

Abstract
Alcohol abuse and dependence in humans causes an extreme shift in metabolism for which the human brain is not evolutionarily prepared. Oxidation of ethanol and acetaldehyde are not regulated, making ethanol a dominating metabolic substrate that prevents the activity of enzymes from oxidizing their usual endogenous substrates. The enzymes required to oxidize ethanol across the variety of affected tissues all produce acetaldehyde which is then converted to acetate by aldehyde dehydrogenases (ALDHs). ALDHs are NAD+-dependent enzymes, and mitochondrial ALDH2 is likely the primary contributor to ethanol-derived acetaldehyde clearance in cells. Metabolism of alcohol has several adverse effects on mitochondria including increased free radical levels, hyperacetylation of mitochondrial proteins, and excessive mitochondrial fragmentation. This review discusses the role of astrocytic and neuronal mitochondria in ethanol metabolism that contributes to the acute and chronic changes in mitochondrial function and morphology, that might promote tolerance, dependence and withdrawal. We also propose potential modes of therapeutic intervention to reduce the toxicity of chronic alcohol consumption.
Keywords: ethanol, brain, mitochondria
INTRODUCTION
Prolonged and excessive alcohol consumption leads to wide-ranging and diverse effects of ethanol on the central nervous system and contributes to the development of brain pathology. The toxic effects of ethanol and its downstream metabolites are associated with cancer, cardiovascular problems, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease [1–4]. Ethanol readily passes through biological membranes and distributes throughout the body into all organs including brain [5]. Since during the evolution of human species there was little alcohol in consumed food, mechanisms for regulation of ethanol metabolism did not evolve, leaving both adult and developing humans vulnerable to ethanol toxicity [5]. Ethanol dependence is now a significant problem leading to pathologic consequences, causing significant medical, social, and economic burdens [3, 4]. Both acute and chronic alcohol exposure produce molecular and cellular neuroadaptations that modulate the activity of discrete brain regions and cell types [3, 6, 7].
After consumption, the elimination of alcohol from blood occurs at a constant rate of about 0.016 g/dl/h for men and slightly higher 0.018 g/dl/h for women [8]. The removal of ethanol by the human body, consumed during social drinking (about 0.9 g/kg of body weight), takes 5 to 7 hours and throughout this time ethanol oxidation is the largest carbon source for energy metabolism [9, 10]. Further intake of ethanol extends the time of increased alcohol levels in blood, but the rate of its elimination does not change. Thus, in individuals consuming alcohol several times a day the blood ethanol levels remain significantly high most of the time, considerably affecting brain energy metabolism, and triggering epigenetic changes associated with development of alcohol use disorder, or addiction.
Enzymes involved in alcohol and acetaldehyde metabolism
Since ethanol readily passes through biological membranes, the concentration in brain tissue approximates that of blood [5]. Oxidation of ethanol in brain is carried out by three enzymes, alcohol dehydrogenase (ADH), catalase, and cytochrome P540 2E1 (CYP2E1) (for review see [9]). However, ethanol metabolism by CYP2E1 and catalase makes only minor contributions [11]. All three enzymes produce acetaldehyde (Figure 1). Conversion of ethanol to acetaldehyde by ADH also reduces NAD+ to NADH. Catalase generates acetaldehyde from ethanol in the presence of hydrogen peroxide, and CYP2E1 requires oxygen and NADPH to convert ethanol to acetaldehyde plus NADP+ and acetate.
Figure 1.
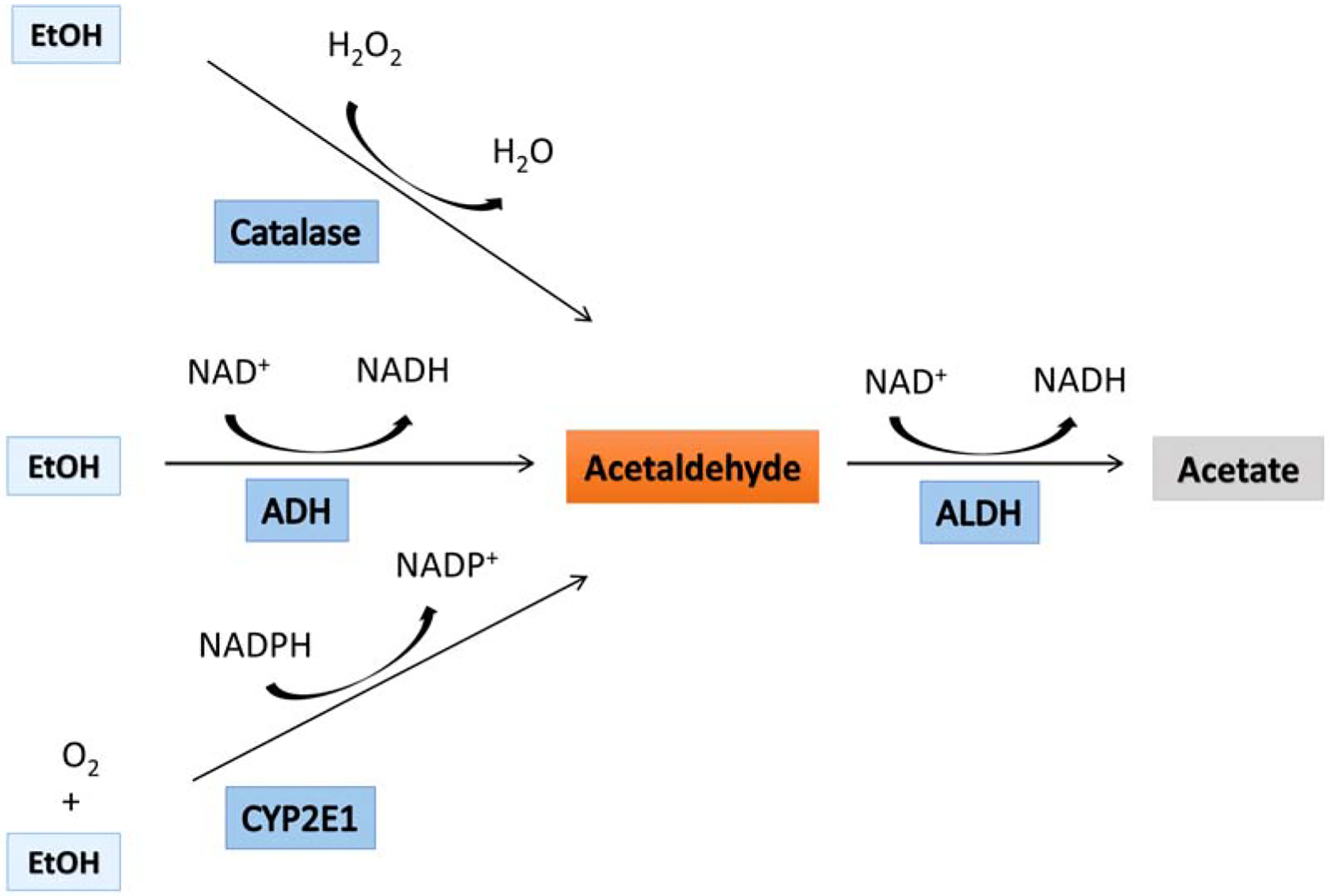
Ethanol-oxidizing enzymes. There are three enzymes that can oxidize ethanol in cells. All three enzymes convert ethanol to acetaldehyde. Alcohol dehydrogenase (ADH) during ethanol oxidation is reducing NAD+ to NADH. Catalase requires a presence of hydrogen peroxide to oxidize ethanol, and Cytochrome p450 2E1 (CYP2E1) uses oxygen and NADPH to convert ethanol to acetaldehyde, and NADP+. Acetaldehyde is oxidized by aldehyde dehydrogenase (ALDH) to acetate with concomitant generation of NADH.
