

Summary
Despite the abundance of multimodal data, suitable statistical models that can improve our understanding of diseases with genetic underpinnings are challenging to develop. Here, we present SparseGMM, a statistical approach for gene regulatory network discovery. SparseGMM uses latent variable modeling with sparsity constraints to learn Gaussian mixtures from multiomic data. By combining coexpression patterns with a Bayesian framework, SparseGMM quantitatively measures confidence in regulators and uncertainty in target gene assignment by computing gene entropy. We apply SparseGMM to liver cancer and normal liver tissue data and evaluate discovered gene modules in an independent single-cell RNA sequencing (scRNA-seq) dataset. SparseGMM identifies PROCR as a regulator of angiogenesis and PDCD1LG2 and HNF4A as regulators of immune response and blood coagulation in cancer. Furthermore, we show that more genes have significantly higher entropy in cancer compared with normal liver. Among high-entropy genes are key multifunctional components shared by critical pathways, including p53 and estrogen signaling.
Graphical abstract
Highlights
-
•
A sparse latent variable model learns gene regulatory network from multiomic data
-
•
HNF4A and PDCD1LG2 robustly regulate clotting and antigen presentation systems in HCC
-
•
PROCR regulates angiogenesis function in normal liver tissue
-
•
Identifies multifunctional components shared by cancer pathways: p53, estrogen pathway
Motivation
Uncovering the structure of biological networks can provide valuable insights into the genetic underpinning of many diseases and opportunities to discover novel drug targets. However, learning the structure of such networks from multimodal genomic data remains challenging. We sought to develop a method for gene regulatory inference and learning from genomic data using a Bayesian approach and apply it to normal and liver cancer data.
Bakr et al. introduce SparseGMM, an algorithm for modeling gene networks using a modular approach. SparseGMM allows genes to be part of multiple modules and outperforms existing methods in identifying novel drug targets for patients with cancer.
Introduction
Many diseases have significant genetic underpinnings that determine both the underlying pathology and potential targets for therapy. One important example where an understanding of the molecular mechanism can aid treatment is cancer. Cancer is a disease of the genome whereby genetic and epigenetic events in certain genes, referred to as driver genes, are causal of a specific cell status that escapes normal physiological regulation and immune surveillance, leading to cancer. Altered driver genes cause dysregulation of biological pathways, downstream changes in gene expression, and cell signaling in a manner that increases cell growth and proliferation. Typically, cancer driver genes fall under the classes of master regulators, such as transcription factors, DNA-damage repair, and cell-cycle genes, among others. With the high rate of genetic mutations in cancer, identifying cancer driver genes represents an important challenge. The introduction of new molecular technologies in the 2010s, such as next-generation sequencing, resulted in a surge in the availability of genomic and transcriptomic data. This increasing availability of multimodal data is exemplified by public projects such as The Cancer Genome Atlas1 (TCGA), a large-scale genome sequencing collaborative effort that aims to accelerate our understanding of the molecular basis of cancer. In TCGA, over 10,000 primary cancer and matched normal samples were characterized, spanning 33 cancer types, generating over 2.5 petabytes of genomic, epigenomic, transcriptomic, and proteomic data. Similarly, the Genotype-Tissue Expression2 (GTEx) is a resource of genetic variation and expression of 54 tissue types in a large population of healthy individuals. Although the GTEx subject population does not contain disease samples, understanding the genetic and genomic variations in healthy tissue can help gain useful insight into genetic diseases and their molecular features. For instance, GTEx data were used to identify the role of a novel coronary artery disease risk gene3 and for detecting pathogenic gene variants related to rare genetic disorders,4 and GTEx data were successfully combined with TCGA data to develop prognostic markers of acute myeloid leukemia (AML).5 Leveraging these large size multimodal datasets to make significant biological discoveries and extract clinically actionable information is only possible through developing suitable statistical models and machine learning algorithms.
Gene regulatory networks (GRNs) are one class of tools that can be applied to genomic data to improve our understanding of systems biology and uncover the molecular basis of disease. Network methods can be used to model gene-level relationships and protein-protein and cell-cell interactions. Several approaches to integrating multiomic data,6 as well as learning GRNs, exist,7 including graph-8,9 and module-based methods.10,11,12 In graph methods, a graph is created based on the expression data, and then the graph is analyzed to extract subnetworks, with hub genes assumed to be regulators of target genes in these subnetworks. Hub genes are a subset of highly connected genes, relative to the other, less connected, downstream targets. Such a scale-free network structure mimics the nature of biological networks. Graph methods were used to discover major gene hubs in human B cells.8 They were also used to identify new molecular targets in glioblastoma.13 GENIE314 and GRNBoost215 build GRNs using variable selection with ensembles of regression trees and gradient-boosting producing and produce directed graphs of regulatory interactions. Module-based methods typically cluster coexpressed genes directly into gene modules and, as a second step, identify regulators of these gene modules. Examples of module-based methods include CONEXIC,16 AMARETTO,17,18 and CaMoDi,12 which have been shown to be more robust and better recapitulate underlying biology than graph-based methods.10 In a previous study, we developed AMARETTO,17,18 a module-based tool that clusters coexpressed genes and assigns each module to its regulators using sparse linear regression. AMARETTO outperforms other methods in its ability to leverage information from copy-number variation and methylation data to improve the discovery of regulators and their assignment to gene modules. The genomic and epigenetic events inform the choice of candidate drive genes, which are used then as features selected by sparse linear regression (LASSO). The resulting modules are functionally annotated using gene set enrichment analysis (GSEA)19 techniques, elucidating the role of driver genes in cancer development and progression. In later work,11 AMARETTO was extended to construct a pancancer module network that confirms the common cancer pathways in different cancer types and uncovers a driver gene of smoking-induced cancers, as well as another driver gene involved in anti-viral immune response exhibited by some cancers. AMARETTO has also been extended to linking genomic and imaging phenotypes from cellular and tissue images.20
In this work, we present SparseGMM, a module network approach in a Bayesian framework, whereby the clustering of target genes and the assignment of regulators are combined in one step, which allows genes to be associated with multiple modules simultaneously. The assignment of a regulator to its modules can be thus calculated with a confidence interval. More specifically, we use Gaussian mixture model (GMM) inference, where the mixture mean is represented as a weighted sparse vector of regulator expression level. This novel framework tackles an important limitation in module-based methods by allowing probabilistic assignments of target genes to modules and significance estimates of individual regulator coefficients. We show an improved performance in sparsity, compared with previous methods, choosing fewer genes as true regulators and confirming biological knowledge of the scale-free nature of gene networks.
We apply this new algorithm to GTEx data from healthy liver tissue, as well as hepatocellular carcinoma (HCC) samples from TCGA liver hepatocellular carcinoma (LIHC). Our algorithm can recover healthy tissue modules such as energy metabolism pathways and cancer-specific modules involved in antigen presentation, immune response, and blood coagulation. We also discover common modules in healthy liver and HCC responsible for inflammation and steroid biosynthesis, among others. Further, we use a publicly available single-cell dataset of CD45+ immune cells21 to evaluate immune-related modules discovered using the bulk sequencing data. The single-cell evaluation of immune modules was able to decouple distinct myeloid and lymphoid biological processes in the HCC micro-environment. Our results demonstrate the ability of our method to represent GRNs as potentially overlapping gene modules as demonstrated on bulk and single-cell RNA sequencing (RNA-seq) data.
Further, contrary to previous methods, the probabilistic assignment approach taken by SparseGMM is potentially superior for modeling genes with multiple biological functions. Thus, we define the entropy of a gene to be the entropy of the estimated module-assignment probability and show that it can then be used as an indicator of a multifunctional biological role based on joint membership to two or more modules. These multifunctional genes could in turn translate to multifunctional proteins having central roles in the crosstalk between two or more pathways in cancer cells and, thus, become attractive targets for overcoming drug resistance through compensation mechanisms. We show that high-entropy genes are more common in cancer samples than in healthy tissue, and we associate them with crosstalk between several pathways including TP53, interferon gammam, and tumor necrosis factor α (TNF-α). Our analysis of high-entropy genes exemplifies ways in which major cancer pathways share key multifunctional components.
Results
Here, we present a new method, SparseGMM, which uses a Bayesian latent variable approach to model the relationship between regulators and downstream target genes (see STAR Methods and Methods S1). To validate our approach, we apply our method to two bulk gene expression liver datasets of normal liver. Gene modules in normal tissue were constructed using publicly available data from GTEx project, while cancer modules were constructed using HCC data from TCGA project (Figure 1). We used community detection methods to screen gene modules for robustness and to uncover shared biology between normal liver tissue and liver cancer. Next, we evaluated these communities in an independent single-cell dataset containing CD45+ immune cells from patients with HCC cancer and analyzed the expression of these communities in different immune cell populations.
