

Abstract
Repeated administration of Δ9-tetrahydrocannabinol (THC), the primary psychoactive constituent of Cannabis sativa, induces profound tolerance that correlates with desensitization and downregulation of CB1 cannabinoid receptors in the CNS. However, the consequences of repeated administration of the endocannabinoid N-arachidonoyl ethanolamine (anandamide, AEA) on cannabinoid receptor regulation are unclear because of its rapid metabolism by fatty acid amide hydrolase (FAAH). FAAH−/− mice dosed subchronically with equi-active maximally effective doses of AEA or THC displayed greater rightward shifts in THC dose-effect curves for antinociception, catalepsy, and hypothermia than in AEA dose-effect curves. Subchronic THC significantly attenuated agonist-stimulated [35S]GTPγS binding in brain and spinal cord, and reduced [3H]WIN55,212-2 binding in brain. Interestingly, AEA-treated FAAH−/− mice showed less CB1 receptor downregulation and desensitization than THC-treated mice. Experiments examining tolerance and cross-tolerance indicated that the behavioral effects of THC, a low efficacy CB1 receptor agonist, were more sensitive to receptor loss than those of AEA, a higher efficacy agonist, suggesting that the expression of tolerance was more affected by the intrinsic activity of the ligand at testing than during subchronic treatment. Additionally, the CB1 receptor antagonist, rimonabant, precipitated a markedly reduced magnitude of withdrawal in FAAH−/− mice treated subchronically with AEA compared to mice treated repeatedly with THC. The findings that repeated AEA administration produces lesser adaptive changes at the CB1 receptor and has reduced dependence liability compared to THC suggest that pharmacotherapies targeting endocannabinoid catabolic enzymes are less likely to promote tolerance and dependence than direct acting CB1 receptor agonists.
Keywords: desensitization, downregulation, [35S]GTPγS autoradiography, endocannabinoid, antinociception, G-protein
Introduction
The endocannabinoid system is comprised of cannabinoid CB1 and CB2 receptors and endogenous ligands including N-arachidonoyl ethanolamine (anandamide, AEA) and 2-arachidonoyl glycerol (2-AG) (Howlett et al., 2002). Cannabinoids, including Δ9-tetrahydrocannabinol (THC), the primary psychoactive component of marijuana, produce a variety of pharmacological effects, including antinociception, motor disturbances, and hypothermia (Hollister, 1986; Compton et al., 1992), which correspond to the widespread distribution of CB1 receptors in the CNS (Herkenham et al., 1991; Glass et al., 1997). AEA was initially shown to produce cannabinoid effects (Smith et al., 1994) that were short-lived due to its rapid degradation (Willoughby et al., 1997). Initial observations on the role of AEA have been extended using genetically modified mice lacking fatty acid amide hydrolase (FAAH), the principal enzyme responsible for degradation of AEA and other fatty acid amides (Cravatt et al., 2001). Administration of AEA to FAAH−/− mice produced antinociception, catalepsy, locomotor inhibition and hypothermia that persisted 6-8 hours. AEA-mediated effects were reversed by the CB1 receptor antagonist SR141716A (rimonabant) (Cravatt et al., 2001) and dual FAAH−/−/CB1 receptor−/− mice were impervious to the analgesic, cataleptic and hypothermic of AEA (Wise et al., 2007), indicating that AEA-induced effects are mediated via CB1 receptors. In addition to this enhanced sensitivity to the pharmacological effects of exogenously administered AEA, FAAH−/−mice were found to exhibit a hypoalgesic phenotype (Cravatt et al., 2001; Lichtman et al., 2004b). Importantly, FAAH−/− mice exhibit normal CB1 receptor expression despite constitutively elevated levels of endogenous fatty acid amides (Cravatt et al., 2001). CB1 receptors belong to the G-protein coupled receptor superfamily and activate primarily Gαi/o, resulting in inhibition of adenylyl cyclase, activation of A-type and inwardly rectifying potassium channels, inhibition of N- and P/Q-type calcium channels and stimulation of MAP kinase (Howlett et al., 2002). Repeated exposure to THC or synthetic cannabinoid agonists (e.g. WIN55,212-2, CP55,940) in rodents produces desensitization of cannabinoid-mediated G-protein activity and inhibition of adenylyl cyclase, as well as CB1 receptor downregulation (Sim-Selley, 2003; Martin et al., 2004). Treatment paradigms that produce these cellular adaptations are also associated with the development of tolerance to in vivo cannabinoid-mediated effects (Martin et al., 2004; Lichtman and Martin, 2005). Rimonabant administration precipitates withdrawal effects in mice treated chronically with cannabinoid receptor agonists (Tsou et al., 1995; Cook et al., 1998) that is accompanied by increased cAMP in the cerebellum (Hutcheson et al., 1998; Rubino et al., 1998; Tzavara et al., 2000). The relevance of these adaptations is suggested by the findings that repeated THC use produces tolerance and dependence in humans (Jones et al., 1976; Jones et al., 1981; Budney and Hughes, 2006) as well as CB1 receptor downregulation in specific brain regions (Villares, 2007). It is not clear whether subchronic AEA administration regulates CB1 receptors in a similar manner as THC and synthetic cannabinoids. AEA administration has been reported to produce an inconsistent and small degree of tolerance and dependence (Pertwee et al., 1993; Aceto et al., 1998), and treatment with AEA or the AEA analog methanandamide either reduced or did not alter levels of cannabinoid receptors and receptor-mediated G-protein activity (Romero et al., 1995; Romero et al., 1999; Rubino et al., 2000). Thus, results to date have been inconclusive and interpretation is complicated by the instability of AEA in the presence of endogenous FAAH (Willoughby et al., 1997).
FAAH−/− mice provide a model in which to examine the effects of repeated AEA administration on CB1 receptor regulation and in vivo activity. Previous studies, while providing variable results, suggest that THC and AEA might differentially regulate CB1 receptors in the CNS. To test this hypothesis, we investigated whether FAAH−/− mice that were repeatedly administered equivalent maximally effective doses of AEA or THC would show differential tolerance to cannabinoid-mediated in vivo effects, signs of rimonabant-precipitated withdrawal, and receptor desensitization and downregulation.
Materials and Methods
Subjects
The subjects were male C57Bl/6J mice (Jackson Laboratory, Bar Harbor, ME) as well as male and female FAAH−/− and FAAH+/+ mice backcrossed for at least 13 generations onto a C57Bl/6J background. Subjects weighed between 20 and 30 g and were housed four mice per cage in a temperature-controlled (20-22°C) facility, with food and water available ad libitum. All animal protocols were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee.
Drugs and Chemicals
[35S]GTPγS (1250 Ci/mmol) and [3H]WIN55,212-2 (52.2 Ci/mmol) were purchased from Perkin Elmer Life Sciences (Boston, MA). THC, rimonabant and AEA for autoradiography were provided by the Drug Supply Program of the National Institute on Drug Abuse. Bovine serum albumin (BSA), GTPγS, GDP and WIN55,212-2 were purchased from Sigma Chemical Company (St. Louis, MO). Econo-1 scintillation fluid was obtained from Fisher Scientific (Norcross, GA). All other chemicals were obtained from Sigma or Fisher. AEA for repeated dosing and in vivo testing was synthesized by the Cravatt laboratory.
Repeated drug administration
All subchronic dosing was administered through the subcutaneous route of administration, given twice daily (08:00 and 16:00 h) with vehicle (emulphor: ethanol: saline; in a ratio of 1:1:18), Δ9-THC (50 mg/kg) or AEA (50 mg/kg) on five consecutive days. On the sixth day, only the 08:00 h injection was administered. These doses of AEA and THC were selected because in preliminary experiments (data not shown), they produced maximal antinociceptive effects in control mice (also see Figures 1A and 3A).
Figure 1.

