

Abstract
The N-terminal caveolin binding motif (CBM) in Na/K-ATPase (NKA) α1 subunit is essential for cell signaling and somitogenesis in animals. To further investigate the molecular mechanism, we have generated CBM mutant human induced pluripotent stem cells (iPSCs) through CRISPR/Cas9 genome editing and examined their ability to differentiate into skeletal muscle (Skm) cells. Compared to the parental wild type human iPSCs, the CBM mutant cells lost their ability of Skm differentiation, which was evidenced by the absence of spontaneous cell contraction, marker gene expression, and subcellular myofiber banding structures in the final differentiated iSkm (induced Skm) cells. Another NKA functional mutant, A420P, which lacks NKA/Src signaling function, did not produce a similar defect. Indeed, A420P mutant iPSCs retained intact pluripotency and ability of Skm differentiation. Mechanistically, the myogenic transcription factor MYOD was greatly suppressed by the CBM mutation. Overexpression of a mouse Myod cDNA through lentiviral delivery restored the CBM mutant cells’ ability to differentiate into Skm. Upstream of MYOD, Wnt signaling was demonstrated from the TOPFlash assay to have a similar inhibition. This effect on Wnt activity was further confirmed functionally by defective induction of the presomitic mesoderm marker genes BRACHYURY (T) and MESOGENIN1 (MSGN1) by Wnt3a ligand or the GSK3 inhibitor/Wnt pathway activator CHIR. Further investigation through immunofluorescence imaging and cell fractionation revealed a shifted membrane localization of β-catenin in CBM mutant iPSCs, revealing a novel molecular component of NKA-Wnt regulation. This study sheds light on a genetic regulation of myogenesis through the CBM of NKA and control of Wnt/β-catenin signaling.
Keywords: caveolin binding motif (CBM), Na/K-ATPase, human induced pluripotent stem cells (iPSCs), Wnt/β-catenin, Src, skeletal muscle cells (Skm)
Graphical Abstract
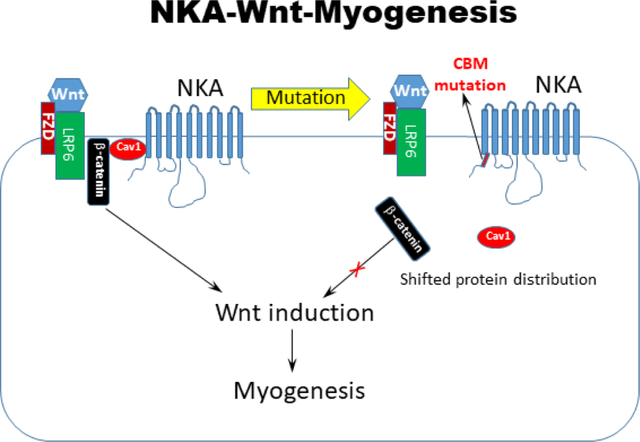
Two amino acid mutation (F97A;F100A) of the caveolin-binding motif in Na/K-ATPase shifted β-catenin localization, blocked Wnt signaling and induction of human induced pluripotent stem cells into skeletal muscle cells. The membrane protein Na/K-ATPase serves as a permissive gate to control cell signaling and muscle cell lineage specification.
Introduction
Na/K-ATPase (NKA), a cell membrane protein, is a member of the P-type ATPase superfamily. It is best known for its enzymatic function, which couples ATP hydrolysis to the transmembrane transport of Na+ and K+ [1,2]. The essential role of this NKA ion transporter in cell lineage-specific properties such as neuronal transmission or muscle contraction has been well recognized since its discovery [3–5].
Reports in model organisms such as C. elegans, Drosophila, or the zebrafish, have pointed to additional lineage-specific roles of NKA during development that are related, at least in part, to non-ion pumping functions of the protein complex [6–11]. Our gene-targeting approach recently revealed that cell lineage specification itself engages a NKA regulatory function that goes beyond its classic ion transport properties during embryonic development [12]. This function, which is critical for the embryonic development of both vertebrates and invertebrates, was revealed through a loss-of-function mutation that we introduced in the N-terminal caveolin-binding motif (CBM) of the NKA α1 subunit. The CBM mutation (mCBM) was chosen because it disrupts NKA’s ability to act as a scaffold and organize the formation of various signalosomes, but does not interfere with the binding and movement of Na+ and K+ ions [12]. The CBM mutation did not affect blastocyst formation, gastrulation, or the initiation of organogenesis, but were arrested in further organ development as evidenced by L1 arrest in C. elegans and embryonic lethality post E9.5 in the mouse. Mechanistically, pharmacological inhibition of the NKA/Src receptor complex did not reproduce the embryogenesis defect observed in the mCBM mouse. Rather, the developmental arrest was attributed to an essential role of NKA α1 in the dynamic operation of Wnt, and timely up-regulation of transcription factors during organogenesis. Hence, NKA has apparently evolved as a dual functional protein that works in concert with Wnt/β-catenin to orchestrate organogenesis in multicellular organisms.
The essence of stem cell biology is the ability of tissue/organ regeneration through a process akin to embryonic organogenesis. Indeed, stem cell differentiation involves both cell lineage specification and development of lineage-specific cellular function. Consistent with the proposed key role of NKA α1 in those processes, we observed that human inducible pluripotent stem cells (iPSC) with CRISPR/CAS9-mediated knock-in mutation of CBM NKA α1 had altered biological properties [12]. Specifically, human iPSCs with mutant CBM NKA α1 had reduced colony formation ability. This was accompanied by a significant reduction in the expression of stemness markers (NANOG and OCT4), and transcription factors controlling germ layer differentiation (MIXL1 and T for mesoderm, OTX2 and SOX1 for ectoderm, and GATA4 and SOX17 for endoderm) [12]. Although fundamentally important and translationally relevant, a functional challenge of the proposed concept of a NKA-mediated and CBM-dependent signaling mechanism in human stem cell differentiation was beyond the scope of our previous study. Critically, whether such mechanism would be 1) Wnt/β-catenin-dependent and 2) NKA/Src-independent as proposed has not been tested.
Accordingly, the stage of developmental arrest (E9.5) in the mouse embryo, combined with previous reports of NKA α1 ion-pumping-independent impact on skeletal muscle biology [13,14] prompted us to use myogenesis as the first lineage-specific process in which to functionally test the hypothesis that NKA/Wnt/β-catenin signaling is a critical determinant of lineage specification in human stem cell differentiation.
Materials and Methods
mCBM Mice.
Animal protocols were approved by the Marshall University Institutional Animal Care and Use Committee (IACUC) according to NIH guidelines. Mutant CBM mice were generated as described [12] and backcrossed to C57/BL6 for at least 6 generations. Female mCBM heterozygous mice (12–16 weeks old) were crossed with male mCBM heterozygous mice. Pregnant female mice at indicated stage were humanely euthanized and embryos at E9.5 were collected as described [12] for histological analysis of somites or quantitation of gene expression.
Culture of human iPSCs. Generation of mCBM and A420P human iPSCs.
Human iPS cells were purchased from iXCells (Cat# 30HU-002) and cultured with TeSR™-E8™ Kit (STEMCELL Technologies Inc, Cat# 05940). The iPSCs were originally derived from the peripheral blood mononuclear cells of a 59-year old Caucasian male, reprogrammed through episomal transduction with OCT4, SOX2, KLF4, LIN28, L-MYC, and TP53 shRNA according to the protocol by Okita et al. [15]. The culture plates were pre-coated with Geltrex from ThermoFisher (Cat# A1413201). Generation of mutant human iPSCs and CBM mutant mice was done as previously reported [12] using CRISPR/CAS9 genome editing. This knockin mutation (F97A;F100A) converted two phenylalanine into alanine at residue sites 97 and 100 of the human ATP1A1 protein, which has been shown to bind caveolin-1 [12,16,17]. The DNA plasmids for Cas9-GFP (Addgene# 44719) and gRNA cloning vector (Addgene# 41824) were from Addgene. sgRNA (single guide RNA) and ssODN (single-stranded oligodeoxynucleotides) were designed according to the published protocol [12]. The DNA sequences of primer oligos and their locations in the genome are presented in Supplemental Figure 1. The cloned Cas9-GFP vector inserted with sgRNA, along with ssODN, was transfected into human iPS cells using a 4D nucleofector device. Single cell sorting and plating were performed using FACS flow cytometry, and the clones were selected and validated by genotyping PCR and DNA sequencing (Supplemental Figure 1). For the A420P-α1 knock in mutant, we used a 150bp ssODN as the homologous arm. Genomic DNA sequences highly similar to the gRNA used, as well as other CBM sequences in ATP1A2, ATP1A3 and ATP1A4 genes were examined (Supplemental Figure 1). No off-target mutations in CBM and A420P mutant iPS cells were detected.
Differentiation of skeletal muscle cells from human iPSCs.
We used the differentiation package from Amsbio (product code: SKM-KITM) including media for three consecutive steps of induction named SKM01, SKM02 and SKM03 (Supplemental Figure 2). These three induction phases gave rise to myogenic precursor cells, myoblast, and myotube cells, respectively. Human iPSCs were dissociated with TrypLE (ThermoFisher, Cat# 12605010) and plated in the Geltrex-coated plates at a density of 5000 cells/cm2. This induction phase was maintained for 6–10 days, with the SKM01 medium changed every other day. The induced myogenic precursor cells were then dissociated and plated at the same density as the first step, and induced with SKM02 medium. This induction phase was maintained for 6–8 days with replacement of the SKM02 medium every other day. Finally, the confluent myoblast cells were cultured with SKM03 medium for about one week to induce differentiation into myotube cells.
Human iPSC-derived adipocytes.
Adipocyte differentiation from human iPSCs was conducted as Supplemental Figure 4 through a modified protocol from Hafner and Dani [18], Tang et al. [19], and Ahfeldt et al. [20]. Human iPSCs with colonies were incubated with MSC (mesenchymal stem cells) differentiation medium (DMEM with 10%FBS and 2.5 ng/mL bFGF) for 7–10 days until a monolayer was formed. The mature MSCs were then transferred to collagen-coated plates at a ratio of 1:3 as passage 1, and maintained in DMEM with 10% FBS. MSCs within 3 passages at 100% confluence were incubated with adipogenesis medium (DMEM containing 15% serum knockout replacement, 0.1% human insulin, 125 nM Dexamethasone, 0.2 mM Indomethacin, 100 μg/mL IBMX, and 5μM Rosiglitazone) for 3 weeks. Adipocyte differentiation was assessed through marker gene expression and oil red O staining described as follows.
Oil Red O Staining.
Oil red O staining for adipocytes was conducted according to the published method [21]. Briefly, adipocytes induced from iPSCs were washed twice with ice-cold PBS, then fixed with 4% formaldehyde for 10 min at room temperature. The fixed cells were stained with 0.21% oil red O in 60% isopropanol for 10 min at room temperature, then washed twice with PBS before imaging. Quantification of oil red O staining was performed after cell imaging. The cells were dried out at room temperature overnight, washed down with 100% isopropanol for 10 min at room temperature. The OD level at 490 nm was measured through Spectramax 190 Microplate Reader (Molecular Devices, Inc).
Histology, immunostaining, confocal and electron microscopy.
We followed our previous protocol to conduct immunohistochemistry in culture cells [22]. Briefly, cultured cells grown on coverslips pre-coated with Geltrex were fixed in prechilled methanol for 15 minutes, rinsed with 1X PBS, blocked with 3% horse serum diluted in PBS, and incubated with the primary antibody at 4°C overnight. After PBS washing, the samples were incubated with the fluorescence-conjugated secondary antibody (ThermoFisher, Cat# A21200) for one hour at room temperature, and counterstained with DAPI. Images were captured under a confocal microscope. The primary antibodies used in this study included anti-sarcomeric alpha actinin (Abcam, Cat# ab28052), anti-alpha skeletal muscle actin (Abcam, Cat# ab9465), Anti-β-Catenin (BD, Cat# 610153). The skeletal muscle fusion index was calculated according to published methods [23,24]. Striation of induced skeletal muscle cells (iSkm) was visualized under electron microscopy (EM) as described [25].
