

Summary
Ancient genomics can directly detect human genetic adaptation to environmental cues. However, it remains unclear how pathogens have exerted selective pressures on human genome diversity across different epochs and affected present-day inflammatory disease risk. Here, we use an ancestry-aware approximate Bayesian computation framework to estimate the nature, strength, and time of onset of selection acting on 2,879 ancient and modern European genomes from the last 10,000 years. We found that the bulk of genetic adaptation occurred after the start of the Bronze Age, <4,500 years ago, and was enriched in genes relating to host-pathogen interactions. Furthermore, we detected directional selection acting on specific leukocytic lineages and experimentally demonstrated that the strongest negatively selected candidate variant in immunity genes, lipopolysaccharide-binding protein (LBP) D283G, is hypomorphic. Finally, our analyses suggest that the risk of inflammatory disorders has increased in post-Neolithic Europeans, possibly because of antagonistic pleiotropy following genetic adaptation to pathogens.
Keywords: ancient DNA, immunity, host defense, natural selection, local adaptation, inflammatory disorders, approximate Bayesian computation, antagonistic pleiotropy, LBP, infectious diseases
Graphical abstract
Highlights
-
•
Ancient genomics studies allow detection of the extent of natural selection over time
-
•
Genetic adaptation in Europe has mainly occurred after the start of the Bronze Age
-
•
Immunity genes have been strongly affected by both positive and negative selection
-
•
Resistance to infection has increased inflammatory disease risk in recent millennia
Kerner et al. analyze ancient human genomes to reconstruct the history of host-pathogen interactions over the last 10,000 years. They found that genetic adaptation has occurred principally after the Bronze Age and show that the risk of inflammatory disorders has increased in post-Neolithic Europeans, following genetic adaptation to infectious diseases.
Introduction
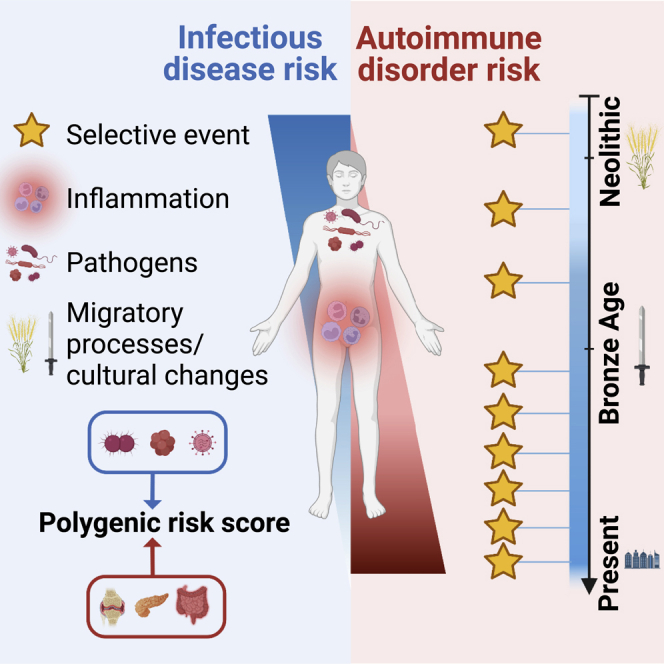
Infectious diseases have been the leading cause of human mortality throughout human history.1,2 Population genetics studies have provided support for the notion that pathogens are among the strongest selective forces faced by humans since the discovery in the 1950s that heterozygosity for red blood cell disorders provides some protection against malaria.3 An increasing number of genes involved in host-pathogen interactions have since been identified as targets of natural selection.4,5,6,7,8 However, major questions remain regarding the evolutionary impact of infectious diseases on human genome diversity. First, little is known about the specific epochs during which humans were most exposed to pathogens and pathogen-mediated selection. It has been suggested that the transition to an agriculture-based lifestyle, which began ∼10,000 years ago (ya), increased exposure to deadly microbes, including density-dependent viruses and zoonoses,9,10,11 but the archaeological and genetic evidence are scarce and even challenge this view.12,13 Second, the extent to which host defenses, including the engagement of leukocytic lineages, qualitative and quantitative disorders of which are associated with common and rare disorders,14,15,16,17 have been affected by such exposure has not been explored. Third, the rising life expectancy in recent centuries18 has contributed to an increase in the prevalence of autoinflammatory and autoimmune disorders, but it has been hypothesized that this increase is also the result of long-term pathogen pressures and antagonistic pleiotropy of the selected gene products.19,20,21
The antagonistic pleiotropy hypothesis is supported by the overlap between loci underlying infectious and inflammatory traits22,23 and the discovery of several pleiotropic variants conferring protection against infectious diseases and susceptibility to some chronic inflammatory or autoimmune conditions.22,23,24,25,26,27 Classic examples are the human leukocyte antigen (HLA) loci, variants of which are thought to be under pathogen-driven positive selection28 and to considerably increase the risk of autoimmune disease (e.g., HLA-B27 and ankylosing spondyloarthritis and HLA-DQ8 and type 1 diabetes).29,30,31,32,33 Another example is the common TYK2 P1104A variant, which protects against autoimmune phenotypes (odds ratio [OR] = 0.1–0.3) but, in the homozygous state, confers a predisposition to mycobacterium-related infectious diseases (OR >10).25,26,34,35,36,37,38,39 Nevertheless, the evidence that antagonistically pleiotropic variants have been selected in humans remains circumstantial.
Detection of a legacy of positive selection (rapid increase in the frequency of a beneficial allele) or negative selection (the removal of a deleterious variant) in human populations has long been limited to statistical inference from patterns of genetic variation in contemporary individuals. In recent years, the increasing availability of ancient DNA (aDNA) data has greatly facilitated the study of human genetic adaptation over time. Direct comparisons of past and current allele frequencies have provided new insight into the genes and functions involved in human adaptation to environmental changes following cultural transitions.40,41,42 A pioneering study based on 230 Eurasian samples across the Holocene period detected 12 positively selected loci associated with diet and skin pigmentation but also host defense against pathogens.43 Another study explored how the arrival of Europeans in the Americas altered the exposure of Native Americans to new pathogens. Comparisons of genomic data from a Canadian First Nation population dating from before and after the first contact with Europeans showed a recent decrease in the frequency of formerly beneficial HLA-DQA1 alleles in the native population.44 These studies highlight how the analysis of ancient genomes can reveal specific genetic variants under selection, but we still lack a comprehensive picture of the selective forces affecting host defense genes during human history and of the times at which these forces operated.
Ancient genomics provides us with a unique opportunity to determine whether resistance to infectious diseases and susceptibility to inflammatory diseases have changed in the recent past. Furthermore, aDNA data can be used to detect variants subject to complex models of selection, such as time-dependent selection, including selection on de novo or standing variation caused by sudden environmental changes (e.g., epidemics). For example, a recent study revealed that negative selection has led to a rapid decrease in the frequency of a 30,000-year-old tuberculosis (TB) risk variant over the last 2,000 years, suggesting that TB has recently imposed a heavy burden in Europeans.35 In this study, we sought to reconstruct the history of host-pathogen interactions by detecting immunity gene (IG) variants affected by natural selection, thereby modulating infectious and/or inflammatory disease risk, over the last 10,000 years. To this end, we explored the strength and timing of both positive and negative selection at the genome-wide scale by analyzing 2,879 ancient and modern European genomes in an ancestry-aware approximate Bayesian computation (ABC) framework.
Results
Inferring the intensity and onset of positive selection from aDNA data
We assembled a genome-wide dataset corresponding to 2,376 ancient and 503 modern individuals of western Eurasian ancestry and computed allele frequency trajectories for 1,233,013 polymorphic sites over a time transect covering the Neolithic period, the Bronze Age, the Iron Age, the Middle Ages, and the present (STAR Methods). Using simulation-based ABC45 and the calculated trajectories, we estimated the selection coefficient (s) and the time of selection onset (T) for each derived allele. We considered a variant to be a candidate for positive selection if its s value was higher than that of 99% of s estimates for simulated neutral variants (psel < 0.01). We used a demographic model35 accounting for (1) the major migratory movements contributing to the genetic diversity of contemporary Europeans, i.e., the arrival of Anatolian farmers ∼8,500 ya46,47 and that of populations associated with the Yamnaya culture around ∼4,500 ya;48,49 (2) the uneven sampling across time, with the matching of simulated data on the observed numbers of ancient and modern DNA data; and (3) ancestry variation across epochs, by matching on observed Anatolian and Pontic Steppe ancestries (STAR Methods).
Our simulations showed that the estimation of s was highly accurate (r2 = 0.93 between simulated and estimated values; Figures S1A–S1E) and that the power to detect selection from s, in the presence of genetic drift and admixture, was >75% regardless of variant frequency and intensity and time of selection onset (Figures S2A–S2C). Furthermore, the detection of selection signals was not spuriously affected by false positives, as measured by type I error rates (Figure S2D). Although the accuracy of estimation was lower for T (r2 = 0.76; Figure S1B), our approach nevertheless distinguished between variants selected before and after the beginning of the Bronze Age, ∼4,500 ya (F1 score = 0.85; Figure S1C). When applying our ABC approach to the empirical data, we replicated the 12 loci previously shown to be subject to positive selection in Europe43 (based on the same criterion of ≥3 candidate variants for positive selection per locus). These loci included genes associated with host defense (HLA and TLR10-TLR1-TLR6), skin pigmentation (SLC45A2 and GRM5), and metabolism (MCM6/LCT, SLC22A4, and FADS1) (Figure 1A; Table S1). The accuracy of our approach is further illustrated by the s estimate for the lactase persistence allele (rs4988235), which was 8.1% (95% confidence interval [CI]: 0.06–0.09) with selection beginning 6,102 ya (3,150–8,683 ya), as previously reported.50,51 More generally, we observed more variants with high s values than expected under neutrality, which supports the presence of true positive signals among loci showing the highest s values (Figure S2E). Our approach expands previous studies by not only identifying known and previously unreported candidates for selection but also providing estimates of the selection parameters, s and T, over the entire genome.
Figure 1.

Genome-wide detection of positive selection
(A) Test for positive selection (−log10(psel)) and selection coefficient estimates (s) for each genomic marker in the aDNA dataset with psel < 0.1. The empirical null distribution was approximated by a beta distribution, and p values correspond to the minimum between the empirical p value and the p value from the beta approximation. The dashed line indicates −log10(psel) = 2. Upward-pointing pink triangles and downward-pointing yellow triangles correspond to the 89 SNPs with the smallest psel at each of the 89 candidate loci selected before and after the beginning of the Bronze Age, respectively. The time of selection onset was estimated as the mean T across all SNPs with psel < 10−2 at each locus. Several candidate genes for positive selection are indicated, and host defense genes are highlighted in bold. Variants with psel < 10−4 outside the 89 enriched loci are colored in gray.
(B and C) Frequency trajectory of the most significant variant at (B) the LCT locus (rs4988235) and (C) the ABO locus (rs8176635). Gray shading around the frequency trajectory indicates the lower and upper bounds of the 95% CI for variant frequency estimation in each epoch.
(D) Mean number of times a single randomly selected candidate SNP per LD group has an estimated T before or after the beginning of the Bronze Age across 1,000 replicates. The error bar for each bar plot indicates the standard deviation (SD) of the distribution obtained. We also included the estimate based on the mean T of all SNPs with psel < 0.01 for each of the 89 LD groups (rectangles), or based on the SNP with the smallest psel for each LD group (circles).
See also Figures S1–S4 and Tables S1, S2, and S3.
Positive selection has pervasively affected host defense genes in Europeans
We identified the strongest signals of time-dependent positive selection by focusing on the top 2.5% of loci with the highest proportions of candidate variants, i.e., regions defined on the basis of linkage disequilibrium (LD) groups containing >7.5% of the candidate positively selected, derived variants (psel < 0.01; STAR Methods). We identified 102 candidate loci for positive selection, 89 of which were non-consecutive. These loci were enriched in Gene Ontology (GO) categories such as “antigen processing and presentation of endogenous antigen” (Bonferroni-corrected padj = 0.01), “viral life cycle” (padj = 0.03), and “positive regulation of leukocyte activation” (padj = 0.05), as well as “transport vesicle,” “vesicle membrane,” “luminal side of endoplasmic reticulum membrane,” “cell surface,” and “vesicle-mediated transport.” These 89 loci were also enriched in a curated list of IGs (STAR Methods), whether we considered all candidate genes (OR = 1.6, p = 8.0 × 10−3) or, to account for gene clustering, a single gene per locus (28/89 loci; OR = 1.6, p = 0.049). These genes included the OAS cluster (OAS1-OAS2-OAS3-RNASEL), which degrades single-stranded RNA (ssRNA) on sensing of cytosolic double-stranded RNA (dsRNA),52 and genes underlying inborn errors of immunity (IEI), such as AICDA, biallelic loss-of-function mutations of which cause antibody deficiencies (MIM: 605258 and 605257), and PLCG2, monoallelic gain-of-function mutations of which underlie autoinflammatory disorders (MIM: 614878 and 614468).