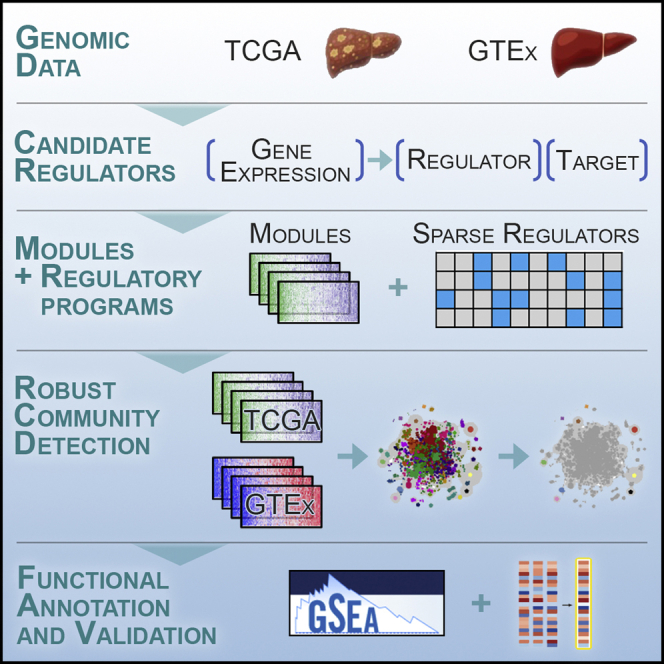
Figure 1.

Overview of study and the SparseGMM method
SparseGMM uses a graph-based Bayesian framework combined with coexpression pattern to connect sparse sets of regulators to their downstream target gene modules. To measure robustness, we ran SparseGMM several times, generating multiple gene networks from each of two datasets with normal liver and liver cancer gene expression profiles. To screen for robust modules and identify normal-cancer shared biology, we ran a community detection algorithm to group robust modules that are consistently discovered in every run. Next, we performed functional gene set enrichment analysis using MSigDB gene collections. Finally, we used publicly available perturbation experiments that identify experimental targets to validate SparseGMM regulators.
Technical validation
For both TCGA1 and GTEx2 data, we compared SparseGMM with AMARETTO17 and showed improved performance in terms of sparsity of regulators for various choices of the regularization parameter values (Figure 2; Table S1). AMARETTO was selected as, to our knowledge, it is the current state-of-the-art method for module-based GRN inference. Analyzing sparsity performance in GTEx and TCGA data, SparseGMM outperforms AMARETTO for all choices of the regularization parameter, lambda, with sparser solutions being more desirable.22 The mean number of regulators per module, with sparsity parameter lambda = 500, is 21.27 for SparseGMM, compared with 84.14 for AMARETTO using GTEx data (p < 0.05, independent t test). Similarly using TCGA data, the mean number of regulators is 38.20 for SparseGMM compared with 196.33 for AMARETTO (p < 0.05, independent t test). For robustness measured using the adjusted Rand index (ARI) on modules from multiple runs on each dataset, both datasets show similar performance with an increasing trend as the regularization parameter increases. On the other hand, both methods show a gradual decrease in R2 with increased regularization for both datasets. SparseGMM performs better than AMARETTO for lower values of lambda. Module size increases with regularization for both datasets. At lambda = 5e3, SparseGMM has larger module sizes than AMARETTO. At values of lambda >500, the module sizes are too large for practical functional annotation and discovery. Overall, the sparsity performance of SparseGMM was superior for all tested values of the regularization parameter. SparseGMM performed consistently when applied to two other datasets from TCGA: lung adenocarcinoma (LUAD) and head and neck squamous cell carcinomas (HNSCs) (Figure S1). Acceptable module sizes and R-squared were seen for lambda = 500 and lower similarly, while acceptable ARI values were seen at 500 and higher. These results dictated the choice of lambda = 500 in subsequent analyses.
Figure 2.

Performance comparison between SparseGMM and AMARETTO at different regularization values
Comparison shown for TCGA HCC (A–D) and GTEx (E–H) liver data.
(A and E) Robustness of clustering is evaluated using adjusted Rand index.
(B and F) Validation of regulators is represented by R-squared.
(C and G) Degree of sparsity is evaluated using statistics on the number of drivers.
(D and H) Module size informs the choice of regularization parameter value.
Further, we compared SparseGMM with GRNBoost215 to evaluate its performance against top existing GRN tools. GRNBoost2 is a gradient-boosting method. It uses an efficient algorithm developed for scaling up regulatory network inference based on the GENIE314 architecture. GENIE3 was the best performer in the DREAM4 In Silico Multifactorial challenge. We compared SparseGMM with GRNBoost2 using several criteria (see STAR Methods). First, we looked at the ability of each method to uncover true regulatory relationships by comparing the number of true regulators discovered by each method in three different TCGA datasets: LUAD, LIHC, and HNSC. Using the Library of Integrated Network-Based Cellular Signatures (LINCS) data, we found that SparseGMM consistently filters for more true regulators by selecting fewer regulators than GRNBoost2. On the other hand, the number of true regulator relationships discovered by SparseGMM is higher than GRNBoost2 for all three datasets. The percentages of true regulators to selected regulators in SparseGMM are 20.17%, 17.14%, and 12.65% for LUAD, HNSC, and LIHC, respectively, while in GRNBoost2, the percentages are 7.99% (p = 7.75e−11), 9.15% (p = 0.078), and 8.09% (p = 0.014), respectively. We also compared the runtime for both methods and found that SparseGMM was consistently superior. We used the average number of target genes per regulator to compare module sizes. GRNBoost2 module sizes were consistently too large for practical functional annotation and discovery. The results of the comparison are summarized in Figure S2.
Liver cancer and healthy livers share an angiogenesis community
From the combined analysis of normal liver and liver cancer tissue, we discovered 72 communities containing normal liver modules, cancer modules, and communities that combined normal and cancer modules (Figure 3). We defined robust communities to be those with an average pairwise Jaccard index ≥0.7 between each two modules. We found 22/72 such communities and were able to reliably identify the biological function of 15/22 robust communities (see STAR Methods; Table S2). We used the LINCS database to validate the uncovered regulatory relationships.
Figure 3.

Sparse GMM module network
Left: a sample module network obtained through community detection algorithm to cancer and normal liver modules after running SparseGMM with different initializations. Right: the community detection clusters robust modules together into distinct subnetworks. Subnetworks at the periphery represent robust modules. Subnetworks are then functionally annotated using gene set enrichments analysis applied to MSigDB gene sets. Highlighted here are robust modules from normal liver and liver cancer, as well as shared communities that contain modules occurring in normal and cancer tissue.
Although many of the regulators (Table S3) do not have corresponding LINCS perturbation experiments, 9 communities (out of 11 non-immune highly robust communities) had at least one regulator validated using LINCS perturbation experiments. For immune communities, we were able to identify known regulators using evidence from previous studies (Table S3). We also used Re-Map, a database of transcriptional regulators peaks derived from DNA-binding sequencing experiments, to validate our robust community regulators. The Re-Map database contained data for 10 regulators from six robust communities.23 The Re-Map results showed that we were able to validate six out of ten regulators with Re-Map data, including HNF4A (Table S4). We hypothesized that SparseGMM could be useful to find communities shared by both HCC and healthy tissues, leading to the identification of highly conserved functions in HCC. We therefore investigated the shared GTEx and TCGA modules, revealing four robust communities enriched in functions important for physiological liver regeneration upon damage and tumor growth, including angiogenesis, cell cycle/DNA replication, ribosome, and sterol biosynthesis (Table S2).
We highlight a shared angiogenesis community that is enriched in gene sets that relate to vasculature development, extension of new blood vessels from existing capillaries into vascular tissues, and movement of an endothelial cell to form an endothelium. LINCS perturbation data confirm 2/2 regulators (Table 1). The first is NPDC1, a neural factor, which downregulates cell proliferation.24 Secondly, PROCR25 is a receptor of activated protein C, which has a documented role in inhibiting metastasis26 and limiting cancer cell extravasation through S1PR1.27 Interestingly, S1PR1 is also a regulator in this community with well-documented roles in angiogenesis and liver fibrosis28,29,30,31 but was not validated due to lack of perturbation experimental data in the LINCS database. PROCR was also shown to induce endothelial cell proliferation and angiogenesis32 and identified as a biomarker of blood vascular endothelial stem cells33 and a potential cancer biomarker.34 Among the shared regulators between cancer and normal samples is LDB2, a transcription factor, which regulates the expression of DLL4,35,36 a notch ligand involved in angiogenesis; DLL4 negatively regulates endothelial cell proliferation and migration and angiogenic sprouting.36
Table 1.
