

Abstract
Reactive sulfur species (RSS) entail a diverse family of sulfur derivatives that have emerged as important effector molecules in H2S-mediated biological events. RSS (including H2S) can exert their biological roles via widespread interactions with metalloproteins. Metalloproteins are essential components along the metabolic route of oxygen in the body, from the transport and storage of O2, through cellular respiration, to the maintenance of redox homeostasis by elimination of reactive oxygen species (ROS). Moreover, heme peroxidases contribute to immune defense by killing pathogens using oxygen-derived H2O2 as a precursor for stronger oxidants. Coordination and redox reactions with metal centers are primary means of RSS to alter fundamental cellular functions. In addition to RSS-mediated metalloprotein functions, the reduction of high-valent metal centers by RSS results in radical formation and opens the way for subsequent per- and polysulfide formation, which may have implications in cellular protection against oxidative stress and in redox signaling. Furthermore, recent findings pointed out the potential role of RSS as substrates for mitochondrial energy production and their cytoprotective capacity, with the involvement of metalloproteins. The current review summarizes the interactions of RSS with protein metal centers and their biological implications with special emphasis on mechanistic aspects, sulfide-mediated signaling, and pathophysiological consequences. A deeper understanding of the biological actions of reactive sulfur species on a molecular level is primordial in H2S-related drug development and the advancement of redox medicine.
Keywords: Hydrogen sulfide, Reactive sulfur species, Metalloprotein, Heme, Oxidative stress
Graphical abstract
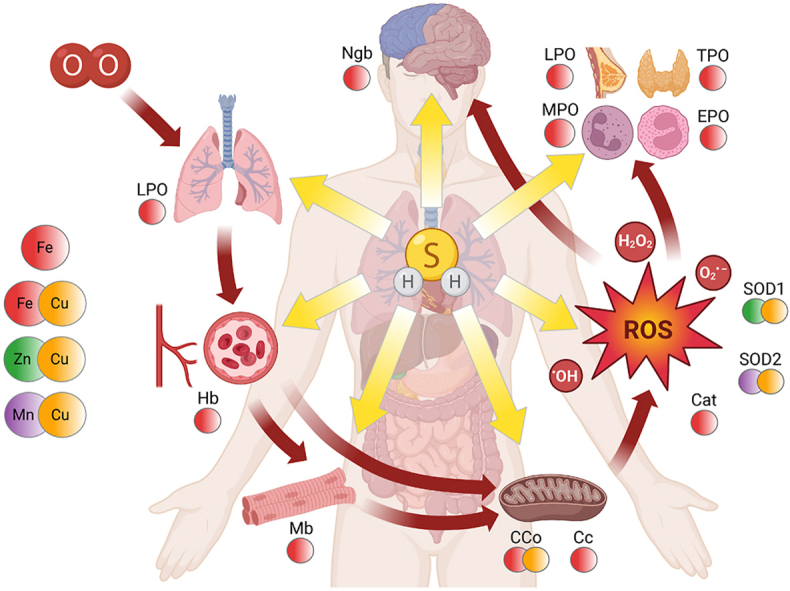
Highlights
-
•
Interactions with metalloproteins is a major pathway of H2S bioactivity.
-
•
Reactive sulfur species (RSS) act as effector molecules in H2S-mediated events.
-
•
Heme proteins are main targets of H2S via coordination and redox exchanges.
-
•
H2S and RSS affect oxygen transport, storage, respiration, and immune defense.
-
•
RSS and metalloproteins cooperate in cytoprotection against oxidative stress.
1. Introduction
In the last two decades, hydrogen sulfide (H2S) has been designated as the third gaseous signaling molecule along with nitric oxide (•NO) and carbon monoxide (CO) [1]. Ever since the discovery of its neuromodulatory effect [2], vast research marks its way from toxic gas to endogenously produced biomolecule with multifaceted roles in health and disease [[3], [4], [5]]. H2S has been associated with a variety of pathophysiological conditions, including cardiovascular diseases [6], inflammation [7], metabolic disorders such as diabetes or obesity [8,9], as well as the initiation and progression of cancer [10]. Increasing efforts have been invested into the detailed understanding of the biomedical significance of H2S, in order to exploit its beneficial properties in therapeutic interventions. Still, to date many questions remain unanswered regarding the molecular mechanisms governing H2S biology.
Most notably, due to the intimate equilibrium network that exists in vivo among a diverse array of biological reactive sulfur species (RSS), it is still not clear whether H2S is the actual effector species in most of its reported (patho)physiological properties. This anomaly is due to the fact that quantitative measurements of H2S and other sulfur species rely on irreversible chemical derivatization, which can alter the dynamic equilibrium network and thereby the speciation of RSS in biological systems [11,12]. Early observations on these grounds introduced the concept of biological sulfide pools, which were proposed to buffer the in vivo concentrations of H2S [13]. However, in many investigated biological systems it is still not clear whether sulfide-binding biomolecules (RSS, e.g. persulfides) or sulfide itself is responsible for the observed effects.
Interpretation of sulfide-mediated biological processes may be aided by a brief overview of the biochemical properties of hydrogen sulfide. More detailed characterization of the presented features is available in previous literature [13]. Sulfide bioavailability relies on its fine-tuned metabolic pathways and the dynamic interconversion equilibria of RSS within the above-mentioned biological sulfide pool. Biogenesis of H2S in the human body occurs via the non-canonical actions of two transsulfuration enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE). Furthermore, the consecutive work of aspartate/cysteine aminotransferase (AAT/CAT) and 3-mercaptopyruvate sulfurtransferase (3-MST) contribute to H2S generation via a protein-cysteine persulfide intermediate species [14,15]. In addition, sulfide is also produced by the reduction of different cysteine persulfide species governed by the thioredoxin and glutathione systems [16,17]. The amino acid cysteine persulfide can be directly produced by CARS2 using cysteine or CBS and CSE using cystine (the disulfide) as substrates and the persulfide moiety can be transferred on other thiols via transpersulfidation reactions [18,19]. Canonical sulfide catabolism takes place in the mitochondria through the so-called sulfide oxidation pathway. The oxidation of sulfide is mediated by an enzymatic system including sulfide:quinone oxidoreductase (SQR), sulfite oxidase (SUOX), persulfide dioxygenase (PDO) also known as ethylmalonic encephalopathy protein 1 (ETHE1), and thiosulfate sulfurtransferase (TST). Ultimately, sulfide is excreted from the cells in the forms of sulfite, sulfate, and thiosulfate [14,15,20,21]. As mentioned above, the bioavailability of sulfide is not limited to its endogenous production and catabolism. In fact, less than 1% of the total available sulfide is present as free sulfide in the human body and the majority is stored in biomolecule-sulfide adducts or sulfide pools with large buffer capacities [22]. Nevertheless, sulfide levels may be modulated exogenously by the introduction of slow-releasing sulfide donor molecules [23].
As a bivalent Brønsted acid, H2S may undergo two subsequent deprotonation steps (eqs. (1), (2)), and the speciation between hydrogen sulfide (H2S), hydrosulfide ion (HS−), and sulfide ion (S2−) is mainly governed by the pH of the environment and Le Chatelier's rule of dynamic equilibria.
| (1) |
| (2) |
Hereinafter, the term "sulfide" refers to each protonation forms collectively. Under physiological conditions (pH = 7.4, T = 37 °C), the dominant form is hydrosulfide anion (HS−), acting as a ligand or redox partner, depending on the reaction context.
Sulfide contains S(-II), the most reduced form of sulfur atom, which may be oxidized all the way to sulfate (S(VI)), through a cascade of enzymatic redox reactions in the body [24]. Sulfur is involved in one- and two-electron redox reactions, via the formation of a multitude of radical-type and closed shell intermediates [25].
Reactive sulfur species (RSS) is an umbrella term analogous to Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS), showing the emerging significance of H2S and its oxidized derivatives as signaling molecules [26]. Primary representatives are the so-called per/polysulfides (RSSnH), where n ≥ 1 and the R group may denote protein cysteine side chains or low molecular weight thiols such as cysteine (Cys-SH) and glutathione (GSH). Protein per/polysulfidation is now recognized as an important posttranslational modification of cysteine residues, due to its relative abundance and described regulatory function on enzymatic activities [16,18,[27], [28], [29]]. A common, yet not an exclusive characteristic of RSS is the presence of sulfane sulfur (S(0)), often responsible for increased reactivity [13,27]. While thiols/thiolates are generally assessed as nucleophiles, per/polysulfides show electrophilic and nucleophilic traits depending on their reaction partners. Persulfide groups have lower pKa than thiols, due to the alpha-effect, making them more susceptible towards oxidation, alkylation, or other electrophilic agents (such as metals) [30]. Thiols and sulfane sulfur-containing species alike are prone to oxidation from S(-II) (thiols or sulfides) to S(VI) (sulfate), the ultimate oxidation product. A number of sulfuroxy (SxOyn−) species are generated during these steps, each belonging to the family of reactive sulfur species with their own distinct functions and signaling potential. Sulfite (SO32−), for example, is a toxic by-product of sulfide oxidation [31] and thiosulfate (S2O32−) is responsible for cyanide detoxification, as substrate of the enzyme rhodanese or thiosulfate sulfurtransferase [32]. These versatile redox and acid-base characteristics substantially contribute to the importance of various RSS in regulation and signaling and allow H2S to exert its biological functions. To our current knowledge, two main platforms of sulfide biology are persulfide formation on protein cysteine residues [[27], [28], [29],[33], [34], [35], [36]] and the intricate cross-talk with NO signaling [26,37,38]. These phenomena represent the bases of increasingly growing individual research fields on their own, discussed extensively in recent literature.
The third important area where sulfide exhibits its biological effects is via extensive interactions with metalloproteins [27,39,40]. Metalloproteins require one or more metal ions for the development of their proper structure and functionality, and they account for 30–40% of the human proteome [41,42]. Metal ions are ubiquitous in the human body and play a vital role in normal physiology. They are involved in the construction and operation of macromolecules, enzymes, organelles, and tissues; maintenance of cellular homeostasis, cellular respiration, metabolism, regulation, signaling, and immune mechanisms. In fact, an individual research field called metallomics entails the study of metal ions in biological systems, creating an interface between inorganic chemistry and life sciences [43,44]. Metallomics utilizes the methods of proteomics and bioinformatics in order to organize and describe the large number of metalloproteins in living organisms [45]. 20 elements are considered essential for humans, of which 10 are metals (Table 1). It is noteworthy that essentiality is still a matter of debate in certain cases, such as chromium or vanadium [46,47].
Table 1.
| Metal | Massa | Main functions | Deficiency symptoms | |
|---|---|---|---|---|
| macro-minerals | Ca | 1.7 kg | bone and dental structure; enzyme cofactor; signal transduction | anomalous skeletal growth |
| K | 140 g | fluid homeostasis; electrolyte components; blood pressure regulation; nerve signaling [48] | various (weakness, arrhythmia, confusion, etc.) | |
| Na | 100 g | various (nausea, seizures, muscle cramps, etc.) | ||
| Mg | 30 g | bone structure; enzyme cofactor – protein and nucleic acid synthesis; ATP hydrolysis; nerve activity in muscles [49] | muscle spasms | |
| Fe | 5 g | electron transfer; O2 transport and storage; sulfur storage (iron-sulfur clusters, ISCs) | anemia, immune disorders, chronic fatigue | |
| Zn | 2 g | acid-base catalysis; structural (zinc fingers); antioxidant (superoxide dismutase 1, SOD1) [50] | growth and immune disorders, skin damage | |
| trace elements | Cu | 100 mg | electron transfer; antioxidant (superoxide dismutase 1, SOD1) | muscle weakness, hair loss, liver disease |
| Mn | 16 mg | mitochondrial superoxide dismutase 2 (SOD2) [51] | infertility, skeletal growth disorders | |
| Mo | 5 mg | Moco enzyme cofactor [52] | MoCD is lethal at early age | |
| Co | 2 mg | B12 – methyl transfer [53] | anemia |
Average amounts in a reference person of 70 kg are indicated.
Metalloproteins perform a wide array of functions from oxygen transport to immune defense, nucleic acid synthesis, or counteracting oxidative stress. Table 2 shows a possible classification of trace metals and related metalloproteins by their main function. Overlaps may occur between the listed groups, i.e. multimetal enzymes like Cu/ZnSODs, which contain active site and stabilizing metals as well. Also noted that electron transport proteins are not fully independent of redox catalysts, although their main function is the easy uptake and donation of electrons.
Table 2.
Classification of trace metal and metalloprotein functions [54].
| Function | Metal | Representative proteins |
|---|---|---|
| Transport and storage of small molecules (mainly O2) | Fe (heme) | hemoglobin (Hb), myoglobin (Mb), hemerythrin (marine worms), hemocyanin (molluscs, crustaceans, spiders) |
| Fe (non-heme) | ||
| Cu | ||
| Catalysis – Metalloenzymes | ||
|
Fe(II)/Fe(III) | catalase (CAT), superoxide dismutase (SOD1), cytochrome c oxidase (CcO), heme peroxidases: myeloperoxidase (MPO), lactoperoxidase (LPO), eosinophil peroxidase (EPO), thyroid peroxidase (TPO) |
| Cu(I)/Cu(II) | ||
| (Mn, Co, Mo) | ||
|
Zn (Ca, Mg, Mn) | carbonic anhydrase (CA), carboxypeptidase A (CPA) |
| Conformational stabilization of macromolecules | ||
|
Zn | alcohol dehydrogenase (ADH5), (1 Zn at the active site and 1 structural), superoxide dismutase (SOD1), prolyl hydroxylase (PHD) |
|
Zn | zinc finger proteins |
| Transport and storage of trace elements | Fe | ferritin, transferrin ceruloplasmin, metallothionein, respective transport proteins of trace elements |
| Cu | ||
| Electron carriers | ||
|
Fe (heme) | cytochrome c, cytochrome P450 (CYPs) |
|
Fe (non-heme) | Ferredoxins, high potential iron-sulfur proteins (HiPIP) |
|
Cu | tyrosinase (human), hemocyanin (see O2 transport) |
Transition metal centers of borderline acid character according to the hard and soft acids and bases principle (Fe(II), Cu(II), Zn(II)) constitute the primary sites of sulfide-metal biochemistry via complex formation and/or electron transfer reactions [39,40,55]. It is noteworthy that due to the ease of sulfur – metal reactions, reactive sulfur species alleviate heavy metal toxicity, as reported for methylmercury or cadmium, among others [56,57]. Metal ions with variable oxidation states play essential roles in oxygen-based life (Table 2), from the transport and storage of oxygen, to the deep involvement of iron and copper proteins in the mitochondrial electron transport chain as well as the detoxification of reactive oxygen species. Furthermore, iron ions are primordial for the immune system, as centers of heme peroxidases. Sulfide and other reactive sulfur species may influence these fundamental events by altering the microenvironment of the metal center. A detailed understanding of such interactions may shed further light on the biological role of endogenous H2S. The present review is aimed to give an overview of the current knowledge on the interactions of reactive sulfur species with a selection of metalloproteins, with special emphasis on mechanistic aspects of metalloprotein-related sulfide signaling and pathophysiological implications.
2. Sulfide storage – iron-sulfur clusters
Iron-sulfur clusters (ISCs) constitute a major group of non-heme iron protein cofactors, abundant in all domains of life (archaea, bacteria, and eukaryota) [58,59]. The prevalence of iron-sulfur clusters across various life-forms implies their evolutionary involvement, and possibly their role in adaptation to early sulfur-rich environments [60,61]. Proteins containing ISCs are commonly called iron-sulfur proteins. Iron-sulfur clusters are multi-metal Fe(II)/Fe(III) structures linked by sulfide bridges in distorted tetrahedral or cubic arrangements, generally coordinated to the protein backbone by cysteine ligands (Fig. 1a–c). Coordinating amino acid ligands might entail aspartic acid (Asp), histidine (His), serine (Ser), or amide-N besides Cys-SH [62]. The most frequent stoichiometries are [2Fe–2S] (Fig. 1b) and [4Fe–4S] (Fig. 1c) ratios [63], although more complex arrangements occur, as well as single-iron centers can directly bind to four cysteines (called rubredoxins; Fig. 1a) [64].
Fig. 1.

Iron-sulfur clusters (ISCs) with different stoichiometries: a) rubredoxin; b) [2Fe–2S] cluster; c) [4Fe–4S] cluster.
Iron-sulfur clusters have versatile biological functions, thoroughly discussed previously [[62], [63], [64], [65], [66], [67]]. Their primary function is electron transfer, due to the variable oxidation state of the iron centers. Fe–S proteins mediating electron transfer events are named ferredoxins. They usually facilitate one-electron reactions, although two-electron transfer may be realized by complex structures ([8Fe–7S]) found in bacterial nitrogenase enzymes [62]. Other functions of Fe–S proteins entail Lewis acid-base catalysis (citrate-isocitrate isomerization by aconitase in the tricarboxylic acid cycle - TAC), regulation of iron metabolism by cytosolic aconitase or otherwise called iron-responsive element binding protein (IRP1), radical generation (the radical S-adenosyl methionine (SAM) superfamily with diverse functions) [68], metal delivery for cofactor synthesis [69], DNA repair, and many more. The mitochondrial electron transport chain in humans heavily relies on ISCs. Complexes I-III contain a number of Fe–S clusters contributing to the electron flow to cytochrome c, and ultimately to molecular oxygen [65].
Iron-sulfur clusters play an integral part in sulfur biochemistry from multiple points of view. They constitute a main fraction of the so-called acid-labile sulfide pool [13,70]. Several large buffer systems mediate circulating sulfide levels in a context-dependent manner, as discussed above [22,27]. Acidification releases a large amount of sulfide from iron-sulfur clusters, causing substantial overestimation of biological sulfide levels in methods performed under acidic conditions, like the methylene blue method [13,70]. Shen et al. showed that the acid-labile sulfide content of human plasma lies in the low-micromolar range, exceeding both free and sulfane sulfur-bound pools [70]. Measurements from a variety of tissue samples and blood also suggested that sulfide concentrations retained in the acid labile pool are much higher compared to free sulfide levels [71]. It was reported that sulfide in ISCs is mobilized below pH 5.4, raising questions regarding the physiological relevance of the acid-labile pool [72]. However, it cannot be excluded that transient local pH drops might cause disruption of Fe–S clusters and subsequent sulfide release, under uncommon circumstances. It should be noted here that all the referred reports related to the acid-labile sulfide levels used conceptionally different methods for H2S measurements. To date, adequate determination of biological sulfide levels is still a major challenge for the field.
Despite tremendous efforts to reconstitute the generation and maturation of iron-sulfur clusters, the exact physiological mechanism is still under scrutiny [59,67,73]. Nevertheless, it is well known that sulfur transfer is a fundamental element for the de novo synthesis of iron-sulfur clusters. In humans, the primary compartments of ISC formation are the mitochondria, although dedicated assembly machineries operate in the cytosol and Fe–S proteins occur in the nucleus as well [59,67]. The early stage of mitochondrial ISC assembly includes similar events between mammalian cells and bacteria [59,62,63,66,67]. The source of sulfur is l-cysteine, converted to alanine (Ala) by a pyridoxal phosphate-dependent cysteine desulfurase, creating a persulfide intermediate on the enzyme [74]. This persulfide equivalent is then transferred to a scaffold protein, where the actual cluster formation takes place from iron and sulfide. The release of sulfide (S2−) from the sulfane sulfur (S0) containing persulfide moiety requires a reduction step and a suitable electron donor. Iron delivery is a matter of debate, so far it is likely that locally available free ferrous (Fe(II)) ions are used since no dedicated donor protein was identified [59]. Frataxin (FXN) was considered as an iron donor protein [66,75], however, this activity was questioned and it has been shown that FXN rather has a rate enhancement role in cluster biogenesis (see below) [73,76]. On the scaffold protein, simpler [2Fe–2S] structures are assembled, which are transported from the mitochondria if necessary and delivered to target apoproteins by dedicated chaperones creating the Fe–S holoproteins or undergo further maturation to generate [4Fe–4S] and more complex clusters [63].
Faulty or disrupted ISC assembly is related to a number of mitochondrial diseases [59,67,77], often linked to iron accumulation or increased sensitivity to oxidative stress. A characteristic example is Friedrich's ataxia (FRDA), an autosomal-recessive hereditary disorder, which is associated with decreased expression of frataxin [77]. FRDA symptoms include walking and speech impairment, heart conditions, diabetes, and difficulties with eyesight or hearing. It is the most common genetic ataxia, with a 1:50,000 occurrence [77,78]. No treatment is currently available, therefore a detailed understanding of the functional role of frataxin is primordial for future therapeutic developments. Initially, it was postulated that FXN acts as an iron storage and delivery protein in ISC assembly, however, more recent data revealed that it is rather involved in sulfur transfer towards the scaffold protein and increases sulfide production [73]. In humans, the general scheme described above is represented by the NFS1/ISD11/ACP–ISCU (cysteine desulfurase/iron-sulfur cluster assembly protein/acyl carrier protein–iron-sulfur cluster assembly enzyme assembly), shortly the NIU complex. In the NIU complex NFS1 acts as cysteine desulfurase, ISD11, and ACP are regulatory factors of the latter, while ISCU is the scaffold protein where the initial assembly takes place [59,73]. Parent et al. used a custom-developed mobility shift-based assay to reveal that FXN coordinates to the NIU complex and accelerates sulfane sulfur transfer from NFS1 to ISCU in a direct step and to low molecular weight thiols like dithiothreitol (DTT), cysteine, and glutathione. They concluded that the rate-limiting step of the system is the sulfide release from NFS1-persulfide. It appeared that ISCU-SSH did not contribute to sulfide production by physiological reductants like Cys-SH or GSH, therefore it was postulated that a dedicated reductase system should be operative to release sulfide for cluster production and that the sulfane sulfur transfer to biological thiols might serve an alternative purpose, i.e. contribution to cellular sulfur trafficking [76]. The same group later revealed that the binding of zinc (Zn2+) ion to ISCU hindered iron binding in earlier reconstitution attempts in vitro and reshuffled persulfide reduction to NFS1-SSH. The replacement of zinc with iron better approximated physiological conditions and allowed ISCU persulfide reduction by ferredoxin 2 (FDX2), an iron-sulfur protein itself, where the electron donors are the coupled ferredoxin reductase (FDXR) and NADPH [73]. This report further corroborated that the role of FXN is the rate enhancement of sulfur transfer from NFS1 to ISCU. The iron vs. zinc mediated assemblies and the respective roles of frataxin are comparatively illustrated in [73].
