

Abstract
Mutations in any of the genes encoding the four subunits of succinate dehydrogenase (SDH), a mitochondrial membrane-bound enzyme complex that is involved in both the tricarboxylic acid cycle and the electron transport chain, can lead to a variety of disorders. Recognized conditions with such mutations include Leigh syndrome and hereditary tumors such as pheochromocytoma and paraganglioma (PPGL), renal cell carcinoma, and gastrointestinal stromal tumor. Tumors appear in SDH mutation carriers with dominant inheritance due to loss of heterozygosity in susceptible cells. Here we describe a mouse model intended to reproduce hereditary PPGL through Cre-mediated loss of SDHC in cells that express tyrosine hydroxylase (TH), a compartment where PPGL is known to originate. We report that while there is modest expansion of TH+ glomus cells in the carotid body upon SDHC loss, PPGL is not observed in such mice, even in the presence of a conditional dominant negative p53 protein and chronic hypoxia. Instead, we report an unexpected phenotype of non-diabetic obesity beginning at about 20 weeks of age. We hypothesize that this obesity is caused by TH+ cell loss or altered phenotype in key compartments of the central nervous system responsible for regulating feeding behavior, coupled with metabolic changes due to loss of peripheral catecholamine production.
Keywords: obesity, mitochondrial disease, succinate dehydrogenase, mouse, familial paraganglioma, tyrosine hydroxylase, catecholamines, dopaminergic cells
Introduction
Nuclear and mitochondrial DNA mutations affecting mitochondrial energy production pathways frequently present as inborn errors of metabolism, leading to a variety of autosomal recessive disorders such as Leigh syndrome (1, 2). Interestingly, in other contexts similar mutations lead to hereditary tumors such as gastrointestinal stromal tumor (GIST) and pheochromocytoma and paraganglioma (PPGL; (3–5)). It has been estimated that ~40% of PPGL tumors result from germline mutations promoting a pseudohypoxic phenotype, including nuclear mutations inactivating the four subunits of succinate dehydrogenase (SDHx) in the tricarboxylic acid (TCA) cycle (6). Among mechanisms proposed to link SDH loss to PPGL tumorigenesis, succinate accumulation as an oncometabolite is a compelling model. Succinate accumulation has been shown to induce multiple downstream pathologies including inhibition of dioxygenases involved in normal degradation of hypoxia-inducible factors, and inhibition of demethylation of histones and DNA (3, 7, 8). The resulting dysregulation of metabolism and epigenetics is tumorigenic in certain tissues (9–11). PPGL tumors arise in neuroendocrine cells, including cells in the carotid bodies (CBs), which originate from neural crest progenitor cells (12). There is evidence that it is the glomus type I neuron-like cells of the CB that are susceptible to PGL tumorigenesis (13). These cells are highly dopaminergic, sharing tyrosine hydroxylase (TH) expression as a common marker (14).
Though there have been multiple attempts to develop mouse models that mimic SDH-related hereditary PPGL, no successes have been reported (3, 15–18). As in humans, homozygous SDH-loss in mice is an embryonic lethal condition. Unlike humans, heterozygosity for SDHx mutations does not predispose mice to PPGL. Conditional SDHx-knockout strategies are therefore reasonable to target complete SDHx loss to certain vulnerable cell populations and developmental stages to test the hypothesis that PPGL tumor initiation can be accelerated. The work reported here was conceived with such a strategy in mind - triggering SDHC loss selectively in susceptible TH+ neuroendocrine cells including cells of the central nervous system, paraganglia, the adrenal medulla, and the glomus cells of the CB. Our design used a TH promoter-driven Cre recombinase to trigger deletion of SDHC in TH+ cells. We introduced two additional tumor promotion strategies (chronic 10% oxygen hypoxia and conditional expression of a dominant negative p53 tumor suppressor protein) to test their impacts on PPGL tumorigenesis. We report the surprising and fascinating observation that none of these conditions resulted in PPGL, but rather SDHC loss in TH+ cells causes an obesity phenotype starting at 20 weeks of age. We hypothesized that this phenotype results from the deletion or altered behavior of SDHC-loss TH+ cells important for dopamine (DA) signaling in feeding behavior and obesity (19, 20). Indeed, our findings suggest that SDHC loss alters such dopaminergic cell populations, decreasing catecholamine production, especially DA, and increasing appetite and food consumption with other alterations to metabolic and activity profiles. Thus, we propose conditional SDH loss in murine TH+ cells as a novel model of obesity driven by catecholamine dysregulation.
Materials and Methods
Ethical treatment of animals
All experimental procedures performed in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Mayo Clinic in accordance with NIH guidelines. Animals were transferred to the experimental protocol at weaning age and observed for a total of 18 m before sacrifice.
Generation of SDHC conditional knockout mice
The SDHC gene-trap line C57BL/6N- Sdhctm1a(EUCOMM)Wtsi was obtained from the European Conditional Mouse Mutagenesis Program at Sanger Center, UK. These animals carrying FLP-FRT recombination signals targeting axon 4 of Sdhc were crossed with flippase recombinase-expressing mice to produce with an Sdhc floxed (fl) allele. These mice were then were crossed with B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J (Jax ID #008601) for two generations to produce Sdhc fl/fl TH-Cre/+ experimental animals and Sdhc fl/+ TH-Cre/+ control animals. PCR primers LJM-4429 (5’-CT2AGA2CTGATC4TGC3) and LJM-4430 (5’-CACTGC3G2CTCATAT3C) were used to confirm Sdhc status while primers LJM-4415 (AC2AGC2AGCTATCA2CTCG), LJM-4416 (T2ACAT2G2TC2AGC2AC2), LJM-4417 (CTAG2C2ACAGA2T2GA3GATCT) and LJM-4418 (GTAG2TG2A3T2CTAGCATCATC2) were used to detect TH-Cre. Sdhc fl/fl TH-Cre/+ animals were also crossed with 129S-Trp53tm2Tyj/J background (Jax ID # 008652) to produce Sdhc fl/fl TH-Cre/+ LSLp53/+ animals carrying a conditional dominant negative p53 allele. In Cre+ cells, deletion of a transcriptional termination sequence (Lox-Stop-Lox) is activated, resulting in dominant negative p53 expression equivalent to a homozygous null p53 mutation. PCR primers LJM-5099 (5’-AGCTAGC2AC2ATG2CT2GAGTCTGCA), LJM-5100 (5’-CT2G2AGACATAGC2ACACTG) and LJM-5101 (5’-T2ACACATC2AGC2TCTGTG2) were used to detect the wild type or LoxP element of p53.
Husbandry
Five or fewer mice were housed with same-sex litter-mates in filter-capped polycarbonate cages supplied with PicoLab Rodent Diet 20 chow and filter-purified tap water ad libitum, in a room with constant temperature (21 ± 2°C) and humidity (45 ± 10%). Animals were exposed to a day-night cycle of 12 h.
Hypoxia
Hypoxic housing of mice was accommodated in a custom-made hypoxia chamber (BioSpherix, Parish, NY), housing 12 cages with 5 mice per cage. Oxygen levels were maintained at 10% using a BioSpherix ProOx P360 oxygen single-chamber controller. Carbon dioxide was similarly held at ≤ 1% using a BioSpherix ProCO2 P120 ppm carbon dioxide single chamber controller. This dual regulation system allowed control of oxygen while modulating total gas flow rate to purge carbon dioxide from the chamber to avoid carbon dioxide toxicity. For hypoxia studies, SDHC (fl/+) and SDHC (fl/fl) mice were introduced to hypoxic or normoxic housing at weaning age (21 d). Survival data were recorded over the span of 18 m and animals that met IACUC humane criteria were euthanized as necessary.
Animal weight, tumorigenesis, and lifespan
Mice were observed three times weekly and euthanized promptly when IACUC humane criteria were met, such as when extreme weight loss, tumor growth or a hunched posture were observed.
Carotid body morphology
For immunohistochemical studies, carotid body and adrenal glands were removed from the carcasses of animals at 72 wk, sacrificed according to IACUC standard protocol for carbon dioxide euthanasia. The abdominal cavity was opened by incision from the groin to the upper neck. Carcasses were then fixed by immersion in 10% neutral buffered formalin solution for 48 h, and transferred to 70% ethanol until processing. The cohort included 9 experimental male mice and 9 controls. For analysis, carotid bifurcations and adrenal glands were cryoprotected overnight with 30% sucrose in PBS, and embedded in OCT (Tissue-Tek). Tissue sections of 10 μm were cut and seriated with a cryostat (Leica, Wetzlar, Germany), and incubated with primary antibodies overnight at 4°C: TH (1:2500 dilution, Novus Biological). Thereafter, the tissues were incubated with fluorescent secondary antibody AlexaFluor 568 (1:400 dilution, Invitrogen,). Nuclei were labeled with 4′,6′-diamidino-2-phenylindole (DAPI). Immunofluorescence images were acquired using an Olympus BX-61 microscope (Olympus, Hamburg, Germany) and carotid body TH+ surfaces in tissue slices were measured with ImageJ software (NIH, USA). The total TH+ surface given in figures (in arbitrary units) is the result of the sum of all the TH+ surfaces obtained from each of the slices in a seriated bifurcation.
