

Abstract
Recently, protein sequence coevolution analysis has matured into a predictive powerhouse for protein structure and function. Direct Coupling Analysis, a method that uses a global statistical model of sequence coevolution, has enabled the prediction of membrane and disordered protein structures, protein complex architectures, and the functional effects of mutations in proteins. The field of membrane protein biochemistry and structural biology has embraced these computational techniques, which provide functional and structural information in an otherwise experimentally-challenging field. Here we review recent applications of protein sequence coevolution analysis to membrane protein structure and function and highlight the promising directions and future obstacles in these fields. We provide insights and guidelines for membrane protein biochemists who wish to apply sequence coevolution analysis to a given experimental system.
Keywords: clustered protocadherin, sequence coevolution analysis, membrane proteins, ABC transporters, protein-protein interactions, conformational changes
Graphical abstract

1. Introduction to sequence coevolution methods
Sequence coevolution analysis methods are based on the principle of compensatory mutations during evolution. Because amino acid side chains pack tightly in a folded protein, the identities of protein residues are dependent on each other, meaning mutations at one position influence evolution at other positions in a protein sequence. One such example would be a phenylalanine residue that is stabilized in a protein core by nearby aliphatic residues (Figure 1A). Random mutation of that position to a polar or charged residue would result in a loss in this stabilizing interaction and could hinder protein function. To regain function, other residues nearby might mutate to a polar or charged residue to restore the interaction, albeit in an alternative form, and compensate for the original chance mutation. Thus, the initial mutation changed the evolutionary trajectory of nearby residues, meaning these residues are evolutionary coupled. Other mechanisms of compensatory evolution have been described in detail [1]. Of note, residues that are in physical proximity are more strongly coupled because the interactions are more direct. In fact, coevolving pairs reflect the thermodynamics of the interaction between the two amino acids [2]. In addition, the role a residue plays in protein function or structure determines how strongly coupled a specific residue may be to other nearby residues. Highly conserved residues may impose strong constraints on the evolutionary trajectory of nearby residues and this recalcitrance to substitution may be the primary factor in identifying coevolving pairs [3], rather than true covariation. Whether or not these theories of coevolution are heuristic in nature is eclipsed by the ability of these methods to successfully predict residue pairs that are close to each other in the three-dimensional structure of proteins.
Figure 1. Compensatory mutations produce evolutionary couplings in sequence alignments that can identify contacting residue pairs in protein structures.
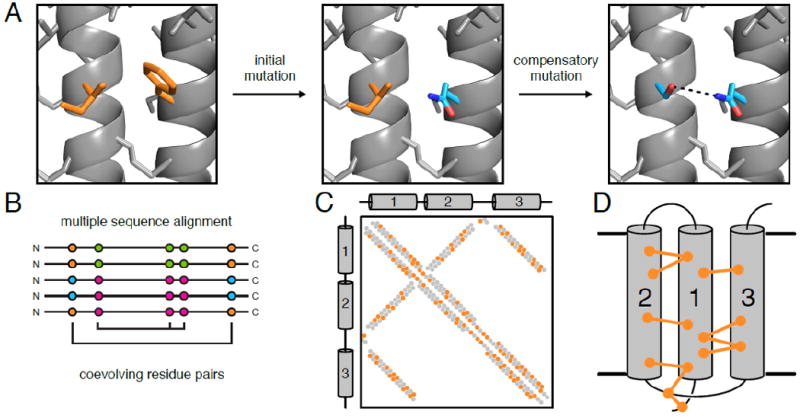
(A) A phenylalanine residue engages in van der Waals interactions with a nearby isoleucine. Random mutation of phenylalanine to glutamine disrupts this interaction, destabilizing the protein. A subsequent mutation of isoleucine to serine restabilizes the interaction through a hydrogen bond (dashed line). (B) Sequence coevolution analysis detects pairs and sets of positions where amino acid identity covaries (e.g. the Phe-Ile to Gln-Ser pairs in orange and cyan, respectively) in a multiple sequence alignment. (C) Coevolving residue pairs can be plotted on a contact map to show overlap of predicted contacts (i.e. coevolving pairs; orange) and true contacts from a high-resolution experimental structure (gray). In α-helical membrane proteins, helical packing results in diagonal signals that can be used to determine which helices contact each other in a parallel (e.g. helices 1 and 3 here) or antiparallel (e.g. helices 1 and 2) interface. (D) Coevolving residue pairs can be used to determine 3-dimensional structures of membrane proteins. Here the coevolving pairs are indicated with dumbbell connections.
Homologous sequences in a protein family serve as a record of coevolution. Algorithms have been developed to identify coevolving pairs or groups of residues from a multiple sequence alignment (MSA) (Figure 1B) by ranking pairs of residues by an evolutionary coupling score. Initially, two methods were used to compute these evolutionary couplings: a substitution correlation approach, where correlations in substitution matrices between pairs of positions are calculated [4], and a mutual information (MI) approach, where the joint probability of finding a specific pair of amino acids in two positions is compared to the probability of finding the amino acids in these two positions independent of each other [5]. These approaches are useful, but overall have low accuracy in predicting true contacts. Further explanation of these methods and their variations are described in an excellent recent review [6].
One limitation of these initial methods was the inability to distinguish direct coevolution from transitive coevolution, where position A effectively coevolves with position C because of the direct coevolution of A and B and of B and C. To overcome the challenge of determining only directly coevolving pairs, a global model of all couplings is used to eliminate transitive couplings. These global models have enormous numbers of parameters that make explicit solutions intractable, and so approximation techniques are used. At first, a Monte Carlo approach was used to explore parameter space randomly [7], but this approach is computationally costly. Since then, various entropy-maximization methods have been implemented to approximate and simplify these coupling parameters [8], including susceptibility propagation [9], mean-field approximation (mfDCA and in the original EVFold) [10-12], multivariate Gaussian modeling [13], pseudo-likelihood maximization (implemented in plmDCA, CCMpred, the new EVFold, and GREMLIN) [14-17], Boltzmann network formalism [18], Bayesian network models [19], and sparse inverse covariance estimation (PSICOV) [20,21]. These methods, which we will call direct methods, have greatly increased the accuracy of sequence coevolution analysis in predicting true contacts, opening up this method to predicting protein structures [11,22-24], protein-protein interactions [25,26], and the effects of mutations [27]. A recent review provides a comparison of these different statistical methods [28]. In addition, multiple direct methods have been integrated to improve prediction through a consensus approach (PconsC, metaPSICOV) [29-32], or through machine-learning algorithms (Raptor-X) [33]. Studies comparing predictions from these various methods found that pseudo-likelihood maximization and consensus methods perform best at predicting true contacts [34,35].
Recent advances in sequence coevolution analysis make it tractable for experimental scientists who wish to use bioinformatics to generate new hypotheses about their proteins of interest. Specifically, we find these methods particularly valuable to studies of membrane proteins because they are often challenging experimental systems for structural and biochemical studies. The relative scarcity of membrane protein structures compared to soluble proteins illustrates the need for new methods to characterize membrane proteins. How useful will sequence coevolution analysis methods be for membrane protein biochemists? While high-profile publications promise to provide high-resolution structures of proteins using sequence information alone, the sample sets are often biased towards well-represented protein families [36]. Here we illustrate the value of sequence coevolution analysis by surveying examples of its application to membrane proteins. These examples show that sequence coevolution can be used to study membrane proteins at many levels, from de novo prediction of protein structure to understanding conformational changes. We could not cover all examples in this fast-growing field and apologize for any overlooked studies. Based on this survey, we provide guidelines to successfully using sequence coevolution analysis.
2. De novo protein structure prediction
One of the promises posited in the first paper on protein sequence coevolution analysis [4] is the de novo prediction of protein structure from sequence information alone. The main concept is to use the coevolving residue pairs as distance constraints – inferring that the paired residues should be close in space in the three-dimensional structure – for structural modeling using methods developed for NMR [37] or computational structure prediction software like Rosetta [38] (Figure 1C, 1D). However, the accuracy of predicted structural contacts only became sufficient to infer protein structure when direct methods developed to discern directly coupled residue pairs from transitively coupled pairs [11,39]. Direct methods were soon applied to α-helical membrane proteins, with some alterations specific for membrane proteins [40]. Specifically, coevolving pairs predicted to be on opposite sides of the membrane based on secondary structure prediction were eliminated from the constraints. The model structures were also scored based on adherence to secondary structure prediction, agreement with coevolution constraints, and agreement with models of which residues are exposed to the lipid membrane. This EVFold_membrane algorithm can produce highly accurate models of α-helical membrane proteins, based on a test set of 25 known membrane protein structures [40]. The root mean squared deviation (RMSD) over Cα atoms for these models and their corresponding experimentally determined structure varies between 2.8 and 5.1 Å. Thus these models are analogous to a reasonable homology model, which is a useful starting point for the membrane protein biochemist who has no other structural information.
EVFold_membrane has since been used to predict structures of other α-helical membrane proteins. One example is the membrane protein insertase YidC, which binds to the ribosome [41]. The EVFold_membrane-derived model of Escherichia coli YidC correctly predicted the transmembrane segment topology and further predicted that the extracellular helical paddle domain (HPD) would lie against the membrane (Figure 2A). A structure of Bacillus halodurans determined concurrently [42] was used to a build a homology model in which the HPD was instead more solvent-accessible [41]. However, in molecular dynamics simulations the HPD stayed against the membrane, indicating that the EVFold_membrane model represents a structurally-accessible conformation of YidC. This suggests that EVFold_membrane could provide information about alternative conformations not yet observed experimentally.
Figure 2. Direct methods provide useful structural insights for membrane proteins with diverse folds.
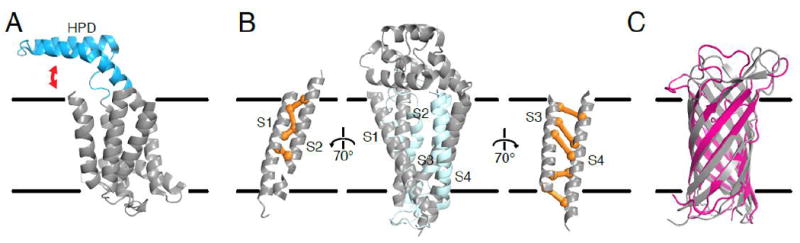
(A) The structure of the membrane protein insertase YidC was predicted using plmDCA [41]. This predicted structure positioned the helical paddle domain (HPD; cyan) at the membrane interface. In combination with a concurrently solved crystal structure (illustrated here; PDB ID: 3WO6 [42]), the predicted structure thus suggested dynamics of the HPD (red arrow). (B) EVFold_membrane accurately models membrane helix topology (light blue) compared to the solved structure (gray; PDB ID: 5TCX). Coevolving pairs (orange) confirmed the unusual membrane topology showing close interactions within transmembrane segment pairs S1-S2 and S3-S4 but relatively few contacts between these pairs [43]. (C) EVFold can predict outer membrane β-barrel protein structures, as shown by the agreement between the predicted structure (magenta) and experimental structure (gray; PDB ID: 1P4T) of the Neisseria meningitidis surface protein A, NspA [23]. Note that although the β barrel is accurately predicted, the loop regions are often sparse of coevolving pairs and thus still challenging to predict.
EVFold_membrane was also used to confirm the unusual structure of ligand-free tetraspanins recently solved by lipidic cubic phase crystallography [43]. In this case, the presence of strong evolutionary couplings only between S1 and S2, and S3 and S4, respectively, corroborated the open conformation of the human tetraspanin CD81 observed in the crystal structure, which shows tight S1-S2 and S3-S4 interactions but little contact between the two helix pairs (Figure 2B). The analysis also suggested that this open conformation might be present in other tetraspanins by using alignments of other tetraspanin subfamilies. In sum, sequence coevolution analysis can provide independent evidence to support unusual or otherwise ambiguous experimental data.
