

Summary
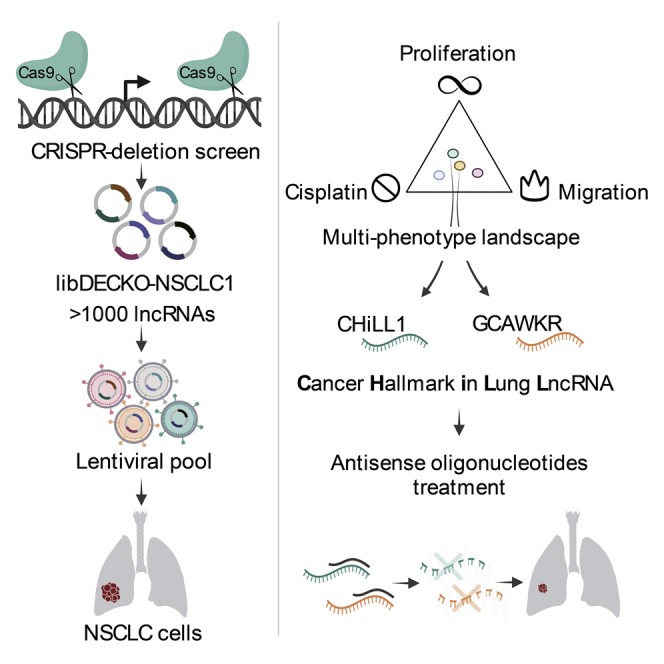
Long noncoding RNAs (lncRNAs) are widely dysregulated in cancer, yet their functional roles in cancer hallmarks remain unclear. We employ pooled CRISPR deletion to perturb 831 lncRNAs detected in KRAS-mutant non-small cell lung cancer (NSCLC) and measure their contribution to proliferation, chemoresistance, and migration across two cell backgrounds. Integrative analysis of these data outperforms conventional “dropout” screens in identifying cancer genes while prioritizing disease-relevant lncRNAs with pleiotropic and background-independent roles. Altogether, 80 high-confidence oncogenic lncRNAs are active in NSCLC, which tend to be amplified and overexpressed in tumors. A follow-up antisense oligonucleotide (ASO) screen shortlisted two candidates, Cancer Hallmarks in Lung LncRNA 1 (CHiLL1) and GCAWKR, whose knockdown consistently suppressed cancer hallmarks in two- and three-dimension tumor models. Molecular phenotyping reveals that CHiLL1 and GCAWKR control cellular-level phenotypes via distinct transcriptional networks. This work reveals a multi-dimensional functional lncRNA landscape underlying NSCLC that contains potential therapeutic vulnerabilities.
Keywords: CRISPR, lncRNA, long noncoding RNA, cancer, non-small cell lung cancer, NSCLC, RNA therapeutics, antisense oligonucleotides
Graphical abstract
Highlights
-
•
CRISPR-based screening pipeline for lncRNAs promoting multiple cancer hallmarks
-
•
Comprehensive, multi-dimensional functional map of lncRNAs in KRAS+ NSCLC
-
•
lncRNA vulnerabilities can be targeted by ASOs in 2D and 3D tumor models
Esposito, Polidori, and colleagues present a CRISPR-based pipeline to comprehensively map functional long noncoding RNAs across disease hallmarks of KRAS+ non-small cell lung cancer. This reveals therapeutic vulnerabilities that could be targeted by potent antisense oligonucleotide inhibitors in a range of tumor models.
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer deaths worldwide,1 and available therapies face a combination of challenges in undruggable mutations, toxicity, and therapy resistance.2, 3, 4 The most common NSCLC subtype, carrying activating KRAS mutations (KRAS+), is routinely treated with cytotoxic platinum chemotherapy, and newly approved targeted therapies only extend life by a few months.5,6
A fertile source for new therapeutic targets is long noncoding RNAs (lncRNAs), with a population likely to exceed 100,000, of which >98% remain uncharacterised.7, 8, 9, 10 Hundreds of lncRNAs have been implicated in the progression of disease hallmarks, as defined by Hanahan and Weinberg,11,12 across cancer types via a variety of mechanisms.13, 14, 15, 16 Examples such as SAMMSON (melanoma) and lncGRS-1 (glioma) have attracted attention as drug targets thanks to tumor cells’ potent and specific sensitivity to their inhibition via antisense oligonucleotide (ASO) therapies.17,18
Considerable evidence points to the central roles of lncRNAs in lung cancer.16 For example, PVT1 and MALAT1 are recurrently overexpressed or amplified in lung tumors, and their manipulation alters cell growth and invasiveness both in vitro and in vivo, making them promising therapeutic targets.19,20 Other examples include LINC00680, which acts via binding to GATA6,21,22 and LINC00511, which promotes NSCLC by binding chromatin-modifying enzyme EZH2 and repressing tumor-suppressor genes, including p57 and LATS2.23 Nonetheless, the extent and nature of lncRNAs promoting the interlocking pathological hallmarks in NSCLC remain unclear.
Efforts to identify lncRNA therapeutic targets have accelerated with the advent of CRISPR-Cas genome editing, which can be used to silence gene expression via targeted genomic deletions or transcriptional inhibition and is readily scaled transcriptome-wide via pooled screening.24 CRISPR screens have revealed scores of lncRNAs promoting disease hallmarks of cell proliferation, pathway activation, and therapy resistance.25, 26, 27
A critical challenge in drug discovery is the poor validation rate of preclinical targets discovered in vitro.28 Highly focused single background/single hallmark screen designs, including those above, are vulnerable to discovering lncRNAs promoting cell-line-specific phenotypes that do not generalize to the disease in question.17 Supporting this, recent CRISPR-inhibition (CRISPRi) screens demonstrated highly specific effects for lncRNAs in cell lines from six distinct cancer types.25 However, the critical question of whether this also affects cell lines from the same cancer type has not been addressed. Thus, to maximize the utility of discovered hits, an ideal screen should prioritize lncRNA targets that are both pleiotropic (impact multiple disease hallmarks) and background independent (effective regardless of cell model).
Here, we comprehensively map the functional lncRNA landscape of KRAS+ NSCLC. We perform 8 disease-specific CRISPR screens for lncRNAs promoting three cancer hallmarks (proliferation, chemoresistance, and invasion) in two KRAS+ NSCLC models (A549 and H460). We reconstruct the functional lncRNA landscape of NSCLC, revealing a catalog of therapeutic vulnerabilities. These lncRNAs connect to cancer hallmarks via complex transcriptional networks and can be targeted by potent, low-toxicity, and on-target ASOs, representing promising future therapeutics.29
Results
A versatile CRISPR screening pipeline for lncRNAs in NSCLC
To identify lncRNAs promoting KRAS+ NSCLC, we adapted the dual-excision CRISPR knockout (DECKO) CRISPR-deletion (CRISPR-del) system to a high-throughput pooled format24,30 (Figure 1A). This approach achieves loss-of-function (LOF) perturbations by deleting target genes’ transcription start sites (TSS) via paired guide RNAs (pgRNAs) and effectively inhibits gene expression.31, 32, 33, 34 This perturbation strategy based on the deletion of TSSs rather than entire genes offers several benefits, including uniform and shorter deletions (which are more efficient)35 and a reduced chance of false positives arising from deleting overlapping DNA regulatory elements.
Figure 1.

Multi-hallmark CRISPR discovery of lncRNAs promoting non-small cell lung cancer
(A) CRISPR-deletion pooled screening strategy for lncRNAs promoting NSCLC hallmarks.
(B) Library design pipeline. lncRNAs from indicated annotations are merged and filtered by TSS proximity to protein-coding genes (<2 kb excluded) and by expression in A549 (<0.1 FPKM excluded). TSS are clustered together if closer than 300 bp, then selected based on three evidence sources: CAGE, ChromHMM, and DNAseI hypersensitivity. TSS candidates are targeted by 10 paired guide RNA (pgRNA) designs using CRISPETa.
(C) Library composition, in terms of targeted regions. Note that some lncRNA loci are represented by >1 targeted transcription start site (TSS).
(D) Fluorescence-activated cell sorting (FACS) was used to sort stable Cas9-expressing cells based on the expression of a blue fluorescent protein (BFP) marker. Boxes indicated the sorted cell populations used in screens.
(E) (Top) One member of the screening library, DNMBP-AS1, was targeted by CRISPR-deletion in A549 cells. The gene locus is shown in the top panel, including genotyping PCR primers (F, R) listed in Data S4, TSS target region (black), and library pgRNAs (red bars). (Bottom) PCR using indicated primers with template genomic DNA (gDNA) from cells transfected with non-targeting pgRNA (control) or DNMBP-AS1 TSS pgRNA_9 (KO). The expected lengths for wild-type and deletion amplicons are indicated.
(F) Quantitative reverse-transcriptase PCR (qRT-PCR) measurement of DNMBP-AS1 RNA after CRISPR-del.
We developed a screening library to comprehensively interrogate the NSCLC lncRNA transcriptome. To avoid the known issues of false-positive results that may occur via unintended off targeting,36 we omitted any target regions <2 kb from the nearest protein-coding exon. We integrated and filtered published10,37,38 and in-house gene annotations39,40 (Figures 1B and S1A), for a final target set of 831 expressed lncRNAs, corresponding to 998 high-confidence TSSs, henceforth named “candidate 1” and so on (Figures 1C and S1B; see STAR Methods). lncRNA candidates with bidirectional promoters or with overlapping TSSs were merged into a single TSS. Taking these cases into account, 95.2% of target regions contain one single lncRNA promoter, while 4.5% and 0.3% contain two and three, respectively (Figure S1D).
To these, we added pgRNAs targeting neutral control loci (not expected to influence cell phenotype) and positive control protein-coding genes (PCGs) (with known roles in cell growth and cisplatin resistance) (Figure 1C).
These targets form the basis for “libDECKO-NSCLC1,” a CRISPR-del library with a depth of 10 unique pgRNAs per target, comprising altogether 12,000 pgRNAs (Figure 1C; Table S1). After cloning into the DECKO backbone (Figure S1C), sequencing revealed high quality in terms of sequence identity (60.8% perfect match across both spacers) and coverage (90th to 10th percentile count ratios: 4.6-fold)41 (Figure S1F).
To identify hits of general relevance to NSCLC, we performed parallel experiments in two widely used KRAS+ NSCLC models, A549 and H460,42,43 Non-clonal cell lines were generated that stably express high levels of Cas9 protein,44 as evidenced by blue fluorescent protein (BFP) (Figures 1D and S1E). Given that the ability of pgRNAs to efficiently generate the desired genomic event has been called into question,45 we first performed extensive validation of library pgRNAs. Targeting known NSCLC-promoting lncRNA DNMBP-AS1 (candidate 331)46 resulted in the deletion of its promoter region and loss of expression (Figures 1E, 1F, and S1G), supporting the effectiveness of the CRISPR-del strategy. To more generally assess the impact of our pgRNA strategy, we randomly cloned another 12 pgRNAs targeting 12 candidates. We observed expected deletion at the target site for 10/12 cases. In terms of impact on target RNA levels, we observed expected downregulations for 7/12, while for three targets we observed no change, and for two we observed upregulation (Figure S3A), in line with previous observations from Shkumatava’s group.47
Multi-phenotype mapping of NSCLC lncRNAs
Cancers thrive via a variety of phenotypic hallmarks, or biological capabilities acquired by cells during tumor progression.11 Previous CRISPR screens have been limited to a single hallmark, either proliferation or drug resistance,17,25,27,48,49 and have been usually focused on a negative “dropout” format, where pgRNAs for genes of interest are depleted.
For a more comprehensive and biomedically relevant vista of NSCLC lncRNAs, we adapted pooled screening to identify causal genes across multiple hallmarks of cancer progression, namely proliferation, chemoresistance, and invasion (Figures 2A–2C). To boost sensitivity, we implemented complementary “positive” screens, where pgRNAs of interest are enriched. Thus, to identify lncRNAs promoting cell fitness and proliferation, we combined (1) a classical negative selection screen, where targets’ pgRNAs become depleted (“dropout”), with (2) a positive selection screen using carboxyfluorescein succinimidyl ester (CFSE) dye to identify growth-promoting lncRNAs by their pgRNAs’ enrichment in slow-growing cells (Figure 2A).50 Complementary screens yielded a modest but consistent benefit in accuracy over pure dropout screens (receiver-operator area under the curve values: A549 proliferation 0.7 versus 0.67, A549 cisplatin 0.77 versus 0.73, respectively); therefore, we decided to include it in subsequent analyses.
Figure 2.

Adapting CRISPR screens to cancer hallmarks
(A) Proliferation: the strategy employs complementary negative (dropout) (growth-promoting lncRNAs’ pgRNAs are depleted) and positive (CFSE dye) (growth-promoting lncRNAs’ pgRNAs are enriched) formats.
(B) Cisplatin sensitivity: another complementary strategy is employed. In the negative (dropout) “survival” screen, cells are exposed to high cisplatin doses (IC30, IC80). Resistance-promoting lncRNAs’ pgRNAs will be depleted in surviving cells. In the positive “death” screen, cells that die in response to low cisplatin concentration (IC20) are collected, and enriched pgRNAs identify resistance-promoting lncRNAs.
(C) Migration: cells that are capable/incapable of migrating through a porous membrane over a given time are separately collected. Migration-promoting lncRNAs are identified via their pgRNAs’ enrichment in migration-impaired cells.
(D) Read counts correlation of the dropout assay in A549. Log2 of the read counts (T3: 3 weeks; T0: time point zero). Z score-transformed log2 values. Statistical significance: Pearson correlation.
(E) lncRNA candidates (black) correlation between biological replicates of the dropout screen in A549 cells. Data presented as Z score-transformed log2FC values (T3/T0). Statistical significance: Pearson correlation.
(F) P-value distribution of lncRNA candidates and neutral and positive controls in A549 dropout screen. Protein-coding genes used as positive controls are analyzed separately using pgRNAs targeting the ORF and TSS. Statistical significance: Wilcoxon test (pairwise comparisons) and Kruskal-Wallis (global differences).
