

The computational tool BipotentR was used to reveal drug targets that can inhibit tumor growth by concurrent immune activation and inhibition of a separate oncogenic pathway, leading to identification of ESRRA as a top immune–metabolic target.
Abstract
Drugs that kill tumors through multiple mechanisms have the potential for broad clinical benefits. Here, we first developed an in silico multiomics approach (BipotentR) to find cancer cell–specific regulators that simultaneously modulate tumor immunity and another oncogenic pathway and then used it to identify 38 candidate immune–metabolic regulators. We show the tumor activities of these regulators stratify patients with melanoma by their response to anti–PD-1 using machine learning and deep neural approaches, which improve the predictive power of current biomarkers. The topmost identified regulator, ESRRA, is activated in immunotherapy-resistant tumors. Its inhibition killed tumors by suppressing energy metabolism and activating two immune mechanisms: (i) cytokine induction, causing proinflammatory macrophage polarization, and (ii) antigen-presentation stimulation, recruiting CD8+ T cells into tumors. We also demonstrate a wide utility of BipotentR by applying it to angiogenesis and growth suppressor evasion pathways. BipotentR (http://bipotentr.dfci.harvard.edu/) provides a resource for evaluating patient response and discovering drug targets that act simultaneously through multiple mechanisms.
Significance:
BipotentR presents resources for evaluating patient response and identifying targets for drugs that can kill tumors through multiple mechanisms concurrently. Inhibition of the topmost candidate target killed tumors by suppressing energy metabolism and effects on two immune mechanisms.
This article is highlighted in the In This Issue feature, p. 517
INTRODUCTION
Cancer treatments designed to target two or more oncogenic pathways simultaneously often provide greater clinical benefits, with the advantage of being effective in distinct clinical populations (1, 2). Such treatments can be designed by either combining multiple drugs or administering a single-agent drug that affects two pathways (bipotent drug). Medicating patients with combinations of multiple drugs is now a common strategy but raises patients’ risk to undesired drug interactions and adverse events such as liver injury (3) and colitis (4).
Bipotent drugs may provide benefits similar to drug combinations without the complications that accompany combining multiple drugs. Few drugs are known to be bipotent. Most notable are CDK4/6 inhibitors (cell-cycle inhibitor and immunomodulatory; ref. 5), immunomodulatory imide drugs (IMiD; antiangiogenic and immunomodulatory; ref. 6), and itaconate (energy metabolism and immunity; ref. 7). Likewise, few bipotent gene regulators have been identified, with examples including HDAC6 (8), CDC7 (1), and PTPN3 (9). Bipotent gene targets may be relatively common but undiscovered, with their clinical impact yet to be fully realized due to the lack of systematic approaches to identify them.
The problem of identifying bipotent regulators of multiple pathways relates to a broader problem of inferring regulators of a single pathway, for which a few algorithms have been recently developed. BartWeb (10) infers likely transcription factor or chromatin regulators (TFCR) of an input gene set by leveraging 13,000 chromatin immunoprecipitation sequencing (ChIP-seq) datasets. RePhine (11) uses regression to ascertain TFCRs underlying response or resistance to an input drug. We previously developed eQTeL (12), a Bayesian multiomics approach, to find regulators of variation of gene transcription within a population. We also recently developed LISA (13), which predicts regulators directly responsible for an input gene set, but the input gene sets should be from differential expression or coregulation analyses. Another limitation of existing approaches is that they cannot handle sample-specific confounding effects of ChIP-seq data. Building on these approaches, we present BipotentR, a computational multiomics method for identifying genes that can simultaneously inhibit tumor growth by activating the immune system and suppressing another oncogenic pathway, such as angiogenesis, growth suppressor evasion, metastasis, immortality, or cancer's energy metabolism (14).
Tumors alter their energy metabolism to meet higher bioenergetic needs and sustain proliferation (14). As a result, they become so dependent on energy metabolism (15, 16) that targeting oncogenic energy metabolism can inhibit their proliferation (17, 18). Oncogenic energy metabolism also helps cancer cells evade anticancer immunity (19, 20). For example, increased glucose uptake by cancer cells (the Warburg effect) limits glucose availability for effector T cells in tumors (21), which dampens immunity (22). Thus, targeting energy metabolism can kill tumors directly (23, 24) or through immune-mediated mechanisms (25). This has led to the emergence of the immunometabolism field (19) and several techniques, including single-cell, cytometry-based, multiomics, and genome-scale modeling approaches, for studying immunometabolism (26). These approaches are almost exclusively focused on the metabolism of immune cells. Complementing these approaches, here we focused on finding targets that regulate metabolism of cancer cells that nonautonomously affect immune cells in tumors.
BipotentR identified 38 immune–metabolic targets, whose knockouts induced metabolic and immune phenotypes. In vitro and in vivo inhibitions of the topmost-ranked candidate, ESRRA, elicited dual anticancer effects, and the inhibition is safe in vivo. Artificial intelligence and machine learning (AI/ML) techniques were then used to show that the activity of bipotent targets in tumors predicts the outcome of melanoma patients to anti–PD-1 treatment. Finally, the broad utility of BipotentR for applications beyond immunometabolism was demonstrated by applying it to angiogenesis and evasion of growth suppressor pathways. BipotentR enables the discovery of new cancer therapies by applying prior regulatory and immunologic knowledge to large bulk and single-cell omics data.
RESULTS
Overview of BipotentR, an Approach to Identify Bipotent Targets
BipotentR consists of two modules: “regulator” and “immune” (Fig. 1A). The regulator module predicts regulators of the input pathway(s) chosen by the user, whereas the “immune” module identifies immunomodulatory TFCRs (Methods). To infer bipotent regulators of energy metabolism and immune response (“immune–metabolic” regulators) from the regulator module, we inputted four energy metabolism pathways with a reported role in immunity (27–32): glycolysis, oxidative phosphorylation (OXPHOS), tricarboxylic acid cycle (TCA cycle), and fatty acid (FA) metabolism. The regulator module estimates the potential of ∼700 individual TFCRs to bind cis-elements near input pathway genes by mining 24,000 ChIP-seq samples (33, 34). For a given TFCR, BipotentR derives its core binding sites by combining all ChIP-seq samples and then estimates its binding potential while controlling for sample-specific confounding effects using a linear mixed model (Methods). BipotentR identified previously known [e.g., ESRRA and BCL3 regulate OXPHOS (35, 36); PPARG and CEBPB regulate FA and glycolysis (37, 38)] and new TFCRs for each pathway (Fig. 1B; Supplementary Table S1). It then prioritized the subset of these regulators that can affect multiple energy metabolism pathways (“master regulators”) by ranking TFCRs according to their average overall binding affinities across pathways. Master regulators were enriched in nuclear receptors (P < 1E−7; Fig. 1C).
Figure 1.

Identification of immune–metabolic regulators. A, Overall schematic of regulation and immune modules of BipotentR. The regulator module identifies regulators of an input pathway using ChIP-seq data. The immune module identifies TFCRs that show immunostimulatory or immunosuppressive properties in bulk tumor transcriptomes and are preferentially active in cancer cells (using single-cell tumor transcriptomes). B, Output of BipotentR regulator module. Potential and significance of regulators to bind cis-regulatory elements of genes in four energy metabolism pathways. Each dot indicates a regulator, colored by individual pathways. OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid cycle. C, The potential of top predicted master regulators to bind energy metabolism genes. Nuclear receptors are displayed in red. D, TFCRs with positive (or negative) associations with proinflammatory signatures are predicted immunostimulators (purple; or immunosuppressors, orange). E, Top TFCRs predicted to be preferentially active in cancer cells (orange; or CD8+ T cells, purple) and their differential activity (estimated from single-cell data). F, Output of BipotentR immune module: combined association with proinflammatory signatures (D, estimate from bulk RNA-seq) and differential activity in cancer cells (E, estimate from single-cell data) are displayed for each TFCR. G, Immune–metabolic regulators identified by BipotentR. Energy regulatory potential (estimated by regulator module) and immune-modulatory potential (estimated by immune module) of TFCRs. Highlighted TFCRs are significant and among the top 15% in both modules. Immunostimulators (purple) and immunosuppressors (orange) are colored. H, Validation of BipotentR-identified targets. Effect of knockout (KO) of target identified by BipotentR on T cell–mediated killing of cancer cells.
Having identified regulators of energy metabolism, we next used the immune module of BipotentR to identify immunomodulatory TFCRs. This module estimates the immunomodulatory potential of ∼700 individual TFCRs from bulk RNA sequencing (RNA-seq) patient tumor data by associating TFCR expression in tumors with levels of a proinflammatory signature. The proinflammatory signature combines 32 key immune response biomarkers, such as mutation burden, neoantigen load, immune infiltration, and IFNγ response (ref. 39; Methods). TFCR immunomodulatory potential was estimated across several cancer types using a linear mixed model that is robust to cancer type–specific immune effects (Supplementary Fig. S1A and S1B) using data from The Cancer Genome Atlas (TCGA; 33 cancer types from 11,000 patients; ref. 39). We evaluated the robustness of immune-module outputs in two datasets comprising nonoverlapping cancer types (akin to 2-fold cross-validation; Methods). The results suggest that inferred TFCRs likely show immunomodulatory properties in several cancer types (R = 0.91, P < 2.2E−16; Supplementary Fig. S1C). TFCRs with the highest inferred immune potential were enriched in immune ontologies, including T-helper differentiation, inflammatory disorders, and viral infection, in addition to carcinogenesis and transcriptional misregulation in cancer (Fig. 1D; Supplementary Fig. S1D). The module predicted well-known regulators of adaptive and innate immunity based on the input signature that was used, in this case, the proinflammatory signature. By changing the input signature, the module can be used to predict regulators of specific immune cells. For example, the module identified macrophage regulators using a macrophage polarization signature as input (Supplementary Note S1).
The immune module also selects candidate TFCRs whose inhibition likely blocks cancer cells without adversely affecting CD8+ T cells, which are essential for antitumor immunity (40). We achieve this by selecting TFCRs that are present and active in cancer cells but not in CD8+ T cells using single-cell RNA-seq (scRNA-seq) and single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq) data. TFCRs are deemed cancer cell–specific if they are differentially active in cancer cells relative to CD8+ T cells across all five scRNA-seq cohorts (refs. 41–45; Methods). The module uses scATAC-seq data (46) to ensure these TFCRs are functional in cancer cells but not in CD8+ T cells. To this end, we examined if target genes (inferred by ChIP-seq) of predicted TFCRs are epigenetically accessible in cancer cells (Methods). We found that target accessibility differences between cancer cells and CD8+ T cells were markedly correlated with scRNA-seq expression differences (Pearson correlation = 0.63, P < 2E−59; Supplementary Fig. S1E), suggesting that predicted TFCRs are active and transcriptionally functional in cancer cells relative to CD8+ T cells. The top predicted TFCRs included SUMO1, SUMO2, and DLX2, genes known to be tumorigenic and highly active in several cancers (Fig. 1E; refs. 47–49).
We next investigated how cancer cell activities of TFCRs (predicted from single-cell data) relate to their immunomodulatory potential (predicted from bulk tumor data). Strikingly, predicted immunosuppressive TFCRs were preferentially active in cancer cells, and immunostimulatory TFCRs were active in CD8+ T cells, evident from a strong Pearson correlation of 0.7 (P < 2E−82) between immunosuppressive potential and cancer-cell activity (Fig. 1F). Thus, inhibiting immunosuppressive TFCRs would likely impact cancer cells but less likely impact CD8+ T cells adversely.
Inhibition of the 38 Candidate Bipotent Immune–Metabolic Regulators Elicits Metabolic and Immune Effects
With the two modules developed and validated, we integrated their outputs to identify 38 TFCRs (30 immunosuppressive and 8 immunostimulatory) with immune–metabolic dual functions (Fig. 1G; Supplementary Table S2). Among these were known immune–metabolic TFCRs such as CDK7, which regulates mitochondrial membrane potential (50) and enhances immune suppression (51), and NFATC1, which regulates energy consumption and CD8+ T-cell effector function (52, 53).
First, we compared how well genetic inhibition of the 38 identified immune–metabolic regulators suppressed transcription of genes in energy metabolism pathways using a published transcriptome dataset comprised of 570 knockdown/knockout experiments for 308 TFCRs (54). We found that inhibition of BipotentR-predicted regulators markedly suppressed energy metabolism pathways (Supplementary Fig. S2A; P < 3.5E−25). Further, inhibition of BipotentR-predicted regulators suppressed energy genes more strongly than other TFCRs (Supplementary Fig. S2B; P < 7.8E−10), indicating preferential regulation of energy metabolism by the identified TFCRs. We benchmarked BipotentR against recently published regulator prediction algorithms (10, 13). We observed that BipotentR-identified regulators (n = 38) held six TFCRs in common with LISA-identified regulators (n = 38, Fisher exact test P < 4E−4, Methods). Despite the overlap, inhibitions of BipotentR-identified regulators suppressed energy genes more strongly than those by LISA-identified regulators (Wilcoxon test P < 1.5E−12; Supplementary Fig. S2C). This and a similar benchmark obtained against BartWeb (Supplementary Fig. S2D; Supplementary Note 2) suggest that BipotentR predicts functional regulators of energy metabolism as accurately as existing approaches.
We next confirmed that inhibition of the identified TFCRs also regulates immunity. We examined a recently published CRISPR screen (55) in which cancer cells were subjected to selection by effector T cells to identify gene knockouts that modulate T cell–mediated killing. CRISPR guide RNAs (gRNA) that knock out BipotentR-predicted immunosuppressive TFCRs were depleted (Fig. 1H; Supplementary Fig. S2E; P < 8E−10, n = 240 and n = 79,481), suggesting that their knockout enhances T cell–mediated killing. In contrast, gRNAs against BipotentR-predicted immunostimulatory TFCRs were enriched, indicating their knockout decreases T cell–mediated killing (Fig. 1H; Supplementary Fig. S2E; P < 1E−3, n = 64 and n = 79,481). Thus, genetic inhibition of immune–metabolic regulators elicited both immune and metabolic effects.
Among 38 Candidate Bipotent Immune–Metabolic Regulator Targets, the Orphan Nuclear Receptor ESRRA Is the Most Highly Ranked
Having evaluated the set of identified bipotent immune–metabolic regulators, we followed up on the topmost bipotent target. An orphan nuclear receptor, ESRRA, was predicted to have the highest immune–metabolic potential. Targeting ESRRA in immunodeficient models has been shown to inhibit tumors by direct cell-intrinsic mechanisms (35, 56), and we hypothesized that targeting ESRRA would also inhibit tumors by immune-mediated mechanisms. To illustrate a proof-of-principle bipotent target, we determined the dual potential and clinical relevance of ESRRA in different cancer types.
Inhibition of ESRRA Stimulates Antitumor Immunity
We evaluated inhibiting ESRRA by small interfering RNA (siRNA) and two structurally similar diaryl ether–based thiazolidinediones, which function as selective ligands against ESRRA (compounds 29 and 39 from Johnson & Johnson, with well-characterized pharmacokinetics; ref. 57). We first tested both small-molecule inhibitors in vitro by mutating the known compound binding site in the ESRRA ligand-binding domain (LBD; C229 site) and showing that the mutation rescued the ESRRA inhibition (Supplementary Fig. S2F and S2G). Next, we investigated siRNA and compound 39 for on-target and off-target effects through RNA-seq (Methods). Both approaches selectively suppressed putative ESRRA gene targets that had been identified from ESRRA ChIP-seq data (Supplementary Fig. S2H). An unbiased prediction of 700 putative regulators of the genes differentially expressed upon the two approaches (13) yielded ESRRA as the top regulator of downregulated genes (topmost for drug inhibition, Supplementary Fig. S2I; second highest for siRNA, Supplementary Fig. S2J). These analyses showed that both siRNA and drug inhibition selectively suppress ESRRA and have limited off-target effects. We chose to pursue drug inhibition of ESRRA (“ESRRAi,” which refers to inhibition by compound 29 or compound 39) because of its translational potential and somewhat superior potency in targeting ESRRA. Compound 29 is more stable metabolically in human microsomes than compound 39 (57), so we used compound 29 for in vivo testing (Methods).
