

ABSTRACT
Changes in the composition of gut-associated microbial communities are associated with many human illnesses, but the factors driving dysbiosis remain incompletely understood. One factor governing the microbiota composition in the gut is bile. Bile acids shape the microbiota composition through their antimicrobial activity and by activating host signaling pathways that maintain gut homeostasis. Although bile acids are host-derived, their functions are integrally linked to bacterial metabolism, which shapes the composition of the intestinal bile acid pool. Conditions that change the size or composition of the bile acid pool can trigger alterations in the microbiota composition that exacerbate inflammation or favor infection with opportunistic pathogens. Therefore, manipulating the composition or size of the bile acid pool might be a promising strategy to remediate dysbiosis.
KEYWORDS: Bile acids, microbiome, intestinal homeostasis, colonization resistance
Introduction
The colonic microbiota contains 100-fold more bacteria than any other microbial community in our body 1. This large microbial community functions in the catabolism of nutrients that are not broken down and absorbed by host enzymes in the upper gastrointestinal tract. Its size and catabolic activity make the colonic microbiota the principal source of microbial metabolites. Since microbiota-derived metabolites affect many aspects of human health, 2–7 changes in the composition of the colonic microbiota are linked to many human diseases.8 Understanding the factors that shape the composition and metabolic activity of the colonic microbiota is therefore a primary objective of microbiome research. The main drivers of the composition and function of the colonic microbiota are the diet and the host environment.9
One important player the host uses to shape the environment of the colonic microbiota is the epithelial lining. By maintaining the colonic epithelium in a state of physiological hypoxia, the host limits the diffusion of oxygen into the intestinal lumen.10 The resulting anaerobiosis drives a dominance of obligately anaerobic bacteria in the colon (reviewed in11). Conditions that abrogate epithelial hypoxia increase the diffusion of oxygen into the lumen of the colon, thereby increasing the abundance of facultatively anaerobic bacteria (reviewed in10), which is a microbial signature of dysbiosis.12,13 A second function of the colonic epithelium is the maintenance of an inner mucus layer that is largely devoid of bacteria, thereby protecting the epithelial surface from the high-density microbial community in the colon.14 Erosion of the inner mucus layer exacerbates colitis, which is attributed to increased penetration of bacteria into crypts.15–17
However, in addition to the local control of microbial growth orchestrated by the colonic epithelium, the environment of the colonic microbiota is also influenced by host digestive functions of the upper gastrointestinal tract. Among these factors, ileal bile absorption has been shown to have a marked impact on the colonic microbiota composition and function.18 Bile is synthesized by the liver and contains as one of its major components the primary bile acids, which are conjugated to either taurine or glycine. After synthesis, bile is stored in the gallbladder before being released in the duodenum during digestion to help in the absorption of dietary lipids and fat-soluble vitamins.19 While about 95% of the conjugated primary bile acids are absorbed in the terminal ileum and transported back to the liver to be recycled, approximately 5% pass into the colon. Here, the conjugated primary bile acids are deconjugated by the microbiota to liberate taurine, glycine, and primary bile acids. The latter are further metabolized by the colonic microbiota into secondary bile acids, thereby modifying the composition of the total bile acid pool.18 Conversely, bile acids regulate the composition of the microbiota, directly via their antimicrobial activity, and indirectly by interacting with nuclear and membrane receptors modulating intestinal homeostasis and immunity.2,20
The complex relationship between bile acids and the gut microbiota plays an important role in shaping the microbiota composition and the defense against enteric pathogens. By regulating the bile acid pool, the microbiota promotes the maintenance and renewal of the intestinal barrier 21 and controls the maturation of hosts' innate and adaptive immune responses.2 Moreover, bile acid-derived microbial metabolites protect against some opportunistic pathogens.22 Infection with enteric bacteria can disrupt ileal bile acids absorption and endocrine regulation of bile acids production.23,24 Furthermore, infection can favor the expansion of bacterial taxa that use bile acid-derived taurine to produce hydrogen sulfide, a metabolite that inhibits respiration of facultatively anaerobic bacteria.25
This review will discuss how the interaction between bile acids and the microbiota shapes the environment in the colon to maintain intestinal barrier function, immune homeostasis, and colonization resistance against enteric pathogens, and how a disturbance in bile metabolism impacts host gut physiology.
Bile acid metabolism and the microbiome
The enterohepatic circulation of bile acids
The enterohepatic circulation of bile acids is a finely regulated process of bile acid production in the liver, conjugation to taurine or glycine, storage in the gallbladder, secretion in the duodenum after a meal, reabsorption in the ileum and transport back to the liver to be recycled. This process happens 4 to 12 times per day in humans and ensures the maintenance of bile acid homeostasis.19
Primary bile acids are synthesized by oxidizing cholesterol, which is catalyzed by cytochrome P450s, with cholesterol 7α-hydroxylase (CYP7A1) being the rate-limiting enzyme (Figure 1).19 The predominant primary bile acids synthesized in human hepatocytes are cholic acid (CA) and chenodeoxycholic acid (CDCA).19 In rodents, the major primary bile acids are CA, as well as α-, β-muricholic acid (MCA) 26 and ursodeoxycholic acid (UDCA)27 synthesized from CDCA. Primary bile acids are then conjugated to taurine or glycine to form tauro- and glyco-conjugated bile salts. Conjugated primary bile acids are stored in the gallbladder, before being secreted into the duodenum following food intake to facilitate the absorption of dietary lipids and fat-soluble vitamins through the formation of micelles. In the gut, conjugated primary bile acids are deconjugated and converted into secondary bile acids by the microbiota, thus further increasing the diversity of the bile acid pool (Figure 2).18 While small amounts of primary bile acids can be absorbed by passive diffusion, effective absorption requires active transport mediated by the apical bile salt transporter (ASBT) expressed in the ileal epithelium.36 Conjugated primary bile acids are mainly transported through enterocytes by the ileal bile acid-binding protein (IBABP).36
Figure 1.
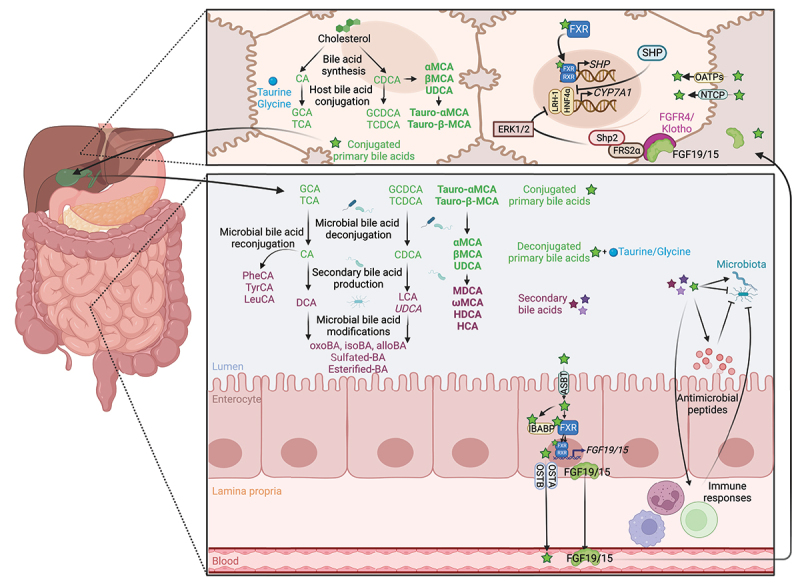
Enterohepatic circulation and bacterial metabolism of bile acids. Primary bile acids, CA and CDCA, are synthesized in the liver from cholesterol.19 In rodents, CDCA is converted to α-MCA, β- MCA 26 and UDCA,27 the latter being considered a secondary bile acid in humans.28,29 Mouse-specific bile acids are represented in bold, and human-specific bile acids are represented in italics. Bile acids are conjugated with glycine or taurine (mainly taurine in rodents) before being mixed to the bile, stored in the gallbladder, and secreted to the gut.19 In the gut, the microbiota deconjugates primary bile acids to release taurine and glycine.30 Conjugated and deconjugated bile acids can be further dehydroxylated,26 oxidated/epimerized,31 esterified,32 sulfated 33 and reconjugated 34 by the microbiota to produce secondary bile acids. Conversely, bile acids shape the gut microbiota composition, both directly via their detergent properties and the ability of the microbiota to metabolize bile acids, and indirectly by modulating hosts antimicrobial peptides production and immune responses.2,20 Primary bile acids are absorbed by the enterocytes via the ASBT transporter,32 transported through the cells by IBABP, and secreted into the blood by the heterodimer OSTα/β.35 Bile acids are transported into hepatocytes by NTCP and OATPs, where they activate FXR, which dimerizes with RXR to induce the transcription of SHP to inhibit CYP7A1 transcription.36 In the gut, bile acids activate FXR/RXR heterodimer to induce the transcription of FGF19/15 that is secreted into the blood to reach the liver. In the liver, FGF15/19 binds to FGFR4/β-Klotho to inhibit CYP7A1 expression via FRS2α, Shp2 and ERK1/2, thus limiting de novo bile acid synthesis.36 Abbreviations: ASBT, apical bile salt transporter; CYP7A1, cholesterol 7α-hydroxylase; ERK1/2, extracellular signal-regulated kinases 1/2; FGF19/15, fibroblast growth factor 19/15; FGFR4/β-Klotho, FGF receptor 4/beta-Klotho; FRS2α, fibroblast growth factor receptor substrate 2α; FXR, farnesoid X receptor; HNF4α, hepatocyte nuclear factor 4α; IBABP, ileal bile acid-binding protein; LRH-1, liver receptor homolog-1; NTCP, sodium taurocholate co-transporting polypeptide; OATPs, organic anion-transporting polypeptides; OSTα/β, organic solute transporter alpha/beta; RXR, retinoid X receptor; SHP, small heterodimer partner; Shp2, Src homology-2 domain-containing protein tyrosine phosphatase-2. Created with BioRender.com.
Figure 2.
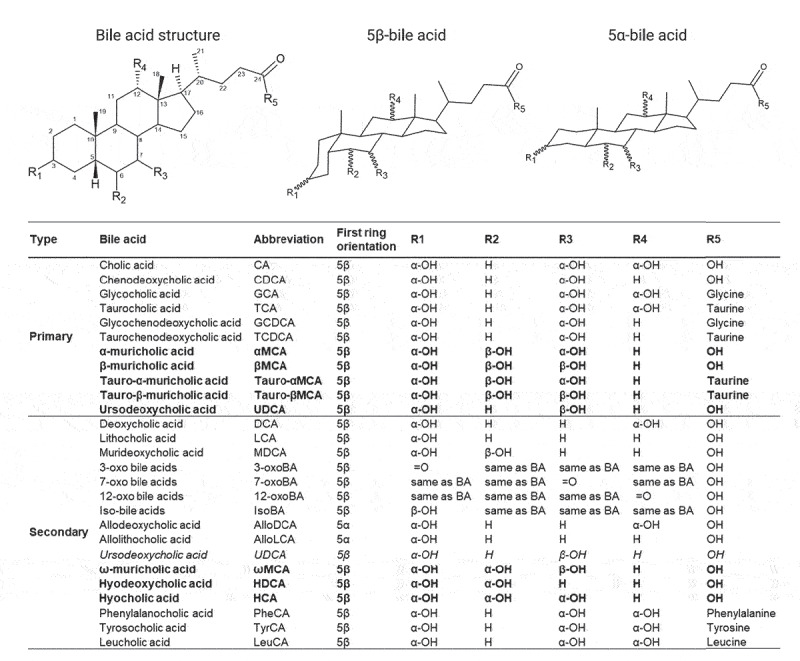
Structure, diversity and metabolism of known human and murine bile acids. Structure and site hydroxylation of the sterol chore for bile acids found in humans and rodents. Hydroxyl groups that are in the α-orientation are located below and are axial to the plane of the sterol chore, while hydroxyl groups in the β-orientation are located above and are equatorial to the plane of the sterol chore. Standard bile acids have the first ring in the β-trans-orientation, yielding 5β-bile acids, while allo-bile acids have this ring in the cis-orientation, yielding 5α-bile acids.37 Mouse-specific bile acids are represented in bold, and human-specific bile acids are represented in italics. Created with ACD/ChemSketch, version 2021.1.2, Advanced Chemistry Development, Inc., (ADC/Labs), Toronto, ON, Canada, www.acdlabs.com.
In enterocytes, primary bile acids bind to the nuclear receptor farnesoid X receptor (FXR), a major regulator of their synthesis, transport, secretion, and absorption.35 Primary bile acids differ in their ability to induce nuclear translocation of FXR.38 FXR dimerizes with the retinoid X receptor (RXR) to activate the transcription of several genes involved in transcellular transport of bile acids and of fibroblast growth factor (FGF) 19 (Fgf15 in mice) (Figure 1).35 The FGF19/15 protein, which is secreted to the portal circulation, travels to the liver where it binds to and activates the FGF receptor 4/β-Klotho (FGFR4/KLB) complex. This complex signals through the docking protein fibroblast growth factor receptor substrate 2 α (FRS2α) and the Src homology-2 domain-containing protein tyrosine phosphatase-2 (Shp2) to stimulate extracellular signal-regulated kinases (ERK) 1/2 phosphorylation, which inhibits activation of CYP7A1 gene expression mediated by the nuclear factors hepatocyte nuclear factor 4α (HNF4α) and liver receptor homolog-1 (LRH-1), thus reducing bile acids synthesis.36 The reabsorbed primary bile acids are mainly exported by the organic solute transporter α/β (OSTα/β) located at the basolateral membrane of the enterocytes into the portal circulation back to the liver.36 In hepatocytes, primary bile acids are transported by the sodium taurocholate co-transporting polypeptide (NTCP) and the organic anion-transporting polypeptides (OATPs).32 Intrahepatic primary bile acids activate hepatic FXR/RXR heterodimer that regulates the expression of several genes involved in primary bile acid synthesis and small heterodimer partner (SHP) that exerts negative feedback on de novo bile acids synthesis by indirectly repressing CYP7A1 expression.35 In total, 95% of the total bile acids are reabsorbed in the ileum and transported back to the liver to be recycled and to regulate their de novo synthesis. The remaining 5% reach the colon, where they are metabolized by the gut microbiota and eliminated in the feces.36
Bile acids as integral part of the gut environment
Bile acids as part of the microbiome
The term “microbiome” can be defined in an ecological context as a micro-ecosystem including the microbiota and its environment.39,40 In the gut, an important component of this environment is bile acids. Conjugated primary bile acids are deconjugated by the microbiota, which yields glycine, taurine, and deconjugated primary bile acids. The latter are further metabolized by the microbiota to form secondary bile acids. As will be discussed below, bile acids and their metabolites shape bacterial growth in the gut. Therefore, to understand what constitutes a healthy microbiome, it is important to consider the status of bile metabolites in addition to analyzing microbiota composition and gene expression.