Building off of direct methods, FILM3 is a fragment-searching approach to membrane protein structure prediction that is combined with coevolving pairs predicted from PSICOV [44]. FILM3 produced similar results to EVFold_membrane [45]. This is likely because the structure of protein fragments, which originate from actual protein structures, are themselves also subject to coevolutionary pressure in their native environment. A study that deconstructed transmembrane helix trimers into representative modes further corroborates this idea that commonly observed protein structure fragments are constrained by coevolution [46]. Coevolution analysis of these representative helix trimer modes using EVFold found that the corresponding sequence motifs were enriched in coevolving pairs. This suggests that the representative structural modes are guided by underlying coevolving pairs, which may partly explain the incredible success of DCA methods in predicting membrane protein structures. This idea is further supported by the use of predicted secondary structure elements and coevolving residue pairs (from PSICOV) to determine overall protein topology [47].
In addition to α-helical membrane proteins, sequence coevolution analysis has been adapted to predict the structure of β-barrel outer membrane proteins [23,48] (Figure 2C). For these predictions, at first only coevolutionary constraints between adjacent β strands were used to ensure correct registry and orientation of the β strands in the barrel, then other coevolutionary constraints were later included in the in silico folding procedure. A unique challenge for these proteins is the lack of observed evolutionary couplings between the first and last β strands of the barrel, which is likely due to a lack in sequence coverage in the MSA for these terminal regions of the proteins. Similar to α-helical membrane proteins, this method can predict β-barrel structures with RMSD of 1.6 to 6.5 Å [23], equivalent to a decent homology model, with much of the inaccuracy often localized in the loop regions. The modeling errors in the loops are again explained by the sparsity of observed coevolving pairs to constrain their predicted structures, because loops tend to be less conserved and the location of numerous insertions and deletions, reducing the effective sequence coverage in the MSA.
As the number of unknown membrane protein structures decrease due to the growing number of membrane protein structures, the need for de novo membrane protein structure prediction becomes less acute. However, the examples above already suggest that sequence coevolution analysis is useful for understanding additional aspects membrane protein structure. Below we highlight a number of studies where these methods have been used to generate hypotheses and confirm experimental results. These illustrate many other ways protein sequence coevolution analysis can be used by the membrane protein biochemist to reveal connections between structure and function.
3. Identification of functional residues
Regardless of whether a structure is known for a particular protein target, sequence coevolution analysis can identify residue pairs and networks that are important for protein structure or function. In fact, the Marks group has recently extended their EVCouplings algorithm to identify sites within a protein that are more sensitive to mutation based on a residue’s evolutionary coupling with other residues in a protein [27]. Any coevolution algorithm can be used to uncover such functional residues, but the accuracy with which they are identified depends on the depth of the MSA and the sophistication of the algorithm.
Mutual information (MI) is a relatively simple sequence coevolution algorithm that compares joint probabilities at two positions to single-site probabilities to infer sites that are probabilistically coupled. MI can reveal residue pairs and networks with functional importance, such as protein-protein interaction interfaces and catalytic sites [49-53]. For example, a structure-based correlated mutation analysis (SCMA) approach, using MI to score coevolving residue pairs, identified the retinal binding site residues in the G-protein coupled receptor rhodopsin [54]. MI also identified which residues were proximal to the lumenal α-helix in the mitochondrial β-barrel porin VDAC [55]. Furthermore, MI has been combined with the evolutionary trace method to identify functional residue pairs in membrane proteins. The evolutionary trace (ET) method identifies specificity-determining positions (SDPs) in proteins by taking into account phylogenetic information in a MSA [56], making it similar to other SDP methods [57]. ET has been used on its own to identify residues networks that determine both superfamily and class-specific functions in GPCRs [58]. When combined with MI, coevolving residue pair predictions are closer in structural space. This combined method was used to identify, and confirm through mutagenesis, allosteric networks and proximity of coevolving pairs in the dopamine D2 receptor [59]. These results corroborate studies showing that reweighting of phylogeny and conservation in MI can improve predictions [52,60,61].
Statistical Coupling Analysis (SCA) is another coevolution method that identifies networks of functional or structural residues in a protein [62]. Typically, each of these networks – termed protein sectors – corresponds to a different protein property, such catalytic activity, substrate binding, or allosteric regulation [63,64]. Residues within sectors are more sensitive to mutation than non-sector residues [65]. SCA was successfully applied to G-protein coupled receptors (GPCRs) and heterotrimeric G proteins to identify allosteric networks of residues involved in ligand binding and activation and GPCR dimerization [64,66-68] (Figure 3A). SCA was also used to reveal the signal transduction network within the outer membrane protein FecA, a TonB-dependent ferric-citrate importer [69]. FecA consists of a 22-strand β-barrel with an internal “plug”, preceded by a small N-terminal periplasmic signaling domain connected to the barrel through the TonB-box, a linker that regulates TonB binding. The identified residue network connects the ferric-citrate binding site to the plug domain, the signaling domain, and the TonB-box. Mutating residues in this network reduced downstream transcriptional activation, demonstrating that these residues are indeed important in signal transduction. It should be noted that while SCA reveals important residue networks within proteins, sequence conservation, rather than covariation, dominates sector determination in proteins with single sectors, meaning that the use of SCA to define sectors is only warranted in systems that have more than one sector [70].
Figure 3. Sequence coevolution can identify functionally important residues.
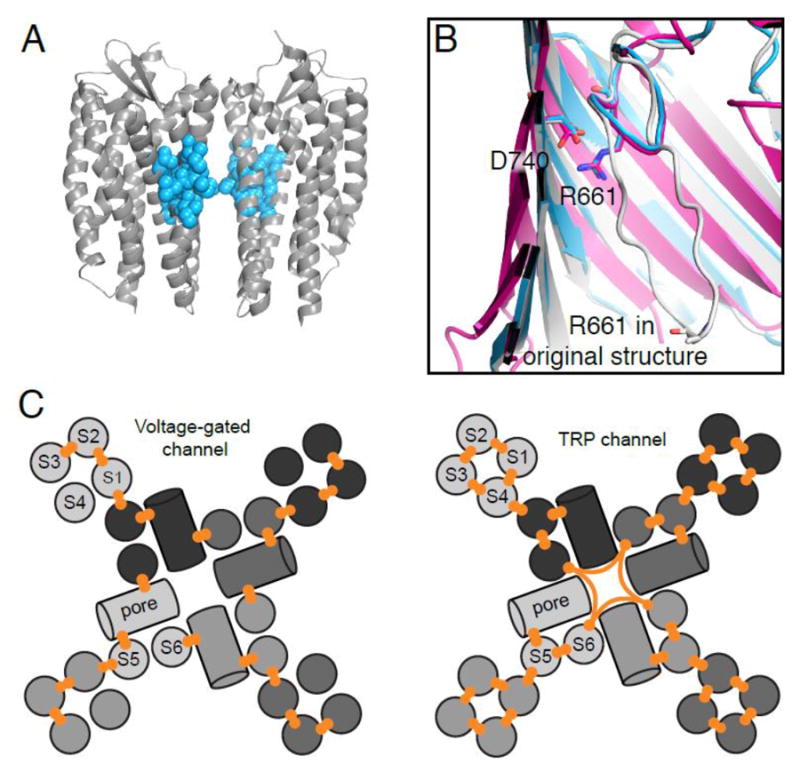
(A) Comparative analysis of dimerizing chemokine GPCRs and non-dimerizing rhodopsin-like GPCRs using SCA reveals a protein sector that is specific to GPCR dimerization (cyan), mapped here on the CXCR4 structure (PDB ID: 3ODU) [66]. (B) mfDCA of BamA identified a coevolving pair (R661-D740) that forms a salt-bridge, as demonstrated in the recent BamA crystal structure (magenta; PDB ID: 4N75). The original structure of the BamA paralog FhaC (gray; PDB ID: 2QDZ), incorrectly modeled the loop containing R661. Reprocessing of the original FhaC dataset with a higher resolution model (cyan; PDB: 4QKY) has the same conformation for this loop as BamA [74,75,77,78]. (C) Differences in helix topology and coevolutionary network between voltage-gated channels and TRP channels, such as the twisting of the S1-S4 domain, the conformational independence of the arginine-bearing S4 of voltage-gated channels, and the allosteric communication between the sensor and pore domains, are supported by coevolving residue pairs [80,83].
As an alternative to SCA, an undirected graphical model approach can uncover networks of functionally important sites [71]. This approach produces a probabilistic model of coevolution, similar to the global models of direct methods, but focuses on strongly correlated pairs of positions (those with high mutual information) and uses far fewer parameters. This approach has been tested on some of the same examples as SCA, namely GPCRs [71], and PDZ domains [72]. Both SCA and an undirected graphical model identified similar networks in GPCRs [64,71].
While SCA and undirected graphical models are specifically designed to identify networks, visualizing an ensemble of coupled residue pairs detected by direct methods can also be used to infer networks of interacting residues in membrane proteins. This approach was used, for example, to identify functional connections between proteins in the bacterial flagellar motor. PSICOV analysis of the flagellar C-ring protein FliM found a strong coevolution network between FliM subunits within the C-ring that is consistent with their role in regulation flagellar motion and may be involved in the switching behavior of flagellar rotation [73].
Another direct method-based coevolution analysis identified functionally important residues on the β-barrel proteins BamA, which is involved in bacterial outer membrane protein assembly, and FhaC, a BamA paralog [74]. At the time of publication, only the structure of FhaC was known [75] (Figure 3B). In a comparison of mfDCA with MI, mfDCA produced a higher true positive rate for contact prediction in FhaC than MI. The top coevolving pair in BamA connected residues in the β-barrel lumen and the L6 extracellular loop. In the FhaC-based BamA homology model, these two residues were surprisingly not in contact with each other. However, screens for rescue of BamA mutants at either position within the top coevolving pair resulted in many of the same suppressor mutants, indicating that the two coevolving residues at least participated in the same functional network. When the BamA structure was determined, these two residues did in fact form a salt-bridge [76,77] (Figure 3B), in contrast to their predicted distance in the homology model. Concomitantly, a higher resolution FhaC structure indicated an incorrect placement of the L6 loop in the original structure, prompting reprocessing of the original data which revealed ambiguities that led to this incorrect modeling [78]. It is likely that sequence coevolution analysis of FhaC before building the original structure could have prevented this incorrect modeling, highlighting the usefulness of sequence coevolution analysis in advancing structural knowledge of proteins.
Observation of networks of coevolving residues from direct methods has also revealed intricacies of ion channel function. Voltage-gated ion channels are tetramers in which each subunit contains a peripheral voltage-sensing domain (transmembrane helices S1-S4) and two additional transmembrane helices with an intervening pore loop (S5-S6) that tetramerize to form the central ion pore [79]. plmDCA on voltage-sensing domains revealed abundant S1-S2 and S2-S3 correlations but a general absence of correlations connecting S4 to the rest of the S1-S4 bundle [80] (Figure 3C). This was at first surprising because S4, containing highly conserved positively-charged arginines, is crucial for voltage-sensing [81]. However, it confirms the known modularity of S4 found in chimera studies [82] and implies that S4, which slides up within the bundle upon channel activation, does not need to coevolve with specific residues in the rest the VSD. Transient receptor potential (TRP) ion channels have the same transmembrane topology as voltage-gated ion channels but lack the voltage-sensing arginines in their S4 helix. In contrast to the voltage-gated channels, the S4 of the TRP channels shows extensive evolutionary coupling with S1 and S3, suggesting a more static structure for these S1-S4 bundles [83] (Figure 3C). These studies, along with SCA studies of voltage-gated potassium channels [84,85], also revealed differences in coevolution between the S1-S4 bundle and the S5-S6 pore of voltage-gated and TRP channels, namely that the pore contacts S1 in voltage-gated potassium channels, but S4 in TRP channels, which is confirmed in the electron microscopy (EM) structure of TRPV1 and TRPA1 [86-88]. Moreover, in comparison to voltage-gated potassium channels, TRP channels have more coevolving pairs at the base of the pore domain, and fewer coevolving pairs between the upper region of the pore domain and the sensor [83], providing interesting evolutionary features that may have functional consequences not revealed by the structure alone. The detailed structural analysis of these ion channels was enabled by expertly curated alignments of ion channel subfamilies and direct methods, showing the ability for sequence coevolution analysis to compare closely related protein families with intricate detail.
