of Dynamic SIL-Glutathione Biosynthesis Using Infrared Matrix-Assisted Laser Desorption Electrospray Ionization. J Proteome Res. [DOI] [PubMed] [Google Scholar]
- Mezza T, Cinti F, Cefalo CMA, Pontecorvi A, Kulkarni RN, and Giaccari A (2019). β-Cell Fate in Human Insulin Resistance and Type 2 Diabetes: A Perspective on Islet Plasticity. Diabetes 68, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Siddiqui N, Katsumura S, Rouya C, Larsson O, Nagashima T, Hekmatnejad B, Takahashi A, Kiyonari H, Zang M, et al. (2019). Hepatic posttranscriptional network comprised of CCR4–NOT deadenylase and FGF21 maintains systemic metabolic homeostasis. Proceedings of the National Academy of Sciences 116, 7973–7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazari M, and Muddiman DC (2015). Cellular-level mass spectrometry imaging using infrared matrix-assisted laser desorption electrospray ionization (IR-MALDESI) by oversampling. Anal Bioanal Chem 407, 2265–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, et al. (2014). Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. The Lancet 384, 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DM, and El-Serag HB (2010). The epidemiology of obesity. Gastroenterol Clin North Am 39, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomer X, Pizarro-Delgado J, Barroso E, and Vázquez-Carrera M (2018). Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol Metab 29, 178–190. [DOI] [PubMed] [Google Scholar]
- Parilla JH, Willard JR, Barrow BM, and Zraika S (2018). A Mouse Model of Beta-Cell Dysfunction as Seen in Human Type 2 Diabetes. J Diabetes Res 2018, 6106051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, and Nolan CJ (2006). Islet β cell failure in type 2 diabetes. The Journal of Clinical Investigation 116, 1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race AM, Styles IB, and Bunch J (2012). Inclusive sharing of mass spectrometry imaging data requires a converter for all. J Proteomics 75, 5111–5112. [DOI] [PubMed] [Google Scholar]
- Rendeiro C, Masnik AM, Mun JG, Du K, Clark D, Dilger RN, Dilger AC, and Rhodes JS (2015). Fructose decreases physical activity and increases body fat without affecting hippocampal neurogenesis and learning relative to an isocaloric glucose diet. Scientific Reports 5, 9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud G, Barry JA, Garrard KP, and Muddiman DC (2013). Infrared matrix-assisted laser desorption electrospray ionization (IR-MALDESI) imaging source coupled to a FT-ICR mass spectrometer. J Am Soc Mass Spectrom 24, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud G, Barry JA, and Muddiman DC (2014). IR-MALDESI mass spectrometry imaging of biological tissue sections using ice as a matrix. J Am Soc Mass Spectrom 25, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Diaz R, Tamayo A, Hara M, and Caicedo A (2019). The Local Paracrine Actions of the Pancreatic α-Cell. Diabetes 69, 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, et al. (2017). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 127, 4059–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, Fujisaka S, Serra D, Herrero L, Willoughby J, et al. (2019). Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab 30, 735–753 e734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis AR, Kennedy NJ, Xin X, Zhou F, Ficarro SB, Yap YS, Matthews BJ, Lauffenburger DA, White FM, Marto JA, et al. (2017). Hepatic Dysfunction Caused by Consumption of a High-Fat Diet. Cell Rep 21, 3317–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Chiles E, Maimouni S, Wondisford FE, Zong WX, and Song C (2020). In-Source CID Ramping and Covariant Ion Analysis of Hydrophilic Interaction Chromatography Metabolomics. Anal Chem 92, 4829–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS, et al. (2016). Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res 44, D463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, and Kushner JA (2005). Very slow turnover of beta-cells in aged adult mice. Diabetes 54, 2557–2567. [DOI] [PubMed] [Google Scholar]
- Tschen S-I, Dhawan S, Gurlo T, and Bhushan A (2009). Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 58, 1312–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger Roger H, and Orci L (2010). Paracrinology of islets and the paracrinopathy of diabetes. Proceedings of the National Academy of Sciences 107, 16009–16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnikrishnan R, Pradeepa R, Joshi SR, and Mohan V (2017). Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 66, 1432–1442. [DOI] [PubMed] [Google Scholar]
- Van Schaftingen E (1989). D-Glycerate kinase deficiency as a cause of D-glyceric aciduria. FEBS Letters 243, 127–131. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang J, Liu M-M, Wang D, Liu Y-Q, Zhao Y, Huang M-M, Liu Y, Sun J, and Dong G-H (2012). Epidemiology of general obesity, abdominal obesity and related risk factors in urban adults from 33 communities of northeast china: the CHPSNE study. BMC Public Health 12, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, DiSalvo M, Gunasekara DB, Dutton J, Proctor A, Lebhar MS, Williamson IA, Speer J, Howard RL, Smiddy NM, et al. (2017). Self-renewing Monolayer of Primary Colonic or Rectal Epithelial Cells. Cellular and molecular gastroenterology and hepatology 4, 165–182.e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehavi Y, Mandel H, Eran A, Ravid S, Abu Rashid M, Jansen EEW, Wamelink MMC, Saada A, Shaag A, Elpeleg O, et al. (2019). Severe infantile epileptic encephalopathy associated with D-glyceric aciduria: report of a novel case and review. Metab Brain Dis 34, 557–563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figure 1 – 4 and S1 – S4.