We next tested if ESRRAi could induce antitumor immunity in two immunosuppressive murine tumor models: 4T1 (triple-negative breast cancer) and B16F10 (melanoma). We treated the 4T1 mice with ESRRAi or vehicle control and surgically resected their tumors. We performed scRNA-seq of CD45+ cells sorted from tumors, clustered and annotated cells using classic markers, and identified major tumor-infiltrating immune cells in both conditions (Fig. 2A; Methods). ESRRA was not expressed in CD45+ cells (Supplementary Fig. S2K). We initially studied immune cells of lymphoid lineage for changes in their fraction by ESRRAi treatment and found higher CD8+ T-cell infiltration with the treatment (Supplementary Fig. S2L). A CD8+ T-cell marker, Cd8a, was the topmost upregulated gene in the lymphoid lineage of ESRRAi-treated tumors compared with controls (Fig. 2B). Markers of activated CD8+ T cells (Fig. 2B), including perforin and granzymes, were also upregulated (P < 3E−5; permutation test), suggesting that infiltrating CD8+ T cells in treated tumors were also activated. We also showed increased infiltration of activated CD8+ T cells with ESRRAi in tumors (P < 2E−3) using fluorescence-activated single-cell sorting (FACS; Fig. 2C). We also analyzed published tumor transcriptomes from 33,000 patients (58) and showed that tumors with the highest levels of immune infiltration, including CD8+ T-cell infiltration, had the lowest ESRRA activity, whereas CD8+ T cell–deficient (or immune-deficient) tumors showed the highest ESRRA activity in multiple cancer cohorts and cancer types (Supplementary Notes S3 and S4). This suggests that T-cell infiltration upon ESRRAi may be clinically relevant in multiple cancer types.
Figure 2.

ESRRA inhibition activates antitumor immunity in 4T1 mice. P values using Wilcoxon rank-sum test unless stated otherwise. A, Uniform manifold approximation and projection (UMAP) display of scRNA-seq of tumor-infiltrating CD45+ cells from ESRRAi- and vehicle-treated mice. NK, natural killer; Treg, regulatory T cell. B, Markers of activated CD8+ T cells in genes differentially expressed by ESRRAi in lymphoid cells from treated mice. Significance of up/downregulation of marker sets estimated using permutation tests. C, Fraction of CD8+ T cells identified by flow cytometry. D, Tumor volume comparisons between ESRRAi and control. E, Tumor volume comparisons between ESRRAi with and without CD8 antibody. F, Markers of Tregs in genes differentially expressed by ESRRAi in lymphoid cells from treated mice. Significance of up/downregulation of marker sets estimated using permutation tests. G, Fraction of Tregs identified by flow cytometry. H, Densities of macrophage polarization toward M1 (i.e., for each macrophage cell, macrophage polarization = average expression of M1 markers − average expression M2 markers); see also Supplementary Fig. S2O and S2P. I–K, Measurements done after tumors were surgically removed in ESRRAi- or vehicle-treated mice comparing tumor regrowth rate (i.e., 1—relapse rate; I), lung metastasis deposits (J), circulating tumor cells in the blood (K). PBMC, peripheral blood mononuclear cell. *, P < 0.05; **, 0.05 < P < 0.01; ***, P < 0.001; ns, P > 0.05.
Next, we asked if ESRRAi-induced infiltrating CD8+ T cells exert an antitumor effect and if ESRRAi also modulates regulatory T-cell (Treg) infiltration. ESRRAi treatment markedly reduced tumor growth (Fig. 2D), which we confirmed in another immune-cold tumor B16F10 mouse model (Supplementary Fig. S2M). Two lines of evidence linked this tumor elimination with CD8+ T cells. First, among ESRRAi-treated mice, those with higher CD8+ T infiltration showed superior tumor elimination (Spearman correlation = −0.62; P < 0.043; Supplementary Fig. S2N). Second, CD8+ T-cell depletion abrogated the antitumor effect of ESRRAi (Fig. 2E; 4T1 tumor growth is shown to be unaltered by CD8+ T depletion alone; ref. 59). Another ESRRAi-induced change in the lymphoid lineage was downregulated (P < 7E−3; permutation test) markers of Tregs (Fig. 2F). Correspondingly, lower Treg infiltration in the ESRRAi condition was observed in single-cell data (Supplementary Fig. S2L), which was further confirmed using FACS (P < 9E−4; Fig. 2G), indicating that ESRRAi treatment suppressed Treg infiltration into tumors. These analyses revealed the specific roles of different T-cell populations in ESRRAi antitumor immunity.
ESRRA-deficient mice in a noncancer context have shown macrophage-mediated inflammation (60). Therefore, we postulated that ESRRAi might affect tumor macrophages. Indeed, monocytes/macrophages were polarized toward proinflammatory M1 in the ESRRAi-treated tumors (Fig. 2H; Supplementary Fig. S2O). In contrast, macrophages were polarized toward protumorigenic M2 in controls (Supplementary Fig. S2P). Moreover, monocytes/macrophages of treated tumors expressed M1 markers (Tnf, Ccl5, Nos2, and Il1a; ref. 61) and downregulated M2 markers (Supplementary Fig. S2Q). Consistent with this result, we found that macrophage polarity was markedly correlated with ESRRA activity in tumors across most cancer types in data from 33,000 patients (ref. 58; Supplementary Fig. S3A; Supplementary Note S3).
Next, we tested the effect of ESRRAi treatment on tumor relapse from minimal residual disease. After surgical removal of 4T1 tumors, ESRRAi-treated mice experienced significantly fewer tumor relapses (Fig. 2I). Moreover, their relapsed tumors had significantly attenuated growth (Supplementary Fig. S2R). We examined incised lungs from treated mice and observed fewer lung metastatic deposits than the control group (Fig. 2J). We also cultured the circulating tumor cells from the blood of treated mice and observed a significant decrease in the number of colonies relative to the control group (Fig. 2K). These data suggest that ESRRAi can prevent the relapse of surgically resected tumors.
Similar ESRRAi antitumor responses were observed in the 4T1 model using two delivery formulations, Solutol and PEG (Supplementary Fig. S5A–S5D; Methods). Thus, our data indicate that ESRRAi polarizes macrophages toward M1 and induces antitumor effects that depend on T cells.
Immune Signaling Pathways Link ESRRAi to Immune Response
We next asked what cell-autonomous immune–metabolic pathways underlie ESRRAi antitumor immunity. We treated a human breast cancer cell line (SKBR3) with ESRRAi and measured transcriptomic changes at three time points. ESRRAi suppressed metabolic genes at all time points, particularly energy metabolic pathway genes (Fig. 3A and B; Supplementary Fig. S6A and S6B), which we confirmed using siRNA (Supplementary Fig. S6C and S6D).
Figure 3.

Signaling induced by ESRRAi in vitro.A, Differentially expressed genes (DEG) between ESRRAi and control in the SKBR3 cell line at three time points. Genes were clustered by K-means. B, Pathway enrichment scores corresponding to clusters of DEGs shown in A. GSVA, gene set variation analysis. C, ESRRA knockout potentiates T-cell killing as observed in CRISPR knockout screens in cancer cells cocultured with T cells. The black line represents the relative position of ESRRA knockout among all gene knockouts ranked from most depleted to least depleted. The significance of ESRRA knockout in screens is also displayed. KO, knockout; SKCM, skin cutaneous melanoma. *, P < 0.05; **, 0.05 < P < 0.01; ***, P < 0.001; ns, P > 0.05.
In contrast, the effect on immune pathways showed a striking temporal trend: The treatment upregulated innate immune signaling at 24 hours, whereas at 72 hours, it upregulated adaptive immune signaling (Fig. 3A and B). The treatment at 24 hours upregulated (Toll-like, Fc-epsilon-RI, Rig-I–like, and NOD-like receptors) receptor signaling that is known to promote antigen presentation and inflammatory cytokine secretion (62). Accordingly, genes involved in antigen presentation (Supplementary Fig. S6E; Fig. 3B) and cytokine interactions, especially macrophage-polarizing cytokines (Supplementary Fig. S6F; Fig. 3B), were upregulated 72 hours after the treatment. This upregulation of macrophage-polarizing cytokines is consistent with macrophage polarization by ESRRAi observed in our in vivo single-cell experiments (Fig. 2H). The ESRRAi treatment also upregulated 20 immunomodulatory TFCRs identified by BipotentR (Supplementary Fig. S6G; Fisher exact test P < 4.7E−12), suggesting that ESRRA is an upstream regulator of other immune regulators.
We next examined if the knockout of ESRRA in cancer cells induces antigen presentation by analyzing data from CRISPR knockout screens (63–66) designed to identify regulators of type-I antigen presentation genes (MHC-I). These screens sort cancer cells transduced with gRNA into low or high MHC-I groups based on their MHC-I protein expression. gRNAs that knock out ESRRA were enriched in high MHC-I and depleted in low MHC-I groups (Supplementary Fig. S7A; Supplementary Note S5), confirming that ESRRA knockout increases MHC-I antigen presentation.
Because increased MHC-I antigen presentation in tumors enhances the ability of T cells to kill cancer cells (55, 67), we hypothesized that ESRRAi would enhance tumor killing by T cells. We tested this hypothesis using published CRISPR screens that coculture cancer cells with T cells to identify which gene knockouts in cancer cells enhance their T cell–mediated killing (55, 68–71). ESRRA knockout potentiated the killing of cancer cells by both patient-derived and engineered effector T cells in various experimental and cell line contexts (Fig. 3C). Because T cell–mediated killing has previously been shown to be enhanced by OXPHOS suppression (27, 55), we asked if OXPHOS targets of ESRRA (derived from ESRRA ChIP-seq; Methods) can explain this effect. Indeed, knockout of COX10, ATP51B, and NDUFA6 alone not only potentiated T cell–mediated killing (Supplementary Table S3), but also increased protein levels of antigen presentation genes (Supplementary Table S4). Thus, OXPHOS suppression by ESRRAi can explain the activation of antigen presentation and T cell–mediated immunity by ESRRAi.
Finally, we generated a signature based on differential expression upon ESRRAi. Using this signature, we divided both 33,000 tumors and 1,000 cell lines from the Cancer Cell Line Encyclopedia data (72) by high and low ESRRA activity. Cell lines or tumors with low ESRRA activity exhibited decreased energy metabolism and upregulated immune pathways, including antigen presentation genes and cytokines that polarize macrophages to M1 (Supplementary Figs. S3A–S3H, S4A–S4G, and S7A–S7G; see Supplementary Notes S3–S5).
ESRRAi Does Not Adversely Affect CD8+ T Cells
Targeting tumor energy metabolism by ESRRAi would be detrimental to patients if it also affects T-cell metabolism (27, 73). To validate predicted cancer cell specificity for ESRRA, we investigated the likely effects of ESRRAi on T cells and cancer cells. We compiled and analyzed 78 single-cell transcriptome datasets from patients with 27 different major cancer types (refs. 74, 75; Methods). We found that ESRRA was expressed at the highest levels in cancer cells but also at lower levels in macrophages and T cells (Supplementary Fig. S8A). Because the functional activity of a nuclear receptor depends not only on its expression but also on its ligands, cofactors, and stimulation, we reasoned that ESRRA might have low functional activity in T cells despite being expressed in T cells. Indeed, ESRRA activity levels, quantified as the expression of ESRRA targets, were lowest in T cells (Fig. 4A; Methods). In contrast, the highest and second-highest levels of ESRRA activity were observed in cancer cells and macrophages (Fig. 4A). Macrophages with high ESRRA activity also expressed M2 markers, suggesting they are M2 macrophages (Supplementary Fig. S8B–S8D).
Figure 4.

ESRRA is activated in immunotherapy-resistant tumors and its inhibition does not adversely affect CD8+ T cells. P values estimated by the Wilcoxon rank-sum test. A, ESRRA activity in cancer and immune cells from 30 scRNA-seq cohorts. ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; BCC, basal cell carcinoma; BRCA, breast cancer; COAD, colon adenocarcinoma; CHOL, cholangiocarcinoma; DC, dendritic cell; EryPro, erythroid progenitor cell; GBM, glioblastoma multiforme; GMP, granulocyte-macrophage progenitor; HNSC, head and neck squamous cell carcinoma; LIHC, liver hepatocellular carcinoma; MB, medulloblastoma; MM, multiple myeloma; NK, natural killer; NSCLC, non–small cell lung cancer; OPC, oligodendrocyte precursor cell; PAAD, pancreatic adenocarcinoma; pDC, plasmacytoid dendritic cell; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UVM, uveal melanoma. B, The chromatin accessibility of ESRRA targets in different cell types from scATAC-seq data of a skin cancer cohort. C, Cancer cell ESRRA activity in patient (skin cancer) tumors with pre– and post–anti–PD-1 treatment for responders and nonresponders. D, Enrichment analysis of an ESRRA-regulated gene set in nonresponding CT26 mice after anti–PD-1 treatment.
Next, we examined ESRRA cell-specific function by comparing the accessibility of its target genes (inferred from ESRRA ChIP-seq; Methods) in different cell types using scATAC-seq data from patients with nonmelanoma skin cancer (46). ESRRA target gene accessibility was highest in cancer cells, second highest in macrophages, and lowest in CD8+ T cells (Fig. 4B), consistent with ESRRA activity distribution in scRNA datasets. The data support a model in which ESRRA has a higher level of activity in cancer cells relative to CD8+ T cells, as measured by gene expression, target transcription, and chromatin accessibility. Thus, ESRRAi likely has a lower impact on the energy metabolism of CD8+ T cells.
To specifically test whether ESRRAi treatment affects the energy metabolism of CD8+ T cells, we analyzed scRNA data from CD45+ cells from our in vivo 4T1 mouse model, in which the antitumor effect of ESRRAi was measured. ESRRA activity in CD8+ T cells was unchanged in ESRRAi-treated mice relative to control (Supplementary Fig. S9A). Macrophages/monocytes were the only CD45+ cells that showed decreased ESRRA activity after treatment. We also evaluated our scRNA data using Augur (76), a method that identifies cell types affected by treatments, which also found no significant cell-intrinsic changes in CD8+ T cells post-ESRRAi (Supplementary Fig. S9B). ESRRAi treatment in 4T1 mice did not significantly change body weights or health parameters (Supplementary Fig. S9C), which we further confirmed in the B16F10 mouse model (Supplementary Fig. S9D), suggesting that ESRRAi treatment was not nonspecifically toxic. These data strongly indicate that ESRRAi enhances CD8+ T-cell recruitment into tumors without adversely affecting CD8+ T cells themselves.
ESRRA Activation in Immunotherapy-Resistant Tumors
Next, we studied the effect of immunotherapy on ESRRA activity. Analysis of a cohort of patients with immunotherapy-resistant melanoma (77) revealed an intriguing trend: Cancer cells from postimmunotherapy tumors had markedly higher ESRRA activity than those from pretreatment tumors (Supplementary Fig. S9E). As the cohort contained only immunotherapy-resistant patients, we asked whether the trend is specific to resistant patients or also present in responders. To that end, we analyzed a nonmelanoma skin cancer scRNA-seq cohort (78) containing both immunotherapy-responder and -resistant patients. Indeed, the trend of immunotherapy-induced ESRRA activity increase was specific to immunotherapy-resistant tumors (Fig. 4C). In fact, in responders, ESRRA activity decreased in cancer cells upon immunotherapy (Fig. 4C). These data are consistent with the hypothesis that immunotherapy-resistant tumors achieve high levels of immune suppression via ESRRA.
We experimentally tested the trend of increased ESRRA activity and resulting immune suppression upon immunotherapy. Specifically, we chose a syngeneic mouse model of colorectal cancer (CT26) known for its heterogeneous immunotherapy (anti–PD-1) response (79), treated the mice with anti–PD-1, and conducted bulk tumor RNA-seq. Target genes of ESRRA were upregulated in immunotherapy-resistant mice (Fig. 4D) but not in responders (Supplementary Fig. S9F), suggesting that ESRRA activity increases in immunotherapy-resistant tumors upon immunotherapy. Increased ESRRA activity was accompanied by immune suppression, including decreased CD8+ T infiltration and increased M2 macrophages in tumors (Supplementary Fig. S9G). This in vivo experiment supports a model in which immune-checkpoint blockade (ICB) increases ESRRA activity, specifically in immunotherapy-resistant tumors. Increased ESRRA activity may also increase the vulnerability of tumors to ESRRA inhibition. The potential vulnerability is supported by our in vivo experiment showing ESRRAi effectiveness in 4T1 and B16F10 models—both of which respond poorly to ICB. Future clinical investigations are required to reveal whether immunotherapy-resistant tumors benefit from the immunostimulatory effect of ESRRA inhibition.