Identifying functional sites in proteins can also aid in the directed evolution or design of proteins with altered or new activity. MI analysis of bacterial two-component signal transduction systems allowed the rewiring of histidine kinases to new response regulators [49]. Similarly, Laub and colleagues used GREMLIN to analyze the toxin-antitoxin system and identify coevolving pairs of residues, which were then subjected to deep mutational scanning to understand evolution of interaction specificity [89]. This approach was also used to identify positions for mutation in a directed evolution of a phosphotriesterase for new organophosphorous compounds that are used as pesticides [90]. While these latter examples are not from membrane proteins, these studies provide a guide to using coevolutionary information to design large mutant libraries for screening purposes.
Biochemists often identify functionally important residues using sequence conservation alone. The examples above demonstrate that sequence coevolution analysis is another method for identifying important residues in proteins based on structural contacts. Coevolution can determine individual residue pairs or networks of coevolving residues and can be used to generate hypotheses about structural features, conformational states of proteins, and allosteric communication through proteins.
4. Protein-protein interactions
The epistatic coevolution of residues across a protein-protein interface enables us to use sequence coevolution analysis to predict and structurally characterize protein-protein interactions. Sequence coevolution analysis can be performed on a potential protein complex similarly to identifying coevolving sites within a protein by concatenating the corresponding sequences from two proteins from the same species in a MSA and identifying coevolving pairs across the two proteins. An important step, which can be a challenging technical hurdle, is that the correct paralogs need to be carefully matched if a genome contains multiple paralogs of either protein. Histidine kinase-response regulator interactions in bacterial two-component signal transduction systems (TCS) were among the first to be elucidated by coevolution methods [9,49,91]. Although bacteria often have many TCS paralogs that generally do not cross-talk with each other, in this case the appropriate pairing of interacting pairs was insured by generally pairing each histidine kinase to the response regulator in its respective operon. Sequence coevolution analysis revealed interacting residue pairs, including those that determine interaction specificity, and allowed researchers to mutate residues to rewire the specificity of many TCS pairs [49,92,93].
Studies of TCS benefited from rich sequence information due to: (i) their ubiquity in bacteria, (ii) the large number of paralogs, and (iii) the many available bacterial genomes. The development of global maximization approaches to coevolution later enabled detection and characterization of protein-protein interactions in a wider range of protein families [25,26]. These seminal papers elucidated a wide array of protein-protein interactions, including many membrane proteins such as ATP-binding cassette (ABC) transporters, tripartite efflux pumps, ATP synthase, ubiquinol synthase, cytochrome c oxidase, and the respiratory complex I. In these first studies, potential protein-protein interactions were chosen based on known structure or the proximity in the genome because bacterial proteins that interact tend to be near each other in the genome (the operon rule alluded to above). However, any potential protein-protein interaction can be tested for coevolution using a large concatenated alignment of the two proteins of interest. Importantly, longer alignments require more sequences for statistical significance [11], thus it is generally more difficult to detect intermolecular coevolving pairs than intramolecular coevolving pairs due to the longer length of the concatenated alignment. Once identified, the coevolving residue pairs across a protein interface can be used to computationally design inhibitors that target these coevolving hotspots [94].
To extend protein-protein interaction prediction to a proteomic level, new algorithms have been devised to systematically identify protein-protein interactions [95,96]. Both studies used TCS as a benchmark case because of their rich sequence information and known interaction pairing. Using a small training set of known interacting histidine kinase-response regulator partners, evolutionary couplings are determined between the two proteins using direct methods. Then, the interaction energy of a randomly selected test protein pair (i.e. a histidine kinase and a response regulator from the same bacterial species that may or may not be a native matched pair) is calculated based on the evolutionary couplings of the training set. In essence, this calculates how well the residue pairs from the test protein pair match the scores from the training set. The best scoring test protein pairs are considered correct and then added to the training set and the process is repeated. This iterative approach is rather robust as it can be done with no training set [95] or without ever recalculating the interaction energy of the training set to throw out outlier matched pairs [96]. These algorithms were extended to subunits of ABC transporters [95] and to members of the tryptophan biosynthesis machinery [96] and they will likely be generally applicable to determine whether two proteins interact, as long as enough sequence information is available.
Homo-oligomers pose unique challenges for investigating protein-protein interactions using coevolution analysis. Unlike heteromers whose intermolecular evolutionary couplings arise from pairs of residues originating from two different proteins, homomeric intermolecular evolutionary couplings come from residues within a single protein, and thus are difficult to distinguish from internal residue contacts (Figure 4A). For example, coevolution analysis of insect odorant receptors led to a model for the odorant receptor protomer, but no homo-multimer model was evident from the evolutionary couplings [97]. However, if we know the structure of a protomer, then evolutionary couplings inconsistent with this monomeric structure can be assigned as a homomer contact [98], as observed with the lipid A exporter MsbA, the methionine transporter MetI, and aquaporin [40]. Similarly, as mentioned above, a structure-based SCA model of the human CXC chemokine receptor type 4 GPCR identified a homodimerization protein sector [66] (Figure 3A).
Figure 4. Coevolution can predict homomer architecture when the protomer structure is known.
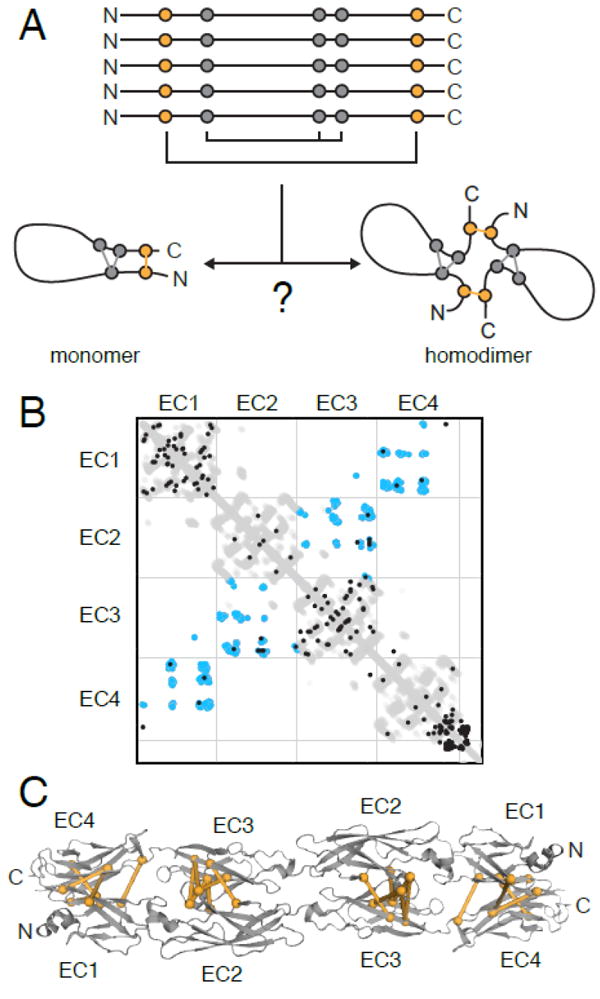
(A) Coevolving pairs can represent intramolecular contacts in a monomeric structure or interacting residues across a homomer interface, which complicates structural modeling. (B) The contact map of the first 4 extracellular cadherin (EC) repeat domains of clustered protocadherins shows the intramolecular contacts (gray), dimerization contacts from crystal structures of various isoforms (blue), and coevolving pairs (black) that overlap with either the intramolecular or dimer contacts. (C) Intermolecular coevolving residue pairs (orange) agree with the PcdhγB3 EC1-4 homodimer structure (PDB ID: 5K8R).
Such sequence coevolution analyses played a predictive role in determining the homodimer interface of the clustered protocadherins. Clustered protocadherins are a large family of membrane proteins (with ~50 paralogous genes, or isoforms, in each mammal genome) which help regulate synapse formation in neurons. In this case, comparison to the contact map built from the intramolecular contacts of the repeat cadherin-domain structure of these proteins made the homodimerization evolutionary couplings evident [99] (Figure 4B). These signals suggested that the first four cadherin repeat domains form an antiparallel oligomer, with the first repeat of one protomer binding the fourth repeat of another protomer, and similarly the second with the third. At the time, a partial structure of the γA1 isoform, consisting of the first three cadherin repeats, showed a crystal-packing interface consistent with the evolutionary couplings [99]. An independently-derived docking model based on mutagenesis data was also consistent [100]. The observation of evolutionary couplings between the first and fourth repeat inspired structural studies of longer constructs containing at least four repeats, which resulted in many structures of different isoforms demonstrating the antiparallel dimer predicted from sequence coevolution analysis [101-103] (Figure 4C). Coevolutionary analysis was extended to hypothesize that this antiparallel dimer would also be present in at least some non-clustered protocadherins [101], which was soon thereafter confirmed with a structure of protocadherin-19 [104].
If a protomer structure is known, several methods have been developed to help build homo-multimer models from evolutionary couplings, either using Structure-Based Models [105] or Rosetta SymDock [106]. However, experimental verification of these models is needed for each specific protein of interest because homomer structures can differ between subfamilies of the same overall fold [98]. Another challenge is that many membrane (and soluble) protein families contain both homomers and heteromers with similar overall folds; for example this is common in ABC transporters [107]. Therefore, there is a continued need for new and more rigorous methods for understanding membrane protein oligomerization.
5. Conformational changes
As the gatekeepers and surveillance systems of the cells, membrane proteins often use conformational changes to regulate transport activity or signaling to soluble messengers. Each conformational state exerts evolutionary pressure on the gene, resulting in evolutionary couplings that we can use to understand multiple conformations of a single protein. Several examples in previous sections already suggested the potential for identifying alternative conformations. Here we review examples of studies that specifically made use of evolutionary couplings to reveal additional conformational states.
Evolutionary couplings of multiple conformations have been described in several types of membrane protein transporters: major facilitator superfamily members GlpT, a bacterial glycerol-3-phosphaste transporter, OCTN1, a human organic cation transporter, and LacY, the bacterial lactose permease [40,108]; Escherichia coli YddG, an aromatic amino acid transporter from the drug/metabolite transporter (DMT) superfamily [109]; and broadly for ABC transporters [110]. In the different conformations of transporters using an alternating-access mechanism [111-113], individual residues alternate between being solvent-accessible and in contact with other residues in the protein. This produces conflicting evolutionary couplings, as residues at one vestibule will coevolve due to their proximity in one conformation, while residues in the other vestibule will coevolve due to their proximity in the other conformation (Figure 5A). When signals from multiple conformations are used to build a de novo model, the conflicting coevolving pairs may enforce an occluded structure where both vestibules are closed, as illustrated for the YddG case in Figure 5B [109]. While occluded structures have been observed experimentally [114,115] and may play a role in the transport cycle of some transporters, determining the structure of inward-open and/or outward-open states requires selection or re-weighting of coevolving pairs, such that some are consistent with single conformations while others may be consistent with multiple conformations. More specifically, coevolving pairs unsatisfied in a known structure of a specific conformation can be used to model other conformations with the aid of molecular dynamics [116-118]. The examples above have provided early successful case studies for the role evolutionary couplings can play in understanding conformational changes in transporters with large structural changes and with residues alternating between buried and solvent-exposed. However, many other transporters and signaling proteins display more subtle structural changes. Other methods, such as one that determines individual residues that play a structural role in multiple conformations by tabulating in how many coevolving pairs each residue participates [119], will be needed to reveal conformational changes in gene families with such subtle structural changes.
Figure 5. Evolutionary couplings of multiple conformations complicates structural modeling of alternating-access transporters.
