

With the current access to the whole genomes of various organisms and the completion of the first draft of the human genome, there is a strong need for a structure-function classification of protein families as an initial step in moving from DNA databases to a comprehensive understanding of human biology. As a result of the explosion in nucleic acid sequence information and the concurrent development of methods for high-throughput functional characterization of gene products, the genomic revolution also promises to provide a new paradigm for drug discovery, enabling the identification of molecular drug targets in a significant number of human diseases. This molecular view of diseases has contributed to the importance of combining primary sequence data with three-dimensional structure and has increased the awareness of computational homology modeling and its potential to elucidate protein function. In particular, when important proteins or novel therapeutic targets are identified—like the family of protein tyrosine phosphatases (PTPs) (reviewed in reference 53)—a structure-function classification of such protein families becomes an invaluable framework for further advances in biomedical science. Here, we present a comparative analysis of the structural relationships among vertebrate PTP domains and provide a comprehensive resource for sequence analysis of phosphotyrosine-specific PTPs.
PTPs are a key group of signal transduction enzymes which, together with protein tyrosine kinases, control the levels of cellular protein tyrosine phosphorylation. Protein tyrosine kinases phosphorylate cellular substrates on tyrosine residues, and much progress has been made over the last 20 years in elucidating their significance in signal transduction (for reviews, see references 26, 30, 31, 33, 71, and 72). However, it is only recently that the complexities of the PTPs have been appreciated. Thus, today it is recognized that the capacity of PTPs to dephosphorylate phosphotyrosine residues selectively on their substrates plays a pivotal role in initiating, sustaining and terminating cellular signaling (for reviews, see references 1, 4, 19, 32, 35, 46, 55, and 83). It has been shown that both the catalytic domain and noncatalytic segments of the PTPs contribute to the definition of substrate specificity in vivo. Whereas noncatalytic domains may target the PTPs to specific intracellular compartments in which the effective local concentration of substrate is high (3, 19, 51), the PTP catalytic domains themselves confer site-selective protein dephosphorylation by recognizing both the phosphotyrosine residue to be dephosphorylated and its flanking amino acids in the substrate. The combination of structural studies, kinetic analysis of PTP domains (37, 74, 76, 90, 91, 96), and studies involving substrate-trapping mutants (20, 23, 89) as well as PTP chimeras (60, 82) has convincingly demonstrated that isolated PTP domains may exhibit exquisite substrate selectivity.
The structurally conserved PTP domain defines membership of the PTP family, and three groups of enzymes are capable of dephosphorylating tyrosine-phosphorylated residues (57): (i) classical PTPs, (ii) dual-specificity PTPs, and (iii) low-molecular-weight PTPs. The dual-specificity PTPs and low-molecular-weight PTPs will not be considered further but have been reviewed (43, 70). The classical PTPs, which are the focus of the present study, encompass both transmembrane receptor-like and nontransmembrane enzymes, and the wide spectrum of protein domains present within this family highlight their diverse cellular functions. Most transmembrane receptor-like PTPs (RPTPs) contain two cytoplasmic PTP domains, a membrane proximal domain (D1) and a membrane distal domain (D2), and in addition have a single transmembrane segment and an extracellular domain.
As the study of PTPs has developed, the availability of an impressive number of X-ray crystal structures (for an updated list, see reference 53) and phylogenetically divergent cDNAs has permitted a detailed structural analysis of the evolutionary relationships among vertebrate PTP domains. Together with numerous enzymological studies revealing insights into the mechanism of PTP catalysis (12, 14, 25, 34, 47, 68, 77, 80, 97–99, 101), this development has permitted us to combine an extensive set of amino acid sequences with representative three-dimensional protein structures to derive new and refined information regarding PTP structure, substrate recognition, and evolutionary conservation. Because perturbed levels of tyrosine phosphorylation are associated with diseases such as cancer, autoimmunity, and allergy, we hope that this comprehensive analysis of the PTP family may assist in providing the structural basis for novel therapeutic strategies involving the development of selective PTP inhibitors.
In the present study, we have compiled a total of 319 vertebrate PTP sequences, including splice variants and partially overlapping sequences. Subsequent analysis narrowed these 319 GenBank entries down to 113 distinct PTP catalytic domain sequences (including non-transmembrane PTPs and domains D1 of RPTPs) and 38 domain D2 sequences from human and other vertebrate species. From this collection of 151 PTP domain sequences, we identified 37 distinct human PTP genes, which we aligned to assist in the identification of conserved regions. This sequence comparison allowed the classification of the vertebrate PTP family into 17 principal subtypes. The motifs identified from our amino acid sequence alignment are reviewed in terms of their location in the tertiary structure and, where relevant, their catalytic function. As a low-resolution and automated homology modeling approach, we applied the methodology of Cα-regiovariation score analysis (10, 11) to identify foci within the PTP domain tertiary structure, where amino acid conservation extended in three dimensions. The conserved foci identified by this approach are discussed including a previously unrecognized conserved cluster of residues located on the face of the molecule opposite the active site.
Collection of unique vertebrate PTP domains.
Following the identification of the first PTP in 1988 (84), intense efforts in the application of PCR and low-stringency screening led to the rapid discovery of a wide variety of other PTP family members. As often happens in rapidly developing research fields, several identical PTPs were independently cloned by different research groups and hence given different names and accession numbers. Consequently, the first step in our structural study was to compile a database of unique PTP domains. A BLAST search (2) of the National Center for Biotechnology Information (NCBI) GenBank database was performed using nucleotide sequences encoding several divergent PTP catalytic domains (PTP1B, SHP1, MEG2, PEST, PTPH1, PTPD1, CD45, RPTPμ, LAR, RPTPα, RPTPγ, RPTPβ, STEP, and the PTP-like protein IA2). This sequence similarity search generated over 3,500 database hits that, following the exclusion of expressed sequence tags and sequences encoding dual specificity and low-molecular-weight phosphatases, identified 319 vertebrate PTP entries. Alignment of the 5′ untranslated regions and the amino acid sequences of these 319 entries revealed a large number of different splice variants, partially overlapping sequences, and duplicate database entries. In total, 113 distinct PTP catalytic domains and 38 domain D2 sequences from tandem domain RPTPs were uncovered. This collection of PTP domains contains 37 human PTP genes and ortholog sequences from vertebrate species (Table 1). Our compilation of PTP-related sequences illustrates the redundancies often observed among GenBank database entries. Moreover, since many of the deposited sequences lack structural or functional annotation, there is a strong requirement for grouping these entries in order to gain access to the combined body of biochemical, structural, and/or functional information known for any given PTP. To this end, we have grouped the entries in Table 1 based on PTP domain sequence similarity (by subtype) and have identified the most likely human orthologs. In addition, to facilitate access to MEDLINE literature for any given PTP of interest, an electronic version of Table 1 can be retrieved (http://science.novonordisk.com/ptp), in which the accession numbers are hyperlinked to the NCBI website and PubMed literature database (http://www.ncbi.nlm.nih.gov). We have also mapped the chromosomal locations of the 37 human PTPs described in this study, allowing a detailed description of their intron and exon structure. Genomic clones, EMBL accession numbers, and the position of these PTPs in the human genome are summarized in Table 2. In addition, we acknowledge that the draft of the human genome contains additional sequences that conform to the PTP consensus motifs, but the expression of these hypothetical proteins has not yet been verified. Only transcripts that have been confirmed from in vitro or in vivo studies are considered in the present structure-function analysis of PTP domains.
TABLE 1.
Compilation of nonredundant set of 113 vertebrate PTPsa
| Name | Subtype | Human ortholog | Full-length name | Synonym(s) | Swiss-Prot | GenBank accession no. |
|---|---|---|---|---|---|---|
| Non-RPTP subtypes | ||||||
| hPTP1B | NT1 | PTP1B | PTP-1, PTP-1B | P18031 | M31724, M33689, | |
| mPTP1B | NT1 | PTP1B | PTP1B | PTP-1, HA2, PTP-HA2 | P35821 | U24700, Z23057, M97590, L40595 |
| rPTP1B | NT1 | PTP1B | PTP1B | PTP-1 | P20417 | M33962 |
| cPTP1B | NT1 | PTP1B | PTP1B | O13016 | U86410 | |
| zPTP1B | NT1 | PTP1B | PTP1B | AF097481, AF097482, AF097483 | ||
| hTCPTP | NT1 | TC-PTP (T-cell phosphatase) | PTP-2 | P17706 | M25393, M81478, M80737 | |
| mTCPTP | NT1 | TCPTP | TC-PTP (T-cell phosphatase) | PTP-2 | O06180 | S52655, M81477, M80739 |
| rTCPTP | NT1 | TCPTP | TC-PTP (T-cell phosphatase) | PTP-2, PTP-S | P35233 | X58828 |
| hSHP1 | NT2 | Src homology domain 2-containing PTP1 | SH-PTP1, SHP, HCP, PTP1C | P29350 | M74903, X62055, M77273, M90388, X82817, X82818 | |
| mSHP1 | NT2 | SHP1 | Src homology domain 2-containing PTP1 | P29351 | M68902, M90389, U65953, U65954, U65955 | |
| rSHP1 | NT2 | SHP1 | Src homology domain 2-containing PTP1 | U77038 | ||
| hSHP2 | NT2 | Src homology domain 2-containing PTP2 | SH-PTP2, SH-PTP3, Syp, PTP-2C, PTP1D | O06124 | D13540, L03535, L07527, X70766, L08807, S78088, S39383 | |
| mSHP2 | NT2 | SHP2 | Src homology domain 2-containing PTP2 | SH-PTP2, Syp | P35235 | L08663, D84372 |
| rSHP2 | NT2 | SHP2 | Src homology domain 2-containing PTP2 | PTP-1D | P41499 | U09307, U05963, D83016 |
| cSHP2 | NT2 | SHP2 | Src homology domain 2-containing PTP2 | SH-PTP2, Syp | U38620 | |
| xSHP2 | NT2 | SHP2 | Src homology domain 2-containing PTP2 | U15287 | ||
| hMEG2 | NT3 | Megakaryocyte-PTP2 | P43378 | M83738 | ||
| mMEG2 | NT3 | MEG2 | Megakaryocyte-PTP2 | AF013490 | ||
| xPTPX1 | NT3 | —b | L33098 | |||
| xPTPX10 | NT3 | — | L33099 | |||
| hPEST | NT4 | Pro, Glu, Ser, Thr-rich PTP | PTP-PEST, PTPG1 | O05209 | D13380, M93425, S69184 | |
| mPEST | NT4 | PEST | Pro, Glu, Ser, Thr-rich PTP | PTPP19 | P35831 | X86781, X63440, S36169 |