(G) A549 dropout screen. The horizontal line indicates the cutoff for hits at FDR <0.2. Previously published lncRNAs in NSCLC and other cancers are labeled in green and blue, respectively.
(H) Comparison of A549 and H460 dropout screens (four points removed for clearer visualization, statistical significance estimated on all the data points using Pearson correlation).
(I) Analysis of lncRNA hits that have potential impacts on the described phenotypes by screens in A549 and H460 cells with FDR <0.2. The light blue bat indicates the fraction of previously discovered “cancer lncRNAs” from the CLC2 database.16
We observed a high correlation between biologically independent replicates at multiple levels of data integration, from raw pgRNA read counts (Figure 2D) to pgRNA fold changes and target-level fold changes (Figures 2E and S2A–S2F). These correlations are observed even when considering lncRNA candidates alone and omitting positive-control targets. Our dropout correlations were comparable to previous CRISPR-del dropout49 (Figure S2F). As expected, pgRNAs for positive-control genes were significantly depleted in dropout screens, while neutral controls were not (Figure 2F). To gauge the LOF efficiency of promoter deletion, pgRNAs targeting positive-control PCGs had been split between two distinct modalities: (1) conventional open reading frame (ORF) mutation, expected to yield maximal LOF, and (2) promoter deletion, similar to lncRNAs. Promoter-deletion pgRNAs displayed a detectable but lower phenotypic impact, indicating that CRISPR-del screens for lncRNAs face intrinsically lower sensitivity compared with ORF-targeting screens for PCGs (Figure 2F).
Using biologically replicated dropout screens, 77 lncRNAs were identified as necessary for proliferation of A549 cells. These include known NSCLC lncRNAs, such as PVT1,20 SBF2-AS1,51 and LINC00511,23 that were also found in H460. In addition, lncRNAs identified in other cancer types include LINC0068021 and LINC0091052 (Figure 2G).
The factors influencing pgRNA deletion efficiency are poorly understood. Using growth phenotype as a proxy for deletion efficiency, we observed the expected correlation between observed and bioinformatically predicted sgRNA efficiency (RuleSet2 algorithm) (Figure S1H). On the other hand, we found no relationship with pgRNA orientation (Figure S1I), and a weak tendency for larger deletions to produce stronger phenotypes, possibly due to a greater impact on lncRNA expression (Figure S1J).
Next, we compared equivalent dropout screens in the two NSCLC backgrounds. There was a significant concordance among identified targets, even after omitting positive controls (Figure 2H). Besides, out of the 109 positive controls included in the library, 37 and 16 scored in the dropout screens of A549 and H460, respectively, of which 10 are common to both (p = 0.011, hypergeometric test). To strengthen these data, we performed complementary CFSE screens in the same cells. As expected, these positive selection screens displayed anti-correlation with dropout results in H460 cells, although not in A549 cells, possibly for technical reasons (Figure S2B).
Patients with KRAS+ tumors are usually treated with cytotoxic platinum-based chemotherapeutics, but tumors frequently evolve resistance.53 To identify lncRNAs promoting chemoresistance, we again employed complementary screens (Figure 2B) at carefully chosen cisplatin concentrations (Figure S2C). As before, correlated results were observed across biological replicates (Figures S2D and S2F). Migration is a key hallmark underlying the invasion and metastasis of tumor cells. By isolating cells with rapid or slow migration through a porous membrane for 48 h (Figures 2C, S2E, and S2G), we screened for migration-promoting lncRNAs. This yielded 65 lncRNAs, of which ten were already associated with migration and invasion in numerous cancer types16 including SNHG654 and SNHG12.55
These data, summarized in Figure 2I and Data S1, represent a resource of functional lncRNAs in NSCLC hallmarks.
Screen hits can be validated and function via RNA products
We next tested the reliability of these results by selecting two lncRNA TSSs for further validation, based on their top ranking and consistency between the two cell lines: candidate 205, identified as a top hit in dropout screens (A549: log2 fold change [FC] = −1.81, false discovery rate [FDR] = 1.2 × 10−7), and candidate 509 in both proliferation and cisplatin (A549: log2FC = −1.15, FDR = 9.78 × 10−7).
Candidate 205 overlaps the TSS of bidirectional antisense GENCODE-annotated genes, LINC00115 and RP11-206L10.11 (Figure 3A). Candidate 509 targets a TSS shared by several BIGTranscriptome lncRNAs (Figure 3A). Supporting the importance of this locus, it contains two additional hits, candidate 507 (LINC00910) and candidate 508 (Figure S2H).
Figure 3.

Validation of screen hits shows reproducible phenotypes
(A) Candidate 205 (left) and candidate 509 (right) loci. Primers and ASO sites are indicated above. The TSS target region and the 10 pgRNAs from the screening library are indicated below.
(B) Log2 fold change of pgRNA count over the course of the dropout screen in A549. T0: time point zero; T2: time point 2 weeks; T3: time point 3 weeks. Arrows indicate the pgRNAs that were randomly chosen and individually cloned for validation.
(C) Competition assay. Fluorescently labeled cells carrying pgRNAs for control AAVS1 locus (green, GFP) or indicated targets (red, mCherry) were measured by flow cytometry over time (days). pgRNAs targeting the ORF of essential ribosomal protein RPS5 were used as a positive control. n = 3, error bars indicate SD statistical significance was estimated by Student’s t test at the last time point.
(D) ASOs were used to separately target the two lncRNAs sharing the TSS at candidate 205. For each, two different ASOs were employed (1 and 2) in A549 cells. Top panels: LINC00115; bower panels: RP11-206L10.11. Cell population (left); RNA expression (right) measured by qRT-PCR after 72 h from the ASO treatment. n = 4; error bars indicate SD significance was estimated by one-tailed Student’s t test.
(E) (Left) Log2 fold change of pgRNAs in migrated and non-migrated cell populations from A549 migration screen. (Right) Validation experiments with A549 cell migration across transwell supports over 24 h. Cells were treated with ASOs targeting indicated lncRNAs or a non-targeting control.
(F) (Left) qRT-PCR in A549 cells treated with two distinct ASOs each for candidates 215, 448, and 489. Expression was normalized to a non-targeting ASO. (Right) Crystal violet quantification normalized to non-targeting ASO. Data are plotted as mean ± SD from three independent biological replicates. Statistical significance was estimated by one-tailed Student’s t test.
To validate the phenotypic effect of these deletions, we tested randomly selected high-scoring pgRNAs (Figure 3B, arrow). Candidate 205 pgRNA efficiently deleted the targeted region (Figure S2I), and while difficulty in designing PCR primers prevented direct testing of deletion by candidate 509, it effectively decreased RNA levels (Figure S2J). Both pgRNAs yielded potent effects on cell fitness: mCherry+ cells expressing pgRNAs were out-competed by control cells (GFP+, expressing pgRNA for AAVS1), with an effect comparable to inactivation of essential ribosomal gene RPS5 (Figures 3C and S2K). Similar results were observed in a conventional growth assay (Figure S2L). Furthermore, the pgRNA for candidate 509 also sensitized cells to cisplatin, consistent with screen results (Figure 3C).
It remained ambiguous which of the two genes overlapping the candidate 205 region is responsible for these effects. Furthermore, genomic deletion cannot distinguish between DNA-dependent (e.g., enhancer) or RNA-dependent mechanisms (e.g., mature lncRNA, or its transcription). To address both questions, we used two gene-specific ASOs to target each gene. This reveals the implication of LINC00115, but not RP11-206L10.11, in driving cell proliferation via an RNA-dependent mechanism56 (Figure 3D). These effects were further corroborated in a three-dimensional (3D) spheroid model (Figure S2M). These results are consistent with previous reports linking LINC00115 to lung cancer proliferation and migration54,57 as well as to other malignancies. For instance, its knockdown suppresses cell proliferation and invasion in breast cancer58 and prostate cancer,59 and it could inhibit colon rectal cancer cells’ metastatic ability and cell growth.60
Similar high validation rates were observed for the migration screens. ASO knockdown of three hits, candidate 215 (AC104024.3), candidate 448 (CECR7), and candidate 489 (MIR23AHG), resulted in dramatic impairment of A549 migration (Figures 3E and 3F).
In summary, these findings support the ability of CRISPR-del screens to identify lncRNA genes that promote cancer hallmarks via RNA-dependent mechanisms.
Multi-hallmark screen integration for target discovery
We next integrated these data into a quantitative and comprehensive map of lncRNAs driving NSCLC hallmarks. To combine diverse screen results while balancing effect size and significance, we created an integrative target prioritization pipeline (TPP) (Figure 4A). TPP can either be run on all screens for a “pan-hallmark” target ranking or for individual hallmarks (“hallmark-specific”; see STAR Methods). The pan-hallmark ranking outperforms common integration methods and individual screens in correctly classifying positive and neutral controls (Figure 4B).
Figure 4.

Functional landscape of lncRNAs in NSCLC
(A) Target prioritization pipeline (TPP) integrates multiple screens to generate a unified hit ranking. TPP employs both effect size (robust rank aggregation [RRA]) and statistical significance (empirical Brown’s method). TPP Pan, TPP target prioritization pipeline integrating the screens together; TPP pheno, target prioritization pipeline that integrates multiple screens of a specific cancer phenotype (i.e., proliferation or cisplatin response).
(B) Comparing performance of screens and integration methods. Performance is measured as the area under the ROC curve (AUC) based on correctly recalling/rejecting library controls (positive and neutral controls, respectively).
(C) Ternary plot contains all significant hits (FDR < 0.2) in pan-hallmark analysis. The three corners represent the hallmarks, indicated by symbols. The proximity to each corner is driven by the TPP significance calculated for each hallmark. The candidates selected for further validations and protein-coding controls are indicated by candidate number or gene name, respectively. Selected lncRNAs previously associated with lung adenocarcinoma (LUAD) are labeled in blue. Petal plots display the hallmark contributions of selected lncRNAs.
(D) The number of hits discovered in pan-hallmark and individual hallmark screens. Right: the target composition of the screening library for comparison. “Lung cancer lncRNAs'' indicate previously published, functionally validated lncRNAs in lung cancer.16 Pro, proliferation; Cis, cisplatin; Mig, migration; Pan, pan-hallmarks, integration of all the screens together.
(E) Scatterplot showing the correlation of the dropout assay hits (left) and the pan-hallmark hits (right) in H460 and A549. The arrows point to the geometric median of the respective group.
(F) Expression of pan-hallmark hits compared with all other screened lncRNAs (non-hits) in the KRAS+ samples from The Cancer Genome Atlas (TCGA) LUAD cohort (n = 101). Statistical significance was estimated by one-tailed Welch’s t test.
(G) Expression of pan-hallmark hits compared with non-hits in an independent cohort of LUAD samples and healthy tissues (87 tumor; 77 normal).61 Statistical significance: pairwise two-tailed Student’s t test.
(H) Pan-cancer recurrent amplifications and deletions, estimated in Pan-Cancer Analysis of Whole Genomes (PCAWG) cohort. Statistical significance: Fisher’s exact test (one-sided).
The hallmark-specific values provide a signature for each lncRNA in three functional dimensions, in the context of overall confidence defined by pan-hallmark ranking (Figure 4C). Pan-hallmark analysis alone identified altogether 80 lncRNA hits (∼8% of those screened; Data S2) (FDR <0.2), of which 73 were not previously linked to NSCLC (Figure 4D). As expected, growth-promoting PCG positive controls and known lung cancer lncRNAs, but not neutral controls, are enriched among hits. This is supported by an independent enrichment score analysis (Figure S3C). Hallmark-specific integration yielded 134 hits (∼13% of those screened) in at least one hallmark, of which 19 are found in two hallmarks, and none in three (Figure S3D).
Previous CRISPR screens have demonstrated that lncRNAs required for cell growth have highly cell-type-specific activities.25 Indeed, comparing lncRNA hits from conventional dropout analysis in A549 and H460 backgrounds revealed a low degree of concordance—lncRNAs tend to only display activity in the cell type where they were discovered (Figure 4E, left). In contrast, pan-hallmark hits display relatively background-independent activity, being active in both cell types (Figure 4E, right), thus supporting the usefulness of an integrative screening strategy combining multiple cell lines for discovering therapeutic targets.
Several additional lines of evidence support oncogenic roles for screen-hit lncRNAs. Cancer-promoting lncRNAs are expected to be upregulated in tumors.62,63 Consistent with this, pan-hallmark hits are significantly higher expressed than non-hits in KRAS+ lung tumors (Figure 4F) and are upregulated in tumors compared with adjacent tissue (Figure 4G). Furthermore, screen hits tend to be amplified, but not depleted, in DNA from tumors (Figures 4H and S3E) and cell lines (Figure S3F).
In summary, the integration of diverse screens yields accurate maps of functional lncRNAs that are enriched for meaningful clinical features and display cell background-independent activity.
RNA therapeutics targeting NSCLC lncRNAs
Multi-hallmark lncRNA maps are a resource of targets for therapeutic ASOs.64, 65, 66 We manually selected nine lncRNAs from top-ranked hits based on criteria of novelty and lack of protein-coding evidence and henceforth referred to as “Tier 1.” Seven are annotated by GENCODE, and two by either FANTOM CAT or BIGTranscriptome (Table S2). For each candidate, we designed a series of ASOs and managed to identify at least two independent ASOs with ≥40% knockdown potency (Figure S4A).
Next, we tested ASOs’ phenotypic effects, in terms of proliferation and cisplatin sensitivity (Figure 5A). For five lncRNAs (Tier 2), we observed reproducible loss of cell proliferation with two distinct ASO sequences, indicating on-target activity.67 To check how broadly applicable these effects are, we retested the ASOs in two other KRAS+ NSCLC cell lines, H460, and H441 (derived from a pericardial effusion metastasis), and observed similar results (Figure 5A). Consistent with their effects on cisplatin sensitivity, Tier 2 genes’ expression is upregulated in response to cisplatin treatment (Figure S4B). Using the expression data from The Cancer Genome Atlas (TCGA) dataset, we noted that Tier 2 lncRNAs are overexpressed in NSCLC tumors, despite this not being a selection criterion (Figure 5B).