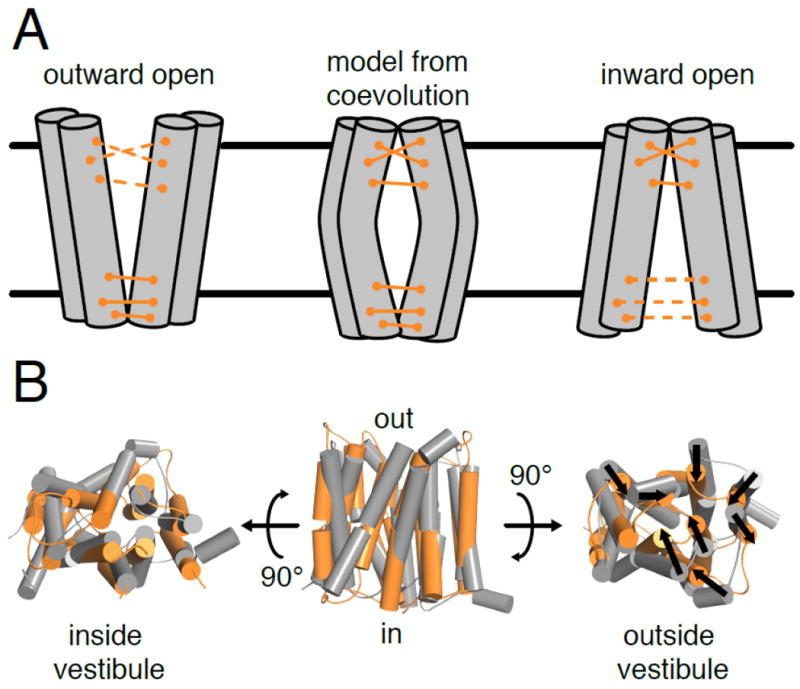
(A) Some coevolving pairs in alternating-access transporters only contact each other in either the outward-open or inward-open conformations. Structural modeling of these transporters using all evolutionary couplings can generate an occluded structure (middle). (B) The outward-open crystal structure of YddG (gray; PDB ID: 5I20) differs from the EVFold_membrane predicted structure (orange) in that the transmembrane segments of the predicted structure are bent inwards (shown with black arrows), closing off the outer vestibule [109].
6. Integrative modeling of membrane protein structure
Experimental structures of membrane proteins are often of lower resolution compared to soluble proteins, because of the difficulty in crystallizing membrane proteins or the overall lower resolution that is currently available from EM. Sequence coevolution analysis can complement experimental studies of membrane protein structure by adding further constraints that can be used in integrative structural modeling that uses experimental structures, biochemical data such as mutagenesis and crosslinking, homology modeling, and other approaches. Such integrative studies have been highly successful in modeling large membrane complex structures as well as individual proteins. Notably, the more accurate contact predictions by direct methods has made it the most successful in integrative modeling applications.
In the simplest cases, coevolving residue pairs are used as additional constraints in homology modeling or Rosetta-based modeling of proteins. This is similar to structural modeling of soluble and membrane proteins in the original Marks et al. (2011) and Hopf et al. (2012) studies [11,40] of soluble and membrane proteins, which use the evolutionary couplings (as well as secondary structure prediction) as constraints in distance geometry algorithms like those used for NMR [37]. An alternative approach is to first build a model using homology modeling or Rosetta, either of which can often generate decent model structures, then use the evolutionary coupling constraints to refine or expand the structural model [120]. This approach has been applied to the human zinc-regulated transporter protein hZIP4, where Rosetta and evolutionary constraints helped build a dimeric model of the transporter [121]. Recently, a structure of a ZIP from the bacterium Bordetella bronchiseptica was solved that has a 4 Å rmsd (over ~200 Cα) with the Rosetta model with evolutionary constraints [122]. While evolutionary couplings provide some evidence for a dimer, the B. bronchiseptica ZIP does not form a parallel dimer in the crystals, leaving this question unresolved. In another example, homology modeling using I-TASSER of the membrane-tethered periplasmic copper-binding protein CopM was refined using coevolving pair constraints from EVFold [123]. Finally, the conformation of specific secondary structure elements of the cone tetrameric cyclic nucleotide-gated (CNG) ion channels that was built from homology models was confirmed through inspection of coevolving residue pairs [124].
Other cases of integrative modeling involve the combination of experimental structural information from NMR, crystallography or EM, as well as mutagenesis and crosslinking. For example, the loop region between two transmembrane domains of a pancreatic ATP-sensitive potassium channel was poorly resolved in a recent EM structure [125], but the authors combined weak density with known topology and evolutionary coupling scores to manually build this region. This method would have helped resolve the L6 loop of the β-barrel protein FhaC in the original crystal structure [74,75], as explained above. In a different study the mitochondrial translocator protein TSPO dimer structure was disputed based on differences in data from NMR [126], EM of helical crystals [127], Rosetta modeling [128], and sequence coevolution analysis [40,129]. A high resolution lipidic cubic phase crystal structure finally resolved the structure of the TSPO dimer [130], which had the topology predicted from sequence coevolution analysis. These examples illustrate the power of leveraging available sequence information and analyses in otherwise mostly experimentally-driven structural studies.
Subunits of large protein complexes are often amenable to sequence coevolution analysis to determine how they interact, especially when low-resolution EM structures are available. The flagellar motor FliM protein interaction network [73] mentioned above provides one example. In another example, one of the first studies using sequence coevolution analysis to predict protein-protein complexes, the 50S ribosomal subunit provided an ideal test case for the method because its atomic structure was known and it is highly conserved in bacteria [26]. GREMLIN successfully identified many covarying residues that were within 8 Å of each other across protein interfaces, demonstrating the utility of the method to help build large protein complexes. Below we detail two fascinating examples in which sequence coevolution analysis was applied to a large protein complex to reveal molecular details absent from low resolution EM or from crystal structures of individual subunits: the rotatory ATP synthases and the Twin Arginine Translocase (TAT) secretion system.
Hopf et al. (2014) initially characterized interactions within subunits of ATP synthase to confirm experimental interactions [25]. These coevolving pairs demonstrated that an interaction between the a and c subunits, which together form the H+ and Na+ ion transport pathway that generates a mechanical rotatory force, could be characterized by sequence coevolution analysis. In the first EM structure of Polytomella sp. mitochondrial F-type ATP synthase four of the a subunit α-helices were surprisingly found packed nearly horizontally within the membrane plane against the vertical helices of the c subunit [131]. Previous biochemical data indicated that H4 and H5 (equivalent to H5 and H6 in the bovine ATP synthase) interact directly with the c subunit [132,133]. However, at the initial resolution of 6.2 Å the a subunit topology could not be determined, and it was thus unclear whether H4 or H5 was more proximal to the mitochondrial matrix. Coevolving residue pairs between the a and c subunits are consistent with H4 being proximal to the mitochondrial matrix [134] (Figure 6A). Furthermore, coevolving residue pairs between H2 and H5, and H3 and H4, support a clockwise topology of H2-H5 in the a subunit (H3-H6 in the bovine ATP synthase). A scoring scheme based on cysteine-crosslinking and coevolution data between the a and c subunits was then used to evaluate thousands of threading models of the a subunit relative to the c subunit. The final model identified a threading (within ~3 amino acids) that minimized the number of distance violations [134]. Sequence coevolution analysis was also used to fit the a subunit into EM density for the bovine mitochondrial ATP synthase [135] (Figure 6A), and the Thermus thermophilus V/A ATP synthase and Saccharomyces cerevisae V ATP synthase [136], all of which were consistent with each other. Thus in the absence of high-resolution information, sequence coevolution analysis was used to build better structural models from EM structures.
Figure 6. Coevolving residue pairs help model large membrane protein complexes through an integrative approach.
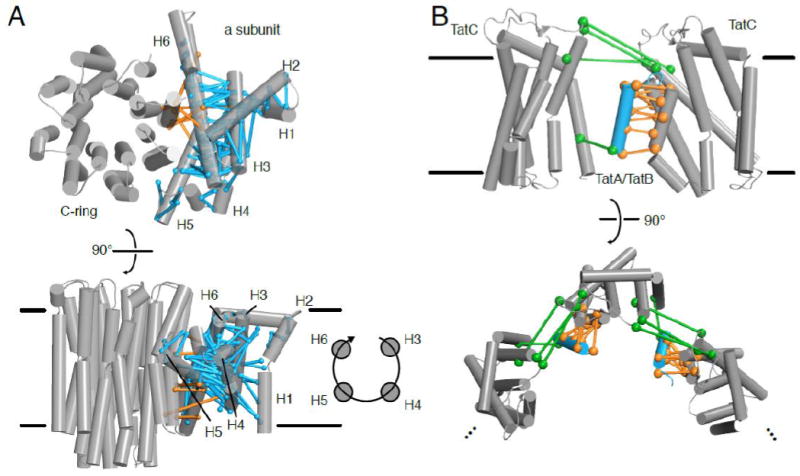
(A) In low resolution EM structures of the ATP synthase, coevolving residue pairs helped determine the registry of the a subunit against the C-ring (orange) and on the clockwise topology of the a subunit helices (cyan) [134,135]. At the top, the structure (the bovine mitochondrial ATP synthase; PDB ID: 5ARA) is viewed from outer side of the membrane, while at the bottom, the structure is view from within the plan of the membrane. (B) Coevolving residue pairs (orange) indicate where TatA or TatB (cyan; PDB ID: 2MN7) dimerizes with TatC (gray; PDB ID: 2HTS). Other coevolving residue pairs (green) suggest oligomerization of the TatA/TatB-TatC complex [145], which, if propagated, may represent formation of the translocation pore, as shown below when the complex is viewed from outside the cell.
The TAT secretion system is a specialized system found in prokaryotes and chloroplast thylakoid membranes that transports fully folded proteins through a pore formed by oligomerization of TatA [137]. The twin-arginine signal sequence at the N-terminus of substrate proteins binds to the six-transmembrane segment protein TatC. Binding of substrate to TatC drives oligomerization of TatA, a small protein with a single transmembrane segment and an amphipathic helix, to form a large pore in the membrane to accommodate substrate transport. Depending on the species, TatC functions either alone or in complex with TatB, a paralog of TatA. The size of the pore depends on the substrate, making it difficult to obtain high resolution EM data, although low resolution information is available [138,139]. In addition, structures of isolated TatA, TatB, and TatC have been obtained using NMR or crystallography [140-144]. Alcock et al. (2016) used direct methods (CCMpred, FreeContact, and PSICOV) to detect interactions between TAT subunits [145]. Because TatA is homologous to TatB, the global alignment of TatA/B homologues was divided into three subalignments to calculate coevolution with TatC: (i) TatA sequences from organisms that only have TatA; (ii) TatA sequences from organisms with both TatA and TatB; and (iii) TatB sequences from these latter organisms. Dividing the TatA/B sequences into these three sub-alignments made it more difficult to obtain statistically significant evolutionary couplings, but strong signals with TM5 and TM6 of TatC were found not only with the alignment of all TatA/B homologues, but also with both subalignments (ii) and (iii), indicating that the same interface is conserved in both TatA and TatB. Furthermore, when the TatA sequences from species with only TatA (subalignment (i)) were combined with the other TatA sequences (subalignment (ii)), these same coevolving pairs were observed as with subalignment (ii) alone, along with additional ones consistent with the same interface, suggesting that subalignment (i) is too small to produce statistically significant results on its own, but does contribute to the evolutionary couplings in the full alignment. The coevolving pairs from all alignments therefore support a helical packing of the TatA transmembrane segment with TM5 and TM6 of TatC (Figure 6B). Gratifyingly, such a packing is also corroborated by observing hydrogen bonds between the three transmembrane segments after computational docking and molecular dynamics simulations using the available structures of the two proteins. These polar residues were found to be essential for TAT secretion. Other coevolving pairs suggested an interaction between TatB and TM1 of TatC that are not present in the TatA only alignments, suggesting a second binding site between TatB and TatC that was confirmed by cysteine crosslinking. Lastly, the combination of a second TatA binding site identified based on the competitive interaction between TatA and TatB at TM5 of TatC, and coevolving pairs in TatC incompatible with the TatC monomeric structures, suggested TatC oligomerization bridged by a TatB molecule simultaneously occupying the binding site on TM5 of one TatC and TM1 of a second TatC (Figure 6B). This TatBC oligomer could be the initial complex triggered by substrate binding to induce TatA oligomerization to translocate substrate. The sequence coevolution analysis on TatA/B/C thus provided a hypothesis for several aspects of the TAT structure and secretion mechanism that are backed up by in vivo and in vitro experiments, and are currently unavailable through experimental structural biology approaches.