Acetaldehyde can react nonenzymatically with other cellular components leading to cytotoxic effects with pathologic consequences for cell function and survival [12]. The concentration of acetaldehyde is kept low by the activity of aldehyde dehydrogenase (ALDH) that is widely distributed in the brain tissue [13, 14]. ALDH oxidizes acetaldehyde to acetate and during this process NAD+ is reduced to NADH. The oxidation of acetaldehyde is extremely efficient keeping the circulating levels of acetaldehyde more than thousand-fold less than ethanol levels in the blood [15, 16]. Furthermore, acetaldehyde does not penetrate the blood vessels into the brain due to the presence of ALDH in the blood-brain barrier [17, 18].
Most of the consumed alcohol is oxidized in liver by alcohol dehydrogenases and then by aldehyde dehydrogenase enzymes. However, the elevated blood alcohol levels during liver-facilitated oxidation will also keep the brain ethanol levels elevated ensuring an effect on brain metabolism and functions. It should be noted that ethanol per se has specific binding sites on several proteins, including neurotransmitter receptors, channel proteins and enzymes that may act directly or indirectly to produce biological effects [3].
In this review we will discuss acute brain ethanol oxidation, its impact on enzymes oxidizing ethanol, on mitochondrial functions, and protein posttranslational modifications.
Initial step in brain alcohol oxidation
There are six main classes of the ADH enzyme, and their expression levels are tissue specific [19]. Originally, it was suggested that only the subtype ADH3 was expressed in the brain [20–22]. However, its contribution to alcohol oxidation was questioned due to very high Km values (about 15 mM) for ethanol [23]. Another group demonstrated the presence of mRNA for ADH1 and ADH4 in rat brain cells [24]. Interestingly, the ADH activity was localized only in specific neurons of cerebral cortex, pyramidal and granule cells of hippocampus, hypothalamus, and cerebellum (granule cells and Purkinje cells), whereas no significant ADH activity was found in whole brain homogenate [24–27]. This suggests that only specific neurons within the brain can significantly contribute to initial ethanol oxidation via ADH activity.
Catalase probably also contributes to conversion of ethanol to acetaldehyde in brain [28]. The finding that inhibitors of catalase were effective in inhibiting the production of acetaldehyde from ethanol in rat brain tissue supports this role [13]. Furthermore, inhibitors of the CYP2E1 or ADH had no significant effect on production of acetaldehyde in experiments using brain homogenate. However, the role of catalase in systemic alcohol metabolism has not been clarified in vivo [29], and the oxidation of ethanol by catalase is also limited by the availability of hydrogen peroxide that is required for the ethanol metabolism.
It should be noted that the endogenous function of ethanol-metabolizing enzymes is not linked directly to ethanol oxidation but rather ADH participates in brain retinol and serotonin metabolism [30, 31], and catalase has an important role in cellular antioxidant defense systems as it detoxifies hydrogen peroxide. Furthermore, ADH3 was suggested to contribute to the defense of the brain against degenerative processes since it is considered a glutathione-dependent formaldehyde dehydrogenase [32], and ADH3 partially regulates nitrosothiol homeostasis by catalyzing the reduction of the endogenous nitrosylating agent S-nitrosoglutathione [33]. The mechanisms that might regulate ADH are not known. However, recently ADP-ribosylation was suggested to be a posttranslational modification affecting the enzymatic activity of ADH [34].
Cytochrome P450 enzymes oxidize ethanol in liver, and have also been implicated in ethanol metabolism in the brain, particularly the isoenzyme CYP2E1 [35]. This isoform of cytochrome P450 was found in both neurons and astrocytes within cerebellum, cerebral cortex, thalamus and hippocampus [35–37]. The biological function of CYP2E1 is to metabolize xenobiotics and endogenous compounds including acetate, fatty acids, and bile acids [38, 39]. Therefore, ethanol intake will lead to competitive inhibition of CYP2E1 activity and reduce the protective oxidation of xenobiotics that can have serious consequences. However, since this enzyme is expressed mostly in liver the ethanol-induced pathologic effect is observed preferentially in this organ [40].
The experiments with catalase, CYP2E1, ADH and ALDH inhibitors in combination with transgenic animal models with a deficiency in ethanol-metabolizing enzymes (catalase and CYP2E1) suggest that the key ethanol-oxidizing enzyme in brain is catalase, which is responsible for about 80% of the ethanol-metabolizing activity, and CYP2E1 which metabolizes about 20% of ethanol [13]. Follow-up studies confirmed that the majority of alcohol oxidation in brain occurs via catalase [41, 42].
Detoxification of acetaldehyde derived from ethanol oxidation
Although, ethanol metabolism and its regulation in brain remains somewhat controversial, the enzymes that process the acetaldehyde derived from ethanol in brain cells have been long known [43, 44]. Aldehyde dehydrogenases (ALDHs) represent a large family of NAD+-dependent dehydrogenases responsible for metabolism of endogenous and exogenous aldehydes produced under many different pathophysiological states or exposure to toxic agents including alcohol. ALDH1 is also essential for formation of retinoic acid from retinal, the oxidation product of retinol (Vitamin A) [45]. Exposure to toxins is usually associated with increased oxidative stress leading to lipid peroxidation and elevated levels of lipid peroxides such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) that are detoxified by ALDH [46]. These aldehydes are highly reactive and lead to modification of cellular macromolecules including DNA and proteins [47, 48].
There are at least 19 ALDH isozymes that have different kinetic parameters including distinct Km values and catalytic activities [49, 50]. Human cytosolic ALDH1A1 isozyme exhibits a high Km value, about 180 mM for acetaldehyde. However, acetaldehyde can hardly be the natural substrate for human cytosolic ALDH1, because the physiological concentrations of acetaldehyde are typically around 2 mM [51, 52]. However, the higher affinity mitochondrial ALDH2 has a Km for acetaldehyde 0.2 mM, a 900-fold lower value when compared to cytosolic ALDH1 [53]. Therefore, the action of the mitochondrial ALDH2 is widely believed to be the major contributor to the clearance of ethanol-derived acetaldehyde in cells [54–56]. This suggests compartmentation of ethanol disposal since most of the ethanol in the brain is oxidized to acetaldehyde by catalase, an enzyme that is localized in microsomes and peroxisomes. To prevent the acetaldehyde accumulation, the acetaldehyde produced needs to be taken up into the mitochondrial matrix for oxidation to acetate by ALDH2. This compartmentation also suggests that after ethanol consumption there are elevated levels of acetaldehyde in microsomes and peroxisomes that show high catalase activity, and in the cytosol compared to mitochondria [13], rendering the cytosolic and nuclear macromolecules vulnerable as the primary targets of acetaldehyde toxicity.
