

Summary
Uncovering the cis-regulatory code that governs when and how much each gene is transcribed in a given genome and cellular state remains a central goal of biology. Here, we discuss major layers of regulation that influence how transcriptional outputs are encoded by DNA sequence and cellular context. We first discuss how transcription factors bind specific DNA sequences in a dosage-dependent and cooperative manner, and then proceed to the cofactors that facilitate transcription factor function and mediate the activity of modular cis-regulatory elements such as enhancers, silencers, and promoters. We then consider the complex and poorly understood interplay of these diverse elements within regulatory landscapes and its relationships with chromatin states and nuclear organization. We propose that a mechanistically informed, quantitative model of transcriptional regulation that integrates these multiple regulatory layers will be the key to ultimately cracking the cis-regulatory code.
eTOC blurb
The cis-regulatory code dictates how DNA sequence controls quantitative transcription levels of each gene depending on cellular context. Kim and Wysocka review progress and challenges in understanding the layers of this code, from transcription factor binding to cofactor recruitment and ultimately cis-regulatory element specificity and function within complex regulatory landscapes.
One of the fundamental goals of biology is to understand how organisms decode genetic information into phenotypes—that is, unique biological characteristics and functions. This decoding occurs over distinct steps; as Francis Crick hypothesized in his Central Dogma1, beginning with the linear sequence information in DNA being transferred into RNA and then into protein. The most archetypal of these steps is encapsulated by the genetic code, which describes how each sequence of nucleotides in a messenger RNA molecule is translated into a string of amino acids2.
Since the seminal discovery that the protein-coding regions of the genome remain largely conserved between humans and chimpanzees, it has long been postulated that phenotypic divergence between and within closely related species arises in large part from quantitative changes to gene expression3. These changes are to a great extent genetically encoded, as evidenced by high heritability of many phenotypic traits in different species and within human populations, suggesting the existence of another DNA-based code that dictates them4. This code is often referred to as the ‘cis-regulatory code’ because gene expression is controlled by the regulatory DNA elements which typically act in ‘cis’, meaning at the same chromosomal allele (and, as discussed below, usually within the same chromosomal neighborhood) as the gene they regulate. In contrast to the genetic code, however, the cis-regulatory code remains ill-defined and difficult to crack. In this review, we compare and contrast the genetic and cis-regulatory codes, discuss features of the cis-regulatory code that make it challenging to comprehensively decipher, outline our current understanding of these features, and consider approaches moving forward.
What is the cis-regulatory code?
The term ‘code’ implies conversion of one form of information to another, akin to the mapping between nucleotides to amino acids in the genetic code. Broadly, the cis-regulatory code can be described as the mapping between DNA sequence and gene expression level. (For simplicity, in this review we will equate the gene expression level with transcriptional activity; nonetheless it is important to note that post-transcriptional regulatory mechanisms also play key roles in gene regulation and in phenotypic divergence between and within species). A solution to the code should be predictive; as the genetic code allows one to predict the amino acid sequence given a messenger RNA sequence, the cis-regulatory code should allow one to predict transcription levels from DNA sequence. However, this definition remains vague: How much DNA sequence is needed to achieve a comprehensive prediction of gene expression? Is it adequate to predict transcription of one gene at a time? And transcription in what cellular contexts? This ambiguity reflects three major challenges of the cis-regulatory code, which contrast three fortuitous features of the genetic code that facilitated efforts to decipher it: the genetic code is universal, modular, and qualitative. In contrast, as we discuss below, the cis-regulatory code is context-dependent, in part modular, but with complex and poorly understood relationships between modules, and quantitative.
A single, universal genetic code is used in all cell types in an organism and among virtually all species, with minor variations in organelles and prokaryotes, reflecting the early evolution of translational machinery2. The cis-regulatory code is instead highly dependent on cellular context, because the transcription factors that directly read DNA sequences differ from one cell type to another5, as do epigenetic marks such as DNA methylation that can modulate transcription factor binding to DNA6. Though it can be tempting to treat the hundreds of major cell types as separate identities, single cell studies have demonstrated how they are linked by uncountable transient intermediate states that can occur naturally in development or only in diseased or experimentally perturbed states. Ultimately, a solved cis-regulatory code would be integrated across the continuum of cell states and be able to predict how perturbations to cell state affect transcription in development and disease.
A second challenge of the cis-regulatory code is the size of the input DNA sequence. In the genetic code, each RNA molecule and the triplet codons within each open reading frame are modular—they are independent (one codon does not affect any others, except stop codons) and non-overlapping. But in the cis-regulatory code, is the entire genome necessary to predict the expression level of all genes? Many have focused on the more operational prediction of the impact of short DNA sequences on the expression of one reporter gene7, as this is experimentally feasible. Indeed, many short cis-regulatory elements such as the 480 bp minimal even-skipped stripe 2 enhancer8 are capable of individually regulating transcription and can often be treated as modular units9. However, such fragments can fail to fully explain the activity of larger genomic regions10, as many cis-regulatory elements interact physically or epistatically, across megabases of DNA (e.g. at the SOX911 and MYC loci12) and even across chromosomes (e.g. during olfactory receptor selection)13. Thus, a complete solution to the cis-regulatory code must be able to predict how large genomic regions, each containing tens or hundreds of cis-regulatory elements with complex relationships among each other, control the expression of multiple genes.
A third twist to the cis-regulatory code is that unlike the qualitative genetic code governing the identity of translated peptides, the cis-regulatory code aims to describe the quantitative levels of transcription. Genes are not simply ‘on’ or ‘off’ but exhibit precise levels of transcription controlled by the frequency of RNA polymerase molecules traversing the gene14. Transcription occurs in ‘bursts’ whose frequency and amplitude are subject to regulation, but which are inherently stochastic15. As a result of this discontinuous process, the two gene alleles within the same cell (even if genetically identical) can have distinct transcriptional status at any given moment in time, whereas within a population of cells of the same ‘type’, transcript levels can vary widely between individual cells. Yet, even relatively subtle changes in gene expression (~2-fold or less) can dramatically impact development or increase risk of disease. For example, modest downregulation of SOX9 causes the craniofacial syndrome Pierre Robin Sequence11, while upregulation of PDGFRA16 or MYC17 accelerate cell proliferation and can drive cancer. These examples underscore the importance of quantitatively precise regulation of transcription. Thus, the ultimate goal of solving the cis-regulatory code requires quantitatively accurate predictions of transcription levels.
Given the aforementioned context-dependent, complex, and quantitative nature of the cis-regulatory code, can we understand how precise transcription levels of more than 20,000 genes across essentially uncountable cell states are encoded in the 3 billion bases of the human genome? Although this goal still remains elusive, the field has made enormous progress in understanding the broad rules by which DNA sequences encode transcriptional outputs. For the purpose of this review, we divide these mechanisms into four levels at which they operate, from smallest to largest in genomic scale: 1) the reading of DNA sequence by transcription factors, 2) the modular function of individual cis-regulatory elements, 3) interactions and compatibility between distal cis-regulatory elements and their target promoters, and 4) relationships among diverse cis-regulatory elements within a complex regulatory landscape (Figure 1). Below, we discuss these four regulatory levels in turn.
Figure 1.
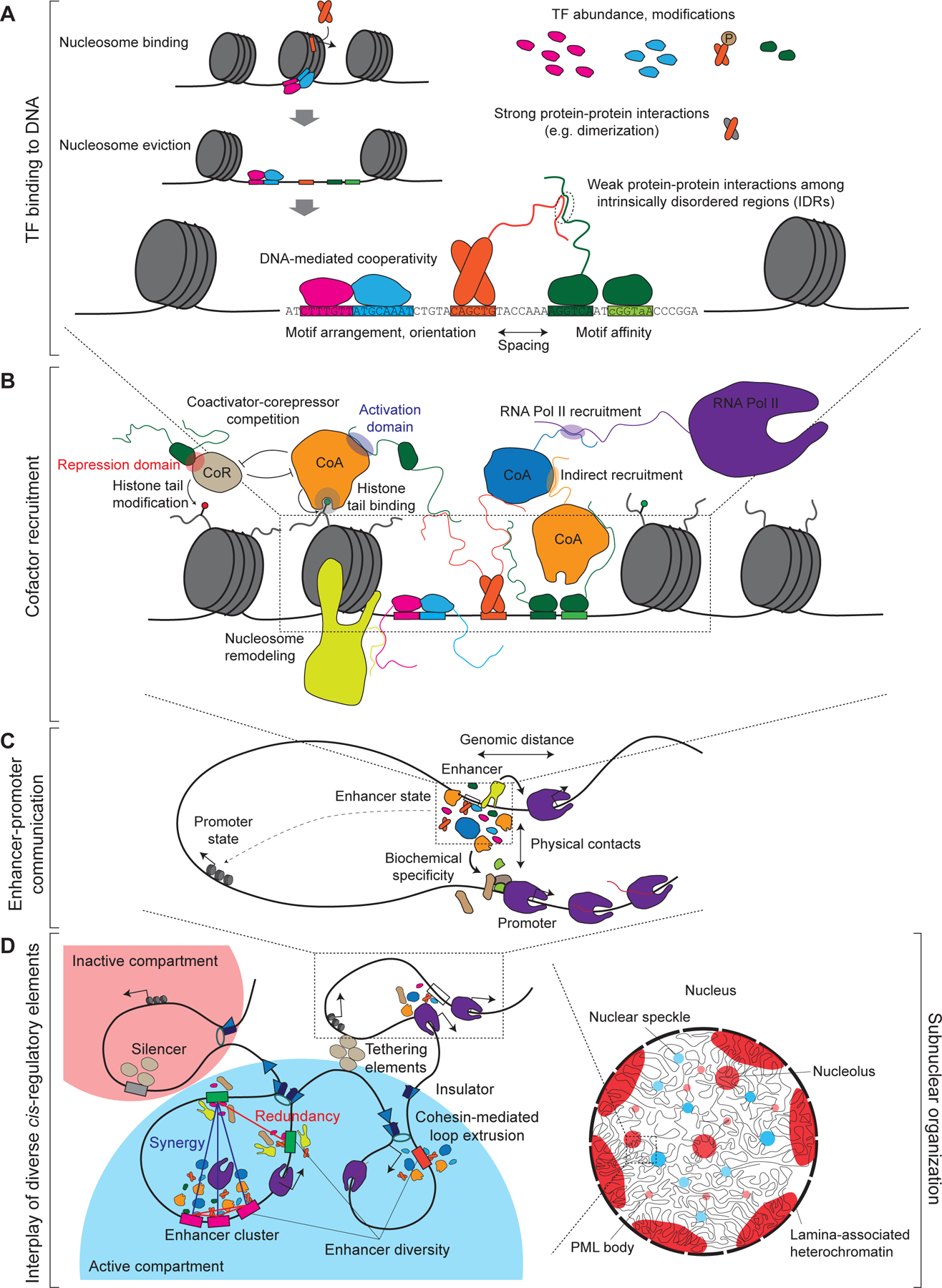
Four layers of the cis-regulatory code. A. TF binding to DNA depends on the sequence recognition and ability to overcome the nucleosomal barrier through direct association with nucleosome and/or cooperative binding of multiple TFs in turn facilitating nucleosome eviction. TF occupancy is dependent on TF levels, posttranslational modifications, protein-protein interactions (which can either be structured or mediated by weak-affinity interactions among the intrinsically disordered regions, IDRs), and is regulated DNA-mediated cooperativity with other TFs, which is itself governed by cis-regulatory features such as motif arrangement, spacing, and affinity. B. TFs function at modular cis-regulatory elements by recruiting cofactors with diverse and sometimes competing functions such as coactivators (CoA) versus corepressors (CoR). Many interactions between TFs and coactivators are mediated by IDRs. C. Enhancers and other distal cis-regulatory elements selectively regulate promoters, depending on multiple features such as genomic distance, enhancer and promoter state, biochemical specificity between enhancer- and promoter-associated proteins and physical contacts. See also Figure 2. D. Diverse regulatory elements, including enhancers, promoters, silencers, insulators and tethering elements interact with each other physically and/or epistatically (e.g. redundancy or synergy) in the context of local chromosomal neighborhood and a spatially organized nucleus.
From DNA sequence to transcription factor (TF) binding
TFs bind a subset of their motifs
The minimal unit of the cis-regulatory code—paralleling the codon of the genetic code—is the transcription factor binding site (TFBS). Transcription factors contain typically structured and evolutionarily conserved DNA binding domains that recognize and bind a 6–12 bp DNA sequence called a TF ‘motif’5. TF motifs are often depicted as a sequence logo or position weight matrix (PWM) that represents the degeneracy in TF binding specificity. The deep conservation of DNA binding domains and high-throughput methods for measuring TF sequence specificities in vitro have enabled the cataloguing of roughly 1600 TFs in humans (though more may exist), and characterizing binding motifs for a large fraction of these known TFs5.