We have a growing number of techniques to understand protein structures, from homology modeling to atomic resolution EM. In most cases, one method is not sufficient to understand all the structural and functional details of a system. These examples show that sequence coevolution analysis can be used to help model structure in combination with other techniques, especially when only low-resolution data is available or when assembling subunits of large membrane protein complexes.
7. Guidelines for using sequence coevolution analysis successfully
Sequence coevolution analysis is a versatile method that complements biochemical and structural studies of membrane proteins. We find it to be as useful as – and complementary to – other commonly used computational methods such as homology modeling, sequence conservation mapping onto structure using Consurf [146], protein interface analysis using PISA [147], and computational alanine scanning [148], and hope that the membrane protein biochemistry field continues to embrace sequence coevolution analysis to guide experimental design. It is important to consider the specific motivation for using sequence coevolution analysis. The examples highlighted here show the breadth of this method, from determining protein structure de novo and revealing functionally important sites, to discovering and characterizing protein-protein interactions, understanding conformational changes, and contributing to the integrative modeling of large membrane protein complexes. The motivation will guide the choice in algorithm and the sequence requirements for the analysis. Some studies, such as de novo structure determination and observation of conformational changes, were only successful after direct methods were pioneered to distinguish direct from transitive evolutionary couples, while functionally important sites and networks have been investigated for a number of years using MI and SCA-based approaches.
The largest determinant of success in sequence coevolution analysis is the MSA used as an input for the algorithm. Sequences are commonly gathered and aligned using HHblits [149] or Jackhmmer [150], which generates a Hidden Markov model (HMM) for the sequence and queries it against a sequence database such as UniProt [151]. Sequences are selected using an E-value cutoff, which indicates the chance of randomly obtaining a sequence with a similar score in the same sized database. Alternatively, bit-scores, which indicate the size of a database needed to randomly obtain a sequence of the same score, are a database-independent metric of scoring sequence similarity in an alignment. Alignments of protein families can also be retrieved from many other general databases, including but not limited to Pfam [152] or UCSC Genome Browser [153], or a more specific database like the GPCRdb [64,154]. Nevertheless, access to diverse sequences is an ongoing challenge for sequence coevolution analysis. Recently, metagenomic sequencing data have been used to generate MSAs to de novo predict protein structures [155]. Large-scale sequencing efforts enabled by next-generation sequencing techniques combined with new strategies to cluster metagenomic data [156] will continue to identify of new protein families and add more sequences to known families with little structural information [157], which will extend the usefulness of coevolution methods to more proteins.
It is important to know and understand the parameters used to generate the MSA and to evaluate the MSA after it is assembled. The MSA is the basis for determining evolutionary couplings, so a low-quality MSA will result in low-quality results, following the “garbage in, garbage out” paradigm. The following review summarizes the considerations for different algorithms for generating MSAs [158], and approaches are continuously being developed to assess MSAs (e.g. [159,160]). After constructing an initial MSA, several steps help ensure successful sequence coevolution analysis.
First, the MSA should be the broadest alignment that is likely to share the same structural or functional features of interest, such that it is large and diverse enough to carry statistical significance. For example, in our study of clustered protocadherins, we removed non-clustered protocadherins from the MSA to ensure the signal originated from our protein family of interest [99]. In a follow up study, we repeated the analysis on the non-clustered protocadherins after removing the clustered protocadherins [101]. Only then could we be assured that evolutionary couplings in the non-clustered protocadherins were not influenced by the presence of these signals in the alignment of the clustered protocadherins. Using phylogenetic trees to manually curate the MSA can both improve the quality of the MSA and confidence in the results [64,69,80,99,101,145].
Another aspect of the MSA that can affect the sequence coevolution analysis results is the presence of alignment columns or sequences with many gaps [11,16,161]. For example, de novo structure prediction of β-barrels was complicated by gapped sequences for the first and last β-strands, making it difficult to determine registry at this seam [23]. Such gapped sequences are also often found in loop regions of proteins. Typically, column positions and sequences above a threshold of gaps (25-50%) are removed from the alignment, when possible (i.e. when the number of sequences is sufficient), to improve MSA quality. Another solution is to add new parameters to the pseudolikelihood maximization model so that gapped sequences can be included in the entropy maximization strategy [162].
Sequence coevolution analysis is biased by the phylogenetics of an alignment. Conservation of sites in a MSA is not only determined by the evolutionary tolerance at that site, but on the evolutionary trajectory of the gene (sequences are sparsely sampled throughout evolution), and experimental sequence sampling bias (certain organisms are more likely to be sequenced than others). Bias in the phylogeny of the alignment can result in poor prediction of evolutionary couplings, and thus deeper alignments, which can have less phylogenetic bias, generally improve predictive power [7,163,164]. In contrast, explicitly modeling phylogeny in an alignment, such as with evolutionary trace methods [56,58,59] and other phylogeny-based covariation methods [57], can help identify gene-wide covariation. Although in sequence coevolution methods phylogeny of the alignment is typically not modeled directly, correcting for the similarity of sequences in an alignment has been used to improve SCA, MI and direct methods [52,60,163], which accounts for experimental sampling bias. Sequences are filtered if they are above a % identity cut-off (90% is common) or the conservation of amino acids in a position of an alignment is weighted by the ‘effective’ number of sequences in the alignment, Meff [165,166]. The overall sparseness of the data leaves undetermined probabilities for many amino acids in sites of the MSA. To account for this, pseudocounts are added to adjust probabilities and make covariance matrices invertible, a necessary step for entropy maximization approaches [10,166].
Many online servers exist for running sequence coevolution analysis, provided by the research groups that have developed these tools, while others are available for download (Table 1). These servers generate alignments for the user using a single input query and/or allow users to input their own MSA. Users should become familiar with the parameters needed to generate a high-quality alignment of their protein of interest. Direct methods servers and software will generate an ordered list of evolutionary couplings using metrics that are algorithm-specific. This list is the basis for contact prediction, with high scoring pairs being more likely to be true contacts. The accuracy of contact prediction can be visualized using a contact map showing the comparison of contacting residues from a known protein structure or by mapping these pairs on a protein structure. For de novo protein structure prediction, these coevolving pairs are used as constraints in structural modeling.
Table 1.
A selection of webserver and downloadable programs for sequence coevolution analysis
The list of coevolving pairs, in order of coupling strength, then needs to be analyzed to determine when the significance of the score is predictive of being a true contact (a true positive). At the onset, it is difficult to know where a score cut-off should be made. Even for fairly stringent score cut-offs, false positive contacts are found [25,26,165], and the origin of these false positives is generally unknown [167]. However, a recent study found that a substantial proportion of these false positive result from structural variation among homologs or from homo-oligomeric contacts, complicating the question of which couplings are true positives [168]. An additional complication is the fact that true-positive rates are dependent on the depth of sequence alignment [36] and on the structural model used to calculate true-positive rates, because it is impossible to tell whether a given coevolving pair is determinative of contact in the model used or could be involved in another conformation. Null hypotheses have been used to better evaluate where cut-offs should be made [163,169]. Both an explicit model of independent site evolution and a normal distribution of coupling scores failed to control the false positive rate against a benchmark set. An empirical model, which simply uses the rank order of the evolutionary couplings as a statistical significance test, mostly controlled the false positive rate when large and deep alignments were analyzed using direct methods [163], although it still does not provide a strict score cut-off or a reasonable probability of an evolutionary coupling being a true contact. In practice, a cut-off value that preserves an 80-90% true-positive rate is commonly used as a benchmark. The number of sequences (N or Meff) per length (L) of the alignment correlates well with the true-positive rate [25,163], and thus in the absence of a known structure it is suggested to have a N/L or Meff/L > 1 to recover true contacts. More work is needed to strengthen sequence coevolution analysis with methods that determine statistical significance, such as shown in a variation of DCA, called Hopfield-Potts model, that allows determination of relative error [166]. In the meantime, researchers should be aware of false positives when generating hypotheses from this analysis.
When preparing this review, we noticed large variations in the details provided about the sequence coevolution analysis used. These disparities make it difficult for others to evaluate the analysis method. We propose that researchers report a few key pieces of information when using sequence coevolution analysis to standardize the field and improve reproducibility. First, the sequence alignment used to generate evolutionary couplings and the list of coevolving residue pairs and their respective scores, depending on the algorithm used, should be provided as supplemental files. Details about the MSA, such as the algorithms, E-value (or bit-score) cutoffs, and databases used to collect sequences and/or generate the alignment, should be reported. Any other curation of the alignment, from either phylogeny or sequence filtering, should be explained. For filtering, it is useful to report the cut-off used to remove sequences and columns with many gaps (typically 30%-50% gaps in columns and sequences are good cut-offs). The number of sequences below a redundancy criterion (typically 90% identity) or effective number of sequence Meff if a weighting algorithm is used should be reported as well. The algorithm used to generate evolutionary couplings should be reported. Lastly, if possible, the accuracy of prediction can be reported as % true contacts in the coevolving pairs considered in the analysis. We have reproduced this information for a few datasets described in the review in a “Table 1” format – familiar to structural biologists – as an example (Table 2).
Table 2.
Suggested information to report when performing sequence coevolution analysis
| Clustered protocadherins [99] | Voltage-gated ion channels [80] | BamA [74] | Insect odorant receptors [97] | |
|---|---|---|---|---|
| Query sequence | mouse PcdhgC3 | Manually curated diverse seed MSA | E. coli BamA | D. melanogaster OR85b or D. melanogaster ORCO |
| Alignment window | 30-670 (640 residues) | S1-S4 (115 residues) | 347-810 (463 residues) | ~430 residues |
| Alignment | Jackhmmer 3.1, 5 iterations, UniRef100; manually curated to remove non-clustered Pcdhs | HMMER 3.0, e-value of 1E-2; manually curated to ensure diverse family representation | HHBlits, 2 iterations, Uniprot20 | Jackhmmer, e-value of 1E-40, of all available insect OR sequences |
| Filtering | 50% gapped columns removed | N/A | 70% positions have no more than 30% gaps; 40% gapped columns removed | 50% gapped columns removed |
| N or Meff | Meff = 3525.48 | 3391 for Nav/Cav; 1832 for Kv | 3073 | 5907 |
| Sequences/L | 5.56 | 28.74 for Nav/Cav; 15.53 for Kv | 6.64 | 13.74 |
| Coevolution algorithm | plmDCA (EVFold) | plmDCA | mean-field DCA | plmDCA (EVFold) |
| Significance cut-off | 80% intramolecular true-positive rate | 87% true-positive rate | 8/8 in the POTRA domain in the top 50 ECs were true-positive | top 200 ECs |
Conclusion
This review surveys the applications of sequence coevolution analysis to studies of membrane protein structures and functions, showcasing the ability of this method to determine membrane protein structure, identify functional sites in proteins, discover and characterize protein-protein interactions, understand conformational changes, and integrate with other structural approaches to reveal the structure of large membrane protein complexes. We have provided guidelines for performing sequence coevolution analysis to promote proper use and increase the chance for successful application of this remarkable and state-of-the-art method. We find such analyses to be tremendously useful in generating hypotheses to test experimentally, and hope biochemists will increasingly use sequence coevolution analysis on their own protein families of interest when enough sequence information can be assembled into a high-quality MSA.
Highlights.