One of the strongest signals identified was close to the ABO gene (smallest psel; Table S2), which encodes the ABO blood group system. We found that alleles tagging the A and B blood groups53 are candidates for positive selection (psel < 0.01; Table S3), suggesting that the frequency of individuals with the A, AB, and, particularly, B groups has increased through positive selection over the last few millennia. The A and B groups have been shown to confer limited protection (OR > 0.94) against childhood ear infections54,55 and mild susceptibility (OR < 1.13) to malaria and coronavirus disease 2019 (COVID-19),53,56 consistent with the action of balancing selection.57 Our analyses indicate that the bulk of recent genetic adaptation in Europe has primarily concerned genes involved in the host response to infection, suggesting that pathogens have imposed strong selective pressures over the last few millennia.
Genetic adaptation has occurred principally since the Neolithic period
We then explored the time at which positive selection began at the 89 candidate loci by determining whether selection occurred before or after the start of the Bronze Age (Figure S1C). For more than 80% of loci, selection was estimated to have begun after the beginning of the Bronze Age (<4,500 ya) (Figures 1B–1D). Indeed, the distribution of T estimates for the top 89 variants was not consistent with a uniform distribution across time (p = 1.3 × 10−4), with a time of selection onset more recent than expected (Tmean = 3,327 ya; Figure S3A; Table S2). This result cannot be explained by differences in detection power because our approach has a higher power for variants with selection beginning at earlier periods (Figures S2B and S2C). Using 100 distributions of T estimates obtained from 89 simulated variants drawn with uniform durations of selection and matched for allele frequency and selection intensity, we also checked that the distributions were not skewed toward T estimates postdating the beginning of the Bronze Age (padj > 0.05 for all 100 distributions; Figure S3A), suggesting no methodological bias because of the nature of the data used. Furthermore, the excess of signals of positive selection detected after the Bronze Age is not due to reduced power to detect short-lived events of selection starting in the Neolithic period (e.g., epidemics caused by extinct pathogens), and the top 89 variants present frequency trajectories consistent with their estimated selection onsets (Figures S3B–S3D). Finally, given that the ancient and modern DNA datasets were processed differently, batch effects could have resulted in a sudden change in allele frequency in the last epoch and, therefore, a bias of T toward more recent times. However, when estimating T for aDNA only, we again obtained a significantly larger fraction of estimates <4,500 ya (n = 67/89; p < 10−3; Figure S3E). Together, these analyses support a scenario in which most of the detected positive selection events have occurred since the Neolithic period.
We next reasoned that the arrival of populations from the Pontic-Caspian region ∼4,500 ya may have brought beneficial variants that continued to be subject to positive selection in the resulting admixed population, a process known as adaptive admixture.58,59 In this case, we would expect to see a positive correlation between Pontic Steppe ancestry and the probability of carrying positively selected alleles. However, no significant differences were observed between positively selected alleles and matched controls (padj > 0.05 for all matched samples; STAR Methods), and no systematic bias was observed in the ancestry of simulated and observed aDNA samples (Figure S4A). Our results therefore rule out adaptive admixture as the main driver of positive selection in post-Neolithic Europe, instead suggesting that there has been selection on standing variation; most of the alleles that became beneficial after the Bronze Age were already present in Neolithic Europe (Figure S4B).
Regulatory elements of IGs have been a prime substrate of positive selection
We then investigated in silico the functional effects of candidate positively selected variants (n = 1,846). We found that these variants were strongly enriched in missense variants (OR = 3.8, p = 8.8 × 10−21) (Figure 2A). Furthermore, IGs harboring missense variants were found to be significantly enriched in positively selected variants relative to the rest of the candidate loci (p = 6.0 × 10−3). At the genome-wide scale, we detected 11 missense variants of IGs with signals of positive selection (MYH9, CHAT, NLRC5, INPP5D, SELE, C3, PSMB8, CFB, PIK3R1, RIPK2, CD300E). Eight of these variants were associated with hematopoietic cell traits and infectious or autoimmune disorders, such as hematopoietic cell counts, viral warts, and type 1 diabetes, in phenome-wide association studies (p < 10−5; Figure 2B). However, given that most positively selected variants (>96%) are non-coding, we investigated whether variation in gene expression could account for the observed selection signals. By pruning variants in LD (r2 > 0.6) from the aDNA dataset and matching derived allele frequencies (DAFs) (STAR Methods), we found that candidate variants were enriched in cis-expression quantitative trait loci (cis-eQTLs) in whole blood, particularly for strong eQTL associations (e.g., OR = 2.9; p = 3.3 × 10−21 for eQTLs with p < 10−50; Figure 2A). For example, the top candidate variant for the OAS cluster, rs1859332>T, was found to be linked (r2 = 0.97) to the OAS1 splice QTL rs10774679>C (isoform p46), which is of Neanderthal origin.60 Interestingly, this isoform has been associated with protection against severe COVID-19,61,62 suggesting that past coronavirus-like viruses may have driven selection at this locus.
Figure 2.

Genetic variants and host defense traits targeted by positive selection
(A) Enrichments in missense, whole blood cis-eQTL, or ASB variants for variants with psel < 10−2 and psel < 10−4, across the genome (“All loci”) or within the 89 candidate positively selected loci (“89 loci”). Only variants of transcripts annotated as “protein coding” were considered for the analysis of variant annotations. Red and black circles indicate significant and non-significant enrichments, respectively, and error bars indicate the boundaries of the 95% CI for the estimated odds ratios. The three eQTL groups were defined on the basis of the statistical significance of the eQTL association, peQTL < 5 × 10−8, peQTL <10−15, or peQTL <10−50.
(B) Significant associations (p < 10−5; https://genetics.opentargets.org) in PheWAS between positively selected missense variants overlapping host defense genes and hematopoietic traits, infections, or inflammatory disorders.
(C) Enrichments in variants associated with the 36 hematopoietic traits studied for variants with psel <10−2, across the genome (“All loci”) or within the 89 candidate positively selected loci (“89 loci”). Red and black circles indicate significant and non-significant enrichments, respectively, and error bars indicate the boundaries of the 95% CI for the estimated odds ratios. Orange and gray triangles indicate significant and non-significant enrichments, respectively, after exclusion of the HLA locus.
(D) Left: mean polygenic score across four equally spaced epochs for each of the hematopoietic traits. Right: −log10(p value) for the significant increases or decreases in each hematopoietic trait over the last 10,000 years (dot), before (upward-pointing triangle) or after (downward-pointing triangle) the beginning of the Bronze Age. Light gray symbols represent non-significant trajectories. The inset shows the decrease in polygenic score over time for platelet counts, which has the highest −log10(p value). The black line is the regression line. (C and D) Traits followed by “_p” indicate the percentage of the cell type considered, whereas other traits are absolute counts.
See also Figure S5A.
Positively selected variants are also enriched in variants affecting transcription factor (TF) binding (OR = 1.75; p = 1.6 × 10−6; Figure 2A), based on allele-specific binding (ASB) events detected in chromatin immunoprecipitation sequencing (ChIP-seq) data covering 1,025 human TFs and 566 cell types (false discovery rate [FDR] < 0.01).63 For example, the positively selected rs3771180>T variant (s = 0.015, T = 2,250, psel = 0.005) at the IL1RL1 locus disturbs the 7-bp binding motif of JUND (OR = 3.07; padj = 5.7 × 10−12) and is associated with a lower risk of asthma64 and higher neutrophil levels.14 Moreover, 17 of the 265 TFs with at least five ASBs have ASB-associated variants enriched in positive selection signals, and the genes closest to these ASBs are enriched in IGs (OR = 1.24; p = 0.04). Collectively, these results provide insight into the regulatory mechanisms underlying recent human adaptation in the context of immunity to infection.
Directional selection has affected leukocytic lineages since the Bronze Age
We analyzed epigenomic data from ENCODE,65 to identify the tissues for which the regulatory elements were the most enriched in positively selected variants. In tests with matched controls and correction for multiple testing, we found an enrichment in positively selected variants at DNase-hypersensitive sites in 24 of the 41 tissues and cell types tested, including monocytes (padj = 6.1 × 10−7), tonsils (padj = 2.1 × 10−6), and blood (padj = 1.0 × 10−5) (STAR Methods). These results, together with the enrichment in ASB in candidate variants close to IGs, led us to investigate whether positively selected variants affect hematopoiesis by altering hematopoietic cell fractions.
Taking LD and DAF into account, we found that variants associated with hematopoietic cell composition in genome-wide association studies (GWASs) were strongly enriched in positively selected variants (OR = 10, p = 7.2 × 10−65) (Figure 2C). For each of the 36 hematopoietic traits, we analyzed the frequency trajectories of all the trait-increasing alleles (pGWAS < 5.0 × 10−8) with a polygenic score derived from GWAS data,40 within the studied candidate positively selected LD groups (Figure S5A; STAR Methods). This score integrates tens or hundreds of generally small effect sizes, which we assessed over hundreds of generations. We found that polygenic scores for platelet and reticulocyte counts had decreased significantly over the last 10,000 years, whereas scores for mean platelet volume and mean corpuscular (red blood cell) volume, as well as the mean mass of hemoglobin per red blood cell, had significantly increased (Figure 2D). Likewise, polygenic scores for the proportion of eosinophils among granulocytes were found to have significantly decreased over the study period, whereas the proportions of lymphocytes and monocytes among white blood cells and neutrophil counts had significantly increased. Importantly, the polygenic scores of seven traits, including traits relating to platelets, reticulocytes, lymphocytes, and monocytes, were found to have significantly increased or decreased in post-Neolithic Europe, whereas no trait other than the number of granulocytes changed significantly during the Neolithic period. These observations are consistent with directional selection acting principally on hematopoietic lineages after the Bronze Age.
Joint temporal increase in resistance to infection and the risk of inflammation
It has been suggested that past selection relating to host-pathogen interactions has favored host resistance alleles that, today, increase the prevalence of chronic inflammatory disorders because of antagonistic pleiotropy.4,19,20,21,23,24 We investigated the evolution of genetic susceptibility to inflammatory disorders and resistance to infectious disease by assembling the results for (1) 40 GWASs of infectious diseases, including severe infections, such as TB, hepatitis B and C, AIDS, and COVID-19 (COVID-19 HGI, release 7), and surrogate phenotypes, such as positive TB test (based on tuberculin skin test) or tonsillectomy; and (2) 30 GWASs of inflammatory/autoimmune disorders, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and inflammatory bowel disease (IBD) (Table S4). For both independent and randomly matched variants, we found that variants significantly associated with both infectious and inflammatory traits (i.e., pleiotropic variants; pGWAS < 5.0 × 10−8) were more prevalent than expected by chance (OR = 132, p = 7.4 × 10−28 and p < 10−3, respectively; STAR Methods), suggesting a shared genetic architecture. Furthermore, that the estimated s values for these pleiotropic variants are stronger than for randomly matched variants (Wilcoxon p = 0.02) supports their adaptive nature.
We used polygenic risk scores (PRSs) to explore changes in the frequency of risk alleles for infectious or inflammatory disorders (pGWAS < 5 × 10−8) over time (STAR Methods). We found that the PRS for the merged set of all inflammatory/autoimmune disorders had significantly increased over the last 10,000 years, whereas that for infectious diseases had significantly decreased (Figure 3A). Furthermore, the absolute median s of PRS variants increases with GWAS effect size, reaching ultimately significant positive and negative median s coefficients for autoimmune and infectious traits, respectively (Figure S5B). Considering the coverage, location, and ancestry of the samples, the risk of Crohn's disease (CD) or of IBD generally has increased significantly, whereas that of severe COVID-19 has decreased significantly, essentially since the Neolithic period (Figures 3A and 3B). Given the recent emergence of SARS-CoV-2, the infectious agent of COVID-19, this finding suggests that other related pathogens have exerted selection pressure in recent European history.
Figure 3.

Resistance to infection and risk of inflammation have increased since the Neolithic period
(A) Polygenic scores for infectious and autoimmune traits as a function of time, over the last 10,000 years. COVID A2 and B2 indicate critical COVID-19 cases versus the general population and hospitalized COVID-19 cases versus the general population, respectively. Dot size scale with the number of SNPs genotyped in the individual. Dark gray lines are the regression lines for a model adjusted for ancestry and geographic location. p values indicate the significance of the regression model, over the last 10 millennia (p), before the Bronze Age (pBefore BA) or since the beginning of the Bronze Age (pAfter BA). Beta values for the regression model considering only samples dating from before (βBefore BA) or after (βAfter BA) the beginning of the Bronze Age are shown. Green, pink, and blue dots indicate individuals with >75% Western hunter-gatherer, Anatolian, or Pontic Steppe ancestry, respectively. Individuals with mixed ancestries are shown in gray.
(B) Polygenic risk score for CD as a function of the geographic location and age of the samples, obtained with the “bleiglas” package of R version 3.6.2. Individuals with >75% Anatolian ancestry are represented by triangles, and the others are represented by circles.
(C) Polygenic risk score for CD as a function of time, after removal of the SNPs most significantly associated with sample age.
(D) Presence or absence of the CD risk allele rs2188962>T as a function of the geographic location and the age of ancient samples.