TH and SDHB western blot analysis
Tissue samples (0.25 mg) were isolated from the hypothalamic region of saline-perfused brains and incubated in ice-cold RIPA buffer for 30 min and subjected to vortex mixing every 5 min. Protein extracts were then quantitated using a Pierce BCA protein assay kit before mixing them with LDS sample buffer and reducing agent. For electrophoresis, 10% Bis-Tris gels were used to analyze protein extracts (10 μg) in 1× MES running buffer at 150 V for 1 h. Following the electrophoresis, blotting was accomplished using a Novex western transfer apparatus and PVDF membrane. Transfer electrophoresis was for 90 min at 30 V and 245 mA in transfer buffer. After checking the quality of the transfer with Ponceau S, the membrane was blocked in 3% nonfat dry milk in TBST buffer. Following 1 h of blocking and three 15-min washes in TBST, the membrane was cut where appropriate. Blots were incubated overnight in 10 mL primary antibody solutions made with 4% BSA in TBST including sodium azide (0.5%) and either 1:2000 anti-SDHB, 1:1000 anti-alpha tubulin or 1:500 anti-tyrosine hydroxylase antibodies (abcam ID# ab113). Membranes were removed from the primary staining solution, washed three times (15 min) in TBST buffer and incubated 1 h in the presence of secondary antibody solution made with 15 mL 3% fat-free milk blocking buffer and 1 μl of the corresponding HRP-conjugated second antibody. Antibody binding was then detected and quantitated using Pierce ECL 2 western blot substrate at room temperature for 15 min prior to imaging with a GE Typhoon Fluorimeter (GE Healthcare Life Sciences, Chicago, IL, USA). Digital quantification was performed using ImageQuant (GE Healthcare Life Sciences) and ImageJ software (National Institutes of Health, Bethesda, MD, USA) normalizing SDHB and TH signals to alpha tubulin upon background subtraction.
Staining of brain sections
Brain tissues were isolated from male mice at 27 wk of age following lethal exposure to carbon dioxide and perfusion with saline and 4% paraformaldehyde (PFA) in PBS solution. Brain tissue was transferred to 4% PFA solution overnight followed by storage in 30% sucrose in PBS at 4°C until the tissue was no longer buoyant. Slides were prepared using 40 μm coronal sections cut in a cryostat. The selected paraventricular nucleus (PVN) sections were then incubated at room temperature in 0.5% Triton in PBS for 15 min followed by 5% BSA in PBS for 4 h prior to staining. Exposure to primary antibodies was done overnight in 4°C using sheep anti-TH (abcam #ab113) and rabbit anti-SDHB (abcam #ab178423). On the following day, primary antibodies solutions were removed and the tissues were washed 3 times (5 min) in PBS containing 0.5% Triton. Secondary antibody solutions were prepared with rabbit anti-sheep IgG H&L Alexa Fluor® 488 (#ab150181) and a donkey anti-Rabbit IgG H&L Alexa Fluor® 647 (#ab150075). Nuclei were stained using DAPI (Sigma #28718-90-3). Fluorescent images were taken using an LSM 510 confocal laser scanning microscope (Carl Zeiss).
Metabolic chamber analysis
A total of 14 experimental male mice (27 wk) were paired with littermate controls and placed in metabolic chambers with locomotor sensors (Oxylet Pro, PanLab). Each pair was allowed to acclimate for 6 h prior to data collection (24 h, covering 12 h of light and 12 h of dark). Animals had ad libitum access to water and food dispensing units which recorded consumption throughout the experiment. Gas exchange and energy expenditure were recorded using an external gas analyzer unit (Panlab, LE 405 Gas Analyzer) reporting to metabolic software (Panlab, Metabolism). Data analysis was conducted in Prism 8.
Open field analysis
Spontaneous locomotor parameters and anxiety-like behavior were assessed by introducing 14 experimental and 14 control males (age 26 wk) to a conventional, brightly lit (500 lux), open field test apparatus consisting of a Plexiglas chamber (41 cm2) placed inside sound-attenuating cubicles equipped with two sets of 16 pulse-modulated infrared photobeams to record X–Y ambulatory movements at a time resolution of 100 ms (Med Associates, Lafayette, IN). The test entailed three 60-min sessions following 30 min of acclimation for three consecutive days during the light cycle.
Insulin, glucose and catecholamine blood panel analysis
Fasting blood serum from 7 experimental mice and a matching number of controls (age 27 wk) was analyzed to determine fasting insulin levels using a commercial ELIZA kit (Novus biological #NBP2–62853). Glucose levels were measured directly from tail blood using a conventional alphaTRAK 2 blood glucose monitoring system. Plasma catecholamines were measured by reversed phase HPLC with electrochemical detection after extraction with activated alumina. Intra-assay catecholamine sensitivities are: norepinephrine 0.7% and 0.63% at 293 and 1234 pg/mL; epinephrine 1.14% and 0.75% at 73.2 and 1019 pg/mL; dopamine 2.9% and 1.62% at 66.8 and 293 pg/mL. Interassay coefficients of variability are: norepinephrine 3.2% and 3.9% at 291 and 1223 pg/mL; epinephrine 4.2% and 4.3% at 76 and 1010 pg/mL; dopamine 8.1% and 7.1% at 64 and 258 pg/mL. Inputs below the detectable level (<10 pg/mL) were calculated as 10/(2)1/2 or 7.1 pg/mL.
Results
Generation of SDHC-loss mice
Inspired by the goal of creating a mouse model of the rare familial neuroendocrine tumor PPGL (16), we utilized a gene trap approach to create a null SDHC allele that, when rearranged by Flp recombination, created a conditional SDHC allele where two LoxP cites flank exon 4 as described (21). When exposed to Cre recombinase, exon 4 is deleted, yielding a mRNA encoding a non-functional, truncated SDHC protein. To avoid the embryonic lethality associated with systematic deletion of SDHC in homozygous floxed animals (16) while knocking out SDHC expression in tissues believed susceptible to PGL tumorigenesis, we generated conditional SDHC mice with one or two copies of the floxed SDHC allele together with Cre recombinase under the control of the TH promoter. We aimed to test the hypothesis that SDHC fl/fl animals would survive SDHC gene loss in TH+ cells and develop PPGL while SDHC fl/+ animals would, like humans, show no SDH haploinsufficiency and serve as controls. As described below, the triggering SDHC loss in TH+ cells, even when combined with p53 loss and/or chronic hypoxia, does not predispose to PGL but rather triggers unexpected obesity.
Carotid body hypertrophy upon SDHC loss in TH+ cells
It has previously been shown that pseudohypoxia driven by PHD2 inactivation and chronic up-regulation of hypoxia-inducible factors (HIFs) in TH+ type I cells of the carotid bodies can lead to hypertrophy, a possible early stage of tumorigenesis (13). SDH loss is accompanied by succinate accumulation, a condition that can similarly inhibit HIF prolylhydroxylases, similarly inducing pseudohypoxia (22). We therefore analyzed carotid body morphology in conditional SDHC-loss mice to test for this effect. TH+ cells of the adrenal medulla were also examined. No evidence of paraganglioma or other tumorigenesis in the adrenal glands or CB of SDHC loss animals was observed in a cohort of 9 experimental and 9 control animals. Interestingly, however, quantitative analysis of TH+ CB cells indicated an increased surface area for these cells in experimental animals as compared to controls, suggesting mild hypertrophy in SDHC-deficient carotid bodies (Fig. 1). This result is consistent with the hypothesis that type I glomus cells of the CB undergo mild expansion in response to pseudohypoxia driven by succinate antagonism of prolylhydroxylase enzymes that normally target HIFs for proteolysis. The result is therefore a milder form of the phenotype reported when PHD2 is inactivated in TH+ cells (13). CB hyperplasia was relatively subtle and preliminary immunohistochemical analysis of HIF2α expression in available specimens did not reveal detectable elevation.
Figure 1.