Figure 5.

Therapeutic targeting of oncogenic lncRNAs CHiLL1 and GCAWKR
(A) (Left) Experimental workflow to test knockdown efficiency and phenotypic effect of ASO transfection. (Right) Summary of all ASO/cell line results. Rows: targeted lncRNAs; columns: ASOs and cell lines. Values reflect the mean log2 fold change in viability following ASO transfection and are normalized to the control non-targeting ASO for proliferation, n > 2. Numbers to the left indicate library candidate identifiers and are grouped into tiers. Each lncRNA is targeted by two independent ASO sequences (1 and 2, below). MALAT1 lncRNA is used as positive control whose knockdown is expected to decrease viability. “Normal” cells are non-transformed cells of lung origin.
(B) Expression of tier 2 lncRNAs—CHiLL1, GCAWKR, candidate 205 (LINC00115), candidate 507 (LINC00910)—in TCGA RNA-seq. LUAD: 513 samples; normal: 59 samples. Statistical significance estimated by Student's t test; ∗∗∗∗p < 0.0001. Data were not available for candidate 215 in the TCGA dataset.
(C) A549 cell growth upon transfection with two independent ASOs. Results are normalized to non-targeting control ASO (n = 4 biological replicates; error bars: SD statistical significance: two-tailed Student’s t test).
(D) Genomic loci encoding CHiLL1 (top panel) and GCAWKR (bottom panel).
(E) (Left) Viability of H441 spheroid cultures 7 days after ASO transfection (25 nM). mTOR ASO was used as a positive control (n = 4 biological replicates; error bars: SD; one-tailed Student’s t test). (Right) Representative images (Leica DM IL LED Tissue Culture Microscope).
(F) Viability of independent replicates of BE874 organoids grown from a single KRAS+ patient-derived xenograft after CHiLL1 ASO transfection. The error bars represent the variability of technical replicates, while the biological replicates are indicated on the bottom (n = 4 biological replicates; n > 3 technical replicates; error bars: SD; one-tailed Student’s t test).
(G) ASO cocktails. (Top) For all experiments, the total amount of ASO did not vary (25 nM). Cocktails were composed of equal proportions of indicated ASOs. (Bottom) A549 cell populations, normalized to non-targeting ASO (n = 4 biological replicates; error bars: SD; statistical significance: one-tailed Mann-Whitney test).
(H) Expression of Tier 2 lncRNAs in KRAS+ LUAD tumors (TCGA, n = 101). Each cell represents a patient and is colored to reflect expression as estimated by RNA-seq (expression defined as >0.5 FPKM). At the bottom is the percentage of patients with at least one lncRNA from the indicated cocktails.
It has been proposed that targeting lncRNAs could cause lower side effects in healthy tissue, although few studies have tested this.17 We evaluated Tier 2 ASOs’ effects on a panel of non-transformed lung-derived cells: HBEC3-KT, MRC5-CV1 (both immortalized), and CCD-12Lu (primary). These cells displayed diminished or absent response, particularly for the first two candidate lncRNAs (Figure 5A). Consequently, we narrowed our focus to these Tier 3 lncRNAs: candidate 42 (ENSG00000253616), henceforth renamed Cancer Hallmark in Lung LncRNA 1 (CHiLL1), and candidate 240 (ENSG00000272808; GCAWKR).68 Both have low protein-coding potential (Figure S4D). Replication experiments confirmed the knockdown potency and phenotypic impacts using both CRISPR-del approach with multiple pgRNAs (Figure S3B) and, more importantly, two independent ASOs for each gene (Figures 5C, S4C, and S4E). ASOs displayed activity in additional KRAS+ and EGFR-mutant cell lines, suggesting subtype-independent activity (Figure S4F). On the other hand, overexpression of neither CHiLL1 nor GCAWKR could rescue ASO-induced phenotypes (Figure S5C). This may be the result of cis-regulatory mechanisms, although we did not observe such an effect for the nearest neighbor of CHiLL1, TNFRSF10B (Figure S5B). An additional plausible explanation is that these lncRNA loci act via functional transcript isoforms that remain unannotated and were not tested here.
CHiLL1 has, to our knowledge, never previously been implicated in cancer. It is located on Chromosome 8 and consists of two annotated isoforms, sharing the first exon (Figure 5D, top panel). The GENCODE annotation may commence slightly downstream of the true TSS, as evidenced by various transcriptomic evidence, but the latter falls within the region targeted by CRISPR-del and hence is unlikely to affect the screening outcome (Figure S5A). It is localized upstream and on the same strand of the PCG TNFRSF10B, previously associated with NSCLC,69 although we find no evidence for readthrough transcription between the two loci as shown by annotated expressed sequence tags (ESTs) (Figure S5A) and RT-PCR (Figure S5B). Supporting its relevance, high expression of CHiLL1 correlates with poor overall survival (Figure S4G).
GCAWKR (Chr15) comprises four isoforms sharing a common TSS (Figure 5D, bottom panel), which is supported by diverse evidence including full-length long-read sequencing70 (Figure S5D). It is associated with poor prognosis in colon cancer71 and, during the preparation of this paper, was reported to be an oncogene in gastric cancer.68 It has a likely ortholog in mouse, the uncharacterized Gm44753 (Figure S5H). In contrast to CHiLL1, the locus of GCAWKR is frequently amplified in cancer genomes (Figure S5F). In TCGA samples, GCAWKR expression is upregulated in the proximal inflammatory (PI) tumor subtype, which is associated with poorer prognosis (Figures S4H and S4I; PI versus PP: p = 4 × 10−5; PI versus TRU: p = 0.008).
3D in vitro models represent a more faithful tumor model compared with monolayer cultures.72,73 We delivered CHiLL1 and GCAWKR ASOs to spheroid cultures of H441 cells and observed a reduction in viability approaching that of the positive control, mTOR (Figures 5E and S4J).
Organoids derived from patient-derived xenografts (PDXs) recapitulate the therapy response of individual patients.74 Delivery of CHiLL1 ASOs resulted in a significant reduction in cell viability of KRAS+ human NSCLC BE874 organoids (Figure 5F).
We were curious whether simultaneous targeting of two or more distinct vulnerabilities, via “cocktails” of ASOs, might offer synergistic benefits. Indeed, a 50:50 cocktail of CHiLL1/GCAWKR ASOs (Tier 3 cocktail) displayed a greater effect on cell viability compared with an equal dose of either ASO alone (Figures 5G and S4K). A five-ASO cocktail for Tier 2 lncRNAs yielded a similar benefit (Figure 5G). Interestingly, cocktails resulted in no additional toxicity for non-cancerous cells (Figure S4L), suggesting that they might represent a therapeutic strategy for increasing potency at no cost of toxicity.
Finally, we asked what fraction of patients might benefit from ASO treatment. RNA sequencing (RNA-seq) data for TCGA tumors indicates that 78% express at least one of CHiLL1 and GCAWKR and might be treated by the Tier 3 cocktail, which rises to 92% for Tier 2 (Figure 5H).
Together, these results demonstrate that Tier 3 lncRNAs can be targeted by potent and low-toxicity ASOs, which may be beneficial alone or in combination for the majority of NSCLC cases (Figure 5H).
CHiLL1 and GCAWKR ASOs regulate cancer hallmarks via distinct modes of action
We next investigated the modes of action linking NSCLC hallmarks to CHiLL1 and GCAWKR. CHiLL1 exons lack obvious functional elements (Figure S5E), whereas GCAWKR contains numerous conserved sequences and structures (Figure 6A), hinting at potential functional elements. Subcellular localization yields important mechanistic clues for lncRNAs.75 Surprisingly, despite their similar oncogenic roles, fluorescence in situ hybridization (FISH) revealed contrasting localization patterns: CHiLL1 is located principally in the cytoplasm, and GCAWKR in the nucleus (Figure 6B). Specificity was validated by knockdown (Figure S5G), and results were further corroborated by cell fractionation (Figure 6C).
Figure 6.

CHiLL1 and GCAWKR drive distinct but overlapping oncogenic pathways
(A) Genomic elements in GCAWKR exons (gray rectangles) and introns. Strand is indicated by element direction, where appropriate. The plot was generated with ezTracks.76
(B) Confocal microscopy images of RNA-FISH performed with CHiLL1 and GCAWKR probe sets in A549 cells. Selected lncRNA foci are arrowed. Nuclear/cytoplasmic (N/C) quantification of CHiLL1 and GCAWKR.
(C) Ratio of concentrations of indicated RNAs as measured by qRT-PCR in nuclear/cytoplasmic fractions of A549 cells. MALAT1 and GAPDH are used as nuclear and cytosolic controls, respectively.
(D) Expression of CHiLL1 as quantified in the three RNA-seq replicates from A549 RNA. The y axis represents the normalized expression (counts) per nucleotide, and the boxplots show the variance of inference using bootstraps generated by Kallisto.
(E) Fold change in gene expression in response to CHiLL1 knockdown by ASO1 and ASO2 in A549 cells. Statistical significance: Pearson correlation coefficient. Trendline depicts regression line. Numbers indicate genes in each quadrant.
(F) Statistical enrichment of KEGG pathways among DEGs resulting from CHiLL1 knockdown. Genes significantly affected in common by ASO1 and ASO2 were included for each cell line. Cancer-relevant pathways that are significant for both cell lines are highlighted in green. Statistical significance: Pearson correlation coefficient.
(G) Annexin-V apoptosis assay 24 h after transfection with ASOs targeting CHiLL1 (left) and GCAWKR (right).
(H) As for (C), for GCAWKR.
(I) As for (D), comparing knockdown of GCAWKR with the same ASO in A549 and H460 cells (p = 2.2 × 10−16; R = 0.8; statistical significance: Pearson correlation coefficient).
(J) MSigDB term enrichment significance for differentially expressed genes in common between A549 and H460 cells. The most significant terms are shown. y axis: adjusted p value (q value).
(K) Cell-cycle assay results after 24 h upon knockdown of CHiLL1 and GCAWKR in A549. (n = 2 biological replicates, error bars: SD statistical significance: two-tailed Student’s t test).
(L) Cell migration across transwell supports in A549 cells. The crystal violet quantification is normalized to non-targeting ASO (n = 4 biological replicates, error bars: SD; statistical significance: one-tailed Student’s t test).
To gain more detailed mechanistic insights, we used molecular phenotyping by RNA-seq to quantify the transcriptome of A549 cells perturbed by CHiLL1 ASOs.71 CHiLL1 expression in control cells was 8.3 transcripts per million (TPM), equivalent to ∼4 molecules per cell and consistent with FISH.77 RNA-seq confirmed ASO knockdown efficiency (Figures 6D and S6A), and resulting transcriptome changes were highly correlated between ASOs, indicating that the majority of effects arise via on-target perturbation of CHiLL1 (Figure 6E). A similar correlation was observed in H460 cells (Figure S6B). The generality of CHiLL1-dependent expression changes was further confirmed by correlated expression changes between A549 and H460 (Figure S6C).
We explored perturbed genes by enrichment analysis. Defining high-confidence target gene subsets from the intersection of both ASOs, we identified enriched KEGG terms78 (Figures 6F and S6D). These underscored disease relevance (e.g., “non-small cell lung cancer”) and also implicated potential mechanistic pathways (MAPK, PI3K-Akt), and the high degree of concordance between the two cell backgrounds again supported the generality of CHiLL1 effects across KRAS+ NSCLC cells.
A similar analysis of the Molecular Signatures Database (MSigDB)79 implicated p53 and mTORC1 signaling (Figure S6E). Numerous transcription factor binding sites (TFBS) are enriched in changing genes, including ZBTB7A (in both A549 and H460)80,81 (Figure S6F). Interestingly, ZBTB7A has been reported as both an oncogene and tumor suppressor that regulates processes including apoptosis and glycolysis.82, 83, 84, 85
Among the cell-type-independent enriched KEGG pathways was “apoptosis” (Figure 6F). Supporting this, knockdown of CHiLL1, but not GCAWKR, resulted in a significant increase of early apoptotic cells (Figure 6G).
Turning to GCAWKR (mean expression 2.5 TPM, ∼1 copy per cell), we observed effective knockdown and cell-type-independent effects using a single ASO (Figures 6H, 6I, and S6G). Gene enrichment analysis revealed partially overlapping terms with CHiLL1 (including p53 pathway, cholesterol homeostasis, and epithelial to mesenchymal transition). Among the GCAWKR-specific terms, we noticed several related to cell-cycle progression, including “G2-M checkpoint” (Figure 6J). Indeed, knockdown of GCAWKR, but not CHiLL1, led to an increase of cells in the G2 phase (Figure 6K). GCAWKR targets are also enriched for migration genes, and knockdown led to impaired cell migratory capability (Figure 6L).
Interestingly, we notice that GCAWKR knockdown resulted in a significant upregulation of CHiLL1, but not vice versa, (Figure S6H), explaining the additive effect on cell viability observed with the CHiLL1/GCAWKR ASO cocktail (Figure 5G).
Together, these data establish that CHiLL1 and GCAWKR promote cancer hallmarks via widespread, non-overlapping downstream gene networks and support the on-target basis for ASO activity.
Discussion
We have mapped the functional lncRNA landscape across hallmarks and cell backgrounds in the most common KRAS+ subtype of NSCLC. This led us to lncRNA targets with pleiotropic oncogenic roles across a range of 2D and 3D models from primary and metastatic tumors, representing a promising foundation for future RNA therapeutics (RNATX).
Pooled CRISPR screening is emerging as a powerful tool for target discovery in RNATX thanks to its practicality and versatility.86 It avoids the capital investment and expertise required for arrayed screening,87 where bespoke ASO libraries would have to be created specifically for lncRNAs expressed in every cell/disease model, would be prohibitively expensive at the current market rates and unable to adapt to dynamically evolving lncRNA annotations. CRISPR also delivers improved perturbation and on-target rates compared with short hairpin RNA (shRNA). CRISPR screening libraries can be rapidly generated, enabling projects to keep pace with evolving lncRNA annotations to identify new, disease-specific targets. By directly identifying lncRNAs via their cell-level function, it represents a welcome addition to widely used, indirect evidence like survival, mutation, differential expression, or evolutionary conservation.19,88 Indeed, it was encouraging to observe concordance between these signatures and screen hits. Nonetheless, key barriers to entry remain, notably the lack of available screening libraries, making the libDECKO-NSCLC1 library a valuable resource for future discovery.
