

Abstract
Oncogenic mutations in RAS or BRAF can drive the inappropriate activation of the ERK1/2. In many cases, tumour cells adapt to become addicted to this deregulated ERK1/2 signalling for their proliferation, providing a therapeutic window for tumour-selective growth inhibition. As a result, inhibition of ERK1/2 signalling by BRAF or MEK1/2 inhibitors is an attractive therapeutic strategy. Indeed, the first BRAF inhibitor, vemurafenib, has now been approved for clinical use, while clinical evaluation of MEK1/2 inhibitors is at an advanced stage. Despite this progress, it is apparent that tumour cells adapt quickly to these new targeted agents so that tumours with acquired resistance can emerge within 6–9 months of primary treatment. One of the major reasons for this is that tumour cells typically respond to BRAF or MEK1/2 inhibitors by undergoing a G1 cell cycle arrest rather than dying. Indeed, although inhibition of ERK1/2 invariably increases the expression of pro-apoptotic BCL2 family proteins, tumour cells undergo minimal apoptosis. This cytostatic response may simply provide the cell with the opportunity to adapt and acquire resistance. Here we discuss recent studies that demonstrate that combination of BRAF or MEK1/2 inhibitors with inhibitors of pro-survival BCL2 proteins is synthetic lethal for ERK1/2-addicted tumour cells. This combination effectively transforms the cytostatic response of BRAF and MEK1/2 inhibitors into a striking apoptotic cell death response. This not only augments the primary efficacy of BRAF and MEK1/2 inhibitors but delays the onset of acquired resistance to these agents, validating their combination in the clinic.
Linked Articles
This article is part of a themed section on Emerging Therapeutic Aspects in Oncology. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-8
Keywords: acquired resistance, apoptosis, cancer, BCL2, BRAF, ERK1/2, MEK1/2, RAS, targeted therapeutics
Introduction
The RAS-regulated RAF-MEK1/2-ERK1/2 signalling cascade is frequently hyperactivated in human cancers due to mutations in BRAF, RAS or receptor tyrosine kinases (RTKs). Activating BRAF mutations, typically BRAFV600E, are found in 60% of melanomas, 30% of thyroid cancers, 10% of colorectal cancers (CRCs) (Davies et al., 2002; Xing, 2005) and almost all hairy cell leukaemias (Tiacci et al., 2011). RAS is the most commonly mutated oncogene in human cancers, being detected in around 90% of pancreatic cancers, 40% of CRC, 20% non-small cell lung cancers (NSCLCs) and 15% of melanomas (Downward, 2003). Tumour cells with mutations that activate ERK1/2 frequently exhibit a high dependence upon, or addiction to, this signalling cascade for proliferation and tumourigenesis (Solit et al., 2006; Davies et al., 2007), and thus targeting components of this pathway is an attractive therapeutic strategy. Accordingly, numerous small molecule inhibitors of RAF and MEK1/2 have been developed. The most advanced of these is the RAF inhibitor vemurafenib (Zelboraf, PLX4032, RG7204), which has been approved for the treatment of melanoma (Bollag et al., 2012). Vemurafenib is effective at inhibiting ERK1/2 in cells expressing mutant BRAF (typically BRAFV600E); however, in cells expressing wild-type BRAF or mutant RAS, RAF inhibitors actually activate ERK1/2 signalling (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Joseph et al., 2010; Poulikakos et al., 2010). Mutant BRAFV600E signals as a RAF-inhibitor sensitive monomer, but conditions that promote RAF dimerization, such as RAS activation, induce ‘paradoxical activation’ of ERK1/2 signalling. Binding of one protomer to a RAF inhibitor within a RAF dimer has been postulated to allosterically transactivate the other protomer, resulting in activation of ERK1/2 (Hatzivassiliou et al., 2010; Poulikakos et al., 2010). This suggests that in BRAFV600E-positive tumours, RAS activity is too low to support the formation of RAF dimers, and consequently BRAFV600E signals as a RAF-inhibitor sensitive monomer (Lito et al., 2012). This selectivity for mutant BRAF means that RAF inhibitors like vemurafenib have a broad therapeutic index but are limited to mutant BRAF-positive cancers. In addition, the activation of ERK1/2 signalling by vemurafenib in cells expressing wild-type BRAF is thought to be responsible for the frequent and often rapid development of previously unsuspected RAS-mutant cutaneous squamous cell carcinoma (SCC) in patients treated with vemurafenib (Su et al., 2012a), as well as recently identified cases of RAS-mutant CRC, colonic adenomas, gastric polyps and leukaemia (Andrews et al., 2012; Callahan et al., 2012; Chapman et al., 2012). These effects are not restricted to vemurafenib and have been observed with other BRAF and RAF inhibitors, such as dabrafenib and sorafenib (Anforth et al., 2012; Arnault et al., 2012). Consequently, ‘paradox breaker’ RAF inhibitors that do not induce paradoxical activation of ERK1/2 in cells with high RAS activity are in development (Ma et al., 2011; Le et al., 2013). One such inhibitor, PLX7904 (or paradox breaker 04), inhibited ERK1/2 as effectively as PLX4720 (a close structural analogue of vemurafenib) in BRAF-mutant melanoma cells, but unlike PLX4720 did not hyperactivate ERK1/2 in melanoma cells expressing mutant NRAS or SCC cells expressing mutant HRAS. In addition, PLX7904 was effective against BRAF-mutant melanoma cells with acquired resistance to vemurafenib mediated by secondary mutation in NRAS (Le et al., 2013).
In contrast to vemurafenib, MEK1/2 inhibitors, such as selumetinib (AZD6244; ARRY-142886), do not induce paradoxical activation of ERK1/2 signalling and are effective against a broader spectrum of tumours, including those driven by mutations in BRAF, KRAS or RTKs. However, the broader action of MEK1/2 inhibitors may result in a narrower therapeutic window when compared with RAF inhibitors that target mutant BRAF only. In contrast to the majority of kinase inhibitors, MEK1/2 inhibitors like selumetinib do not compete with ATP, but instead bind to an allosteric pocket within MEK1/2 (Davies et al., 2007). This is thought to account for the exquisite selectivity of selumetinib for MEK1/2 in preclinical studies, in which ∼40 other kinases (including the closely related MEK5) were not substantially inhibited at 10 μM (Yeh et al., 2007).
Clinical trials have been conducted, or are ongoing, with several MEK1/2 inhibitors, including selumetinib. Phase II trials assessing the efficacy of selumetinib in patients with advanced NSCLC, CRC and pancreatic cancer who had failed first line therapy showed little or no advantage to selumetinib monotherapy versus standard treatment course (Hainsworth et al., 2010; Bennouna et al., 2011; Bodoky et al., 2012). In addition, no significant difference in progression free survival was observed in patients with chemotherapy-naïve advanced melanoma undergoing treatment with selumetinib versus temozolomide (Kirkwood et al., 2012). However, this study and a study of the efficacy of selumetinib in iodine-refractory papillary thyroid cancer indicated that future trials should focus on patients with BRAF mutations (Hayes et al., 2012). Greater success has been achieved using selumetinib in combination with other agents. In a phase II trial of KRAS-mutant advanced NSCLC, selumetinib in combination with docetaxel improved median overall and progression free survival relative to placebo plus docetaxel by approximately twofold (Jänne et al., 2012). There is also evidence that the clinical sensitivity of BRAF-mutant metastatic melanoma is enhanced when selumetinib is combined with either conventional chemotherapeutics or targeted drugs (Patel et al., 2013). Thus, meaningful clinical responses to MEK1/2 inhibitors such as selumetinib are likely to depend on the development of effective combination therapies.
The enrolment criterion for phase II and phase III trials with the BRAF inhibitor vemurafenib included selecting patients with treatment-naïve BRAF-mutant metastatic melanoma. Both the phase II trial and interim analysis of the phase III trial comparing vemurafenib to dacarbazine reported confirmed clinical responses in around 50% of patients, compared with 5% for dacarbazine (Chapman et al., 2011; Sosman et al., 2012). Interim analysis of the phase III trial also reported superior median progression free survival with vemurafenib compared with dacarbazine (5.3 vs. 1.6 months). With these interim results and the conclusion of the phase II trial both demonstrating the substantial clinical efficacy of vemurafenib, the United States Food and Drug Administration recommended that the phase III trial be revised to assume the greater efficacy of vemurafenib over dacarbazine and independent data and safety monitoring board recommended patient crossover from the dacarbazine control arm to vemurafenib (Chapman et al., 2011). This rapid clinical development led to the approval of vemurafenib for the treatment of BRAF-mutant metastatic melanoma in the United States in August 2011 and in the European Union in February 2012.