Data Availability Statement
Metabolomics data have been deposited at NIH Common Fund’s National Metabolomics Data Repository (NMDR) and are publicly available as of the date of publication (Sud et al., 2016). Accession number is listed in the key resources table. Original western blot images and analyzed data are available as Data S1. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse Glut5 | NSJ Bioreagents | Cat# R30827 |
| Anti-Human GLUT5 | NSJ Bioreagents | Cat# R31175 |
| Anti-Khk | Thermo Fisher Scientific | Cat# PA5-29004, RRID: AB_2546480 |
| Anti-Aldob | Thermo Fisher Scientific | Cat# PA5-30218, RRID: AB_2547692 |
| Anti-β-Actin | Cell Signaling Technology | Cat# 12262, RRID: AB_2566811 |
| Anti-Insulin (mouse) | Thermo Fisher Scientific | Cat# 53-9769-82, RRID: AB_2574469 |
| Anti-Insulin (rabbit) | Cell Signaling Technology | Cat# 9016, RRID: AB_2797688 |
| Anti-Cleaved Caspase-3 | Cell Signaling Technology | Cat# 9664, RRID: AB_2070042 |
| Anti-Glucagon | Cell Signaling Technology | Cat# 2760, RRID: AB_659831 |
| Anti-Proinsulin | R and D Systems | Cat# MAB13361, RRID: AB_2126534 |
| Anti-Chromogranin A | Proteintech | Cat# 10529-1-AP, RRID: AB_2081122 |
| Anti-Glucagon (mouse) | Novus Biologicals | Cat# NBP2-21803AF647 |
| Biological samples | ||
| Human jejunum tissue | Duke Transplant Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Glyceric Acid | TCI | D0602 |
| D-(−)-Fructose | Sigma | F0127 |
| D-(+)-Glucose | Sigma | G8270 |
| Streptozocin | Sigma | S0130 |
| U-13C fructose | Sigma | 587621 |
| DL-Glyceric-2,3,3-d3 acid calcium salt dihydrate | Sigma | 616672 |
| D-Glucose-6,6-d2 | Sigma | 488720 |
| D-Fructose-1,1,3,4,5,6,6-d7 | Sigma | 729051 |
| PowerUp™ SYBR™ Green Master Mix | Thermo Fisher Scientific | A25742 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | 89900 |
| Halt™ Protease Inhibitor Cocktail | Thermo Fisher Scientific | 87786 |
| GlutaMAX™ | Thermo Fisher Scientific | 35050061 |
| HEPES | Thermo Fisher Scientific | 15630080 |
| Primocin® | InvivoGen | ant-pm-1 |
| B-27™ | Thermo Fisher Scientific | 12587010 |
| N-Acetyl-L-cysteine | Fisher | ICN19460305 |
| Murine EGF | PeproTech | AF-315-09 |
| Nicotinamide | Sigma | N0636 |
| Gastrin | AnaSpec | AS-64149 |
| Prostaglandin E2 (PGE2) | Cayman Chemicals | 14010 |
| A 83-01 | Sigma | SML0788 |
| SB202190 | LC Laboratories | S-1700 |
| Y27632 | ApexBio | A3008-200 |
| JAG-1 peptide | AnaSpec | AS-61298 |
| Sucrose | Fisher | BP220-1 |
| Sorbitol | Fisher | BP439-500 |
| EDTA | Thermo Fisher Scientific | 15575-038 |
| 1,4-Dithiothreitol (DTT) | Sigma | 10197777001 |
| Matrigel® Growth Factor Reduced Basement Membrane Matrix | Corning | 354230 |
| Rat Tail Collagen I | Corning | 354236 |
| Sodium palmitate | Sigma | P9767 |
| Sodium oleate | Sigma | O7501 |
| Bovine Albumin Fraction V (7.5% solution) | Thermo Fisher Scientific | 15260037 |
| Insulin human | Sigma | 1342106 |
| Collagenase P | Millipore Sigma | 11213865001 |
| Fluoro-Gel II Mounting Medium | EMS | 17985-50 |
| Critical Commercial assays | ||
| Quick-RNA Miniprep Kit | ZYMO | R1055 |
| RNAqueous™-Micro Total RNA Isolation Kit | Thermo Fisher Scientific | AM1931 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368814 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 |
| STELLUX® Chemi Rodent Insulin ELISA kit | ALPCO | 80-INSMR-CH01 |
| Cell Counting Kit 8 | Abcam | ab228554 |
| Deposited data | ||
| Metabolomics data | This paper | http://dx.doi.org/10.21228/M84127 |
| Raw and unprocessed data | This paper | Data S1 |
| Experimental models: Cell lines | ||
| L-WRN | ATCC | CRL-3276™ |
| NIT1 | ATCC | CRL-2055™ |
| βTC-6 | ATCC | CRL-11506™ |
| Experimental models: Organisms/strains | ||
| C57BL6/J | Jackson lab | 000664 |
| Khk-ciKO | Cholsoon Jang lab | N/A |
| Oligonucleotides | ||
| qPCR primers | This paper | Table S2 |
| Software and algorithms | ||
| Fiji 2.0.0 | Schindelin et al., 2012 | https://imagej.net |
| R version 4.0.3 (2020-10-10) | R Foundation | https://www.r-project.org |
| EI-MAVEN v0.11.0 | Agrawal et al., 2019 | https://www.elucidata.io/el-maven |
| MSiReader v1.03c | Bokhart et al., 2018 | https://msireader.wordpress.ncsu.edu/ |
| Other | ||
| High fat diet (HFD) | Research diet | D12492 |
| Control diet (CD) | Research diet | D12450J |