Cumulative dose-response curves of THC vs. repeated vehicle injections in FAAH−/− or FAAH+/+ mice in the tail immersion test for antinociception (top panel), bar test for catalepsy (center panel), or hypothermia (bottom panel). Data are presented as mean ± SEM, n= 6-8 mice/condition.
Figure 3.

Cumulative dose-response curves of AEA in FAAH−/− that treated subchronically with vehicle, THC, or AEA. Subjects were evaluated in the tail immersion test for antinociception (top panel), bar test for catalepsy (center panel), or hypothermia (bottom panel). Data are presented as mean ± SEM, n= 6-8 mice/condition.
In Vivo Measures
Nociceptive behavior was assessed in the tail withdrawal test using a 52° C water bath, a temperature that does not produce FAAH−/− phenotypic hypoalgesic responses (Cravatt et al., 2001). The tail withdrawal data were expressed as a percentage of maximal possible effect [%MPE = (withdrawal latency-baseline withdrawal) / (10 s-baseline withdrawal]. Body temperature was obtained by inserting a digital thermometer (Fisher Scientific, Pittsburgh, PA) 2 cm into the anus and recorded to the nearest 0.1°C. Catalepsy was scored as positive when mice maintained a rigid posture for 10 s in the bar test (Cravatt et al., 2001).
Cumulative Dose Response Curves
Twenty-four h after the final subchronic injection, a cumulative dosing regimen was employed to determine the dose-response relationships for antinociception, hypothermia, and hypomotility. Subjects received increasing doses of THC or AEA every 40 min with endpoints assessed 30 min after each injection, as specified in the results. Several experiments were conducted that included control mice that received repeated vehicle injections every 40 min, with endpoints assessed 30 min to ensure that no vehicle effects occurred.
Rimonabant-Precipitated Withdrawal
Subjects (n=36) were treated with vehicle, AEA, or Δ9-THC according to the repeated administration schedule described above. On Day 6, mice were injected with the final subchronic treatment, and placed in a Plexiglas observation chamber (21.5 × 21.5 × 15 cm). Thirty min later, subjects were administered rimonabant (10mg/kg; i.p.) and scored for paw tremors (shaking of one or both paws) and head shaking/twitching (minimum of two quick, successive head movements in a counterclockwise/clockwise fashion and righted to the original position). Mice were observed for 1 h and scored in 5- min bins, separated by 5-min break periods.
Comparison of THC Brain Levels Between Bolus and Cumulative Dosing Procedures
Acute THC brain and blood levels were determined 30 min following single bolus administration of 3, 10, or 30 mg/kg THC or 30 min after the final injection of cumulative dosing of 10, 30, or 56 mg/kg THC. The mice were decapitated 20 min after drug administration and the blood was collected in heparinized (Elkins-Sinn, Inc., Cherry Hill, NJ) tubes. The THC extraction procedure and quantification procedure were conducted as previously described (Wilson et al., 2006). Fifty ng of deuterated THC (Radian Corporation, Austin, TX) was added to the blood sample, brain homogenate, and calibrators (blank mouse whole blood and homogenized brain) as an internal standard. Following an equilibration period, 2.5 ml cold acetonitrile (HPLC grade, Fisher Scientific, Raleigh NC) was added drop-wise while vortexing. The samples were centrifuged at 2500 rpm for 15 min to pelletize solids and stored in a freezer (−20 °C) overnight to separate the acetonitrile and aqueous layers. The acetonitrile layer was then removed and evaporated to dryness under nitrogen. The THC /deuterated THC was resolublized in 0.1 ml methanol (HPLC grade, Fisher Scientific) and quantified via LC-MS (Quattro II). Ions analyzed in single ion monitoring mode were 315 and 118 for THC deuterated THC, respectively. A calibration curve was constructed for each assay based on linear regression using the peak-area ratios of THC to deuterated THC of the extracted calibration samples.
Agonist-Stimulated [35S]GTPγS Autoradiography
Mice were treated subchronically with vehicle, AEA or THC as described above. Twenty-four h after the final injection, brains were removed and frozen in isopentane (−30°C). Coronal sections (20 μm) in regions of interest were cut on a cryostat (−20°C) and collected on subbed slides. Slides were desiccated overnight at 4°C and stored at −80°C. Assays were conducted as published (Sim-Selley and Martin, 2002). Slides were incubated in TME buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl) for 10 minutes, then transferred to TME buffer + 0.5% BSA containing 2 mM GDP and 10 mU/ml adenosine deaminase for 20 minutes. Sections were then incubated for 2 h in 0.04 nM [35S]GTPγS, 2 mM GDP, 10 mU/ml adenosine deaminase, and maximally effective concentrations of WIN55,212-2 (10 μM) or AEA (20 μM) in TME buffer + BSA at 25°C. Slides were rinsed in Tris buffer (2 × 2 min at 4°C, 50 mM, pH 7.4) and ddH2O (30 s, 4°C), dried and placed in cassettes with 14C microscales and Kodak Biomax MR film for 24 hours.
[3H]WIN55,212-2 Autoradiography
[3H]WIN55,212-2 binding was conducted as published (Breivogel et al., 1999). Sections were collected as described above. Slides were incubated in assay buffer (20 mM HEPES, 1 mM MgCl2, and 0.5% w/v BSA, pH 7.0) for 20 min at 30°C. Sections were then incubated in 1 nM [3H]WIN55,212-2 in assay buffer (80 min, 30°C) in the absence or presence of 1 μM unlabeled WIN55,212-2 to assess total and nonspecific binding, respectively. Slides were washed in assay buffer (4 × 10 minutes each, 25°C) and dipped in ddH20 (4°C). Slides were dried and exposed to Kodak Biomax MS film for 6 weeks with 3H microscales.
Agonist-Stimulated [35S]GTPγS Binding in Membranes
Assays were conducted as previously published (Sim-Selley et al., 2006). Spinal cords were removed, rapidly frozen and stored at −80°C. For each experiment, tissue was thawed and homogenized in membrane buffer (Tris-HCl, pH 7.4, 3 mM MgCl2 and 1 mM EDTA). Homogenates were centrifuged at 50,000 x g for 10 min at 4°C, the supernatant was discarded and the pellet re-suspended in membrane buffer. Centrifugation was repeated, the pellet re-suspended in TME buffer and protein concentration determined. Membranes were pretreated with adenosine deaminase (10 mU/ml) for 15 min at 30°C prior to assay. Membrane protein (10 μg) was incubated in TME with 0.1% BSA, 30 μM GDP, 0.1 nM [35S]GTPγS and varying concentrations of WIN55,212-2 or AEA for 2 hr at 30°C. Non-specific binding was determined with 10 μM unlabeled GTPγS and basal binding was determined in the absence of agonist. The incubation was terminated by rapid filtration through GF/B glass fiber filters and three rinses with ice-cold Tris-HCl, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry after extraction of filters in ScintiSafe Econo-1 scintillation fluid.
Data Analysis
All in vivo data were analyzed using one or two-way analyses of variance (ANOVA), followed by Tukey post hoc tests when appropriate. Because no significant sex differences were observed in any of the studies, this factor was collapsed in all analyses. P-values of less than 0.05 were considered significant. The ED50 values with 95% confidence intervals (CI) were calculated using standard linear regression analysis of the dose–response curve. Potency ratios with 95% confidence intervals were calculated by comparing the potency between two treatments, as described by Colquhoun (1971). Treatments that had potency ratios that did not include a ratio of 1 in the 95% confidence intervals were considered significant.