It has been suggested that frataxin plays a role in the coping mechanisms against oxidative stress. A Drosophila model overexpressing frataxin was found to exhibit elevated antioxidant protection and life expectancy [79]. The above-described sulfur transfer promoting capacity of FXN might contribute to this protective effect by stimulation of sulfur trafficking in the cells, and the increased generation of Cys-SSH, GSSH, and H2S. These species are closely related to protein persulfidation by dynamic equilibria and recent discoveries strongly support the cytoprotective effects of low molecular weight and protein-bound persulfides [18,30,[80], [81], [82]]. On the other hand, iron-sulfur clusters are themselves extremely vulnerable to oxidative stress, iron accumulation and elevated Fenton-type ROS production are often linked to oxidative damage [66,83]. Typical intracellular antioxidant systems such as glutathione and glutaredoxins are involved in the delivery and later assembly of Fe–S clusters [83]. Therefore, it can be concluded that an intricate balance connects the dedicated redox systems of the cells with iron and sulfur homeostasis and the detailed characterization of the underlying molecular mechanisms may allow their modulation, thus getting us closer to more efficient treatments of redox diseases, perhaps even cancer therapies [66,77,83,84].
3. Sulfide interactions with heme proteins
Growing evidence support that ligation and redox interactions between heme proteins and reactive sulfur species play a signaling and/or regulatory role in the human body [[85], [86], [87], [88], [89], [90], [91]]. In the current review, we present reported interactions of RSS with heme proteins concerning their primary function, such as oxygen transport and storage, mitochondrial electron transport, and antioxidant capacity.
Heme proteins are among the most widely researched subfamilies of metalloproteins. Their heme prosthetic group consists of a porphyrin (Por) scaffold coordinated to an iron center, occupying four of six coordination sites of the metal ion. In hexa-coordinated heme proteins, the remaining two available sites of the iron are occupied by the side chains of the respective protein. Meanwhile, in penta-coordinated proteins, the polypeptide chain is attached to the metal center by occupying only its fifth coordination site, typically through a histidine residue (proximal ligand), leaving one unsaturated position (distal site) for ligand binding. Under physiological conditions, a water molecule is coordinated to this site [86]. Heme prosthetic groups are classified by the nature of the side chains of the porphyrin ring (Fig. 2).
Fig. 2.

Structure of major biologically relevant heme prosthetic groups (examples of associated proteins are indicated): a) heme a (cytochrome c oxidase) b) heme b (hemoglobin, myoglobin, neuroglobin), and c) heme c (cytochrome c).
The redox activity of the heme prosthetic group relies on the various oxidation states of the iron center between the ferrous (Fe(II)), ferric (Fe(III)), and ferryl ((Fe(IV)) forms. From a mechanistic point of view, redox reactions between sulfide and the heme group may occur via outer-sphere or inner-sphere electron transfer. In the latter case sulfide binding to the iron center precedes the electron transfer. Theoretically, both ferric and ferrous forms can bind sulfide, although the affinity of sulfide towards the ferrous heme is lower, due to its higher electron density. Thus, the formation of Fe(III)–H2S/Fe(III)-HS− complexes is physiologically more relevant [87]. One-electron reduction by sulfide results in the formation of HS• radical, opening the way for subsequent redox reactions and the production of diverse oxidized sulfur and reactive oxygen species [92]. This is consistent with the concept that sulfide-related biological activity might be connected to associated derivatives rather than sulfide directly. Another common interaction between sulfide and heme proteins is sulfheme formation, which represents sulfur incorporation into one of the pyrrole rings of the porphyrin frame (Fig. 3), and the formation of heme-chlorine-type structures.
Fig. 3.

Pyrrol structure a) in heme proteins and b-d) in identified pyrrole structures in sulfheme derivatives of myoglobin. Sulfur incorporation into the porphyrin frame by the saturation of the Cβ – Cβ bond in one of the pyrrole ring results in the formation of chlorin-type structures such as b) episulfide; c) ring opened episulfide or d) thiochlorin isoforms [91].
This modification leads to altered protein function, which phenomenon was reviewed in detail by Rios-Gonzalez et al. [91]. The underlying mechanism is still unclear, though presumably the involvement of high-valent ferryl species, Compound II ([Fe(IV) O] or [Fe(IV)–OH]) is pivotal [93,94]. Hydrogen peroxide is needed for the formation of these ferryl species, thus oxidative stress may facilitate sulfheme development. To date, two mechanisms have been proposed for sulfheme formation (Fig. 4). Compound I (+•Por-Fe(IV) O) is formed in the reaction of the ferric heme with H2O2 through a ferric hydroperoxide complex (Fe(III)–OOH−) intermediate and subsequent one electron reduction results in the production of Compound II. H2S may form an intermediate complex with Fe(III)–OOH− (Fig. 4, mechanism I) or Compound II (Fig. 4, mechanism II) with the assistance of the histidine ligand in the distal location of the heme pocket. The following reaction results in the conversion of the heme center into Compound II or Fe(III) respectively, accompanied by H2O elimination and HS• production. HS• has been identified as the reactive agent, which readily attacks the generated form of heme in each case.
Fig. 4.

Proposed mechanisms of sulfheme (sulfPor-Fe*-L) formation. (Fe*: oxidation state of the iron center is variable in the sulfheme derivatives; L: ligand that occupies the sixth coordination site of the iron center in sulfheme) [91].
The described possible interactions are partially governed by external factors such as pH, sulfide concentration, or specific structural characteristics of the proteins, such as surface accessibility, polarity, and local protein environment of the active site. Firstly, the intracellular pH mainly affects the protonation equilibria of sulfide (eqs. (1), (2))) and thus the relative accessibility of the interacting species. For example, the reaction rate between ferric hemoglobin and sulfide increases with decreasing pH and consequently with increasing H2S ratio [95]. Secondly, the significance of the H2S concentration is best demonstrated through its interaction with cytochrome c oxidase (CcO), the last enzyme of the mitochondrial electron transport chain. At high levels, sulfide irreversibly inhibits the activity of CcO and blocks cellular respiration. At low concentrations, sulfide can donate an electron to CcO and support mitochondrial energy production [40]. Thirdly, in penta-coordinated heme proteins, the type of protein chain residues surrounding the active site nearby the distal binding position has a pivotal role. Non-polar or low-polarity active centers with amino acid residues capable of forming hydrogen bonds favor the stabilization of a heme-H2S complex. The presence of strong proton acceptor groups promotes the deprotonation of the sulfide ligand [86,89,96]. The decisive role of the existence and orientation of a distal His was proposed based on the fact that many heme proteins with this residue form sulfheme derivatives (hemoglobin - Hb [97,98], myoglobin - Mb [[98], [99], [100], [101]], catalase - CAT [101], lactoperoxidase - LPO [102]). In addition, HbI from Lucina pectinata possesses a distal glutamine (Gln) residue, which did not form a sulfheme. But mutagenesis of the Gln residue to histidine in HbI enabled sulfheme development [98]. However, the mere presence of a His side chain is not sufficient for sulfheme formation. For instance, myeloperoxidase with His in the distal location can catalyze sulfide oxidation without the formation of a sulfheme [103]. In the case of hexa-coordinated heme proteins, sulfide can compete for the sixth coordination site of the iron center with the associated protein side chain.
Undoubtedly, the study of the interaction between heme proteins and sulfide is a fascinating but challenging area, fraught with unanswered questions. Still, there are plenty of theories for the physiological roles of these interactions based on experimental evidence. To date, several proteins and protein complexes have been investigated in this regard, with the findings presented below. Furthermore, numerous studies focus on the investigation of heme model porphyrins [86], but since the heme pocket environment significantly affects the outcome of the sulfide – heme protein interactions, a detailed discussion of these model systems is outside the scope of this review.
4. Sulfide in oxygen transport and storage
Hemoglobin (Hb), myoglobin (Mb), and neuroglobin (Ngb), representatives of the five human globins, besides cytoglobin (Cygb) and androglobin (Adgb), are primarily responsible for oxygen transport, storage, and sensing in the body. The exact physiological function of the recently discovered remaining two human globins, Cygb and Adgb, is currently unclear [104]. Generally, the reduced, ferrous forms of Hb, Mb, and Ngb are capable of reversible oxygen binding through their heme group, which is the most abundant heme b type. However, their oxidized, ferric (Fe(III)) forms, namely methemoglobin (metHb), metmyoglobin (metMb), and metneuroglobin (metNgb) are unsuitable for oxygen transport. The physiological functions of these globins primarily rely on their diverse protein structure and active site environment. Hemoglobin is the main oxygen supplier in almost all vertebrates and is the most abundant protein present in red blood cells (RBCs), at about 5 mM concentration. It transports oxygen from the lungs to the tissues through the vascular system and also carries a part of the respiratory product carbon dioxide (CO2) back to the lungs. Regarding the structure of Hb, it consists of four subunits, each containing a polypeptide chain and connected heme groups. Its quaternary structure enables the cooperative oxygen binding mechanism for optimal operation. Mb is located in the cardiac and skeletal muscle tissue and its fundamental functions are oxygen storage and release during oxygen deprivation. Mb is a single polypeptide chain associated with one heme group. It needs no conformational change to bind O2 compared to Hb, and there is no cooperative oxygen binding either. While Hb and Mb are penta-coordinated heme proteins, neuroglobin, the globin of the brain, is hexa-coordinated with proximal and distal His ligands. Therefore, oxygen binding to the monomeric Ngb is accompanied by the dissociation of the distal His ligand from the heme center and consequent conformational change. Although it possesses a higher oxygen affinity than Hb, its biological function is not yet clarified. Current research implies that the functions of Ngb are highly concentration-dependent. Its oxygen transport and buffer roles seem to be relevant in specific cell types where it is expressed in larger quantities (∼100 μM), like in the retinal cells and neurons in the hypothalamus. Signaling function through its enzymatic activity is more likely to be dominant at low Ngb levels (∼1 μM) in other parts of the central nervous system [[105], [106], [107]]. Besides their canonical functions, growing evidence supports that all three of these globins (Hb, Mb, Ngb) are involved in additional processes, including sulfide biochemistry [85,104]. Nevertheless, the interaction between sulfide and Cygb or Adgb may be an attractive area for future research as well.
4.1. Hemoglobin
Hemoglobin is the predominant protein in RBCs as stated above. Under basal conditions, it is present mostly in ferrous form (deoxyHb or oxyHb) and 1–3% as methemoglobin (metHb), due to autoxidation (Fig. 5) [108,109]. The oxygenation state is denoted by the “deoxy” and “oxy” prefixes referring to the absence or presence of bound O2 to the Fe(II) center, respectively. Cytochrome-b5 reductase is primarily responsible for the maintenance of ferrous Hb levels, thus it is also called methemoglobin reductase [110]. To the best of our knowledge, H2S does not directly affect the transport of oxygen, as it does not interact with oxyHb [111]. However, its interaction with metHb is in the focus of vast research. MetHb is historically considered as an inert or pathological compound, given that it does not participate in oxygen transport, but its elevated levels cause methemoglobinemia, a disease associated with symptoms like headache, nausea, cyanosis, and even coma or death in severe cases (metHb levels above 70%) [112,113]. Nowadays, vital physiological roles have been attributed to metHb concerning sulfide chemistry.
Fig. 5.

Summarized scheme of the proposed possible interactions of sulfide with hemoglobin, myoglobin, and neuroglobin including 1) the autoxidation of ferrous heme center to the ferric derivative; 2) suggested reversible binding of sulfide to the ferric center; and 3) reduction of the ferric center to the oxygen binding ferrous form, followed by the formation of oxidized sulfide species through the two proposed mechanism: 3a) homolytic cleavage and 3b) iron center bound sulfide oxidation pathways [108,122,126].
The first evidence for the formation of the metHb-H2S complex originates from 1933 [114]. Based on multiple studies, H2S seems to be the main reactant, although HS− is the most abundant species under physiological conditions (pH = 7.4, T = 37 °C) [55,[115], [116], [117]]. A global mechanism was proposed for the coordination of H2S to the penta-coordinated heme center, comprising the following steps:
-
•
dissociation of the bound water molecule from the distal position;
-
•
migration of the H2S molecule to the heme pocket;
-
•
reversible H2S binding and complex stabilization;
-
•
or possible deprotonation of the bound H2S and stabilization of the Fe(III)-HS− complex depending on the local protein environment [86,90,118].
Hence, one of the potential functions of heme proteins is the storage and transport of sulfide, which role is well documented in the case of the extensively studied seashell Lucina pectinata [[119], [120], [121]]. It has been demonstrated that one of its three hemoglobins, HbI, has an outstanding affinity toward H2S and it is responsible for the transport of H2S to endosymbiotic bacteria [115,116]. Experiments with purified human protein showed rapid and reversible coordination of H2S to the ferric iron center of human Hb, which indicated that metHb may contribute to the H2S distribution in the circulatory system by concentration-dependent sulfide binding and release. Experimental data also suggest that the metHb-H2S intermediate is relatively stable, while consecutive sulfide oxidation is a slow process, and only occurs after prolonged (1–2 h) incubation [95,122]. In contrast, when intact human RBCs are treated with sulfide, oxidized sulfide products were detectable after a few minutes [111]. With all of this in mind, it is unlikely that metHb plays a dominant role as an H2S transport protein, similarly to Lucina pectinata HbI. It is more reasonable to assume that metHb principally facilitates non-canonical catabolism of H2S in RBCs, which are lacking mitochondria, the canonical sites of sulfide catabolism [108]. Thus, metHb may act as a line of defense against the accumulation of H2S and the associated toxicity in the blood [108,111,123]. Structural differences between HbI of Lucina pectinata and Hb of RBCs may explain these functional variations. A flexible glutamine residue in the distal position and a tight aromatic heme pocket in HbI, provided by four phenylalanine residues, ensure the stabilization of the sulfide ligand, while in the distal pocket of human Hb, the histidine and the flexible side chains of surrounding valine (Val) and leucine (Leu) residues create appropriate conditions for sulfide oxidation pathways.
Significant evidence support that Hb-Fe(III)-HS− is the first Hb catalyzed sulfide oxidation intermediate [111,124]. However, the fate of the Fe(III)-HS− complex is more intriguing. The hydrosulfide anion is able to transfer an electron to the metal center resulting in the reduction of the ferric iron to ferrous form and thiyl radical (HS•) formation. The evolution of HS• opens the way to per- and polysulfide (RSSH and RSSnH; n > 1) or thiosulfate (S2O32−) production, although the mechanism of this process is poorly established. On the one hand, combined experimental and computational data demonstrated that interactions of ferric porphyrin model compounds with sulfide induce the formation of free HS• radicals [125]. According to this, the homolytic cleavage of Fe(III)-HS− may take place involving HS• release and consecutive reactions leading to the final oxidized products (Fig. 5, pathway ‘a’) [87,111]. On the other hand, it was suggested that the formation of these oxidized sulfide derivatives occurs in a manner where the oxidation products remain bound to the Fe center (Fig. 5, pathway ‘b’) [108,126].
Despite the identified Hb-Fe(III)-SH− species [124], it was also raised that Fe(III) reduction by H2S may take part in a bimolecular electron transfer without the formation of Hb-Fe(III)–SH2 complex (Fe(III)–OH2 + H2S → Fe(II) + H2O + H+ + HS•) [122]. Nevertheless, the produced oxidized polysulfide derivatives can significantly increase the antioxidant activity of RBCs and may take part in sulfide signaling through additional protein persulfidation reactions [33,87,124]. In vitro studies imply that iron-bound polysulfides are not released from the heme prosthetic group into the solution, but are reduced by Cys-SH or GSH during the formation of H2S and the corresponding protein persulfides, Cys-SSH and GSSH, which are likely involved in protein persulfidation via transpersulfidation reactions [124]. In another study, extra- and intracellular polysulfide species were identified after sulfide treatment of intact RBCs and the characteristic spectrum of metHb-HS− complex remained unchanged in the presence of reduced thiols such as cysteine or glutathione [111]. Evidently, the exact mechanisms of Hb-mediated polysulfide formation and release are not yet fully elucidated and the controversial results need to be resolved.
From another perspective, H2S may possess cytochrome b5 reductase-like activity by supporting the maintenance of low metHb levels and thus preserving the oxygen-carrying capacity of the RBCs [111]. Additionally, possible cross-talk between H2S and nitric oxide (•NO) in RBCs has also been proposed. Ferrous Hb is responsible for the inactivation of endothelium-derived •NO and so for the regulation of •NO levels in the vascular system. The ferrous iron center of deoxyHb reacts with •NO or NO2− (the autoxidation product of •NO) to ultimately produce nitrate (NO3−) while being converted to metHb [87]. In vivo experiments showed that Na2S treatment of rats increased nitrosyl-Hb levels in the RBCs, indicating a link between H2S and •NO signaling pathways. Early studies implied that this phenomenon may also provide an opportunity to treat sulfide poisoning by nitrate-induced methemoglobinemia (elevated metHb levels in the blood), which also alleviates cytochrome c oxidase inhibition by H2S [37,127]. Moreover, under physiological conditions, complex reactions between H2S and •NO may lead to the formation of bioactive intermediates like nitrosopersulfide (SSNO−) and probably unstable HSNO/ONS− species, polysulfides (Sn2−) and so-called SULFI/NO (ON(NO)SO3−). The cross-talk between the two gasotransmitters is not limited to RBCs, but H2S may also promote or hinder the endothelial nitric oxide synthase (eNOS) •NO signaling-related activity [37,38].
Sulfhemoglobin (sulfHb) formation is another characteristic interaction between sulfide and hemoglobin, although sulfHb is practically absent (less than 2%) in blood under normal physiological conditions. The incorporation of sulfur into the porphyrin scaffold dramatically reduces the oxygen-binding affinity of Hb [128]. Sulfhemoglobinemia, i.e. higher levels of sulfHb in blood, is a potentially life-threatening condition if sulfHb levels exceed 60% and cause cyanosis and organ damage at lower levels. It is a relatively rare, typically drug-induced pathological condition that usually co-occurs with methemoglobinemia. Its diagnosis is hampered by the fact that the two pathophysiological conditions have similar symptoms [129]. Unlike methemoglobinemia, which can be treated with methylene blue therapy, to date, no antidote is known for sulfhemoglobinemia. SulfHb excretion from the body is driven by the lifespan of RBCs (100–120 days), thus blood transfusion is the only clinical means currently available in the treatment of serious cases [[130], [131], [132], [133]].
It has been shown by various groups that oxidized Hb derivatives have diverse pathological effects that promote inflammation, calcification, tissue remodeling and oxidative damage in the vasculature [[134], [135], [136]]. In atherosclerosis, different forms of Hb (oxyHb, metHb, and ferryl Hb) and fragmentation of globin were detected in atherosclerotic plaques, especially complicated hemorrhagic lesions [[136], [137], [138]]. Both metHb and ferryl Hb release heme moieties [135,139] provoking heme-mediated vascular damage [[140], [141], [142]] and some of these detrimental effects arise from lipoprotein oxidation [141,143]. Suppression of heme-mediated oxidation of low-density lipoprotein and subsequent endothelial reactions by H2S was observed [144]. It was demonstrated that hydrogen sulfide has protective effects in atherosclerosis as well as vascular and heart valve calcification and some of these benefits were attributed to its interactions with Hb [[145], [146], [147], [148]]. It has been shown that sulfide reduces the ferryl Hb species in vitro, ultimately generating sulfHb [147]. H2S also inhibits the production of Hb oligomers and polymers and prevents Hb-lipid interactions in atherosclerotic plaques [147]. These reactions contribute to the attenuation of the production of lipid peroxides and oxidative damage, thus subsequently reducing the pathological endothelial functions and injury as well as macrophage polarization by ferryl Hb species.
4.2. Myoglobin
Myoglobin (Mb) is located in cardiac and skeletal muscle tissues in the ferrous (Fe(II)) form. Deoxy/oxyMb species are distinguished by the absence/presence of bound oxygen similar to Hb. Normally the level of its ferric counterpart, metmyoglobin (metMb), is not detectable in the human body, in contrast to metHb [149]. Even though autoxidation of Mb to metMb is spontaneous [150,151] and it has been proposed that Mb may be involved in •NO homeostasis accompanied by metMb generation. Metmyoglobin reductase is responsible for its enzymatic recovery [152]. Considering the shared physiological role and active center of Mb and Hb, it is reasonable to assume that metMb shows similar activity to metHb with regard to sulfide biochemistry. To this end, the interaction of sulfide (H2S/HS−) with holoprotein metMb and a model compound, namely ferric bis-N-acetyl-microperoxidase 11 (NAcMP11-Fe(III)) has been compared [153]. It was concluded that the association of HS− to the heme center was favored over H2S coordination when the protein matrix was lacking. In contrast, metMb interaction with HS− was negligible. Presumably, hydrophobic migration channels of the active center of the heme protein hinder the accessibility of the active site for the charged HS− anion. Thus, H2S ligand binding and subsequent deprotonation are also proposed for metMb (Fig. 5). Not surprisingly, experimental evidence supports the possibility of the metMb mediated H2S oxidation route in muscle tissues [122,126]. However, the exact mechanism is still unclear. Based on extensive spectroscopic and mass spectrometric experimental results, accompanied by density functional theory calculations, Bostelaar et al. further promoted the generation of a heme-bound oxidized sulfur compound [126]. On the other hand, Jensen and Fago suggested the homolytic cleavage mechanism assumed from spectrometric equilibrium and kinetic measurements similar to metHb [122]. The proof of the biological relevance of metMb in H2S metabolism requires more investigation considering its vanishingly low level under physiological conditions. However, like Hb, Mb was also shown to form the sulfheme-containing derivative sulfmyoglobin (sulfMb) in vitro. As sulfMb has three orders of magnitude lower affinity to oxygen than Mb, its endogenous formation may cause hypoxic conditions in muscle tissues [99,154].
