

Abstract
Temporarily discontinuing the use of antibiotics has been proposed as a means to eliminate resistant bacteria by allowing sensitive clones to sweep through the population. In this study, we monitored a tetracycline-sensitive subpopulation that emerged during experimental evolution of E. coli K12 MG1655 carrying the multiresistance plasmid pB10 in the absence of antibiotics. The fraction of tetracycline-sensitive mutants increased slowly over 500 generations from 0.1 to 7%, and loss of resistance could be attributed to a recombination event that caused deletion of the tet operon. To help understand the population dynamics of these mutants, three mathematical models were developed that took into consideration recurrent mutations, increased host fitness (selection), or a combination of both mechanisms (full model). The data were best explained by the full model, which estimated a high mutation frequency (λ = 3.11 × 10−5) and a significant but small selection coefficient (σ = 0.007). This study emphasized the combined use of experimental data, mathematical models, and statistical methods to better understand and predict the dynamics of evolving bacterial populations, more specifically the possible consequences of discontinuing the use of antibiotics.
TODAY, numerous pathogens have developed resistance to one or multiple antibiotics (Cohen 1992), to such an extent it is called “an epidemic of microbial resistance” (Gibbons 1992). Aside from the constant search for new antibiotics that can be used against multidrug-resistant pathogens, another strategy is to lower or discontinue the use of a specific antibiotic against which resistance is widespread (Levy 1994; OTA 1995; WHO 1995, 2003; Lenski 1997). This approach assumes that antibiotic resistance imposes a fitness cost to the host (Spratt 1996; Andersson and Levin 1999; Bjorkman and Anderson 2000; Normark and Normark 2002) high enough for sensitive strains to have a selective advantage relative to resistant ones in the absence of the drug. Therefore, ending the use of a specific antibiotic would allow these sensitive strains to sweep through and replace the resistant population (Levin et al. 1997). An important mechanism by which bacteria acquire antibiotic resistance genes is through conjugative transfer of plasmids on which they are encoded. Many authors have observed that the carriage of an antibiotic resistance plasmid does indeed reduce the fitness of the host in the absence of antibiotics (Godwin and Slater 1979; Helling et al. 1981; Bouma and Lenski 1988; Modi and Adams 1991; Turner et al. 1998; Dahlberg and Chao 2003). Therefore, when plasmid-free segregants emerge, they might be expected to replace the resistant plasmid-containing population in the absence of selection for the plasmid. It has also been shown that the elimination of resistance genes from a plasmid can lower the burden of that plasmid. During an evolution experiment under glucose-limited conditions, Godwin and Slater (1979) observed that an initial population of Escherichia coli K12 that carried a multiple antibiotic resistance plasmid was replaced by a tetracycline-sensitive population that still carried a plasmid encoding the other resistance determinants. Also Dahlberg and Chao (2003) and Turner et al. (1998) detected different types of sensitive subpopulations in a multiresistant parent population. In a similar experiment with E. coli JA104 (pBR322Δ5), a plasmid variant that possessed a 2.25-kb deletion encompassing the tetracycline resistance operon arose, which resulted in a 10–20% fitness increase for the host compared to carriage of the wild-type plasmid (Modi et al. 1991). The population carrying this mutated plasmid increased steadily from 5 to 99% between the 380th and 560th generation, representing a clear selective sweep. These studies suggest that drug-sensitive mutants could replace the resistant population after ending antibiotic treatment. In contrast, other reports have clearly shown that coevolution between plasmid-encoded genes and the host (Lenski et al. 1994) or compensatory mutations (Schrag et al. 1997; Andersson and Levin 1999) may result in resistant strains being equally as fit as or even fitter than their sensitive counterparts.
Mutations in bacteria that result in loss of resistance to a particular antibiotic are typically infrequent. The frequency of spontaneous mutations has been estimated to be in the range of 10−9–10−10 per base pair and per generation (Drake 1991; Andersson and Hughes 1996; Drake et al. 1998) and only a fraction of these mutations will convert a resistant phenotype into a sensitive one. Due to these low frequencies and the lack of direct detection of antibiotic-sensitive clones, such clones that arise within a resistant population are usually observed only once they sweep through the population due to a fitness advantage in the absence of the antibiotic.
In this study, we monitored the loss of antibiotic resistance in E. coli K12 populations that carried the multiresistance IncP1-β plasmid pB10 during a long-term evolution experiment in a medium without antibiotics. Our goal was to identify the molecular basis of the tetracycline sensitivity observed in the mutant population and to develop a mathematical model based on the data that helped explain the observed population dynamics, taking into account the associated mutation frequency and selection coefficient. This model was further used to generate predictions concerning the effects of cycling antibiotics on the decline of resistance levels.
MATERIALS AND METHODS
Experimental procedures
Media:
Luria-Bertani broth (LB) medium and M9 minimal medium were prepared according to Sambrook and Russel (2001). Unless otherwise stated, antibiotics were used in the following concentrations: 10 mg/liter tetracycline (Tc), 50 mg/liter kanamycin (Km), 75 mg/liter amoxicillin (Amx), and 40 mg/liter streptomycin (Sm).
Bacterial strains and plasmid:
The bacterial strains used were E. coli K12 MG1655 (ATCC 47076) and a Km-resistant variant (K12::Km) obtained by conjugation of E. coli K12 with E. coli S17/1 λpir (pUTminiTn5Km) and selection on M9 + 0.4% lactose + Km. The 64.5-kb plasmid pB10 was isolated from the bacterial community of a wastewater treatment plant in Germany (Dröge et al. 2000) and has been sequenced (Schlüter et al. 2003). It has been identified as a self-transmissible broad-host-range IncP-1β plasmid and it mediates resistance against the antibiotics tetracycline, amoxicillin, sulfonamide, and streptomycin and against mercury ions. The plasmid was transferred from its original host E. coli DH5α to K12 and K12::Km through conjugation and selection on BBL MacConkey Agar supplemented with 15 mg/liter Sm and LBKmTc, respectively.
Evolution experiment:
Experimental evolution of 10 independent lineages was accomplished by serial batch culture of each lineage for 500 generations in 10 ml M9 with 2 g/liter glucose as sole carbon source and 5 ml/liter stock E (Stanier et al. 1966) in 50-ml flasks, which were shaken at 225 rpm at 30°. This medium supported a stationary-phase density of ∼4 × 109/ml. Five lineages were founded from E. coli K12(pB10) and five from E. coli K12::Km(pB10). Every 24 hr, 39 μl of each population was transferred to fresh media. The regime permitted 8 generations of binary fission every day. At 100-generation intervals a 1-ml sample of each population was mixed with 0.3 ml glycerol and archived at −80°. The founding populations were archived in the same way.
Screening for tetracycline-sensitive clones:
At weekly intervals from day 14 on, a sample from three lineages of each K12(pB10) and K12::Km(pB10) was plated on LB and colonies were picked onto LBTc, LBAmx, and LB. Colonies that scored Tc−Amx+Sm+ were archived at −80°. From both archived start cultures, E. coli K12(pB10) and E. coli K12::Km(pB10), a sample was plated on LB, and 1000 colonies were picked onto LBTc, LBAmx, and LB. Samples from the frozen cultures that were archived every 100 generations were plated onto LB and colonies (see Table 1 for sample size) were picked onto LBTc, LBAmx, and LB.
TABLE 1.