The catalytic activity of ALDH isozymes is modified by several post-translational modifications (for review see [57]). Alcohol consumption induces oxidative modifications of cysteine residue (Cys302) at the active site of ALDH1 and ALDH2 [58], leading to inhibition of their activity [59–61]. Other studies show that S-nitrosylated ALDH2 found in alcohol-exposed animals also has lower activity [59, 62]. Similarly, phosphorylation of ALDH2 by JNK kinase at Ser463 residue results in lower activity of this enzyme [63], thereby decreasing cellular defense capacity against aldehyde toxicity. Suppression of ALDH2 activity triggers an increase in reactive lipid peroxide levels and the subsequent accumulation of these toxic molecules will damage cellular macromolecules including microsomal and mitochondrial proteins, causing mitochondrial dysfunction and eventually cell death [48, 64–66].
In contrast to JNK-dependent phosphorylation, protein kinase Cε (PKCε)-mediated phosphorylation of ALDH2 at Thr185, Thr412 or Ser279 leads to stimulation of ALDH2 activity and has protective effects against ischemia-induced pathology in heart [67–69]. Furthermore, phosphorylation by inositol-3-kinase also leads to stimulation of ALDH2 [70].
Since ALDH2 is a mitochondrial protein, the inhibition of this enzyme can have detrimental effects, leading to the intramitochondrial accumulation of toxic aldehydes that consequently trigger mitochondrial dysfunction and compromise cell survival.
Acetylation, mitochondria and brain ethanol metabolism
Acetylation is one of the most common post-translational modifications of proteins, and changes in the acetylation levels are linked mainly to regulation of cellular metabolism [71, 72]. In particular, acetylation has significant impact on the function of mitochondrial enzymes. The end product of ethanol metabolism is acetate generated by ALDH from acetaldehyde. Acetate is an energy substrate that can be converted to acetyl-CoA by Acetyl-CoA synthetase in the presence of coenzyme A (CoA) and ATP. Acetyl-CoA in mitochondria can enter the TCA cycle where it condenses with oxaloacetate to form citrate which is oxidized in the cycle for energy. Alternatively, mitochondrial acetyl-CoA can serve as a substrate for the acetyl transferase enzyme (KAT) to acetylate lysine residues on target proteins (Figure 2). Therefore, it is not surprising that many cytosolic and mitochondrial proteins including cytosolic ALDH1A1 and mitochondrial ALDH2 are hyper-acetylated in alcohol-exposed animals [73]. Hyperacetylation of ALDH2 leads to increased activity, and therefore facilitates the detoxification of acetaldehyde or other toxins [74–76].
Figure 2.
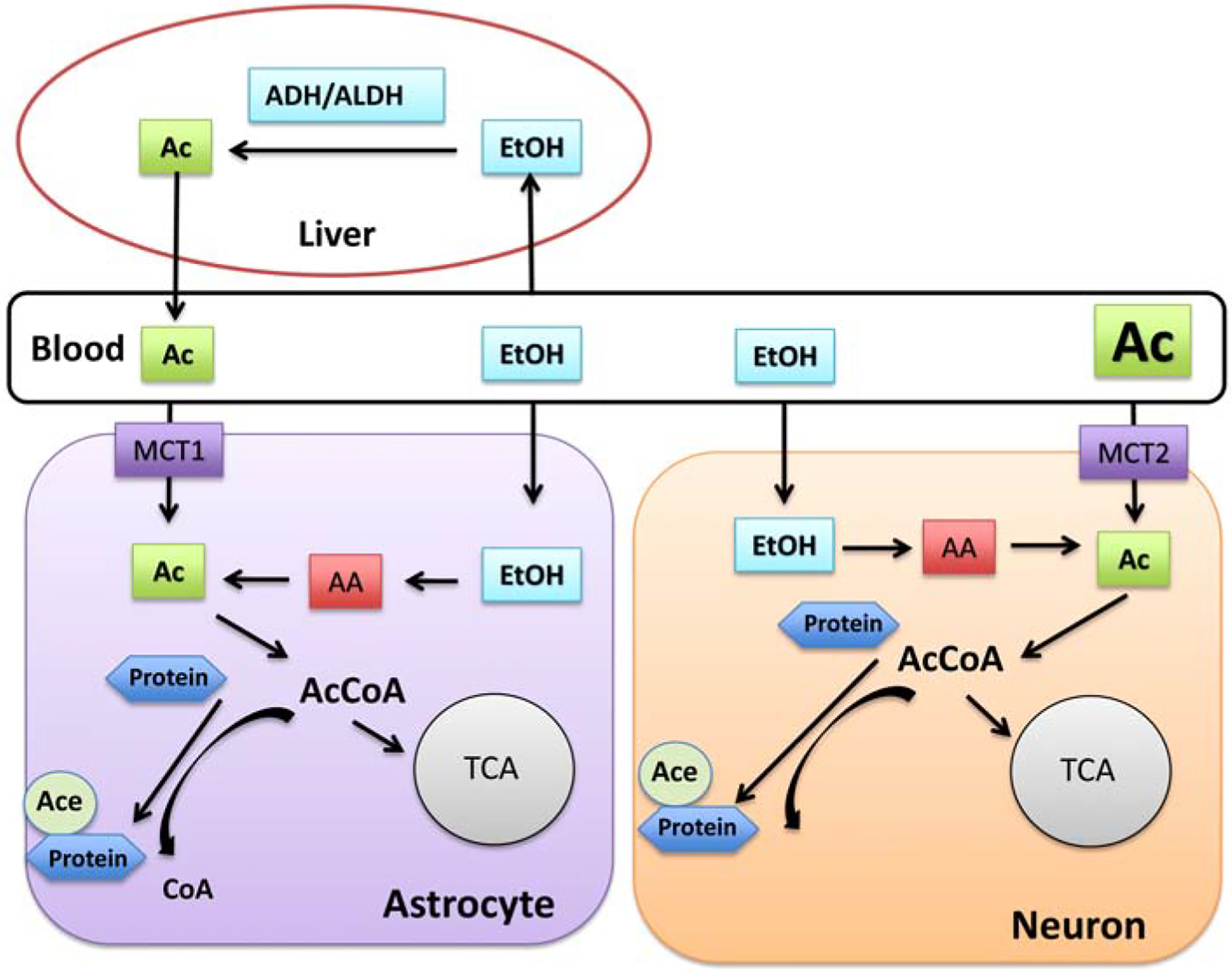
Effects of ethanol oxidation on cellular metabolism in brain. Since ethanol can pass cellular membranes, it enters both liver and brain cells where it is converted to acetate. Acetate generated by liver via reactions of alcohol dehydrogenase (ALD) and acetaldehyde dehydrogenase (ALDH) enters the blood and it is transported into brain cells by monocarboxylase transporters (MCT1 and MCT2). The high capacity, low affinity MCT1 transporter is expressed on astrocytes and the low capacity, higher affinity transporter MCT2 is localized on neurons. Acetate after entering the cytoplasm is a substrate for acetyl-CoA synthetases that form Acetyl-CoA (AcCoA). In cells AcCoA is serving as a substrate for lipid and sterols metabolism and is required by acetyl transferases to acetylate histone and non-histone proteins. AcCoA formed in mitochondria can either enter mitochondrial TCA cycle or can be used for acetylation of mitochondrial proteins.