It is a widely accepted concept today that inorganic (HSnSH, n ≥ 1) and protein (protein-SnSH, n ≥ 1) per/polysulfide species, rather than H2S itself, play the role of active agent in sulfide-mediated signaling and regulatory processes [27]. To extend the understanding of the interactions between heme proteins and hydropersulfides, the reaction of metMb with in situ, enzymatically generated Cys-SSH has been studied. Cysteine persulfide was generated from Cys-SH using cystathionine γ-lyase. MetMb reduction to Mb-Fe(II) and sulfmyoglobin (sulfMb) formation were observed in the reaction, which cannot be unequivocally assigned to the action of the persulfide species, given that CSE-mediated cystine breakdown may also produce H2S [18,126,155]. The interaction of Mb in different oxidation states (Mb-Fe(III) or metMb, Mb-Fe(II)–O2 or oxyMb, Mb-Fe(IV) or ferryl Mb) with a novel hydropersulfide donor, namely S-methoxycarbonyl penicillamine disulfide (MCPD) [156], has been also investigated [155,157,158]. The uniqueness of the reactant lies in the fact that it transforms to MCP-SSH (N-methoxycarbonyl penicillamine persulfide) at physiological pH (7.4), meaning that it can be used as an H2S-free hydropersulfide donor [156]. Under anaerobic conditions, the reduction of metMb by MCP-SSH occurs with the concomitant generation of deoxyMb (Mb-Fe(II)). It was pointed out that human Hb reacts similarly under such conditions, but the experimental results have not been published [155]. Under aerobic conditions, MCP-SSH readily converts metMb to Mb-Fe(II) in a one-electron reduction step and subsequent ligation of O2 leads to oxyMb (Mb-Fe(II)–O2) formation [155,157]. Important to note that analogous thiol N-acetyl penicillamine does not show this kind of reaction with metMb [158]. Furthermore, Alvarez et al. proposed that oxyMb reacts with MCP-SSH in two possible ways [157]. On the one hand, O2 reduction and Mb-Fe(III) formation occur. It has been speculated that this reaction may take place analogously to the well-known interaction of oxyMb with ascorbate or hydroxylamine, accompanied by H2O2 or H2O formation, respectively. On the other hand, the detection of sulfMb in the reaction between oxyMb and MCP-SSH implies that hydropersulfides can also induce sulfheme evolution. Both MCP-SSH and Cys-SSH were found to be suitable for the reduction of ferryl Mb (Mb-Fe(IV)) to metMb (Mb-Fe(III)) in a rapid, yet complex manner. Then metMb was slowly transformed into oxyMb, the final product under aerobic conditions. Furthermore, the sulfMb by-product has also been identified in accordance with other studies [155]. It is worth mentioning that H2S is not capable of the reduction of ferryl Mb to Mb, only forms sulfMb [100] and so the engagement of Cys-SSH in the reduction of hypervalent Mb-Fe(IV) to the Mb-Fe(II) form foreshadows the possible role of per/polysulfides in sulfide mediated Mb regulation [155]. The biological relevance of these results is currently unknown, but further strengthens the concept of sulfide-mediated redox signaling and regulatory functions of per- and polysulfides [157,158].
As it was previously mentioned, non-canonical oxidation of sulfide by hemoglobin is important in RBCs that are lacking mitochondria [108]. In other tissues, the impairment of mitochondrial sulfide oxidation pathways due to ETHE1 deficiency leads to the elevation of tissue sulfide concentration and the development of a disorder called ethylmalonic encephalopathy. Toxic sulfide levels induce severe cytochrome c deficiency particularly in the brain and muscle tissue [159]. Bostelaar and colleagues speculate that the damaged sulfide catabolic pathway facilitates the interactions between sulfide and myoglobin. It is possible that the binding and oxidation of sulfide by myoglobin negatively affects cytochrome c activity and stability and hinders mitochondrial function [126].
4.3. Neuroglobin
Neuroglobin has been identified in 2000 as the first representative of the globin superfamily in the vertebrate nervous system [160]. The discovery boosted plenty of researchers to reveal the biological activity and functions of Ngb. Despite the increased attention and extensive results, the exact functions of Ngb are still unclear, but many theories have emerged about its possible roles like its cytoprotective activity and the related results have been regularly overviewed [[105], [106], [107]].
Oxygen binding and redox properties of human Ngb show similarities to the previously discussed two globins, although to a lesser extent as Hb and Mb relative to each other. Due to the hexa-coordinated nature of the heme prosthetic group in Ngb, molecules have to compete for the sixth coordination site of Fe(II) with the distal His(64), which results in a significant difference in its ligand binding properties compared to the penta-coordinated Hb and Mb. Reduction of the disulfide bridge between cysteine(46) and cysteine(55) of human Ngb diminishes the dissociation rate of His(64). Thus, cellular redox conditions highly affect its ligand binding affinity. Ferrous Ngb is capable of reversible oxygen binding [161] and autoxidation of the Ngb-Fe(II)–O2 complex (oxyNgb) readily occurs, leading to the formation of ferric metNgb. This presumes the existence of a metNgb reductase enzyme in the nervous system [162]. Interaction between H2S and neuroglobin has been investigated since H2S is not only present at high levels in the brain, but it is also considered as a neuromodulator by its involvement in N-methyl-d-aspartate receptor activation in neurons [2,[163], [164], [165]]. Furthermore, the possible formation of metNgb under physiological conditions ensures a place for H2S ligation to the ferric heme center. The study of the interaction between H2S and Ngb is challenging because, in the presence of H2S, it is difficult to preserve the disulfide bridge in the Ngb structure in vitro. The reaction conditions in recent studies assumed the existence of the relevant Cys-SH residues in thiol form, stabilizing the hexa-coordinated structure of Ngb, thus hindering its reactivity towards exogenous H2S. Brittain et al. showed that human metNgb forms a stable Ngb-Fe(III)-SH2 complex both at alkaline (8.0) and acidic (5.5) pH and no complex dissociation has been detected based on UV-VIS spectroscopic data [166]. The biphasic time course of H2S binding to Ngb has been interpreted as a faster H2S concentration-dependent initial step and a second, slower phase, independent of H2S concentration. Considering the dependencies on H2S concentration and the fact that the dissociation of the distal histidine ligand has been shown to be two orders of magnitude faster than the slower reaction in question, it is most likely that the initial time course, that is related to H2S binding, is followed by structural change. Interestingly, low pH is favored for complex formation in contrast to the observations in the case of Mb and Hb. For neuroglobin, it has been proposed that small molecule ligation to the heme center is affected by the pH-dependent ionization properties of the distal histidine residue. From a physiological perspective, it has been speculated that the cytotoxic actions of stroke-induced elevated H2S levels may be alleviated by the activity of metNgb on H2S sink in neurons and retinal cells [166]. However, this observation could not be reproduced in a recent study by Ruetz et al. [167]. The contradiction has been explained by the different H2S donors (NaSH and Na2S) in the two independent studies, based on the work of Greiner et al. [168]. They previously demonstrated that freshly made aqueous NaSH (used by Brittain et al.) solution contains polysulfide impurities in large amounts. Thus, it is conceivable that the actions attributed to H2S actually belong to its oxidized derivatives [168,169]. The involvement of per- and/or polysulfides in the concerned reaction is also supported by the similar changes in the UV-VIS spectrum during the reaction of metNgb with Cys-SSH observed by Ruetz et al. Based on extended spectroscopic and mass spectrometric measurements, Ngb mediated H2S oxidation may occur in vitro, though to a substantially lesser extent. From a mechanistic point of view, it is more likely that an outer-sphere electron transfer reaction arises in the heme pocket involving the Fe(III) center and oxygen. This interaction mainly results in the limited formation of HSO•, HSO4− and HS2O3−. The formation of a Ngb-Fe(III)-SH2 complex has not been completely ruled out, yet it is undoubtedly not dominant. The interaction of metNgb and Cys-SSH leads to the accumulation of the ferrous Ngb form under anaerobic conditions. Investigation of the penta-coordinated metNgb His64Ala mutant revealed that the hexa-coordinated character of the heme center in wild type metNgb is responsible for the different reactivity towards H2S compared to Hb and Mb [167]. It is difficult to predict the physiological significance of these reactions, since intracellular redox conditions affect the redox state of cysteine residues forming disulfide bridge, and thus the affinity of metNgb towards small molecule ligands. However, MetNgb in the dithiol form seems not to be relevant in H2S oxidation.
5. Effect of RSS on mitochondrial electron transport
Heme proteins are involved in oxygen metabolism as members of the mitochondrial electron transport chain (ETC). ETC is composed of five multi-subunit protein complexes, namely: NADH-ubiquinone oxidoreductase (Complex I), succinate dehydrogenase (Complex II), ubiquinone - cytochrome c reductase (Complex III), cytochrome c oxidase (Complex IV or CcO) and ATP synthase (Complex V), embedded within the inner membrane of the mitochondria. Two additional mobile electron carriers are associated with the ETC: ubiquinone (coenzyme Q, abbreviated as Q) located in the inner membrane and a peripheral membrane protein, cytochrome c (Cc). The electron and proton flow in the ETC are depicted in Fig. 6. The citric acid cycle supplies 2 pairs of electrons to Complexes I and II through NADH and FADH2, which are used to reduce Q to ubiquinol (QH2). Complex III then re-oxidizes QH2 and donates one electron at a time to cytochrome c. The latter delivers the electrons to Complex IV where molecular oxygen is reduced to water. The electron flow from Complex I, III and IV results in the simultaneous pumping of 10 protons through the inner membrane to create the proton gradient used by Complex V in the production of ATP (Fig. 6).
Fig. 6.

Mitochondrial electron transport chain. (1: cytosol; 2: outer membrane; 3: intermembrane space; 4: inner membrane; 5: mitochondrial matrix; ADP: adenosine diphosphate; ATP: adenosine triphosphate; CAC: citric acid cycle; Cc: cytochrome c, where subscripts “ox” and “red” refer to the oxidation state of the heme proteine's iron center Fe(III) and Fe(II), respectively; FADH2: dihydroflavine-adenine dinucleotide; NADH: nicotinamide adenine dinucleotide; Q: ubiquinone; QH2 ubiquinol. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Historically, hydrogen sulfide was long known as a toxic substance that can ultimately cause respiratory failure by the inhibition of Complex IV [170,171]. However, the complex biological role of H2S is best demonstrated by the fact that the enzymes of the canonical sulfide oxidation pathway also support the ETC by donating electrons to the mobile electron carriers. Sulfide quinone oxidoreductase catalyzes the two-electron oxidation of H2S and in return the reduction of Q to QH2 [172]. Moreover, the two-electron oxidation of sulfite (SO32−) to sulfate (SO42−) by sulfite oxidase is accompanied by Cc-Fe(III) reduction to Cc-Fe(II) [173]. The combined presence of sulfide catabolism and various ETC-related heme centers in the mitochondria has prompted several studies to investigate their possible interactions.
5.1. Cytochrome c
Cytochromes are redox active proteins in the ETC, classified into four groups (a-d) based on the type of their heme cofactor. Cytochrome c (Cc) contains heme c, which is covalently bound to the polypeptide chain via a thioether bridge with a cysteine (Cys) residue. Histidine and methionine (Met) residues are axially bound to the hexa-coordinated low-spin Fe(III) center. The Fe(III)-Met bond is relatively weak and the substitution of methionine with lysine (Lys) or histidine opens the way to other functions. The His-Fe(III)-Met coordinated cytochrome c has a redox potential of 0.25 V, which enables its electron carrier activity in ETC. Cc also engages in the detoxification of reactive oxygen species which are generated during the normal operation of ETC in healthy cells [174]. Furthermore, it plays a key role in apoptosis through interaction with cardiolipin, a phospholipid component of the mitochondrial membrane [175].
As discussed above, Cc-Fe(III) is reduced to Cc-Fe(II) by Complex III and carries electrons to Complex IV. Sulfide and inorganic polysulfides are also capable of the reduction of Cc-Fe(III) to Cc-Fe(II) in vitro, under both anaerobic and aerobic conditions [171,176,177]. However, the reaction mechanism is far from clear. Considering the redox potentials of S•−, 2H+/H2S (E◦’ = +0.92 V) and Cc-Fe(III)/Cc-Fe(II) (E◦’ = + 0.25 V) couples, the outer-sphere reduction of the heme center is thermodynamically not favorable [108,177]. Inner-sphere reduction could be possible, while sulfide coordination to the heme center could shift the redox potentials and thereby allow the electron transfer [177]. Notwithstanding, the Cc-Fe(III)-HS− complex has not been identified with Cryogenic electrospray ionization - mass spectrometry (Cryo-ESI MS) [176], which method was successfully used to capture heme-Fe(III)-HS− intermediate in the case of myoglobin [126]. So, it is not clarified yet, if the outer- or the inner sphere reduction occurs in Cc the case of Cc reduction by sulfide.
Kinetic measurements on Cc-Fe(III) reduction by sub-stochiometric and stochiometric Na2S revealed an initial lag phase in the process, which may indicate a slow reaction step e.g. ligand exchange between His to H2S. But it was also demonstrated that Cc-Fe(III) reduction is faster with Na2S2 than with Na2S, and the initial lag phase is also missing. To explain the acceleration of the process in the reaction of Cc-Fe(III) with Na2S, another theory has been also discussed. The thermodynamically unfavored outer-sphere electron transfer, accompanied by S•−/HS• formation was proposed, followed by various reactive sulfur species formation. Three possibilities have been discussed concerning the accelerating reactive sulfur species formation: 1) reactive HS2− formation through the dimerization of the accumulated (S•−/HS• radicals; 2) S•−/HS• reaction with O2 may lead to the production of SO2•− a strong reducing agent; and 3) S•−/HS• reaction with HS− may lead to the formation of H2S2•−(E◦’ (HS2−, H+/H2S2•−) = - 1.13 V). But neither theory explains the measured results unequivocally. The accumulation of S•−/HS• is unlikely. The outer-sphere electron transfer between HS2− and the heme center is not probable either, based on the redox potential of the S2•−, H+/HS2− couple (E◦’ = + 0.27 V). However, biphasic oxygen consumption during the reaction of Cc-Fe(III) with excess sulfide was observed, followed by the accumulation of sulfite (HSO3−), sulfite radical (SO3•−), sulfate (HSO4−) and thiosulfate (S2O3−). These results support the idea that electron transfer occurs between SO2•− and Cc-Fe(III) resulting in SO2 and Cc-Fe(II) formation. And the subsequent oxidation of the produced SO2 (in the presence of sulfur donor) could be responsible for the production of the detected derivatives (HSO3−, SO3•−, HSO4−, S2O3−). But the extent of Cc-Fe(III) reduction was found to be independent of the presence of oxygen [176]. In the case of excess sulfide, the reaction rate was 6 times higher under anaerobic than under aerobic conditions, which suggests that oxygen is not essential for sulfide-mediated Cc-Fe(III) reduction. The lower reaction rate under aerobic conditions has been explained by the possibility of parallel, oxygen-dependent, sulfide-consuming reactions [177]. It can be concluded that further experiments are required to explore the mechanism of sulfide-mediated Cc-Fe(III) reduction.
The Cc-Fe(III) reducing ability of glutathione (GSH) and glutathione persulfide (GSSH) has also been compared to that of sulfide, using Na2S as a sulfide donor. GSSH showed a 20-fold faster reduction rate than GSH, and a 5-fold higher reaction rate compared to sulfide. It has been pointed out that GSSH was formed in the reaction of oxidized glutathione (GSSG) with Na2S. Then the GSSH level in the reducing reagent was determined as 30% of the total GSH after 15 min reaction time. However, the reducing effect attributed to GSSH could also result from the activity of other species, considering the complex equilibrium in such systems [178]. Further studies are required to confirm the Cc-Fe(III) reducing ability of protein persulfides.
As mentioned above, the generation of S•−/HS• opens the way for protein persulfidation [103]. In this regard, Cc-Fe(III) induced protein persulfidation has been investigated in vitro [176]. The Cc-Fe(III)/H2S system was proved to be suitable for inducing Cys persulfidation on human serum albumin (HSA) as a model protein, ETHE1 and adenosyltransferase (ATR), which are mitochondrial proteins. Furthermore, recombinant murine procaspase 9, which triggers a protease cascade after activation by apoptotic protease factors in the apoptosome, was also found persulfidated in the presence of Cc. In vitro studies have revealed the possibility that in biological systems, in case of the dysfunction of the canonical sulfide oxidation pathway, Cc may contribute to H2S removal from the mitochondria and H2S may donate electrons to the ETC through the reduction of Cc-Fe(III). Moreover, the Cc-mediated sulfide oxidation pathway may be a possible source of various other reactive sulfur species which can be responsible for protein persulfidation in mitochondria and thereby exert protective, regulatory, or signaling effects.
5.2. Cytochrome c oxidase
Cytochrome c oxidase (CcO) or Complex IV belongs to the terminal heme-copper oxidases superfamily. Terminal oxidases are the last members of the electron transfer chain, they catalyze O2 reduction to water, while "heme-copper" refers to the active center of the protein. As described above, CcO catalyzes the reduction of oxygen in the mitochondria as the final electron transporter of the ETC. CcO is a protein complex and shows extremely high diversity across various organisms, including the number of subunits or the type of its heme group. However, the catalytic core, which is involved in oxidative phosphorylation, consists of the three largest subunits (I-III), regardless of the CcO origin. In mammals, additional ten subunits are tightly bound to the core and play a regulatory role. CcO contains three redox-active metal centers. A binuclear CuA (2CuA) and a hexa-coordinated low spin heme a (Fea) are located in Subunit II, whose primary function is the transport of electrons to the catalytic center. The third redox active metal center is a hetero binuclear center (BNC) consisting of CuB and a penta-coordinated high spin heme a3 (CuB/Fea3), located in Subunit I. BNC and a tyrosine (Tyr) residue represent the catalytic center of CcO, while subunit III is responsible for proton pumping. Mammalian cytochrome c oxidase is often called cytochrome aa3, indicating the related types of heme centers. Redox active centers allow the development of diverse net CcO oxidation states, from the fully oxidized (2CuA(II) Fea(III) Fea3(III) CuB(II)) to the fully reduced (2CuA(I) Fea(II) Fea3(II) CuB(I)) forms [174,179]. As discussed by Moody, the nomenclature of the CcO redox forms is not harmonized in the literature, so here we place special emphasis on consistent wording [180].
More than 95% of the oxygen consumption in mammals is associated with CcO activity and related ATP production. The mechanism of oxygen reduction by CcO has been widely studied, yet the exact process and the nature of the generated intermediates still remain elusive. CcO-mediated O2 reduction is briefly outlined here, in accordance with previous detailed reviews [[180], [181], [182], [183], [184]]. Initially, an oxygen molecule binds to the fully reduced hetero binuclear center (BNC) and CuB(I)/Fea3(II)–O2 is generated. Electron donation from the reduced metal centers and the tyrosine side chain to the oxygen molecule leads to the splitting of the O–O double bond. Subsequent electron transfer from the ETC through 2CuA and heme a to the site of oxygen reduction is accompanied by the protonation of the bound oxygen atoms. This process leads to water formation and release from the active center. In turn, recovery of the Tyr-OH side chain and binding of another oxygen molecule to the reduced CuB(I)/Fea3(II) center occur. During the catalytic cycle, intermediates with partially and fully oxidized CuB/Fea3 centers are involved, in which the Fea3 center exists in ferrous (Fea3(II), ferric (Fea3(III)), or in ferryl (Fea3(IV)) state and CuB in a cuprous (CuB(I)) or cupric (CuB(II)) state.
It should be noted here that studying of isolated CcO is not a straightforward exercise. It is complicated by the fact that during isolation and purification completely oxidized CcO (2CuA(II) Fea(III) Fea3(III) CuB(II)) can be obtained, which shows heterogeneity. In terms of reactivity, “slow” and “fast” forms of fully oxidized CcO can be distinguished. The slow form is not part of the catalytic cycle of CcO turnover and it possesses different spectral and ligand-binding properties than the catalytically active fast species. Presumably, the slow form develops during enzyme purification and storage, i.e. it has no physiological role. The fast form is the catalytically active intermediate which is part of the catalytic cycle of CcO turnover. It reacts fast with inhibitors, but it is metastable and spontaneously transforms to the slow form in vitro [180,185]. Based on the fast and slow form ratio in the isolate, the terms “resting” and “pulsed” characterize the CcO preparations. Resting CcO mainly contains the slow form, while pulsed CcO consists of mostly the fast form. To investigate the catalytic activity of CcO, the resting enzyme (“slow form”) should be converted to the pulsed enzyme (“fast form”) in reduction – re-oxidation cycles. In addition, oxygenated or oxygen-pulsed CcO sample is also distinguished, which contains partially reduced CcO beside the fully oxidized fast form. Oxygenated CcO can be also obtained from the resting enzyme in reduction – re-oxidation cycles, if the catalytic cycle of CcO is activated and catalytic intermediates are generated during the process [180].
Owing to the known inhibitory effect of H2S on CcO activity and thereby on mitochondrial respiration, its interaction with isolated CcO has been intensively studied in vitro since the 1970s, principally by UV-VIS spectroscopy [171,[186], [187], [188], [189], [190]] and electron paramagnetic resonance (EPR) [185,186,[191], [192], [193]]. Important to note that it has been poorly studied whether H2S or HS− is the reactive species in the case of CcO inhibition, thus H2S or sulfide will be used to denote the differently protonated forms in this section. These studies revealed pieces of evidence regarding the characteristic interactions between redox active metal centers (2CuA, Fea, Fea3, CuB) of CcO and H2S. Fea3(III) heme center of CcO shows high affinity towards H2S as expected in the case of penta-coordinated heme proteins. Experimental studies support the possibility of H2S ligation to the Fea3(III) center and electron transfer to Fea3(III) from H2S accompanied by the postulated formation of a Fea3(II)-HS• intermediate [185,187,188]. To the best of our knowledge, no data has been reported on H2S (or other ligands) binding to the other, hexa-coordinated Fea center, presumably due to the fully occupied nature of this heme center [185,191]. Sulfheme formation has not been observed in the case of CcO [187,188]. Importantly, in the binuclear active center, CuB is also capable of H2S binding besides Fea3 and it has been also demonstrated that both redox active metal centers can be reduced by H2S. Based on these findings, H2S appears to be a potent reductant and binding ligand for the enzyme. Considering the H2S oxidation activity of CcO, its direct involvement in H2S metabolism is conceivable. One-electron oxidation of sulfide by CcO can be assumed to be associated with the formation of HS• or related sulfide oxidation products such as elemental sulfur, per-, and polysulfides. Moreover, taking into account the oxygen consumption during sulfide oxidation by CcO, the formation of reactive oxygen species is also an alternative [171]. Still, the detailed characterization of these compounds remains a challenge to date. Investigating the underlying mechanism of the interactions between H2S and CcO is highly demanding, due to the complexity of CcO, the heterogeneity of the isolated enzyme, and the marked dependence on the concentration of H2S - considering the toxic threshold.