Fraction of tetracycline-sensitive mutants found in 10 parallel and independent lineages over 504 generations, as well as the predicted fractions under the three models
| Observed fractions
|
Predicted fractions
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Generation: lk |
Sample size: Dk |
WT1: s1k |
WT2: s2k |
WT3: s3k |
WT4: s4k |
WT5: s5k |
Km1: s6k |
Km2: s7k |
Km3: s8k |
Km4: s9k |
Km5: s10k |
Mean | M & S | S | M |
| 0 | 1000 | 0.002 | 0.001 | 0.0015 | 0.0013 | 0.0029 | 0.0010 | ||||||||
| 104 | 208 | 0.010 | 0.000 | 0.000 | 0.010 | 0.005 | 0.005 | 0.010 | 0.000 | 0.010 | 0.005 | 0.0053 | 0.0063 | 0.0059 | 0.0107 |
| 200 | 104 | 0.010 | 0.019 | 0.019 | 0.000 | 0.010 | 0.010 | 0.010 | 0.019 | 0.010 | 0.000 | 0.0106 | 0.0137 | 0.0113 | 0.0196 |
| 304 | 67 | 0.015 | 0.060 | 0.075 | 0.015 | 0.030 | 0.015 | 0.030 | 0.015 | 0.090 | 0.030 | 0.0373 | 0.0266 | 0.0229 | 0.0292 |
| 400 | 52 | 0.058 | 0.058 | 0.000 | 0.000 | 0.058 | 0.096 | 0.077 | 0.038 | 0.058 | 0.038 | 0.0481 | 0.0453 | 0.0436 | 0.0379 |
| 504 | 44 | 0.045 | 0.136 | 0.091 | 0.023 | 0.068 | 0.000 | 0.023 | 0.114 | 0.114 | 0.068 | 0.0682 | 0.0766 | 0.0853 | 0.0473 |
Each independent lineage is labeled as either WT1–5 [E. coli K12(pB10) lineages] or Km1–5 [E. coli K12::Km(pB10) lineages]. k denotes the cycle and l the number of generations per cycle (l = 8). Dk is the number of clones screened and sik (i = 1, 2, 3, … 10) is the observed fraction of mutants for each replicate i at cycle k. M & S, mutation and selection model; S, selection-alone model; M, mutation-alone model.
Plasmid extraction and restriction enzyme analysis:
Plasmids were extracted from cells grown in LBTc or LBAmx using the QIAprep spin miniprep kit according to the manufacturer's instructions (QIAGEN, Valencia, CA) and digested with HindIII or NotI (Invitrogen, Carlsbad, CA). Samples were loaded on a 0.8% agarose gel and run for ∼16.5 hr at 30 V. The gels were stained with ethidium bromide and the bands were visualized by UV light.
PCR and sequencing:
PCR was used to assess the occurrence of recombination between the direct repeats that flank the tetracycline-resistance operon. These primers (forward, 5′-GACGGCGGCCTGGAGACAAGTC; reverse, 5′-TTTGCTCGGTGCCCTTTCGGGTAA) targeted the regions on the plasmid (Schlüter et al. 2003) just outside the direct repeat sequences as described previously (Flores et al. 2000). If no recombination has taken place and the tetracycline-resistance operon is still present in the plasmid, the annealed primers would be separated by an ∼6.4-kb stretch and the PCR reaction would not continue. If recombination has occurred and the tetracycline-resistance genes have been lost, the primers would be separated by ∼900 bp and a PCR product of that size would be formed. PCR products were cleaned with the MiniElute PCR purification kit according to the manufacturer's instructions (QIAGEN).
To sequence the 900-bp fragment, 2 μl of PCR product was mixed with 1 μl of either the forward or the reverse primer and 3 μl ddH2O and put at 95° for 5 min, after which it was cooled down to 50°. Then, 4 μl of BigDye Terminator cycle sequencing ready reaction mix (Version 3.0; Perkin-Elmer, Foster City, CA) was added. Cycle sequencing conditions (Peltier Thermal Cycler-200; MJ Research, Reno, NV) were as follows: 95° for 30 sec, 50° for 20 sec, 60° for 4 min, with a total of 44 cycles performed. The sequencing products were cleaned with the DyeEx 2.0 spin kit (QIAGEN) for dye-terminator removal, followed by vacuum centrifuging for 10 min, after which 10 μl formamide was added and the samples were put at 96° for 3 min. They were kept on ice before running on a POP6 polymer on a 3100 DNA automated capillary sequencer (Applied Biosystems, Foster City, CA).
Electroporation:
To obtain ancestral strains carrying evolved, mutated pB10 plasmids, E. coli K12 cells were prepared for electroporation according to Enderle and Farwell (1998). Approximately 200 ng of plasmid DNA was added to each of the cell suspensions and the mixtures were brought into electroporation cuvettes (1 mm gap, 100 μl volume; Eppendorf, Hamburg, Germany) and electroporated at 2.5 kV (25 μF, 200 Ω). The suspensions were then immediately transferred to SOC medium (Sambrook and Russel 2001), incubated for 1 hr at 37°, and plated on selective media after which the presence of the appropriate plasmid was confirmed.
Competition experiments:
Competition experiments were performed by mixing overnight cultures of the two competitors grown in M9 + 2 g/liter glucose + 5 ml/liter stock E in a 1/100 ratio. For each experiment, six to eight replicate cultures were tested. The cultures were transferred every 24 hr into fresh media (39 μl inocula into 10 ml of fresh media). After two transfers, the ratio of the two competitors was determined by selective plating. Using these data, the relative fitness (W) was calculated as described previously (Lenski et al. 1991). Subsequently, the selection coefficient, s = W − 1 (Lenski et al. 1991), was calculated and a t-test was performed to determine if the s-value was significantly different from zero. A t-test was also performed to detect significant differences between different s-values.
Mathematical models and statistical methodology
Modeling the experimental evolutionary process:
The experimental evolutionary process described above consisted of daily growth periods, wherein the populations multiplied in a neutral environment for l generations (l = 8), interspersed by daily bottlenecks of 39 μl/10,000 μl. We define a cycle as the combination of a growth period and a bottleneck and a neutral environment as one that does not contain any antibiotics. The growth period of any cycle might encompass the event of a deletion mutation occurring at random and resulting in a plasmid without tet operon. We refer to cells with a mutated plasmid as mutants.
We assumed the mutation process to be unidirectional; i.e., the cells can lose the Tc resistance but cannot gain it. We denote the average number of mutants at a certain time t by mt and the average of the remaining wild-type cells in period t by nt. Therefore, the total population at time t is the sum of mt and nt. At any generation, mutants increase due to (i) growth of the mutants from the previous generation, which occurs at a rate 21+σ with σ being the selection coefficient and (ii) mutation of wild-type cells at rate λ. The following recursive equation captures the average number of mutants at time t:
 |
1 |
Using the no-back-mutation assumption, the average number of nonmutants at any time t is given by
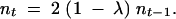 |
2 |
The average fraction of mutants available at any time t is
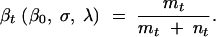 |
3 |
β0 represents the fraction of mutants that might already be available at the start of the experiment.