Autoradiographic analysis was performed using NIH ImageJ software. Data are reported as mean ± S.E.M. of triplicate sections from 4-6 (study 1) or 8-12 (study 2) brains/group. Net [35S]GTPγS binding is defined as (agonist-stimulated [35S]GTPγS binding – basal [35S]GTPγS binding). Percent stimulation is defined as (net [35S]GTPγS binding / basal [35S]GTPγS binding) × 100%. Specific [3H]WIN55,212-2 binding is defined as (total [3H]WIN55,212-2 binding-nonspecific [3H]WIN55,212-2 binding). In study 1, a two-way ANOVA and post-hoc Tukey tests were used to compare the three FAAH −/− treatment groups (Vehicle-, THC- and AEA-treated) across genotype; in study 2, a two-way ANOVA examined an overall effect of treatment across brain region, while one-way ANOVAs (with post-hoc Dunnett’s tests) compared the FAAH −/− treatment groups within each region. Concentration-effect curves in membranes were fit by non-linear regression analysis to obtain Emax and EC50 values using GraphPad Prism 4.0 software. The significance of concentration-effect curves was determined by two-way ANOVA; significant differences in Emax and EC50 values were determined by one-way ANOVA with a post-hoc Dunnett’s test.
Results
Behavioral Measures
Cumulative dosing of THC produces identical dose-response curves in naïve FAAH −/− and FAAH +/+ mice
In initial experiments, we evaluated the feasibility of evaluating the dose-response relationship of THC using a cumulative dosing regimen in which mice were dosed with increasing amounts of drug and tested repeatedly across the same session. The data presented in Table 1 compare the resulting blood and brain levels of THC between mice subjected to cumulative dosing and single bolus dosing. Both types of injection regimens led to equivalent levels of THC in both blood and brain, as indicated by a lack of significance between injection regimens. Next, we compared the dose-response relationship of THC following cumulative dosing between FAAH +/+ and FAAH−/− mice. As previously reported using separate groups of mice (Cravatt et al., 2001), THC was equipotent and elicited similar dose-response profiles for antinociception (potency ratio (95% CI)) values: 1.1 (0.8-1.5); Figure 1, top panel), catalepsy (1.1 (0.8-1.6); Figure 1, center panel), and hypothermia (0.8 (0.7-1.1); Figure 1, bottom panel) in both genotypes. Importantly, FAAH +/+ and FAAH−/− mice given repeated vehicle injections did not display any alterations in any of the three dependent measures. In order to reduce the number of mice needed as well as the quantity of drugs required for the tolerance and dependence experiments, all dose-response relationships were assessed using a cumulative dosing regimen.
Table 1.
Comparison of blood and brain THC levels between single bolus and cumulative dosing in C57Bl/6J mice
| Blood (ng/ml) | Brain (ng/ml) | |||
|---|---|---|---|---|
|
| ||||
| THC (mg/kg) | Single bolus | Cumulative | Single bolus | Cumulative |
|
| ||||
| 3 | 97 ± 14 | ---------- | 77 ± 5 | ---------- |
| 10 | 209 ± 28 | 179 ± 23 | 212 ± 19 | 225 ± 32 |
| 30 | 456 ± 83 | 438 ± 102 | 520 ± 79 | 641 ± 134 |
| 56 | 1054 ± 184 | 1315 ± 386 | 1072 ± 156 | 1294 ± 178 |
Values represent mean ± SEM concentration of THC (n = 5-8 mice/group).
Evaluation of subchronic dosing of THC and AEA on the dose-response relationship of THC in FAAH −/− and FAAH +/+ mice
Repeated administration of two daily injections of THC (50 mg/kg) for 5.5 days elicited profound tolerance to its antinociceptive (Figure 2, top panel), cataleptic (Figure 2, center panel), and hypothermic (Figure 2, bottom panel) effects in both FAAH +/+ and FAAH−/− mice, accompanied by a decrease in efficacy for antinociception and catalepsy. Interestingly, repeated administration of THC led to increased tolerance to the hypothermic effects in FAAH −/− mice compared to FAAH +/+ mice, as indicated by a significant interaction between genotype and dose (F7,84 = 2.6, p < 0.05), although no significant differences were found between the genotypes at any specific dose. In addition, FAAH −/− mice treated subchronically with AEA displayed rightward shifts in THC dose-response curves for all three measures, with concomitant decreases in efficacy for antinociception and catalepsy. The potency of THC in FAAH −/− and FAAH+/+ mice treated repeatedly with THC or vehicle, as well as its potency following repeated AEA administration in FAAH −/− mice are represented in Table 2.
Figure 2.

Cumulative dose-response curves of THC in FAAH−/− or FAAH+/+ mice that were treated subchronically with either THC or AEA. Also included for comparison were naïve FAAH−/− or FAAH+/+ mice (data also presented in Figure 1). Subjects were evaluated in the tail immersion test for antinociception (top panel), bar test for catalepsy (center panel), or hypothermia (bottom panel). Data are presented as mean ± SEM, n= 6-8 mice/condition.
Table 2.
Comparison of THC potency in FAAH−/− vs FAAH+/+ mice that were naïve or treated repeatedly with THC, as well as FAAH−/− mice treated with AEA
| Subchronic Treatment |
Antinociception | Catalepsy | Hypothermia | |||
|---|---|---|---|---|---|---|
| −/− | +/+ | −/− | +/+ | −/− | +/+ | |
| Naive | 22 (17-30) | 19 (14-26) | 21 (15-28) | 18 (13-25) | 22 (16-32) | 18 (15-22) |
| THC | 25 + 4%MPE @ 300 mg/kg |
44 + 16%MPE @ 300 mg/kg |
67% @ 300 mg/kg |
62% @ 300 mg/kg |
224 (190- 266) |
109 (62- 190) |
| AEA | 42 + 8%MPE @ 300 mg/kg |
n.d | 25% @ 300 mg/kg |
n.d. | 133 (106- 166) |
n.d. |
Data represent ED50 values (95% CI) or maximum effect ± SEM if efficacy < 70%. (n.d. = not determined), and are represented in mg/kg.
Evaluation of subchronic dosing of THC and AEA on the dose-response relationship of AEA in FAAH −/− mice
Repeated administration of two daily injections of AEA (50 mg/kg) for 5.5 days resulted in rightward shifts in the dose-response relationships to the antinociceptive (Figure 3, top panel), cataleptic (Figure 3, center panel), and hypothermic (Figure 3, bottom panel) effects of AEA in FAAH −/− mice. Additionally, subchronic administration of THC led to cross-tolerance to the pharmacological effects of AEA. Interestingly, the efficacy of AEA in eliciting catalepsy was profoundly decreased in FAAH −/− mice treated subchronically with THC. The potencies of AEA in FAAH −/− mice treated repeatedly with vehicle, AEA, or THC are presented in Table 3.
Table 3.
Comparison of THC potency in FAAH−/− mice that were treated repeatedly with vehicle, THC, or AEA
| Subchronic Treatment | Antinociception | Catalepsy | Hypothermia |
|---|---|---|---|
| Vehicle | 20 (16-24) | 16 (13-19) | 17 (15-20) |
| THC | 76 (58-99) | 50% @ 150 mg/kg | 32 (27-38) |
| AEA | 55 (44-69) | 62 (56-69) | 50 (40-55) |
Data represent ED50 values (95% CI) or maximum effect ± SEM if efficacy < 70%, and are represented in mg/kg.
Dependence
Rimonabant (Rim) precipitated a similar magnitude of withdrawal responses in FAAH−/− and FAAH+/+ mice treated subchronically with THC (Figure 4). A two-way ANOVA, with genotype (FAAH−/− vs. FAAH+/+) and treatment (subchronic vehicle-challenge vehicle, subchronic vehicle-challenge rimonabant, subchronic THC- challenge rimonabant) as between subject factors, revealed main effects of treatment for both head twitches (F2,29 = 48, p < 0.0001) and paw flutters (F2,29 = 65, p < 0.0001). Post hoc analyses revealed that rimonabant challenge elicited significant increases in both headshakes and paw tremors in mice treated with subchronic THC, but not subchronic vehicle. Interestingly rimonabant challenge precipitated paw flutters in FAAH−/− mice treated subchronically with AEA (Figure 4A; t10 = 5.3, p = 0.001), but produced no changes in the number of head twitches (Figure 4B). Additionally, the magnitude of the rimonabant precipitated paw flutters following subchronic AEA administration was smaller than following subchronic THC (AEA mean ± SEM = 74 ± 13; THC mean ± SEM = 222 ± 41).