We then searched for the specific genetic variants making the largest contribution to the changes over time in the risks of IBD/CD and severe COVID-19. Taking multiple testing into account, we found that four risk alleles for IBD/CD and one protective allele for COVID-19 had significantly increased in frequency over time (Figure 3C; Table S5). The risk alleles concerned were the IBD- and/or CD-associated rs1456896>T, rs2188962>T, rs11066188>A, and rs492602>G alleles and the COVID-19-associated rs10774679>C allele. The first three of these variants increase the expression of IRF1, SH2B3, and IKZF1, respectively, in blood (rs2188962, s = 0.022, T = 8,075; rs11066188, s = 0.016, T = 4,045; rs1456896>C, s = 0.007, T = 3,264; Figure 3D). These genes have been shown to protect against several infectious agents,27,66,67 suggesting that increases in their expression may be beneficial, even if they slightly increase autoimmunity risk. The fourth IBD-associated risk allele, rs492602>G (s = 0.0015, T = 6,823), is a FUT2 variant in complete LD (r2 = 0.99) with the null allele rs601338>A, which confers monogenic resistance to infections with intestinal norovirus68 and respiratory viruses.69 Finally, the COVID-19-associated variant rs10774679>C (s = 0.013, T = 3,617) is linked (r2 = 0.9) to the OAS1 splice variant rs10774671>G, which is also associated with slightly higher susceptibility to IBD (OR = 1.08; p = 1.4 × 10−3; FINNGEN_R5_K11_IBD). Overall, these analyses provide support for a role of selection in increasing autoimmune disease risk over recent millennia, particularly for gastrointestinal inflammatory traits, probably because of antagonistic pleiotropy.
Searching for the footprints of time-dependent negative selection
Given the observed links between candidate selected variants and host defense, we then used our approach to identify variants that may increase infectious disease risk. Specifically, we searched for variants of genes involved in host-pathogen interactions that have been under time-dependent negative selection, that is, variants that have become deleterious since the Neolithic period. A typical example is TYK2 P1104A, which we have previously shown to underlie clinical TB34,39 and to have evolved under negative selection over the last 2,000 years, probably because of an increase in the pressure imposed by Mycobacterium tuberculosis.35 We removed the 89 positively selected candidate regions and loci presenting positive selection signals in contemporary Europeans (STAR Methods) and then searched for candidate deleterious variants displaying significant decreases in DAF in the rest of the genome (93% of all LD groups).
We first checked that the s and T estimates for alleles under negative selection are as accurate as those for positively selected alleles (Figures S1D, S1E, S2B, and S2F). We confirmed that variants with a low DAF (<5% across epochs) are strongly enriched in candidate negatively selected variants (OR = 4.8, p = 2.7 × 10−210), as opposed to positively selected alleles (OR = 0.64, p = 4.2 × 10−7) (Table S6). We found that negative selection had a broader impact across the genome; 25% of LD groups harbor candidate negatively selected variants (psel < 10−4), whereas only 3% harbor positively selected alleles (Figure 4). By focusing on negatively selected missense variants at conserved positions (genomic evolutionary rate profiling GERP score > 4; n = 50; Table S7), we found that the vast majority of them (41/50; 82%) had an estimated onset of selection <4,500 ya, as for the positively selected variants (Figure 4D). Focusing on IGs, we identified six negatively selected missense variants: LBP (lipopolysaccharide [LPS]-binding protein) D283G (rs2232607), TNRC6A P788S (rs3803716), C1S R119H (rs12146727), IL23R R381Q (rs11209026), TLR3 L412F (rs3775291), and TYK2 P1104A (rs34536443). These time-series analyses provide compelling evidence for negative selection against variants of host defense genes in recent millennia.
Figure 4.

Genome-wide detection of negative selection
(A) Test for negative selection (−log10(p)) and selection coefficient estimates (s) for each genomic marker in the capture dataset as in Figure 1A. Red and yellow triangles indicate missense variants at conserved positions (GERP score > 4) under negative selection, starting before or after the beginning of the Bronze Age, respectively. The dashed line indicates a −log10(psel) = 2. The triangles for the six candidate negatively selected variants of immunity genes (IGs) are outlined in black.
(B) Frequency trajectory for the strongest candidate negatively selected variant, LBP D283G.
(C) Frequency trajectory for the candidate negatively selected missense variant, IL23R R381Q.
(D) Number of candidate variants under negative selection beginning before or after the beginning of the Bronze Age.
Negatively selected variants have a functional impact on immune functions
Finally, we investigated the biochemical or functional impact of several of these negatively selected variants on immune phenotypes. We first focused on the LBP D283G variant, which has the strongest negative selection signal (s = −0.018, T = 3,084). LBP encodes the LBP, which senses LPS, a major component of the outer membrane of Gram-negative bacteria, and initiates immune responses that prime host defense mechanisms against further infection.70 We investigated whether this variant, which has decreased in frequency from 6% to 1.2% over the last 5,000 years, had a biochemical impact on LBP expression and/or function. We transiently transfected HEK293T cells with plasmids encoding the ancestral or the D283G LBP cDNAs, including the previously reported P333L hypomorphic variant71 for comparison purposes. We confirmed that transfection efficiency was the same for all plasmids by qRT-PCR (Figure 5A). The ancestral and LBP D283G proteins were detected at the expected molecular weight by western blot (∼60 kDa) with an LBP-specific antibody and an antibody against the C-terminal tag (Figure 5B). However, D283G levels were lower than ancestral protein levels, whereas P333L was not detectable. The low levels of D283G protein were confirmed by ELISA (Figure 5C), suggesting that this variant may be less stable when secreted. We showed that the D283G and ancestral proteins have similar LPS-binding capacities (Figure 5D), demonstrating that this variant does not affect this function. These findings suggest that D283G alters LBP stability, resulting in lower LBP protein levels.
Figure 5.

Functional impact of negatively selected variants
(A–D) Functional study of the LBP D283G variant. (A) qRT-PCR for LBP on cDNA from HEK293T cells non-transfected (NT) or transfected with an empty plasmid (EV), or plasmids encoding ancestral or derived LBP. Dots indicate three independent experiments, and the height of each bar their mean values. (B) Western blot of whole-cell lysates or cell culture supernatants from HEK293T cells either left NT, transfected with an EV, or C-terminally tagged plasmids expressing ancestral or derived LBP forms. LBP was detected with a polyclonal anti-LBP antibody and an antibody against the C-terminal DDK tag. An antibody against GADPH was used as loading control. The results shown are representative of three independent experiments. (C) LBP concentration in cell culture supernatants from transfected HEK293T cells as measured by ELISA. Dots indicate three independent experiments, and the height of each bar their mean values. (D) Binding of ancestral or derived LBP collected from cell culture supernatant at increasing concentrations to LPS and Pam2CSK4 assessed by a binding assay. Each point represents the mean of two biological replicates ± SD.
(E and F) Functional study of the IL23R R381Q variant. (E) T cell blasts from seven healthy controls homozygous for the ancestral variant, seven patients homozygous for the derived IL23R R381Q variant, and an IL-12Rβ1-deficient patient were left unstimulated or were stimulated with IL-23 (ranging from 1 to 100 ng/mL) or IFN-α2b as a positive control. STAT3 phosphorylation was evaluated by flow cytometry. A Wilcoxon test was used to assess whether observed control and IL23R R381Q-derived values belong to the same population. ∗p < 0.05, ∗∗p < 0.01. (F) T cell blasts from nine healthy controls homozygous for the ancestral IL23R variant, two individuals homozygous for IL23R R381Q, two homozygous for TYK2 P1104A, one TYK2−/− patient, and one IL-12Rβ1−/− patient were stimulated with IL-1β for 24 h and with IL-12 or IL-23 for 6 h. Then, RNA sequencing was performed. The impact of the IL23R R381Q on the IFN-γ network was dissected by performing gene set enrichment analysis (GSEA: fgsea) using hallmark gene sets (http://www.gsea-msigdb.org/) and a mean log2 fold-change (log2FC) in each condition. Dot heatmaps are shown for the 10 most strongly affected gene sets in healthy controls on IL-23 plus IL-1β stimulation when compared with IL-1β stimulation.
(G–I) Functional study of the TLR3 L412F variant. (G) Principal-component analysis (PCA) of RNA sequencing-quantified gene expression for two controls homozygous for the ancestral TLR3 variant, four TLR3 L412F homozygotes, and one complete TLR3-deficient primary fibroblast cell left non-stimulated (NS) or treated with poly(I:C) for 6 h. (H) Scatterplots of average log2FCs in RNA sequencing-quantified gene expression following poly(I:C) stimulation for 6 h, in primary fibroblast from four individuals homozygous for the TLR3 L412F variant versus two non-homozygous controls, and from one TLR3-deficient individual versus the same two controls. Each point represents a single gene. (I) Heatmaps of RNA sequencing-quantified gene induction (log2FC) relative to non-stimulated conditions, for the two controls, the four TLR3 L412F homozygotes, and the TLR3-deficient individual, treated with poly(I:C) + Lipofectamine (Lipo) for 8 h (left panel) or with poly(I:C) for 6 h (right panel). All transcripts with a Log2FC > 2 in controls are shown.
See also Table S11.
We then assessed the impact of the IL23R R381Q variant on protein function by stimulating T cell blasts from healthy controls homozygous for the ancestral allele (n = 7), IL23R R381Q homozygotes (n = 7), and one interleukin (IL)-12Rβ1-deficient patient, whose cells are unresponsive to both IL-12 and IL-23,72 with IL-23 or interferon (IFN)-α2b as a control. T cell blasts from controls responded to IL-23 by phosphorylating STAT3, whereas T cell blasts from IL23R R381Q homozygotes displayed an impaired STAT3 phosphorylation (Figure 5E). We next tested if reduced STAT3 phosphorylation would lead to decreased transcription of IL-23-dependent genes. We stimulated T cell blasts from IL23R R381Q homozygotes (n = 2), healthy controls homozygous for the ancestral allele (n = 9), individuals homozygous for TYK2 P1104A (n = 2), whose cells selectively respond poorly to IL-23,34 and autosomal recessive complete TYK2-deficient (n = 1) or IL-12Rβ1-deficient (n = 1) patients, with IL-12, IL-23, IL-1β, and IL-1β plus IL-23 for 6 h, and performed RNA sequencing. Cells from patients with complete TYK2 deficiency were previously shown to respond poorly to IL-10, IL-12, IL-23, and type I IFNs.73,74 We used IL-1β as an IL-23-sensitizing agent as previously reported.74 We found that although T cell blasts from IL23R R381Q homozygotes responded normally to IL-12 and IL-1β, their response to IL-23 alone or IL-23 plus IL-1β was impaired, as measured by reduced overall gene expression of the IFN-γ network (Figure 5F). Overall, T cell blasts from individuals homozygous for the IL23R R381Q variant displayed an impaired response to IL-23, in terms of both STAT3 phosphorylation and target gene induction.
Lastly, we focused on the TLR3 L412F variant, which has been previously tested experimentally by gene transfer, with different in vitro functional impacts of the exogenously expressed variant protein in different cell types and experimental conditions.75,76,77 Here, we assessed the impact of the endogenous protein. We performed RNA sequencing in primary fibroblasts from individuals homozygous for the ancestral allele (n = 2) or homozygous for TLR3 L412F (n = 4) on stimulation with extracellular polyinosinic-polycytidylic acid (poly(I:C)), which activates the endosomal TLR3 pathway. The average TLR3-dependent gene induction of TLR3 L412F homozygotes was significantly decreased (mean Δ log2 fold-change = 0.93; Wilcoxon p < 10−16), when compared with that of cells with the ancestral allele, with TLR3-deficient cells used as negative control (Figures 5G–5I). Conversely, their responses to intracellular poly(I:C) stimulation, which activates pathways other than TLR3, were similar to those of cells harboring the ancestral allele. These results suggest that TLR3 L412F homozygosity decreases endosomal poly(I:C) responsiveness in fibroblasts.
Discussion
Based on a time-series of human aDNA data and extensive computer simulations, we have delineated the genomic regions under the strongest selective pressure over the last 10,000 years of European history. Our results indicate that host defense genes are enriched in positive selection signals, with selected variants primarily involved in regulatory functions. We also found evidence suggesting that directional selection has operated on at least four leukocytic traits (|r| <0.6; Figure S5A) in recent millennia. Leukocytic lineages show evidence of having undergone positive selection, with the exception of eosinophils, which have decreased in proportion among granulocytes, possibly reflecting an evolutionary trade-off in favor of neutrophils, consistent with the apparently less essential role of eosinophils in immunity to infection.78 Conversely, non-leukocytic lineages, such as red blood cells, seem to have undergone changes in efficacy, given the observed decrease in the number of reticulocytes but also the increase in their mean size and in the concentration of hemoglobin per red blood cell. These results suggest that recent positive selection has targeted the regulatory machinery underlying immune cell variation, possibly as a result of temporal changes in pathogen exposure.35,79,80
The estimated times of onset of selection highlight the importance of the post-Neolithic period in the adaptive history of Europeans because most selection events detected, both positive and negative, postdate the beginning of the Bronze Age (<4,500 ya) and cannot result from reduced power to detect old short-lived selection events. Furthermore, our results support a history of selection preferentially targeting variants that were already segregating in Europe before the arrival of Pontic-Caspian groups.81 The increase in adaptation following the Neolithic period may be caused by the population growth that followed the “Neolithic decline,”82 with higher selection efficacy.83 Alternatively, selection pressures may have increased during the Bronze Age; the expansion of urban communities, greater human mobility, animal husbandry,84 and environmental changes85 may have favored the spread of epidemics, such as plague, as suggested by archeological,13 ancient microbial,79,80 and more recently, ancient human data.86
Our analyses also provide several lines of evidence supporting a significant contribution of antagonistic pleiotropy to the emergence of modern chronic diseases. First, selection over the last 10,000 years, particularly since the beginning of the Bronze Age, has likely led to a higher genetic risk of inflammatory gastrointestinal disorders.87 Second, the main risk variants for IBD/CD are located close to key IGs (IRF1, IKZF1, FUT2, and SH2B3), for which monogenic lesions confer susceptibility or resistance to infectious diseases.27,66,68 Third, we found that pleiotropic variants underlying infectious and inflammatory phenotypes are overrepresented among primary targets of positive selection in recent millennia.