| rRKPTP | NT4 | PEST | Rat kidney PTP | D38072 | ||
| hLyPTP | NT4 | Lymphoid phosphatase | LyP1, LyP2 | AF001846, AF001847, AF077031, AF150732 | ||
| mPEP | NT4 | LyPTP | Hematopoietic cell PTP | P29352 | M90388 | |
| hBDP1 | NT4 | Brain-derived phosphatase 1 | X79568 | |||
| rPTP20 | NT4 | BDP1 | U69673 | |||
| mPTPK1 | NT4 | BDP1 | PTPFLP1 (fetal liver phosphatase 1) | U35124, U52523, U49853 | ||
| hMEG1 | NT5 | Megakaryocyte-PTP1 | PTPG1, PTPF36-15 | P29074 | M68941, AAB26477 | |
| mPTPtep | NT5 | MEG1 | Testis-enriched phosphatase | PTPMEG | AF106702 | |
| zPTPH1 | NT5 | MEG1 | AF097477, AF097478, AF097479, AF097480 | |||
| hPTPH1 | NT5 | PTPH1 | P26045 | M64572, S39392 | ||
| mPTPRL10 | NT6 | PTPD1 | Q62136 | D37801, D83072 | ||
| rPTP2E | NT6 | PTPD1 | PTPD1 | Q62728 | U17971, U18293 | |
| hPTPD1 | NT6 | PTPD1 | PTPD1 | Q16825 | X79510 | |
| hPTPD2 | NT6 | PTPD2 | PEZ (phosphatase ezrin-like) | Q15678 | X82676 | |
| mPTP36 | NT6 | PTPD2 | PTPD2 | Q62130 | D31842 | |
| hPTPBAS | NT7 | FAS-associated PTP1 | BAS, PTP1E, PTPE1, FAP-1, PTPL1, CD95 | Q12923 | X80289, U12128, D21209, D21210, D21211, U81561, X79676 | |
| mPTPBL | NT7 | PTPBAS | DPZPTP, PTPRIP | D28529, Z32740, D83966 | ||
| bPTPBA14 | NT7 | PTPBAS | U20807 | |||
| hPTPTyp | NT8 | Testis-specific tyrosine phosphatase | Typ | AL050040 | ||
| mPTPTyp | NT8 | PTPTyp | Testis-specific tyrosine phosphatase | Typ | D64141 | |
| hHDPTP | NT9 | His domain-containing PTP | HD-PTP, PTPTD14 | T14756, AB025194, AB040904, AL110210, AF169350 | ||
| rPTPTD14 | NT9 | HDPTP | AF077000 | |||
| RPTP subtypes | ||||||
| hCD45 | R1/R6 | Cluster of differentiation 45 | Leukocyte common antigen (LCA), T200, PTPRC | P08575 | Y00638, Y00062 | |
| mCD45 | R1/R6 | CD45 | Cluster of differentiation 45 | LCA, T200, Ly5 | P06800 | M14342, M92933, M33482 |
| rCD45 | R1/R6 | CD45 | Cluster of differentiation 45 | Leukocyte common antigen | P04157 | M10072, Y00065, M25820, M25821, M25822, M25823, K03039 |
| cPTPlambda | R1/R6 | CD45 | PTPlambda | L13285, Z21960 | ||
| xCD45 | R1/R6 | CD45 | Cluster of differentiation 45 | AF024438 | ||
| hPTPlambda | R2A | RPTPlambda | PCP2, PTPomicron, PTPfmi, PTPpi, PTPi, PTPRO | Q92729 | U60289, X97198, U73727, U71075, X95712, AL049570 | |
| mPTPlambda | R2A | PTPλ | RPTPlambda | PTPftp1, PTPpsi | U55057, D88187 | |
| rPTPpsi | R2A | PTPI | RPTPpsi | U66566 | ||
| hPTPkappa | R2A | RPTPkappa | Q15262 | L77886, Z70660 | ||
| mPTPkappa | R2A | PTPκ | RPTPkappa | P35822 | L10106 | |
| hPTPmu | R2A | RPTPmu | P28827 | X58288 | ||
| mPTPmu | R2A | PTPm | RPTPmu | P28828 | X58287 | |
| hPTPrho | R2A | RPTPrho | AF043644, AL024473, AL022239, Z93942 | |||
| mPTPrho | R2A | PTPr | RPTPrho | AF152556 | ||
| xPTPrho | R2A | PTPr | RPTPrho | AF173857 | ||
| hLAR | R2B | LCA-related PTPc | PTP-LAR | P10586 | Y00815 | |
| mLAR | R2B | LAR | LCA-related PTP | PTP-LAR | Z37988 | |
| rLAR | R2B | LAR | LCA-related PTP | PTP-LAR | L11586, U00477, X83546, X83505 | |
| xLAR | R2B | LAR | LCA-related PTP | PTP-LAR | AF197945 | |
| hPTPdelta | R2B | RPTPdelta | P23468 | X54133, L38929 | ||
| mPTPdelta | R2B | PTPδ | RPTPdelta | D13903 | ||
| cLAR | R2B | PTPδ | CRYPalpha | L32780 | ||
| xPTPdelta | R2B | PTPδ | RPTPdelta | AF197944 | ||
| hPTPsigma | R2B | RPTPsigma | U35234, U40317, U41725, AC005788, S78080, S78086 | |||
| rPTPsigma | R2B | PTPς | RPTPsigma | LAR-PTP2, PTP-PS, PTP-P1 | L11587, AF073999 | |
| mPTPNU3 | R2B | PTPς | PTPsigma, PTPT9a, PTPT9b | X82288, D28530, D28531 | ||
| xCRYPalpha | R2B | PTPς | AF198450 | |||
| hPTPS31 | R3 | I32038, I32036, I32037, I32035, I32039 | ||||
| rPTPGMC | R3 | PTPS31 | Glomerular mesangial cell receptor | PTPRQ, PTPGMC1 | AF063249 | |
| hGLEPP1 | R3 | Glomerular epithelial protein 1 | PTPU2, PTProt | U20489, Z48541 | ||
| mPTPphi | R3 | GLEPP1 | PTP-BK, PTP-ro, mGLEPP1 | U37465, U37466, U37467, AF295638 | ||
| rPTPBEM1 | R3 | GLEPP1 | Brain-enriched membrane-associated PTP1 | PTPD30, BSM-1 | D45412, U28938 | |
| rabPTPoc | R3 | GLEPP1 | Osteoclastic PTP | U32587 | ||
| cPTPcryp2 | R3 | GLEPP1 | CRYP-2 | U65891 | ||
| hPTPbeta | R3 | RPTPβ | P23467 | X54131 | ||
| mPTPbeta | R3 | PTPβ | Vascular endothelial PTP (VE-PTP) | X58289, AF157628 | ||
| hDEP1 | R3 | Density-enhanced PTP | PTPeta, CD148, F-36-12 | Q12913 | U10886, D37781, AAB26475 | |
| mPTPBYP | R3 | DEP1 | RPTPbeta-like PTP | PTPeta | Q64455 | D45212 |
| rDEP1 | R3 | DEP1 | Density enhanced PTP | Vascular PTP-1 | U40790 | |
| hSAP1 | R3 | Stomach cancer-associated PTP | hPTPH | D15049, AAF91411 | ||
| rPTPBEM2 | R3 | SAP1 | Brain-enriched membrane-associated PTP2 | D45413 | ||
| mPTPesp | R3 | — | Embryonic stern cell PTP | OST-PTP | P70289 | U36488, AF300701 |
| rOSTPTP | R3 | — | Osteotesticular PTP | O64612 | L36884 | |
| hPTPalpha | R4 | RPTPalpha | P18433 | M34668, X54130, X54890, X53364 | ||
| mPTPalpha | R4 | PTPα | RPTPalpha | LCA-related PTP | P18052 | M36033, M33671, M36034 |
| rPTPalpha | R4 | PTPα | RPTPalpha | Q03348 | L01702 | |
| cPTPalpha | R4 | PTPα | RPTPalpha | Z32749, L22437, | ||
| xPTPalpha | R4 | PTPα | RPTPalpha | U09135 | ||
| hPTPepsilon | R4 | RPTPepsilon | P23469 | X54134 | ||
| mPTPepsilon | R4 | PTPɛ | RPTPepsilon | P49446 | U35368, U36758, D83484, U62387, U40280 | |
| rPTPepsilon | R4 | PTPɛ | RPTPepsilon | D78610, D78613 | ||
| hPTPgamma | R5 | RPTPgamma | P23470 | L09247, X54132 | ||
| mPTPgamma | R5 | PTPγ | RPTPgamma | Q05909 | L09562 | |
| cPTPgamma | R5 | PTPγ | RPTPgamma | Q98936 | U38349 | |
| cPTPzeta | R5 | RPTPzeta | L27625 | |||
| hPTPzeta | R5 | PTPζ | RPTPzeta | P23471 | M93426, X54135, U88967 | |
| rPTPzeta | R5 | PTPζ | RPTPzeta | Q62656 | U09357 | |
| hPCPTP1 | R7 | PC12-derived PTP | PTPch1g, PTPCOM1, hCh1PTPα, PTPEC | D64053, U77916, U77917, U42361, X82635, Z79693 | ||
| rPCPTP1 | R7 | PCPTP | PC 12-derived PTP | PC12-PTP1, CBPTP | D38292, D64050, U14914 | |
| mPTPSL | R7 | PCPTP | PTPBR7, PTP-SL, PC 12-PTP1 | Z30313, AF041866, D31898 | ||
| hSTEP | R7 | Striatum-enriched phosphatase | P54829 | U27831 | ||
| mSTEP61 | R7 | STEP | Striatum-enriched phosphatase | P54830 | U28217, S80329, U28216 | |
| rSTEP | R7 | STEP | Striatum-enriched phosphatase | P35234 | S49400 | |
| hHePTP | R7 | Hematopoetic PTP | Leucocyte PTP | P35236 | M64322, D11327 | |
| rLCPTP | R7 | HePTP | Leukocyte PTP | Hematopoetic PTP | P49445 | U28356 |
| IRL subtyped | ||||||
| hPTPIA2 | R8 | Islet cell antigen | Islet cell antigen, ICA-512 | Q16849 | L18983, Z48226, X62899 | |
| mPTPIA2 | R8 | IA2 | Islet cell antigen | PTP35 | Q60673 | U11812, X74438 |
| rPTPIA2 | R8 | IA2 | Islet cell antigen | BEM-3, PTPN, ICA105, PTPLP | Q63259 | D45414, X92563, D38222, U40652 |
| bPTPIA2 | R8 | IA2 | Islet cell antigen | ICA512 | P56722 | AF075170 |
| hPTPIA2beta | R8 | PTP-IA-2beta | IAR, RPTPX | Q92932 | U65065, AF007555, L76258, U81561, AB002385 | |
| mPTPNP | R8 | IA2β | Nervous system and pancreatic PTP | IA2beta, RPTPX, PTPNP-2 | P80560 | U57345 |
| macPTPIA2beta | R8 | IA2β | IA2beta | O02695 | U91574 | |
| rPTPNE6 | R8 | IA2β | IA2beta, phogrin | Q63475 | U73458, Z50735 |
Summary of vertebrate PTP database entries showing nomenclature and accession numbers assigned to each PTP. The PTP name is that most frequently used in the PubMed literature database. The letters preceding the PTP name represent the following: b, bovine; c, chicken; h, human; m, mouse; mac, monkey; r, rat; rab, rabbit; x, xenopus; z, zebra fish. Entries (either Swiss-Prot or GenBank) were used for the primary amino acid sequence alignment of PTP domains (Fig. 1). The PTP entries are grouped according to catalytic domain sequence similarity and have been classified into 17 PTP subtypes: nine nontransmembrane (NT1 to NT9) and eight receptor types (R1/R6, R2a, R2b, R3, R4, R5, R7, and R8). The basis for this classification is detailed in the legend to the homology tree in Fig. 2. The existing nomenclature for RPTP subtypes was used since these have previously been classified into almost identical groups based on extracellular domain similarity (8). Ortholog pairs of proteins were identified from the phylogentic analysis (Fig. 2). Note that RPTP subtypes R3, R4, and R7 include both transmembrane and cytoplasmic enzymes (see text for details). An electronic version of this table can be retrieved (http://science.novonordisk.com/ptp), in which the accession numbers are hyperlinked to the NCBI website and PubMed literature database (http://www.ncbi.nim.nih.gov).
—, no obvious human ortholog identified.
LCA, leukocyte common antigen.
IRL, IA2 receptor-like.
TABLE 2.
Chromosomal locations of human PTP genes and their genomic clonesa
| Protein name | Chr | Band | Ensembl gene ID | Genomic clones (EMBL accession no.) |
|---|---|---|---|---|
| hLyPTP | 1 | p13.1 | ENSG00000081021 | AL365321, AL137856 |
| hLAR | 1 | p34.2 | —c | AL158083 |
| hPTPlambda | 1 | p35.2 | ENSG00000060656 | AL049570 |
| hCD45 | 1 | q32.1 | ENSG00000081237 | AL355988 |
| hHePTP | 1 | q32.1b | — | — |
| hPTPD2 | 1 | q41 | ENSG00000065995 | AC026065, AC068586 |
| hMEG1 | 2 | q14.2 | — | AC016691 |
| hBDP1 | 2 | q21.2 | ENSG00000072135 | AC068137 |
| hPTPIA2 | 2 | q35 | — | AC60820 |
| hPTPgamma | 3 | p14.2 | — | AC024885 |
| pHDPTP | 3 | p25.1 | — | AC023230 |
| hPTPBAS | 4 | q22.1 | — | AC007525, AC079237 |
| hPTPkappa | 6 | q22.33 | — | AL035465 |
| hPEST | 7 | q11.23 | ENSG00000127947 | AC006451, AC090421 |
| hPTPzeta | 7 | q31.33 | ENSG00000106278 | AC073471, AC006020 |
| hPTPIA2beta | 7 | q36.3 | ENSG00000002748 | AC005481, AC006372, AC006321 |
| hPTPdelta | 9 | p23 | ENSG00000099228 | AC026466 |
| hPTPH1 | 9 | q32 | — | AL359963, AL450025, AC013568 |
| hPTPTyp | 10 | q11.22 | ENSG00000126542 | AL358791 |
| hPTPepsilon | 10 | q26.2 | ENSG00000132334 | AL390236 |
| hDEP1 | 11 | p11.2 | — | AC026975 |
| hSTEP | 11 | p15.1 | ENSG0000110786 | AC016750 |
| hGLEPP1 | 12 | p12.3 | ENSG00000084474 | AC007542 |
| hSHP1 | 12 | p13.31 | ENSG00000111679 | AC006512, U47924, M86525, U72506 |
| hPCPTP1 | 12 | q15 | ENSG00000111585 | AC055123, AC083809, AC015544, AC090676, AC090670 |
| hPTPbeta | 12 | q15 | — | AC015544, AC083809, AC011053, AC025569 |
| hPTPS31 | 12 | q21.31 | ENSG00000091041 | AC078825, AC074031 |
| hSHP2 | 12 | q24.13 | ENSG00000089131 | AC004086, AC004216 |
| hPTPD1 | 14 | q31.3 | — | AL353786, AL162171, AL049834 |
| hMEG2 | 15 | q23 | — | AC009712 |
| hTCPTP | 18 | p11.21 | ENSG00000128772 | AP001077, AC007734, AP002449 |
| hPTPmu | 18 | p11.22 | ENSG00000069927 | AC006566, AC021310, AP001094, AC069097, AC023663 |
| hPTPsigma | 19 | p13.3 | ENSG00000105426 | AC005338, AC005788 |
| hSAP1 | 19 | q13.42 | ENSG00000080031 | AC010327, AC010619 |
| hPTPalpha | 20 | p13 | ENSG00000037980 | AL121905, AL138803 |
| hPTPrho | 20 | q12 | ENSG00000087530 | AL024473 |
| hPTP1B | 20 | q13.13 | ENSG00000063920 | AL133230, AL034429 |
The PTP amino acid sequences of the proteins were searched against those in the human genome databases (www.genome.ucsc.edu and www.ensembl.org) in order to determine their chromosomal locations (chromosome number [Chr] and band). Genomic clones for the human PTP genes (annotated genes in Ensembl and EMBL accession numbers) are hyperlinked to their respective databases (see http://science.novonordisk.com/ptp for the electronic version of the table).