Effect of ethanol on cellular bioenergetics
Ethanol oxidation has a significant impact on cellular bioenergetic metabolism since it leads to an unregulated increase in intramitochondrial and cytosolic NADH [77]. Furthermore, the downstream acetyl-CoA generated from ethanol-derived acetate can lead to hyperacetylation of intracellular proteins including mitochondrial proteins [9, 78]. There are two sources of acetate that supply acetate to brain cells after ethanol intake. One is the acetate released from liver into the blood and then transported into brain and astrocytes via the high capacity, low affinity monocarboxylate transporter (MCT1) with reported Km values between 1.6–9.3 mM, which is expressed mostly in astrocytes [79, 80]. The neuronal, higher affinity, lower capacity monocarboxylate transporter (MCT2; Km value 2.6 mM) also transports acetate (Figure 2); however, at high concentrations this system would be saturated allowing for more uptake into astrocytes [80]. The other source of acetate is intramitochondrial production. In mitochondria, acetate is formed by mitochondrial ALDH2 from acetaldehyde (see above). Both cytosolic and mitochondrial acetate serve as a substrate for Acetyl-CoA synthetase 1 and 2 (AceCS1 and AceCS2) leading to production of acetyl-CoA [72, 80–82]. Interestingly, acetate from both sources will end up mostly in astrocytes since MCT1, which has a high capacity for uptake [83] is expressed preferentially in astrocytes and ALDH2 is expressed in astrocytic mitochondria [84]. Thus, one could expect that a significant hyperacetylation of astrocytic proteins will be detected after ethanol consumption [85]. Furthermore, due to localization of AceCS1 in cytosol and nucleus with its attachment to the chromatin, AceCS1 will be able to directly regulate histone acetylation [86, 82]. The mitochondrial AceCS2 enzyme apart from generating Acetyl-CoA for the TCA cycle, will also support the acetylation of mitochondrial proteins [87, 88] by supplying acetyl-CoA to mitochondrial acetyl transferase GCN5L1 [89]. Additionally, both AceCS isoforms are found in neurons and astrocytes, and are subjected to post-translational regulation via acetylation under control of NAD+-dependent deacetylases, Sirtuins (SIRTs). AceCS1 is deacetylated by cytosolic SIRT1 and AcesCS2 is deacetylated by mitochondrial SIRT3 [90].
Ethanol metabolism-driven increases in redox pressure reflected in an elevated NADH/NAD+ ratio will increase superoxide production by respiratory complexes [91] and reduce the activity of mitochondrial NAD+-dependent deacetylase SIRT3 due to low NAD+ levels [92–95]. Thus, the hyperacetylation of mitochondrial proteins will be further accelerated due to SIRT3 inhibition. As a result, the activity of the pyruvate dehydrogenase complex, Electron Transport Chain respiratory complex I, and TCA cycle enzymes including citrate synthase, isocitrate dehydrogenase 2, α-ketoglutarate dehydrogenase and malate dehydrogenase will be reduced [92, 96, 97].
One can envisage that due to differential capacity of MCT1 and MCT2 for acetate uptake, moderate drinking will cause acetylation changes preferentially in astrocytes. However, heavy drinkers or repeated intake of ethanol will lead to profound changes in neuronal protein acetylation, alterations in their mitochondrial and cellular metabolism with subsequent epigenetic effects leading to alcohol use disorder and addiction.
Ethanol metabolism-driven mitochondrial free radical generation
It is generally accepted that ethanol metabolism increases free radical production that contributes to its pathophysiological effects [98, 99]. However, the initial cause of higher reactive oxygen species (ROS) generation due to ethanol consumption is not completely understood. Mitochondria are a main source of free radicals in the cell and possess an active antioxidant system comprised of enzymes that detoxify free radicals [100]. Superoxide, mostly generated by the mitochondrial respiratory complexes [101, 102], is converted to hydrogen peroxide by mitochondrial superoxide dismutase 2 (SOD2) [100, 103]. In the next step, hydrogen peroxide is then detoxified by glutathione peroxidase to water. The mitochondrial ROS levels are determined by the rates of superoxide production and of its detoxification. Ethanol consumption results in hyperacetylation of mitochondrial proteins that has an inhibitory effect on the activity of some of the detoxification enzymes [92, 96, 97]. The hyperacetylation of SOD2 leads to increased levels of ROS due to reduced rate of superoxide detoxification by this enzyme [104, 105]. The ethanol-induced high ROS levels further impair the mitochondrial antioxidant mechanisms by decreasing the mitochondrial glutathione content [106, 107]. The significance of mitochondria-generated ROS as a main source of free radicals after ethanol administration was confirmed by the protective effects of the mitochondria-targeted antioxidant, lipophilic ubiquinone (MitoQ), which ameliorated alcohol-induced oxidative stress, reversed the ethanol-induced inhibition of ALDH2 activity and increased acetaldehyde clearance [108]. Furthermore, ROS generated during EtOH metabolism in mitochondria will inhibit the activity of aconitase, an enzyme which is downstream of citrate leading to intramitochondrial accumulation and transport of citrate from the mitochondrial matrix into the cytosol. The increase in cytosolic citrate will stimulate generation of cytosolic acetyl-CoA by ATP citrate lyase (ACLY) and further contribute to hyperacetylation of cytosolic proteins and histones [109]. ACLY is mostly localized to neurons [110] and is stabilized by acetylation due to blocking of acetylated ACLY ubiquitylation and degradation [111]. Thus, ethanol-induced changes in mitochondrial TCA cycle metabolism can indirectly modulate histone acetylation and alter gene expression via ACLY activity.
Effects of ethanol on mitochondrial dynamics
Mitochondrial response to stress is reflected not only in modification of their function but also in their structure and morphology. To efficiently respond to cellular metabolic demands, mitochondria possess the unique capability as an organelle that can go through cycles of fission and fusion [112–114]. This process of dynamic change in the mitochondrial morphology is controlled by several proteins, the activities of which are regulated by post-translational modifications (Figure 3) [115]. Fission and fragmentation of mitochondria are triggered by various stress conditions including elevated free radical levels [115, 116]. Excessive mitochondrial fission is associated with both acute brain injury and neurodegenerative diseases [117–120]. The main fission controlling enzyme, dynamin-related protein (Drp1), is activated by several post-translational modifications [115, 121–123]. The S-nitrosylation, phosphorylation at Ser616, SUMOylation, and acetylation increase the translocation of Drp1 to the mitochondria and activate the fission process [115]. In vitro studies showed that ethanol treatment increased mitochondrial fission and this process was inhibited by the mitochondria-targeted antioxidant MitoQ [124]. Similarly, ethanol treatment of neuroblastoma cells caused an increase in ROS levels and phosphorylation of Drp1 by activation of calmodulin-dependent protein kinase 2 (Camk2) which led to increased mitochondrial fragmentation [125]. Pretreatment of cells with antioxidants or using KN-93 a Camk2 inhibitor attenuated Drp1 phosphorylation and subsequent mitochondrial fission [125]. In addition, acetylation of Drp1 at lysine 642 (K642) was associated with Drp1 phosphorylation at Ser616, and its translocation to mitochondria followed by mitochondrial fission [122]. Acetaldehyde’s toxicity to neuronal cell lines is accompanied by excessive mitophagy and a drastic reduction in mitochondrial mass [126]. Mitophagy was attenuated by treatment with N-acetyl-L-cysteine, highlighting the oxidative stress induced by ethanol [126].