AI/ML-Based Aggregation of Bipotent Target Activity in Tumors Predicts Patient Response to Anti–PD-1
The FDA recently approved using tumor mutation burden as a diagnostic biomarker to select patients with solid tumors for an anti–PD-1 treatment (80). However, tumor mutational burden and other ICB biomarkers fail to identify which patients will best respond to ICB therapy (81, 82), and complementary biomarkers are needed. Adopting the strategy from Wang and colleagues (83), we evaluated bipotent target activity in tumors as a potential ICB biomarker. We focused on anti–PD-1 treatment in melanoma because of the availability of multiple melanoma cohorts with RNA-seq (84–88), making it possible to evaluate biomarkers rigorously. Our algorithm first learned a risk score (Fig. 5A; see Methods) that combined tumor activity of bipotent targets through a multivariate Cox hazards model (89): 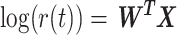 . Here, X is the expression of bipotent targets in tumors, r(t) is risk score parameter, and W is weights learned using 465 immunotherapy-naïve melanoma patients from the TCGA (Methods). We refer to the risk scores as bipotent target activity scores (BTAS).
. Here, X is the expression of bipotent targets in tumors, r(t) is risk score parameter, and W is weights learned using 465 immunotherapy-naïve melanoma patients from the TCGA (Methods). We refer to the risk scores as bipotent target activity scores (BTAS).
Figure 5.

Machine learning evaluation of immune–metabolic targets in predicting patient response to anti–PD-1. A, Overview of the estimation of bipotent target activity score in tumors using a linear function (BTAS). B, Kaplan–Meier plots showing PFS and OS differences for patients receiving anti–PD-1 between the low-risk and high-risk groups defined by the median value of BTAS. C, Survival stratification performance of BTAS versus seven other previously published ICB biomarkers for patients with melanoma receiving ICB. Significance of survival was estimated using log-rank P value (−log10). D–K, Kaplan–Meier plots similar to B, with the low-risk and high-risk groups defined by the median value of the signatures. Signatures displayed are TIDE (D; ref. 97), T-cell inflamed (E; ref. 96), immune (F; ref. 90), cytotoxic (G; ref. 91), IFNG (H; ref. 92), melanocytic plasticity (I; ref. 98), immune checkpoint (J), and PD-L1 expression (K; ref. 96). L, Overview of the estimation of BTAS in tumors using a nonlinear function (deepBTAS). M, Kaplan–Meier plots similar to B with the low-risk and high-risk groups defined by the median deepBTAS. N, Performance improvement (OS and PFS) with the addition of deepBTAS to biomarkers (rows). An improvement quantified as (P value of) increase in the likelihood of multivariate model containing a biomarker and deepBTAS over a model containing the biomarker alone. LRT P, P value from likelihood ratio test. O, Kaplan–Meier plots similar to M done separately on tumors with high (left) and low (right) immune signature.
Without additional training on ICB cohorts, we used the model learned on TCGA to quantify the BTAS for each patient in ICB cohorts. We found that the BTAS stratified patients into likely ICB responders and nonresponders. In particular, when we stratified patients into the low-risk and high-risk groups based on median BTAS, the groups showed significant differences in progression-free survival (PFS; log-rank P < 1.7E−3; Cox P < 3.6E−4) and overall survival (OS; log-rank P < 5.1E−3; Cox P < 5.7E−3; Fig. 5B). To show that such stratification is unlikely by random chance, we used an empirical sampling procedure. We randomly sampled a gene set (from 20,000 protein-coding genes) of the same size as bipotent targets (N = 38), combined their activity using a similar procedure used to learn BTAS, and calculated its predictive power for stratifying anti–PD-1 outcome in terms of PFS and OS. We repeated this procedure for 10,000 randomly sampled gene sets and observed that only 27 sets showed higher predictive power than BTAS (i.e., empirical P < 2.7E−3; Fig. 5C). We also asked whether the higher predictive power in ICB cohorts of BTAS could be explained by its predictive power within TCGA survival relative to random gene sets. However, out of 5,888 gene sets with higher TCGA predictive power than BTAS, only 21 gene sets outperformed BTAS in ICB cohorts (i.e., empirical P < 3.6E−3). These observations suggest that BTAS has predictive power to stratify ICB responders and nonresponders.
How does the predictive power of BTAS compare with other biomarkers? We collected seven other previously published ICB biomarkers (90–98) and evaluated them in our ICB cohort collection. The highest predictive power was displayed by immune (90, 94), T cell–inflamed (95, 96), and cytotoxic (91, 95) signatures (Fig. 5C–K). Importantly, BTAS achieved a predictive power comparable with these best performing ICB biomarkers. We then separately evaluated biomarkers on patients who received anti–PD-1 alone and patients who received the combination of anti–PD1 and anti-CTLA4. For anti–PD-1 alone, the immune signature (90) achieved the best survival stratification (Supplementary Fig. S10A–S10J), whereas, for the combination, the T cell–inflamed signature (96) performed the best (Supplementary Fig. S11A–S11J). BTAS showed comparable performances in both therapy regimens (Supplementary Figs. S10B and S11B), albeit at lower significance levels, highlighting the importance of larger cohorts for assessing the robustness of biomarkers. Note that to make biomarker estimates comparable across cohorts, we normalized the expression uniformly across cohorts, which would differ when normalization is done within a single cohort, as was the case in original publications. We also used immune-checkpoint genes, generated their tumor activity scores similar to BTAS (Methods), and showed that resulting activity scores have predictive power comparable to BTAS (Fig. 5J), further supporting the role of bipotent targets in determining ICB response.
BTAS captures likely ICB response through a linear relationship between bipotent TFCRs, but TFCRs could interact nonlinearly (99). Therefore, nonlinear functions of bipotent targets might capture ICB responses more accurately than linear functions. To test this, we fit a nonlinear model (Fig. 5L): 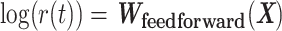 of bipotent target expression X. Here to capture nonlinear relationships between targets, we modeled
of bipotent target expression X. Here to capture nonlinear relationships between targets, we modeled 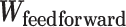 using a feedforward neural network (see Methods; Fig. 5L) with two hidden layers (with rectified linear activations). To learn the parameters of the feedforward network, we used 85% of 465 TCGA patients with melanoma for training and the rest, 15%, for testing. We optimized the parameters that minimized the partial log-likelihood (100) using stochastic gradient descent and stopped the training when the testing error started to increase to avoid overfitting (Supplementary Fig. S10K). We refer to the risk scores inferred from the feedforward network as deepBTAS.
using a feedforward neural network (see Methods; Fig. 5L) with two hidden layers (with rectified linear activations). To learn the parameters of the feedforward network, we used 85% of 465 TCGA patients with melanoma for training and the rest, 15%, for testing. We optimized the parameters that minimized the partial log-likelihood (100) using stochastic gradient descent and stopped the training when the testing error started to increase to avoid overfitting (Supplementary Fig. S10K). We refer to the risk scores inferred from the feedforward network as deepBTAS.
Without additional training on ICB cohorts, we used the trained feedforward network to quantify the deepBTAS score in patients from ICB cohorts. deepBTAS score stratified patients into likely ICB responders markedly more accurately than BTAS in both PFS (log-rank P = 8.3E−4) and OS (log-rank P = 2.3E−05; Fig. 5M). We further studied whether deepBTAS and BTAS can distinguish partial/complete responders from those with stable/progressive disease based on their tumor radiology. The deepBTAS classified ICB patients with an accuracy measure area under the curve (AUC) of 0.603, whereas BTAS AUC was 0.585 (Supplementary Fig. S12A). These classification accuracies were similar to those of the top performing ICB biomarker, immune signature, with an AUC of 0.602. deepBTAS outperformed BTAS when we evaluated biomarkers separately for patient anti–PD1 monotherapy and anti-CTLA4 + anti–PD-1 (Supplementary Figs. S10C and S11C). We, therefore, conclude that a nonlinear function of bipotent target activity captures ICB response more accurately than a linear function.
We also investigated whether BTAS/deepBTAS can improve the predictive power of current ICB biomarkers. First, even when we accounted for differences in levels of other ICB biomarkers in tumors, BTAS/deepBTAS showed a significant association with ICB survival outcomes (Supplementary Fig. S12B–S12E; Methods), suggesting our AI/ML models learned information that is nonredundant with other biomarkers. Next, we combined BTAS/deepBTAS with current biomarkers and assessed whether the addition improved the biomarker performance (Methods). The addition of either BTAS or deepBTAS improved the performance of all ICB biomarkers in predicting PFS and OS (Fig. 5N; Supplementary Fig. S12F). Importantly, deepBTAS markedly improved the best ICB biomarker, immune signature (Fig. 5N). Interestingly, this improvement was observed in tumors with high immune signature (likely responders) but not in tumors with low immune signature (likely nonresponders; Fig. 5O; Supplementary Fig. S13A). This suggests that deepBTAS improves the biomarker by correctly identifying tumors that were misidentified as responders by the immune signature. deepBTAS also correctly identified tumors that were misidentified as responders by other top performing ICB markers: T cell–inflamed signature and cytotoxic signature (Supplementary Fig. S13B–S13E). These analyses indicate that our AI/ML models improve the performance of current ICB biomarkers.
Both Bulk and Single-Cell Submodules Contribute to BipotentR Predictive Power
We assessed contributions from bulk RNA and scRNA submodules. We removed the single-cell submodule from BipotentR and evaluated the identified TFCRs (Supplementary Fig. S14A; Methods). The identified TFCRs showed predictive power in in vitro CRISPR screens but failed to stratify patients by ICB response (Supplementary Fig. S14B–S14D), indicating that the scRNA submodule is critical for predicting ICB response in patients. The fact that predictive power was observed in the in vitro screen but not in patient response may be because gene knockouts in the in vitro screen (55) were performed on cancer cells and not immune cells.
Next, we removed the bulk RNA-seq submodule from BipotentR. This version of BipotentR was still able to produce significant predictive power but to a lesser extent compared with the full BipotentR (Supplementary Fig. S15A–S15D; Methods). Together, these evaluations suggest that both submodules contribute to BipotentR and both are required for maximal predictive power.
Application of BipotentR to Angiogenesis and Growth Suppressor Evasion
To demonstrate the utility of BipotentR using a different input pathway, we identified 14 bipotent TFCRs that simultaneously regulate angiogenesis and immune response (Supplementary Table S5) and 14 TFCRs that regulate evasion of growth suppressors and immune response (Supplementary Table S6), using these pathways as inputs to BipotentR (Methods). This included TBX21 and histone demethylase JMJD1C; whereas TBX21 is known to regulate angiogenesis and Tregs (101, 102), JMJD1C is known to regulate angiogenesis (103–105) and can polarize macrophages (106). We also evaluated bipotent targets for angiogenesis and growth suppressor evasion analogously as was done for immunometabolism targets. Using existing CRISPR datasets (55), we tested if genetic inhibitions of these 28 identified TFCRs can elicit dual antitumor efficacy. CRISPR knockouts of these TFCRs markedly improved the killing of cancer cells by T cells (Fig. 6A; P < 1.3E−6). Knockdown or knockout of the identified TFCRs suppressed genes involved in angiogenesis or evasion of growth suppressors (Supplementary Fig. S16A, P < 8.1E−24), and the suppressed genes were preferentially regulated by the bipotent TFCRs (Supplementary Fig. S16B, P < 9E−10).
Figure 6.

Evaluation of bipotent regulators of angiogenesis or growth suppressor evasion. A, Effect of knockout of 28 bipotent targets on T cell–mediated killing of cancer cells. B–M, Performance of BTAS and deepBTAS versus other previously published ICB biomarkers. C, Survival stratification performance of BTAS score versus other ICB biomarkers. Significance of survival was estimated using log-rank P value (−log10). B, D, and E, Kaplan–Meier plots show PFS and OS between the four equal quartiles of risk groups divided based on the following signatures: BTAS (B), immune (D; ref. 90), and cytotoxic (E; ref. 91). F–J, Kaplan–Meier plots show PFS and OS between the four equal quartiles of risk groups divided based on the following signatures: IFNG (F; ref. 92), T-cell inflamed (G; ref. 96), melanocytic plasticity (H; ref. 98), PD-L1 (I; ref. 96), and TIDE (J; ref. 97). K and L, Kaplan–Meier plots show PFS and OS between the four equal quartiles of risk groups divided based on the following signatures: immune checkpoint (K) and deepBTAS (L). M, Performance evaluation of ICB biomarkers in terms of AUC.
Finally, we evaluated whether tumor activity of bipotent targets (angiogenesis and growth suppressor evasion) can predict patient response to anti–PD-1. Analogous to immunometabolism targets, we first fit Cox hazard models to learn a BTAS function that quantifies the activity of angiogenesis and growth suppressor evasion bipotent targets in a tumor (Methods). BTAS showed a significant association with the OS of patients with melanoma treated with anti–PD-1 (Cox regression P < 1.1E−3), but it failed to stratify patients into two groups with significant OS differences (Supplementary Fig. S16C). Dividing patients into 4 equal quartiles of the BTAS score resulted in patient stratification with marked survival differences in both PFS (log-rank P < 2.2E−4) and OS (log-rank P < 3E−3; Fig. 6B). We showed that such patient stratification is unlikely by random chance: Only 16 out of 10,000 randomly sampled gene sets showed higher predictive power than BTAS (Fig. 6C, i.e., empirical P < 1.6E−3). The predictive power of BTAS in stratifying ICB patients was comparable to other top ICB biomarkers (Fig. 6D–L). We also trained a nonlinear version of BTAS using a feedforward network of the same architecture used for immune metabolism and learned deepBTAS scores for angiogenesis and growth-suppressor bipotent targets but observed only a marginal improvement over BTAS in patient stratification (Fig. 6M).
Together, the data suggest that bipotent gene targets such as ESRRA are relatively common, and their discovery could enable therapies that kill cancer through multiple mechanisms simultaneously.
DISCUSSION
This study introduces a new computational tool, BipotentR, that identifies targets that can inhibit tumors by activating immunity and suppressing a second user-defined oncogenic pathway. BipotentR leverages public databases to link the user-defined pathway with immunity and find targets with previously unrecognized functions. Using BipotentR, we predicted its 38 bipotent regulators whose inhibition induced metabolic and immune phenotypes. Tumor activity of bipotent targets can stratify melanoma patients’ responses to anti–PD-1, which we showed by applying an AI/ML approach. We validated the topmost implicated target, ESRRA, and uncovered its two antitumor immune mechanisms (Supplementary Fig. S17A). ESRRA inhibition is safe in vivo, and its effects on energy metabolism are focused on cancer cells.
ESRRA regulates OXPHOS and proliferation in tumors of breast (107), prostate (108), and endometroid (109) cancers. ESRRA is amplified in endometrial cancers and associated with immune infiltration (109). We observed that ESRRA was active in immunosuppressive and immunoresistant tumors (Fig. 4A–C; Supplementary Figs. S3A and S3B, and S4A and S4B), suggesting potential clinical relevance in several tumor types. Inhibiting ESRRA stunted tumors by stimulating antigen presentation and cytokines that recruited effector T cells to tumors and polarized macrophages. These findings are supported by studies showing OXPHOS suppression alters both T cells (27, 55) and macrophages (110–112) because ESRRA regulates OXPHOS.
Nonetheless, we note several limitations of our study. First, because our approach aims to discover new gene targets and drugs to improve cancer therapy, particularly immunotherapy, it requires users to have basic cancer and immunology expertise. However, our group previously did not have metabolism expertise but was led by the algorithm prediction to investigate ESRRA, thus BipotentR can be of utility to a wide range of clinicians and researchers.
Second, because metabolic dependencies are often affected by their context in the tumor microenvironment, our study relied on immunocompetent mouse models (4T1, B16F10, and CT26). To supplement these experiments, we performed in vitro experiments to uncover the signaling underlying antitumor immune functions of ESRRA. Previously, Yuk and colleagues showed in a noncancer context that ESRRA-deficient mice induce the cytokines TNFα, IL6, and IL1β (60). In the cancer context, we show targeting ESRRA induced these cytokines, which in turn polarized macrophages toward proinflammatory states. CRISPR or drug targeting of ESRRA stimulated antigen presentation genes (Supplementary Figs. S6A and S7A) and recruited effector CD8+ T cells to tumors (Fig. 2B and C), which in turn enhanced tumor elimination by T cells (Fig. 2D and E). ESRRA has also been shown to function in autophagy (113), which is a regulator of antigen presentation genes (114). Consistent with this, we found that ESRRA suppression activated autophagy-related genes (Supplementary Fig. S6H), particularly at 72 hours after the suppression, which is consistent with the activation of antigen presentation at 72 hours. Autophagy also regulates macrophage polarization (115), consistent with our findings (Fig. 2H; Supplementary Fig. S3A). Finally, we observed that ESRRA regulates other immune regulators, including a bipotent target, FLI1 (Supplementary Fig. S18A), suggesting that regulators of bipotent targets are more likely to be bipotent themselves.