Yet, TF binding motifs are insufficient to predict DNA binding in vivo for most TFs. While most TF binding sites contain at least an imperfect match for their preferred motifs, most TFs bind only a small fraction of their motif matches in the genome18. While efforts to better encapsulate TF specificity by including nucleotides flanking the core motif19 or using more complex representations of sequence preference (e.g. dinucleotide motifs, DNA shape20) can improve predictions of genomic binding for some TFs, the best predictor of in vivo binding for most TFs is chromatin accessibility as measured by DNase-seq21,22 or ATAC-seq23. This observation has largely been attributed to the occupancy of DNA by nucleosomes that must be outcompeted or evicted either by so-called ‘pioneer’ factors that possess a special capacity to bind nucleosomal DNA24 or combinations of co-bound TFs25,26.
TFs bind DNA in nucleosomal context
The idea of pioneer factors was conceived as an explanation for TFs that bound enhancer sequences in a closed chromatin state preceding subsequent activation during development27. Thus, in their original definition pioneer factors are able to both access nucleosome-bound DNA and facilitate enhancer competence for binding of other TFs28. Consequently, pioneer factors are associated with TF cocktails capable of inducing cellular reprogramming upon overexpression, such as the OSKM (Oct4, Sox2, Klf4, c-Myc) pluripotency factors29, as their nucleosome-binding ability30 is thought to allow them to function outside of their native cell state. TFs clearly vary in their capacity to bind nucleosomal DNA in vitro31,32 and closed chromatin in vivo33, with variation even among paralogs with conserved DNA binding domains.
Although pioneering activity is often described as a binary trait of TFs that allows binding to any cognate motif regardless of nucleosome occupancy, multiple lines of evidence suggest that pioneering is both mechanistically diverse and highly context-dependent. First, pioneer factors span many TF families (i.e. protein folds) and can bind nucleosomal DNA with different positional preferences (e.g. near nucleosomal ends, at the dyad, or periodically along the exposed sections of the DNA major groove)31,32,34. Second, the catalog of pioneer vs. non-pioneer TFs remains far from complete, and the precise distinction remains unclear. A recent study showed that a well-characterized pioneer factor, FOXA1, and a TF described as a non-pioneer factor, HNF4A, were equally capable of opening chromatin upon overexpression in a context in which neither is normally expressed, to a degree dependent on TF expression level35. While pioneering mechanisms often invoke direct functions such as bending DNA to unwrap it from histones34, pioneer TFs can also recruit nucleosome remodelers36. Whether pioneering activity is dependent on remodeler recruitment and whether remodelers might allow non-nucleosome-binding TFs to open chromatin remains largely unexplored. However, a recent in vitro study on the hematopoietic TF PU.1 provided an early proof-of-principle that pioneer factors can in fact facilitate activity of nucleosome remodelers at specific DNA sites—in this case, in a manner dependent not on the DNA binding domain, but on the intrinsically disordered region of PU.137. And third, pioneer factor activity is context-dependent: even pioneer TFs like OCT4 or SOX2 usually bind distinct sites in different cell types, reflecting cooperativity with other TFs that is required at least for stable binding38,39. Thus, the mechanism and extent of nucleosomal DNA binding appears to be one of several regulatory layers controlling which DNA sequences TFs can bind.
TF cooperate to bind DNA
A key (and importantly, non-exclusive) alternative to the pioneer factor model is that multiple TFs cooperate to outcompete nucleosomes together by binding within the same nucleosomal distance of ~150 bp25. While the idea of cooperativity among multiple co-bound TF molecules is not new, the precise nature of this cooperativity remains debated. Initial studies of the INFβ enhancer suggested that an array of different TFs must bind in tight spacing to allow protein-protein interactions that mediate cooperativity40. However, subsequent studies revealed that different enhancers have diverse sets of bound activators in different arrangements, in line with the more flexible nucleosome-mediated cooperativity. Even in the original enhanceosome, TFs make few protein-protein contacts41; instead many TFs have been shown to cooperate via DNA-mediated cooperativity42, in which specific TFs bind adjacently in certain arrangements and spacings to stabilize joint DNA binding.
A major challenge for the field has been to assess the relative extents of these forms of cooperativity (nucleosome-mediated, protein-protein interactions, DNA-mediated) among natively functioning enhancers and other cis-regulatory elements. Analysis of TF motif positions within nucleosome-depleted regions showed that different TFs tend to be at different positions, e.g. with known nucleosome-binding factors enriched at the edges of the nucleosome depleted area, suggesting that TFs play distinct functional roles43. Recent technological advances have enabled improved inference of cooperative interactions without relying on finding enrichment of specific motif arrangements or spacings. Convolutional neural networks and other deep learning models can learn cooperative interactions de novo, in an interpretable manner, from training on quantitative TF binding44, accessibility45,46, or enhancer reporter assay7 data.
These studies, which systematically tested the distance dependence of cooperativity in silico, suggest that TF cooperative interactions are diverse and common. TFs can prefer proximal binding with certain partners, with cooperativity decaying either sharply or slowly with distance, or instead prefer to have some distance between them7. Most cooperativity appears to be constrained to the ~150 bp span of nucleosomes (translational position), sometimes with periodicity at the ~10.5 bp scale of the turns of the DNA double helix (rotational position). In addition to varying cooperative modes, the extent of cooperativity can vary across TFs. TFs with generally reduced cooperativity may be more potent DNA binders or have stronger pioneering activity that enable their binding independent of cooperative partners47, while the weakest binders (e.g. Nanog of the often weakly binding homeodomain family) are most reliant on cooperativity44. These suggest that while few TFs must be in a certain position or orientation to function, diverse forms of TF cooperativity quantitatively modulate binding and function.
Although deep learning approaches are powerful for systematically detecting cooperativity, they are mechanistically agnostic. In some cases, the distance scales suggest certain mechanisms, e.g. ~10.5 bp periodicity suggests that TFs co-bind the same face of the DNA double helix, but whether those TFs rely on protein-protein interactions requires further experiments to resolve. While some TFs can form strong protein-protein interactions, such as obligate DNA-independent dimerization among family members of basic helix-loop-helix (bHLH), bZIP, and AP-2 factors5, most other interactions are weaker and more challenging to accurately identify. Disordered regions of TFs, which are less conserved48 and less straightforward to disrupt genetically49, are likely to be involved in longer-range cooperativity and more broadly DNA specificity beyond the binding motif50. Experiments swapping these unstructured regions between TFs suggest that they can play roles in determining loose cooperativity with other TFs (i.e. enrichments of other TFs near the core motif47,51) or genomic domain preference that may restrict which subset of motif instances to bind49.
New experimental methods are also facilitating a mechanistic understanding of TF cooperation. Single-molecule footprinting methods26,52–55 exploit unnatural DNA methylases to determine both nucleosome and TF binding states at single-molecule resolution, which demonstrated that simultaneous co-binding to DNA can be common even for TFs that do not bind immediately adjacent positions26,55. Proximity labeling approaches like BioID56 offer alternatives to the more traditional immunoprecipitation and can detect more transient interactions57.
TF activity is a function of concentration and modifications
For simplicity, our discussion thus far has treated TF binding to DNA as a mostly binary event occurring at a subset of motif matches. However, unlike tRNAs, which overwhelmingly bind their cognate codon and rarely bind others, TF binding is less well-defined. In addition to binding high-affinity sites that closely match their motif, TFs also bind many imperfect matches to their cognate motifs, termed low-affinity binding sites58. This variation in binding site affinity is thought to quantitatively affect TF occupancy (the percentage of the time the TF is at that site) and dwell time (duration of an individual binding event). Low affinity sites can still function, either through cooperation among multiple weak binding sites59,60 or through optimal cooperativity (spacing) with other TFBSs61.
In fact, the low affinity of these sites can be important for their proper specificity of function, as making them higher affinity can decrease tissue specificity of the cognate cis-regulatory element61–63. This observation supports the idea that binding site affinity is a major mechanism by which cells respond differentially to effective TF concentrations, with high-affinity sites bound by TFs even at low concentrations but low-affinity sites requiring higher TF levels. Similarly, post-translational modifications that reduce TF-DNA binding strength (such as acetylation64, phosphorylation65, or SUMOylation66) can in some cases selectively reduce binding to low-affinity sites, counterintuitively increasing transcriptional activity at high-affinity sites64. This quantitative model of TF activity as a function of precise levels and states is supported by recent work experimentally titrating a TF to varying dosages, in which the most dosage-sensitive accessible genomic regions were the highest affinity binding sites67.
While individual cases of functional low-affinity sites have been described, it remains challenging to determine how often and in which cases they function. One potential challenge is that disrupting even high-affinity sites individually often results in marginal phenotypes68; thus, detecting low-affinity site function may require a whole-organism readout rather than expression levels in a single cell type. For example, systematic mutagenesis of a Drosophila embryonic enhancer revealed that most bases within the enhancer—including those outside the strong TF motif matches—caused a detectable phenotype upon mutation69. Another challenge is that creating or destroying TF binding sites necessarily affects overlapping motifs, which can have stronger effects than the created or destroyed motif70. Weak binding sites may individually have small effects and yet collectively play important roles in gene regulation.
Note that while TF binding to DNA is the primary mechanism of cis regulation, sequences outside of strong TF binding sites may also contribute to transcriptional regulation without directly impacting TF binding affinity. For example, sequence properties can affect intrinsic nucleosome positioning71 or affect RNA Polymerase (Pol) II elongation rate72.
It also remains unclear what level of TF abundance is sufficient for function (and which functions, e.g. binding to low vs high affinity sites). TF expression is most often measured by RNA levels, which are further modulated by translation, protein stability, and sometimes protein localization or modification73. One potential scalable approach for identifying relevant TF RNA expression levels is comparing TF levels and motif enrichments in open chromatin across cell types74; however, this approach cannot resolve co-expressed paralogous TFs that may bind the same motif75, or other interaction partners that can modulate TF conformation and DNA binding76,77. Meanwhile, target gene-based inference of TF activity remains limited by the dearth of curated target genes78. Ultimately, more precise titrations of TF levels and states will be instructive as to the dependence of TF function on quantitative TF abundance67,79.
TF binding to DNA constitutes the first layer of the cis-regulatory code, with the TF-to-binding site code defining the key ‘words’ that are arranged into clusters called cis-regulatory elements. The next layer of the code determines the transcriptional functions of these elements—the meaning of these ‘sentences’.
From TF binding to cis-regulatory element function
TF binding site clusters as modular cis-regulatory elements
The nucleosomal barrier to TF binding ensures that TF occupancy on the genome is concentrated at clusters of TF binding sites where several different TFs converge80. These cis-regulatory elements are classified based on their position relative to genes (transcription start site-proximal promoters or distal elements) and their effect on transcription (enhancers that upregulate transcription, silencers that downregulate transcription, or insulators that separate regulatory domains). Promoters can be readily identified by sequencing RNA 5’ ends, but identifying distal elements is often less straightforward. Enhancers, the best studied class of distal elements, were originally defined as short sequences that autonomously drive transcription of a promoter independent of distance or orientation9. Nowadays, candidate enhancers active in a given cell type can be comprehensively mapped using chromatin marks81,82 and/or massively-parallel reporter assays68,83, but systematic genetic perturbations to validate their activity and define contributions of individual enhancers to the target gene expression remain challenging84.
Compared to promoters and enhancers, silencers have been much less studied. However, approaches analogous to those used to map enhancers have begun to yield catalogs of silencers. Some groups have used a chromatin mark of facultative repression, H3K27me3, combined with 3D interactions with promoters to identify candidate silencers85,86, while others used high-throughput reporter assays to test open chromatin regions lacking active chromatin marks for capacity to silence a strong promoter87,88. Many silencers appear to function as enhancers in other cell types, at least in Drosophila89. Each cis-regulatory element can serve as a superimposed set of binding sites for different sets of TFs across cellular contexts. Thus, the same element might be bound by repressive TFs and function as a silencer in one cell type and be bound by activating TFs and function as an enhancer in another.
Cis-regulatory elements integrate biological information to achieve spatiotemporal specificity
The organization of TF binding sites into cis-regulatory elements allows integration of multiple signals to achieve spatiotemporal specificity90. For example, an enhancer might be bound by and take input from a lineage-specific TF and a signaling effector, only becoming active in cells with both signals. From a spatiotemporal perspective, each input TF has a pattern of activity in the developing embryo; each cis-regulatory element combines these patterns according to the cooperativity or competition among these input TFs to generate its expression pattern. For example, an enhancer requiring three input TFs to be expressed would be active only in the intersection of the input TFs’ expression ranges, while an enhancer that is inhibited by a repressor would be restricted to regions where the repressor is absent. These simplified examples imply logic based on binary absence vs. presence of TFs, but these relationships are often quantitative91.
Based on the TF binding code that indicates which TFs regulate each cis-regulatory element, can we predict enhancer spatiotemporal specificities80,92? Or conversely, can we design synthetic enhancers with a desired specificity93? For some cell types, combinatorial binding of a few highly specifically expressed TFs is sufficient to produce cell type-specific expression. However, predicting activity patterns from arbitrary DNA sequences92,93 remains more challenging than classifying genomic enhancers80, and the difficulty of prediction depends on the cell type92.