Finally, our work highlights the value of adopting an evolutionary genomics approach, not only to determine the legacy of past epidemics in human genome diversity but also to identify negatively selected variants that potentially increase infectious disease risk. Besides the known TB risk TYK2 P1104A,39 candidate variants for negative selection include the hypomorphic LBP D283G altering protein expression, the IL23R R381Q impairing responses to IL-23, and the TLR3 L412F altering responses to TLR3-dependent viral stimuli. The functional impact on immune molecular phenotypes shown here for LBP D283G, IL23R R381Q, and TLR3 L412F suggests that they may increase risk for bacterial, mycobacterial, and viral diseases, respectively. Interestingly, TLR3 L412F has been associated with mild protection against autoimmune thyroid disease (OR = 0.93, p = 7.0 × 10−12),88 IL23R R381Q with decreased IBD risk (OR = 0.49),89 and TYK2 P1104A with protection against autoimmune/autoinflammatory disorders (ORs ranging from 0.1 to 0.3).39 These findings collectively support a model of antagonistic pleiotropy between infectious and autoimmune disorders.
In conclusion, this study shows that natural selection has targeted host defense genes over the last 10 millennia of European history, particularly since the start of the Bronze Age, and has probably contributed to present-day disparities in infectious and inflammatory disease risk.
Limitations of the study
This study assumes population continuity since the Bronze Age, but fine-scale migrations have probably affected the European gene pool in modern times. Nevertheless, aDNA data suggest that no major population turnover has occurred over the last two millennia,43,90,91,92 indicating that our results should be largely robust to unmodeled discontinuity. Furthermore, the spatially heterogeneous nature of the aDNA dataset used here reduces the power to detect loci undergoing local adaptation. Likewise, the array used to generate ancient genomes, originally designed for demographic purposes, does not capture most rare variants, particularly those that became very rare, or even extinct, as a result of negative selection, providing a partial view of the evolutionary past of west Eurasians. Finally, we caution that PRSs were not used here to predict the actual disease genetic risk of ancient individuals because their predictive accuracy can be decreased in the presence of allelic turnover93 or gene-by-environment interactions.94 Given that environmental exposures may have differed in nature and intensity, during and beyond the studied time frame, larger and denser sequence-based aDNA datasets, together with methodological improvements in population genetics, are required to replicate these findings and detect more subtle, region-specific selection events.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody produced in mouse | Merck | Cat#A8592; RRID:AB_439702 |
| Human LBP Antibody | R&D Systems | Cat#AF870-SP; RRID:AB_2138317 |
| GAPDH-HRP antibody | Santa Cruz biotechnology | Cat#sc-47724; RRID:AB_627678 |
| mouse anti-goat IgG-HRP | Santa Cruz biotechnology | Cat#sc-2354; RRID:AB_628490 |
| PE-conjugated anti-STAT3-pY705 antibody | BD Biosciences | Cat#612569; RRID:AB_399860 |
| Bacterial and virus strains | ||
| NEB® 10-beta Competent E. coli (High Efficiency) | New England Biolabs | Cat#C3019H |
| Chemicals, peptides, and recombinant proteins | ||
| PfuUltra II Fusion HotStart DNA Polymerase | Agilent technologies | Cat#600674 |
| DpnI | New England Biolabs | Cat#R0176L |
| X-tremeGENE™ 9 DNA Transfection Reagent | Merck | Cat#6365809001 |
| PowerUp™ SYBR™ Green Master Mix | ThermoFisher Scientific | Cat#A25742 |
| DTT, 1M | ThermoFisher Scientific | Cat#P2325 |
| Phenylmethylsulfonyl fluoride (PMSF) | Merck | Cat#10837091001 |
| PhosSTOP™ | Merck | Cat#4906837001 |
| 10% Criterion™ TGX™ Precast Midi Protein Gel, 18 well, 30 μL | BIO-RAD | Cat#5671034 |
| Clarity Western ECL Substrate, 500 mL | BIO-RAD | Cat#1705061 |
| LPS, E. Coli O111:B4 | Merck | Cat#LPS25 |
| Pam2CSK4 (Synthetic diacylated lipopeptide; TLR2/TLR6 agonist) | InvivoGen | Cat#tlrl-pm2s-1 |
| Trans-Blot Turbo Midi 0.2 μm PVDF Transfer Packs | BIO-RAD | Cat#1704157 |
| cOmplete™ ULTRA Tablets, Mini, EASYpack Protease Inhibitor Cocktail | Merck | Cat#5892970001 |
| Critical commercial assays | ||
| HiSpeed Plasmid Maxi Kit (25) | Qiagen | Cat#12663 |
| Quick-RNA Microprep Kit | Zymo research | Cat#R1051 |
| High-Capacity RNA-to-cDNA™ Kit | ThermoFisher Scientific | Cat#4387406 |
| Human LBP DuoSet ELISA | R&D Systems | Cat#DY870-05 |
| ImmunoCultTM Human CD3/CD28/CD2 T-cell activator | Stemcell | Cat#10970 |
| Human IL-2 Recombinant Protein | Gibco Thermo | Cat#PHC0023 |
| Recombinant Human IL-23 Protein | R&D/Bio techne | Cat#1290-IL-010 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34966 |
| Intron A | Merk | N/A |
| Lipofectamine™ 2000 Transfection Reagent | Invitrogen | Cat#11668019 |
| Polyinosinic-polycytidylic acid (poly(I:C)) | Tocris | Cat#4287 |
| Deposited data | ||
| Ancient genomes | https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data (v44.2) | Table S8 of this paper |
| Modern genomes | 1000 Genomes Project data;95; https://www.internationalgenome.org/ | Population label EUR |
| RNA-sequencing for IL23R R381Q | This paper | GEO: GSE216458 |
| RNA-sequencing for TLR3 L412F | This paper | GEO: GSE216304 |
| Original western blot for LBP D283G | This paper | Mendeley Data: http://doi.org/10.17632/kwdmpxw3np.1 |
| Original code | This paper | Mendeley Data: https://doi.org/10.17632/38d7vv96m9.1 |
| Experimental models: Cell lines | ||
| Human: HEK293T cells | ATCC | ATCC CRL-3216 |
| TLR3 E746X/P554S Human fibroblasts | Guo et al.77 | N/A |
| TLR3 L412F/L412F HOM1 Human fibroblasts | This paper | N/A |
| TLR3 L412F/L412F HOM2 Human fibroblasts | This paper | N/A |
| TLR3 L412F/L412F HOM3 Human fibroblasts | This paper | N/A |
| TLR3 L412F/L412F HOM4 Human fibroblasts | This paper | N/A |
| Control 1 Human fibroblasts | This paper | N/A |
| Control 2 Human fibroblasts | This paper | N/A |
| Oligonucleotides | ||
| See Table S11 for Primer sequences for the analysis of LBP D283G | N/A | |
| Recombinant DNA | ||
| LBP (NM_004139) Human Tagged ORF Clone pCMV6-Entry | OriGene | Cat#RC221961 |
| Software and algorithms | ||
| Online Mendelian Inheritance in Man | http://www.omim.org | OMIM |
| eQTLGen | https://www.eqtlgen.org | eQTLs |
| GWAS atlas | https://atlas.ctglab.nl/PheWAS | N/A |
| Protein Atlas | https://www.proteinatlas.org/ | N/A |
| ADASTRA | https://adastra.autosome.ru | ASB |
| Open Targets Genetics | https://genetics.opentargets.org | N/A |
| GWAS catalog | https://www.ebi.ac.uk/gwas/home | N/A |
| Gene set enrichment analysis (fgsea) | Korotkevich et al.96 | http://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp?collection=H |
| Gene ontology | Young et al.97 | GO |
| Selink | https://github.com/h-e-g/selink | iHs, iHH, nSL, DIND |
| Ldsc | Bulik-Sullivan et al.98 | Genetic correlations (Figure S5A) |
| ggplot2 v2.2.1 | http://ggplot2.org/; RRID:SCR_014601 | N/A |
| DESeq2 | Love MI et al.99 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ComplexHeatmapv1.17.1 | Gu et al.100 | 10.18129/B9.bioc.ComplexHeatmap |
| Other | ||
| ImmunoCult™-XF T Cell Expansion Medium | Stemcell | Cat#10981 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing | Takara | Cat#634888 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC-131-1024 |
| Immunity Genes | Deschamps et al.101 | IG |
| Epigenomic data from ENCODE | Davis et al.65 | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lluis Quintana-Murci (quintana@pasteur.fr).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Ancient samples
We analyzed 2,632 aDNA genomes (Table S8) (i) originating from burial sites in western Eurasia (−9< longitude (°) <42.5 and 36 < latitude (°) <70.1), (ii) genotyped for 1,233,013 polymorphic sites by ‘1240k capture’,43 (iii) showing genotype calls for at least 50,000 polymorphisms, and (iv) retrieved from the V44.3: January 2021 release at https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data. Metadata for these individuals was downloaded from this website and is available in Table S8. All individuals were treated as pseudohaploid (i.e., hemizygotes for either the reference or the alternative allele). Using READ,102 we manually removed 95 samples that were either annotated as duplicated or found to correspond to first-degree relatives of at least one other individual with higher coverage in the dataset.
Samples used for experimental analyses
For the experimental work conducted on cells from human subjects (see details below), informed consent was obtained from all the individuals participating to this project. The studies were approved by France-IDF II RB comité: C10-13 (N° IDRCB 2010-A00634-35) and C10-16 (N° IDRCB 2010-A00650-35).
Method details
Variant filtering
We removed variants for which the derived allele was present in fewer than five aDNA samples from the analysis. We also excluded variants absent from gnomAD v2.1.1 (ref. 103) and the 1,000 Genomes Project95 or for which the ancestral allele was not annotated in the 1,000 Genomes Project. Finally, we included only variants with the ‘PASS’ flag in gnomAD v2.1.1.
We controlled for potential artifacts due to undetected technical problems in the ‘1240k capture’ dataset, by comparing the allele frequencies obtained with those from shotgun sequencing data. We processed 952 available published aDNA shotgun data (FASTQ files) with a published pipeline91 to obtain pseudohaploid data comparable with those of the capture dataset. We combined all samples and retrieved 4,620,071 variants after filtering, accounting for 95% of those present in the ‘1240k capture’ dataset. Strikingly, nine of the top 10 variants in the capture dataset, ranked by s value, had a frequency trajectory different from that of the shotgun dataset, with a single, strong change in frequency in the present generation, suggesting a misestimation of frequency in either the ancient or modern dataset. For these variants, we found that, when both genotypes were called in the capture and shotgun datasets, they were consistent between datasets, but there was a high percentage of missingness in the capture dataset when shotgun-sequenced individuals were called homozygotes for the alternative allele (Table S9). This was not the case for genotypes called homozygous for the reference allele in the shotgun dataset, generating a frequency bias at the population level. We found that, whereas only 3% of all variants in the capture dataset were within or close to an indel (within 20 bp) with a frequency >5% in European populations, 90 and 44% of our top 10 and top 100 variants from the capture dataset, ranked by s value, respectively, were located in such a position. We therefore conservatively removed all SNPs close to (<50 bp) an indel with a frequency >5% in the European population from the capture dataset, and variants for which the increase (or decrease) in frequency over the last 1,000 years exceeded 10% (a highly unlikely scenario), thereby excluding 28,493 variants in total. Our final dataset consisted of 933,781 SNPs from 2,537 ancient genomes.
Allele frequency trajectories across time transects
For each SNP, we used the ancient genomes and 503 modern European genomes from the 1,000 Genomes Project (CEU, GBR, FIN, TSI and IBR populations),104 to compute time-series data corresponding to the trajectory of allele frequencies across various time transects. The ancient genomes were grouped by well-characterized historical periods.35 We considered the Neolithic (8,500-5,000 ya; n = 729 for the capture dataset), the Bronze Age (5,000–2,500 ya; n = 893), the Iron Age (2,500-1,250 ya; n = 319) and the Middle Ages (1,250–750 ya; n = 435). We excluded 190 samples dated to before 8,500 ya because the number of individuals for this long time period is relatively small. The genetic information for the variants in ancient and modern genomes was summarized in a five-dimensional frequency vector.
Ancestry estimation
We used factor analysis105 to estimate ancestry proportions at the individual level on a merged dataset consisting of 363 present-day European individuals from the 1,000 Genomes Project (IBS, TSI, GBR and FIN, available at the V42.4: March 1 2020 release) and all ancient samples (see above). Before ancestry analysis, samples were imputed with the ‘LEA’ R v3.6 package, as recommended elsewhere.105 We imputed individuals with high levels of genome coverage (>795,475 SNPs covered in the 1240k dataset; n = 421) first, to prevent bias due to the inclusion of low-coverage samples. The remaining individuals were then imputed one-by-one and added to the imputed set of high-coverage samples. Factors are interpreted as the principal components of a principal component analysis (PCA), but with temporal correction for present-day and ancient samples. We set the drift parameter so as to remove the effect of time on the Kth factor (K = 3 here), where K is the number of ancestral groups considered. As source populations, we used 41 samples annotated as Mesolithic hunter-gatherers, 25 as Anatolian farmers, and 17 as Yamnaya herders. After filtering, 143,081 high-quality SNPs were used to obtain ancestry proportion estimations.