RNA analysis using RNA-seq, reverse transcription and real-time RT-PCR
We used RNeasy Mini Kit (Qiagen, Cat# 74104) for isolation of total RNA from mouse embryos and TRIzol reagent (ThermoFisher, Cat# 15596018) for human iPSCs and induced skeletal muscle cells (iSkm). RNA-seq was carried out by Novogene (Durham, NC, USA) to compare mRNA expression between the parental iPSCs-mCBM and iPSCs-WT, as well as the myogenic precursor cells of mCBM and wild type, named SKM01-mCBM and SKM01-WT. Each group included triplicate samples. Total RNA samples (1 μg) with a concentration greater than 20 ng/μl were examined for quality control, including OD260/280>2.0, and RIN (RNA Integrity Number)>6.8. cDNA libraries consisting of 250–300bp fragments were sequenced with Illumina platform and 20 M paired-end reads were generated. Transcript expression was normalized with RPKM (Reads Per Kilobase of exon model per Million mapped reads). For differential expression analysis, an adjusted P value < 0.05 in False Discovery Rate (FDR) was considered to be significant. Cytoscape was used to generate the heatmap of the altered genes.
cDNA synthesis was performed using the SuperScript® III First-Strand Synthesis system (ThermoFisher, Cat# 11752–050). Total RNA (1 μg) was mixed with 2 μl Enzyme Mix, 10 μl 2X Reaction Mix which includes 2.5 μM oligo(dT)20, 2.5 ng/μL random hexamers, 10 mM MgCl2, and dNTPs, and H2O till the total volume of 20 μl. The reverse transcription reaction was performed in a PCR machine under the following program: primer annealing at 25°C for 10 min, cDNA synthesis at 50°C for 20 min, inactivation of enzymes at 85°C for 5 min, and termination of the reaction at 4°C. The RNA template was digested with 1μL E. coli RNase H (2U) at 37°C for 20 minutes. The final products were used immediately or stored at −80°C until use. Real-time PCR was carried out as previously described [26]. The reaction included 2μl of the first strand cDNA synthesized as above, 10μl SYBR Green master mix (Roche Cat# 4887352001), 1μl forward and reverse primers (10μM, each) for the specific gene, and nuclease-free H2O to a total volume 20 μl. cDNA amplification was performed in a 384-microwell plate under LightCycler® 480 Real-Time PCR System. PCR conditions included an initial step of denaturation at 95°C for 10 minutes, followed by 45 cycles of amplification program consisting of 95°C for 10 seconds, 60°C for 10 seconds, and 68°C for 30 seconds. Average Ct (cycle threshold) values from the triplicate PCR reactions for a target gene were normalized against the Ct values for the internal control, Hypoxanthine-guanine phosphoribosyltransferase 1 gene (HPRT1) from the same cDNA sample. Three repeat samples were included for each gene. The relative expression was calculated according to the 2(−ΔΔCt) method [27,28]. The primer sequences are listed in Table 1.
Table 1.
Primer sequences
| Primer Name | Primer Sequence (5'→3') |
|---|---|
| human HPRT1 Forward | TGGACAGGACTGAACGTCTT |
| human HPRT1 Reverse | TCCAGCAGGTCAGCAAAGAA |
| Human MYOD Forward | CGACGGCATGATGGACTACA |
| Human MYOD Reverse | GGCAGTCTAGGCTCGACAC |
| Human MYOG Forward | CCAGGGGATCATCTGCTCACG |
| Human MYOG Reverse | GGAAGGCCACAGACACATCT |
| Human MYH8 Forward | GCAGACAGAAGCGGGTGAAT |
| Human MYH8 Reverse | GCTGAGTAGATGCTTGCTTGC |
| Human MYH3 Forward | GAGGCTGGTGAGCTGAGTC |
| Human MYH3 Reverse | GCTGCCTCTTGAGCTCTTCT |
| Human TNNT1 Forward | ACCTGGTCAAGGCAGAACAG |
| Human TNNT1 Reverse | TGTCCAGAGGCTTCTTACGC |
| Human CAV3 Forward | ACCTTCTGCAACCCACTCTTC |
| Human CAV3 Reverse | GAGCAGTCCCTGGCTTTAGAC |
| Human MYF5 Forward | TGAGAGAGCAGGTGGAGAACT |
| Human MYF5 Reverse | ACATTCGGGCATGCCATCAGA |
| Human MSGN1 Forward | TGTTGGACCCACCAGAACAC |
| Human MSGN1 reverse | TTGCAAAGGATGAGCCTCCC |
| Human PAX3 Forward | CAAGCCCAAGCAGGTGACAA |
| Human PAX3 reverse | TCGGATTTCCCAGCTGAACA |
| Human MIXL1 Forward | AGTCCAGGATCCAGGTATGGT |
| Human MIXL1 Reverse | GGCCTAGCCAAAGGTTGGAA |
| Human CTNNB1 Forward | GGCTACTCAAGCTGATTTGATGG |
| Human CTNNB1 Reverse | AAGACTGTTGCTGCCAGTGA |
| Human TBXT Forward | GGGTACTCCCAATCCTATTCTGAC |
| Human TBXT Reverse | ACTGACTGGAGCTGGTAGGT |
| Human NANOG Forward | CAATGGTGTGACGCAGGGAT |
| Human NANOG Reverse | TGCACCAGGTCTGAGTGTTC |
| Human OCT4 Forward | CGAGAAGGATGTGGTCCGAG |
| Human OCT4 Reverse | GAGACAGGGGGAAAGGCTTC |
| Human PPARG Forward | GATACACTGTCTGCAAACATATCACAA |
| Human PPARG Reverse | CCACGGAGCTGATCCCAA |
| Human FASN Forward | TCGTGGGCTACAGCATGGT |
| Human FASN Reverse | GCCCTCTGAAGTCGAAGAAGAA |
| Human ADIPOQ Forward | TCTGCCTTCCGCAGTGTAGG |
| Human ADIPOQ Reverse | GGTGTGGCTTGGGGATACGA |
| Human FABP4 Forward | GCTTTGCCACCAGGAAAGTG |
| Human FABP4 Reverse | ATGGACGCATTCCACCACCA |
| Mouse Myog Forward | GTCCCAACCCAGGAGATCATT |
| Mouse Myog Reverse | AGTTGGGCATGGTTTCGTCT |
| Mouse Myod1 Forward | TGGCATGATGGATTACAGCGG |
| Mouse Myod1 Reverse | GGTGGTGCATCTGCCAAAAG |
| mouse Myh3 Forward | CTCTGTCACAGTCAGAGGTGT |
| mouse Myh3 Reverse | CTTTTCCGACTTGCGGAGGA |
| mouse Myh8 Forward | CTGAGGAGGCTGAGGAACAATC |
| mouse Myh8 Reverse | CCTCCTTCCTCTGCAAGATGT |
| Mouse Actb Forward | GGCTGTATTCCCCTCCATCG |
| Mouse Actb Reverse | CCAGTTGGTAACAATGCCATGT |
Lentivirus-mediated gene delivery
Lentivirus expression vector for the mouse Myod1 gene (pLv-CMV-Myod) was from Addgene (Plasmid #26808). Virus packaging and transfection were carried out as previously described [26]. After one week of virus transduction, infected cells were used for Skm differentiation. To serve as a negative control, an empty vector with the CMV promoter (pLenti-CMV-GFP, Addgene plasmid# 17446) was used to generate CBM mutant iPSCs carrying CMV-GFP (iPSCs-mCBM-CMV-GFP). The resulting cells were subjected to the same Skm differentiation protocol. The induction of myogenic markers was compared with that in iPSCs-WT and iPSCs-mCBM.
Western blot and cell fractionation
Cell extracts isolation, caveolin-rich membrane fraction, protein concentration measurement, and Immunoblot analysis were performed as described [12,17]. After rapidly washing with ice-cold PBS, the iPSCs were lysed in modified ice-cold RIPA buffer (radioimmune precipitation assay buffer containing 1% Nonidet P-40, 1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1 mM NaF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 50 mM Tris-HCl, pH 7.4). After the cell lysates were centrifuged at 14,000g for 15 min, the supernatants with protein samples were separated by SDS-PAGE and transferred to the Optitran nitrocellulose membranes. After blocking with milk, the membranes were probed with the specific antibodies listed below. The films were developed with an ECL kit.
The antibodies used were Na/K-ATPase α1 (Developmental Studies Hybridoma Bank, Cat# a6f), Na/K-ATPase β−1 (Millipore Sigma, Cat# 05–382), caveolin-1 (Cell Signaling Technology, Cat# 3267S), β-catenin (BD biosciences, Cat# 610153), Phospho-β-Catenin (Ser33/37/Thr41) antibody (Cell Signaling Technology, Cat# 9561), GSK-3β (Cell Signaling Technology, Cat# 12456S), alpha-tubulin (Sigma-Aldrich, Cat# T5168).
Wnt activity by TOPFlash luciferase assay
The assay was conducted as we have previously described [12]. Wnt-3A conditioned media was prepared according to the instructions provided by ATCC (Cat# CRL-2647™). Briefly, Wnt-3A producing cells were grown in T-75 flask with 10ml DMEM medium containing 0.4 mg/ml G-418. Confluent cells were split at a ratio of 1:10 and subcultured in DMEM without G-418 for 4 days. Upon confluence, cell culture media were collected and replenished with 10ml flesh DMEM without G-418 for another 3 days. Combine culture media collected with the first batch. This Wnt3a conditioned medium was cleaned with 0.2 μm syringe filter and aliquots were stored at −20 °C. Human iPSCs were cultured on pre-coated 6-wells plates and transfected with 1 μg TOPFlash (Addgene# 12456) DNA plasmid, as well as 100 ng pRenilla (Addgene# 38235) as the reporter control using Lipofectamine 2000 (Invitrogen, Cat# 11668–027). After eight hours, transfected cells were washed with fresh culture medium and cultured overnight. Then, cells were stimulated with either Wnt-3A conditioned medium or control medium for 6 hours. The luciferase assay was performed with the dual luciferase assay reporter kit according to manufacturer’s recommendations (Promega Cat# E1910).
Statistical Analysis
All data are expressed as mean+/− standard error (SE). Student’s t-test was used to compare two groups and ANOVA followed by multiple t-tests was used to compare more than two groups. *, **, *** indicates p<0.05, 0.01 and 0.001 respectively.
Results
The CBM of NKA α1 is essential for somite development and myogenesis in the mouse embryo.
Segmentation initiates very early in vertebrate embryonic development. It is first observed when embryos develop somites, the precursors of several segmented organs such as the axial skeleton, body skeletal muscles, and part of the dermis. Somites are formed during the highly regulated process of somitogenesis from the unsegmented presomitic mesoderm (PSM). Normally, the posterior PSM cells remain in an undetermined and immature state and become anteriorly displaced as new cells migrate to the posterior PSM. When PSM cells arrive to a prescribed position in the anterior PSM, they undergo a dramatic transition in gene expression driven by signaling networks dependent of β-catenin/Wnt, Notch, and FGF[29]. This initiates the segmentation program and somitogenesis, leading to the formation of the dermomyotome, myotome, and sclerotome. It is within the somites that skeletal myogenesis is initiated at around E8 in the mouse, with the specification of premyogenic progenitors and skeletal myoblasts [30–32].
As shown in Figure 1B, somitogenesis is arrested in mCBM homozygous mouse embryos at E9.5. Consistent with a defect in skeletal myogenesis, we detected a significant decrease in mRNA levels of the skeletal muscle marker genes myogenin (Myog), myosin heavy chain 3 (Myh3), and myosin heavy chain 8 (Myh8) as shown in Figure 1C.
Figure 1.