In 1984, Hill et al. presented experimental evidence on sulfide-inhibited CcO, where the CuB center was shown to exist in previously reduced form and to bind a hydrosulfide anion (−SH-CuB(I)) [186]. This observation overwrote the previously proposed CcO inhibition mechanisms. They suggested a CcO reduction and inhibition mechanism by high levels of H2S, considering the recent literature and their pioneering experimental results [170,171,187,191,192]. Complex experiments on the interaction of excess sulfide and isolated bovine heart CcO in fully oxidized (2CuA(II) Fea(III) Fea3(III) CuB(II)) state under aerobic conditions revealed that sulfide initially reduces the Fea(III), CuA and CuB centers. Presumably, Fea(III) and CuA reduction take place at the canonical cytochrome c reductive site of CcO and H2S is capable of the direct reduction of CuB(II) center. Complete inhibition of CcO requires 3 mol of H2S per mole of enzyme. It is noteworthy that the enzyme eventually oxidizes all the excess sulfide, which confirms the heterogeneity of isolated CcO and indicates the existence of at least one CcO species with a conformational state that possess different reactivity towards sulfide. The suggested fundamental mechanism of inhibition is shown in Fig. 7a. The first molecule of sulfide reduces CuB(II) to CuB(I) and then a second sulfide equivalent coordinates to this reduced CuB(I) center. The reactivity of the formed intermediate is not known, but a third sulfide molecule is needed for its complete inhibition, which binds to the Fea3(III) center [186].
Fig. 7.

Concentration-dependent interaction of sulfide with the binuclear active center (BNC; Fea3 – CuB) of cytochrome c oxidase (CcO). At high sulfide levels: a) inhibition mechanism [186]; b) nuanced supplemented inhibition mechanism [194]. Low sulfide levels: direct electron donation to the electron transport chain through the BNC of CcO [194]. The exact products of one-electron sulfide oxidation by CcO remained unclear so far, thus HS• evolution indicates not identified sulfide oxidation products (possibly HS• itself).
However, this proposed inhibition mechanism has been nuanced in 2013 by Nicholls and coworkers based on their results [194] and comprehensive evaluation of the relevant data available in the literature [170,171,[186], [187], [188], [189], [190],195] (Fig. 7b). In addition, they extended the mechanism with the possible transfer of the coordinated sulfide between the two metal centers of the mixed valence BNC (Fea3(III)–CuB(I)). On the other hand, they have proposed another possible route in which the first sulfide molecule ligates to the oxidized CuB(II) center and then is transferred to the Fea3(III). The consequent reduction of CuB(I) by sulfide leads to the formation of the mixed valence BNC and inhibition occurs via the coordination of the third sulfide molecule to CuB(I) (or Fea3(III) if ligand transfer occurs between the metal centers before the binding of the third sulfide). It is noteworthy that all mechanisms assume that sulfide initially interacts with CuB(II) in the case of the BNC. From a kinetic point of view, Petersen et al. showed that H2S is a non-competitive inhibitor towards oxygen and cytochrome c with an inhibition constant of 0.2 μM [195].
In contrast, at low sulfide levels, CcO is not initially inhibited. Instead, the intermediate evolution of the catalytic oxygen reduction cycle of CcO implies that H2S donates electrons to CcO (Fig. 7c). Thus, H2S is possibly involved in mitochondrial respiration through direct reaction with redox active metal centers of CcO. It has been proposed that at low sulfide levels two molecules of sulfide bind to the fully oxidized BNC and the reduction of both metal centers leads to the formation of oxygen binding CcO intermediate with fully reduced BNC [194].
In conclusion, the toxicity of H2S at high levels mainly relies on its inhibitory effect on CcO, by coordinatively blocking the binuclear (Fea3-CuB) active center. However, at low levels, H2S is theoretically able to donate electrons to the electron transport chain in three ways: i) through the canonical sulfide oxidation pathway; ii) as a substrate, via direct reduction of the redox active metal centers of CcO and iii) by direct reduction of Cc, the physiological substrate of CcO (Fig. 8.)
Fig. 8.

Possible electron donation to the electron transport chain by H2S. Notation: Cc: cytochrome c, where subscripts “ox” and “red” refer to the oxidation state of the heme proteine's iron center Fe(III) and Fe(II), respectively; CcO: cytochrome c oxidase; ETHE1: ethylmalonic encephalopathy 1 protein; GSH: glutathione; GSSH: glutathione persulfide; Q: ubiquinon; QH2: ubiquinol; SQR: sulfide quinone oxidoreductase; SUOX: sulfite oxidase. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
5.3. Cellular aspects
Investigation of potential sulfide-related interactions with members of ETC in the cellular milieu is rather challenging, but the effort invested in this field has yielded substantial results. Based on these experiments two main phenomena have been attributed to the joint effect of Cc and sulfide: 1) affecting the global persulfidation level [17,176] and 2) anti-apoptotic effect [175,176,196,197]. Furthermore, results of experiments conducted in cellular environments support the idea, that sulfide affects mitochondrial respiration (characterized by the oxygen consumption rate) [176]. Finally, the potential protective effect of exogenous sulfide under hypoxic conditions has been demonstrated in animal models [198,199].
The effect of Cc activity on global protein persulfidation has been studied in cells. Wedmann et al. revealed, that a) under normoxic conditions, slightly less endogenous protein persulfide has been detected in Cc silenced HeLa cells compared to the wild-type cells (WT), using the Cy3-CN-based tag switch method; b) the mitochondria-targeted H2S donor, AP39 induced protein persulfidation has been significantly decreased after silencing Cc; and c) under hypoxic conditions, increased endogenous sulfide levels caused elevated protein persulfidation, which was also diminished by Cc silencing [17]. These data suggest Cc-assisted protein persulfidation in the cellular environment, which was also studied in Complex III and/or Complex IV inhibited yet functional yeast (S. cerevisiae) mitochondria. Inhibition of Complex III resulted in increased protein persulfidation, which is consistent with the hypothesis that suppression of the H2S oxidation pathway by the inhibition of Complex III leads to H2S accumulation and increased persulfidation, presumably induced by Cc. In Complex IV inhibited mitochondria the formation of protein persulfides decreased due to the absence of the re-oxidation path of Cc-Fe(II) by Complex IV [176].
Moreover, the anti-apoptotic effect of sulfide has been linked to Cc-mediated persulfidation. As mentioned above, Cc release to the cytosol activates apoptosis. Cc and deoxyadenosine triphosphate (dATP) binding to the apoptotic protease activating factor-1 enables the formation of the apoptosome, a protein oligomer constructed from seven Apaf-1 monomers. Apoptosome binds and cleaves procaspase-9 and generates active caspase-9. This activation triggers the enzymatic cascade through the subsequent activation of caspase-3 and caspase-7. This process ultimately leads to cell death [175,196]. In this regard, the joint antiapoptotic action of sulfide and Cc has been investigated. Caspase 9 activity has been increased after Cc addition to HeLa cell lysate, but the simultaneous addition of Cc and H2S suppressed the action of Cc [176]. Inhibition of Complex III in HeLa cells with antimycin A treatment also induced apoptosis by blocking mitochondrial respiration. 16 h of antimycin A treatment induced caspase-3 activation, which has been partially suppressed by 2 h of preincubation of the cells with the slow-releasing H2S donor, GYY4137. Additionally, procaspase-9 persulfidation has been also investigated in cells. HeLa cells were treated with GYY4137 before or 2 h after staurosporine-induced apoptosis and the relative amount of procaspase-9 persulfides has been determined after 6 h. Complex experiments showed that procaspase-9 persulfidation increased in the case of joint GYY4137 and staurosporine treatment [176]. In intact Jurkat cells, staurosporine-induced Cc release from the mitochondria [197] and consequent activation of caspase-9 have been partly or completely diminished in 6 h upon fast-releasing (ammonium tetrathiomolybdate) or a slow-releasing H2S donor (GYY4137) treatment, respectively. Based on these evidence, it has been speculated that the antiapoptotic effect of H2S originates from Cc-mediated various reactive other sulfur species generation and subsequent protein persulfidation [176].
The effect of sulfide on mitochondrial respiration has also been studied in cellular environments. The interactions of sulfide with Complex III inhibited HepG2 and HT29 cells have been also investigated. Complex III inhibition by antimycin A resulted in an almost complete reduction of oxygen consumption rate (OCR) in both cell types (to 2.5% and 5% respectively). While the addition of a moderate amount of H2S increased OCR by 30% (HepG2) and 95% (HT29) in comparison to Complex III inhibited cells. It has been speculated that the complete reduction of OCR by Complex IV inhibition with KCN in Complex III inhibited cells, supports the concept that the increment of OCR induced by exogenous H2S is linked to mitochondrial respiration [176]. Although, based on the experimental design and the discussions, it appears that Complex IV inhibition was implemented without the addition of exogenous H2S, which was responsible for the OCR increase. Thus, the linkage between sulfide-induced oxygen consumption and mitochondrial respiration in these experiments remains questionable.
As it was mentioned above, exogenously given hydrogen sulfide interacts with Complex IV and cytochrome c, possibly altering cellular respiration and the metabolic rate [171]. The physiological relevance of these reactions apart from toxicity has been demonstrated by Blackstone and colleagues. Exposure to 80 ppm hydrogen sulfide induced a suspended animation-like state in mice. The metabolic rate and the core body temperature of the animals dropped by 90% after the 6h exposure. These effects were completely reversible without any short, or long-term health complications or changes in function or behavior. It was implicated that lowering the metabolic demand in case of trauma can be used to reduce physiological damage [198]. Further investigation revealed that exposure to 150 ppm sulfide induced a suspended animation-like state in 20 min without lasting physiological damage and protected mice from subsequent exposure to severe hypoxia (5% O2). It was also speculated however that inhibition of cyclooxygenase by sulfide plays a major part in these protective actions [198,199].
6. Interactions of sulfide with heme peroxidases
Heme peroxidases represent a large group of heme proteins responsible for the oxidation of several biological substrates using endogenously generated H2O2 as an oxidant. Heme peroxidases, as their names imply, contain a heme prosthetic group in their active site. Their active ferric form contains penta-coordinated Fe(III) ions. The sixth coordination site of the metal center is occupied by a water molecule at the distal position (similarly to Hb or Mb), which is replaced by H2O2 upon peroxidase activation [200]. A novel classification of heme peroxidases was recently suggested based on structural and functional similarities, rather than their phylogenetic origin [201]. This gave place for the definition of the peroxidase-cyclooxygenase superfamily of proteins, comprising myeloperoxidase (MPO), eosinophil peroxidase (EPO), lactoperoxidase (LPO), and thyroid peroxidase (TPO), among others, previously termed mammalian peroxidases [202]. Other sources may refer to this group as XPO or haloperoxidase enzymes. Although, the main characteristic of the prosthetic group in heme peroxidases is very similar, an important structural difference is that in the case of LPO, EPO, and TPO two ester bonds attach the heme group to the protein. By contrast, in the MPO structure, an additional thioether sulfonium bond also exists between the heme group and the protein frame. This phenomenon may also contribute to differences in reactivity. In general, these proteins catalyze either one- or two-electron oxidation reactions, in the peroxidase cycle and halogenation cycle respectively, as illustrated in Fig. 9. In both cycles, native peroxidase (Por-Fe(III)) reduces H2O2 and Compound I is formed during water elimination. In the peroxidative cycle, firstly, Compound I oxidizes an appropriate substrate molecule (AH2) accompanied by radical (•AH) release and Compound II formation. In a second one-electron oxidation reaction, Compound II oxidizes another substrate molecule while the native form of the enzyme is restored. In the halogenation cycle, Compound I oxidizes an appropriate halogenic ion (X−) while hypohalous acid (HOX) is formed and the native enzyme is reconstituted. Generally speaking, Compound I and Compound II forms of the peroxidases are involved in their catalytic turnover in the peroxidase and the halogenation cycle [200,[202], [203], [204], [205], [206]]. Important to note that alternative turnover pathways may also occur in the case of all peroxidases (MPO, LPO, EPO, TPO), involving their characteristic and/or alternative substrates, their ferrous form and/or the Compound I, Compound II and Compound III intermediates of the enzymes. Moreover, interconversion between the Compound II and III forms of the peroxidases is also possible (not shown in Fig. 9.) [207,208].
Fig. 9.

General and sulfide related reaction mechanism of peroxidases. Proposed general mechanism of the peroxidase cycle (reactions (1), (2a), (3); green dotted arrows); Proposed general mechanism of the halogenation cycle (reactions (1), (4); grey dashed arrows). Alternative turnover mechanisms of myeloperoxidase (MPO) by H2S (facilitated by superoxide dismutase (SOD) or ascorbate) suggested by Garai et al. [103] (reaction (1), (2b), (5), (6), (7) and reaction (8), (9), (6), (7); orange dash-dotted arrows); Reaction of lactoperoxidase-Compound II (LPO-Compound II) and H2S (reactions (10 …); purple dash dot arrows) [209].) AH2 and •AH denote organic substrates and their radical-type oxidation products and X− refers to halides and pseudohalides (Cl−, Br−, I−, SCN−), all naturally occurring in humans. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The generated radical species (•AH) and hypohalous acids (HOX) are strong oxidants effectively used by mammalian organisms to kill pathogens, therefore these peroxidases are vital contributors to the immune system. The substrate specificity and redox characteristics of mammalian heme peroxidases, and their significance in health and disease has been previously reviewed [200,202,203,205,206,210]. Actually, metHb and metMb also possess peroxidase activity, but this has primarily pathological relevance. In their cellular environment, the abundance of metHb is relatively low and metMb is negligible, due to their enzymatically controlled recovery to the active ferrous form. However, in the case of pathological conditions, where Hb or Mb are released from their cellular milieu, they can cause serious damage as a result of their peroxidase activity [211]. Moreover, as mentioned before, the peroxidase activity of Cc plays a key role in apoptosis [175]. These latter enzymes have been defined as pseudo-peroxidases, while their primer function does not rely on their peroxidase activity [200].
6.1. Myeloperoxidase
Myeloperoxidase (MPO) is a unique member of the peroxidase-cyclooxygenase superfamily, the most abundant enzyme of neutrophil granulocytes located in the primary granules [103,212,213]. One of the main enzymatic functions of MPO is the halogenation reaction (Fig. 9), that mainly yields hypochlorous acid from chloride ions. The production of hypochlorous acid mainly happens in the phagosome for the elimination of bacteria and other pathogens. In the peroxidase cycle of the enzyme various biological substrates are turned over, including tyrosine to generate tyrosyl radicals and thus contributing to protein cross-linking [214,215]. It was demonstrated that hydrogen sulfide showed anti-inflammatory properties in various conditions, such as rheumatoid arthritis [216], reperfusion injury [217] or atherosclerosis [146]. The detrimental effect of MPO in these particular conditions have also been confirmed [[218], [219], [220]], therefore our group hypothesized that the observed protective effects of H2S may be attributed to its local interactions with MPO [213].
Spectroscopic evidence revealed that under anaerobic conditions in the absence of H2O2, sulfide coordinates to ferric MPO-Fe(III) to form a low-spin MPO-Fe(III)–hydrosulfido complex that is then slowly reduced to the MPO-Fe(II)–H2S complex (Fig. 9). In case of Compound I and II forms of MPO the detailed kinetic analysis by conventional and sequential stopped-flow spectroscopy and peroxidase assays showed that both intermediates rapidly oxidize hydrogen sulfide ultimately yielding ferrous MPO-Fe(II) and the MPO-Fe(II)–H2S complex, which was also generated in the absence of peroxide (Fig. 9). The high second-order rate constants with Compound I and II imply that sulfide may act as a substrate of MPO and sulfide radicals including thiyl radicals might be generated. The coordination of hydrogen sulfide to MPO effectively inhibits the peroxidase activity with an LC50 value of ∼1 μM, in both steady-state enzyme kinetic experiments and in biological samples (rat tissue and human neutrophil homogenates). This inhibitory effect was fully reversible with a dilution step in our experimental settings, however, under physiological conditions, the sulfide radicals generated by Compound I and II could damage the protein structure thus a loss of activity occurs [213].
In the presence of molecular oxygen, we gathered further knowledge of the mechanism governing the MPO/H2O2/sulfide/O2 system and corroborated the potential implications of these interactions in sulfide-mediated redox signaling as well as antioxidant protection [103]. Our data showed that the reaction of ferric MPO with H2S under aerobic conditions brings about the formation of an MPO redox state with a distinct absorption peak at 625 nm (thereby named "the 625 nm species"). A detailed discussion is given in [103] whether this species could be assigned as a sulfheme intermediate or the Compound III enzyme form. On the other hand, MPO did not exhibit the typical characteristics of a sulfheme intermediate when reacted with sulfide. The most compelling argument to support this notion was that the 625 nm species (and the corresponding UV–vis band) appeared even in the absence of sulfide when MPO was reacted with hydralazine with similar kinetic and spectroscopic features. Several lines of evidence confirmed that the 625 nm species is indeed Compound III, which is a resonance form of oxygenated ferrous MPO (MPO-Fe(II)–O2) and ferric-superoxide-like complex (MPO-Fe(III)–O2•−).
The formation of Compound III in the presence of H2S is a key phenomenon that distinguishes MPO from related heme peroxidases. According to our findings, the generation of Compound III gives rise to the superoxide dismutase (SOD) or ascorbate-facilitated turnover of MPO and subsequent oxidation of sulfide, in the absence of initial H2O2. SOD and ascorbate produce ferric MPO and H2O2 by dismutation and reduction, respectively, thus initiating the normal peroxidase cycle and processing sulfide, as described above. We showed that this sulfide oxidation leads to the slow release of sulfane sulfur species (HS•) from the cycle promoting protein persulfidation [103]. We believe that this discovery may have important implications in persulfide-mediated regulation of redox signaling events [27] as well as in the protection against oxidative stress. The cytoprotective capacity of protein persulfides based on their increased nucleophilicity has been hypothesized earlier [30,168,221] and we recently reported detailed experimental evidence supporting this concept [80], as discussed above.
Unlike other related peroxidases, MPO has unique structural features that prevent the formation of sulfheme and ultimately the irreversible inactivation of the enzyme. The main reason is illustrated in [103], namely that related peroxidases covalently bind the heme cofactor to the protein backbone via 2 ester groups with aspartate and glutamate residues (which makes the structure more rigid than in globins), whilst MPO has an additional sulfonium ion (R1R2R3-S+) linkage to the respective heme by its methionine residue 243 [202]. Consequently, the heme pocket of MPO is distorted into a bow-shaped conformation and displaces the iron center out-of-plane towards the proximal histidine, significantly changing the catalytic properties of the enzyme. The single mutation of methionine residue 243 to valine (Met243Val mutant) corroborated the significance of this sulfonium linkage. Compound III formation was prevented in Met243Val mutant, which hindered SOD or ascorbate-facilitated enzyme turnover and the inhibition by sulfide was complete and irreversible.
The effect of hydrogen sulfide treatment on MPO activity in vivo has been extensively studied in various conditions. In mouse model of burn injury and porcine model of myocardial ischemia/reperfusion injury, MPO treatment with hydrogen sulfide significantly lowered MPO activity in the affected tissues. These results suggest that inhibition of MPO could be an important part of the anti-inflammatory functions of sulfide [222,223].
6.2. Lactoperoxidase
Lactoperoxidase (LPO) is present in mucosal surfaces, in exocrine secretions such as milk, tears, and saliva, as well as in nasal and lung airway fluids. The two main physiological substrates of the LPO halogenation cycle (Fig. 9) are iodide and thiocyanate. Due to the significantly lower occurrence of iodide in the body compared to thiocyanate, LPO catalyzed formation of HOSCN and its antimicrobial effect is dominant [200,224]. The interactions of LPO and sulfide have been investigated by Nakamura et al. [209]. Their primary motivation was to thoroughly study the proposed inhibitory effect of an antithyroid drug, 2-mercapto-1-methylimidazole (MMI) on LPO activity. They investigated the interactions of LPO-Compound II (LPO-Fe(IV) O) with MMI and with sulfhydryl model substances, namely Na2S, cysteine (Cys), and DTT, to reveal the role of the thiol functional group in the inhibition mechanism. It has been concluded that each sulfhydryl compounds react with LPO-Fe(IV) O along two parallel reaction pathways with a different probability. H2S preferably forms ferrous sulfheme species (sulfLPO(FeII)) which is slowly oxidized to its ferric sulfLPO-Fe(III) form. Cys and sulfide show similar properties in this regard, however, the ferric cysteine LPO adduct was not observed. MMI and DTT are both capable of ferrous and ferric adduct formation, although, they preferably participate in one-electron reduction of LPO-Fe(IV) O to its native LPO-Fe(III) form. Interestingly, in the case of excess sulfide, the characteristic spectrum of LPO-Fe(IV) O transformed into a native LPO-Fe(III) like spectrum accompanied by recovered LPO activity [209]. These results indicate the possible biological relevance of LPO interactions with endogenous sulfide.
6.3. Thyroid peroxidase
Thyroid peroxidase (TPO) is a membrane-bound enzyme found in the thyroid follicle cells. As a peroxidase, TPO mainly reacts with iodide resulting in HOI formation. Moreover, TPO is essential in the synthesis of thyroid gland hormones [200,225]. Regarding sulfide biochemistry, Zhao et al. showed that H2S plays important role in the protection of thyroid functions. Among others, they demonstrated that NaSH treatment of human primary thyrocytes enhances the expression of molecules that are essential for thyroid hormone synthesis and secretion. Moreover, TPO displayed increased activity upon NaSH treatment, but the exact mechanism is unknown [226]. TPO inactivation by MMI has been also proposed similarly to the case of LPO. The formation of TPO-MMI adduct likely occurs with the involvement of the TPO-Compound II intermediate [227]. The TPO-MMI adduct shows similar spectral properties that the LPO-sulfhydryl adducts and sulfmyoglobin [225]. In light of these findings, interactions of TPO with sulfide or other sulfhydryl compounds are probable and could be a fascinating research area in the future.
