

SUMMARY
Nonalcoholic fatty liver disease (NAFLD) is the leading chronic liver disease worldwide. Its more advanced subtype, nonalcoholic steatohepatitis (NASH), connotes progressive liver injury that can lead to cirrhosis and hepatocellular carcinoma. Here we provide an in-depth discussion of the underlying pathogenetic mechanisms that lead to progressive liver injury, including the metabolic origins of NAFLD, the effect of NAFLD on hepatic glucose and lipid metabolism, bile acid toxicity, macrophage dysfunction, and hepatic stellate cell activation, and consider the role of genetic, epigenetic, and environmental factors that promote fibrosis progression and risk of hepatocellular carcinoma in NASH.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is emerging as the leading chronic liver disease worldwide (Loomba and Sanyal, 2013; Younossi et al., 2016). NAFLD is viewed as the hepatic manifestation of metabolic syndrome and is commonly associated with metabolic risk factors, including obesity, dyslipidemia, hypertension, and diabetes (Chalasani et al., 2018; Neuschwander-Tetri et al., 2010; Samuel and Shulman, 2018). The rising rates of obesity and type 2 diabetes worldwide are paralleled by a rise in the global prevalence of NAFLD.
In this review, we describe the emerging data regarding the epidemiology and natural history of NAFLD and its progression tononalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma (HCC) and summarize data regarding new mechanistic insights into metabolic drivers of the transition from NAFL (nonalcoholic fatty liver), the non-progressive subtype of NAFLD, to NASH, the progressive subtype of NAFLD, and pathogenetic mechanisms of fibrosis progression and HCC in NASH.
Epidemiology of NAFLD
NAFLD is estimated to afflict one billion individuals globally and may be present in approximately 25% of the world population (Younossi et al., 2019b). There is considerable variability in the prevalence of NAFLD across the various geographic regions in the world. The Middle East and South America have the highest and Africa the lowest prevalence of NAFLD. In the United States, up to 80 million individuals may have NAFLD (Estes et al., 2018).
Globally, the rates of NAFLD in non-obese individuals average ~10%–30% in Western and Eastern countries (Kim and Kim, 2017). There are also gender and racial/ethnic differences in the prevalence of non-obese NAFLD; the prevalence of NAFLD is much higher in non-obese South Asian men than in non-obese South Asian women and in men and women of other ethnic groups (Petersen et al., 2006), and the prevalence of NAFLD is higher in Hispanic and white than in Black individuals (Browning and Horton, 2004). Non-obese subjects with NAFLD also have a markedly higher cardiovascular risk (Yoshitaka et al., 2017).
Over the last 3 decades, the prevalence of NAFLD in the United States has risen from 20% in 1988–1994 to 32% in 2012–2016. Although the prevalence of viral hepatitis has been declining in the United States, there has been a tremendous increase in NAFLD. These rising trends in the prevalence and incidence of NAFLD are also noted in children and adolescents in the United States. Similar rises are seen elsewhere in the world, including Europe, China, India, and other regions. Therefore, NAFLD has become a global health problem that imposes a significant socioeconomic burden.
Definition of NAFLD
NAFLD is diagnosed using either an imaging or a liver biopsy assessment in individuals with at least 5% of hepatocytes infiltrated with steatosis (or a magnetic resonance imaging proton density fat fraction [MRI-PDFF] of 5% or higher or magnetic resonance spectroscopy liver fat content of more than 5.56% (Szczepaniak et al., 2005) in individuals who consume little or no alcohol and without any secondary causes of hepatic steatosis, such as other metabolic liver diseases (e.g., Wilson’s disease, congenital or acquired lipodystrophy) or drugs (e.g., amiodarone, steroids, tamoxifen, valproic acid, etc.) (Chalasani et al., 2018). NAFLD is broadly classified into two subtypes: nonalcoholic fatty liver (NAFL), the non-progressive form of NAFLD, and NASH, the progressive form of NAFLD (Figure 1; Singh et al., 2015). Diagnosis of NASH requires histological assessment in the context of suspected NAFLD and is typically characterized by the presence of steatosis, lobular inflammation, and ballooning with or without perisinusoidal fibrosis (Kleiner et al., 2005). A subset of individuals with NASH may develop progressive liver injury that advances to cirrhosis and HCC (Dulai et al., 2017).
Figure 1. Histologic difference between NAFLD and NASH.

Shown is the liver histology of NAFLD with an example of an individual with nonalcoholic fatty liver (NAFL), the non-progressive form of NAFLD, showing macrovesicular steatosis and mild lobular inflammation but no ballooning, and nonalcoholic steatohepatitis (NASH), the progressive form of NAFLD, showing ballooned hepatocytes (arrows) in addition to steatosis and lobular inflammation.
Clinical presentation
The majority of individuals with NAFLD are asymptomatic, and the disease may remain silent until it has progressed to cirrhosis (Spengler and Loomba, 2015). The most common symptoms noted at initial referral among individuals with NAFLD include right upper quadrant pain and fatigue. Affected people may have an echogenic liver on ultrasound or evidence of liver fat based on an imaging test that is noted incidentally or as part of a workup for right upper quadrant pain (Chalasani et al., 2018). Liver-related serum tests typically reflect a hepatocellular pattern of enzyme elevations with serum alanine aminotransferase (ALT) higher than serum aspartate aminotransferase (AST)(Torres et al., 2012). When advanced fibrosis, cirrhosis, and portal hypertension have developed, the platelet count declines gradually over years (Loomba and Adams, 2019). Decreased serum albumin and an increased prothrombin time is noted in individuals with cirrhosis who manifest evidence of synthetic dysfunction (Loomba and Adams, 2019).
Natural history of NAFLD
Recent data from pooled analyses and paired biopsy studies suggest that the transition from NAFL to NASH is quite dynamic. Fibrosis progression is significantly slower in NAFL than NASH, requiring 14 years per stage of fibrosis, whereas in NASH, each stage progresses over 7 years (Singh et al., 2015). More importantly, approximately 20% of individuals with NASH may be classified as “rapid progressors,” in whom each stage progresses in less than 7 years. Early identification of rapid progressors is among the greatest unmet needs in the field. Predictors of rapid progression have not been fully quantified but include higher serum ALT, presence of diabetes, family history of cirrhosis in first-degree relatives, and possibly genetic susceptibility (Caussy et al., 2017; Rafiq et al., 2009; Stender and Loomba, 2020).
It is estimated that only 20% of individuals with NAFLD have NASH, and 20% of these individuals may progress to cirrhosis over 3–4 decades (Singh et al., 2015). People with NASH-related cirrhosis carry an ~1.5%–2% per year risk of developing incident HCC (Figure 2; Loomba et al., 2020; see below). Therefore, among individuals with NASH-related cirrhosis, HCC screening and surveillance are routinely recommended by the American Association for the Study of Liver Diseases, with ultrasound with or without alpha-fetoprotein (AFP) every 6 months. Although HCC has been reported in non-cirrhotic individuals with NAFLD at a higher rate than for other chronic liver disease etiologies, the incidence rate of HCC is still too low to recommend routine HCC screening or surveillance among those with NAFLD unless there is advanced fibrosis or cirrhosis.
Figure 2. Natural history of NAFLD.

NAFLD afflicts up to 80 million Americans, and its progressive form, NASH, affects approximately 20% of individuals with NAFLD. People with NASH are at a high risk of developing liver fibrosis, and a small fraction, approximately 15%, develop cirrhosis. Individuals with NASH cirrhosis are at increased risk of developing HCC and require HCC screening and surveillance; they are at increased risk of liver decompensation and may require liver transplantation. The risk of liver-related mortality is increased significantly in individuals with cirrhosis. Overall, among people with NAFLD, cardiovascular disease is the leading cause of death, followed by cancer and liver-related death.
In individuals with NAFLD, cardiovascular disease is the leading cause of mortality, followed by cancer and then liver disease (Adams et al., 2005). In a Japanese cohort, the adjusted hazard ratio for cardiovascular disease was ~10-fold higher in nono-bese individuals with NAFLD compared with those without NAFLD (Yoshitaka et al., 2017). However, when cirrhosis has developed, liver disease becomes the dominant risk of mortality. NASH is now the second leading indication for liver transplantation in the United States and is likely to become the leading indication within a decade. NASH-related HCC is also increasing, and over the last decade it has been the etiology with the greatest increase in HCC incidence requiring liver transplantation (Charlton et al., 2011; Younossi et al., 2019a). In the next section, we discuss pathogenetic mechanisms of NAFLD and NASH and their progression to cirrhosis and HCC to contextualize the clinical presentation and natural history of NAFLD and NASH.