Through its integrative strategy, this work sets new standards. Previous studies typically screened a single cell line with a single hallmark (often proliferation) and in a single format (often dropout).25,27,89 Internal benchmarking here highlighted the risks of this approach, in identifying hits whose activity is specific to that cell background alone. We mitigated this with parallel screens in distinct but matched cell backgrounds, and the resulting hits could be validated in several additional models and mutational subtypes. To identify pleiotropic hits, we performed parallel screens in multiple phenotypic dimensions and both positive and negative formats. The latter produced marginal improvements in accuracy, although this benefit should be considered in light of the extra resources required. The resulting challenge of integrating diverse screen data was solved by developing the simple yet robust TPP pipeline that balances effect size and significance. Overall, the combination of multiple screens with TPP yielded improved performance compared with conventional approaches.
The result is a unique functional panorama of lncRNAs in a single cancer type. Given the paucity of global-level functional lncRNA maps,25,71 this dataset represents an invaluable resource for understanding both the basic biology of lncRNAs and their roles in cancer. Overall, our data implicate approximately 8% of lncRNAs with cell-level functions, comparable to Liu’s and Zhu’s estimates.25,49 The conceptual and experimental configuration will have a broad application for other diseases and biological systems.
This resource enabled us to narrow down nine lncRNAs for ASO development, from which we identified a pair of oncogenic lncRNAs, CHiLL1 and GCAWKR, with particularly promising characteristics as therapeutic targets. Firstly, replication experiments using distinct ASO sequences and in different cell backgrounds strongly suggested that observed phenotypic and molecular effects occur on target (via the intended lncRNA).90 Second, ASOs were effective in both monolayer and 3D KRAS+ NSCLC backgrounds, in addition to several EGFR-mutant cell lines, raising hope for a more general utility. Future efforts will be required to further refine ASO sequences and chemistry and to effectively deliver them in vivo. Third, those phenotypic effects were diminished in non-transformed cells, pointing to reduced non-specific toxicity in vivo. This raises hopes for reduced toxicity in healthy cells, allowing not only higher doses but also combination therapies to suppress therapy resistance.91 Finally, CHiLL1 or GCAWKR are detected in the majority of KRAS+ NSCLC tumors, suggesting that the majority of patients might benefit from eventual treatment.
Mechanistically, the concordance of CRISPR and ASO phenotypes indicates that both genes act via an RNA transcript or at least the production thereof.58,92 Both have profound effects on the cellular transcriptome, affecting hundreds of target genes that converge on many shared, oncogenic pathways, which yielded experimentally verifiable predictions. Interestingly, however, these effects are mediated by very different immediate molecular mechanisms, as evidenced by their distinct subcellular localization, non-overlapping target genes, and the fact that mixing their ASOs yielded greater than additive effects.
We have shown how ASO cocktails boosted efficacy without increasing toxicity compared with equal doses of single ASOs. Our findings open the possibility of using either fixed cocktails or cocktails tailored specifically to a patient’s tumor transcriptome, for potent, enduring, low-toxicity, and personalized cancer treatment.
Limitations of the study
One important caveat of CRISPR-del, in common with CRISPRi, is the risk of false-positive hits arising from unintentional targeting of overlapping cis- or trans-acting elements, such as DNA regulatory regions or opposite-strand lncRNA genes. Fortunately, these cases can be resolved by ASO validation, which can assign correct-strand RNA-mediated mechanisms for observed phenotypes. Our careful ASO validation indicated that while the majority of screen hits arise via the intended lncRNA-dependent mechanism, there were instances of likely false positives, i.e., candidates 408/316 (Figure 5A). A similar strategy could also narrow down LINC00115 as the functional lncRNA in the bidirectional locus candidate 205 (Figure 3A). Overall, we could validate 8/12 (67%) lncRNA hits using ASOs (9 for proliferation/cisplatin, 3 for migration), suggesting that around one-third of screen hits may act via DNA-dependent mechanisms.
Nonetheless, several issues remain to be addressed in the future. This study identified known and new lncRNAs promoting NSCLC but is clearly not comprehensive in terms of excluding that other lncRNAs play important disease roles. Despite targeting an extensive catalog comprising 831 lncRNAs detected at a permissive expression cutoff in A549 cells, it is likely that we overlooked many valuable targets due to the relative inefficiency of CRISPR-del as a perturbation compared with ORF mutation47 and also due to ongoing annotation incompleteness, resulting in lncRNAs that were either not screened or whose TSS was incorrectly targeted.70 For example, we could correctly identify some lncRNAs known to be involved in lung cancer pathogenesis16 like PVT1, LINC00680, and PCAT7 (Figure 2H) but, surprisingly, not MALAT1 or MIAT.16 The reason for the latter false negatives is unclear, although it may reflect their unusually high expression and/or else the presence of alternative TSSs that compensate for the loss of the targeted promoter. This may be overcome in the future by employing complementary direct RNA-targeting CRISPR perturbations, such as Cas13.93,94 We screened in two monolayer cell backgrounds; however, future screens should be performed in parallel across the widest panel of mutationally matched 2D and 3D models.73 Finally, our screens discovered scores of lncRNAs that could not be followed up here and hopefully will provide a fertile source of new targets in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| LentiCas9-Blast | Addgene | Cat#52962 |
| pDECKO_mCherry | Addgene | Cat#78534 |
| Electrocompetent EnduraTM cells | Lucigen | Cat#60242-2 |
| Chemicals, peptides, and recombinant proteins | ||
| Blasticidin | ThermoFisher | Cat#A1113903 |
| Cytofix/Cytoperm Fixation | BD | Cat#51-2090KZ |
| DAPI | Roche | Cat#10236276001 |
| Lipofectamine 2000 | Thermofisher | Cat#11668019 |
| Ki-67 Antibody | ThermoFisher | Cat#12-5698-82) |
| Matrigel® Matrix GFR, LDEV-free | Corning | Cat#356231 |
| PE Annexin | ThermoFisher | Cat#L34960 |
| Perm/Wash | BD | Cat#51-2091KE |
| Puromycin | ThermoFisher | Cat#A1113803 |
| Trypan Blue Solution (100mL) | Sigma-Aldrich | Cat#T8154 |
| Viability dye | ThermoFisher | Cat#35111 |
| Critical commercial assays | ||
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63880 |
| Blood & Cell Culture DNA Midi | Qiagen | Cat#13343 |
| Blood & Cell Culture DNA Mini | Qiagen | Cat#13323 |
| CellTrace™ CFSE Cell Proliferation Kit | ThermoFisher | Cat#C34570 |
| CellTiter-Glo 2.0 | Promega | Cat#G9242 |
| CellTiter-Glo® 3D Cell Viability Assay | Promega | Cat#G9682 |
| GoScript™ Reverse Transcriptase | Promega | Cat#A5003 |
| GoTaq | Promega | Cat#M3001 |
| GoTaq® qPCR Master Mix kit | Promega | Cat#A6002 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| Qubit™ dsDNA HS Assay Kit | ThermoFisher | Cat#Q32854 |
| Quick-RNA™ kit from (#) | ZymoResearch | Cat#R1055 |
| Z-Competent E.coli | ZymoResearch | Cat#T3001 |
| Deposited data | ||
| Pooled CRISPR screens in A549 and NCI-H460 | This paper | GEO: GSE207228 |
| RNA-Seq of A549 and NCI-H460 upon ASO treatment | This paper | GEO: GSE207227 |
| Experimental models: Cell lines | ||
| Human: A549 (male) | N/A | N/A |
| Human: CCD-16Lu (female) | N/A | N/A |
| Human: HEK293T (female) | N/A | N/A |
| Human: H441 (male) | N/A | N/A |
| Human: NCI-H460 (male) | N/A | N/A |
| Human: MRC-5 (male) | N/A | N/A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG® mice) | Charles River Laboratories | N/A |
| Oligonucleotides | ||
| LibDECKO-NSCLC1 (see File S1 for sequences) | This paper | N/A |
| ASOs (see Table S1 for sequences) | Qiagen | N/A |
| NGS sequencing primers (see Table S2 for sequences) | Sigma-Aldrich | N/A |
| Primers for genomic deletion and Primers RT-qPCR (see File S5) | Sigma-Aldrich | N/A |
| Software and algorithms | ||
| CRISPETa | 92 | N/A |
| FlowJo 10 | FlowJo | N/A |
| Graphpad Prism | https://www.graphpad.com/scientific-software/prism/ | N/A |
| R Statistical Software | https://www.r-project.org/ | N/A |
| Target Prioritisation Pipeline (TPP) | https://doi.org/10.5281/zenodo.6912510 | N/A |
| Stellaris probe designer | www.biosearchtech.com | N/A |
| Other | ||
| ChemiDoc Imaging System | BIO RAD | N/A |
| FACSAria™ III Cell Sorter | BD® | N/A |
| LSR II Flow Cytometer | BD® | N/A |
| Tecan Infinite® 200 Pro | N/A | N/A |
| Cell counter | Sigma-Aldrich | Cat#Z169021-1EA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dr. Rory Johnson, rory.johnson@ucd.ie.
Materials availability
Plasmids and cell lines generated in this study are available from the corresponding author R.J. upon request. This study did not generate new unique reagents.
Experimental model and subject details
HEK293T (female), A549 (male), NCI-H460 (male), H441 (male), CCD-16Lu (female) cell lines were a kind gift by the groups of Adrian Ochsenbein and Renwang Peng (University Hospital of Bern, Switerland). MRC-5 (male) cells were provided by the group of Ronald Dijkmanthe (Institute of Virology and Immunology, University of Bern, Switzerland). HBEC3-KT (female) bronchial epithelial human cells were purchased from the American Type Culture Collection (ATCC; http://www.atcc.org). All the cell lines were authenticated using Short Tandem Repeat (STR) profiling (Microsynth Cell Line Typing) and tested negative for mycoplasma contamination.
A549 and HEK293T cells were maintained in DMEM, MRC-5 in EMEM, NCI-H460, H441 as well as CCD-16Lu in RPMI-1640 medium, all supplemented with 10% Fetal Bovine Serum, 1% L-Glutamine, 1% Penicillin-Streptomycin. HBEC3-KT cells were maintained in Airway Epithelial Cell Basal Medium (ATCC #PCS-300-030) supplemented with Bronchial Epithelial Cell Growth Kit (ATCC #PCS-300-040).
All cells were passed every 2–3 days and maintained at 37°C in a humid atmosphere with 5% CO2.
Mouse experiments were approved by the local experimental animal committee of the Canton of Bern and performed according to Swiss laws for animal protection (BE76/17). Analysis of samples was approved by the Bern local ethical committee (KEK 2018-01801). Written informed consent was collected from all patients involved in the study.
Method details
Lentivirus and stable cell line production
The plasmids used in this paper are listed in Data S3. Lentivirus production was carried out by co-transfecting HEK293T cells with 12.5 μg of Cas9 plasmid with blasticidin resistance (Addgene #52962), 7.5 μg psPAX2 plasmid, and 4 μg of the packaging pVsVg plasmids, using Lipofectamine2000. 24 h before the transfection, 2.5 × 106 HEK293T cells were seeded in a 10 cm dish coated with Poly-L-Lysine (Sigma #P4832) (diluted 1:5 in 1X PBS). The supernatant containing viral particles was harvested 24, 48, and 72 h after transfection. Viral particles were then concentrated 100-fold by adding 1 volume of cold PEG-it Virus Precipitation Solution (BioCat #LV810A-1-SBI) to every four volumes of supernatant. After 12 h at 4°C, the supernatant/PEG-it mixture was centrifuged at 1,500 × g for 30 min at 4°C, resuspended in 1X PBS, and stored at −80°C until use.
For the generation of stable Cas9-expressing cell lines, A549 and H460 were incubated for 24 h with a culture medium containing concentrated viral preparation carrying pLentiCas9-T2A-BFP and 8 μg/mL polybrene. Infected cells were selected for at least five days with blasticidin (8 μg/mL) and then were FACS-sorted two times, to have at least 60% BFP-positive cells.
DECKO and lentiviral production
For the design and cloning of DECKO plasmids, we used our previously-described protocol30,92 (http://crispeta.crg.eu/).
All the pgRNA guides for CRISPR-del validations are reported in Data S4.
To produce lentivirus carrying the pDECKO plasmid, we followed the same protocol. After infection with pDECKO plasmid-carrying viruses, cells were selected with puromycin (2 μg/mL) for at least three days.
Library design
We downloaded Gene Transfer Format (GTF) annotations from the following sources: i) GENCODE annotation release 19 (GRCh37) from gencodegenes.org; ii) BIGTranscriptome annotation37 from http://big.hanyang.ac.kr/CASOL/; iii) FANTOM CAT.10 We alsma, lo generated a novel transcriptome assembly of A549 RNA-seq39,40 using StringTie,95 version 1.3. All 4 annotations were merged into a final annotation using the StringTie ‘-merge’ option.
All lncRNAs from the merged annotation were filtered thus: First, those with transcription start sites (TSS) overlapping or <2kb up or downstream of any protein-coding gene exon included in the GENCODE annotation were removed. Second, expression was calculated with RSEM v1.397, using the A549 RNA-seq data used to generate the novel transcriptome assembly, and transcripts with FPKM <0.1 were removed. The remaining TSSs within 300 bp from each other were clustered into a single TSS cluster. TSS clusters were intersected with ENCODE evidence source specific to A549 cells: Cap Analysis of Gene Expression (CAGE), DNAse I hypersensitivity sites, and ChromHMM marks: Active TSS, Flanking TSS, Promoter Downstream TSS, Flanking TSS Downstream, Genic enhancer1, Genic enhancer2, Active Enhancer 1, Active Enhancer 2, Weak Enhancer and Bivalent-Poised TSS.39,40,96 Candidate TSS clusters were prioritised by the number of evidence sources.