Figure 4.

Evaluation of rimonabant (Rim; 10 mg/kg) precipitated withdrawal in FAAH−/− mice treated subchronically with THC (50 mg/kg twice daily for 5.5. days) or AEA (50 mg/kg twice daily for 5.5. days), and FAAH +/+ mice treated subchronically with THC (50 mg/kg twice daily for 5.5. days). Top Panel. Rim-precipitated significant increases in paw tremors in mice treated repeatedly with THC, regardless of genotype. Rim also precipitated significant increases in paw tremors in FAAH−/− mice treated repeatedly with AEA, though the magnitude of this effect was considerably less than in mice treated repeatedly with THC. Bottom Panel. Rim precipitated significant increases in head twitches in mice treated repeatedly with THC, regardless of genotype. Rim did not elicit any differences in head twitches between FAAH−/− mice treated subchronically with vehicle or AEA. ***p < 0.001 versus corresponding subchronic vehicle-vehicle challenge group of the same genotype. ### p < 0.001 versus corresponding subchronic vehicle-rimonabant challenge group of the same genotype. Data are presented as mean ± SEM; n= 6 mice/condition.
Agonist-stimulated [35S]GTPγS binding
Desensitization of cannabinoid-stimulated G-protein activity usually occurs following repeated cannabinoid treatment regimens that produce tolerance (Sim-Selley, 2003). Therefore, agonist-stimulated [35S]GTPγS binding was conducted to assess cannabinoid receptor-mediated G-protein activity between FAAH +/+ and FAAH−/− mice treated repeatedly with vehicle, THC, or AEA in the representative regions of caudate-putamen, hippocampus, and cerebellum. In these regions, WIN55,212-2-stimulated [35S]GTPγS binding appeared similar between the FAAH +/+ and FAAH−/− genotypes in autoradiograms from vehicle-treated mice (data not shown). Similarly, in both genotypes, WIN55,212-2-stimulated [35S]GTPγS binding in brains from THC-treated mice was visibly reduced, whereas activity in brains from AEA-treated mice appeared similar to vehicle-treated animals. Densitometric analysis of caudate-putamen, hippocampus, and cerebellum (Figure 5) revealed that WIN55,212-2-stimulated [35S]GTPγS binding did not differ between FAAH−/− and FAAH+/+ mice for any treatment. Two-way ANOVAs revealed that THC treatment significantly reduced WIN55,212-2-stimulated [35S]GTPγS binding in both genotypes compared to appropriate vehicle control in all analyzed regions including caudate putamen (F1,25 = 9.06, p<0.01), hippocampus (F1,25 = 8.04, p<0.01), and cerebellum (F1,25 = 5.35, p<0.05). In contrast, WIN55,212-2-stimulated [35S]GTPγS binding following AEA treatment was not different from vehicle control in either FAAH +/+ or FAAH −/− mice in any of the analyzed regions.
Figure 5.

Subchronic THC reduces CB1 receptor-mediated G-protein activity in both FAAH+/+ and FAAH−/− mice, but subchronic AEA does not significantly alter CB1 receptor-mediated activity. Densitometric analysis of net WIN55,212-2-stimulated [35S]GTPγS binding in FAAH+/+ and FAAH−/− mice treated with vehicle (VEH), THC, or anandamide (AEA) in CB1 receptor-containing brain regions: caudate putamen (CPu), hippocampus (Hip) and cerebellum (Cblm). No significant genotype differences were observed regardless of treatment or brain region. Values represent mean net [35S]GTPγS binding (nCi/g) ± SEM, n= 4-6 mice/group.
To investigate this finding further, FAAH−/− mice received the same drug treatment regimens and then underwent a more extensive regional analysis. Because WIN55,212-2-stimulated [35S]GTPγS binding did not differ statistically between genotypes in any region, only FAAH−/− mice, in which AEA would be stable, were used for this study. Representative autoradiograms illustrate WIN55,212-2-stimulated [35S]GTPγS binding in the three treatment groups of FAAH−/− mice at several coronal levels, including caudate putamen, globus pallidus, periaqueductal gray, and cerebellum (Figure 6). Visual inspection revealed an obvious reduction in WIN55,212-2-stimulated [35S]GTPγS binding in brains from THC-treated FAAH−/− mice, whereas only a modest attenuation was noted in sections from AEA-treated FAAH−/− mice. Densitometric analysis confirmed these observations (Table 4). Because the magnitude of agonist-stimulated [35S]GTPγS binding varied substantially between regions, separate one-way ANOVAs with appropriate post-hoc Dunnett’s tests were used to compare the three FAAH −/− treatment groups within each brain region. Treatment of FAAH−/− mice with THC significantly reduced WIN55,212-2-stimulated [35S]GTPγS binding compared to vehicle-treated mice in all regions analyzed, including caudate putamen, globus pallidus, hippocampus, substantia nigra, periaqueductal gray, and cerebellum (Table 4). The percent loss of WIN55,212-2-stimulated [35S]GTPγS binding varied by region, with relatively less reduction in globus pallidus and substantia nigra and the greatest reduction in hippocampus (Table 4; Figure 8), as previously reported (Sim-Selley, 2003). In contrast to results in THC-treated mice, WIN55,212-2-stimulated [35S]GTPγS binding did not significantly differ between AEA- and vehicle-treated FAAH −/− mice in any region analyzed except cerebellum, although a modest reduction was usually observed (Table 4). This was confirmed when a two-way ANOVA with region and treatment as factors revealed a significant overall effect of treatment (F2,166= 34.4, p<0.001), whereby AEA produced an overall intermediate effect that was significantly different than both vehicle- and THC- treated mice (post hoc Tukey test, p<0.05). Assays were also conducted to examine AEA-stimulated [35S]GTPγS binding (data not shown). AEA-stimulated [35S]GTPγS binding in brains from vehicle-treated FAAH−/− mice was approximately 50% less than produced by WIN55,212-2, consistent with reports that AEA is a partial agonist for G-protein activation (Breivogel et al., 1998). Results obtained using AEA-stimulated [35S]GTPγS binding also revealed decreased cannabinoid-stimulated G-protein activity in brains from THC-treated mice, with significant reductions in globus pallidus, PAG and cerebellum. These results indicate that subchronic treatment with THC produced significant CB1 receptor desensitization in multiple brain regions of FAAH−/− mice. In contrast, AEA treatment produced only a modest reduction in CB1 receptor-mediated G-protein activity that was intermediate between that seen after subchronic vehicle or THC treatment, and not statistically different from vehicle in any region except cerebellum.
Figure 6.

Representative autoradiograms illustrating WIN55,212-2 stimulated [35S]GTPγS binding in brains from vehicle, THC- and AEA-treated FAAH−/− mice. WIN55,212-2-stimulated [35S]GTPγS binding is visibly reduced in FAAH−/− mice treated with THC in nearly all regions including caudate-putamen (top panels), globus pallidus (top center panels), PAG (bottom center panels), and cerebellum (bottom panels). AEA-treated FAAH−/− mice had intermediate levels of WIN55,212-2-stimulated [35S]GTPγS binding between vehicle and THC-treated mice. Data from densitometric analysis are presented in Table 4.
Table 4.
Net WIN55,212-2-stimulated [35S]GTPγS binding in brain sections from vehicle-, THC-, and AEA-treated FAAH−/− mice
| REGION | Vehicle | THC | Anandamide |
|---|---|---|---|
| Caudate-Putamen | 218 ± 34 | 97 ± 16* | 152 ± 28 |
| Globus Pallidus | 774 ± 30 | 539 ± 41* | 639 ± 60 |
| Hippocampus | 190 ± 27 | 65 ± 14* | 132 ± 24 |
| Substantia Nigra | 673 ± 30 | 506 ± 32* | 592 ± 49 |
| PAG | 119 ± 21 | 55 ± 13* | 79 ± 19 |
| Cerebellum | 258 ± 20 | 138 ± 21* | 178 ± 23* |
Brain sections were processed as described in Methods.