No genomic clones were found. Chromosomal localization was determined by fluorescent in situ hybridization (93).
—, no gene annotation in Ensembl has been available so far.
Primary sequence alignment of PTP domains.
To provide a platform for classification of members of the PTP family and for the identification of conserved residues, a multiple-sequence alignment was constructed using the entire set of vertebrate PTPs identified above (Table 1). In Fig. 1, we have reduced the alignment to a sequence comparison between the 37 human PTP domains, but the extended version used in our analysis can be retrieved from the World Wide Web (http: //science.novonordisk.com/ptp) and includes all 113 vertebrate PTP catalytic domains. To enable the assessment of both the level of conservation and the degree of sequence variation, the alignment is color coded according to amino acid identity (see legend to Fig. 1). The N- and C-terminal boundaries for this alignment correspond to residues 1 to 279 in PTP1B and encompass all invariant residues and structurally conserved elements. In the past, the PTP domain has been described to consist of ∼250 amino acids, but the extensive set of PTPs included in this multiple-sequence alignment, combined with structural knowledge and secondary-structure prediction algorithms, has permitted us to identify conservation at the N terminus of the PTP domain comprising the α1′ and α2′ helices of PTP1B (37) (Fig. 1). We now define the PTP domain as comprising ∼280 residues. In Fig. 1, an alignment of domain D2 of RPTPs is included, but these domains were not used in the definition of the PTP consensus sequence.
FIG. 1.



Sequence comparison of human PTP domains. Shown is an amino acid sequence alignment of 37 human PTP domains (from nontransmembrane PTP and RPTP domains D1) (above) and comparison with domain D2 sequences of RPTPs (below). Amino acids are numbered according to the residue position in human PTP1B. The locations of α-helices and β-strands (based on the X-ray crystal structure of PTP1B [7]) are shown at the top of the alignment. Twenty-two invariant residues (underscored) and 42 highly conserved residues (>80% identity) are indicated at the bottom of the alignment. The PTP consensus motifs (M1 to M10) are detailed in Table 2. Amino acids are color coded according to their degree of conservation, as indicated below the alignment. Nonconserved residues involved in the definition of substrate selectivity-determining regions are boxed with black lines (see text and Fig. 9). The four-residue conserved linker in tandem RPTP enzymes is boxed in yellow (above) and corresponds to encircled area 1 in Fig. 8. Sequences were aligned using the Clustalw algorithm and the Genetics Computer Group PileUp software (version 8.1) by applying the BLOSUM 62 scoring matrix together with default gap creation and extension penalty. Alignment of the N termini of the PTP domains was guided by crystallographic structural data and secondary structure predictions (nnpredict at http://www.cmpharm.ucsf.edu). The complete alignment of all vertebrate PTP domains can be retrieved (http://science.novonordisk.com/ptp) in several standard GCG formats, including MSF, TFA, and ALN.
PTP family can be classified into 17 subtypes.
Since phylogenetic analysis of sequence alignments serves as a useful tool for the classification of homologous proteins, we derived a phylogenetic tree from our alignment of 113 PTP catalytic domains (Fig. 2). The clustering of sequences into divergent branches of this tree provided a basis for subdivision of PTP family members (see figure legend for details). In total, 17 principal PTP subtypes were identified as indicated in Fig. 2. In addition, all PTP domain D1 sequences from tandem domain RPTPs clustered into one major trunk of the phylogenetic tree, allowing the definition of a PTP supertype encompassing these five RPTP subtypes (R1/R6, R2A, R2B, R4, and R5). The high intrasubtype sequence identity of 60 to 80% among domain D1 of these RPTPs, compared to 45 to 60% among PTP domains of the RPTPβ-like subtype (R3), which contain only one PTP domain, supports earlier suggestions that during evolution, intragenic catalytic domain duplication (i.e., duplication of the PTP domain within an ancestral PTP gene) preceded gene duplication (88). Consistent with this concept, all domain D2 sequences also clustered into one separate branch of the phylogenetic tree (data available at http://science.novonordisk.com/ptp), suggesting structural, and perhaps functional, conservation among these PTP domains. The result of the present classification system, together with a diagram of the overall domain structure of a representative member of each PTP subtype, is presented in Fig. 3.
FIG. 2.

Classification of family of PTPs into 17 subtypes. Shown is an unrooted tree derived from the alignment of 113 vertebrate PTP domain sequences (residue positions 1 to 279 in human PTP1B). The tree was drawn by the neighbor-joining method (73). The horizontal distance indicates the degree of sequence divergence, and the scale at the top corner represents the number of substitution events (10 per 100 amino acids). Seventeen PTP domain subtypes were identified from the phylogram: nine nontransmembrane subtypes (NT1 to NT9), five tandem receptor-like subtypes (R1/R6, R2A, R2B, R4, and R5), and three single-domain RPTP subtypes (R3, R7, and R8 [subtype R8 is believed to be catalytically inactive]). As a statistical test of the significance of sequence similarity within PTP subtypes, bootstrap values were calculated (values are at the dendogram node). With the exception of the RPTPβ-like subtype (R3) and the tandem PTP domain supertype, all subdivisions were assigned based on maximal bootstrap values (1,000). (A tree including the PTP domain D2 sequences can be viewed [http://science.novonordisk.com/ptp], and the raw data files can also be retrieved in several standard GCG formats).
FIG. 3.

Schematic representation of PTP family members. Determination of sequence similarity among PTP catalytic domains (Fig. 2) was used to classify the PTP family of enzymes into nine nontransmembrane PTP subtypes (NT) and eight RPTP subtypes (R). Only the human PTPs are listed, and a representative member of each subtype is shown. Synonyms and classifications of all vertebrate PTPs are given in Table 1. PTPs having closely related catalytic domains also tend to be similar in overall structural topology.
Sequence similarity between PTP domains can be used for overall structural classification of PTP family.
A major finding from the phylogenetic analysis of the alignment is the very close relationship between PTP domain sequence similarity and the presence of similar structural and functional domains in the full-length proteins (Fig. 3). Thus, the RPTPs, previously classified by their extracellular domains into nine distinct subtypes (8), are categorized into virtually identical groups based solely on catalytic domain sequence homology (hence the use of the existing nomenclature for the RPTPs) (8). However, one difference lies with chicken PTPλ. Based on its unique extracellular segment, this PTP was previously assigned to its own subtype (R6). but the present classification system suggests that it is the avian homologue of CD45. Therefore, we have included it within the CD45 subtype and defined it as R1/R6.
For the nontransmembrane PTPs, the nine subtypes defined from the phylogenetic analysis of PTP domains also correlated with the presence of particular regulatory and/or targeting domains. Thus, the SH2 domain-containing PTPs, SHP1 and SHP2, are classified as one PTP subtype (NT2), and the three PTPs containing a carboxy-terminal PEST-like domain (viz. human BDP, PEST, and LyPTP) are categorized as another distinct subtype (NT4) (Fig. 2). However, it should be noted that the FERM domain-containing PTPs, which vary in their central segments and contain distinct numbers of PDZ domains, fall into three distinct subtypes (NT5, NT6, and NT7). Although HDPTP (85) and PTPTyp (58) contain segments with a high content of proline, glutamate, serine, and threonine residues (PEST-like domains), they are categorized as distinct subtypes (NT8 and NT9). Since PEST-like sequence annotation is subjective and these sequences do not correspond to conserved protein domains in the Pfam and Interpro databases (5), the functions of these PEST-like segments are most likely unrelated.
Another important observation from the phylogenetic mapping of PTP domains relates to the traditional classification of this protein family into two broad classes: transmembrane RPTPs and intracellular nontransmembrane PTPs. Although we have maintained this conceptual subdivision for the classifications shown in Fig. 2 and 3, it is significant that several of the PTP subtypes (R3, R4, and R7) contain both transmembrane and nontransmembrane enzymes. Thus, the PCPTP1-like subtype (R7) contains both the receptor-like enzyme PCPTP1 (mouse PTP-SL) and two cytoplasmic enzymes STEP and HePTP (mouse LCPTP), for which no transmembrane isoforms have been identified so far. For the RPTP subtype R3, alternative splicing of GLEPP1 mRNA (mouse PTPφ) generates either a cytoplasmic or transmembrane form of the enzyme (67), and for PTPɛ (subtype R4), the alternate usage of isoform specific 5′ exons and promoters generates either a cytoplasmic or transmembrane form of the enzyme (18). Since the above examples illustrate that the classical subdivision of PTP family members, based on the presence or absence of an extracellular and transmembrane segment may be ambiguous, a novel classification system based on catalytic domain sequence similarity, as described here, was considered appropriate. We have made the phylogenetic tree available (http://science.novonordisk.com/ptp), and we hope it will serve as a useful tool for the classification of novel PTPs discovered in the postgenomic area.
Ten conserved motifs define family of PTPs.
Another application of the present PTP sequence alignment is the identification of conserved motifs that define this class of signal-transducing enzymes. In particular, the definition of consensus amino acid sequences—either for the PTP family as a whole or for functional and therapeutically interesting PTP subtypes—will help to probe genomes of other organisms for the presence of PTP orthologs and thereby identify relevant model organisms for rapid genetic analysis of the involvement of PTPs in control of fundamental cellular functions.
In the present study, we have defined a conserved motif as a stretch of three or more amino acids in which two of three of the residues are at least 80% conserved by amino acid similarity (substitution groups are specified in Table 3). Based on amino acid identity, 10 discrete and highly conserved motifs (M1 to M10) were identified from the alignment of PTP domains (Table 3). In addition, outside these motifs, seven single conserved residues were found (Glu19, Glu115, Arg156, Arg169, Leu192, Arg254, and Arg257; residues are numbered according to the numbering of human PTP1B) (Table 4). Several of the conserved residues identified have previously been reviewed (6, 95), and their functions have been studied extensively by site-directed mutagenesis (20, 78) and X-ray crystallography (13, 95). However, the existing PTP consensus sequences in the literature have been defined from a much smaller number of aligned sequences, and some important structural (noncatalytic) motifs have received less attention or, until now, have remained undisclosed (Table 3). Therefore, an overview of all motifs and their proposed function together with an evaluation of their degree of conservation in three-dimensional space is provided below.
TABLE 3.