We designed neutral control pgRNAs in genomic regions not expected to affect cell phenotype. We retrieved 10 regions in the AAVS1 gene loci from the publication of Zhu and colleagues.49 To this set, we added a set of 65 randomly selected intergenic regions (>10 kb distant from nearest gene annotation) and 25 intronic regions (for introns >5 kb in length). Moreover, 53 positive (promoting cell growth) and 50 negative (opposing cell growth) protein-coding gene (PCG) controls, with known roles in promoting/opposing cancer cell growth and cisplatin resistance were added. These were manually selected from literature and retrieved from the paper of Zhu and colleagues.49 The complete list of genes contained in the library is available in Table S1.
10 unique pgRNAs were generated for each candidate TSS cluster region with CRISPETa92 using the following parameters: -eu 0 -ed 0 -du 1000 -dd 1000 -si 0.2 -t 0,0,0,x,x -v 0.4 -c DECKO. During design, the CRISPETa software’s off-targeting filters were employed. We performed repeated designs with decreasing stringency until 10 pgRNAs for each target were identified. Specifically, in the first run, only pgRNAs without mismatches were allowed. If < 10 pgRNAs per candidate were identified, the parameters were subsequently relaxed: in the second round one off-target with 2 mismatches was allowed; in the third round, the design region was repeatedly increased in size. A summary can be found in Data S4.
The final library design comprised 12,000 unique sequences of length 165/166 bp, with overhangs compatible with cloning into the pDECKO plasmid.30,92 The median distance between the pgRNAs is shown in Figure S1D.
Library cloning
The library was synthesised as single-stranded oligonucleotides by Twist Bioscience (USA), and upon arrival resuspended in nuclease-free low Tris-EDTA (TE) buffer (10 mM Tris-HCl, pH 8.0 and 0.1 mM EDTA) to a final concentration of 10 ng/μL. This was PCR amplified using the Oligo-Fw: 5′-ATCTTGTGGAAAGGACGAAA-3′ and Oligo-Rev: GCCTTATTTTAACTTGCTATTTC (PCR reaction ssDNA to dsDNA detailed in Data S4) with the following conditions: 95 °C × 1 min; 10 cycles of (95 °C × 1 min, 53 °C × 20 s, 72 °C × 1 min); 72 °C × 10 min. The amplification product was purified using the QIAquick PCR Purification Kit (Qiagen #28104) according to the manufacturer’s instructions. The correct amplicon size was checked on a 2% agarose gel at 100V for 40 min.
The steps of cloning follow the low-throughput protocol described in.30 In the first step, the pDECKO_mCherry plasmid was digested following the pDECKO_backbone plasmid digestion conditions listed in Data S4. The amplified library was inserted into the digested plasmid, using Gibson Assembly mix (obtained from ‘Biomolecular Screening & Protein Technologies’ Unit at CRG, Barcelona) at 50°C for 1 h (200 ng of pDECKO_mCherry plasmid, 20 ng amplified library, H2O up to 10 μL, 10 μL of Gibson mix 10 μL). 1 μL of the Gibson reaction was delivered to 25 μL of electrocompetent EnduraTM cells (Lucigen #60242-2) using Gene Pulser/MicroPulser Electroporation Cuvettes, 0.1 cm gap (Biorad #16520891). The library coverage of 66.7X was estimated by counting the number of obtained bacterial colonies divided by the total number of different sequences in the designed library (12,000). The intermediate plasmid obtained in this step contains the pgRNA variable sequences, but still lacks the constant part of the first sgRNA and the H1 promoter (Figure S1B).30
In the second step of cloning, the intermediate plasmid was digested by BmsbI enzyme (ThermoFisher #ER0451). After purification, the constant insert was assembled by ligation, by using PAGE purified and 5′ phosphorylated long oligos (Long oligos to generate pDECKO constant part listed in Data S4), as explained in Pulido-Quetglas C. et al.92 Afterwards, 5 μL of the ligation product was transformed and used for the electroporation of electrocompetent Endura cells as described above. Clones were tested by colony PCR and by Sanger sequencing using colony PCR primers (Data S4). The colony PCR conditions are listed in Data S4. The overall library quality was evaluated by NGS sequencing. Briefly, the plasmid containing the pgRNAs was PCR amplified with Primers for NGS sequencing (listed in Table S3), and purified using Agencourt AMPure XP beads (Beckman Coulter #A63880), according to the manufacturer’s protocol. The purified product was sequenced by Illumina at a depth of 20M PE125 reads. The reads were aligned to the pgRNAs library and the read distribution of each pgRNA was determined using the Ineq package in R (version 3.5.3) to calculate both the Lorenz-curve and Gini-coefficient (Figure S1C).
Lentiviral titer calculation and infection
To achieve the desired MOI of 0.3–0.4, a titration experiment in A549 and H460 cells was performed. 2 × 106 cells were plated in each well of a 12-well plate and supplemented with 8 μg/mL polybrene. Each well was treated with viruses ranging from 2.5 to 50 μL and transduced via spin-infection as previously described.97 After centrifugation, the media was replaced with complete fresh media without polybrene and incubated overnight. The following day, cells were counted and each well was split into two equal aliquots, of which one was treated with 2 μg/mL puromycin. After 72 h, the MOI was calculated by dividing the number of surviving cells in the puromycin well, by the number in the puromycin-free well. The MOI of 0.3 was used for all screening experiments. For large-scale screens, 120M cells were seeded in 12-well plates with a density of 2M per well for spin-infection. The following day, cells were pooled together and a fresh puromycin-containing (2 μg/mL) medium was added. Puromycin selection was maintained for six days until phenotypic screens began.
CRISPR screens
One week after infection (Time point 0 or T0), cells were counted and the reference sample was collected (T0, 16M cells corresponding to a library coverage >1,000x). For all screens, cells were cultured in 150 mm culture-treated dishes and passed every 2-3 days.
Proliferation
Dropout screens: at T0 16M of cells were plated and passaged to maintain a coverage >1,000X (defined as the number of cells divided by the number of unique library sequences). Cells were harvested at 14 and 21 days for gDNA extraction. CFSE screens: At T7, 16M cells were seeded and starved for 24 h with media lacking FBS. Then cells were stained using CellTrace CFSE Cell Proliferation Kit (ThermoFisher #C34570) following the manufacturer’s instructions. One aliquot of stained cells was immediately analyzed by flow cytometry, while the rest were plated with normal media. Five days later (T5), cells were sorted into two populations: 20% brightest (slow-growing) and 20% least bright (fast-growing). The two populations were plated separately and, five days later (T10) subjected to another round of staining and sorting.
Cisplatin screen
Optimal cisplatin working concentrations were established via dose-response and cell doubling time (Figure S2C). In the dose-response, 3,000 A549 and H460 cells were plated in 96-well plates and treated with a range of cisplatin concentrations (0.5;1; 2; 2.5; 5; 6.5; 8; 12.5; 25; 50μM). After 72 h, CellTiter-Glo 2.0 (Promega #G9242) was added to the media (1:1), and luminescence was recorded. For the cell doubling time, 1M cells were plated in 10 cm plates. Different cisplatin concentrations were added at indicated concentrations, and living cells were counted every 2-3 days up to 14 days. Cisplatin survival screen: 48 and 96M cells were plated at T0 and treated with 6.5 and 25 μM of Cisplatin for A549 cells, corresponding to IC30 and IC80, respectively. Two different cisplatin concentrations were used to increase the dynamic range of the screens and account for diverging effects of different doses: a mild concentration (IC30) treatment decreases cell proliferation by about 30%. On the other hand, high concentration treatments induce proliferative arrest or cell death.98 Cell pellets were collected after 14 and 21 days. The death screen was carried out as follows: 144M cells were seeded and treated with cisplatin at 2 and 1 μM (IC20) for A549 (Figure S2C). Every 24 h, for five days, floating (dead) cells were collected and pooled together for gDNA extraction.
Migration screen
To test the optimal conditions, the following set-up experiment was performed. 0.5 M A549 cells/well were seeded in 5 Boyden chambers (Corning, PC Membrane, 8.0 μm, 6.5 mm #3422-COR). Each migration assay was stopped at a different time point (ranging from 5 h up to 48 h; Figure S2G). 48 h was selected as the time point for the following experiment. At T0 infected cells (∼16M) were divided and seeded in the upper part of 32 transwell inserts (0.5 M cells/transwell). The upper part of transwell inserts was filled with media lacking FBS, and the lower part with media containing 10% FBS. After 48 h cells in the upper part of the chamber (impaired migration) and lower part (accelerated migration) (Figure 2C) were trypsinized and plated separately for 48 h, after this time, cells were counted and collected for gDNA extraction. Control cells that did not undergo the migration assay were harvested at the same time as a reference population.
Genomic DNA preparation and sequencing
Genomic DNA (gDNA) was isolated using the Blood & Cell Culture DNA Midi (5e6–3e07 cells) (Qiagen #13343), or Mini (<5 × 106 cells) Kits (Qiagen #13323) as per the manufacturer’s instructions. The gDNA concentrations were quantified by Nanodrop.
For PCR amplification, gDNA was divided into 100 μL reactions such that each well had at most 4 μg of gDNA. Each well consisted of 66.5 μL gDNA plus water, 23.5 μL PCR master mix (20 μL Buffer 5X, 2 μL dNTPs 10 μM, 1.5 μL GoTaq; Promega #M3001), and 5 μL of the forward universal primer, and 5 μL of a uniquely barcoded P7 primer (both stock at 10 μM concentration). PCR cycling conditions: an initial 2 min at 95°C; followed by 30 s at 95°C, 40 s at 60°C, 1 min at 72°C, for 22 cycles; and a final 5 min extension at 72°C. Forward and reverse oligos for PCR and NGS of the library are listed in Table S3. PCR products were purified with Agencourt AMPure XP SPRI beads according to the manufacturer’s instructions (Beckman Coulter #A63880). Purified PCR products were quantified using the Qubit dsDNA HS Assay Kit (ThermoFisher #Q32854). Samples were sequenced on a HiSeq2000 (Illumina) with paired-end 150 bp reads at coverage of 40M reads/sample.
Screen hit identification and prioritization
The raw sequencing reads from individual screens were analyzed by using CASPR.99 The quality of all NGS experiments was monitored across several metrics using the CASPR analysis pipeline. Overall, 36% of the reads were usable, while recombination events affected a low rate (typically <3%) of reads. After the mapping step, the obtained counts per million (cpm) for each pgRNA were filtered to remove sequences with 3 > cpm>666. Low scoring guides were removed by GuideScan,100 and a batch effect correction was applied using MageckFlute.101 After all the corrections, the table count was provided to CASPR to calculate log2-Fold Change and FDR corrected p-values at a target level.
Replicate correlation analysis was performed based on CASPR intermediate files providing normalized read counts for each replicate per sample. Pearson correlation was calculated for log2-transformed read counts of all pgRNAs (guide level) targeting lncRNA regions excluding controls, guide level log2-transformed fold change (log2FC) (treatment sample ‘trt’/control sample ‘ctr) and gene level log2-transformed fold change. Gene level (replicate specific) log2FC was derived as the median of log2FC values from all pgRNAs (filtered as described) targeting the same genomic region according to CASPR and MAGeCK’s method97 or the average of the top 3 guides similar to previous studies.25 Log2FC values were also Z score scaled, based on the mean and SD of the neutral control pgRNAs.
At the single-screen level, we selected the hits with FDR<0.2 and LFC>0 for the positive screens (i.e. CFSE, death, and migration) and FDR<0.2 and LFC<0 for the dropout and cisplatin screens.
To integrate multiple screens an integrative target prioritization pipeline (TPP) was designed, applying two different approaches in parallel: the Robust Rank Aggregation (RRA)102 to compute a ranking based on the effect size (CASPR log2FC) across screens, and an empirical adaptation of Brown’s method (EBM)103 to combine the significance values (CASPR p-value) of each candidate across screens. The RRA-scores were converted to exact p-values using the rho-score correction from the same R package. Subsequently, the harmonic mean p-value (HMP)104 was calculated using the two significance scores from RRA and EBM. These p-values were corrected for multiple hypothesis testing using the Benjamini & Hochberg method, and a cutoff of FDR<0.2 was used to define hits. The code is available at https://doi.org/10.5281/zenodo.6912510.
Enrichment scores and nominal p-values (GSEA simulation, n = 10,000) of positive and neutral control genes were used as an indication of the quality of the ranking, as well as the fraction of detected genes previously linked to lung cancer.49 Positive and neutral control genes were also used as “true positives/false negatives” and “false positives/true negatives” respectively to calculate Receiver Operating Characteristics (ROC) curves and associated statistical metrics.
Statistical analysis and data visualization was performed using R software version 4.1.2 and the following R packages: ggplot2 (3.3.5), ggpubr (0.4.0), ggtern (3.3.5), ggrepel (0.9.1), ggbeeswarm (0.6.0), GGally (2.1.2), Gmedian (1.2.6), GSEABase (1.54.0), clusterProfiler (4.0.5), harmonicmeanp (3.0), metap (1.8), Hmisc (4.6-0), precrec (0.12.8), MASS (7.3-55), car (3.0-12), eulerr (6.1.1), SuperExactTest (1.1.0), UpSetR (1.4.0), metaRNASeq (1.0.7).
Public RNA-sequencing data
101 KRAS+ Lung Adenocarcinoma (LUAD) RNA-seq samples were downloaded from TCGA (gdc.cancer.gov), applying the following filters: Adenocarcinoma - not treated - KRAS mutated, and the expression of target genes was estimated using HTSeq.105 Another independent cohort of LUAD RNA-seq ex vivo data, containing 87 tumor and 77 adjacent normal tissue samples, was obtained from the TANRIC.61,106
PCR amplification from genomic DNA
gDNA was extracted with GeneJET Genomic DNA Purification Kit (ThermoFisher #K0702) from pDECKO-transduced A549-Cas9-expressing cells. The PCR was done with genomic primers flanking the deleted region (Data S4) as shown in Figure 1E, using the Phusion High-Fidelity DNA Polymerase (2 U/μl) (ThermoFisher #F-530S). The product was run on a 1% agarose gel.