Demographic model
The model used includes demographic parameters, such as divergence times, effective population sizes, migration rates and exponential growth rates of continental populations (ancestral African population, and West and East Eurasians), that are described in detail elsewhere35 and summarized here (Table S10). This model also accounts for the two major migratory movements that shaped the genetic diversity of current Europeans: the arrival of Anatolian farmers in Europe around 8,500 ya, admixing with the local Mesolithic hunter-gatherers, and that of populations of Yamnaya culture from the north of the Caucasus around 4,500 ya. We accounted for the over/under representation of a particular ancestry at selected epochs, by matching simulated ancestry proportions to the mean ancestry proportions of the observed samples, as described in ref. 35, and by performing the analyses at the SNP level taking into account their coverage in each individual.
Forward-in-time simulations
Computer simulations of an allele evolving under the aforementioned demographic model were performed with SLiM 3,106 as described elsewhere.35 Briefly, for each simulation, three main evolutionary parameters were randomly sampled from uniform prior distributions: the age of the mutation Tage ∼ U(1000, 106) ya, the time of selection onset T ∼ U(1000, 10,000) ya and selection strength, as measured by the selection coefficient s ∼ U(-0.05, 0) for negative selection and s ∼ U (0, 0.1) for positive selection, under an additive model (h = 0.5)107. The age of the mutation was defined as the point at which the mutation was introduced into the model in a randomly chosen population. Each observed ancient genome was randomly sampled from simulated diploid individuals at the generation corresponding to its calibrated, radiocarbon-based age, so that all aDNA samples (i.e., 729 Neolithic, 893 Bronze Age, 319 Iron Age, 453 Middle Ages), as well as the 503 modern samples, are simulated with each simulation. For each simulated ancient individual and each polymorphic site, we randomly sampled one allele to generate pseudohaploid data, mirroring the observed pseudohaploid empirical aDNA data used. Simulated present-day European individuals were randomly drawn from the last generation of the simulated population. Of note, variants were simulated one at a time, so that a single simulated allele frequency trajectory was obtained from each simulation.
ABC estimation
We applied the ABC approach45 to each of the genetic variants studied, as previously used to estimate the age (Tage), strength (s) and the time of onset (T) of selection for TYK2 P1104A.35 Parameter estimates were obtained from 400,000 simulations (for positive or negative selection) with underlying parameters drawn from predefined uniform prior distributions. Parameters were estimated from computer simulations best fitting the observed time series data for allele frequencies. Simulated and empirical time series data were described by a vector of K allele frequencies over K epochs, used as the summary statistics to fit the observed data. For each parameter, posterior distributions, point estimates (i.e., posterior mode) and the 95% CIs were obtained from the parameter values of the 1,000 simulations with simulated summary statistics the closest to the empirical ones (‘abc’ R package, method = “Loclinear”).
Simulation-based evaluation of the ABC approach
To evaluate the performance of our ABC approach, we simulated pseudo-empirical data incorporating the effects of drift and admixture characterizing recent European history, closely reproducing the pseudohaploid empirical aDNA data used in this study. Based on these pseudo-empirical data, we then assessed the accuracy of our ABC estimations by comparing parameter estimates to simulated values through cross-validation (Figure S1). We also assessed power to detect selection (Figures S2B and S2C) and type I error rates (Figure S2D). This simulation-based evaluation was performed by grouping simulated ancient individuals into the same time periods used to analyze empirical aDNA data (Neolithic, Bronze Age, Iron Age, Middle Ages and present).
Detection of selection
We used the ABC estimates of the selection coefficient and their 95% CI (under a negative or positive selection model) to detect selection acting on specific genetic variants. The empirical threshold for rejecting neutrality (i.e., type I error estimation) was determined by simulating ∼500,000 neutral alleles (s = 0) evolving under the same demographic model. We then estimated selection coefficients and their 95% CIs for each simulated neutral variant to obtain the distribution of s under neutrality (i.e., the null distribution). Simulated neutral SNPs were resampled such that the simulated and observed allele frequency spectra were identical. Finally, we determined empirical thresholds at the 1% nominal level, by calculating the 99th quantile (Q99) of the resulting s distributions. Rather than using the distribution of s to determine these thresholds, we used the empirical distribution of the lower bound of the 95% CI of the selection coefficient (sl), as a more conservative approach to provide empirical p values (psel) for each SNP. This approach, like all methods for detecting selection at the variant level, is not designed to infer the distribution of fitness effects (DFE)107 due to the loss of true selection signals at the high conservative thresholds used to detect robust selection candidates.
Empirical p value computation
The significance threshold varied with variant frequency, as expected given that the power to detect selected variants depends on allele frequency. We thus calculated the null distribution of the lower bound of the confidence interval of the selection coefficient (sl) by frequency bin, to obtain bin-dependent significance thresholds. This was performed for each frequency bin by computing the distribution of the estimated sl from allele frequency-matched (to the empirical aDNA dataset) simulated neutral variants belonging to the study bin, and then deriving quantiles at nominal values from it.
For the analysis of positive selection, we used the following allele frequency bins: [0.025–0.2]; [0.2–0.6] and [0.6–0.8], whereas for negative selection, we used: [0.025–0.05]; [0.05–0.1]; [0.1–0.2] and [0.2–0.8]. We showed that the method is robust to the choice of frequency bin boundaries (Figure S2C). The rationale for using frequency bins that differ between negative and positive selection is that the estimation accuracy of the s parameter and, therefore, the power to detect selected alleles depends on allele frequency and the mode of selection (Figures S1 and S2). Low-frequency variants under negative selection are hard to identify as they can be confused with low-frequency neutral variants decreasing in frequency just by chance. To calibrate bin boundaries, we therefore used cross-validation to assess the accuracy of the estimation of s (Figure S1) and verified that accuracy was good (high correlation between true and estimated values, r2 > 0.8). We also excluded variants in the [0–0.025] frequency bin, because estimation accuracy was poor for this bin, in both selection scenarios. Then, for each frequency bin, we computed the distribution of sl under the null hypothesis from simulated neutral variants. The number of allele frequency-matched, simulated neutral variants was approximately even across the bins used to compute the distribution of sl under the null hypothesis. We identified the bin to which a variant belongs by calculating, for each variant, the CI for allele frequency estimation at each epoch, according to an approximation to the normal distribution of the 95% binomial proportion CI. We obtained the maximum for the lower bound of these CI for each SNP. Finally, if this maximum lays between 2.5 and 5%, the variant was considered to belong to the bin [0.025–0.05]. The same rationale was used for the rest of the bins. We excluded from the analysis of positive selection, any variant for which the minimum higher bound of the 95% CI of the DAF was >80%, as such variants poorly matched the simulated data.
As the lowest level of empirical significance depends on the number of neutral simulations in each frequency bin, we approximated the empirical null distribution with a known theoretical distribution, to improve discrimination between very small p values. Given the shape of the empirical null distribution, we compared the null distribution to a gamma, a beta and a lognormal distribution, for which parameters were estimated with a maximum likelihood approach (R packages ‘fitdistrplus’ and ‘EnvStats’ (v. 3.6.0)). We generated a Cullen and Frey graph (kurtosis vs. skewness) with the R package ‘fitdistrplus’ (v. 3.6.0), to distinguish between our options, and obtained p values for the beta distribution that best adjusted the null empirical distribution.
Time of selection onset for positively selected loci
We evaluated the shape of the distribution of the onset of selection estimated for the top 89 positively selected variants (Figure S3A; Table S2, with mean T estimate of 3,327 ya), by simulating sets of 89 independent variants matching the allele frequency and the selection coefficient of the most significant variant for each positively selected LD group, and the estimated onset of selection. We investigated whether the frequency trajectories based on both ancient and modern DNA samples resulted in biased T estimations, due to differences in genotype calling between datasets, by re-estimating T values for the variant with the smallest psel at each of the 89 candidate positively selected loci using frequencies from aDNA only (Figure S3E). We thus repeated the ABC estimation for frequency trajectories, but we excluded the last epoch corresponding to current frequencies.
To investigate the occurrence of adaptive admixture, we averaged the Pontic Steppe proportion of all the carriers of each of the selected alleles (for different psel thresholds) or that of 89,000 random alleles (Figure S4B). We also checked that Pontic Steppe ancestry was similar between the carriers of the variant with smallest psel at each of the 89 candidate loci and the simulated carriers of the 1,000 simulated variants used for each estimation of the evolutionary parameters of such variants (Figure S4A).
LD grouping
We took LD into account, by using an LD map for Europeans constructed from whole-genome sequence data.108 This map displays additive “linkage disequilibrium unit” (LDU) distances, which can be used to define genetic units in which variants are in strong LD. Genomic windows were then defined as non-overlapping regions of 15 LDUs, referred to as LD groups. This grouping generated genomic units with a mean size of 660 kb, consistent with previous studies.43
Enrichment analyses for positively selected loci
We calculated enrichment for genes (IGs or GO annotations), variants (eQTLs, ASBs, GWAS variants) and variant annotations (e.g., “missense”), with 2 × 2 contingency tables with two predefined categories (e.g. variants with psel <0.01 vs. variants with psel ≥ 0.01; missense variants vs. others), from which we calculated ORs, 95% CIs and Fisher exact test p values (for cells with counts <20) or Chi-squared p values, with the “oddsratio” function of the R (v 3.6.0) package “epitools”. We used independent variants to determine enrichment, by pruning variants in LD with the plink command --indep-pairwise 100 10 0.6 --maf 0.01, on our aDNA dataset, thus removing variants with r2 > 0.6 in 100 kb windows, using sliding windows of 10 variants. For the HLA region, which is located between hg19 coordinates 27,298,200 and 34,036,446 of chromosome 6, we used a more conservative LD pruning method that considers 1,000 kb rather than 100 kb windows (plink command --indep-pairwise 1000 100 0.6 --maf 0.01), for variants with a minor allele frequency (MAF) > 1%. Where indicated, we also matched the DAF distribution of the pruned dataset to that of the studied group of variants (e.g., eQTLs or GWAS variants), using 5% frequency bins.
We calculated enrichment in IGs101 or GO annotations,97 by considering, for each LD group with >9 variants (4,134 LD groups), a binary variable, indicating whether the locus included an immunity gene or a gene with a given GO annotation, respectively. This was done to eliminate spurious enrichments due to the presence of gene clusters in a given LD group. For eQTL analyses, we used data from a meta-analysis of whole-blood cis-eQTLs. Briefly, whole blood cis-eQTL statistics were retrieved from the eQTLGen consortium data,109 which incorporates 37 whole blood gene expression datasets and provides Z score and p values for significant SNP-gene associations. We also used ENCODE data65 and DNase hypersensitive sites110 to estimate enrichment in positively selected variants for ENCODE tissues. Finally, for the study of hematopoietic traits, we retrieved GWAS data for counts or proportions of different heamtopoietic cell types (36 hematopoietic traits) from UK Biobank and INTERVAL study data.14
Calculations of polygenic scores
For the analysis of infectious, inflammatory and hematological traits, we calculated genetic values for each ancient individual as proposed elsewhere.40 Specifically, we weighted the presence/absence status of the most significant GWAS Bonferroni-significant variant (p < 5.0 × 10−8) by the GWAS-estimated effect size, for each LD group. Coverage was variable across ancient samples, and some SNPs were not present in all samples. We accounted for missing information by calculating a weighted proportion in which the estimated score was divided by the maximum possible score given the SNPs present in the sample. We then used weighted (on coverage) linear regression to investigate the association between polygenic score and ancient sample age. We included ancestry as a covariate in the model, by including the first four Factor components, and the geographic location of each sample (latitude and longitude). We compared the full model to a nested model without sample age, by performing a likelihood ratio test to obtain a p value [‘ANOVA(nested model, full model, test = ‘Chisq’)’]. For the stratified analysis, we divided the dataset into mutually exclusive ancestry groups. We categorized individuals as western European hunter-gatherers, Anatolian farmers, or Pontic steppe herders if they carried over 75% of the estimated respective ancestry component (for steppe individuals, we also required the individual to be < 5,000 years old). We also conducted this analysis for individuals classified as being from before or after the beginning of the Bronze Age. For the analyses of hematological traits, genetic correlations between all 36 traits were estimated using ‘ldsc’ (ref. 98) (Figure S5). For all analyses, we checked that, despite the consideration of only one variant per LD group, none of the variants were in LD with each other.
Note that the PRS used in this study, obtained from modern GWAS, may poorly estimate actual PRS across time because (i) causal loci may vary in number and frequency between GWAS on present-day cohorts and ancient populations, and (ii) effect sizes of the most significant GWAS hits may differ from the true effect sizes in ancient times. Thus, PRS were not used here to estimate the PRS for prehistoric populations, but rather to test whether infectious and autoimmune disorders have been under directional selection since the Neolithic.