Figure 3.
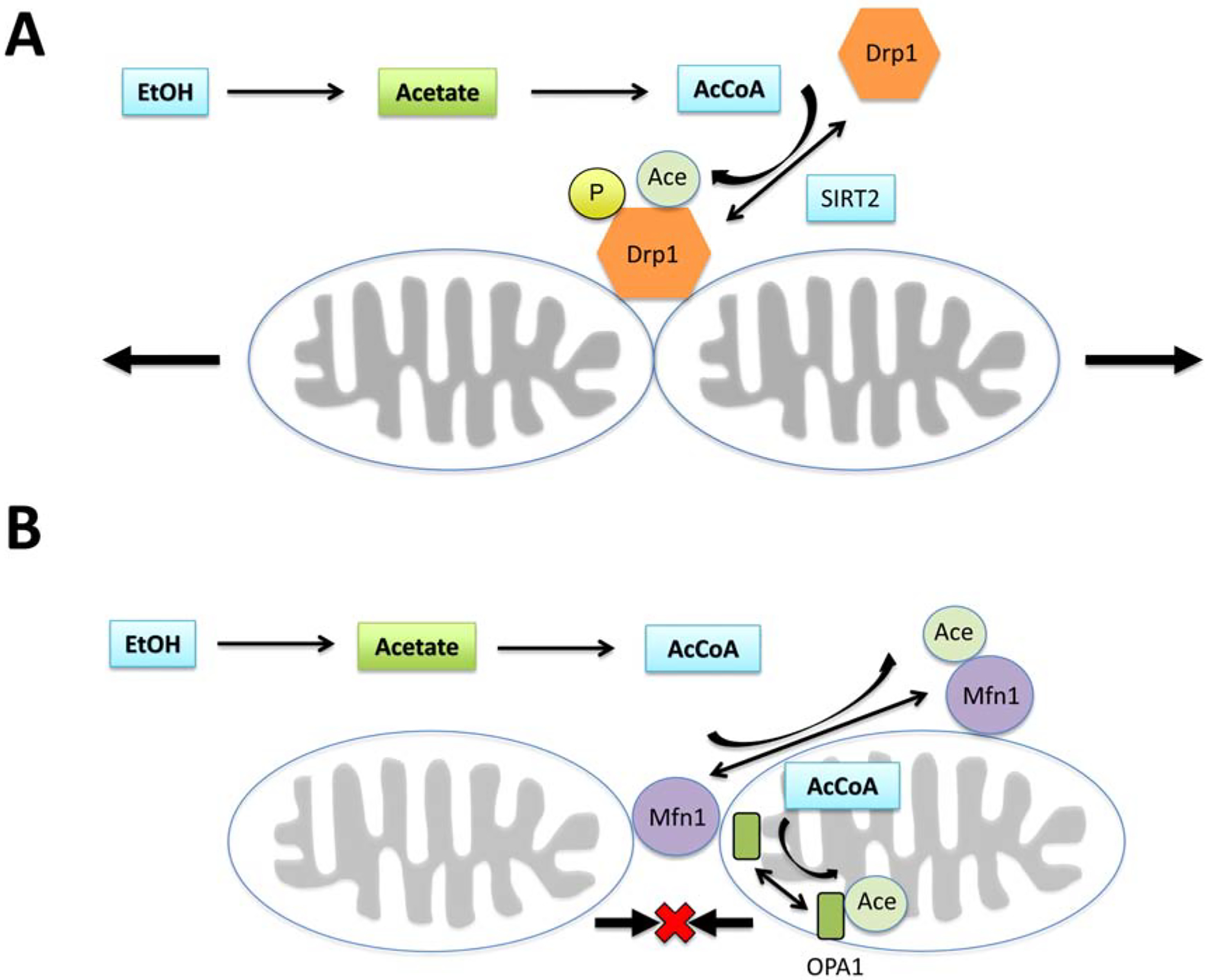
Modulation of mitochondrial dynamics by ethanol oxidation. Activity of both fission and fusion-controlling proteins are altered by acetylation. (A) Elevated levels of AcCoA due to ethanol metabolism can lead to acetylation of Drp1 and its translocation to mitochondrial membrane and initiation of the fission process. Similarly, acetylation of Mfn1 and Opa1 will inhibit fusion, thus maintaining the mitochondria in fragmented state.
The opposite process to fragmentation is a mechanism that carries out fusion of the small organelles into larger mitochondrion [115, 127, 128]. The process of fusion is controlled by the intramitochondrial optic atrophy 1 protein (OPA1) enzyme, responsible for inner membrane fusion and mitofusin1 and 2 (Mfn1, Mfn2) that regulate the fusion of the mitochondrial outer membrane [127]. Similar to the enzymes regulating the fission process, the activity of fusion-controlling proteins is also modulated by post-translational modifications. Hyperacetylation of OPA1 reduces its activity, thus inhibiting the fusion and subsequently leading to increased mitochondrial fragmentation [129]. Hyperacetylation of Mfn1 will also cause inhibition of fusion, as reflected in high levels of small mitochondrial organelles [130].
Apart from stress or pathologic conditions, mitochondria fragmentation is also initiated by energy requirements in distal part of the cells where smaller mitochondria organelles are required to move to provide energy [131]. A recent study demonstrated that changes in spine morphology and the trafficking of mitochondria to synapses is a critical step in the adverse effects of exposure to ethanol. Mitochondrial trafficking was also determined as necessary for acquisition of alcohol-induced place preference [132]. Thus, changes in mitochondrial dynamics in response to ethanol are similar to those observed in memory formation, where they fuel the synapse to support translation [131].
Alcohol metabolism has several adverse effects on mitochondria including an increase in free radical levels, hyperacetylation of mitochondrial proteins, and excessive fragmentation with pathologic dysregulation of oxidative energy metabolism. Since activation of Sirtuins can reverse these adverse effects, a therapeutic approach that will increase cellular and mitochondrial NAD+ levels and activate Sirtuins could be beneficial in preventing posttranslational changes leading to addiction or associated with ethanol-induced damage. This notion is supported by the data showing that animal exposure to running wheels prevents ethanol rewarding effects via activation of SIRT1 and SIRT2 [133].
CONCLUSION
Ethanol metabolism represents a complex process that includes several downstream pathways and has tissue-type specific characteristics. Generally, the normal functions of the enzymes that oxidize alcohol are not to process ethanol, but rather they are part of specific metabolic pathways in cells that are not directly related to ethanol oxidation. Since there was no evolutionary pressure for developing ethanol-specific oxidizing enzymes in humans, there are no intrinsic control mechanisms that could reduce the toxic and adverse effects of increased ethanol consumption.