Mechanisms and heritability of NAFLD and NASH
Metabolic mechanisms responsible for development of NAFLD
The metabolic mechanisms leading to NAFLD reflect an imbalance of energy metabolism in the liver: excess energy, mostly in the form of carbohydrates and fat, entering the liver relative to the ability of the liver to oxidize this energy to CO2 or export it as very-low-density lipoproteins (VLDLs) (Figure 3). The result is a net accumulation of energy in the liver as triglycerides, which can explain the widespread presence of NAFLD in obese individuals and in those with lipodystrophy, where surplus energy is stored in the liver because of the deficiency in white adipose tissue lipid storage (Petersen et al., 2002; Shulman, 2000; Wehmeyer et al., 2016). Although excess consumption of any food can lead to development of NAFLD, mono- and disaccharides, especially fructose, sucrose, and high-fructose corn syrup, which is ubiquitous in processed foods, can activate programs of hepatic de novo lipogenesis that further exacerbate NAFLD. Furthermore, fructose is metabolized almost exclusively by the liver, and therefore dietary fructose is funneled into the liver and mostly metabolized to triglycerides by de novo lipogenesis (DNL) (Herman and Samuel, 2016; Petersen et al., 2001; Samuel and Shulman, 2018). Skeletal muscle insulin resistance, one of the earliest defects associated with metabolic syndrome and prediabetes (Perseghin et al., 1996; Rothman et al., 1995), can also promote development of NAFLD through increased hepatic DNL and hypertriglyceridemia by diverting ingested glucose away from skeletal muscle glycogen synthesis and into the liver for DNL (Flannery et al., 2012; Petersen et al., 2007; Rabøl et al., 2011). Development of hepatic insulin resistance, where insulin activation of glycogen synthase is impaired (Petersen and Shulman, 2018), would also be expected to redirect glucose into lipogenic pathways and further promote NAFLD. Consistent with this hypothesis, mice lacking hepatic glycogen synthase manifest hepatic insulin resistance but increased hepatic lipogenesis and NAFLD (Irimia et al., 2017).
Figure 3. Metabolic causes of NAFLD.
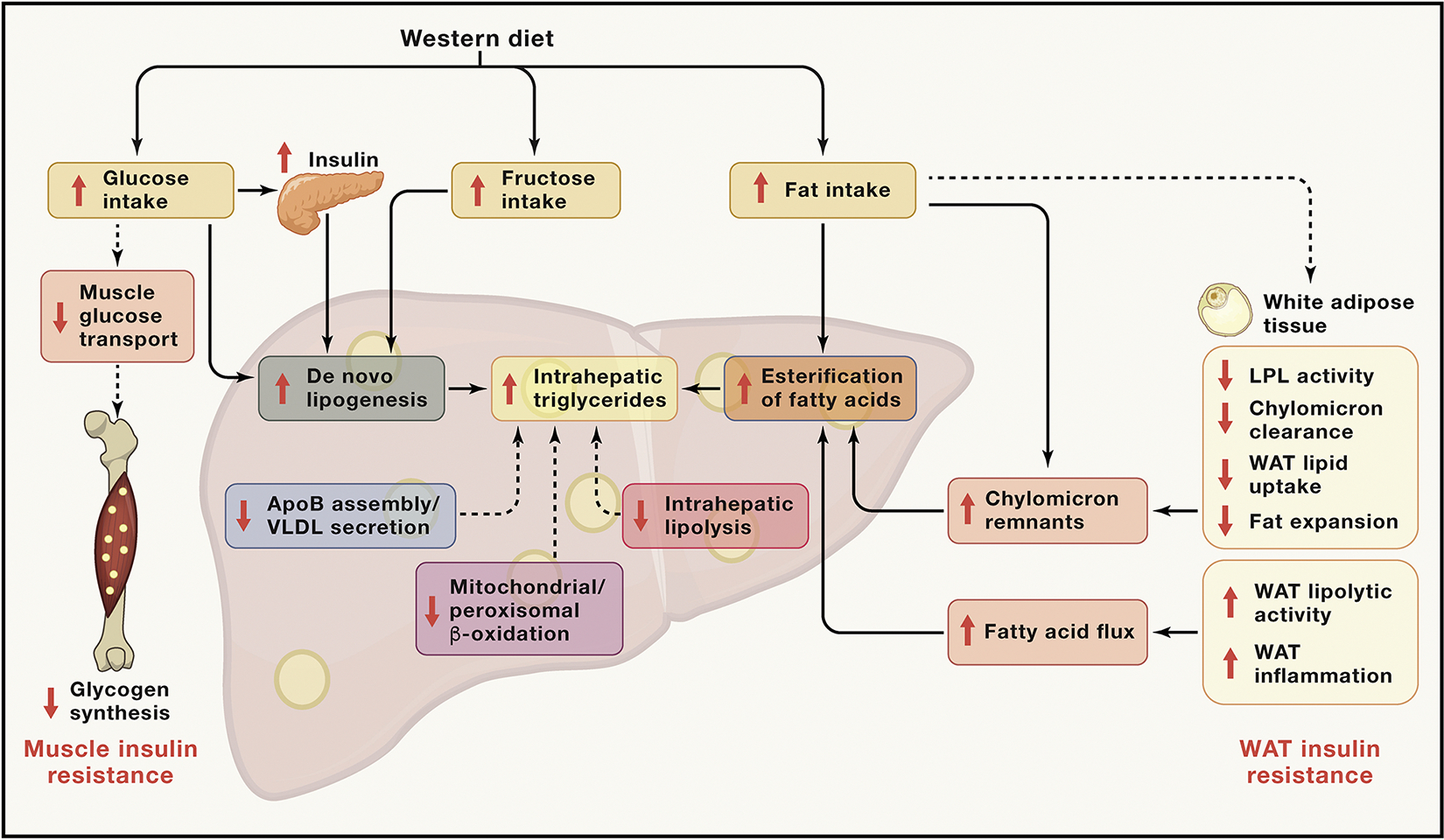
Increased energy intake, particularly with a Western diet rich in sucrose, high-fructose corn syrup, and saturated fat, leads to NAFLD and increased ectopic lipid deposition in skeletal muscle. Increases in intramyocellular lipid content, which typically occurs prior to onset of NAFLD, causes muscle insulin resistance, resulting in inhibition of insulin signaling and decreased insulin-stimulated glucose transport and muscle glycogen synthesis. Because ingested glucose cannot be stored properly as muscle glycogen, it is redirected to the liver, where, in combination with compensatory portal vein hyperinsulinemia because of muscle insulin resistance, it stimulates SREBP1c, which, in turn, promotes increased expression of key hepatic enzymes that regulate DNL, resulting in increased VLDL production, hypertriglyceridemia, and NAFLD. Monosaccharides can also promote development of NAFLD by recruiting other transcription factors, including ChREBP, PPARγ coactivator 1-β, and LXR to activate hepatic lipogenesis. WAT insulin resistance and/or WAT inflammation results in increased rates of lipolysis and increased fatty acid delivery to the liver, which promotes increased fatty acid esterification into hepatic triglycerides and NAFLD in a substrate-dependent and mostly insulin-independent manner. Triglyceride rich lipoproteins (TRLs) are cleared from the circulation by peripheral lipoprotein lipase (Lpl) activity. Endogenous Lpl inhibitors (e.g. ApoC3, ANGPTL3/8 complex, and ANGPTL4) attenuate peripheral triglyceride clearance, increasing hepatic triglyceride uptake from TRLs (e.g. chylomicron remnants). Reduced fat mass because of acquired and congenital forms of lipodystrophy/partial lipodystrophy also result in increased ectopic lipid storage in the liver and skeletal muscle because of lipid spillover from WAT to the liver. Other metabolic causes of NAFLD and NASH, which are most often genetically related, include (1) defects in intrahepatic lipolysis (e.g., decreased ATGL/CGI-58 activity); (2) defects in triglyceride export (e.g., defective Apo-B 100, MTTP activity); (3) increased glucokinase activity, resulting in increased hepatic DNL; and (4) reductions in hepatic mitochondrial/peroxisomal β-oxidation. Other genetic causes that confer an increased risk (PNPLA3, TM6SF2, and MBOAT7) or decreased risk (HSD17B13) of NAFLD are also likely to be linked metabolically through unknown mechanisms.