Overlap between infectious and autoimmune GWAS variants
We searched for significant overlaps between aDNA variants tagging lead GWAS SNPs for infectious and autoimmune/inflammatory diseases or traits, by retrieving summary statistics for (i) 40 GWAS of infectious diseases and (ii) 30 GWAS of inflammatory/autoimmune phenotypes. We identified lead infectious or autoimmune disease-associated SNPs by retaining the variant with the highest GWAS effect size (OR) in consecutive 200 kb genomic windows (Table S4). We indicate in column ‘EXTRACTION’ of Table S4 whether the corresponding lead SNP was retrieved either from GWAS catalog (‘GWAS CATALOG’; for which we only have access to genome-wide significant SNPs) using as key the corresponding listed phenotype or from the full GWAS data of Tian et al.55 (‘TIAN’) for the corresponding listed infectious phenotype and GWAS atlas (‘GWAS ATLAS’; version v20191115) for IBD,89 CD,89 Ulcerative colitis89 and RA.111 We then used aDNA variants to tag (r2 > 0.6) the lead GWAS-significant variants (usually absent from the aDNA array) with the plink command: plink --show-tags aDNA_variant_list --list-all --tag-r2 0.6 --tag-kb 1000. We obtained a list of aDNA variants tagging infectious and/or autoimmune disease-associated lead SNPs. We then performed 1,000 samplings of random SNPs, matched for the DAF and number of LD groups of aDNA variants tagging either infectious or autoimmune traits. For each replicate, we calculated the number of overlapping variants and found that none was higher than the observed overlap (p < 10−3). We also used a different approach to test for an enrichment in pleiotropic variants, based on the pruning of aDNA-tagged SNPs (r2 = 0.6 and windows of 1 Mb) and the calculation of a classic OR for the resulting list of independent tagged SNPs. We assessed the enrichment in selection signals in the observed overlapping variants, by performing a Wilcoxon test to compare the s distribution of the observed overlapping variants to that of the 1,000 randomly sampled controls. Finally, the PRS and respective p and OR of the regression model were obtained for the lead GWAS SNPs present in the aDNA data, either for all infectious or for all autoimmune diseases considered together. PRS and the corresponding p and OR for CD, IBD, COVID A2 and COVID B2 were obtained for all GWAS-significant variants, because this information was available for such phenotypes. Of note, whereas no significant genetic correlations were found between non-overlapping infectious phenotypes (e.g., COVID A2 and COVID B2 were considered as overlapping) nor between non-overlapping inflammatory phenotypes, formal testing for genetic correlation, as done for hematopoietic traits, was not possible since, for the majority of traits, we only had access to the lead GWAS-significant SNPs.
Detection of negatively selected variants
We excluded positively selected variants from our list of candidate negatively selected variants, by calculating four haplotype-based statistics used to detect recent positive selection: iHS,112 iHH,113 nSL114 and DIND.115 This was done for all SNPs with a DAF >0.2 in Europeans of the 1,000 Genomes Project,104 with selink (http://github.com/h-e-g/selink) and a 100 kb genomic window. We computed the 99% quantiles (Q99) of each of the four distributions and the proportion of SNPs with scores higher than the respective Q99, in 200 kb sliding windows. Only windows with >9 variants were considered. We then retrieved all the genomic windows enriched for at least one of the four selection statistics, i.e., those with proportions on the top 1% of at least one of the four distributions. LD groups overlapping, completely or partially, at least one of these windows were then removed from the analysis of negative selection.
Functional analyses
LBP D283G
Site-directed mutagenesis and transient transfection for LBP
Site-directed mutagenesis was performed on a pCMV6 plasmid (#RC221961, OriGene) containing the LBP ancestral sequence, with appropriate primers (Table S11) and the Pfu Ultra II Fusion HS DNA (#600674, Agilent) polymerase, followed by digestion with DpnI (#R0176L, New England Biolabs). Plasmids were amplified in NEB-10 β competent E. coli (#C3019H, New England Biolabs) and purified with the HiSpeed Plasmid Maxi Kit (#12663, Qiagen). Transient transfection was carried out in HEK293T cells transfected with 1 μg of plasmid DNA in the presence of X-tremeGene9 DNA transfection reagent (#6365809001, Merck), according to the manufacturer’s instructions. Transfected cells were cultured at 37°C, under an atmosphere containing 5% CO2, in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. After 48 h, supernatants and whole-cell lysates were collected for subsequent experiments.
RNA isolation and RT-qPCR
Total RNA was extracted with the Quick-RNA MicroPrep Kit (#R1051, Zymo) according to the manufacturer’s instructions. Residual genomic DNA was removed by in-column DNase I digestion. We reverse-transcribed 1 μg of RNA with the High-Capacity RNA-to-cDNA Kit (#4387406, Thermo Fisher Scientific), and performed quantitative qPCR with PowerUp SYBR Green Master Mix (#A25742, Thermo Fisher Scientific) and the ViiA7 system (Thermo Fisher Scientific) with primers for LBP (PrimerBank ID 31652248c1 and 31652248c2) and GAPDH (PrimerBank ID 378404907c1) obtained from the PrimerBank database.116 Normalization of LBP mRNA was performed for each sample with GAPDH (ΔcT) and values are expressed as 2- ΔcT.
Protein isolation and western blotting
Whole-cell protein lysates were extracted in modified radioimmunoprecipitation assay buffer supplemented with protease inhibitors (#5892970001, Merck) and phosphatase inhibitor cocktail (#4906837001, Merck), 0.1 mM dithiothreitol (DTT; Life Technologies), and 1 mM PMSF (#10837091001, Merck). Protein extracts and supernatants were resolved by electrophoresis in Criterion TGX 10% precast gels (Bio-Rad), with the resulting bands transferred onto PVDF membranes (#1704157, Bio-Rad) with the Trans-blot turbo system (Bio-Rad). Membranes were probed by incubation for 1 h at room temperature with antibodies against LBP (#AF870-SP, R&D Systems, 1:2,000), DDK (#A8592, Merck, 1:10,000) and GAPDH (#sc-47724, Santa Cruz Biotechnology, 1:5,000). Proteins were detected by chemiluminescence with Clarity Western ECL substrate (#1705061, Bio-Rad) reagents.
ELISA for LBP and bacterial ligand binding
Supernatants from transfected HEK293T cells were analyzed for their LBP content by Human LBP DuoSet ELISA (#DY870–05, R&D Systems) according to the manufacturer’s instructions. Bacterial ligand binding was assessed on microtiter plates coated with 30 μg/mL LPS derived from E. coli O 111:B4 (#LPS25, Merck) or Pam2CSK4 (#tlrl-pm2s-1, InvivoGen) in 100 mM Na2CO3 (pH 9.6) for 18 h at 4°C. The plates were blocked by incubation with 0.005% Tween and 1% bovine serum albumin in PBS for 1 h at room temperature. Plates were then incubated with cell supernatants at various concentrations. Bound LBP was detected with the detection antibodies from the Human LBP DuoSet ELISA kit. Absorbance was read at 450 nm with a Victor X4 plate reader (Perkin Elmer).
IL23R R381Q
T cell blast culture
T-cell blasts were induced from 2 million peripheral blood mononuclear cells (PBMCs; fresh or cryopreserved) in 2 mL ImmunoCult™-XF T Cell Expansion Medium (StemCell Technologies, 10981) supplemented with 10 ng/mL IL-2 (Gibco, PHC0023) and 1:40 diluted ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator (StemCell Technologies, 10970). T-cell blasts were then cultured in ImmunoCult Medium with IL-2 for 2–3 weeks, before they were re-stimulated in the same manner.
Assessment of STAT3 phosphorylation by flow cytometry
Levels of pSTAT3 in T-cell blasts were determined in serum-starved cells. Approximately 1 × 106 cells were stained with Aqua Dead cell marker (Thermo Fisher Scientific) before being incubated for 30 min with IL-23 (100 ng/mL, R&D), IL-12 (50 ng/mL, R&D), or IFN-α2b (105 IU/mL, Intron A, Merk). Cells were then incubated at 37°C in Fix buffer I (1:1 vol, BD Biosciences), permeabilized by incubation at room temperature in Perm buffer III (BD Biosciences) and stained at 4°C with anti-STAT3-pY705 antibody (BD Biosciences, Cat: 612569, Clone: 4/P-STAT3, 1:25). The cells were analyzed on a Gallios flow cytometer (Beckman Coulter), and the results were analyzed with ‘FlowJo’ software.
T cell blast cytokine stimulation and RNA sequencing
T-cell blasts were starved off IL-2 over-night. Then, they were stimulated with 50 ng/mL IL-12 (R&D Systems, CAT: 219-IL-005) or 100 ng/mL IL-23 (R&D Systems, Cat: 1290-IL) for 6 h or with 2.5 ng/mL IL1β (R&D Systems, CAT: 201-LB-005)- for 24 h. Double stimulated cells were stimulated with IL-1β for 18 h and then with IL-23 and IL-1β for 6 h. Total RNA was isolated from cells using the Quick-RNA MicroPrep spin column kit (Zymogen, R1050), following manufacturer’s protocol. RNA was then submitted for RNA sequencing library preparation and deep-sequencing to Rockefeller University Genomics Core Facility.
RNA sequencing data analysis
RNA-sequencing was performed on the Illumina NovaSeq platform, with a read length of 100bp and read depth of 30–40 million reads. All FASTQ files passed QC and were aligned to reference genome GRCh38 using STAR (2.6.1day). BAM files were converted to a raw count’s expression matrix using ‘featurecount’. Raw count data was normalized using DEseq2. The Ensembl IDs targeting multiple genes were collapsed (average), and a final data matrix gene was generated for downstream analysis. Gene set enrichment analysis (GSEA) was conducted based on the fold-change (pairwise analysis of stimulated vs non-stimulated sample) ranking against the Hallmark (HM) gene sets (http://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp?collection=H).
TLR3 L412F
Cell culture and stimulation
Primary human fibroblasts were established from skin biopsy specimens from individuals that carry the homozygous TLR3 ancestral or derived L412F variation, or from a previously published patient with complete TLR3 deficiency.77 Fibroblasts were cultured in DMEM medium (GIBCO) with 10% fetal bovine serum (FBS) (GIBCO). 450.000 cells/well were seeded in 6-well plates and incubated for 16h. For intracellular poly(I:C) stimulation, cells were treated with Lipofectamine 2000: Poly(I:C) ratio of 1:5, for 8h with 10 μg/mL of poly(I:C). For Poly(I:C) extracellular stimulation, cells were treated for 6 h with 10 μg/mL of Poly(I:C). Non-stimulated cells were left untreated and harvested at the same time as intracellular or extracellular Poly(I:C) stimulated cells.
RNA sequencing data analysis
Total RNA extraction, library preparation, RNA sequencing and alignment/read counts quantification were performed as explained above. Differential gene expression analyses were conducted by contrasting the extracellular Poly(I:C)-stimulated samples or the intracellular Poly(I:C)-stimulated samples, with the non-stimulated samples. Genes displaying significant differences in expression were selected according to the following criteria: FDR ≤0.05 and |log2(FoldChange)| ≥ 1. Differential gene expression was plotted as a heatmap with ‘ComplexHeatmap’, and genes and samples were clustered based on complete linkage and the Euclidean distances of gene expression Z score values.
Acknowledgments
We thank all members of the Human Evolutionary Genetics Laboratory at Institut Pasteur, Paris, and Nicolas Rascovan, Bertrand Boisson, and Iain Mathieson for data sharing and helpful discussions. We acknowledge the help of the HPC Core Facility of Institut Pasteur for this work. This work was supported by the Institut Pasteur; the Collège de France; the Centre Nationale de la Recherche Scientifique (CNRS); the Agence Nationale de la Recherche (ANR) grants LIFECHANGE (ANR-CE-1712-0018-02), CNSVIRGEN (ANR-19-CE15-0009-02), and MORTUI (ANR-19-CE35-0005); the French Government’s Investissement d’Avenir program; Laboratoires d’Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR-10-LABX-62-IBEID) and “Milieu Intérieur” (ANR-10-LABX-69-01); the Fondation pour la Recherche Médicale (Equipe FRM DEQ20180339214); the Fondation Allianz-Institut de France; and the Fondation de France (00106080). G.K. was supported by a Pasteur-Roux-Cantarini fellowship.
Author contributions
G.K., E.P., G.L., and L.Q.-M. conceived and designed the study. G.K. was the lead analyst, with important contributions from E.P. and G.L. A.-L.N. performed the functional analyses of the LBP D283G variant. Q.P. and J.B. performed the functional analyses of the IL23R R381Q variant. D.R. performed the RNA sequencing analyses for the IL23R R381Q and TLR3 L412F variants. N.K. performed the functional analysis of the TLR3 L412F variant. S.-Y.Z. supervised the work on the TLR3 L412F variant, and A.P. and S.B.-D. supervised that on the IL23R R381Q variant. E.P., G.L., and L.Q.-M. oversaw the study. G.K., E.P., G.L., and L.Q.-M. wrote the manuscript with substantial contributions from A.-L.N., Q.P., J.B., N.K., L.A., and J.-L.C.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100248.
Contributor Information
Gaspard Kerner, Email: gakerner@pasteur.fr.
Lluis Quintana-Murci, Email: quintana@pasteur.fr.