LINCS validation of robust normal liver and liver cancer communities
| Community | Main pathway/gene set | Jaccard index | LINCS-validated regulators |
|---|---|---|---|
| Normal communities | |||
| 61 | complement pathway | 0.7 | FGB |
| 11 | REACTOME _DIGESTION | 1 | GFOD1 |
| 62 | myofibril formation | 0.8 | DLX3 |
| 71 | hepatic differentiation and metabolism | 0.8 | BPHL, BDH1, HADH, HSD17B8, KLHDC9 |
| Cancer communities | |||
| 72 | blood coagulation | 0.7 | ABCG5, HNF4A, SLC25A13, EPB41L4B, BHMT2, PDXP |
| Shared Communities | |||
| 15 | GO polysomal ribosome | 0.8 | IRAK1 |
| 17 | angiogenesis | 0.8 | PROCR, NPDC1 |
| 21 | cell cycle/DNA replication | 0.9 | BRCA1, CDC20, CDCA8, CDK2, CEP55, HSF2, CDK6, ALDH4A1, MCM7 |
| 23 | sterol biosynthesis | 0.9 | SREBF2, ACAT2, NSDHL |
Robust communities were defined by having a Jaccard index ≥0.7. Main pathway of each community was revealed through gene set enrichment analysis of SparseGMM modules in GTEx and TCGA data against MSigDB collections. Validation of regulators is established with an adjusted p <0.05. See also Tables S2 and S3.
Antigen presentation and blood coagulation are robust communities revealed by SparseGMM in HCC
After applying SparseGMM only to HCC gene expression data, we discovered five robust communities enriched in pathways with important roles in the interaction between hepatocytes and the immune system: antigen presentation, interferon signaling, myeloid and CD4 and CD8 T cells, and blood coagulation (Figure 3; Table S2).
The antigen presentation community included 34 target genes that are directly involved in the process of antigen processing and presentation by the HLA complex to the T cell receptor (TCR) present on the surface of immune cells (Figure S3). This community is regulated by the PDCD1LG2 gene, encoding PD-L2, an immune checkpoint receptor of PD-1, and a recently adopted revolutionary immunotherapy drug target in patients with HCC. In our analyses, PDCD1LG2 appeared as one of the regulators of the myeloid community, while PDCD1 regulated the T cell community (Table S3; Figure S3).
Next, we highlight the community enriched in pathways related to components of the blood coagulation system and the clotting cascade (Figure 4B). This community is also enriched in processes involved in the maintenance of an internal steady state of lipid and sterol, which interact with the coagulation system.37,38 Of the 31 regulators in this community, LINCS experimental data were available for 13 genes, and 6 (46%) genes were validated (Table 1). Among these, HNF4A is the main transcriptional regulator in hepatocytes and regulates multiple coagulation genes.39,40,41,42,43 Other validated regulators of this community include EPB41L4B, which promotes cellular adhesion, migration, and motility in vitro and is reported to play a role in wound healing.44,45 SparseGMM also correctly identified SERPINC1 as a regulator of this community. While there are no LINCS perturbation experiments for SERPINC1, the regulatory role of this member of the serpin family in blood coagulation cascade has been well documented in previous studies.46,47 These results show that the clotting system is robustly regulated in HCC. While the impact of impaired liver function on blood coagulation is evident, the specific role of this pathway in HCC progression is largely unexplored.
Figure 4.

Heatmap of coexpression patterns in target genes of sample modules and graph of regulatory relationships
Regulator genes are shown in red and target genes are shown in green.
(A) Shared communtiy between HCC and normal liver: PROCR and NPDC1 regulate target genes of the angiogenesis community.
(B) Liver cancer: HNF4A and other regulators control coagulation factors and apolipoproteins involved in blood coagulation community.
(C) Normal liver community: BDH1 and HADH regulate a group of Acyl-CoA dehydrogenases and a group of cytochrome P450 enzymes involved in hepatic differentiation and metabolism.
SparseGMM identifies potential modules of hepatic differentiation and metabolism in healthy livers
We found six communities that highlight important normal liver functions. GSEA results reveal six distinct functions: hepatic differentiation and metabolism; lipid and protein catabolism; complement; cancer and vesicle trafficking; myofibril formation; and FGFR1 signaling. For example, we highlight the hepatic differentiation and metabolism community, an important pathway capturing the liver’s unique metabolic functions. Specifically, LINCS perturbation experiments validated 50% of regulators (5 out of 10 with available LINCS data) in this community (Table 1). Confirmed regulators in this community include two enzymes: BDH1, a short-chain dehydrogenase that catalyzes the interconversion of ketone bodies produced during fatty acid catabolism,48 and HADH, which is responsible for the oxidation of straight-chain 3-hydroxyacyl-coenzyme As (CoAs) as part of the β-oxidation pathway49,50,51 (Figure 4C). Five target genes in this community were reported as part of a transcriptomic signature of obesity-related steatosis in rat hepatocytes, with functions related to mitochondrial and peroxisomal oxidation of fatty acids, and detoxification.52 Additionally, it was shown that Bdh1-mediated β-hydroxybutyrylation potentiates propagation of HCC stem cells53 and that deletion of Bdh1 causes low ketone body level and fatty liver during fasting.54 Moreover, Bdh1 overexpression ameliorates hepatic injury in a metabolic-associated fatty liver disorder (MAFLD) mouse model.55 These findings point to SparseGMM-identified hepatic differentiation and metabolism genes as potential bona fide transcriptional biomarkers of hepatic differentiation and metabolism in healthy livers.
SparseGMM decouples distinct myeloid and lymphoid biological processes in HCC micro-environment, blood, and normal liver
We next evaluated the highly robust communities in an independent singe-cell RNA dataset of CD45+ immune cells for patients with HCC from five immune-relevant sites: tumor, adjacent liver, hepatic lymph node (LN), blood, and ascites.21 We used Seurat to cluster the cells and compared markers of Seurat clusters with markers of various immune cells to identify the different cell types in the tumor samples of three patients (Figure S4; see STAR Methods). Overall, we found that 4 out of 9 communities expressed in the single-cell dataset were cell-type specific (Figures 5A, 5B, and S4). These communities were the CD4 and CD8 T cell community, myeloid community, cell-cycle community (specific to T cells and dendritic cells), and community 60 (specific to T cells).
Figure 5.

Single-cell evaluation of highly robust communities
(A) Top, left to right: average expression of the T cell, myeloid, and cell-cycle community and cell types. Bottom, left to right: number of genes expressed in T cell, myeloid, and cell-cycle community in their corresponding cell type versus average number of genes expressed in other cell types.
(B) Top: cell-type annotation. Bottom: most significant gene set enrichments for the three communities.
(C) Heatmap of target genes of T cell and myeloid communities in different single-cell populations.
(D) Boxplot of cell cycle phase versus expression of cell-cycle community target genes. Higher expression of cell-cycle genes corresponds to proliferative G2M and S phases.
See also Figures S4 and S5.
The expression of target genes from communities 67 and 68 distinguished CD4 and CD8 T cells from myeloid cells, respectively (Figures 5A–5C) with a similar expression pattern in immune cells from blood and normal liver tissue (Figure S5). CD4 and CD8 T cells (myeloid cells) expressed a significantly larger number of genes from the CD4 and CD8 T cell community (myeloid cell community) than other cell types (adjusted p < 0.05, chi-squared test), confirming that the communities are cell-type specific. Additionally, we observed a subset of T cells that specifically express genes from the cell-cycle community (adjusted p < 0.05 chi-squared test). As expected, the cell-cycle community gene expression was lower (p < 2.22e−16, independent t test) in the G1 phase than in the proliferating G2M and S phases (Figure 5D). When comparing this community’s average expression in cells from different environments, we found a higher level of cell-cycle gene expression in tumor-derived immune cells than in normal immune cells (p < 2.22e−16, independent t test; Figure S5).
Finally, the percentages of variance explained in average target gene expression by regulator expression (R2) were 0.53%, 0.80%, and 0.80% in CD4 and CD8 T cell, myeloid, and cell-cycle communities, respectively, demonstrating the accuracy of the inferred regulatory programs. These results further support the robustness of communities identified in bulk RNA-seq data.