Activating BRAF mutations occur in around 8–10% of CRCs and correlate with poor prognosis (Richman et al., 2009; Tol et al., 2009; Fariña-Sarasqueta et al., 2010). In comparison to the striking clinical responses seen in melanoma, the response to vemurafenib in a phase I study of patients with BRAF-mutant metastatic CRC was modest (Kopetz et al., 2010). This is consistent with reports that vemurafenib treatment does not lead to sustained suppression of ERK1/2 phosphorylation in BRAFV600E-mutant CRC cells due to epidermal growth factor receptor (EGFR)-mediated reactivation of ERK1/2 (Corcoran et al., 2012; Prahallad et al., 2012).
Despite these advances, the efficacy of BRAF and MEK1/2 inhibitors is limited by the development of acquired resistance that typically results in disease progression 6–7 months after treatment initiation (reviewed in Little et al., 2013). Acquired resistance to BRAF inhibitors can arise through multiple mechanisms, such as switching to ARAF or CRAF (Montagut et al., 2008; Villanueva et al., 2010); up-regulation of alternative MEK1/2 activators (Johannessen et al., 2010); activating MEK1 mutation (Wagle et al., 2011; Greger et al., 2012); expression of BRAFV600E splice variants that preferentially dimerize (Poulikakos et al., 2011); BRAF amplification (Shi et al., 2012); and activating RAS mutations (Nazarian et al., 2010; Greger et al., 2012; Su et al., 2012b) or up-regulation/activation of RTKs (Nazarian et al., 2010; Villanueva et al., 2010; Yadav et al., 2012; Girotti et al., 2013). Interestingly, mutations in BRAF itself, such as those encoding ‘gatekeeper’ mutations that block drug binding, have not been observed in cell lines or patients with acquired resistance to BRAF inhibitors, despite the observation that engineering such mutations within BRAF can confer resistance in vitro (Whittaker et al., 2010). Acquired resistance to MEK1/2 inhibitors may arise through amplification of BRAFT1799A (encodes BRAFV600E) (Corcoran et al., 2010; Little et al., 2011), amplification of KRASG38A (encodes KRASG13D) (Little et al., 2011) or mutations within MEK1 or MEK2 (Emery et al., 2009; Wang et al., 2011; Hatzivassiliou et al., 2012). The majority of these mechanisms serve to reinstate ERK1/2 signalling and cell proliferation in the presence of drug (reviewed in Little et al., 2013). Interestingly, we observed that acquired resistance to selumetinib resulting from BRAF amplification was reversible (Little et al., 2011). Culturing these resistant cells in the absence of drug for several months led to their complete re-sensitization to the anti-proliferative effects of selumetinib, with BRAF expression and phospho-ERK1/2 levels returning to those observed in selumetinib-naïve cells. The mechanisms underlying resistance reversibility are currently unclear and are under investigation, but these results nevertheless suggest that staggered treatment strategies may delay or overcome acquired resistance in this context. A recent study using primary human BRAFV600E-positive melanoma cells that acquire resistance to vemurafenib through elevated BRAF expression has supported this hypothesis (Das Thakur et al., 2013). Dosing these cells intermittently with vemurafenib in xenograft models markedly stalled tumour growth relative to continual treatment. Together, these studies (Little et al., 2011; Das Thakur et al., 2013) suggest that such ‘drug holiday’ strategies with ERK1/2 pathway inhibitors may warrant evaluation in clinical trials.
Inhibition of ERK1/2 signalling in tumour cells addicted to this pathway typically results in G1 cell cycle arrest (Davies et al., 2007; Sale and Cook, 2013). This reflects the pivotal role that ERK1/2 signalling plays in cell cycle progression (Figure 1). Through the activation of AP1 and ETS transcription factors, ERK1/2, in conjunction with ERK1/2-dependent ribosomal protein S6 kinase (RSK) activation, drives transcription of CCND1 (cyclin D1) during the G1 phase of the cell cycle (Meloche and Pouysségur, 2007). CCND1 binds to and promotes activation of CDK4 and CDK6, which in turn phosphorylate and inactivate retinoblastoma protein (RB). RB inactivation alleviates repression of E2F-mediated transcription, thereby permitting expression of many genes important for entry into, and progression through, S phase (Cobrinik, 2005). In addition, ERK1/2-mediated phosphorylation stabilizes MYC (Sears et al., 2000), which in turn increases expression of cyclin D2, CDK4 and CDC25A, and may repress transcription of the cyclin-dependent kinase (CDK) inhibitor p21CIP1, all of which act to promote progression into S phase (Galaktionov et al., 1996; Bouchard et al., 1999; Hermeking et al., 2000; Gartel et al., 2001). Finally, ERK1/2 and RSK may down-regulate the expression and/or promote inactivation of the CDK-inhibitor p27KIP1 (Rivard et al., 1999; Delmas et al., 2001; Fujita et al., 2003). Thus, blockade of ERK1/2 signalling in tumour cells that are addicted to this pathway for proliferation typically promotes G1 cell cycle arrest through the down-regulation of positive cell cycle regulators and the accumulation of negative cell cycle regulators.
Figure 1.
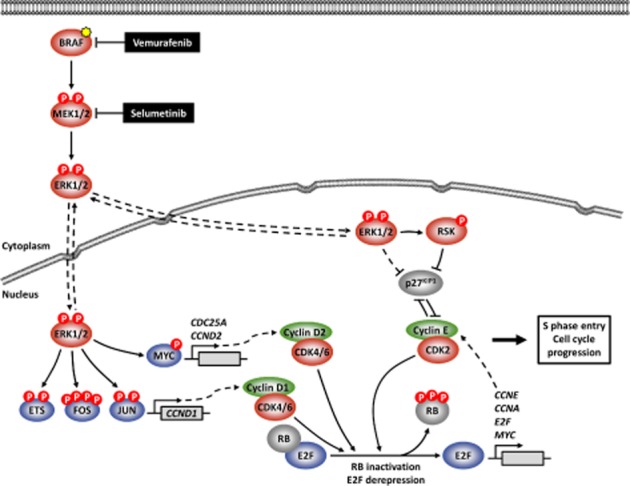
ERK1/2-mediated regulation of the G1-S phase transition. Nuclear ERK1/2 phosphorylates and stabilizes/activates members of ETS and AP1 transcription factor families, which can induce transcription of CCND1 (cyclin D1). The transcription factor MYC is stabilized by ERK1/2-mediated phosphorylation, and MYC can up-regulate the expression of cell cycle regulators such as cyclin D2 CCND2 (cyclin D2) and CDC25A. CCND1 and CCND2 bind to and activate CDK4 and CDK6 (CDK4/6). Phosphorylation of RB by CDK4/6 frees the E2F transcription factors from RB-mediated repression, allowing E2F-induced transcription of genes such as CCNE (cyclin E), CCNA (cyclin A) and MYC. Newly synthesized CCNE binds and activates CDK2, which can also phosphorylate RB in a feedforward loop. p27KIP1, an endogenous inhibitor of CDK2, is down-regulated and inactivated during cell cycle entry by a variety of mechanisms mediated by cyclin E-CDK2, ERK1/2 and RSK. CDK2, initially in complex with CCNE and later with CCNA, and the E2F transcription factors regulate many target factors to drive progression into, and through, S phase. ERK1/2 signalling is frequently hyperactivated in tumour cells as a result of mutations in RTKs, RAS or BRAF (shown, yellow star), providing validation for selective inhibitors of mutant BRAF (e.g. vemurafenib) or MEK1/2 (e.g. selumetinib).
ERK1/2-mediated regulation of the BCL2 protein family
ERK1/2 signalling has been implicated in the regulation of many members of the BCL2 protein family. This regulation typically promotes tumour cell survival through the up-regulation of pro-survival factors and down-regulation of pro-apoptotic BCL2 family members. Consequently, inhibition of ERK1/2 signalling using MEK1/2 or RAF inhibitors generally induces expression of pro-apoptotic BCL2 proteins in tumour cells.