Adipocyte dysfunction also influences development of NAFLD (Lotta et al., 2017; Petersen and Shulman, 2018; Shulman, 2000). Severe NAFLD/NASH is a complication of congenital lipodystrophies, where the absence of adipose tissue forces the liver to store excess fatty acids, leading to severe insulin resistance (Kim et al., 2000; Petersen et al., 2002). Recent genetic studies further support the role of adipose dysfunction in the pathogenesis of metabolic diseases. Genetic loci linked to insulin resistance and dyslipidemia are also associated with lower adipose tissue mass, suggesting impaired adipose tissue expansion (Lotta et al., 2017). Some loci are in genes that alter lipoprotein lipase (Lpl) activity, whereas other loci are in genes encoding components of the insulin signaling pathway. Furthermore, factors such as ApoC3, the ANGPTL3/8 complex, and ANGPTL4, which can inhibit Lpl activity, can promote hepatic steatosis by promoting increased hepatic triglyceride uptake of chylomicron remnants, whereas factors that activate LpL activity in white adipocytes, such as adiponectin, protect against development of NAFLD in high-fat diet (HFD)-fed rodents (Petersen and Shulman, 2018). Specific changes in adipose tissue gene expression are also associated with development of NASH (du Plessis et al., 2015). Other metabolic causes of hepatic steatosis, which are most often genetically driven, include (1) defects in intrahepatic lipolysis (e.g., decreased ATGL/CGI-58 activity; Schweiger et al., 2009), (2) defects in triglyceride export (e.g., defective Apo-B 100, MTTP activity; Iqbal et al., 2020), (3) increased glucokinase activity resulting in increased hepatic DNL, and (4) reductions in hepatic mitochondrial/peroxisomal β-oxidation (Camporez et al., 2017; Chennamsetty et al., 2016; Reddy, 2001). Additional genetic causes of NAFLD and NASH that are likely to be linked metabolically will be discussed under Heritability of NAFLD.
Increases in visceral adipose tissue (VAT) have been associated with hyperlipidemia (Després et al., 1990), insulin resistance (Couillard et al., 1999), and NAFLD (Kelley et al., 2003). A prevailing view has been that VAT is itself causally related to the pathogenesis of NAFLD and its associated metabolic disturbances because of higher lipolytic rates (Nielsen et al., 2004; Rebuffé-Scrive et al., 1990), which may be driven in part by interleukin-6 (IL-6) (Wueest et al., 2016). This results in an increase in fatty acid delivery to the liver, which, in turn, promotes hepatic steatosis, insulin resistance, and dyslipidemia. However, it is also possible that VAT simply serves as another ectopic site for lipids when subcutaneous adipose tissue capacity is exceeded (Cuthbertson et al., 2017; Jensen, 2008; Smith, 2015) and would therefore not play a primary causal role in metabolic dysregulation but, instead, represents a sequela of abnormal subcutaneous white adipose tissue (WAT) biology. Thus, increases in VAT may not be a true independent risk factor for NAFLD but, instead, serve as a more visible marker of NAFLD. Evidence in support of this hypothesis stems from studies showing that, although the rates of VAT lipolysis are increased in obesity, subcutaneous WAT lipolysis from the upper body quantitively accounts for the majority of fatty acids delivered to the liver (Jensen, 2008; Nielsen et al., 2004). Furthermore intrahepatic triglyceride content, not VAT mass, correlates better with measures of hepatic insulin sensitivity (Fabbrini et al., 2009), even in individuals with marked obesity (Magkos et al., 2010). Moreover, removal of VAT alone (Fabbrini et al., 2010) does not significantly affect key metabolic parameters (Andersson et al., 2017; Fabbrini et al., 2010). Although Asians and South Asians have proportionally more liver fat (Petersen et al., 2006) and VAT (Kadowaki et al., 2006; Nazare et al., 2012) than other races at any given BMI, the association between VAT and hepatic lipid content is similar across races (Nazare et al., 2012). Aging also impairs subcutaneous adipose tissue function, possibly because of accumulation of senescent adipocytes, impaired pre-adipocyte development (Palmer and Kirkland, 2016), and reduced mitochondrial activity (Lee et al., 2010; Petersen et al., 2003), which is associated with increases in ectopic lipid accumulation in VAT, liver, and skeletal muscle and insulin resistance. In a group of weight-stable individuals followed over 10 years, an increase in VAT mass was associated with development of a metabolically unhealthy phenotype (Hwang et al., 2015). Impaired expansion of subcutaneous adipose tissue may also account for non-obese subjects with NAFLD.
Insulin is the master regulator of hepatic glucose and lipid metabolism through direct and indirect mechanisms. Following a meal, the relatively high concentrations of insulin in the portal vein, which is ~3-fold higher than peripheral concentrations, directly activates hepatic insulin receptor tyrosine kinase (IRTK) activity, initiating a coordinated relay of intracellular signals (Petersen and Shulman, 2018). These include 3-phosphoinositide-dependent kinase-1 (PDK1) and mammalian target of rapamycin complex 2 (mTORC2) (Guertin and Sabatini, 2007), which converge on Akt phosphorylation (Alessi et al., 1997; Guertin and Sabatini, 2007; Stephens et al., 1998). This model has been used to account for the mechanisms by which insulin directly suppresses hepatic glucose production through activation of the hepatic insulin receptor kinase, leading to (1) activation/translocation of glucokinase to the cytoplasm (Nozaki et al., 2020), (2) activation of hepatic glycogen synthase (Nozaki et al., 2020; Petersen et al., 1998), and (3) reduction of the expression of gluconeogenic enzymes via phosphorylation and nuclear exclusion of FOXO1 (Nakae et al., 2001). However, this transcriptional regulation does not fully account for the acute changes in hepatic gluconeogenesis following an increase in portal vein insulin concentrations after a meal; the changes in hepatic protein expression of gluconeogenic enzymes lag behind the changes in mRNA and hepatic glucose production that occur after an acute insulin stimulus (Petersen and Shulman, 2018).
Levine and Fritz (1956) proposed that insulin inhibits hepatic glucose production indirectly via action at peripheral sites. Subsequent studies (Lewis et al., 1996; Rebrin et al., 1996) postulated that these indirect effects of insulin to suppress hepatic glucose production were mediated by suppression of WAT lipolysis. Perry et al. showed that insulin suppression of WAT lipolysis, which occurs within a few minutes, results in immediate reductions in hepatic acetyl-coenzyme A (CoA) content, an allosteric activator of pyruvate carboxylase (PC), which, in turn, reduces PC activity and PC flux (Perry et al., 2015). Inhibiting WAT lipolysis also curtails glycerol delivery to the liver, reducing conversion of glycerol to glucose by a substrate push mechanism (Perry et al., 2014; Previs et al., 1999).
A new model of insulin action on hepatic glucose metabolism can be proposed, where insulin has acute direct effects on hepatic glucose metabolism that are mediated through the hepatic IRTK/AKT2 pathway and affect mostly hepatic glycogen metabolism and acute indirect effects mediated through the WAT IRTK/AKT2 pathway that, in turn, affect WAT lipolysis, altering fatty acid/glycerol flux to the liver, and subsequently regulate hepatic gluconeogenesis through allosteric and substrate push mechanisms, respectively (Perry et al., 2015). Although insulin can also regulate hepatic gluconeogenesis through FOXO1-mediated transcriptional/translational events, these effects do not occur acutely and more likely set the capacity for hepatic gluconeogenic flux. The direct effects of insulin on hepatic glycogen metabolism and glucose production would be expected to predominate in a hepatic glycogen-replete state following short-term fasting. Conversely, the indirect effects of insulin on hepatic glucose metabolism (i.e., suppression of WAT lipolysis and inhibition of hepatic gluconeogenesis) would be expected to predominate in regulation of hepatic glucose production in hepatic glycogen-depleted states, when gluconeogenesis is the major contributor to hepatic glucose production.
Insulin regulation of hepatic lipid metabolism also involves direct hepatic and indirect extrahepatic pathways (although multiple hormonal, neural, and metabolic signals modulate hepatic lipid metabolism). Direct hepatic insulin action activates SREBP1c by increasing mRNA expression and proteolytic cleavage of the SREBP1c precursor protein into a mature nuclear transcription factor (Brown and Goldstein, 2008). Insulin signaling also increases the activity of mTORC1, which regulates SREBP1c mRNA expression (Li et al., 2010) and processing (Peterson et al., 2011). SREBP1c can be regulated independent of insulin, as demonstrated with induction of SREBP1c after feeding in mice lacking hepatic insulin receptors (Haas et al., 2012). Other factors also influence hepatic DNL. For example, monosaccharides also recruit other transcription factors, including ChREBP (Erion et al., 2013; Uyeda and Repa, 2006), PPARγ coactivator 1-β (Nagai et al., 2009), and liver X receptor (Bindesbøll et al., 2015), to activate hepatic lipogenesis.