Third, standardized metrics are lacking to benchmark compound multiomics approaches such as ours. Prediction of gene targets is an emerging area in which computational approaches have been published recently (10–13). Another fast-emerging area is system immunometabolism. To study system immunometabolism, experimental and computational approaches have been proposed including single-cell (116), cytometry-based (117, 118), multiomics (119), and genome-scale modeling approaches (26). Although these approaches study metabolism within immune cells, BipotentR focuses on regulators of cancer cell adaptations that alter the immune microenvironment. Thus, these immunometabolism approaches differ fundamentally from BipotentR. For evaluating BipotentR predictions, we needed a multipronged strategy. (i) We benchmarked BipotentR against competing or simpler alternatives. First, BipotentR identified regulators of energy metabolism at least as accurately as regulator prediction approaches LISA and BARTWeb (Supplementary Fig. S2C and S2D). Because Rephine focuses on predicting drug response, we could not compare its output with BipotentR output. Second, BipotentR accurately predicted immune regulators when compared with an alternative approach focused on regulators of macrophage polarization (Supplementary Note S1; Supplementary Fig. S18B). Third, BipotentR showed a comparable statistical power in detecting regulators against alternatives of using modules and submodules serially in BipotentR (Supplementary Note S6; Supplementary Fig. S18C). Fourth, BipotentR's (regulation module) robustness to the class imbalance problem was comparable to Firth's regression (Supplementary Note S7). (ii) We deconvolved contributions of bulk RNA and scRNA submodules and showed both submodules are essential for BipotentR performance. (iii) We observed knockout of the set of immune–metabolic regulators potentiated CD8+ T cells in the CRISPR screen. In this screen, gene knockouts are done only on cancer cells and not on immune cells. To overcome this limitation, we presented a complementary evaluation of BipotentR predictions focusing on melanoma. Specifically, we showed using AI/ML approaches that the activity of bipotent targets predicted the outcome of patients with melanoma to anti–PD-1 treatment. Importantly, combining these AI/ML approaches with current ICB biomarkers markedly improved biomarkers’ ability to predict patient outcomes. (iv) We experimentally followed up on BipotentR's top predicted candidate (ESRRA) and showed its antitumor functions. Experimental validations are needed to establish the dual anticancer potential of the other candidates identified by BipotentR.
Although this work focused on targets for immune metabolism, BipotentR can be applied to prioritize drug targets for any given pathway. We used BipotentR to identify immunotherapeutic targets that regulate angiogenesis and growth-suppressor pathways. Overall, these data demonstrate the utility of BipotentR in the discovery of a single drug that suppresses one pathway in selected cell types while simultaneously stimulating an immune response.
METHODS
Analyses and datasets included in the article, also available as a part of the “BipotentR” R package, are summarized in Supplementary Fig. S17B and Supplementary Table S7. It provides comprehensive resources to researchers and clinicians working on novel agents to explore the potential clinical relevance of agents using more than 200 cancer cohorts (33,000 patients), 80 cohorts of scRNA experiments (2.5 million cells), and several immunologic CRISPR screens curated.
Tumor Cell Lines
The 4T1 cells were obtained from the Steele Lab (Massachusetts General Hospital) and routinely tested for Mycoplasma using the Lonza MycoAlert Mycoplasma Detection Kit. The B16F10 cells were purchased from ATCC, which carries out authentication prior to shipment. Mycoplasma testing was also carried out routinely using the Lonza MycoAlert Mycoplasma Detection Kit.
Mouse Models
Female Balb-c mice were purchased from Harlan, and female C57BL/6 mice were obtained from Vivo Bio-Tech (under license from Taconic). All protocols used in this study have been approved by the Axis Bioservices Animal Welfare and Ethical Review Committee. All procedures were carried out under the guidelines of the Animal (Scientific Procedures) Act 1986.
Wild-type Balb/c recipient mice were purchased from the Charles River Laboratory. All mice were housed in a standard cage in the Dana-Farber Cancer Institute Animal Resources Facility (ARF). All animal procedures were carried out under the ARF Institutional Animal Care and Use Committee (IACUC) protocol and were in accordance with the IACUC standards for the welfare of animals (CT26).
In Vitro Treatment Procedures
siRNA Treatment of Cells.
SK-BR-3 cells were seeded in a T75 flask (Greiner Bio-One, #658175) and transfected 4 days later with 25 nmol/L siRNA-ESRRA (Dharmacon; #L-03403) using dharmafect 1 (Dharmacon, #T-2001). As a negative control, a 25 nmol/L siRNA-nontargeting pool (Dharmacon, #D-0018) was used. The transfected cells were reseeded 48 hours after transfection into 6-well plates (Greiner Bio-One, #657160) at 5,000 cells per well. After 0, 3, and 5 days, RNA was isolated using QIAzol Lysis Reagent (Qiagen, #79306) according to the manufacturer's protocol.
Compound Treatment of Cells.
We study two known structurally similar inhibitors (compounds 29 and 39) of ESRRA. The compounds’ efficacy against ESRRA was first analyzed using the AlphaScreen assay. Compounds were tested for their ability to bind to the ESRRA-LBD protein using AlphaScreen Technology (PerkinElmer). ESRRA-LBD protein was expressed in Escherichia coli as a 6xHis Small Ubiquitin-like Modifier (SUMO) fusion. This fusion bacterial protein (i.e., 6xHis-SUMO-ESRRA-LBD) was purified using affinity chromatography. All experiments were performed at room temperature in 384-well, white nonbinding plates (Greiner) using 50 mmol/L Tris-HCl pH 7.5, 100 mmol/L NaCl, 0.1% Pluronic F-127, 0.05% BSA, and 5 mmol/L TCEP as the buffer. The final DMSO concentration was 1% in the assay. Compounds were assayed in triplicate and incubated with 0.81 nmol/L ESRRA-LBD protein and 10 μg/mL streptavidin donor beads and 10 μg/mL Ni-chelate acceptor beads for 1 hour at room temperature, followed by a 2-hour incubation with 15 nmol/L biotin-PGC1α-3 peptide (QRRPCSELLKYLTTNDDPP) corresponding to amino acids 202 to 220. The AlphaScreen signal was measured using an Envision Xcite plate reader (PerkinElmer). Although both inhibitors were bound to the LBD, compound 39 (AlphaScreen log-IC50 = −8.124 M) was more potent than compound 29 (AlphaScreen log-IC50 = −6.349 M). Hence, compound 39 was used for conducting RNA-seq.
RNA-seq.
SKBR3 cells were seeded in 6-well plates (Greiner Bio-One, #657160). Two different cell concentrations were used; 4E5 cells/well were seeded for the 24-hour samples, and 2.7E5 cells/well were seeded for the 48- and 72-hour samples. The next day, medium was refreshed, and 1 or 5 μmol/L compound 39 in culture medium was added to the cells. After 24, 48, and 72 hours of incubation with compound 39, RNA was isolated using QIAzol Lysis Reagent (Qiagen, #79306) according to the manufacturer's protocol (n = 3 per condition).
Total RNA concentration was measured using a NanoDrop (Thermo Scientific), and 1.5 μg was sent for sequencing. Clustering and DNA sequencing using the Illumina NovaSeq 6000, paired-end 150 bp, were performed according to the manufacturer's protocols. NovaSeq control software NCS v1.6 was used. Image analysis, base calling, and a quality check were performed with the Illumina data analysis pipeline RTA3.4.4 and Bcl2fastq v2.20. Ensembl GRCh37.75 was chosen as the reference transcriptome sequence for the alignment of the reads. Data were analyzed as described below.
In Vivo Treatment Procedures
4T1 Mouse Model.
A total of 24 female Balb-c mice ages 6 to 8 weeks and weighing approximately 17 to 22 g were used for the study. The 4T1 cells (5 × 104 in Matrigel) were implanted orthotopically into the left inguinal mammary fat pad of mice using a 25-gauge needle. The compound was formulated in 15% vitamin E-TPGS:30% PEG400:55% PBS. Mice were treated with compound 39 (30 mg/kg; n = 12) and vehicle (n = 12) once a day from day −3 (implantation day = 0). Once tumors reached a mean volume of 150 mm³, the tumors were surgically removed under general anesthesia (Ketamine:xylazine; 100 mg/kg:10 mg/kg). Treatment continued for 21 days after surgery. At the end of the study, the lungs were removed and surface macroscopic nodules were counted using a dissecting microscope.
A cardiac puncture was performed, and the blood was plated into 10-cm cell culture dishes with media containing 6-thioguanine to select for circulating tumor cells. Seven to 10 days after initial plating, any colonies that grew were counted. The colonies that formed were counted, and the cells were collected for RNA extraction.
Postsurgery and FACS Analysis of 4T1 Mice.
Following surgery, all tumors from each compound 39 treatment group and 3 each of size and age-matched controls were digested into a single-cell suspension using collagenases. The cells were then fixed before further staining and analyzed on the FACSCanto II flow cytometer. Different populations of cells were quantified and expressed as a percentage:
Alloreactive T cells: CD8+/CD137+ cells (% of CD8 cells)
Tregs: FoxP3+/CD25+ (% of CD4+ cells)
ESSRAi in CD8+ T-Depleted 4T1 Mice.
Mice were split into two groups (n = 10) to receive ESSRAi alone or in combination with CD8 depletion using an anti-CD8 antibody [InVivoMAb anti-mouse CD8β (Lyt 3.2), Bio X Cell]. The ESSRAi-alone group also received control antibody (InVivoMAb rat IgG1 isotype control, anti–horseradish peroxidase). Anti-CD8 (100 μg), or isotype control, was injected intraperitoneally at days −6, −3, 0, 3, 6, and 9. At day −3, ESSRAi was delivered by oropharyngeal gavage daily (M–F) in 100 μL at 30 mg/kg until the study endpoint. Day 0 marked the day of tumor implantation, where 5 × 105 4T1 cells were injected into the inguinal mammary fat pad of BALBc mice (in 50 μL volume). Tumors were measured every 2 to 3 days until sizes reached >1,000 mm3, and mice were then euthanized. Spleens were isolated from mice of each group to verify CD8 depletion efficacy.
Single-Cell Sequencing of ESRRAi-Treated 4T1 Mice.
Mice were split into two groups (n = 3 each group) to receive ESSRAi alone or vehicle only (DMSO 10%, Solutol 20%, water 70%). On day −3, ESSRAi was delivered by oropharyngeal gavage daily (M–F) in 100 μL at 30 mg/kg until study day 14. Day 0 marked the day of tumor implantation, where 5 × 105 4T1 cells were injected into the inguinal mammary fat pad of BALBc mice (in 50 μL volume). Tumors were measured every 2 to 3 days. On day 14, tumors were harvested for scRNA-seq.
Tumors from mice were harvested and broken down into smaller fragments. Then, each sample was digested with reagents from the Mouse Tumor Dissociation Kit (Miltenyi, cat. # 130-096-730) according to the manufacturer's instructions using gentleMACS Octo Dissociator from Miltenyi, and cell suspensions were passed through a 70-μm filter twice before staining. Single tumor cells were washed with ice-cold PBS containing 2% FBS and stained with CD45.2-APC/Cy7 antibody (BioLegend, cat. #109823) at 4°C for 45 minutes. All cells were then washed and resuspended in ice-cold PBS with 2% FBS. Live CD45.2-positive cells were sorted with BD Aria after staining. Each tumor from the same group was processed individually and mixed according to the same cell number. After cell collection, immune cells were resuspended at 1 × 106 cells/mL in PBS with 0.04% BSA (Thermo Fisher Scientific, cat. #AM2616). Single-cell suspensions of all samples were then barcoded with a 10X Chromium Controller (10X Genomics). RNA from the barcoded cells for each sample was subsequently reverse-transcribed, and sequencing libraries were constructed with reagents from a Chromium Single-Cell 30 v2 reagent kit (10X Genomics, cat. #PN-120267) according to the manufacturer's instructions. Sequencing was performed with Illumina HiSeq according to the manufacturer's instructions (Illumina).
B16F10 Mouse Model.
Experiments were performed in 20 female C57BL/6 mice weighing approximately 18 to 20 g. B16F10 cells (0.1 million) were suspended in 50 μL of serum-free medium and mixed at a 1:1 ratio with Matrigel before implanting subcutaneously into the right flank of mice using a 1-mL BD syringe attached to a 24-gauge needle. When the average tumor volume reached around 57 mm³, animals were dosed after randomization into different treatment groups keeping tumor volume and number of animals so that the average tumor volume of each group remained the same across the groups. Compound 39 (30 mg/kg; n = 10) and vehicle (n = 10) were performed by mouth once a day. Body weights were recorded on day 0 (animal randomization) and thrice weekly thereafter until study termination. Changes in percent body weight (% BW Change) were calculated according to: % BW change = (BWFinal – BWInitial)/(BWInitial) × 100. Growth of B16F10 allografts was recorded on days 1, 4, 6, 8, 11, and 13. The length and width of tumors were measured by caliper on the day of animal randomization based on tumor volume (day 1) and thrice weekly thereafter until study termination. Tumor dimensions were measured on day 0 (animal randomization based on tumor volume) and thrice weekly thereafter until study termination. Tumor volumes were calculated using the formula b2 × l × 0.52, where l = length, b = breadth.
CT26 Mouse Model.
We transplanted 2.5 × 105 CT26 cells subcutaneously into the left and right flanks of 6- to 8-week-old female Balb/c mice (Charles River Laboratory). When the tumors became palpable (day 7 after transplantation), we began treatment with control IgG (clone 2A3) or anti–PD-1 (clone 1A12) by intraperitoneal injection (200 μg per mouse in 200 μL HBSS buffer) every 3 days for a total of 5 times. We identified “responder” or “nonresponder” tumors in the anti–PD-1 cohort based on their size at the end of treatment. The top 25% were classified as nonresponders, and the bottom 25% were classified as responders. Three days after the last treatment, we harvested tumors for RNA-seq. RNA extraction was performed using the AllPrep DNA/RNA Mini Kit (Qiagen, #80204) following the manufacturer's protocol. Total RNA was submitted to Novogene Inc. for sequencing. A standard mRNA library preparation kit was used for library preparation. Paired-end 150 bp sequencing was done on an Illumina HiSeq 2500. Sequencing reads were mapped to the mm10 genome by RSEM. Statistics for differentially expressed genes (DEG) were calculated by DESeq2.
Calculation of Regulatory Potential Using Epigenetic Data
We adopt the definition of regulation potential from MARGE (120). MARGE defines regulatory potential (RP) of a ChIP-seq sample 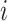 of a TFCR at a gene j as the sum of ChIP-seq signals weighted by genomic distance from the gene transcription start site (
of a TFCR at a gene j as the sum of ChIP-seq signals weighted by genomic distance from the gene transcription start site (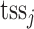 ), that is,
), that is, 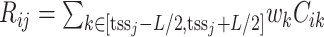 .
. 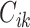 is signal of ChIP-seq sample i at genomic position k, which is weighted by
is signal of ChIP-seq sample i at genomic position k, which is weighted by 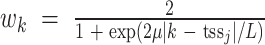 and aggregated within the window of genomic region L = 200 kb around
and aggregated within the window of genomic region L = 200 kb around 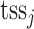 . μ is the decay rate of weights, which is set to −ln10/3. Analogous to RP for ChIP-seq data, RP can also be estimated for (both bulk and single-cell) chromatin accessibility data.
. μ is the decay rate of weights, which is set to −ln10/3. Analogous to RP for ChIP-seq data, RP can also be estimated for (both bulk and single-cell) chromatin accessibility data.
Description of BipotentR
The BipotentR integrative framework is composed of a regulation module and an immune module.
Regulation Module.
The regulation module identifies the regulator of an input pathway. Here we describe bipotent identification for energy metabolism as input. To identify putative energy metabolic regulators, we selected four major energy metabolic pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database as input: FA metabolism, glycolysis, OXPHOS, and TCA. The regulation module of BipotentR leveraged the Cistrome Data Browser (Cistrome DB) dataset (33), which comprises human and mouse ChIP-seq, DNase I hypersensitive sites sequencing, and ATAC-seq chromatin profiling assays. Low-quality samples were filtered out, adopting our previously reported strategy (33), resulting in 21,046 ChIP-seq of 672 TFCRs. Then, putative regulators of each of the four energy metabolism pathways were analyzed separately as follows.