Protein sequence coevolution detects interacting residues pairs in membrane proteins
Coevolving pairs can be used to predict protein structures and interactions
Coevolving pairs can reveal conformational changes in membrane transport proteins
Coevolving pairs can serve as constraints in integrative protein structure modeling
Coevolution algorithm usage guidelines will improve data quality and reproducibility
Acknowledgments
We would like to thank Niels Bradshaw, Sriram Srikant and the rest of the Gaudet Lab for thoughtful discussions. We would like to thank Debora Marks, Charlotta Schärfe and Anna Green for sharing their expertise in sequence coevolutionary analysis. Lastly, we would like to thank the reviewers of this article for providing additional detail and insight. This work was funded in part by grants from the American Heart Association (16GRNT27250119) and the National Institutes of Health (R01GM120996) to R.G., and a National Defense Science and Engineering Graduate Fellowship to J.M.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ivankov DN, Finkelstein AV, Kondrashov Fa. A structural perspective of compensatory evolution. Curr Opin Struct Biol. 2014;26C:104–112. doi: 10.1016/j.sbi.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coucke A, Uguzzoni G, Oteri F, Cocco S, Monasson R, Weigt M. Direct coevolutionary couplings reflect biophysical residue interactions in proteins. J Chem Phys. 2016;145:1–17. doi: 10.1063/1.4966156. [DOI] [PubMed] [Google Scholar]
- 3.Talavera D, Lovell SC, Whelan S. Covariation is a poor measure of molecular coevolution. Mol Biol Evol. 2015;32:2456–2468. doi: 10.1093/molbev/msv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Göbel U, Sander C, Schneider R, Valencia A. Correlated Mutations and Residue Contacts in Proteins. Proteins Struct Funct Genet. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 5.Korber BTM, Farber M, Robert, Wolpert DH, Lapedes AS. Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein : An information theoretic analysis. Proc Natl Acad Sci U S A. 1993;90:7176–7180. doi: 10.1073/pnas.90.15.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Juan D, Pazos F, Valencia A. Emerging methods in protein co-evolution. Nat Rev Genet. 2013;14:249–61. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 7.Lapedes A, Giraud BG, Liu L, Stormo GD. Correlated Mutations in Models of Protein Sequences : Phylogenetic and Structural Effects A. Stat Mol Biol. 1999;33:236–256. [Google Scholar]
- 8.Stein RR, Marks DS, Sander C. Inferring Pairwise Interactions from Biological Data Using Maximum-Entropy Probability Models. PLoS Comput Biol. 2015;11:1–22. doi: 10.1371/journal.pcbi.1004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weigt M, White Ra, Szurmant H, Hoch Ja, Hwa T. Identification of direct residue contacts in protein-protein interaction by message passing. Proc Natl Acad Sci U S A. 2009;106:67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M. PNAS Plus: Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaján L, Hopf TA, Marks DS, Rost B. FreeContact : fast and free software for protein contact prediction from residue co-evolution. 2014:1–6. doi: 10.1186/1471-2105-15-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldassi C, Zamparo M, Feinauer C, Procaccini A, Zecchina R, Weigt M, Pagnani A. Fast and accurate multivariate Gaussian modeling of protein families: Predicting residue contacts and protein-interaction partners. PLoS One. 2014;9:1–12. doi: 10.1371/journal.pone.0092721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekeberg M, Lövkvist C, Lan Y, Weigt M, Aurell E. Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models. Phys Rev E - Stat Nonlinear Soft Matter Phys. 2013;87:1–16. doi: 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- 15.Balakrishnan S, Kamisetty H, Carbonell JG, Lee S-I, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79:1061–1078. doi: 10.1002/prot.22934. [DOI] [PubMed] [Google Scholar]
- 16.Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci U S A. 2013;110:1–6. doi: 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seemayer S, Gruber M, Söding J. CCMpred - Fast and precise prediction of protein residue-residue contacts from correlated mutations. Bioinformatics. 2014;30:3128–3130. doi: 10.1093/bioinformatics/btu500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lapedes A, Giraud B, Jarzynski C. Using Sequence Alignments to Predict Protein Structure and Stability With High Accuracy. bioRxiv. 2012:1–29. [Google Scholar]
- 19.Burger L, Van Nimwegen E. Disentangling direct from indirect co-evolution of residues in protein alignments. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones DT, Buchan DWA, Cozzetto D, Pontil M. PSICOV : precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–190. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 21.Ma J, Wang S, Wang Z, Xu J. Protein contact prediction by integrating joint evolutionary coupling analysis and supervised learning. Bioinformatics. 2015;31:3506–3513. doi: 10.1093/bioinformatics/btv472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopf TA, Colwell LJ, Sheridan R, Rost B, Sander C, Marks DS. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayat S, Sander C, Marks DS, Elofsson A. All-atom 3D structure prediction of transmembrane β-barrel proteins from sequences. Proc Natl Acad Sci U S A. 2015;112:5413–8. doi: 10.1073/pnas.1419956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toth-Petroczy A, Palmedo P, Ingraham J, Hopf TA, Berger B, Sander C, Marks DS. Structured States of Disordered Proteins from Genomic Sequences Article Structured States of Disordered Proteins from Genomic Sequences. Cell. 2016;167:158–170. doi: 10.1016/j.cell.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hopf TA, Schärfe CPI, Rodrigues JPGLM, Green AG, Sander C, Bonvin AMJJ, Marks DS. Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife. 2014;3:e03430. doi: 10.7554/eLife.03430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife. 2014;3:e02030. doi: 10.7554/eLife.02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CPI, Springer M, Sander C, Marks DS. Mutation effects predicted from sequence co-variation. Nat Biotechnol. 2017;35:128–135. doi: 10.1038/nbt.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cocco S, Feinauer C, Figliuzzi M, Weigt M. Inverse Statistical Physics of Protein Sequences: A Key Issues Review. arXiv. 2017:1–18. doi: 10.1088/1361-6633/aa9965. [DOI] [PubMed] [Google Scholar]
- 29.Jones DT, Singh T, Kosciolek T, Tetchner S. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics. 2015;31:999–1006. doi: 10.1093/bioinformatics/btu791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skwark MJ, Abdel-Rehim A, Elofsson A. PconsC: Combination of direct information methods and alignments improves contact prediction. Bioinformatics. 2013;29:1815–1816. doi: 10.1093/bioinformatics/btt259. [DOI] [PubMed] [Google Scholar]
- 31.Skwark MJ, Raimondi D, Michel M, Elofsson A. Improved Contact Predictions Using the Recognition of Protein Like Contact Patterns. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michel M, Hayat S, Skwark MJ, Sander C, Marks DS, Elofsson A. PconsFold: Improved contact predictions improve protein models. Bioinformatics. 2014;30:482–488. doi: 10.1093/bioinformatics/btu458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. 2017 doi: 10.1371/journal.pcbi.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henrique S, De Oliveira P, Shi J, Deane CM. Comparing co-evolution methods and their application to template-free protein structure prediction. Bioinformatics. 2017;33:373–381. doi: 10.1093/bioinformatics/btw618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wuyun Q, Zheng W, Peng Z, Yang J. A large-scale comparative assessment of methods for residue-residue contact prediction. Brief Bioinform. 2016 doi: 10.1093/bib/bbw106. bbw106. [DOI] [PubMed] [Google Scholar]
- 36.Orlando G, Raimondi D, Vranken WF. Observation selection bias in contact prediction and its implications for structural bioinformatics. Nat Publ Gr. 2016:1–8. doi: 10.1038/srep36679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr Sect D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Simons KT, Strauss C, Baker D. Prospects for ab initio protein structural genomics. J Mol Biol. 2001;306:1191–1199. doi: 10.1006/jmbi.2000.4459. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs A, Martin-Galiano AJ, Kalman M, Fleishman S, Ben-Tal N, Frishman D. Co-evolving residues in membrane proteins. Bioinformatics. 2007;23:3312–3319. doi: 10.1093/bioinformatics/btm515. [DOI] [PubMed] [Google Scholar]
- 40.Hopf Ta, Colwell LJ, Sheridan R, Rost B, Sander C, Marks DS. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickles S, Singharoy A, Andreani J, Seemayer S, Bischoff L, Berninghausen O, Soeding J, Schulten K, van der Sluis EO, Beckmann R. A structural model of the active ribosome-bound membrane protein insertase YidC. Elife. 2014:e03035. doi: 10.7554/eLife.03035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumazaki K, Chiba S, Takemoto M, Furukawa A, Nishiyama K, Sugano Y, Mori T, Dohmae N, Hirata K, Nakada-Nakura Y, Maturana AD, Tanaka Y, Mori H, Sugita Y, Ito K, Ishitani R, Tsukazaki T, Nureki O. Structural basis of Sec-independent membrane. Nature. 2014;509:516–520. doi: 10.1038/nature13167. [DOI] [PubMed] [Google Scholar]
- 43.Zimmerman B, Kelly B, McMillan BJ, Seegar TCM, Dror RO, Kruse AC, Blacklow SC. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell. 2016;167:1041–1051. e11. doi: 10.1016/j.cell.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nugent T, Jones DT. Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc Natl Acad Sci U S A. 2012;109:E1540–7. doi: 10.1073/pnas.1120036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marks DS, Hopf TA, Sander C. Protein structure prediction from sequence variation. Nat Biotechnol. 2012;30:1072–80. doi: 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng X, Barth P. A topological and conformational stability alphabet for multipass membrane proteins. Nat Chem Biol. 2016;12:167–173. doi: 10.1038/nchembio.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor WR, Jones DT, Sadowski MI. Protein topology from predicted residue contacts. Protein Sci. 2012;21:299–305. doi: 10.1002/pro.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Wang H, Yan L, Su L, Xu D. OMPcontact: An Outer Membrane Protein Inter-Barrel Residue Contact Prediction Method. J Comput Biol. 2016;24 doi: 10.1089/cmb.2015.0236. cmb.2015.0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. Rewiring the Specificity of Two-Component Signal Transduction Systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aguilar D, Oliva B, Buslje CM. Mapping the Mutual Information Network of Enzymatic Families in the Protein Structure to Unveil Functional Features. PLoS One. 2012;7:1–12. doi: 10.1371/journal.pone.0041430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buslje CM, Teppa E, Di Doménico T, Delfino JM, Nielsen M. Networks of High Mutual Information Define the Structural Proximity of Catalytic Sites : Implications for Catalytic Residue Identification. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colwell LJ, Brenner MP, Murray AW. Conservation Weighting Functions Enable Covariance Analyses to Detect Functionally Important Amino Acids. PLoS One. 2014;9:e107723. doi: 10.1371/journal.pone.0107723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pelé J, Moreau M, Abdi H, Rodien P, Castel H, Chabbert M. Comparative analysis of sequence co-variation methods to mine evolutionary hubs : Examples from selected GPCR families. Proteins Struct Funct Bioinforma. 2014:1–16. doi: 10.1002/prot.24570. [DOI] [PubMed] [Google Scholar]
- 54.Park K, Kim D. Structure-based rebuilding of coevolutionary information reveals functional modules in rhodopsin structure. Biochim Biophys Acta. 2012;1824:1484–1489. doi: 10.1016/j.bbapap.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 55.Bay DC, Hafez M, Young MJ, Court DA. Phylogenetic and coevolutionary analysis of the β-barrel protein family comprised of mitochondrial porin (VDAC) and Tom40. Biochim Biophys Acta - Biomembr. 2012;1818:1502–1519. doi: 10.1016/j.bbamem.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 56.Lichtarge O, Bourne HR, Cohen FE. An Evolutionary Trace Method Defines Binding Surfaces Common to Protein Families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 57.De Juan D, Pazos F, Valencia A. Emerging methods in protein co-evolution. Nat Publ Gr. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 58.Madabushi S, Gross AK, Philippi A, Meng EC, Wensel TG, Lichtarge O. Evolutionary Trace of G Protein-coupled Receptors Reveals Clusters of Residues That Determine Global and Class-specific Functions. 2004;279:8126–8132. doi: 10.1074/jbc.M312671200. [DOI] [PubMed] [Google Scholar]