Competition assay
A549 cells were infected with DECKO lentiviruses expressing fluorescent proteins. Viruses expressing control pgRNAs targeting AAVS1 also expressed GFP protein (pgRNAs-AASV1-GFP+), while the pgRNAs targeting candidate lncRNAs expressed mCherry. After infection, and seven days of puromycin (2 μg/mL) selection, GFP and mCherry cells were mixed 1:1 in a six-well plate (150,000 cells). Cell counts were analyzed by LSR II SORP instrument (BD Biosciences) and analyzed by FlowJo software (Treestar).
Patient-derived xenograft organoids
The KRAS+ patient-derived organoid BE874 was derived in the following way. Small pieces (∼1–2 cm3) of lung cancer tissue (provided by the Institute of Pathology, University of Bern) were taken from the surgically resected lung cancer specimen with patients’ informed consent. Parts of the sample (pieces of around 5 mm) were separated and implanted subcutaneously into the flanks of 6 weeks old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (purchased from Charles River Laboratories) for cancer engraftment.107 After successful engraftment, tumor-bearing mice were euthanized and tumors were resected. Single cells were isolated through mechanical and enzymatic tissue disruption for the generation of BE874 organoids. Genotyping of BE874 organoids was performed at the Institute of Pathology, University of Bern, using KRAS targeted sanger sequencing. KRAS c.34G>T (p.Gly12Cys) mutation was detected in both BE874 organoids and the corresponding primary cancer. Analysis of samples was approved by the Bern local ethical committee (KEK 2018-01801). Written informed consent was collected from all patients involved in the study.
NSG mice were housed under specific pathogen-free conditions in isolated ventilated cages on a regular 12-h/12-h cycle of light and dark. Mice were fed ad libitum and were regularly monitored for pathogens. Mouse experiments were approved by the local experimental animal committee of the Canton of Bern and performed according to Swiss laws for animal protection (BE76/17).
Antisense oligonucleotides
Locked nucleic acid ASOs were designed using the Qiagen custom LNA oligonucleotides designer (www.qiagen.com). Per each target, we designed from 3 to 5 different ASOs. On the day of the transfection, 300,000 cells were counted and plated on a 6-well plate. ASOs were transfected into the cells still in suspension, using Lipofectamine3000 (ThermoFisher #L3000015) with a final 25 nM in 2 mL media for A549, H460, NCI-H441, and MRC5-CV1 and 10 nM in 2 mL for HBEC3-KT and CCD-16LU, following the manufacturer’s instructions.
For cocktail experiments, the final concentration of the ASOs mix was kept at 25 nM. The media was refreshed 24 h post-transfection and cells were harvested to check the efficiency of gene knockdown or sub-cultured for cell viability experiments. The ASOs target sequences are listed in Table S2. We checked ASOs penetration in cells employing the 5′-FAM-labelled control ASO A provided by Qiagen (Figure S4J).
2D cell viability assay
Cell viability assay was carried out in 2D cell lines by using CellTiter-Glo 2.0 (Promega #G9242). The assays were performed according to the corresponding manufacturer’s protocol. 24 h after the transfection, A549, H460, NCI-H441, H1975, H157, WVU-Ma-0005A, H820, and H1650 cells were harvested, counted and 3,500 cells/well were seeded in triplicate in 96 well plates. For Mrc5-SV1, HBEC3-KT, and CCD-16LU 3,000, 3,500, and 1,000 cells/well were seeded, respectively. The number of viable cells was estimated after 24, 48, 72, 96, and/or 144 h. On the day of the measurement, a mix of 1:1 media and CellTiter-Glo was added to the plates and the luminescence was recorded with Tecan Infinite 200 Pro. The Student’s t test was used to evaluate significance (p < 0.05).
3D cell viability assay
NCI-H441 cells were detached, counted, and 200,000 cells were plated in 24 well plates. The ASO-Lipofectamine3000 mix was delivered to the cells in suspension as described above. After 24 h, the cells are detached, counted, and seeded onto 96-well Black/Clear Round Bottom Ultra-Low Attachment Surface Spheroid Microplate (Corning #4520) in 20 μL domes of Matrigel Matrix GFR, LDEV-free (Corning #356231) and RPMI-1640 growth medium (1:1) with a density of 20,000 cells per dome. Matrigel containing the cells was allowed to solidify for an hour in the incubator at 37°C before adding DMEM-F12 (Sigma #D6421) media on top of the wells (40 and 80 μL for the wells intended for the first and second-time point, respectively. The spheroids were allowed to grow in the incubator at 37°C in a humid atmosphere with 5% CO2. After 4 h the number of viable cells in the 3D cell culture was recorded as time point 0 (T0), CellTiter-Glo 3D Cell Viability Assay (Promega #G9682) was added to the wells, following the manufacturer’s instructions, and the contents transferred into a Corning 96-well Flat Clear Bottom White (Corning #3610) for the reading with the Tecan Infinite 200 Pro. After one week the measurement was repeated.
BE874 organoids were generated and expanded using a special composition of lung cancer organoids (LCO) medium (Data S4).
BE874 organoids were transfected with ASOs as described for the NCI-H441. 24 h after transfection the cells were detached, counted, and seeded onto Corning 96-well Flat Clear Bottom White (Corning #3610) in 20 μL domes of LCO growth medium and Matrigel (1:1) with a density of 20,000 cells per dome. The Matrigel domes containing PDX-organoids were allowed to solidify for an hour in the incubator at 37°C before adding 80 μL LCO growth media on top. The organoids were allowed to grow in the incubator at 37°C in a humid atmosphere with 5% CO2. After 24 h, 100 μL of CellTiter-Glo 3D Cell Viability Assay (Promega #G9682) were added to the wells intended for the T0, and the luminescence was recorded with Tecan Infinite 200 Pro. After three days the 80 μL of LCO media were added to the wells to keep them from drying out. After one week, the media was aspirated and replenished with fresh 80 μL, before proceeding with the measurement with CellTiter-Glo 3D.
Rescue experiments
To overexpress CHiLL1 and GCAWKR, the full-length sequences of these genes (ENST00000520840.1; ENST00000558254.5, respectively) were synthesized by Gene Universal Inc. and cloned into pcDNA3.1 vector to generate the overexpressing plasmids. And the empty vector pcDNA3.1 was considered as a control. Cell transfection for A549 cells was performed using Lipofectamine 3000 (Invitrogen, CA). Successfully transfected cells were selected with puromycin (2 μg/mL) for 96 h. The transfection efficiency was confirmed by RT-qPCR and cells were collected for the subsequent experiments.
Apoptosis assay
Annexin V and viability dye were used to detect early apoptotic and dead cells, respectively. 24 h after the transfection cells were counted and 150,000 cells were suspended in 100 μL of 1X PBS. The viability dye (ThermoFisher #35111) was added (1:5,000) in 100 μL of 1X PBS and cells were incubated for 30 min at 4°C. Cells were then washed once with 1X PBS and suspended in 100 μL of Annexin buffer PH 7, added PE Annexin (1:200; ThermoFisher #L34960), and incubated for 30 min at 4°C. After a wash with 1X PBS, cells were resuspended in 300 μL of Annexin buffer and underwent the flow analysis by using the LSR Fortessa instrument (BD Biosciences). Unstained cells were used as control.
Cell cycle assay
Cells were transfected with ASOs using Lipofectamine3000 according to the manufacturer’s instructions. After 24 h cells were harvested and fixed with 100 μL of BD Cytofix/Cytoperm Fixation (BD Biosciences #51-2090KZ) for 30 min at room temperature. The cells were then washed with 200 μL of 1X BD Perm/Wash (BD Biosciences #51-2091KE) and resuspended in 100 μL of 1X PBS. The Ki-67 Antibody (ThermoFisher #12-5698-82) was added (1:100) and incubated for 30 min at 4°C. Wash again with 1X BD perm/wash and stain with DAPI (Roche #10236276001) was added (1:10,000) in 100 μL of 1X PBS. Incubate 5′ at room temperature and wash with 1X PBS. Acquire the data with the Fortessa flow cytometer. Data analysis was performed using FlowJo, and the different cell cycle phases were determined according to the Dean-Jett Fox (DJF) model.
Low-throughput migration assay
Migration assay was performed as previously described.108 24 h after ASOs transfection, A549 cells were counted and seeded in the upper part of Boyden chambers with a density of 35,000 cells/transwell. The upper part of transwell inserts was filled with media without FBS, while the lower part with media supplemented with 10% FBS to induce the directional movement of cells. After 24 h the cells were washed three times with 1X PBS and stained using 300 μL of crystal violet 1% for 30 min. Three washes with 1X PBS followed. The cells in the upper part of the membrane were removed by using a cotton swab. The chambers are left to dry overnight. The day after, the crystal violet was solubilized in 1X PBS containing 1% SDS and the absorbance at 595 nm was recorded by using the Tecan Infinite 200 Pro.
RNA isolation and RT–qPCR
To purify total RNAs from cultured cells, a Quick-RNA kit from ZymoResearch (#R1055) was used according to the manufacturer’s protocol. RNAs were reverse transcribed to produce cDNAs by using the GoScript Reverse Transcription System Kit (Promega #A5003). The cDNAs were then used for qPCR to evaluate gene expression, using the GoTaq qPCR Master Mix kit (Promega #A6002). The expression of HPRT1 was used as an internal control for normalization. All the RT-qPCR primers are listed in Data S4.
RNA-sequencing and analysis
24 h after ASO transfection, A549, and H460 cells were harvested and the total RNA was extracted as explained before and samples’ quality was checked at Bioanalyzer. Libraries were prepared using the NEBNext Ultra RNA Library Prep Kit and sequenced in paired-end 150 format to a depth of 30M reads/sample.
Transcript quantification was performed using Kallisto v0.46.0110 against GENCODE v36.38 Gene level expression was inferred by aggregating the counts of the individual isoforms. Differential expression analysis was performed using Sleuth v0.30111. Genes with a q-value <0.2 were considered significant. For CHiLL1, the genes that were significantly up- and down-regulated with two different ASO were selected. For GCAWKR, we selected the pool of common genes deregulated in A549 and H460.
Visualization of the results was produced in R 4.0.0 (R: The R Project for Statistical Computing, n.d.) using ggplot2 package v3.3.2 (Ggplot2 - Elegant Graphics for Data Analysis | Hadley Wickham | Springer, n.d.). Functional enrichment analysis was performed through the enrichR package v2.1.109
FISH and cell fractionation
Fluorescent In-Situ Hybridization (FISH) was performed on A549 cell lines, according to the Stellaris protocol (https://www.biosearchtech.com/support/resources/stellaris-protocols). For detection of CHiLL1 and GCAWKR at a single-cell level, pools of 25 and 48 FISH probes respectively were designed using the Stellaris probe designer software (www.biosearchtech.com). Cells were grown on round coverslip slides (ThermoFisher, 18 mm), fixed in 3.7% formaldehyde, and permeabilized in ethanol 70% overnight. Hybridization was carried out overnight at 37°C in the hybridization buffer from Stellaris. Cells were counterstained with DAPI and visualized using the DeltaVision microscope.
Nuclear and cytoplasmic fractionation was carried out in A549 cells as described previously.80 The pipeline used to analyze the data was adapted from CellProfiler,110 and it is named 'SpeckleCounting'.
ezTracks visualization of RNA elements
Genomic tracks were retrieved for the hg38 human genome assembly from their original publications: predicted neo-functionalized fragments of transposable elements, also known as RIDLs (repeat insertion domains of lncRNA),75 RNA structures conserved in vertebrates (CRS),111 and ENCODE candidate cis-regulatory elements.108 In addition, the following tracks were downloaded from the UCSC Genome Browser112: repeat-masked CpG islands, phastCons conserved elements in 7, 20, 30, and 100-way multiple alignments,113 and repeat families from the RepeatMasker annotation (http://www.repeatmasker.org).114
The comprehensive gene annotations for CHiLL1 (ENSG00000253616) and GCAWKR (ENSG00000272808 and ENSG00000232386) loci were extracted from the GENCODE v36 GTF file.38 Then, all the exons corresponding to each locus were collapsed into a meta-transcript and output as separate GTF files using BEDTools115 merge. Third, configuration files for each locus were prepared to draw the meta-transcript annotation alongside the genomic tracks using the program.76
Copy number status of pan-hallmark candidates
The copy number status of A549 and H460 cells was retrieved from the CCLE (https://portals.broadinstitute.org/ccle/data). Then, we intersected the hg19 coordinates of the pgRNA with the hg19 coordinates of the CCLE copy number data.
TCGA-LUAD copy number data were downloaded as ‘log2 ratio segment means’ using the R package TCGAbiolinks and converted to hg19 coordinates using liftOver. The values of each candidate were averaged across all TCGA-LUAD samples.
The pan-cancer recurrently amplified or deleted genomic regions were downloaded from the ICGC Data Portal (https://dcc.icgc.org/releases/PCAWG/consensus_cnv/GISTIC_analysis; all_lesions.conf_95.rmcnv.pt_170207.txt.gz). Then, we searched for overlaps between each candidate and the recurrent copy number altered regions (“Wide Peak Limits”). The differences in the proportions of amplified, deleted, or non-copy number altered hits versus non-hits were tested using Fisher’s exact tests.
Quantification and statistical analysis
Statistical analysis, quantification and visualisation was performed using Graphpad Prism (https://www.graphpad.com/scientific-software/prism/), R Statistical Software (https://www.r-project.org/; versions 3.5.3, 4.0.0 and 4.1.2) and the following R packages: ggplot2 (3.3.2, 3.3.5), ggpubr (0.4.0), ggtern (3.3.5), ggrepel (0.9.1), ggbeeswarm (0.6.0), GGally (2.1.2), Gmedian (1.2.6), GSEABase (1.54.0), clusterProfiler (4.0.5), harmonicmeanp (3.0), metap (1.8), Hmisc (4.6-0), TCGAbiolinks, liftOver, precrec (0.12.8), MASS (7.3-55), car (3.0-12), eulerr (6.1.1), SuperExactTest (1.1.0), UpSetR (1.4.0), metaRNASeq (1.0.7), enrichR (2.1).