Proposed roles of conserved residues in vertebrate PTP domainsa
| Motif (residues in PTP1B) | Conservation | Conservation in 3D | Proposed roles of residues |
|---|---|---|---|
| Motif 1 | |||
| 40–46 | NXXKNRY | Medium | pTyr-recognition loop: restricts substrate specificity to pTyr (Asn44, coordinates Asn68 which links Arg257; Arg45, putative substrate binding site, electrostatic attraction of ligand; Tyr46, hydrophobic packing with phosphotyrosine residue of substrate) |
| 40–46 | NXX(K/R)NRY | ||
| Motif 2 | |||
| 53–59 | DXXRVXL | Low | Conserved secondary structure (β1 sheet), surface exposed (Arg56, H bonds to Asp65; Ile57, hydrophobic core cluster [residues 57, 67, 69, 82, 98]; Leu59, hydrophobic core) |
| 53–59 | DXXR(V/I)XL | ||
| Motif 3 | |||
| 65–69 | DYINA | Medium | Core structure (Tyr66, coordinates Asn44 through hydrogen bonding; Ile67, hydrophobic core cluster [residues 57, 67, 69, 82, 98]; Asn68, H bonds with Arg257; Ala69, hydrophobic core cluster [residues 57, 67, 69, 82, 98]) |
| 65–70 | DYINA(N/S) | ||
| Motif 4 | |||
| 82–87 | IAXQGP | High | Core structure surrounding PTP loop (Ile82, hydrophobic core cluster [residues 57, 67, 69, 82, 98]; Ala83, packs or surrounds the PTP loop; Gln85, H bonds with highly buried water molecule; Gly86, packs or surrounds the PTP loop; Pro87, packs or surrounds PTP loop) |
| 81–87 | (F/Y)(I/V)AXQGP | ||
| Motif 5 | |||
| 91–100 | TXXDFWXMXW | Medium | Conserved secondary structure (α2 helix) (Asp94, contributes to conserved subdomain at the “back side”; Phe95, energetically favored T-stacking arrangement with invariant Trp96; Trp96, H bonds to backbone of invariant Tyr124; Met98, hydrophobic core cluster [residues 57, 67, 69, 82, 98]; Trp100, contributes to conserved subdomain at the back side) |
| 91–101 | TXXDFWX(M/L/V)X(W)(E/Q) | ||
| Motif 6 | |||
| 107–111 | IVMXT | Medium | Hydrophobic core structure (Ile107, hydrophobic core structure packs with invariant Trp96; Val108, hydrophobic core structure packs with invariant Trp96; Met109, packs with invariant Trp125; Thr111, packs with PTP loop) |
| 107–111 | (I/L/V)(V/I)MXT | ||
| Motif 7 | |||
| 120–126 | KCXXYWP | Low | Hydrophobic core structure (Lys120, interacts with Asp181 [ligand induced]; Tyr124, H bonds with His214, stabilizing T-stacking arrangement with Trp125; Trp125, favored T arrangement of aromatic ring system with Tyr124) |
| 120–126 | KCXXYWP | ||
| Motif 8 | |||
| 179–185 | WPDXGXP | Low | WPD loop, surface exposed, movable, contains general acid (Trp179, center of movable WPD loop, mediating motion of loop; Pro180, H bonds to NH2 of Arg221, mediating motion of loop; Asp181, general acid catalyst; Gly183, energetically favorable in loop motion [acts as hinge]; Pro185, energetically favorable in loop movement [no backbone H bonding]) |
| 176–185 | (Y/F)XXWPDXGXP | ||
| Motif 9 | |||
| 210–223 | PXXVHCSAGXGRTG | High | PTP loop surrounding active site Cys where seven successive main-chain nitrogens coordinate three phosphate oxyanions (Pro210, structural hydrophobic core; His214, lowers pKa of Cys215; Cys215, nucleophile; Ser216, H bonds with Tyr46 stabilizing its interaction with substrate; Ala217, phosphotyrosine binding, nonpolar interaction with substrate phenyl; Gly218, phosphotyrosine binding; Gly220, phosphotyrosine binding; Arg221, H bonds with phosphate oxygens [transition-state stabilization]; Thr222, lower pKa of Cys215) |
| 210–223 | PXX(V/I)HCSAGXGR(T/S)G | ||
| Motif 10 | |||
| 262–269 | QTXXQYXF | Low | The Q loop: interaction with active site water molecule (Gln262, H bonds with scissile oxygen and active site water molecule; Gln266, H bonds with active site water molecule; Tyr267, defines α6′ helix structure; Phe269, defines α6′ helix structure) |
| 261–269 | (V/I/L)QTXXQYXF |
Motifs are numbered (M1 to M10) in order of appearance in the primary amino acid sequence alignment (Fig. 1). Amino acids are numbered according to human PTP1B. Conserved residues were identified from our multiple-sequence alignment of 113 vertebrate PTP domains (available at http://www.science.nononordisk.com /ptp), and domain D2 sequences were not used in these evaluations. Underscored bold letters represent invariant amino acids. Residues in bold letters are conserved in ≥90% of the sequences, and nonbold letters represent ≥80% conservation. Sequence conservation of each motif was calculated according to (i) amino acid identity (top sequence) and (ii) amino acid similarity (bottom sequence), where X represents any amino acid (Fig. 1). Consensus substitution groups are defined as follows: 1, DN; 2, EQ; 3, ST; 4, KR; 5, FYW; 6, LIVM. Residue conservation in the immediate three-dimensional (3D) surroundings was determined using Cα-regiovariation score analysis (Fig. 5).
TABLE 4.
Proposed roles of single conserved residues in vertebrate PTP domains that reside outside the 10 PTP motifsa
| Amino acid in PTP1B | Conserved by amino acid identity | Proposed roles of the residues |
|---|---|---|
| Ile19 | E (>80%) | Definition of α2′ helix structure |
| Glu115 | E (100%) | Conserved H bonds with Arg221 |
| Arg156 | R (>80%) | Definition of β10-sheet |
| Arg169 | R (>80%) | Definition of β11-sheet |
| Leu192 | L (>80%) | Definition of the α3-helix structure |
| Arg254 | R (>90%) | H bonds with PTP loop |
| Arg257 | R (100%) | H bonds with PTP loop lowering pKa of Cys215 |
Conserved residues were identified from our multiple-sequence alignment of 113 vertebrate PTP domains. Underscored bold letters represent invariant residues. Residues in bold letters are conserved in ≥90% of the sequences, and non-bold letters represent ≥80% conservation.
Superimposition of PTP domains reveals conserved Cα-backbone trace that allows evaluation of the multiple sequence alignment in 3D space.
To date, X-ray crystallographic structures are available for seven different PTP catalytic domains, including the nontransmembrane enzymes (PTP1B, Yop51, SHP1, and SHP2) (7, 27, 79, 92) and RPTP domains (PTPμ, PTPα, and LAR) (29, 50, 56). When the crystal structures of vertebrate PTP domains were superimposed, we observed a conserved fold and a consistent Cα-backbone trace (Fig. 4). This striking conservation of tertiary structure allowed us to quantify the degree of conservation of each amino acid residue in three dimensional space (i.e., relative to the conservation of neighboring residues). In brief, such low-resolution homology modeling, the so-called Cα-regiovariation score analysis (10, 11), uses the information in a set of aligned sequences and calculates the average degree of conservation which has occurred within a given “sphere of influence” for each residue position along the folded polypeptide backbone of a representative tertiary structure. The method has previously identified interactive sites for cytochrome c, the pancreatic trypsin inhibitor family of proteinases, and carboxypeptidases A and B (10, 11). To avoid bias towards catalytic domains that are represented by a large number of ortholog sequences, we selected a nonredundant set of 37 aligned human PTP catalytic domains (Fig. 1). In agreement with Cα-regiovariation score analyses of other protein families (10, 11), we observed that a 6- to 8-Å sphere of influence provides an optimal signal-to-noise ratio and yields consistent results for different PTP templates (not shown). All figures in the present work were produced with a sphere of influence of 7 Å.
FIG. 4.
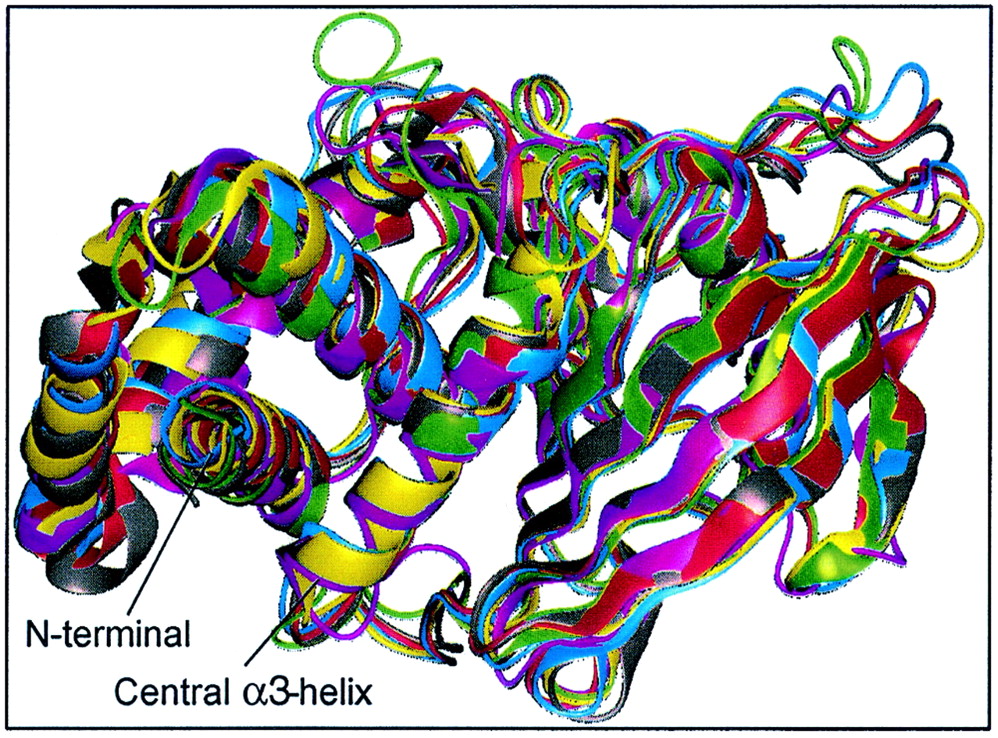
Crystal structures of vertebrate PTP domains show conserved fold and consistent Cα-backbone trace. PTP1B (magenta), RPTPα (gray), RPTPμ (red), LAR (blue), SHP1 (green), and SHP2 (yellow) were aligned and superimposed using Quanta (Molecular Simulations Inc.). For clarity, residues 280 to 298 (C terminal) of PTP1B, 250 to 281 (N terminal) and 522 to 532 (C terminal) of SHP1, and 2 to 218 (N terminal) of SHP2 were omitted from the figure, as well as D2 of LAR. The calculated RMS deviations between all Cα atoms between PTP1B and other PTPs are as follows: PTPα, 1.35 Å; RPTPμ, 2.72 Å; LAR D1, 2.78 Å; SHP1, 3.14 Å; and SHP2, 2.74 Å. For comparison, the RMS deviation between domains D1 and D2 of LAR is 1.3 Å. The X-ray structures are compared in their native open conformation.
Structural motifs make up the most highly conserved regions in PTP structure.
The score values for the Cα-regiovariation analysis are shown in Fig. 5. Conserved residues in conserved surroundings are identified as peaks. Hydrophobic segments in the primary amino acid sequence alignment make up the most highly conserved microenvironments in the PTP structure. Thus, the structural motifs TXXDFWXMXW (M5), IVMXT, (M6) and KCXXYWP (M7) (Table 3) together form a densely packed hydrophobic core with energetically favored T stacking (52) of their aromatic ring systems (Phe95, Trp96, Tyr124, and Trp125). Extensive hydrophobic interactions were also observed between the stretch of amino acids DYINAS (M3) and [F/Y]IAXQGP (M4), which packed together in the PTP crystal structure by arrangement in parallel and anti-parallel β-sheets (Fig. 6). Hydrophobic packing is important for protein structures to gain stability (16). In agreement with this concept, thermosensitive variants of LAR, TC-PTP, and PTP1B (54, 86) were found to contain mutations in the hydrophobic motifs above (M2 to M7), indicating a critical role of these residues in stabilizing the secondary structure of the PTP domain. Moreover, projection of the secondary structure of PTP1B onto the alignment (Fig. 1) and Cα-regiovariation score values (Fig. 5) revealed that the β-sheets and α-helixes in the structural motifs M2 to M6 are dominated by conservative amino acid substitutions, whereas nonconservation mutations frequently have been accepted in the regions flanking these secondary structures. To visualize the conservation of the core of the PTP structure, conserved residues are indicated on the Cα-backbone (Fig. 6).
FIG. 5.

The HCSAGXGR and IAXQGP motifs reside within the most highly conserved microenvironment of the PTP structure. Residues located within a highly conserved three dimensional space of the PTP structure are identified by peaks. The Cα-regiovariation score was calculated using the alignment information in Fig. 1 and the tertiary structure of PTP1B as template. Neighboring residues were defined using a three-dimensional 7-Å sphere of influence. Similar results were obtained for a 5- to 8-Å sphere and when using PTPα, PTPμ, or SHP2 as templates for Cα-regiovariation score analysis (results not shown).
FIG. 6.
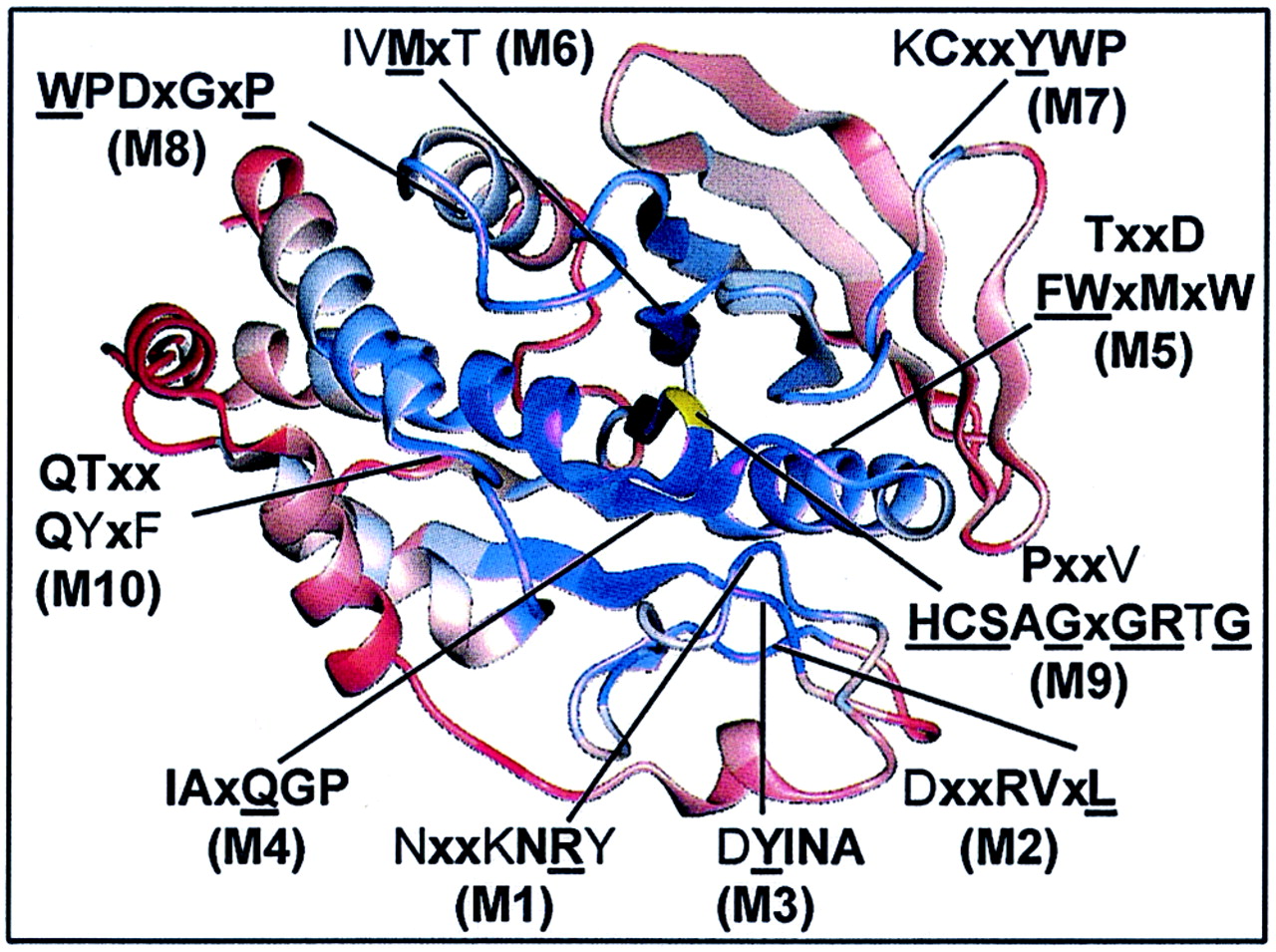
Core structures within the PTP domain are highly conserved and surface loops between secondary structure elements are least conserved. Shown is a ribbon diagram indicating the positions of conserved motifs (M1 to M10) within the tertiary structure. The degree of conservation was determined from Cα-regiovariation score analysis of 37 aligned human PTP catalytic domains (see Fig. 5). Areas of conservation (blue, most conserved; red, least conserved) are illustrated using the PTP1B catalytic domain as the representative tertiary structure. Shown is the front view of PTP1B looking into the active site. The catalytically essential Cys215 residue is shown in yellow.