Peripheral insulin action indirectly regulates hepatic lipid synthesis. Hepatic fatty acids may be derived from triglyceride in lipid particles (e.g., chylomicron remnants, low-density lipoprotein [LDL]), which occur with increased plasma concentrations of ApoC3, as well as from circulating fatty acids, a major source for hepatic triglyceride synthesis (Donnelly et al., 2005). Insulin influences each of these inputs via extrahepatic actions. For example, insulin activates LpL at peripheral sites (e.g., WAT), promoting hydrolysis of triglycerides, mostly in the form of chylomicrons and VLDL particles, and promotes uptake of fatty acids into WAT. Insulin also suppresses WAT lipolysis and influences the amount of fatty acyl-CoA available for esterification into hepatic triglyceride. Vatner et al. (2015) demonstrated that hepatic triglyceride synthesis by hepatic fatty acid esterification is driven primarily by fatty acid availability independent of changes in hepatic insulin signaling. In contrast, de novo hepatic lipogenesis is insulin dependent and reduced in rats with defective hepatic insulin signaling (ter Horst et al., 2020).
Mechanisms by which NAFLD causes dysregulated hepatic glucose and lipid metabolism
Although multiple models have been proposed for development of hepatic insulin resistance in NAFLD (reviewed in Samuel and Shulman, 2012), a substantial body of work, building on earlier studies of muscle, have also demonstrated a key role of the plasma membrane sn-1,2-diacylglycerol-protein kinase Cε-insulin receptor kinase threonine1160 phosphorylation pathway in mediating hepatic insulin resistance.
In rats and mice, short-term high-fat diets lead to hepatic steatosis and hepatic insulin resistance without muscle lipid accumulation or peripheral insulin resistance within a few days (Samuel et al., 2004). These studies demonstrate that ectopic lipid in the liver is (1) associated specifically with hepatic insulin resistance; (2) disassociates hepatic insulin resistance from obesity and visceral adiposity, similar to lipodystrophy; and (3) is an excellent animal model to examine the molecular mechanisms of lipid-induced hepatic insulin resistance.
In high fat diet rodent models of NAFLD, the increase in hepatic sn-1,2-DAG content activates protein kinase C ε (PKCε) (Samuel et al., 2004), which is the primary nPKC isoform in the liver (Qu et al., 1999; Samuel et al., 2004). DAG-mediated PKCε activation is associated with decreased insulin activation of IRTK activity (Samuel et al., 2007). Lowering PKCε expression in the liver protects rats from lipid-induced hepatic insulin resistance and preserves IRTK activity despite increases in liver lipid content (Samuel et al., 2007). Similarly, Prkce−/− mice are protected from HFD diet-induced hepatic insulin resistance following 1 week of high-fat diet feeding despite increases in liver lipid content (Raddatz et al., 2011). Recently, Petersen et al. (2016) identified Thr1160 (the Thr1150 homolog in mice) as a PKCε phosphorylation site located in the catalytic loop of IRTK (Petersen et al., 2016). Moreover, these T1150A mice are protected from high-fat diet-induced reductions in activation of glucokinase and hepatic glycogen synthesis, reflecting the importance of direct hepatic insulin signaling in regulating hepatic glycogen metabolism (Petersen et al., 2016). These data support a model of hepatic insulin resistance in which accumulation of hepatic DAG activates PKCε and impairs IRTK activation, demonstrating that PKCε activation is necessary for lipid-induced hepatic insulin resistance.
To determine the specific lipid metabolite and cellular compartment that mediates lipid-induced hepatic insulin resistance, Lyu et al. (2020) developed a subcellular fractionation method to quantify DAG stereoisomers (sn-1,2-DAGs, sn-2,3-DAGs, and sn-1,3-DAGs) and ceramides in the endoplasmic reticulum, mitochondria, plasma membrane, lipid droplets, and cytosol. Acute liver-specific knockdown of diacylglycerol acyltransferase-2, using an antisense oligonucleotide, induced hepatic insulin resistance in rats. This could be attributed to increased plasma membrane sn-1,2-DAGs, which led to PKCε activation, insulin receptor kinase Thr1160 phosphorylation, and reduced IRTK Y1162 phosphorylation. Liver-specific PKCε knockdown abrogated HFD-induced hepatic insulin resistance by decreasing IRTK T1160 phosphorylation, whereas liver-specific overexpression of constitutively active PKCε induced hepatic insulin resistance by increasing IRTK T1160 phosphorylation in rats. These data identify plasma membrane sn-1,2 DAGs as the key cellular compartment and lipid species that activate PKCε and establish that hepatic PKCε is necessary and sufficient for mediating hepatic insulin resistance (Lyu et al., 2020). The same mechanism underlies lipid-induced insulin resistance in WAT (Lyu et al., 2021) and skeletal muscle (Song et al., 2020a).
This mechanism for lipid-induced hepatic insulin resistance has been translated to humans. Kumashiro et al. (2011) studied determinants of insulin resistance in a cohort of people undergoing bariatric surgery. Hepatic DAG content and PKCε activation were the strongest predictors of hepatic insulin resistance in individuals with NAFLD. Magkos et al. (2012) also demonstrated that hepatic DAG content, but not hepatic ceramide content, was the best predictor of hepatic insulin resistance in obese people with NAFLD. Luukkonen et al. (2016b) performed a lipidomics analysis of liver samples from people undergoing bariatric surgery and reported increases in specific lipid metabolites (species of fatty acids, ceramides, and TAGs), including specific diacylglycerol species in association with hepatic insulin resistance. Consistent with these results, ter Horst et al. (2017) have shown that liver diacylglycerol content and increased PKCε activity are related to hepatic insulin sensitivity in obese individuals undergoing bariatric surgery, and Lyu et al. (2020) found that liver plasma membrane sn-1,2-DAG content and IRTK T1160 phosphorylation were increased in obese people with hepatic insulin resistance and NAFLD. In contrast, there was no consistent association between hepatic insulin resistance and hepatic ceramides in any hepatocellular compartment. This plasma membrane sn-1,2-DAG-PKCε-IRTK T1160 phosphorylation hypothesis also explains the disassociation between hepatic steatosis and hepatic insulin resistance under conditions of reduced intrahepatic lipolysis (e.g., CGI-58, ATGL deficiencies) and hepatic triglyceride export (e.g., microsomal triglycedride transfer protein deficiency, apolipoprotein B-100 deficiency), where sn-1,2-DAGs accumulate in the lipid droplet fraction and not in the plasma membrane (Abulizi et al., 2020; Cantley et al., 2013).
Heritability of NAFLD
NAFLD and NASH have a heritable component that is estimated to be between 35%–61% (Eslam et al., 2018; Sookoian and Pirola, 2019). Strong family clustering with a heightened risk of advanced disease among first-degree relatives of individuals with NASH and increased risk among monozygotic twins (Loomba et al., 2015b) reinforce this hereditary risk but also reflect a shared microbiome among members of the same household (Caussy et al., 2019). Twin studies also reveal overlapping genetic effects of hepatic steatosis and fibrosis (Cui et al., 2016). One example is a variant in the TMPO gene, which encodes the lamina-associated polypeptide-2 (LAP-2) protein, generating a truncation mutant that promotes fat accumulation in the Huh7 HCC cell line (Brady et al., 2018).
Epigenetic alterations have also been implicated in heritability of NAFLD (Eslam et al., 2018; Lee et al., 2017; Stols-Gonçalves et al., 2019), and differences in their expression explain, in part, the higher concordance of risk between monozygotic and dizygotic twins (Zarrinpar et al., 2016). Among microRNAs (miRNAs), miR-331–3p and miR-30c are heritable and predicted to share similar target genes, suggesting that they are functionally linked (Zarrinpar et al., 2016). Additional miRNAs implicated in NAFLD include miR-122 (Cheung et al., 2008), miR-21 (Benhamouche-Trouillet and Postic, 2016), miR-124a (Fang et al., 2019), miR-23b (Borji et al., 2019), miR451a (Zeng et al., 2018), and miR-34a (Ding et al., 2015), among others.