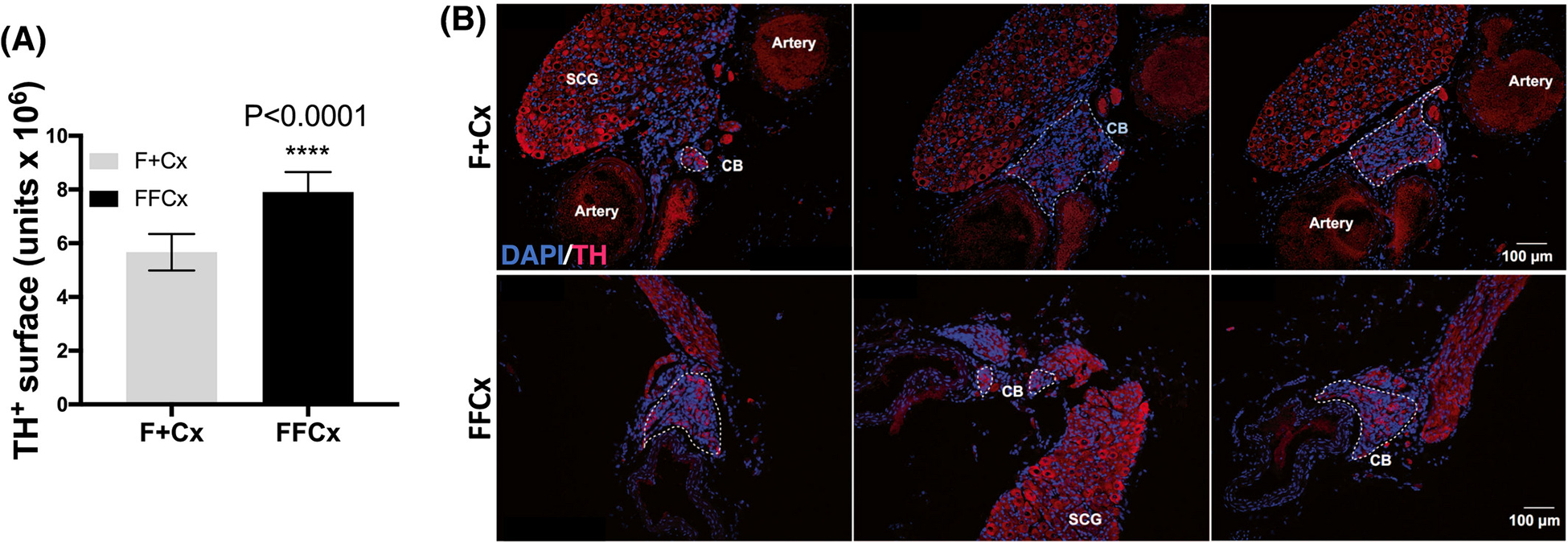
Quantitative assessment of TH expression in the CB of mice at 72 wk. A. TH+ area (arbitrary units) in carotid bifurcations based on analysis of 9 TH-Cre experimental (FFCx) and 9 control (F+Cx) mice. Statistical analysis used a Student’s t-test where significance is reported for P-values below 0.05. Bars represent mean ± SEM. B. Example immunofluorescent staining shows TH (red) and DAPI (blue) where the discontinuous white line outlines the CB regions in each section.
Adrenal medulla pathology upon SDHC loss in TH+ cells
Interestingly, while the relative size of the adrenal medulla was not overtly altered in SDHC-loss obese mice (Supporting Information Fig. S1), a striking inflammatory infiltrate was consistently observed in all experimental adrenal medulla tissues (Supporting Information Fig. S2) suggesting a change in chromaffin cell physiology upon SDHC loss.
Absence of PPGL tumorigenesis upon SDHC loss in TH+ cells
Three cohorts of 7 experimental and 7 control mice were dissected shortly after CO2 euthanasia at IACUC humane end-point criteria, or at age 78 wk. An initial assessment of tumors present at autopsy was recorded before whole carcasses were sent for gross pathology analysis after fixation in 10% neutral buffered formalin containing methanol for 48 h. A more detailed histopathology analysis was conducted for major tissues including brain, heart, liver, adrenal gland, spleen, stomach, small intestine, and colon. No PPGL tumors were observed. Both experimental and control animals displayed age-related liver necrosis, lympho-histiocytic and other neoplasms. Two experimental females showed signs of adrenal spindle cell hyperplasia, moderate and vascular ectasia, and cortical/medullary junction unrelated to PGL formation.
Absence of PGL tumorigenesis upon SDHC loss in TH+ cells with p53 loss and/or chronic hypoxia
There is literature precedent for the concept that chronic hypoxia is an inducing or promoting factor in parasympathetic PPGL (6, 23). Likewise, p53 loss is believed to enhance tumorigenesis in many cancers. Although p53 loss has not been found to be a common driver of PPGL in humans (24, 25), we investigated the potential of promoting PPGL by p53 loss and hypoxia. Conditional SDHC-loss mice were bred with mice carrying a conditional dominant negative p53 allele, producing FFCxp53 and F+Cxp53 animals (where the shorthand nomenclature F refers to the SDHC conditional floxed allele, + refers to an SDHC wild type allele, Cx refers to at least one Cre allele, and p53 refers to a dominant negative p53 allele). These and FFCx and F+Cx animals were housed under chronic 10% oxygen conditions from weaning age. As shown in Fig. 2, conditional SDHC-loss animals had lower life expectancies, though no PPGL tumorigenesis was observed by autopsy or histopathology. Among causes of premature death was an inconsistent trend in tissue necrosis and neoplasms.
Figure 2.
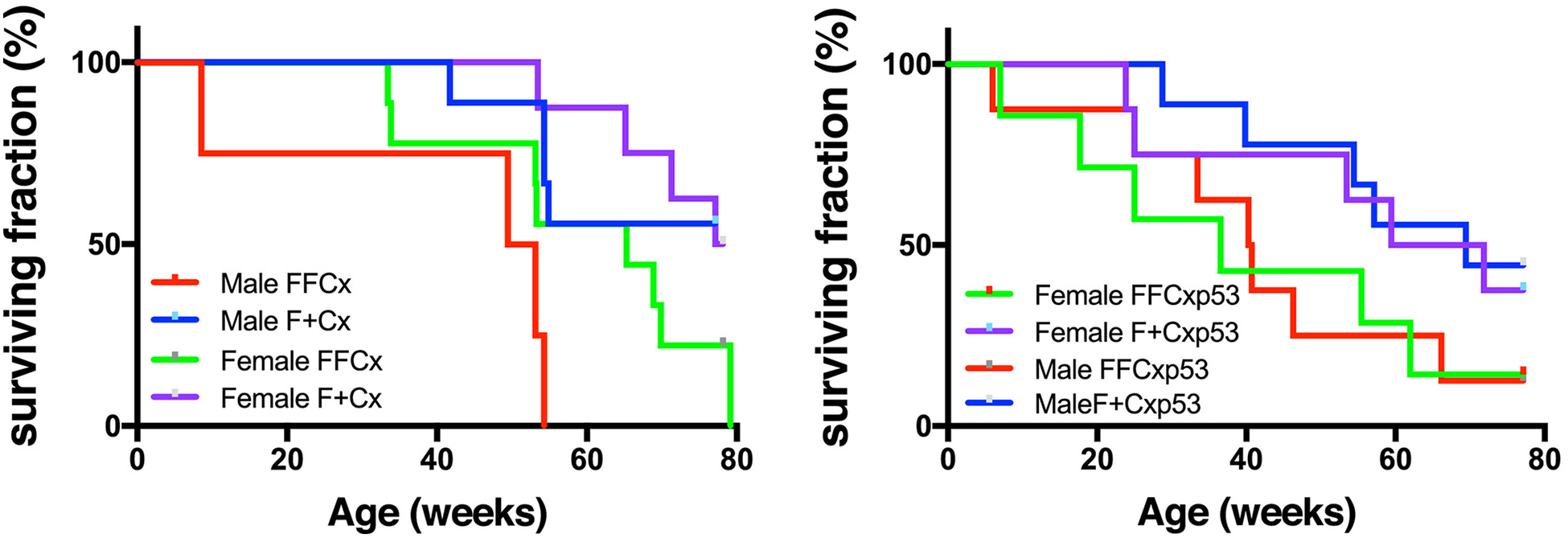
Survival of mice of the indicated genotypes maintained under chronic hypoxia (10% O2). A. Lifespan data for mice of the indicated SDHC TH-Cre genotypes over 80 wk. Data represent 23 FFCx and 18 F+Cx males; 16 FFCx and 16 F+Cx females. B. Lifespan data for mice of the indicated genotypes expressing a dominant negative p53 allele in TH+ cells over 80 wk. Data represent 14 FFCx and 14 F+Cx males; 15 FFCx and 14 females. Data analysis was conducted in Prism 8.
Obesity upon SDHC loss in TH+ cells
Weight data were recorded weekly for experimental and control mice between 10 – 78 wk of age, initially simply as a means to monitor health and tumor formation. Strikingly, conditional SDHC-loss mice became obese starting ~20 wk of age (Fig. 3A). Lean and fat mass body composition analysis revealed a higher body fat composition in experimental mice compared to controls (Fig. 3B,C). This phenotype did not strongly reduce life expectancy (Fig. 3D).
Figure 3.