Data shown are mean net [35S]GTPγS binding values (nCi/g) ± SEM from 8-12 mice per group
p<0.05 different from vehicle-treated group by one-way ANOVA with a post-hoc Dunnett’s test.
Figure 8.
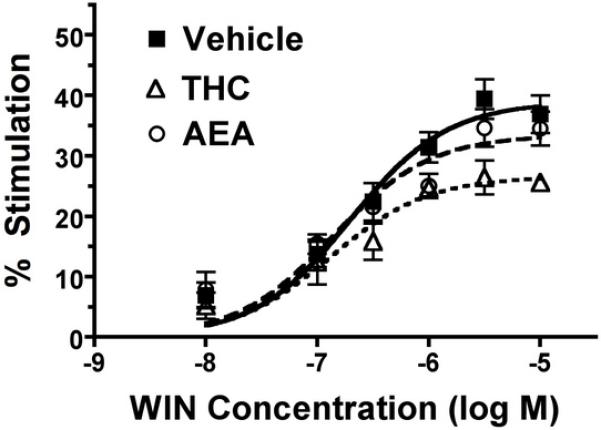
Effect of subchronic THC or AEA treatment on WIN-stimulated [35S]GTPγS binding in spinal cord. Spinal cord membranes from THC- or AEA-treated mice were incubated 0.1 nM [35S]GTPγS, 30 μM GDP and varying concentrations of WIN55,212-2 (WIN), as described in Methods. Data are mean percent stimulation ± SEM (n= 7).
[3H]WIN55,212-2 binding
Cannabinoid treatment also produces receptor downregulation in the brain following administration paradigms that produce desensitization and tolerance (Sim-Selley, 2003). Thus, CB1 receptor binding was assessed in near-adjacent brain sections from FAAH−/− mice treated as described above using [3H]WIN55,212-2. Visual inspection of brain sections revealed reduced [3H]WIN55,212-2 binding in brains from THC-treated mice, whereas binding in AEA-treated mice appeared only slightly reduced compared to vehicle-treated mice. Representative autoradiograms of [3H]WIN55,212-2 binding in comparison to WIN55,212-2-stimulated [35S]GTPγS binding at the level of the substantia nigra and hippocampus are shown in Figure 7. Densitometric analysis of the autoradiograms confirmed these observations (Table 5). [3H]WIN55,212-2 binding was significantly reduced in brains from THC- compared to vehicle-treated mice in all regions examined (Table 5). These results are similar to those found using agonist-stimulated [35S]GTPγS binding, indicating that the apparent desensitization that occurs in FAAH−/− mice with THC treatment is at least in part a result of CB1 receptor downregulation. In contrast to results in THC-treated mice, [3H]WIN55,212-2 binding did not differ between brains from AEA- and vehicle-treated mice in any region examined, consistent with results obtained using agonist-stimulated [35S]GTPγS binding. However, a two-way ANOVA revealed an overall effect of treatment (F5,176=27.6, p<0.001), and AEA-treated animals exhibited significantly reduced [3H]WIN55,212-2 binding when compared with vehicle, but significantly greater [3H]WIN55,212-2 binding when compared with THC (post-hoc Tukey test, p<0.05).
Figure 7.
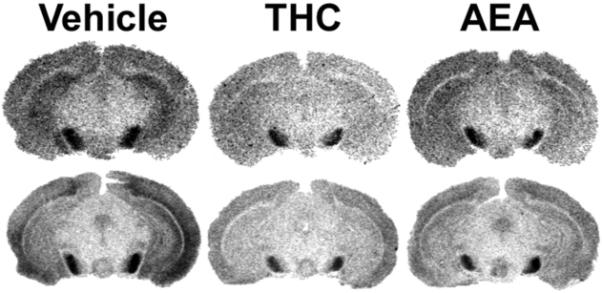
Representative autoradiograms illustrating [3H]WIN55,212-2 binding (top row) compared with WIN55,212-2-stimulated [35S]GTPγS binding (bottom row) in brains from FAAH−/− mice treated subchronically with vehicle, THC or AEA at the level of the hippocampus and substantia nigra. [3H]WIN55,212-2 and WIN55,212-2-stimulated [35S]GTPγS binding are both visibly reduced in THC-treated mice, whereas brain sections from AEA-treated mice show intermediate levels of [3H]WIN55,212-2 and WIN55,212-2-stimulated [35S]GTPγS binding between vehicle- and THC-treated mice. Data from densitometric analysis are presented in Table 5.
Table 5.
[3H]WIN55,212-2 binding in brain sections from vehicle-, THC-, and AEA-treated FAAH−/− mice
| REGION | Vehicle | THC | Anandamide |
|---|---|---|---|
| Caudate-Putamen | 1.3 ± 0.13 | 0.82 ± 0.11* | 0.95 ± 0.09 |
| Globus Pallidus | 2.69 ± 0.28 | 1.55 ± 0.12* | 2.31 ± 0.18 |
| Hippocampus | 1.08 ± 0.10 | 0.49 ± 0.07* | 0.88 ± 0.06 |
| Substantia Nigra | 3.61 ± 0.44 | 2.25 ± 0.20* | 3.11 ± 0.29 |
| PAG | 0.76 ± 0.10 | 0.43 ± 0.07* | 0.57 ± 0.06 |
| Cerebellum | 1.79 ± 0.16 | 1.19 ± 0.12* | 1.69 ± 0.18 |
Brain sections were assayed as described in Methods.
Data shown are mean specific [3H]WIN55,212-2 binding values (pCi/g) ± SEM from 8-12 mice per group
p<0.05 vs. vehicle treated animals, one-way ANOVA with a post-hoc Dunnett’s test.
Agonist-Stimulated [35S]GTPγS Binding in Spinal Cord Membranes
To determine the effect of subchronic THC or AEA administration on cannabinoid receptor-mediated G-protein activation in spinal cord, a CNS region that is relevant for antinociception, agonist-stimulated [35S]GTPγS binding was examined using WIN55,212-2 and AEA in isolated membrane preparations. Significant concentration-dependent stimulation of [35S]GTPγS binding was observed with both WIN55,212-2 (2-way ANOVA, F98=10.01, p < 0.0001, Figure 8) and AEA (2-way ANOVA, F86=24.52, p < 0.0001, data not shown) in spinal cord membranes. Prior treatment with THC, but not AEA, decreased stimulation of [35S]GTPγS binding by WIN55,212-2 (2-way ANOVA, F68=19.44, p < 0.0001, Figure 8) or AEA (2-way ANOVA, F41=12.96, p=0.0009, data not shown). These findings were confirmed by non-linear regression analysis of data (Table 6). The Emax values of WIN55,212-2 in spinal cord membranes from THC- or AEA-treated mice were 72% (p <0.05 by ANOVA, F=4.97, df=2, with post-hoc Dunnett’s test) and 85% (non-significant trend), respectively, of that obtained in vehicle-treated mice. Similar results were observed with AEA-simulated [35S]GTPγS binding, where Emax values of AEA were 75% (p <0.05 by ANOVA, F=6.61, df=2, with post-hoc Dunnett’s test) and 104% (not significant), respectively, of that obtained in vehicle treated mice. There were no significant differences in WIN55,212-2 or AEA EC50 values between THC- or AEA-treated mice and vehicle control. Although prior treatment with AEA appeared to increase the AEA EC50 value, this apparent increase was not statistically significant (p=0.124 by ANOVA, F=2.50, df=2). Thus, these results indicate that subchronic administration of THC, but not AEA, produced significant CB1 receptor desensitization in the spinal cord, similar to results obtained using autoradiography in brain, where subchronic THC produced more significant desensitization than AEA.