Developmental exposure contributes to the risk of NAFLD (Wesolowski et al., 2017). Evidence in animals and humans links maternal BMI to neonatal hepatic fat content (Bayol et al., 2010; Brumbaugh et al., 2013; Dudley et al., 2011) and lipotoxicity (McCurdy et al., 2009). In addition, maternal insulin resistance, hyperglycemia, and hyperlipidemia are drivers of neonatal adiposity (Lawlor et al., 2012; Wesolowski et al., 2017). Placental factors may also promote NAFLD, including enhanced lipid and glucose transport, oxidative stress, and inflammation (Wesolowski et al., 2017). These changes can promote fatty liver disease in neonates, characterized by oxidative stress, lipid accumulation, and altered immunity, with the latter primarily through effects on macrophage phenotype and by changes in the microbiome conferred by the diet of the mother and child. In mice, obesogenic diets early in life can durably affect DNA methylation, which, in turn, is dependent on specific gene variants of ribosomal DNA (Holland et al., 2016).
An increasing number of single-nucleotide polymorphisms (SNPs) has been linked to risk of NAFLD, as summarized in several recent reviews (Sookoian and Pirola, 2019; Trépo and Valenti, 2020). Discovery of genetic variants not only facilitates prediction of disease risk but, more importantly, can unearth biologic drivers of disease not considered or understood previously. SNP discovery can also implicate shared mechanisms across different liver diseases. For example, discovery of a SNP in the patatin-like phospholipase domain-containing 3 (PNPLA3) gene uncovered a role of this previously unappreciated enzyme in regulation of hepatocyte lipid homeostasis (Romeo et al., 2008). The discovery also led to the finding that the I148M variant, which is associated with enhanced NAFLD risk, is resistant to ubiquitylation and proteasomal degradation, disrupting triglyceride mobilization to promote its accumulation (BasuRay et al., 2017). The SNP also confers a heightened, independent risk of fibrosis through its effect on hepatic stellate cells, a fibrogenic cell type in the liver. In these cells, PNPLA3 normally hydrolyzes retinyl esters to promote extracellular retinol release, an activity that is reduced when the I148M variant is present (Pirazzi et al., 2014), leading to enhanced stellate cell fibrogenesis (Bruschi et al., 2017). These effects of the I148M PNPLA3 SNP, and possibly additional mechanisms, contribute to an ~12-fold greater risk of HCC in individuals with NAFLD and the homozygous I148M variant compared with a healthy population (Figure 4; Liu et al., 2014). Risks associated with the I148M PNPLA3 SNP have been replicated in multiple independent cohorts (Stender and Loomba, 2020). Discovery of the PNPLA3 risk allele also strongly implicates a shared pathogenesis between alcoholic and non-alcoholic fatty liver disease because the I148M variant is associated with severity of alcoholic liver disease as well (Tian et al., 2010). Finally, the higher prevalence of the I148M variant among Hispanics explains, in part, the higher risk of NAFLD and liver disease in this ethnic group (Betancourt-Garcia et al., 2017; Palmer et al., 2013).
Figure 4. A gene-environment nexus drives the risk of cirrhosis and HCC in NASH.

The PNPLA3 gene is a major driver of NAFLD, NASH, cirrhosis, and HCC risk in individuals who have metabolic risk factors such as obesity, diabetes, and metabolic syndrome. Other genetic factors are also involved in NASH progression. Most genetic factors manifest in the setting of metabolic risk factors, including obesity, diabetes, and metabolic syndrome, as well as other environmental factors, such as alcohol and smoking. The gut microbiome is altered by diet and alcohol and in concert with changes in bile acid and metabolic dysfunction, including lipotoxicity. These alteration promote disease progression to cirrhosis and HCC in a susceptible host. Therefore, gene-environment interaction drives the risk of NASH cirrhosis and HCC.
PNPLA3 may have dual functions where it can operate as a triacylglycerol hydrolase and as an acylglycerol transacylase (Kumari et al., 2012), but the mechanism by which this polymorphism, which attenuates hydrolase activity (Pingitore et al., 2014), increases liver fat is still unclear (Mitsche et al., 2018). Overexpression of mutant PNPLA3 in mice had mixed effects, including a decrease in hepatocyte TAG hydrolysis and an increase in enzymes regulating DNL (Li et al., 2012), although humans with this isoform do not have increases in DNL (Mancina et al., 2015). Genetic deletion of this enzyme fails to induce NAFLD in mice (Basantani et al., 2011; Chen et al., 2010), whereas a PNPLA3 ASO decreases hepatic fatty acid esterification and protects fat-fed rats from NAFLD, PKCε activation, and hepatic insulin resistance (Kumashiro et al., 2013). Thus, although the association between PNPLA3 and NAFLD is clear, the mechanism by which PNPLA3 causes NAFLD requires further study.
Other genetic loci have also been linked to NAFLD. Polymorphisms in transmembrane 6 superfamily member 2 (rs58542926) have also been associated with NAFLD (Mahdessian et al., 2014). The protein may regulate incorporation of polyunsaturated fatty acids into hepatic triglyceride (Luukkonen et al., 2017) as well as cholesterol metabolism (Fan et al., 2016; Smagris et al., 2016) and impaired hepatic secretion of large triglyceride-rich VLDL (Borén et al., 2020).
A polymorphism (rs641738) that decreases expression of membrane bound O-acyltransferase domain containing 7-transmembrane channel-like 4 (MBOAT7-TMC4) is associated with increased liver lipid content and worsening liver histology (Luukkonen et al., 2016a; Mancina et al., 2016). MBOAT7-TMC4 is localized to intracellular membranes, such as the endoplasmic reticulum, mitochondria, and lipid droplets, and is thought to function as a lysophospholipid acyltransferase, specifically regulating incorporation of arachidonic acid into phosphatidylinositol (Gijón et al., 2008). This polymorphism is also associated with fibrosis in alcoholic liver disease (Buch et al., 2015) and chronic hepatitis C (Thabet et al., 2016), suggesting that components of this pathway could represent therapeutic targets for several liver diseases. Deletion of Mboat7 in hepatocytes in a mouse model leads to heightened fibrosis with no effect on inflammation in a NASH model resembling humans with the risk allele (Thangapandi et al., 2021). Mboat7 promotes degradation of lysophosphatidylinositol (LPI), and its accumulation in Mboat7 KO mice promotes features of NASH (Helsley et al., 2019), at least in part, through activation of the G-protein coupled receptor GPR55 (Fondevila et al., 2021).
Several polymorphisms in ApoC3 (Petersen et al., 2010, Peter et al., 2012 and Zhang et al., 2016c) associated with NAFLD may increase plasma ApoC3 concentrations, which, in turn, inhibits LpL activity and promotes increased plasma concentrations of triglyceride-rich lipoproteins (e.g., chylomicron remnants) which result in increaed receptor-mediated hepatic uptake of triglyceride, predisposing lean individuals to fasting and postprandial hypertriglyceridemia as well as NAFLD and insulin resistance. Consistent with these findings, knockdown of hepatic Apo C3 expression, using an antisense oligonucleotide, reduces hepatic steatosis and insulin resistance in a HFD-fed mouse model of NAFLD (Valladolid-Acebes et al., 2021), whereas overexpression of human Apo C3 predisposes mice to diet-induced NAFLD and hepatic insulin resistance (Lee et al., 2011). Less common genetic causes have also been identified in pathways that regulate glucose metabolism (e.g., rs1260326 in glucokinase regulatory protein; Santoro et al., 2012). Polymorphisms that decrease expression of the monocarboxylate transporter SCL16A11 have been linked to type 2 diabetes in Mexican and Latin American people (Williams et al., 2014a), and, in vitro, inhibition of SCL16A11 in hepatocytes promotes accumulation of lipid metabolites, including diacylglycerol (Rusu et al., 2017).