Gene entropy identifies key elements of cancer pathway crosstalk
Both in liver physiology and liver cancer, functional crosstalk, defined as the interaction between two pathways belonging to different cell processes, is a natural way of responding to new environmental challenges. Previous studies reported crosstalk between major cancer pathways such as p53 and nuclear factor κB (NF-κB)/TNF-α56,57 and p53 and estrogen.58 Furthermore, this crosstalk between pathways represents compensation mechanisms by which a cancer cell can generate resistance to the blockage of a specific gene or pathway.59,60 We hypothesized that gene entropy (Methods S1), which is a measure of uncertainty in its assignment to a gene module, could be interpreted as a proxy for multiple module membership and thus be used to unveil the elements of hidden crosstalk in cancer.
We calculated the average entropy of each target gene over multiple runs of SparseGMM on TCGA and GTEx samples from the genes’ posterior probability (see STAR Methods). We set an entropy threshold of 1, which corresponds to the maximum possible value of entropy between two modules, to identify genes with uncertainty in module assignment. We found that for target genes with entropy >1, TCGA target genes showed significantly higher degree of entropy when compared with GTEx (p < 2.22e−16, independent t test; Figure 6A). This difference in entropy distribution reflects the heterogeneity of cancer tissue compared with normal healthy tissue.
Figure 6.

Analysis of high-entropy genes
(A) Boxplot showing difference in mean entropy distribution for high-entropy target genes in GTEx and TCGA, reflecting heterogeneity of cancer samples. Entropy is calculated from the posterior probability of target genes in each dataset, and the mean is calculated over several runs of SparseGMM on each dataset.
(B) Distribution of communities of high-entropy genes.
(C–E) Expression of communities with high-entropy genes.
We then analyzed the distribution of community membership among high-entropy genes in TCGA. Interestingly, genes with high entropy clustered in a few communities such as p53-related networks, NF-κB/TNF-α response, response to interferon-γ, estrogen response, and bile acid metabolism (Figure 6B). p53 harbors a loss-of-function mutation in around one-third of patients with HCC.61 Most HCCs originate in an inflammatory liver background such as hepatitis C or B chronic infection or non-alcoholic steatohepatitis (NASH),62 and bile acid composition has been related to HCC.63 Finally, estrogen signaling has been studied in liver cancer as a potential protective factor and as one of the reasons HCC is more frequently seen in males than in females.64 Altogether, these results suggest an unbiased efficient capturing of clinically relevant pathway crosstalk by SparseGMM. If multifunctional, the genes captured by our method in each crosstalk could be important for identifying key targets for an efficient therapeutic disruption of cancer growth.
Next, we studied in detail the detected highly entropic genes within crosstalk. We found 15 high-entropy genes that were assigned to both estrogen-mediated signaling and p53 communities. One of these genes, GREB1 is an estrogen-regulated gene that is expressed in estrogen receptor α (ERα)-positive breast cancer cells modulating its function and promoting cancer cell proliferation.65 The expression of GREB1 is controlled by a p53 target.66 Similarly, IGFALS is another high-entropy gene with assignment to both p53 and estrogen signaling communities. This is consistent with the fact that IGFALS interacts with a p53 target67,68 and has a role in regulating ERs in breast cancer.69,70,71,72 Additionally, we examined more closely the crosstalk between p53 and NF-κB/TNF-α pathways. PAX8, a transcription factor expressed in 90% of high-degree serous carcinoma,73 is among the highly entropic genes identified by SparseGMM as participating in both p53- and TNF-α/NF-κB1-related signaling. Interestingly, we found a significant correlation between average target gene expression of p53 and NF-κB/TNF-α pathways (Figure 6C; Pearson correlation = 0.46 confidence interval [CI] [0.37–0.53], p < 2.2e−16). Previous studies also showed that p53 and NF-κB/TNF-α coregulate proinflammatory gene responses in human macrophages.74 We observed significant correlation between the TNF-α-induced NF-κB community and the myeloid community (Figure 6D; Pearson correlation = 0.48, CI [0.41–0.56], p < 2.2e−16). The p53-NF-κB/TNF-α crosstalk is also implicated in increased invasiveness.75 We found a significant correlation between the NF-κB/TNF-α community and the epithelial-to-mesenchymal transition (EMT) and cancer stemness community (Figure 6E; Pearson correlation = 0.53, CI [0.45–0.60], p < 2.2e−16). Accordingly, SparseGMM is able not only to infer key regulators and their downstream gene modules but also potentially identify key multifunctional components shared by critical cancer pathways based on their entropy.
Discussion
SparseGMM has a unique capability to infer GRN relationships from bulk RNA-seq data while assigning target genes to multiple gene modules and sparse sets of regulators to their respective modules. As a result, SparseGMM accurately models the scale-free nature of biological networks, as well as the molecular heterogeneity of biological tissue and the versatile roles of a subset of genes in different biological pathways.
SparseGMM accomplishes this goal by combining coexpression- and graph-based approaches in a Bayesian setting to model the relationships between downstream target genes and their regulator genes. SparseGMM enforces a sparsity constraint, which reduces overfitting and increases the interpretability of regulatory relationships by restricting the number of regulators in each module. This sparsity in regulator selection achieves an improvement in prioritizing potential therapeutic targets and discovering new regulator genes.
We demonstrated the utility and reliability of SparseGMM by applying it to datasets of normal and cancerous liver tissue and employing community detection methods to screen gene modules for robustness and for shared biology between normal liver tissue and liver cancer. We then used GSEA to functionally annotate highly robust modules. Despite the complex physiology of the liver, from metabolism and immunity to protein synthesis, SparseGMM recovered important physiological functions that are active in healthy and cancerous liver tissue. SparseGMM also recovered the molecular similarities and differences between the biological processes in healthy and cancer tissues. This has implications on our understanding of the mechanisms of cancer development and progression. Further, SparseGMM was able to identify new and known regulators of normal liver physiology and hepatocarcinogenesis such as BDH1 and HNF4A. To validate the resulting new associations between regulators and downstream targets, we used experimental genetic perturbation data from the LINCS. While our analysis of the LINCS datasets validates results in liver cells, different experimental models are required to validate the identified immune regulatory relationships. Although we do not validate the immune modules in this work, the discovery of several communities with immune function and their subsequent verification in an independent single-cell dataset reflects the ability of SparseGMM to decouple biological processes related to distinct immune cell populations.
Thanks to the Bayesian nature of the proposed algorithm, SparseGMM can probabilistically assign each target gene to multiple modules and the uncertainty in gene assignment can be measured using the information theoretic measure of entropy as a proxy for a gene’s versatile functions and capturing potential crosstalk between biological pathways. In our results, SparseGMM identified GREB1 and IGFALS as high-entropy genes that were assigned to both p53 and estrogen signaling communities. The plausibility of a GREB1-p53 interaction is supported by the fact that the phosphorylation of PBX homeobox interacting protein 1 (HPIP), a target of p53, is necessary for estrogen-mediated GREB1 expression.66 On the other hand, IGFALS was previously reported to form a complex with IGFBP-3, a well-known target of p53,67,68 which has growth inhibitory and pro-apoptotic properties.76 IGFALS was also reported to regulate ERs in breast cancer.69,70,71,72 Importantly, we also examined crosstalk between p53 and another significant pathway, NF-κB/TNF-α. Previous studies have shown that TNF-induced, NF-κB-directed gene expression relies on p53,57 but the significance and specific mechanisms of this interaction are not fully explained. We identified PAX8, a high-entropy gene that belongs to both pathways and that encodes a transcription factor. While PAX8 binding is inhibited by TNF-α,77 its pro-proliferative role relies on p53-p21.78
SparseGMM was able to identify PROCR as a regulator of an angiogenesis community shared between normal liver and liver cancer. In liver cancer tissue, SparseGMM recovered an antigen processing and presentation community, regulated by PDCD1LG2, that encodes a key immunotherapy drug target in HCC and an immune checkpoint receptor of PD-1. Interestingly, PDCD1LG2 was also a regulator of the myeloid community identified by SparseGMM. In normal liver, SparseGMM identified a hepatic differentiation and metabolism community regulated by BDH1, a short-chain dehydrogenase, which catalyzes the interconversion of ketone bodies produced during fatty acid catabolism.48 Lastly, the discovery of several communities with immune function and their subsequent verification in single-cell data reflect the ability of SparseGMM to decouple biological processes related to distinct immune cell populations.