Apoptosis is regulated by the BCL2 protein family
The mitochondrial pathway of apoptosis is regulated by members of the BCL2 protein family (Chipuk et al., 2010). Interactions between these factors ultimately control the integrity of the outer mitochondrial membrane (OMM), thereby determining whether a cell survives or commits to apoptosis. Pro-apoptotic signals converge to promote mitochondrial outer membrane permeabilization (MOMP), which allows the release of soluble proteins resident in the intermembrane space into the cytosol. Most notably, cytochrome c released from mitochondria binds to APAF1, promoting its oligomerization and assembly into the apoptosome. The apoptosome acts as a caspase activation platform by first recruiting pro-caspase-9 and promoting its activation. Active caspase-9 is then able to cleave and activate the executioner caspases, caspase-3 and caspase-7, which cleave a large number of cellular substrates resulting in apoptosis (Tait and Green, 2010).
BCL2 family members are classified as either pro-apoptotic or pro-survival. A1/BFL1, BCL2, BCL-w, BCL-XL and MCL1 are the major pro-survival (or anti-apoptotic) members, and contain four BCL2-homology domains (BH1–4). They largely associate with the OMM and act to inhibit apoptosis by binding to pro-apoptotic factors (Chipuk et al., 2010). The pro-apoptotic members of the BCL2 family are further divided into the BH3-only proteins and the effector proteins. BH3-only proteins are induced by a variety of cellular stresses and include BAD, BID, BIK, BIM, BMF, HRK, NOXA and PUMA. Upon induction, these bind to and inhibit their target pro-survival BCL2 family members. Whereas BIM and PUMA are thought to bind to and inhibit all five major pro-survival factors, most other BH3-only proteins exhibit more restricted binding preferences (Chipuk et al., 2010). The pro-survival proteins are also able to bind and inhibit the effector proteins BAX and BAK. Thus, by binding to the pro-survival factors, BH3-only proteins can displace the effector molecules BAX and BAK, which are then free to undergo further activation events that lead to MOMP. In addition to inhibiting the action of pro-survival factors, certain BH3-only proteins such as BIM and BID may directly activate BAX and BAK (Kuwana et al., 2005; Chipuk et al., 2010). Once activated, BAX and BAK induce MOMP by homo-oligomerizing to form pores within the OMM, thereby allowing the release of soluble factors such as cytochrome c and subsequent formation of the apoptosome (Tait and Green, 2010).
ERK1/2-mediated regulation of pro-apoptotic BH3-only proteins
ERK1/2 signalling is a prominent regulator of apoptosis, and influences the expression and/or activity of many members of the BCL2 protein family (Figure 2; Balmanno and Cook, 2009). At least six of the BH3-only proteins have been proposed to be regulated by ERK1/2 signalling. The potent BH3-only protein BIM, in particular the most abundant extra-long isoform, BIMEL, is an important target of ERK1/2 signalling. Phosphorylation of BIMEL on multiple sites by ERK1/2 targets it for ubiquitination and subsequent proteasome-dependent degradation (Ley et al., 2003). In addition, BIM transcription is positively regulated by FOXO3 (Dijkers et al., 2000), which is itself a target of ERK1/2. ERK1/2-mediated phosphorylation of FOXO3 promotes its MDM2-dependent ubiquitination and degradation by the proteasome, thereby repressing BIM transcription (Yang et al., 2008). Phosphorylation of BIMEL by ERK1/2 has also been shown to rapidly disrupt preformed BIM:BCL-XL and BIM:MCL1 complexes, with dissociated BIMEL then being more rapidly turned over (Ewings et al., 2007). Thus, ERK1/2 activation represses expression of BIMEL protein and mRNA, and promptly impairs its pro-apoptotic activity by preventing association with, and promoting dissociation from, pro-survival factors. Indeed, tumour cells with BRAF mutations are addicted to ERK1/2 signalling for repression of BIM (Wickenden et al., 2008). Consequently, pharmacological inhibition of ERK1/2 signalling induces BIM expression in many contexts. Notably, pretreatment BIM expression levels may be predictive biomarkers for tumour cell responses to some kinase inhibitors (Faber et al., 2011).
Figure 2.
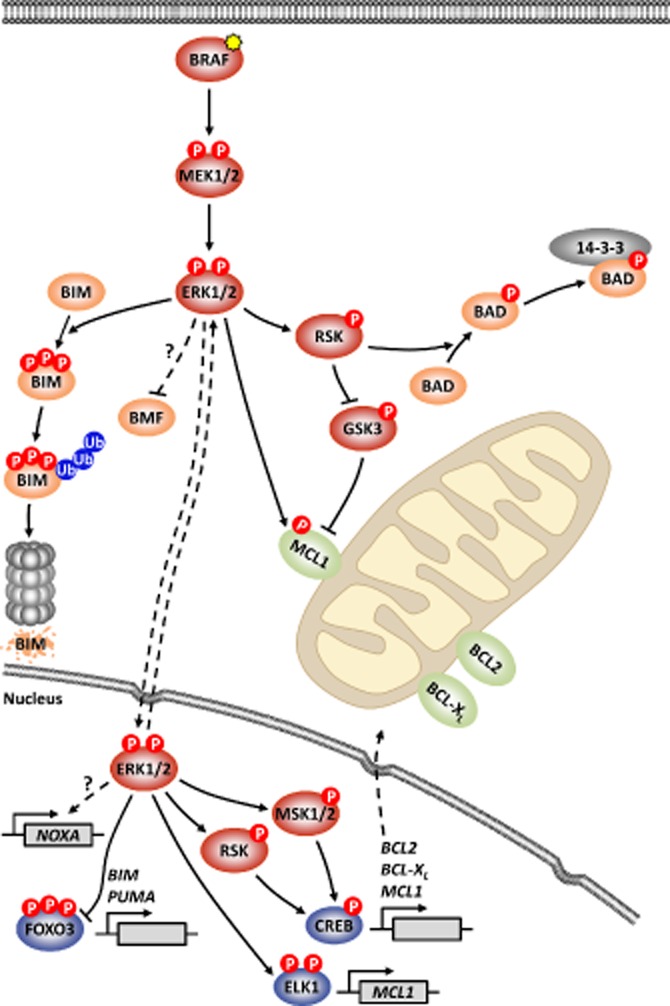
ERK1/2-mediated regulation of the BCL2 protein family. ERK1/2 activation by RTK signalling, mutant RAS or mutant BRAF (shown, yellow star) has pleiotropic effects on the expression and/or activity of BCL2 family members. The BH3-only protein BIM is phosphorylated by ERK1/2 on at least three sites, marking BIM for ubiquitination and subsequent degradation by the 26S proteasome. ERK1/2 also negatively regulates the expression and/or activity of BMF through incompletely understood mechanisms. Activation of RSK by ERK1/2 promotes BAD phosphorylation, which creates a 14-3-3 binding site and sequesters BAD away from the mitochondria. MCL1 stability is subject to reciprocal regulation by the actions of ERK1/2 and GSK3. Phosphorylation of MCL1 by ERK1/2 stabilizes MCL1, whereas GSK3-mediated phosphorylation promotes MCL1 degradation. In the nucleus, ERK1/2 influences the transcription of BCL2 family members. Activation of ERK1/2-dependent RSK and MSK1/2 activates CREB, which promotes transcription of the pro-survival genes BCL2, BCL-XL and MCL1. ELK1 activation by ERK1/2 may also augment MCL1 transcription. ERK1/2 promotes the degradation of FOXO3, thereby inhibiting FOXO3-dependent transcription of pro-apoptotic BIM and PUMA. In contrast to other pro-apoptotic BH3-only proteins, ERK1/2 signalling induces the expression of NOXA mRNA and protein. Thus, with the exception of NOXA, tumour cell ERK1/2 signalling typically promotes the expression of pro-survival factors, and represses the expression and/or activity pro-apoptotic BCL2 family members.
We recently demonstrated that inhibition of ERK1/2 in both KRAS- and BRAF-mutant CRC cells leads to a striking up-regulation of the BH3-only protein BMF (Sale and Cook, 2013). The mechanisms by which ERK1/2 signalling represses BMF expression are unclear, and form the focus of current efforts. ERK2 was recently shown to phosphorylate BMF on two sites; however, it is currently unclear how, or indeed whether, these phosphorylation events influence the function or properties of BMF (Shao and Aplin, 2012). In addition, ERK1/2 signalling can regulate expression of BMF mRNA and BMF localization (VanBrocklin et al., 2009; Shao and Aplin, 2010).
Inhibition of MEK1/2 frequently leads to increased expression of the BH3-only protein PUMA (Wang et al., 2007; Sale and Cook, 2013). PUMA transcription is known to be positively regulated by FOXO3 in response to growth factor or cytokine withdrawal (You et al., 2006). Thus, ERK1/2-dependent modulation of FOXO3 expression may contribute to this up-regulation of PUMA.