Further software used for quantification and statistical analysis were StringTie95 (1.3), RSEM95 (1.3), CRISPETa,92 CASPR,99 GuideScan,100 MageckFlute,101 Kallisto116 (0.46.0), Sleuth117 (0.30), bedtools115 and Excel (Microsoft).
For all analyses, p ≤ 0.05 is considered statistically significant unless otherwise specified. Specific details concerning statistical tests for individual experiments are noted in the relevant Figure legends. For detailed statistical analysis of the computational analysis see Method Details.
Acknowledgments
We thank members of the GOLD Lab, including Joana Carlevaro-Fita, Núria Bosch-Guiteras, Michela Coan, Antonio Tarruell, and Tina Uroda, for insightful discussions. We also thank Roderic Guigó (CRG Barcelona, Spain) for insightful discussions. Thomas Marti and Renwang Peng (Department of BioMedical Research, DBMR, Bern, Switzerland) generously donated cell lines and advice on functional assays. We thank Basak Ginsbourger (DMBR) for administrative support and Willy Hofstetter and Patrick (DMBR) for logistical support. All computations were performed on the Bern Interfaculty Bioinformatics Unit computing cluster maintained by Rémy Bruggmann and Pierre Berthier. This work was funded by the Swiss National Science Foundation through the National Centre of Competence in Research (NCCR) “RNA & Disease” (51NF40-182880), project funding “The elements of long noncoding RNA function” (31003A_182337), Sinergia project “Regenerative strategies for heart disease via targeting the long noncoding transcriptome” (173738); by the Medical Faculty of the University and University Hospital of Bern; by the Helmut Horten Stiftung, Swiss Cancer Research Foundation (4534-08-2018); and by Science Foundation Ireland through Future Research Leaders award 18/FRL/6194.
Author contributions
T.P., R.E., and R.J. conceived and designed the experiment procedure and performed data analysis and interpretation. D.F.M. developed the TPP pipeline and performed most of the bioinformatics analysis. C.P.-Q. designed the libDECKO-NSCLC1 with the help of S.H. P.C. analyzed the RNA-seq data. S.F. and M.R. established the patient-derived organoids. P.S., A.K., G.B., and J.S. generated the stably expressing Cas9 cell line lines and helped with the validation. E.S.R., I.M., and P.C.M. validated the results in additional cell lines. D.H. performed the analysis on the FISH images. L.Z. and X.W. performed the survival analysis from TCGA data. A.V. provided the set of known cancer genes in NSCLC. A.A., H.A.G.-R., P.P.M., and A.L. contributed to the bioinformatics analysis. I.C. prepared the libraries for the NGS. S.H., C.R., and A.F.O. provided key inputs and tools. T.P., R.E., and R.J. wrote the manuscript with input from all the authors.
Declaration of interests
T.P., R.E., and R.J. have filed a patent application (21202363.4) relating to the therapeutic targeting of lncRNAs identified here. Applicant: University of Bern. R.J. is a paid consultant for NextRNA (USA).
Published: August 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100171.
Supplemental information
This file contains, per each target, the sequence of the pgRNAs and the target gene.
Data and code availability
-
•
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO)118 and are publicly available as of the date of publication at GEO: GSE207229. Accession numbers are listed in the key resources table.
-
•
The code related to the target prioritisation pipeline (TPP) for multiple screens integration has been deposited at Zenodo: 6912511 and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Gridelli C., Rossi A., Carbone D.P., Guarize J., Karachaliou N., Mok T., Petrella F., Spaggiari L., Rosell R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015;11:1–16. doi: 10.1038/nrdp.2015.9. [DOI] [PubMed] [Google Scholar]
- 3.Salgia R., Pharaon R., Mambetsariev I., Nam A., Sattler M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC) Cell Rep. Med. 2021;2:100186. doi: 10.1016/j.xcrm.2020.100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rotow J., Bivona T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer. 2017;17:637–658. doi: 10.1038/nrc.2017.84. [DOI] [PubMed] [Google Scholar]
- 5.Hong D.S., Fakih M.G., Strickler J.H., Desai J., Durm G.A., Shapiro G.I., Falchook G.S., Price T.J., Sacher A., Denlinger C.S., et al. KRASG12C inhibition with Sotorasib in Advanced solid tumors. N. Engl. J. Med. 2020;383:1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 7.Uszczynska-Ratajczak B., Lagarde J., Frankish A., Guigó R., Johnson R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018;19:535–548. doi: 10.1038/s41576-018-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma L., Cao J., Liu L., Du Q., Li Z., Zou D., Bajic V.B., Zhang Z. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019;47:2699–D134. doi: 10.1093/nar/gky960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volders P.J., Anckaert J., Verheggen K., Nuytens J., Martens L., Mestdagh P., Vandesompele J. Lncipedia 5: towards a reference set of human long non-coding rnas. Nucleic Acids Res. 2019;47:D135–D139. doi: 10.1093/nar/gky1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hon C.-C., Ramilowski J.A., Harshbarger J., Bertin N., Rackham O.J.L., Gough J., Denisenko E., Schmeier S., Poulsen T.M., Severin J., et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017;543:199–204. doi: 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan D., Weinberg R.A., Ouzounis C.A.C., López-Bigas N., Hanahan D., Weinberg R.A.R., Vogelstein B., Kinzler K., Friedberg E., Brentani H., et al. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 13.Statello L., Guo C.J., Chen L.L., Huarte M. 2021. Gene Regulation by Long Non-coding RNAs and its Biological Functions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montes M., Lubas M., Arendrup F.S., Mentz B., Rohatgi N., Tumas S., Harder L.M., Skanderup A.J., Andersen J.S., Lund A.H. The long non-coding RNA MIR31HG regulates the senescence associated secretory phenotype. Nat. Commun. 2021;12:2459. doi: 10.1038/s41467-021-22746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee B., Sahoo A., Marchica J., Holzhauser E., Chen X., Li J.L., Seki T., Govindarajan S.S., Markey F.B., Batish M., et al. The long noncoding RNA SPRIGHTLY acts as an intranuclear organizing hub for pre-mRNA molecules. Sci. Adv. 2017;3:e1602505. doi: 10.1126/sciadv.1602505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vancura A., Lanzós A., Bosch-Guiteras N., Esteban M.T., Gutierrez A.H., Haefliger S., Johnson R., Vancura A., Lanzós A., Bosch-Guiteras N., et al. Cancer LncRNA Census 2 (CLC2): an enhanced resource reveals clinical features of cancer lncRNAs. NAR Cancer. 2021;3:zcab013. doi: 10.1093/narcan/zcab013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S.J., Malatesta M., Lien B.V., Saha P., Thombare S.S., Hong S.J., Pedraza L., Koontz M., Seo K., Horlbeck M.A., et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020;21:83. doi: 10.1186/s13059-020-01995-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leucci E., Vendramin R., Spinazzi M., Laurette P., Fiers M., Wouters J., Radaelli E., Eyckerman S., Leonelli C., Vanderheyden K., et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531:518–522. doi: 10.1038/nature17161. [DOI] [PubMed] [Google Scholar]
- 19.Gutschner T., Hämmerle M., Eissmann M., Hsu J., Kim Y., Hung G., Revenko A., Arun G., Stentrup M., Gross M., et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tseng Y.Y., Moriarity B.S., Gong W., Akiyama R., Tiwari A., Kawakami H., Ronning P., Reuland B., Guenther K., Beadnell T.C., et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., Feng L., Zheng Y., Li W., Liu L., Xie S., Zhou Y., Chen C., Cheng D. Linc00680 promotes the progression of non-small cell lung cancer and functions as a sponge of mir-410-3p to enhance hmgb1 expression. OncoTargets Ther. 2020;13:8183–8196. doi: 10.2147/OTT.S259232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun X., Wang R., Tan M., Tian X., Meng J. LncRNA LINC00680 promotes lung adenocarcinoma growth via binding to GATA6 and canceling GATA6-mediated suppression of SOX12 expression. Exp. Cell Res. 2021;405:112653. doi: 10.1016/j.yexcr.2021.112653. [DOI] [PubMed] [Google Scholar]
- 23.Sun C.C., Li S.J., Li G., Hua R.X., Zhou X.H., Li D.J. Long intergenic noncoding RNA 00511 acts as an oncogene in non-small-cell lung cancer by binding to EZH2 and suppressing p57. Mol. Ther. Nucleic Acids. 2016;5:e385. doi: 10.1038/mtna.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esposito R., Bosch N., Lanzós A., Polidori T., Pulido-Quetglas C., Johnson R. Hacking the cancer genome: profiling therapeutically-actionable long noncoding RNAs using CRISPR-Cas9 screening. Cancer Cell. 2019;35:545–557. doi: 10.1016/j.ccell.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 25.Liu S.J., Horlbeck M.A., Cho S.W., Birk H.S., Malatesta M., He D., Attenello F.J., Villalta J.E., Cho M.Y., Chen Y., et al. CRISPRi-based genome-scale identification of functional long non-coding RNA loci in human cells. Science. 2017;355:aah7111. doi: 10.1126/science.aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bester A.C., Lee J.D., Chavez A., Lee Y.R., Nachmani D., Vora S., Victor J., Sauvageau M., Monteleone E., Rinn J.L., et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell. 2018;173:649–664.e20. doi: 10.1016/j.cell.2018.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hay M., Thomas D.W., Craighead J.L., Economides C., Rosenthal J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- 29.MacLeod A.R., Crooke S.T. RNA therapeutics in oncology: advances, challenges, and future directions. J. Clin. Pharmacol. 2017;57:S43–S59. doi: 10.1002/jcph.957. [DOI] [PubMed] [Google Scholar]
- 30.Aparicio-Prat E., Arnan C., Sala I., Bosch N., Guigó R., Johnson R., Guigó R., Johnson R. DECKO: single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genom. 2015;16:846. doi: 10.1186/s12864-015-2086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furlan G., Gutierrez Hernandez N., Huret C., Galupa R., van Bemmel J.G., Romito A., Heard E., Morey C., Rougeulle C. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Mol. Cell. 2018;70:462–472.e8. doi: 10.1016/j.molcel.2018.03.024. [DOI] [PubMed] [Google Scholar]
- 32.George M.R., Duan Q., Nagle A., Kathiriya I.S., Huang Y., Rao K., Haldar S.M., Bruneau B.G. Minimal in vivo requirements for developmentally regulated cardiac long intergenic non-coding RNAs. Development. 2019;146:dev185314. doi: 10.1242/dev.185314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry R.B.T., Hezroni H., Goldrich M.J., Ulitsky I. Regulation of Neuroregeneration by long noncoding RNAs. Mol. Cell. 2018;72:553–567.e5. doi: 10.1016/j.molcel.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H.J., Gopalappa R., Sunwoo H., Choi S.W., Ramakrishna S., Lee J.T., Kim H.H., Nam J.W. En bloc and segmental deletions of human XIST reveal X chromosome inactivation-involving RNA elements. Nucleic Acids Res. 2019;47:3875–3887. doi: 10.1093/nar/gkz109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canver M.C., Bauer D.E., Dass A., Yien Y.Y., Chung J., Masuda T., Maeda T., Paw B.H., Orkin S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014;289:21312–21324. doi: 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goyal A., Myacheva K., Groß M., Klingenberg M., Duran Arqué B., Diederichs S., Groß M., Klingenberg M., Duran Arqué B., Diederichs S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017;45:e12. doi: 10.1093/nar/gkw883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You B.-H., Yoon S.-H., Nam J.-W. High-confidence coding and noncoding transcriptome maps. Genome Res. 2017;27:1050–1062. doi: 10.1101/gr.214288.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K., et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.ENCODE Project Consortium. Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imkeller K., Ambrosi G., Boutros M., Huber W. Modelling asymmetric count ratios in CRISPR screens to decrease experiment size and improve phenotype detection. bioRxiv. 2019 doi: 10.1101/699348. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brower M., Carney D.N., Oie H.K., Gazdar A.F., Minna J.D. Growth of cell lines and clinical specimens of human non-small cell lung cancer in a Serum-free defined medium. Cancer Res. 1986;46:798–806. [PubMed] [Google Scholar]
- 43.Giard D.J., Aaronson S.A., Todaro G.J., Arnstein P., Kersey J.H., Dosik H., Parks W.P. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 44.Stojic L., Lun A.T.L., Mangei J., Mascalchi P., Quarantotti V., Barr A.R., Bakal C., Marioni J.C., Gergely F., Odom D.T. Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res. 2018;46:5950–5966. doi: 10.1093/nar/gky437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horlbeck M.A., Liu S.J., Chang H.Y., Lim D.A., Weissman J.S. Fitness effects of CRISPR/Cas9-targeting of long noncoding RNA genes. Nat. Biotechnol. 2020;38:573–576. doi: 10.1038/s41587-020-0428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ota T., Suzuki Y., Nishikawa T., Otsuki T., Sugiyama T., Irie R., Wakamatsu A., Hayashi K., Sato H., Nagai K., et al. Complete sequencing and characterization of 21, 243 full-length human cDNAs. Nat. Genet. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- 47.Lavalou P., Eckert H., Damy L., Constanty F., Majello S., Bitetti A., Graindorge A., Shkumatava A. Strategies for genetic inactivation of long noncoding RNAs in Zebrafish. RNA. 2019 doi: 10.1261/rna.069484.118. rna.069484.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu S., Li W., Liu J., Chen C.H., Liao Q., Xu P., Xu H., Xiao T., Cao Z., Peng J., et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 2016;34:1279–1286. doi: 10.1038/nbt.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beermann J., Kirste D., Iwanov K., Lu D., Kleemiß F., Kumarswamy R., Schimmel K., Bär C., Thum T., Kleemiss F., et al. A large shRNA library approach identifies lncRNA Ntep as an essential regulator of cell proliferation. Cell Death Differ. 2018;25:307–318. doi: 10.1038/cdd.2017.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lv J., Qiu M., Xia W., Liu C., Xu Y., Wang J., Leng X., Huang S., Zhu R., Zhao M., et al. High expression of long non-coding RNA SBF2-AS1 promotes proliferation in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2016;35:75. doi: 10.1186/s13046-016-0352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y., Cao Z., Wang Y., Guo Y., Xu P., Yuan P., Liu Z., He Y., Wei W. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 2018;36:1203–1210. doi: 10.1038/nbt.4283. [DOI] [PubMed] [Google Scholar]