We found that the functional motif defined by the PTP signature sequence, VHCSXGXGR[T/S]G (M9), together with the structural motif [F/Y]IAxQGP (M4), constitutes the most highly conserved area within the PTP tertiary structure (Fig. 5). Importantly, the C-terminal stretch of residues QGP in motif M4 leads to the termination of a β-sheet and is involved in a bend situated very near to the catalytic cysteine (Cys215) (Fig. 6). Intriguingly, the conserved proline in the [F/Y]IAXQGP motif (Pro87 in PTP1B) is replaced by a cysteine in SHP1 and SHP2, which is likely to result in a more flexible main chain with a greater configurational entropy (27, 92). Whereas the structural motifs are detailed in Table 3, we will discuss further the role of conserved residues in the four motifs (M1 and M8 to M10) that define the catalytic functionality of PTP domains.
PTP signature motif or phosphate-binding loop (motif 9).
The active site sequence VHCSXGXGR[T/S]G (residues 213 to 223 in PTP1B) defines the PTP family and is often referred to as the PTP signature motif or the “PTP loop.” Residues in this motif (M9) form the phosphate-binding loop, which is located at the base of the active site cleft. The cysteine in the PTP signature motif acts as a nucleophile and accepts phosphate transiently during catalysis (25), and the invariant Arg221 is involved in both substrate binding and in the stabilization of the phosphoenzyme intermediate (99). Our Cα-regiovariation score analysis identified two conserved polar residues (Glu115 and Arg257) in a microenvironment of the PTP structure which otherwise has accommodated many amino acid substitutions during evolution (Fig. 5). Importantly, these two residues form hydrogen bonds with the PTP loop, with the invariant Glu115 determining the position of Arg221 through a conserved salt bridge between the carboxy and guanidinium groups. Their invariance among human PTPs highlights their principal role in defining the architecture and function of the phosphate-binding loop. The close proximity of the catalytic Cys215 residue to main-chain amide groups of the PTP loop and hydrogen bonding with both the side chain of Arg221 and the hydroxyl group of Ser222 stabilizes the thiolate (deprotonated) form of the cysteine, favoring its function as a nucleophile (7, 94, 97). Moreover, theoretical investigations revealed that Arg257 may also contribute to stabilizing the nucleophilic nature of the active site cysteine (65). Mutation of the catalytic Cys215 to serine or alanine abrogates all enzyme activity while maintaining affinity for substrates in vitro, a feature that has been successfully utilized to obtain structures of PTPs in complex with phosphotyrosine peptide substrates (37, 74, 75, 91).
Phosphotyrosine recognition loop (motif 1).
Whereas the phosphate group in the substrate phosphotyrosine residue is surrounded by residues corresponding to the PTP signature motif (37, 99), aromatic (Tyr46 and Phe182) and nonpolar (Val49, Ala217, and lle219) amino acids pack with the phenyl ring of the phosphotyrosine and delineate the boundaries of the active site binding pocket (37). The fact that these five residues are conserved by amino acid similarity suggests that the mechanism for recognition of the phosphotyrosine moiety of the substrate is similar among all tyrosine-specific PTPs. Collectively, the residues KNRY (Lys43 to Tyr46) are known as the phosphotyrosine recognition loop (37) since this element (M1) defines the depth of the active site crevice and hence creates selectivity for phosphotyrosine by excluding the hydrolysis of the shorter phosphoserine or phosphothreonine residues in target proteins (37, 91).
WPD loop (motif 8).
The binding of phosphopeptides to the PTP loop promotes a major conformational change in the catalytic site surface loop (residues 179 to 187) that moves several angstroms to close the active site pocket and trap the bound phosphotyrosine (37, 80). The amino acid sequence of this surface loop is quite diverse, except for the WPDXGXP motif that contains a general acid-base catalyst (Asp181) (98). The presence of two proline residues (which do not support hydrogen bonding) and a glycine in the hinge bend region of this segment is critical for the dynamics of the WPD loop motion (63, 64). Comparison of the structures of ligand-free form of the Yersinia PTP (Yop51) and the enzyme complexed with oxyanions (77, 80) has revealed that an interaction between the invariant tryptophan in the WPD loop (Trp354, equivalent to Trp179 in PTP1B) and the above-mentioned arginine in the PTP loop (Arg409, equivalent to Arg221 in PTP1B) plays an important role in closure of the WPD loop. Enzyme kinetic analyses of this PTP has confirmed that mutation of the hinge Trp179 disables catalysis (28, 42). Closure of the WPD loop is critical for phosphoester hydrolysis, since it positions Asp181 close to the scissile oxygen of the tyrosyl substrate, allowing it to donate a proton to the phenolate leaving group (reviewed in references 22 and 95). Consistent with its role as a general acid catalyst, the substitution of Asp181 for an alanine allows phosphorylated substrates to form stable complexes with the enzyme. This “substrate-trapping” mutation has been used to enable isolation and identification of PTP substrates in vitro and in vivo (20, 23, 89, 94). The Asp181-to-alanine mutation creates a more efficient substrate trap than the mutation in which the active site Cys215 is changed to serine or alanine, possibly because the former mutation promotes the hydrophobic properties of the active site cleft and removes the potential for electrostatic repulsion between the Asp181 and the phosphate moiety of the substrate (23, 94).
As can be seen from the alignment in Fig. 1, the WPD loop is strictly conserved among PTP domains but not among domains D2 of RPTPs. Of the RPTPs with a single PTP domain, only PTP-IA2 and PTP-IA2β have accepted non conservative substitutions within the WPD motif, and the substrate trapping alanine mutation (20) occurs naturally in PTP-IA2. Since catalytic activity has not been shown for IA2 in vitro, it has been suggested that its biological function is to compete with catalytically active PTPs for specific substrates preventing their dephosphorylation (49). The protein scaffold of PTP-IA2 could be compatible with protein binding since two point mutations in IA2 (Ala877Asp and Asp911Ala), which in PTP1B is equivalent to the restoration of the general acid Asp181 in the WPD loop and the canonical Ala217 in the PTP loop, is sufficient to reconstitute catalytic activity towards myelin basic protein phosphorylated on tyrosine (49). Another intriguing variation of the WPD loop is observed in four human PTP catalytic domains (PTPD1, PTPS31, PTPλ, and HDPTP) where a longer glutamate residue replaces the general acid Asp181. This is noteworthy because (i) the WPE loop variation is the hallmark of domain D2 sequences, which usually account for less than 0.1% of the total activity of the full-length enzyme (24, 38, 68, 78, 90), and (ii) this replacement in PTP1B leads to a reduction of up to 3 orders of magnitude in catalytic efficiency (20, 90). It will be interesting to see whether these four PTPs have diminished enzyme activity compared to enzymes containing a general acid aspartate residue. However, reconstitution of the WPD motif in domain D2 of RPTPα is not sufficient to increase its catalytic activity to a level comparable to that of domain D1, indicating that there are structural differences other than the general acid-base among PTP domain D1 and D2 (90) (see below).
Catalytic-water motif or Q loop (motif 10).
In the QTXXQYXF motif (M10), two glutamine residues (Gln262 and Gln266) and two conserved arginine residues (Arg254 and Arg257) N terminal to this motif form crucial hydrogen bonds with interacting residues of the PTP loop and its amide backbone. In particular, Gln262 positions and activates an active site water molecule involved in the second hydrolysis step of the phosphocysteine enzyme complex (61, 100). Enzyme kinetic analysis of the Yersinia PTP combined with site-directed mutagenesis have revealed that Gln446 (equivalent to Gln262 in PTP1B) and, to a lesser extent, Gln450 (equivalent to Gln266 in PTP1B) is responsible for restricting phosphoryl transfer from the phosphoenzyme intermediate to water and not to other nucleophile acceptors (i.e., preventing the phosphoenzyme intermediate from acting as a kinase phosphorylating undesirable substrates [100]). Notably, within this highly variable area of the PTP structure (Fig. 5), only these arginine and glutamine residues are invariant, consistent with their involvement in catalysis (74, 100) and critical hydrogen bonding with residues of the PTP loop (80).
Conservation of surface-exposed amino acids in vicinity of active site: comparison between cytoplasmic PTPs and RPTP domains D1 and D2.
In an attempt to reveal novel structure-function relationships, we performed the Cα-regiovariation score analysis on three subsets of PTP domains: (i) nontransmembrane PTPs, (ii) receptor-like D1, and (iii) receptor-like D2 (Fig. 7A, B, and C). The crystal structure of PTP1B was used as a template for the intracellular PTPs, whereas the molecular surface conservation among the aligned RPTP domain D1 and D2 sequences was illustrated using the X-ray crystal structure of PTPα domain D1 (50). The interchangeable use of the PTP catalytic domains of PTP1B and PTPα for the calculation of the Cα-regiovariation score values is justified by the excellent overlay of their tertiary structure, with a root mean square (RMS) deviation of 1.35 Å between the two domains (the RMS deviations between other PTP domain tertiary structures are given in the legend to Fig. 4). Although conserved residues converged around the active site for the intracellular PTPs and for domain D1 of RPTPs (Fig. 7A and B, respectively), the domain D2 sequences exhibited a much greater variation in the vicinity of the active site (Fig. 7C).
FIG. 7.

PTP domains from cytoplasmic PTPs and RPTP domains D1 and D2 show significant differences in their conservation of surface-exposed amino acids. Shown is surface conservation (blue, most conserved; red, least conserved) of PTP domains from nontransmembrane PTPs (A), RPTP domains D1 (B), and RPTP domains D2 (C). Shown is the front view looking into the active site. Cα-regiovariation score values for the cytoplasmic PTPs are illustrated using the X-ray crystal structure of PTP1B with the catalytically essential Cys215 (yel low) and epidermal growth factor receptor-derived peptide (green) bound within the active site (closed conformation). For ease of comparison, Cα-regiovariation score values among RPTP domains D1 and D2 sequences are illustrated using the X-ray crystal structure of RPTPα domain D1 (50). The EGFR peptide (green) is modeled in the active site of RPTPα for orientation using only a closed conformation of the X-ray crystal structures. Amino acids are labeled according to the residue position in human PTP1B with the equivalent residues in RPTPα given in brackets (A and B). The conserved four-residue structural linker located at the N terminus of domain D2 (encircled area 1 in panel C), and which constrains the relative orientation of tandem PTP domains in LAR, is compared to the corresponding nonconserved area for the RPTP domain D1 sequences (encircled area 1 in panel B). The amino acid residues defining this conserved linker are boxed and colored yellow in the alignment in Fig. 1.
Our analysis of domain D2 sequences revealed several intriguing aspects of tandem domain RPTPs. Thus, the domains D2 align extremely well with the catalytically active PTP sequences (with CD45 accommodating an acidic insert of 20 amino acids; Fig. 1), yet all the domains D2 are phylogenetically distinct from domains D1 (i.e., D2 sequences do not cluster together with D1 sequences in the phylogenetic tree but define a separate subfamily of PTP domains (data available at http://science.novonordisk.com/ptp). In fact, the sequence similarity between domains D2 of LAR, RPTPς, and RPTPδ (subtype R2B) is even higher than that between the corresponding domain D1 sequences (45). Since phylogenetic analyses have shown that PTP domain duplication (occurring in five out of nine RPTP subfamilies) happened very early in evolution (59) it can be argued that there must be a separate function of the membrane distal domain in order for these amino acids to be conserved at the present level. Noticeably, both regulatory (38) and substrate-binding (78) functions have been proposed for these domains.