Although most of the ethanol oxidation is carried out in the liver, the increased blood levels of acetate and elevated blood ethanol concentrations have significant impact on brain metabolism and the functions of specific neuronal subpopulations leading to alcohol use disorder and alcohol addiction. There are several adverse effects induced by ethanol metabolism in brain cells. First, the function of enzymes participating in ethanol oxidation will be inhibited leading to perturbed metabolism of their endogenous substrates. Second, the intracellular redox change due to an increased ratio of NADH to NAD+ will contribute to the increased generation of superoxide by the mitochondrial respiratory complexes and alter the metabolism of carbohydrates in cytosol and mitochondria in brain. Third, the rise in production of acetyl-CoA due to uptake of acetate from blood and the acetate intracellularly generated from ethanol oxidation will lead to hyperacetylation of cellular proteins, and modification of histones, which can lead to epigenetic consequences.
Since alcohol addiction and alcohol-triggered brain pathology is tightly associated with alterations of functions in specific neuronal subpopulations, most research in this field has focused on adverse effects of ethanol consumption on these specific cells/brain regions. However, considering that the acetate generated from ethanol oxidation in liver enters brain and is taken up by astrocytes and converted to acetaldehyde by the enzyme ALDH2, which is selectively expressed in astrocytic mitochondria, more research should be focused on the role of astrocytes and their mitochondria in processes leading to addiction and alcohol use pathology.
ACKNOWLEDGEMENT
This research was funded by NIH NINDS R01NS122777 to J. W., NIH NINDS R01NS119275 and Veteran’s Affairs Merit Review Award BX004895 to T. K., and NIH NICHD P01 HD085928 to M. C. M.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
REFERENCES
- 1.Yu HS, Oyama T, Isse T, Kitagawa K, Pham TT, Tanaka M and Kawamoto T 2010, Chem. Biol. Interact, 188, 367. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Lin LL, Guo JM, Cheng YQ, Qian J, Mehta JL, Su DF, Luan P and Liu AJ 2015, Int. J. Stroke, 10, 1261. [DOI] [PubMed] [Google Scholar]
- 3.Abrahao KP, Salinas AG and Lovinger DM 2017, Neuron, 96, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamal H, Tan GC, Ibrahim SF, Shaikh MF, Mohamed IN, Mohamed RMP, Hamid AA, Ugusman A and Kumar J 2020, Front Cell Neurosci, 14, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egervari G, Siciliano CA, Whiteley EL and Ron D 2021, Trends Neurosci, 44, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandey SC, Kyzar EJ and Zhang H 2017, Neuropharmacology, 122, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ron D and Barak S 2016, Neuroscience, 17, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dettling A, Fischer F, Bohler S, Ulrichs F, Skopp G, Graw M and Haffner HT 2007, Alcohol, 41, 415. [DOI] [PubMed] [Google Scholar]
- 9.Wilson DF and Matschinsky FM 2020, Med. Hypotheses, 140, 109638. [DOI] [PubMed] [Google Scholar]
- 10.Volkow ND, Kim SW, Wang GJ, Alexoff D, Logan J, Muench L, Shea C, Telang F, Fowler JS, Wong C, Benveniste H and Tomasi D 2013, Neuroimage, 64, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurley TD The search for individualized therapies. The pharmocogenomics of alcpholism, Licinio J and Wong Ma-Li (Eds.), Weinheim, Germany, 2002; pp. 417. [Google Scholar]
- 12.Zakhari S 2006, Alcohol Res. Health, 29, 245. [PMC free article] [PubMed] [Google Scholar]
- 13.Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ and Deitrich RA 2006, Alcohol Clin. Exp. Res, 30, 1500. [DOI] [PubMed] [Google Scholar]
- 14.Zimatkin SM, Rout UK, Koivusalo M, Buhler R and Lindros KO 1992, Alcohol Clin. Exp. Res, 16, 1162. [DOI] [PubMed] [Google Scholar]
- 15.Harada S, Agarwal DP, Goedde HW and Takagi S 1983, Pharmacology, Biochemistry, and Behavior, 18(Suppl. 1), 139. [DOI] [PubMed] [Google Scholar]
- 16.Mizoi Y, Yamamoto K, Ueno Y, Fukunaga T and Harada S 1994, Alcohol Alcohol 29, 707. [PubMed] [Google Scholar]
- 17.Petersen DR 1985, Alcohol, 2, 79. [DOI] [PubMed] [Google Scholar]
- 18.Tampier L, Cariz S and Quintanilla ME 1993, Alcohol, 10, 203. [DOI] [PubMed] [Google Scholar]
- 19.Edenberg HJ and McClintick JN 2018, Alcohol Clin. Exp. Res, 42, 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beisswenger TB, Holmquist B and Vallee BL 1985, Proc. Natl. Acad. Sci. USA, 82, 8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerr JT, Maxwell DS and Crabb DW 1989, Alcohol Clin. Exp. Res, 13, 730. [DOI] [PubMed] [Google Scholar]
- 22.Giri PR, Linnoila M, O’Neill JB and Goldman D 1989, Brain Res, 481, 131. [DOI] [PubMed] [Google Scholar]
- 23.Pares X and Vallee BL 1981, Biochem. Biophys. Res. Commun, 98, 122. [DOI] [PubMed] [Google Scholar]
- 24.Martinez SE, Vaglenova J, Sabria J, Martinez MC, Farres J and Pares X 2001, Eur. J. Biochem, 268, 5045. [PubMed] [Google Scholar]
- 25.Galter D, Carmine A, Buervenich S, Duester G and Olson L 2003, Eur. J. Biochem, 270, 1316. [DOI] [PubMed] [Google Scholar]
- 26.Buhler R, Pestalozzi D, Hess M and Von Wartburg JP 1983, Pharmacology, Biochemistry, and Behavior, 18(Suppl. 1), 55. [DOI] [PubMed] [Google Scholar]
- 27.Zimatkin SM and Deitrich RA 1997, Addict. Biol, 2, 387. [DOI] [PubMed] [Google Scholar]
- 28.Gill K, Menez JF, Lucas D and Deitrich RA 1992, Alcohol Clin. Exp. Res, 16, 910. [DOI] [PubMed] [Google Scholar]
- 29.Weiner ML, Freeman C, Trochimowicz H, de Gerlache J, Jacobi S, Malinverno G, Mayr W and Regnier JF 2000, Food Chem. Toxicol, 38, 607. [DOI] [PubMed] [Google Scholar]
- 30.Svensson S, Some M, Lundsjo A, Helander A, Cronholm T and Hoog JO 1999, Eur. J. Biochem, 262, 324. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi J, Palmer TD and Gage FH 1999, J. Neurobiol, 38, 65. [PubMed] [Google Scholar]
- 32.Danielsson O and Jornvall H 1992, Proc. Natl. Acad. Sci. USA, 89, 9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson CM, Ceder R and Grafstrom RC 2010, Toxicol. Lett, 193, 1. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita S, Tanaka M, Nodono H, Hamada A, Hamada T, Hasegawa M, Nishi Y, Moss J and Miwa M 2019, Biochem. Pharmacol, 167, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansson T, Tindberg N, Ingelman-Sundberg M and Kohler C 1990, Neuroscience, 34, 451. [DOI] [PubMed] [Google Scholar]