Supplemental information
Table S2. The 89 variants with the smallest psel at each of the 89 candidate positively selected loci, related to Figure 1
Table S4. Lead GWAS SNPs of all infectious and autoimmune traits, related to Figure 3
Table S7. Candidate negatively selected missense variants at conserved positions, related to Figure 4
Data and code availability
-
•
RNA-sequencing data for the IL23R R381Q and the TLR3 L412F variants have been deposited at GEO and are publicly available as of the date of publication from GEO: GSE216458 and GEO: GSE216304, respectively. Accession numbers are also listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication from Mendeley Data: http://doi.org/10.17632/kwdmpxw3np.1. The DOI is also listed in the key resources table.
-
•
The code for reproducing main analyses and figures of the paper is available from Mendeley Data: https://doi.org/10.17632/38d7vv96m9.1 and is also listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Casanova J.-L., Abel L. Inborn errors of immunity to infection : the rule rather than the exception. J. Exp. Med. 2005;202:197–201. doi: 10.1084/jem.20050854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cairns J. Princeton University Press; 1997. Matters of Life and Death. [Google Scholar]
- 3.Allison A.C. Protection afforded by sickle-cell trait against subtertian malareal infection. Br. Med. J. 1954;1:290–294. doi: 10.1136/bmj.1.4857.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barreiro L.B., Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet. 2010;11:17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- 5.Quintana-Murci L. Human immunology through the lens of evolutionary genetics. Cell. 2019;177:184–199. doi: 10.1016/j.cell.2019.02.033. [DOI] [PubMed] [Google Scholar]
- 6.Fumagalli M., Sironi M. Human genome variability, natural selection and infectious diseases. Curr. Opin. Immunol. 2014;30:9–16. doi: 10.1016/j.coi.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Karlsson E.K., Kwiatkowski D.P., Sabeti P.C. Natural selection and infectious disease in human populations. Nat. Rev. Genet. 2014;15:379–393. doi: 10.1038/nrg3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintana-Murci L., Clark A.G. Population genetic tools for dissecting innate immunity in humans. Nat. Rev. Immunol. 2013;13:280–293. doi: 10.1038/nri3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamond J. Evolution, consequences and future of plant and animal domestication. Nature. 2002;418:700–707. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- 10.Key F.M., Posth C., Esquivel-Gomez L.R., Hübler R., Spyrou M.A., Neumann G.U., Furtwängler A., Sabin S., Burri M., Wissgott A., et al. Emergence of human-adapted Salmonella enterica is linked to the Neolithization process. Nat. Ecol. Evol. 2020;4:324–333. doi: 10.1038/s41559-020-1106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfe N.D., Dunavan C.P., Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper K.N., Armelagos G.J. Genomics, the origins of agriculture, and our changing microbe-scape: time to revisit some old tales and tell some new ones. Am. J. Phys. Anthropol. 2013;152:135–152. doi: 10.1002/ajpa.22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs K., Rinne C., Drummer C., Immel A., Krause-Kyora B., Nebel A. Infectious diseases and Neolithic transformations: evaluating biological and archaeological proxies in the German loess zone between 5500 and 2500 BCE. Holocene. 2019;29:1545–1557. doi: 10.1177/0959683619857230. [DOI] [Google Scholar]
- 14.Astle W.J., Elding H., Jiang T., Allen D., Ruklisa D., Mann A.L., Mead D., Bouman H., Riveros-Mckay F., Kostadima M.A., et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167:1415–1429.e19. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao E.L., Cheng A.N., Sankaran V.G. The genetics of human hematopoiesis and its disruption in disease. EMBO Mol. Med. 2019;11 doi: 10.15252/emmm.201910316. e10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liggett L.A., Sankaran V.G. Unraveling hematopoiesis through the lens of genomics. Cell. 2020;182:1384–1400. doi: 10.1016/j.cell.2020.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notarangelo L.D., Bacchetta R., Casanova J.-L., Su H.C. Human inborn errors of immunity: an expanding universe. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abb1662. eabb1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riley J.C. Cambridge University Press; 2001. Rising Life Expectancy: A Global History. [DOI] [Google Scholar]
- 19.Barreiro L.B., Quintana-Murci L. Evolutionary and population (epi)genetics of immunity to infection. Hum. Genet. 2020;139:723–732. doi: 10.1007/s00439-020-02167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benton M.L., Abraham A., LaBella A.L., Abbot P., Rokas A., Capra J.A. The influence of evolutionary history on human health and disease. Nat. Rev. Genet. 2021;22:269–283. doi: 10.1038/s41576-020-00305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sironi M., Clerici M. The hygiene hypothesis: an evolutionary perspective. Microbes Infect. 2010;12:421–427. doi: 10.1016/j.micinf.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Fodil N., Langlais D., Gros P. Primary immunodeficiencies and inflammatory disease: a growing genetic intersection. Trends Immunol. 2016;37:126–140. doi: 10.1016/j.it.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langlais D., Fodil N., Gros P. Genetics of infectious and inflammatory diseases: overlapping discoveries from association and exome-sequencing studies. Annu. Rev. Immunol. 2017;35:1–30. doi: 10.1146/annurev-immunol-051116-052442. [DOI] [PubMed] [Google Scholar]
- 24.Fumagalli M., Pozzoli U., Cagliani R., Comi G.P., Riva S., Clerici M., Bresolin N., Sironi M. Parasites represent a major selective force for interleukin genes and shape the genetic predisposition to autoimmune conditions. J. Exp. Med. 2009;206:1395–1408. doi: 10.1084/jem.20082779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsoi L.C., Spain S.L., Knight J., Ellinghaus E., Stuart P.E., Capon F., Ding J., Li Y., Tejasvi T., Gudjonsson J.E., et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhernakova A., Elbers C.C., Ferwerda B., Romanos J., Trynka G., Dubois P.C., de Kovel C.G.F., Franke L., Oosting M., Barisani D., et al. Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection. Am. J. Hum. Genet. 2010;86:970–977. doi: 10.1016/j.ajhg.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prugnolle F., Manica A., Charpentier M., Guégan J.F., Guernier V., Balloux F. Pathogen-driven selection and worldwide HLA class I diversity. Curr. Biol. 2005;15:1022–1027. doi: 10.1016/j.cub.2005.04.050. [DOI] [PubMed] [Google Scholar]
- 29.Chen H., Hayashi G., Lai O.Y., Dilthey A., Kuebler P.J., Wong T.V., Martin M.P., Fernandez Vina M.A., McVean G., Wabl M., et al. Psoriasis patients are enriched for genetic variants that protect against HIV-1 disease. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002514. e1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gough S.C.L., Simmonds M.J. The HLA region and autoimmune disease: associations and mechanisms of action. Curr. Genomics. 2007;8:453–465. doi: 10.2174/138920207783591690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matzaraki V., Kumar V., Wijmenga C., Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017;18:76. doi: 10.1186/s13059-017-1207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ritari J., Koskela S., Hyvärinen K., FinnGen, Partanen J. HLA-disease association and pleiotropy landscape in over 235, 000 Finns. Hum. Immunol. 2022;83:391–398. doi: 10.1016/j.humimm.2022.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Ryder L.P., Svejgaard A., Dausset J. Genetics of HLA disease association. Annu. Rev. Genet. 1981;15:169–187. doi: 10.1146/annurev.ge.15.120181.001125. [DOI] [PubMed] [Google Scholar]
- 34.Boisson-Dupuis S., Ramirez-Alejo N., Li Z., Patin E., Rao G., Kerner G., Lim C.K., Krementsov D.N., Hernandez N., Ma C.S., et al. Tuberculosis and impaired IL-23-dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant. Sci. Immunol. 2018;3 doi: 10.1126/sciimmunol.aau8714. eaau8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerner G., Laval G., Patin E., Boisson-Dupuis S., Abel L., Casanova J.-L., Quintana-Murci L. Human ancient DNA analyses reveal the high burden of tuberculosis in Europeans over the last 2, 000 years. Am. J. Hum. Genet. 2021;108:517–524. doi: 10.1016/j.ajhg.2021.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dendrou C.A., Cortes A., Shipman L., Evans H.G., Attfield K.E., Jostins L., Barber T., Kaur G., Kuttikkatte S.B., Leach O.A., et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aag1974. 363ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diogo D., Bastarache L., Liao K.P., Graham R.R., Fulton R.S., Greenberg J.D., Eyre S., Bowes J., Cui J., Lee A., et al. TYK2 protein-coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non-autoimmune complex traits. PLoS One. 2015;10 doi: 10.1371/journal.pone.0122271. e0122271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.International Genetics of Ankylosing Spondylitis Consortium IGAS. Cortes A., Hadler J., Pointon J.P., Robinson P.C., Karaderi T., Leo P., Cremin K., Pryce K., Harris J., et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerner G., Ramirez-Alejo N., Seeleuthner Y., Yang R., Ogishi M., Cobat A., Patin E., Quintana-Murci L., Boisson-Dupuis S., Casanova J.-L., Abel L. Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc. Natl. Acad. Sci. USA. 2019;116:10430–10434. doi: 10.1073/pnas.1903561116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ju D., Mathieson I. The evolution of skin pigmentation-associated variation in West Eurasia. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2009227118. e2009227118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Key F.M., Fu Q., Romagné F., Lachmann M., Andrés A.M. Human adaptation and population differentiation in the light of ancient genomes. Nat. Commun. 2016;7:10775. doi: 10.1038/ncomms10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathieson I. Limited evidence for selection at the FADS locus in native American populations. Mol. Biol. Evol. 2020;37:2029–2033. doi: 10.1093/molbev/msaa064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathieson I., Lazaridis I., Rohland N., Mallick S., Patterson N., Roodenberg S.A., Harney E., Stewardson K., Fernandes D., Novak M., et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindo J., Huerta-Sánchez E., Nakagome S., Rasmussen M., Petzelt B., Mitchell J., Cybulski J.S., Willerslev E., DeGiorgio M., Malhi R.S. A time transect of exomes from a Native American population before and after European contact. Nat. Commun. 2016;7:13175. doi: 10.1038/ncomms13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beaumont M.A., Rannala B. The Bayesian revolution in genetics. Nat. Rev. Genet. 2004;5:251–261. doi: 10.1038/nrg1318. [DOI] [PubMed] [Google Scholar]
- 46.Lazaridis I., Patterson N., Mittnik A., Renaud G., Mallick S., Kirsanow K., Sudmant P.H., Schraiber J.G., Castellano S., Lipson M., et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skoglund P., Malmström H., Raghavan M., Storå J., Hall P., Willerslev E., Gilbert M.T.P., Götherström A., Jakobsson M. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336:466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- 48.Allentoft M.E., Sikora M., Sjögren K.G., Rasmussen S., Rasmussen M., Stenderup J., Damgaard P.B., Schroeder H., Ahlström T., Vinner L., et al. Population genomics of Bronze age Eurasia. Nature. 2015;522:167–172. doi: 10.1038/nature14507. [DOI] [PubMed] [Google Scholar]
- 49.Haak W., Lazaridis I., Patterson N., Rohland N., Mallick S., Llamas B., Brandt G., Nordenfelt S., Harney E., Stewardson K., et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522:207–211. doi: 10.1038/nature14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burger J., Link V., Blöcher J., Schulz A., Sell C., Pochon Z., Diekmann Y., Žegarac A., Hofmanová Z., Winkelbach L., et al. Low prevalence of lactase persistence in Bronze age Europe indicates ongoing strong selection over the last 3, 000 years. Curr. Biol. 2020;30:4307–4315.e13. doi: 10.1016/j.cub.2020.08.033. [DOI] [PubMed] [Google Scholar]
- 51.Segurel L., Guarino-Vignon P., Marchi N., Lafosse S., Laurent R., Bon C., Fabre A., Hegay T., Heyer E. Why and when was lactase persistence selected for? Insights from Central Asian herders and ancient DNA. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000742. e3000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kristiansen H., Scherer C.A., McVean M., Iadonato S.P., Vends S., Thavachelvam K., Steffensen T.B., Horan K.A., Kuri T., Weber F., et al. Extracellular 2'-5' oligoadenylate synthetase stimulates RNase L-independent antiviral activity: a novel mechanism of virus-induced innate immunity. J. Virol. 2010;84:11898–11904. doi: 10.1128/JVI.01003-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shelton J.F., Shastri A.J., Ye C., Weldon C.H., Filshtein-Sonmez T., Coker D., Symons A., Esparza-Gordillo J., 23andMe COVID-19 Team. Aslibekyan S., et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021;53:801–808. doi: 10.1038/s41588-021-00854-7. [DOI] [PubMed] [Google Scholar]