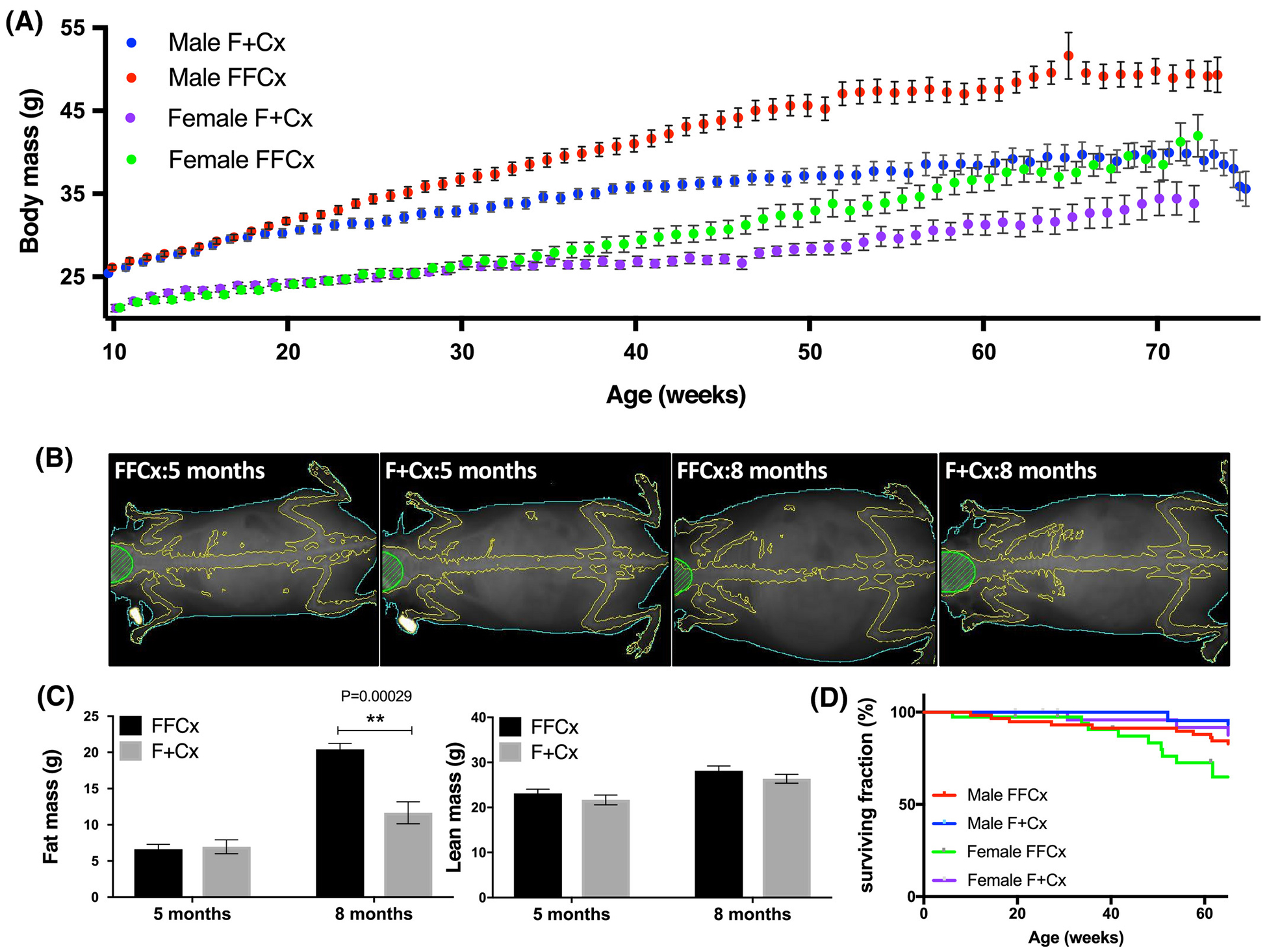
Obesity in experimental mice upon SDHC loss in TH+ cells. A. Body weight over 78 wk. Numbers of mice for each genotype were: 36 FFCx and 30 F+Cx males; 28 FFCx and 30 F+Cx females. B. X-ray imaging of representative mice at 20 (pre-obese) and 32 (obese) wk using densitometry with dual energy X-ray absorptiometry (DEXA) in a Lunar PIXImus scanner. C. Fat and lean mass for 7 experimental and 7 control mice at 20 and 32 wk. D. Survival of mice represented in panel A. Statistical analyses were performed using Prism 8 with bars indicating mean ± SD. Fat and lean mass data were analyzed by 2-way ANOVA with Tukey’s correction test over genotype and time.
Characterization of pre-obese and obese SDHC-loss mice
We sought to understand the basis for obesity in animals lacking SDHC in TH+ cells. We considered behavioral and physiological factors. We used a conventional open field apparatus to measure spontaneous locomotor function and anxiety-like behavior in obese males at 27 wk of age. Our results (Fig. 4) suggest that while total distance traveled did not vary significantly across genotypes (Fig. 4C), experimental animals moved less during the first 10 min of a habituated open field session, possibly suggesting a higher stress relative to control mice (Fig. 4A). On the other hand, there was no difference between experimental and control animals for time spent in the center of the field, or in average velocity. Interestingly, obese animals displayed reduced rearing behavior, suggesting a lack of explorational drive in the open field (Fig. 4B). Visual representations of representative open field habituated sessions are shown in Fig. 4D.
Figure 4.
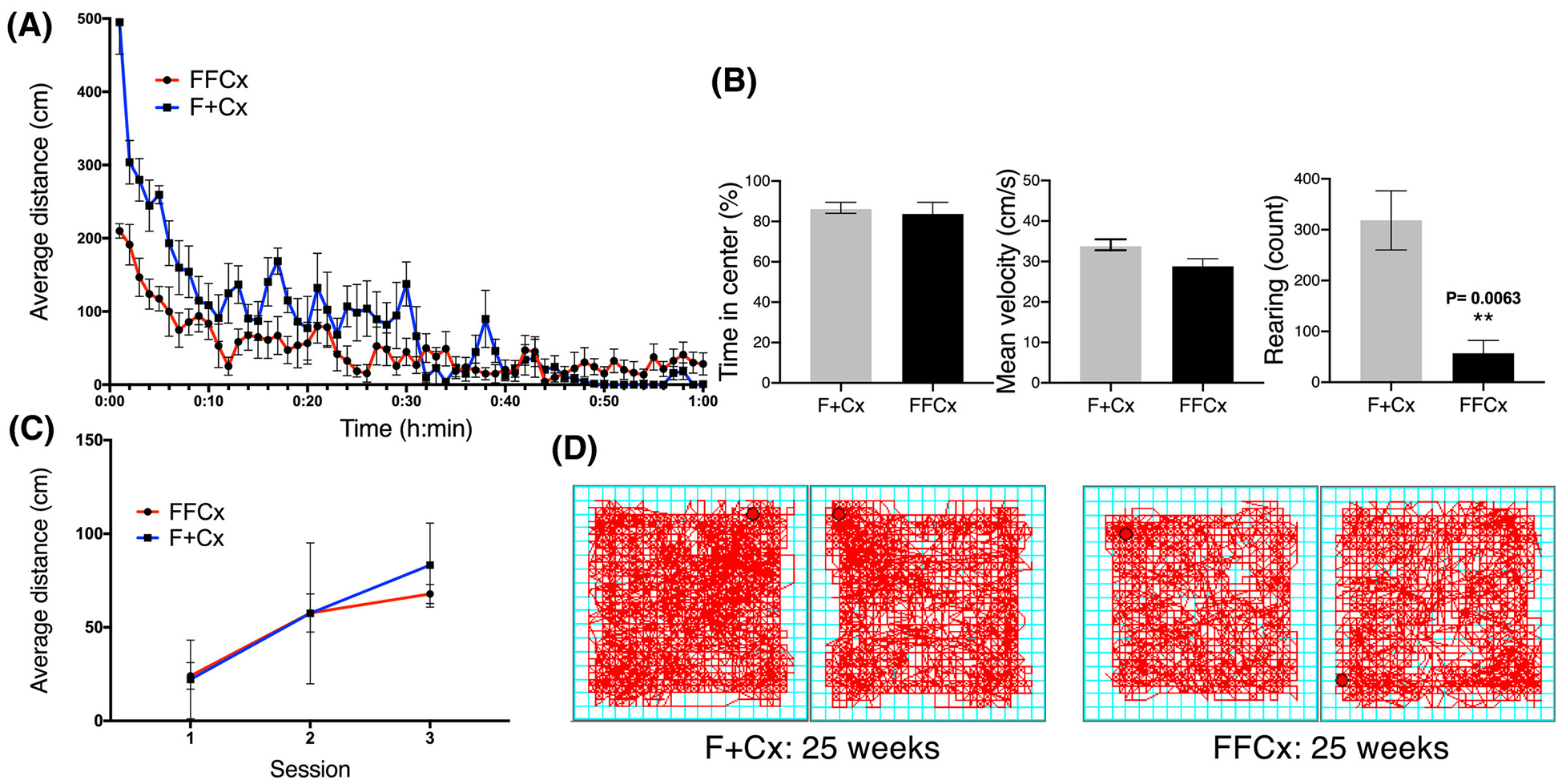
Assessment of behavior and locomotor activity in open field test for male mice of the indicated genotypes at 24 wk. Data are for representative control and experimental pairs habituated in the chamber for 3 consecutive d in 60-min sessions during daylight. A. Distance traveled by habituated mice during third session. B. Percentage of time spent in the center, mean velocity, and rearing behavior in habituated mice over 1 h. C. Average distance traveled distance over all 3 d of testing. D. Representative motion tracings for mice of the indicated genotypes. Average distance traveled during a 60-min session, time in the center, mean velocity and rearing count were analyzed using a Student’s t-test. Average distance across 3 sessions was analyzed using 2-way ANOVA followed by Tukey’s correction test. Bars represent mean ± SE. 14 animals of each genotype were used to complete this test.