In summary, SparseGMM employs a coexpression-based GRN inference approach in a Bayesian framework from bulk transcriptomic data and achieves superior performance compared with state-of-the-art module-based GRN inference methods and identifies important biological pathways, and corresponding gene regulators, as exemplified by application to human liver healthy and diseased tissue.
Limitations of the study
In framing this work, we note limitations mainly with respect to biological validation. First, while we were able to leverage single-cell data to verify immune communities, the validation of regulatory relationships in these communities cannot be performed using cancer cell line data such as the perturbation experiment datasets used in this study. Future directions include biological validation of regulatory relationships in immune communities. Second, while the regulatory evidence from the cell line perturbation experiment data used presents strong evidence for the regulatory relationships discovered in normal and cancer liver cell types, relevant functional assays are still required to confirm the nature of regulatory relationships uncovered by our method.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| TCGA RNA-Seq data | Cancer Genome Atlas Research Network. | https://doi.org/10.1016/j.cell.2017.05.046 |
| TCGA GTEx RNA-Seq data | Consortium, G. T. et al | https://doi.org/10.1038/ng.2653 |
| Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma | Gene Expression Omnibus - NCBI | GSE140228 |
| Software and algorithms | ||
| Data analysis was done using R 3.6.1 | R | N/A |
| Data analysis was done using Python | Python | |
| Analysis code for R, and Python is available at the Zenodo code repository:https://doi.org/10.5281/zenodo.7418960 | https://doi.org/10.5281/zenodo.7418960 | |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Olivier Gevaert (ogevaert@stanford.edu).
Materials availability
-
•
This study did not generate new unique reagents.
Method details
Data preprocessing
Gene modules in normal tissue were constructed using publicly available data from GTEx project, while cancer modules were constructed using HCC data from TCGA project.The datasets were preprocessed and provided in,79 in which they provided reference RNA-seq expression levels from healthy human tissue that can be compared with the expression levels found in human cancer tissue.
A list of candidate genes was obtained from previously generated AMARETTO17 data objects extracted using the TFutils R package.80 In addition, genes whose gene expression can be explained using changes in copy number variation81 or methylation82 status from the TCGA dataset were extracted using the AMARETTO package. The combined list of genes was used as an initial candidate regulator gene list. Next, the top 75% varying genes were identified to each dataset separately. Of the top 75%, the top 2000 genes that are also present in the candidate regulator genes list were used to build the regulator gene matrix, the rest were regarded as target genes. The gene expression data matrix was centered to mean 0 and standard deviation 1 and then split into a regulator gene matrix and a target gene matrix. A similar approach was used to preprocess and build the input data matrices from the GTEx dataset. Overall, the TCGA contained 8017 protein-coding genes including 2000 candidate drivers, while the GTEx network contains 10804 protein-coding genes including 1800 candidate drivers.
Implementation and technical validation
We implemented SparseGMM (Methods S1) in Python. SparseGMM was run 5 times on each data set with different seeds to evaluate the robustness of the method. We first split both data sets into training (70%) and test (30%) sets. Four different metrics were employed to validate the performance of SparseGMM: 1) Adjusted index (ARI) to measure robustness, 2) R-Squared to measure goodness of fit, 3) Number of selected regulators to measure sparsity, and 4) Size of module to evaluate the sensitivity of module size to the regularization parameter lambda. The values of lambda above 5000 produced very large modules and were excluded from further analysis. Values below 50 were also excluded due to low adjusted rand index. Lambda values examined were 50, 275, 500, 2750 and 5000 were used. The output of different seeds was also used to filter the generated modules using cAMARETTO as explained below. The input number of clusters used was 150 as it resulted an average size of 60 genes per cluster and reduced false positive results in downstream functional gene set enrichments (Figures 2D and 2H).
Comparison of SparseGMM to existing GRN methods
We compared the performance of SparseGMM to GRNBoost2 in three data sets LUAD, LUHC and HNSC using several criteria: 1) the number and percentage of true regulators validated against LINCS data, 2) runtime 3) the size of modules, and 4) sparsity. To measure the ability of each method to uncover true regulatory relationships we performed GSEA of SparseGMM modules for each regulator against corresponding the downstream targets in LINCS data. Similarly, we compared downstream targets of GRNBoost2 to LINCS downstream of each regulator. The types of LINCS perturbation experiment data used were 1) Consensus signature from shRNAs targeting the same gene and 2) cDNA for overexpression of wild-type gene. The Fast Gene Set Enrichment Analysis tool was used to test for significance in enrichment. For each TCGA data set, the corresponding cell lines used are shown in Figure S2. We used a p value <0.05 as a threshold for validated regulators and used the Bonferroni method for multiple hypothesis correction.83 To compare the number of validated regulators in both methods we used a chi-squared test. Since GRNBoost2 does not build sparsely regulated gene modules, we used the number of regulators per target gene as a proxy for sparsity and the number of target gene per regulator as a proxy for module size and compared the distribution of these metrics (Figure S2).
Robust module recovery via community detection
To detect robust modules, we used the community-AMARETTO (cAMARETTO) package20 to build communities among modules discovered by running SparseGMM with different seeds on the same data set. cAMARETTO identifies gene modules and their regulators that are shared and distinct across multiple regulatory networks. Specifically, cAMARETTO takes as input multiple networks inferred using the sparseGMM algorithm. cAMARETTO can learn communities or subnetworks from regulatory networks derived from multiple cohorts, diseases, or biological systems. To do this cAMARETTO uses the Girvan-Newman “edge betweenness community detection” algorithm.84 The cAMARETTO algorithm consists of 1) constructing a master network composed of multiple regulatory networks followed by 2) detecting groups (communities) of modules that are shared across systems, as well as highlighting modules that are system-specific and distinct. By applying cAMARETTO to modules discovered by running SparseGMM with different seeds on the same data set, modules that are consistently discovered by SparseGMM will be grouped in the same subnetwork or community, i.e., copies of the same module will be clustered in a distinct community. cAMARETTO parameters used were p value = 0.01 and intersection = 10. When running cAMARETTO on a single data set (either GTEx or TCGA), we filtered for communities of size 5, one from each run and further narrowed down results by Jaccard index ≥0.7. In contrast, for communities with both TCGA and GTEx modules, communities of size 10 were selected. The selected communities were used as input to the GSEA function of cAMARETTO.
Single cell transcriptomic evaluation
We evaluated the highly robust communities in an independent singe cell RNA data set with samples from immune-relevant sites in five HCC patients: tumor, adjacent liver, hepatic lymph node (LN), blood, and ascites (Accession number: GSE140228, Gene Expression Omnibus).21 This data set contains only purified CD45+ immune cells and no other cell types. We focused on expression in tumor core to evaluate our communities, which was available from three patients. Preprocessing procedure was as follows: single cells were processed through the GemCode Single Cell Platform using the GemCode Gel Bead, Chip and Library Kits (10× Genomics, Pleasanton) as per the manufacturer’s protocol.85 The cells were partitioned into Gel Beads in Emulsion in the GemCode instrument, where cell lysis and barcoded reverse transcription of RNA occurred, followed by amplification, shearing and 30 adaptor and sample index attachment. Libraries were sequenced on an Illumina Hiseq 4000. We used Seurat to analyze the data set.86 To perform quality control of the data,87 we filtered genes that were expressed in less than 40 cells and cells that had fewer than 1000, greater than 5000 genes, and cells with a proportion of transcripts mapping to mitochondrial genes greater than 5%.86,88,89 We then scale the data and apply PCA, then clustering and UMAP using the top 10 PCA dimensions. We use the resulting Seurat clusters and PanglaoDB cell markers to identify cell marker expression.90 We compared the average expression of cell type markers to annotate cells and compared Seurat clusters to PanglaoDB annotations to assign an immune cell type to each cluster in the core tumor samples. To evaluate the expression of our communities, and the cell-type specificity of each community, we compared the number of genes expressed from each community in each cell type to the average number of genes expressed in other cell types using a chi-squared test. We used Seurat to score the cell cycle phase of cells.
Quantification and statistical analysis
Gene set enrichment analysis
We then applied GSEA using MSigDB collections (Hallmarks and C1-5) to functionally annotate each of the communities. A p value<1e-5, adjusted for testing of multiple hypotheses using the Benjamini-Hochberg method, was selected to filter enriched datasets.