Microbiota changes the bile acid pool
Directly by metabolizing bile acids. Microbial transformation of bile acids involves four major pathways: deconjugation, dehydroxylation, oxidation, and epimerization. Moreover, bile acids can be sulfated, esterified, and even reconjugated by the microbiota, thus increasing the diversity of the bile acid pool (Figures 1 and 2).
Deconjugation of tauro- and glyco-conjugated bile salts is the primary step of all subsequent modifications and consists in the cleavage of the glycine or taurine residues to release unconjugated primary bile acids. This reaction mainly occurs in the small intestine and is catalyzed by the microbial bile salt hydrolase (BSH) encoded by the bsh gene and found in all major gut microbiota phyla, including Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria and across gut archaea (Figure 1).30
Deconjugated primary bile acids can be dehydroxylated at position C-7 of the cholesterol chore by 7α/β-dehydroxylases. The enzymes responsible for this synthesis are encoded by the polycistronic bile acid inducible (bai) operon carried by a few bacterial species belonging to the Clostridium cluster XIV.41 This process converts CA and CDCA into secondary deoxycholic acid (DCA) and lithocholic acid (LCA), respectively, which account for the most abundant secondary bile acids in humans (Figure 1). In rodents, deconjugation and dehydroxylation of the primary bile acids αMCA and βMCA result in the formation of murideoxycholic acid (MDCA).26
The bile acid pool can be further diversified by the intestinal microbiota by oxidation and subsequent epimerization at the C-3, C-6, C-7, and C-12 positions of the sterol chore.31 These reactions are catalyzed by stereochemically distinct 3-, 6-, 7-, or 12- α-hydroxysteroid dehydrogenases (HSDHs) and β-HSDHs to form oxo- and iso-epimers of these bile acids (Figure 1). They can be performed by an individual bacterial species expressing both α-HSDHs and β-HSDHs or by two species, one expressing an α-HSDH and the other a β-HSDH.31 These enzymes have been observed in numerous gut bacteria, including Bacteroides, Clostridium, Escherichia, Eggerthella, Eubacterium, Peptostreptococcus, and Ruminococcus genera.31,42 While UDCA is the primary bile acid in mice,27 the production of this bile acid in humans results from the 7α/β oxidation and epimerization of CDCA, performed by Clostridium absonum, and therefore is considered a secondary bile acid.28,29 Moreover, some human gut bacteria expressing 5α-reductases can transform the classic 5β-bile acid molecules into allo- (or 5α-) bile acid epimers by acting on the C-5 position of the sterol chore.37,43 Compared to classical bile acids with a bend structure, these allo-epimers exhibit a planar structure, which is associated with a modification of their physiological effects.37 Finally, in mice, 6β-epimerization of the primary bile acid βMCA yields ωMCA,44 while subsequent 7β-dehydroxylation or 7β-epimerization of ωMCA yields hyodeoxycholic acid (HDCA) or hyocholic acid (HCA; also known as γMCA), respectively.45,46
Other microbiota bile acids metabolism pathways can further increase bile acids pool diversity and regulate bile acids excretion. Sulfation, performed by intestinal bacteria belonging to the genera Clostridium, Peptococcus, Fusobacterium, and Pseudomonas, is a major metabolic pathway to detoxify and eliminate bile acids by increasing their solubility, decrease their toxicity and intestinal absorption, and enhance their fecal and urinary excretions.33 Esterification of bile acids, an activity detected in Bacteroides, Eubacterium, and Lactobacillus, may increase their hydrophobicity and reduce their solubility (Figure 1).32 While the physiological significance of such modifications remains to be investigated, it could minimize their intestinal reabsorption and their toxicity for the microbiota. Recently, a novel set of bile acids has been identified. Instead of taurine and glycine, CA was conjugated with the amino-acids tyrosine, phenylalanine, and leucine (Figure 1).34 Studies showed that these compounds are produced by the bacterium Enterocloster bolteae, formerly Clostridium bolteae, by a mechanism that remains to be elucidated. Although further investigations are needed, these modifications may have a significance in bile acids mediated-intracellular signaling.34 Together, these pathways lead to a large diversification of the bile acid pool and regulate bile acids elimination.
Disruption of the microbiota in the case of intestinal diseases impairs bile acid homeostasis. Dysbiosis in the fecal microbiota characterized by an expansion of the Proteobacteria (with a strong increase in Enterobacteriaceae, including Escherichia coli) and a reduced abundance of Firmicutes (including Clostridiales), is observed in patients with inflammatory bowel disease (IBD) compared to healthy subjects.47 This change in the microbiota composition impairs microbial metabolism involved in bile acid biotransformation, leading to an increase in luminal primary bile acids, elevated sulfated bile acids, and a decrease in luminal secondary bile acids.48–52 The decrease in secondary bile acids has been associated with reduced relative abundance of members of the microbiota harboring bsh genes, in particular Bifidobacterium and Clostridium clusters IV and XIVb.53–55 This is consistent with the dysbiosis observed in IBD patients 47 and shows an association between the abundance of bsh genes in the microbiome, the composition of the bile acid pool, and the potential repercussion on intestinal health, because a reduced abundance of Firmicute-derived bsh gene sequences in the microbiota is associated with colorectal cancer.56
Indirectly by regulating hepatic biosynthesis. By modifying the composition of the bile acid pool, the microbiota influences FXR activation and thus bile acids enterohepatic circulation and hepatic production. Bile acids differ in their ability to activate FXR and follow the order of CDCA > DCA > LCA > CA.38 Conjugated versions of these bile acids have an even lower potential to activate FXR, while tauro-αMCA tauro-βMCA, αMCA, βMCA, and UDCA are FXR antagonists.38 Gut microbiota depletion in rodents, induced either by antibiotic treatment or germ-free condition, has been associated with an increase in the total bile acid pool compared to conventionally raised animals.27,57 Mechanistically, microbiota-depleted mice exhibit an increased proportion of tauro-αMCA, tauro-βMCA, and UDCA. 27,57 This leads to a decreased expression and activation of the ileal FXR and its downstream target genes in the ileum, among which Fgf15, thus increasing hepatic Cyp7a1 expression level and de novo bile acids synthesis.27,57 Gut microbiota depletion also increases bile acid uptake in the colon.57 Conversely, the presence of intact microbiota results in a bile acid pool that more potently activates FXR, thereby increasing the ileal expression of Fgf15 and inhibiting hepatic Cyp7a1.27,57 In conclusion, whereas an expansion of the bile acid pool results in a feedback repression of de novo bile acid synthesis, this regulatory loop is impaired in mice with disrupted microbiota, demonstrating that the microbiota plays an important role in regulating hepatic synthesis of bile acids.
Modulation of gut microbiota composition with chemical compounds or probiotics in mice influences FXR/FGF15/CYP7A1-mediated bile acids synthesis. Administration of tempol, an antioxidant, reduces the abundance of the genus Lactobacillus and its bsh activity, leading to the accumulation of tauro-βMCA to inhibit FXR signaling.58 Similarly, the administration of the VSL#3 commercial probiotic mixture enhances bile acids deconjugation and fecal excretion, and represses the FXR/FGF15 axis, thus increasing hepatic Cyp7a1 expression and de novo bile acids synthesis.45 Although these results cannot be directly extrapolated to humans who lack MCA, in vitro and in vivo studies recently revealed that the abnormal composition of the bile acid pool also limits FXR activation in humans.59
Bile acids shape the intestinal microbiota composition
The composition and density of bacterial communities is governed by their environment.9 Bile acid pool size and composition regulate gut microbial ecology by exerting direct antimicrobial effects on intestinal microbes via their detergent properties and indirect effects by inducing the production of antimicrobial peptides and regulating host immunity (Figure 1).2,20 The antimicrobial activity of bile acids is demonstrated in rodent models of biliary obstruction and liver injury, which results in small intestinal bacterial overgrowth (SIBO) that can be reversed by administration of bile acids.60–62 Rodents fed bile acids and healthy individuals receiving a bile acid analog exhibit significant changes in gut microbiota composition. Administration of taurocholic acid (TCA) or tauro-βMCA to neonate mice shifts the gut microbiota toward a more adult-like composition compared to control mice, demonstrating that bile acids mediate gut microbiota maturation.63 In adult rats, the administration of CA-supplemented feed results in a marked expansion of Firmicutes, from 54% of the microbiome in control animals to between 93% and 98% in CA-fed animals.64 Among Firmicutes, Clostridium spp. expanded from 39% to 70% in CA-fed animals.64 Conversely, supplementation of mice with DCA decreases bsh activity 65 as well as the abundance of Firmicutes, while increasing the proportion of Bacteroidetes. At the genus level, Parabacteroides and Bacteroides abundance is increased, whereas the abundance of the BSH producers Lactobacillus, Clostridium XI, and Clostridium XIV is decreased in DCA-fed animals compared to the control group.66 In healthy human subjects, the synthetic FXR agonist obeticholic acid (or INT-747) suppresses the synthesis of endogenous bile acids and induces the proliferation of Gram-positive bacteria, including Streptococcus thermophilus, Lactobacillus casei, Lactobacillus paracasei, Bifidobacterium breve, and Lactococcus lactis, suggesting that FXR activation shapes the intestinal microbiota community composition via bile acid-dependent mechanisms.67
Consequences of microbiota-bile acid interactions for gut health
The interaction between the microbiota and bile acids impacts the maintenance of intestinal barrier function, regulates innate and adaptive immunity, and modulates colonization resistance. Effects of bile acids on host cells are mainly mediated by membrane-associated and nuclear bile acid receptors, including FXR, membrane G-protein bile acid-activated receptor (GPBAR)-1, also known as Takeda G protein-coupled receptor 5 (TGR5), nuclear receptor pregnane X receptor (PXR), and vitamin D receptor (VDR). FXR and TGR5 are highly expressed in the liver, the distal ileum, and the colon, in epithelial, endothelial, and immune cells. In addition to the role of FXR in regulating bile acid synthesis, both receptors are essential for maintaining intestinal barrier integrity and limiting inflammation. Bile acids differ in their ability to activate TGR5 and follow the order of LCA > DCA > CDCA > UDCA > CA. PXR is highly expressed in tissues exposed to bile, while VDR is expressed in most tissues, and both preferentially bind LCA.68
Microbiota-bile acids interactions modulate intestinal barrier function
The ability of intestinal epithelial cells to form tight junctions is critical to the formation and maintenance of the intestinal barrier. Several in vivo studies supported the role of bile acids in the regulation of tight junction functions. In mice and rats fed a high fat diet, the increased intestinal permeability and decreased expression of tight junction proteins are associated with an alteration of cecal and plasma bile acid concentration, with an increase in the total bile acid pool and of secondary bile acids.69,70 Conversely, administration of the primary bile acid CDCA to prematurely weaned piglets improves intestinal barrier function by inducing zona occludens ZON-1 expression and limiting TNFA (tumor necrosis factor alpha), IL6 (interleukin 6), and IL10 gene expression.71 Bile acid modulation of intestinal epithelial integrity is mediated by their ability to activate receptors. In mouse and rat models of bile flow obstruction via bile duct ligation, the absence of endogenous FXR ligands increases gut permeability and bacterial translocation and reduces expression of the tight junction proteins occludin and claudin 2 (Figure 3).60,72 Similar results are observed in mice with a normal bile flow but FXR deficiency.60 Conversely, administration of the FXR agonist obeticholic acid to bile duct-ligated rats decreases the severity of intestinal inflammation by increasing tight junction protein expression.72 Similarly, in mouse models of chemically induced colitis, activation of FXR limits epithelial barrier permeability and prevents intestinal inflammation.73 The role of FXR in intestinal epithelial homeostasis is mediated through FGF proteins. Mice fed a DCA supplemented diet develop dysbiosis, which reduces bile acid deconjugation, thus limiting the activation of the FXR-FGF15 axis and impairing mucosal barrier functions.66 When mice on a DCA supplemented diet are treated with the synthetic FXR agonist feraxamine, intestinal injury is reduced, whereas the FXR-FGF15 axis and bile acid homeostasis are restored.66 In a mouse model of colitis induced by dextran sulfate sodium (DSS), the administration of an engineered FGF19 protein represses de novo bile acid synthesis, modulates the microbiota and the bile acid pool size and composition, favors the maintenance of the intestinal epithelial barrier integrity, and protects from intestinal inflammation.82 The anti-inflammatory effects of FGF19 administration are abrogated in DSS-treated mice that are deficient for FXR.82 Thus, FXR is required for the protective effects of the FXR-FGF15/19 axis against intestinal inflammation and epithelial disruption, while FGF15/19 complements FXR-mediated maintenance of the intestinal homeostasis by modulating the circulating bile acid pool. In addition, TGR5-deficient mice display an altered expression of tight junctions, an increased intestinal permeability and are more susceptible to chemically induced colitis compared to wild-type mice, suggesting the role of this bile acid receptor in the maintenance of the intestinal barrier.74
Figure 3.