6.4. Eosinophil peroxidase
Eosinophil peroxidase (EPO or EPX) is an enzyme of the innate immune cells of humans and mammals and it is concentrated in the secretory granules of eosinophils. Under normal conditions, its main action is the catalysis of bromide and thiocyanide oxidation to HOBr and HOSCN, respectively [200,228]. However, interactions of EPO with sulfide or polysulfides have not been studied yet, but it has been shown that EPO is capable of catalyzing SO32− oxidation to SO3•− radicals in the presence of H2O2 in the peroxidative cycle. Subsequent reaction of SO3•− with oxygen leads to the formation of –O3SOO• which can generate SO4•− radicals in the presence of SO3−. These radicals are involved in protein oxidation and may cause oxidative damage [229]. It is important to note that sulfite interaction with heme peroxidases and heme globins is a known phenomenon, but its detailed discussion is out of the scope of the current review. Maiti et al. currently provided an up-to-date summary on this topic [230]. Due to the close structural and functional similarities of MPO, LPO, TPO, and EPO, the biological relevance of interactions between EPO and sulfide or related compounds cannot be ruled out.
7. Interactions of reactive sulfur species with antioxidant metalloproteins
In the previous sections we discussed how sulfide and other reactive sulfur species affect fundamental redox-related life events, such as the delivery of O2, cellular respiration, and immune defense by strong oxidizing agents. The accumulation of oxidizing power may lead to a state called oxidative stress, defined as impaired redox balance in favor of oxidants, negatively affecting redox signaling and causing molecular damage [231]. Oxidative stress has been associated with a large number of pathological conditions including aging, neurodegenerative processes, diabetes, cancer, brain damage, and infarction [[232], [233], [234]], therefore counteracting its harmful effects and the maintenance of redox homeostasis is crucial to our health.
The initial cause of oxidative stress is an elevated level of ROS, especially superoxide (O2•−) and hydrogen peroxide (H2O2). Superoxide is endogenously generated by several sources including electron leakage from the mitochondrial ETC, the activities of NADPH oxidase (NOX) and dual oxidase (DUOX) enzyme clusters or autoxidation of hemoglobin, and may arise from external factors such as radiation or drugs [235,236]. H2O2 is produced by superoxide dismutases (SODs) in eq. (3). Certain NADPH oxidases are capable of further reduction of superoxide to hydrogen peroxide by donation of a second electron [232].
| 2 O2•−+ 2 H+ → H2O2 + O2 | (3) |
Superoxide and hydrogen peroxide do not pose an immediate threat to living cells, given the kinetic constraint of their direct reactions with biomolecules [236]. Moreover, H2O2 is widely recognized today as a signaling molecule, playing a substantial role in redox regulation, i.e. the modulation of thiol enzymes by posttranslational modifications [233,[237], [238], [239]]. However, secondary radicals and more potent oxidants are derived from O2•− and H2O2 which can inflict serious damage on cells. Transition metal ions catalyze the formation of harsh oxidants, such as hydroxyl radicals (•OH) in Fenton-type reactions (eq. (4)). It was also shown in bacterial systems, that superoxide further supports this type of chemistry by disruption of iron-sulfur clusters and subsequent iron release [235].
| H2O2 + Fe2+ → OH− + Fe3+ + •OH | (4) |
The hydroxyl radical is a universal oxidant reacting with all biological macromolecules, leading to alkoxyl radicals (RO•), peroxyl radicals (ROO•), organic hydroperoxides (ROOH) and corresponding nucleic acid, protein and lipid oxidation [232,236]. Furthermore, hydrogen peroxide and/or superoxide promote the formation of other reactive species, such as peroxynitrite (ONOO−) (eq. (5)) and related RNS, peroxynitrous acid (ONOOH) and nitrogen dioxide (•NO2), strong and unspecific oxidants themselves [232,240].
| O2•−+ •NO → ONOO− | (5) |
As discussed above, the halogenation cycles of mammalian heme peroxidases produce hypohalogenic acids (HOX) (Fig. 9), creating a harsh oxidizing environment to destroy pathogens. Although highly useful for immune defense, the accumulation of these species is harmful to healthy tissue as well. These examples well demonstrate how and why elevated levels of H2O2 and O2•− may lead to serious oxidative stress and become a dangerous biological risk.
It is noteworthy that extracellular hemoglobin can also be a significant source of oxidative stress. Hb may appear in the extracellular matrix by the destruction of RBCs or exogenous infusion of hemoglobin-based oxygen carriers (HBOCs). Inside healthy RBCs, Hb oxidation state is under strict enzymatic control and ROS are neutralized by effective antioxidant systems (see below). However, as the extracellular space lacks these machineries, Hb-Fe(II) may induce oxidative stress in several ways. Its autoxidation to Hb-Fe(III) produces O2•− as shown in Fig. 5. Oxidation of ferrous Hb by H2O2 may result in a Fenton-type reaction (eq. (4)) or in the formation of an unstable ferryl Hb (Hb-Fe(IV) O) species. The latter subsequently decomposes into heme degradation products and free iron ions, accompanied by further ROS generation. Finally, the reaction of H2O2 or lipid peroxidases with Hb-Fe(III) may generate ferryl Hb and/or highly reactive oxyferryl Hb radical (•Hb-Fe(IV) O). However, haptoglobin (Hp) and hemopexin (Hpx) are proteins responsible for the binding of extracellular Hb and free heme, respectively, counteracting their deleterious effects. These Hb-related phenomena can be associated with many pathophysiological conditions, if the elevated levels of Hb and free heme exceed the binding capacities of Hp and Hpx [134,241,242]. As discussed above, sulfide exerts a protective effect against such pathological events through its interactions with Hb derivatives [[145], [146], [147], [148]].
In humans, numerous enzymatic and non-enzymatic systems operate to overcome excess oxidation and/or eliminate detrimental oxidants. Metalloproteins are widely represented in antioxidant protection. Superoxide is processed by 3 isoforms of superoxide dismutases in humans, all three contain transition metal centers [243]. H2O2 arising from the dismutation reaction (eq. (3)) is captured by three groups of proteins, peroxiredoxins (Prxs), catalase (CAT), and glutathione peroxidases (Gpxs), of which catalase belongs to the heme protein family [236]. Reported interactions between SODs and catalase with reactive sulfur species are discussed in more detail below. It should be noted that the term "antioxidant proteins" is not a strictly defined group. In a broader sense, the discussed heme peroxidases (MPO, LPO, EPO, and TPO) can be considered antioxidant proteins as well, since H2O2 is consumed in their catalytic cycle [233].
7.1. Superoxide dismutase
Superoxide (O2•−) is an abundant reactive oxygen species and a major player in oxidative stress. Superoxide dismutases (SODs) are a group of crucial antioxidant systems that provide cellular defense by catalyzing eq. (3) [244,245].
Mammals have three isoforms of SODs: cytoplasmic SOD1, mitochondrial SOD2, and extracellular SOD3, allowing compartmentalized redox signaling [246]. SOD1 and 3 contain copper and zinc, while SOD2 is a manganese protein. Cu/ZnSODs are composed of a 32 kDa homodimer of two identical subunits, consisting of a Greek key beta-barrel composed of eight antiparallel β-strands [247]. The Cu and Zn ions are positioned in the active site channel of the protein, which is formed by two other major external loops outside of the beta-barrel. Cu plays a catalytic role and is bound by four histidines, His46, 48, 63, and 120, in a distorted tetrahedral geometry. The zinc ion, which is bound by His63, 71, 80 and Asp83 provides structural stability. His63 bridges the two metal centers [248]. This scaffold supports dimer or tetramer formation, as well as electrostatic guidance and electron transfer at the catalytic site.
SOD-catalyzed dismutation takes place in two steps. The first half-reaction begins with the substrate binding to Cu(II), which is then reduced to Cu(I), while O2•− is oxidized to molecular oxygen (eq. (6)):
| Cu2+-SOD + O2•−→ Cu+-SOD + O2 | (6) |
This results in the cleavage of the bond, bridging the Cu ion to His63, leaving this histidine residue protonated. In the second half reaction, a proton from His63 and an electron from Cu(I) are donated to another superoxide equivalent, resulting in the formation of hydrogen peroxide, whereas Cu(I) is oxidized to Cu(II) (eq. (7)). The copper bound to the bridging histidine is then restored [249]:
| Cu+-SOD + O2•−+ 2H+ → Cu2+-SOD + H2O2 | (7) |
MnSOD or SOD2 is composed of a 96 kDa homotetramer and is localized in the mitochondrial matrix [250]. Its Mn(III) ion at the active site catalyzes the disproportionation of superoxide to oxygen and hydrogen peroxide in two half-reactions (eqs. (8), (9))), in a similar fashion as Cu/ZnSODs [251]:
| Mn3+-SOD + O2•−→ Mn2+-SOD + O2 | (8) |
| Mn2+-SOD + O2•− + 2H+ → Mn3+-SOD + H2O2 | (9) |
Since 90% of cellular ROS are generated in the mitochondria and mitochondrial DNA is particularly susceptible to oxidative damage, this catalytic activity is critically required for antioxidant protection.
Early studies by Searcy et al. indicated that the combination of sulfide and bovine erythrocyte Cu/ZnSOD resulted in a two-fold increase of the superoxide scavenging activity [252]. Their spectral and EPR observations suggested that the effector species is HS−, which directly interacts with the catalytic site. Stoichiometric studies suggested that the hydrosulfide anion is a substrate for SOD, it can rapidly and tightly bind to Cu(II), forming a Cu(II)-HS– complex before the appearance of superoxide, as shown by optical and EPR spectra.
The products formed during the enzymatic turnover were H2O2 and sulfane sulfur, such as elemental sulfur or polysulfide. This suggests that sulfide detoxification is a second possible role of SOD in addition to O2•− scavenging via the proposed reaction (eq. (10)):
| (10) |
Sulfide was also found to increase the activities of Cu/Zn and MnSODs in cardiomyocytes under ischemia/reperfusion conditions [253]. Further studies indicated that the enhanced activity of MnSOD is a result of its upregulated expression, but Cu/ZnSOD expression was not affected by hydrogen sulfide. The direct interaction between Cu/ZnSOD and H2S was proposed and confirmed by isothermal titration calorimetry (ITC) assays, where heat was released upon the interaction of Cu/ZnSOD with H2S, suggesting that sulfide can bind to the enzyme and allosterically activate it.
The production of various reactive sulfur species by SOD through the metabolism of hydrogen sulfide was also demonstrated by Olson et al. [254]. However, they obtained different results during the investigation of the interaction between Cu/ZnSOD and MnSOD with H2S. According to their findings, aerobic addition of Cu/ZnSOD to H2S resulted in rapid sulfide consumption, which was proportional to the concentration of the enzyme. Using the polysulfide-specific fluorescent probe SSP4, they corroborated the formation of polysulfides by SOD-mediated metabolism of H2S. This polysulfide production was also found to be dependent on SOD concentration over a range of H2S concentrations, while hypoxic studies indicated that molecular oxygen is also required for this process. Although Searcy et al. proposed increased SOD activity in the presence of sulfide, Olson et al. demonstrated the inhibitory effect of H2S on SOD oxidation above 750 μM, which was found to be complete at 1.75 mM [252,254]. They also obtained different results during the experiments performed to determine whether H2S or HS− was the preferred substrate for SOD. Based on their findings, they not only suggested that the enzyme preferentially reacts with dissolved H2S rather than the hydrosulfide anion, but paradoxically, HS− was found to directly inhibit SOD oxidation of H2S.
Regarding the nature of the polysulfides produced, persulfide (H2S2) was the major species detected but longer-chain polysulfides (H2S3 and H2S5) were also identified, which could have been the result of persulfide recombination during the detection protocol [11]. These processes appeared to be dependent on both SOD and O2 concentrations; however, the longer-chain polysulfides produced were not further metabolized by the enzyme. It is clearly stated that additional studies are required to identify the mechanisms involved but the following steps were proposed to explain the observations (eq. (11), (12), (13), (14), (15))):
| Cu2+-SOD + H2S → HS•Cu+-SOD + H+ | (11) |
| HS•Cu+-SOD → HS• + Cu +-SOD | (12) |
| 2 HS• →H2S2 | (13) |
| Cu+-SOD + O2 → Cu2+-SOD + O2•– | (14) |
| 2 Cu2+-SOD + 2 O2•– + 2 H+ → Cu2+-SOD + O2 + H2O2 | (15) |
Experiments performed with MnSOD suggested similar sulfide/sulfur metabolism to that of Cu/ZnSOD, which might be especially critical in dealing with mitochondrial hydrogen sulfide. Further experiments indicated that polysulfide production by SOD oxidation of H2S is not only regulated by H2S itself but also by its metabolites, suggesting that their target is not the reactive Cu/Zn motif, but rather SOD cysteines.
Although the biological relevance of H2S metabolism by SOD remains to be clarified, numerous studies have suggested that sulfide exerts a protective role by enhancing the activity of antioxidant enzymes, which results in decreased ROS levels. Both activation and inhibition appear to be considerable, since impaired SOD activity was found to be directly linked to neurodegenerative diseases, such as familial amyotrophic lateral sclerosis, a lethal neurodegenerative disorder [255]. Furthermore, an in vivo study demonstrated the fundamental mitochondrial function of MnSOD, as the lack of the enzyme lead to the accumulation of hepatic lipids, mitochondrial defects, dilated cardiomyopathy as well as early neonatal death [256,257].
These results not only highlight the importance of adequate SOD functions in biological systems but also suggest that SOD and its mimetics may have therapeutic potential in combating oxidative stress and associated diseases.
7.2. Catalase
Catalase is one of the most ancient and well-studied antioxidant enzymes, responsible for the breakdown of H2O2 in eq. (16). Technically, eq. (16) is also a disproportionation or otherwise called dismutation reaction, yet the term "dismutation" is traditionally used for SOD activity (eq. (3)) in biochemical literature.
| 2 H2O2 → 2 H2O + O2 | (16) |
Catalases are present in most aerobic cells and possess the highest turnover rate for H2O2 among antioxidant enzymes. Human and mammalian catalases belong to the group of monofunctional heme catalases or so-called ‘"true catalases". Additionally, two other types of catalases are distinguished based on their functions: catalase-peroxidases which are heme proteins, and non-heme catalases, which have manganese in their active center. The current review focuses on the first group and the term catalases (CATs) will be used to denote monofunctional heme catalases. Typically, CATs are tetrameric proteins and each identical subunit contains a heme prosthetic group. The iron center of the native enzyme is in the ferric form of the native enzyme. In its catalytic cycle, CAT-Fe(III) reacts with a H2O2 molecule in a two-electron oxidation reaction, resulting in Compound I and water formation. Recovery of the native enzyme occurs in a two-electron reduction reaction between Compound I and H2O2 accompanied by water and oxygen release (Fig. 10) [243].
Fig. 10.

Canonical catalytic cycle of monofunctional heme catalases.
In contrast to heme peroxidases, Compound II is not a part of the catalytic cycle of CAT. Although at low H2O2 concentrations, CAT may be trapped in Compound I form and in the presence of electron donors, it can be reduced to Compound II in a one-electron reaction. It is a temporarily inactive state of the enzyme which can be further reduced to the native form only in slow reactions [258].
Early studies suggested the inhibition of catalase by sulfide. It has been proposed that inhibition occurs in at least two possible ways [259]. On the one hand, reversible H2S ligation to the Fe(III) center of CAT results in activity loss. On the other hand, the formation of sulfcatalase (sulfCAT) may also occur, which is a catalytically inactive form of the enzyme. SulfCAT seems to be less stable than other sulfheme proteins and can be regenerated by oxidizing agents e.g. by oxygen [101]. CAT inactivation by biologically relevant thiol compounds (RSHs), namely cysteine (Cys-SH), homocysteine (HCys-SH), and glutathione (GSH) has also been observed in vitro. Although the IC50 values significantly exceeded normal physiological levels of these thiols. Interestingly, under mimicked pathological conditions (in the presence of iron), physiological concentrations of Cys-SH and GSH led to reversible Compound II formation, while HCys induced sulfCAT generation, in the absence of sulfide. In the proposed mechanism of HCys mediated sulfCAT formation, an initial S-oxygenation occurs by direct oxygen transfer from Compound II (Por-Fe(IV) O) to HCys-SH. In further steps, the interaction of HCys-SOH with the ferric heme center (Por-Fe(III)) results in the incorporation of the sulfur atom to one of the pyrrol rings of the porphyrin frame accompanied by stable sulfonium ion formation. The stable sulfCAT-Fe(III) species is generated in following reactions.
Interestingly, IC50 has been dropped into the physiological/pathological range of these RSHs in the presence of iron in the reaction mixture. While perturbation of transition metal (e.g. iron) homeostasis is characteristic of various forms of cancer and neurodegenerative diseases, it was suggested that RSH and transition metal ions may mediate catalase inhibition under these conditions.
As mentioned above, catalase is an ancient enzyme dating back to when sulfur prevailed over oxygen in the atmosphere. A detailed study was carried out by Olson et al. based on the premise that RSS predate ROS, therefore catalase might be able to metabolize the former to some extent [260]. Indeed, it was found that catalase could use sulfur species as substrates in an oxygen-dependent manner. In the presence of oxygen, catalase removed mixed polysulfides and H2S from buffered solution. The ultimate products remain to be identified. On the other hand, in hypoxia, catalase could release H2S from various sulfur-containing molecules, DTT, garlic oil, diallyl-trisulfide, and thioredoxin. NADPH promoted sulfide cleavage from thioredoxin (Trx), which is a major antioxidant protein, so this reaction may have important implications in redox regulation. The authors argue that increased H2S production by catalase under hypoxia may be beneficial to the kidneys in case of ischemia-reperfusion injury. The exact mechanisms of sulfide oxidation or release by catalase are not discussed in the study, although it is shown that CO inhibits the reductase function, therefore the heme iron is involved in the process. It is also concluded that H2S may be the preferred substrate over HS− anion, and the latter may inhibit sulfide metabolism by catalase. The paper mentions the inhibitory effect of H2S on catalase activity, in a non-canonical oxidase reaction (the oxidation of dichlorofluorescein). According to Olson et al., their findings describe the first known reductase function of catalase, which may shift its notion from classical antioxidant towards a redox-regulatory enzyme, intricately involved in hypoxia response.
It has been previously suggested that H2S is directly involved in antioxidant protection by chemically reducing the above-mentioned oxidizing species. However, kinetic considerations raised doubts about the physiological relevance of this pathway, as we discussed in [80]. First, the dedicated peroxidases and catalase are highly abundant proteins and react with H2O2 with rate constants of ∼107 M−1s−1 order of magnitude [236]. On the other hand, the concentration of free sulfide falls in the nanomolar range, quite low compared to other biological reductants, such as Cys-SH or GSH. Finally, the rate constants of the reactions of H2S with the oxidants discussed above (O2•−, H2O2, peroxynitrite etc.) do not justify the importance of their direct elimination by sulfide. Consequently, the reported protective effects of H2S should be implemented by indirect means. The interactions of H2S and other RSS with metalloproteins presented in the current review may contribute to the protection against oxidative stress in multiple ways. From a protective point of view, the effect of the inhibition of MPO and potentially other heme peroxidases is twofold. The production of the harsh oxidizing agent, hypochlorite and the highly reactive ferryl intermediates is diminished by sulfide and the H2O2 independent turnover of sulfide releases sulfane sulfur in the process, aiding protein persulfidation. We and others recently demonstrated the cytoprotective capacity of protein persulfidation [30,80,82]. A persulfide group may act as a temporary switch under elevated oxidative conditions, which are reverted to the reduced form by the NADPH-dependent Trx and GSH disulfide reductase systems when the stress is over [16,80,261]. Furthermore, increased SOD activity induced by H2S is a contributing factor to antioxidant protection, due to diminished ROS production [252,253]. Although catalase inhibition by sulfCAT formation might counteract the canonical antioxidant capacity of the enzyme, catalase-mediated various RSS production under hypoxia is an intriguing aspect of alleviating ischemia reperfusion-related insults [260]. Altogether, the presented findings support the beneficial effects of reactive sulfur species on cellular health. Future endeavors in hydrogen sulfide-based drug development aim to exploit the cytoprotective potential of sulfide and derivatives in therapeutics of redox diseases.
8. Future directions
The family of reactive sulfur species is widely recognized as important regulatory and signaling molecules in human physiology. Part of their bioactivity may be manifested via interactions with metalloproteins, with significant implications in health and disease. Metalloproteins and metalloenzymes are essential pillars of oxygen-based life, therefore modulation of their activities by RSS influences vital physiological functions, such as respiration or immune defense. The interplay between bioinorganic chemistry and hydrogen sulfide research is a dynamically growing area of redox biology, yet there are plenty of obstacles to overcome. First, the mere number and variety of metalloproteins pose a challenge, given that one-third of the human proteome contains one or more metal centers. A helpful approach may be the classification of proteins and investigation of the effect of RSS on frequent motifs, as was done here with heme groups. Reactive sulfur species also compose a diverse and not tightly defined family with versatile biochemical properties, thus it is important to keep a meticulous and chemically accurate viewpoint in order to identify actual reaction partners and mechanistic details. The examination of metalloproteins requires a combination of analytical, spectroscopical, and biomedical techniques, with potential applications of theoretical and computational means. With harmonized efforts of interdisciplinary nature, targeting metal sites and the exploitation of reactive sulfur species-mediated signaling pathways is a viable approach to consider in hydrogen sulfide-based drug development.
Author contributions
All authors contributed to the preparation of the manuscript.
Declaration of competing interest
The authors declare no competing interest.