Every cycle k encompasses l generations. Joyce et al. (2005) showed that for the purposes of statistical analysis one can assume that the fraction of mutants at the end of each cycle, βlk, is deterministic and unaffected by the bottleneck. [While the results of Joyce et al. (2005) were derived for the special case where σ = 0, it can be shown both analytically and by simulation that the effect of the bottleneck on the variation of the process is strongest when σ = 0. So even in the case where the highest amount of variation due to the bottleneck is expected, the effect is still negligible.] Time can be measured using lk to reflect the cyclic behavior of the system. Equation 3 becomes
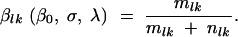 |
4 |
Solving this recursion (appendix B) results in the following general equation:
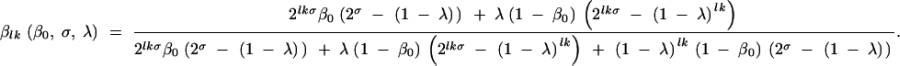 |
5 |
Equation 5 assumes that both mutation (at rate λ) and selection of mutants (represented by the selection coefficient σ) affect the population dynamics. This equation is readily interpretable. The denominator is composed of three parts: 2lkσβ0(2σ − (1 − λ)) represents the growth of initial mutants, λ(1 − β0)(2lkσ − (1 − λ)lk) represents the formation and growth of new mutants during evolution, and (1 − λ)lk(1 − β0)(2σ − (1 − λ)) represents the growth of TcR cells. The numerator involves the first two terms and describes the formation and growth of mutants. When there is no fitness difference between the mutants and the wild type we can set the selection coefficient σ to zero. Equation 5 becomes
 |
6 |
Then the increase in fraction of mutants in the population is due solely to mutation. When there is no mutation effect, λ can be set equal to zero, and the relative increase in mutants is modeled only by the selective growth advantage:
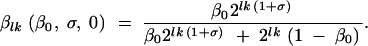 |
7 |
Modeling the sampling process:
Samples were taken every 100 generations and diluted and plated to screen for tetracycline-sensitive clones. The used sample sizes, from 42 to 1000 colonies, were very small compared to the bottleneck imposed (∼107 transferred cells) and can result in a magnification of stochastic effects (Joyce et al. 2005); i.e., the numbers of mutants observed on the basis of such a sampling method might fluctuate merely due to the randomness of this sampling process. We call such an effect “observational error” and use a stochastic model in the analysis of this process. A stochastic model that fits such a process is a binomial sampling process. In a sample of size Dk, taken at the kth day, each individual has a probability βlk of being a mutant and 1 − βlk of being a wild type. If βlk is small we can approximate this model to a Poisson model with parameter Dkβlk, representing the expected number of mutants in a sample. Letting this number of mutants from culture i at the end of day k, Sik, be a random variable, then Sik is modeled by
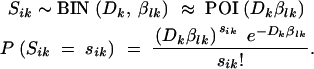 |
8 |
Statistical analysis:
Equation 8 presents the main model used in our data analysis and provides the link between the mathematical modeling and the statistical analysis, which proceeded in three stages. First, the model was fitted to the data using a maximum-likelihood approach. We estimated the mutation rate λ under the mutation-alone model and estimated the selection coefficient σ under the selection-alone model. Both parameters (λ, σ) were jointly estimated under the general (mutation and selection) model. Next, we determined whether any of the models adequately described the data using an absolute goodness-of-fit test and a parametric bootstrap approach. Finally, we assessed the relative fit of the model by comparing the simpler cases (mutation alone, selection alone) to the general model by using a likelihood-ratio test and a parametric bootstrap approach.
Parameter estimation:
To estimate the parameters λ, σ, and β0, we used the method of maximum likelihood (Bain and Engelhardt 1991; Rice 1995). The probability of a data set S = {sik} given the model M is referred to as the likelihood of that data set under the model. Using the Poisson model approximation (Equation 8) and assuming that we have rk independent replicates per cycle k and that these replicates are independent across cycles, the likelihood of the data set S given the model is
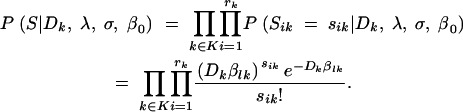 |
9 |
Multiple methods are available for maximizing the likelihood of the data. We used the Nelder-Mead algorithm (Nelder and Mead 1965) implemented in both MatLab6.5 and R1.6.2 (GNU license). The resulting estimated parameters are referred to as the maximum-likelihood estimates (MLEs). Programs, using MatLab6.5 and R1.6.2 (http://www.r-project.org), were developed for estimation. These programs are available through the web under http://www.webpages.uidaho.edu/~joyce/Labpage/Evo-x.html.
Absolute goodness of fit:
The absolute goodness-of-fit test for each of the three models considered is based on the null hypothesis that the data are Poisson distributed with E(Sik) = Dkβlk against the alternative that each Sik is Poisson distributed with a different mean μik (Rice 1995). The associated likelihood-ratio test (LRT) statistic Λ is derived in appendix C and given by
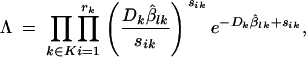 |
10 |
where β̂lk is the estimated proportion of mutants at the end of the kth cycle, computed by replacing the null-model parameters by the MLEs in the solution of Equation 4. The solution equation needed changes depending on which model is tested (see Equations 5–7). By the invariance principle of the MLEs (Rice 1995), β̂lk is also a MLE. Therefore, this estimate is different for each of the three absolute goodness-of-fit tests. Taking the natural logarithm of the likelihood-ratio statistic Λ results in the following familiar format:
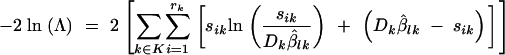 |
11 |
We used a parametric bootstrap (Efron and Tibshirani 1993) to estimate the distribution of this −2 ln Λ test statistic. For each case, 2000 data sets were simulated under the Poisson distribution of the null model, taking its MLEs to be the true parameter values. Then we estimated the MLEs for each of the simulated data sets, thus generating a sampling distribution for these MLEs. This sampling distribution was used to construct confidence intervals and confidence regions for the MLEs associated with the actual data. Applying Equation 11 to each of the simulated data sets and their associated MLEs we generated a distribution of the −2 ln Λ test statistic. The proportion of times that the simulated −2 ln Λ values were greater than the −2 ln Λ calculated using the actual data set resulted in a P-value. This proportion is an estimate of the probability that a LRT value greater than or equal to the one observed would actually occur given that the hypothesized model is true.
It is interesting to note that if the above model assumptions are accurate, then one can gain the same amount of information from a single replicate with samples of size r × n at various time points as by taking samples of size n for each of r replicated experiments. Since replicating the experiment is more time consuming and expensive than simply taking larger samples, one might think that our article argues against replication. However, one has a much better chance of detecting violations of model assumptions if one replicates the experiment. Our program can be downloaded at the website http://www.webpages.uidaho.edu/~joyce/Labpage/Evo-x.html. The user's manual that can be downloaded along with the program provides detailed information on the effect of departures from model assumptions and how to test for these departures.
Comparing the different models:
We compared the mutation-alone and the selection-alone models to the full model. The likelihood-ratio test statistic needed to compare the mutation-alone model to the full model is given by
 |
12 |
where L is the likelihood function as defined in Equation 9, (β̂0, σ̂, λ̂) are the MLEs, and ΛM(β̂0, λ̂) and Λ(β̂0, σ̂, λ̂) are the absolute goodness-of-fit likelihood test statistics (Equation 10) for the mutation-alone and the full models, respectively. Taking the natural log the following equation was obtained:
 |
13 |
A similar setup for the comparison of the selection-alone model to the full model results in
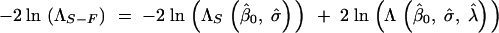 |
14 |
A 2000-replicates parametric bootstrap was also used to generate the distribution of the LRT. MatLab6.5 and R1.6.2 (http://www.r-project.org) programs in the S language were also developed for this analysis and are also available at http://www.webpages.uidaho.edu/~joyce/Labpage/Evo-x.html.