- 36.Sohda T, Shimizu M, Kamimura S and Okumura M 1993, Alcohol Alcohol, Suppl 1B, 69. [PubMed] [Google Scholar]
- 37.Upadhya SC, Tirumalai PS, Boyd MR, Mori T and Ravindranath V 2000, Arch. Biochem. Biophys, 373, 23. [DOI] [PubMed] [Google Scholar]
- 38.Lu D, Ma Y, Zhang W, Bao D, Dong W, Lian H, Huang L and Zhang L 2012, Hypertension, 60, 81. [DOI] [PubMed] [Google Scholar]
- 39.Xu J, Ma HY, Liang S, Sun M, Karin G, Koyama Y, Hu R, Quehenberger O, Davidson NO, Dennis EA, Kisseleva T and Brenner DA 2017, Hepatol. Commun, 1, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang HF, Wang HH, Gao N, Wei JY, Tian X, Zhao Y, Fang Y, Zhou J, Wen Q, Gao J, Zhang YJ, Qian XH and Qiao HL 2016, The Journal of Pharmacology and Experimental Therapeutics, 358, 83. [DOI] [PubMed] [Google Scholar]
- 41.Jamal M, Ameno K, Uekita I, Kumihashi M, Wang W and Ijiri I 2007, Neurotoxicology, 28, 1245. [DOI] [PubMed] [Google Scholar]
- 42.Correa M, Manrique HM, Font L, Escrig MA and Aragon CM 2008, Psychopharmacology (Berl.), 200, 455. [DOI] [PubMed] [Google Scholar]
- 43.Deitrich RA 1966, Biochem. Pharmacol, 15, 1911. [DOI] [PubMed] [Google Scholar]
- 44.Erwin VG and Deitrich RA 1966, J. Biol. Chem, 241, 3533. [PubMed] [Google Scholar]
- 45.Duester G 2008, Cell, 134, 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deza-Ponzio R, Herrera ML, Bellini MJ, Virgolini MB and Herenu CB 2018, Neurotoxicology, 68, 19. [DOI] [PubMed] [Google Scholar]
- 47.Esterbauer H, Schaur RJ and Zollner H 1991, Free Radic. Biol. Med, 11, 81. [DOI] [PubMed] [Google Scholar]
- 48.LoPachin RM, Gavin T, Petersen DR and Barber DS 2009, Chem. Res. Toxicol, 22, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sladek NE 2003, J. Biochem. Mol. Toxicol, 17, 7. [DOI] [PubMed] [Google Scholar]
- 50.Marchitti SA, Brocker C, Stagos D and Vasiliou V 2008, Expert Opin. Drug Metab. Toxicol, 4, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harada S, Agarwal DP and Goedde HW 1981, Lancet, 2, 982. [DOI] [PubMed] [Google Scholar]
- 52.Hatake K, Taniguchi T, Ouchi H, Sakaki N, Hishida S and Ijiri I 1990, Pharmacology, Biochemistry, and Behavior, 35, 437. [DOI] [PubMed] [Google Scholar]
- 53.Klyosov AA 1996, Biochemistry, 35, 4457. [DOI] [PubMed] [Google Scholar]
- 54.Smolen TN, Smolen A and van de Kamp JL 1990, Alcohol, 7, 69. [DOI] [PubMed] [Google Scholar]
- 55.Huang M and Lindahl R 1990, Arch. Biochem. Biophys, 277, 296. [DOI] [PubMed] [Google Scholar]
- 56.Mitchell DY and Petersen DR 1991, Hepatology 13, 728. [DOI] [PubMed] [Google Scholar]
- 57.Song BJ, Abdelmegeed MA, Yoo SH, Kim BJ, Jo SA, Jo I and Moon KH 2011, J. Proteomics, 74, 2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farres J, Wang TT, Cunningham SJ and Weiner H 1995, Biochemistry, 34, 2592. [DOI] [PubMed] [Google Scholar]
- 59.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD and Song BJ 2006, Hepatology, 44, 1218. [DOI] [PubMed] [Google Scholar]
- 60.Song BJ, Moon KH, Olsson NU and Salem N Jr. 2008, J. Hepatol, 49, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim BJ, Hood BL, Aragon RA, Hardwick JP, Conrads TP, Veenstra TD and Song BJ 2006, Proteomics, 6, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Finnerty N, O’Riordan SL, Klamer D, Lowry J and Palsson E 2015, Nitric Oxide, 47, 52. [DOI] [PubMed] [Google Scholar]
- 63.Moon KH, Lee YM and Song BJ 2010, Free Radic. Biol. Med, 48, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chaudhary P, Sharma R, Sharma A, Vatsyayan R, Yadav S, Singhal SS, Rauniyar N, Prokai L, Awasthi S and Awasthi YC 2010, Biochemistry, 49, 6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raza H and John A 2006, Toxicol. Appl. Pharmacol, 216, 309. [DOI] [PubMed] [Google Scholar]
- 66.Petersen DR and Doorn JA 2004, Free Radic. Biol. Med, 37, 937. [DOI] [PubMed] [Google Scholar]
- 67.Chen CH, Gray MO and Mochly-Rosen D 1999, Proc. Natl. Acad. Sci. USA, 96, 12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD and Mochly-Rosen D 2008, Science, 321, 1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Churchill EN, Disatnik MH and Mochly-Rosen D 2009, J. Mol. Cell Cardiol, 46, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lagranha CJ, Deschamps A, Aponte A, Steenbergen C and Murphy E 2010, Circ. Res, 106, 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choudhary C, Weinert BT, Nishida Y, Verdin E and Mann M 2014, Molecular Cell Biology, 15, 536. [DOI] [PubMed] [Google Scholar]
- 72.Klimova N, Long A, Scafidi S and Kristian T 2019, Biochim. Biophys. Acta Mol. Basis Dis, 1865, 2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shepard BD, Tuma DJ and Tuma PL 2010, Alcohol Clin. Exp. Res, 34, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu Z, Bourdi M, Li JH, Aponte AM, Chen Y, Lombard DB, Gucek M, Pohl LR and Sack MN 2011, EMBO Reports, 12, 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xue L, Xu F, Meng L, Wei S, Wang J, Hao P, Bian Y, Zhang Y and Chen Y 2012, FEBS Lett, 586, 137. [DOI] [PubMed] [Google Scholar]
- 76.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP and Verdin E 2010, Cell Metabolism, 12, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sugano T, Handler JA, Yoshihara H, Kizaki Z and Thurman RG 1990, J. Biol. Chem, 265, 21549. [PubMed] [Google Scholar]
- 78.Shepard BD and Tuma PL 2009, World J. Gastroenterol, 15, 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deelchand DK, Shestov AA, Koski DM, Ugurbil K and Henry PG 2009, J. Neurochem, 109(Suppl. 1), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rae C, Fekete AD, Kashem MA, Nasrallah FA and Broer S 2012, Neurochem. Res, 37, 2541. [DOI] [PubMed] [Google Scholar]
- 81.Starai VJ and Escalante-Semerena JC 2004, Cell Mol. Life Sci, 61, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ariyannur PS, Moffett JR, Madhavarao CN, Arun P, Vishnu N, Jacobowitz DM, Hallows WC, Denu JM and Namboodiri AM 2010, Journal of Comparative Neurology, 518, 2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tildon JT, McKenna MC, Stevenson J and Couto R 1993, Neurochem. Res, 18, 177. [DOI] [PubMed] [Google Scholar]