TFs recruit cofactors to cis-regulatory elements
Transcription factors do not modulate transcription directly, but instead serve as adaptors that bring coactivators or corepressors (often jointly referred to as cofactors) to their binding sites. How do these cofactors in turn modulate transcription? This topic is too extensive to cover in full and instead we direct the reader to excellent reviews94–100. Here, we briefly outline major classes of cofactors and our current understanding of their functions.
Cofactors can be broadly grouped into three major and likely universal classes 1) the Mediator complex94, 2) nucleosome remodelers97, and 3) histone modifiers101, plus other scaffold or adaptor proteins that bridge TFs and cofactors102. The major cofactors are typically organized in large multi-subunit complexes, some with variable composition, but have key catalytic subunits. A combination of cryo-electron microscopy103,104, conditional protein depletion technologies105, and catalytic inhibitors have revealed new insights into how these complexes function.
The Mediator complex plays a central role in phosphorylating and activating the pre-initiation complex at most promoters94, while nucleosome remodelers actively maintain chromatin accessibility needed for promoter and distal enhancer activity106–108. Furthermore, although these core coactivators are needed for most transcription, acute depletion can exhibit surprisingly selective consequences due to feedback mechanisms109. For example, Mediator depletion leads to loss of transcription primarily at cell type-specific genes due to a compensatory increase in Pol II pause release at other genes109. By contrast, histone modifiers have competing activating and repressing roles, with coactivators depositing modifications enriched at enhancers like H3K4me and H3K27ac, and corepressors either removing those activating marks or adding repressive marks like H3K27me3 and H3K9me3. At least in some biological contexts coactivators like CBP and P300, which deposit H3K27ac, are limited in abundance compared to corepressors such as histone deacetylases that remove H3K27ac110. This observation is consistent with the rapid genome-wide changes in H3K27ac and transcription upon acute chemical inhibition of P300/CBP catalytic activity111. It is also important to note that while these modifications can modulate transcription per se112, they often represent only a part of these cofactors’ function, with a growing number of studies documenting major non-enzymatic roles of the histone-modifying coactivator proteins101,113–115, including recruiting Pol II to enhancers114 and promoting Pol II pausing at Polycomb response elements113. Histone modifications likely serve to stabilize the relatively weak recruitment of coactivators, many of which bind the same histone marks they deposit116.
Each transcription factor can recruit multiple cofactors, sometimes even both coactivators and corepressors117. The TF residues involved in cofactor recruitment are often unstructured (though they can form structures upon binding)118, and the interactions between TFs and their cofactors can be relatively weak. Furthermore, cofactors can recruit additional cofactors either directly119 or via modifications to histone tails that modulate cofactor binding116. Despite these complexities, recent work has begun to make substantial progress toward dissecting TF-cofactor recruitment, largely by measuring transcriptional consequences of TF and cofactor recruitment. The core assay is the activator bypass assay120, in which a reporter gene is controlled by a minimal promoter with binding sites for an orthogonal DNA-binding domain (DBD) that in turn recruits a fused TF, cofactor, or fragments of thereof, followed by measurements of reporter activity. High-throughput variants of this assay have been used to identify minimal fragments of TFs that are necessary and sufficient for activating or repressing transcription, called activation or repression domains, respectively121,122.
These studies have illustrated how TFs and cofactors not only differ in the quantitative strength of activation or repression122, but also how their transcriptional output depends on context—i.e. presence or absence of other TFs or cofactors within the cis-regulatory element. TFs clustered with different cofactors in their regulatory activity at diverse genomic contexts (i.e. DNA sequence flanking the TF/cofactor domain recruitment sites)123, suggesting that this distinct context-dependence is mediated by each TF recruiting specific cofactors, combined with differential ‘need’ for certain limiting coactivators in each genomic context124. Consistent with this model, proximity labeling of TFs and coactivators confirmed that different TFs can preferentially associate with different families of coactivators122. While such cofactor dependence has been thus far assayed in the reporter context, it could be potentially even more pronounced in the native genomic context—for example, certain coactivator combinations may be required for enhancer function over long distances or in different chromatin environments, and they could also mediate preferential enhancer-promoter contacts, as discussed below.
Dissecting the cofactor recruitment code
If TFs each recruit distinct sets of cofactors, we would expect each cofactor to be recruited by a different class of activation or repression domain with distinguishable features. However, until recently only a small fraction of TFs had any well-defined activation or repression domains, precluding efforts to glean generalizable rules or subclassifications into different mechanistic classes125.
Recent studies have vastly expanded the lexicon of activation and repression domains117,121,122, with now a majority of TFs and cofactors having at least one annotated effector domain, but surprisingly, activation domains lack obvious subclasses. Although activation domains have previously been noted for their different amino acid compositions, such as glutamine-rich or proline-rich domains, these enriched amino acids often are not the critical residues needed for domain function. Instead, the presence of hydrophobic residues and their interspersed position among certain other residues appears to be the main feature driving activation domain function117,118.
How can we reconcile the contrast between functional inference of distinct TF:coactivator recruitment and the scarcity of well-defined, functionally diverse subsets of activation domains? For one, the aforementioned recruitment screens, which typically measure the effect of a single candidate regulatory domain on the reporter activity, may miss activation domains that are too weak or dependent on other coactivators to function in isolation. In addition, the tested domains were fragments of up to 80 amino acids, so if recruitment of different cofactors is mediated by multiple distributed domains, these domains would not have been detected. Consistent with this idea, weak activation domains tend to synergize and activate transcription more strongly when fused together126.
In contrast to activation domains, known repression domains appear to be more diverse. Short linear motifs known to recruit different families of corepressors are critical for many repressor domains, as are SUMOylation sites, SUMO binding domains, and various structured domains including several DNA binding (zinc finger, homeodomain, bHLH) or protein interaction domains117. Notably, some TFs may be repressive only through recruiting other repressive TFs that in turn recruit corepressors, suggesting how TFs may alter their transcriptional function depending on the cellular context (e.g. repressive TF partner abundance).
Determining total functional output of TFs and cis-regulatory elements
As we compile maps of cofactor recruitment domains within TFs, these maps should also enable quantitative and context-specific predictions of the overall functional output of a TF, and an understanding of how multiple TFs collectively shape cis-regulatory element function. How do the different effector domains in a TF ‘add up’? With increasing numbers of either activation or repression domains present within a given TF, one might expect a wider array of coactivators or corepressors, respectively, recruited by this TF, and thus higher activity and less context-dependent function. Recruitment screens using whole open reading frames suggests wide variation in activation strength122; whether this is due to greater numbers and diversity of activation domains remains to be seen. Furthermore, given the relatively weak nature of cofactor recruitment, multivalency is thought to mediate phase separation of transcriptional regulators127 that leads to more robust activation128. Formation of macromolecular condensates may provide an additional layer of regulatory specificity, whereby cofactors coalesce in a manner that is dependent on multivalent interactions among specific coactivators and can be further regulated by post-translational modifications or RNAs (recently reviewed in refs 129–132).
When a TF contains both activation and repression domains, does one dominate, and under what conditions? In yeast, several TFs are known to contain both activation and repression domains, with intramolecular interactions masking one of the two depending on the presence of ligands133,134. In rare cases, the same short amino acid sequence can function as both an activation domain and a repression domain even at the same promoter117. a recent combinatorial recruitment screen found that in human cells, fusions of repressor domains with activator or dual-function domains tend to function as repressors126, potentially indicating a generally dominant role of repressor domains. However, further studies will be needed to resolve potential regulatory domains that might toggle activator vs. repressor function across genomic or cellular contexts.
Currently, it remains unknown how variable TF function is across cell types and binding sites. Many TFs have been reported to be both activating and repressing in different cellular or genomic contexts, but many of these observations may be indirect effects of TF loss105. Mutations of binding sites, e.g. in reporter assays, can resolve site-specific functions, but may be confounded by alteration of binding sites for other TFs70. The number and affinity of motifs and presence of certain other TFs nearby have been implicated in variation in TF function across sites135, but the mechanism remains unclear. One could envision that a TF has both activation and repression domains of differing affinities, such that its increased occupancy at a cis-regulatory element leads to saturation of the stronger recruitment domain and allows the weaker domain to catch up. Another possibility is that post-translational modifications can modify both TF binding strength and interaction with cofactors136 such that TF function varies with binding site affinity. Or, in the ‘limiting coactivator’ model, increasing recruitment of one coactivator might increase transcription up to a point, but once it is no longer limiting, it might crowd out other needed coactivators.
TF function may also be modulated by interactions among TFs at the same cis-regulatory element. While cooperative binding to DNA is the best characterized of such interactions, other mechanisms may exist. For example, TF-mediated repression could arise through one TF outcompeting and preventing binding by other more strongly activating TFs that share overlapping sites69. TFs binding the same cis-regulatory element could also impact each other by working together to more efficiently recruit the same coactivators. Yet, the diversity of TFs binding a cis-regulatory element has been proposed to be correlated with activation strength137, suggesting that TFs may collaborate by recruiting complementary coactivators. Such interactions might underlie parts of cis-regulatory grammar that can’t be explained by DNA binding cooperativity7. It remains an important task for the field to decipher TF function in their cellular and genomic contexts (e.g. to understand the consequences of non-coding genetic variation), and a mechanistic framework of TF-cofactor recruitment would add useful insights toward this goal.
From cis-regulatory elements to promoter transcription
While cis-regulatory elements such as enhancers are sequence- and cellular context-dependent modular ‘units’ of transcriptional regulation, the ultimate output of the cis-regulatory code is gene transcription. In complex genomes of multicellular organisms, enhancers often function at distances of tens or hundreds of kilobases away from their regulated promoters; in humans, there are known examples of enhancers regulating genes over a megabase away11,138,139. Such long-range function can result in regulatory domains—genomic regions where genes (or inserted reporter genes) have similar expression patterns140. Yet, in other cases, enhancers appear to exhibit promoter specificity, skipping nearby genes to regulate more distal genes138. Understanding how genomic distance, contact frequency between enhancers and promoters, biochemical compatibility, and chromatin state influence gene expression is another regulatory layer that has to be unpacked in the cis-regulatory code.
Generating a map of all the connections between the hundreds of thousands of enhancers and tens of thousands of promoters is a massive challenge. Recent efforts to systematically and experimentally determine these connections by individually perturbing enhancers with CRISPRi and measuring changes in transcription have yielded on the order of hundreds of connections141,142. Given how enhancer-promoter linkages serve as a critical link for interpreting how chromatin or genetic changes affect transcription and downstream phenotypes, various heuristics for approximating these linkages have been used to improve predictions in the absence of experimentally validated connections.
Genomic distance
The simplest heuristic for associating enhancers with promoters is the linear genomic distance between the enhancer and promoter (Figure 2A). In such an approach, enhancers are assumed either 1) to regulate the closest gene promoter, or 2) to regulate all genes within a distance limit, such as 100 kb. Despite the simplicity of this approach, it is remarkably accurate—functionally validated enhancer-promoter connections are greatly enriched at the closest distances, with substantial but gradual fall-off by 100 kb142. Similarly, highly transcribed randomly integrated promoter constructs were enriched near enhancers143. A more recent study tested random local insertions of a strong enhancer around a reporter gene and found that insertions closest to the promoter drove the strongest activation144, highlighting the quantitative dependence of enhancer-mediated transcriptional activation on genomic distance.
Figure 2.