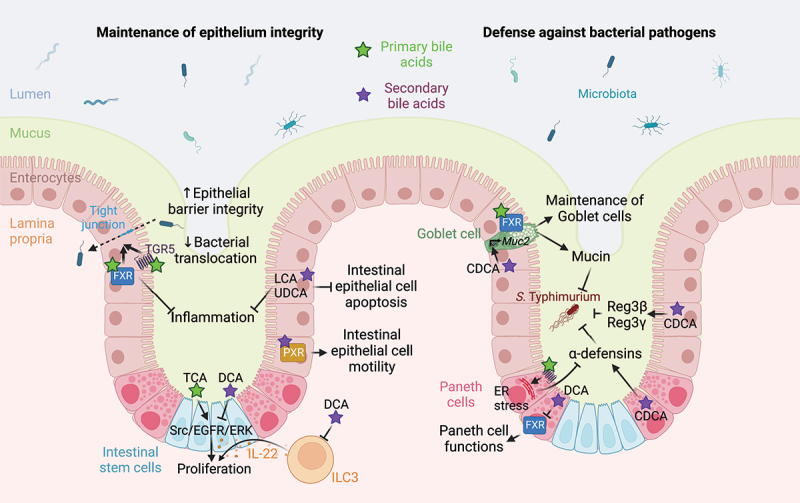
Bile acid-mediated regulation of intestinal barrier function. Bile acids favor the maintenance of epithelial integrity. Indeed, bile acid-mediated activation of TGR5 and FXR receptors in intestinal epithelial cells increases the expression of tight junction proteins, thus improving epithelial barrier integrity and limiting bacterial translocation and inflammation.60, 72–74 LCA and UDCA prevents intestinal epithelial cells apoptosis and limit inflammation.75 LCA-mediated activation of PXR promotes intestinal epithelial cell motility,76 while TCA and DCA modulates Src/EGFR/ERK pathway activation to regulate intestinal stem/epithelial cell proliferation.77 DCA also indirectly modulates intestinal stem cells proliferation and differentiation by modulating IL-22-producing type 3 innate lymphoid cells.78 Bile acids also promote defense against bacterial pathogens by preventing loss of goblet cells in an FXR-dependent manner 73 and favoring Muc2 expression and mucin production in an FXR-independent manner.79 In Paneth cells, primary bile acid-mediated TGR5 activation induces an endoplasmic reticulum stress that limits α-defensins production,80 while the secondary bile acid CDCA increases the release of α-defensins by Paneth cells and of Reg3α and Reg3γ by IECs.79 However, DCA inhibits FXR in Paneth cells, thus impairing their function.81 Abbreviations: CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; ER, endoplasmic reticulum; FXR, farnesoid X receptor; IL-22, interleukin 22; ILC3, type 3 innate lymphoid cells; LCA, lithocholic acid; PXR, pregnane X receptor; Src/EGFR/ERK Src-mediated epidermal growth factor receptor and extracellular signal-regulated kinases activation; TCA, taurocholic acid; TGR5, Takeda G protein-coupled receptor 5; UDCA, ursodeoxycholic acid. Created with BioRender.com.
Bile acids also induce the proliferation of intestinal epithelial cells and limit apoptosis. In mice, the secondary bile acids LCA and UDCA protect from DSS-induced intestinal inflammation and limit epithelial cell apoptosis (Figure 3).75 Bile acids promote epithelial regeneration by acting on the TGR5 receptor in intestinal stem cells.83 In vitro, PXR stimulation by a chemical agonist increases intestinal epithelial cell motility and wound closure.76 TCA induces proliferation of intestinal epithelial cells in vitro via Src-mediated epidermal growth factor receptor (EGFR) and ERK activation, while DCA inhibits cell proliferation by an FXR-dependent mechanism that may inactivate the Src/EGFR/ERK pathway.77 Finally, an increase in DCA induced by a high-fat diet reduces intestinal stem cells proliferation and differentiation by reducing the number of IL-22-producing type 3 innate lymphoid cells, which results in fewer Paneth and goblet cells.78
Bile acids also modulate the formation and composition of the mucus layer, which is composed of mucins immersed in antimicrobials, such as defensins. In mouse models of chemically induced colitis, activation of FXR prevents loss of mucin-producing goblet cells.73 Mice fed a diet supplemented by CDCA exhibit an increased expression of α-defensins by Paneth cells, elevated transcription of Muc2 (mucin 2-encoding gene) by goblet cells, and enhanced synthesis of the type-C lectins Reg3β and Reg3γ by the ileal epithelium (Figure 3).79 These consequences of CDCA supplementation are linked to an FXR-independent enhancement of resistance against infection with bile-resistant pathogens, including Salmonella enterica serovar Typhimurium (S. Typhimurium) and Citrobacter rodentium.79 Conversely, consumption of a high-fat, high-sugar diet increases production of DCA by Clostridium spp. in the gut, which in turn inhibits Paneth cell function by acting on FXR and increasing type I IFN signaling.81 Similarly, high-fat feeding, by increasing bile acid production and TGR5 expression, induces endoplasmic reticulum stress in the Paneth cells and leads to a decrease in α-defensin 5 and 6, contributing to gut dysbiosis.80 The secondary bile acid DCA stimulates, while UCDA inhibits the expression and secretion of human β-defensin-1 and −2 in vitro,84 which may have implications in the maintenance of intestinal homeostasis.
Finally, VDR may exert a protective function against colon cancer by favoring the detoxification of LCA, a potential enteric carcinogen, in the liver and intestine. However, it is not clear whether this protection is due to vitamin D, LCA, or both.85
Microbiota-bile acids interactions modulate immune homeostasis
Bile acids produced by the microbiota regulate different aspects of immunity, including the induction of inflammatory genes to the recruitment of innate and adaptive immune cells.
The bile acid receptors FXR, TGR5, and PXR modulate pro-inflammatory gene expression. In chemically induced colitis mouse models, FXR-deficiency worsens, while treatment with the FXR agonist obeticholic acid protects from mucosal inflammation and promotes the expression of antibacterial genes.73 Obeticholic acid-mediated activation of FXR decreases TNF-α production in several immune cell populations ex vivo.73 Mechanistically, FXR regulates the expression of pro-inflammatory genes in macrophages in an SHP-dependent manner by inducing SHP transcription. SHP exerts its regulatory effect by inhibiting nuclear factor-kappa B (NF-κB)-mediated activation of the pro-inflammatory gene expression (Figure 4).88 Moreover, FXR can regulate the expression of pro-inflammatory genes in an SHP-independent manner by directly binding the promoter of pro-inflammatory genes, including NOS2 (nitric oxide synthase 2), TNFA, and IL1B. Binding of FXR to promoter regions stabilizes the nuclear receptor corepressor 1 (NCor1) complex, thus repressing gene expression.89 Activation of Toll-like receptor 4 (TLR4) by pathogen-associated molecular patterns leads to the release of NCor1 from the promoters of pro-inflammatory genes, allowing their transcriptional activation.89 Finally, PXR and VDR directly inhibit NF-κB signaling, thus reducing pro-inflammatory responses.90,91
Figure 4.
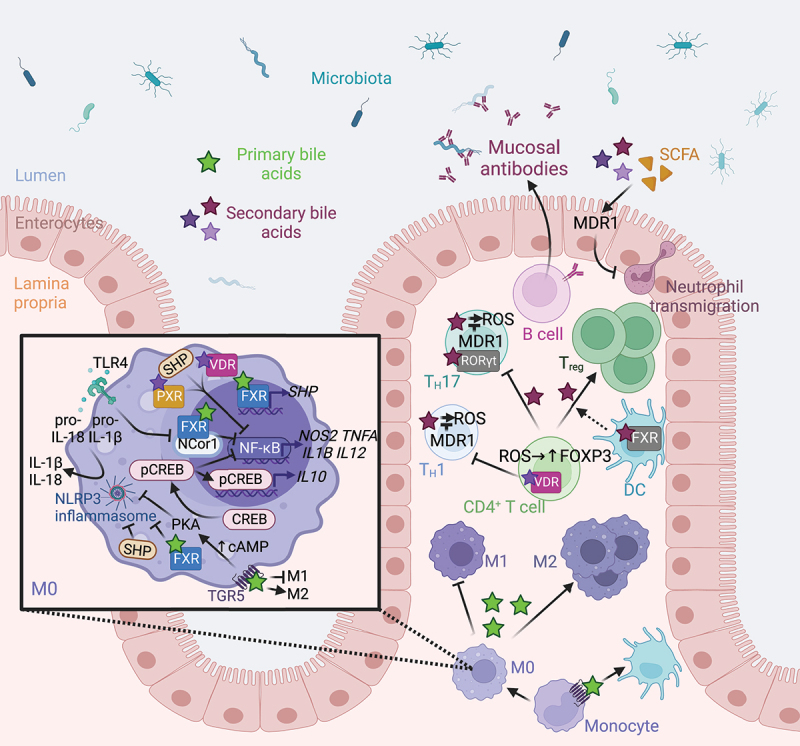
Microbiome-derived bile acids modulates intestinal innate and adaptive immunity. TGR5 stimulation with bile acids promotes the differentiation of monocytes into tolerogenic dendritic cells.86 Bile acids promote the polarization of M0 macrophages toward an anti-inflammatory M2 phenotype by stimulating nuclear and membrane receptors.87 Indeed, FXR inhibits NF-κB both indirectly by increasing SHP expression 88 and directly by binding to the NCor1 complex,89 while PXR and VDR receptors directly repress NF-κB-mediated pro-inflammatory gene expression.90,91 However, TLR4 activation promotes the release of NCor1 and the activation of NF-κB-mediated gene transcription. FXR, SHP 92,93 and the TGR5-cAMP-PKA signaling pathway 94,95 repress inflammasome assembly and activation, thus limiting IL-1B and IL-18 production. Finally, TGR5-cAMP-PKA signaling pathway activates CREB phosphorylation, nuclear translocation, and IL-10 production 87,96 and inhibits NF-κB-mediated pro-inflammatory gene transcription.96 In CD4+ T cells, VDR activation inhibits TH1 differentiation.97 Secondary bile acid epimers promote the differentiation of Treg by blocking FXR in dendritic cells,98 and by activating VDR and promoting the formation of ROS in naïve CD4+ T cells.99 Conversely, bile acid epimers act as an inverse agonist of RORγt in TH17 to limit their differentiation. 42,100,101 TH1 and TH17 cell exposure to bile acids drives oxidative stress, which is modulated by MDR1.102 Moreover, bile acids and SCFA regulates MDR1 expression to suppress neutrophil transmigration.103 Finally, B cells produce mucosal antibodies shaping the gut microbiota.104 Abbreviations: cAMP, Cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; FXR, farnesoid X receptor; IL, interleukin; MDR1, Multidrug Resistance Protein 1; NF-κB, nuclear factor-kappa B; NLRP3, Nod-like receptor family pyrin domain containing 3; PKA, protein kinase A; PXR, pregnane X receptor; RORγt, retinoic acid-related orphan receptor gamma t; ROS, reactive oxygen species; SCFA, short-chain fatty acids; SHP, small heterodimer partner; TGR5, Takeda G protein-coupled receptor 5; TH, T helper cell; TLR4, Toll-like receptor 4; Treg, regulatory T cell; VDR, vitamin D receptor. Created with BioRender.com.
Bile acids also limit Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation. FXR and SHP suppress the assembly of NLRP3 inflammasome by physically interacting with NLRP3 and caspase-1 (Figure 4),92,93 while activation of the TGR5-Cyclic adenosine monophosphate (cAMP) pathway blocks NLRP3 inflammasome activation by inducing its ubiquitination, which ultimately limits the production of IL-1β and IL-18.94,95 Rectal administration of DCA and LCA to various murine colitis models mitigates inflammation, in part by acting on the TGR5 receptor.105 Thus, dysbiosis-induced deficiency of secondary bile acids in UC patients may promote inflammation, which could be alleviated by restoring secondary bile acid levels.105 Conversely, another study reported that DCA administration in the colon activates the NLRP3 inflammasome in part by stimulating cathepsin B release, which increases IL-1β secretion by macrophages and exacerbates DSS-induced colitis.106 Similarly, conflicting results are observed during bacterial infection, where FXR- or TGR5-deficiency decreases inflammasome activation, thereby impairing clearance of Listeria monocytogenes or Escherichia coli.107 Thus, FXR and TGR5 enhance host resistance to bacterial infection by promoting inflammasome-mediated antimicrobial responses in an inflammatory context. However, given the opposite effects reported for secondary bile acids in colitis models, further work is needed to better understand their role during intestinal inflammation.
Bile acids direct the recruitment and differentiation of various immune cells. Compared to wild-type mice, FXR-deficient mice exhibit reduced recruitment of inflammatory cells during DSS-induced colitis.89 Mice receiving CDCA supplementation exhibit decreased recruitment of monocytes/macrophages and neutrophils to the intestinal mucosa, whereas the relative amount of B cells is elevated during infection with S. Typhimurium and C. rodentium.79 In macrophages stimulated with lipopolysaccharide (LPS), binding of bile acids agonists to TGR5 activates protein kinase A that phosphorylates cAMP response element-binding protein (CREB). CREB acts as a transcriptional activator of the anti-inflammatory gene IL10 by directly binding to its promoter,87,96 and as a transcriptional inhibitor of pro-inflammatory genes, such as IL12, IL1B, or TNFA, by reducing NF-κB translocation (Figure 4).96 In a mouse colitis model, TGR5 activation shifts the intestinal macrophages from a classical activation status (M1) to an alternative activation state (M2).87 In human monocytes, TGR5 activation promotes the differentiation of monocytes into tolerogenic dendritic cells that secrete low levels of TNF-α and IL-12, two cytokines inducing pro-inflammatory T helper (TH) 1 responses commonly elevated in IBD patients.86 The activation of VDR by LCA reduces TH1 cell differentiation in vitro by limiting the expression of TH1 genes and reducing the production of the TH1 cytokines TNF-α and interferon (IFN) γ.97 Obeticholic acid-mediated activation of FXR increases the retention of dendritic cells to the spleen, limits splenic regulatory T cells (Treg) differentiation, and increases the level of plasma IL-10, thus favoring the amelioration of DSS-induced murine colitis.108 Recent studies showed that oxo-, iso-, and allo-epimers of secondary bile acids modulate T cell differentiation. In vivo, microbiota-derived isoDCA increases the immunostimulatory properties of dendritic cells by limiting FXR activity, thus indirectly promoting the differentiation of colonic Treg.98 Furthermore, oxo-, iso-, and allo-epimers of secondary bile acids promote the differentiation of Treg by inducing the production of mitochondrial reactive oxygen species 100 and by acting on VDR.99 Polymorphisms in the Vdr gene significantly increase the risk for developing IBD.109 Oxo-, iso-, and allo-epimers of secondary bile acids can also directly bind to and act as inverse agonists of the retinoid-related orphan receptor (ROR) γt, a nuclear receptor mainly expressed by the pro-inflammatory TH17 cells,101 to limit their differentiation.42,100 In mice, oxo-, iso-, and allo-epimers of secondary bile acids alleviate colitis induced by either DSS treatment or by an adoptive transfer of CD4+ T cells by modulating the balance between TH17 and Treg cells.99,100 Numerous studies reported that an imbalance between TH17 and Treg compartments contributes to IBD.110 The levels of oxo-, iso-, and allo-epimers of secondary bile acids, as well as the levels of microbiome genes required for their synthesis are significantly reduced in the fecal microbiota of IBD patients.42,43 Therefore, dysbiosis-induced reduction in the levels of oxo-, iso-, and allo-epimers of secondary bile acids and Vdr genetic variants associated with IBD 109 might both affect disease susceptibility by altering the TH17/Treg balance.