Most of the invariant amino acids in domain D1, which show considerable substitution in domains D2, converge around the active site. Therefore, it is noteworthy that only two point mutations (which restore the equivalent of Tyr46 within the NXXKNRY motif) and the general acid equivalent of Asp181 (within the WPD loop) are sufficient to confer a robust PTP activity back to domain D2 of some RPTPs, including PTPs (48), PTPα (9, 47), and LAR (56). However, domains D2 of other RPTPs, such as CD45, PTPζ, and RPTPγ, have additional critical substitutions in several amino acids in the PTP signature motif and, therefore, are most likely to be truly inactive (48) (Fig. 1). Nevertheless, the structural architecture of the active site signature motif of domains D2 may still be sufficiently preserved to retain the capacity to bind phosphotyrosine-containing proteins. Thus, the function of domain D2, at least for some PTPs, may be similar to that of other tyrosine-phosphate recognition units, such as SH2 domains (62) and phoshotyrosine binding domains (21). In this regard, the Cα-regiovariation score analysis of the domain D2 sequences (Fig. 7C), with the highly variable molecular surface area surrounding the phosphate-binding site, would signify the preference for divergent and probably highly selective protein binding partners. Such a potential is illustrated by domain D2 of CD45, which has been shown to be critical for interleukin-2 secretion and substrate recruitment of TCR-ζ (41).
Identification of conserved interface between domains D1 and D2 of RPTPs.
When comparing the RPTP domains D1 and D2 sequences by using the X-ray crystal structure of RPTPα domain D1 as template (79), the encircled area 1 in Fig. 7 was found to be highly conserved among RPTP domain D2 sequences (Fig. 7C) but not among RPTP domain D1 sequences (Fig. 7B). Significantly, the residues in this area (Fig. 1, boxed in yellow) were recently identified as the structural linker that constrains the relative orientation of the two PTP domains in LAR (56). Although this is so far the only report describing the X-ray crystal structure of the tandem arrangement of PTP domains, our alignment reveals that this structural linker is highly conserved among all tandem domain-containing RPTPs, suggesting conservation in function. The consensus motif for this four-residue linker is G[D/E]TE (Fig. 1, highlighted in yellow).
In addition to the structural linker, our Cα-regiovariation score analysis of RPTP domains D1 and D2 identified additional conserved residues (Fig. 8B and C, encircled area II), which were confined to the region of the PTP tertiary structure that correlates with the interdomain interface revealed by the X-ray crystal structure of LAR (56). Again, the Cα-regiovariation score analysis predicted the exact location of the interface in domain D2, including hydrogen bonding polar residues and conserved hydrophobic residues responsible for the extensive van der Waals interactions and tight complementary fit described for the interface between domains D1 and D2 of LAR (56). For the PTP domains in Fig. 8B, the center of conservation is less focused on the exact residues involved in this interface, but when the single-domain RPTPs (subtypes R3, R7, and R8) were removed from the Cα-regiovariation score analysis, the conservation of this area in domain D1 became even more apparent (not shown). Significantly, the lack of conservation of this region for the nontransmembrane PTPs (Fig. 8A) illustrates that this interface is unique to tandem domain RPTPs (Fig. 8B and C).
FIG. 8.

Identification of novel conserved area on surface of PTP domain opposite active site. Shown are surface conservation (Cα-regiovariation score values) among nontransmembrane PTPs (A), RPTP domains D1 (B), and RPTP domains D2 (C). The tertiary structure is rotated 180° compared to structures in Fig. 7, showing the surface of the molecule opposite the active site. Encircled area II (B and C) corresponds to the interface for domains D1 and D2 as revealed in the X-ray crystal structure of LAR (56). Encircled area III is a novel putative interactive site, which appears to be conserved in all three subsets of PTP domain sequences. Amino acids are labeled according to the residue positions in PTP1B (A) and RPTPα (B and C).
Identification of novel conserved pocket on surface of PTP domain opposite active site.
In addition to the identification of conserved regions surrounding the active site and at the interface between domains D1 and D2 of RPTPs, the Cα-regiovariation score analysis identified another focus of conservation that was confined to the area of the molecule opposite the active site (Fig. 8A, B, and C, encircled area III). This surface-accessible area of conservation extends above a shallow hydrophobic pocket formed by residues IIe57, Ala69, IIe82, and Met98. These four residues have adopted a configuration that accommodates extensive van der Waals interactions and only one PTP (the Xenopus, mouse, and human orthologs of MEG2) has accepted nonconservative changes to residues within this pocket. In addition, residues from motif 2 (DXXRVXL) and motif 5 (TXXDFWXMXW) contribute to this conserved microenvironment, which explains why this cluster of conservation can be identified only when the PTP chain fold is considered.
Size and physiochemical nature of conserved pocket is consistent with recognition site for protein-protein interaction.
Statistical examination of protein-protein associations suggests a central role for hydrophobic residues at interfacial regions (40, 67). In terms of amino acid composition at protein-protein domain interfaces, it has been noted that there is a preference for larger, nonpolar residues, particularly aromatic amino acids, as well as a few key basic or acidic residues (87). Indeed, from the alignment analysis, we observed that the conserved residues residing on the rim and within the shallow pocket (Fig. 8, encircled area III) are more hydrophobic and aromatic than the remainder of the surface of the PTP molecule, with several charged residues surrounding the hydrophobic pocket. Therefore, the amino acid composition in this region of the PTP molecule is consistent with the identification of a possible novel site of interaction. The size of the solvent-accessible area that appears conserved (i.e., the dark blue area) is ∼250 Å2. Usually, the area of protein-protein interaction surfaces are larger (∼700 Å2), typically constituting from 7 to 30% of the total surface area of a monomer (39, 87). However, extensive additional surface area can be included in this putative interaction site of the PTP domain if the adjacent less-conserved residues (i.e., the white area in Fig. 8) are considered in the evaluation.
In conclusion, the regiovariation score analysis has led to the identification of the catalytic active site, the conserved linker between domains D1 and D2 as revealed by the X-ray crystal structure of LAR, as well as the approximate surface area of interaction between domain D1 and D2. In addition, our analysis highlights a focus of conservation on the surface of the PTP domain opposite the active site. Although the conservation of this pocket may be of structural importance, it is tempting to speculate the existence of additional roles for this site in effector interactions with other protein domains or signaling molecules. However, mutational and functional studies of appropriate PTP mutants will be necessary to corroborate the significance of residues in this area of the PTP domain.
Identification of nonconserved residues surrounding active site—implications for substrate recognition and inhibitor design.
In addition to the identification of three-dimensionally conserved regions, the Cα-regiovariation score analysis offers a unique opportunity to identify areas in a protein family that are less well conserved and therefore might indicate a specialized function. As an example, analysis of areas in the proximity of the active site of enzymes, here PTPs, may lead to the identification of putative substrate-binding pockets. Furthermore, such analysis in conjunction with primary sequence alignments may allow the identification of unique combinations of amino acid residues that can be addressed in a structure-based design of selective inhibitors.
At present, two PTPs have been cocrystallized with peptide substrates, PTP1B (37, 74, 75) and SHP1 (91). Although significant differences in the binding modes were observed (see below), both studies show peptide binding to residues defined by the α1/β1 loop and the M10 motif (α5-loop-α6) (Fig. 1). Some of these residues, such as Tyr46 and Gln262, are highly conserved and hence likely to be involved in the binding or catalysis of all phosphotyrosine substrates. However, other residues are quite variable and are potentially responsible for defining substrate selectivity. Significantly, our structural alignment analysis identifies at least four areas in proximity to the active site which are nonconserved and for which involvement in the recognition of peptides and small molecule ligands have been documented in biochemical and crystallographic studies (Fig. 9). Although no single residue in these areas appears to be a unique hallmark of any particular PTP, the combination of residues in these areas is unique and could consequently represent a selectivity-determining region. This is probably most apparent for the region defined by residues 47, 48, 258, and 259 of PTP1B (Fig. 9). In agreement with this, we have recently shown that residue 259 is a key determinant in substrate recognition and catalysis (66). Thus, the residue at position 259 in PTP1B is a glycine, which forms the bottom of an open cleft that creates access to a second binding pocket adjacent to the active site. This structural feature in PTP1B, together with the plasticity conferred by Arg47 in accommodating either acidic residues at the P-1 and P-2 positions in the substrate (as illustrated in the EGFR988-993 peptide) (37) or hydrophobic residues at P-1 (75), explains why PTP1B is able to accommodate a broad range of artificial peptide substrates in vitro. In contrast, PTPs with bulky residues at the position equivalent to 259 in PTP1B including RPTPα and LAR show more limited peptide recognition capacity in vitro. We have shown recently by kinetic and X-ray crystallographic studies that replacing the bulky Gln259 residue in PTPα with a glycine converts PTPα into a PTP1B-like enzyme and vice versa (66). However, in a physiological context, the presence of a glycine at position 259 in PTP1B allows for high-affinity binding of substrates, such as the activation loop of the insulin receptor, which contain two adjacent phosphotyrosine residues (74). Thus, the simultaneous engagement of the substrate phosphotyrosine residue in the active site and the adjacent phosphotyrosine residue at a second substrate-binding pocket may make an important contribution to the substrate recognition by PTP1B in vivo.
FIG. 9.
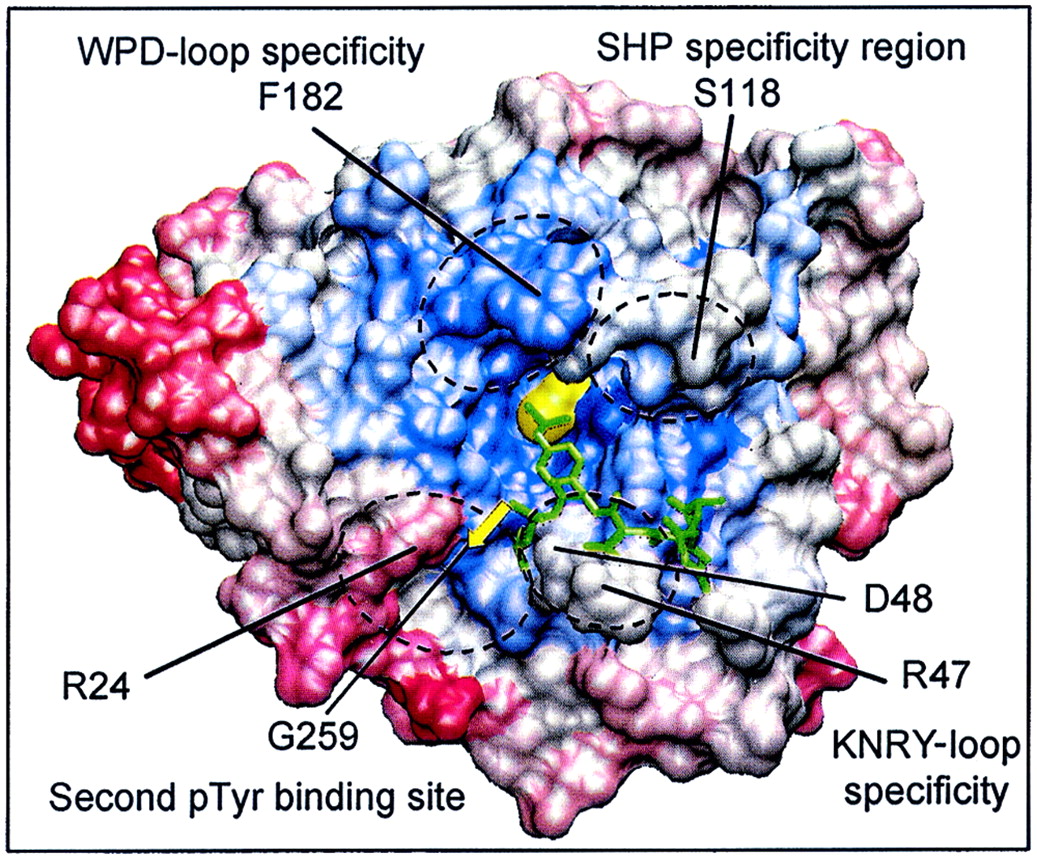
Nonconserved amino acids in the proximity of the PTP active site are involved in the recognition of PTP substrates and nonpeptide PTP inhibitors. Shown is the visualization of four selectivity-determining regions on the molecular surface of PTP1B. Areas of conservation (blue, most conserved; red, least conserved) represent the Cα-regiovariation score values of 37 aligned human PTP catalytic domains (values from Fig. 5). The amino acids involved in defining these four selectivity-determining regions are indicated (boxed) in the alignment in Fig. 1.
The most important residues in this second phosphotyrosine binding site of PTP1B appear to be Arg24 and Arg254. Although Arg254 is a highly conserved residue, the presence of Arg24 and Gly259 seems to be unique to PTP1B and TC-PTP. The tethering together of a ligand that simultaneously occupies the active site with a ligand that interacts with residues of this second phosphotyrosine binding pocket has been suggested as a paradigm for PTP1B inhibitor design (69) resulting in remarkably selective bis(aryldifluorophosphonate) inhibitors of PTP1B (81). Furthermore, and consistent with this paradigm, Ramachadran and colleagues have recently reported that peptides containing two nonhydrolyzable analogs of phosphotyrosine [difluoro(phospho)methyl-phenylalanine] were potent and specific inhibitors of PTP1B, illustrating that exquisite substrate and inhibitor selectivity exists in close vicinity to the active sites of PTPs (15).