Another BH3-only protein, BAD, is phosphorylated at three distinct sites, each acting to inhibit its pro-apoptotic activity. Growth factor-induced phosphorylation of Ser112 was initially demonstrated to be MEK1/2 dependent (Fang et al., 1999; Scheid et al., 1999), and subsequently found to be catalysed by the ERK1/2-regulated kinases RSK and MSK1 (Bonni et al., 1999; Tan et al., 1999; Shimamura et al., 2000; She et al., 2002). Phosphorylation of this site, as well as phosphorylation of Ser136 by PKB (also known as Akt) (Zha et al., 1996; Datta et al., 1997; del Peso et al., 1997), is proposed to inhibit BAD by facilitating binding to 14-3-3 proteins that sequester BAD in the cytosol away from pro-survival BCL2 proteins at the mitochondria (Zha et al., 1996; Tan et al., 1999).
Recently, ERK1/2 was proposed to regulate the stability of the BH3-only protein BIK, in a manner analogous to BIM (Lopez et al., 2012). Direct phosphorylation of BIK on Thr124 by ERK1/2 was suggested to promote ubiquitination and subsequent proteasome-mediated degradation of BIK. Consistent with these observations, the authors demonstrated that MEK1/2 inhibition in tumour cells with BRAF and RAS mutations caused a striking up-regulation of BIK protein. However, we and others have observed little change in BIK expression upon perturbation of ERK1/2 signalling in tumour cells, and the reason for this discrepancy is currently unclear (Sheridan et al., 2008; Sale and Cook, 2013).
In contrast to the above examples, the BH3-only protein NOXA is induced rather than repressed by ERK1/2 signalling (Sheridan et al., 2010; Elgendy et al., 2011; Basile and Aplin, 2012). Inhibition of ERK1/2 down-regulates NOXA protein and mRNA expression, but the underlying mechanisms are unknown (Basile and Aplin, 2012). Why NOXA should exhibit this opposing reciprocal regulation by ERK1/2 signalling is unclear but it may be relevant to the onset of autophagy during oncogene-induced senescence. Strong ERK1/2 signalling induced by conditional overexpression of mutant HRAS increased NOXA expression, which appeared to be involved in the induction of autophagy under these conditions (Elgendy et al., 2011).
ERK1/2-mediated regulation of pro-survival BCL2 family members
The pro-survival factor MCL1 has a short half-life due to the presence of a PEST [peptide sequence that is rich in proline (P), glutamic acid (E), serine (S) and threonine (T)] domain. MCL1 turnover is regulated by phosphorylation of sites within the PEST domain. ERK1/2-mediated phosphorylation of Thr163 within this domain is proposed to stabilize MCL1 (Domina et al., 2004) whereas GSK3-catalysed phosphorylation within the PEST domain enhances MCL1 turnover (Maurer et al., 2006). Thus, inactivation of GSK3 by the ERK1/2-dependent RSK kinases is a further mechanism by which ERK1/2 may promote MCL1 expression. In addition, MCL1 transcription is postulated to be positively regulated by ERK1/2-mediated activation of the transcription factor ELK1 (Townsend et al., 1999; Vickers et al., 2004; Booy et al., 2011).
cAMP responsive element binding protein (CREB) plays a central role in promoting transcription of BCL2, BCL-XL and MCL1 in response to ERK1/2 signalling (Wilson et al., 1996; Wang et al., 1999; Boucher et al., 2000). This is likely mediated by the ERK1/2-dependent kinases RSK and MSK, which can phosphorylate and activate CREB (Bonni et al., 1999).
Tumour cell responses to ERK1/2 pathway inhibitors are typically cytostatic
Broadly, with the exception of NOXA, tumour cell ERK1/2 signalling promotes cell survival by increasing expression of pro-survival proteins while repressing and inactivating pro-apoptotic proteins. Inhibition of ERK1/2 with MEK1/2 or BRAF inhibitors invariably promotes the expression or activation of multiple BH3-only proteins. However, despite strong induction of even the most potent pro-apoptotic proteins such as BIM and PUMA, the predominant response of tumour cells in culture or xenografts to ERK1/2 pathway inhibition is cytostatic rather than cytotoxic; that is, cells undergo G1 cell cycle arrest rather than apoptosis (Davies et al., 2007; Sale and Cook, 2013). This is exemplified by the observation that CCND1 overexpression alone can confer resistance to ERK1/2 pathway inhibitors (Smalley et al., 2008). Pro-survival BCL2 proteins are frequently up-regulated in tumour cells (Kirkin et al., 2004) and, in comparison to the regulation of pro-apoptotic factors such as BIM or BMF, ERK1/2 pathway inhibition often has only modest effects on pro-survival protein levels and/or activity (Sale and Cook, 2013). Thus, in this setting, pro-survival proteins may provide residual buffering capacity against the BH3-only proteins that accumulate upon ERK1/2 inhibition. This provides a rationale for co-targeting ERK1/2 signalling and the pro-survival BCL2 family members, in an attempt to push tumour cells over the threshold required for considerable (i.e. clinically relevant) apoptosis. Over the past 10–15 years, several agents have been developed that inhibit the pro-survival BCL2 family members, with varying degrees of success.
Targeting pro-survival BCL2 family members in cancer
Given that the deregulation of apoptosis in tumour cells typically occurs upstream of BAX and BAK, altering the balance between pro-apoptotic and pro-survival BCL2 family members is an attractive strategy for promoting tumour cell apoptosis. Several molecules have been developed along these lines (reviewed in Chonghaile and Letai, 2008) including the antisense oligonucleotide oblimersen that targets pro-survival BCL2 (Klasa et al., 2002), BH3 peptides, and small molecule mimics of the pro-apoptotic BH3 domain (BH3 mimetics). Oblimersen and the BH3 mimetics obatoclax (GX-15–070) and ABT-263 are the most clinically advanced.
Despite promising preclinical and early clinical trial data, the performance of oblimersen in phase III multiple myeloma and chronic lymphocytic leukaemia (CLL) clinical trials was unconvincing (Chanan-Khan et al., 2009; O'Brien et al., 2009b). These results have been attributed to the inability of oblimersen to reduce BCL2 protein expression in tumours in vivo.
Obatoclax is a putative BH3 mimetic small molecule that exhibits activity against all the pro-survival BCL2 family members, but has modest potency (Zhai et al., 2006). Obatoclax can induce cell death through various mechanisms, some of which are consistent with BH3 mimetic activity but some of which do not require BAX and BAK (Konopleva et al., 2008). Thus, the principal mechanism by which obatoclax induces cell death is uncertain. Obatoclax has shown modest clinical benefit in early stage trials of CLL and small-cell lung cancer (SCLC) (O'Brien et al., 2009a; Paik et al., 2010). However, a phase II study of obatoclax plus topotecan in SCLC showed no improvement in overall response rate compared with that observed with topotecan monotherapy (Paik et al., 2011).
ABT-737 and ABT-263 (navitoclax) are small molecule BH3 mimetics developed by Abbott Laboratories (Abbott Park, IL, USA) which inhibit BCL2, BCL-XL and BCL-w (Oltersdorf et al., 2005; Tse et al., 2008). Although used in the majority of preclinical studies, ABT-737 is not orally bioavailable and so the closely related molecule ABT-263 is being evaluated in clinical trials. ABT-263 is orally bioavailable and has similar affinities and specificities for the pro-survival proteins as ABT-737. In contrast to other putative BH3 mimetics, ABT-737/263-induced cell death absolutely requires BAX or BAK (van Delft et al., 2006), and ABT-737/263 can displace pro-apoptotic factors from pro-survival proteins (Del Gaizo Moore et al., 2007), indicating that ABT-737 and ABT-263 act ‘on target’. In addition, these compounds exhibit 100–10 000-fold greater affinity for their target BCL2 family members than other small molecule inhibitors (Oltersdorf et al., 2005; Zhai et al., 2006; Tse et al., 2008). However, ABT-737 and ABT-263 have low affinity for MCL1, A1/BFL1 and BCL-B. Thus, although there are exceptions, these small molecules are typically most effective at killing cells expressing higher BCL2 and BCL-XL, whereas tumour cells expressing higher MCL1 exhibit intrinsic resistance (Konopleva et al., 2006; van Delft et al., 2006). Consistent with this, acquired resistance to ABT-737 can arise through increased expression of MCL1 and/or A1 (Yecies et al., 2010).