Acknowledgement
The project was implemented with the support from the National Research, Development and Innovation Fund of the Ministry of Culture and Innovation, under the National Laboratories Program (National Tumor Biology Laboratory (2022–2.1.1-NL-2022-00010)) and the Hungarian Thematic Excellence Program (under project TKP2021-EGA-44) based on Grant Agreements with the National Research, Development and Innovation Office.
P.N. acknowledges support from the Eötvös Loránd Research Network – ATE – Laboratory of Redox Biology (grant 15002) from the Hungarian Academy of Sciences. P.N. and É.D. are supported by the Hungarian National Research, Development and Innovation Office under K_129286 and PD_132082 grants.
J.B. and Gy.B. acknowledge financial support from the Eötvös Loránd Research Network (grant 11003) from the Hungarian Academy of Science and K_132828, TKP2020-NKA-04, and TKP2021-EGA-18 projects which have been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund. TKP grants were financed under the 2020–4.1.1-TKP2020 and TKP2021-EGA funding schemes.
The graphical abstract was created with biorender.com.
List of abbreviations
- BNC
hetero binuclear center
- CAT
catalase
- Cc
cytochrome c
- CcO
cytochrome c oxidase
- Cys-SH
cysteine
- Cys-SSH
cysteine persulfide
- CSE
cystathionine-γ-lyase
- DTT
dithiothreitol
- EPO
eosinophil peroxidase
- EPR
electron paramagnetic resonance spectrometry
- ETC
electron transport chain
- ETHE1
ethylmalonic encephalopathy protein 1
- FXN
frataxin
- GSH
glutathione
- H2S
hydrogen sulfide
- Hb
hemoglobin
- HCys-SH
homocysteine
- His
histidine
- ISC
iron-sulfur cluster
- LPO
lactoperoxidase
- Mb
myoglobin
- Met
methionine
- metHb
methemoglobin
- metMb
metmyoglobin
- metNgb
metneuroglobin
- MMI
2-mercapto-1-methylimidazole
- MPO
myeloperoxidase
- Ngb
neuroglobin
- Por
porphyrin
- Q
coenzyme Q; ubiquinone
- QH2
ubiquinol
- RBC
red blood cell
- RNS
Reactive Nitrogen Species
- ROS
Reactive Oxygen Species
- RSS
Reactive Sulfur Species
- SOD
superoxide dismutase
- SSNO−
nitrosopersulfide
- sulfCAT
sulfcatalase
- sulfHb
sulfhemoglobin
- sulfMb
sulfmyoglobin
- TPO
thyroid peroxidase
Data availability
Original data have not been generated for this review article.
References
- 1.Szabo C. A timeline of hydrogen sulfide (H2S) research: from environmental toxin to biological mediator. Biochem. Pharmacol. 2018;149:5–19. doi: 10.1016/j.bcp.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe K., Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996;16(3):1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szabo C. Gaseotransmitters: new frontiers for translational science. Sci. Transl. Med. 2010;2(59):59ps54. doi: 10.1126/scitranslmed.3000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace J.L., Wang R. Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015;14(5):329–345. doi: 10.1038/nrd4433. [DOI] [PubMed] [Google Scholar]
- 5.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012;92(2):791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 6.Lefer D.J. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 2007;104(46):17907–17908. doi: 10.1073/pnas.0709010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zanardo R.C.O., Brancaleone V., Distrutti E., Fiorucci S., Cirino G., Wallace J.L. Hydrogen sulfide is an endogenous modulator of leukocyte‐mediated inflammation. Faseb. J. 2006;20(12):2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 8.Hine C., Harputlugil E., Zhang Y., Ruckenstuhl C., Lee B.C., Brace L., Longchamp A., Trevino-Villarreal J.H., Mejia P., Ozaki C.K., Wang R., Gladyshev V.N., Madeo F., Mair W.B., Mitchell J.R. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell. 2015;160(1–2):132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hine C., Zhu Y., Hollenberg A.N., Mitchell J.R. Dietary and endocrine regulation of endogenous hydrogen sulfide production: implications for longevity. Antioxidants Redox Signal. 2018;28(16):1483–1502. doi: 10.1089/ars.2017.7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016;15(3):185–203. doi: 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogdandi V., Ida T., Sutton T.R., Bianco C., Ditroi T., Koster G., Henthorn H.A., Minnion M., Toscano J.P., van der Vliet A., Pluth M.D., Feelisch M., Fukuto J.M., Akaike T., Nagy P. Speciation of reactive sulfur species and their reactions with alkylating agents: do we have any clue about what is present inside the cell? Br. J. Pharmacol. 2019;176(4):646–670. doi: 10.1111/bph.14394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy P., Doka E., Ida T., Akaike T. Measuring reactive sulfur species and thiol oxidation states: challenges and cautions in relation to alkylation-based protocols. Antioxidants Redox Signal. 2020;33(16):1174–1189. doi: 10.1089/ars.2020.8077. [DOI] [PubMed] [Google Scholar]
- 13.Nagy P., Palinkas Z., Nagy A., Budai B., Toth I., Vasas A. Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim. Biophys. Acta. 2014;1840(2):876–891. doi: 10.1016/j.bbagen.2013.05.037. [DOI] [PubMed] [Google Scholar]
- 14.Kabil O., Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxidants Redox Signal. 2014;20(5):770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yadav P.K., Yamada K., Chiku T., Koutmos M., Banerjee R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 2013;288(27):20002–20013. doi: 10.1074/jbc.M113.466177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doka E., Pader I., Biro A., Johansson K., Cheng Q., Ballago K., Prigge J.R., Pastor-Flores D., Dick T.P., Schmidt E.E., Arner E.S., Nagy P. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016;2(1) doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wedmann R., Onderka C., Wei S., Szijártó I.A., Miljkovic J.L., Mitrovic A., Lange M., Savitsky S., Yadav P.K., Torregrossa R., Harrer E.G., Harrer T., Ishii I., Gollasch M., Wood M.E., Galardon E., Xian M., Whiteman M., Banerjee R., Filipovic M.R. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016;7(5):3414–3426. doi: 10.1039/C5SC04818D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N.O., Xian M., Fukuto J.M., Akaike T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 2014;111(21):7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akaike T., Ida T., Wei F.Y., Nishida M., Kumagai Y., Alam M.M., Ihara H., Sawa T., Matsunaga T., Kasamatsu S., Nishimura A., Morita M., Tomizawa K., Nishimura A., Watanabe S., Inaba K., Shima H., Tanuma N., Jung M., Fujii S., Watanabe Y., Ohmuraya M., Nagy P., Feelisch M., Fukuto J.M., Motohashi H. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017;8(1):1177. doi: 10.1038/s41467-017-01311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melideo S.L., Jackson M.R., Jorns M.S. Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry. 2014;53(28):4739–4753. doi: 10.1021/bi500650h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szabo C., Ransy C., Modis K., Andriamihaja M., Murghes B., Coletta C., Olah G., Yanagi K., Bouillaud F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014;171(8):2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ditroi T., Nagy A., Martinelli D., Rosta A., Kozich V., Nagy P. Comprehensive analysis of how experimental parameters affect H2S measurements by the monobromobimane method. Free Radic. Biol. Med. 2019;136:146–158. doi: 10.1016/j.freeradbiomed.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Whiteman M., Winyard P.G. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expet Rev. Clin. Pharmacol. 2011;4(1):13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- 24.Kozich V., Schwahn B.C., Sokolova J., Krizkova M., Ditroi T., Krijt J., Khalil Y., Krizek T., Vaculikova-Fantlova T., Stiburkova B., Mills P., Clayton P., Barvikova K., Blessing H., Sykut-Cegielska J., Dionisi-Vici C., Gasperini S., Garcia-Cazorla A., Haack T.B., Honzik T., Jesina P., Kuster A., Laugwitz L., Martinelli D., Porta F., Santer R., Schwarz G., Nagy P. Human ultrarare genetic disorders of sulfur metabolism demonstrate redundancies in H2S homeostasis. Redox Biol. 2022;58 doi: 10.1016/j.redox.2022.102517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuto J.M., Carrington S.J., Tantillo D.J., Harrison J.G., Ignarro L.J., Freeman B.A., Chen A., Wink D.A. Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012;25(4):769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cortese-Krott M.M., Koning A., Kuhnle G.G.C., Nagy P., Bianco C.L., Pasch A., Wink D.A., Fukuto J.M., Jackson A.A., van Goor H., Olson K.R., Feelisch M. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxidants Redox Signal. 2017;27(10):684–712. doi: 10.1089/ars.2017.7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy P. Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol. 2015;554:3–29. doi: 10.1016/bs.mie.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 28.Mustafa A.K., Gadalla M.M., Sen N., Kim S., Mu W., Gazi S.K., Barrow R.K., Yang G., Wang R., Snyder S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009;2(96) doi: 10.1126/scisignal.2000464. ra72-ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paul B.D., Snyder S.H. H₂S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 30.Ono K., Akaike T., Sawa T., Kumagai Y., Wink D.A., Tantillo D.J., Hobbs A.J., Nagy P., Xian M., Lin J., Fukuto J.M. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications of their possible biological activity and utility. Free Radic. Biol. Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellis A.T., Misko A.L., Arjune S., Liang Y., Erdelyi K., Ditroi T., Kaczmarek A.T., Nagy P., Schwarz G. The role of glutamate oxaloacetate transaminases in sulfite biosynthesis and H2S metabolism. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Libiad M., Motl N., Akey D.L., Sakamoto N., Fearon E.R., Smith J.L., Banerjee R. Thiosulfate sulfurtransferase-like domain-containing 1 protein interacts with thioredoxin. J. Biol. Chem. 2018;293(8):2675–2686. doi: 10.1074/jbc.RA117.000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filipovic M.R., Zivanovic J., Alvarez B., Banerjee R. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 2018;118(3):1253–1337. doi: 10.1021/acs.chemrev.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuto J.M., Ignarro L.J., Nagy P., Wink D.A., Kevil C.G., Feelisch M., Cortese-Krott M.M., Bianco C.L., Kumagai Y., Hobbs A.J., Lin J., Ida T., Akaike T. Biological hydropersulfides and related polysulfides - a new concept and perspective in redox biology. FEBS Lett. 2018;592(12):2140–2152. doi: 10.1002/1873-3468.13090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura H. Hydrogen sulfide (H2S) and polysulfide (H2Sn) signaling: the first 25 years. Biomolecules. 2021;11(6):896. doi: 10.3390/biom11060896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadav P.K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M.R., Banerjee R. Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 2016;138(1):289–299. doi: 10.1021/jacs.5b10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortese-Krott M.M., Kuhnle G.G., Dyson A., Fernandez B.O., Grman M., DuMond J.F., Barrow M.P., McLeod G., Nakagawa H., Ondrias K., Nagy P., King S.B., Saavedra J.E., Keefer L.K., Singer M., Kelm M., Butler A.R., Feelisch M. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. U.S.A. 2015;112(34):E4651–E4660. doi: 10.1073/pnas.1509277112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kevil C., Cortese-Krott M.M., Nagy P., Papapetropoulos A., Feelisch M., Szabo C. In: Nitric Oxide. third ed. Ignarro L.J., Freeman B.A., editors. Academic Press; 2017. Chapter 5 - cooperative interactions between NO and H2S: chemistry, biology, physiology, pathophysiology; pp. 57–83. [DOI] [Google Scholar]
- 39.Maiti B.K., Maia L.B., Moura J.J.G. Sulfide and transition metals - a partnership for life. J. Inorg. Biochem. 2022;227 doi: 10.1016/j.jinorgbio.2021.111687. [DOI] [PubMed] [Google Scholar]
- 40.Woods J.J., Wilson J.J. In: Encyclopedia of Inorganic and Bioinorganic Chemistry. Scott R.A., editor. John Wiley & Sons, Ltd.; 2021. Bioinorganic chemistry of hydrogen sulfide: detection, delivery, and interactions with metalloproteins; pp. 1–22. [DOI] [Google Scholar]
- 41.Lothian A., Hare D.J., Grimm R., Ryan T.M., Masters C.L., Roberts B.R. Metalloproteomics: principles, challenges and applications to neurodegeneration. Front. Aging Neurosci. 2013;5:35. doi: 10.3389/fnagi.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maret W. Metallomics: the science of biometals and biometalloids. Adv. Exp. Med. Biol. 2018;1055:1–20. doi: 10.1007/978-3-319-90143-5_1. [DOI] [PubMed] [Google Scholar]
- 43.Banci L., Bertini I. Metallomics and the cell: some definitions and general comments. Met. Ions Life Sci. 2013;12:1–13. doi: 10.1007/978-94-007-5561-1_1. [DOI] [PubMed] [Google Scholar]
- 44.Maret W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics. 2010;2(2):117–125. doi: 10.1039/b915804a. [DOI] [PubMed] [Google Scholar]
- 45.Andreini C., Bertini I., Rosato A. Metalloproteomes: a bioinformatic approach. Acc. Chem. Res. 2009;42(10):1471–1479. doi: 10.1021/ar900015x. [DOI] [PubMed] [Google Scholar]
- 46.Maret W. The metals in the biological periodic system of the elements: concepts and conjectures. Int. J. Mol. Sci. 2016;17(1) doi: 10.3390/ijms17010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoroddu M.A., Aaseth J., Crisponi G., Medici S., Peana M., Nurchi V.M. The essential metals for humans: a brief overview. J. Inorg. Biochem. 2019;195:120–129. doi: 10.1016/j.jinorgbio.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 48.Pohl H.R., Wheeler J.S., Murray H.E. Sodium and potassium in health and disease. Met. Ions Life Sci. 2013;13:29–47. doi: 10.1007/978-94-007-7500-8_2. [DOI] [PubMed] [Google Scholar]
- 49.Al Alawi A.M., Majoni S.W., Falhammar H. Magnesium and human health: perspectives and research directions. Int. J. Endocrinol. 2018;2018 doi: 10.1155/2018/9041694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anzellotti A.I., Farrell N.P. Zinc metalloproteins as medicinal targets. Chem. Soc. Rev. 2008;37(8):1629–1651. doi: 10.1039/b617121b. [DOI] [PubMed] [Google Scholar]
- 51.Borrelli A., Schiattarella A., Bonelli P., Tuccillo F.M., Buonaguro F.M., Mancini A. The functional role of MnSOD as a biomarker of human diseases and therapeutic potential of a new isoform of a human recombinant MnSOD. BioMed Res. Int. 2014;2014 doi: 10.1155/2014/476789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johannes L., Fu C.Y., Schwarz G. Molybdenum cofactor deficiency in humans. Molecules. 2022;27(20) doi: 10.3390/molecules27206896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toohey J.I. Possible involvement of hydrosulfide in B12-dependent methyl group transfer. Molecules. 2017;22(4) doi: 10.3390/molecules22040582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiss T., Gajda T., Gyurcsik B. 2007. Bevezetés a Bioszervetlen Kémiába (Introduction to Bioinorganic Chemistry) and References Therein; Textbook in Hungarian for Higher Education Tankönyvkiadó. Budapest, Hungary. [Google Scholar]
- 55.Pluth M.D., Tonzetich Z.J. Hydrosulfide complexes of the transition elements: diverse roles in bioinorganic, cluster, coordination, and organometallic chemistry. Chem. Soc. Rev. 2020;49(12):4070–4134. doi: 10.1039/c9cs00570f. [DOI] [PubMed] [Google Scholar]
- 56.Mostofa M.G., Rahman A., Ansary M.M., Watanabe A., Fujita M., Tran L.S. Hydrogen sulfide modulates cadmium-induced physiological and biochemical responses to alleviate cadmium toxicity in rice. Sci. Rep. 2015;5 doi: 10.1038/srep14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Unoki T., Akiyama M., Shinkai Y., Kumagai Y., Fujimura M. Spatio-temporal distribution of reactive sulfur species during methylmercury exposure in the rat brain. J. Toxicol. Sci. 2022;47(1):31–37. doi: 10.2131/jts.47.31. [DOI] [PubMed] [Google Scholar]
- 58.Beinert H., Holm R.H., Munck E. Iron-sulfur clusters: nature's modular, multipurpose structures. Science. 1997;277(5326):653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 59.Braymer J.J., Freibert S.A., Rakwalska-Bange M., Lill R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim. Biophys. Acta Mol. Cell Res. 2021;1868(1) doi: 10.1016/j.bbamcr.2020.118863. [DOI] [PubMed] [Google Scholar]
- 60.Crichton R.R. In: Biological Inorganic Chemistry. second ed. Crichton R.R., editor. Elsevier; Oxford: 2012. Chapter 4 - biological ligands for metal ions; pp. 69–89. [DOI] [Google Scholar]
- 61.Meyer J. Iron-sulfur protein folds, iron-sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 2008;13(2):157–170. doi: 10.1007/s00775-007-0318-7. [DOI] [PubMed] [Google Scholar]
- 62.Johnson D.C., Dean D.R., Smith A.D., Johnson M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 63.Braymer J.J., Lill R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017;292(31):12754–12763. doi: 10.1074/jbc.R117.787101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lovenberg W. In: Microbial Iron Metabolism. Neilands J.B., editor. Academic Press; 1974. Chapter 8 – ferredoxin and rubredoxin; pp. 161–185. [DOI] [Google Scholar]
- 65.Read A.D., Bentley R.E., Archer S.L., Dunham-Snary K.J. Mitochondrial iron-sulfur clusters: structure, function, and an emerging role in vascular biology. Redox Biol. 2021;47 doi: 10.1016/j.redox.2021.102164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rydz L., Wróbel M., Jurkowska H. Sulfur administration in Fe–S cluster homeostasis. Antioxidants. 2021;10(11) doi: 10.3390/antiox10111738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stehling O., Lill R. The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harbor Perspect. Biol. 2013;5(8):a011312. doi: 10.1101/cshperspect.a011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frey P.A., Hegeman A.D., Ruzicka F.J. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008;43(1):63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 69.Dos Santos P.C., Dean D.R. A newly discovered role for iron-sulfur clusters. Proc. Natl. Acad. Sci. U.S.A. 2008;105(33):11589–11590. doi: 10.1073/pnas.0805713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen X., Peter E.A., Bir S., Wang R., Kevil C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012;52(11–12):2276–2283. doi: 10.1016/j.freeradbiomed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levitt M.D., Abdel-Rehim M.S., Furne J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxidants Redox Signal. 2011;15(2):373–378. doi: 10.1089/ars.2010.3525. [DOI] [PubMed] [Google Scholar]
- 72.Ishigami M., Hiraki K., Umemura K., Ogasawara Y., Ishii K., Kimura H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxidants Redox Signal. 2009;11(2):205–214. doi: 10.1089/ARS.2008.2132. [DOI] [PubMed] [Google Scholar]
- 73.Gervason S., Larkem D., Mansour A.B., Botzanowski T., Muller C.S., Pecqueur L., Le Pavec G., Delaunay-Moisan A., Brun O., Agramunt J., Grandas A., Fontecave M., Schunemann V., Cianferani S., Sizun C., Toledano M.B., D'Autreaux B. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019;10(1):3566. doi: 10.1038/s41467-019-11470-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng L., White R.H., Cash V.L., Jack R.F., Dean D.R. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1993;90(7):2754–2758. doi: 10.1073/pnas.90.7.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qi W., Cowan J.A. Structural, Mechanistic and coordination chemistry of relevance to the biosynthesis of iron-sulfur and related iron cofactors. Coord. Chem. Rev. 2011;255(7–8):688–699. doi: 10.1016/j.ccr.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parent A., Elduque X., Cornu D., Belot L., Le Caer J.P., Grandas A., Toledano M.B., D'Autreaux B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015;6:5686. doi: 10.1038/ncomms6686. [DOI] [PubMed] [Google Scholar]
- 77.Rouault T.A. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 2012;5(2):155–164. doi: 10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Damasio J., Sardoeira A., Araujo M., Carvalho I., Sequeiros J., Barros J. Rare occurrence of severe blindness and deafness in Friedreich ataxia: a case report. Cerebellum Ataxias. 2021;8(1):17. doi: 10.1186/s40673-021-00140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Runko A.P., Griswold A.J., Min K.T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008;582(5):715–719. doi: 10.1016/j.febslet.2008.01.046. [DOI] [PubMed] [Google Scholar]