For an input pathway p and given m ChIP-seq samples of a TCFR T, we infer the potential of 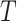 to bind to genes j in p using the logistic generalized linear mixed model (LMM). For generalized LMM formulation, we extend the notation of Zhang (121):
to bind to genes j in p using the logistic generalized linear mixed model (LMM). For generalized LMM formulation, we extend the notation of Zhang (121):
The matrix containing RPs of N protein-coding genes in m ChIP-seq samples was flattened to generate the vector R of size Nm. 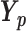 (size Nm) is an incidence vector that relates genes in R to pathway p, that is, it contains indicator variables that equal 1 for genes in the target pathway
(size Nm) is an incidence vector that relates genes in R to pathway p, that is, it contains indicator variables that equal 1 for genes in the target pathway 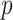 and 0 otherwise. The number of pathway and nonpathway genes, respectively, is 32 and 25,573 for TCA, 135 and 25,470 for OXPHOS, 42 and 25,563 for FA metabolism, 62 and 25,543 for glycolysis (Supplementary Fig. S18D).
and 0 otherwise. The number of pathway and nonpathway genes, respectively, is 32 and 25,573 for TCA, 135 and 25,470 for OXPHOS, 42 and 25,563 for FA metabolism, 62 and 25,543 for glycolysis (Supplementary Fig. S18D). 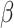 is unknown fixed effect,
is unknown fixed effect, 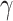 is a vector of size m for unknown random sample–specific confounding effects having distribution
is a vector of size m for unknown random sample–specific confounding effects having distribution 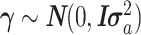 , and Z (size n × t) is an incidence matrix of 1s and 0s relating genes in R to samples.
, and Z (size n × t) is an incidence matrix of 1s and 0s relating genes in R to samples. 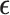 is a residual effect vector having distribution
is a residual effect vector having distribution 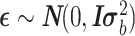 . I is the identity matrix,
. I is the identity matrix, 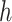 is an inverse logistic link function (i.e.,
is an inverse logistic link function (i.e., 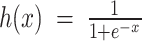 ), and
), and 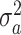 and
and 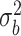 are unknown variances.
are unknown variances.
Parameters of the model were estimated by maximizing the log-likelihood L(β, 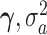 ,
, 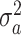 |
|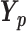 ). To calculate P value, we test the null hypothesis
). To calculate P value, we test the null hypothesis 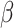 = 0 using t test assuming the t-distribution with degrees of freedom calculated through Satterthwaite's approximation method (122). Parameters were estimated using lmer (123) and lmerTest (122). The P value from the coefficient of
= 0 using t test assuming the t-distribution with degrees of freedom calculated through Satterthwaite's approximation method (122). Parameters were estimated using lmer (123) and lmerTest (122). The P value from the coefficient of 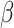 was used as the significance of binding potential for the TFCR T. P values were adjusted for multiple hypotheses for a total number of TFCRs tested using the Benjamini–Hochberg correction (124). We denote this final adjusted P value (
was used as the significance of binding potential for the TFCR T. P values were adjusted for multiple hypotheses for a total number of TFCRs tested using the Benjamini–Hochberg correction (124). We denote this final adjusted P value (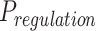 ). We also show
). We also show 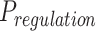 is robust to class imbalance by benchmarking against Firth's regression, which is an approach developed to mitigate class imbalance (See Supplementary Note 7).
is robust to class imbalance by benchmarking against Firth's regression, which is an approach developed to mitigate class imbalance (See Supplementary Note 7).
For the TFCR 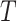 , we estimated the effect size of its binding potential to a pathway as the predictive power of TFCR RP to discriminate genes in the pathway p from other genes. We estimate a single binding potential of TFCR
, we estimated the effect size of its binding potential to a pathway as the predictive power of TFCR RP to discriminate genes in the pathway p from other genes. We estimate a single binding potential of TFCR 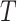 that combines all its ChIP-seq samples. In particular, for each
that combines all its ChIP-seq samples. In particular, for each 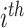 TFCR ChIP-seq sample, we first calculate area under curve
TFCR ChIP-seq sample, we first calculate area under curve 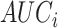 and its confidence interval
and its confidence interval 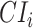 for predictor
for predictor  (using “pROC” (125)). Then
(using “pROC” (125)). Then 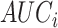 estimates from all ChIP-seq samples of same TFCR T are combined as:
estimates from all ChIP-seq samples of same TFCR T are combined as: 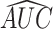 =
=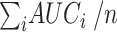 and
and 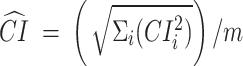 , where
, where 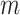 is the number of ChIP-seq of the TFCR. The final binding potential (
is the number of ChIP-seq of the TFCR. The final binding potential (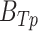 ) of T for pathway p was defined as
) of T for pathway p was defined as 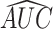 and at 95% of
and at 95% of  .
.
To integrate potential of energy metabolism pathways, we calculate the binding potential (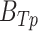 ) and their significance for each of the four metabolic pathways p separately. Then we combined the
) and their significance for each of the four metabolic pathways p separately. Then we combined the 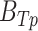 of four pathways by taking their average to get the final binding potential of energy metabolism as:
of four pathways by taking their average to get the final binding potential of energy metabolism as: 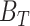 =
= 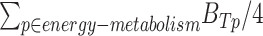 To obtain significance of energy metabolism binding potential (
To obtain significance of energy metabolism binding potential (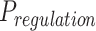 ), we combined the P values of each of the four pathways (Supplementary Fig. S18E) by using Fisher methods (R package “MetaRNaseq”).
), we combined the P values of each of the four pathways (Supplementary Fig. S18E) by using Fisher methods (R package “MetaRNaseq”).
Immune Module.
The immune pipeline is divided into bulk-level identification and single cell–level identification.
Identification of immunomodulatory TFCRs using bulk tumor transcriptome:
Signatures of immune response were collected from Thorsson and colleagues (39), which included biomarkers such as mutation burden, immune infiltration, IFNγ response. Normal samples were excluded, and missing values were imputed (using missMDA; ref. 126). BipotentR first controls for cancer type–specific effects by regressing out cancer type–specific effects from immune signatures (using R package “remef”) and then derives their independent principal components to control confounding effect of cross-correlation between signatures and cancer type–specific effects. Because the resulting first principal component (PC1) in an unbiased manner gave high weights to proinflammatory signatures (Supplementary Fig. S1B), it was used to estimate TFCR association with proinflammatory signatures as follows.
For given expression E of any TFCR T in TCGA samples (n = 10,000), its association with PC1 (P) was performed using a LMM while controlling for confounding effects of cancer type–specific transcription. Expression was quantified using transcript per million from TCGA bulk RNA-seq (obtained from Xena browser; http://xena.ucsc.edu/public/). We use LMM formulation similar to LMM used in the regulation module:
where E is a vector of size n (number of samples) containing log-transformed expression of the TFCR T; P is the vector (size n) of a PC1 in TCGA samples; 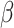 is unknown fixed effect representing the association of PC1 with E; and
is unknown fixed effect representing the association of PC1 with E; and 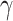 is a vector of size t (number of cancer types, t = 32) for unknown random cancer type–specific transcription effects having distribution
is a vector of size t (number of cancer types, t = 32) for unknown random cancer type–specific transcription effects having distribution 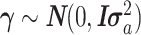 . Z (size n × t) is an incidence matrix of 1s and 0s relating samples to cancer types.
. Z (size n × t) is an incidence matrix of 1s and 0s relating samples to cancer types.  is a residual effect vector having distribution
is a residual effect vector having distribution 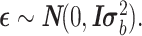 I is the identity matrix, and
I is the identity matrix, and 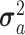 and
and 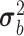 are unknown variances.
are unknown variances.
Similar to the regulation module, parameters and their confidence interval were estimated by maximizing the log-likelihood L(β,  ,
, 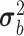 |E). A 95% confidence interval of
|E). A 95% confidence interval of 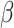 was assumed at 1.96 standard deviation. P-value testing of the null hypothesis
was assumed at 1.96 standard deviation. P-value testing of the null hypothesis 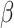 = 0 was done using t test (122), and P value (
= 0 was done using t test (122), and P value (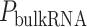 ) was adjusted for multiple hypotheses (number of TFCR tested) using the Benjamini–Hochberg correction (124). Immunomodulatory potential
) was adjusted for multiple hypotheses (number of TFCR tested) using the Benjamini–Hochberg correction (124). Immunomodulatory potential 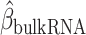 of the TFCR was defined
of the TFCR was defined 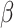 at 95% confidence. TFCRs with the significant (adjusted P < 0.05) positive (negative) potential were predicted as putative immune-stimulatory (immune-suppressive) TFCRs.
at 95% confidence. TFCRs with the significant (adjusted P < 0.05) positive (negative) potential were predicted as putative immune-stimulatory (immune-suppressive) TFCRs.
Identification of TFCRs preferentially active in cancer cells using single-cell data:
To identify TFCRs that are differentially active in cancer cells relative to CD8 T cells, we collected scRNA from five cancer cohorts (GSE103322, GSE72056, EMTAB6149, GSE117570, and GSE127465). Data preprocessing and cell annotation of each cohort were done by MAESTRO (127). A challenge with read counts of TFCRs in single-cell experiments is that they are prone to high dropout rates, especially due to their low basal expression in cells. Therefore, their read counts are an unreliable measure of their activity. To mitigate this effect, we impute activity TCFRs. In particular, for a TFCR T we define its activity 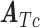 of a cell c as:
of a cell c as: 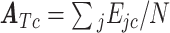 , where
, where 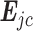 is the normalized cell expression (TPM) of jth gene targets of T. Here, we used the top 15% direct targets of T from Cistrome Cancer (128). Next, we determine if the TFCR T is differentially active in cancer cells relative to CD8− T cells. We perform differential activity analysis across scRNA cohorts while accounting for patient-specific differences in the cohorts by adopting an LMM-based strategy from (129) as:
is the normalized cell expression (TPM) of jth gene targets of T. Here, we used the top 15% direct targets of T from Cistrome Cancer (128). Next, we determine if the TFCR T is differentially active in cancer cells relative to CD8− T cells. We perform differential activity analysis across scRNA cohorts while accounting for patient-specific differences in the cohorts by adopting an LMM-based strategy from (129) as:
where  is a vector of size n (total number of cancer and CD8− T cells in five scRNA cohorts) containing cell activity of the TFCR T; X is an incidence vector that relates cells to their annotation of cancer or CD8− T cells (cancer cells were coded as 1, CD8 T cells as 0);
is a vector of size n (total number of cancer and CD8− T cells in five scRNA cohorts) containing cell activity of the TFCR T; X is an incidence vector that relates cells to their annotation of cancer or CD8− T cells (cancer cells were coded as 1, CD8 T cells as 0);  is unknown fixed effect representing a differential expression of T in cancer cells relative to CD8− T cells; and
is unknown fixed effect representing a differential expression of T in cancer cells relative to CD8− T cells; and  is a vector of size P ( total number of patients in five scRNA cohorts) for unknown random patient-specific confounding effects having distribution
is a vector of size P ( total number of patients in five scRNA cohorts) for unknown random patient-specific confounding effects having distribution 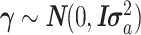 , Z (size n × P) is an incidence matrix of 1s and 0s relating cells in T to patients.
, Z (size n × P) is an incidence matrix of 1s and 0s relating cells in T to patients.  is a residual effect vector having distribution
is a residual effect vector having distribution 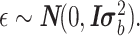 I is the identity matrix, and
I is the identity matrix, and 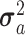 and
and 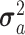 are unknown variances.
are unknown variances.
Parameters and their confidence interval and P values were estimated using a similar procedure used for bulk RNA-seq pipeline. P values (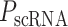 ) were adjusted for the number of TFCR tested using the Benjamini–Hochberg correction (124).
) were adjusted for the number of TFCR tested using the Benjamini–Hochberg correction (124).
Integration of bulk and single-cell estimates:
We define integrated immune-modulatory potential as 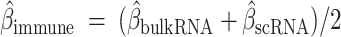 , and P value as
, and P value as 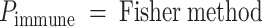
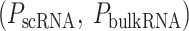 (Supplementary Fig. S18F; ref. 130). Note,
(Supplementary Fig. S18F; ref. 130). Note, 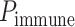 does not need correction for the number of TFCRs analyzed because
does not need correction for the number of TFCRs analyzed because 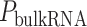 and
and  had been corrected for multiple hypotheses.
had been corrected for multiple hypotheses.
Model Integration.
TFCRs were selected as immune–metabolic TFCRs if their 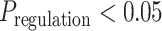 ,
, 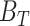 > 85% quantile of all
> 85% quantile of all 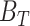 and
and 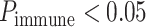 ,
,  > 85% quantile < 15% quantile of all
> 85% quantile < 15% quantile of all  . A more conservative threshold set at 95% (and 5%) failed to identify any TFCR. The procedure identified 38 metabolic TFCRs.
. A more conservative threshold set at 95% (and 5%) failed to identify any TFCR. The procedure identified 38 metabolic TFCRs.
Validation of BipotentR Predictions
Validation of 38 Immune–Metabolic TFCRs Identified by BipotentR.
Validation of metabolic roles:
TFCRs’ metabolic roles were studied using transcriptomic changes induced by their knockdown/knockout. Transcriptomic changes for 38 TFCRs were obtained from Feng and colleagues (54), comprising 570 experiments. Each experiment measures transcriptomes in cell lines with and without TFCR inhibition and then quantifies levels of induced transcriptome changes by the TFCR inhibition using differential expression between inhibition and control conditions. We investigated the suppression of genes in immune–metabolic pathways by testing two hypotheses. The first hypothesis tested whether BipotentR-identified TFCRs knockdowns/knockouts suppress the expression of energy metabolism genes (relative to other genes). Specifically, we tested whether expression fold changes induced by inhibiting identified TFCRs were significantly lower for energy metabolism genes than those in other genes using the Wilcoxon rank-sum test. The second hypothesis tested whether BipotentR-identified TFCRs preferentially regulate energy metabolism genes as compared with other TFCRs. Specifically, for energy metabolism genes, fold changes induced in their expression by inhibiting BipotentR-identified TFCRs were compared with those induced by other TFCRs.
Validation of immune roles:
Regulation of immune response by BipotentR-identified TFCRs was studied by examining how their knockout in cancer cells affected killing by T cells. A CRISPR screen was obtained from ref. 55, in which cancer cells (B16F10) induced with gRNAs were cocultured with (Pmel) T cells in the presence of IFNγ. Increase (or decrease) in T cell–mediated killing by a gene knockout was estimated by fold changes of depletion (or enrichment) of its guides (comparing with and without T-cell coculture), calculated using MAGeCK (131). For BipotentR-identified immunosuppressive TFCRs, we tested whether fold changes of their guides were significantly lower than those of other genes using the Wilcoxon rank-sum test. Similarly, for BipotentR-identified immunostimulatory TFCRs, we tested whether fold changes were significantly greater than those of other genes.
Validation of Metabolic TFCRs Using scATAC-seq Data.
TFCRs that were differentially active in cancer cells were evaluated for target accessibility in cancer cells versus CD8 T cells. Target accessibility in a cell was determined using an scATAC-seq dataset of skin cancer (46). The scATAC-seq was processed by our MAESTRO pipeline (127). Around 200,000 single cells from patients with basal cell carcinoma passed the default filtering criteria of MAESTRO, and the RPs of each cell at each gene were calculated. Then, given T, and the set of targets of the T from Cistrome Cancer (128), we define the target accessibility of T as: 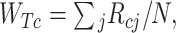 where
where 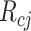 is the RP of jth gene targets of T (top 15% targets of T from Cistrome Cancer), and N is the total number of targets. Differential target accessibility of the TFCR
is the RP of jth gene targets of T (top 15% targets of T from Cistrome Cancer), and N is the total number of targets. Differential target accessibility of the TFCR 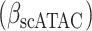 in cancer cells relative to CD8 T cells was determined using:
in cancer cells relative to CD8 T cells was determined using: 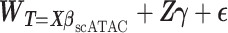 , where
, where 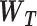 is a target accessibility vector of T across cancer and CD8− T cells; X is an incidence vector that relates cells to their annotation of cancer or CD8− T cells; and
is a target accessibility vector of T across cancer and CD8− T cells; X is an incidence vector that relates cells to their annotation of cancer or CD8− T cells; and 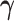 and Z are defined similar to single-cell module of BipotentR.
and Z are defined similar to single-cell module of BipotentR. 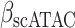 is an unknown fixed effect representing differential accessibility.
is an unknown fixed effect representing differential accessibility. 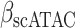 (at 95% confidence interval) were compared with
(at 95% confidence interval) were compared with 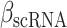 for each TFCR.
for each TFCR.