ABT-263 has shown efficacy in early stage clinical trials, particularly for the treatment of CLL. In a phase I study of relapsed or refractory lymphoid malignancies (including CLL) 22% of patients had partial responses (Wilson et al., 2010), and in a phase I study of relapsed or refractory CLL 35% of patients exhibited partial responses (Roberts et al., 2012). In contrast, the results of trials targeting SCLC and other solid tumours have been modest. In a phase II trial assessing the efficacy of ABT-263 against advanced or recurrent SCLC, 1 patient of 39 had a partial response (Rudin et al., 2012). This suggests that, for solid tumours at least, ABT-263 may be best employed as combination therapy with other agents.
Harnessing the apoptotic potential of ERK1/2 pathway inhibitors using BH3 mimetics
Response duration to targeted cancer monotherapies is often short, with acquired resistance rapidly emerging. This is reminiscent of early strategies that targeted rapidly mutating or heterogeneous infectious diseases such as human immunodeficiency virus (HIV) or tuberculosis, in which responses to monotherapies were transient and quickly gave rise to resistant variants (Bock and Lengauer, 2012; Glickman and Sawyers, 2012). However, the use of targeted combination therapies has changed this: antibiotic combinations cure tuberculosis, and treatment of HIV with anti-viral combinations indefinitely suppresses the virus to a chronic low level (Fox et al., 1999; Clavel and Hance, 2004). HIV combination therapy works by rapidly and synergistically reducing viral load relative to single agent therapies, thereby minimizing the pool of viruses from which resistance can develop (Clavel and Hance, 2004; Bock and Lengauer, 2012). In the case of tuberculosis, similar initial responses are achieved regardless of whether treated with a single antibiotic or combination (Fox et al., 1999); importantly, however, combination therapy halts the emergence of resistance that occurs with single agents. Clearly, human cancer biology is far more complex than that of HIV and tuberculosis, but based on this experience, combinations of targeted cancer therapeutics that achieve rapid tumour elimination and suppress the development of acquired resistance will be required for durable clinical responses.
ABT-263 synergizes with ERK1/2 pathway inhibitors to induce substantial tumour cell apoptosis
As discussed above, despite the induction of potent pro-apoptotic BH3-only proteins such as BIM, BMF and PUMA, MEK1/2 and BRAF inhibitors typically induce minimal apoptosis with the predominant response being a G1 cell cycle arrest. However, the BH3-only proteins induced by ERK1/2 inhibition may prime tumour cells for apoptosis. Thus, combining an ERK1/2 pathway inhibitor with other agents that take advantage of this priming event may tip the balance of pro-apoptotic and pro-survival factors in favour of commitment to apoptosis. One such approach is to combine an ERK1/2 pathway inhibitor, such as selumetinib or vemurafenib, with the BH3 mimetic ABT-737 or ABT-263. Scott and colleagues first demonstrated the potential for MEK inhibitors to combine synergistically with BH3 mimetics and induce tumour cell apoptosis (Cragg et al., 2008). Recent studies have now extended these findings using the MEK1/2-selective inhibitor selumetinib in both BRAF- and RAS-mutant cancers, and the BRAF selective inhibitor PLX4720 in BRAF-mutant cancers. Results from our work demonstrated that while treating a variety of CRC tumour cells with selumetinib or ABT-263 alone, induced little cell death (typically < 20% at 48 h post-treatment), strong synergistic cell death and inhibition of clonogenic survival were observed upon treatment with combined selumetinib and ABT-263 (∼60–80% cell death at 48 h). We observed similar results in melanoma cell lines expressing mutant RAS or BRAF, and others have demonstrated that combined MEK1/2 and BCL2/BCL-XL inhibition is frequently effective against KRAS-mutant lung and pancreatic tumour cells (Corcoran et al., 2013; Tan et al., 2013). In all cases examined, cell death required caspase activity and was confined to tumour cells addicted to ERK1/2 signalling; tumour cells with high PI3K-PKB signalling known to be intrinsically resistant to MEK1/2 inhibitors also exhibited resistance to this combination (Cragg et al., 2008; Balmanno et al., 2009; Corcoran et al., 2013; Sale and Cook, 2013). Several reports have demonstrated that PI3K/mTOR inhibitors can also interact synergistically with BH3 mimetics to induce apoptosis in certain contexts (Qian et al., 2009; Spender and Inman, 2012; Rahmani et al., 2013). Thus, combining such agents may be a valid approach for tumour cells with high PI3K-PKB activity and intrinsic resistance to MEK1/2 inhibitors. Furthermore, a recent study has shown that while the novel MEK1/2 inhibitor G-963 synergized to promote apoptosis in the majority of NSCLC and pancreatic cancer cell lines tested, these effects were enhanced by the addition of the PI3K inhibitor GDC-0941 (Tan et al., 2013).
Engelman and colleagues observed that sensitivity to selumetinib plus ABT-263 correlated with the expression of markers of epithelial differentiation, such as E-cadherin (Corcoran et al., 2013). Furthermore, knock-down of ZEB1 in the mesenchymal KRAS-mutant lung cancer cell line A549 promoted expression of epithelial markers and sensitized these cells to both ABT-263 and selumetinib plus ABT-263. This is consistent with many reports demonstrating that epithelial-mesenchymal transition (EMT) promotes resistance to various apoptotic stimuli (Vega et al., 2004; Robson et al., 2006; Arumugam et al., 2009), which has frequently been attributed to PKB activation (Escrivà et al., 2008; Tiwari et al., 2012).
The BRAF inhibitor PLX4720, a close preclinical analogue of vemurafenib (PLX4032), also combined synergistically with ABT-737/263 to induce caspase-dependent cell death and inhibit clonogenic survival of colorectal and melanoma tumour cell lines (Sale and Cook, 2013; Wroblewski et al., 2013). This combination was also effective against cells established from patients with BRAF inhibitor-naïve melanoma (Wroblewski et al., 2013). In addition to these observations in solid tumour types, ERK1/2 pathway inhibitors have also been shown to combine synergistically with ABT-737 to induce the death of acute myeloid leukaemia cells (Zhang et al., 2008; Konopleva et al., 2012).
Combining a MEK1/2 inhibitor and BH3 mimetic also shows potent in vivo efficacy. Xenograft models of the KRAS-mutant CRC cell lines HCT116, SW620 and SW1463 yielded similar results: selumetinib treatment in isolation slowed tumour growth considerably relative to control but did not induce regressions; ABT-263 treatment had little effect on tumour expansion relative to control but combination of selumetinib with ABT-263 caused marked tumour regressions that were sustained for the entirety of the 21–27 day experiments (Corcoran et al., 2013). In addition, generating true in vivo tumours using a genetically engineered KRAS-driven lung cancer mouse model revealed that while selumetinib or ABT-263 led to average tumour regressions of around 30–40% after 2 weeks of treatment, the combination caused 70–80% tumour regression (Corcoran et al., 2013).
ERK1/2 pathway inhibitors combine with ABT-263 to induce BAX- and BIM-dependent apoptosis
The interplay between the pro-survival BCL2 proteins and pro-death BH3-only proteins ultimately serves to regulate the activation of the pro-death effector proteins BAX and BAK. Using isogenic HCT116 cells lacking BAK, BAX or both [double knockout (DKO)] (Wang and Youle, 2012), we found that those lacking BAX (BAX−/− and BAK−/−BAX−/− DKO) were almost completely resistant to apoptosis induced by selumetinib, ABT-263 or selumetinib plus ABT-263, whereas those lacking only BAK remained sensitive. Thus, at least in HCT116 cells, apoptosis in response to these agents absolutely required BAX, whereas BAK was dispensable (Sale and Cook, 2013). Consistent with this, selumetinib and ABT-263 combined to synergistically activate BAX (Sale and Cook, 2013).
Analysing the expression of upstream regulators of BAX in COLO205 cells revealed that selumetinib (alone or in combination with ABT-263) strongly induced the expression of the pro-apoptotic BH3-only proteins BIM and BMF. In these cells, RNAi-mediated knock-down of BIM inhibited apoptosis induced by selumetinib plus ABT-263 by 50–70%, demonstrating that cell death in response to this combination was in large part BIM dependent (Sale and Cook, 2013). However, in other cell types, including HCT116 (KRASG13D), the picture was more complex. As with COLO205 cells, MEK1/2 inhibition in HCT116 cells strongly induced BIM and BMF expression, but in this case PUMA expression also increased and robust knock-down of BIM and/or PUMA did not inhibit apoptosis induced by selumetinib and ABT-263 (Sale and Cook, 2013). Given that cell death was absolutely dependent on BAX, it is likely that alternative BH3-only proteins, such as BMF or BAD, or the reduced expression of pro-survival factors acts in a redundant fashion to promote apoptosis. Knock-down of BIM in the BRAFV600E-positive melanoma cell lines Mel-RMu and SK-MEL-28 inhibited apoptosis induced by the combination of PLX4720 plus ABT-263 by 50–70% (Wroblewski et al., 2013). In these cells, PLX4720 plus ABT-263 also promoted PUMA expression and altered the abundance of pro-survival factors. These effects, in addition to possible up-regulation of BMF, which was not assessed, may contribute to apoptosis in these melanoma cells.