We undertook metabolic chamber studies to evaluate the metabolic characteristics of pre-obese and obese mice in terms of dietary intake, gas exchange, energy expenditure and light cycle-dependent locomotor activity. Consistent with what had been observed for locomotor behavior in the open field, experimental mice displayed intervals of low motor activity throughout the day, more evident during the dark cycle (Fig. 5A), though oxygen consumption was comparable to matched controls (Fig. 5B). Experimental mice also presented an altered pattern of energy expenditure, also more evident during the dark cycle (Fig. 5C). Daily food consumption, but not water consumption, was found to be significantly higher for experimental animals compared to controls (Fig. 5D,E).
Figure 5.
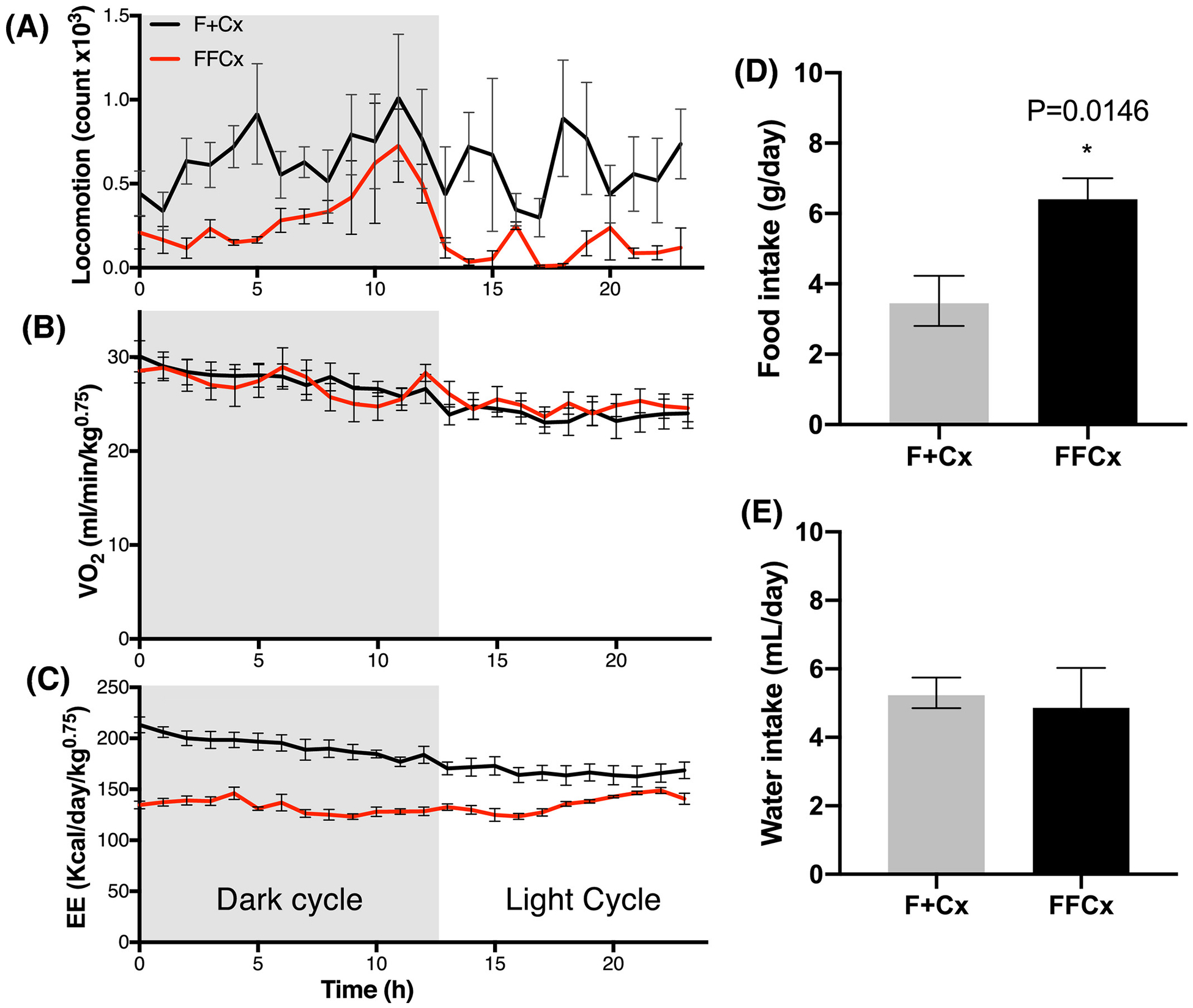
Metabolic energy profile and dietary intake for male mice at 24 wk. Experimental (FFCx) mice were placed in a metabolic cage and compared with control littermates in another cage for each trial over one d with the indicated dark/light cycle. A. Locomotion activity (arbitrary units). B. Normalized oxygen consumption rate. C. Energy expenditure. Food intake (D) and water intake (E) were also measured over 24 h. Statistical analysis was by Student’s t-test, with only P-values expressing significance below 0.05 being reported. Bars represent mean ± SE. Numbers of animals used for each genotype: FFCx (14) and F+Cx (14).
Brain morphology after SDHC loss in TH+ cells
We exploited western blotting and immunohistochemistry to investigate morphological effects of SDHC loss in TH+ cells in key areas of the central nervous system responsible for regulating feeding behavior and metabolism. Particularly, we focused on the paraventricular nucleus (PVN) of the hypothalamus, considered as a crucial region regulating physiological feeding response and metabolic balance (26). Hypothalamus tissue and sections were analyzed for TH and SDHB protein. SDHB is a convenient surrogate for SDHC, as the loss of SDHC is known to trigger loss of SDHB (27). Digital quantification of western blot signal was normalized to alpha tubulin and reported as a ratio (Fig. 6A). The results suggest a significant decrease in TH protein expression across 4 experimental samples compared to controls (P=0.001). This loss of TH protein raises the possibility that at least some TH+ cells of the hypothalamus are deleted upon loss of SDHC expression. SDHB reduction was not detectable by western blot, consistent with the notion that the majority of cells in this tissue are TH− with unaffected levels of SDH proteins.
Figure 6.
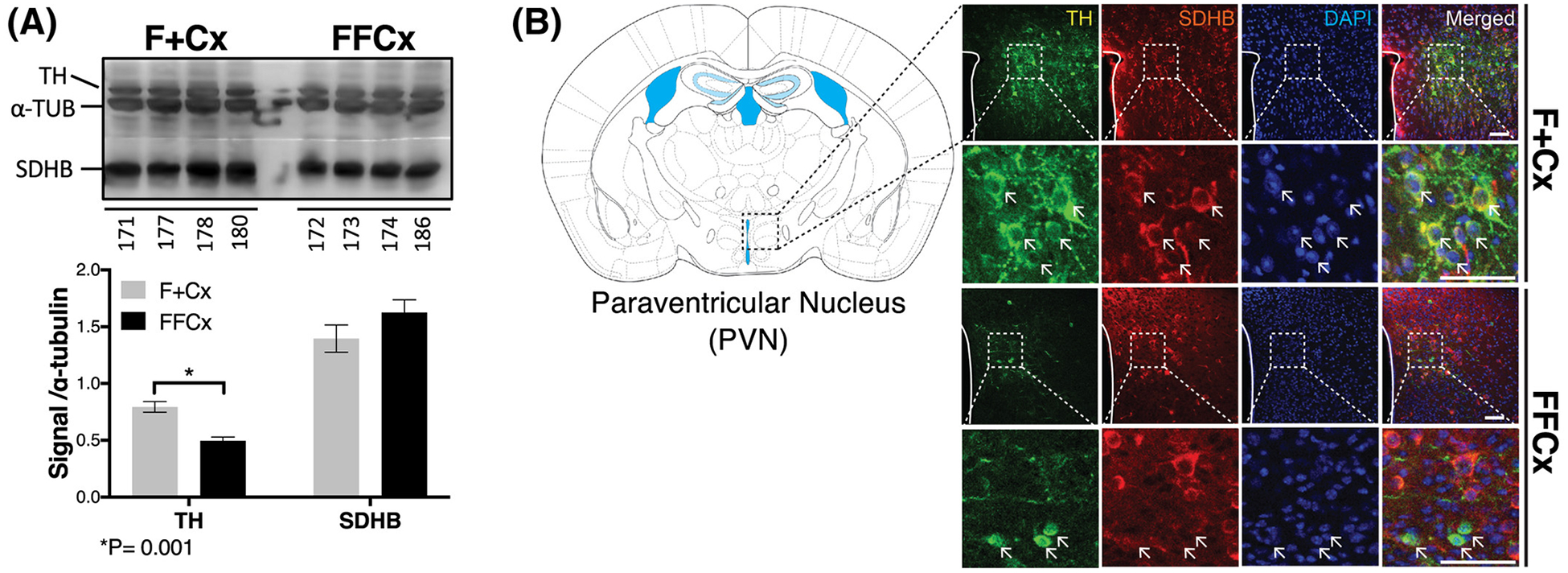
Changes in TH+ cells in key regions of the hypothalamus important for feeding behavior. A. Western blot analysis of TH and SDH (represented by SDHB) in hypothalamic preoptic tissue for the indicated 4 representative animals. Western blot signal quantitation statistics were analyzed using a Student’s t-test with bars indicating mean ± sd. B. Fluorescent immunohistochemical staining analysis of the PVN monitoring TH (green), SDHB (red) and DAPI (blue) Arrows indicate example cells showing contrasting features. Scale bar: 50 μm.