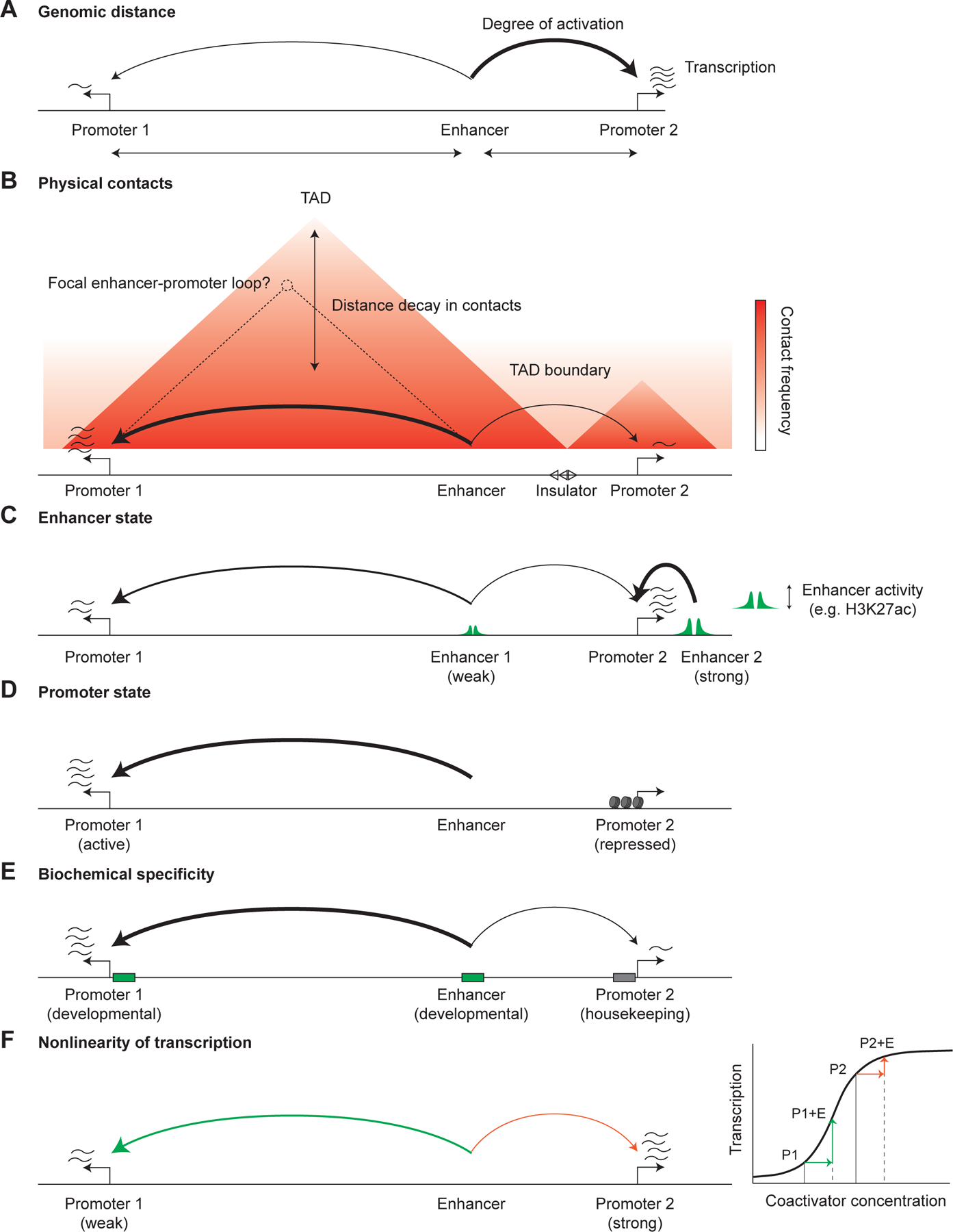
Potential mechanisms underlying enhancer-promoter specificity. A. Enhancers tend to preferentially activate promoters at closer genomic distances. B. Enhancers typically activate promoters within the same topologically associating domain (TAD), though not exclusively and not necessarily to equal extents. This preference has been ascribed to the increased frequency of physical contacts of genomic regions within, as compared to between, TADs. In rare cases, focal enhancer-promoter loops are also observed on the contact frequency maps, especially at promoters and enhancers overlapping a CTCF binding site. C. Differences in relative enhancer activity levels (that can be estimated by quantitative levels of H3K27ac or other enhancer chromatin features) can create apparent promoter specificity as an enhancer must contribute a significant fraction of the total activation at a promoter to detectably regulate it. D. Promoters can be in repressed states unresponsive to enhancer activation. E. Promoters and enhancers can be grouped into classes (such as developmental and housekeeping) with biochemical specificity (albeit quantitative) for each other, resulting in preferential activation. F. The nonlinearity of transcription as a function of coactivator concentration can create apparent specificity, where weak promoters are more responsive to activation by enhancers.
Physical contacts
Nevertheless, genomic distance fails to explain how some enhancers skip genes to regulate only more distal promoters, and how some enhancers can function at very long range while others do not. Upon the discovery of sequence-specific 3D organization of the genome, physical contacts between enhancers and promoters were proposed as a mechanism for long-range enhancer action145 (Figure 2B). Two key observations and associated hypotheses emerged. First, most enhancer-promoter connections appeared to lie within the same topologically associating domain (TAD), regions of self-interaction with insulation from neighboring regions146,147, suggesting that TAD boundaries might prevent inappropriate enhancer-promoter activation and that within a TAD all enhancers can activate all promoters. This notion was inspired by correlated expression of genes within the same TAD146 and case studies that highlighted mis-regulation resulting from loss of domain boundaries146,148. Indeed, TADs often correspond to regions in which integrated reporter genes exhibit similar expression patterns140. However, this heuristic fails to account for 1) the somewhat arbitrary definition of TADs, which often contain multiple sub-TADs, and 2) the overwhelming role of genomic distance in contact probability, in which very closely located regions inevitably form frequent contacts (simply through polymer physics), at much higher rates than more distal regions within (or outside) of a TAD. Consistent with this, global loss of the main TAD boundary factor, CTCF, leads to relatively minor changes in transcription, mostly at promoters with CTCF binding149,150.
Second, at least some enhancers and promoters appeared to form frequent loops, suggesting that identifying these loops might be a scalable approach to identify enhancer-promoter functional connections. Various modified versions of the chromosome conformation capture assay (capture-C151, promoter-capture Hi-C152, HiChIP153, micro-C154,155) have been used to prioritize candidate enhancer-promoter connections, but face two challenges. First, genome-wide assays like micro-C require large amounts of sequencing to achieve the resolution to resolve individual enhancers, while targeted approaches can be challenging to normalize and distinguish focally enriched interactions. Second, due to the high background of random interactions between nearby genomic regions, these approaches are often unable to detect enhancers at <~20 kb where many enhancers lie. However, the common focus on statistically enriched interactions may be unnecessary, as apparently ‘random’ interactions may still be functional and as a whole line up with the general decay in enhancer function with increasing distance144. This suggests that a diversity of mechanisms, including CTCF/cohesin-mediated loop extrusion, TF oligomerization (such as YY1156), Polycomb complex-mediated clustering157,158, sharing the same TAD, and even simple genomic proximity all generally serve to modulate largely equivalent physical contacts.
Enhancer state
A seemingly obvious element of enhancer-promoter connectivity is that the enhancer must be in an active state to regulate any promoters. Accounting for the relative activity levels of enhancers, together with genomic distance or contact probability, can predict enhancer-promoter connections reasonably well. The Activity-by-Contact (ABC) model uses a simple product of enhancer activity (a geometric mean of chromatin accessibility and active enhancer mark abundance) and contact probability with each promoter to estimate each enhancer’s contribution to that promoter’s transcription level141. This mirrors the experimental setup of most functional validation, where a single enhancer is deleted or perturbed, and the resulting change in transcription must be large enough to be detectable. Thus, a promoter surrounded by many strong enhancers may not be significantly regulated by a weaker and more distal enhancer, even though that same enhancer may be the strongest and closest enhancer for another promoter lacking other enhancers (Figure 2C). Notably, the model performed nearly as well using a power law scaling of the genomic distance in place of measured contact frequencies, in line with the role of specific 3D genome conformations primarily in a subset of more distal enhancer-promoter connections.
Promoter state
Similarly, in order to be activated by any enhancers, a promoter must be in a responsive state (Figure 2D). While the idea that some promoters may be unresponsive to enhancers in certain cell states is intuitive (and was effectively incorporated into the ABC model141, which only considers actively transcribed gene promoters), we lack a framework for predicting which promoters are responsive and which are not—which will likely need to incorporate chromatin state as well as absolute transcription levels. Mechanistically, a promoter might be in a repressed chromatin state (e.g. H3K9me2/3-marked, minimal chromatin accessibility, CpG methylated) that the coactivators recruited by the enhancer cannot overcome. For example, tissue-specific CpG methylation of the Zfp42 promoter renders it unresponsive to the Fat1 enhancer159. In another example, the Hoxa cluster requires promoter retinoic acid receptor elements (RAREs) for basal transcription levels that the enhancers then amplify; without the RAREs, there is minimal activation of Hoxa genes160.
Biochemical compatibility
Biochemical specificity or compatibility between enhancers and promoters has been proposed as another explanation for enhancers only activating certain promoters, especially for promoter-skipping (Figure 2E). Indeed, in Drosophila, both core promoters and enhancers can be classified as housekeeping or developmental108,123,161–163, with different sets of sequence motifs, cofactor dependencies, and relative positions. Housekeeping ‘enhancers’ are in direct proximity to their promoters, and perhaps are not enhancers per se, but should be simply considered as akin to Upstream Regulatory Sequences (UAS) in yeast164. Regardless, a similar division is also believed to exist in mammals, but recent studies that combinatorically tested many enhancers and promoters in an episomal reporter assay concluded that while these groups exist, they mostly exhibit more minor quantitative differences in activation strength compared to the effect of intrinsic promoter and enhancer strength165–167. We note that the Drosophila studies were done with minimal core promoters, while the mammalian studies were done with larger promoters that may include proximal enhancers or UAS—which more closely resembles the genomic context but may be confounded by synergy/antagonism between the enhancers.
Nonlinearity of transcription
While enhancer-promoter specificity is often considered in a binary manner (an enhancer does or does not regulate a given promoter), transcription is quantitative. By default, enhancers and promoters are assumed to behave linearly (often additive in log scale), but in reality the process of transcription is nonlinear—which may create the illusion of enhancer-promoter specificity (Figure 2F). From the perspective of an enhancer, strong promoters may be at a saturated level of transcription and therefore be unresponsive to gain or loss of a weaker enhancer, while inactive promoters may require sufficiently strong enhancers for increased transcription to be detectable.
This nonlinearity manifests in the quantitative relationship between contact frequency and transcription144, and might be explained by a probabilistic and multi-step process168. Consistently, imaging studies tracking both enhancer and promoter loci plus nascent transcription showed that contacts are perhaps weakly enriched preceding transcriptional bursts169,170, but contacts (as measurable by microscopy) clearly have an imperfect correspondence to transcription171, suggesting a time delay and probabilistic function168. That is, an enhancer-promoter contact may increase the probability of a cascade of events culminating in transcription172—but exactly what these events are remains unclear. One intriguing proposed mechanism is that enhancer recruitment of p300/CBP leads to transient acetylation and activation of transcription factors that can diffuse to and activate transcription at physically proximal promoters173. Further, the presence of multiple enhancers and promoters may contribute to additional non-linearity, as we discuss below.
These explanations of enhancer-promoter specificity are not mutually exclusive. Instead, an integrated quantitative model (like the ABC model) accounting for all these features, together with more extensive quantitative measurements of enhancer-promoter relationships in diverse endogenous and synthetic contexts, should lead us toward a comprehensive enhancer-promoter code.
From regulatory connections to regulatory landscapes
Thus far, we have focused on individual enhancer-promoter relationships, but in the genome, many genes have multiple, simultaneously active enhancers and promoters nearby that can cooperate or compete with one another. Furthermore, what constitutes an individual enhancer rather than parts of a larger enhancer? While short sequences (even < 200 bp) can exhibit reporter activity174, histone marks associated with enhancers can stretch for 1 kb or more175. Similarly, as discussed above, promoters contain core promoters and UAS-like proximal regulatory sequences. Zooming out further, these enhancers and promoters function in a broader genomic context, with 3D localization at the nuclear lamina176, nucleoli, or nuclear speckles177.
Diversity of interaction modes
One common operational framework for interpreting this complexity is epistasis. Epistasis is defined by deviation from a null expectation of independence, in which perturbing one enhancer should have the same effect regardless of the presence of other cis-regulatory elements (Figure 3). Redundancy occurs when loss of only one of a pair of enhancers has little effect as each enhancer can confer most of the transcriptional output, whereas their combined loss dramatically reduces transcription. Conversely, synergy occurs when all such enhancers are required for full output and each individual enhancer has much less activity on its own. However, the operational definition of epistasis has ambiguities—should the expectation be additive or multiplicative? On what scale (e.g. allele-specific expression vs. total expression)? This definition also focuses on a single cellular context—but the same pair of developmental enhancers might interact in different ways across cell types (Figure 3B).
Figure 3.