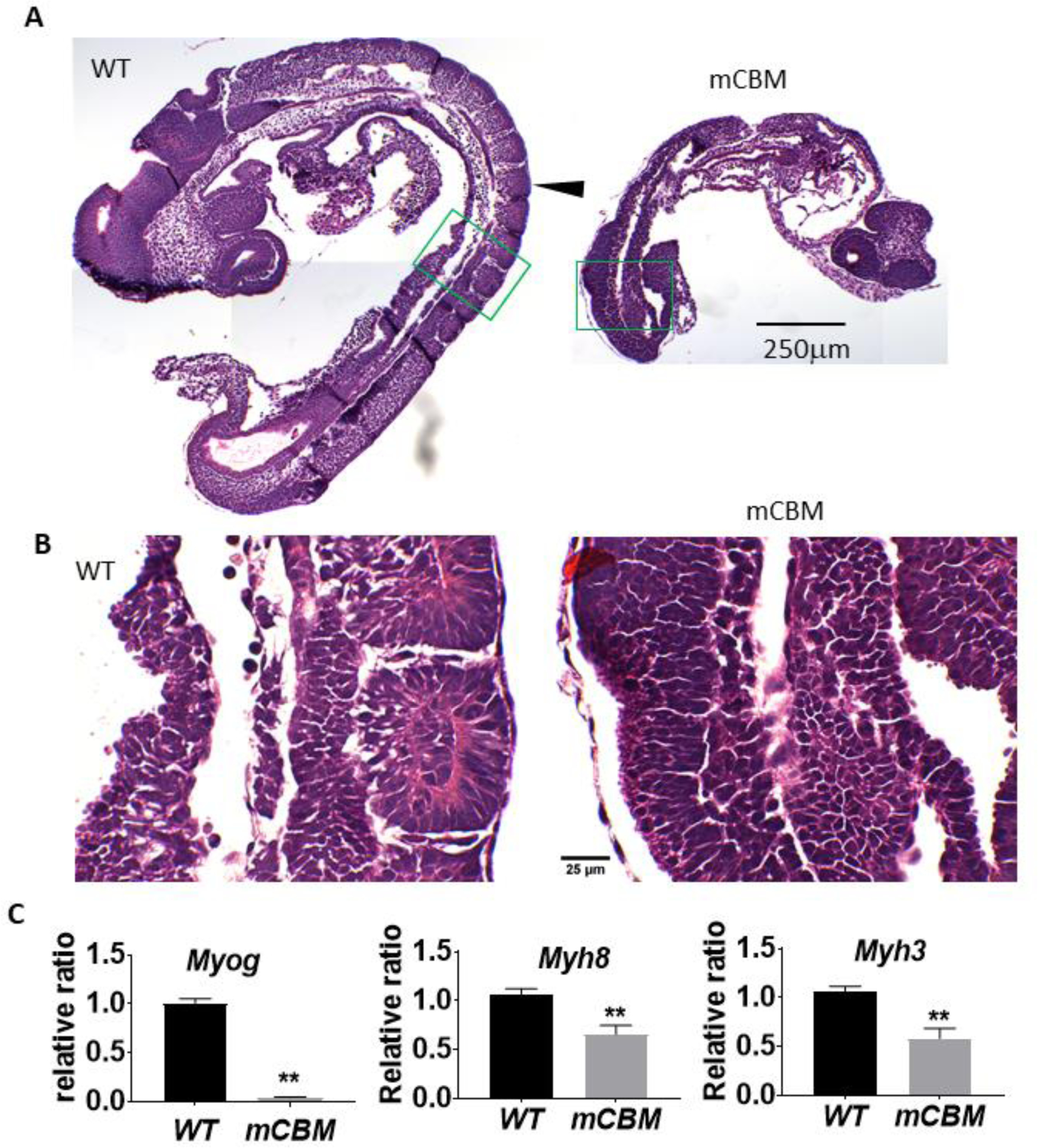
The CBM mutation (mCBM) inhibits somite growth in the mouse embryo.
Sagittal sections of mouse embryos (E9.5) show altered somites (arrowhead pointed in A) in the CBM mutant compared to the segmented structure in wild type (WT). Green framed regions are shown in B with a higher magnification. mCBM embryos have reduced mRNA expression of muscle marker genes Myog, Myh8 and Myh3 (C). Triplicate samples were used for each group. Bar graphs in all figures are mean ± standard error. Student’s t-test was used to compare the difference between wild type and mCBM. ** indicates p<0.01. Images in panel A share the same scale bar, so do the images in panel B.
The CBM mutation in NKA α1 inhibits skeletal muscle differentiation of human induced pluripotent stem cells (iPSCs)
The above observations provided an impetus to determine whether the NKA α1 CBM-mediated control of Wnt/β-catenin revealed by our recent report [12] is a key molecular determinant of skeletal muscle cell specification and differentiation. To test this functionally, we used an approach based on human iPSC differentiation in vitro, which allowed us to transpose early signaling events of mesoderm specification in the vertebrate embryo to a system that is more amenable to genetic and pharmacological interventions [30,33–35].
The differentiation system from AMSBIO includes three steps of induction as illustrated in Supplemental Figure 2A. Step I (SKM01 medium) gives rise to myogenic precursors. Step II (SKM02 medium) induces myoblasts. Step III (SKM03 medium) leads to myotubes, henceforth referred to as induced skeletal muscle cells (iSkm). Gene expression analyses confirmed the expected specific transcription activity pattern during differentiation from WT human iPSCs, which recapitulates muscle development during early embryogenesis (Supplemental Figure 2B–G). This includes an early induction of PAX3 by SKM01, followed by an increase of myogenic transcription factors MYOD and MYOG by SKM02, and the late induction of mature Skm markers MYH8, CAV3 and TNNT1.
Morphologically, the expected sequential differentiation from smaller and round-shaped cells to long and neatly aligned myotube cells was observed (Figure 2A). Moreover, the differentiated iSkm were able to twitch spontaneously (Supplemental Video 1). Subcellular imaging by electron microscopy (EM) revealed the expected striations and banding structures around glycogen granules (Figure 2C). Positive immunostaining with antibodies against sarcomeric alpha actinin and alpha skeletal muscle actin further validated the identity of skeletal muscle cells (iSkm, Figure 2E and 2G).
Figure 2.
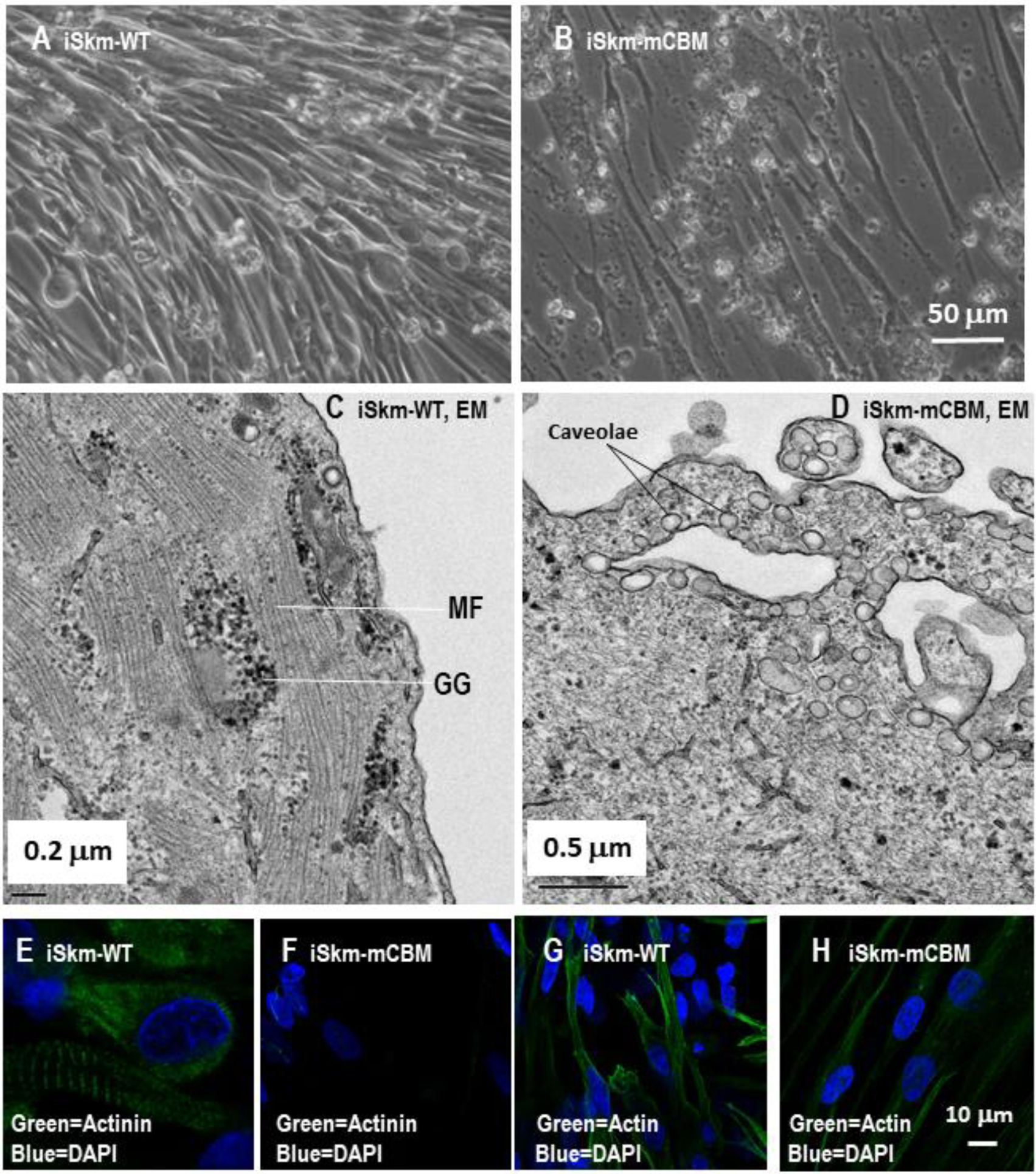
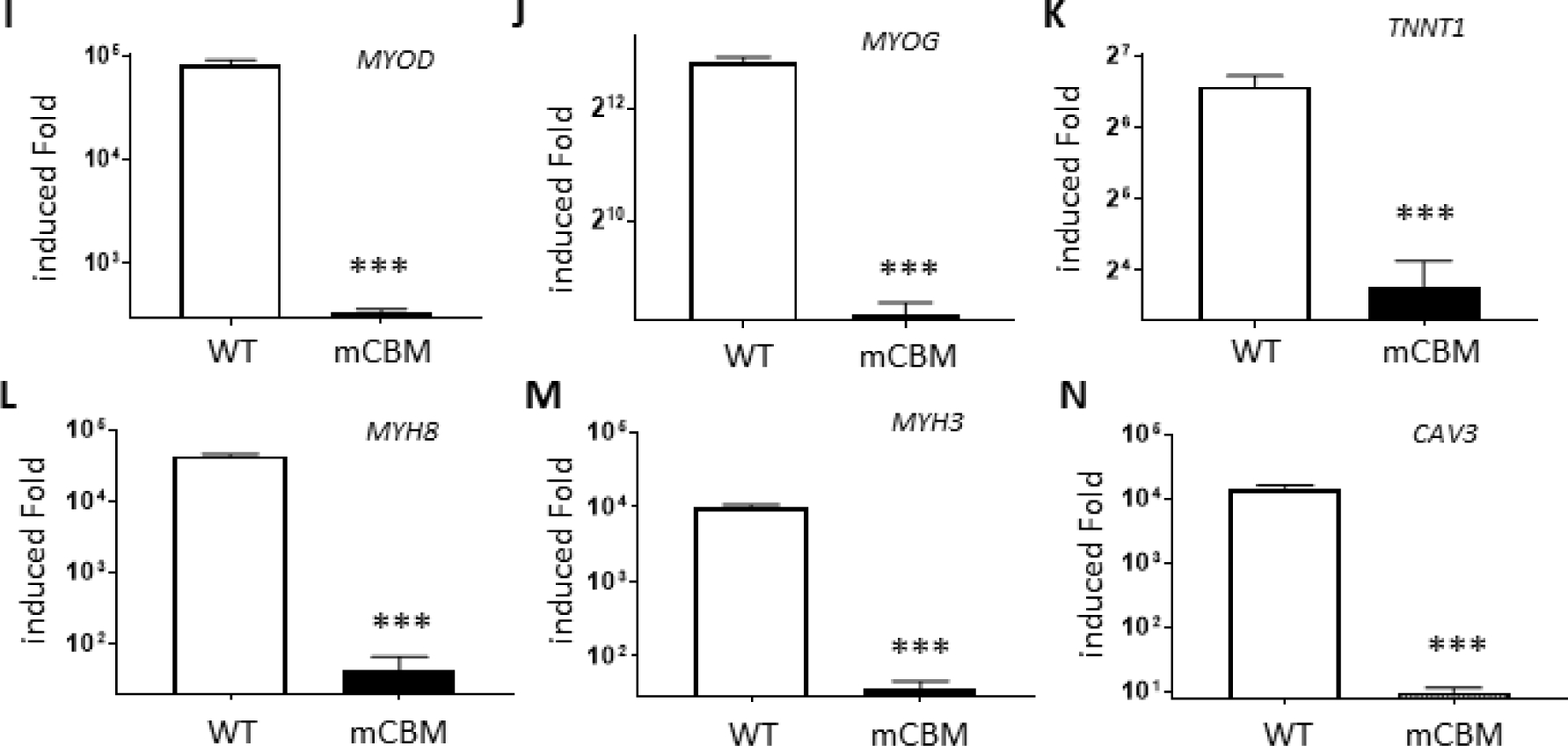
CBM mutation inhibits Skm differentiation in human iPSCs.