Similar to PNPLA3, a new disease mechanism has been suggested by discovery of a variant of 17-beta hydroxysteroid dehydrogenase 13, encoded by the HSD17B13 gene, a poorly characterized enzyme present in hepatocytes (Abul-Husn et al., 2018). The rs72613567:TA variant confers protection from NAFLD by generating a truncated, inactive form of the enzyme, which implies that the non-truncated product of this enzyme contributes to disease. The protective variant also attenuates liver injury associated with the PNPLA3 I148M risk variant (Abul-Husn et al., 2018). The enzyme has retinol dehydrogenase activity, but in contrast to PNPLA3, it is not expressed in hepatic stellate cells (Ma et al., 2019), so the phenotype associated with this variant may be attributable solely to its effects in hepatocytes through an unknown mechanism. HSD17B13 is associated with increased steatosis and decreased inflammation, ballooning, and Mallory-Denk bodies upon liver histology (Ma et al., 2019). Despite the lack of clarity about how truncation of the HSD17B13 protein and reduced activity protect against NAFLD, this discovery has already led to therapeutic programs intended to mimic the effect of the protective allele by small interfering RNA (siRNA) knockdown of the mRNA or small-molecule antagonism of the protein. The approach replicates the successful strategy of antagonizing PCSK9 following the discovery that variants of this gene that attenuate its activity confer protection from coronary artery disease (Cohen et al., 2006).
Several other SNPs have also been linked to NAFLD risk, including those encoding proteins that regulate glucose (e.g., GKCR) and lipid metabolism (e.g., PNPLA3, TM6SF2), oxidative stress (e.g., uncoupling protein 2), lipophagy (Lin et al., 2016), myokine secretion (Metwally et al., 2019), immune responses (e.g., interferon lambda; Petta et al., 2017), and fibrosis (e.g., Kruppel-like factor 6). Children with NAFLD have additional genetic risk factors distinct from adults (Wattacheril et al., 2017).
Environmental and other non-genetic factors may amplify the genetic risk conferred by SNPs. For example, adiposity heightens the risk of liver disease associated with polymorphisms in PNPLA3, TM6SF2, and GCKR (Stender et al., 2017). There is also a significant effect of concurrent alcohol use, which has provoked a recent suggestion to consider alcoholic fatty liver disease and NAFLD as a single entity within the same spectrum (Eslam and George, 2019). Although some risk polymorphisms are shared between alcoholic fatty liver disease and NAFLD (e.g., PNPLA3 I148M), others are not. For example, there are polymorphisms in the alcohol- and acetaldehyde-dehydrogenase genes that uniquely confer a risk of alcoholic liver disease (reviewed in Anstee et al., 2016).
Despite the growing list of genetic variants linked to NAFLD, this information has not yet led to generation of a genetic predictive risk score of the type that was robustly validated for cirrhosis risk in individuals with hepatitis C infection (Huang et al., 2007). The difficulty of predicting NAFLD risk through genetic markers alone, in contrast to hepatitis C virus (HCV) infection, may reflect the significant contributions of environmental and non-genetic factors, in particular the microbiome. It may also reflect the possibility that NASH has multiple subtypes, each of which has unique drivers and therapeutic targets. Large-scale genetic studies are beginning to hint at this prospect (Liu et al., 2020).
Role of the gut microbiota in development of NAFLD and NASH
The gut microbiome may also play an important role in the pathogenesis of NAFLD. A link between NAFLD/NASH and the microbiome is supported by several studies (Caussy et al., 2019; Da Silva et al., 2018; Hoyles et al., 2018; Loomba et al., 2017; Oh et al., 2020; Raman et al., 2013; Schwimmer et al., 2019; Sharpton et al., 2019). As NAFLD progresses and fibrosis worsens, gut microbial communities in affected individuals become less diverse and exhibit a higher abundance of Streptococcus and Gram-negative bacteria (Hoyles et al., 2018). Specific genera (e.g., Bacteroides and Ruminococcus) have also been associated with steatohepatitis and fibrosis, respectively (Boursier et al., 2016). A metagenomic analysis of stool samples from 56 obese individuals with steatosis found enrichment of genes related to lipid metabolism, endotoxin biosynthesis, and hepatic inflammation and evidence of dysregulated aromatic and branched-chain amino acid metabolism (Hoyles et al., 2018). These studies suggest that the gut microbiota may contribute to NAFLD through several mechanisms: (1) altered energy harvest and decreased microbial diversity; (2) increased branched-chain and aromatic amino acid levels; (3) increased microbial production of metabolites such as phenylacetic acid and ethanol, which can increase hepatic lipid accumulation in vitro and in vivo; and (4) increased microbial endotoxins, which may contribute to inflammation. Using a well-characterized cohort of individuals with biopsy-proven NAFLD, a gut microbiome-based metagenomic signature has been described that can differentiate between mild/moderate and advanced fibrosis in people with NAFLD (Loomba et al., 2017). In a subsequent study, using 16S rRNA analysis, Caussy et al. (2019) demonstrated that a microbiome signature can accurately detect NAFLD cirrhosis. However, given the influence of host and environmental factors on the gut microbiome (Li et al., 2014), the universal applicability of microbiome-based diagnostic signatures was not known. To address this question, a prospective study was conducted using quantitative metagenomic analysis of stool samples from a cohort of 27 individuals with NAFLD-cirrhosis and 54 non-NAFLD control individuals (Oh et al., 2020). Random forest modeling identified a gut microbiome signature for cirrhosis that included 19 core microbial species. These data were then validated in two independent cohorts of people from China and Italy with cirrhosis because of diverse etiologies. These data suggest that there is a universal gut microbiome signature for cirrhosis. Increased proteobacteria, E. coli and Negativicutes, and decreased Firmicutes have been seen consistently in advanced fibrosis in NAFLD. In individuals with cirrhosis, oralization of the gut microbiome has been reported, and, in particular, an increase in Veillonella has been noted consistently. Further studies are needed to determine whether the microbiome is associated causally with fibrosis progression or a consequence of fibrosis progression. As noted above, it is possible that not all individuals with NAFLD harbor the same disease drivers; for example, in some people, disease may result primarily from lipid dysregulation, in others from enhanced inflammation or insulin resistance or from an interaction between host genetics and the microbiome or other determinants.
Cell Signaling, Inflammation, and Injury in NAFLD
Bile acid and nuclear receptor signaling.
The recognition that bile acids regulate liver homeostasis and contribute to parenchymal liver diseases beyond cholestatic disorders has ignited interest in their biology and therapeutic potential in NAFLD (Arab et al., 2017). Specifically, the discovery of the bile acid receptors farnesoid X receptor (FXR), Takeda G-coupled protein receptor 5 (TGR5; GPBAR1, M-BAR), pregnane X receptor (PXR), sphingosine-1-phosphate receptor 2 (S1PR2), the muscarinic receptors M2/3, and the vitamin D receptor have established bile acids as multifunctional signaling molecules (Chavez-Talavera et al., 2017; Chiang, 2013). Among these, FXR, PXR, and VDR are ligand-activated nuclear receptors, and TGR5 is a G-protein-coupled receptor. FXR has been studied most intensively because of ongoing clinical development of small-molecule FXR ligands (Neuschwander-Tetri et al., 2015). Specific TGR5 agonists (Thomas et al., 2009) and dual TGR5-FXR agonists (Carino et al., 2017) reduce hepatic steatosis. The therapeutic benefit of FXR agonism has been attributed to induction of its downstream effector, FGF15/19. FXR activation favorably affects lipid and glucose homeostasis, energy metabolism, hepatic inflammation (Gai et al., 2018), and cellular stress (Han et al., 2018) and promotes intestinal bile acid uptake, although some effects are discordant between rodent models and humans (Adorini et al., 2012; Arab et al., 2017; McIlvride et al., 2019). These species differences might be attributable in part to varied interaction with the gut microbiome (Chen et al., 2019), and, FXR agonism in the intestine may contribute to its overall therapeutic benefit (Fang et al., 2015). TGR5 agonism also has pleiotropic benefits through signaling in hepatic stellate cells, sinusoidal endothelium, cholangiocytes, and macrophages (Keitel and Häussinger, 2018); it also promotes glucagon like peptide-1 (GLP-1) secretion from the intestine (Chavez-Talavera et al., 2017).
Other nuclear receptors besides bile acid receptors have also been implicated in NAFLD. Liver X receptor (LXR) signaling has distinct effects on glucose and lipid homeostasis; loss of LXR in mice enhances insulin sensitivity, but these mice are more glucose intolerant (Beaven et al., 2013). However, LXR agonism increases hepatic fat; thus, LXR agonists are not appropriate therapies for metabolic syndrome (Beaven and Tontonoz, 2006). Additional nuclear receptors implicated in hepatic lipid homeostasis include PPARα, PPARδ, PPARγ, PXR, constitutive androstane receptor (CAR), RORγ, and REV-ERBβ (reviewed in Fuchs et al., 2016).