ABT-263 promotes redistribution of selumetinib-induced BH3-only proteins from BCL-XL to MCL1 resulting in stronger inhibition of pro-survival BCL2 proteins
To further define the mechanisms by which selumetinib and ABT-263 synergize to induce apoptosis, we immunoprecipitated BCL-XL and MCL1 from cells treated with selumetinib, ABT-263 or the combination to assess their interaction with BH3-only binding partners. In COLO205 cells, MEK1/2 inhibition strongly promoted accumulation of BIM and BMF and their binding to both BCL-XL and MCL1 (Sale and Cook, 2013). However, in the presence of the BH3 mimetic ABT-263, BIM and BMF no longer bound to BCL-XL, but instead exhibited greater binding to MCL1. This is consistent with the high affinity of ABT-263 for BCL-XL preventing binding of BIM and BMF to BCL-XL, but not MCL1. Very similar effects were observed in HCT116 cells, except in this case the combination of selumetinib and ABT-263 promoted the redistribution of PUMA as well as BIM and BMF onto MCL1 (Sale and Cook, 2013). Thus, while ABT-263 alone can efficiently inhibit BCL-XL but not MCL1, in the presence of selumetinib it promotes the redistribution of the BH3-only proteins BIM, BMF and PUMA from BCL-XL to MCL1, resulting in greater overall inhibition of the pro-survival BCL2 proteins (Figure 3). In addition, while selumetinib only induced partial displacement BAX from BCL-XL (we did not observe BAX binding to MCL1), ABT-263 robustly disrupted this interaction. Thus, in the presence of selumetinib plus ABT-263, BAX may undergo more efficient direct activation by any residual BIM freed up from BCL-XL.
Figure 3.

Selumetinib and ABT-263 synergize to inhibit pro-survival BCL2 family proteins and activate BAX in ERK1/2-addicted tumour cell lines. Treatment of tumour cells with a MEK1/2 inhibitor such as selumetinib invariably induces strong expression of BH3-only proteins such as BIM and BMF (A, top). PUMA expression may also be induced. These BH3-only proteins then bind to pro-survival factors such as BCL-XL and MCL1 (A, bottom). Despite this, little cell death occurs with MEK1/2 inhibition alone, likely due to residual pro-survival activity, including BCL-XL and MCL1 (A, bottom). Addition of the BH3 mimetic ABT-263 (red triangle; B, top) causes a redistribution of selumetinib-induced BIM and BMF from ABT-263 sensitive BCL-XL to ABT-263-resistant MCL1 (B, bottom). Thus, although ABT-263 cannot directly target MCL1, its combination with selumetinib results in indirect inhibition of MCL1 and consequently greater inhibition of pro-survivals (C, top). In addition, whereas selumetinib only resulted in partial displacement of BAX from BCL-XL, ABT-263 efficiently disrupted this interaction. BAX could then potentially be directly activated by any residual BIM freed up from BCL-XL (C, top). Activated BAX can subsequently oligomerize and insert into the outer mitochondrial membrane, resulting in MOMP (C, bottom). This allows cytochrome c release from the intermembrane space, followed by apoptosome formation, caspase activation and consequent apoptosis. (Reproduced with permission from Sale and Cook, 2013, The Biochemical Journal, 450, 285-294 © the Biochemical Society)
ABT-263 can delay acquired resistance to ERK1/2 pathway inhibitors
Tumour cells chronically exposed to ERK1/2 pathway inhibitors evolve to circumvent the G1 arrest induced by these agents, thereby acquiring resistance to their anti-proliferative effects (Little et al., 2013). We reasoned that the weak cell death responses to ERK1/2 pathway inhibitors may simply allow tumour cells greater opportunity to adapt and evolve acquired resistance. Thus, using ABT-263 to boost primary cell death responses to ERK1/2 pathway inhibitors might minimize the residual population of cells from which resistant clones could emerge and/or increase the number of mutations required before a strong selective advantage is gained, in a manner analogous to drug combinations that rapidly reduce HIV viral load and inhibit acquired resistance (Clavel and Hance, 2004; Bock and Lengauer, 2012).
We tested this hypothesis in CRC cells with RAS and BRAF mutations. Treating CRC cells transiently (72 h) with selumetinib plus ABT-263 inhibited the frequency of colonies that subsequently developed acquired resistance to selumetinib over 4–6 weeks by 90–95% (Sale and Cook, 2013). Continual treatment with selumetinib plus ABT-263 over a period of 2 weeks similarly inhibited the frequency of resistant colonies by 95%. This was observed in both KRAS- and BRAF-mutant tumour cells that otherwise rapidly develop 100-fold resistance to selumetinib (Little et al., 2011). These results are consistent with the KRAS-mutant CRC xenograft models discussed above, in which selumetinib plus ABT-263 induced sustained tumour regressions over the course of these 3–4 week experiments (Corcoran et al., 2013). In addition, durable regressions of up to 7 weeks were observed in mutant KRAS-driven lung cancer mouse models (Corcoran et al., 2013).
Thus, these studies provide substantial support for the clinical use of ERK1/2 pathway inhibitors in conjunction with ABT-263. This combination has the potential to augment primary responses and delay acquired resistance, while importantly continuing to harness tumour cell addiction to ERK1/2 signalling.
ABT-263 can overcome established acquired resistance to ERK1/2 pathway inhibitors in some cases
We also examined the effect of ABT-263 treatment on BRAF- or KRAS-mutant CRC cells that had already acquired resistance to selumetinib. In these cells, acquired resistance had arisen through an amplification of the driving oncogene, which acts to reinstate ERK1/2 signalling in the presence of drug (Little et al., 2011). Under their normal growth conditions (in the presence of selumetinib), selumetinib-resistant COLO205 and HT29 cells (driven by BRAF amplification) remained sensitive to ABT-263. In fact, these resistant cells exhibited greater sensitivity to ABT-263 than parental COLO205 or HT29 cells by ∼1.5–2-fold, suggesting they may be ‘primed’ for ABT-263-induced apoptosis. In the absence of selumetinib, however, these cells were resistant to ABT-263 treatment relative to parental cells. Selumetinib removal results in a rapid and sustained hyperactivation of ERK1/2 (Little et al., 2011) which appears to abolish the priming event and diminish sensitivity to ABT-263. In contrast, HCT116 cells with acquired resistance to selumetinib (KRAS amplification) were cross-resistant to ABT-263, regardless of whether selumetinib was present. KRAS amplification in these cells activates PI3K-PKB signalling (Little et al., 2011), which could potentially provide survival signals to promote ABT-263 cross-resistance. However, even when PI3K-PKB and ERK1/2 signalling in selumetinib-resistant HCT116 cells was inhibited to levels seen in parental HCT116, these cells remained less sensitive to ABT-263 than parental HCT116 cells; thus, KRAS amplification conferred resistance to ABT-263 under all conditions. This is consistent with our observations that alternative pathways contribute to acquired resistance in this setting (Little et al., 2011) and underline the challenge faced by KRAS amplification as a mechanism of resistance.
Using melanoma cell lines established from patients pre- and post-vemurafenib treatment, Wroblewski et al. demonstrated that while vemurafenib-naïve cells underwent apoptosis in response to PLX4720 plus ABT-263, cells harvested post-treatment were refractory to this combination. It is currently unclear why some tumour cells with acquired resistance to ERK1/2 pathway inhibitors remain sensitive to ABT-263, while others are resistant. Nevertheless, ABT-263 has the potential to overcome acquired resistance to ERK1/2 pathway inhibitors in some settings.