Table 6.
Emax and EC50 values of WIN- and AEA-stimulated [35S]GTPγS binding in spinal cord membranes from vehicle-, THC- or AEA-treated FAAH−/− mice
| Treatment: | Vehicle | THC | Anandamide |
|---|---|---|---|
| WIN Emax (% Stim) | 39 ± 3.1 | 27 ± 1.4* | 33 ±3.2 |
| WIN EC50 (nM) | 229 ± 81 | 182 ± 39 | 162 ± 64 |
| AEA Emax (% Stim) | 34 ± 1.6 | 25 ± 2.3* | 35 ± 3.3 |
| AEA EC50 (nM) | 559 ± 164 | 599 ± 84 | 1633 ± 603 |
Membranes were incubated with 0.1% BSA, 30μM GDP, 0.1nM [35S]GTPγS and varying concentrations of WIN55,212-2 (WIN) or AEA and concentration-effect curves were analyzed by non-linear regression, as described in Methods.
Data are mean values ± SE from 7 (WIN) or 5 (AEA) separate experiments
p < 0.05 different from vehicle control by one-way ANOVA with a post-hoc Dunnett’s test.
In summary, WIN55,212-2-stimulated [35S]GTPγS and [3H]WIN55,212-2 binding were significantly decreased throughout the brain in FAAH−/− animals treated with THC, whereas AEA treatment minimally affected receptor levels or receptor-mediated G-protein activity. This conclusion is clearly illustrated in Figure 9, in which data from THC- and AEA-treated mouse brains are expressed as % vehicle control. These results are also similar to those obtained in the spinal cord, suggesting that THC and AEA differentially regulate cannabinoid receptors throughout the CNS.
Figure 9.
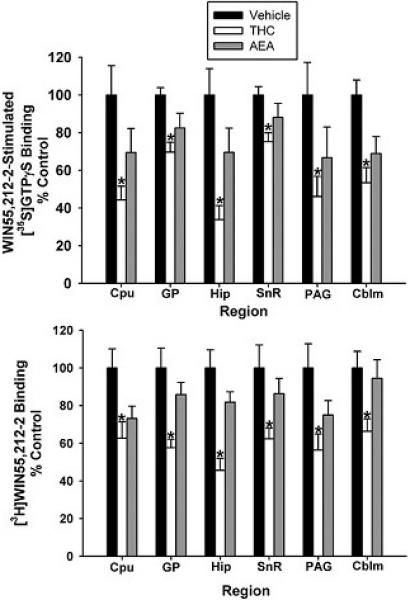
Subchronic THC attenuates CB1 receptor binding and G-protein activity, whereas subchronic AEA produces minimal CB1 receptor adaptation. Net WIN55,212-2-stimulated [35S]GTPγS binding (top) and [3H]WIN55,212-2 binding (bottom) in THC- and AEA-treated FAAH −/− mice are expressed as a percentage of control (vehicle-treated) mice (n= 8-12 mice/group, * p<0.05 different from vehicle-treated group by one-way ANOVA with a post-hoc Dunnett’s test).
Discussion
The results of this study revealed that repeated administration of AEA in FAAH−/− mice produced different neuroadaptations and in vivo consequences as compared to THC administration in these animals. Repeated administration of THC significantly reduced cannabinoid-stimulated G-protein activity and receptor binding levels in all regions examined, which was associated with tolerance to in vivo effects and antagonist-precipitated withdrawal. In contrast, repeated administration of AEA only slightly reduced receptor levels or receptor-mediated G-protein activity in FAAH−/− mice to levels intermediate between vehicle- or THC-treated FAAH−/− mice. In vivo data that were consistent with these effects include the observation that rimonabant elicited a significantly reduced magnitude of withdrawal effects in FAAH−/− mice treated repeatedly with AEA compared to FAAH−/− or FAAH+/+ mice that received subchronic THC treatment. The relationship between the expression of in vivo tolerance and CB1 receptor adaptations following repeated THC or AEA administration is complex and appeared to vary by effect. Profound rightward shifts of THC dose-response curves were found in both genotypes following subchronic THC administration as well as for FAAH−/− mice treated subchronically with AEA. In fact, THC produced considerably less catalepsy in FAAH−/− mice treated repeatedly with AEA compared to FAAH−/− or FAAH+/+ mice treated subchronically with THC (see Figure 2B and Table 2). In contrast, AEA dose-response curves for antinociception and hypothermia were shifted only three fold in FAAH−/− mice, regardless of whether the mice were treated subchronically with either THC or AEA. However, the AEA dose-response curve for catalepsy in FAAH−/− mice was shifted further to the right following repeated administration of THC than repeated administration of AEA. Thus, in vivo tolerance was found in FAAH−/− mice treated repeatedly with AEA or THC, but the magnitude of tolerance expressed depended more on the test drug than the drug administered subchronically, although catalepsy appears to be an exception.
As previously reported (Cravatt et al., 2001), FAAH −/− and FAAH+/+ mice displayed similar sensitivity to the acute effects of THC. Here, using the cumulative dosing procedure yielded similar ED50 values for antinociception, catalepsy, and hypothermia as previously reported when separate groups of mice were given a single bolus injection. Additionally, cumulative dosing and single bolus injections led to similar brain and blood levels of THC (see Table 1). Collectively, these findings indicate the following two conclusions: 1) cumulative dosing and single bolus dosing lead to similar blood and brain levels of THC at the time of in vivo testing; and 2) the THC cumulative dose-response curves for antinociception, catalepsy, and hypothermia are virtually identical between FAAH +/+ and FAAH−/− mice. Thus, the impact of repeated THC or AEA administration on tolerance was evaluated by using cumulative dosing procedure to derive dose-response relationships.
Previous studies showed that brains from FAAH−/− and FAAH +/+ mice exhibited similar levels of CB1 receptors as assessed in membrane homogenates prepared from whole brain (Cravatt et al., 2001). The current study extended those findings using autoradiography to show that the levels of CB1 receptors and receptor-mediated activity do not differ between FAAH−/− and FAAH+/+ mice in several regions that express CB1 receptors and mediate cannabinoid effects. Moreover, THC treatment attenuated receptor binding and receptor-mediated G-protein activity similarly in FAAH−/− and FAAH +/+ mice. These findings suggest that elevated AEA levels do not affect acute CB1 receptor activity or regulation in response to chronic THC. The present results differ somewhat from a previous study showing that 15 day AEA administration in FAAH-competent rats produced desensitization of cannabinoid-mediated G-protein activity in the absence of alterations in receptor binding (Rubino et al., 2000). Differences in both species and treatment duration could have contributed to the discrepancy with the present results.
Subchronic administration of AEA to FAAH −/− mice led to approximately 3-fold decreases in the potency of its antinociceptive, cataleptic, and hypothermic effects. In contrast, a much more profound tolerance was found following repeated THC treatment in FAAH−/− and FAAH+/+ mice. Both genotypes developed a similar degree of tolerance to the analgesic and cataleptic effects of THC, but FAAH−/− mice exhibited a greater magnitude of tolerance to the hypothermic effects of THC than FAAH +/+ mice. This pattern of findings is consistent with the hypothesis that adaptation of CB1 receptors and/or downstream signaling responses following repeated cannabinoid administration plays a predominant role in the dampening of cannabinoid-mediated in vivo effects rather than via effects on endocannabinoids directly.