RESULTS
Experimental results:
Detection of tetracycline-sensitive clones:
We sought to monitor the loss of antibiotic resistance in an E. coli K12 population carrying the multiresistance plasmid pB10 during a long-term evolution experiment in a medium without antibiotics. To do this, 10 independent lineages were evolved for ∼500 generations in serial batch cultures. Loss of resistance to tetracycline or amoxicillin was monitored weekly in 6 of the 10 lines starting from day 14, as well as in the 2 founding cultures. Tetracycline sensitive (TcS) clones were detected in the founding cultures (0.15%) and their fraction gradually increased on average over 500 generations to ∼6% (Joyce et al. 2005). All but one of the TcS clones were still resistant to amoxicillin and streptomycin, indicating a very high plasmid maintenance. Since the frequency of the TcS clones was rather variable, probably due to the small sample size (maximum of 52 clones per culture per time point), a new sampling scheme that fixed the relative error rate at 0.21 was derived (Joyce et al. 2005). This allowed us to more precisely determine the fraction of the TcS mutants in all 10 available cultures that were archived every 100th generation. As shown in Table 1, the fractions of mutants observed in these new samples showed a clear increasing trend over time. All TcS clones obtained were still resistant to amoxicillin, indicating that they had not lost the entire plasmid.
Characterization of the plasmids of tetracycline-sensitive mutants:
To examine whether the loss of tetracycline resistance was due to deletions or rearrangements in the plasmid, restriction fragment length polymorphisms (RFLPs) of the plasmids were determined. Comparison of these RFLPs with the theoretical restriction map of plasmid pB10 (Schlüter et al. 2003) showed that the plasmids of all 73 sensitive clones examined lacked a 7057-bp HindIII fragment, whereas the patterns of randomly picked TcR clones were not different from that of the ancestral plasmid (Figure 1). A NotI restriction analysis performed on a few TcS clones showed that the 13,920-bp NotI fragment was not present in their plasmids. Analysis of the plasmid sequence revealed that the differences in RFLPs arose as the result of deletion of a 5.5-kb region that included the tetracycline-resistance operon (tetA, tetR) and was flanked by two direct repeats of 863 bp. This suggests that the sensitivity to tetracycline in the mutants was due to deletion of the tet operon by recombination between the direct repeats. To further analyze this deletion event, this region of plasmids from 18 mutants from six different lineages was amplified using primers described above and sequenced. All PCR products obtained from the sensitive clones showed identical DNA sequences, in which only one of the direct repeats was still present and the sequence in between the repeats was absent. This indicated that the deletion of the tetracycline-resistance operon was caused by a recombination event between the two flanking direct repeats.
Figure 1.—

HindIII RFLP patterns of ancestral and evolved plasmids. Lanes 1 and 7, 1-kb extended ladder, 5 μg and 10 μg, respectively; lane 2, ancestral pB10; lanes 3 and 4, plasmid DNA from TcS mutants; lanes 5 and 6, plasmid DNA from evolved TcR clones. Arrow indicates missing band in TcS mutants. Colors were inverted and brightness and contrast were adjusted to enhance the quality of the image.
Competition experiments:
To investigate the effects of the mutated plasmid on host fitness, the ancestral host K12::Km was transformed with a mutated plasmid isolated from a clone obtained at generation 500 [yielding K12::Km(pM57)], and this strain was competed against the ancestral host with ancestral plasmid K12(pB10). The selection coefficient (s-value) was 0.0273, which was not significantly different (P = 0.974) from the s-value (0.0271) of the control experiment that detected fitness differences due to the chromosomal marker [K12::Km(pB10) vs. K12(pB10)]. The results of these pairwise competition experiments suggest that deletion of the plasmid fragment containing the tet operon did not measurably increase the host fitness. Therefore it appeared that the increase in frequency of the TcS mutants could not be attributed to a selective advantage.
Modeling and statistical analysis:
Parameter estimates and confidence intervals:
A mathematical model was developed to capture the observed dynamics of the TcS mutants in the population. Overall, the data analysis strongly indicated that both selection and frequency of mutation, as described by the full model, seemed to affect the population dynamics of the mutants. Neither selection alone nor mutation alone could explain the observed patterns of population increase as well as the full model did. Using the full model, the selection coefficient σ was estimated to be 0.00699 and the rate of mutation λ was estimated to be 3.11 × 10−5/generation (Table 2). The MLEs and their confidence intervals were significantly distinct from zero in each of the three cases. Further corroboration of this conclusion is indicated in Figure 2, which presents the bootstrapped joint 94.8% confidence region for the mutation rate λ and the selection coefficient σ of the full model. This region excludes zero, indicating that the parameter estimates are significant positive quantities. The joint confidence region can be thought of as an inverted likelihood-ratio test: it is the set of all the parameter values consistent with the hypothesized model. The MLE of the selection coefficient increased by 142% in the selection-alone model compared to its estimated value using the full model. This increase compensates for the absence of mutation in this model. When considering mutation alone in the model, the MLE of the mutation rate increased by 303% from its value calculated using the full model. The third parameter, the initial fraction of mutants β0, was estimated under each model and also found to be significantly different from zero (Table 2).
TABLE 2.
Maximum-likelihood parameter estimators and their 95% confidence intervals for each model
| Selection and mutation
|
Selection alone
|
Mutation alone
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | 2.5% | MLE | 97.5% | 2.5% | MLE | 97.5% | 2.5% | MLE | 97.5% |
| β0 | 4.23 × 10−10 | 1.29 × 10−3 | 3.00 × 10−3 | 1.69 × 10−3 | 2.88 × 10−3 | 4.36 × 10−3 | 1.47 × 10−10 | 9.95 × 10−4 | 2.48 × 10−3 |
| σ | 3.48 × 10−3 | 6.99 × 10−3 | 1.02 × 10−2 | 8.20 × 10−3 | 9.95 × 10−3 | 1.20 × 10−2 | — | 0.00 | — |
| λ | 2.91 × 10−6 | 3.11 × 10−5 | 6.22 × 10−5 | — | 0.00 | — | 7.39 × 10−5 | 9.41 × 10−5 | 1.14 × 10−4 |
The lower and upper confidence limits are found by taking the 2.5 and 97.5% percentiles, respectively, of the bootstrap distribution under each model. MLE, maximum-likelihood parameter estimator; β0, initial fraction of mutants; σ, selection coefficient; λ, mutation rate.
Figure 2.—

Plot of the joint 94.8% bootstrapped confidence region for both the mutation rate λ and the selection coefficient σ. The solid diamonds represent the joint parameter estimate of the mutation rate λ and the selection coefficient σ.
Testing the models:
Utilizing the MLEs and each of the three models, we calculated the average proportion of the mutants at the end of each of the sampled generations (Table 1). These predictions were used to construct Figure 3. The closer the dispersion of the circles around the curves in Figure 3A or the diagonal solid line in Figure 3B, the better the fit. On the basis of this, both the mutation and the selection and the selection-alone models seem to be fitting the data better than the mutation-alone model, which drastically underestimated the mean mutant proportions toward the end of the experiment. Table 3 introduces the bootstrapped likelihood-ratio results for both the absolute goodness-of-fit tests and the model-comparison tests. The P-value of the absolute goodness-of-fit tests confirmed that the proposed full and selection-alone models fit the data quite well, while the mutation-alone model (P-value = 0.0065) does not. The P-values associated with testing the selection-alone and the mutation-alone models against the full model were 0.0025 and 0, respectively. This provides clear statistical evidence that the full model is significantly better than the other two simple models. This important result suggests that both selection and mutation, together, played a significant role in the observed evolutionary process. Figure 4 provides a visual examination of the absolute goodness-of-fit results.