Exposure to bile acids in the lamina propria drives oxidative stress in effector TH1 and TH17 cells. Effector T cells adapt upon migration to the ileum by upregulating the expression of the xenobiotic transporter Multidrug Resistance Protein 1 (MDR1, alternatively called P-glycoprotein), to limit bile acid-driven oxidative stress (Figure 4).102 Bile acids and microbiota-derived short-chain fatty acids (SCFA) act in concert to regulate the expression of Mdr1 to suppress neutrophil transmigration, thus limiting intestinal inflammation.103 A loss of function mutation in MDR1 has been observed in Crohn’s disease (CD) patients,102 while ulcerative colitis (UC) patients display a reduced capacity of the microbiota to induce MDR1 expression and diminished MDR1 levels, thus supporting the role of the microbiome in regulating mucosal immunity and protecting against intestinal diseases.103
Mucosal antibody responses shape microbiota composition, which in turn shape the bile acid pool. B cell deficiency perturbs ileal bsh activity of the microbiota, thus altering the bile acid composition in the gut and favoring the development of an ileal enteropathy (Figure 4).104
In summary, the ability of the microbiome to produce secondary bile acids constitutes an important factor in the modulation of inflammation and in the recruitment, differentiation, and activation of innate and adaptive immune cells. Conversely, adaptive immunity modulates the microbiota and the production of secondary bile acids. Therefore, maintenance of a balance between these factors is necessary to maintain intestinal homeostasis.
Microbiota-bile acids interactions modulate colonization resistance
Microbiota confers protection against opportunistic infections through competition for resources and the production of metabolites, such as short-chain fatty acids, that limit bacterial growth.111 Microbiota-derived metabolites that limit bacterial growth could be viewed as habitat filters that select for metabolic traits best suited for the environment. Interestingly, the microbiota prevents gut colonization by opportunistic pathogens by cooperating with the host. Specifically, the production of primary bile acids by the host and of secondary bile acids by the microbiota constitutes a habitat filter that strengthens colonization resistance.
Microbiota-mediated metabolism of bile acids increases defense against pathogens
The regulation of the bile acid pool by the microbiota plays a role in protecting the host against pathogenic infections. In humans, a higher abundance and activity of bsh in the gut microbiota confers an increased resistance against Vibrio cholerae infection by degrading the primary bile acid TCA that activates the expression of the pathogen’s virulence genes.112 Similarly, Bifidobacterium bifidum, by metabolizing GDCA, TDCA, and CA into DCA inhibits the activity of V. cholerae type VI protein secretion system (T6SS), a system that injects toxins into competing bacteria, thereby preventing the killing of commensal E. coli by the pathogen.113 The presence of commensal Escherichia coli confers a resistance to S. Typhimurium infection by competing for critical resources, such as oxygen,114 nitrate 115, or iron.116
Commensal bacteria converting primary bile acids into secondary bile acids provide colonization resistance against Clostridioides difficile. Whereas primary bile acids can induce C. difficile spore germination, secondary bile acids are toxic to vegetative cells (Figure 5).118 Commensal Clostridia encoding the bai operon protects against C. difficile infection by producing secondary bile acids that inhibit C. difficile germination, growth, and toxin production.130 Moreover, the α-dehydroxylating gut bacteria, Clostridium scindens and Clostridium sordellii, secrete natural antibiotics that inhibit C. difficile growth in the presence of secondary bile acids.119 Therefore, disruption of the gut microbiota following antibiotic use constitutes a key risk factor for C. difficile infection by altering the bile acid pool.131 Antibiotic use also increases the susceptibility to Candida albicans colonization in the gut. In susceptible mice, antibiotic treatment favors C. albicans growth by altering the gut microbiome and metabolome, including primary and secondary bile acids.132 In vitro, the secondary bile acids LCA and DCA possess direct antifungal activity against C. albicans.117 Conversely, the administration of the primary bile acid TCA to antibiotic-treated mice favors the colonization and dissemination of C. albicans by altering the microbiota composition and decreasing the number of intestinal mononuclear phagocytes and TH17 cells.133,134 Thus, modulation of the bile acid pool by the microbiota regulates colonization resistance, either directly by inhibiting pathogen growth or indirectly by modulating mucosal innate and adaptive responses.
Figure 5.
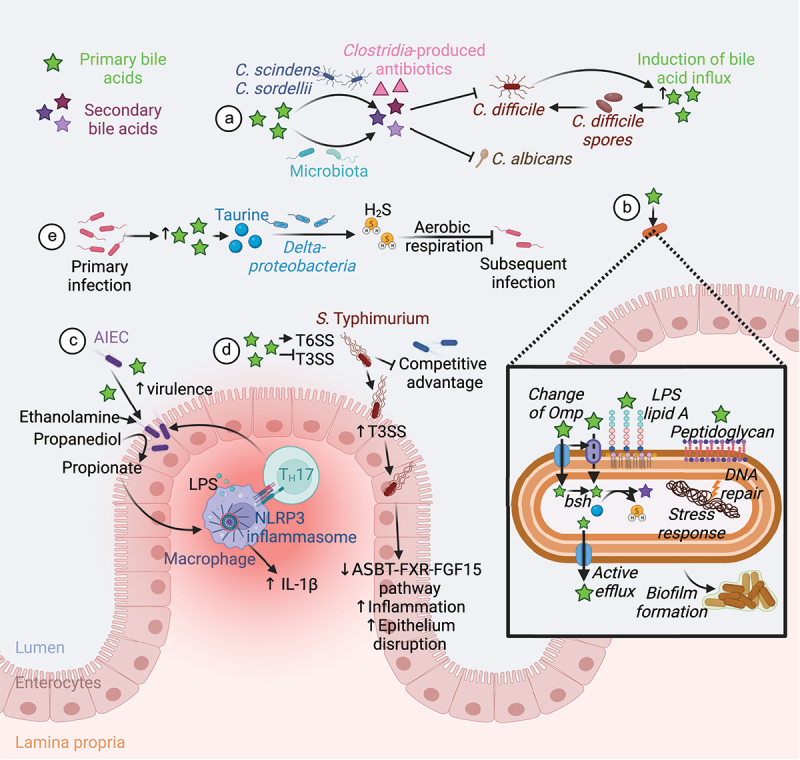
Microbiota-mediated bile acid metabolism and defense against pathogens. (A) Conversion of primary to secondary bile acid by the microbiota confers a colonization resistance against various pathogens. Secondary bile acids exert a direct antifungal activity against C. albicans.117 The gut members C. scindens and C. sordellii produce secondary bile acids and antibiotics that act together to inhibit C. difficile growth. However, C. difficile induces secretion of primary bile acid in the gut to favor its spore germination.118–120 (B) Intestinal bacteria and pathogens resist to bile acids by changing the structure of their membrane components (Omp, Lipid A, peptidoglycan), using bile acids as a source of nutrients, actively eliminating bile acids, repairing DNA damage, promoting stress responses, and forming biofilms.121 (C) Bile acids promote the expression of AIEC virulence genes 122–124 and the use of ethanolamine and propanediol as a nitrogen and a carbon source respectively.125,126 Propionate generated from propanediol synergize with LPS to trigger IL-1β production and TH17 cell activation, thus promoting intestinal inflammation.127 (D) Luminal bile acids increase S. Typhimurium T6SS activity and represses its T3SS until the bacteria can reach the epithelium.128,129 This confers a competitive advantage to the pathogen in a bile acid-rich lumen.128,129 Infection with S. Typhimurium decreases the ASBT-FXR-FGF15 pathway and increases inflammation and epithelial disruption.23,24 (E) Finally, primary bacterial infection increases the abundance of primary bile acids in the gut. Deltaproteobacteria metabolize bile acid-derived taurine to hydrogen sulfide that block pathogen aerobic respiration, thus conferring a resistance to subsequent bacterial infection.25 Abbreviations: AIEC, Adherent-invasive E. coli; ASBT, apical bile salt transporter; FGF15, fibroblast growth factor 15; FXR, farnesoid X receptor; H2S, hydrogen sulfide; IL, interleukin; LPS, lipopolysaccharide; NLRP3, Nod-like receptor family pyrin domain containing 3; Omp, outer membrane porins; T3SS, type III protein secretion system; T6SS, type VI protein secretion system; TH, T helper cell. Created with BioRender.com.
Microbial strategies to resist bile acids
Gut commensals and enteric pathogens inevitably encounter bile acids. Mechanistically, their amphipathic character and detergent properties directly disrupt bacterial membrane lipid composition, impair macromolecule stability by causing RNA and DNA damages, favor protein misfolding, and limit bacterial access to iron and calcium.20 Thus, bile resistance mechanisms, which consist of bile acid metabolism, bile exclusion and extrusion, repair and defense against damages, and modulation of virulence, are crucial to these microbes (Figure 5).121
The most important strategy to resist bile acid toxicity is the ability to deconjugate bile acids and reduce bile acid-derived amino-acids. 135,136 Some bacteria, such as the pathogens Brucella abortus 137 and Listeria monocytogenes 138 express bsh as virulence factors. By deconjugating bile acids, BSHs decrease their solubility and emulsifying capacities, thus reducing their toxicity. Probiotic strains of Lactobacillus encode multiple distinct BSHs that have different substrate preferences to adapt to specific host niches in the gut depending on bile acid availability.139,140 Amino acids arising from deconjugation of bile are metabolized by the gut microbiota. While bacterial metabolism of both glycine and taurine leads to the production of ammonia and carbon dioxide, taurine catabolism results in the additional release of hydrogen sulfide (Figure 5). These products constitute a source of carbon, nitrogen, and sulfur for some bacterial species. In vitro, taurine supplementation promotes the growth of bsh-expressing Clostridium species.141 The reduction of CA-derived taurine to hydrogen sulfide favors Bacteroides growth by stimulating 7α-dehydroxylation of CA, suggesting that taurine metabolism further increases bile acid degradation.142 Consumption of taurine and TCA boosts the expansion of the sulfide producer Bilophila wadsworthia143,144 and promotes colitis in genetically susceptible IL-10−/− mouse model.143
Other mechanisms of resistance are shared between commensals and pathogens. The structure of the bacterial cell envelope constitutes a barrier against bile acid entry, which is used by Gram-negative bacteria to increase their tolerance to bile acids.145 For example, certain modifications in the lipid A moiety of lipopolysaccharide are important for the bile resistance of S. Typhimurium and E. coli (Figure 5).146,147 Furthermore, the bile acid tolerance of S. Typhimurium involves changes in the peptidoglycan structure 148 and expression of very long O antigen chains.149 Even though the bacterial envelope limits bile acid uptake, these metabolites can reach the bacterial cytosol through outer membrane porins (Omp), which have different diameters and little substrate specificity. Thus, changes in Omp synthesis can limit bile acids influx. In S. Typhimurium and E. coli, bile acid exposure reduces OmpF synthesis while increasing OmpC synthesis. This shift in porin synthesis limits bile acid entry, because OmpF is a larger diffusion channel than OmpC.150 A similar mechanism is observed in V. cholerae, which expresses the narrower porin OmpU and represses the wider porin OmpT upon bile acid exposure.151 The rapid exchange of porins and other surface components that is needed when the pathogen transits from the stomach to the small intestine is facilitated by outer membrane vesiculation.152
Once bile acids have entered through the outer membrane or when encountering gram-positive bacteria, they reach the inner membrane and diffuse through it.153 Thus, active efflux mechanisms are needed to remove intracellular bile acids (Figure 5). The multidrug efflux pump AcrAB–TolC, expressed by numerous Enterobacteriaceae, including Salmonella enterica and E. coli, is the more extensively studied and is essential for resistance to various toxic molecules, including bile acids.154 Intracellular bile acids promote the expression of the transcriptional regulator RamA, which activates the transcription of acrAB and tolC.155 In Campylobacter jejuni, a homolog of AcrAB–TolC, CmeABC, confers resistance to various bile acids,156 while in V. cholerae, the efflux pump VexCD has an increased susceptibility to DCA and VexAB has a broad substrate specificity.157
Cytosolic bile acids can induce DNA damage. Therefore, DNA repair mechanisms, such as direct mismatch repair mediated by the DNA adenine methylase,158 nucleotide excision repair, and base excision repair 159 are essential for tolerating bile acids (Figure 5). By inflicting damage to the bacterial cell, the presence of bile acids activates stress responses. In S. enterica, the RpoS-dependent general stress response is required for bile resistance,160 while, in E. coli, the SOS response gene dinF protects against bile acids in reducing oxidative stress.161 In Salmonella enterica serovar Typhi (S. Typhi), exposure to bile acids leads to the production of reactive oxygen species. In return, the quorum-sensing system of S. Typhi increases the production of anti-oxidative enzymes.162
Finally, bacteria can shield themselves from bile by forming biofilms (Figure 5). Bile acids can induce or enhance the formation of biofilms in enteric pathogens, such as V. cholerae, opportunistic pathogens, such as C. difficile,163,164 or commensal bacteria, such as Bacteroides fragilis 165 or Bifidobacterium.166 By converting CA into DCA, the commensal C. scindens enhances biofilm formation by C. difficile, thus favoring its persistence and potentially increasing the risk of relapse.164 TCA disperses ingested mature V. cholerae biofilm in the gut, prior to activating the expression of virulence genes, thus favoring its colonization.167
Bile can act as an environmental signal for pathobionts and pathogens
Since bile acids and their metabolites are keystone features of the gut environment, many opportunists and pathogens use these cues to regulate expression of virulence factors that are needed for gut colonization.