- 59.Sung Y, Wilkins AD, Rodriguez GJ, Wensel TG, Lichtarge O. Intramolecular allosteric communication in dopamine D2 receptor revealed by evolutionary amino acid covariation. Proc Natl Acad Sci U S A. 2016;113:3539–3544. doi: 10.1073/pnas.1516579113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dunn SD, Wahl LM, Gloor GB. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- 61.Fodor AA, Aldrich RW. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins Struct Funct Genet. 2004;56:211–221. doi: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 62.Lockless SW, Ranganathan R. Evolutionarily Conserved Pathways of Energetic Connectivity in Protein Families. Science (80-) 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 63.Halabi N, Rivoire O, Leibler S, Ranganathan R. Theory Protein Sectors : Evolutionary Units of Three-Dimensional Structure. Cell. 2009;138:774–786. doi: 10.1016/j.cell.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Mol Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 65.Mclaughlin RN, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;490:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nichols SE, Hern CX, Wang Y, Mccammon JA. Structure-based network analysis of an evolved G protein-coupled receptor homodimer interface. Protein Sci. 2013;22:745–754. doi: 10.1002/pro.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sayar K, Özlem U, Liu T, Hilser VJ, Onaran O. Exploring allosteric coupling in the α -subunit of Heterotrimeric G proteins using evolutionary and ensemble-based approaches. BMC Struct Biol. 2008;14:1–14. doi: 10.1186/1472-6807-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dima RI, Thirumalai D. Determination of network of residues that regulate allostery in protein families using sequence analysis. Protein Sci. 2006;15:258–268. doi: 10.1110/ps.051767306.of. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferguson AD, Amezcua CA, Halabi NM, Chelliah Y, Rosen MK, Ranganathan R, Deisenhofer J. Signal transduction pathway of TonB-dependent transporters. Proc Natl Acad Sci U S A. 2006;104:513–518. doi: 10.1073/pnas.0609887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teşileanu T, Colwell LJ, Leibler S. Protein Sectors: Statistical Coupling Analysis versus Conservation. PLoS Comput Biol. 2015;11:1–20. doi: 10.1371/journal.pcbi.1004091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas J, Ramakrishnan N, Bailey-Kellogg C. Graphical Models of Residue Coupling in Protein Families, IEEE/ACM Trans. Comp Biol Bioinf. 2008;5:183–197. doi: 10.1109/TCBB.2007.70225. [DOI] [PubMed] [Google Scholar]
- 72.Thomas J, Ramakrishnan N, Bailey-Kellogg C. Graphical models of protein-protein interaction specificity from correlated mutations and interaction data. Proteins Struct Funct Bioinforma. 2009;76:911–929. doi: 10.1002/prot.22398. [DOI] [PubMed] [Google Scholar]
- 73.Pandini A, Kleinjung J, Rasool S, Khan S. Coevolved mutations reveal distinct architectures for two core proteins in the bacterial flagellar motor. PLoS One. 2015;10:1–28. doi: 10.1371/journal.pone.0142407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dwyer RS, Ricci DP, Colwell LJ, Silhavy TJ, Wingreen NS. Predicting functionally informative mutations in Escherichia coli BamA using evolutionary covariance analysis. Genetics. 2013;195:443–455. doi: 10.1534/genetics.113.155861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clantin B, Delattre A-SS, Rucktooa P, Saint N, Méli AC, Locht C, Françoise J-D, Villeret V. Structure of the membrane protein FhaC: a member of the Omp85-TpsB transporter superfamily. Science (80-) 2007;317:957–961. doi: 10.1126/science.1143860. [DOI] [PubMed] [Google Scholar]
- 76.Ni D, Wang Y, Yang X, Zhou H, Hou X, Cao B, Lu Z, Zhao X, Yang K, Huang Y. Structural and functional analysis of the β-barrel domain of BamA from Escherichia coli. FASEB J. 2014;28:2677–2685. doi: 10.1096/fj.13-248450. [DOI] [PubMed] [Google Scholar]
- 77.Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. Structural insight into the biogenesis of β-barrel membrane proteins. Nature. 2013;501:385–90. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maier T, Clantin B, Gruss F, Dewitte F, Delattre A-S, Jacob-Dubuisson F, Hiller S, Villeret V. Conserved Omp85 lid-lock structure and substrate recognition in FhaC. Nat Commun. 2015;6:7452. doi: 10.1038/ncomms8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Catterall WA, Wisedchaisri G, Zheng N. The chemical basis for electrical signaling. Nat Chem Biol. 2017;13:455–463. doi: 10.1038/nchembio.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palovcak E, Delemotte L, Klein ML, Carnevale V. Evolutionary imprint of activation : The design principles of VSDs. J Gen Physiol. 2014;143:145–156. doi: 10.1085/jgp.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Catterall WA. Ion Channel Voltage Sensors: Structure, Function, and Pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alabi AA, Bahamonde MI, Jung HJ, IlKim J, Swartz KJ. Portability of paddle motif function and pharmacology in voltage sensors. Nature. 2007;450:370–375. doi: 10.1038/nature06266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Palovcak E, Delemotte L, Klein ML, Carnevale V. Comparative sequence analysis suggests a conserved gating mechanism for TRP channels. J Gen Physiol. 2015;146:37–50. doi: 10.1085/jgp.201411329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee S, Banerjee A, Mackinnon R. Two Separate Interfaces between the Voltage Sensor and Pore Are Required for the Function of Voltage-Dependent K+ Channels. PLoS Biol. 2009;7:0676–0686. doi: 10.1371/journal.pbio.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pless SA, Niciforovic AP, Galpin JD, Nunez J, Kurata HT, Ahern CA. A novel mechanism for fine-tuning open-state stability in a voltage-gated potassium channel. Nat Commun. 2013;4:1711–1784. doi: 10.1038/ncomms2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. 2013 doi: 10.1038/nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cao E, Liao M, Cheng Y, Julius D. TRPV1 structures in distinct conformations reveal mechanisms of activation. 2013 doi: 10.1038/natu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Paulsen CE, Armache J, Gao Y, Cheng Y, Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;520:511–517. doi: 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aakre CD, Herrou J, Phung TN, Perchuk BS, Crosson S, Laub MT. Evolving New Protein-Protein Interaction Specificity through Promiscuous Intermediates. Cell. 2015;163:594–606. doi: 10.1016/j.cell.2015.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goldsmith M, Eckstein S, Ashani Y, Greisen P, Leader H, Sussman JL, Aggarwal N, Ovchinnikov S, Tawfik DS, Baker D, Thiermann H, Worek F. Catalytic efficiencies of directly evolved phosphotriesterase variants with structurally different organophosphorus compounds in vitro. Arch Toxicol. 2016;90:2711–2724. doi: 10.1007/s00204-015-1626-2. [DOI] [PubMed] [Google Scholar]
- 91.White RA, Szurmant H, Hoch JA, Hwa T. Features of Protein-Protein Interactions in Two-Component Signaling Deduced from Genomic Libraries. Methods Enzymol. 2007;422:75–101. doi: 10.1016/S0076-6879(06)22004-4. [DOI] [PubMed] [Google Scholar]
- 92.Cheng RR, Morcos F, Levine H, Onuchic JN. Toward rationally redesigning bacterial two-component signaling systems using coevolutionary information. Proc Natl Acad Sci U S A. 2014;111:E563–71. doi: 10.1073/pnas.1323734111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Procaccini A, Lunt B, Szurmant H, Hwa T, Weigt M. Dissecting the specificity of protein-protein interaction in bacterial two-component signaling: Orphans and crosstalks. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bai F, Morcos F, Cheng RR, Jiang H, Onuchic JN. Elucidating the druggable interface of protein – protein interactions using fragment docking and coevolutionary analysis. Proc Natl Acad Sci. 2016:8051–8058. doi: 10.1073/pnas.1615932113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bitbol A-F, Dwyer RS, Colwell LJ, Wingreen NS. Inferring interaction partners from protein sequences. Proc Natl Acad Sci U S A. 2016;113:50732. doi: 10.1101/050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gueudré T, Baldassi C, Zamparo M, Weigt M, Pagnani A. Simultaneous identification of specifically interacting paralogs and interprotein contacts by direct coupling analysis. Proc Natl Acad Sci U S A. 2016;113:12186–12191. doi: 10.1073/pnas.1607570113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hopf TA, Morinaga S, Ihara S, Touhara K, Marks DS, Benton R. Amino acid coevolution reveals three-dimensional structure and functional domains of insect odorant receptors. Nat Commun. 2015;6:6077. doi: 10.1038/ncomms7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Uguzzoni G, John S, Oteri F, Schug A, Szurmant H, Weigt M. Large-scale identification of coevolution signals across homo-oligomeric protein interfaces by direct coupling analysis. Proc Natl Acad Sci. 2017 doi: 10.1073/pnas.1615068114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nicoludis JM, Lau S, Schärfe C, Marks DS, Weihofen WA, Gaudet R. Structure and Sequence Analyses of Clustered Protocadherins Reveal Antiparallel Interactions that Mediate Homophilic Specificity. Structure. 2015;23:2087–2098. doi: 10.1016/j.str.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rubinstein R, Thu CA, Goodman KM, Wolcott HN, Bahna F, Mannepalli S, Ahlsen G, Chevee M, Halim A, Clausen H, Maniatis T, Shapiro L, Honig B. Molecular Logic of Neuronal Self-Recognition through Protocadherin Domain Interactions Article Molecular Logic of Neuronal Self-Recognition through Protocadherin Domain Interactions. Cell. 2015;163:1–14. doi: 10.1016/j.cell.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicoludis JM, Vogt BE, Green AG, Schärfe CPI, Marks DS, Gaudet R. Antiparallel protocadherin homodimers use distinct affinity-and specificity-mediating regions in cadherin repeats 1-4. Elife. 2016;5:1–14. doi: 10.7554/eLife.18449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goodman KM, Rubinstein R, Thu CA, Bahna F, Mannepalli S, Ahlsén G, Rittenhouse C, Maniatis T, Honig B, Shapiro L. Structural Basis of Diverse Homophilic Recognition by Clustered α- and β-Protocadherins. Neuron. 2016;90:709–23. doi: 10.1016/j.neuron.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goodman KM, Rubinstein R, Thu CA, Shapiro L, Maniatis T, Honig B. γ-Protocadherin structural diversity and functional implications. Elife. 2016:1–25. doi: 10.1002/ecy.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cooper SR, Jontes JD, Sotomayor M. Structural determinants of adhesion by protocadherin-19 and implications for its role in epilepsy. Elife. 2016;5:1–22. doi: 10.7554/eLife.18529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dos Santos RN, Morcos F, Jana B, Andricopulo AD, Onuchic JN. Dimeric interactions and complex formation using direct coevolutionary couplings. Sci Rep. 2015;5:13652. doi: 10.1038/srep13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang Y, Lü Q, Zeng J. Using evolutionary couplings to optimize dimer prediction with Rosetta SymDock. Proc - 2013 IEEE Int Conf Bioinforma Biomed; IEEE BIBM 2013; 2013. pp. 65–68. [DOI] [Google Scholar]
- 107.Procko E, Mara MLO, Bennett WFD, Tieleman DP, Gaudet R. The mechanism of ABC transporters : general lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 2009;23:1287–1302. doi: 10.1096/fj.08-121855. [DOI] [PubMed] [Google Scholar]
- 108.Stansfeld PJ. Computational studies of membrane proteins: from sequence to structure to simulation. Curr Opin Struct Biol. 2017;45:133–141. doi: 10.1016/j.sbi.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 109.Tsuchiya H, Doki S, Takemoto M, Ikuta T, Higuchi T, Fukui K, Usuda Y, Tabuchi E, Nagatoishi S, Tsumoto K, Nishizawa T, Ito K, Dohmae N, Ishitani R, Nureki O. Structural basis for amino acid export by DMT superfamily transporter YddG. Nature. 2016;534:417–420. doi: 10.1038/nature17991. [DOI] [PubMed] [Google Scholar]