Assessment of the Robustness of the Bulk RNA-seq Immune Pipeline Using Cross-Validation.
TCGA cancer types were randomly divided into two equal (nonoverlapping) subsets, TCGA samples were split into two groups based on their cancer-type assignment to the subsets, and the immune pipeline was applied separately to each group. The immune RP estimates were compared using Pearson correlation in the two groups.
Differential Expression Analysis
An ER− breast cancer cell line, SKBR3, was treated with ESRRA inhibitor compound 39 at two concentrations (1 and 5 μmol/L), and the vehicle-only treatment was used as a control. Three replicates were collected for each treatment and control, and RNA-seq was performed 24, 48, and 72 hours after treatment. DEGs at each time point were also identified by analyzing the corresponding time point sample using DESeq2 (132). In the analysis, differential effects of drug concentration were also controlled by defining a model matrix = treatment + concentration. The significance of DEGs was estimated using a likelihood ratio test between the above model with the NULL model (i.e., model matrix = concentration). The DEGs were selected with Benjamini–Hochberg adjusted P < 0.05 and absolute log2 fold change (logFC) >0.5. Because the largest transcriptome changes were observed at 72 hours, it was chosen to identify the ESRRA activity signature.
DEGs across all time points were also identified in an analogous manner by defining a model matrix = treatment + concentration + time point and the NULL model matrix = concentration + time point.
Comparison of ESRRAi by Drug or siRNA in Terms of Off-Target Effects
The comparison of inhibition approaches was done through the following two methods:
Cistrome DB.
Downregulated gene sets from the two inhibition approaches were compared for ESRRA potential to regulate these gene sets using ESRRA ChIP-seq data from Cistrome DB. Genome-wide ESRRA RP (defined as the sum of ChIP-seq peaks weighted by genomic distance to the target gene transcription start site) was downloaded from Cistrome DB (34). RP values of downregulated genes were compared between two inhibition approaches as well as with background genes (using one-sided Wilcoxon test).
LISA.
Putative regulators of the DEGs upon two inhibition approaches were identified unbiasedly using LISA (13). LISA is a tool to identify transcription factors that underlie a query gene set. Predicted regulators by LISA were then compared between the two inhibition approaches.
Estimation of ESRRA Activity
To estimate ESRRA activity in cell lines, bulk tumors, or single-cell datasets, we adopted a strategy from ref. 97. ESRRA activity in a sample from a dataset was estimated from the sample transcriptome using the ESRRA activity signature. The expression of each gene was first normalized by dividing its average across all samples in the dataset. Then, ESRRA activity of a sample was estimated as the correlation of ESRRA signature genes between their normalized sample expression and their differential expression by ESRRA inhibition, that is,
where differential expression = −log10(Padj) × sign(fold change) was used.
Pathway Enrichment in Samples with Low ESRRA Activities
Pathway enrichment was conducted by comparing the top 10% with the bottom 10% samples based on ESRRA activity.
Association of Expression of Antigen Presentation Genes and ESRRA Activity
The association of ESRRA activity with antigen presentation gene expression in CCLE and TCGA datasets was evaluated using the Spearman correlation. The association was estimated in each cancer type separately. In TCGA, cancer types with sample sizes > 150 were considered for the analysis.
Analysis of Immune-Related CRISPR Screens
We collected 18 published immune-related CRISPR screens, including 6 MHC-I screens from refs. 63–66 and 8 T-cell coculture screens from refs. 55 and 68–71. In MHC-I screens, to identify genes that regulate MHC-I expression, cancer cells transduced with genome-wide single-guide RNA (sgRNA) had been sorted and classified into MHC-I–high or –low groups based on MHC-I protein expression. The logFC of depletion of each guide (comparing the sorted MHC-I population vs. the unsorted population) were calculated using MAGeCK (131). Then, we estimated knockout's significance and effect size of each gene in increasing MHC-I expression using CRISPhieRmix (133). CRISPhieRmix compares the logFC of all gene guides with the background and uses a hierarchical mixture model of CRISPR screens that leverage two group models to compute P value and were corrected for multiple hypothesis testing (133).
In coculture screens, cancer cells transduced with genome-wide sgRNA were subjected to T cell–mediated killing. LogFC was estimated for both for each guide by comparing experiments with and without coculture using MAGeCK. LogFC and significance were inferred for each gene from its guides’ LogFC using a similar approach used for MHC-I screen analyses described above.
Analysis of Immune Infiltration in Tumors
Abundances of immune cells in all TCGA samples were estimated using CIBERSORT (134), which estimates the abundance of 22 immune cells in bulk RNA-seq using default settings. Myeloid-derived suppressor cell abundance and the M2 polarization score (M2/M1 ratio) were estimated in a TCGA sample from its expression using the approach used previously (97).
For each immune infiltration estimate, its association with ESRRA activity was determined in each cancer type separately by two alternative methods: (i) Wilcoxon test comparing estimates between high and low ESRRA activity groups divided based on the median ESRRA activity, and (ii) Spearman correlations between the estimated infiltration and ESRRA activity (displayed using the R package “corrplot”).
Immune Feature Associations with ESRRA Activity
Cytokine–cytokine interaction pathway and antigen presentation pathway were collected from the KEGG database (135). The expression level of each pathway is estimated by the single-sample gene set enrichment analysis (GSEA; ref. 136) in each dataset using the “GSVA” R package (137). Spearman correlations were calculated between ESRRA activities and these immune features across all patients from TCGA and PRECOG (138) databases.
Bladder Cancer Cohort Analysis
The RNA-seq data and clinical annotation were collected from the bladder cancer patient cohort (139). Patients were clustered using cytokine expressions (genes in KEGG_CYTOKINE_CYTOKINE_INTERACTIONS pathway) by uniform manifold approximation and projection (UMAP) dimension reduction using “uwot” with parameters n_neighbors = 40; learning_rate = 0.1; min_dist = 0.001; number of PC = 10. The clustering resulted in two clusters. DEGs between two clusters were identified by the R package “Seurat.” Finally, ESRRA activities were compared among UMAP clusters, immune phenotypes, and Lund2 classification of bladder cancer (140). The significance of comparisons was determined using the Wilcoxon test for pairwise comparison and the Kruskal–Wallis test for group-wise comparison.
Analysis of the CT26 Mouse Model
Genes significantly suppressed by ESRRA drug inhibition were defined as ESRRA-regulated genes. Enrichment analysis of ESRRA-regulated genes was conducted using GSEA (141), which calculates P values using a permutation test (100 permutations) across all KEGG pathways and adjusts multiple testing using the Benjamini–Hochberg correction. The immune cell-type abundance of mice was estimated using CIBERSORT, based on the mouse immune cell signature from ImmuCC (142). The Spearman correlations of ESRRA activity with immune cell infiltration were calculated across all mice.
Single-Cell Analysis of Public Datasets
Single-cell cancer cohorts were compiled from the Tumor Immune Single-Cell Hub (TISCH) database (74). TISCH consists of single-cell transcriptomic profiles of nearly 2 million cells from 76 cohorts across 27 cancer types. The data from all cohorts were processed using our standardized MAESTRO workflow (127), which included quality control, batch effect removal, clustering, and cell-type annotation. Thirty cohorts that contained annotated cancer cells, macrophages, T and B cells, as well as transcriptomes from at least 1,500 single cells were further selected for the analysis. scATAC-seq data were downloaded from ref. 78 and were processed using a standardized pipeline that uses the RP to quantify chromatin accessibility of a genomic region (127).
The calculation of ESRRA activity for each cell was similar to the activity calculation for a bulk RNA-seq sample. The score was defined as the Spearman correlation between the normalized expression and differential expression (by ESSRAi). DEGs between clusters were found by FindMarkers with default parameters. Pathway enrichment was conducted using ClusterProfiler (143). KEGG terms with adjusted P < 0.05 were selected as significant.
scRNA-seq Analysis of the 4T1 Mouse Model
Alignment, filtering, and counting were done using Cell Ranger with the human reference GRCh38. The filtered gene expression matrix was further analyzed in R using the standard workflow of Seurat v.3.0. Briefly, after removing low-quality cells (<200 or >2,500 genes, or >5% genes from the mitochondrial genome), normalization, principal component analysis of highly variable genes, and clustering were performed using default Seurat parameters (144). Marker genes were identified for each cluster using differential expression analysis. Matching identified markers of each cluster with known canonical markers (145), each cluster was assigned putative cell types: CD4+ T cells (Cd3e, Cd4), monocytes/macrophages (Fcgr2b, C1qa, Mmp12, and Thbs1), B cells (Ms4a1, and Iglv3), CD8+ T cells (Cd8a, Cd3e, Cd3g, and Gzmb), Tregs (Cd4, Ctla4, and Foxp3), natural killer (NK) cells (Ncr1, Nkg7, and Klri2), and neutrophils (Cd15, Cd16, Mmp9, and Csf3r). This annotation was also further tested using SingleR (146) and SciBet (147).
Analysis of Macrophage Polarization
Adopting the strategy of scoring a gene set at a single-cell level (42), the average expression of M1 and M2 marker gene sets (61) was used to evaluate macrophage polarization (Supplementary Fig. S19). This was done using the “AddModuleScore” function in Seurat.
Estimating Significance of Differential Expression of a Gene Set
To calculate the significance of differential expression of any given gene set M of size N, empirical distribution was determined. Specifically, differential expression 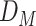 of the set M was calculated as the average of the differential expression of genes of the set M, where the differential expression of each gene was quantified as:
of the set M was calculated as the average of the differential expression of genes of the set M, where the differential expression of each gene was quantified as:  , which accounts for both fold change and P value of the differential expression. Then, a random gene set R of size N was sampled, and
, which accounts for both fold change and P value of the differential expression. Then, a random gene set R of size N was sampled, and 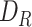 (i.e., average differential expression of genes in the set R) was calculated. Repeating this procedure 1E7 times, we constructed the empirical distribution of
(i.e., average differential expression of genes in the set R) was calculated. Repeating this procedure 1E7 times, we constructed the empirical distribution of 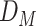 . P value of the significance of upregulation of M was calculated as
. P value of the significance of upregulation of M was calculated as 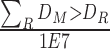 , and significance of downregulation of M was calculated as
, and significance of downregulation of M was calculated as 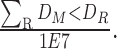
We compared cells of lymphoid lineage (i.e., CD4+ T, CD8+ T, B, and NK cells) between ESRRAi treatment and control (vehicle). The significance of the differential expression of a marker set is determined by the empirical distribution procedure described above.
Identification and Validation of Candidate Bipotent Regulators of Angiogenesis and Evasion of Growth Suppressors
To identify bipotent regulators that simultaneously modulate angiogenesis and tumor immunity, a gene set of angiogenesis (148) was given as input to BipotentR. This identified 14 bipotent regulators. Bipotent regulators of growth suppressor evasion were also identified analogously. Validation of these candidate regulators was done similarly as was done for immune–metabolic bipotent regulators (see “Validation of metabolic roles” and “Validation of immune roles”).
Prediction of Melanoma Response to Anti–PD-1 Using Tumor Activity of Bipotent Targets
Following an input set of bipotent targets, we followed BTAS and deepBTAS procedures described below to estimate bipotent target activity in tumors. We applied the procedure separately for (i) immune-metabolism bipotent targets and (ii) a set of bipotent targets of angiogenesis and growth suppressor evasion.
ICB Data and Preprocessing.
We focused on melanoma cohorts with tumor RNA-seq from patients receiving anti–PD-1 alone or in combination with anti-CTLA4. The RNA-seq and outcomes (OS, PFS, and RECIST) response were obtained from cohorts (84–88). We considered anti–PD-1 alone and anti–PD-1 + anti-CTLA4 as separate cohorts. Patient cohorts with fewer than 10 samples with RNA-seq and outcome were excluded. This resulted in samples from 296 patients (182 anti–PD-1 and 114 anti–PD-1 + anti-CTLA4) with RNA-seq and outcome information. Gene expression was quantile-normalized to normal distribution across all samples in cohorts.
BTAS Details.
To learn a risk score combining tumor activity of bipotent targets through a Cox proportional hazards model (using notation from ref. 100)
Here, 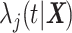 and
and 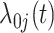 are hazard and baseline-hazard functions of
are hazard and baseline-hazard functions of 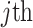 strata (e.g., cohort or different outcomes within a cohort). X is the activity (normalized expression) of bipotent targets (vector of size: number of bipotent targets) in the tumor of a patient, W is the weights to be estimated, and our BTAS is risk score r(t) =
strata (e.g., cohort or different outcomes within a cohort). X is the activity (normalized expression) of bipotent targets (vector of size: number of bipotent targets) in the tumor of a patient, W is the weights to be estimated, and our BTAS is risk score r(t) =  which is effect of bipotent target activity on baseline hazard of the patient, which we defined as our BTAS.
which is effect of bipotent target activity on baseline hazard of the patient, which we defined as our BTAS.
deepBTAS Architecture.
We used a feedforward full network to model  . Specifically, three stacked fully connected layers Fc1, Fc2, and Fc3, parameterized by Y1 ∈ ℝn × 8, b1 ∈ ℝ8, and Y2 ∈ ℝ8× 4, b2 ∈ ℝ4 , and Y3 ∈ ℝ4× 1, b3 ∈ ℝ1, each followed by rectified linear unit (ReLU) activation and batch normalization (BN), allowed us to model hazard function that aligns with outcomes. n is the number of inputs.
. Specifically, three stacked fully connected layers Fc1, Fc2, and Fc3, parameterized by Y1 ∈ ℝn × 8, b1 ∈ ℝ8, and Y2 ∈ ℝ8× 4, b2 ∈ ℝ4 , and Y3 ∈ ℝ4× 1, b3 ∈ ℝ1, each followed by rectified linear unit (ReLU) activation and batch normalization (BN), allowed us to model hazard function that aligns with outcomes. n is the number of inputs.
 |
where T(x) = BN(ReLU(x)).
Training Procedure.
We learned the weights W for both BTAS and deepBTAS using expression, OS, and PFS from TCGA melanoma samples (N = 465). Normal samples were removed from TCGA samples. To leverage both OS and PFS during the training, we used those two different outcomes as two different strata in our training and validation procedure. W were estimated by minimization of the Cox partial likelihoods. Partial likelihoods for linear and feedforward weights were adopted from Katzman and colleagues (100) as: 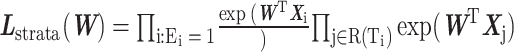 . Here,
. Here, 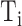 i,
i, 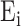 , and
, and 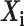 are the event time, event indicator, and bipotent target expression vector data for the ith patient.
are the event time, event indicator, and bipotent target expression vector data for the ith patient. 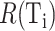 is a set of the patient risk at time
is a set of the patient risk at time 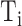 . Likelihoods were estimated separately for PFS and OS as strata, and the final likelihood was defined
. Likelihoods were estimated separately for PFS and OS as strata, and the final likelihood was defined  .
.
For learning, linear weights were estimated by minimizing  using the R package “survival coxph.” For feedforward
using the R package “survival coxph.” For feedforward 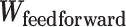 , we divided TCGA melanoma samples (N = 465) into 85% training and the rest 15% validation. We minimized the likelihood to update the model parameters using the Adam optimizer a learning rate of 1 × E−3. To avoid overfitting, we used dropout with P = 0.4 after every hidden layer, and early stopping that was triggered when the likelihood is not decreased for 20 consecutive epochs, and selected the model with the highest validation likelihood (Supplementary Fig. S10K).
, we divided TCGA melanoma samples (N = 465) into 85% training and the rest 15% validation. We minimized the likelihood to update the model parameters using the Adam optimizer a learning rate of 1 × E−3. To avoid overfitting, we used dropout with P = 0.4 after every hidden layer, and early stopping that was triggered when the likelihood is not decreased for 20 consecutive epochs, and selected the model with the highest validation likelihood (Supplementary Fig. S10K).
ICB Evaluation.
The activity (normalized expression) of bipotent targets in ICB cohorts was used as input X to get BTAS and deepBTAS scores from the learned models described above. Note, the parameters of models were not updated using data from the ICB cohort. The BTAS and deepBTAS scores were individually used for patient stratification and response prediction. For patient stratification, patients were divided into either two (using median score) or four equal groups (in <25, 25–50, 50–75, and >75 percentiles). Kaplan–Meier plots were plotted using the stratifications, and the significance of survival difference was estimated using the log-rank test. For response prediction, we calculate accuracy (AUC) for classifying radiologic-based (partial/complete) responders and nonresponders (stable/progressive disease).
Evaluation of Bulk RNA and scRNA Submodule Contributions to BipotentR
BipotentR without the scRNA Submodule.