Conclusions
Oncogene addiction and the therapeutic window that this can provide for tumour selective intervention holds a great promise for the development of new targeted anti-cancer agents. This has been most evident in the success of agents such as imatinib for the treatment of chronic myelogenous leukaemia (CML) (Capdeville et al., 2002), gefitinib and erlotinib, for the treatment of NSCLC with mutant EGFR (Sequist and Lynch, 2008) and vemurafenib, and for the treatment of BRAFV600E-positive melanoma (Chapman et al., 2011). However, acquired resistance to these therapies is a major problem; indeed, the exquisite selectivity of some new targeted agents may actually provide a strong and very focused selection pressure for the rapid emergence of resistance through pathway remodelling (Little et al., 2013). This may be exacerbated by a general failure to sufficiently engage or activate pro-death signalling pathways, including the BCL2 proteins. Indeed, the predominant response to BRAF and MEK1/2 inhibitors is cytostatic rather than cytotoxic. However, addiction to ERK1/2 signalling provides one arm of a synthetic lethal pair that, when combined with BH3 mimetics, results in a strong synergistic tumour cell death that is only observed in ERK1/2-addicted cells. This greatly improves primary efficacy and inhibits and delays the onset of acquired resistance. This strategy is also effective in tumours addicted to breakpoint cluster region/Abelson murine leukaemia viral oncogene and EGFR (Kuroda et al., 2006; Cragg et al., 2007), and so may merit more general consideration as a drug combination that can harness oncogene addiction and transform it into tumour cell-specific cell death.
Acknowledgments
This work was supported by a sponsored research collaboration between AstraZeneca and the Babraham Institute in which AstraZeneca provided selumetinib, AZ628 and AZ12321046 for no charge. M. S. was supported by a BBSRC studentship.
Glossary
- A1
BCL2-related protein A1
- AP1
activator protein 1
- APAF1
apoptotic peptidase activating factor 1
- ARAF
v-raf murine sarcoma 3611 viral oncogene homologue
- BAD
BCL-XL/BCL2-associated death promoter
- BAK
BCL2 homologous antagonist/killer
- BAX
BCL2-associated x protein
- BCL2
B-cell lymphoma 2
- BCL-XL
B-cell lymphoma extra large
- BH3
BCL2 homology domain 3
- BID
BH3 interacting domain death agonist
- BIK
BCL2-interacting killer
- BIM
BCL2-interacting mediator of cell death
- BMF
BCL2-modifying factor
- BRAF
v-raf murine sarcoma viral oncogene homologue B1
- CCND1
cyclin D1
- CDC25A
cell division cycle 25 homologue A
- CDK
cyclin-dependent kinase
- CIP1
CDK-interacting protein 1
- CRAF
v-raf-1 murine leukaemia viral oncogene homologue 1
- CREB
cAMP responsive element binding protein
- DKO
double knockout
- EGFR
epidermal growth factor receptor
- CRC
colorectal cancer
- ELK1
ETS-like gene 1
- EMT
epithelial-mesenchymal transition
- ETS
v-ets erythroblastosis virus E26 oncogene homologue
- FOXO3
forkhead box O 3
- G1
growth phase 1
- GSK3
glycogen synthase kinase 3
- HIV
human immunodeficiency virus
- HRAS
v-Ha-ras Harvey rat sarcoma viral oncogene homologue
- HRK
harakiri, BCL2 interacting protein (contains only BH3 domain)
- KIP1
kinase inhibitory protein 1
- KRAS
v-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue
- MCL1
myeloid cell leukaemia 1
- MDM2
Mdm2, p53 E3 ubiquitin protein ligase homologue (mouse)
- MEK
MAPK or ERK kinase
- MOMP
mitochondrial outer membrane permeabilization
- MSK
mitogen- and stress-activated protein kinase
- mTOR
mammalian target of rapamycin
- MYC
v-myc myelocytomatosis viral oncogene homologue (avian)
- NRAS
neuroblastoma RAS viral (v-ras) oncogene homologue
- NSCLC
non-small cell lung cancer
- OMM
outer mitochondrial membrane
- PDGFR
platelet-derived growth factor receptor
- PI3K
phosphoinositide 3-kinase
- PUMA
p53-up-regulated modulator of apoptosis
- RAS
rat sarcoma virus oncogene
- RB
retinoblastoma protein
- RNAi
RNA interference
- RSK
ribosomal protein S6 kinase
- RTK
receptor tyrosine kinase
- SCLC
small cell lung cancer
- ZEB1
zinc finger E-box binding homeobox 1
Conflict of interest
Payments from AZ were confined to laboratory consumables and associated overheads. Neither S. C. nor M. S. received any personal financial remuneration of any sort from AstraZeneca.
References
- Andrews M, Behren A, Chiohn F, Tebbutt N, Do H, Dobrovic A, et al. Colorectal cancer promoted in a melanoma patient receiving dabrafenib (GSK2118436) in combination with MEK1/2 inhibitor trametinib (GSK1120212) Pigment Cell Melanoma Res. 2012;25:842. [Google Scholar]
- Anforth RM, Blumetti TC, Kefford RF, Sharma R, Scolyer RA, Kossard S, et al. Cutaneous manifestations of dabrafenib (GSK2118436): a selective inhibitor of mutant BRAF in patients with metastatic melanoma. Br J Dermatol. 2012;167:1153–1160. doi: 10.1111/j.1365-2133.2012.11155.x. [DOI] [PubMed] [Google Scholar]
- Arnault JP, Mateus C, Escudier B, Tomasic G, Wechsler J, Hollville E, et al. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res. 2012;18:263–272. doi: 10.1158/1078-0432.CCR-11-1344. [DOI] [PubMed] [Google Scholar]
- Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–377. doi: 10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer. 2009;125:2332–2341. doi: 10.1002/ijc.24604. [DOI] [PubMed] [Google Scholar]
- Basile KJ, Aplin AE. Downregulation of Noxa by RAF/MEK inhibition counteracts cell death response in mutant B-RAF melanoma cells. Am J Cancer Res. 2012;2:726–735. [PMC free article] [PubMed] [Google Scholar]
- Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011;29:1021–1028. doi: 10.1007/s10637-010-9392-8. [DOI] [PubMed] [Google Scholar]
- Bock C, Lengauer T. Managing drug resistance in cancer: lessons from HIV therapy. Nat Rev Cancer. 2012;12:494–501. doi: 10.1038/nrc3297. [DOI] [PubMed] [Google Scholar]
- Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30:1216–1223. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Booy EP, Henson ES, Gibson SB. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene. 2011;30:2367–2378. doi: 10.1038/onc.2010.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher MJ, Morisset J, Vachon PH, Reed JC, Lainé J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-xL, and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–369. [PubMed] [Google Scholar]
- Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316–2321. doi: 10.1056/NEJMoa1208958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Chanan-Khan AA, Niesvizky R, Hohl RJ, Zimmerman TM, Christiansen NP, Schiller GJ, et al. Phase III randomised study of dexamethasone with or without oblimersen sodium for patients with advanced multiple myeloma. Leuk Lymphoma. 2009;50:559–565. doi: 10.1080/10428190902748971. [DOI] [PubMed] [Google Scholar]
- Chapman P, Metz D, Sepulveda A, Uehara T, Rustgi A, Nathanson KL, et al. Development of colonic adenomas and gastric polyps in BRAF mutant melanoma patients treated with vemurafenib. Pigment Cell Melanoma Res. 2012;25:847. [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonghaile TN, Letai A. Mimicking the BH3 domain to kill cancer cells. Oncogene. 2008;27:S149–S157. doi: 10.1038/onc.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. Plos Med. 2007;4:1681–1689. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118:3651–3659. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/ pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–2219. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas C, Manenti S, Boudjelal A, Peyssonnaux C, Eychene A, Darbon JM. The p42/p44 mitogen-activated protein kinase activation triggers p27Kip1 degradation independently of CDK2/cyclin E in NIH 3T3 cells. J Biol Chem. 2001;276:34958–34965. doi: 10.1074/jbc.M101714200. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the proapoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23:5301–5315. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escrivà M, Peiró S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentís B, et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol. 2008;28:1528–1540. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, et al. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 2007;26:2856–2867. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Yu S, Eder A, Mao M, Bast JRC, Boyd D, et al. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]
- Fariña-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VE, Rutten HJ, et al. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol. 2010;21:2396–2402. doi: 10.1093/annonc/mdq258. [DOI] [PubMed] [Google Scholar]
- Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3:S231–S279. [PubMed] [Google Scholar]
- Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278:49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaktionov K, Chen XC, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A. 2001;98:4510–4515. doi: 10.1073/pnas.081074898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–167. doi: 10.1158/2159-8290.CD-12-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MS, Sawyers CL. Converting cancer therapies into cures: lessons from infectious diseases. Cell. 2012;148:1089–1098. doi: 10.1016/j.cell.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–920. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5:1630–1636. doi: 10.1097/JTO.0b013e3181e8b3a3. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Liu B, O'Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11:1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18:2056–2065. doi: 10.1158/1078-0432.CCR-11-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2012;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107:14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, Joos S, Zörnig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta. 2004;1644:229–249. doi: 10.1016/j.bbamcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–567. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasa RJ, Gillum AM, Klem RE, Frankel SR. Oblimersen Bcl-2 antisense: facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002;12:193–213. doi: 10.1089/108729002760220798. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:193–213. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax) Cancer Res. 2008;68:3413–3420. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778–787. doi: 10.1038/leu.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Lee RJ, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28 Abstract 3534. [Google Scholar]