Wild type human iPSCs were differentiated into twitching skeletal muscle cells (A and Supplemental Video 1) with typical striation structure in electron microscope images (C) and positively immunostained with anti-sarcomeric alpha actinin antibody (E) and anti-alpha skeletal muscle actin antibody (G). Compared to the parental iPSCs, these induced skeletal muscle cells (iSkm) had dramatically increased mRNA expression of myogenic marker genes (panels I-N). RT-qPCR results are shown as mean±SE. Student’s t-test was used to compare the difference between wild type and mCBM. *** p<0.001, n=3. In contrast, mCBM cells lost this potential and failed to differentiate into contracting myocytes, as evidenced by morphology (B, D), immunostaining (F and H) and marker gene expression (I-N). GG in panel C stands for glycogen granules and MF for myofilaments. Panel A shares the same scale bar as Panel B, and so do the panels E-H.
In parallel, this differentiation protocol was applied to mCBM iPSCs obtained previously by recombination mediated-CRISPR/Cas9 genome editing to knockin the mutations (F97A;F100A) of the DNA sequences encoding the consensus caveolin-1 binding motif in human ATP1A1 protein. The replacement of these two key amino acids has been shown to disrupt the interaction between ATP1A1 and caveolin-1 [12,16,17]. Strikingly, no visible cell contraction or twitching was observed in the differentiated cells at the final step of SKM03 (data not shown). Morphologically, the induced mCBM iSkm cells were very thin and sparse (Figure 2B) with a poor alignment. EM failed to detect any banding structures, and instead revealed numerous thin, disorganized fibers randomly distributed and poorly aligned (Figure 2D). The CBM mutant iSkm did not accumulate glycogen granules, but an increased number of caveolae structures was noted close to the cell surface (4.5±0.79 in iSkm-mCBM vs 0.38±0.02 in iSkm-WT, p<0.01, Figure 2D).
Contrary to wild type (WT) iSkm, the CBM mutant iSkm cells were weakly stained with anti-sarcomeric alpha actinin and anti-alpha skeletal muscle actin (Figure 2F & 2H vs 2E & 2G, respectively). Consistent with these morphological observations, the induction of marker gene expression for skeletal muscle differentiation was drastically reduced compared to WT iSkm (Figure 2 I–N). These defects were also observed in another mCBM iPSC clone (Supplemental Figure 3). All together, these observations revealed an inability of CBM mutant iPSCs to differentiate into skeletal muscle cells. In contrast, mCBM-iPSCs retained the ability to differentiate into adipocytes, as evidenced by marker gene expression including PPARG (peroxisome proliferator activated receptor gamma), FASN (fatty acid synthase), ADIPOQ (adiponectin), FABP4 (fatty acid binding protein 4, Supplemental Figure 4, panel B-E), as well as positive staining for oil red O (Supplemental Figure 4, panel A and F). Of note, although mutant adipocytes (Adi-mCBM) expressed more ADIPOQ and FABP4 RNA than Adi-WT, Adi-mCBM had a weaker oil red O staining.
Disrupting NKA/Src through A420P mutation of NKA α1 does not phenocopy the myogenic defect of CBM NKA α1 mutation in iPSCs
By disrupting NKA scaffolding function, the CBM mutation is known to impair NKA/Src kinase receptor function [12]. Therefore, we next tested whether the observed myogenic defect in the mCBM iPSC was due to the defective NKA/Src receptor function. To this end, a well characterized NKA/Src loss of function mutation, A420P [36,37], was introduced in the NKA α1 sequence of human iPSCs using CRISPR-Cas9 genome editing (Supplemental Figure 1). Like the wild type iPSCs, A420P grew typical stem cell colonies (Figure 3A&B) and highly expressed pluripotency marker genes NANOG and OCT4 (Figure 3C). Additionally, A420P mutant iPSCs retained a Skm differentiation potential comparable to WT iPSCs. This was evidenced by the induction of gene expression of skeletal muscle markers (Figure 3D), immunostaining with antibodies against myosin heavy chain (MHC, Figure 3F), sarcomeric alpha actinin and alpha skeletal muscle actin (Supplemental Figure 5), as well as spontaneous contraction of the differentiated iSkm A420P cells (Supplemental Video 2). Overall, mild differences in induction fold for myogenic marker genes (Supplemental Figure 5) were noted in WT vs A420P. Accordingly, it was concluded that the observed inability of mCBM iPSC to differentiate into skeletal muscle cells was not recapitulated by the A420P mutation.The NKA/Src receptor function was not the primary contributor to the mCBM defect.
Figure 3.
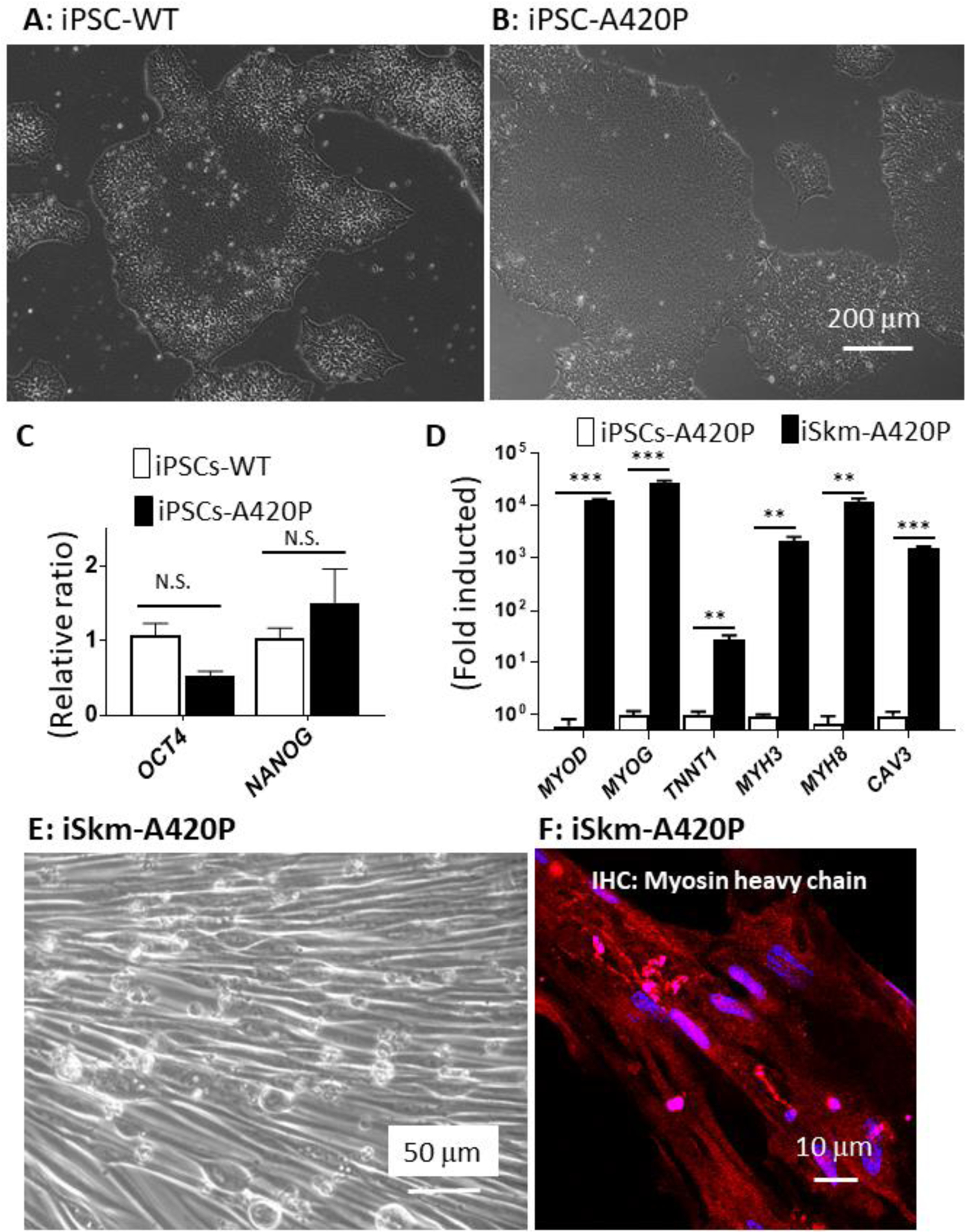
A420P mutant iPSCs retain iSkm differentiation potential.
A420P mutant iPSCs, defective in NKA mediated Src signaling, formed typical colonies (A and B) and expressed pluripotency marker genes OCT4 and NANOG (C), as observed in WT-iPSCs. These mutant iPSCs were capable of differentiating into Skm with normal gross morphology (E), increased expression of Skm marker genes (D) and positively immunostained with the myosin heavy chain antibody (red, F). RT-qPCR results in panel D were expressed as mean±SE (n=3, **p<0.01, ***p<0.001, N.S.: not significant by Student’s t-test analysis).
Lentivirus-mediated expression of MYOD rescues the expression of downstream myogenesis marker genes in mCBM iPSCs
The basic helix-loop-helix (bHLH) transcription factors MYF5 and MYOD control early phases of mammalian myogenesis. Either one of these myogenic regulatory factors is reportedly capable of driving muscle cell differentiation in vitro [38–40]. We observed that mRNA of MYOD, but not MYF5, was significantly induced in our WT Skm differentiation system (Supplemental Figure 6). To test whether late markers of differentiation (i.e., TNNT1, MYH8 and CAV3) in mCBM iSkm could be restored by expression of MYOD in mCBM iPSC, we used lentiviral delivery of mouse Myod cDNA (mMyod1). Successful overexpression of mMyod1 was verified at the mRNA level in mCBM iSkm (Supplemental Figure 6). mCBM iSkm cells expressing mMyod1 transgene (Figure 4B), had the morphological features of WT iSkm cells under transmission light microscopy. They also presented an ultrastructural organization comparable to that of WT iSkm cells, including banding structures (Figure 4C). Moreover, the number of caveolae structures in iSkm-CBM was reduced to the level of iSkm-WT upon expression of the Myod transgene (0.19±0.029 in iSkm-mCBM-Myod1 vs 0.38±0.02 in iSkm-WT, p>0.05). In constrast to mCBM iPSCs expressing the empty vector, mMyod1-expressing mCBM iPSCs regained sensitivity to the Skm differentiation media in terms of their induced expression of myogenic marker genes (Figure 4D), reaching levels similar to, or even higher than those measured in iSkm derived from WT iPSCs. Immunofluorescence labeling of MHC further validated the differentiation mCBM iPSCs toward skeletal muscle upon expression of exogenous mMyod1 (Figure 4A), albeit with a defect in fusion capacity as revealed by morphometric analyses and scored fusion index (Supplemental Figure 6). Of note, spontaneous twitching was not observed in the mMyod1-rescue iSkm (data not shown). Taken together, these data indicated a functional defect upstream of MYOD due to the CBM mutation in NKA α1.
Figure 4.