Figure 3.—

Population dynamics of the tetracycline-sensitive mutants. (A) Time course of the observed (•) and predicted (—) average fractions of mutants under each of the three mathematical models. (B) Ratios of observed vs. predicted fractions of mutants under each model. The solid lines indicate identical observed and predicted values. All replicate and average values are reported in Table 1.
TABLE 3.
Parametric bootstrap maximum-likelihood-ratio tests
| Model | −2 ln(Λ) | P-value |
|---|---|---|
| Mutation and selection | 59.169 | 0.3395 |
| Selection alone | 63.551 | 0.2440 |
| Mutation alone | 73.848 | 0.0650 |
| Selection vs. mutation and selection | 4.381 | 0.0025 |
| Mutation vs. mutation and selection | 14.678 | 0.0000 |
In the first three rows, the P-value is an estimate of the probability that a −2 ln Λ value greater than or equal to the one observed would actually occur given that the hypothesized model is true. For the last two rows, it gives an estimate of the probability that a difference in model scores would be as large as or larger than the one actually seen if the data came from the selection-alone and mutation-alone models, respectively.
Figure 4.—

Pictorial representation of the bootstrap likelihood-ratio test (LRT) outcomes for each mathematical model. The plotted density is the bootstrap sampling distribution of the LRT, −2 ln Λ. The P-value (pBoot) is the area under the curve to the right of the dashed vertical lines (corresponding to the −2 ln Λ observed value). It represents the probability that a LRT value greater than or equal to the one observed would actually occur given that the hypothesized model is true.
The bootstrap vs. the asymptotic theory:
Under certain regularity conditions that guarantee that the MLEs are asymptotically normally distributed, the −2 ln(Λ) test statistic (plotted in Figure 4 for each model) is χ2 distributed in the limit as the sample size increases (Bickel and Doksum 1977; Self and Liang 1987; Bain and Engelhardt 1991). Accordingly, it is tempting to use the asymptotic theory rather than the bootstrap in hypothesis testing; the χ2 distribution is readily tabulated and only degrees of freedom have to be known to find the P-values. However, our simulations definitely showed that the asymptotic χ2 distribution was not valid (results not shown). The use of the χ2 distribution resulted in overly conservative incorrect P-values and hence a higher tendency to reject the null when true (higher type I error). Moreover, the selection-alone model lends itself to a special boundary problem when the mutation rate is set to zero as the mutation rate cannot be negative. The asymptotic distribution in this case is not the regular χ2 distribution under the general theory but a tighter mixed distribution (Self and Liang 1987). The parametric bootstrap approach does not rely on asymptotic theory and provides the correct sampling distributions under the model assumptions.
DISCUSSION
We observed a gradual increase in the frequency of a tetracycline-sensitive subpopulation during experimental evolution of E. coli K12(pB10) in the absence of antibiotics and showed that this phenotypic change was caused by deletion of the plasmid-encoded tet operon. The extent to which these tetracycline-sensitive mutants sweep through a population and replace the residing resistant clones was determined by the mutation rate and the selection coefficient associated with this phenotypic and genotypic change. Therefore we estimated the values of these two parameters by mathematically modeling the dynamics of the tetracycline-sensitive subpopulation. Such a modeling approach has not been used previously in similar studies (Godwin and Slater 1979; Turner et al. 1998; Dahlberg and Chao 2003), where sweeps of antibiotic-sensitive mutants were observed during experimental evolution. We report here a very high mutation frequency in agreement with the nature of the mutation and a low selection coefficient that could not be detected by means of competition experiments. Rejecting both the mutation-alone and selection-alone models with high statistical significance in favor of the full model (mutation and selection) highlighted the strong impact of both mutation and selection together on the dynamics of the sensitive subpopulation.
As confirmed by sequencing, the loss of tetracycline resistance in all sensitive clones examined can be attributed to a recombination event between two direct repeats present on the plasmid that are flanking the tet operon. According to the parameter estimates of the “mutation and selection” model, the frequency of the deletion of the tet operon was determined to be 3.11 × 10−5/generation. This value corresponds fairly well with the estimate we obtained experimentally by determining the fraction of TcS mutants in a colony grown overnight on LB agar, founded by a cell from an overnight culture of K12(pB10) in LBTc. Out of 941 clones screened within the colony, 3 had lost the tet operon after ∼26 generations [= log2(108)], yielding a deletion frequency of ∼1.2 × 10−4/generation. This high mutation frequency is due to the specific nature of the mutation, namely a recombination event. In general, recombination frequencies have been reported from as high as 10−3 to as low as 10−11 (Dianov et al. 1991; Mazin et al. 1991; Lovett et al. 1994; Bi and Liu 1996) depending on the length of the direct repeats and the distance between them. Considering the long direct repeats (863 bp) flanking the tet operon and the large fragment being deleted (5.5 kb), the estimated high mutation frequency of 3.11 × 10−5/generation is in agreement with the range of values previously described. Thus, in the absence of antibiotics, this fragment containing the tet operon was systematically lost at a fairly high rate, causing a tetracycline-sensitive subpopulation to emerge and to expand initially according to a linear pattern. In the absence of selection, continuous deletion events would result in a steady increase of the TcS population as described by the “mutation-alone” model (Figure 3A).
In addition to deletions of resistance genes from a plasmid, the loss of the entire plasmid itself could also cause resistance to decline in a bacterial population. Although the fitness cost of carrying the plasmid pB10 was estimated to be 0.02 (our unpublished data), only one plasmid-free segregant out of 7086 screened clones was detected during the 500 generations of evolution. This indicates that the plasmid was very stably maintained, which is a known characteristic for IncP-1β plasmids (Thomas 2004), or that plasmid-free segregants were rapidly reinfected by plasmid-carrying cells (Thomas 2004). A similar observation has recently been made by Dahlberg and Chao (2003), where during 1100 generations no plasmid-free cells were ever detected in R1- or RP4-bearing populations under antibiotic-free conditions, although a large fitness cost for carrying the plasmid had been determined. These results suggest that even though resistance plasmids confer a cost to their host, plasmid-free segregants will not necessarily form and thus cannot sweep through the population, thereby lowering the level of resistance. It also follows that the loss of one resistance gene on a multiresistance plasmid such as pB10 may still leave the host with other plasmid-encoded resistance determinants.
In competition experiments, we did not detect a significant fitness advantage for the ancestral E. coli K12 carrying a deleted evolved plasmid compared to carrying the ancestral plasmid. On the other hand, the parameter estimate of the selection coefficient under the mutation and selection model was determined to be 0.007 and statistically significantly different from zero. Selection coefficients of <1% are considered to be quite low and difficult or even impossible to detect with statistical significance in a competition experiment (Levin et al. 1997; Andersson and Levin 1999). This is probably due to experimental error and stochasticity, which introduces considerable variation between replicate competition experiments, lowering the statistical power of the test. Ideally, competition experiments are conducted only for one growth cycle (Lenski et al. 1991; Dahlberg and Chao 2003), although an increased sensitivity can be obtained by measuring fitness over several cycles (Dahlberg and Chao 2003). However, in the latter case, mutations, not just those at the tet locus but also in the chromosome, could be a confounding factor that can influence the competition process in the long run. Both the mutant and ancestral populations would continue to evolve during a long competition experiment and one would have a difficult time trying to demonstrate that the differences in fitness were due to the original mutation, i.e., the tet deletion. In our study competition experiments were already run for three growth cycles, or 24 generations, and extending this period is thus not recommended. In our view the strength of the modeling effort lies in its ability to produce a better way of detecting small fitness differences between evolved and ancestral strains compared to a competition experiment. This was achieved by reproducing the observed patterns on the basis of hypothesized processes (mutation and selection acting together) and provided appropriate explanations and predictions. Furthermore, the proposed processes were adequately tested statistically.