Adherent invasive E. coli (AIEC) is a distinct pathotype of resident mucosa-associated bacteria that is enriched in CD patients compared to control subjects.168 AIEC take advantage of a specific intestinal environment to increase their replication and induce inflammation. In the lumen, bile acids promote the expression of AIEC virulence genes such as the flagellin fliC favoring bacterial persistence in the gut, as well as the long polar fimbriae lpf and the gipA factor facilitating bacterial interaction with and growth in Peyer’s patches (Figure 5).122–124 Moreover, the presence of bile salts activates secondary metabolic pathways allowing AIEC to use ethanolamine as a nitrogen source and propanediol as a carbon source, thus conferring a competitive advantage of these strains over other commensal bacteria.125,126 The utilization of propanediol by AIEC generates propionate that increases their virulence,169 but also synergizes with lipopolysaccharide to trigger IL-1β production and TH17 cell activation, thus promoting T cell-dependent intestinal inflammation.127 The OmpR transcriptional regulator, required for adhesion and invasion of AIEC in intestinal epithelial cells in vitro,170 is also involved in the resistance of AIEC to DCA.171
S. Typhimurium exposure to bile increases the activity of its type VI protein secretion system (T6SS) to deliver effector proteins with an antibacterial activity into adjacent cells, thus killing commensal bacteria and successfully colonizing the gut (Figure 5).128 Conversely, the expression of Salmonella pathogenicity island (SPI) 1 encoding the type III protein secretion system (T3SS) that injects effector proteins required for intestinal invasion and induction of ileitis, is repressed in presence of bile.129 The difference in regulation of these two secretion systems may favor bacterial infection by conferring a competitive advantage to S. Typhimurium against commensal enterobacteria in the bile acid-rich lumen, while delaying the expression of virulence genes required for intestinal invasion until the pathogen can reach the epithelium.128,129 In S. Typhi, the presence of bile upregulates the expression of genes encoded within SPI-1, thus increasing the invasion of the gallbladder epithelium.172 While the differences in regulation of SPI-1 between S. Typhimurium and S. Typhi are not fully resolved, they might be explained by the difference in stability of the dominant SPI-1 regulator, hilD, between these two strains in the presence of bile.172,173
In V. cholerae, primary bile acids increase virulence and motility,174,175 thus representing a signal for the bacteria to colonize the ileum. Finally, C. difficile induces a rapid influx of bile acids into the intestine during host colonization, which facilitates spore germination and outgrowth (Figure 5).120
The microbiota remembers past infections to better resist in the future
Infection with enteric pathogens disrupts the ileal absorption of bile acids and endocrine regulation of bile acids production (Figure 5).23,24 Oral or intravenous infection of mice with S. Typhimurium or L. monocytogenes respectively significantly reduces the expression of Fgf15 in the ileum and its hepatic receptor components (Fgfr4 and Klb).23 These changes are associated with hepatic pathophysiology and colonization of the hepatobiliary tract.23 In the porcine intestine, S. Typhimurium infection downregulates the expression of genes in the FXR pathway, reduces ASBT expression, upregulates the expression of NF-κB-dependent genes, and alters the expression of tight junction genes. These changes indicate a disruption of bile acid absorption.24 Thus, infection with invasive pathogens might alter FXR-FGF15 signaling and increase the flow of bile into the large intestine.
Interestingly, recent evidence suggests that the microbiota “remembers” prior infections to increase colonization resistance.25 Laboratory mice exhibit increased colonization resistance to Klebsiella pneumoniae several weeks after surviving an infection with the enteric pathogen Yersinia pseudotuberculosis.25 The underlying mechanism is that infection with Y. pseudotuberculosis, an enteric pathogen that invades ileal Peyer’s patches, increases the abundance of Deltaproteobacteria in the gut microbiota (Figure 5).25 Deltaproteobacteria are a class of bacteria metabolizing bile acid-derived taurine.144 Taurine consumption by Deltaproteobacteria results in the release of hydrogen sulfide,176 a gas that inhibits the growth of K. pneumoniae by aerobic respiration.25,177 Taurine supplementation mimics the effect of Y. pseudotuberculosis infection, suggesting that ileitis caused by the pathogen increases the flow of bile into the large intestine to promote growth of Deltaproteobacteria. An increased abundance of Deltaproteobacteria also enhances colonization resistance against C. rodentium,25 a pathogen that requires oxygen to grow in the gut environment.178 Supplementation with bismuth subsalicylate, a known sulfide sequestrant,179 impairs colonization resistance against K. pneumoniae in mice, 25 suggesting that microbiota-derived hydrogen sulfide limits growth of facultatively anaerobic opportunistic pathogens.
Although an increase in bile acid concentrations in the colon might be beneficial to enhance colonization resistance, excessive bile acid concentrations are associated with inflammatory disorders and colorectal cancer.180 Thus, a fine regulation of bile acid metabolism is required to enhance colonization resistance while limiting deleterious effects on the host.
Bile acid malabsorption in CD
While in CD inflammation predominantly affects the terminal ileum and the colon but can occur anywhere in the gastrointestinal tract from the mouth to the anus, UC is characterized by a chronic inflammation restricted to the colon and the rectum.181
The terminal ileum constitutes the main site of active reabsorption of bile acids. Diarrhea due to malabsorption of bile acids and nutrients in the ileum is common in CD patients, while in UC patients, diarrhea results from a limited absorption of water and electrolytes through the damaged colonic mucosa.182 Vantrappen and coauthors were the first to demonstrate that CD patients, but not UC patients, present a reduced bile acid pool size compared to healthy subjects and that this decrease was inversely correlated with the Colitis Disease Activity Index.183 Similarly, Rutgeerts and coauthors revealed an increased turnover of primary bile acids in CD patients with ileal dysfunction and demonstrated that the severity of CA loss correlated with the extent of ileal disease, but also that affection of both ileum and colon lead to a total depletion of secondary bile acids.184 Over the years, several other studies confirmed that bile acid malabsorption occurs in CD patients with ileocolic disease, which is more severe after surgical resection of the distal ileum.185–187 The resulting increased flow of bile into the colon increases the bile acid pool, which can influence the gut microbiota composition.
Fecal samples of IBD patients exhibit dysbiosis compared to healthy subjects,47 which is associated with altered bacterial metabolism. Metagenomic analysis show that 12% of the microbial metabolic pathways changed between IBD patients and healthy subjects.188 Specifically, dysbiosis in IBD impairs microbial metabolism related to bile acids biotransformation, leading to an increase in primary bile acids and in sulfated bile acids and a decrease in secondary bile acids compared to healthy subjects.49–51 Interestingly, fecal dysbiosis and metabolite profiles also differ between CD and UC patients.34,50,51 Ileal CD patients exhibit a difference in microbiota composition, as well as a higher abundance of significantly altered microbial metabolic pathways and biological processes than CD patients without ileal involvement and UC patients.188 These variations may be explained by the different ability of the ileum to efficiently absorb bile acids in patients with ileal CD compared to IBD patients without ileal involvement, resulting in a different composition of the bile acid luminal pool reaching the colon, thus influencing the microbiota composition and its ability to produce various metabolites, among which bile acids.
In conclusion, bile acid malabsorption occurring in ileal CD may explain the differences observed by numerous studies in fecal dysbiosis, metabolomics, and inflammatory profiles between ileal CD patients and other IBD patients.
Future directions and conclusion
Changes in the composition and function of the fecal microbiota have been linked to many non-communicable human diseases.189 Dynamic changes in the composition of gut-associated microbial communities reflect changes in microbial growth conditions.190 One of the key factors controlling microbial growth in the gut is the host environment.191 Bile is an important host-derived component of the gut environment. Bile acids and microbiota-derived bile metabolites influence the gut environment directly, by inhibiting the growth of some microbes, thereby contributing to colonization resistance against opportunistic pathogens. Furthermore, bile acids moderate the gut environment indirectly by maintaining mucosal epithelial barrier function and modulating innate and adaptive immune responses.
Consequently, changes in the composition or size of the bile acid pool disrupt gut homeostasis. For example, antibiotic-mediated depletion of microbes with 7-α-dehydroxylase activity changes the composition of the bile acid pool by decreasing the concentration of secondary bile acids, which favors an expansion of the opportunistic pathogen C. difficile in the fecal microbiota. A condition that changes the size of the bile acid pool is malabsorption in the ileum, which increases the flow of bile into the colon, and may explain the differences observed in fecal dysbiosis and metabolomic composition between ileal CD patients and UC patients. In turn, an increase in the bile acid pool in the colon provides Deltaproteobacteria with elevated amounts of bile-derived taurine to produce hydrogen sulfide, a metabolite that can exacerbate colitis.143
Thus, modulating the size and composition of the bile acid pool constitutes an interesting therapeutic approach to modulate intestinal inflammation and colonization resistance. For instance, fecal microbiota transplant is a key strategy in the treatment of recurrent C. difficile infection. However, the efficacy of microbiome reconstruction partly depends on the transplant's ability to restore BSH and 7-α-dehydroxylase functionalities and has been associated with an increased level of secondary bile acids and an increased signaling in the bile acid-FXR-FGF19 pathway.192 The bile acid-FXR-FGF pathway, as well as the bile acid-TGR5 pathway, play a major role in the maintenance of intestinal homeostasis. Agonists of these receptors may be of potential interest in the treatment of IBD. In mice, several FXR and TGR5 agonists showed an ability to improve symptoms of chemically induced colitis by reducing intestinal barrier permeability and inflammation.73,74 The fluoroquinolone ciprofloxacin, an antibiotic used in the treatment of IBD, is of potential interest because of its antibiotic and TGR5 agonist properties.74 Several FXR and TGR5 agonists are currently being evaluated in animal models and in clinical trials for treatment of hepatic and metabolic disorders.193,194 However, given the broad effect of bile acid receptors on the organism and contradicting reports on the role of secondary bile acids on inflammatory processes, further in vivo and clinical studies are needed to evaluate the potential benefits in the treatment of IBD. Finally, FXR-FGF15 signaling pathway is also modulated following bacterial infection of the gastrointestinal tract with bile-acid resistant pathogens.23,24 The recent discovery that bile acid-derived taurine can boost colonization resistance against opportunistic pathogens, such as Klebsiella pneumoniae, suggests that a bile metabolite-based approach could be an interesting strategy to enhance colonization resistance against various infections.25
In conclusion, the size and composition of the bile acid pool is an important modulator of gut associated microbial communities and could be targeted by approaches to remediate dysbiosis. Immunomodulation through precision edition of the microbiota, modulation of bile acid receptors using synthetic ligands, and microbiota-derived metabolites may become a powerful tool to restore intestinal homeostasis, improve FMT outcome, strengthen colonization resistance, and limit the recurrence of infection.