Previous studies have shown that AEA-treated mice exhibit cross-tolerance to the cataleptic, hypolocomotor, analgesic and hypothermic effects of THC (Fride, 1995). In contrast, animals treated with THC exhibit cross tolerance to only certain AEA-mediated effects, for example antinociception (Welch, 1997), but not hypothermia (Pertwee et al., 1993). In fact, an assessment of cross-tolerance between THC and AEA and its analogs revealed that cross-tolerance was dependent on the task as well as the particular anandamide analog tested (Wiley et al., 2005). The present study used FAAH−/− mice, to examine cross-tolerance in both THC and AEA-treated mice. FAAH −/− mice treated subchronically with AEA displayed profound cross-tolerance to THC, with the greatest decrease in efficacy or potency to THC-mediated cataleptic effects. On the other hand, FAAH −/− mice treated repeatedly with THC displayed cross-tolerance to the behavioral effects of AEA, with approximately 3-fold decreases in the potency to its antinociceptive and hypothermic effects, although a greater decrease in the potency or efficacy of AEA-mediated cataleptic effects was found. These findings taken together suggest that the expression of tolerance is more greatly affected by the specific ligand used during testing than the ligand administered throughout the development of tolerance. In other words, testing with a low efficacy ligand, such as THC, revealed greater tolerance and cross-tolerance than a higher efficacy ligand (e.g., AEA) and therefore perhaps greater sensitivity to receptor loss, similarly to previous findings at the level of CB1 receptor signaling (Breivogel et al., 2003; Selley et al., 2004). A similar relationship between efficacy of the test ligand and sensitivity to cross-tolerance has been reported for mu opioid receptor-mediated antinociception (Paronis and Holtzman, 1992). However, further experiments with multiple agonists differing in intrinsic efficacy will be required to definitively determine whether this relationship holds true for cannabinoid receptors. Nonetheless, the present results demonstrate that subchronic administration of AEA produces less tolerance to the effects of subsequently administered AEA, compared to the magnitude of tolerance observed with subchronic THC administration and subsequent testing with THC.
Subchronic administration of maximally effective doses of THC produced similar magnitudes of CB1 receptor desensitization and downregulation in FAAH+/+ and FAAH−/− mice, as previously reported in various rodent strains using a variety of treatment paradigms (Sim-Selley, 2003). The THC brain levels resulting from 50 mg/kg THC were 1072 ± 156 ng/gram of tissue or roughly equivalent to those following 5 mg/kg of intravenously administered THC (Wilson et al., 2006). However, an equi-active dose of AEA administered subchronically did not alter CB1 receptors or activity in FAAH+/+ mice, which was predicted due to the instability of AEA in these mice. Surprisingly, in FAAH−/− mice, AEA treatment did not significantly reduce either CB1 receptor levels or receptor-mediated mediated G-protein activity in the CNS regions evaluated. The mechanism underlying differential cannabinoid receptor regulation by THC versus AEA is not clear. Both THC and AEA are partial agonists, although AEA has higher intrinsic efficacy compared to THC (Breivogel et al., 1998). Previous studies showed that although treatment with equi-active doses of THC or WIN55,212-2, a full agonist, produced desensitization and downregulation, THC treatment produced significantly greater desensitization than WIN55,212-2 in a number of regions (Sim-Selley and Martin, 2002). This finding is consistent with the present results and suggests that low efficacy agonists might produce greater receptor adaptation because they must occupy a higher percentage of total receptors to produce equivalent effects. However, comparison of repeated treatments with multiple agonists of varying intrinsic efficacies would be necessary to confirm this hypothesis. It is also possible that different agonists can differentially induce receptor interaction with β-arrestin to produce varying levels of desensitization, as suggested in previous studies (Bohn et al., 2004; Breivogel et al., 2008).
Rimonabant-precipitated withdrawal is used to assess cannabinoid dependence (Lichtman and Martin, 2002). In the present study, rimonabant elicited profound somatic withdrawal signs in FAAH −/− and +/+ mice treated subchronically with THC, whereas FAAH −/− mice treated with AEA displayed a greatly reduced withdrawal response. The role of cannabinoid receptor desensitization and/or downregulation in withdrawal is not clear, but the results are consistent with the general finding that repeated AEA treatment did not produce significant adaptation whereas THC treatment produced both desensitization and downregulation of CB1 receptors. Withdrawal following rimonabant administration in cannabinoid-treated animals is often associated with increased cAMP activity (Hutcheson et al., 1998; Rubino et al., 1998; Tzavara et al., 2000), suggesting that fewer downstream adaptations also occur following AEA as compared to THC treatment, although this hypothesis will have to be confirmed in future studies. Nevertheless, diminished dependence elicited by subchronic AEA in FAAH−/− mice is consistent with lack of dependence liability of the FAAH inhibitor, URB597 (Schlosburg et al., 2009).
Cannabinoids have generated interest for treatment of a number of disorders, but their therapeutic potential is limited by unwanted side effects and the development of tolerance and dependence with repeated use. An alternate approach is to increase levels of endogenous cannabinoids by administering inhibitors of their degradative enzymes. Piomelli and colleagues initially reported the development of URB532 and URB597 (Kathuria et al., 2003), FAAH inhibitors that produced anxiolytic and analgesic effects in the absence of catalepsy, hypothermia or appetite stimulation. Subsequent studies have shown that several FAAH inhibitors produce analgesia in a variety of pain models (Lichtman et al., 2004a; Jayamanne et al., 2006), further generating interest in the therapeutic potential of these compounds. The current results indicate that increasing AEA produces less cellular adaptation and associated dependence and less tolerance to its own effects than administration of THC, and further support the possible clinical utility of FAAH inhibitors.
Acknowledgements
These studies were supported by NIH grants DA014277, P01DA017259, R01DA15197, R01DA03672, R01DA02396, R01DA015683, P50DA005274, P01DA009789, T32DA007027, and F31DA026279 and The Institute of International Education/ Scholar Rescue Funds/ New York (RA).
Disclosure/Conflict of Interest: This research has been supported solely by the National Institutes of Health (NIH). The following authors: KWF, JES, RAA, THS, DES and LJS declare that, except for income received from the primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. AHL, BFC and AJT report no conflicts of interest with the work presented in this manuscript. AJT was employed by Abbott labs after completion of this work and is currently employed by AMAG Pharmaceuticals. AHL declares that over the past three years he has received compensation from Pfizer, Ironwood Pharmaceuticals, and Allergan. In addition, AHL has received funding from Pfizer and Ironwood for contracts unrelated to the research presented in this paper. BFC declares that over the past three years he has received compensation from Pfizer, Activx Biosciences, and aTyr Pharma.