- 80.Doka E., Ida T., Dagnell M., Abiko Y., Luong N.C., Balog N., Takata T., Espinosa B., Nishimura A., Cheng Q., Funato Y., Miki H., Fukuto J.M., Prigge J.R., Schmidt E.E., Arner E.S.J., Kumagai Y., Akaike T., Nagy P. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020;6(1):eaax8358. doi: 10.1126/sciadv.aax8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Erdelyi K., Ditroi T., Johansson H.J., Czikora A., Balog N., Silwal-Pandit L., Ida T., Olasz J., Hajdu D., Matrai Z., Csuka O., Uchida K., Tovari J., Engebraten O., Akaike T., Borresen Dale A.L., Kasler M., Lehtio J., Nagy P. Reprogrammed transsulfuration promotes basal-like breast tumor progression via realigning cellular cysteine persulfidation. Proc. Natl. Acad. Sci. U.S.A. 2021;118(45) doi: 10.1073/pnas.2100050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zivanovic J., Kouroussis E., Kohl J.B., Adhikari B., Bursac B., Schott-Roux S., Petrovic D., Miljkovic J.L., Thomas-Lopez D., Jung Y., Miler M., Mitchell S., Milosevic V., Gomes J.E., Benhar M., Gonzalez-Zorn B., Ivanovic-Burmazovic I., Torregrossa R., Mitchell J.R., Whiteman M., Schwarz G., Snyder S.H., Paul B.D., Carroll K.S., Filipovic M.R. Selective persulfide detection reveals evolutionarily conserved antiaging effects of s-sulfhydration. Cell Metabol. 2019;30(6):1152–1170. doi: 10.1016/j.cmet.2019.10.007. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muhlenhoff U., Braymer J.J., Christ S., Rietzschel N., Uzarska M.A., Weiler B.D., Lill R. Glutaredoxins and iron-sulfur protein biogenesis at the interface of redox biology and iron metabolism. Biol. Chem. 2020;401(12):1407–1428. doi: 10.1515/hsz-2020-0237. [DOI] [PubMed] [Google Scholar]
- 84.Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 85.Bilska-Wilkosz A., Iciek M., Gorny M., Kowalczyk-Pachel D. The role of hemoproteins: hemoglobin, myoglobin and neuroglobin in endogenous thiosulfate production processes. Int. J. Mol. Sci. 2017;18(6) doi: 10.3390/ijms18061315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boubeta F.M., Bieza S.A., Bringas M., Palermo J.C., Boechi L., Estrin D.A., Bari S.E. Hemeproteins as targets for sulfide species. Antioxidants Redox Signal. 2020;32(4):247–257. doi: 10.1089/ars.2019.7878. [DOI] [PubMed] [Google Scholar]
- 87.Cortese-Krott M.M. Red blood cells as a "central hub" for sulfide bioactivity: scavenging, metabolism, transport, and cross-talk with nitric oxide. Antioxidants Redox Signal. 2020;33(18):1332–1349. doi: 10.1089/ars.2020.8171. [DOI] [PubMed] [Google Scholar]
- 88.Gall T., Petho D., Nagy A., Balla G., Balla J. Therapeutic potential of carbon monoxide (CO) and hydrogen sulfide (H2S) in hemolytic and hemorrhagic vascular disorders-interaction between the heme oxygenase and H2S-producing systems. Int. J. Mol. Sci. 2020;22(1) doi: 10.3390/ijms22010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murphy B., Bhattacharya R., Mukherjee P. Hydrogen sulfide signaling in mitochondria and disease. Faseb. J. 2019;33(12):13098–13125. doi: 10.1096/fj.201901304R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pietri R., Roman-Morales E., Lopez-Garriga J. Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxidants Redox Signal. 2011;15(2):393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rios-Gonzalez B.B., Roman-Morales E.M., Pietri R., Lopez-Garriga J. Hydrogen sulfide activation in hemeproteins: the sulfheme scenario. J. Inorg. Biochem. 2014;133:78–86. doi: 10.1016/j.jinorgbio.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxidants Redox Signal. 2013;18(13):1623–1641. doi: 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kwon H., Basran J., Casadei C.M., Fielding A.J., Schrader T.E., Ostermann A., Devos J.M., Aller P., Blakeley M.P., Moody P.C.E., Raven E.L. Direct visualization of a Fe(IV)-OH intermediate in a heme enzyme. Nat. Commun. 2016;7 doi: 10.1038/ncomms13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moody P.C.E., Raven E.L. The nature and reactivity of ferryl heme in compounds I and II. Acc. Chem. Res. 2018;51(2):427–435. doi: 10.1021/acs.accounts.7b00463. [DOI] [PubMed] [Google Scholar]
- 95.Jensen B., Fago A. A novel possible role for met hemoglobin as carrier of hydrogen sulfide in the blood. Antioxidants Redox Signal. 2020;32(4):258–265. doi: 10.1089/ars.2019.7877. [DOI] [PubMed] [Google Scholar]
- 96.Pietri R., Lewis A., León R.G., Casabona G., Kiger L., Yeh S.-R., Fernandez-Alberti S., Marden M.C., Cadilla C.L., López-Garriga J. Factors controlling the reactivity of hydrogen sulfide with hemeproteins. Biochemistry. 2009;48(22):4881–4894. doi: 10.1021/bi801738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michel H.O. A study of sulfhemoglobin. J. Biol. Chem. 1938;126(1):323–348. doi: 10.1016/S0021-9258(18)73923-9. [DOI] [Google Scholar]
- 98.Roman-Morales E., Pietri R., Ramos-Santana B., Vinogradov S.N., Lewis-Ballester A., Lopez-Garriga J. Structural determinants for the formation of sulfhemeprotein complexes. Biochem. Biophys. Res. Commun. 2010;400(4):489–492. doi: 10.1016/j.bbrc.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berzofsky J.A., Peisach J., Blumberg W.E. Sulfheme proteins. II. The reversible oxygenation of ferrous sulfmyoglobin. J. Biol. Chem. 1971;246(23):7366–7372. doi: 10.1016/S0021-9258(19)45895-X. [DOI] [PubMed] [Google Scholar]
- 100.Libardi S.H., Pindstrup H., Cardoso D.R., Skibsted L.H. Reduction of ferrylmyoglobin by hydrogen sulfide. Kinetics in relation to meat greening. J. Agric. Food Chem. 2013;61(11):2883–2888. doi: 10.1021/jf305363e. [DOI] [PubMed] [Google Scholar]
- 101.Nicholls P. The formation and properties of sulphmyoglobin and sulphcatalase. Biochem. J. 1961;81(2):374–383. doi: 10.1042/bj0810374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nakamura S., Nakamura M., Yamazaki I., Morrison M. Reactions of ferryl lactoperoxidase (compound II) with sulfide and sulfhydryl compounds. J. Biol. Chem. 1984;259(11):7080–7085. doi: 10.1016/S0021-9258(17)39840-X. [DOI] [PubMed] [Google Scholar]
- 103.Garai D., Rios-Gonzalez B.B., Furtmuller P.G., Fukuto J.M., Xian M., Lopez-Garriga J., Obinger C., Nagy P. Mechanisms of myeloperoxidase catalyzed oxidation of H2S by H2O2 or O2 to produce potent protein Cys-polysulfide-inducing species. Free Radic. Biol. Med. 2017;113:551–563. doi: 10.1016/j.freeradbiomed.2017.10.384. [DOI] [PubMed] [Google Scholar]
- 104.Keppner A., Maric D., Correia M., Koay T.W., Orlando I.M.C., Vinogradov S.N., Hoogewijs D. Lessons from the post-genomic era: globin diversity beyond oxygen binding and transport. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Exertier C., Montemiglio L.C., Freda I., Gugole E., Parisi G., Savino C., Vallone B. Neuroglobin, clues to function and mechanism. Mol. Aspect. Med. 2022;84 doi: 10.1016/j.mam.2021.101055. [DOI] [PubMed] [Google Scholar]
- 106.De Simone G., Sbardella D., Oddone F., Pesce A., Coletta M., Ascenzi P. Structural and (pseudo-)enzymatic properties of neuroglobin: its possible role in neuroprotection. Cells. 2021;10(12) doi: 10.3390/cells10123366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ascenzi P., di Masi A., Leboffe L., Fiocchetti M., Nuzzo M.T., Brunori M., Marino M. Neuroglobin: From structure to function in health and disease. Mol. Aspect. Med. 2016;52:1–48. doi: 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]
- 108.Vitvitsky V., Yadav P.K., Kurthen A., Banerjee R. Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 2015;290(13):8310–8320. doi: 10.1074/jbc.M115.639831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Winterbourn C.C. Free-radical production and oxidative reactions of hemoglobin. Environ. Health Perspect. 1985;64:321–330. doi: 10.1289/ehp.8564321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Elahian F., Sepehrizadeh Z., Moghimi B., Mirzaei S.A. Human cytochrome b5 reductase: structure, function, and potential applications. Crit. Rev. Biotechnol. 2014;34(2):134–143. doi: 10.3109/07388551.2012.732031. [DOI] [PubMed] [Google Scholar]
- 111.Bianco C.L., Savitsky A., Feelisch M., Cortese-Krott M.M. Investigations on the role of hemoglobin in sulfide metabolism by intact human red blood cells. Biochem. Pharmacol. 2018;149:163–173. doi: 10.1016/j.bcp.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 112.Haymond S., Cariappa R., Eby C.S., Scott M.G. Laboratory assessment of oxygenation in methemoglobinemia. Clin. Chem. 2005;51(2):434–444. doi: 10.1373/clinchem.2004.035154. [DOI] [PubMed] [Google Scholar]
- 113.Umbreit J. Methemoglobin—it's not just blue: a concise review. Am. J. Hematol. 2007;82(2):134–144. doi: 10.1002/ajh.20738. [DOI] [PubMed] [Google Scholar]
- 114.Keilin D. On the combinations of methæmoglobin with H2S. Proc. Roy. Soc. Lond. B. 1933;113(784):393–404. doi: 10.1098/rspb.1933.0056. [DOI] [Google Scholar]
- 115.Kraus D.W., Wittenberg J.B. Hemoglobins of the Lucina pectinata/bacteria symbiosis. I. Molecular properties, kinetics and equilibria of reactions with ligands. J. Biol. Chem. 1990;265(27):16043–16053. doi: 10.1016/S0021-9258(17)46185-0. [DOI] [PubMed] [Google Scholar]
- 116.Kraus D.W., Wittenberg J.B., Lu J.F., Peisach J. Hemoglobins of the Lucina pectinata/bacteria symbiosis. II. An electron paramagnetic resonance and optical spectral study of the ferric proteins. J. Biol. Chem. 1990;265(27):16054–16059. doi: 10.1016/S0021-9258(17)46186-2. [DOI] [PubMed] [Google Scholar]
- 117.Olson K.R., Straub K.D. The role of hydrogen sulfide in evolution and the evolution of hydrogen sulfide in metabolism and signaling. Physiology (Bethesda, Md. 2016;31(1):60–72. doi: 10.1152/physiol.00024.2015. [DOI] [PubMed] [Google Scholar]
- 118.Boubeta F.M., Bari S.E., Estrin D.A., Boechi L. Access and binding of H2S to hemeproteins: the case of HbI of Lucina pectinata. J. Phys. Chem. B. 2016;120(36):9642–9653. doi: 10.1021/acs.jpcb.6b06686. [DOI] [PubMed] [Google Scholar]
- 119.Collazo E., Pietri R., De Jesús W., Ramos C., Del Toro A., León R.G., Cadilla C.L., López-Garriga J. Functional characterization of the purified holo form of hemoglobin I from Lucina pectinata overexpressed in Escherichia coli. Prot. J. 2004;23(4):239–245. doi: 10.1023/B:JOPC.0000027848.40972.97. [DOI] [PubMed] [Google Scholar]
- 120.León R.G., Munier-Lehmann H., Barzu O., Baudin-Creuza V., Pietri R., López-Garriga J., Cadilla C.L. High-level production of recombinant sulfide-reactive hemoglobin I from Lucina pectinata in Escherichia coli. High yields of fully functional holoprotein synthesis in the BLi5 E. coli strain. Protein Expr. Purif. 2004;38(2):184–195. doi: 10.1016/j.pep.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 121.Ramirez E., Cruz A., Rodriguez D., Uchima L., Pietri R., Santana A., López-Garriga J., López G.E. Effects of active site mutations in haemoglobin I from Lucina pectinata: a molecular dynamic study. Mol. Simulat. 2008;34(7):715–725. doi: 10.1080/08927020802144114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jensen B., Fago A. Reactions of ferric hemoglobin and myoglobin with hydrogen sulfide under physiological conditions. J. Inorg. Biochem. 2018;182:133–140. doi: 10.1016/j.jinorgbio.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 123.Smith R.P., Gosselin R.E. On the mechanism of sulfide inactivation by methemoglobin. Toxicol. Appl. Pharmacol. 1966;8(1):159–172. doi: 10.1016/0041-008x(66)90112-8. [DOI] [PubMed] [Google Scholar]
- 124.Vitvitsky V., Yadav P.K., An S., Seravalli J., Cho U.S., Banerjee R. Structural and mechanistic insights into hemoglobin-catalyzed hydrogen sulfide oxidation and the fate of polysulfide products. J. Biol. Chem. 2017;292(13):5584–5592. doi: 10.1074/jbc.M117.774943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mittra K., Singha A., Dey A. Mechanism of reduction of ferric porphyrins by sulfide: identification of a low spin Fe(III)-SH intermediate. Inorg. Chem. 2017;56(7):3916–3925. doi: 10.1021/acs.inorgchem.6b02878. [DOI] [PubMed] [Google Scholar]
- 126.Bostelaar T., Vitvitsky V., Kumutima J., Lewis B.E., Yadav P.K., Brunold T.C., Filipovic M., Lehnert N., Stemmler T.L., Banerjee R. Hydrogen sulfide oxidation by myoglobin. J. Am. Chem. Soc. 2016;138(27):8476–8488. doi: 10.1021/jacs.6b03456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ng P.C., Hendry-Hofer T.B., Witeof A.E., Brenner M., Mahon S.B., Boss G.R., Haouzi P., Bebarta V.S. Hydrogen sulfide toxicity: mechanism of action, clinical presentation, and countermeasure development. J. Med. Toxicol. 2019;15(4):287–294. doi: 10.1007/s13181-019-00710-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Carrico R.J., Blumberg W.E., Peisach J. The reversible binding of oxygen to sulfhemoglobin. J. Biol. Chem. 1978;253(20):7212–7215. doi: 10.1016/S0021-9258(17)34486-1. [DOI] [PubMed] [Google Scholar]
- 129.Baranoski G.V., Chen T.F., Kimmel B.W., Miranda E., Yim D. On the noninvasive optical monitoring and differentiation of methemoglobinemia and sulfhemoglobinemia. J. Biomed. Opt. 2012;17(9) doi: 10.1117/1.Jbo.17.9.097005. [DOI] [PubMed] [Google Scholar]
- 130.Aravindhan N., Chisholm D.G. Sulfhemoglobinemia presenting as pulse oximetry desaturation. Anesthesiology. 2000;93(3):883–884. doi: 10.1097/00000542-200009000-00040. [DOI] [PubMed] [Google Scholar]
- 131.George A., Goetz D. A case of sulfhemoglobinemia in a child with chronic constipation. Respir. Med. Case Rep. 2017;21:21–24. doi: 10.1016/j.rmcr.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gharahbaghian L., Massoudian B., Dimassa G. Methemoglobinemia and sulfhemoglobinemia in two pediatric patients after ingestion of hydroxylamine sulfate. West. J. Emerg. Med. 2009;10(3):197–201. doi: not available. [PMC free article] [PubMed] [Google Scholar]
- 133.Gopalachar A.S., Bowie V.L., Bharadwaj P. Phenazopyridine-induced sulfhemoglobinemia. Ann. Pharmacother. 2005;39(6):1128–1130. doi: 10.1345/aph.1E557. [DOI] [PubMed] [Google Scholar]
- 134.Gáll T., Nagy P., Garai D., Potor L., Balla G.J., Balla G., Balla J. Overview on hydrogen sulfide-mediated suppression of vascular calcification and hemoglobin/heme-mediated vascular damage in atherosclerosis. Redox Biol. 2022;57 doi: 10.1016/j.redox.2022.102504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Balla J., Jacob H.S., Balla G., Nath K., Eaton J.W., Vercellotti G.M. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. U.S.A. 1993;90(20):9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Potor L., Hendrik Z., Patsalos A., Katona É., Méhes G., Póliska S., Csősz É., Kalló G., Komáromi I., Combi Z., Posta N., Sikura K., Pethő D., Oros M., Vereb G., Tóth C., Gergely P., Nagy L., Balla G., Balla J. Oxidation of hemoglobin drives a proatherogenic polarization of macrophages in human atherosclerosis. Antioxidants Redox Signal. 2021;35(12):917–950. doi: 10.1089/ars.2020.8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nagy E., Eaton J.W., Jeney V., Soares M.P., Varga Z., Galajda Z., Szentmiklósi J., Méhes G., Csonka T., Smith A., Vercellotti G.M., Balla G., Balla J. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010;30(7):1347–1353. doi: 10.1161/atvbaha.110.206433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Posta N., Csősz É., Oros M., Pethő D., Potor L., Kalló G., Hendrik Z., Sikura K., Méhes G., Tóth C., Posta J., Balla G., Balla J. Hemoglobin oxidation generates globin-derived peptides in atherosclerotic lesions and intraventricular hemorrhage of the brain, provoking endothelial dysfunction. Lab. Invest. 2020;100(7):986–1002. doi: 10.1038/s41374-020-0403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kassa T., Jana S., Meng F., Alayash A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio. 2016;6(9):876–884. doi: 10.1002/2211-5463.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Balla G., Vercellotti G.M., Muller-Eberhard U., Eaton J., Jacob H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Invest. 1991;64(5):648–655. doi: not available. [PubMed] [Google Scholar]
- 141.Jeney V., Balla J., Yachie A., Varga Z., Vercellotti G.M., Eaton J.W., Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100(3):879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 142.Balla J., Vercellotti G.M., Jeney V., Yachie A., Varga Z., Jacob H.S., Eaton J.W., Balla G. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxidants Redox Signal. 2007;9(12):2119–2137. doi: 10.1089/ars.2007.1787. [DOI] [PubMed] [Google Scholar]
- 143.Balla G., Jacob H.S., Eaton J.W., Belcher J.D., Vercellotti G.M. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. Vasc. Biol. 1991;11(6):1700–1711. doi: 10.1161/01.atv.11.6.1700. [DOI] [PubMed] [Google Scholar]
- 144.Jeney V., Komódi E., Nagy E., Zarjou A., Vercellotti G.M., Eaton J.W., Balla G., Balla J. Supression of hemin-mediated oxidation of low-density lipoprotein and subsequent endothelial reactions by hydrogen sulfide (H(2)S) Free Radic. Biol. Med. 2009;46(5):616–623. doi: 10.1016/j.freeradbiomed.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zavaczki E., Jeney V., Agarwal A., Zarjou A., Oros M., Katkó M., Varga Z., Balla G., Balla J. Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 2011;80(7):731–739. doi: 10.1038/ki.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mani S., Li H., Untereiner A., Wu L., Yang G., Austin R.C., Dickhout J.G., Lhoták Š., Meng Q.H., Wang R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;127(25):2523–2534. doi: 10.1161/circulationaha.113.002208. [DOI] [PubMed] [Google Scholar]
- 147.Potor L., Nagy P., Méhes G., Hendrik Z., Jeney V., Pethő D., Vasas A., Pálinkás Z., Balogh E., Gyetvai Á., Whiteman M., Torregrossa R., Wood M.E., Olvasztó S., Nagy P., Balla G., Balla J. Hydrogen sulfide abrogates hemoglobin-lipid interaction in atherosclerotic lesion. Oxid. Med. Cell. Longev. 2018;2018 doi: 10.1155/2018/3812568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sikura K., Potor L., Szerafin T., Oros M., Nagy P., Méhes G., Hendrik Z., Zarjou A., Agarwal A., Posta N., Torregrossa R., Whiteman M., Fürtös I., Balla G., Balla J. Hydrogen sulfide inhibits calcification of heart valves; implications for calcific aortic valve disease. Br. J. Pharmacol. 2020;177(4):793–809. doi: 10.1111/bph.14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hagler L., Coppes R.I., Jr., Herman R.H. Metmyoglobin reductase. Identification and purification of a reduced nicotinamide adenine dinucleotide-dependent enzyme from bovine heart which reduces metmyoglobin. J. Biol. Chem. 1979;254(14):6505–6514. doi: 10.1016/S0021-9258(18)50397-5. [DOI] [PubMed] [Google Scholar]
- 150.Brantley R.E., Jr., Smerdon S.J., Wilkinson A.J., Singleton E.W., Olson J.S. The mechanism of autooxidation of myoglobin. J. Biol. Chem. 1993;268(10):6995–7010. doi: 10.1016/S0021-9258(18)53138-0. [DOI] [PubMed] [Google Scholar]
- 151.Sugawara Y., Shikama K. Autoxidation of native oxymyoglobin. Thermodynamic analysis of the pH profile. Eur. J. Biochem. 1980;110(1):241–246. doi: 10.1111/j.1432-1033.1980.tb04861.x. [DOI] [PubMed] [Google Scholar]
- 152.Hendgen-Cotta U.B., Kelm M., Rassaf T. Myoglobin functions in the heart. Free Radic. Biol. Med. 2014;73:252–259. doi: 10.1016/j.freeradbiomed.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 153.Boubeta F.M., Bieza S.A., Bringas M., Estrin D.A., Boechi L., Bari S.E. Mechanism of sulfide binding by ferric hemeproteins. Inorg. Chem. 2018;57(13):7591–7600. doi: 10.1021/acs.inorgchem.8b00478. [DOI] [PubMed] [Google Scholar]
- 154.Román-Morales E., López-Alfonzo E., Pietri R., López-Garriga J. Sulfmyoglobin conformational change: a role in the decrease of oxy-myoglobin functionality. Biochem. Biophys. Rep. 2016;7:386–393. doi: 10.1016/j.bbrep.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Galardon E., Huguet F., Herrero C., Ricoux R., Artaud I., Padovani D. Reactions of persulfides with the heme cofactor of oxidized myoglobin and microperoxidase 11: reduction or coordination. Dalton Trans. 2017;46(24):7939–7946. doi: 10.1039/c7dt01638g. [DOI] [PubMed] [Google Scholar]
- 156.Artaud I., Galardon E. A persulfide analogue of the nitrosothiol SNAP: formation, characterization and reactivity. Chembiochem. 2014;15(16):2361–2364. doi: 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]
- 157.Alvarez L., Suarez Vega V., McGinity C., Khodade V.S., Toscano J.P., Nagy P., Lin J., Works C., Fukuto J.M. The reactions of hydropersulfides (RSSH) with myoglobin. Arch. Biochem. Biophys. 2020;687 doi: 10.1016/j.abb.2020.108391. [DOI] [PubMed] [Google Scholar]
- 158.Bianco C.L., Chavez T.A., Sosa V., Saund S.S., Nguyen Q.N.N., Tantillo D.J., Ichimura A.S., Toscano J.P., Fukuto J.M. The chemical biology of the persulfide (RSSH)/perthiyl (RSS•) redox couple and possible role in biological redox signaling. Free Radic. Biol. Med. 2016;101:20–31. doi: 10.1016/j.freeradbiomed.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M.D., Prelle A., Fagiolari G., Rimoldi M., Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009;15(2):200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 160.Burmester T., Weich B., Reinhardt S., Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407(6803):520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 161.Hamdane D., Kiger L., Dewilde S., Green B.N., Pesce A., Uzan J., Burmester T., Hankeln T., Bolognesi M., Moens L., Marden M.C. The redox state of the cell regulates the ligand binding affinity of human neuroglobin and cytoglobin. J. Biol. Chem. 2003;278(51):51713–51721. doi: 10.1074/jbc.M309396200. [DOI] [PubMed] [Google Scholar]
- 162.Dewilde S., Kiger L., Burmester T., Hankeln T., Baudin-Creuza V., Aerts T., Marden M.C., Caubergs R., Moens L. Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J. Biol. Chem. 2001;276(42):38949–38955. doi: 10.1074/jbc.M106438200. [DOI] [PubMed] [Google Scholar]
- 163.Kimura H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 2000;267(1):129–133. doi: 10.1006/bbrc.1999.1915. [DOI] [PubMed] [Google Scholar]
- 164.Kimura H. Hydrogen sulfide as a neuromodulator. Mol. Neurobiol. 2002;26(1):13–19. doi: 10.1385/mn:26:1:013. [DOI] [PubMed] [Google Scholar]