Biological validation
To experimentally validate regulators of the discovered communities, we interrogated the robust regulators, defined as regulators consistently associated with the same community by SparseGMM across all runs, against publicly available genetic perturbation studies in the Library of Integrated Network-Based Cellular Signatures (LINCS) database. In this validation experiment we leveraged the HEPG2 liver cell line data. The types of perturbation experiments used were 1) Consensus signature from shRNAs targeting the same gene and 2) cDNA for overexpression of wild-type gene. The Fast Gene Set Enrichment Analysis tool was used to test for significance in enrichment. To empirically derive p values, we permuted 1000 lists of genes of the same size as the community target sets for each community and for each regulator. Regulator-gene set pairs which had a corresponding p value<0.05, adjusted for testing of multiple hypotheses using the Benjamini-Hochberg method, were considered validated cellular signatures in either of the two signature types.
We also used Re-Map,23 a database of transcriptional regulators peaks derived curated from DNA-binding sequencing experiments to validate our robust community regulators. We restricted our analysis to experiments on the HEPG2 liver cell line data. We used a hypergeometric test91 to test for significance between Re-Map data and our data. We used the Bonferroni method83 to correct for multiple comparisons.
Acknowledgments
We thank M. Nabian for his support in using the community AMARETTO package and S. Napel for his advice on study design. We also thank Stanford University’s Quantitative Sciences Unit and in particular A. Gentles for their advice on statistical methods. This work was supported by the National Cancer Institute Office of Cancer Genomics Cancer Target Discovery and Development (CTD^2) initiative, as well as the National Cancer Institute (NCI) under awards R01 CA260271, U01 CA217851, and U01 CA199241.
Author contributions
S.B. initiated and designed the study with the guidance of O.G., M.H., and K.B.; designed and developed the statistical model with the guidance of M.H. and O.G.; performed all bulk and single-cell data analyses; performed all biological validation analyses; and wrote the manuscript and generated all figures and data visualizations. S.B. and J.A. performed entropy GSEA. All other authors reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interests
Published: January 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100392.
Contributor Information
Mikel Hernaez, Email: mhernaez@unav.es.
Olivier Gevaert, Email: ogevaert@stanford.edu.
Supplemental information
(a) Functional enrichments for normal liver modules using MSigDB collections. (b) Functional enrichments for cancer modules using MSigDB collections. (c) Functional enrichments for shared modules using MSigDB collections.
(a) Regulator Genes of robust modules (average Jaccard Index=>0.7 in module subnetwork). (b) Target Genes in robust modules (average Jaccard Index=>0.7 in module subnetwork). (c) Regulator and target genes for the EMT and stemness community.
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for thedatasets are listed in the key resources table.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Cancer Genome Atlas Research Network Electronic address wheeler@bcmedu. Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GTEx Consortium. Laboratory, Data Analysis & Coordinating Center (LDACC)—Analysis Working Group. Statistical Methods groups-Analysis Working Group. Enhancing GTEx (eGTEx) groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu S., Xu Y., Liu P., Zhang S., Liu H., Slavin S., Kumar S., Koroleva M., Luo J., Wu X., et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur. Heart J. 2019;40:2398–2408. doi: 10.1093/eurheartj/ehz303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohammadi P., Castel S.E., Cummings B.B., Einson J., Sousa C., Hoffman P., Donkervoort S., Jiang Z., Mohassel P., Foley A.R., et al. Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science. 2019;366:351–356. doi: 10.1126/science.aay0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J.D., Zhou H.S., Tu X.X., He Y., Liu Q.F., Liu Q., Long Z.J. Prediction of competing endogenous RNA coexpression network as prognostic markers in AML. Aging (Albany NY) 2019;11:3333–3347. doi: 10.18632/aging.101985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu M., Yi H., Ma S. Vertical integration methods for gene expression data analysis. Brief. Bioinform. 2021;22:bbaa169. doi: 10.1093/bib/bbaa169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh M., Park S., Kim S., Chae H. Machine learning-based analysis of multi-omics data on the cloud for investigating gene regulations. Brief. Bioinform. 2021;22:66–76. doi: 10.1093/bib/bbaa032. [DOI] [PubMed] [Google Scholar]
- 8.Basso K., Margolin A.A., Stolovitzky G., Klein U., Dalla-Favera R., Califano A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 9.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernaez M., Blatti C., Gevaert O. Comparison of single and module-based methods for modeling gene regulatory networks. Bioinformatics. 2020;36:558–567. doi: 10.1093/bioinformatics/btz549. [DOI] [PubMed] [Google Scholar]
- 11.Champion M., Brennan K., Croonenborghs T., Gentles A.J., Pochet N., Gevaert O. Module Analysis Captures Pancancer Genetically and Epigenetically Deregulated Cancer Driver Genes for Smoking and Antiviral Response. EBioMedicine. 2018;27:156–166. doi: 10.1016/j.ebiom.2017.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manolakos A., Ochoa I., Venkat K., Goldsmith A.J., Gevaert O. CaMoDi: a new method for cancer module discovery. BMC Genomics. 2014;15:S8. doi: 10.1186/1471-2164-15-S10-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horvath S., Zhang B., Carlson M., Lu K.V., Zhu S., Felciano R.M., Laurance M.F., Zhao W., Qi S., Chen Z., et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl. Acad. Sci. USA. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5:e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moerman T., Aibar Santos S., Bravo González-Blas C., Simm J., Moreau Y., Aerts J., Aerts S. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019;35:2159–2161. doi: 10.1093/bioinformatics/bty916. [DOI] [PubMed] [Google Scholar]
- 16.Akavia U.D., Litvin O., Kim J., Sanchez-Garcia F., Kotliar D., Causton H.C., Pochanard P., Mozes E., Garraway L.A., Pe'er D. An integrated approach to uncover drivers of cancer. Cell. 2010;143:1005–1017. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gevaert O., Villalobos V., Sikic B.I., Plevritis S.K. Identification of ovarian cancer driver genes by using module network integration of multi-omics data. Interface Focus. 2013;3 doi: 10.1098/rsfs.2013.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gevaert O., Plevritis S. Identifying master regulators of cancer and their downstream targets by integrating genomic and epigenomic features. Pac. Symp. Biocomput. 2013:123–134. [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gevaert O., Nabian M., Bakr S., Everaert C., Shinde J., Manukyan A., Liefeld T., Tabor T., Xu J., Lupberger J., et al. Imaging-AMARETTO: An Imaging Genomics Software Tool to Interrogate Multiomics Networks for Relevance to Radiography and Histopathology Imaging Biomarkers of Clinical Outcomes. JCO Clin. Cancer Inform. 2020;4:421–435. doi: 10.1200/CCI.19.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q., He Y., Luo N., Patel S.J., Han Y., Gao R., Modak M., Carotta S., Haslinger C., Kind D., et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell. 2019;179:829–845.e20. doi: 10.1016/j.cell.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Leclerc R.D. Survival of the sparsest: robust gene networks are parsimonious. Mol. Syst. Biol. 2008;4:213. doi: 10.1038/msb.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammal F., de Langen P., Bergon A., Lopez F., Ballester B. ReMap 2022: a database of Human, Mouse, Drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res. 2022;50:D316–D325. doi: 10.1093/nar/gkab996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sansal I., Dupont E., Toru D., Evrard C., Rouget P. NPDC-1, a regulator of neural cell proliferation and differentiation, interacts with E2F-1, reduces its binding to DNA and modulates its transcriptional activity. Oncogene. 2000;19:5000–5009. doi: 10.1038/sj.onc.1203843. [DOI] [PubMed] [Google Scholar]