Funding Statement
Work in A.J.B.’s laboratory was supported by Crohn’s and Colitis Foundation of America Award 650976 and by Public Health Service Grants AI044170, AI096528, AI112445, AI112949, AI146432, and AI153069.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Sender R, Fuchs S, Milo R.. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. Available from: https://pubmed.ncbi.nlm.nih.gov/27541692/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levy M, Blacher E, Elinav E. Microbiome, metabolites and host immunity. Curr Opin Microbiol. 2017;35:8–27. Available from: https://pubmed.ncbi.nlm.nih.gov/27883933/. [DOI] [PubMed] [Google Scholar]
- 3.Koh A, de Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell [Internet] 2016; 165:1332–1345. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27259147 [DOI] [PubMed] [Google Scholar]
- 4.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol [Internet] 2014; 12:661–672. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25198138. [DOI] [PubMed] [Google Scholar]
- 5.McCarville JL, Chen GY, Cuevas VD, Troha K, Ayres JS. Microbiota metabolites in health and disease. Annu Rev Immunol [Internet] 2020; 38:147–170. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32340573. [DOI] [PubMed] [Google Scholar]
- 6.Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol [Internet] 2016; 16:341–352. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27231050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong Y, Marion T, Schett G, Luo Y, Liu Y. Microbiota and metabolites in rheumatic diseases. Autoimmun Rev [Internet] 2020; 19:102530. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32240855. [DOI] [PubMed] [Google Scholar]
- 8.Cani PD. Gut microbiota-at the intersection of everything? Nat Rev Gastroenterol Hepatol. 2017;14:321–322. [DOI] [PubMed] [Google Scholar]
- 9.Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients [Internet] 2014; 7:17–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25545101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litvak Y, MX Byndloss, AJ Bäumler. Colonocyte metabolism shapes the gut microbiota. Science [Internet] 2018; 362. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30498100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J-Y, Tsolis RM, Bäumler AJ. The microbiome and gut homeostasis. Science [Internet] 2022; 377:eabp9960. Available from: http://www.ncbi.nlm.nih.gov/pubmed/35771903 [DOI] [PubMed] [Google Scholar]
- 12.Litvak Y, Byndloss MX, Tsolis RM, Bäumler AJ. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol [Internet] 2017; 39:1–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28783509 [DOI] [PubMed] [Google Scholar]
- 13.Shin N-R, Whon TW, Bae J-W. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol [Internet] 2015; 33:496–503. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26210164 [DOI] [PubMed] [Google Scholar]
- 14.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A [Internet] 2008; 105:15064–15069. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18806221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ME v Johansson, JK Gustafsson, KE Sjöberg, Petersson J, Holm L, Sjövall H, GC Hansson. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLoS One [Internet]. 2010;5. e12238. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20805871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Post S, Jabbar KS, Birchenough G, Arike L, Akhtar N, Sjovall H, Johansson ME, Hansson GC. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut [Internet] 2019; 68:2142–2151. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30914450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johansson ME, Gustafsson JK, Holmén-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjövall H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut [Internet] 2014; 63:281–291. Available from: https://gut.bmj.com/lookup/doi/10.1136/gutjnl-2012-303207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staley C, Weingarden AR, Khoruts A, Sadowsky MJ. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl Microbiol Biotechnol [Internet] 2017; 101:47–64. Available from: http://link.springer.com/10.1007/s00253-016-8006-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang JYL. Bile acid metabolism and signaling. Compr Physiol [Internet] 2013; 3:1191–1212. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23897684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bustos AY, Font de Valdez G, Fadda S, Taranto MP. New insights into bacterial bile resistance mechanisms: the role of bile salt hydrolase and its impact on human health. Food Res Int [Internet] 2018; 112:250–262. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30131136 [DOI] [PubMed] [Google Scholar]
- 21.Ghosh S, Whitley CS, Haribabu B, Jala VR. Regulation of intestinal barrier function by microbial metabolites. Cell Mol Gastroenterol Hepatol [Internet] 2021; 11:1463–1482. Available from http://www.ncbi.nlm.nih.gov/pubmed/33610769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang P. Chemical mechanisms of colonization resistance by the gut microbial metabolome. ACS Chem Biol [Internet] 2020; 15:1119–1126. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31895538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romain G, Tremblay S, Arena ET, Antunes LCM, Covey S, Chow MT, Finlay BB, Menendez A. Enterohepatic bacterial infections dysregulate the FGF15-FGFR4 endocrine axis. BMC Microbiol [Internet] 2013; 13:238.Available from: http://www.ncbi.nlm.nih.gov/pubmed/24165751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uribe JH, Collado-Romero M, Zaldívar-López S, Arce C, Bautista R, Carvajal A, Cirera S, Claros MG, Garrido JJ. Transcriptional analysis of porcine intestinal mucosa infected with Salmonella Typhimurium revealed a massive inflammatory response and disruption of bile acid absorption in ileum. Vet Res [Internet] 2016; 47:11.Available from: http://www.veterinaryresearch.org/content/47/1/11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stacy A, Andrade-Oliveira V, McCulloch JA, Hild B, Oh JH, Perez-Chaparro PJ, Sim CK, Lim AI, Link VM, Enamorado M, et al. Infection trains the host for microbiota-enhanced resistance to pathogens. Cell [Internet] 2021; 184:615–627. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33453153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Dawson PA. Animal models to study bile acid metabolism. Biochim Biophys Acta Mol Basis Dis [Internet] 2019; 1865:895–911. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29782919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab [Internet] 2013; 17:225–235. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23395169 [DOI] [PubMed] [Google Scholar]
- 28.Macdonald IA, White BA, Hylemon PB. Separation of 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activities from clostridium absonum ATCC# 27555 and cellular response of this organism to bile acid inducers. J Lipid Res [Internet] 1983; 24:1119–1126. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6579144 [PubMed] [Google Scholar]
- 29.Ridlon JM, Bajaj JS. The human gut sterolbiome: bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm Sin B [Internet] 2015; 5:99–105. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26579434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones B, Begley M, Hill C, Gahan CGM, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proceedings of the National Academy of Sciences [Internet] 2008; 105:13580–13585. Available from: https://pnas.org/doi/full/10.1073/pnas.0804437105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doden HL, Ridlon JM. Microbial Hydroxysteroid dehydrogenases: from alpha to omega. Microorganisms [Internet] 2021; 9:469.Available from: https://www.mdpi.com/2076-2607/9/3/469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res [Internet] 2015; 56:1085–1099. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25210150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alnouti Y. Bile Acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci [Internet] 2009; 108:225–246. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19131563 [DOI] [PubMed] [Google Scholar]
- 34.Quinn RA, Melnik A, Vrbanac A, Fu T, Patras KA, Christy MP, Bodai Z, Belda-Ferre P, Tripathi A, Chung LK, et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature [Internet] 2020; 579:123–129. Available from http://www.ncbi.nlm.nih.gov/pubmed/32103176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR-FGF15/19 pathway. Dig Dis [Internet] 2015; 33:327–331. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26045265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiorucci S, Carino A, Baldoni M, Santucci L, Costanzi E, Graziosi L, Distrutti E, Biagioli M. Bile Acid signaling in inflammatory bowel diseases. Dig Dis Sci [Internet] 2021; 66:674–693. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33289902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiffka SJ, Kane MA, Swaan PW. Planar bile acids in health and disease. Biochimica et Biophysica Acta (BBA) - Biomembranes [Internet] 2017; 1859:2269–2276. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0005273617302699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang L, Zhang H, Xiao D, Wei H, Chen Y. Farnesoid X receptor (FXR): structures and ligands. Comput Struct Biotechnol J [Internet] 2021; 19:2148–2159. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2001037021001410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tipton L, Darcy JL, Hynson NA. A developing symbiosis: enabling cross-talk between ecologists and microbiome scientists. Front Microbiol. [Internet] 2019; 10. Available from https://www.frontiersin.org/article/10.3389/fmicb.2019.00292/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berg G, Rybakova D, Fischer D, Cernava T, Vergès M-CC, Charles T, Chen X, Cocolin L, Eversole K, Corral GH, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome [Internet] 2020; 8:103.Available from: http://www.ncbi.nlm.nih.gov/pubmed/32605663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ridlon JM, Harris SC, Bhowmik S, Kang D-J, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes [Internet] 2016; 7:22–39. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26939849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paik D, Yao L, Zhang Y, Bae S, D’Agostino GD, Zhang M, Kim E, Franzosa EA, Avila-Pacheco J, Bisanz JE, et al. Human gut bacteria produce ΤΗ17-modulating bile acid metabolites. Nature [Internet] 2022; 603:907–912. Available from: http://www.ncbi.nlm.nih.gov/pubmed/35296854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Hang S, Fang Y, Bae S, Zhang Y, Zhang M, Wang G, McCurry MD, Bae M, Paik D, et al. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe [Internet] 2021; 29:1366–1377. Available from: http://www.ncbi.nlm.nih.gov/pubmed/34416161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eyssen H, de Pauw G, Stragier J, Verhulst A. Cooperative formation of omega-muricholic acid by intestinal microorganisms. Appl Environ Microbiol [Internet] 1983; 45:141–147. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6824314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep [Internet] 2014; 7:12–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24656817 [DOI] [PubMed] [Google Scholar]
- 46.Madsen D, Beaver M, Chang L, Bruckner-Kardoss E, Wostmann B. Analysis of bile acids in conventional and germfree rats. J Lipid Res [Internet] 1976; 17:107–111. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1270929 [PubMed] [Google Scholar]
- 47.Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol [Internet] 2018; 11:1–10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29285689 [DOI] [PubMed] [Google Scholar]
- 48.Duboc H, Rainteau D, Rajca S, Humbert L, Farabos D, Maubert M, Grondin V, Jouet P, Bouhassira D, Seksik P, et al. Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil [Internet] 2012; 24:513–520. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22356587 [DOI] [PubMed] [Google Scholar]
- 49.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert M-A, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut [Internet] 2013; 62:531–539. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22993202 [DOI] [PubMed] [Google Scholar]
- 50.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol [Internet] 2019; 4:293–305. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30531976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature [Internet] 2019; 569:655–662. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31142855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacobs JP, Goudarzi M, Singh N, Tong M, McHardy IH, Ruegger P, Asadourian M, Moon B-H, Ayson A, Borneman J, et al. A Disease-associated microbial and metabolomics state in relatives of pediatric inflammatory bowel disease patients. Cell Mol Gastroenterol Hepatol [Internet] 2016; 2:750–766. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28174747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Gao X, Zhang X, Xiao F, Hu H, Li X, Dong F, Sun M, Xiao Y, Ge T, et al. Microbial and metabolic features associated with outcome of infliximab therapy in pediatric Crohn’s disease. Gut Microbes [Internet] 2021; 13:1–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33430702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Labbé A, Ganopolsky JG, Martoni CJ, Prakash S, Jones ML. Bacterial bile metabolising gene abundance in Crohn’s, ulcerative colitis and type 2 diabetes metagenomes. PLoS One [Internet] 2014; 9:e115175. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25517115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia B, Park D, Hahn Y, Jeon CO. Metagenomic analysis of the human microbiome reveals the association between the abundance of gut bile salt hydrolases and host health. Gut Microbes [Internet] 2020; 11:1300–1313. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32329665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst [Internet] 2013; 105:1907–1911. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24316595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selwyn FP, Csanaky IL, Zhang Y, Klaassen CD. Importance of large intestine in regulating bile acids and glucagon-like peptide-1 in germ-free mice. Drug Metab Dispos [Internet] 2015; 43:1544–1556. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26199423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun [Internet] 2013; 4:2384. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24064762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson A, Almousa A, Teft WA, Kim RB. Attenuation of bile acid-mediated FXR and PXR activation in patients with Crohn’s disease. Sci Rep [Internet] 2020; 10:1866. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32024859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A [Internet] 2006; 103:3920–3925. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16473946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lorenzo-Zúñiga V, Bartolí R, Planas R, Hofmann AF, Viñado B, Hagey LR, Hernández JM, Mañé J, Alvarez MA, Ausina V, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology [Internet] 2003; 37:551–557. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12601352 [DOI] [PubMed] [Google Scholar]
- 62.Clements WD, Parks R, Erwin P, Halliday MI, Barr J, Rowlands BJ. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut [Internet] 1996; 39:587–593. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8944570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Best N, Rolle-Kampczyk U, Schaap FG, Basic M, Swm OD, Bleich A, Savelkoul PHM, von Bergen M, Penders J, Hornef MW. Bile acids drive the newborn’s gut microbiota maturation. Nat Commun [Internet] 2020; 11:3692. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32703946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile Acid Is a Host Factor That Regulates the Composition of the Cecal Microbiota in Rats. Gastroenterology [Internet] 2011; 141:1773–1781. Available from: https://linkinghub.elsevier.com/retrieve/pii/S001650851101081X [DOI] [PubMed] [Google Scholar]
- 65.Tian Y, Gui W, Koo I, Smith PB, Allman EL, Nichols RG, Rimal B, Cai J, Liu Q, Patterson AD. The microbiome modulating activity of bile acids. Gut Microbes [Internet] 2020; 11:979–996. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32138583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu M, Cen M, Shen Y, Zhu Y, Cheng F, Tang L, Hu W, Dai N. Deoxycholic acid-induced gut dysbiosis disrupts bile acid enterohepatic circulation and promotes intestinal inflammation. Dig Dis Sci [Internet] 2021; 66:568–576. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32198567 [DOI] [PubMed] [Google Scholar]
- 67.Friedman ES, Li Y, Shen T-CD, Jiang J, Chau L, Adorini L, Babakhani F, Edwards J, Shapiro D, Zhao C, et al. FXR-dependent modulation of the human small intestinal microbiome by the bile acid derivative obeticholic acid. Gastroenterology [Internet] 2018; 155:1741–1752. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30144429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile Acids activated receptors regulate innate immunity. Front Immunol [Internet] 2018; 9:1853. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30150987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suzuki T, Hara H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr Metab (Lond) [Internet] 2010; 7:19.Available from: http://nutritionandmetabolism.biomedcentral.com/articles/10.1186/1743-7075-7-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murakami Y, Tanabe S, Suzuki T. High-fat diet-induced intestinal hyperpermeability is associated with increased bile acids in the large intestine of mice. J Food Sci [Internet] 2016; 81:H216–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26595891 [DOI] [PubMed] [Google Scholar]