Inflammatory and immune signaling.
An interdependent web of inflammatory and immune signaling pathways has been implicated in NAFLD and NASH pathogenesis, including inflammasome activation; release of hepatocyte-derived signals; altered innate immune signaling; altered macrophage, T cell, platelet, and neutrophil responses; and changes in cytokine, adipokine, and chemokine biology. As with other elements of NASH pathogenesis, their relative importance and at which stage they contribute to the disease are not yet clear.
Activation of the inflammasome occurs in multiple cell types in NASH, including resident and infiltrating cells. This multiprotein complex responds to danger signals, typically damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), through activation of caspase-1 to cleave and activate IL-1β (Szabo and Iracheta-Vellve, 2015). IL-1β and its subfamily members IL-1α, IL-18, and IL-33 promote features of NASH, including ICAM-1 induction on sinusoidal endothelium, metabolic syndrome, and hepatic stellate cell activation and fibrosis (Mirea et al., 2018).
Among the different types of inflammasomes, the NLRP3 complex has been characterized most extensively in NASH, and its inhibition in myeloid cells attenuates inflammation and fibrosis in a rodent model (Mridha et al., 2017). The AIM2 inflammasome, which senses DNA, is also induced in a dietary model of NASH (Csak et al., 2014). In addition to myeloid cells, inflammasome activation drives disease responses in hepatocytes, macrophages, and hepatic stellate cells (Alegre et al., 2017; Watanabe et al., 2009), but the quantitative contribution of inflammasome activation in each cell type to overall disease pathology is not clear.
Downstream of inflammasome activation, gasdermin-D, a substrate of inflammatory caspases, induces pyroptosis (inflammatory cell death) of hepatocytes, which amplifies the features of steatohepatitis in humans and rodent models (see below; Xu et al., 2018).
Macrophage contributions to NASH pathogenesis have been characterized most extensively compared with other inflammatory cell types in the liver, in part because of their phenotypic diversity (i.e., polarization), and prominence in the histopathology of the disease. Pathways driven by macrophages include not only inflammation and fibrosis but also steatosis through secretion of IL-1β, tumor necrosis factor alpha (TNF-α), the CCL2 chemokine, and MCP-1 (Krenkel and Tacke, 2017).
Pathways regulating macrophage polarization in the liver are incompletely understood, but during injury, the cells undergo metabolic reprogramming upon activation, characterized by inflammasome activation and increased generation of lactate and reactive oxygen species. Their role is reinforced by a study showing that inhibition of monocyte-derived macrophage recruitment through administration of a small-molecule CCR2/CCR5 antagonist attenuates steatosis and fibrosis in an experimental model of NASH (Krenkel et al., 2018). More recently, single-cell RNA sequencing of non-parenchymal cells in a murine model of NASH has highlighted an increase in TREM-2+ macrophages (Xiong et al., 2019), which are normally reparative (Deczkowska et al., 2020) but whose role in NASH needs to be clarified. Macrophages in experimental NASH acquire a distinct inflammatory phenotype that can be recapitulated by ex vivo incubation with fatty acids (Krenkel et al., 2020).
Several other regulators of macrophage activation in NASH have been described. Levels of the transmembrane protein 173 (TMEM173 or STING), a potent inducer of type I interferon immunity, are increased in human NASH, and genetic loss of this mediator attenuates experimental NASH (Luo et al., 2018). In contrast, RORα in macrophages has a protective effect on steatohepatitis by inducing the transcription factor Kruppel-like factor 4, which drives polarization of macrophages toward an anti-inflammatory, tissue-remodeling “M2” phenotype (Han et al., 2017). A newly uncovered ligand for RORα, maresin-1, a docosahexaenoic acid metabolite, is hepatoprotective by promoting polarization of macrophages toward a tissue-resolving M2 phenotype while further inducing RORα in an autoregulatory loop (Han et al., 2019). Digoxin, a cardiac glycoside, is hepatoprotective by inhibiting transcription of the HIF-1α pathway through direct binding to the pyruvate kinase M2, which alters chromatin structure (Ouyang et al., 2018), highlighting digoxin’s potential as a therapeutic prospect in NASH. TRIF-4 (TIR-domain containing adaptor inducing interferon-α), a Toll-like receptor 4 (TLR4) adaptor protein, has a unique profile of promoting lipid accumulation while inhibiting injury, inflammation, and fibrosis (Yang et al., 2017). Finally, lipotoxic hepatocyte-derived extracellular vesicles enriched with β-integrin promote infiltration and adhesion of monocytes, which promote inflammation (Guo et al., 2019).
Progression of NASH is also associated with altered T cell responses (Nati et al., 2016). TH17 cells, which secrete IL-17, contribute to inflammation and metabolic features of NASH (Gomes et al., 2016). There is a higher frequency of Th17 cells in the liver of people with NASH, associated with a lower frequency of resting Tregs (rTregs; CD4+ CD45RA+CD25++) and higher frequencies of IFN-γ+ and/or IL-4+ cells in the circulation of individuals with NASH (Rau et al., 2016). Interestingly, these changes are reversed 1 year later in people undergoing bariatric surgery. Increased hepatic natural killer (NK) T cells are also present in the liver of people with NASH (Syn et al., 2010), and their depletion attenuates disease features in experimental NASH (Wu et al., 2012).
Neutrophils have been overlooked until recently; however, their emergence as mediators of tissue inflammation and fibrosis resolution underscores their newfound importance in NASH pathogenesis (Musso et al., 2018; Schuster et al., 2018). The cells are a component of the specialized proresolving mediator (SPM) system, which involves four key steps: (1) termination of chronic injury, (2) shifting the balance from inflammation to resolution, (3) deactivation of myofibroblasts, and (4) extracellular matrix degradation (Musso et al., 2018; Serhan, 2017). Several enzymatic steps involving metabolism of arachidonic acid, eicosapentanoic acid, docosapentaneoic acid, and docosahexaenoic acid promote generation of a series of lipid mediators known as resolvins, protectins, and maresins (Musso et al., 2018). NASH has been viewed as a disorder of defective proresolution, which is especially intriguing because the same lipid mediators implicated in SPM have also been identified as potential biomarkers that can distinguish NAFL from NASH and define the stage of fibrosis (Loomba et al., 2015a; Puri et al., 2009; Sanyal and Pacana, 2015).
Contributions by neutrophils to NASH have been underscored by recent studies. Mice lacking two mitogen-activated kinases, p38γ and p38δ, in myeloid cells display attenuated features of advanced NASH in a diet-induced model, and the protection is attributed to impaired migration of p38γ/δ-deficient neutrophils into the damaged liver (González-Terán et al., 2016). Consistent with a potential role in human NASH, p38γ and p38δ are elevated in the liver of individuals with NASH (González-Terán et al., 2016). Neutrophils have also been implicated in fibrosis resolution through their ability to promote polarization of macrophages into a restorative, matrix-degrading phenotype (Calvente et al., 2019). This effect is mediated by delivery of miR-223 from neutrophils to macrophages carried by extracellular vesicles, which attenuates expression of mRNA encoding the Nlrp3 inflammasome scaffold protein. Interactions between neutrophils and macrophages are further promoted by circulating lipocalin-2, a member of a family of small circulating proteins that are induced in inflammation, in part through induction of the CXCR2 receptor (Ye et al., 2016).
Platelets may amplify inflammation and hepatocellular damage in part through activity of GPIbα, a membrane receptor for von Willebrand factor (Malehmir et al., 2019). Recruitment of platelets in NASH is dependent on resident macrophages (Kupffer cells), and their activity in promoting NASH also requires thrombin or fibrinogen (Kopec et al., 2017; Malehmir et al., 2019). Interestingly, platelet activation and adhesion, but not platelet aggregation, contribute to NASH pathogenesis in animal models (Malehmir et al., 2019). These observations raise the prospect of using available platelet antagonists as a prophylactic or disease therapy for NASH and NASH-related HCC.
Liver sinusoidal endothelial cells (LSECs) are also dysregulated in NASH (Hammoutene and Rautou, 2019; Lafoz et al., 2020). In particular, capillarization or development of a thickened subendothelial basement membrane and loss of LSEC fenestrations may contribute to steatosis by obstructing the passage of chylomicron remnants to hepatocytes, driving compensatory synthesis of triglycerides and cholesterol. The cells also contribute to oxidant stress and inflammatory signaling to other resident cell types as well as infiltrating macrophages.