- 25.Mohan Rao L.V., Esmon C.T., Pendurthi U.R. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. 2014;124:1553–1562. doi: 10.1182/blood-2014-05-578328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bezuhly M., Cullen R., Esmon C.T., Morris S.F., West K.A., Johnston B., Liwski R.S. Role of activated protein C and its receptor in inhibition of tumor metastasis. Blood. 2009;113:3371–3374. doi: 10.1182/blood-2008-05-159434. [DOI] [PubMed] [Google Scholar]
- 27.Van Sluis G.L., Niers T.M.H., Esmon C.T., Tigchelaar W., Richel D.J., Buller H.R., Van Noorden C.J.F., Spek C.A. Endogenous activated protein C limits cancer cell extravasation through sphingosine-1-phosphate receptor 1-mediated vascular endothelial barrier enhancement. Blood. 2009;114:1968–1973. doi: 10.1182/blood-2009-04-217679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang L., Yue S., Yang L., Liu X., Han Z., Zhang Y., Li L. Sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis is involved in liver fibrosis-associated angiogenesis. J. Hepatol. 2013;59:114–123. doi: 10.1016/j.jhep.2013.02.021. [DOI] [PubMed] [Google Scholar]
- 29.Ben Shoham A., Malkinson G., Krief S., Shwartz Y., Ely Y., Ferrara N., Yaniv K., Zelzer E. S1P1 inhibits sprouting angiogenesis during vascular development. Development. 2012;139:3859–3869. doi: 10.1242/dev.078550. [DOI] [PubMed] [Google Scholar]
- 30.Balaji Ragunathrao V.A., Anwar M., Akhter M.Z., Chavez A., Mao D.Y., Natarajan V., Lakshmikanthan S., Chrzanowska-Wodnicka M., Dudek A.Z., Claesson-Welsh L., et al. Sphingosine-1-Phosphate Receptor 1 Activity Promotes Tumor Growth by Amplifying VEGF-VEGFR2 Angiogenic Signaling. Cell Rep. 2019;29:3472–3487.e4. doi: 10.1016/j.celrep.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cartier A., Leigh T., Liu C.H., Hla T. Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc. Natl. Acad. Sci. USA. 2020;117:3157–3166. doi: 10.1073/pnas.1906246117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchiba M., Okajima K., Oike Y., Ito Y., Fukudome K., Isobe H., Suda T. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ. Res. 2004;95:34–41. doi: 10.1161/01.RES.0000133680.87668.FA. [DOI] [PubMed] [Google Scholar]
- 33.Yu Q.C., Song W., Wang D., Zeng Y.A. Identification of blood vascular endothelial stem cells by the expression of protein C receptor. Cell Res. 2016;26:1079–1098. doi: 10.1038/cr.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ducros E., Mirshahi S., Azzazene D., Camilleri-Broët S., Mery E., Al Farsi H., Althawadi H., Besbess S., Chidiac J., Pujade-Lauraine E., et al. Endothelial protein C receptor expressed by ovarian cancer cells as a possible biomarker of cancer onset. Int. J. Oncol. 2012;41:433–440. doi: 10.3892/ijo.2012.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi H.J., Rho S.S., Choi D.H., Kwon Y.G. LDB2 regulates the expression of DLL4 through the formation of oligomeric complexes in endothelial cells. BMB Rep. 2018;51:21–26. doi: 10.5483/BMBRep.2018.51.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brütsch R., Liebler S.S., Wüstehube J., Bartol A., Herberich S.E., Adam M.G., Telzerow A., Augustin H.G., Fischer A. Integrin cytoplasmic domain-associated protein-1 attenuates sprouting angiogenesis. Circ. Res. 2010;107:592–601. doi: 10.1161/CIRCRESAHA.110.217257. [DOI] [PubMed] [Google Scholar]
- 37.Nemerson Y. The role of lipids in the tissue factor pathway of blood coagulation. Adv. Exp. Med. Biol. 1975;63:245–253. doi: 10.1007/978-1-4684-3258-9_16. [DOI] [PubMed] [Google Scholar]
- 38.Marcus A.J. The role of lipids in blood coagulation. Adv. Lipid Res. 1966;4:1–37. [PubMed] [Google Scholar]
- 39.Inoue Y., Peters L.L., Yim S.H., Inoue J., Gonzalez F.J. Role of hepatocyte nuclear factor 4alpha in control of blood coagulation factor gene expression. J. Mol. Med. 2006;84:334–344. doi: 10.1007/s00109-005-0013-5. [DOI] [PubMed] [Google Scholar]
- 40.Safdar H., Cheung K.L., Vos H.L., Gonzalez F.J., Reitsma P.H., Inoue Y., van Vlijmen B.J.M. Modulation of mouse coagulation gene transcription following acute in vivo delivery of synthetic small interfering RNAs targeting HNF4alpha and C/EBPalpha. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Safdar H., Inoue Y., van Puijvelde G.H., Reitsma P.H., van Vlijmen B.J.M. The role of hepatocyte nuclear factor 4alpha in regulating mouse hepatic anticoagulation and fibrinolysis gene transcript levels. J. Thromb. Haemost. 2010;8:2839–2841. doi: 10.1111/j.1538-7836.2010.04080.x. [DOI] [PubMed] [Google Scholar]
- 42.DeLaForest A., Di Furio F., Jing R., Ludwig-Kubinski A., Twaroski K., Urick A., Pulakanti K., Rao S., Duncan S.A. HNF4A Regulates the Formation of Hepatic Progenitor Cells from Human iPSC-Derived Endoderm by Facilitating Efficient Recruitment of RNA Pol II. Genes (Basel) 2018;10:21. doi: 10.3390/genes10010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qu M., Duffy T., Hirota T., Kay S.A. Nuclear receptor HNF4A transrepresses CLOCK:BMAL1 and modulates tissue-specific circadian networks. Proc. Natl. Acad. Sci. USA. 2018;115:E12305–E12312. doi: 10.1073/pnas.1816411115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakajima H., Tanoue T. Lulu2 regulates the circumferential actomyosin tensile system in epithelial cells through p114RhoGEF. J. Cell Biol. 2011;195:245–261. doi: 10.1083/jcb.201104118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bosanquet D.C., Ye L., Harding K.G., Jiang W.G. Expressed in high metastatic cells (Ehm2) is a positive regulator of keratinocyte adhesion and motility: The implication for wound healing. J. Dermatol. Sci. 2013;71:115–121. doi: 10.1016/j.jdermsci.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Szabo R., Netzel-Arnett S., Hobson J.P., Antalis T.M., Bugge T.H. Matriptase-3 is a novel phylogenetically preserved membrane-anchored serine protease with broad serpin reactivity. Biochem. J. 2005;390:231–242. doi: 10.1042/BJ20050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubin H. Serine protease inhibitors (SERPINS): where mechanism meets medicine. Nat. Med. 1996;2:632–633. doi: 10.1038/nm0696-632. [DOI] [PubMed] [Google Scholar]
- 48.Adami P., Duncan T.M., McIntyre J.O., Carter C.E., Fu C., Melin M., Latruffe N., Fleischer S. Monoclonal antibodies for structure-function studies of (R)-3-hydroxybutyrate dehydrogenase, a lipid-dependent membrane-bound enzyme. Biochem. J. 1993;292:863–872. doi: 10.1042/bj2920863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett M.J., Russell L.K., Tokunaga C., Narayan S.B., Tan L., Seegmiller A., Boriack R.L., Strauss A.W. Reye-like syndrome resulting from novel missense mutations in mitochondrial medium- and short-chain l-3-hydroxy-acyl-CoA dehydrogenase. Mol. Genet. Metab. 2006;89:74–79. doi: 10.1016/j.ymgme.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Clayton P.T., Eaton S., Aynsley-Green A., Edginton M., Hussain K., Krywawych S., Datta V., Malingre H.E., Berger R., van den Berg I.E. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J. Clin. Invest. 2001;108:457–465. doi: 10.1172/JCI11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barycki J.J., O'Brien L.K., Bratt J.M., Zhang R., Sanishvili R., Strauss A.W., Banaszak L.J. Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry. 1999;38:5786–5798. doi: 10.1021/bi9829027. [DOI] [PubMed] [Google Scholar]
- 52.Buqué X., Martínez M.J., Cano A., Miquilena-Colina M.E., García-Monzón C., Aspichueta P., Ochoa B. A subset of dysregulated metabolic and survival genes is associated with severity of hepatic steatosis in obese Zucker rats. J. Lipid Res. 2010;51:500–513. doi: 10.1194/jlr.M001966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang H., Chang Z., Qin L.N., Liang B., Han J.X., Qiao K.L., Yang C., Liu Y.R., Zhou H.G., Sun T. MTA2 triggered R-loop trans-regulates BDH1-mediated beta-hydroxybutyrylation and potentiates propagation of hepatocellular carcinoma stem cells. Signal Transduct. Target. Ther. 2021;6:135. doi: 10.1038/s41392-021-00464-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Otsuka H., Kimura T., Ago Y., Nakama M., Aoyama Y., Abdelkreem E., Matsumoto H., Ohnishi H., Sasai H., Osawa M., et al. Deficiency of 3-hydroxybutyrate dehydrogenase (BDH1) in mice causes low ketone body levels and fatty liver during fasting. J. Inherit. Metab. Dis. 2020;43:960–968. doi: 10.1002/jimd.12243. [DOI] [PubMed] [Google Scholar]