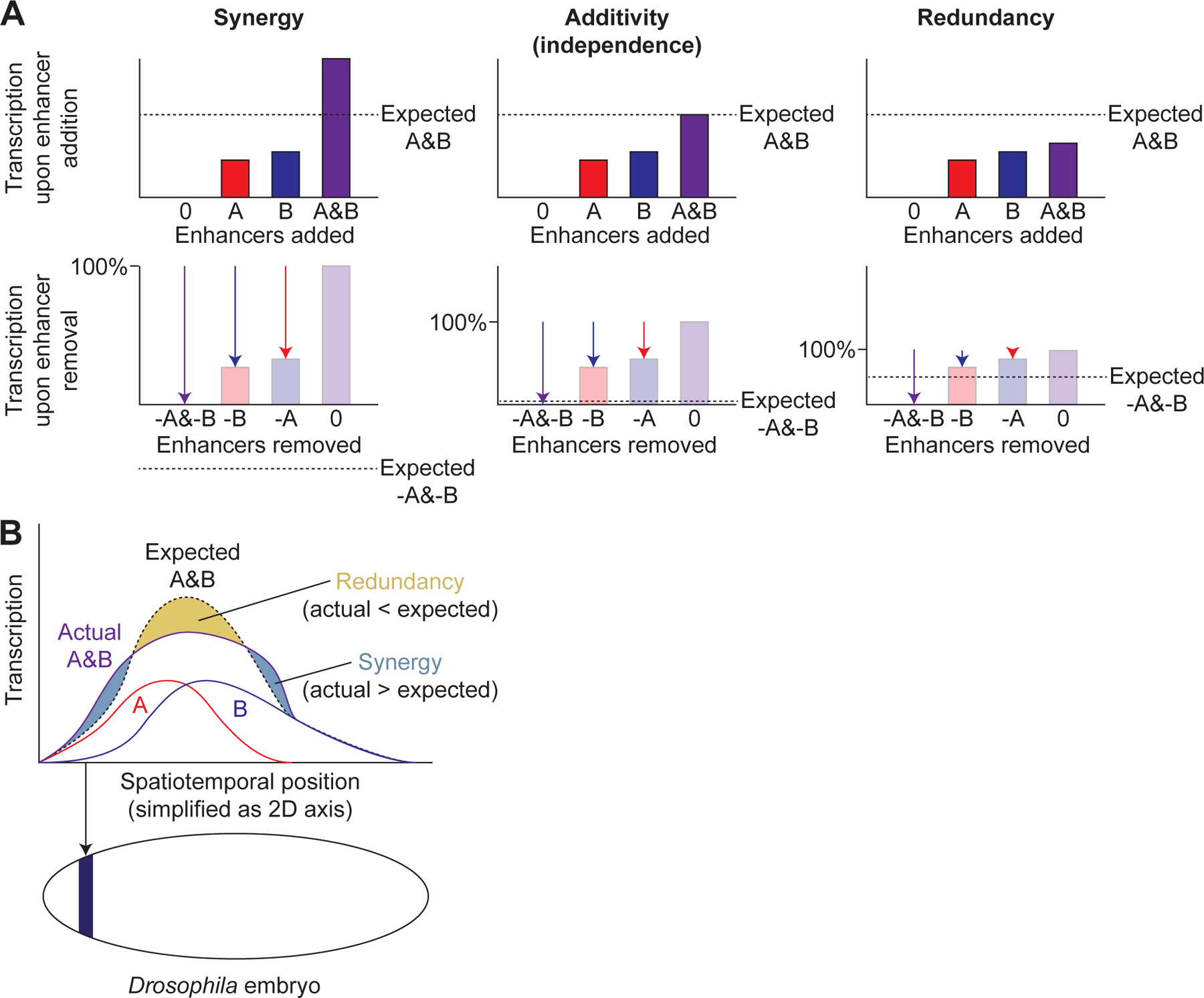
Enhancer epistasis. A. Examples of synergy, additivity, and redundancy between enhancers A and B, shown both from the perspective of enhancer addition (top) and enhancer removal (bottom) for the same examples. Dashed horizontal lines indicate the expected transcriptional output of adding both enhancers A and B (top) or of removing both enhancers (bottom). Bars indicate transcriptional output upon addition of enhancers while arrows indicate the change upon removal of enhancers. B. Illustration of how a single pair of enhancers, A and B, can exhibit multiple types of epistatic interaction across spatiotemporal positions (e.g. Drosophila embryonic anterior-posterior position as shown below x-axis, with dark blue color indicating one such position), simplified here as 2D axis.
Numerous studies of interactions among enhancers, parts of enhancers, and promoters have largely failed to find universal rules. Enhancers that possess similar tissue-specific activity and regulate the same promoter are a widespread feature of metazoan genomes178–180. These so-called ‘shadow enhancers’ are thought to be largely redundant. Yet, shadow enhancers are often conserved evolutionarily, suggesting that in addition to enabling cis-regulatory evolution by buffering the consequences of genetic variation, their overlapping activity serves important functions. Possibilities include buffering against stressful conditions181 or transcriptional noise182, or generating more precise expression patterns91. Notably, these sharper expression patterns can involve regions where an individual enhancer activates transcription but not the shadow enhancers together, due to repressor activity91. In addition, the prevalence of enhancer redundancy remains debated; a recent study of enhancer epistasis found a mix of additivity and synergy, rather than prevalent redundancy183.
Interactions among dense groups or clusters of enhancers that lie within tens of kilobases of DNA, called super-enhancers184,185, stretch enhancers175, or enhancer clusters186, have garnered particular attention, even as some studies have questioned the distinction187. These clusters, which are typically defined by high levels of H3K27ac, Mediator binding, or chromatin accessibility within extended genomic windows, have been postulated to exhibit unique properties that distinguish them from ‘typical’ enhancers, such as frequent 3D interactions188 that seed or associate with phase-separated condensates of coactivators and transcriptional machinery that make them particularly potent189. However, whether this implies redundancy or synergy is unclear, and both have been observed190 in addition to additivity187.
How do promoters interact with one another in genomic context? Do they compete for enhancers191, or can multiple promoters be simultaneously activated by a single enhancer192? Or do promoters serve as distal enhancers for other promoters193? All these scenarios have been observed, indicating that there is no single rule for promoter interactions, but it remains unclear when and where each condition applies. These conditions need not be mutually exclusive—for example, even when an enhancer can activate multiple promoters simultaneously, it may activate a single promoter more strongly (thus demonstrating competition).
Mechanisms underlying epistasis
Despite the caveats of interpreting epistatic interactions, epistasis reflects multi-step biological pathways and can help suggest mechanisms. For example, one pervasive observation is that within enhancers and enhancer clusters, a small portion or subset commonly drives the majority of transcription194–196, to the point that many regions appear individually dispensable or insufficient for autonomous enhancer activity, even though combined loss with other enhancers has measurable effects. This has often been used to invoke different classes or hierarchies of enhancers, including predominant vs. supporting sites, or ‘amplifier’ enhancers194,197–199 that differ in TF motifs or chromatin state features.
Given the diversity of observed epistatic interactions, is there a unifying mechanistic explanation? One potentially general principle is that different classes of enhancers may recruit different sets of coactivators that modulate different steps of the transcriptional process, such as Pol II recruitment vs phosphorylation. Thus, enhancers that only regulate non-limiting steps in the endogenous context may have little phenotype upon individual loss, but may become more important when those steps become limiting. Biochemical diversity could also underlie promoter interactions, e.g. competing promoters may be limited by the same coactivator.
Physical contacts can also mediate enhancer epistasis, but the relationship is complicated. TFs and coactivators recruited to one enhancer can spread to other enhancers in spatial proximity199; this spreading may allow more ‘dominant’ enhancers to further activate nearby supporting enhancers. A recent study proposed that enhancers that are very distally located and infrequently contact each other may buffer each other’s loss12. Yet, enhancers that form 3D hubs, including super-enhancers, have also been proposed to seed condensates or hubs that confer robust transcription of cell type identity genes189.
Impact of chromosome organization
In addition to specific interactions among nearby enhancers and promoters, the function of insulators and tethering elements, together with the broader organization of the domain within the nucleus, can also impact transcription.
Insulator elements—usually binding sites for the transcription factor CTCF200, which can form a directional barrier for loop extrusion by cohesin—serve to insulate adjacent TADs or loop domains. As with enhancers and silencers, strong boundary elements consist of clusters of TF binding sites201. These boundaries can impact transcription across broad domains by preventing spreading of heterochromatin202. Furthermore, the process of loop extrusion by cohesin shapes transcription beyond creating TAD boundaries, by increasing contacts between distal enhancers and promoters within the same TAD203, particularly for promoters with nearby CTCF sites150, or by broadly increasing contacts across the domain via promoting formation of multi-loop structures204. In addition to insulator elements, recently described ‘tethering elements’ can function independently of CTCF and loop extrusion not only to link specific enhancers and promoters205 but also to facilitate temporally coordinated transcription206.
Zooming out further, chromosomes are organized into two major types of ‘compartments’ — A compartments that tend to be enriched for active genes, and B compartments that tend to contain silenced heterochromatin (each of which can be subcategorized further). These compartments tend to self-associate within their own type. The compartment-level organization tends to be broadly similar across cell types, but with shifted boundary locations correlated with changes in chromatin state207, suggesting it is an emergent property shaped by chromatin state and transcription, and loop extrusion208. Some of these silenced regions tend to localize at the nuclear periphery176, while clusters of actively transcribed genes associate with nuclear speckles209, nuclear regions enriched in splicing factors. While the causality of these associations are tricky to untangle, experiments targeting genomic regions to nuclear bodies have demonstrated that nuclear environment can impact transcription210. The interplay between individual elements being influenced by domain-level effects versus shaping the organization of the domain remains unclear, but it is important to note that these domains can be heterogeneous and dynamic176.
Outlook
Deciphering the cis-regulatory code is a monumental challenge. Unlike the genetic code, which had a satisfyingly universal, modular, and deterministic solution, the cis-regulatory code is elusively context-specific, complex, combinatorial, and replete with redundancies and quantitative effects. It also consists of several interdependent tiers, starting from transcription factors binding DNA, to interacting with one another and cofactors at cis-regulatory elements, to complex regulatory domains and landscapes. Ultimately, understanding cis regulation will require understanding how the thousands of proteins involved in transcription—a substantial fraction of the proteome—work with each other and much of the genome. As Sydney Brenner said of the challenge of deciphering how embryonic development is encoded in the genome, “What is going to be difficult is the immense amount of detail that will have to be subsumed”211.
Nevertheless, our rapidly advancing tools are enabling experiments and interpretation at unprecedented scales and resolution. A complete catalog of cell types and their transcriptional profiles is within reach, with other epigenomic features to follow. We can test tens to hundreds of thousands of cis-regulatory elements or transcriptional regulators in reporter assays83,117. Even some of the largest protein complexes involved in transcription can be now structurally resolved, yielding unparalleled mechanistic insights103,104. Novel deep learning approaches are detecting previously unseen patterns in genomic data212,213. New tools for high-throughput super-resolution chromatin tracing and live-imaging of regulatory elements and transcripts provide a glimpse into genomic topology and kinetics underlying transcription at single-cell and single-allele level214–216. Our key remaining challenges are 1) to complement our currently scalable nucleic acid assays with improved technologies for measuring levels, localization, and dynamics of proteins at high resolution and throughput, 2) to integrate our catalogs of multi-scale measurements into a quantitative and mechanistic (interpretable) model of how transcription works at each of the regulatory levels discussed here, and 3) accurately predict gene expression patterns from DNA sequence under defined protein concentrations. By addressing these challenges, we may finally approach a complete cis-regulatory code, and be able to readily interpret noncoding genomic sequences in the context of evolution, development, and disease.
Acknowledgements
We thank K. Xue, L. Ichino, and L.-F. Chen for their comments and discussion. This work was supported by HHMI-Damon Runyon Cancer Research Foundation Fellowship (DRG-2420–21) to S.K., and NIH grant R35 GM131757, funding from the Howard Hughes Medical Institute, a Lorry Lokey endowed professorship, and a Stinehart Reed award to J.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
J.W. is paid scientific advisory board member at Camp4 and Paratus Sciences. J.W. is an advisory board member at Cell Press journals, including Cell, Molecular Cell and Developmental Cell.