The small cost imposed by the tetracycline-resistance determinant encoded on plasmid pB10 may be explained by the presence of the repressor TetR, which represses expression of tetA in the absence of tetracycline. This is in agreement with previous studies, which concluded that deletion of a constitutively expressed plasmid-encoded tet operon drastically increased host fitness in the absence of tetracycline (Modi et al. 1991), but that carrying an inducible tetracycline-resistance operon imposes essentially no burden (Lee and Edlin 1985; Nguyen et al. 1989).
The rigorous combination of mathematical modeling, statistical methods, and biological experiments, applied in this work, is rarely seen in our field. Lenski et al. (1991) used a regression framework to fit an evolutionary model to their data, including only selection, and compared the differences between the fitnesses of their bacterial lineages using ANOVA. They also introduced models to explain the dynamics of their experiments that included the mutation rate although did not estimate parameters for these models using the data. Instead, they used their fitness estimate and an estimate of the effective population size to infer the mutation rate associated with some of these models. Austin et al. (1999) have fit a model to data on occurrence of resistance levels in hospitals and estimated the corresponding parameters, using weighted least squares, but they did not thoroughly evaluate its goodness of fit, nor did they test the significance of their parameters. In contrast, we used the method of maximum likelihood (Fisher 1922) to fit our models and estimated the associated parameters on the basis of the experimental data. Likelihood-ratio tests and the parametric bootstrap were used to evaluate the goodness of fit and to quantify our confidence in these models. Many studies present mathematical models that describe evolutionary processes involving bacteria (Bonhoeffer et al. 1997; Levin et al. 1997; Otto and Whitlock 1997; Bergstrom et al. 2000; Wahl and Krakauer 2000; Wahl and Gerrish 2001; Johnson and Barton 2002; Martiel 2002; Wahl et al. 2002). In general, such studies focus more on presenting the model itself and often do not fit experimental data to the model. In the few cases where these studies involve data (Levin et al. 1997; Wahl and Krakauer 2000; Wahl et al. 2002), these are usually used to compare against the results of the suggested model. This is quite different from our approach where we formally fitted the model to the data.
In response to the widespread emergence of multiresistant pathogens, a number of measures have been proposed to reverse this trend, including cycling different antibiotics and reductions of antibiotic use (Levy 1994; OTA 1995; WHO 1995; Bonhoeffer et al. 1997; Lenski 1997; WHO 2003). It is believed that this will allow sensitive clones with a selective advantage to sweep through the population and thereby eliminate their resistant counterpart (Levin et al. 1997) as seen by Modi et al. (1991) in evolution experiments. Our study shows that such elimination of drug resistance proceeds very slowly when the cost of resistance is very small. Yet even such a small selective advantage is of great importance in the long run, as shown by comparison of predictions made by the mutation-alone and the full model (Table 4). The mutation-alone model predicts that even with the high estimated mutation rate, replacing 99.9% of the resistant population with TcS mutants would take between 21 and 32 years at eight generations per day. However, when including the small drug-resistance cost (σ = 0.007) in the mutation and selection model, such a replacement of 99.9% would take only between 0.55 and 1.83 years. To reach the same replacement level in 5 weeks on average, the resistance cost should be at least 0.06, which is realistic for a constitutively expressed tetracycline operon on a high-copy-number plasmid (Modi et al. 1991), but not for an induced resistance on a low-copy-number broad-host-range plasmid. Our findings thus indicate once again that the cost of resistance is the key factor to successfully displace antibiotic-resistant populations with sensitive ones (Spratt 1996; Lenski 1997; Levin 2001), and that the time required for this, after ending the drug treatment, may be very long. Moreover, even with 0.0001% of the resistant population still present in the environment, which would take ∼1.3 years (Table 4), the reintroduction of the antibiotic will cause the resistant bacteria to ascend to high frequencies again, at a much higher pace than the original decline (Levin et al. 1997; Austin et al. 1999; Heinemann et al. 2000). Thus, as a guideline for designing new antibiotics, our results support the statement made earlier (Bjorkman and Anderson 2000) that the cost for bacteria to acquire and maintain resistance to drugs should be as high as possible, and that models can be used to estimate this cost.
TABLE 4.
Long-term predictions for the decline of the tetracycline-resistant population
| Mutation alone
|
Mutation and selection
|
||||||
|---|---|---|---|---|---|---|---|
| Time (yr) to replace
|
Time (yr) to replace
|
||||||
| λ | 99.9% | 99.9999% | λ | σ | 99.9% | 99.9999% | |
| 1st CL | 7.39 × 10−5 | 32.01 | 64.02 | 2.91 × 10−6 | 0.00348 | 1.83 | 2.81 |
| MLE | 9.40 × 10−5 | 25.14 | 50.28 | 3.10 × 10−5 | 0.00698 | 0.83 | 1.31 |
| 2nd CL | 1.14 × 10−4 | 20.75 | 41.50 | 6.22 × 10−5 | 0.01020 | 0.55 | 0.88 |
Predicted time required to displace the resistant population to 1/1000 (99.9% sensitive) and to 1/1,000,000 (99.9999% sensitive) under the mutation-alone model and the mutation and selection model is shown. 1st CL and 2nd CL represent the limits of a 95% confidence interval. MLE is the maximum-likelihood estimator.
We realize that our model simulates the population dynamics only under the conditions of the evolution experiment, and that predictions made for time points far away from our experimental data should be interpreted with caution. However, as stated by Bruce Levin (Lenski 1997, p. 149), “mathematical models with empirical estimates of their parameters” are the only way “to predict the rate of descent of resistance genes and plasmids” and “should be employed to develop antibiotics use policy” instead of “irresponsible non-quantitative yak-yak.” Our modeling approach with empirical estimates of parameters is a step in this direction that is unique among the studies that have observed sweeps of antibiotic-sensitive mutants under laboratory conditions (Godwin and Slater 1979; Modi et al. 1991; Turner et al. 1998; Dahlberg and Chao 2003).
We aim to expand our methodology of modeling and testing to study other experimental evolutionary systems. To do so we will consider other factors that may affect the evolutionary process and are unaccounted for in our current modeling, such as compensatory mutations (Böttger et al. 1998; Bjorkman and Anderson 2000; Levin et al. 2000; Normark and Normark 2002), background selection on the host and/or on the plasmid, plasmid loss, and horizontal plasmid transfer. With our current model these effects would likely add noise across lines in other experiments. A statistical test to detect this added noise is included in the user manual for our program.