- 55.Xu B.T., Teng F.Y., Wu Q., Wan S.R., Li X.Y., Tan X.Z., Xu Y., Jiang Z.Z. Bdh1 overexpression ameliorates hepatic injury by activation of Nrf2 in a MAFLD mouse model. Cell Death Discov. 2022;8:49. doi: 10.1038/s41420-022-00840-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Webster G.A., Perkins N.D. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell. Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider G., Henrich A., Greiner G., Wolf V., Lovas A., Wieczorek M., Wagner T., Reichardt S., von Werder A., Schmid R.M., et al. Cross talk between stimulated NF-kappaB and the tumor suppressor p53. Oncogene. 2010;29:2795–2806. doi: 10.1038/onc.2010.46. [DOI] [PubMed] [Google Scholar]
- 58.Berger C., Qian Y., Chen X. The p53-estrogen receptor loop in cancer. Curr. Mol. Med. 2013;13:1229–1240. doi: 10.2174/15665240113139990065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delou J.M.A., Souza A.S.O., Souza L.C.M., Borges H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells. 2019;8:1013. doi: 10.3390/cells8091013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellis L.M., Hicklin D.J. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin. Cancer Res. 2009;15:7471–7478. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 61.Schulze K., Imbeaud S., Letouzé E., Alexandrov L.B., Calderaro J., Rebouissou S., Couchy G., Meiller C., Shinde J., Soysouvanh F., et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang J.D., Hainaut P., Gores G.J., Amadou A., Plymoth A., Roberts L.R. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019;16:589–604. doi: 10.1038/s41575-019-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma C., Han M., Heinrich B., Fu Q., Zhang Q., Sandhu M., Agdashian D., Terabe M., Berzofsky J.A., Fako V., et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalra M., Mayes J., Assefa S., Kaul A.K., Kaul R. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol. 2008;14:5945–5961. doi: 10.3748/wjg.14.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghosh M.G., Thompson D.A., Weigel R.J. PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res. 2000;60:6367–6375. [PubMed] [Google Scholar]
- 66.Shostak K., Patrascu F., Göktuna S.I., Close P., Borgs L., Nguyen L., Olivier F., Rammal A., Brinkhaus H., Bentires-Alj M., et al. MDM2 restrains estrogen-mediated AKT activation by promoting TBK1-dependent HPIP degradation. Cell Death Differ. 2014;21:811–824. doi: 10.1038/cdd.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baxter R.C. IGF binding proteins in cancer: mechanistic and clinical insights. Nat. Rev. Cancer. 2014;14:329–341. doi: 10.1038/nrc3720. [DOI] [PubMed] [Google Scholar]
- 68.Grimberg A., Coleman C.M., Burns T.F., Himelstein B.P., Koch C.J., Cohen P., El-Deiry W.S. p53-Dependent and p53-independent induction of insulin-like growth factor binding protein-3 by deoxyribonucleic acid damage and hypoxia. J. Clin. Endocrinol. Metab. 2005;90:3568–3574. doi: 10.1210/jc.2004-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shao Z.M., Sheikh M.S., Ordonez J.V., Feng P., Kute T., Chen J.C., Aisner S., Schnaper L., LeRoith D., Roberts C.T., Jr., et al. IGFBP-3 gene expression and estrogen receptor status in human breast carcinoma. Cancer Res. 1992;52:5100–5103. [PubMed] [Google Scholar]
- 70.Rocha R.L., Hilsenbeck S.G., Jackson J.G., Lee A.V., Figueroa J.A., Yee D. Correlation of insulin-like growth factor-binding protein-3 messenger RNA with protein expression in primary breast cancer tissues: detection of higher levels in tumors with poor prognostic features. J. Natl. Cancer Inst. 1996;88:601–606. doi: 10.1093/jnci/88.9.601. [DOI] [PubMed] [Google Scholar]
- 71.Figueroa J.A., Jackson J.G., McGuire W.L., Krywicki R.F., Yee D. Expression of insulin-like growth factor binding proteins in human breast cancer correlates with estrogen receptor status. J. Cell. Biochem. 1993;52:196–205. doi: 10.1002/jcb.240520211. [DOI] [PubMed] [Google Scholar]
- 72.Yu H., Levesque M.A., Khosravi M.J., Papanastasiou-Diamandi A., Clark G.M., Diamandis E.P. Associations between insulin-like growth factors and their binding proteins and other prognostic indicators in breast cancer. Br. J. Cancer. 1996;74:1242–1247. doi: 10.1038/bjc.1996.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nonaka D., Chiriboga L., Soslow R.A. Expression of pax8 as a useful marker in distinguishing ovarian carcinomas from mammary carcinomas. Am. J. Surg. Pathol. 2008;32:1566–1571. doi: 10.1097/PAS.0b013e31816d71ad. [DOI] [PubMed] [Google Scholar]
- 74.Lowe J.M., Menendez D., Bushel P.R., Shatz M., Kirk E.L., Troester M.A., Garantziotis S., Fessler M.B., Resnick M.A. p53 and NF-kappaB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014;74:2182–2192. doi: 10.1158/0008-5472.CAN-13-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Minin G., Bellazzo A., Dal Ferro M., Chiaruttini G., Nuzzo S., Bicciato S., Piazza S., Rami D., Bulla R., Sommaggio R., et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell. 2014;56:617–629. doi: 10.1016/j.molcel.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 76.Neumann O., Kesselmeier M., Geffers R., Pellegrino R., Radlwimmer B., Hoffmann K., Ehemann V., Schemmer P., Schirmacher P., Lorenzo Bermejo J., Longerich T. Methylome analysis and integrative profiling of human HCCs identify novel protumorigenic factors. Hepatology. 2012;56:1817–1827. doi: 10.1002/hep.25870. [DOI] [PubMed] [Google Scholar]
- 77.Ohmori M., Harii N., Endo T., Onaya T. Tumor necrosis factor-alpha regulation of thyroid transcription factor-1 and Pax-8 in rat thyroid FRTL-5 cells. Endocrinology. 1999;140:4651–4658. doi: 10.1210/endo.140.10.7021. [DOI] [PubMed] [Google Scholar]
- 78.Ghannam-Shahbari D., Jacob E., Kakun R.R., Wasserman T., Korsensky L., Sternfeld O., Kagan J., Bublik D.R., Aviel-Ronen S., Levanon K., et al. PAX8 activates a p53-p21-dependent pro-proliferative effect in high grade serous ovarian carcinoma. Oncogene. 2018;37:2213–2224. doi: 10.1038/s41388-017-0040-z. [DOI] [PubMed] [Google Scholar]
- 79.Wang Q., Armenia J., Zhang C., Penson A.V., Reznik E., Zhang L., Minet T., Ochoa A., Gross B.E., Iacobuzio-Donahue C.A., et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data. 2018;5 doi: 10.1038/sdata.2018.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stubbs B.J., Gopaulakrishnan S., Glass K., Pochet N., Everaert C., Raby B., Carey V. TFutils: Data structures for transcription factor bioinformatics. F1000Res. 2019;8:152. doi: 10.12688/f1000research.17976.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gevaert O. MethylMix: an R package for identifying DNA methylation-driven genes. Bioinformatics. 2015;31:1839–1841. doi: 10.1093/bioinformatics/btv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bland J.M., Altman D.G. Multiple significance tests: the Bonferroni method. BMJ. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Newman M.E.J., Girvan M. Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004;69 doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- 85.Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q., et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 86.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luecken M.D., Theis F.J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019;15 doi: 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lukassen S., Bosch E., Ekici A.B., Winterpacht A. Single-cell RNA sequencing of adult mouse testes. Sci. Data. 2018;5 doi: 10.1038/sdata.2018.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mercer T.R., Neph S., Dinger M.E., Crawford J., Smith M.A., Shearwood A.M.J., Haugen E., Bracken C.P., Rackham O., Stamatoyannopoulos J.A., et al. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Franzen O., Gan L.M., Bjorkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019;2019:baz046. doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kachitvichyanukul V., Schmeiser B. Computer generation of hypergeometric random variates. J. Stat. Comput. Simul. 1985;22:127–145. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Functional enrichments for normal liver modules using MSigDB collections. (b) Functional enrichments for cancer modules using MSigDB collections. (c) Functional enrichments for shared modules using MSigDB collections.
(a) Regulator Genes of robust modules (average Jaccard Index=>0.7 in module subnetwork). (b) Target Genes in robust modules (average Jaccard Index=>0.7 in module subnetwork). (c) Regulator and target genes for the EMT and stemness community.
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for thedatasets are listed in the key resources table.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.