References
- 1.Crick F (1970). Central Dogma of Molecular Biology. Nature 227, 561–563. 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 2.Koonin EV, and Novozhilov AS (2017). Origin and Evolution of the Universal Genetic Code. Annu. Rev. Genet 51, 45–62. 10.1146/annurev-genet-120116-024713. [DOI] [PubMed] [Google Scholar]
- 3.King MC, and Wilson AC (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116. 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 4.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 337, 1190–1195. 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. 10.1016/j.cell.2018.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239. 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Almeida BP, Reiter F, Pagani M, and Stark A (2022). DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet 54, 613–624. 10.1038/s41588-022-01048-5. [DOI] [PubMed] [Google Scholar]
- 8.Small S, Blair A, and Levine M (1992). Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J 11, 4047–4057. 10.1002/j.1460-2075.1992.tb05498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerji J, Rusconi S, and Schaffner W (1981). Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308. 10.1016/0092-8674(81)90413-X. [DOI] [PubMed] [Google Scholar]
- 10.Crocker J, and Stern DL (2017). Functional regulatory evolution outside of the minimal even-skipped stripe 2 enhancer. Development 144, 3095–3101. 10.1242/dev.149427. [DOI] [PubMed] [Google Scholar]
- 11.Long HK, Osterwalder M, Welsh IC, Hansen K, Davies JOJ, Liu YE, Koska M, Adams AT, Aho R, Arora N, et al. (2020). Loss of Extreme Long-Range Enhancers in Human Neural Crest Drives a Craniofacial Disorder. Cell Stem Cell 27, 765–783.e14. 10.1016/j.stem.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin X, Liu Y, Liu S, Zhu X, Wu L, Zhu Y, Zhao D, Xu X, Chemparathy A, Wang H, et al. (2022). Nested epistasis enhancer networks for robust genome regulation. Science 0, eabk3512. 10.1126/science.abk3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monahan K, Horta A, and Lomvardas S (2019). LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453. 10.1038/s41586-018-0845-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuda NJ, Ardehali MB, and Lis JT (2009). Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186–192. 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez J, and Larson DR (2020). Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu. Rev. Biochem 89, 189–212. 10.1146/annurev-biochem-011520-105250. [DOI] [PubMed] [Google Scholar]
- 16.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, and Bernstein BE (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, et al. (2018). Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412.e22. 10.1016/j.cell.2018.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. (2012). Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22, 1798–1812. 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordân R, Shen N, Dror I, Zhou T, Horton J, Rohs R, and Bulyk ML (2013). Genomic Regions Flanking E-Box Binding Sites Influence DNA Binding Specificity of bHLH Transcription Factors through DNA Shape. Cell Rep 3, 1093–1104. 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohs R, West SM, Sosinsky A, Liu P, Mann RS, and Honig B (2009). The role of DNA shape in protein–DNA recognition. Nature 461, 1248–1253. 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, and Crawford GE (2008). High-Resolution Mapping and Characterization of Open Chromatin across the Genome. Cell 132, 311–322. 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaret KS (2020). Pioneer Transcription Factors Initiating Gene Network Changes. Annu. Rev. Genet 54, 367–385. 10.1146/annurev-genet-030220-015007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirny LA (2010). Nucleosome-mediated cooperativity between transcription factors. Proc. Natl. Acad. Sci 107, 22534–22539. 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sönmezer C, Kleinendorst R, Imanci D, Barzaghi G, Villacorta L, Schübeler D, Benes V, Molina N, and Krebs AR (2021). Molecular Co-occupancy Identifies Transcription Factor Binding Cooperativity In Vivo. Mol. Cell 81, 255–267.e6. 10.1016/j.molcel.2020.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, and Zaret KS (2002). Opening of Compacted Chromatin by Early Developmental Transcription Factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289. 10.1016/S1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 28.Zaret KS, and Carroll JS (2011). Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25, 2227–2241. 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi K, and Yamanaka S (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126, 663–676. 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 30.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer Transcription Factors Target Partial DNA Motifs on Nucleosomes to Initiate Reprogramming. Cell 161, 555–568. 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez Garcia M, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, and Zaret KS (2019). Structural Features of Transcription Factors Associating with Nucleosome Binding. Mol. Cell 75, 921–932.e6. 10.1016/j.molcel.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, et al. (2018). The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81. 10.1038/s41586-018-0549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desanlis I, Kherdjemil Y, Mayran A, Bouklouch Y, Gentile C, Sheth R, Zeller R, Drouin J, and Kmita M (2020). HOX13-dependent chromatin accessibility underlies the transition towards the digit development program. Nat. Commun 11, 2491. 10.1038/s41467-020-16317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michael AK, Grand RS, Isbel L, Cavadini S, Kozicka Z, Kempf G, Bunker RD, Schenk AD, Graff-Meyer A, Pathare GR, et al. (2020). Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science 368, 1460–1465. 10.1126/science.abb0074. [DOI] [PubMed] [Google Scholar]
- 35.Hansen JL, Loell KJ, and Cohen BA (2022). A test of the pioneer factor hypothesis using ectopic liver gene activation. eLife 11, e73358. 10.7554/eLife.73358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King HW, and Klose RJ (2017). The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife 6, e22631. 10.7554/eLife.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frederick MA, Williamson KE, Fernandez Garcia M, Ferretti MB, McCarthy RL, Donahue G, Luzete Monteiro E, Takenaka N, Reynaga J, Kadoch C, et al. (2022). A pioneer factor locally opens compacted chromatin to enable targeted ATP-dependent nucleosome remodeling. Nat. Struct. Mol. Biol 10.1038/s41594-022-00886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, and Wysocka J (2014). Reorganization of Enhancer Patterns in Transition from Naive to Primed Pluripotency. Cell Stem Cell 14, 838–853. 10.1016/j.stem.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lodato MA, Ng CW, Wamstad JA, Cheng AW, Thai KK, Fraenkel E, Jaenisch R, and Boyer LA (2013). SOX2 Co-Occupies Distal Enhancer Elements with Distinct POU Factors in ESCs and NPCs to Specify Cell State. PLOS Genet 9, e1003288. 10.1371/journal.pgen.1003288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thanos D, and Maniatis T (1995). Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100. 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 41.Panne D, Maniatis T, and Harrison SC (2007). An Atomic Model of the Interferon-β Enhanceosome. Cell 129, 1111–1123. 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, and Taipale J (2015). DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388. 10.1038/nature15518. [DOI] [PubMed] [Google Scholar]
- 43.Grossman SR, Engreitz J, Ray JP, Nguyen TH, Hacohen N, and Lander ES (2018). Positional specificity of different transcription factor classes within enhancers. Proc. Natl. Acad. Sci 115, E7222–E7230. 10.1073/pnas.1804663115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avsec Ž, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, et al. (2021). Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet 53, 354–366. 10.1038/s41588-021-00782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim DS, Risca VI, Reynolds DL, Chappell J, Rubin AJ, Jung N, Donohue LKH, Lopez-Pajares V, Kathiria A, Shi M, et al. (2021). The dynamic, combinatorial cis-regulatory lexicon of epidermal differentiation. Nat. Genet 53, 1564–1576. 10.1038/s41588-021-00947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minnoye L, Taskiran II, Mauduit D, Fazio M, Aerschot LV, Hulselmans G, Christiaens V, Makhzami S, Seltenhammer M, Karras P, et al. (2020). Cross-species analysis of enhancer logic using deep learning. Genome Res 30, 1815–1834. 10.1101/gr.260844.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee QY, Mall M, Chanda S, Zhou B, Sharma KS, Schaukowitch K, Adrian-Segarra JM, Grieder SD, Kareta MS, Wapinski OL, et al. (2020). Pro-neuronal activity of Myod1 due to promiscuous binding to neuronal genes. Nat. Cell Biol 22, 401–411. 10.1038/s41556-020-0490-3. [DOI] [PubMed] [Google Scholar]
- 48.Brown CJ, Johnson AK, Dunker AK, and Daughdrill GW (2011). Evolution and Disorder. Curr. Opin. Struct. Biol 21, 441–446. 10.1016/j.sbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brodsky S, Jana T, Mittelman K, Chapal M, Kumar DK, Carmi M, and Barkai N (2020). Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol. Cell 79, 459–471.e4. 10.1016/j.molcel.2020.05.032. [DOI] [PubMed] [Google Scholar]
- 50.Staller MV (2022). Transcription factors perform a 2-step search of the nucleus. Genetics, iyac111. 10.1093/genetics/iyac111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fonseca GJ, Tao J, Westin EM, Duttke SH, Spann NJ, Strid T, Shen Z, Stender JD, Sakai M, Link VM, et al. (2019). Diverse motif ensembles specify non-redundant DNA binding activities of AP-1 family members in macrophages. Nat. Commun 10, 414. 10.1038/s41467-018-08236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Battaglia S, Dong K, Wu J, Chen Z, Najm FJ, Zhang Y, Moore MM, Hecht V, Shoresh N, and Bernstein BE (2022). Long-range phasing of dynamic, tissue-specific and allele-specific regulatory elements. Nat. Genet, 1–10. 10.1038/s41588-022-01188-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stergachis AB, Debo BM, Haugen E, Churchman LS, and Stamatoyannopoulos JA (2020). Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368, 1449–1454. 10.1126/science.aaz1646. [DOI] [PubMed] [Google Scholar]
- 54.Abdulhay NJ, McNally CP, Hsieh LJ, Kasinathan S, Keith A, Estes LS, Karimzadeh M, Underwood JG, Goodarzi H, Narlikar GJ, et al. (2020). Massively multiplex single-molecule oligonucleosome footprinting. eLife 9, e59404. 10.7554/eLife.59404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao S, Ahmad K, and Ramachandran S (2021). Cooperative binding between distant transcription factors is a hallmark of active enhancers. Mol. Cell 81, 1651–1665.e4. 10.1016/j.molcel.2021.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Göös H, Kinnunen M, Salokas K, Tan Z, Liu X, Yadav L, Zhang Q, Wei G-H, and Varjosalo M (2022). Human transcription factor protein interaction networks. Nat. Commun 13, 766. 10.1038/s41467-022-28341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanay A (2006). Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res 16, 962–972. 10.1101/gr.5113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shahein A, López-Malo M, Istomin I, Olson EJ, Cheng S, and Maerkl SJ (2022). Systematic analysis of low-affinity transcription factor binding site clusters in vitro and in vivo establishes their functional relevance. Nat. Commun 13, 5273. 10.1038/s41467-022-32971-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F, et al. (2015). Low Affinity Binding Site Clusters Confer Hox Specificity and Regulatory Robustness. Cell 160, 191–203. 10.1016/j.cell.2014.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, and Levine MS (2015). Suboptimization of developmental enhancers. Science 350, 325–328. 10.1126/science.aac6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jindal GA, Bantle AT, Solvason JJ, Grudzien JL, D’Antonio-Chronowska A, Lim F, Le SH, Larsen RO, Klie A, Frazer KA, et al. (2022). Affinity-optimizing variants within cardiac enhancers disrupt heart development and contribute to cardiac traits. Preprint at bioRxiv 10.1101/2022.05.27.493636. [DOI] [Google Scholar]
- 63.Lim F, Ryan GE, Le SH, Solvason JJ, Steffen P, and Farley EK (2022). Affinity-optimizing variants within the ZRS enhancer disrupt limb development. Preprint at bioRxiv 10.1101/2022.05.27.493789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Louphrasitthiphol P, Siddaway R, Loffreda A, Pogenberg V, Friedrichsen H, Schepsky A, Zeng Z, Lu M, Strub T, Freter R, et al. (2020). Tuning Transcription Factor Availability through Acetylation-Mediated Genomic Redistribution. Mol. Cell 79, 472–487.e10. 10.1016/j.molcel.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jantz D, and Berg JM (2004). Reduction in DNA-binding affinity of Cys2His2 zinc finger proteins by linker phosphorylation. Proc. Natl. Acad. Sci. U. S. A 101, 7589–7593. 10.1073/pnas.0402191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosonina E, Akhter A, Dou Y, Babu J, and Sri Theivakadadcham VS (2017). Regulation of transcription factors by sumoylation. Transcription 8, 220–231. 10.1080/21541264.2017.1311829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Naqvi S, Kim S, Hoskens H, Matthews HS, Spritz RA, Klein OD, Hallgrímsson B, Swigut T, Claes P, Pritchard JK, et al. (2022). Precise modulation of transcription factor levels reveals drivers of dosage sensitivity. Preprint at bioRxiv 10.1101/2022.06.13.495964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee S-I, Cooper GM, et al. (2012). Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol 30, 265–270. 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fuqua T, Jordan J, van Breugel ME, Halavatyi A, Tischer C, Polidoro P, Abe N, Tsai A, Mann RS, Stern DL, et al. (2020). Dense and pleiotropic regulatory information in a developmental enhancer. Nature 587, 235–239. 10.1038/s41586-020-2816-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Boer CG, Vaishnav ED, Sadeh R, Abeyta EL, Friedman N, and Regev A (2020). Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol 38, 56–65. 10.1038/s41587-019-0315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. (2009). The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366. 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 3, e02407. 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Filtz TM, Vogel WK, and Leid M (2014). Regulation of transcription factor activity by interconnected, post-translational modifications. Trends Pharmacol. Sci 35, 76–85. 10.1016/j.tips.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qiu C, Cao J, Martin BK, Li T, Welsh IC, Srivatsan S, Huang X, Calderon D, Noble WS, Disteche CM, et al. (2022). Systematic reconstruction of cellular trajectories across mouse embryogenesis. Nat. Genet 54, 328–341. 10.1038/s41588-022-01018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Ho TD, Buchler NE, and Gordan R (2021). Competition for DNA binding between paralogous transcription factors determines their genomic occupancy and regulatory functions. Genome Res, gr.275145.120. 10.1101/gr.275145.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bourgeois B, Gui T, Hoogeboom D, Hocking HG, Richter G, Spreitzer E, Viertler M, Richter K, Madl T, and Burgering BMT (2021). Multiple regulatory intrinsically disordered motifs control FOXO4 transcription factor binding and function. Cell Rep 36. 10.1016/j.celrep.2021.109446. [DOI] [PubMed] [Google Scholar]