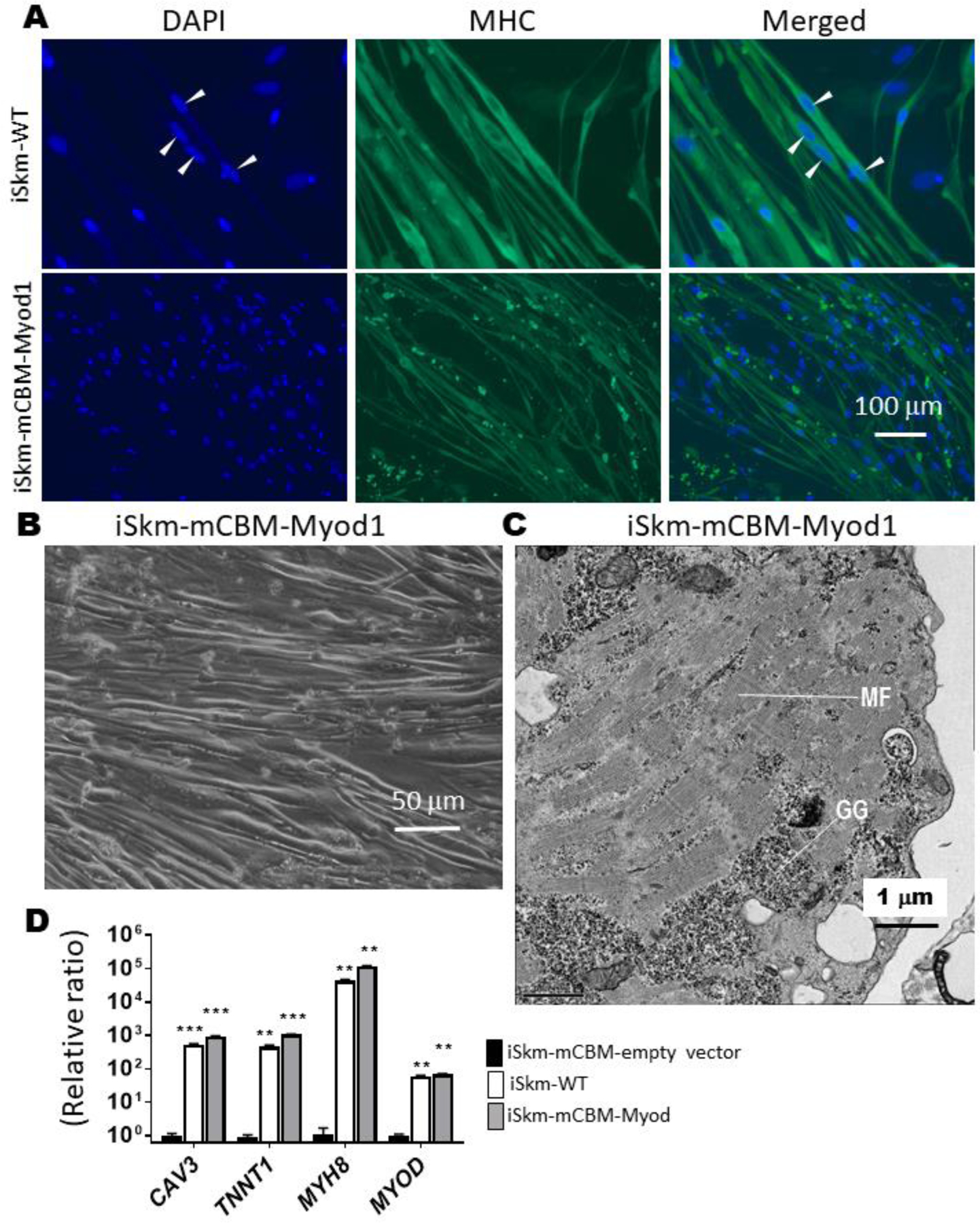
Rescue of Skm differentiation in mCBM cells with a mouse Myod1 transgene.
Myod1-rescued iSkm were positively stained with myosin heavy chain antibody (Panel A), developed typical striations (panel C, GG-glycogen granules, MF-myofilaments), and expressed myogenic marker genes, at levels equivalent to, or even higher than iSkm from wild type iPSCs (D). In panel D, an empty vector (pLenti-CMV-GFP, Addgene plasmid# 17446) was used as the negative control. RT-qPCR results were shown as mean±SE and analyzed by multiple t-tests in ANOVA (n=3, ** p<0.01, *** p<0.001, all compared to the empty vector control). Arrow heads in panel A points the multiple nuclei in one muscle fiber.
β-catenin/Wnt signaling relays CBM-NKA regulation of myogenesis
In addition to its importance for the regulation of NKA/Src receptor function, we have recently uncovered an essential role of NKA’s CBM for the dynamic operation of Wnt in mammalian epithelial cells and the regulation of target transcription factors, which likely contributed to defective organogenesis of the mCBM mouse [12]. Functionally, a TOPFlash assay revealed that the addition of Wnt3a strongly increased TCL/LEF-firefly luciferase activity in WT iPSCs, but not in CBM mutant cells (Figure 5A), exposing a blunted response to Wnt stimulation similar to the one observed in the mammalian cell CBM mutant [12]. CBM regulation on Wnt signaling was further supported by RNA-seq in the present studies. We compared mRNA expression between the parental wild type and mCBM iPSCs, as well as the SKM01-induced myogenic precursors (Supplemental Table 1 and 2). Altered expression of Wnt signaling members by CBM mutation was observed in both parental iPSCs and SKM01-induced precursors (Supplemental Figure 7). Meanwhile, all known pluripotency markers were reduced in mCBM mutant iPSCs. We also found mCBM-regulated nuclear transcription factors (e.g. MED12, PAF1, PYGO1/2, RUVB1/2, SMARCA4) mediating in β-catenin transcriptional activation of downstream targets (Supplemental Figure 7). Finally, another cell signaling important in myogenesis, the TGF-β pathway, was altered by the CBM mutation (Supplemental Figure 7). To a certain degree, these alterations might have contributed to the defective phenotype in myogenesis.
Figure 5.
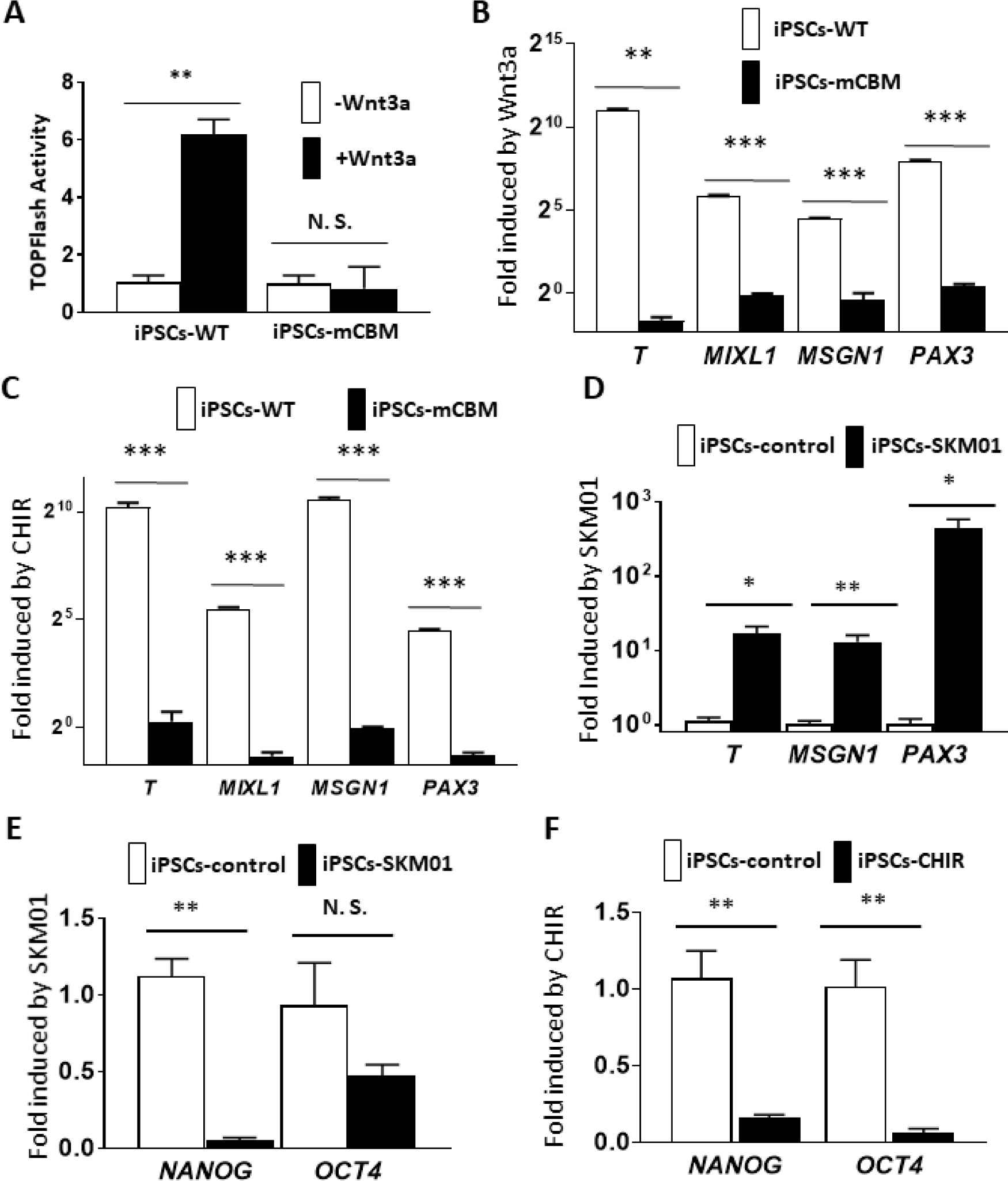
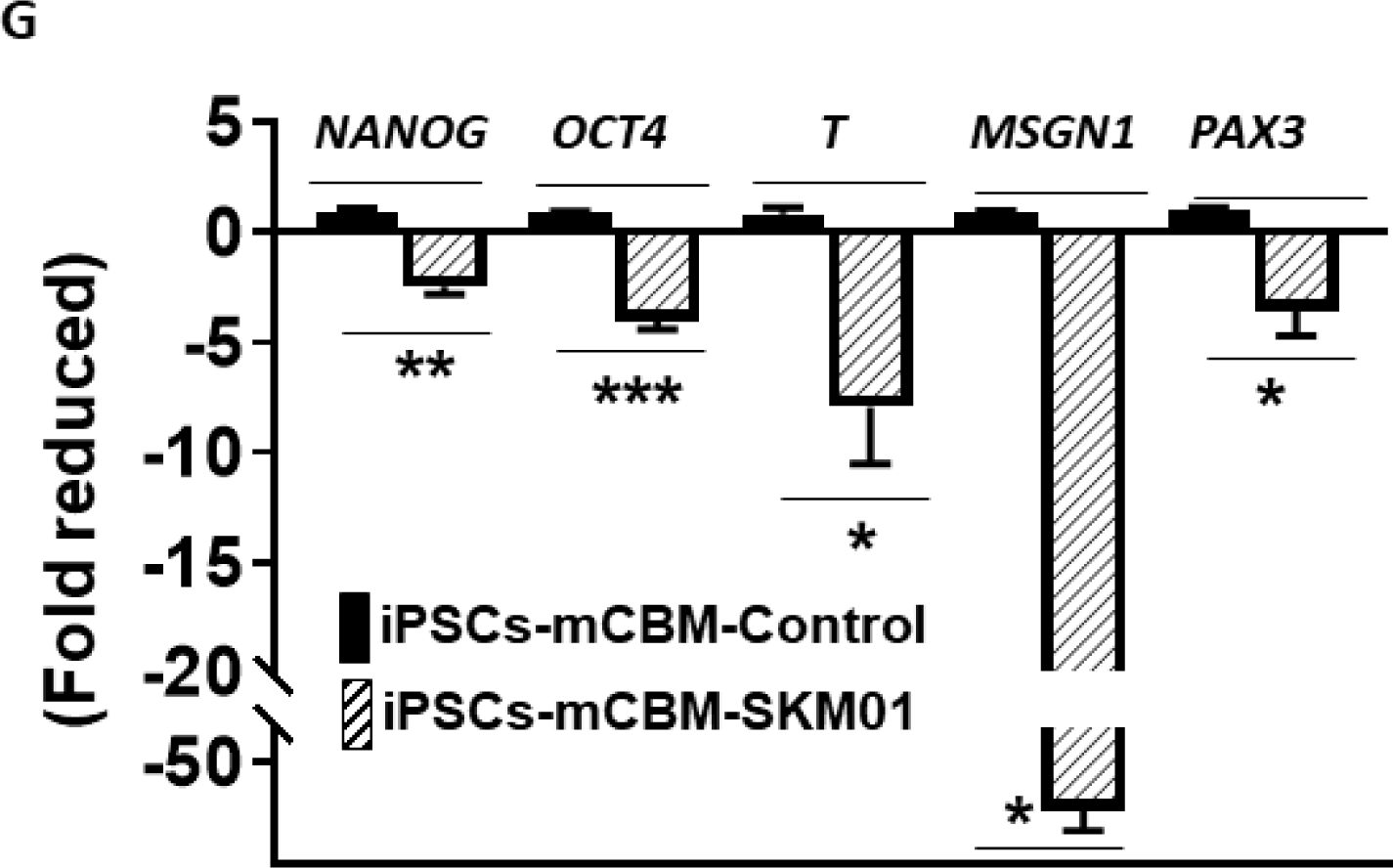
Defective Wnt induction in CBM mutant cells.
Wnt activity was measured with the TOPFlash assay, showing the increased luciferase in wild type human iPSCs by Wnt3a stimulation. In contrast, a blunted response was observed in CBM mutant cells (A). This inhibition of Wnt signaling was further reflected by the blocked induction of mRNA expression for the mesoderm marker genes T and MIXL1, presomitic marker gene MSGN1 and somatic marker gene PAX3, with either Wnt3a ligand (5 days for PAX3 and 3 days for T, MIXL1, and MSGN1, panel B) or the GSK-3β inhibitor CHIR (10 μM, 4 days for PAX3 and 2 days for T, MIXL1 and MSGN1, panel C). Moreover, this induction was mimicked by SKM01 treatment in wild type human iPSCs (3 days, panel D). Both SKM01 (panel E) and CHIR (panel F) treatment in wild type human iPSCs reduced mRNA expression of pluripotency markers NANOG and OCT4. However, SKM01 treatment (3 days) in mCBM iPSCs reduced all marker gene expression (panel G). All data were expressed as mean±SE (n=3, *p<0.05, **p<0.01, ***p<0.001, N.S.: not significant, by Student’s t-test).