- 165.Łowicka E., Bełtowski J. Hydrogen sulfide (H2S) - the third gas of interest for pharmacologists. Pharmacol. Rep. 2007;59(1):4–24. doi: not available. [PubMed] [Google Scholar]
- 166.Brittain T., Yosaatmadja Y., Henty K. The interaction of human neuroglobin with hydrogen sulphide. IUBMB Life. 2008;60(2):135–138. doi: 10.1002/iub.16. [DOI] [PubMed] [Google Scholar]
- 167.Ruetz M., Kumutima J., Lewis B.E., Filipovic M.R., Lehnert N., Stemmler T.L., Banerjee R. A distal ligand mutes the interaction of hydrogen sulfide with human neuroglobin. J. Biol. Chem. 2017;292(16):6512–6528. doi: 10.1074/jbc.M116.770370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Greiner R., Palinkas Z., Basell K., Becher D., Antelmann H., Nagy P., Dick T.P. Polysulfides link H2S to protein thiol oxidation. Antioxidants Redox Signal. 2013;19(15):1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hughes M., Centelles M., Moore K. Making and working with hydrogen sulfide the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic. Biol. Med. 2009;47:1346–1353. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 170.Keilin D. Cytochrome and respiratory enzymes. Proc. Roy. Soc. Lond. B. 1929;104(730):206–252. doi: 10.1098/rspb.1929.0009. [DOI] [Google Scholar]
- 171.Nicholls P., Kim J.K. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem. 1982;60(6):613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 172.Landry A.P., Ballou D.P., Banerjee R. Hydrogen sulfide oxidation by sulfide quinone oxidoreductase. Chembiochem. 2021;22(6):949–960. doi: 10.1002/cbic.202000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Borisov V.B., Forte E. Impact of hydrogen sulfide on mitochondrial and bacterial bioenergetics. Int. J. Mol. Sci. 2021;22(23) doi: 10.3390/ijms222312688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Melin F., Hellwig P. Redox properties of the membrane proteins from the respiratory chain. Chem. Rev. 2020;120(18):10244–10297. doi: 10.1021/acs.chemrev.0c00249. [DOI] [PubMed] [Google Scholar]
- 175.Santucci R., Sinibaldi F., Cozza P., Polticelli F., Fiorucci L. Cytochrome c: an extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019;136:1237–1246. doi: 10.1016/j.ijbiomac.2019.06.180. [DOI] [PubMed] [Google Scholar]
- 176.Vitvitsky V., Miljkovic J.L., Bostelaar T., Adhikari B., Yadav P.K., Steiger A.K., Torregrossa R., Pluth M.D., Whiteman M., Banerjee R., Filipovic M.R. Cytochrome c reduction by H2S potentiates sulfide signaling. ACS Chem. Biol. 2018;13(8):2300–2307. doi: 10.1021/acschembio.8b00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wedmann R., Bertlein S., Macinkovic I., Böltz S., Miljkovic J., Muñoz L.E., Herrmann M., Filipovic M.R. Working with "H2S": facts and apparent artifacts. Nitric Oxide. 2014;41:85–96. doi: 10.1016/j.niox.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 178.Francoleon N.E., Carrington S.J., Fukuto J.M. The reaction of H(2)S with oxidized thiols: generation of persulfides and implications to H2S biology. Arch. Biochem. Biophys. 2011;516(2):146–153. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 179.Kadenbach B., Hüttemann M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion. 2015;24:64–76. doi: 10.1016/j.mito.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 180.Moody A.J. 'As prepared' forms of fully oxidised haem/Cu terminal oxidases. Biochem. Biophys. Acta. 1996;1276(1):6–20. doi: 10.1016/0005-2728(96)00035-7. [DOI] [PubMed] [Google Scholar]
- 181.Konstantinov A.A. Cytochrome c oxidase: intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett. 2012;586(5):630–639. doi: 10.1016/j.febslet.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 182.Reed C.J., Lam Q.N., Mirts E.N., Lu Y. Molecular understanding of heteronuclear active sites in heme–copper oxidases, nitric oxide reductases, and sulfite reductases through biomimetic modelling. Chem. Soc. Rev. 2021;50(4):2486–2539. doi: 10.1039/D0CS01297A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rich P.R. Mitochondrial cytochrome c oxidase: catalysis, coupling and controversies. Biochem. Soc. Trans. 2017;45(3):813–829. doi: 10.1042/bst20160139. [DOI] [PubMed] [Google Scholar]
- 184.Yoshikawa S., Shimada A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015;115(4):1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 185.Shaw R.W., Hansen R.E., Beinert H. Responses of the a3 component of cytochrome c oxidase to substrate and ligand addition. Biochim. Biophys. Acta. 1978;504(1):187–199. doi: 10.1016/0005-2728(78)90017-8. [DOI] [PubMed] [Google Scholar]
- 186.Hill B.C., Woon T.C., Nicholls P., Peterson J., Greenwood C., Thomson A.J. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem. J. 1984;224(2):591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nicholls P. The effect of sulphide on cytochrome aa3. Isosteric and allosteric shifts of the reduced alpha-peak. Biochim. Biophys. Acta. 1975;396(1):24–35. doi: 10.1016/0005-2728(75)90186-3. [DOI] [PubMed] [Google Scholar]
- 188.Nicholls P. Inhibition of cytochrome c oxidase by sulphide. Biochem. Soc. Trans. 1975;3(2):316–319. doi: 10.1042/bst0030316. [DOI] [PubMed] [Google Scholar]
- 189.Nicholls P., Kim J.K. Oxidation of sulphide by cytochrome aa3. Biochem. Biophys. Acta. 1981;637(2):312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- 190.Nicholls P., Petersen L.C., Miller M., Hansen F.B. Ligand-induced spectral changes in cytochrome c oxidase and their possible significance. Biochem. Biophys. Acta. 1976;449(2):188–196. doi: 10.1016/0005-2728(76)90132-8. [DOI] [PubMed] [Google Scholar]
- 191.Seiter C.H., Angelos S.G. Cytochrome oxidase: an alternative model. Proc. Natl. Acad. Sci. U.S.A. 1980;77(4):1806–1808. doi: 10.1073/pnas.77.4.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wever R., Van Gelder B.F., Dervartanian D.V. Biochemical and biophysical studies on cytochrome c oxidase. XX. Reaction with sulphide. Biochim. Biophys. Acta. 1975;387(2):189–193. doi: 10.1016/0005-2728(75)90102-4. [DOI] [PubMed] [Google Scholar]
- 193.Wilson D.F., Erecińska M., Owen C.S. Some properties of the redox components of cytochrome c oxidase and their interactions. Arch. Biochem. Biophys. 1976;175(1):160–172. doi: 10.1016/0003-9861(76)90495-1. [DOI] [PubMed] [Google Scholar]
- 194.Nicholls P., Marshall Doug C., Cooper Chris E., Wilson Mike T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013;41(5):1312–1316. doi: 10.1042/BST20130070. [DOI] [PubMed] [Google Scholar]
- 195.Petersen L.C. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochem. Biophys. Acta. 1977;460(2):299–307. doi: 10.1016/0005-2728(77)90216-x. [DOI] [PubMed] [Google Scholar]
- 196.Li P., Zhou L., Zhao T., Liu X., Zhang P., Liu Y., Zheng X., Li Q. Caspase-9: structure, mechanisms and clinical application. Oncotarget. 2017;8(14):23996–24008. doi: 10.18632/oncotarget.15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Luetjens C.M., Kögel D., Reimertz C., Düßmann H., Renz A., Schulze-Osthoff K., Nieminen A.-L., Poppe M., Prehn J.H.M. Multiple kinetics of mitochondrial cytochrome c release in drug-induced apoptosis. Mol. Pharmacol. 2001;60(5):1008. doi: 10.1124/mol.60.5.1008. [DOI] [PubMed] [Google Scholar]
- 198.Blackstone E., Morrison M., Roth M.B. H2S induces a suspended animation-like state in mice. Science. 2005;308(5721):518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 199.Blackstone E., Roth M.B. Suspended animation-like state protects mice from lethal hypoxia. Shock. 2007;27(4):370–372. doi: 10.1097/SHK.0b013e31802e27a0. [DOI] [PubMed] [Google Scholar]
- 200.Vlasova I.I. Peroxidase activity of human hemoproteins: keeping the fire under control. Molecules. 2018;23(10):2561. doi: 10.3390/molecules23102561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zamocky M., Jakopitsch C., Furtmuller P.G., Dunand C., Obinger C. The peroxidase-cyclooxygenase superfamily: reconstructed evolution of critical enzymes of the innate immune system. Proteins. 2008;72(2):589–605. doi: 10.1002/prot.21950. [DOI] [PubMed] [Google Scholar]
- 202.Furtmuller P.G., Zederbauer M., Jantschko W., Helm J., Bogner M., Jakopitsch C., Obinger C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006;445(2):199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 203.Arnhold J., Monzani E., Furtmüller P.G., Zederbauer M., Casella L., Obinger C. Kinetics and thermodynamics of halide and nitrite oxidation by mammalian heme peroxidases. Eur. J. Inorg. Chem. 2006;19:3801–3811. doi: 10.1002/ejic.200600436. 2006. [DOI] [Google Scholar]
- 204.Davies M.J., Hawkins C.L., Pattison D.I., Rees M.D. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxidants Redox Signal. 2008;10(7):1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 205.O'Brien P.J. Peroxidases. Chem. Biol. Interact. 2000;129(1):113–139. doi: 10.1016/S0009-2797(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 206.Davies M.J. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 2011;48(1):8–19. doi: 10.3164/jcbn.11-006FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Furtmüller P.G., Jantschko W., Zederbauer M., Jakopitsch C., Arnhold J., Obinger C. Kinetics of interconversion of redox intermediates of lactoperoxidase, eosinophil peroxidase and myeloperoxidase. Jpn. J. Infect. Dis. 2004;57(5):S30–S31. doi: not available. [PubMed] [Google Scholar]
- 208.Kohler H., Taurog A., Dunford H.B. Spectral studies with lactoperoxidase and thyroid peroxidase: interconversions between native enzyme, Compound II, and Compound III. Arch. Biochem. Biophys. 1988;264(2):438–449. doi: 10.1016/0003-9861(88)90309-8. [DOI] [PubMed] [Google Scholar]
- 209.Nakamura S., Nakamura M., Yamazaki I., Morrison M. Reactions of ferryl lactoperoxidase (compound II) with sulfide and sulfhydryl compounds. J. Biol. Chem. 1984;259(11):7080–7085. doi: 10.1016/S0021-9258(17)39840-X. [DOI] [PubMed] [Google Scholar]
- 210.Davies M.J., Hawkins C.L., Pattison D.I., Rees M.D. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxidants Redox Signal. 2008;10(7):1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 211.Wilson M.T., Reeder B.J. The peroxidatic activities of Myoglobin and Hemoglobin, their pathological consequences and possible medical interventions. Mol. Aspect. Med. 2022;84 doi: 10.1016/j.mam.2021.101045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Garai D., Palinkas Z., Balla J., Kettle A.J., Nagy P. Measurements for sulfide-mediated inhibition of myeloperoxidase activity. Methods Mol. Biol. 2007:179–203. doi: 10.1007/978-1-4939-9528-8_14. 2019. [DOI] [PubMed] [Google Scholar]
- 213.Palinkas Z., Furtmuller P.G., Nagy A., Jakopitsch C., Pirker K.F., Magierowski M., Jasnos K., Wallace J.L., Obinger C., Nagy P. Interactions of hydrogen sulfide with myeloperoxidase. Br. J. Pharmacol. 2015;172(6):1516–1532. doi: 10.1111/bph.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Heinecke J.W., Li W., Francis G.A., Goldstein J.A. Tyrosyl radical generated by myeloperoxidase catalyzes the oxidative cross-linking of proteins. J. Clin. Invest. 1993;91(6):2866–2872. doi: 10.1172/JCI116531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Marquez L.A., Dunford H.B. Kinetics of oxidation of tyrosine and dityrosine by myeloperoxidase compounds I and II. Implications for lipoprotein peroxidation studies. J. Biol. Chem. 1995;270(51):30434–30440. doi: 10.1074/jbc.270.51.30434. [DOI] [PubMed] [Google Scholar]
- 216.Whiteman M., Haigh R., Tarr J.M., Gooding K.M., Shore A.C., Winyard P.G. Detection of hydrogen sulfide in plasma and knee-joint synovial fluid from rheumatoid arthritis patients: relation to clinical and laboratory measures of inflammation. Ann. N. Y. Acad. Sci. 2010;1203:146–150. doi: 10.1111/j.1749-6632.2010.05556.x. [DOI] [PubMed] [Google Scholar]
- 217.Elrod J.W., Calvert J.W., Morrison J., Doeller J.E., Kraus D.W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C.-W., Lefer D.J. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 2007;104(39):15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Heinecke J.W. Mechanisms of oxidative damage of low density lipoprotein in human atherosclerosis. Curr. Opin. Lipidol. 1997;8(5):268–274. doi: 10.1097/00041433-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 219.Nicholls S.J., Hazen S.L. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005;25(6):1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 220.Stamp L.K., Khalilova I., Tarr J.M., Senthilmohan R., Turner R., Haigh R.C., Winyard P.G., Kettle A.J. Myeloperoxidase and oxidative stress in rheumatoid arthritis. Rheumatology. 2012;51(10):1796–1803. doi: 10.1093/rheumatology/kes193. [DOI] [PubMed] [Google Scholar]
- 221.Millikin R., Bianco C.L., White C., Saund S.S., Henriquez S., Sosa V., Akaike T., Kumagai Y., Soeda S., Toscano J.P., Lin J., Fukuto J.M. The chemical biology of protein hydropersulfides: studies of a possible protective function of biological hydropersulfide generation. Free Radic. Biol. Med. 2016;97:136–147. doi: 10.1016/j.freeradbiomed.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sodha N.R., Clements R.T., Feng J., Liu Y., Bianchi C., Horvath E.M., Szabo C., Stahl G.L., Sellke F.W. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J. Thorac. Cardiovasc. Surg. 2009;138(4):977–984. doi: 10.1016/j.jtcvs.2008.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zeng J., Lin X., Fan H., Li C. Hydrogen sulfide attenuates the inflammatory response in a mouse burn injury model. Mol. Med. Rep. 2013;8(4):1204–1208. doi: 10.3892/mmr.2013.1610. [DOI] [PubMed] [Google Scholar]
- 224.Boots J.W., Floris R. Lactoperoxidase: from catalytic mechanism to practical applications. Int. Dairy J. 2006;16(11):1272–1276. doi: 10.1016/j.idairyj.2006.06.019. [DOI] [Google Scholar]
- 225.Ohtaki S., Nakagawa H., Nakamura M., Kotani T. Thyroid peroxidase: experimental and clinical integration. Endocr. J. 1996;43(1):1–14. doi: 10.1507/endocrj.43.1. [DOI] [PubMed] [Google Scholar]
- 226.Zhao X., Cao Y., Jin H., Wang X., Zhang L., Zhang Y., Yu Y., Huang Y., Gao Y., Zhang J. Hydrogen sulfide promotes thyroid hormone synthesis and secretion by upregulating sirtuin-1. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.838248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ohtaki S., Nakagawa H., Nakamura M., Yamazaki I. Reactions of purified hog thyroid peroxidase with H2O2, tyrosine, and methylmercaptoimidazole (goitrogen) in comparison with bovine lactoperoxidase. J. Biol. Chem. 1982;257(2):761–766. doi: 10.1016/S0021-9258(19)68261-X. [DOI] [PubMed] [Google Scholar]
- 228.Acharya K.R., Ackerman S.J. Eosinophil granule proteins: form and function. J. Biol. Chem. 2014;289(25):17406–17415. doi: 10.1074/jbc.R113.546218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ranguelova K., Chatterjee S., Ehrenshaft M., Ramirez D.C., Summers F.A., Kadiiska M.B., Mason R.P. Protein radical formation resulting from eosinophil peroxidase-catalyzed oxidation of sulfite. J. Biol. Chem. 2010;285(31):24195–24205. doi: 10.1074/jbc.M109.069054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Maiti B.K. Cross-talk between (hydrogen)sulfite and metalloproteins: impact on human health. Chemistry. 2022;28(23) doi: 10.1002/chem.202104342. [DOI] [PubMed] [Google Scholar]
- 231.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Forman H.J., Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021;20(9):689–709. doi: 10.1038/s41573-021-00233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sies H., Berndt C., Jones D.P. Oxidative stress. Annu. Rev. Biochem. 2017;86(1):715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 234.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 235.Winterbourn C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008;4(5):278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 236.Winterbourn C.C. In: Encyclopedia of Radicals in Chemistry, Biology and Materials. Chatgilialoglu C., Studer A., editors. Wiley; 2012. Biological chemistry of reactive oxygen species. [DOI] [Google Scholar]
- 237.Antunes F., Brito P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017;13:1–7. doi: 10.1016/j.redox.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Stocker S., Van Laer K., Mijuskovic A., Dick T.P. The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxidants Redox Signal. 2018;28(7):558–573. doi: 10.1089/ars.2017.7162. [DOI] [PubMed] [Google Scholar]
- 239.Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008;45(5):549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 240.Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc. Natl. Acad. Sci. U.S.A. 2018;115(23):5839–5848. doi: 10.1073/pnas.1804932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rifkind J.M., Mohanty J.G., Nagababu E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2015;5 doi: 10.3389/fphys.2014.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Quaye I.K. Extracellular hemoglobin: the case of a friend turned foe. Front. Physiol. 2015;6 doi: 10.3389/fphys.2015.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bartosz G. In: Reactions, Processes: Oxidants and Antioxidant Defense Systems. Grune T., editor. Springer Berlin Heidelberg; Berlin, Heidelberg: 2005. Superoxide dismutases and catalase; pp. 109–149. [DOI] [Google Scholar]
- 244.McCord J.M., Fridovich I. Superoxide dismutase: the first twenty years (1968-1988) Free Radic. Biol. Med. 1988;5(5–6):363–369. doi: 10.1016/0891-5849(88)90109-8. [DOI] [PubMed] [Google Scholar]
- 245.McCord J.M., Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J. Biol. Chem. 1969;244(22):6049–6055. doi: 10.1016/S0021-9258(18)63504-5. [DOI] [PubMed] [Google Scholar]
- 246.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants Redox Signal. 2011;15(6):1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Richardson J.S. beta-Sheet topology and the relatedness of proteins. Nature. 1977;268(5620):495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 248.Tainer J.A., Getzoff E.D., Beem K.M., Richardson J.S., Richardson D.C. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J. Mol. Biol. 1982;160(2):181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 249.Elchuri S., Oberley T.D., Qi W., Eisenstein R.S., Jackson Roberts L., Van Remmen H., Epstein C.J., Huang T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24(3):367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 250.Weisiger R.A., Fridovich I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973;248(10):3582–3592. doi: 10.1016/S0021-9258(19)43969-0. [DOI] [PubMed] [Google Scholar]
- 251.Azadmanesh J., Borgstahl G.E.O. A review of the catalytic mechanism of human manganese superoxide dismutase. Antioxidants. 2018;7(2) doi: 10.3390/antiox7020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Searcy D.G., Whitehead J.P., Maroney M.J. Interaction of Cu,Zn superoxide dismutase with hydrogen sulfide. Arch. Biochem. Biophys. 1995;318(2):251–263. doi: 10.1006/abbi.1995.1228. [DOI] [PubMed] [Google Scholar]
- 253.Sun W.H., Liu F., Chen Y., Zhu Y.C. Hydrogen sulfide decreases the levels of ROS by inhibiting mitochondrial complex IV and increasing SOD activities in cardiomyocytes under ischemia/reperfusion. Biochem. Biophys. Res. Commun. 2012;421(2):164–169. doi: 10.1016/j.bbrc.2012.03.121. [DOI] [PubMed] [Google Scholar]
- 254.Olson K.R., Gao Y., Arif F., Arora K., Patel S., DeLeon E.R., Sutton T.R., Feelisch M., Cortese-Krott M.M., Straub K.D. Metabolism of hydrogen sulfide (H2S) and production of reactive sulfur species (RSS) by superoxide dismutase. Redox Biol. 2018;15:74–85. doi: 10.1016/j.redox.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Perry J.J., Shin D.S., Getzoff E.D., Tainer J.A. The structural biochemistry of the superoxide dismutases. Biochem. Biophys. Acta. 2010;1804(2):245–262. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Li Y., Huang T.T., Carlson E.J., Melov S., Ursell P.C., Olson J.L., Noble L.J., Yoshimura M.P., Berger C., Chan P.H., Wallace D.C., Epstein C.J. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 257.Melov S., Coskun P., Patel M., Tuinstra R., Cottrell B., Jun A.S., Zastawny T.H., Dizdaroglu M., Goodman S.I., Huang T.T., Miziorko H., Epstein C.J., Wallace D.C. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 1999;96(3):846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Goyal M.M., Basak A. Human catalase: looking for complete identity. Protein Cell. 2010;1(10):888–897. doi: 10.1007/s13238-010-0113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Beers R.F., Sizer I.W. Sulfide inhibition of catalase. Science. 1954;120(3105):32–33. doi: 10.1126/science.120.3105.32. [DOI] [PubMed] [Google Scholar]
- 260.Olson K.R., Gao Y., DeLeon E.R., Arif M., Arif F., Arora N., Straub K.D. Catalase as a sulfide-sulfur oxido-reductase: an ancient (and modern?) regulator of reactive sulfur species (RSS) Redox Biol. 2017;12:325–339. doi: 10.1016/j.redox.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Miller C.G., Holmgren A., Arner E.S.J., Schmidt E.E. NADPH-dependent and -independent disulfide reductase systems. Free Radic. Biol. Med. 2018;127:248–261. doi: 10.1016/j.freeradbiomed.2018.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data have not been generated for this review article.