In addition, other areas in the proximity of the active site show considerable variability and could potentially be involved in defining substrate specificity (Fig. 9). One such area is defined by β5-loop-β6 (i.e., between the M6 and M7 motifs). Whereas no substrate binding has been observed in this region in PTP1B, a study of SHP1 that had cocrystallized with two different synthetic peptides revealed significant interaction within this region, in one case due to salt bridge formation between AspP-4 in the peptide substrate and Arg360 in SHP-1 (corresponding to S118 in PTP1B) (91). Interestingly, this region is quite different in SHP2. Therefore, these X-ray crystallographic studies provide structural support for the observed different substrate specificities of these closely related SH2 domain-containing PTPs, as demonstrated in two elegant catalytic-domain-swapping experiments (60, 82). Although no direct interaction has been observed in the crystal structure of PTP1B complexed with peptide substrates, computational studies suggest a similar role of this region in substrate recognition by PTP1B (64). However, for the SHP1 cocrystal, it should be noted that binding of peptide substrate to the PTP catalytic domain did not bring the WPD loop into its closed conformation (91), thus raising the question of whether it is a catalytically competent complex.
Since such unique aspects of the structure in the vicinity of the active site contribute to substrate specificity, we investigated whether selective nonphosphonate, nonpeptide inhibitors of PTP1B could be obtained by addressing one of these regions. Our attention was directed to Asp48 and Arg47 in PTP1B. As a starting point, we used a general PTP inhibitor, 2-(oxalylamino)-benzoic acid, which we had identified by high-throughput screening of the Novo Nordisk compound library. We reasoned that a correctly positioned basic nitrogen in the inhibitor would be able to form a salt bridge with the side chain of Asp48 in PTP1B, whereas an asparagine, which is found in the equivalent position in many other PTPs, would cause repulsion (Fig. 1). Indeed, a low-molecular-weight, nonphosphorus compound containing such a basic nitrogen displayed a remarkable selectivity for PTP1B (36). Recently, studies with PTP1B knockout mice (17, 44) and PTP1B antisense oligonucleotides have provided compelling evidence that inhibition of PTP1B may be an effective approach for the treatment of diabetes and obesity (53). The identification of selectivity-determining regions suggests that it may be possible to generate specific inhibitors of PTP1B for use in this context. Furthermore, it is now becoming apparent that the inhibition of other members of the PTP family may offer novel strategies for therapeutic interaction in various human diseases. We hope that the analysis presented here will not only assist in further characterization of the PTP family but also may contribute to the development of selective inhibitors of other potential drug targets within the PTP family.
ACKNOWLEDGMENTS
This work was supported by an industrial Ph.D. fellowship from the Danish Academy of Technical Sciences (J.N.A) and grants from the NIH (RO1 CA53840 and GM 55989) and the Mellam Family Foundation (N.K.T).
We thank Yu Shen for helpful discussions on the human genome databases.
REFERENCES
- 1.Alexander D R. The CD45 tyrosine phosphatase: a positive and negative regulator of immune cell function. Semin Immunol. 2000;12:349–359. doi: 10.1006/smim.2000.0218. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Andersen J N, Elson A, Lammers R, Romer J, Clausen J, Moller K B, Moller N P H. Comparative study of protein tyrosine phosphatase epsilon isoforms: membrane localization confers specificity in cellular signalling. Biochem J. 2001;354:581–590. doi: 10.1042/0264-6021:3540581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angers-Loustau A, Cote J F, Tremblay M L. Roles of protein tyrosine phosphatases in cell migration and adhesion. Biochem Cell Biol. 1999;77:493–505. [PubMed] [Google Scholar]
- 5.Apweiler R, Attwood T K, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning M D, Durbin R, Falquet L, Fleischmann W, Gouzy J, Hermjakob H, Hulo N, Jonassen I, Kahn D, Kanapin A, Karavidopoulou Y, Lopez R, Marx B, Mulder N J, Oinn T M, Pagni M, Servant F. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barford D, Das A K, Egloff M P. The structure and mechanism of protein phosphatases—insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 7.Barford D, Flint A J, Tonks N K. Crystal-structure of human protein-tyrosine-phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 8.Brady-Kalnay S M, Tonks N K. Protein-tyrosine phosphatases as adhesion receptors. Curr Opin Cell Biol. 1995;7:650–657. doi: 10.1016/0955-0674(95)80106-5. [DOI] [PubMed] [Google Scholar]
- 9.Buist P, Zhang Y L, Keng Y F, Wu L, Zhang Z Y, den Hertog J. Restoration of potent protein-tyrosine phosphatase activity into the membrane-distal domain of receptor protein-tyrosine phosphatase alpha. Biochemistry. 1999;38:914–922. doi: 10.1021/bi981936b. [DOI] [PubMed] [Google Scholar]
- 10.Cardle L, Dufton M J. Identification of important functional environs in protein tertiary structures from the analysis of residue variation in 3D—application to cytochromes-c and carboxypeptidase-a and carboxypeptidase-b. Protein Eng. 1994;7:1423–1431. doi: 10.1093/protein/7.12.1423. [DOI] [PubMed] [Google Scholar]
- 11.Cardle L, Dufton M J. Foci of amino-acid residue conservation in the 3D structures of proteinase-inhibitors—how do variants from snake-venom differ. Protein Eng. 1997;10:131–136. doi: 10.1093/protein/10.2.131. [DOI] [PubMed] [Google Scholar]
- 12.Cho H, Ramer S E, Itoh M, Kitas E, Bannwarth W, Burn P, Saito K, Walsh C T. Catalytic domains of LAR and CD45 protein tyrosine phosphatases from Escherichia coli expression systems: purification and characterization for specificity and mechanism. Biochemistry. 1992;31:133–138. doi: 10.1021/bi00116a019. [DOI] [PubMed] [Google Scholar]
- 13.Denu J M, Lohse D L, Vijayalakshmi J, Saper M A, Dixon J E. Visualization of intermediate and transition-state structures in protein-tyrosine-phosphatase catalysis. Proc Natl Acad Sci USA. 1996;93:2493–2498. doi: 10.1073/pnas.93.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denu J M, Tanner K G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 15.Desmarais S, Friesen R W, Zamboni R, Ramachandran C. [Difluro(phosphono)methyl]phenylalanine-containing peptide inhibitors of protein tyrosine phosphatases. Biochem J. 1999;337:219–223. [PMC free article] [PubMed] [Google Scholar]
- 16.Dill K A. Polymer principles and protein folding. Protein Sci. 1999;8:1166–1180. doi: 10.1110/ps.8.6.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy A L, Normandin D, Cheng A, Himms-Hagen J, Chan C C, Ramachandran C, Gresser M J, Tremblay M L, Kennedy B P. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 18.Elson A, Leder P. Identification of a cytoplasmic, phorbol ester-inducible isoform of protein-tyrosine-phosphatase epsilon. Proc Natl Acad Sci USA. 1995;92:12235–12239. doi: 10.1073/pnas.92.26.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer E H. Cell signaling by protein tyrosine phosphorylation. Adv Enzyme Regul. 1999;39:359–369. doi: 10.1016/s0065-2571(98)00014-4. [DOI] [PubMed] [Google Scholar]
- 20.Flint A J, Tiganis T, Barford D, Tonks N K. Development of substrate-trapping mutants to identify physiological substrates of protein-tyrosine phosphatases. Proc Natl Acad Sci USA. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forman-Kay J D, Pawson T. Diversity in protein recognition by PTB domains. Curr Opin Struct Biol. 1999;9:690–695. doi: 10.1016/s0959-440x(99)00031-7. [DOI] [PubMed] [Google Scholar]
- 22.Frisch S M, Vuori K, Ruoslahti E, Chanhui P Y. Control of adhesion-dependent cell-survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garton A, Flint A J, Tonks N K. Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gebbink M F B G, Verheijen M H G, Zondag G C M, Vanetten I, Moolenaar W H. Purification and characterization of the cytoplasmic domain of human receptor-like protein-tyrosine-phosphatase RPTPμ. Biochemistry. 1993;32:13516–13522. doi: 10.1021/bi00212a017. [DOI] [PubMed] [Google Scholar]
- 25.Guan K L, Dixon J E. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J Biol Chem. 1991;266:17026–17030. [PubMed] [Google Scholar]
- 26.Hanks S K, Quinn A M. Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- 27.Hof P, Pluskey S, Dhepaganon S, Eck M J, Shoelson S E. Crystal-structure of the tyrosine phosphatase shp-2. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 28.Hoff R H, Hengge A C, Wu L, Keng Y F, Zhang Z Y. Effects on general acid catalysis from mutations of the invariant tryptophan and arginine residues in the protein tyrosine phosphatase from Yersinia. Biochemistry. 2000;39:46–54. doi: 10.1021/bi991570i. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmann K M V, Tonks N K, Barford D. The crystal-structure of domain-1 of receptor protein-tyrosine-phosphatase-μ. J Biol Chem. 1997;272:27505–27508. doi: 10.1074/jbc.272.44.27505. [DOI] [PubMed] [Google Scholar]
- 30.Hubbard S R, Till J H. Protein tyrosine kinase structure and function Annu. Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 31.Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 32.Hunter T. Protein-kinases and phosphatases—the yin and yang of protein-phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 33.Hunter T. The Onian-lecture 1997—the phosphorylation of proteins on tyrosine—its role in cell-growth and disease. Philos Trans R Soc Lond Biol Sci. 1998;353:583–605. doi: 10.1098/rstb.1998.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser M J, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 35.Ibarra-Sanchez M, Simoncic P D, Nestel F R, Duplay P, Lapp W S, Tremblay M L. The T-cell protein tyrosine phosphatase. Semin Immunol. 2000;12:379–386. doi: 10.1006/smim.2000.0220. [DOI] [PubMed] [Google Scholar]
- 36.Iversen L F, Andersen H S, Branner S, Mortensen S B, Peters G H, Norris K, Olsen O H, Jeppesen C B, Lundt B F, Ripka W, Moller K B, Moller N P H. Structure-based design of a low molecular weight, nonphosphorus, nonpeptide, and highly selective inhibitor of protein-tyrosine phosphatase 1B. J Biol Chem. 2000;275:10300–10307. doi: 10.1074/jbc.275.14.10300. [DOI] [PubMed] [Google Scholar]
- 37.Jia Z C, Barford D, Flint A J, Tonks N K. Structural basis for phosphotyrosine peptide recognition by protein- tyrosine-phosphatase 1B. Science. 1996;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 38.Johnson P, Ostergaard H L, Wasden C, Trowbridge I S. Mutational analysis of CD45, a leukocyte-specific protein tyrosine phosphatase. J Biol Chem. 1992;267:8035–8041. [PubMed] [Google Scholar]
- 39.Jones S, Thornton J M. Protein-protein interactions: a review of protein dimer structures. Prog Biophys Mol Biol. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 40.Jones S, Thornton J M. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kashio N, Matsumoto W, Parker S, Rothstein D M. The second domain of the CD45 protein tyrosine phosphatase is critical for interleukin-2 secretion and substrate recruitment of TCR-zeta in vivo. J Biol Chem. 1998;273:33856–33863. doi: 10.1074/jbc.273.50.33856. [DOI] [PubMed] [Google Scholar]
- 42.Keng Y F, Wu L, Zhang Z Y. Probing the function of the conserved tryptophan in the flexible loop of the Yersinia protein-tyrosine phosphatase. Eur J Biochem. 1999;259:809–814. doi: 10.1046/j.1432-1327.1999.00090.x. [DOI] [PubMed] [Google Scholar]
- 43.Keyse S M. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 44.Klaman L D, Boss O, Peroni O D, Kim J K, Martino J L, Zabolotny J M, Moghal N, Lubkin M, Kim Y B, Sharpe A H, Stricker-Krongrad A, Shulman G I, Neel B G, Kahn B B. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krueger N X, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990;9:3241–3252. doi: 10.1002/j.1460-2075.1990.tb07523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Dixon J E. Form, function, and regulation of protein tyrosine phosphatases and their involvement in human diseases. Semin Immunol. 2000;12:75–84. doi: 10.1006/smim.2000.0209. [DOI] [PubMed] [Google Scholar]
- 47.Lim K L, Kolatkar P R, Neg K P, Neg C H, Pallen C J. Interconversion of the kinetic identities of the tandem catalytic domains of receptor-like protein-tyrosine phosphatase PTP alpha by two point mutations is synergistic and substrate-dependent. J Biol Chem. 1998;273:28986–28993. doi: 10.1074/jbc.273.44.28986. [DOI] [PubMed] [Google Scholar]