- 53.Hanna N., Johnson D., Temin S., Baker S., Brahmer J., Ellis P.M., Giaccone G., Hesketh P.J., Jaiyesimi I., Leighl N.B., et al. Systemic therapy for stage IV non–small-cell lung cancer: American Society of clinical oncology clinical practice guideline update. J. Clin. Oncol. 2017;35:3484–3515. doi: 10.1200/JCO.2017.74.6065. [DOI] [PubMed] [Google Scholar]
- 54.Shao L., Yu Q., Lu X., Zhang X., Zhuang Z. Downregulation of LINC00115 inhibits the proliferation and invasion of lung cancer cells in vitro and in vitro. Ann. Transl. Med. 2021;9:1256. doi: 10.21037/atm-21-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang P., Chen D., Ma H., Li Y. LncRNA SNHG12 contributes to multidrug resistance through activating the MAPK/Slug pathway by sponging miR-181a in non-small cell lung cancer. Oncotarget. 2017;8:84086–84101. doi: 10.18632/oncotarget.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai F., Damle S.S., Ling K.K., Rigo F. Directed RNase H cleavage of Nascent transcripts causes transcription termination. Mol. Cell. 2020;77:1032–1043.e4. doi: 10.1016/j.molcel.2019.12.029. [DOI] [PubMed] [Google Scholar]
- 57.Wu B., Xue X., Lin S., Tan X., Shen G. LncRNA LINC00115 facilitates lung cancer progression through miR-607/ITGB1 pathway. Environ. Toxicol. 2022;37:7–16. doi: 10.1002/tox.23367. [DOI] [PubMed] [Google Scholar]
- 58.Yuan C., Luo X., Duan S., Guo L. Long noncoding RNA LINC00115 promotes breast cancer metastasis by inhibiting miR-7. FEBS Open Bio. 2020;10:1230–1237. doi: 10.1002/2211-5463.12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peng N., Zhang Z., Wang Y., Yang M., Fan J., Wang Q., Deng L., Chen D., Cai Y., Li Q., et al. Down-regulated LINC00115 inhibits prostate cancer cell proliferation and invasion via targeting miR-212-5p/FZD5/Wnt/β-catenin axis. J. Cell Mol. Med. 2021;25:10627–10637. doi: 10.1111/jcmm.17000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng W., Li B., Wang J., Zhang H., Liu Y., Xu D., Cheng K., Zhuang J. Long non-coding RNA LINC00115 contributes to the progression of colorectal cancer by targeting miR-489-3p via the PI3K/AKT/mTOR pathway. Front. Genet. 2020;11:567630. doi: 10.3389/fgene.2020.567630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seo J.S., Ju Y.S., Lee W.C., Shin J.Y., Lee J.K., Bleazard T., Lee J., Jung Y.J., Kim J.O., Shin J.Y., et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marín-Béjar O., Mas A.M., González J., Martinez D., Athie A., Morales X., Galduroz M., Raimondi I., Grossi E., Guo S., et al. The human lncRNA LINC-PINT inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 2017;18:202. doi: 10.1186/s13059-017-1331-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huarte M. Nature Publishing Group; 2015. The Emerging Role of lncRNAs in Cancer. [DOI] [PubMed] [Google Scholar]
- 64.Blokhin I., Khorkova O., Hsiao J., Wahlestedt C. Developments in lncRNA drug discovery: where are we heading? Expert Opin. Drug Discov. 2018;13:837–849. doi: 10.1080/17460441.2018.1501024. [DOI] [PubMed] [Google Scholar]
- 65.Dhuri K., Bechtold C., Quijano E., Pham H., Gupta A., Vikram A., Bahal R. Antisense oligonucleotides: an emerging area in drug discovery and development. J. Clin. Med. 2020;9:2004. doi: 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaczmarek J.C., Kowalski P.S., Anderson D.G. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60. doi: 10.1186/s13073-017-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsui M., Corey D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017;16:167–179. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma M., Zhang Y., Weng M., Hu Y., Xuan Y., Hu Y., Lv K. lncRNA GCAWKR promotes gastric cancer development by scaffolding the chromatin modification factors WDR5 and KAT2A. Mol. Ther. 2018;26:2658–2668. doi: 10.1016/j.ymthe.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y., Guo S., Li D., Tang Y., Li L., Su L., Liu X. YIPF2 promotes chemotherapeutic agent-mediated apoptosis via enhancing TNFRSF10B recycling to plasma membrane in non-small cell lung cancer cells. Cell Death Dis. 2020;11:242. doi: 10.1038/s41419-020-2436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lagarde J., Uszczynska-Ratajczak B., Carbonell S., Pérez-Lluch S., Abad A., Davis C., Gingeras T.R., Frankish A., Harrow J., Guigo R., Johnson R. High-throughput annotation of full-length long noncoding RNAs with Capture Long-Read Sequencing. Nat. Genet. 2017;49:1731–1740. doi: 10.1038/ng.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramilowski J.A., Yip C.W., Agrawal S., Chang J.-C., Ciani Y., Kulakovskiy I.V., Mendez M., Ooi J.L.C., Petri A., Roos L., et al. Functional annotation of human long non-coding RNAs via molecular phenotyping. bioRxiv. 2019 doi: 10.1101/gr.254219.119. Preprint at. [DOI] [Google Scholar]
- 72.Unger C., Kramer N., Walzl A., Scherzer M., Hengstschläger M., Dolznig H. Modeling human carcinomas: physiologically relevant 3D models to improve anti-cancer drug development. Adv. Drug Deliv. Rev. 2014;79-80:50–67. doi: 10.1016/j.addr.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 73.Han K., Pierce S.E., Li A., Spees K., Anderson G.R., Seoane J.A., Lo Y.H., Dubreuil M., Olivas M., Kamber R.A., et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature. 2020;580:136–141. doi: 10.1038/s41586-020-2099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386.e10. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 75.Carlevaro-Fita J., Polidori T., Das M., Navarro C., Zoller T.I., Johnson R. Ancient exapted transposable elements promote nuclear enrichment of human long noncoding RNAs. Genome Res. 2019;29:208–222. doi: 10.1101/gr.229922.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guillen-Ramirez H.A., Johnson R. 2021. ezTracks v0.1.0. [Google Scholar]
- 77.Kellis M., Wold B., Snyder M.P., Bernstein B.E., Kundaje A., Marinov G.K., Ward L.D., Birney E., Crawford G.E., Dekker J., et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Apostolopoulou K., Pateras I.S., Evangelou K., Tsantoulis P.K., Liontos M., Kittas C., Tiniakos D.G., Kotsinas A., Cordon-Cardo C., Gorgoulis V.G. Gene amplification is a relatively frequent event leading to ZBTB7A (Pokemon) overexpression in non-small cell lung cancer. J. Pathol. 2007;213:294–302. doi: 10.1002/path.2222. [DOI] [PubMed] [Google Scholar]
- 81.Zhijun Z., Jingkang H. MicroRNA-520e suppresses non-small-cell lung cancer cell growth by targeting Zbtb7a-mediated Wnt signaling pathway. Biochem. Biophys. Res. Commun. 2017;486:49–56. doi: 10.1016/j.bbrc.2017.02.121. [DOI] [PubMed] [Google Scholar]
- 82.Berkers C.R., Maddocks O.D.K., Cheung E.C., Mor I., Vousden K.H. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617–633. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maeda T., Hobbs R.M., Merghoub T., Guernah I., Zelent A., Cordon-Cardo C., Teruya-Feldstein J., Pandolfi P.P. Role of the proto-oncogene Pokemon in cellular transformation and ARF repression. Nature. 2005;433:278–285. doi: 10.1038/nature03203. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y.Q., Xiao C.X., Lin B.Y., Shi Y., Liu Y.P., Liu J.J., Guleng B., Ren J.L. Silencing of pokemon enhances caspase-dependent apoptosis via fas- and mitochondria-mediated pathways in hepatocellular carcinoma cells. PLoS One. 2013;8:e68981. doi: 10.1371/journal.pone.0068981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Constantinou C., Spella M., Chondrou V., Patrinos G.P., Papachatzopoulou A., Sgourou A. The multi-faceted functioning portrait of LRF/ZBTB7A. Hum. Genomics. 2019;13:66. doi: 10.1186/s40246-019-0252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doench J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018;19:67–80. doi: 10.1038/nrg.2017.97. [DOI] [PubMed] [Google Scholar]
- 87.Agrotis A., Ketteler R. A new age in functional genomics using CRISPR/Cas9 in arrayed library screening. Front. Genet. 2015;6:300. doi: 10.3389/fgene.2015.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hosono Y., Niknafs Y.S., Prensner J.R., Iyer M.K., Dhanasekaran S.M., Mehra R., Pitchiaya S., Tien J., Escara-Wilke J., Poliakov A., et al. Oncogenic role of THOR , a conserved cancer/Testis non-coding RNA. Cell. 2017;171:1559–1572.e20. doi: 10.1016/j.cell.2017.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Joung J., Engreitz J.M., Konermann S., Abudayyeh O.O., Verdine V.K., Aguet F., Gootenberg J.S., Sanjana N.E., Wright J.B., Fulco C.P., et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548:343–346. doi: 10.1038/nature23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gagnon K.T., Corey D.R. Guidelines for experiments using antisense oligonucleotides and double-stranded RNAs. Nucleic Acid Ther. 2019;29:116–122. doi: 10.1089/nat.2018.0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bayat Mokhtari R., Homayouni T.S., Baluch N., Morgatskaya E., Kumar S., Das B., Yeger H. Combination therapy in combating cancer. Oncotarget. 2017;8:38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pulido-Quetglas C., Aparicio-Prat E., Arnan C., Polidori T., Hermoso T., Palumbo E., Ponomarenko J., Guigo R., Johnson R. Scalable design of paired CRISPR guide RNAs for genomic deletion. PLoS Comput. Biol. 2017;13:e1005341. doi: 10.1371/journal.pcbi.1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Konermann S., Lotfy P., Brideau N.J., Oki J., Shokhirev M.N., Hsu P.D. Transcriptome engineering with RNA-targeting type VI-D CRISPR Effectors. Cell. 2018;173:665–676.e14. doi: 10.1016/j.cell.2018.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J., Verdine V., Cox D.B.T., Kellner M.J., Regev A., et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ernst J., Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang B., Krall E.B., Aguirre A.J., Kim M., Widlund H.R., Doshi M.B., Sicinska E., Sulahian R., Goodale A., Cowley G.S., et al. ATXN1L, CIC, and ETS transcription factors Modulate sensitivity to MAPK pathway inhibition. Cell Rep. 2017;18:1543–1557. doi: 10.1016/j.celrep.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bergadà-Pijuan J., Pulido-Quetglas C., Vancura A., Johnson R. CASPR, an analysis pipeline for single and paired guide RNA CRISPR screens, reveals optimal target selection for long non-coding RNAs. Bioinformatics. 2020;36:1673–1680. doi: 10.1093/bioinformatics/btz811. [DOI] [PubMed] [Google Scholar]
- 100.Perez A.R., Pritykin Y., Vidigal J.A., Chhangawala S., Zamparo L., Leslie C.S., Ventura A. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 2017;35:347–349. doi: 10.1038/nbt.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang B., Wang M., Zhang W., Xiao T., Chen C.H., Wu A., Wu F., Traugh N., Wang X., Li Z., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019;14:756–780. doi: 10.1038/s41596-018-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kolde R., Laur S., Adler P., Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poole W., Gibbs D.L., Shmulevich I., Bernard B., Knijnenburg T.A. Bioinformatics. Oxford University Press; 2016. Combining dependent P-values with an empirical adaptation of Brown’s method; pp. i430–i436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wilson D.J. The harmonic mean p-value for combining dependent tests. Proc. Natl. Acad. Sci. USA. 2019;116:1195–1200. doi: 10.1073/pnas.1814092116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anders S., Pyl P.T., Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li J., Han L., Roebuck P., Diao L., Liu L., Yuan Y., Weinstein J.N., Liang H. TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015;75:3728–3737. doi: 10.1158/0008-5472.CAN-15-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J., et al. Human lymphoid and myeloid cell development in NOD/LtSz- scid IL2R γ null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 108.ENCODE Project Consortium. Purcaro M.J., Epstein C.B., Adrian J., Davis C.A., Kaul R., Van Nostrand E.L., Gorkin D.U., He Y., Pauli-Behn F., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jawaid W. 2021. Provides an R Interface to “Enrichr” [R Package enrichR Version 3.0] [Google Scholar]
- 110.McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D., et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Seemann S.E., Mirza A.H., Hansen C., Bang-Berthelsen C.H., Garde C., Christensen-Dalsgaard M., Torarinsson E., Yao Z., Workman C.T., Pociot F., et al. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 2017;27:1371–1383. doi: 10.1101/gr.208652.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smit A.F.A., Hubely R. RepeatMasker home page. RepeatModeler open-1.0. 2008. http://www.repeatmasker.org/
- 115.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 117.Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017;14:687–690. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 118.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains, per each target, the sequence of the pgRNAs and the target gene.
Data Availability Statement
-
•
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO)118 and are publicly available as of the date of publication at GEO: GSE207229. Accession numbers are listed in the key resources table.
-
•
The code related to the target prioritisation pipeline (TPP) for multiple screens integration has been deposited at Zenodo: 6912511 and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.