- 71.de Diego-Cabero N, Mereu A, Menoyo D, Holst JJ, Ipharraguerre IR. Bile acid mediated effects on gut integrity and performance of early-weaned piglets. BMC Vet Res [Internet] 2015; 11:111.Available from: http://www.ncbi.nlm.nih.gov/pubmed/25972097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verbeke L, Farre R, Verbinnen B, Covens K, Vanuytsel T, Verhaegen J, Komuta M, Roskams T, Chatterjee S, Annaert P, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol [Internet] 2015; 185:409–419. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25592258 [DOI] [PubMed] [Google Scholar]
- 73.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, Klomp LWJ, Siersema PD, Schipper MEI, Danese S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut [Internet] 2011; 60:463–472. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21242261 [DOI] [PubMed] [Google Scholar]
- 74.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, Baldelli F, Donini A, Fiorucci S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One. 2011;6(10):e25637. https://pubmed.ncbi.nlm.nih.gov/22046243/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lajczak-McGinley NK, Porru E, Fallon CM, Smyth J, Curley C, McCarron PA, Tambuwala MM, Roda A, Keely SJ. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol Rep [Internet] 2020; 8:e14456. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32562381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Terc J, Hansen A, Alston L, Hirota SA. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur J Pharm Sci [Internet] 2014; 55:12–19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24486481 [DOI] [PubMed] [Google Scholar]
- 77.Dossa AY, Escobar O, Golden J, Frey MR, Ford HR, Gayer CP. Bile acids regulate intestinal cell proliferation by modulating EGFR and FXR signaling. Am J Physiol Gastrointest Liver Physiol [Internet] 2016; 310:G81–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26608185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu J, Huang D, Xu X, Wu X, Liu L, Niu W, Lu L, Zhou H. An elevated deoxycholic acid level induced by high-fat feeding damages intestinal stem cells by reducing the ileal IL-22. Biochem Biophys Res Commun [Internet] 2021; 579:153–160. Available from http://www.ncbi.nlm.nih.gov/pubmed/34601200 [DOI] [PubMed] [Google Scholar]
- 79.Tremblay S, Romain G, Roux M, X-L Chen, Brown K, DL Gibson, Ramanathan S, Menendez A. Bile Acid Administration Elicits an Intestinal Antimicrobial Program and Reduces the Bacterial Burden in Two Mouse Models of Enteric Infection. Infect Immun [Internet] 2017; 85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28348052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou H, S-Y Zhou, Gillilland M, J-Y Li, Lee A, Gao J, Zhang G, Xu X, Owyang C. Bile acid toxicity in Paneth cells contributes to gut dysbiosis induced by high-fat feeding. JCI Insight [Internet] 2020; 5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33055426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu T-C, Kern JT, Jain U, Sonnek NM, Xiong S, Simpson KF, VanDussen KL, Winkler ES, Haritunians T, Malique A, et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe [Internet] 2021; 29:988–1001. Available from: http://www.ncbi.nlm.nih.gov/pubmed/34010595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gadaleta RM, Garcia-Irigoyen O, Cariello M, Scialpi N, Peres C, Vetrano S, Fiorino G, Danese S, Ko B, Luo J, et al. Fibroblast Growth Factor 19 modulates intestinal microbiota and inflammation in presence of Farnesoid X Receptor. EBioMedicine [Internet] 2020; 54:102719. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32259714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sorrentino G, Perino A, Yildiz E, El Alam G, Bou SM, Gioiello A, Pellicciari R, Schoonjans K. Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology [Internet] 2020; 159:956–968. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32485177 [DOI] [PubMed] [Google Scholar]
- 84.Lajczak NK, Saint‐Criq V, O’Dwyer AM, Perino A, Adorini L, Schoonjans K, Keely SJ. Bile acids deoxycholic acid and ursodeoxycholic acid differentially regulate human β‐defensin‐1 and ‐2 secretion by colonic epithelial cells. The FASEB J [Internet] 2017; 31:3848–3857. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1096/fj.201601365R [DOI] [PubMed] [Google Scholar]
- 85.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science [Internet] 2002; 296:1313–1316. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12016314. [DOI] [PubMed] [Google Scholar]
- 86.Ichikawa R, Takayama T, Yoneno K, Kamada N, Kitazume MT, Higuchi H, Matsuoka K, Watanabe M, Itoh H, Kanai T, et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology [Internet] 2012; 136:153–162. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22236403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Biagioli M, Carino A, Cipriani S, Francisci D, Marchianò S, Scarpelli P, Sorcini D, Zampella A, Fiorucci S. The bile acid receptor GPBAR1 regulates the M1/M2 Phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J Immunol [Internet] 2017; 199:718–733. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28607110 [DOI] [PubMed] [Google Scholar]
- 88.Yang Z, Koehler AN, Wang L. A novel small molecule activator of nuclear receptor SHP inhibits HCC Cell migration via suppressing Ccl2. Mol Cancer Ther. [Internet] 2016; 15:2294301. Available from: .2294301. Available from: .http://www.ncbi.nlm.nih.gov/pubmed/27486225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol [Internet] 2009; 183:6251–6261. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19864602 [DOI] [PubMed] [Google Scholar]
- 90.Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol [Internet] 2007; 292:G1114–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17170021 [DOI] [PubMed] [Google Scholar]
- 91.Liu W, Chen Y, Golan MA, Annunziata ML, Du J, Dougherty U, Kong J, Musch M, Huang Y, Pekow J, et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J Clin Invest [Internet] 2013; 123:3983–3996. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23945234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, Wang G, Gonzalez FJ. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab [Internet] 2017; 25:856–867. Available from http://www.ncbi.nlm.nih.gov/pubmed/28380377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang C-S, Kim J-J, Kim TS, Lee PY, Kim SY, Lee H-M, Shin D-M, Nguyen LT, Lee M-S, Jin HS, et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun [Internet] 2015; 6:6115. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25655831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity pInternet] 2016; 45:802–816. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27692610 [DOI] [PubMed] [Google Scholar]
- 95.Chen Y, Le TH, Du Q, Zhao Z, Liu Y, Zou J, Hua W, Liu C, Zhu Y. Genistein protects against DSS-induced colitis by inhibiting NLRP3 inflammasome via TGR5-cAMP signaling. Int Immunopharmacol [Internet] 2019; 71:144–154. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30901677 [DOI] [PubMed] [Google Scholar]
- 96.Haselow K, Bode JG, Wammers M, Ehlting C, Keitel V, Kleinebrecht L, Schupp A-K, Häussinger D, Graf D. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J Leukoc Biol [Internet] 2013; 94:1253–1264. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23990628. [DOI] [PubMed] [Google Scholar]
- 97.Pols TWH, Puchner T, Korkmaz HI, Vos M, Soeters MR, de Vries CJM. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS One. 2017;12(5):e0176715. https://pubmed.ncbi.nlm.nih.gov/28493883/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin W-B, Guo C-J, Violante S, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature [Internet] 2020; 581:475–479. Available from:. ;(7809):. http://www.ncbi.nlm.nih.gov/pubmed/32461639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature [Internet] 2020; 577:410–415. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31875848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature [Internet] 2019; 576:143–148. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31776512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mickael ME, Bhaumik S, Basu R. Retinoid-related orphan receptor RORγt in CD4+ T-cell–mediated intestinal homeostasis and inflammation. Am J Pathol [Internet] 2020; 190:1984–1999. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32735890 [DOI] [PubMed] [Google Scholar]
- 102.Cao W, Kayama H, Chen ML, Delmas A, Sun A, Kim SY, Rangarajan ES, McKevitt K, Beck AP, Jackson CB, et al. The xenobiotic transporter Mdr1 enforces T Cell homeostasis in the presence of intestinal bile acids. Immunity [Internet] 2017; 47:1182–1196. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29262351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Foley SE, Tuohy C, Dunford M, Grey MJ, de Luca H, Cawley C, Szabady RL, Maldonado-Contreras A, Houghton JM, Ward D, et al. Gut microbiota regulation of P-glycoprotein in the intestinal epithelium in maintenance of homeostasis. Microbiome [Internet] 2021; 9:183.Available from: http://www.ncbi.nlm.nih.gov/pubmed/34493329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mohammed AD, Mohammed Z, Roland MM, Chatzistamou I, Jolly A, Schoettmer LM, Arroyo M, Kakar K, Tian Y, Patterson A, et al. Defective humoral immunity disrupts bile acid homeostasis which promotes inflammatory disease of the small bowel. Nat Commun [Internet] 2022; 13:525.Available from: http://www.ncbi.nlm.nih.gov/pubmed/35082296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, Sim D, Jarr K, Spear ET, Singh G, et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe [Internet] 2020; 27:659–670. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32101703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao S, Gong Z, Du X, Tian C, Wang L, Zhou J, Xu C, Chen Y, Cai W, Wu J. Deoxycholic Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Exacerbates DSS-Induced Colitis through Promoting Cathepsin B Release. J Immunol Res [Internet]. 2018:2481418. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29854830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kang J-H, Kim M, Yim M. FXR/TGR5 mediates inflammasome activation and host resistance to bacterial infection. Biochem Biophys Rep [Internet] 2021; 27:101051. Available from: https://linkinghub.elsevier.com/retrieve/pii/S240558082100145X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Massafra V, Ijssennagger N, Plantinga M, Milona A, Ramos Pittol JM, Boes M, van Mil SWC. Splenic dendritic cell involvement in FXR-mediated amelioration of DSS colitis. Biochim Biophys Acta [Internet] 2016; 1862:166–173. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26554605 [DOI] [PubMed] [Google Scholar]
- 109.Xue L-N, Xu K-Q, Zhang W, Wang Q, Wu J, Wang X-Y. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn's disease. Inflamm Bowel Dis [Internet] 2013; 19:54–60. Available from: https://academic.oup.com/ibdjournal/article/19/1/54-60/4602987 [DOI] [PubMed] [Google Scholar]
- 110.Yan J, Luo M, Chen Z, He B. The function and role of the Th17/Treg cell balance in inflammatory bowel disease. J Immunol Res [Internet] 2020; 2020:1–8. Available from: https://www.hindawi.com/journals/jir/2020/8813558/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rogers AWL, Tsolis RM, Bäumler AJ. Salmonella versus the Microbiome. Microbiol Mol Biol Rev [Internet] 2021; 85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33361269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alavi S, Mitchell JD, Cho JY, Liu R, Macbeth JC, Hsiao A. Interpersonal gut microbiome variation drives susceptibility and resistance to cholera infection. Cell [Internet] 2020; 181:1533–1546. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32631492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bachmann V, Kostiuk B, Unterweger D, Diaz-Satizabal L, Ogg S, Pukatzki S, Picardeau M. Bile salts modulate the mucin-activated type VI secretion system of pandemic Vibrio cholerae. PLoS Negl Trop Dis [Internet] 2015; 9:e0004031. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26317760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Velazquez EM, Nguyen H, Heasley KT, Saechao CH, Gil LM, Rogers AWL, Miller BM, Rolston MR, Lopez CA, Litvak Y, et al. Endogenous Enterobacteriaceae underlie variation in susceptibility to Salmonella infection. Nat Microbiol [Internet] 2019; 4:1057–1064. Available from: http://www.nature.com/articles/s41564-019-0407-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liou MJ, Miller BM, Litvak Y, Nguyen H, Natwick DE, Savage HP, Rixon JA, Mahan SP, Hiyoshi H, Rogers AWL, et al. Host cells subdivide nutrient niches into discrete biogeographical microhabitats for gut microbes. Cell Host Microbe [Internet] 2022; 30:836–847. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1931312822002189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, Raffatellu M. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity [Internet] 2014; 40:262–273. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24508234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guinan J, Villa P, Thangamani S. Secondary bile acids inhibit Candida albicans growth and morphogenesis. Pathog Dis [Internet] 2018; 76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29648597 [DOI] [PubMed]
- 118.Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol [Internet] 2016; 14:609–620. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27573580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kang JD, Myers CJ, Harris SC, Kakiyama G, Lee I-K, Yun B-S, Matsuzaki K, Furukawa M, Min H-K, Bajaj JS, et al. Bile acid 7α-dehydroxylating gut bacteria secrete antibiotics that inhibit Clostridium difficile: role of secondary bile acids. Cell Chem Biol [Internet] 2019; 26:27–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30482679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wexler AG, Guiberson ER, Beavers WN, Shupe JA, Washington MK, Lacy DB, Caprioli RM, Spraggins JM, Skaar EP. Clostridioides difficile infection induces a rapid influx of bile acids into the gut during colonization of the host. Cell Rep [Internet] 2021; 36:109683. Available from https://linkinghub.elsevier.com/retrieve/pii/S221112472101130X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sistrunk JR, Nickerson KP, Chanin RB, Rasko DA, Faherty CS. Survival of the fittest: how bacterial pathogens utilize bile to enhance infection. Clin Microbiol Rev [Internet] 2016; 29:819–836. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27464994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chassaing B, Etienne-Mesmin L, Bonnet R, Darfeuille-Michaud A. Bile salts induce long polar fimbriae expression favouring Crohn’s disease-associated adherent-invasive Escherichia coli interaction with Peyer’s patches. Environ Microbiol [Internet] 2013; 15:355–371. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22789019 [DOI] [PubMed] [Google Scholar]
- 123.Vazeille E, Chassaing B, Buisson A, Dubois A, de Vallée A, Billard E, Neut C, Bommelaer G, Colombel J-F, Barnich N, et al. GipA factor supports colonization of peyer’s patches by crohn’s disease-associated Escherichia coli. Inflamm Bowel Dis [Internet] 2016; 22:68–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26512715 [DOI] [PubMed] [Google Scholar]
- 124.Sevrin G, Massier S, Chassaing B, Agus A, Delmas J, Denizot J, Billard E, Barnich N. Adaptation of adherent-invasive E. coli to gut environment: impact on flagellum expression and bacterial colonization ability. Gut Microbes [Internet] 2020; 11:364–380. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29494278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ormsby MJ, Logan M, Johnson SA, McIntosh A, Fallata G, Papadopoulou R, Papachristou E, Hold GL, Hansen R, Ijaz UZ, et al. Inflammation associated ethanolamine facilitates infection by Crohn’s disease-linked adherent-invasive Escherichia coli. EBioMedicine [Internet] 2019; 43:325–332. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31036531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Delmas J, Gibold L, Faïs T, Batista S, Leremboure M, Sinel C, Vazeille E, Cattoir V, Buisson A, Barnich N, et al. Metabolic adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci Rep [Internet] 2019; 9:2175. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30778122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Viladomiu M, Metz ML, Lima SF, Jin W-B, Chou L, JRI Live Cell Bank, Guo C-J, Diehl GE, Simpson KW, Scherl EJ, et al. Adherent-invasive E. coli metabolism of propanediol in Crohn’s disease regulates phagocytes to drive intestinal inflammation. Cell Host Microbe. [Internet] 2021; 29:607–619. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33539767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sana TG, Flaugnatti N, Lugo KA, Lam LH, Jacobson A, Baylot V, Durand E, Journet L, Cascales E, Monack DM. Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc Natl Acad Sci U S A [Internet] 2016; 113:E5044–51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27503894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Prouty AM, Gunn JS. Salmonella enterica serovar Typhimurium invasion is repressed in the presence of bile. Infect Immun [Internet] 2000; 68:6763–6769. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11083793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reed AD, Nethery MA, Stewart A, Barrangou R, Theriot CM. Strain-dependent inhibition of Clostridioides difficile by commensal Clostridia carrying the bile acid-inducible (bai) operon. J Bacteriol [Internet] 2020; 202. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32179626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Theriot CM, Bowman AA, Young VB. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere. 2016;1(1): e00045–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gutierrez D, Weinstock A, VC Antharam, Gu H, Jasbi P, Shi X, Dirks B, Krajmalnik-Brown R, Maldonado J, Guinan J, et al. Antibiotic-induced gut metabolome and microbiome alterations increase the susceptibility to Candida albicans colonization in the gastrointestinal tract. FEMS Microbiol Ecol [Internet] 2020; 96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31769789 [DOI] [PMC free article] [PubMed]