Given the essential role of Wnt signaling in vertebrate myogenesis upstream from MyoD [41–43] and early stages of myocyte lineage specification [44], we next examined the functional impact of the mCBM defect in the early initiation of muscle cell fate from iPSCs, including the stages of mesoderm and presomitic mesoderm (PSM). We assessed the response of iPSCs to Wnt3a ligand and CHIR99021 (CHIR), a small molecule inhibitor of GSK3β that prevents the degradation of β-catenin and has been widely used in the protocols of in vitro differentiation of iSkm from iPSCs [24,45–50]. As shown in Figure 5B&C, the expected induction of marker genes was observed in WT but not mCBM iPSCs, confirming that the disrupted Wnt signaling is a key reason for the defective Skm differentiation in CBM mutant cells. As shown in Figures 5E&F, incubation of WT iPSC with CHIR (10 μM) significantly decreased pluripotency markers NANOG and OCT4, comparable to the effect of three days of incubation with the SKM01 incubation medium. Similar to Wnt3a ligand and CHIR, mRNA expression of mesoderm markers T and MIXL1, PSM markers MSGN1, and somitic marker PAX3 was upregulated in WT iPSCs exposed to SKM01 medium (Figure 5D). In contrast, in mCBM iPSCs, mRNA expression of T, MIXL1, MSGN1 and PAX3 by Wnt3a, CHIR or SKM01 was blunted, or even down-regulated (Figure 5B, C, and G).
CBM-NKA integrates protein complex of Wnt signalosome
Wnt signaling relies on dynamic cellular trafficking of β-catenin proteins from the cell membrane to the cytoplasm/nucleus, which is profoundly perturbed in porcine epithelial cells when WT NKA is replaced by a CBM mutant NKA [12]. Using confocal microscopy following immunofluorescence staining, we observed that β-catenin was similarly distributed away from the plasma membrane in mCBM iPSCs (Figure 6A). Consistent with this finding, western blot analysis of β-catenin in subcellular fractions of iPSCs revealed a redistribution away from the low-density membrane fractions, particularly fractions 4/5 typically enriched with caveolae (Figure 6C,F), besides the equal amount of total β-catenin (Figure 6B, E). This abnormal distribution in mCBM iPSCs was consistent with the defect observed in mCBM myogenic precursor cells (human iPSCs induced with SKM01 for one week, Supplemental Figure 8), and mammalian epithelial cells expressing the CBM mutant form of NKA [12]. Additional similarities between the epithelial and iPSCs models further included the increased caveolin-1, and unchanged NKA α1 and β1 total protein contents in the CBM mutants compared to WT (Figure 6B, E).
Figure 6.
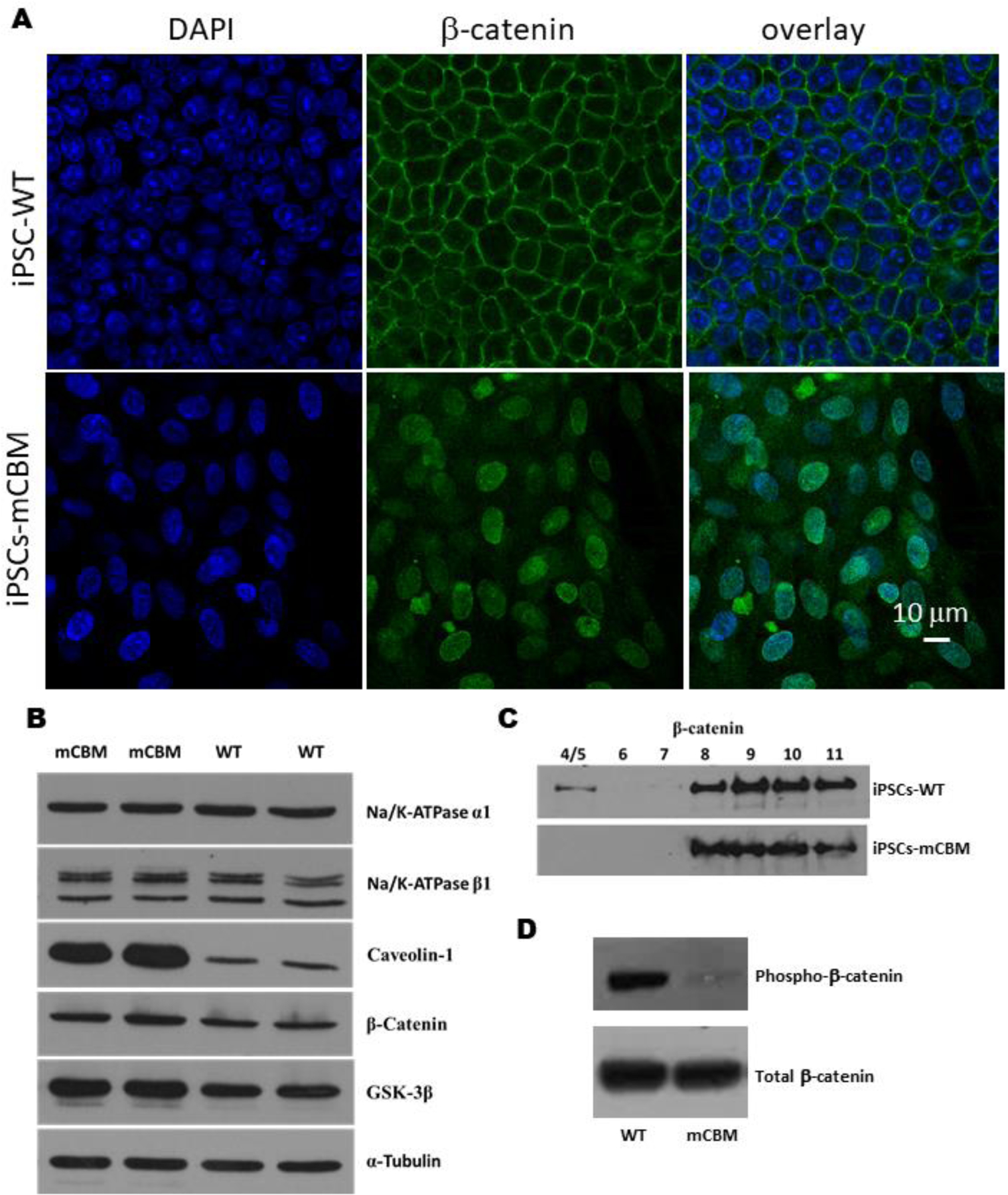
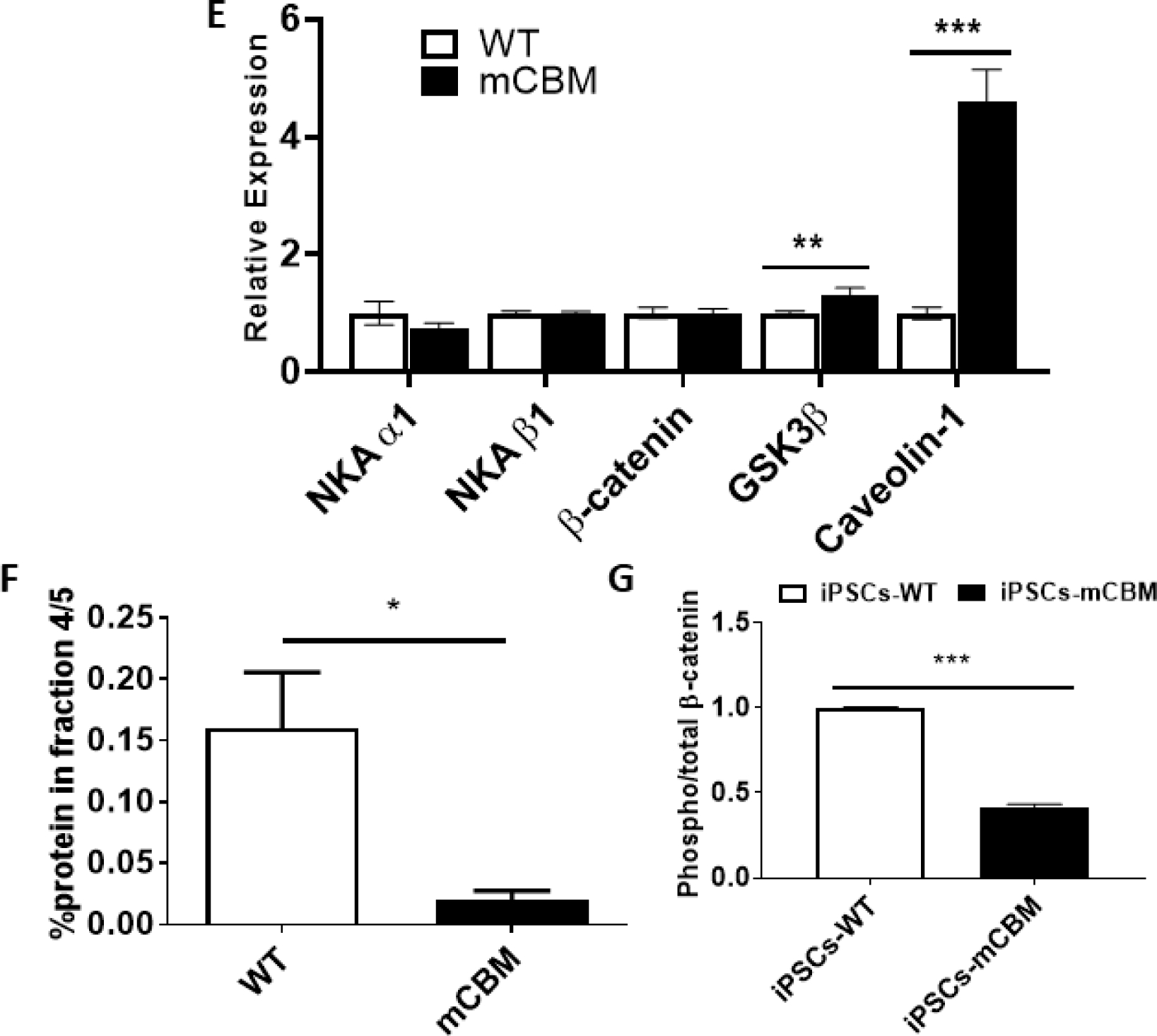
Disrupted protein distribution and trafficking of β-catenin by CBM mutation.
Immunostaining under confocal imaging shows shifted localization of β-catenin from cell membrane to nucleus/cytosol in mCBM iPSCs (A), although the total protein of β-catenin was not significantly changed (B, western blot). This alteration was further confirmed through a cell fractionation assay (C), showing the absence of β-catenin blot signal in caveolin-enriched fractions 4/5. Finally, CBM mutation decreased phosphorylation (Ser33/37/Thr41) of β-catenin in iPSCs (D, quantification for panel B, C and D is shown in panel E, F and G, respectively. n=2–4 in panel A, n=6–8 in panel B, n=3 in panel C and D). Data are shown as mean ± SE. *P < 0.05 by student’s t-test.
Nuclear accumulation of β-catenin is normally associated with activation of Wnt signaling. Without ligand stimulation, β-catenin is phorphorylated by casein kinase 1 (CK1) and GSK-3β and subsequently degraded. Ligand binding with Wnt receptor triggers aggregation of protein complex inside the cell membrane-associated vesicles, including LRP6 and GSK-3β, which sequester cytosolic β-catenin from proteasome degradation. Stabilized β-catenin goes into the nucleus to activate gene transcription of Wnt downstream targets [51]. Consistently, the abnormal accumulation of β-catenin in the nucleus was accompanied by a significant decrease of phospho-β-catenin in mCBM-iPSCs, evidenced by western blot using antibodies for phospho-β-Catenin (Ser33/37/Thr41) and total β-Catenin (Figure 6D,G). GSK-3β phosphorylates β-catenin at three residues (Ser33, Ser37 and Thr47), which targets β-catenin for ubiquitination and proteosomal degradation [12]. Accumulation of β-catenin that is specifically non-phosphorylated at these GSK-3β sites is known to be critical for β-catenin-mediated transcription [13][14].This premature activation, correlated with increased MSGN1 and PAX3 expression in mCBM-iPSCs (Supplemental Figure 9), likely accounts for the blunted induction with either Wnt ligand, CHIR or SKM01 medium observed in the CBM mutant cells (Figure 5).