We assessed how BipotentR would have performed without the scRNA submodule. TFCRs were selected that passed the filter 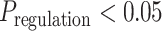 ,
,  > 85% quantile of all
> 85% quantile of all 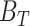 and
and  , and
, and 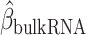 85% quantile < 15% quantile of all
85% quantile < 15% quantile of all 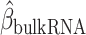 . Among the selected, 38 TFCRs with the largest score =
. Among the selected, 38 TFCRs with the largest score = 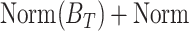 (
(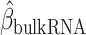 ) were selected for comparison with BipotentR-predicted regulators. Here
) were selected for comparison with BipotentR-predicted regulators. Here  . The 38 predicted TFCRs were evaluated in a similar manner as was done for BipotentR-predicted regulators in the sections “Validation of immune roles” and “Prediction of melanoma response to anti–PD-1 using tumor activity of bipotent targets.”
. The 38 predicted TFCRs were evaluated in a similar manner as was done for BipotentR-predicted regulators in the sections “Validation of immune roles” and “Prediction of melanoma response to anti–PD-1 using tumor activity of bipotent targets.”
BipotentR without the Bulk RNA Submodule.
We assessed how BipotentR would have performed without the bulk RNA submodule. TFCRs were selected that passed the filter  ,
, 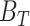 > 85% quantile of all
> 85% quantile of all 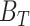 and
and 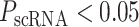 , and
, and 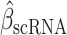 85% quantile < 15% quantile of all
85% quantile < 15% quantile of all 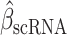 . Among the selected, 38 TFCRs with the largest score =
. Among the selected, 38 TFCRs with the largest score = 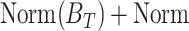 (
(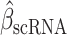 ) were selected for comparison with BipotentR-predicted regulators. Here
) were selected for comparison with BipotentR-predicted regulators. Here 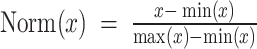 . The 38 predicted TFCRs were evaluated in a similar manner as was done for BipotentR-predicted regulators in sections “Validation of immune roles” and “Prediction of melanoma response to anti–PD-1 using tumor activity of bipotent targets.”
. The 38 predicted TFCRs were evaluated in a similar manner as was done for BipotentR-predicted regulators in sections “Validation of immune roles” and “Prediction of melanoma response to anti–PD-1 using tumor activity of bipotent targets.”
Benchmarking BipotentR against LISA
Using (union of) genes set within the four energy metabolism pathways as input to LISA, we obtained LISA-predicted TFCRs. The top 38 LISA-predicted TFCRs were then evaluated against BipotentR-predicted TFCRs (n = 38). For evaluating these two TFCR sets, we compared how strongly their knockout suppressed energy metabolism genes. The suppression of energy metabolism for a given TFCR knockout was evaluated using the KnockTF database (54) similar to the procedure described in section “Validation of metabolic roles.”
Evaluating Improvement of Predictive Power of Current ICB Biomarkers by BTAS/deepBTAS
Three analyses were done to assess the improvement by BTAS/deepBTAS on current biomarkers. These analyses were done separately for PFS and OS.
BTAS/deepBTAS Association with ICB Survival Outcomes after Controlling for ICB Biomarker.
The survival association of either BTAS or deepBTAS score was determined by controlling for levels for each ICB biomarker separately. Levels of a biomarker were controlled through a multivariate Cox proportional hazards model: 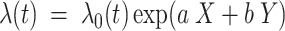 . Here,
. Here,  and
and  are hazard and baseline-hazard functions.
are hazard and baseline-hazard functions. 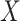 and
and 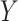 are the BTAS (or deepBTAS) and biomarker levels of a patient tumor, respectively.
are the BTAS (or deepBTAS) and biomarker levels of a patient tumor, respectively. 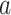 and
and 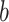 are the coefficients of association. The significance of association (
are the coefficients of association. The significance of association (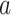 and
and 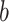 ) is determined using the Wald test. Because association was estimated separately for each biomarker, multiple hypothesis correction is not required.
) is determined using the Wald test. Because association was estimated separately for each biomarker, multiple hypothesis correction is not required.
Performance Improvement by the Addition of BTAS/deepBTAS to Biomarkers.
The improvement was quantified as an increase in the likelihood of the Cox model containing BTAS/deepBTAS and biomarker (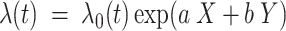 ) over the model containing biomarker alone (
) over the model containing biomarker alone (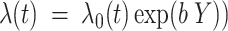 . The significance of the improvement was assessed using the likelihood ratio test with χ2 test with a degree of freedom 1, which accounted for increased model complexity.
. The significance of the improvement was assessed using the likelihood ratio test with χ2 test with a degree of freedom 1, which accounted for increased model complexity.
BTAS/deepBTAS Survival Association Separately on Tumors with High and Low Biomarker Levels.
For each biomarker, tumors were divided into high and low tumor subcohorts (N = 148 each) using the median level of the biomarker. In these two subcohorts, Kaplan–Meier analyses for association with BTAS/deepBTAS were performed separately.
Statistical Analysis
The tests used for statistical analyses are described in the legends of each concerned figure and have been performed using R v3.4. For each experimental group, n represents the number of subjects within each group. Wilcoxon rank-sum test was used for two-group comparison. Correlations were calculated using Spearman correlation analysis. A two-tailed P value < 0.05 was considered statistically significant. Symbols for significance: ns, nonsignificant; *, <0.05, **, <0.01; ***, <0.001; ****, <0.0001. P values were adjusted for multiple hypotheses using the Benjamini–Hochberg correction (124) throughout the article wherever applicable.
Data Availability Statement
BipotentR is available at http://bipotentr.dfci.harvard.edu/. The R package “BipotentR” is available at (https://github.com/vinash85/TRIM). For reproducibility, data and code are shared publicly at the Zenodo repository that is linked to the BipotentR website at http://bipotentr.dfci.harvard.edu/. The website also provides access to bulk and single RNA-seq data generated from the current study. AI/ML approaches developed from the study are accessible using a publicly available Web browser (https://rconnect.dfci.harvard.edu/BTAS/) and a stand-alone software tool, which users can use to predict patient response to immunotherapy in any input melanoma cohort.
Supplementary Material
S1: TFCRs of energy metabolism S2: Bipotent TFCRs S3: Log fold change of T-cell mediated killing upon knockout of OXPHOS genes S4: Log fold change of MHC-I protien expression upon knockout of OXPHOS genes S5: Bipotent TFCRs of angiogenesis S6: Bipotent TFCRs of evasion of growth repressors S7: Datasets included in Methods sections.
The description and validation of the BipotentR immune module
ESRRA inhibition induces antitumor immunity in vivo.
ESRRA inhibition is associated with immune infiltrations and proinflammatory signaling in patient tumors.
ESRRA inhibition correlated with immune infiltrations and proinflammatory signaling in patient tumors.
Robust in vivo antitumor elimination by ESRRA inhibition.
Energy metabolism and immune signaling by targeting ESRRA in vitro.
Energy metabolism and immune signaling by targeting ESRRA in vitro.
The effect of ESRRA inhibition on tumor microenvironments at single-cell resolutions.
ESRRAi does not affect CD8+T T-cells, and ESRRA activity increases post-immunotherapy in resistant tumors.
Machine learning evaluation of immune-metabolic targets in predicting patient response to anti-PD1 monotherapy.
Machine learning evaluation of immune-metabolic targets in predicting patient response to anti-PD1 and anti-CTLA4 combination therapy.
deepBTAS improve the performance of current ICB biomarkers.
Kaplan–Meier plots showing progression-free survival and overall survival differences for patients receiving anti-PD1 between the low-risk and high-risk groups defined by the median value of deepBTAS.
Evaluation of bulk-BipotentR (BipotentR without single-cell sub-module).
Evaluation of scRNA-BipotentR (BipotentR without bulk-tumor sub-module).
Validation of bipotent regulators of angiogenesis or growth-suppressor.
Overall design.
Evaluation of BipotentR.
Expression of markers of M1 and M2 in scRNA data of ESRRAi and Vehicle treated mice.
We detailed a comparison of BipotentR with competing and alternative approaches in Supplementary notes 1, 2, 6, and 7. Supplementary Note 3-5 describes the in vitro findings of ESRRA, the potential clinical relevance of ESRRA across cancer-types, and the association of tumor activity of ESRRA with immune infiltration.
Acknowledgments
A. Sahu was supported by the NIH/NCI (K99CA248953) and the Human Vaccines Project (MP19-02-190). D.E. Fisher was supported by the NIH/NCI (R01AR072304, R01AR043369, P01CA163222, and R01CA222871). K.T. Flaherty and D.E. Fisher were supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. X. Wang was supported by the China Scholarship Council (201806210422). C. Tokheim was supported by the Damon Runyon Cancer Research Foundation (DRQ-04-20). N.U. Nair was supported in part by the Intramural Research Program of the NIH, NCI, and the Center for Cancer Research. The authors from Lead Pharma, including A. Oubrie, J.P.G. Klomp, J.A.P. Rens, M. Bourajjaj, B. Jansen, I. Leenders, J. Lemmers, M. Musters, S. van Zanten, and L. van Zelst, were supported by Eurostars (Eureka, PREVAIL 11590). We thank Rafael A. Irizarry and Myles A. Brown for their valuable suggestions.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
A. Sahu reports nonfinancial support from Lead Pharma during the conduct of the study, as well as nonfinancial support from C-Reveal Therapeutics and Checkmate Pharmaceuticals outside the submitted work. J.P.G. Klomp reports grants from Eurostars during the conduct of the study; other support from Lead Pharma outside the submitted work; and a patent for WO2021001453 pending and a patent for WO2021074365 pending. S.S. Gu reports a Sara Elizabeth O'Brien Trust Fellowship outside the submitted work. C. Tokheim reports grants from the Damon Runyon Cancer Research Foundation during the conduct of the study. M. Bourajjaj reports grants from Eurostars during the conduct of the study, as well as other support from Lead Pharma outside the submitted work. B. Jansen reports grants from Eurostars during the conduct of the study, as well as other support from Lead Pharma outside the submitted work. I. Leenders reports grants from Eurostars during the conduct of the study; other support from Lead Pharma outside the submitted work; and a patent for WO2021001453 pending and a patent for WO2021074365 pending. J. Lemmers reports grants from Eurostars during the conduct of the study; other support from Lead Pharma outside the submitted work; and a patent for WO2021001453 pending and a patent for WO2021074365 pending. S. van Zanten reports grants from Eurostars during the conduct of the study, as well as other support from Lead Pharma outside the submitted work. L. van Zelst reports grants from Eurostars during the conduct of the study, as well as other support from Lead Pharma outside the submitted work. J. Worthington reports grants from Eurostars during the conduct of the study. D. Juric reports grants and personal fees from Novartis, Genentech, Syros, Eli Lilly, and Eisai, grants from Pfizer, Amgen, InventisBio, Arvinas, Takeda, Blueprint, Ribon, and Infinity, and personal fees from Relay, PIC Therapeutics, Vibliome, and Mapkure outside the submitted work. A. Oubrie reports grants from Eurostars during the conduct of the study; other support from Lead Pharma outside the submitted work; and a patent for WO2021001453 pending and a patent for WO2021074365 pending. X.S. Liu conducted the work while on the faculty at Dana-Farber Cancer Institute and is currently a board member for and CEO of GV20 Therapeutics. D.E. Fisher reports grants from the NIH, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and Lead Pharma during the conduct of the study; grants from the NIH, Lead Pharma, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation outside the submitted work; and a financial interest in Soltego, a company developing salt inducible kinase inhibitors for topical skin-darkening treatments that might be used for a broad set of human applications. The interests of D.E. Fisher were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. K.T. Flaherty reports personal fees from Clovis Oncology, Strata Oncology, Checkmate Pharmaceuticals, Kinnate Biopharma, Scorpion Therapeutics, PIC Therapeutics, Apricity, Tvardi, xCures, Monopteros, Vibliome, ALX Oncology, Fog Pharma, Soley Therapeutics, Nextech, Takeda, Transcode Therapeutics, Oncoceutics, OMRx, Quanta Therapeutics, Lilly, and Genentech, and grants from Novartis and Sanofi during the conduct of the study. No disclosures were reported by the other authors.
Authors’ Contributions
A. Sahu: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, methodology, writing–original draft, project administration, writing–review and editing. X. Wang: Data curation, software, formal analysis, visualization, methodology, writing–review and editing. P. Munson: Validation, writing–review and editing. J.P.G. Klomp: Resources, writing–review and editing. X. Wang: Data curation. S.S. Gu: Data curation, writing–review and editing. Y. Han: Data curation. G. Qian: Data curation. P.B. Nicol: Software. Z. Zeng: Resources. C. Wang: Data curation. C. Tokheim: Data curation. W. Zhang: Data curation. J. Fu: Data curation. J. Wang: Data curation. N.U. Nair: Data curation. J.A.P. Rens: Data curation. M. Bourajjaj: Data curation. B. Jansen: Data curation. I. Leenders: Data curation. J. Lemmers: Data curation. M. Musters: Data curation. S. van Zanten: Data curation. L. van Zelst: Data curation. J. Worthington: Data curation. J.S. Liu: Project administration, writing–review and editing. D. Juric: Data curation. C.A. Meyer: Writing–review and editing. A. Oubrie: Resources, data curation. X.S. Liu: Conceptualization, supervision, writing–review and editing. D.E. Fisher: Conceptualization, supervision, project administration, writing–review and editing. K.T. Flaherty: Conceptualization, supervision, project administration, writing–review and editing.