- 48.Lim K L, Neg C H, Pallen C J. Catalytic activation of the membrane distal domain of protein tyrosine phosphatase epsilon, but not CD45, by two point mutations. Biochim Biophys Acta. 1999;1434:275–283. doi: 10.1016/s0167-4838(99)00189-2. [DOI] [PubMed] [Google Scholar]
- 49.Magistrelli, G., S. Toma, and A. Isacchi. Substitution of 2 variant residues in the protein-tyrosine phosphatase-like PTP35/IA-2 sequence reconstitutes catalytic activity. Biochem. Biophys. Res. Commun. 227:581–588. [DOI] [PubMed]
- 50.Majeti R, Bilwes A M, Noel J P, Hunter T, Weiss A. Dimerization-induced inhibition of receptor protein-tyrosine-phosphatase function through an inhibitory wedge. Science. 1998;279:88–91. doi: 10.1126/science.279.5347.88. [DOI] [PubMed] [Google Scholar]
- 51.Mauro L J, Dixon J E. Zip codes direct intracellular protein-tyrosine phosphatases to the correct cellular address. Trends Biochem. 1994;19:151–155. doi: 10.1016/0968-0004(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 52.McGaughey G B, Gagne M, Rappe A K. π-Stacking interactions. Alive and well in proteins. J Biol Chem. 1998;273:15458–15463. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 53.Moller N P H, Iversen L F, Andersen H S, McCormack J J. Protein tyrosine phosphatases (PTPs) as drug targets: inhibitors of PTP-1B for the treatment of diabetes. Curr Opin Drug Discov Dev. 2000;3:527–540. [PubMed] [Google Scholar]
- 54.Mulse E S, Vrielink A, Ennis M A, Lemieux N H, Tremblay M L. Thermosensitive mutants of the mPTP1B and hPTP1B protein-tyrosine phosphatases—isolation and structural-analysis. Protein Sci. 1996;5:604–613. doi: 10.1002/pro.5560050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mustelin T, Brockdorff J, Rudbeck L, Gjorloff-Wingren A, Han S, Wang X, Tailor P, Saxena M. The next wave: protein tyrosine phosphatases enter T cell antigen receptor signalling. Cell Signal. 1999;11:637–650. doi: 10.1016/s0898-6568(99)00016-9. [DOI] [PubMed] [Google Scholar]
- 56.Nam H J, Poy F, Krueger N X, Saito H, Frederick C A. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97:449–457. doi: 10.1016/s0092-8674(00)80755-2. [DOI] [PubMed] [Google Scholar]
- 57.Neel B G, Tonks N K. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 58.Ohsugi M, Kuramochi S, Matsuda S, Yamamoto T. Molecular-cloning and characterization of a novel cytoplasmic protein-tyrosine-phosphatase that is specifically expressed in spermatocytes. J Biol Chem. 1997;272:33092–33099. doi: 10.1074/jbc.272.52.33092. [DOI] [PubMed] [Google Scholar]
- 59.Ono K, Suga H, Iwabe N, Kuma K, Miyata T. Multiple protein tyrosine phosphatases in sponges and explosive gene duplication in the early evolution of animals before the parazoan-eumetazoan split. J Mol Evol. 1999;48:654–662. doi: 10.1007/pl00006509. [DOI] [PubMed] [Google Scholar]
- 60.O'Reilly A M, Neel B G. Structural determinants of SHP-2 function and specificity in Xenopusmesoderm induction. Mol Cell Biol. 1998;18:161–177. doi: 10.1128/mcb.18.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pannifer A D, Flint A J, Tonks N K, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J Biol Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 62.Pawson T. Protein modules and signaling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 63.Peters G H, Frimurer T M, Andersen J N, Olsen O H. Molecular dynamics simulations of protein-tyrosine phosphatase 1B. I. Ligand-induced changes in the protein motions. Biophys J. 1999;77:505–515. doi: 10.1016/S0006-3495(99)76907-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peters G H, Frimurer T M, Andersen J N, Olsen O H. Molecular dynamics simulations of protein-tyrosine phosphatase 1B. II. Substrate-enzyme interactions and dynamics. Biophys J. 2000;78:2191–2200. doi: 10.1016/S0006-3495(00)76768-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peters G H, Frimurer T M, Olsen O H. Electrostatic evaluation of the signature motif (H/V)CX5R(S/T) in protein-tyrosine phosphatases. Biochemistry. 1998;37:5383–5387. doi: 10.1021/bi971187i. [DOI] [PubMed] [Google Scholar]
- 66.Peters G H, Iversen L F, Branner S, Andersen H S, Mortensen S B, Olsen O H, Moller K B, Moller N P H. Residue 259 is a key determinant of substrate specificity of protein-tyrosine phosphatases 1B and alpha. J Biol Chem. 2000;275:18201–18209. doi: 10.1074/jbc.M910273199. [DOI] [PubMed] [Google Scholar]
- 67.Pixley F J, Lee P S W, Dominguez M G, Einstein D B, Stanley E R. A heteromorphic protein-tyrosine-phosphatase, PTPphi, is regulated by CSF-1 in macrophages. J Biol Chem. 1995;270:27339–27347. doi: 10.1074/jbc.270.45.27339. [DOI] [PubMed] [Google Scholar]
- 68.Pot D A, Woodford T A, Remboutsika E, Haun R S, Dixon J E. Cloning, bacterial expression, purification, and characterization of the cytoplasmic domain of rat LAR, a receptor-like protein tyrosine phosphatase. J Biol Chem. 1991;266:19688–19696. [PubMed] [Google Scholar]
- 69.Puius Y A, Zhao Y, Sullivan M, Lawrence D S, Almo S C, Zhang Z Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc Natl Acad Sci USA. 1997;94:13420–13425. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramponi G, Stefani M. Structural, catalytic, and functional-properties of low molecular weight phosphotyrosine protein phosphatases—evidence of a long evolutionary history. Int J Biochem Cell Biol. 1997;29:279–292. doi: 10.1016/s1357-2725(96)00109-4. [DOI] [PubMed] [Google Scholar]
- 71.Robertson S C, Tynan J A, Donoghue D J. RTK mutations and human syndromes when good receptors turn bad. Trends Genet. 2000;16:265–271. doi: 10.1016/s0168-9525(00)02077-1. [DOI] [PubMed] [Google Scholar]
- 72.Robinson D R, Wu Y M, Lin S F. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 73.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 74.Salmeen A, Andersen J N, Myers M P, Tonks N K, Barford D. Molecular basis for recognition and dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 75.Sarmiento M, Puius Y A, Vetter S W, Keng Y F, Wu L, Zhao Y, Lawrence D S, Almo S C, Zhang Z Y. Structural basis of plasticity in protein tyrosine phosphatase 1B substrate recognition. Biochemistry. 2000;39:8171–8179. doi: 10.1021/bi000319w. [DOI] [PubMed] [Google Scholar]
- 76.Sarmiento M, Zhao Y, Gordon S J, Zhang Z Y. Molecular-basis for substrate-specificity of protein-tyrosine-phosphatase 1B. J Biol Chem. 1998;273:26368–26374. doi: 10.1074/jbc.273.41.26368. [DOI] [PubMed] [Google Scholar]
- 77.Schubert H L, Fauman E B, Stuckey J A, Dixon J E, Saper M A. A ligand-induced conformational change in the Yersinia protein-tyrosine-phosphatase. Protein Sci. 1995;4:1904–1913. doi: 10.1002/pro.5560040924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Streuli M, Krueger N X, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J. 1990;9:2399–2407. doi: 10.1002/j.1460-2075.1990.tb07415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stuckey J A, Schubert H L, Fauman E B, Dixon J E, Saper M A. The crystal-structure of the Yersinia protein-tyrosine-phosphatase yop51. FASEB J. 1994;8:18780–18788. [Google Scholar]
- 80.Stuckey, J. A., H. L. Schubert, E. B. Fauman, Z. Y. Zhang, J. E. Dixon, and M. A. Saper. Crystal-structure of Yersinia protein-tyrosine-phosphatase at 2.5 angstrom and the complex with tungstate. Nature 370:571–575. [DOI] [PubMed]
- 81.Taing M, Keng Y F, Shen K, Wu L, Lawrence D S, Zhang Z Y. Potent and highly selective inhibitors of the protein tyrosine phosphatase 1B. Biochemistry. 1999;38:3793–3803. doi: 10.1021/bi9813781. [DOI] [PubMed] [Google Scholar]
- 82.Tenev T, Keilhack H, Tomic S, Stoyanov B, Steingerlach M, Lammers R, Krivtsov A V, Ullrich A, Bohmer F D. Both SH2 domains are involved in interaction of shp-1 with the epidermal growth-factor receptor but cannot confer receptor-directed activity to shp-1/shp-2 chimera. J Biol Chem. 1997;272:5966–5973. doi: 10.1074/jbc.272.9.5966. [DOI] [PubMed] [Google Scholar]
- 83.Tonks N K, Neel B G. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 84.Tonks N K, Diltz C D, Fischer E H. Purification of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. 1988;263:6722–6730. [PubMed] [Google Scholar]
- 85.Toyooka S, Ouchida M, Jitsumori Y, Tsukuda K, Sakai A, Nakamura A, Shimizu N, Shimizu K. HD-PTP: a novel protein tyrosine phosphatase gene on human chromosome 3p21.3. Biochem Biophys Res Commun. 2000;278:671–678. doi: 10.1006/bbrc.2000.3870. [DOI] [PubMed] [Google Scholar]
- 86.Tsai A Y, Itoh M, Streuli M, Thai T, Saito H. Isolation and characterization of temperature-sensitive and thermostable mutants of the human receptor-like protein tyrosine phosphatase LAR. J Biol Chem. 1991;266:10534–10543. [PubMed] [Google Scholar]
- 87.Tsai C J, Lin S L, Wolfson H J, Nussinov R. Studies of protein-protein interfaces: a statistical analysis of the hydrophobic effect. Protein Sci. 1997;6:53–64. doi: 10.1002/pro.5560060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Huijsduijnen R H. Protein tyrosine phosphatases: counting the trees in the forest. Gene. 1998;225:1–8. doi: 10.1016/s0378-1119(98)00513-7. [DOI] [PubMed] [Google Scholar]
- 89.Walchli S, Curchod M L, Gobert R P, Arkinstall S, Hooft van Huijsduijnen R. Identification of tyrosine phosphatases that dephosphorylate the insulin receptor. A brute force approach based on “substrate-trapping” mutants. J Biol Chem. 2000;275:9792–9796. doi: 10.1074/jbc.275.13.9792. [DOI] [PubMed] [Google Scholar]
- 90.Wu L, Buist A, den Hertog J, Zhang Z Y. Comparative kinetic-analysis and substrate-specificity of the tandem catalytic domains of the receptor-like protein-tyrosine-phosphatase-alpha. J Biol Chem. 1997;272:6994–7002. doi: 10.1074/jbc.272.11.6994. [DOI] [PubMed] [Google Scholar]
- 91.Yang J, Cheng Z, Niu T, Liang X, Zhao Z J, Zhou G W. Structural basis for substrate specificity of protein-tyrosine phosphatase SHP-1. J Biol Chem. 2000;275:4066–4071. doi: 10.1074/jbc.275.6.4066. [DOI] [PubMed] [Google Scholar]
- 92.Yang J, Liang X S, Niu T Q, Meng W Y, Zhao Z Z, Zhou G W. Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem. 1998;273:28199–28207. doi: 10.1074/jbc.273.43.28199. [DOI] [PubMed] [Google Scholar]
- 93.Zanke B, Squire J, Griesser H, Henry M, Suzuki H, Patterson B, Minden M, Mak T W. A hematopoietic protein-tyrosine-phosphatase (heptp) gene that is amplified and overexpressed in myeloid malignancies maps to chromosome Iq32.1. Leukemia. 1994;8:236–244. [PubMed] [Google Scholar]
- 94.Zhang Y L, Yao Z J, Sarmiento M, Wu L, Burke J, Zhang Z Y. Thermodynamic study of ligand binding to protein tyrosine phosphatase 1B and its substrate-trapping mutants. J Biol Chem. 2000;275:34205–34212. doi: 10.1074/jbc.M004490200. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Z Y. Protein-tyrosine phosphatases—biological function, structural characteristics, and mechanism of catalysis. Crit Rev Biochem Mol Biol. 1998;33:1–52. doi: 10.1080/10409239891204161. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Z Y, Maclean D, Roeske A M, Roeske R W, Dixon J E. A continuous spectrophotometric and fluorimetric assay for protein tyrosine phosphatase using phosphotyrosine-containing peptides. Anal Biochem. 1993;211:7–15. doi: 10.1006/abio.1993.1224. [DOI] [PubMed] [Google Scholar]
- 97.Zhang Z Y, Palfey B A, Wu L, Zhao Y. Catalytic function of the conserved hydroxyl group in the protein tyrosine phosphatase signature motif. Biochemistry. 1995;34:16389–16396. doi: 10.1021/bi00050a020. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Z Y, Wang Y, Dixon J E. Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc Natl Acad Sci USA. 1994;91:1624–1627. doi: 10.1073/pnas.91.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Z Y, Wang Y, Wu L, Fauman E B, Stuckey J A, Schubert H L, Saper M A, Dixon J E. The Cys(x)(5)Arg catalytic motif in phosphoester hydrolysis. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
- 100.Zhao Y, Wu L, Noh SJ, Guan KL, Zhang ZY. Altering the nucleophile specificity of a protein-tyrosine phosphatase-catalyzed reaction. Probing the function of the invariant glutamine residues. J Biol Chem. 1998;273:5484–5492. doi: 10.1074/jbc.273.10.5484. [DOI] [PubMed] [Google Scholar]
- 101.Zhao Y, Zhang Z Y. Reactivity of alcohols toward the phosphoenzyme intermediate in the protein-tyrosine phosphatase-catalyzed reaction—probing the transition-state of the dephosphorylation step. Biochemistry. 1996;35:11797–11804. doi: 10.1021/bi960471r. [DOI] [PubMed] [Google Scholar]