Immunohistochemistry with fluorescent detection was used to assess tissue morphology and SDHB and TH levels in the PVN. The results (Fig. 6B) also indicate reduction, but not complete ablation, of TH+ cells in the PVN (Fig. 6A, compare first column in upper two rows vs. lower two rows), consistent with western blotting results. The strong reduction in yellow color in the merged data (Fig. 6B, right column) indicate that few cells show both TH and SDH expression as expected. Altered morphology of TH+ neurons in PVN sections was also noted (arrows in Fig. 6, rows 2 and 4), with TH expression throughout the cytoplasm in controls but strikingly perinuclear upon SDHC loss. TH+ SDHC-loss cells are also diminished in size (Fig 6B, bottom row). In summary, we find evidence of SDH loss specific to TH+ cells in the PVN, and evidence that at least some SDH-loss cells survive, but with altered morphology. These results support the hypothesis that SDHC loss in TH+ neurons impacts brain structure and function, with potential consequences for feeding behavior.
Diabetic status of experimental mice
We wished to rule out a role for diabetes in the etiology of obesity in conditional SDHC-loss mice. Blood glucose was found to be significantly lower in experimental animals compared to controls in the tested cohort (Fig. 7A) while insulin levels were similar between experimental mice and controls (Fig. 7B). Neither of these parameters suggest diabetes as an underlying condition in mice lacking SDHC in TH+ cells.
Figure 7.
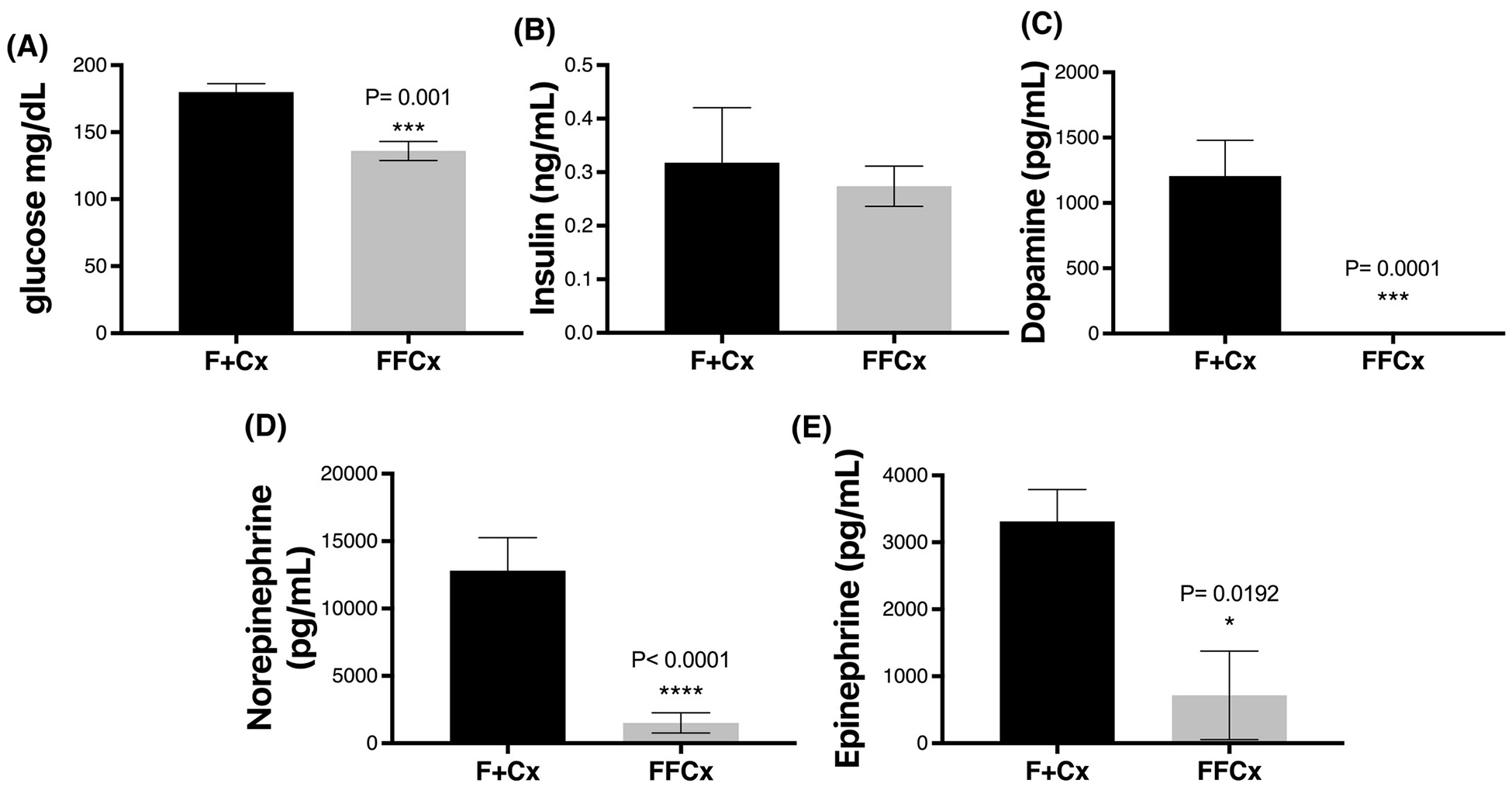
Blood glucose, insulin and catecholamines for male mice at 24 wk. Mice were fasted for 4 h prior to serum collection. A. Tail vein whole blood was used to monitor glucose using an AlphaTRAK 2 monitoring kit. B-E. Serum was analyzed to measure insulin and the indicated hormones. Statistical analysis was by Student’s t-tests, with only P values below 0.05 indicated and measurements below the detection limit were substituted with the lowest detectable level of each assay divided by 21/2, as described (Ogden, 2010). Bars indicate mean ± SE. Data represent 7 male mice per group.
Systemic catecholamine status after SDHC loss in TH+ cells
Because tyrosine hydroxylase is the first and rate-limiting enzyme in the pathway of catecholamine biosynthesis (28), we were curious if SDHC loss in TH+ cells might decrease the viability and/or hormone production of these cells, impacting catecholamine activities such as satiety related to dopamine levels. Remarkably, catecholamine analysis showed all experimental animals in a cohort of 7 to have serum dopamine and norepinephrine below the detection limit (< 10 pg/mL), and epinephrine levels were also found to be significantly lower in experimental animals compared to controls (Fig. 7, C–E).
Discussion
We originally set out to develop a mouse model of familial PPGL triggered by loss of SDH. The field has been plagued by the lack of such mouse models of tumorigenesis, and limited cell line models (15, 16, 18, 29–31) though recent progress has been reported in rat (32). It is possible that the time required for spontaneous LOH in heterozygous SDH-loss mice is too long for development of slow-growing PGL tumors given the short lifespan of the animal. It is therefore reasonable to drive complete conditional SDH loss in tissues of interest. Whole-body SDH loss is an embryonic lethal condition, though, remarkably, we have shown that conditional whole-body SDH-loss mice can survive in hypoxia (21). As there is evidence that TH+ cells are the stem cells of chromaffin tumors (13, 33–35) we created a conditional model to enhance loss of SDHC expression in TH+ cells. We monitored SDHC-loss animals for tumorigenesis, and attempted to increase the penetrance of a tumor phenotype by triggering expression of a dominant negative p53 protein in TH+ cells, and exposing mice to chronic 10% oxygen hypoxia, perhaps contributing other tumor growth signals (23, 24, 36).