References
- 1. Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019;574:268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017;171:1678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benesic A, Jalal K, Gerbes AL. Drug-drug combinations can enhance toxicity as shown by monocyte-derived hepatocyte-like cells from patients with idiosyncratic drug-induced liver injury. Toxicol Sci 2019;171:296–302. [DOI] [PubMed] [Google Scholar]
- 4. Nahar KJ, Rawson RV, Ahmed T, Tattersall S, Sandanayake N, Kiely CJ, et al. Clinicopathological characteristics and management of colitis with anti–PD-1 immunotherapy alone or in combination with ipilimumab. J Immunother Cancer 2020;8:e001488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petroni G, Formenti SC, Chen-Kiang S, Galluzzi L. Immunomodulation by anticancer cell cycle inhibitors. Nat Rev Immunol 2020;20:669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22. [DOI] [PubMed] [Google Scholar]
- 7. O'Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 2019;19:273–81. [DOI] [PubMed] [Google Scholar]
- 8. Moreno-Gonzalo O, Jr MF, Sánchez-Madrid F. HDAC6 at crossroads of infection and innate immunity. Trends Immunol 2018;39:591–5. [DOI] [PubMed] [Google Scholar]
- 9. Koga S, Onishi H, Masuda S, Fujimura A, Ichimiya S, Nakayama K, et al. PTPN3 is a potential target for a new cancer immunotherapy that has a dual effect of T cell activation and direct cancer inhibition in lung neuroendocrine tumor. Transl Oncol 2021;14:101152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma W, Wang Z, Zhang Y, Magee NE, Feng Y, Shi R, et al. BARTweb: a web server for transcriptional regulator association analysis. NAR Genom Bioinform 2021;3:lqab022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X, Zhang Z, Qin W, Liu S, Liu C, Genchev GZ, et al. RePhine: an integrative method for identification of drug response-related transcriptional regulators. Genomics Proteomics Bioinformatics 2021;19:534–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das A, Morley M, Moravec CS, Tang WHW, Hakonarson H, MAGN Consortium, et al. Bayesian integration of genetics and epigenetics detects causal regulatory SNPs underlying expression variability. Nat Commun 2015;6:8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol 2020;21:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 15. Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab 2019;29:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov 2017;7:716–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med 2017;23:1234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med 2018;24:1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prado-García H, Sánchez-García FJ. Editorial: immuno-metabolism in tumor microenvironment. Front Immunol. 2017;8:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015;43:435–49. [DOI] [PubMed] [Google Scholar]
- 21. KO Alfarouk. Tumor metabolism, cancer cell transporters, and microenvironmental resistance. J Enzyme Inhib Med Chem 2016;31:859–66. [DOI] [PubMed] [Google Scholar]
- 22. Chang C-H, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015;162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE, et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov 2019;9:628–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ye Y, Hu Q, Chen H, Liang K, Yuan Y, Xiang Y, et al. Characterization of hypoxia-associated molecular features to aid hypoxia-targeted therapy. Nat Metab 2019;1:431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim SH, Li M, Trousil S, Zhang Y, Pasca di Magliano M, Swanson KD, et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J Invest Dermatol 2017;137:1740–8. [DOI] [PubMed] [Google Scholar]
- 26. Purohit V, Wagner A, Yosef N, Kuchroo VK. Systems-based approaches to study immunometabolism. Cell Mol Immunol 2022;19:409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019;366:1013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol 2017;8:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scharping NE, Delgoffe GM. Tumor microenvironment metabolism: a new checkpoint for anti-tumor immunity. Vaccines (Basel) 2016;4:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 2020;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS, et al. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest 2006;116:3015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Calder PC. The relationship between the fatty acid composition of immune cells and their function. Prostaglandins Leukot Essent Fatty Acids 2008;79:101–8. [DOI] [PubMed] [Google Scholar]
- 33. Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, et al. Cistrome Data Browser: a data portal for ChIP-seq and chromatin accessibility data in human and mouse. Nucleic Acids Res 2017;45:D658–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome data browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 2019;47:D729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deblois G, Giguère V. Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim Biophys Acta 2011;1812:1032–40. [DOI] [PubMed] [Google Scholar]
- 36. Yang J, Williams RS, Kelly DP. Bcl3 interacts cooperatively with peroxisome proliferator-activated receptor gamma (PPARgamma) coactivator 1alpha to coactivate nuclear receptors estrogen-related receptor alpha and PPARalpha. Mol Cell Biol 2009;29:4091–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta 2011;1812:1007–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab 2018;28:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med 2007;204:345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song Q, Hawkins GA, Wudel L, Chou PC, Forbes E, Pullikuth AK, et al. Dissecting intratumoral myeloid cell plasticity by single-cell RNA-seq. Cancer Med 2019;8:3072–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 2019;50:1317–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24:1277–89. [DOI] [PubMed] [Google Scholar]
- 46. Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 2019;37:925–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer 2017;17:184–97. [DOI] [PubMed] [Google Scholar]
- 48. Yilmaz M, Maass D, Tiwari N, Waldmeier L, Schmidt P, Lehembre F, et al. Transcription factor Dlx2 protects from TGFβ-induced cell-cycle arrest and apoptosis. EMBO J 2011;30:4489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang P, Huang H, Chang J, Zhao GF, Lu ML, Wang Y. Increased expression of DLX2 correlates with advanced stage of gastric adenocarcinoma. World J Gastroenterol 2013;19:2697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morishita J, Kang MJ, Fidelin K, Ryoo HD. CDK7 regulates the mitochondrial localization of a tail-anchored proapoptotic protein, Hid. Cell Rep. 2013;5:1481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN, et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat Commun 2017;8:14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, et al. NFATc1 controls the cytotoxicity of CD8 T cells. Nat Commun 2017;8:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vaeth M, Feske S. NFAT control of immune function: new frontiers for an abiding trooper. F1000Res 2018;7:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feng C, Song C, Liu Y, Qian F, Gao Y, Ning Z, et al. KnockTF: a comprehensive human gene expression profile database with knockdown/knockout of transcription factors. Nucleic Acids Res 2020;48:D93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Park S, Chang CY, Safi R, Liu X, Baldi R, Jasper JS, et al. ERRα-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep 2016;15:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Patch RJ, Searle LL, Kim AJ, De D, Zhu X, Askari HB, et al. Identification of diaryl ether-based ligands for estrogen-related receptor α as potential antidiabetic agents. J Med Chem 2011;54:788–808. [DOI] [PubMed] [Google Scholar]
- 58. Fu J, Li K, Zhang W, Wan C, Zhang J, Jiang P, et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med 2020;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 2012;18:1224–31. [DOI] [PubMed] [Google Scholar]
- 60. Yuk JM, Kim TS, Kim SY, Lee HM, Han J, Dufour CR, et al. Orphan nuclear receptor ERRα controls macrophage metabolic signaling and A20 expression to negatively regulate TLR-induced inflammation. Immunity 2015;43:80–91. [DOI] [PubMed] [Google Scholar]
- 61. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018;174:1293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamada S, Shimojima M, Narita R, Tsukamoto Y, Kato H, Saijo M, et al. RIG-I-like receptor and toll-like receptor signaling pathways cause aberrant production of inflammatory cytokines/chemokines in a severe fever with thrombocytopenia syndrome virus infection mouse model. J Virol 2018;92:e02246–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gu SS, Zhang W, Wang X, Jiang P, Traugh N, Li Z, et al. Therapeutically increasing MHC-I expression potentiates immune checkpoint blockade. Cancer Discov 2021;11:1524–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019;36:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pech MF, Fong LE, Villalta JE, Chan LJ, Kharbanda S, O'Brien JJ, et al. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. Elife 2019;8:e47362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Freeman AJ, Vervoort SJ, Ramsbottom KM, Kelly MJ, Michie J, Pijpers L, et al. Natural killer cells suppress T cell-associated tumor immune evasion. Cell Rep 2019;28:2784–94. [DOI] [PubMed] [Google Scholar]
- 67. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity 2011;35:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell 2020;180:404–5. [DOI] [PubMed] [Google Scholar]
- 69. Decker CE, Young T, Pasnikowski E, Chiu J, Song H, Wei Y, et al. Genome-scale CRISPR activation screen uncovers tumor-intrinsic modulators of CD3 bispecific antibody efficacy. Sci Rep 2019;9:20068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020;135:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol 2018;3:eaar3451. [DOI] [PubMed] [Google Scholar]
- 72. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Patsoukis N, Bardhan K, Weaver J, Herbel C, Seth P, Li L, et al. The role of metabolic reprogramming in T cell fate and function. Curr Trends Immunol 2016;17:1–12. [PMC free article] [PubMed] [Google Scholar]
- 74. Sun D, Wang J, Han Y, Dong X, Ge J, Zheng R, et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res 2021;49:D1420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021;374:abe6474. [DOI] [PubMed] [Google Scholar]
- 76. Skinnider MA, Squair JW, Kathe C, Anderson MA, Gautier M, Matson KJE, et al. Cell type prioritization in single-cell data. Nat Biotechnol 2021;39:30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 2019;25:1251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A 2014;111:11774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 2020;21:1353–65. [DOI] [PubMed] [Google Scholar]
- 81. McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol 2021;32:661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021;184:596–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang K, Patkar S, Lee JS, Gertz EM, Robinson W, Schischlik F, et al. Deconvolving clinically relevant cellular immune cross-talk from bulk gene expression using CODEFACS and LIRICS stratifies patients with melanoma to anti–PD-1 therapy. Cancer Discov 2022;12:1088–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti–PD-1 monotherapy and anti–PD-1/anti-CTLA-4 combined therapy. Cancer Cell 2019;35:238–55. [DOI] [PubMed] [Google Scholar]
- 85. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti–PD-1 therapy in metastatic melanoma. Cell 2017;168:542. [DOI] [PubMed] [Google Scholar]
- 86. Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 2019;25:1916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Freeman SS, Sade-Feldman M, Kim J, Stewart C, Gonye ALK, Ravi A, et al. Combined tumor and immune signals from genomes or transcriptomes predict outcomes of checkpoint inhibition in melanoma. Cell Rep Med 2022;3:100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cox DR. Regression models and life-tables. J R Stat Soc 1972;34:187–202. [Google Scholar]
- 90. Ribas A, Robert C, Hodi FS, Wolchok JD, Joshua AM, Hwu WJ, et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J Clin Oncol 33:15s, 2015(suppl; abstr 3001). [Google Scholar]
- 91. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016;387:1837–46. [DOI] [PubMed] [Google Scholar]
- 92. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Aguiar PN Jr, De Mello RA, Hall P, Tadokoro H, Lima Lopes Gd. PD-L1 expression as a predictive biomarker in advanced non-small-cell lung cancer: updated survival data. Immunotherapy 2017;9:499–506. [DOI] [PubMed] [Google Scholar]
- 94. Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun 2017;8:1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Steiniche T, Rha SY, Chung HC, Georgsen JB, Ladekarl M, Nordsmark M, et al. T-cell–inflamed gene expression profile (GEP) and PD-L1 expression in patients (pts) with esophageal cancer (EC). J Clin Oncol 37:4s, 2019(suppl; abstr 26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24:1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pérez-Guijarro E, Yang HH, Araya RE, El Meskini R, Michael HT, Vodnala SK, et al. Multimodel preclinical platform predicts clinical response of melanoma to immunotherapy. Nat Med 2020;26:781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zuin J, Roth G, Zhan Y, Cramard J, Redolfi J, Piskadlo E, et al. Nonlinear control of transcription through enhancer-promoter interactions. Nature 2022;604:571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Katzman JL, Shaham U, Cloninger A, Bates J, Jiang T, Kluger Y. DeepSurv: personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med Res Methodol 2018;18:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haybar H, Rezaeeyan H, Shahjahani M, Shirzad R, Saki N. T-bet transcription factor in cardiovascular disease: Attenuation or inflammation factor? J Cell Physiol 2019;234:7915–22. [DOI] [PubMed] [Google Scholar]
- 102. Di Giovangiulio M, Rizzo A, Franzè E, Caprioli F, Facciotti F, Onali S, et al. Tbet expression in regulatory T cells is required to initiate Th1-mediated colitis. Front Immunol 2019;10:2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang S, Lu Y, Jiang C. Inhibition of histone demethylase JMJD1C attenuates cardiac hypertrophy and fibrosis induced by angiotensin II. J Recept Signal Transduct Res 2020;40:339–47. [DOI] [PubMed] [Google Scholar]
- 104. Choi SH, Ruggiero D, Sorice R, Song C, Nutile T, Vernon Smith A, et al. Six novel loci associated with circulating VEGF levels identified by a meta-analysis of genome-wide association studies. PLoS Genet 2016;12:e1005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Boeckel JN, Guarani V, Koyanagi M, Roexe T, Lengeling A, Schermuly RT, et al. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc Natl Acad Sci U S A 2011;108:3276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhong C, Tao B, Yang F, Xia K, Yang X, Chen L, et al. Histone demethylase JMJD1C promotes the polarization of M1 macrophages to prevent glioma by upregulating miR-302a. Clin Transl Med 2021;11:e424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Park SA, Sung NJ, Choi BJ, Kim W, Kim SH, Surh YJ. Gremlin-1 augments the oestrogen-related receptor α signalling through EGFR activation: implications for the progression of breast cancer. Br J Cancer 2020;123:988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen Y, Zhou Y, Han F, Zhao Y, Tu M, Wang Y, et al. A novel miR-1291-ERRα-CPT1C axis modulates tumor cell proliferation, metabolism and tumorigenesis. Theranostics 2020;10:7193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang S, Huo X. Comprehensive analysis of ESRRA in endometrial cancer. Technol Cancer Res Treat 2021;20:1533033821992083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep 2016;17:684–96. [DOI] [PubMed] [Google Scholar]
- 111. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Koo SJ, Garg NJ. Metabolic programming of macrophage functions and pathogens control. Redox Biol 2019;24:101198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kim SY, Yang CS, Lee HM, Kim JK, Kim YS, Kim YR, et al. ESRRA (estrogen-related receptor α) is a key coordinator of transcriptional and post-translational activation of autophagy to promote innate host defense. Autophagy 2018;14:152–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol 2009;182:3335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014;10:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, et al. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 2015;163:1413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Palmer CS, Anzinger JJ, Butterfield TR, McCune JM, Crowe SM. A simple flow cytometric method to measure glucose uptake and glucose transporter expression for monocyte subpopulations in whole blood. J Vis Exp 2016;114:54255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019;569:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, et al. Coupled scRNA-seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell 2020;182:872–85. [DOI] [PubMed] [Google Scholar]
- 120. Wang S, Zang C, Xiao T, Fan J, Mei S, Qin Q, et al. Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res 2016;26:1417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet 2010;42:355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kuznetsova A, Brockhoff PB, Christensen RHB. lmerTest package: tests in linear mixed effects models. J Stat Softw 2017;82:1–26. [Google Scholar]
- 123. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw 2015;67:1–48. [Google Scholar]
- 124. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med 1990;811–8. [DOI] [PubMed] [Google Scholar]
- 125. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Josse J, Husson F. missMDA: a package for handling missing values in multivariate data analysis. J Stat Softw 2016;70:1–31. [Google Scholar]
- 127. Wang C, Sun D, Huang X, Wan C, Li Z, Han Y, et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol 2020;21:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mei S, Meyer CA, Zheng R, Qin Q, Wu Q, Jiang P, et al. Cistrome Cancer: a web resource for integrative gene regulation modeling in cancer. Cancer Res 2017;77:e19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. He L, Kulminski AM. NEBULA: a fast negative binomial mixed model for differential expression and co-expression analyses of large-scale multi-subject single-cell data. Commun Biol 2021;4:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Brown MB. 400: A method for combining non-independent, one-sided tests of significance. Biometrics 1975;31:987. [Google Scholar]
- 131. Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Love MI, Anders S, Huber W. Analyzing RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Daley TP, Lin Z, Lin X, Liu Y, Wong WH, Qi LS. CRISPhieRmix: a hierarchical mixture model for CRISPR pooled screens. Genome Biol 2018;19:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45:D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015;21:938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lerner SP, McConkey DJ, Hoadley KA, Chan KS, Kim WY, Radvanyi F, et al. Bladder cancer molecular taxonomy: summary from a consensus meeting. Bladder Cancer 2016;2:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chen Z, Huang A, Sun J, Jiang T, Qin FXF, Wu A. Inference of immune cell composition on the expression profiles of mouse tissue. Sci Rep 2017;7:40508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020;583:590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019;20:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Li C, Liu B, Kang B, Liu Z, Liu Y, Chen C, et al. SciBet as a portable and fast single cell type identifier. Nat Commun 2020;11:1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang D, Huo D, Xie H, Wu L, Zhang J, Liu L, et al. CHG: a systematically integrated database of cancer hallmark genes. Front Genet 2020;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1: TFCRs of energy metabolism S2: Bipotent TFCRs S3: Log fold change of T-cell mediated killing upon knockout of OXPHOS genes S4: Log fold change of MHC-I protien expression upon knockout of OXPHOS genes S5: Bipotent TFCRs of angiogenesis S6: Bipotent TFCRs of evasion of growth repressors S7: Datasets included in Methods sections.
The description and validation of the BipotentR immune module
ESRRA inhibition induces antitumor immunity in vivo.
ESRRA inhibition is associated with immune infiltrations and proinflammatory signaling in patient tumors.
ESRRA inhibition correlated with immune infiltrations and proinflammatory signaling in patient tumors.
Robust in vivo antitumor elimination by ESRRA inhibition.
Energy metabolism and immune signaling by targeting ESRRA in vitro.
Energy metabolism and immune signaling by targeting ESRRA in vitro.
The effect of ESRRA inhibition on tumor microenvironments at single-cell resolutions.
ESRRAi does not affect CD8+T T-cells, and ESRRA activity increases post-immunotherapy in resistant tumors.
Machine learning evaluation of immune-metabolic targets in predicting patient response to anti-PD1 monotherapy.
Machine learning evaluation of immune-metabolic targets in predicting patient response to anti-PD1 and anti-CTLA4 combination therapy.
deepBTAS improve the performance of current ICB biomarkers.
Kaplan–Meier plots showing progression-free survival and overall survival differences for patients receiving anti-PD1 between the low-risk and high-risk groups defined by the median value of deepBTAS.
Evaluation of bulk-BipotentR (BipotentR without single-cell sub-module).
Evaluation of scRNA-BipotentR (BipotentR without bulk-tumor sub-module).
Validation of bipotent regulators of angiogenesis or growth-suppressor.
Overall design.
Evaluation of BipotentR.
Expression of markers of M1 and M2 in scRNA data of ESRRAi and Vehicle treated mice.
We detailed a comparison of BipotentR with competing and alternative approaches in Supplementary notes 1, 2, 6, and 7. Supplementary Note 3-5 describes the in vitro findings of ESRRA, the potential clinical relevance of ESRRA across cancer-types, and the association of tumor activity of ESRRA with immune infiltration.
Data Availability Statement
BipotentR is available at http://bipotentr.dfci.harvard.edu/. The R package “BipotentR” is available at (https://github.com/vinash85/TRIM). For reproducibility, data and code are shared publicly at the Zenodo repository that is linked to the BipotentR website at http://bipotentr.dfci.harvard.edu/. The website also provides access to bulk and single RNA-seq data generated from the current study. AI/ML approaches developed from the study are accessible using a publicly available Web browser (https://rconnect.dfci.harvard.edu/BTAS/) and a stand-alone software tool, which users can use to predict patient response to immunotherapy in any input melanoma cohort.