Taken together, these data strongly suggested a defect of Wnt/β-catenin signaling secondary to the CBM mutation in iPSCs. The molecular characteristics of the defect (unchanged NKA pumping function and expression, but abnormal distribution of β-catenin and blunted Wnt response) were comparable to what we have observed in terminally differentiated epithelial cells expressing the CBM NKA mutant [12]. This genetic cascade is conserved in different mammalian cells, and likely among different species, demanding further investigation for consolidation.
Discussion
A cell lineage-specific-, ATPase-independent role of the ubiquitous membrane NKA protein complex during development in metazoans has long been suspected [6–11]. Using a gene-targeting approach, we recently established a direct link between cell lineage specification and NKA beyond its classic ATPase-dependent ion transport properties during mouse embryonic development [12]. This function is mediated by two key phenylalanine residues (F97 and F100) in one of the two potential Caveolin-1 Binding Motifs (CBM) of the NKA α1 polypeptide (encoded by ATP1A1). Mutation of those residues, which both belong to the N-terminal CBM of NKA α1 [52,53], does not modify the enzyme’s ion-transport properties but disrupts the scaffolding function of NKA α1, and leads to embryonic lethality in C. elegans and mice [12]. In human iPSCs, the present study provides evidence for a lineage-specific and CBM-dependent role of ATP1A1 in cell differentiation. This new mechanism has important biological implications discussed as follows. It should also be mentioned that, as the number of reports of disease-causing mutations in ATP1A1 continues to grow [54,55], clinical links to this gene may accrue.
Somitogenesis, genetic manipulation of Atp1a1, and Wnt/β-catenin signaling
Unlike the global knockout of Atp1a1 in the mouse, which blocks embryonic development early at the preimplantation stage of blastocyst [56], the CBM mutation appears to be compatible with life at early embryonic stages. However, the CBM mutation causes developmental arrest and lethality at the ensuing gastrulation stage, which is highly controlled by cell signaling. Consistently, RNA-seq analysis revealed the impact of CBM mutation on multiple signaling pathways responsible for organogenesis, including the Wnt/β-catenin pathway which is critical for myogenesis [12,41,42,57,58]. Examination of the gross morphology of WT and mCBM homozygous mouse embryos at E9.5 revealed a truncation of the PSM (Figure 1), a phenotype reminiscent of the loss of β-catenin signaling. Specifically, gene targeting on Wnt ligands or other members of this pathway gives rise to somite or muscle growth arrest [59]. On the other hand, a number of mCBM homozygous embryos failed to turn [12], a phenotype similar to the gain of β-catenin signaling [60]. The present study reveals a mCBM phenotype in iPSCs exposed to Skm differentiation that is consistent with a loss of the dynamic regulation of the β-catenin pathway, which may explain these apparently paradoxical defects but warrant further analysis in the embryo.
The myogenesis defect in vitro is a lineage-specific phenotype, not a general loss of CBM iPSC stemness
Myogenesis involves multiple steps of differentiation and lineage specification that are tightly controlled by cell signaling. The iPSCs modeling system recapitulates myocyte differentiation during embryonic development, including the sequential induction of pluripotent stem cells into mesoderm stem cells, presomatic mesoderm stem cells, premyogenic stem cells, myoblast and myotube cells. Each cell lineage is marked by different gene expression, including NANOG and OCT4 (pluripotent stem cells), T/TBXT and MIXL1 (mesoderm stem cells), MSGN1 (presomitic mesoderm), PAX3/PAX7 (somatic stem cells), MYOD/MYF5/MYOG/MRF4 (myoblast) and MYH3, MYH8, TNNT1 and CAV3 (final differentiated muscle cells)[30]. Recently developed single-cell RNA-seq techniques have further consolidated and enriched the characterization of these lineages and has also demonstrated the essential roles of cell signaling, including Wnt, in these early cell fate specification processes [33,61,62]. In our initial study, we observed that human iPSCs with CRISPR/CAS9-induced knock-in mutation of CBM NKA α1 had altered biological properties. Specifically, human iPSCs with defective mCBM NKA α1 had reduced colony formation ability, accompanied by a significant reduction in the expression of stemness markers (NANOG and OCT4), and transcription factors controlling germ layer differentiation (MIXL1 and T for mesoderm, OTX2 and SOX1 for ectoderm, and GATA4 and SOX17 for endoderm) [12]. The present study revealed that the mCBM iPSCs were unable to differentiate into iSkm, unless they were rescued by the exogenous transgene of mouse Myod. This observation was consistent with a crucial role of Wnt/β-catenin signaling in the induction of MYOD expression (40,41). This is also consistent with a lineage-specific defect due to NKA/Wnt signaling defect, rather than a general inability to differentiate due to a complete loss of stemness. In fact, we have observed that mCBM iPSCs retain the ability to differentiate into non-skeletal muscle lineages, such as adipocytes (Supplemental figure 4).
Linkage of NKA CBM with Wnt/β-catenin signaling through caveolin-1
The CBM mutation disrupts protein localization of CAV1 and β-catenin, indicating an essential role in cell membrane integrity[12]. As a scaffold protein, NKA has been proposed to provide a docking site securing the proper location and function of membrane proteins, as well as signal transduction[16,63].
The up-regulation of Cav1 expression and disruption of the caveolae network in mCBM iPSCs may be key to the molecular mechanism that leads to defective Wnt signaling and myogenesis. Indeed, caveolin-1 has been shown to negatively regulate Wnt signaling through binding of β-catenin [64]. Further, genetic manipulation of Cav1 expression altered β-catenin transcriptional activity [65–68]. Interestingly, Cav1 is highly expressed in quiescent adult muscle stem cells (satellite cells) [69], and down-regulated in activated satellite cells. Finally, overexpression of Cav1 inhibited muscle regeneration [70,71]. Hence, increased CAV1 expression in mCBM iPSCs may partially contribute to the decreased Wnt activity, since CBM mutation also shifts CAV1 protein distribution within the cell, evidenced by our previous assay using cell fractionation [12]. In this study, we also discovered the altered protein distribution of β-catenin (Figure 6). In response to Wnt ligand stimulation, LRP6, located in lipid rafts on the membrane, is internalized to the cytosol and recycled back to the membrane for reuse [72]. This endocytosis process is controlled by CAV1 [73,74]. We surmise that altered expression and distribution of CAV1 protein in the CBM mutant alters cellular endocytosis, which subsequently affects β-catenin degradation, as well as Wnt signaling.
Significance in the adult skeletal muscle
In the adult skeletal muscle, canonical Wnt signaling regulates the differentiation/fusion of muscle stem cells (satellite cells) [75,76], whereas non-canonical Wnt7a signals working through planar cell polarity pathway controls the self-renewal of satellite stem cells and the growth of muscle fibers [77,78]. We have previously observed an impact of a moderate global genetic reduction of Atp1a1 on the size of oxidative muscles such as the soleus in mice [13]. More recently, we have observed a dramatic impact of a muscle-specific, Myod-iCre mediated ablation of Atp1a1 in the skeletal muscle in this species [79]: the Cre/LoxP model points to a profound role of NKA/Src in skeletal muscle structure and function at developmental stages after MyoD expression. Of note, our previous study did not reveal any apparent effect of the NKA/Src blocking peptide pNaktide on organogenesis including muscle development [12]. This is corroborated in the present study by the differentiation of NKA/Src null mutant iPSC (A420P) into iSkm (Figure 4), although mild but significant changes noted between iSkm-WT and iSkm-A420P (Supplemental Figure 5A) do not completely exclude a minor contribution of this mutation to skeletal muscle differentiation. However, this study does not exclude a role of the CBM/β-catenin in the adult skeletal muscle downstream from MyoD and related to Src, which could be involved in the fusion defect of iSkm-mCBM-Myod (Figure 4, Supplemental Figure 6).
In a broader context, the evolutionary nature of the CBM and Src binding sites on the NKA α1 polypeptide is important to mention. Indeed, while CBM was evolved at the origin of animal kingdom, the Src-binding sequence was acquired and conserved only in mammals. Thus, while CBM appears to have a vital role in organogenesis in metazoans, the NKA/Src receptor complex could represent a more specialized mechanism important for the regulation of muscle metabolism in mammals [13].
Significance in Organogenesis
Cell signaling controls lineage specification, a process breaking from the homogeneity of unicellular organisms to the cellular heterogeneity of multicellular organisms. Developmental biologists continue to elucidate the complex molecular machinery that allows a single egg to turn into a body structure with diverse and specialized cell types. Our discovery revealed an important connection between NKA and Wnt/β-catenin signaling, one of the ancient pathways conserved in multicellular organisms that is essential for embryo development and tissue morphogenesis [80,81]. NKA is present in all eukaryotes, while the CBM sequence appeared in NKA with multi-cellular species [12]. Conceptually, this co-evolution between CBM domain of NKA and cell signaling is consistent with the proposed connection between this membrane protein and cell differentiation.
Much has been learned about the role of secreted and membrane-bound signaling molecules such as Wnt, Shh, and FGF in driving cell lineage specification during segmentation, somitogenesis and embryonic myogenesis [82–84]. The functional cross-talk among these signaling pathways, for both positive (e.g. Wnt and Shh) and negative (e.g. BMP) specification have also been extensively characterized [85]. It is clear that these receptor-mediated signaling events regulate cell lineage specification, and consequently cell lineage-specific cellular activity such as specific ion transport for muscle contraction and membrane structure [86–88]. It is also clear that cell lineage-specific activity must communicate with the signaling network in order to ensure temporospatial coordination of the development of various embryonic structures (e.g. somites) and genetic commitment (e.g. formation of myotome) [89,90]. However, knowledge of how the regulation of these signaling events by membrane transporters and structural proteins may occur is lacking. In this context, our findings provided evidence that the non-ion pumping function of NKA α1 could serve as an integrator for Wnt signaling in lineage-specific mechanism.
Supplementary Material
Acknowledgement
This manuscript is dedicated to the memory of Dr. Zi-Jian Xie. A pioneer of NKA non-ion pumping function, he was a driving force behind this project from its inception. This work was supported by the Marshall Institute for Interdisciplinary Research (MIIR), as well as AR060042, AR071976 and AR72644 (CF), AR067766 (HZ), and HL138757 (DW).
Footnotes
Disclosures
The authors declared no potential conflicts of interest.
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.