In conclusion, we have shown that a tetracycline-sensitive subpopulation of E. coli K12(pB10) emerged during evolution in an antibiotic-free medium due to deletion of the tet operon. A mathematical model helped explain the population dynamics, estimating a high mutation rate and a low but significant selection coefficient, and allowed us to make long-term predictions regarding the elimination of antibiotic resistance in the population. The mathematical modeling and statistical analyses thus worked in perfect symbiosis with the experimental aspects of this study. The predictions call into question the general effectiveness of drug cycling as a strategy to eradicate resistance. Although we do not want our observations to be interpreted as arguments for the futility of reduced and more prudent drug use, we caution against too much optimism regarding eliminating resistance to antibiotics by drug cycling.A
Acknowledgments
We thank two anonymous reviewers for useful comments and suggestions. We thank Holger Heuer for the primer design and Mayee Wong for help with sequencing. Furthermore, we thank Stacey Poler, Whitney Weibler, Erin Quinn, and Monica Flory for their technical assistance. This publication was made possible by National Institutes of Health (NIH) grant P20 RR 16448 from the Centers of Biomedical Research Excellence Program of the National Center for Research Resources. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH. Paul Joyce is also funded by the National Science Foundation (NSF DEB-0089756 and NSF DMS-0072198).
APPENDIX A
IMPORTANT RECURSION
appendix B relies heavily on the solution of the following recursion in deriving the models used in this article:
 |
A1 |
It follows by induction that
 |
implying that
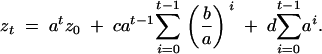 |
Using a geometric series identity (Ross 1991, p. 69, Equation 1) results in
 |
A2 |
APPENDIX B
DERIVATION OF EQUATIONS 6, 7, AND 8
Let the average number of mutants at a certain time t be mt and the average of the remaining nonmutant individuals in period t be nt. Therefore, the total population at time t is the sum of mt and nt. We relist Equations 1, 2, and 3 for convenience:
 |
1 |
 |
2 |
 |
3 |
To solve Equation 3, which represents the proportion of mutants at time t, we need to solve the recursions in Equations 1 and 2. The solution of Equation 2 is obvious:
 |
B1 |
To solve Equation 1 we start by substituting Equation B1 in it to get
 |
B2 |
Note that this is the same format as that of Equation A1 with a = 21+σ, b = 2(1 − λ), and c = 2λn0. Therefore, we use Equation A2 to solve Equation B2:
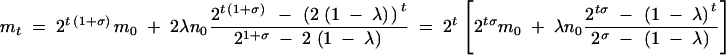 |
B3 |
Using Equations B1 and B3 we can now solve Equation 3. We first note that
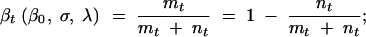 |
B4 |
hence,
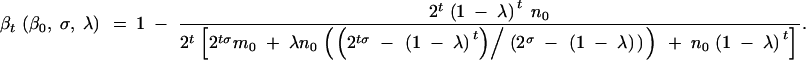 |
Rearranging,
 |
Noting that β0 = m0/(m0 + n0), by definition, results in
 |
Rearranging again,
 |
B5 |
which is equivalent to Equation 6 when replacing t with lk.
APPENDIX C:
DERIVATION OF THE ABSOLUTE GOODNESS-OF-FIT LIKELIHOOD-RATIO TEST STATISTIC
The absolute-best Poisson model fits the data exactly and, hence, exactly predicts the number of mutants in a sample. Such a model has a parameter for each replicate, i, and at each cycle we sample, k. We call this parameter μik, corresponding to the expected number of mutants in the current replicate i at the kth cycle. The likelihood-ratio test statistic Λ associated with this absolute goodness-of-fit test is
 |
C1 |
 |
C2 |
C2 holds as the maximum for the numerator is attained at the maximum-likelihood estimate β̂lk, and the maximum for the denominator holds when the data are predicted exactly. Some algebraic manipulation results in Equation 10:
 |
10 |
References
- Andersson, D. I., and D. Hughes, 1996. Muller's ratchet decreases fitness of a DNA-based microbe. Proc. Natl. Acad. Sci. USA 93: 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, D. I., and B. R. Levin, 1999. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2: 489–493. [DOI] [PubMed] [Google Scholar]
- Austin, D. J., K. G. Kristinsson and R. M. Anderson, 1999. The relationship between the volume of antimicrobial consumption in human communities and the frequency of resistance. Proc. Natl. Acad. Sci. USA 96: 1152–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain, L. J., and B. Engelhardt, 1991 Introduction to Probability and Mathematical Statistics. Duxbury Press, Belmont, CA.
- Bergstrom, C. T., M. Lipsitch and B. R. Levin, 2000. Natural selection, infectious transfer and the existence conditions for bacterial plasmids. Genetics 155: 1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, X., and L. F. Liu, 1996. A replication model for DNA recombination between direct repeats. J. Mol. Biol. 256: 849–858. [DOI] [PubMed] [Google Scholar]
- Bickel, P. J., and K. A. Doksum, 1977 Mathematical Statistics: Basic Ideas and Selected Topics. Holden-Day, Oakland, CA.
- Bjorkman, J., and D. I. Anderson, 2000. The cost of antibiotic resistance from a bacterial perspective. Drug Resist. Upd. 3: 237–245. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer, S., M. Lipsitch and B. R. Levin, 1997. Evaluating treatment protocols to prevent antibiotic resistance. Proc. Natl. Acad. Sci. USA 94: 12106–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttger, E. C., B. Springer, M. Pletschette and P. Sander, 1998. Fitness of antibiotic-resistant microorganisms and compensatory mutations. Nat. Med. 4: 1343–1344. [DOI] [PubMed] [Google Scholar]
- Bouma, J. E., and R. E. Lenski, 1988. Evolution of a bacteria/plasmid association. Nature 335: 351–352. [DOI] [PubMed] [Google Scholar]
- Cohen, M. L., 1992. Epidemiology of drug resistance: implications for a post-antimicrobial era. Science 257: 1050–1055. [DOI] [PubMed] [Google Scholar]
- Dahlberg, C., and L. Chao, 2003. Amelioration of the cost of conjugative plasmid carriage in Escherichia coli K12. Genetics 165: 1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov, G. L., A. V. Kuzminov, A. V. Mazin and R. I. Salganik, 1991. Molecular mechanisms of deletion formation in Escherichia coli plasmids. I. Deletion formation mediated by long direct repeats. Mol. Gen. Genet. 228: 153–159. [DOI] [PubMed] [Google Scholar]
- Drake, J. W., 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 88: 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake, J. W., B. Charlesworth, D. Charlesworth and J. F. Crow, 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge, M., A. Pühler and W. Selbitschka, 2000. Phenotypic and molecular characterization of conjugative antibiotic resistance plasmids isolated from bacterial communities of activated sludge. Mol. Gen. Genet. 263: 471–482. [DOI] [PubMed] [Google Scholar]
- Efron, B., and R. J. Tibshirani, 1993 An Introduction to the Bootstrap. Chapman & Hall, New York.