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Le K, Blomain E, Rodeck U, Aplin AE. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013 doi: 10.1111/pcmr.12092. doi: 10.1111/pcmr.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little AS, Balmanno K, Sale MJ, Newman S, Dry JR, Hampson M, et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal. 2011;4:ra17. doi: 10.1126/scisignal.2001752. [DOI] [PubMed] [Google Scholar]
- Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. 2013;32:1207–1215. doi: 10.1038/onc.2012.160. [DOI] [PubMed] [Google Scholar]
- Lopez J, Hesling C, Prudent J, Popgeorgiev N, Gadet R, Mikaelian I, et al. Src tyrosine kinase inhibits apoptosis through the Erk1/2-dependent degradation of the death accelerator Bik. Cell Death Differ. 2012;19:1459–1469. doi: 10.1038/cdd.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Zhang C, Habets G, Burton EA, Wong B, Nguyen H, et al. BRAF inhibitors upregulate EGFR ligands: a molecular link to RAF inhibitor-induced cutaneous squamous cell carcinomas. Mol Cancer Ther. 2011;10:Abstract A229. [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009a;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki AB, Koziner B, et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without Oblimersen. J Clin Oncol. 2009b;27:5208–5212. doi: 10.1200/JCO.2009.22.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Paik PK, Rudin CM, Brown A, Rizvi NA, Takebe N, Travis W, et al. A phase I study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol. 2010;66:1079–1085. doi: 10.1007/s00280-010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik PK, Rudin CM, Pietanza MC, Brown A, Rizvi NA, Takebe N, et al. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer. 2011;74:481–485. doi: 10.1016/j.lungcan.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR, et al. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013;119:799–805. doi: 10.1002/cncr.27790. [DOI] [PubMed] [Google Scholar]
- del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Qian J, Zou Y, Rahman JS, Lu B, Massion PP. Synergy between phosphatidylinositol 3-kinase/Akt pathway and Bcl-xL in the control of apoptosis in adenocarcinoma cells of the lung. Mol Cancer Ther. 2009;8:101–109. doi: 10.1158/1535-7163.MCT-08-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Aust MM, Attkisson E, Williams DC, Jr, Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013;73:1340–1351. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–5937. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- Rivard N, Boucher MJ, Asselin C, L'Allemain G. MAP kinase cascade is required for p27 downregulation and S phase entry in fibroblasts and epithelial cells. Am J Physiol. 1999;277:C652–C664. doi: 10.1152/ajpcell.1999.277.4.C652. [DOI] [PubMed] [Google Scholar]
- Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson EJ, Khaled WT, Abell K, Watson CJ. Epithelial-to-mesenchymal transition confers resistance to apoptosis in three murine mammary epithelial cell lines. Differentiation. 2006;74:254–264. doi: 10.1111/j.1432-0436.2006.00075.x. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale MJ, Cook SJ. The BH3-mimetic ABT-263 synergises with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem J. 2013;450:285–294. doi: 10.1042/BJ20121212. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Schubert KM, Duronio V. Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J Biol Chem. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Lynch TJ. EGFR tyrosine kinase inhibitors in lung cancer: an evolving story. Annu Rev Med. 2008;59:429–442. doi: 10.1146/annurev.med.59.090506.202405. [DOI] [PubMed] [Google Scholar]
- Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Aplin AE. ERK2 phosphorylation of serine 77 regulates Bmf pro-apoptotic activity. Cell Death Dis. 2012;3:e253. doi: 10.1038/cddis.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She QB, Ma WY, Zhong S, Dong Z. Activation of JNK1, RSK2, and MSK1 is involved in serine 112 phosphorylation of Bad by ultraviolet B radiation. J Biol Chem. 2002;277:24039–24048. doi: 10.1074/jbc.M109907200. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Brumatti G, Martin SJ. Oncogenic B-RafV600E inhibits apoptosis and promotes ERK-dependent inactivation of Bad and Bim. J Biol Chem. 2008;283:22128–22135. doi: 10.1074/jbc.M800271200. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Brumatti G, Elgendy M, Brunet M, Martin SJ. An ERK-dependent pathway to Noxa expression regulates apoptosis by platinum-based chemotherapeutic drugs. Oncogene. 2010;29:6428–6441. doi: 10.1038/onc.2010.380. [DOI] [PubMed] [Google Scholar]
- Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Ballif BA, Richards SA, Blenis J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr Biol. 2000;10:127–135. doi: 10.1016/s0960-9822(00)00310-9. [DOI] [PubMed] [Google Scholar]
- Smalley KS, Lioni M, Dalla Palma M, Xiao M, Desai B, Egyhazi S, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7:2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spender LC, Inman GJ. Phosphoinositide 3-kinase/AKT/mTORC1/2 signaling determines sensitivity of Burkitt's lymphoma cells to BH3 mimetics. Mol Cancer Res. 2012;10:347–359. doi: 10.1158/1541-7786.MCR-11-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012a;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012b;72:969–978. doi: 10.1158/0008-5472.CAN-11-1875. [DOI] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tan N, Wong M, Nannini MA, Hong R, Lee LB, Price S, et al. Bcl-2/Bcl-xL inhibition increases the efficacy of Mek inhibition alone and in combination with PI3 Kinase inhibition in lung and pancreatic tumor models. Mol Cancer Ther. 2013 doi: 10.1158/1535-7163.MCT-12-0949. doi: 10.1158/1535-7163.MCT-12-0949. [DOI] [PubMed] [Google Scholar]
- Tan Y, Ruan H, Demeter MR, Comb MJ. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J Biol Chem. 1999;274:34859–34867. doi: 10.1074/jbc.274.49.34859. [DOI] [PubMed] [Google Scholar]
- Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol. 2012;22:194–207. doi: 10.1016/j.semcancer.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–99. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- Townsend KJ, Zhou P, Qian L, Bieszczad CK, Lowrey CH, Yen A, et al. Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J Biol Chem. 1999;274:1801–1813. doi: 10.1074/jbc.274.3.1801. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- VanBrocklin MW, Verhaegen M, Soengas MS, Holmen SL. Mitogen-activated protein kinase inhibition induces translocation of Bmf to promote apoptosis in melanoma. Cancer Res. 2009;69:1985–1994. doi: 10.1158/0008-5472.CAN-08-3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers ER, Kasza A, Kurnaz IA, Seifert A, Zeef LA, O'donnell A, et al. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol. 2004;24:10340–103451. doi: 10.1128/MCB.24.23.10340-10351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Youle RJ. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1's inhibitory effect on Bak. Oncogene. 2012;31:3177–3189. doi: 10.1038/onc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B, et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011;71:5535–5545. doi: 10.1158/0008-5472.CAN-10-4351. [DOI] [PubMed] [Google Scholar]
- Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13:4934–4942. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2:35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- Wickenden JA, Jin H, Johnson M, Gillings AS, Newson C, Austin M, et al. Colorectal cancer cells with the BRAF(V600E) mutation are addicted to the ERK1/2 pathway for growth factor-independent survival and repression of BIM. Oncogene. 2008;27:7150–7161. doi: 10.1038/onc.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B, Mochon E, Boxer L. Induction of bcl-2 expression by phosphorylated CREB proteins during B- cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski D, Mijatov B, Mohana-Kumaran N, Lai F, Gallagher SJ, Haass NK, et al. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis. 2013;34:237–247. doi: 10.1093/carcin/bgs330. [DOI] [PubMed] [Google Scholar]
- Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12:245–262. doi: 10.1677/erc.1.0978. [DOI] [PubMed] [Google Scholar]
- Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem. 2012;287:28087–28098. doi: 10.1074/jbc.M112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Zong CS, Xia WY, Yamaguchi H, Ding QQ, Xie XM, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological Characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 Inhibitor. Clin Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006;13:1419–1421. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- Zhang W, Konopleva M, Ruvolo VR, McQueen T, Evans RL, Bornmann WG, et al. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008;22:808–818. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]