We observed subtle CB hyperplasia among TH+ cells upon SDHC loss. This effect is consistent with the more profound CB hyperplasia observed in mice lacking prolylhydroxylase PHD2 in type I cells (36). It is thus evident that chronic hypoxic signaling due to low oxygen or the inability to downregulate hypoxia inducible factors is sufficient to promote mitogenesis in CB tissue. We hypothesize that SDHC loss in type I cells of the CB causes succinate accumulation, partially inhibiting dioxygenases such as PHD2, thus creating a weak phenocopy of PHD2 loss, with pseudohypoxic consequences.
Despite this evidence for mild CB hyperplasia upon SDHC loss in TH+ cells, the animals did not go on to develop detectable PPGL in CB, adrenal medulla, or other locations. Results were similar even upon conditional p53 loss in the same cells, and with additional chronic hypoxia. We conclude that SDHC loss triggered by developmental TH expression in mouse development is insufficient for PGL tumorigenesis. This result might be due to multiple factors. It is possible that mice require loss of additional PGL tumor suppressor genes compared to humans. It is possible that tumor growth is too slow for sensitive detection in mice. Our hypothesis in designing this work was that complete loss of any SDH subunit has the same tumorigenic consequences as loss of any other SDH subunit. However, it is also possible that SDHC loss is not as penetrant as loss of other SDH subunits, e.g. SDHB (37) for reasons yet to be understood. Similarly, lack of SDHD is reported to induce death of CB and adrenal medulla cells (17). Finally, it is possible that the timing of SDHC loss, coinciding with onset of TH expression, is inappropriate for PPGL tumorigenesis.
While monitoring the physiology of mice with SDHC loss in TH+ cells we made the unexpected observation that these animals are prone to obesity relative to SDHC +/− littermates (Fig. 3). This mystery suggested that altered physiology upon SDH loss in TH+ cells influences mouse bioenergetics or behavior to alter feeding and/or energy expenditure. Interestingly, the observed non-diabetic obesity occurred in adulthood and did not cause significant morbidity or premature mortality. These results are consistent with reports that elevated weight, per se, has only a minor effect on lifespan when obesity is reached later in life (38). Moreover, other conditional obesity models have reported mild phenotypes and normal lifespan when the causative mutation was triggered after maturity (39).
DA signaling pathways play key roles in regulating feeding. Pharmacogenetic alterations to DA signaling have been linked to obesity. However, the precise role of DA in regulating food intake remains poorly understood (20). It has been proposed that the dopaminergic functions activated in TH+ cells modulate feeding behavior through regulation of sympathetic neurons associated with reward control, sensation, and conditioning (40). This implies that vulnerability to overfeeding is influenced by the ability to regulate craving and reward-driven behaviors. Catecholamines produced during the intermediate steps of TH-dependent pathways also influence energy metabolism through chemical signaling and control biological processes in response to stress stimuli (41). For example, epinephrine and norepinephrine play critical roles in the production of ketone bodies through lipolysis (41, 42) implying that interference with catecholaminergic pathways could impose changes upon homeostatic circuits. The result may be impairment of metabolism and DA-dependent behaviors such as stress response, physical activity, energy expenditure as well as obesity (43). On this basis, we propose that inadequate catecholamine-mediated (especially DA-mediated) energy homeostasis is the likely mechanistic basis for the striking obese phenotype observed in this SDHC conditional loss model. We show clear evidence of altered morphology of TH+ dopaminergic cells in the hypothalamus and the carotid bodies, suggesting smaller cells with altered subcellular TH localization in the former, and expansion in the latter (Figs. 1, 6). Western blot analysis confirmed a significant decrease in TH expression within the hypothalamic region (Fig. 6A). Most strikingly, a serum catecholamine panel showed profound decreases in serum epinephrine, norepinephrine and dopamine upon SDHC loss in TH+ cells, supporting the hypothesis that SDHC loss triggers loss of dopaminergic functions. Our finding of suspicious adrenal medulla histology (normal adrenal medulla size but inflammatory infiltrate suggestive of chromaffin cell degeneration or dysfunction) suggests a defect in catecholamine synthesis by TH+ cells. Chemical denervation could be used in future studies to test this possibility. Likewise, understanding the balance between adrenergic defects in the central nervous system vs. the periphery could be addressed in future studies by analysis of altered gene expression in the adrenal medulla of obese SDHC-deficient mice.
Analysis of blood glucose and insulin argues against diabetes in our model. Mutations in the leptin signaling pathway are often associated with diabetes, but diabetes is not observed in most chemically- or metabolically-induced obesity models. The Tubby model is an example of metabolically-induced animal obesity that does not present hyperglycemia or insulin sensitivity (39). The metabolic profile of the obese mice in our work also reveals altered food consumption where experimental genotypes were prone to overeating (Fig. 5D), showed subtle alteration of locomotion (Fig. 4A, 5A) and a lower energy expenditure (Fig. 5C), all confirming abnormal metabolism and behavior.
In summary, our results suggest that reduced systemic catecholamine production and dopaminergic signaling can trigger obesity. Ironically, PPGL tumorigenesis, the original motivation behind this conditional SDHC loss model, would have been expected to produce the opposite effect, with catecholamine overproduction by expanding tumor cells a common feature of functional sympathetic PPGL tumors (44). The fact that PPGL patients with catecholamine elevation do not generally demonstrate a corresponding increased metabolism with weight loss may also reflect receptor downregulation. We have not discovered reports of obesity associated with other mouse models utilizing the TH-Cre driver, suggesting that SDHC loss in TH+ cells is a specific effect. For example, deletion of Bcl-x in TH+ cells generate smaller mice with lower brain mass (45). Thus, the model described here may be useful in future studies characterizing the relationship between catecholamine production and obesity.
Supplementary Material
Acknowledgements
This work was supported by the Mayo Clinic, the Paradifference Foundation (LJM), National Institute on Alcohol Abuse and Alcoholism (K01 AA027773 to SK, R01 AA018779 to DSC). PO-S and JL-B are supported by grants from the Spanish Ministries of Science and Innovation and Health (SAF2016-74990-R) and the European Research Council (ERC-ADGPRJ201502629), The technical assistance of Daniel Lindberg is acknowledged.
Funding: Paradifference Foundation (FAK, MNH, LJM); NIH NIAAA AA018779 (SK, D-SC); NIH NIAAA AA027773 (SK); Spanish Ministries of Science and Innovation and Health SAF2016-74990-R (PO-S, JL-B); European Research Council ERC-ADGPRJ201502629 (PO-S, JL-B).
Nonstandard abbreviations
- CLAMS
Comprehensive Lab Animal Monitoring System
- Cre
Cre recombinase
- dox
doxycycline
- GIST
gastrointestinal stromal tumor
- LOH
loss of heterozygosity
- PPGL
pheochromocytoma and paraganglioma
- RER
respiratory exchange ratio
- SDH
succinate dehydrogenase
- TCA
tricarboxylic acid
Footnotes
Conflicts of interests
D.-S. Choi is a scientific advisory board member to Peptron Inc.