- Enderle, P. J., and M. A. Farwell, 1998. Electroporation of freshly plated Escherichia coli and Pseudomonas aeruginosa cells. BioTechniques 25: 954–958. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A., 1922. On the mathematical foundations of theoretical statistics. Philos. Trans. R. Soc. Lond. Ser. A 222: 309–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores, M., P. Mavingui, X. Perret, W. J. Broughton, D. Romero et al., 2000. Prediction, identification, and artificial selection of DNA rearrangements in Rhizobium: toward a natural genomic design. Proc. Natl. Acad. Sci. USA 97: 9138–9143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons, A., 1992. Exploring new strategies to fight drug-resistant microbes. Science 257: 1036–1038. [DOI] [PubMed] [Google Scholar]
- Godwin, D., and J. H. Slater, 1979. The influence of the growth environment on the stability of a drug resistance plasmid in Escherichia coli K12. J. Gen. Microbiol. 111: 201–210. [DOI] [PubMed] [Google Scholar]
- Heinemann, J. A., R. G. Ankenbauer and C. F. Amabile-Cuevas, 2000. Do antibiotics maintain antibiotic resistance? Drug Discov. Today 5: 195–204. [DOI] [PubMed] [Google Scholar]
- Helling, R. B., T. Kinney and J. Adams, 1981. The maintenance of plasmid-containing organisms in populations of Escherichia coli. J. Gen. Microbiol. 123: 129–141. [DOI] [PubMed] [Google Scholar]
- Johnson, T., and N. H. Barton, 2002. The effect of deleterious alleles on adaptation in asexual populations. Genetics 162: 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce, P., Z. Abdo, J. Ponciano, L. De Gelder, L. J. Forney et al., 2005. Modeling the impact of periodic bottlenecks, unidirectional mutation, and observational error in experimental evolution. J. Math. Biol. (in press). [DOI] [PubMed]
- Lee, S. W., and G. Edlin, 1985. Expression of tetracycline resistance in pBR322 derivatives reduces the reproductive fitness of plasmid-containing Escherichia coli. Gene 39: 173–180. [DOI] [PubMed] [Google Scholar]
- Lenski, R. E., 1997 The cost of antibiotic resistance—from the perspective of the bacterium, pp. 131–151 in Antibiotic Resistance: Origins, Evolution, Selection and Spread, edited by S. B. Levy. John Wiley & Sons, Chichester, UK.
- Lenski, R. E., M. R. Rose, S. C. Simpson and S. C. Tadler, 1991. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am. Nat. 138: 1315–1341. [Google Scholar]
- Lenski, R. E., S. C. Simpson and T. T. Nguyen, 1994. Genetic analysis of a plasmid-encoded, host genotype-specific enhancement of bacterial fitness. J. Bacteriol. 176: 3140–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, B. R., 2001. Minimizing potential resistance: a population dynamics view. Clin. Infect. Dis. 33(Suppl 3): S161–S169. [DOI] [PubMed] [Google Scholar]
- Levin, B. R., M. Lipsitch, V. Perrot, S. J. Schrag, R. Antia et al., 1997. The population genetics of antibiotic resistance. Clin. Infect. Dis. 24(Suppl 1): S9–S16. [DOI] [PubMed] [Google Scholar]
- Levin, B. R., V. Perrot and N. Walker, 2000. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154: 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, S. B., 1994. Balancing the drug-resistance equation. Trends Microbiol. 2: 341–342. [DOI] [PubMed] [Google Scholar]
- Lovett, S. T., T. J. Gluckman, P. J. Simon, V. A. Sutera, Jr. and P. T. Drapkin, 1994. Recombination between repeats in Escherichia coli by a recA-independent, proximity-sensitive mechanism. Mol. Gen. Genet. 245: 294–300. [DOI] [PubMed] [Google Scholar]
- Martiel, J. L., 2002. Transposable elements and fitness of bacteria. Theor. Popul. Biol. 61: 509–518. [DOI] [PubMed] [Google Scholar]
- Mazin, A. V., A. V. Kuzminov, G. L. Dianov and R. I. Salganik, 1991. Molecular mechanisms of deletion formation in Escherichia coli plasmids. II. Deletions mediated by short direct repeats. Mol. Gen. Genet. 228: 209–214. [DOI] [PubMed] [Google Scholar]
- Modi, R. I., and J. Adams, 1991. Coevolution in bacterial-plasmid populations. Evolution 45: 656–667. [DOI] [PubMed] [Google Scholar]
- Modi, R. I., C. M. Wilke, R. F. Rosenzweig and J. Adams, 1991. Plasmid macro-evolution: selection of deletions during adaptation in a nutrient-limited environment. Genetica 84: 195–202. [DOI] [PubMed] [Google Scholar]
- Nelder, J. A., and R. Mead, 1965. A simplex algorithm for function minimization. Comput. J. 7: 308–313. [Google Scholar]
- Nguyen, T. N. M., Q. G. Phan, L. P. Duong, K. P. Bertrand and R. E. Lenski, 1989. Effects of carriage and expression of the Tn10 tetracycline-resistance operon on the fitness of Escherichia coli K12. Mol. Biol. Evol. 6: 213–225. [DOI] [PubMed] [Google Scholar]
- Normark, B. H., and S. Normark, 2002. Evolution and spread of antibiotic resistance. J. Intern. Med. 252: 91–106. [DOI] [PubMed] [Google Scholar]
- OTA, 1995 Impacts of Antibiotic-Resistant Bacteria. Office of Technology Assessment, United States Congress, Washington, DC.
- Otto, S. P., and M. C. Whitlock, 1997. The probability of fixation in populations of changing size. Genetics 146: 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, J. A., 1995 Mathematical Statistics and Data Analysis. Duxbury Press, Belmont, CA.
- Ross, K. A., 1991 Elementary Analysis: The Theory of Calculus. Springer-Verlag, New York.
- Sambrook, J., and D. W. Russel, 2001 Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schlüter, A., H. Heuer, R. Szczepanowski, L. J. Forney, C. M. Thomas et al., 2003. The 64,508 bp IncP-1B antibiotic multiresistance plasmid pB10 isolated from a wastewater treatment plant provides evidence for recombination between members of different branches of the IncP-1B group. Microbiology 149: 3139–3153. [DOI] [PubMed] [Google Scholar]
- Schrag, S. J., V. Perrot and B. R. Levin, 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc. R. Soc. Lond. Ser. B 264: 1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self, S. G., and K. Liang, 1987. Asymptotic properties of maximum likelihood estimation and likelihood tests under nonstandard conditions. J. Am. Stat. Assoc. 398: 605–610. [Google Scholar]
- Spratt, B. G., 1996. Antibiotic resistance: counting the cost. Curr. Biol. 6: 1219–1221. [DOI] [PubMed] [Google Scholar]
- Stanier, R. Y., N. J. Palleroni and M. Doudoroff, 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43: 159–271. [DOI] [PubMed] [Google Scholar]
- Thomas, C. M., 2004 Evolution and population genetics of bacterial plasmids, pp. 509–528 in Plasmid Biology, edited by B. E. Funnell and G. J. Phillips. ASM Press, Washington, DC.
- Turner, P. E., V. S. Cooper and R. E. Lenski, 1998. Tradeoff between horizontal and vertical modes of transmission in bacterial plasmids. Evolution 52: 315–329. [DOI] [PubMed] [Google Scholar]
- Wahl, L. M., and P. J. Gerrish, 2001. The probability that beneficial mutations are lost in populations with periodic bottlenecks. Evolution 55: 2606–2610. [DOI] [PubMed] [Google Scholar]
- Wahl, L. M., and D. C. Krakauer, 2000. Models of experimental evolution: the role of genetic chance and selective necessity. Genetics 156: 1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, L. M., P. J. Gerrish and I. Saika-Voivod, 2002. Evaluating the impact of population bottlenecks in experimental evolution. Genetics 162: 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 1995 Scientific working group on monitoring and management of bacterial resistance of antimicrobial agents. WHO/CDS/BVI 7, World Health Organization, Geneva. [DOI] [PMC free article] [PubMed]
- WHO, 2003 Impact of antimicrobial growth promoter termination in Denmark. WHO/CDS/CPE/ZFK/2003.1, World Health Organization, Geneva.