- 77.Jen Y, Weintraub H, and Benezra R (1992). Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev. 6, 1466–1479. 10.1101/gad.6.8.1466. [DOI] [PubMed] [Google Scholar]
- 78.Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, and Saez-Rodriguez J (2019). Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res 29, 1363–1375. 10.1101/gr.240663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noviello G, Gjaltema RAF, and Schulz E (2022). CasTuner: a degron and CRISPR/Cas-based toolkit for analog tuning of endogenous gene expression. Preprint at bioRxiv 10.1101/2022.10.05.511019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zinzen RP, Girardot C, Gagneur J, Braun M, and Furlong EEM (2009). Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462, 65–70. 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 81.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, and Stark A (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 84.Gasperini M, Tome JM, and Shendure J (2020). Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet 21, 292–310. 10.1038/s41576-019-0209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cai Y, Zhang Y, Loh YP, Tng JQ, Lim MC, Cao Z, Raju A, Lieberman Aiden E, Li S, Manikandan L, et al. (2021). H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun 12, 719. 10.1038/s41467-021-20940-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang D, Petrykowska HM, Miller BF, Elnitski L, and Ovcharenko I (2019). Identification of human silencers by correlating cross-tissue epigenetic profiles and gene expression. Genome Res 29, 657–667. 10.1101/gr.247007.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doni Jayavelu N, Jajodia A, Mishra A, and Hawkins RD (2020). Candidate silencer elements for the human and mouse genomes. Nat. Commun 11, 1061. 10.1038/s41467-020-14853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pang B, and Snyder MP (2020). Systematic identification of silencers in human cells. Nat. Genet 52, 254–263. 10.1038/s41588-020-0578-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gisselbrecht SS, Palagi A, Kurland JV, Rogers JM, Ozadam H, Zhan Y, Dekker J, and Bulyk ML (2020). Transcriptional Silencers in Drosophila Serve a Dual Role as Transcriptional Enhancers in Alternate Cellular Contexts. Mol. Cell 77, 324–337.e8. 10.1016/j.molcel.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Buecker C, and Wysocka J (2012). Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. TIG 28, 276–284. 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perry MW, Boettiger AN, and Levine M (2011). Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc. Natl. Acad. Sci 108, 13570–13575. 10.1073/pnas.1109873108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pennacchio LA, Loots GG, Nobrega MA, and Ovcharenko I (2007). Predicting tissue-specific enhancers in the human genome. Genome Res 17, 201–211. 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taskiran II, Spanier KI, Christiaens V, Mauduit D, and Aerts S (2022). Cell type directed design of synthetic enhancers. Preprint at bioRxiv 10.1101/2022.07.26.501466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richter WF, Nayak S, Iwasa J, and Taatjes DJ (2022). The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol, 1–18. 10.1038/s41580-022-00498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Näär AM, Lemon BD, and Tjian R (2001). Transcriptional coactivator complexes. Annu. Rev. Biochem 70, 475–501. 10.1146/annurev.biochem.70.1.475. [DOI] [PubMed] [Google Scholar]
- 96.Li B, Carey M, and Workman JL (2007). The role of chromatin during transcription. Cell 128, 707–719. 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 97.Clapier CR, Iwasa J, Cairns BR, and Peterson CL (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol 18, 407–422. 10.1038/nrm.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brahma S, and Henikoff S (2020). Epigenome Regulation by Dynamic Nucleosome Unwrapping. Trends Biochem. Sci 45, 13–26. 10.1016/j.tibs.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Perissi V, Jepsen K, Glass CK, and Rosenfeld MG (2010). Deconstructing repression: evolving models of co-repressor action. Nat. Rev. Genet 11, 109–123. 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- 100.Chen Y-JC, Koutelou E, and Dent SYR (2022). Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell 82, 716–727. 10.1016/j.molcel.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morgan MAJ, and Shilatifard A (2020). Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet 52, 1271–1281. 10.1038/s41588-020-00736-4. [DOI] [PubMed] [Google Scholar]
- 102.Matkar S, Thiel A, and Hua X (2013). Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci 38, 394–402. 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hanske J, Sadian Y, and Müller CW (2018). The cryo-EM resolution revolution and transcription complexes. Curr. Opin. Struct. Biol 52, 8–15. 10.1016/j.sbi.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Osman S, and Cramer P (2020). Structural Biology of RNA Polymerase II Transcription: 20 Years On. Annu. Rev. Cell Dev. Biol 36, 1–34. 10.1146/annurev-cellbio-042020-021954. [DOI] [PubMed] [Google Scholar]
- 105.Jaeger MG, and Winter GE (2021). Fast-acting chemical tools to delineate causality in transcriptional control. Mol. Cell 81, 1617–1630. 10.1016/j.molcel.2021.02.015. [DOI] [PubMed] [Google Scholar]
- 106.Schick S, Grosche S, Kohl KE, Drpic D, Jaeger MG, Marella NC, Imrichova H, Lin J-MG, Hofstätter G, Schuster M, et al. (2021). Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat. Genet 53, 269–278. 10.1038/s41588-021-00777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iurlaro M, Stadler MB, Masoni F, Jagani Z, Galli GG, and Schübeler D (2021). Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat. Genet 53, 279–287. 10.1038/s41588-020-00768-w. [DOI] [PubMed] [Google Scholar]
- 108.Hendy O, Serebreni L, Bergauer K, Muerdter F, Huber L, Nemčko F, and Stark A (2022). Developmental and housekeeping transcriptional programs in Drosophila require distinct chromatin remodelers. Mol. Cell 10.1016/j.molcel.2022.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jaeger MG, Schwalb B, Mackowiak SD, Velychko T, Hanzl A, Imrichova H, Brand M, Agerer B, Chorn S, Nabet B, et al. (2020). Selective Mediator dependence of cell-type-specifying transcription. Nat. Genet 52, 719–727. 10.1038/s41588-020-0635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gillespie MA, Palii CG, Sanchez-Taltavull D, Shannon P, Longabaugh WJR, Downes DJ, Sivaraman K, Espinoza HM, Hughes JR, Price ND, et al. (2020). Absolute Quantification of Transcription Factors Reveals Principles of Gene Regulation in Erythropoiesis. Mol. Cell 78, 960–974.e11. 10.1016/j.molcel.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J, Satpathy S, Liebner T, Hamilton WB, Maskey E, et al. (2021). Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol. Cell 81, 2166–2182.e6. 10.1016/j.molcel.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 112.Policarpi C, Munafo M, Tsagkris S, Carlini V, and Hackett JA (2022). Systematic Epigenome Editing Captures the Context-dependent Instructive Function of Chromatin Modifications. Preprint at bioRxiv 10.1101/2022.09.04.506519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hunt G, Boija A, and Mannervik M (2022). p300/CBP sustains Polycomb silencing by non-enzymatic functions. Mol. Cell 82, 3580–3597.e9. 10.1016/j.molcel.2022.09.005. [DOI] [PubMed] [Google Scholar]
- 114.Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD, Garcia BA, Adelman K, and Wysocka J (2017). Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 66, 568–576.e4. 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rickels R, Herz H-M, Sze CC, Cao K, Morgan MA, Collings CK, Gause M, Takahashi Y-H, Wang L, Rendleman EJ, et al. (2017). Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet 49, 1647–1653. 10.1038/ng.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang T, Cooper S, and Brockdorff N (2015). The interplay of histone modifications – writers that read. EMBO Rep 16, 1467–1481. 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.DelRosso N, Tycko J, Suzuki P, Andrews C, Aradhana, Mukund A, Liongson I, Ludwig C, Spees K, Fordyce P, et al. (2022). Large-scale mapping and systematic mutagenesis of human transcriptional effector domains. Preprint at bioRxiv 10.1101/2022.08.26.505496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sanborn AL, Yeh BT, Feigerle JT, Hao CV, Townshend RJ, Lieberman Aiden E, Dror RO, and Kornberg RD (2021). Simple biochemical features underlie transcriptional activation domain diversity and dynamic, fuzzy binding to Mediator. eLife 10, e68068. 10.7554/eLife.68068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang Z-Q, Li J, Sachs LM, Cole PA, and Wong J (2003). A role for cofactor–cofactor and cofactor–histone interactions in targeting p300, SWI/SNF and Mediator for transcription. EMBO J 22, 2146–2155. 10.1093/emboj/cdg219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ptashne M, and Gann A (1997). Transcriptional activation by recruitment. Nature 386, 569–577. 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 121.Tycko J, DelRosso N, Hess GT, Aradhana, Banerjee A, Mukund A, Van MV, Ego BK, Yao D, Spees K, et al. (2020). High-Throughput Discovery and Characterization of Human Transcriptional Effectors. Cell 183, 2020–2035.e16. 10.1016/j.cell.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alerasool N, Leng H, Lin Z-Y, Gingras A-C, and Taipale M (2022). Identification and functional characterization of transcriptional activators in human cells. Mol. Cell 82, 677–695.e7. 10.1016/j.molcel.2021.12.008. [DOI] [PubMed] [Google Scholar]
- 123.Stampfel G, Kazmar T, Frank O, Wienerroither S, Reiter F, and Stark A (2015). Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature 528, 147–151. 10.1038/nature15545. [DOI] [PubMed] [Google Scholar]
- 124.Bell CC, Talarmain L, Scolamiero L, Lam EY, Ang C-S, Gilan O, and Dawson MA (2022). Transcription factors use a unique combination of cofactors to potentiate different promoter-dependent steps in transcription. Preprint at bioRxiv 10.1101/2022.10.25.513774. [DOI] [Google Scholar]
- 125.Soto LF, Li Z, Santoso CS, Berenson A, Ho I, Shen VX, Yuan S, and Bass JIF (2022). Compendium of human transcription factor effector domains. Mol. Cell 82, 514–526. 10.1016/j.molcel.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mukund AX, Tycko J, Allen SJ, Robinson SA, Andrews C, Ludwig CH, Spees K, Bassik MC, and Bintu L (2022). High-throughput functional characterization of combinations of transcriptional activators and repressors. Preprint at bioRxiv 10.1101/2022.12.20.521091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Trojanowski J, Frank L, Rademacher A, Mücke N, Grigaitis P, and Rippe K (2022). Transcription activation is enhanced by multivalent interactions independent of phase separation. Mol. Cell 82, 1878–1893.e10. 10.1016/j.molcel.2022.04.017. [DOI] [PubMed] [Google Scholar]
- 129.Sabari BR (2020). Biomolecular Condensates and Gene Activation in Development and Disease. Dev. Cell 55, 84–96. 10.1016/j.devcel.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 130.Sharp PA, Chakraborty AK, Henninger JE, and Young RA (2022). RNA in formation and regulation of transcriptional condensates. RNA N. Y. N 28, 52–57. 10.1261/rna.078997.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Strom AR, and Brangwynne CP (2019). The liquid nucleome - phase transitions in the nucleus at a glance. J. Cell Sci 132, jcs235093. 10.1242/jcs.235093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lyon AS, Peeples WB, and Rosen MK (2021). A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol 22, 215–235. 10.1038/s41580-020-00303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Polish JA, Kim J-H, and Johnston M (2005). How the Rgt1 Transcription Factor of Saccharomyces cerevisiae Is Regulated by Glucose. Genetics 169, 583–594. 10.1534/genetics.104.034512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sze J-Y, Woontner M, Jaehning JA, and Kohlhaw GB (1992). In Vitro Transcriptional Activation by a Metabolic Intermediate: Activation by Leu3 Depends on α-Isopropylmalate. Science 258, 1143–1145. 10.1126/science.1439822. [DOI] [PubMed] [Google Scholar]
- 135.White MA, Kwasnieski JC, Myers CA, Shen SQ, Corbo JC, and Cohen BA (2016). A Simple Grammar Defines Activating and Repressing cis-Regulatory Elements in Photoreceptors. Cell Rep 17, 1247–1254. 10.1016/j.celrep.2016.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marceau AH, Brison CM, Nerli S, Arsenault HE, McShan AC, Chen E, Lee H-W, Benanti JA, Sgourakis NG, and Rubin SM (2019). An order-to-disorder structural switch activates the FoxM1 transcription factor. eLife 8, e46131. 10.7554/eLife.46131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Singh G, Mullany S, Moorthy SD, Zhang R, Mehdi T, Tian R, Duncan AG, Moses AM, and Mitchell JA (2021). A flexible repertoire of transcription factor binding sites and a diversity threshold determines enhancer activity in embryonic stem cells. Genome Res 10.1101/gr.272468.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lettice LA, Horikoshi T, Heaney SJH, van Baren MJ, van der Linde HC, Breedveld GJ, Joosse M, Akarsu N, Oostra BA, Endo N, et al. (2002). Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl. Acad. Sci 99, 7548–7553. 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354, 769–773. 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, and Spitz F (2014). Functional and topological characteristics of mammalian regulatory domains. Genome Res 24, 390–400. 10.1101/gr.163519.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, et al. (2019). A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 377–390.e19. 10.1016/j.cell.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, Berns A, Wessels LFA, van Lohuizen M, and van Steensel B (2013). Chromatin Position Effects Assayed by Thousands of Reporters Integrated in Parallel. Cell 154, 914–927. 10.1016/j.cell.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 144.Zuin J, Roth G, Zhan Y, Cramard J, Redolfi J, Piskadlo E, Mach P, Kryzhanovska M, Tihanyi G, Kohler H, et al. (2022). Nonlinear control of transcription through enhancer–promoter interactions. Nature 604, 571–577. 10.1038/s41586-022-04570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Popay TM, and Dixon JR (2022). Coming full circle: On the origin and evolution of the looping model for enhancer–promoter communication. J. Biol. Chem 298. 10.1016/j.jbc.2022.102117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 161, 1012–1025. 