- 54.Pickrell J.K., Berisa T., Liu J.Z., Ségurel L., Tung J.Y., Hinds D.A. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 2016;48:709–717. doi: 10.1038/ng.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian C., Hromatka B.S., Kiefer A.K., Eriksson N., Noble S.M., Tung J.Y., Hinds D.A. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 2017;8:599. doi: 10.1038/s41467-017-00257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malaria Genomic Epidemiology Network. Band G., Le Q.S., Clarke G.M., Kivinen K., Hubbart C., Jeffreys A.E., Rowlands K., Leffler E.M., Jallow M., et al. Insights into malaria susceptibility using genome-wide data on 17, 000 individuals from Africa, Asia and Oceania. Nat. Commun. 2019;10:5732. doi: 10.1038/s41467-019-13480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ségurel L., Thompson E.E., Flutre T., Lovstad J., Venkat A., Margulis S.W., Moyse J., Ross S., Gamble K., Sella G., et al. The ABO blood group is a trans-species polymorphism in primates. Proc. Natl. Acad. Sci. USA. 2012;109:18493–18498. doi: 10.1073/pnas.1210603109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cuadros-Espinoza S., Laval G., Quintana-Murci L., Patin E. The genomic signatures of natural selection in admixed human populations. Am. J. Hum. Genet. 2022;109:710–726. doi: 10.1016/j.ajhg.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davy T., Ju D., Mathieson I., Skoglund P. Hunter-gatherer admixture facilitated natural selection in Neolithic European farmers. bioRxiv. 2022 doi: 10.1101/2022.09.05.506481. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeberg H., Pääbo S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2026309118. e2026309118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huffman J.E., Butler-Laporte G., Khan A., Pairo-Castineira E., Drivas T.G., Peloso G.M., Nakanishi T., COVID-19 Host Genetics Initiative. Ganna A., Verma A., et al. Multi-ancestry fine mapping implicates OAS1 splicing in risk of severe COVID-19. Nat. Genet. 2022;54:125–127. doi: 10.1038/s41588-021-00996-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou S., Butler-Laporte G., Nakanishi T., Morrison D.R., Afilalo J., Afilalo M., Laurent L., Pietzner M., Kerrison N., Zhao K., et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med. 2021;27:659–667. doi: 10.1038/s41591-021-01281-1. [DOI] [PubMed] [Google Scholar]
- 63.Abramov S., Boytsov A., Bykova D., Penzar D.D., Yevshin I., Kolmykov S.K., Fridman M.V., Favorov A.V., Vorontsov I.E., Baulin E., et al. Landscape of allele-specific transcription factor binding in the human genome. Nat. Commun. 2021;12:2751. doi: 10.1038/s41467-021-23007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreira M.A.R., Mathur R., Vonk J.M., Szwajda A., Brumpton B., Granell R., Brew B.K., Ullemar V., Lu Y., Jiang Y., et al. Genetic architectures of childhood- and adult-onset asthma are partly distinct. Am. J. Hum. Genet. 2019;104:665–684. doi: 10.1016/j.ajhg.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K., et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nunes-Santos C.J., Kuehn H.S., Rosenzweig S.D. IKAROS family zinc finger 1-associated diseases in primary immunodeficiency patients. Immunol. Allergy Clin. North Am. 2020;40:461–470. doi: 10.1016/j.iac.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Panda D., Gjinaj E., Bachu M., Squire E., Novatt H., Ozato K., Rabin R.L. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front. Immunol. 2019;10:1019. doi: 10.3389/fimmu.2019.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lindesmith L., Moe C., Marionneau S., Ruvoen N., Jiang X., Lindblad L., Stewart P., LePendu J., Baric R. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003;9:548–553. doi: 10.1038/nm860. [DOI] [PubMed] [Google Scholar]
- 69.Raza M.W., Blackwell C.C., Molyneaux P., James V.S., Ogilvie M.M., Inglis J.M., Weir D.M. Association between secretor status and respiratory viral illness. BMJ. 1991;303:815–818. doi: 10.1136/bmj.303.6806.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park B.S., Lee J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eckert J.K., Kim Y.J., Kim J.I., Gürtler K., Oh D.-Y., Sur S., Lundvall L., Hamann L., van der Ploeg A., Pickkers P., et al. The crystal structure of lipopolysaccharide binding protein reveals the location of a frequent mutation that impairs innate immunity. Immunity. 2013;39:647–660. doi: 10.1016/j.immuni.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 72.Filipe-Santos O., Bustamante J., Chapgier A., Vogt G., de Beaucoudrey L., Feinberg J., Jouanguy E., Boisson-Dupuis S., Fieschi C., Picard C., Casanova J.L. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin. Immunol. 2006;18:347–361. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 73.Kreins A.Y., Ciancanelli M.J., Okada S., Kong X.F., Ramírez-Alejo N., Kilic S.S., El Baghdadi J., Nonoyama S., Mahdaviani S.A., Ailal F., et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J. Exp. Med. 2015;212:1641–1662. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ogishi M., Arias A.A., Yang R., Han J.E., Zhang P., Rinchai D., Halpern J., Mulwa J., Keating N., Chrabieh M., et al. Impaired IL-23–dependent induction of IFN-γ underlies mycobacterial disease in patients with inherited TYK2 deficiency. J. Exp. Med. 2022;219 doi: 10.1084/jem.20220094. e20220094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Croci S., Venneri M.A., Mantovani S., Fallerini C., Benetti E., Picchiotti N., Campolo F., Imperatore F., Palmieri M., Daga S., et al. The polymorphism L412F in TLR3 inhibits autophagy and is a marker of severe COVID-19 in males. Autophagy. 2022;18:1662–1672. doi: 10.1080/15548627.2021.1995152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gorbea C., Makar K.A., Pauschinger M., Pratt G., Bersola J.L.F., Varela J., David R.M., Banks L., Huang C.H., Li H., et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010;285:23208–23223. doi: 10.1074/jbc.M109.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo Y., Audry M., Ciancanelli M., Alsina L., Azevedo J., Herman M., Anguiano E., Sancho-Shimizu V., Lorenzo L., Pauwels E., et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011;208:2083–2098. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gleich G.J., Klion A.D., Lee J.J., Weller P.F. The consequences of not having eosinophils. Allergy. 2013;68:829–835. doi: 10.1111/all.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andrades Valtueña A., Neumann G.U., Spyrou M.A., Musralina L., Aron F., Beisenov A., Belinskiy A.B., Bos K.I., Buzhilova A., Conrad M., et al. Stone Age Yersinia pestis genomes shed light on the early evolution, diversity, and ecology of plague. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2116722119. e2116722119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rascovan N., Sjögren K.G., Kristiansen K., Nielsen R., Willerslev E., Desnues C., Rasmussen S. Emergence and spread of basal lineages of Yersinia pestis during the Neolithic decline. Cell. 2019;176:295–305.e10. doi: 10.1016/j.cell.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 81.Skoglund P., Mathieson I. Ancient genomics of modern humans: the first decade. Annu. Rev. Genomics Hum. Genet. 2018;19:381–404. doi: 10.1146/annurev-genom-083117-021749. [DOI] [PubMed] [Google Scholar]
- 82.Kristiansen K., Fowler C., Harding J., Hofmann D. The decline of the Neolithic and the rise of bronze age society. 2014. Oxford Handbooks Online. [DOI]
- 83.Charlesworth B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009;10:195–205. doi: 10.1038/nrg2526. [DOI] [PubMed] [Google Scholar]
- 84.Scott A., Reinhold S., Hermes T., Kalmykov A.A., Belinskiy A., Buzhilova A., Berezina N., Kantorovich A.R., Maslov V.E., Guliyev F., et al. Emergence and intensification of dairying in the Caucasus and Eurasian steppes. Nat. Ecol. Evol. 2022;6:813–822. doi: 10.1038/s41559-022-01701-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Racimo F., Woodbridge J., Fyfe R.M., Sikora M., Sjögren K.G., Kristiansen K., Vander Linden M. The spatiotemporal spread of human migrations during the European Holocene. Proc. Natl. Acad. Sci. USA. 2020;117:8989–9000. doi: 10.1073/pnas.1920051117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klunk J., Vilgalys T.P., Demeure C.E., Cheng X., Shiratori M., Madej J., Beau R., Elli D., Patino M.I., Redfern R., et al. Evolution of immune genes is associated with the Black Death. Nature. 2022;611:312–319. doi: 10.1038/s41586-022-05349-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Song W., Shi Y., Wang W., Pan W., Qian W., Yu S., Zhao M., Lin G.N. A selection pressure landscape for 870 human polygenic traits. Nat. Hum. Behav. 2021;5:1731–1743. doi: 10.1038/s41562-021-01231-4. [DOI] [PubMed] [Google Scholar]
- 88.Saevarsdottir S., Olafsdottir T.A., Ivarsdottir E.V., Halldorsson G.H., Gunnarsdottir K., Sigurdsson A., Johannesson A., Sigurdsson J.K., Juliusdottir T., Lund S.H., et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature. 2020;584:619–623. doi: 10.1038/s41586-020-2436-0. [DOI] [PubMed] [Google Scholar]
- 89.de Lange K.M., Moutsianas L., Lee J.C., Lamb C.A., Luo Y., Kennedy N.A., Jostins L., Rice D.L., Gutierrez-Achury J., Ji S.-G., et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017;49:256–261. doi: 10.1038/ng.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brunel S., Bennett E.A., Cardin L., Garraud D., Barrand Emam H., Beylier A., Boulestin B., Chenal F., Ciesielski E., Convertini F., et al. Ancient genomes from present-day France unveil 7, 000 years of its demographic history. Proc. Natl. Acad. Sci. USA. 2020;117:12791–12798. doi: 10.1073/pnas.1918034117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Margaryan A., Lawson D.J., Sikora M., Racimo F., Rasmussen S., Moltke I., Cassidy L.M., Jørsboe E., Ingason A., Pedersen M.W., et al. Population genomics of the Viking world. Nature. 2020;585:390–396. doi: 10.1038/s41586-020-2688-8. [DOI] [PubMed] [Google Scholar]
- 92.Olalde I., Mallick S., Patterson N., Rohland N., Villalba-Mouco V., Silva M., Dulias K., Edwards C.J., Gandini F., Pala M., et al. The genomic history of the Iberian Peninsula over the past 8000 years. Science. 2019;363:1230–1234. doi: 10.1126/science.aav4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carlson M.O., Rice D.P., Berg J.J., Steinrücken M. Polygenic score accuracy in ancient samples: quantifying the effects of allelic turnover. PLoS Genet. 2022;18 doi: 10.1371/journal.pgen.1010170. e1010170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mostafavi H., Harpak A., Agarwal I., Conley D., Pritchard J.K., Przeworski M. Variable prediction accuracy of polygenic scores within an ancestry group. Elife. 2020;9 doi: 10.7554/eLife.48376. e48376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korotkevich G., Sukhov V., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2019 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 97.Young M.D., Wakefield M.J., Smyth G.K., Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.-R., ReproGen Consortium. Psychiatric Genomics Consortium. Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3. Duncan L., et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 101.Deschamps M., Laval G., Fagny M., Itan Y., Abel L., Casanova J.-L., Patin E., Quintana-Murci L. Genomic signatures of selective pressures and introgression from archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 2016;98:5–21. doi: 10.1016/j.ajhg.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Monroy Kuhn J.M., Jakobsson M., Günther T. Estimating genetic kin relationships in prehistoric populations. PLoS One. 2018;13 doi: 10.1371/journal.pone.0195491. e0195491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fairley S., Lowy-Gallego E., Perry E., Flicek P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020;48:D941–D947. doi: 10.1093/nar/gkz836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.François O., Jay F. Factor analysis of ancient population genomic samples. Nat. Commun. 2020;11:4661. doi: 10.1038/s41467-020-18335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haller B.C., Messer P.W. SLiM 3: forward genetic simulations beyond the wright-fisher model. Mol. Biol. Evol. 2019;36:632–637. doi: 10.1093/molbev/msy228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gutenkunst R.N., Hernandez R.D., Williamson S.H., Bustamante C.D. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000695. e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vergara-Lope A., Jabalameli M.R., Horscroft C., Ennis S., Collins A., Pengelly R.J. Linkage disequilibrium maps for European and African populations constructed from whole genome sequence data. Sci. Data. 2019;6:208. doi: 10.1038/s41597-019-0227-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S., et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schmidt E.M., Zhang J., Zhou W., Chen J., Mohlke K.L., Chen Y.E., Willer C.J. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics. 2015;31:2601–2606. doi: 10.1093/bioinformatics/btv201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Okada Y., Wu D., Trynka G., Raj T., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Yoshida S., et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grossman S.R., Shlyakhter I., Karlsson E.K., Byrne E.H., Morales S., Frieden G., Hostetter E., Angelino E., Garber M., Zuk O., et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327:883–886. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- 114.Ferrer-Admetlla A., Liang M., Korneliussen T., Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol. Biol. Evol. 2014;31:1275–1291. doi: 10.1093/molbev/msu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Barreiro L.B., Ben-Ali M., Quach H., Laval G., Patin E., Pickrell J.K., Bouchier C., Tichit M., Neyrolles O., Gicquel B., et al. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000562. e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang X., Spandidos A., Wang H., Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. The 89 variants with the smallest psel at each of the 89 candidate positively selected loci, related to Figure 1
Table S4. Lead GWAS SNPs of all infectious and autoimmune traits, related to Figure 3
Table S7. Candidate negatively selected missense variants at conserved positions, related to Figure 4
Data Availability Statement
-
•
RNA-sequencing data for the IL23R R381Q and the TLR3 L412F variants have been deposited at GEO and are publicly available as of the date of publication from GEO: GSE216458 and GEO: GSE216304, respectively. Accession numbers are also listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication from Mendeley Data: http://doi.org/10.17632/kwdmpxw3np.1. The DOI is also listed in the key resources table.
-
•
The code for reproducing main analyses and figures of the paper is available from Mendeley Data: https://doi.org/10.17632/38d7vv96m9.1 and is also listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.