References
- 1.Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJH, Mayatepek E, Morava E, and Distelmaier F (2014) A guide to diagnosis and treatment of Leigh syndrome. Journal of Neurology, Neurosurgery & Psychiatry 85, 257–265 [DOI] [PubMed] [Google Scholar]
- 2.Ruhoy IS, and Saneto RP (2014) The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl Clin Genet 7, 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Her YF, and Maher LJ (2015) Succinate Dehydrogenase Loss in Familial Paraganglioma: Biochemistry, Genetics, and Epigenetics. Int J Endocrin 2015, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang Y, Liu Y, Pacak K, and Yang C (2019) Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers (Basel) 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buffet A, Burnichon N, Favier J, and Gimenez-Roqueplo AP (2020) An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab, 101416. [DOI] [PubMed] [Google Scholar]
- 6.Kluckova K, and Tennant DA (2018) Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res 372, 367–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85 [DOI] [PubMed] [Google Scholar]
- 8.Smith EH, Janknecht R, and Maher LJ 3rd. (2007) Succinate Inhibition of {alpha}-KetoglutarateDependent Enzymes in a Yeast Model of Paraganglioma. Hum Mol Genet 16, 3136–3148 [DOI] [PubMed] [Google Scholar]
- 9.Millán-Uclés Á, Díaz-Castro B, García-Flores P, Báez A, Pérez-Simón JA, López-Barneo J, and Piruat JI (2014) A Conditional Mouse Mutant in the Tumor Suppressor SdhD Gene Unveils a Link between p21WAF1/Cip1 Induction and Mitochondrial Dysfunction. PLoS ONE 9, e85528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jochmanova I, Zhuang Z, and Pacak K (2015) Pheochromocytoma: Gasping for Air. Horm Cancer 6, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin A, Goncalves J, Moog S, Castro-Vega LJ, Job S, Buffet A, Fontenille MJ, Woszczyk J, Gimenez-Roqueplo AP, Letouze E, and Favier J (2020) TET-Mediated Hypermethylation Primes SDH-Deficient Cells for HIF2alpha-Driven Mesenchymal Transition. Cell Rep 30, 4551–4566 e4557 [DOI] [PubMed] [Google Scholar]
- 12.Pardal R, Ortega-Saenz P, Duran R, and Lopez-Barneo J (2007) Glia-like stem cells sustain physiologic neurogenesis in the adult mammalian carotid body. Cell 131, 364–377 [DOI] [PubMed] [Google Scholar]
- 13.Fielding JW, Hodson EJ, Cheng X, Ferguson DJP, Eckardt L, Adam J, Lip P, MatonHowarth M, Ratnayaka I, Pugh CW, Buckler KJ, Ratcliffe PJ, and Bishop T (2018) PHD2 inactivation in Type I cells drives HIF-2alpha-dependent multilineage hyperplasia and the formation of paraganglioma-like carotid bodies. J Physiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez-Barneo J, Gonzalez-Rodriguez P, Gao L, Fernandez-Aguera MC, Pardal R, and Ortega-Saenz P (2016) Oxygen sensing by the carotid body: mechanisms and role in adaptation to hypoxia. Am J Physiol Cell Physiol 310, C629–642 [DOI] [PubMed] [Google Scholar]
- 15.Bayley JP, van Minderhout I, Hogendoorn PC, Cornelisse CJ, van der Wal A, Prins FA, Teppema L, Dahan A, Devilee P, and Taschner PE (2009) Sdhd and SDHD/H19 knockout mice do not develop paraganglioma or pheochromocytoma. PLoS ONE 4, e7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maher LJ, Smith EH, Rueter EM, Becker NA, Bida JP, Nelson-Holte M, Paloma JIP, Garcia-Flores P, Lopez-Barneo J, and van Deursen J (2011) Mouse Models of Human Familial Paraganglioma In Pheochromocytoma - A new view of the old problem (Martin JF, ed), IntechOpen [Google Scholar]
- 17.Diaz-Castro B, Pintado CO, Garcia-Flores P, Lopez-Barneo J, and Piruat JI (2012) Differential impairment of catecholaminergic cell maturation and survival by genetic mitochondrial complex II dysfunction. Mol Cell Biol 32, 3347–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lussey-Lepoutre C, Buffet A, Morin A, Goncalves J, and Favier J (2018) Rodent models of pheochromocytoma, parallels in rodent and human tumorigenesis. Cell Tissue Res 372, 379–392 [DOI] [PubMed] [Google Scholar]
- 19.Preite NZ, Nascimento BP, Muller CR, Americo AL, Higa TS, Evangelista FS, Lancellotti CL, Henriques FS, Batista ML Jr., Bianco AC, and Ribeiro MO (2016) Disruption of beta3 adrenergic receptor increases susceptibility to DIO in mouse. J Endocrinol 231, 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boekhoudt L, Roelofs TJM, de Jong JW, de Leeuw AE, Luijendijk MCM, Wolterink-Donselaar IG, van der Plasse G, and Adan RAH (2017) Does activation of midbrain dopamine neurons promote or reduce feeding? Int J Obes (Lond) 41, 1131–1140 [DOI] [PubMed] [Google Scholar]
- 21.Al Khazal F, Holte MN, Bolon B, White TA, LeBrasseur N, and Maher LJ 3rd. (2019) A conditional mouse model of complex II deficiency manifesting as Leigh-like syndrome. FASEB J 33, 13189–13201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tretter L, Patocs A, and Chinopoulos C (2016) Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1857, 1086–1101 [DOI] [PubMed] [Google Scholar]
- 23.Amorim-Pires D, Peixoto J, and Lima J (2016) Hypoxia Pathway Mutations in Pheochromocytomas and Paragangliomas. Cytogenet Genome Res 150, 227–241 [DOI] [PubMed] [Google Scholar]
- 24.van Nederveen FH, Dannenberg H, Sleddens HF, de Krijger RR, and Dinjens WN (2003) p53 alterations and their relationship to SDHD mutations in parasympathetic paragangliomas. Mod Pathol 16, 849–856 [DOI] [PubMed] [Google Scholar]
- 25.Castro-Vega LJ, Letouze E, Burnichon N, Buffet A, Disderot PH, Khalifa E, Loriot C, Elarouci N, Morin A, Menara M, Lepoutre-Lussey C, Badoual C, Sibony M, Dousset B, Libe R, Zinzindohoue F, Plouin PF, Bertherat J, Amar L, de Reynies A, Favier J, and Gimenez-Roqueplo AP (2015) Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun 6, 6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill JW (2012) PVN pathways controlling energy homeostasis. Indian J Endocrinol Metab 16, S627–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, Richardson AL, Sidhu SB, Robinson BG, and Clifton-Bligh RJ (2010) Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Human pathology 41, 805–814 [DOI] [PubMed] [Google Scholar]
- 28.Daubner SC, Le T, and Wang S (2011) Tyrosine hydroxylase and regulation of dopamine synthesis. Archives of biochemistry and biophysics 508, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lepoutre-Lussey C, Thibault C, Buffet A, Morin A, Badoual C, Benit P, Rustin P, Ottolenghi C, Janin M, Castro-Vega LJ, Trapman J, Gimenez-Roqueplo A-P, and Favier J (2015) From Nf1 to Sdhb knockout: Successes and failures in the quest for animal models of pheochromocytoma. Molecular and Cellular Endocrinology, 1–9 [DOI] [PubMed] [Google Scholar]
- 30.Smestad J, Erber L, Chen Y, and Maher LJ 3rd. (2018) Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smestad J, Hamidi O, Wang L, Nelson Holte M, Al Khazal F, Erber L, Chen Y, and Maher LJ (2018) Characterization and metabolic synthetic lethal testing in a new model of SDH-loss familial pheochromocytoma and paraganglioma. Oncotarget 9, 5864–5882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powers JF, Cochran B, Baleja JD, Sikes HD, Pattison AD, Zhang X, Lomakin I, Shepard-Barry A, Pacak K, Moon SJ, Langford TF, Stein KT, Tothill RW, Ouyang Y, and Tischler S (2020) A xenograft and cell line model of SDH-deficient pheochromocytoma derived from Sdhb+/− rats. Endocr Relat Cancer 27, 337–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hockman D, Adameyko I, Kaucka M, Barraud P, Otani T, Hunt A, Hartwig AC, Sock E, Waithe D, Franck MCM, Ernfors P, Ehinger S, Howard MJ, Brown N, Reese J, and Baker CVH (2018) Striking parallels between carotid body glomus cell and adrenal chromaffin cell development. Developmental Biology, 1–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sobrino V, Gonzalez-Rodriguez P, Annese V, Lopez-Barneo J, and Pardal R (2018) Fast neurogenesis from carotid body quiescent neuroblasts accelerates adaptation to hypoxia. EMBO Rep 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bishop T, and Ratcliffe PJ (2020) Genetic basis of oxygen sensing in the carotid body: HIF2alpha and an isoform switch in cytochrome c oxidase subunit 4. Sci Signal 13 [DOI] [PubMed] [Google Scholar]
- 36.Bishop T, Talbot NP, Turner PJ, Nicholls LG, Pascual A, Hodson EJ, Douglas G, Fielding JW, Smith TG, Demetriades M, Schofield CJ, Robbins PA, Pugh CW, Buckler KJ, and Ratcliffe PJ (2013) Carotid body hyperplasia and enhanced ventilatory responses to hypoxia in mice with heterozygous deficiency of PHD2. The Journal of Physiology 591, 3565–3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, and Eng C (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. Jama 292, 943–951 [DOI] [PubMed] [Google Scholar]
- 38.Wagener A, Muller U, and Brockmann GA (2013) The age of attaining highest body weight correlates with lifespan in a genetically obese mouse model. Nutr Diabetes 3, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lutz TA, and Woods SC (2012) Overview of animal models of obesity Curr Protoc Pharmacol Chapter 5, Unit5 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volkow ND, Wang G-J, and Baler RD (2011) Reward, dopamine and the control of food intake: implications for obesity. Trends in Cognitive Sciences 15, 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ushiama S, Ishimaru Y, Narukawa M, Yoshioka M, Kozuka C, Watanabe N, Tsunoda M, Osakabe N, Asakura T, Masuzaki H, and Abe K (2016) Catecholamines Facilitate Fuel Expenditure and Protect Against Obesity via a Novel Network of the Gut-Brain Axis in Transcription Factor Skn-1deficient Mice. EBioMedicine 8, 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bahnsen M, Burrin JM, Johnston DG, Pernet A, Walker M, and Alberti KG (1984) Mechanisms of catecholamine effects on ketogenesis. Am J Physiol 247, E173–180 [DOI] [PubMed] [Google Scholar]
- 43.Dadalko OI, Niswender K, and Galli A (2015) Impaired mTORC2 signaling in catecholaminergic neurons exaggerates high fat diet-induced hyperphagia. Heliyon 1, e00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr., and Endocrine S (2014) Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99, 1915–1942 [DOI] [PubMed] [Google Scholar]
- 45.Savitt JM, Jang SS, Mu W, Dawson VL, and Dawson TM (2005) Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci 25, 6721–6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.