References
- Aceto MD, Scates SM, Razdan RK, Martin BR. Anandamide, an endogenous cannabinoid, has a very low physical dependence potential. J Pharmacol Exp Ther. 1998;287:598–605. [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ. Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptor-activated G-proteins in brain. Journal of Neurochemistry. 1999;73:2447–2459. doi: 10.1046/j.1471-4159.1999.0732447.x. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Lambert JM, Gerfin S, Huffman JW, Razdan RK. Sensitivity to delta9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2 −/− mice. Behav Pharmacol. 2008;19:298–307. doi: 10.1097/FBP.0b013e328308f1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel CS, Scates SM, Beletskaya IO, Lowery OB, Aceto MD, Martin BR. The effects of delta9-tetrahydrocannabinol physical dependence on brain cannabinoid receptors. Eur J Pharmacol. 2003;459:139–150. doi: 10.1016/s0014-2999(02)02854-6. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Selley DE, Childers SR. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPγS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. Journal of Biological Chemistry. 1998;273:16865–16873. doi: 10.1074/jbc.273.27.16865. [DOI] [PubMed] [Google Scholar]
- Budney AJ, Hughes JR. The cannabis withdrawal syndrome. Curr Opin Psychiatry. 2006;19:233–238. doi: 10.1097/01.yco.0000218592.00689.e5. [DOI] [PubMed] [Google Scholar]
- Compton DR, Johnson MR, Melvin LS, Martin BR. Pharmacological profile of a series of bicyclic cannabinoid analogs: classification as cannabimimetic agents. Journal of Pharmacology and Experimental Therapeutics. 1992;260:201–209. [PubMed] [Google Scholar]
- Cook SA, Lowe JA, Martin BR. CB1 receptor antagonist precipitates withdrawal in mice exposed to Delta9-tetrahydrocannabinol. J Pharmacol Exp Ther. 1998;285:1150–1156. [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fride E. Anandamides: tolerance and cross-tolerance to delta 9-tetrahydrocannabinol. Brain Res. 1995;697:83–90. doi: 10.1016/0006-8993(95)00790-w. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RLM. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. Journal of Neuroscience. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister LE. Health Aspects of Cannabis. Pharmacological Reviews. 1986;38:1–20. [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacological Review. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Hutcheson DM, Tzavara ET, Smadjs C, Valjent E, Roques BP, Hanoune J, Maldonado R. Behavioural and biochemical evidence for signs of abstinence in mice chronically treated with Δ9-tetrahydrocannabinol. British Journal of Pharmacology. 1998;125:1567–1577. doi: 10.1038/sj.bjp.0702228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RT, Benowitz N, Bachman J. Clinical studies of cannabis tolerance and dependence. Ann N Y Acad Sci. 1976;282:221–239. doi: 10.1111/j.1749-6632.1976.tb49901.x. [DOI] [PubMed] [Google Scholar]
- Jones RT, Benowitz NL, Herning RI. Clinical relevance of cannabis tolerance and dependence. Journal of Clinical Pharmacology. 1981;21:143S–152S. doi: 10.1002/j.1552-4604.1981.tb02589.x. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004a;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Martin BR. Marijuana withdrawal syndrome in the animal model. J Clin Pharmacol. 2002;42:20S–27S. doi: 10.1002/j.1552-4604.2002.tb05999.x. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Martin BR. Cannabinoid tolerance and dependence. Handb Exp Pharmacol. 2005:691–717. doi: 10.1007/3-540-26573-2_24. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004b;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Martin BR, Sim-Selley LJ, Selley DE. Signaling pathways invovled in the development of cannabinoid tolerance. Trends in Pharmacological Sciences. 2004;25:325–330. doi: 10.1016/j.tips.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Paronis CA, Holtzman SG. Development of tolerance to the analgesic activity of mu agonists after continuous infusion of morphine, meperidine or fentanyl in rats. Journal of Pharmacology and Experimental Therapeutics. 1992;262:1–9. [PubMed] [Google Scholar]
- Pertwee RG, Stevenson LA, Griffin G. Cross-tolerance between delta-9-tetrahydrocannabinol and the cannabimimetic agents, CP 55,940, WIN 55,212-2 and anandamide. British Journal of Pharmacology. 1993;110:1483–1490. doi: 10.1111/j.1476-5381.1993.tb13989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero J, Berrendero F, Garcia-Gil L, Lin SY, Makriyannis A, Ramos JA, Fernandez-Ruiz JJ. Cannabinoid receptor and WIN-55,212-2-stimulated [35S]GTPgammaS binding and cannabinoid receptor mRNA levels in several brain structures of adult male rats chronically exposed to R-methanandamide. Neurochemistry International. 1999;34:473–482. doi: 10.1016/s0197-0186(99)00020-0. [DOI] [PubMed] [Google Scholar]
- Romero J, Garcia L, Fernandez-Ruiz JJ, Cebeira M, Ramos JA. Changes in rat brain cannabinoid binding sites after acute or chronic exposure to their endogenous agonist, anandamide, or to delta 9-tetrahydrocannabinol. Pharmacol Biochem Behav. 1995;51:731–737. doi: 10.1016/0091-3057(95)00023-p. [DOI] [PubMed] [Google Scholar]
- Rubino T, Patrini G, Massi P, Fuzio D, Vigano D, Giognoni G, Parolaro D. Cannabinoid-precipitated withdrawal: A time-course study of the behavioral aspect and its correlation with cannabinoid receptors and G protein expression. Journal of Pharmacology and Experimental Therapeutics. 1998;285:813–819. [PubMed] [Google Scholar]
- Rubino T, Vigano D, Costa B, Colleoni M, Parolaro D. Loss of cannabinoid-stimulated guanosine 5′-O-(3-[(35)S]Thiotriphosphate) binding without receptor down-regulation in brain regions of anandamide-tolerant rats. Journal of Neurochemistry. 2000;75:2478–2484. doi: 10.1046/j.1471-4159.2000.0752478.x. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Carlson BL, Ramesh D, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH. Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. Aaps J. 2009;11:342–352. doi: 10.1208/s12248-009-9110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley DE, Cassidy MP, Martin BR, Sim-Selley LJ. Long-term administration of delta9-tetrahydrocannabinol desensitizes CB1-, adenosine A1-, and GABAB-mediated inhibiton of adenylyl cyclase in mouse cerebellum. Molecular Pharmacology. 2004;66:1275–1284. doi: 10.1124/mol.104.000604. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ. Regulation of cannabinoid CB1 receptors in the central nervous system by chronic cannabinoids. Critical Reviews in Neurobiology. 2003;15:91–119. doi: 10.1615/critrevneurobiol.v15.i2.10. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Martin BR. Effect of chronic administration of WIN 55,212-2 or Δ9-tetrahydrocannabinol on cannabinoid receptor adaptation in mice. Journal of Pharmacology and Experimental Therapeutics. 2002;303:36–44. doi: 10.1124/jpet.102.035618. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Schechter NS, Rorrer WK, Dalton GD, Hernandez J, Martin BR, Selley DE. Prolonged recovery rate of CB1 receptor adaptation after cessation of long-term cannabinoid administration. Mol Pharmacol. 2006;70:986–996. doi: 10.1124/mol.105.019612. [DOI] [PubMed] [Google Scholar]
- Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R, Martin BR. The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J Pharmacol Exp Ther. 1994;270:219–227. [PubMed] [Google Scholar]
- Tsou K, Patrick SL, Walker JM. Physical withdrawal in rats tolerant to Δ9-tetrahydrocannabinol precipitated by a cannabinoid receptor antagonist. European Journal of Pharmacology. 1995;280:R13–R15. doi: 10.1016/0014-2999(95)00360-w. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Valjent E, Firmo C, Mas M, Beslot F, Defer N, Roques BP, Hanoune J, Maldonado R. Cannabinoid withdrawal is dependent upon PKA activation in the cerebellum. European Journal of Neuroscience. 2000;12:1038–1046. doi: 10.1046/j.1460-9568.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- Villares J. Chronic use of marijuana decreases cannabinoid receptor binding and mRNA expression in the human brain. Neuroscience. 2007;145:323–334. doi: 10.1016/j.neuroscience.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Welch SP. Characterization of anandamide-induced tolerance: comparison to delta-9-THC-induced interactions with dynorphinergic systems. Drug and Alcohol Dependence. 1997;45:39–45. doi: 10.1016/s0376-8716(97)01342-2. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Smith FL, Razdan RK, Dewey WL. Task specificity of cross-tolerance between Delta9-tetrahydrocannabinol and anandamide analogs in mice. Eur J Pharmacol. 2005;510:59–68. doi: 10.1016/j.ejphar.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Willoughby KA, Moore SF, Martin BR, Ellis EF. The biodisposition and metabolism of anandamide in mice. J Pharmacol Exp Ther. 1997;282:243–247. [PubMed] [Google Scholar]
- Wilson DM, Varvel SA, Harloe JP, Martin BR, Lichtman AH. SR 141716 (Rimonabant) precipitates withdrawal in marijuana-dependent mice. Pharmacol Biochem Behav. 2006;85:105–113. doi: 10.1016/j.pbb.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Wise LE, Shelton CC, Cravatt BF, Martin BR, Lichtman AH. Assessment of anandamide’s pharmacological effects in mice deficient of both fatty acid amide hydrolase and cannabinoid CB1 receptors. Eur J Pharmacol. 2007;557:44–48. doi: 10.1016/j.ejphar.2006.11.002. [DOI] [PubMed] [Google Scholar]