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22. 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kubo N, Ishii H, Xiong X, Bianco S, Meitinger F, Hu R, Hocker JD, Conte M, Gorkin D, Yu M, et al. (2021). Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat. Struct. Mol. Biol 28, 152–161. 10.1038/s41594-020-00539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Downes DJ, Smith AL, Karpinska MA, Velychko T, Rue-Albrecht K, Sims D, Milne TA, Davies JOJ, Oudelaar AM, and Hughes JR (2022). Capture-C: a modular and flexible approach for high-resolution chromosome conformation capture. Nat. Protoc 17, 445–475. 10.1038/s41596-021-00651-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre B-M, Nagano T, Katsman Y, Sakthidevi M, Wingett SW, et al. (2015). The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res 25, 582–597. 10.1101/gr.185272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hsieh T-HS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, and Darzacq X (2020). Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol. Cell 78, 539–553.e8. 10.1016/j.molcel.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Krietenstein N, Abraham S, Venev SV, Abdennur N, Gibcus J, Hsieh T-HS, Parsi KM, Yang L, Maehr R, Mirny LA, et al. (2020). Ultrastructural Details of Mammalian Chromosome Architecture. Mol. Cell 78, 554–565.e7. 10.1016/j.molcel.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588.e28. 10.1016/j.cell.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eagen KP, Aiden EL, and Kornberg RD (2017). Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci 114, 8764–8769. 10.1073/pnas.1701291114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kraft K, Yost KE, Murphy SE, Magg A, Long Y, Corces MR, Granja JM, Wittler L, Mundlos S, Cech TR, et al. (2022). Polycomb-mediated genome architecture enables long-range spreading of H3K27 methylation. Proc. Natl. Acad. Sci 119, e2201883119. 10.1073/pnas.2201883119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ringel AR, Szabo Q, Chiariello AM, Chudzik K, Schöpflin R, Rothe P, Mattei AL, Zehnder T, Harnett D, Laupert V, et al. (2022). Repression and 3D-restructuring resolves regulatory conflicts in evolutionarily rearranged genomes. Cell 185, 3689–3704.e21. 10.1016/j.cell.2022.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pinglay S, Bulajić M, Rahe DP, Huang E, Brosh R, Mamrak NE, King BR, German S, Cadley JA, Rieber L, et al. (2022). Synthetic regulatory reconstitution reveals principles of mammalian Hox cluster regulation. Science 377, eabk2820. 10.1126/science.abk2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Haberle V, Arnold CD, Pagani M, Rath M, Schernhuber K, and Stark A (2019). Transcriptional cofactors display specificity for distinct types of core promoters. Nature 570, 122–126. 10.1038/s41586-019-1210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Neumayr C, Haberle V, Serebreni L, Karner K, Hendy O, Boija A, Henninger JE, Li CH, Stejskal K, Lin G, et al. (2022). Differential cofactor dependencies define distinct types of human enhancers. Nature, 1–8. 10.1038/s41586-022-04779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, and Stark A (2015). Enhancer–core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518, 556–559. 10.1038/nature13994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Guarente L, and Hoar E (1984). Upstream activation sites of the CYC1 gene of Saccharomyces cerevisiae are active when inverted but not when placed downstream of the “TATA box”. Proc. Natl. Acad. Sci 81, 7860–7864. 10.1073/pnas.81.24.7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bergman DT, Jones TR, Liu V, Ray J, Jagoda E, Siraj L, Kang HY, Nasser J, Kane M, Rios A, et al. (2022). Compatibility rules of human enhancer and promoter sequences. Nature, 1–3. 10.1038/s41586-022-04877-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Martinez-Ara M, Comoglio F, van Arensbergen J, and van Steensel B (2022). Systematic analysis of intrinsic enhancer-promoter compatibility in the mouse genome. Mol. Cell 82, 2519–2531.e6. 10.1016/j.molcel.2022.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sahu B, Hartonen T, Pihlajamaa P, Wei B, Dave K, Zhu F, Kaasinen E, Lidschreiber K, Lidschreiber M, Daub CO, et al. (2022). Sequence determinants of human gene regulatory elements. Nat. Genet, 1–12. 10.1038/s41588-021-01009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xiao JY, Hafner A, and Boettiger AN (2021). How subtle changes in 3D structure can create large changes in transcription. eLife 10, e64320. 10.7554/eLife.64320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen H, Levo M, Barinov L, Fujioka M, Jaynes JB, and Gregor T (2018). Dynamic interplay between enhancer–promoter topology and gene activity. Nat. Genet 50, 1296–1303. 10.1038/s41588-018-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, and Boettiger AN (2019). Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54. 10.1038/s41586-019-1035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Alexander JM, Guan J, Li B, Maliskova L, Song M, Shen Y, Huang B, Lomvardas S, and Weiner OD (2019). Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. eLife 8, e41769. 10.7554/eLife.41769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Reimer A, Alamos S, Westrum C, Turner MA, Talledo P, Zhao J, and Garcia HG (2021). Minimal synthetic enhancers reveal control of the probability of transcriptional engagement and its timing by a morphogen gradient. Preprint at bioRxiv 10.1101/2021.07.10.451524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Karr JP, Ferrie JJ, Tjian R, and Darzacq X (2021). The transcription factor activity gradient (TAG) model: contemplating a contact-independent mechanism for enhancer–promoter communication. Genes Dev 10.1101/gad.349160.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Klein JC, Agarwal V, Inoue F, Keith A, Martin B, Kircher M, Ahituv N, and Shendure J (2020). A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods 17, 1083–1091. 10.1038/s41592-020-0965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, NISC Comparative Sequencing Program, et al. (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci 110, 17921–17926. 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Leemans C, van der Zwalm MCH, Brueckner L, Comoglio F, van Schaik T, Pagie L, van Arensbergen J, and van Steensel B (2019). Promoter-Intrinsic and Local Chromatin Features Determine Gene Repression in LADs. Cell 177, 852–864.e14. 10.1016/j.cell.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757.e24. 10.1016/j.cell.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kvon EZ, Waymack R, Gad M, and Wunderlich Z (2021). Enhancer redundancy in development and disease. Nat. Rev. Genet 22, 324–336. 10.1038/s41576-020-00311-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hong J-W, Hendrix DA, and Levine MS (2008). Shadow Enhancers as a Source of Evolutionary Novelty. Science 321, 1314–1314. 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243. 10.1038/nature25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Frankel N, Davis GK, Vargas D, Wang S, Payre F, and Stern DL (2010). Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493. 10.1038/nature09158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Waymack R, Fletcher A, Enciso G, and Wunderlich Z (2020). Shadow enhancers can suppress input transcription factor noise through distinct regulatory logic. eLife 9, e59351. 10.7554/eLife.59351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Choi J, Lysakovskaia K, Stik G, Demel C, Söding J, Tian TV, Graf T, and Cramer P (2021). Evidence for additive and synergistic action of mammalian enhancers during cell fate determination. eLife 10, e65381. 10.7554/eLife.65381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-Enhancers in the Control of Cell Identity and Disease. Cell 155, 934–947. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhou HY, Katsman Y, Dhaliwal NK, Davidson S, Macpherson NN, Sakthidevi M, Collura F, and Mitchell JA (2014). A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev 28, 2699–2711. 10.1101/gad.248526.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Moorthy SD, Davidson S, Shchuka VM, Singh G, Malek-Gilani N, Langroudi L, Martchenko A, So V, Macpherson NN, and Mitchell JA (2017). Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res 27, 246–258. 10.1101/gr.210930.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ing-Simmons E, Seitan VC, Faure AJ, Flicek P, Carroll T, Dekker J, Fisher AG, Lenhard B, and Merkenschlager M (2015). Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res 25, 504–513. 10.1101/gr.184986.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Farooq U, Saravanan B, Islam Z, Walavalkar K, Singh AK, Jayani RS, Meel S, Swaminathan S, and Notani D (2021). An interdependent network of functional enhancers regulates transcription and EZH2 loading at the INK4a/ARF locus. Cell Rep 34, 108898. 10.1016/j.celrep.2021.108898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Oh S, Shao J, Mitra J, Xiong F, D’Antonio M, Wang R, Garcia-Bassets I, Ma Q, Zhu X, Lee J-H, et al. (2021). Enhancer release and retargeting activates disease-susceptibility genes. Nature 595, 735–740. 10.1038/s41586-021-03577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fukaya T, Lim B, and Levine M (2016). Enhancer Control of Transcriptional Bursting. Cell 166, 358–368. 10.1016/j.cell.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nguyen TA, Jones RD, Snavely AR, Pfenning AR, Kirchner R, Hemberg M, and Gray JM (2016). High-throughput functional comparison of promoter and enhancer activities. Genome Res 26, 1023–1033. 10.1101/gr.204834.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Carleton JB, Berrett KC, and Gertz J (2017). Multiplex Enhancer Interference Reveals Collaborative Control of Gene Regulation by Estrogen Receptor α-Bound Enhancers. Cell Syst 5, 333–344.e5. 10.1016/j.cels.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tippens ND, Liang J, Leung AK-Y, Wierbowski SD, Ozer A, Booth JG, Lis JT, and Yu H (2020). Transcription imparts architecture, function and logic to enhancer units. Nat. Genet 52, 1067–1075. 10.1038/s41588-020-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen LLP, Kassouf MT, Oudelaar AM, Sharpe JA, Suciu MC, et al. (2016). Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet 48, 895–903. 10.1038/ng.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Blayney JW, Kassouf M, Francis H, Camellato BR, Stolper R, Higgs DR, Mitchell L, and Boeke J (2022). Super-enhancers require a combination of classical enhancers and novel facilitator elements to drive high levels of gene expression. Preprint at bioRxiv 10.1101/2022.06.20.496856. [DOI] [Google Scholar]
- 198.Thomas HF, Kotova E, Jayaram S, Pilz A, Romeike M, Lackner A, Penz T, Bock C, Leeb M, Halbritter F, et al. (2021). Temporal dissection of an enhancer cluster reveals distinct temporal and functional contributions of individual elements. Mol. Cell 81, 969–982.e13. 10.1016/j.molcel.2020.12.047. [DOI] [PubMed] [Google Scholar]
- 199.Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, and Reddy TE (2016). Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281.e19. 10.1016/j.cell.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Huang H, Zhu Q, Jussila A, Han Y, Bintu B, Kern C, Conte M, Zhang Y, Bianco S, Chiariello AM, et al. (2021). CTCF mediates dosage- and sequence-context-dependent transcriptional insulation by forming local chromatin domains. Nat. Genet 53, 1064–1074. 10.1038/s41588-021-00863-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, and Reinberg D (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. 10.1126/science.1262088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Calderon L, Weiss FD, Beagan JA, Oliveira MS, Georgieva R, Wang Y-F, Carroll TS, Dharmalingam G, Gong W, Tossell K, et al. (2022). Cohesin-dependence of neuronal gene expression relates to chromatin loop length. eLife 11, e76539. 10.7554/eLife.76539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chen L-F, Long HK, Park M, Swigut T, Boettiger AN, and Wysocka J (2022). Structural elements facilitate extreme long-range gene regulation at a human disease locus. Preprint at bioRxiv 10.1101/2022.10.20.513057. [DOI] [PubMed] [Google Scholar]
- 205.Pachano T, Sánchez-Gaya V, Ealo T, Mariner-Faulí M, Bleckwehl T, Asenjo HG, Respuela P, Cruz-Molina S, Muñoz-San Martín M, Haro E, et al. (2021). Orphan CpG islands amplify poised enhancer regulatory activity and determine target gene responsiveness. Nat. Genet 53, 1036–1049. 10.1038/s41588-021-00888-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Levo M, Raimundo J, Bing XY, Sisco Z, Batut PJ, Ryabichko S, Gregor T, and Levine MS (2022). Transcriptional coupling of distant regulatory genes in living embryos. Nature 605, 754–760. 10.1038/s41586-022-04680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fortin J-P, and Hansen KD (2015). Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol 16, 180. 10.1186/s13059-015-0741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Haarhuis JHI, Weide R.H. van der, Blomen VA, Yáñez-Cuna JO, Amendola M, Ruiten M.S. van, Krijger PHL, Teunissen H, Medema RH, Steensel B. van, et al. (2017). The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707.e14. 10.1016/j.cell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Quinodoz SA, Jachowicz JW, Bhat P, Ollikainen N, Banerjee AK, Goronzy IN, Blanco MR, Chovanec P, Chow A, Markaki Y, et al. (2021). RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790.e30. 10.1016/j.cell.2021.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wang H, Xu X, Nguyen CM, Liu Y, Gao Y, Lin X, Daley T, Kipniss NH, La Russa M, and Qi LS (2018). CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 175, 1405–1417.e14. 10.1016/j.cell.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Judson HF (1996). The eighth day of creation: makers of the revolution in biology Expanded ed (CSHL Press; ). [Google Scholar]
- 212.Ching T, Himmelstein DS, Beaulieu-Jones BK, Kalinin AA, Do BT, Way GP, Ferrero E, Agapow P-M, Zietz M, Hoffman MM, et al. (2018). Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 15, 20170387. 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Li Y, Chen C-Y, Kaye AM, and Wasserman WW (2015). The identification of cis-regulatory elements: A review from a machine learning perspective. Biosystems 138, 6–17. 10.1016/j.biosystems.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 214.Boettiger A, and Murphy S (2020). Advances in Chromatin Imaging at Kilobase-Scale Resolution. Trends Genet. TIG 36, 273–287. 10.1016/j.tig.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hafner A, and Boettiger A (2022). The spatial organization of transcriptional control. Nat. Rev. Genet, 1–16. 10.1038/s41576-022-00526-0. [DOI] [PubMed] [Google Scholar]
- 216.Brandão HB, Gabriele M, and Hansen AS (2021). Tracking and interpreting long-range chromatin interactions with super-resolution live-cell imaging. Curr. Opin. Cell Biol 70, 18–26. 10.1016/j.ceb.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]