- 133.Datta A, Hernandez-Franco JF, Park S, Olson MR, HogenEsch H, Thangamani S. Bile acid regulates mononuclear phagocytes and T Helper 17 Cells to control Candida albicans in the intestine. J Fungi [Internet] 2022; 8:610.Available from: https://www.mdpi.com/2309-608X/8/6/610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Thangamani S, Monasky R, Lee JK, Antharam V, HogenEsch H, Hazbun TR, Jin Y, Gu H, Guo GL. Bile acid regulates the colonization and dissemination of Candida albicans from the gastrointestinal tract by controlling host defense system and microbiota. J Fungi [Internet] 2021; 7:1030. Available from: https://www.mdpi.com/2309-608X/7/12/1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lebertz H, Andreesen JR. Glycine fermentation by Clostridium histolyticum. Arch Microbiol. [Internet] 1988. [cited 2022 Jul 5]; 150:11–14. Available from: http://link.springer.com/10.1007/BF00409710 [Google Scholar]
- 136.Laue H, Denger K, Cook AM. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol [Internet] 1997; 63:2016–2021. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9143131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Delpino MV, Marchesini MI, Estein SM, Comerci DJ, Cassataro J, Fossati CA, Baldi PC. A bile salt hydrolase of Brucella abortus contributes to the establishment of a successful infection through the oral route in mice. Infect Immun [Internet] 2007; 75:299–305. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17088355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dussurget O, Cabanes D, Dehoux P, Lecuit M, Buchrieser C, Glaser P, Cossart P. European Listeria Genome Consortium. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol Microbiol [Internet] 2002; 45:1095–1106. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12180927 [DOI] [PubMed] [Google Scholar]
- 139.O’Flaherty S, Briner Crawley A, CM Theriot, Barrangou R. The Lactobacillus Bile Salt Hydrolase Repertoire Reveals Niche-Specific Adaptation. mSphere [Internet] 2018; 3. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29848760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.MH Foley, O’Flaherty S, Allen G, AJ Rivera, AK Stewart, Barrangou R, Theriot CM. Lactobacillus bile salt hydrolase substrate specificity governs bacterial fitness and host colonization. Proc Natl Acad Sci U S A [Internet] 2021; 118. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33526676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Huijghebaert SM, Eyssen HJ. Specificity of bile salt sulfatase activity from Clostridium sp. strains S1. Appl Environ Microbiol [Internet] 1982; 44:1030–1034. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7181500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van Eldere J, Celis P, de Pauw G, Lesaffre E, Eyssen H. Tauroconjugation of cholic acid stimulates 7 alpha-dehydroxylation by fecal bacteria. Appl Environ Microbiol [Internet] 1996; 62:656–661. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8593067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature [Internet] 2012; 487:104–108. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22722865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, Schleheck D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc Natl Acad Sci U S A [Internet] 2019; 116:3171–3176. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30718429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Begley M, Gahan CGM, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev [Internet] 2005; 29:625–651. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16102595 [DOI] [PubMed] [Google Scholar]
- 146.Froelich JM, Tran K, Wall D. A pmrA constitutive mutant sensitizes Escherichia coli to deoxycholic acid. J Bacteriol [Internet] 2006; 188:1180–1183. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16428424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Murata T, Tseng W, Guina T, Miller SI, Nikaido H. PhoPQ-mediated regulation produces a more robust permeability barrier in the outer membrane of Salmonella enterica serovar Typhimurium. J Bacteriol [Internet] 2007; 189:7213–7222. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17693506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hernández SB, Cava F, Pucciarelli MG, García-Del Portillo F, de Pedro MA, Casadesús J. Bile-induced peptidoglycan remodelling in Salmonella enterica. Environ Microbiol [Internet] 2015; 17:1081–1089. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24762004 [DOI] [PubMed] [Google Scholar]
- 149.Crawford RW, Keestra AM, Winter SE, Xavier MN, Tsolis RM, Tolstikov V, Bäumler AJ. Very long O-antigen chains enhance fitness during Salmonella-induced colitis by increasing bile resistance. PLoS Pathog [Internet] 2012; 8:e1002918. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23028318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev [Internet] 2003; 67:593–656. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14665678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Provenzano D, Klose KE. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A [Internet] 2000; 97:10220–10224. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10944196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zingl FG, Kohl P, Cakar F, Leitner DR, Mitterer F, Bonnington KE, Rechberger GN, Kuehn MJ, Guan Z, Reidl J, et al. Outer membrane vesiculation facilitates surface exchange and in vivo adaptation of Vibrio cholerae. Cell Host Microbe [Internet] 2020; 27:225–237. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1931312819306316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Donovan JM, Jackson AA. Transbilayer movement of fully ionized taurine-conjugated bile salts depends upon bile salt concentration, hydrophobicity, and membrane cholesterol content. Biochemistry [Internet] 1997; 36:11444–11451. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9298964 [DOI] [PubMed] [Google Scholar]
- 154.Weston N, Sharma P, Ricci V, Piddock LJ. Regulation of the AcrAB-TolC efflux pump in Enterobacteriaceae. Res Microbiol [Internet] 2018; 169:425–431. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29128373 [DOI] [PubMed] [Google Scholar]
- 155.Baucheron S, Nishino K, Monchaux I, Canepa S, Maurel M-C, Coste F, Roussel A, Cloeckaert A, Giraud E. Bile-mediated activation of the acrAB and tolC multidrug efflux genes occurs mainly through transcriptional derepression of ramA in Salmonella enterica serovar Typhimurium. J Antimicrob Chemother [Internet] 2014; 69:2400–2406. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24816212 [DOI] [PubMed] [Google Scholar]
- 156.Lin J, Sahin O, Michel LO, Zhang Q. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect Immun [Internet] 2003; 71:4250–4259. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12874300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch Microbiol [Internet] 2006; 186:171–181. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16804679 [DOI] [PubMed] [Google Scholar]
- 158.Prieto AI, Ramos-Morales F, Casadesús J. Bile-induced DNA damage in Salmonella enterica. Genetics [Internet] 2004; 168:1787–1794. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15611156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Prieto AI, Ramos-Morales F, Casadesús J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics [Internet] 2006; 174:575–584. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16888329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hernández SB, Cota I, Ducret A, Aussel L, Casadesús J. Adaptation and preadaptation of Salmonella enterica to Bile. PLoS Genet [Internet] 2012; 8:e1002459. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22275872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rodríguez-Beltrán J, Rodríguez-Rojas A, Guelfo JR, Couce A, Blázquez J. The Escherichia coli SOS gene dinF protects against oxidative stress and bile salts. PLoS One [Internet] 2012; 7:e34791. Available from http://www.ncbi.nlm.nih.gov/pubmed/22523558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Walawalkar YD, Vaidya Y, Nayak V. Response of Salmonella Typhi to bile-generated oxidative stress: implication of quorum sensing and persister cell populations. Pathog Dis. [Internet] 2016; 74. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27609462 [DOI] [PubMed] [Google Scholar]
- 163.Hung DT, Zhu J, Sturtevant D, Mekalanos JJ. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol Microbiol [Internet] 2006; 59:193–201. Available from: https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2958.2005.04846.x [DOI] [PubMed] [Google Scholar]
- 164.Dubois T, Tremblay YDN, Hamiot A, Martin-Verstraete I, Deschamps J, Monot M, Briandet R, Dupuy B. A microbiota-generated bile salt induces biofilm formation in Clostridium difficile. NPJ Biofilms Microbiomes [Internet] 2019; 5:14.Available from: http://www.ncbi.nlm.nih.gov/pubmed/31098293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pumbwe L, Skilbeck CA, Nakano V, Avila-Campos MJ, Piazza RMF, Wexler HM. Bile salts enhance bacterial co-aggregation, bacterial-intestinal epithelial cell adhesion, biofilm formation and antimicrobial resistance of Bacteroides fragilis. Microb Pathog [Internet] 2007; 43:78–87. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0882401007000472 [DOI] [PubMed] [Google Scholar]
- 166.Kelly SM, Lanigan N, O’Neill IJ, Bottacini F, Lugli GA, Viappiani A, Turroni F, Ventura M, van Sinderen D. Bifidobacterial biofilm formation is a multifactorial adaptive phenomenon in response to bile exposure. Sci Rep [Internet] 2020; 10:11598. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32665665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hay AJ, Zhu J. Host intestinal signal-promoted biofilm dispersal induces Vibrio cholerae colonization. Infect Immun [Internet] 2015; 83:317–323. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25368110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology [Internet] 2004; 127:412–421. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15300573 [DOI] [PubMed] [Google Scholar]
- 169.Ormsby MJ, Johnson SA, Carpena N, Meikle LM, Goldstone RJ, McIntosh A, Wessel HM, Hulme HE, McConnachie CC, Connolly JPR, et al. Propionic Acid promotes the virulent phenotype of Crohn’s disease-associated adherent-invasive Escherichia coli. Cell Rep [Internet] 2020; 30:2297–2305. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32075765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rolhion N, Carvalho FA, Darfeuille-Michaud A. OmpC and the sigma(E) regulatory pathway are involved in adhesion and invasion of the Crohn’s disease-associated Escherichia coli strain LF82. Mol Microbiol [Internet] 2007; 63:1684–1700. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17367388 [DOI] [PubMed] [Google Scholar]
- 171.Lucchini V, Sivignon A, Pieren M, Gitzinger M, Lociuro S, Barnich N, Kemmer C, Trebosc V. The role of OmpR in bile tolerance and pathogenesis of adherent-invasive Escherichia coli. Front Microbiol [Internet] 2021; 12. Available from: https://www.frontiersin.org/articles/10.3389/fmicb.2021.684473/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Johnson R, Ravenhall M, Pickard D, Dougan G, Byrne A, Frankel G. Comparison of Salmonella enterica Serovars Typhi and Typhimurium Reveals Typhoidal Serovar-Specific Responses to Bile. Infect Immun. [Internet] 2018; 86. Available from:.http://www.ncbi.nlm.nih.gov/pubmed/29229736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Eade CR, Hung C-C, Bullard B, Gonzalez-Escobedo G, Gunn JS, Altier C. Bile acids function synergistically to repress invasion gene expression in Salmonella by destabilizing the invasion regulator hilD. Infect Immun [Internet] 2016; 84:2198–2208. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27185788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A [Internet] 2013; 110:2348–2353. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23341592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gupta S, Chowdhury R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun [Internet] 1997; 65:1131–1134. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9038330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Barton LL, Ritz NL, Fauque GD, Lin HC. Sulfur cycling and the intestinal microbiome. Dig Dis Sci [Internet] 2017; 62:2241–2257. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28766244 [DOI] [PubMed] [Google Scholar]
- 177.Forte E, Borisov VB, Falabella M, Colaço HG, Tinajero-Trejo M, Poole RK, Vicente JB, Sarti P, Giuffrè A. The terminal oxidase cytochrome bd promotes sulfide-resistant bacterial respiration and growth. Sci Rep [Internet] 2016; 6:23788. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27030302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lopez CA, Miller BM, Rivera-Chávez F, Velazquez EM, Byndloss MX, Chávez-Arroyo A, Lokken KL, Tsolis RM, Winter SE, Bäumler AJ. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science [Internet] 2016; 353:1249–1253. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27634526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Suarez FL, Furne JK, Springfield J, Levitt MD. Bismuth subsalicylate markedly decreases hydrogen sulfide release in the human colon. Gastroenterology [Internet] 1998; 114:923–929. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9558280. [DOI] [PubMed] [Google Scholar]
- 180.Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol [Internet] 2018; 15:111–128. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29018272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang Y-Z, Li Y-Y. Inflammatory bowel disease: pathogenesis. World J Gastroenterol [Internet] 2014; 20:91–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24415861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tiratterra E. Role of bile acids in inflammatory bowel disease. Ann Gastroenterol [Internet] 2018; 31:266–272. Available from: http://www.annalsgastro.gr/files/journals/1/earlyview/2018/ev-02-2018-08-AG3388-REV-0239.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Vantrappen G, Ghoos Y, Rutgeerts P, Janssens J. Bile acid studies in uncomplicated Crohn’s disease. Gut [Internet] 1977; 18:730–735. Available from: http://www.ncbi.nlm.nih.gov/pubmed/604194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rutgeerts P, Ghoos Y, Vantrappen G. Kinetics of primary bile acids in patients with non-operated Crohn’s disease. Eur J Clin Invest [Internet] 1982; 12:135–143. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6807684 [DOI] [PubMed] [Google Scholar]
- 185.Gothe F, Beigel F, Rust C, Hajji M, Koletzko S, Freudenberg F. Bile acid malabsorption assessed by 7 alpha-hydroxy-4-cholesten-3-one in pediatric inflammatory bowel disease: correlation to clinical and laboratory findings. J Crohns Colitis [Internet] 2014; 8:1072–1078. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24666974 [DOI] [PubMed] [Google Scholar]
- 186.Nyhlin H, Merrick M, Eastwood MA. Bile acid malabsorption in Crohn’s disease and indications for its assessment using SeHCAT. Gut [Internet] 1994; 35:90–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8307458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lenicek M, Duricova D, Komarek V, Gabrysova B, Lukas M, Smerhovsky Z, Vitek L. Bile acid malabsorption in inflammatory bowel disease: assessment by serum markers. Inflamm Bowel Dis [Internet] 2011; 17:1322–1327. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21058331. [DOI] [PubMed] [Google Scholar]
- 188.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward D, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol [Internet] 2012; 13:R79.Available from: http://www.ncbi.nlm.nih.gov/pubmed/23013615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Byndloss MX, Bäumler AJ. The germ-organ theory of non-communicable diseases. Nat Rev Microbiol [Internet] 2018; 16:103–110. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29307890 [DOI] [PubMed] [Google Scholar]
- 190.Winter SE, Lopez CA, Bäumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep [Internet] 2013; 14:319–327. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23478337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Byndloss MX, Pernitzsch SR, Bäumler AJ. Healthy hosts rule within: ecological forces shaping the gut microbiota. Mucosal Immunol [Internet] 2018; 11:1299–1305. Available from: https://www.nature.com/articles/s41385-018-0010-y [DOI] [PubMed] [Google Scholar]
- 192.Mullish BH, Allegretti JR. The contribution of bile acid metabolism to the pathogenesis of Clostridioides difficile infection. Therap Adv Gastroenterol [Internet] 2021; 14:175628482110177. Available from: http://journals.sagepub.com/doi/10.1177/17562848211017725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Panzitt K, Zollner G, Marschall H-U, Wagner M. Recent advances on FXR-targeting therapeutics. Mol Cell Endocrinol [Internet] 2022; 552:111678. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0303720722001265 [DOI] [PubMed] [Google Scholar]
- 194.Hodge RJ, Nunez DJ. Therapeutic potential of Takeda-G-protein-receptor-5 (TGR5) agonists. Hope or hype? Diabetes Obes Metab [Internet] 2016; 18:439–443. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26818602 [DOI] [PubMed] [Google Scholar]