- 110.Gulyás-Kovács A. Integrated Analysis of Residue Coevolution and Protein Structure in ABC Transporters. PLoS One. 2012;7:e36546. doi: 10.1371/journal.pone.0036546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 112.Radestock S, Forrest LR. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol. 2011;407:698–715. doi: 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 113.Forrest LR, Krämer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta - Bioenerg. 2011;1807:167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 114.Hirai T, Subramaniam S. Structure and Transport Mechanism of the Bacterial Oxalate Transporter OxlT. Biophys J. 2004;87:3600–3607. doi: 10.1529/biophysj.104.049320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the Multidrug Transporter EmrD from Escherichia coli. Science (80-) 2011;312:741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sutto L, Marsili S, Valencia A, Gervasio FL. From residue coevolution to protein conformational ensembles and functional dynamics. Proc Natl Acad Sci. 2015;112:13567–13572. doi: 10.1073/pnas.1508584112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sfriso P, Duran-Frigola M, Mosca R, Emperador A, Aloy P, Orozco M. Residues Coevolution Guides the Systematic Identification of Alternative Functional Conformations in Proteins. Structure. 2016;24:116–126. doi: 10.1016/j.str.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 118.Morcos F, Jana B, Hwa T, Onuchic JN. Coevolutionary signals across protein lineages help capture multiple protein conformations. Proc Natl Acad Sci U S A. 2013;110:20533–8. doi: 10.1073/pnas.1315625110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jeon J, Nam HJ, Choi YS, Yang JS, Hwang J, Kim S. Molecular evolution of protein conformational changes revealed by a network of evolutionarily coupled residues. Mol Biol Evol. 2011;28:2675–2685. doi: 10.1093/molbev/msr094. [DOI] [PubMed] [Google Scholar]
- 120.Baker D, Sali A. Protein Structure Prediction and Structural Genomics. Science (80-) 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 121.Antala S, Ovchinnikov S, Kamisetty H, Baker D, Dempski RE. Computation and functional studies provide a model for the structure of the zinc transporter hZIP4. J Biol Chem. 2015;290:17796–17805. doi: 10.1074/jbc.M114.617613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang T, Liu J, Fellner M, Zhang C, Sui D, Hu J. Crystal structures of a ZIP zinc transporter reveal a binuclear metal center in the transport pathway. Sci Adv. 2017;3:e1700344. doi: 10.1126/sciadv.1700344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abriata LA. An homology- and coevolution-consistent structural model of bacterial copper- tolerance protein CopM supports function as a “metal sponge” and suggests regions for metal-dependent interactions with other proteins. bioRxiv. 2015:0–4. [Google Scholar]
- 124.Gofman Y, Schärfe C, Marks DS, Haliloglu T, Ben-Tal N. Structure, Dynamics and Implied Gating Mechanism of a Human Cyclic Nucleotide-Gated Channel. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li N, Wu J, Ding D, Li N, Wu J, Ding D, Cheng J, Gao N, Chen L. Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell. 2017;168:101–110. e10. doi: 10.1016/j.cell.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 126.Jaremko L, Jaremko M, Giller K, Becker S, Zweckstetter M. Structure of the Mitochondrial Translocator Protein in Complex with a Diagnostic Ligand. Science (80-) 2014;1363:1363–1367. doi: 10.1126/science.1248725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Korkhov VM, Sachse C, Short JM, Tate CG. Three-Dimensional Structure of TspO by Electron Cryomicroscopy of Helical Crystals. Structure. 2010;18:677–687. doi: 10.1016/j.str.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li F, Xia Y, Meiler J, Ferguson-Miller S. Characterization and modeling of the oligomeric state and ligand binding behavior of purified translocator protein 18 kDa from Rhodobacter sphaeroides. Biochemistry. 2013;52:5884–5899. doi: 10.1021/bi400431t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hinsen K, Vaitinadapoule A, Ostuni MA, Etchebest C, Lacapere JJ. Construction and validation of an atomic model for bacterial TSPO from electron microscopy density, evolutionary constraints, and biochemical and biophysical data, Biochim. Biophys Acta - Biomembr. 2015;1848:568–580. doi: 10.1016/j.bbamem.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 130.Li F, Liu J, Zheng Y, Garavito RM, Ferguson-Miller S. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science (80-) 2015;347:555–558. doi: 10.1126/science.1260590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Allegretti M, Klusch N, Mills DJ, Vonck J, Kühlbrandt W, Davies KM. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature. 2015;521:237–240. doi: 10.1038/nature14185. [DOI] [PubMed] [Google Scholar]
- 132.Jiang W, Fillingame RH. Interacting helical faces of subunits a and c in the F1Fo ATP synthase of Escherichia coli defined by disulfide cross-linking. Proc Natl Acad Sci U S A. 1998;95:6607–12. doi: 10.1073/pnas.95.12.6607. VL - 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fillingame RH, Steed PR. Half channels mediating H+ transport and the mechanism of gating in the Fo sector of Escherichia coli F1Fo ATP synthase. Biochim Biophys Acta - Bioenerg. 2014;1837:1063–1068. doi: 10.1016/j.bbabio.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 134.Leone V, Faraldo-Gómez JD. Structure and mechanism of the ATP synthase membrane motor inferred from quantitative integrative modeling. J Gen Physiol. 2016;148 doi: 10.1085/jgp.201611679. jgp.201611679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhou A, Rohou A, Schep DG, Bason JV, Montgomery MG, Walker JE, Grigorieffniko N, Rubinstein JL. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife. 2015;4:1–15. doi: 10.7554/eLife.10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schep DG, Zhao J, Rubinstein JL. Models for the a subunits of the Thermus thermophilus V/A-ATPase and Saccharomyces cerevisiae V-ATPase enzymes by cryo-EM and evolutionary covariance. Proc Natl Acad Sci. 2016;113:3245–3250. doi: 10.1073/pnas.1521990113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Palmer T, Berks BC. The twin-arginine translocation Tat protein export pathway. Nat Rev Microbiol. 2012;10:483–96. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- 138.Gohlke U, Pullan L, Mcdevitt CA, Porcelli I, De Leeuw E, Palmer T, Saibil HR, Berks BC. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Na. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tarry MJ, Scäfer E, Chen S, Buchanan G, Greene NP, Lea SM, Palmer T, Saibil HR, Berks BC. Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc Natl Acad Sci U S A. 2009;106:13284–13289. doi: 10.1073/pnas.0901566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hu Y, Zhao E, Li H, Xia B, Jin C. Solution NMR structure of the TatA component of the twin-arginine protein transport system from gram-positive bacterium bacillus subtilis. J Am Chem Soc. 2010;132:15942–15944. doi: 10.1021/ja1053785. [DOI] [PubMed] [Google Scholar]
- 141.Rollauer SE, Tarry MJ, Graham JE, Jääskeläinen M, Jäger F, Johnson S, Krehenbrink M, Liu S-M, Lukey MJ, Marcoux J, McDowell MA, Rodriguez F, Roversi P, Stansfeld PJ, Robinson CV, Sansom MSP, Palmer T, Högbom M, Berks BC, Lea SM. Structure of the TatC core of the twin-arginine protein transport system. Nature. 2012;492:210–214. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rodriguez F, Rouse SL, Tait CE, Harmer J, De Riso A, Timmel CR, Sansom MSP, Berks BC, Schnell JR. Structural model for the protein-translocating element of the twin-arginine transport system. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1219486110. DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1219486110. [DOI] [PMC free article] [PubMed]
- 143.Zhang Y, Wang L, Hu Y, Jin C. Solution structure of the TatB component of the twin-arginine translocation system. Biochim Biophys Acta - Biomembr. 2014;1838:1881–1888. doi: 10.1016/j.bbamem.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 144.Ramasamy S, Abrol R, Suloway CJM, Clemons WM. The glove-like structure of the conserved membrane protein TatC provides insight into signal sequence recognition in twin-arginine translocation. Structure. 2013;21:777–788. doi: 10.1021/nl061786n.Core-Shell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Alcock F, Stansfeld PJ, Basit H, Habersetzer J, Matthew AB. Assembling the Tat protein translocase. Elife. 2016;5:e20718. doi: 10.7554/eLife.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Glaser F, Pupko T, Paz I, Bell RE, Bechor D, Martz E. ConSurf: identification of functional regions in proteins by surface mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- 147.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 148.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 150.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 151.Consortium TU. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Isberg V, Mordalski S, Munk C, Rataj K, Harpsøe K, Hauser AS, Vroling B, Bojarski AJ, Vriend G, Gloriam DE. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2016;44:D356–D364. doi: 10.1093/nar/gkv1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ovchinnikov S, Park H, Varghese N, Huang P-S, Pavlopoulos GA, Kim DE, Kamisetty H, Kyrpides NC, Baker D. Protein structure determination using metagenome sequence data. Science (80-) 2017;355:294–298. doi: 10.1126/science.aah4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Steinegger M, Soding J. Linclust: clustering billions of protein sequences per day on a single server. bioRxiv. 2017:1–5. doi: 10.1101/104034. [DOI] [Google Scholar]
- 157.Godzik A. Metagenomics and the protein universe. Curr Opin Struct Biol. 2011;21:398–403. doi: 10.1016/j.sbi.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Do CB, Katoh K. Protein Multiple Sequence Alignment. Funct Proteomics. 2008:379–413. doi: 10.1007/978-1-59745-398-1. [DOI] [PubMed] [Google Scholar]
- 159.Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015;43:W7–W14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wolfsheimer S, Hartmann A, Rabus R, Nuel G. Computing posterior probabilities for score-based alignments using ppALIGN. Stat Appl Genet Mol Biol. 2012;11 doi: 10.1515/1544-6115.1702. [DOI] [PubMed] [Google Scholar]
- 161.Fuchs A, Martin-Galiano AJ, Kalman M, Fleishman S, Ben-Tal N, Frishman D. Co-evolving residues in membrane proteins. Bioinformatics. 2007;23:3312–9. doi: 10.1093/bioinformatics/btm515. [DOI] [PubMed] [Google Scholar]
- 162.Feinauer C, Skwark MJ, Pagnani A, Aurell E. Improving Contact Prediction along Three Dimensions. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Avila-Herrera A, Pollard KS. Coevolutionary analyses require phylogenetically deep alignments and better null models to accurately detect inter-protein contacts within and between species. BMC Bioinformatics. 2015;16:268. doi: 10.1186/s12859-015-0677-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dutheil JY. Detecting coevolving positions in a molecule: Why and how to account for phylogeny. Brief Bioinform. 2012;13:228–243. doi: 10.1093/bib/bbr048. [DOI] [PubMed] [Google Scholar]
- 165.Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cocco S, Monasson R, Weigt M. Inference of Hopfield-Potts patterns from covariation in protein families: calculation and statistical error bars. J Phys Conf Ser. 2013;473:12010. doi: 10.1088/1742-6596/473/1/012010. [DOI] [Google Scholar]
- 167.van Nimwegen E. Inferring Contacting Residues within and between Proteins: What Do the Probabilities Mean? PLoS Comput Biol. 2016;12:1–10. doi: 10.1371/journal.pcbi.1004726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Anishchenko I, Ovchinnikov S, Kamisetty H, Baker D. Origins of coevolution between residues distant in protein 3D structures. Proc Natl Acad Sci. 2017:201702664. doi: 10.1073/pnas.1702664114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rivas E, Clements J, Eddy SR. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods. 2016;14:1–8. doi: 10.1038/nmeth.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jeong CS, Kim D. Reliable and robust detection of coevolving protein residues. Protein Eng Des Sel. 2012;25:705–713. doi: 10.1093/protein/gzs081. [DOI] [PubMed] [Google Scholar]
- 171.Simonetti FL, Teppa E, Chernomoretz A, Nielsen M, Marino Buslje C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013;41:8–14. doi: 10.1093/nar/gkt427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gouveia-Oliveira R, Roque FS, Wernersson R, Sicheritz-Ponten T, Sackett PW, Mølgaard A, Pedersen AG. InterMap3D: Predicting and visualizing co-evolving protein residues. Bioinformatics. 2009;25:1963–1965. doi: 10.1093/bioinformatics/btp335. [DOI] [PubMed] [Google Scholar]
- 173.Ackerman SH, Tillier ER, Gatti DL. Accurate Simulation and Detection of Coevolution Signals in Multiple Sequence Alignments. PLoS One. 2012;7 doi: 10.1371/journal.pone.0047108. [DOI] [PMC free article] [PubMed] [Google Scholar]