REFERENCES
- 1.Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 1957;23(2):394–401. [DOI] [PubMed] [Google Scholar]
- 2.Morth JP, Pedersen BP, Buch-Pedersen MJ et al. A structural overview of the plasma membrane Na+,K+-ATPase and H+-ATPase ion pumps. Nat Rev Mol Cell Biol 2011;12(1):60–70. [DOI] [PubMed] [Google Scholar]
- 3.Pirkmajer S, Chibalin AV. Na,K-ATPase regulation in skeletal muscle. Am J Physiol Endocrinol Metab 2016;311(1):E1–E31. [DOI] [PubMed] [Google Scholar]
- 4.Pivovarov AS, Calahorro F, Walker RJ. Na(+)/K(+)-pump and neurotransmitter membrane receptors. Invert Neurosci 2018;19(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glitsch HG. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev 2001;81(4):1791–1826. [DOI] [PubMed] [Google Scholar]
- 6.Davis MW, Somerville D, Lee RY et al. Mutations in the Caenorhabditis elegans Na,K-ATPase alpha-subunit gene, eat-6, disrupt excitable cell function. J Neurosci 1995;15(12):8408–8418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doi M, Iwasaki K. Na+/K+ ATPase regulates the expression and localization of acetylcholine receptors in a pump activity-independent manner. Mol Cell Neurosci 2008;38(4):548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruaud AF, Bessereau JL. The P-type ATPase CATP-1 is a novel regulator of C. elegans developmental timing that acts independently of its predicted pump function. Development 2007;134(5):867–879. [DOI] [PubMed] [Google Scholar]
- 9.Paul SM, Palladino MJ, Beitel GJ. A pump-independent function of the Na,K-ATPase is required for epithelial junction function and tracheal tube-size control. Development 2007;134(1):147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genova JL, Fehon RG. Neuroglian, Gliotactin, and the Na+/K+ ATPase are essential for septate junction function in Drosophila. J Cell Biol 2003;161(5):979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellertsdottir E, Ganz J, Durr K et al. A mutation in the zebrafish Na,K-ATPase subunit atp1a1a.1 provides genetic evidence that the sodium potassium pump contributes to left-right asymmetry downstream or in parallel to nodal flow. Dev Dyn 2006;235(7):1794–1808. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Cai L, Xie JX et al. A caveolin binding motif in Na/K-ATPase is required for stem cell differentiation and organogenesis in mammals and C. elegans. Sci Adv 2020;6(22):eaaw5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kutz LC, Mukherji ST, Wang X et al. Isoform-specific role of Na/K-ATPase alpha1 in skeletal muscle. Am J Physiol Endocrinol Metab 2018;314(6):E620–E629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dostanic-Larson I, Lorenz JN, Van Huysse JW et al. Physiological role of the alpha1- and alpha2-isoforms of the Na+-K+-ATPase and biological significance of their cardiac glycoside binding site. Am J Physiol Regul Integr Comp Physiol 2006;290(3):R524–528. [DOI] [PubMed] [Google Scholar]
- 15.Okita K, Matsumura Y, Sato Y et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 2011;8(5):409–412. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Haas M, Liang M et al. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem 2004;279(17):17250–17259. [DOI] [PubMed] [Google Scholar]
- 17.Cai T, Wang H, Chen Y et al. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol 2008;182(6):1153–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hafner AL, Dani C. Human induced pluripotent stem cells: A new source for brown and white adipocytes. World J Stem Cells 2014;6(4):467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang W, Zeve D, Suh JM et al. White fat progenitor cells reside in the adipose vasculature. Science 2008;322(5901):583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahfeldt T, Schinzel RT, Lee YK et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat Cell Biol 2012;14(2):209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sodhi K, Maxwell K, Yan Y et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv 2015;1(9):e1500781. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Liang M, Cai T, Tian J et al. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J Biol Chem 2006;281(28):19709–19719. [DOI] [PubMed] [Google Scholar]
- 23.van der Wal E, Herrero-Hernandez P, Wan R et al. Large-Scale Expansion of Human iPSC-Derived Skeletal Muscle Cells for Disease Modeling and Cell-Based Therapeutic Strategies. Stem Cell Reports 2018;10(6):1975–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi IY, Lim H, Estrellas K et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep 2016;15(10):2301–2312. [DOI] [PubMed] [Google Scholar]
- 25.Cai L, Das B, Brown DD. Changing a limb muscle growth program into a resorption program. Dev Biol 2007;304(1):260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner EC, Huang CL, Sawhney N et al. A Novel Selectable Islet 1 Positive Progenitor Cell Reprogrammed to Expandable and Functional Smooth Muscle Cells. Stem Cells 2016;34(5):1354–1368. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- 28.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3(6):1101–1108. [DOI] [PubMed] [Google Scholar]
- 29.Pourquie O. Vertebrate somitogenesis. Annu Rev Cell Dev Biol 2001;17:311–350. [DOI] [PubMed] [Google Scholar]
- 30.Chal J, Pourquie O. Making muscle: skeletal myogenesis in vivo and in vitro. Development 2017;144(12):2104–2122. [DOI] [PubMed] [Google Scholar]
- 31.Tam PP. The control of somitogenesis in mouse embryos. J Embryol Exp Morphol 1981;65 Suppl:103–128. [PubMed] [Google Scholar]
- 32.Guibentif C, Griffiths JA, Imaz-Rosshandler I et al. Diverse Routes toward Early Somites in the Mouse Embryo. Dev Cell 2021;56(1):141–153 e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xi H, Fujiwara W, Gonzalez K et al. In Vivo Human Somitogenesis Guides Somite Development from hPSCs. Cell Rep 2017;18(6):1573–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiwlawat N, Lynch E, Jeffrey J et al. Current Progress and Challenges for Skeletal Muscle Differentiation from Human Pluripotent Stem Cells Using Transgene-Free Approaches. Stem Cells Int 2018;2018:6241681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abujarour R, Valamehr B. Generation of skeletal muscle cells from pluripotent stem cells: advances and challenges. Front Cell Dev Biol 2015;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai F, Madan N, Ye Q et al. Identification of a mutant alpha1 Na/K-ATPase that pumps but is defective in signal transduction. J Biol Chem 2013;288(19):13295–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Ye Q, Liu C et al. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic Biol Med 2014;71:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braun T, Buschhausen-Denker G, Bober E et al. A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J 1989;8(3):701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987;51(6):987–1000. [DOI] [PubMed] [Google Scholar]
- 40.Weintraub H, Tapscott SJ, Davis RL et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A 1989;86(14):5434–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tajbakhsh S, Borello U, Vivarelli E et al. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development 1998;125(21):4155–4162. [DOI] [PubMed] [Google Scholar]
- 42.Brunelli S, Relaix F, Baesso S et al. Beta catenin-independent activation of MyoD in presomitic mesoderm requires PKC and depends on Pax3 transcriptional activity. Dev Biol 2007;304(2):604–614. [DOI] [PubMed] [Google Scholar]
- 43.Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 2005;433(7023):317–322. [DOI] [PubMed] [Google Scholar]
- 44.von Maltzahn J, Chang NC, Bentzinger CF et al. Wnt signaling in myogenesis. Trends Cell Biol 2012;22(11):602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borchin B, Chen J, Barberi T. Derivation and FACS-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent stem cells. Stem Cell Reports 2013;1(6):620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shelton M, Metz J, Liu J et al. Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Reports 2014;3(3):516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shelton M, Kocharyan A, Liu J et al. Robust generation and expansion of skeletal muscle progenitors and myocytes from human pluripotent stem cells. Methods 2016;101:73–84. [DOI] [PubMed] [Google Scholar]
- 48.Chal J, Al Tanoury Z, Hestin M et al. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat Protoc 2016;11(10):1833–1850. [DOI] [PubMed] [Google Scholar]
- 49.Chal J, Oginuma M, Al Tanoury Z et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol 2015;33(9):962–969. [DOI] [PubMed] [Google Scholar]
- 50.Caron L, Kher D, Lee KL et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl Med 2016;5(9):1145–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng Q, Gao N. Keeping Wnt signalosome in check by vesicular traffic. J Cell Physiol 2015;230(6):1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collins BM, Davis MJ, Hancock JF et al. Structure-based reassessment of the caveolin signaling model: do caveolae regulate signaling through caveolin-protein interactions? Dev Cell 2012;23(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byrne DP, Dart C, Rigden DJ. Evaluating caveolin interactions: do proteins interact with the caveolin scaffolding domain through a widespread aromatic residue-rich motif? PLoS One 2012;7(9):e44879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biondo ED, Spontarelli K, Ababioh G et al. Diseases caused by mutations in the Na(+)/K(+) pump alpha1 gene ATP1A1. Am J Physiol Cell Physiol 2021;321(2):C394–C408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clausen MV, Hilbers F, Poulsen H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front Physiol 2017;8:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barcroft LC, Moseley AE, Lingrel JB et al. Deletion of the Na/K-ATPase alpha1-subunit gene (Atp1a1) does not prevent cavitation of the preimplantation mouse embryo. Mech Dev 2004;121(5):417–426. [DOI] [PubMed] [Google Scholar]
- 57.Munsterberg AE, Kitajewski J, Bumcrot DA et al. Combinatorial signaling by Sonic hedgehog and Wnt family members induces myogenic bHLH gene expression in the somite. Genes Dev 1995;9(23):2911–2922. [DOI] [PubMed] [Google Scholar]
- 58.Borello U, Berarducci B, Murphy P et al. The Wnt/beta-catenin pathway regulates Gli-mediated Myf5 expression during somitogenesis. Development 2006;133(18):3723–3732. [DOI] [PubMed] [Google Scholar]
- 59.Shaw G, Weber K. Differential expression of neurofilament triplet proteins in brain development. Nature 1982;298(5871):277–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dunty WC Jr., Biris KK, Chalamalasetty RB et al. Wnt3a/beta-catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development 2008;135(1):85–94. [DOI] [PubMed] [Google Scholar]
- 61.Xi H, Langerman J, Sabri S et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell 2020;27(1):158–176 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi IY, Lim H, Cho HJ et al. Transcriptional landscape of myogenesis from human pluripotent stem cells reveals a key role of TWIST1 in maintenance of skeletal muscle progenitors. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cui X, Xie Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017;22(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galbiati F, Volonte D, Brown AM et al. Caveolin-1 expression inhibits Wnt/beta-catenin/Lef-1 signaling by recruiting beta-catenin to caveolae membrane domains. J Biol Chem 2000;275(30):23368–23377. [DOI] [PubMed] [Google Scholar]
- 65.Lu Z, Ghosh S, Wang Z et al. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 2003;4(6):499–515. [DOI] [PubMed] [Google Scholar]
- 66.Wang X, Lu B, Dai C et al. Caveolin-1 Promotes Chemoresistance of Gastric Cancer Cells to Cisplatin by Activating WNT/beta-Catenin Pathway. Front Oncol 2020;10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torres VA, Tapia JC, Rodriguez DA et al. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J Cell Sci 2006;119(Pt 9):1812–1823. [DOI] [PubMed] [Google Scholar]
- 68.Torres VA, Tapia JC, Rodriguez DA et al. E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor of apoptosis protein survivin via reduced beta-catenin-Tcf/Lef-dependent transcription. Mol Cell Biol 2007;27(21):7703–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gnocchi VF, White RB, Ono Y et al. Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS One 2009;4(4):e5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Volonte D, Liu Y, Galbiati F. The modulation of caveolin-1 expression controls satellite cell activation during muscle repair. FASEB J 2005;19(2):237–239. [DOI] [PubMed] [Google Scholar]
- 71.Baker N, Tuan RS. The less-often-traveled surface of stem cells: caveolin-1 and caveolae in stem cells, tissue repair and regeneration. Stem Cell Res Ther 2013;4(4):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakane H, Yamamoto H, Kikuchi A. LRP6 is internalized by Dkk1 to suppress its phosphorylation in the lipid raft and is recycled for reuse. J Cell Sci 2010;123(Pt 3):360–368. [DOI] [PubMed] [Google Scholar]
- 73.Yamamoto H, Komekado H, Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of beta-catenin. Dev Cell 2006;11(2):213–223. [DOI] [PubMed] [Google Scholar]
- 74.Bilic J, Huang YL, Davidson G et al. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007;316(5831):1619–1622. [DOI] [PubMed] [Google Scholar]
- 75.Otto A, Schmidt C, Luke G et al. Canonical Wnt signalling induces satellite-cell proliferation during adult skeletal muscle regeneration. J Cell Sci 2008;121(Pt 17):2939–2950. [DOI] [PubMed] [Google Scholar]
- 76.Brack AS, Conboy IM, Conboy MJ et al. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2008;2(1):50–59. [DOI] [PubMed] [Google Scholar]
- 77.Le Grand F, Jones AE, Seale V et al. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell 2009;4(6):535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bentzinger CF, Wang YX, von Maltzahn J et al. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013;12(1):75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kutz LC, Cui X, Xie JX et al. The Na/K-ATPase alpha1/Src interaction regulates metabolic reserve and Western diet intolerance. Acta Physiol (Oxf) 2021:e13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Croce JC, McClay DR. Evolution of the Wnt pathways. Methods Mol Biol 2008;469:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holstein TW. The evolution of the Wnt pathway. Cold Spring Harb Perspect Biol 2012;4(7):a007922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Sinha T, Wynshaw-Boris A. Wnt signaling in mammalian development: lessons from mouse genetics. Cold Spring Harb Perspect Biol 2012;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wodarz A, Nusse R. Mechanisms of Wnt signaling in development [in eng]. Annu Rev Cell Dev Biol 1998;14:59–88. [DOI] [PubMed] [Google Scholar]
- 84.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science 2000;287(5458):1606–1609. [DOI] [PubMed] [Google Scholar]
- 85.Hubaud A, Pourquie O. Signalling dynamics in vertebrate segmentation. Nat Rev Mol Cell Biol 2014;15(11):709–721. [DOI] [PubMed] [Google Scholar]
- 86.Zhao C, Yu Y, Zhang Y et al. beta-Catenin Controls the Electrophysiologic Properties of Skeletal Muscle Cells by Regulating the alpha2 Isoform of Na(+)/K(+)-ATPase. Front Neurosci 2019;13:831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liang W, Cho HC, Marban E. Wnt signalling suppresses voltage-dependent Na(+) channel expression in postnatal rat cardiomyocytes. J Physiol 2015;593(5):1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De A Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai) 2011;43(10):745–756. [DOI] [PubMed] [Google Scholar]
- 89.Levin M. Molecular bioelectricity in developmental biology: new tools and recent discoveries: control of cell behavior and pattern formation by transmembrane potential gradients. Bioessays 2012;34(3):205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Webb SE, Miller AL. Ca2+ signaling during vertebrate somitogenesis. Acta Pharmacol Sin 2006;27(7):781–790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.