- 84.Jin S, Cao Q, Yang F, Zhu H, Xu S, Chen Q, Wang Z, Lin Y, Cinar R, Pawlosky RJ, Zhang Y, Xiong W, Gao B, Koob GF, Lovinger DM and Zhang L 2021, Nat. Metab, 3, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang J, Du H, Jiang L, Ma X, de Graaf RA, Behar KL and Mason GF 2013, Proc. Natl. Acad. Sci. USA, 110, 14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mews P, Donahue G, Drake AM, Luczak V, Abel T and Berger SL 2017, Nature, 546, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jung ME and Metzger DB 2015, Journal of Pharmacology and Experimental Therapeutics, 352, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ, Garcia BA and Berger SL 2019, Nature, 574, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scott I, Webster BR, Li JH and Sack MN 2012, Biochem. J, 443, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hirschey MD, Shimazu T, Capra JA, Pollard KS and Verdin E 2011, Aging, 3, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoek JB, Cahill A and Pastorino JG 2002, Gastroenterology, 122, 2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Weissman S, Verdin E and Schwer B 2007, Molecular and Cellular Biology, 27, 8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirschey MD, Shimazu T, Huang JY, Schwer B and Verdin E 2011, Cold Spring Harbor Symposia on Quantitative Biology, 76, 267. [DOI] [PubMed] [Google Scholar]
- 94.Onyango P, Celic I, McCaffery JM, Boeke JD and Feinberg AP 2002, Proc. Natl. Acad. Sci. USA, 99, 13653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fritz KS, Galligan JJ, Hirschey MD, Verdin E and Petersen DR 2012, J. Proteome Res, 11, 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anderson KA and Hirschey MD 2012, Essays in Biochemistry, 52, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sol EM, Wagner SA, Weinert BT, Kumar A, Kim HS, Deng CX and Choudhary C 2012, PLoS One, 7, e50545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Henderson GI, Chen JJ and Schenker S 1999, Frontiers in Bioscience: A Journal and Virtual Library, 4, D541. [DOI] [PubMed] [Google Scholar]
- 99.Jung ME and Metzger DB 2010, Molecules, 15, 4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Klimova N, Fearnow A and Kristian T 2020, Brain Sci, 10, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, Filenko OF, Kalinina NI, Kapelko VI, Kolosova NG Kopnin BP, Koeshunova GA, Lichinitser MR, Obukhova LA, Pasyukova EG, Pisarenko OI, Roginsky VA, Ruuge EK, Senin II, Severina II, Skulachev MV, Spivak IM, Tashlicky VN, Tkachuk VA, Vyssokikh MY, Yaguzhinsky LS and Zorov DB 2009, Biochim Biophys Acta 1787, 437. [DOI] [PubMed] [Google Scholar]
- 102.Han D, Canali R, Rettori D and Kaplowitz N 2003, Mol. Pharmacol, 64, 1136. [DOI] [PubMed] [Google Scholar]
- 103.Fukai T and Ushio-Fukai M 2011, Antioxidants & Redox Signaling, 15, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, Bohr VA and Mattson MP 2016, Cell Metabolism, 23, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klimova N, Fearnow A, Long A and Kristian T 2020, Exp. Neurol, 325, 113144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Almansa I, Fernandez A, Garcia-Ruiz C, Muriach M, Barcia JM, Miranda M, Fernandez-Checa JC and Romero FJ 2009, J. Physiol. Biochem, 65, 305. [DOI] [PubMed] [Google Scholar]
- 107.Reddy VD, Padmavathi P, Kavitha G, Gopi S and Varadacharyulu N 2011, J. Med. Food, 14, 62. [DOI] [PubMed] [Google Scholar]
- 108.Hao L, Sun Q, Zhong W, Zhang W, Sun X and Zhou Z 2018, Redox Biology, 14, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CB 2009, Science, 324, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Szutowicz A and Lysiak W 1980, J. Neurochem, 35, 775. [DOI] [PubMed] [Google Scholar]
- 111.Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y and Lei QY 2013, Mol. Cell, 51, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kraus F, Roy K, Pucadyil TJ and Ryan MT 2021, Nature, 590, 57. [DOI] [PubMed] [Google Scholar]
- 113.Lee H and Yoon Y 2016, Biochem. Soc. Trans, 44, 1725. [DOI] [PubMed] [Google Scholar]
- 114.Youle RJ and van der Bliek AM 2012, Science, 337, 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Klimova N, Long A and Kristian T 2018, Transl. Stroke Res, 9, 223. [DOI] [PubMed] [Google Scholar]
- 116.Chen H and Chan DC 2009, Human Molecular Genetics, 18, R169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Owens K, Park JH, Gourley S, Jones H and Kristian T 2015, J. Bioenerg. Biomembr, 47, 13. [DOI] [PubMed] [Google Scholar]
- 118.Kumari S, Anderson L, Farmer S, Mehta SL and Li PA 2012, Transl. Stroke Res, 3, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z and Lipton SA 2009, Science, 324, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS and Schuh RA 2015, BMC Neurol, 15, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chang CR and Blackstone C 2010, Annals of the New York Academy of Sciences, 1201, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu Q, Zhang H, Gutierrez Cortes N, Wu D, Wang P, Zhang J, Mattison JA, Smith E, Bettcher LF, Wang M, Lakatta EG, Sheu SS and Wang W 2020, Circ. Res, 126, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT, Kitajewski J, Rehman J, Yoon Y, Cho J, Fukai T and Fukai MU 2018, Cell Reports, 23, 3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bonet-Ponce L, Saez-Atienzar S, da Casa C, Flores-Bellver M, Barcia JM, Sancho-Pelluz J, Romero FJ, Jordan J and Galindo MF 2015, Biochim. Biophys. Acta, 1852, 1400. [DOI] [PubMed] [Google Scholar]
- 125.Lim JR, Lee HJ, Jung YH, Kim JS, Chae CW, Kim SY and Han HJ 2020, Cell Commun. Signal, 18, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yan T, Zhao Y, Jiang Z and Chen J 2022, Mol Neurobiol, 59, 3933. [DOI] [PubMed] [Google Scholar]
- 127.van der Bliek AM, Shen Q and Kawajiri S 2013, Cold Spring Harbor Perspectives in Biology, 5, a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Waddell J, Banerjee A and Kristian T 2021, Cells, 10, 3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC and Gupta MP 2014, Molecular and Cellular Biology, 34, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee JY, Kapur M, Li M, Choi MC, Choi S, Kim HJ, Kim I, Lee E, Taylor JP and Yao TP 2014, Journal of Cell Science, 127, 4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rajgor D, Welle TM and Smith KR 2021, Front. Cell Dev. Biol, 9, 711446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Knabbe J, Protzmann J, Schneider N, Berger M, Dannehl D, Wei S, Strahle C, Tegtmeier M, Jaiswal A, Zheng H, Kruger M, Rohr K, Spanagel R, Bilbao A, Engelhardt M, Scholz H and Cambridge SB 2022, Proc. Natl. Acad. Sci. USA, 119, e2122477119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Conto MB, Dos Santos NB, Munhoz CD, Marcourakis T, D’Almeida V and Camarini R 2021, Neuroscience, 469, 125. [DOI] [PubMed] [Google Scholar]