Paracrine and inter-organ interactions
Hepatocytes, as the main target of cell injury in NASH, also elicit key mediators that promote the disease through a number of pathways. The transcriptional regulator TAZ (also known as WWTR1) is an effector of the Hippo pathway that typically pairs with the YAP protein to promote cell growth and has been implicated in tumorigenesis by transactivating growth-promoting target genes. However, TAZ is elevated in human NASH (Chong et al., 2019; Wang et al., 2016) and has been uncovered as a driver of inflammation, hepatocyte death, and fibrosis in animal models of the disease (Planas-Paz et al., 2019; Wang et al., 2016). These effects are attributable to transcriptional induction by TAZ of Indian hedgehog (Ihh), which functions as a paracrine activator of injury and fibrosis. Recent findings indicate that plasma membrane cholesterol activates soluble adenylyl cyclase (sAC; ADCY10) to stimulate a calcium-RhoA-mediated pathway that suppresses TAZ degradation (Wang et al., 2020). However, these findings also support studies implicating other hedgehog ligands as fibrogenic stimuli toward hepatic stellate cells (Xie et al., 2013) and products of ballooned hepatocytes (Rangwala et al., 2011), a histologic hallmark of NASH.
Selective activation of Notch in hepatocytes correlates with disease severity in human NASH and HFD-induced NASH (but not in a methionine-choline deficient diet), as indicated by selective upregulation of Notch target genes (Zhu et al., 2018). Fibrosis in response to Notch is attributed to Sox-9-dependent induction of osteopontin, which activates hepatic stellate cells (Zhu et al., 2018). The findings are especially intriguing because a robust ductular reaction (i.e., amplification of biliary ductal cells) correlates with fibrosis in NASH (Machado et al., 2015a; Williams et al., 2014b), and osteopontin has been linked independently to the ductular reaction (Wang et al., 2014). Osteopontin may also be produced by infiltrating NK T cells, which are increased in NASH (Syn et al., 2010). However, the ductular reaction correlates only with fibrosis but not with regeneration (Rókusz et al., 2017; Sato et al., 2019), consistent with the impaired regeneration associated with NASH. These findings underscore the need to more precisely characterize the ductular reaction’s contribution to hepatic homeostasis, regeneration, and response to injury, specifically distinguishing between ductular cells that contribute to fibrosis and those that promote liver cell repopulation in regeneration and injury by trans-differentiation to hepatocytes (Sato et al., 2019).
The oxidative state of hepatocytes in NASH can also influence intracellular signaling. Damage to mitochondria generates DAMPs that can activate stellate cells (An et al., 2020). In humans and animal models of NASH, obesity is associated with inactivation in hepatocytes of the JAK/STAT protein tyrosine phosphatase, leading to increased STAT-1 and STAT-3 signaling (Grohmann et al., 2018). The effects of STAT-1 and STAT-3 in this setting are dissociated, with STAT-1 driving T cell infiltration and fibrosis, whereas STAT-3 is linked to increased cancer development. Signaling by nuclear factor of activated T cells (NFATc4) similarly downregulates PPARα, preventing fatty acid oxidation, which leads to secretion of osteopontin 1, which, in turn, can activate stellate cells (Du et al., 2020; Song et al., 2020b).
Hepatocytes may also contribute to systemic effects of obesity, especially inflammation and insulin resistance. Their secretion of dipeptidyl peptidase 4 (DPP4) promotes inflammation of visceral adipose and insulin resistance by activating inflammatory signaling in macrophages, underscoring the critical role of cross-talk between hepatocytes and adipose tissue (Ghorpade et al., 2018). Similarly, β-Klotho, the co-receptor for FGF15/19 and FGF21, regulates brown fat conversion by altering hepatic bile acid secretion into the gut, leading to changes in the microbiome that alter signaling through the intestinal bile acid receptor TGR-5, which regulates thermogenesis (Somm et al., 2017). In obesity-induced inflammation, hepatocyte expression of protein tyrosine phosphatase receptor gamma (PTPR-γ) correlates with inflammation and antagonizes insulin action, linking hepatic injury to systemic features of metabolic syndrome (Brenachot et al., 2017). Conversely, adipose tissue-derived signals may influence hepatic function and disease. For example, neuregulin-4, an epidermal growth factor (EGF)-like family member secreted by adipose tissue, protects hepatocytes from cell death (Guo et al., 2017). Finally, endoplasmic reticulum stress in the circumventricular subfornical region (SFO) of the brain through HFD administration or activation of the unfolded protein response in mice may promote steatosis in the liver and can be ameliorated by administration of a chaperone protein to the SFO (Horwath et al., 2017). Mediators linking altered brain function with hepatic fat homeostasis have not yet been identified. Conversely, NAFLD has been associated with a lower brain volume in otherwise healthy adults, suggesting that the disease is linked to brain aging through unknown mechanisms (Weinstein et al., 2018). A recent DNA methylation study provided additional clues regarding accelerated aging in individuals with advanced fibrosis because of NASH and its association with the hepatic collagen content (Loomba et al., 2018).
Epigenetic regulators in hepatocytes may also contribute to NASH pathogenesis. In addition to miRNAs, as described above, G-protein pathway suppressor 2 (GPS-2), a co-factor in the HDAC3 repressor complex, promotes experimental NASH because its genetic deletion attenuates disease through derepression of PPARα (Liang et al., 2019). These modifications could be potential therapeutic targets (Bayoumi et al., 2020).
Multicellular interactions in NASH, although critical to pathogenesis, have been difficult to model. Substantial progress in generating organoid models derived from pluripotent stem cells (Collin de l’Hortet et al., 2019; Ouchi et al., 2019; Ramli et al., 2020) primary liver organoids (Elbadawy et al., 2020), microtissues (Feaver et al., 2016; Vacca et al., 2020), or spheroids using cell lines (Pingitore et al., 2019) offers the prospect of dissecting these interactions and establishing a platform for high-throughput testing of candidate therapeutic agents.
Hepatocyte injury and death
Mechanisms of hepatocyte injury and death are central to the pathogenesis of NASH and explain why hepatocyte ballooning, a key histologic hallmark of severe hepatocellular injury, is so closely linked to disease severity. These ballooned, so-called “undead cells” harbor a specialized form of cellular degeneration that may amplify inflammatory and fibrotic signaling in the pericellular milieu (Ibrahim et al., 2018). A gradient of cell death mechanisms elicits a range of responses and mediators in progressive NASH (Hirsova and Gores, 2015; Schuster et al., 2018). These mechanisms include apoptosis, necroptosis, and pyroptosis. Sublethal lipotoxic hepatocyte injury has also been proposed as a mechanism that cripples hepatocyte function and promotes disease but does not lead to complete cell death (Ibrahim et al., 2018). In addition, autophagy, a homeostatic response to preserve cellular energy homeostasis, may culminate in cell death when it is insufficient to maintain cell viability.
Several mediators of apoptosis have been described in human and experimental NASH; prominent among these are apoptosis signaling-regulated kinase 1 (ASK-1), TRAIL, caspase-2, caspase-8, and FADD-like apoptosis regulator (CFLAR) (Schwabe and Luedde, 2018). Although initial studies focused on TNF-α-dependent programmed cell death, other pathways are increasingly implicated in hepatocyte cell death. ASK-1 induces activity of the downstream effectors p38 and mitogen-activated protein kinase (MAPK) and is induced in the liver of individuals with NASH and ameliorated by the deubiquitinating enzyme TNFAIP3 (Zhang et al., 2018). CFLAR directly targets ASK-1, and a CFLAR-mimicking peptide that blocks ASK-1 dimerization attenuates NASH in rodents and non-human primates (Wang et al., 2017). A deubiquitinating enzyme, TNF-α-induced protein 3 (TNFAIP3), has been identified as a critical pathway of ASK-1 inactivation and endogenous suppressor of steatohepatitis (Zhang et al., 2018). These and related findings have generated enthusiasm for therapeutic inhibition of ASK-1 in NASH, but clinical trials using a small-molecule antagonist have been negative to date. The death receptor TRAIL contributes to cell injury because its deletion abrogates liver inflammation in an animal model (Hirsova et al., 2017; Zhu et al., 2018) but, interestingly, has no effect in adipose tissue. TRAIL receptor 2 (also known as DR5) mediates release of extracellular vesicles from hepatocytes in response to toxic lipids and proteins, which stimulates macrophages to become pro-inflammatory (Hirsova et al., 2016). Other cargo within these hepatocyte-derived vesicles may also amplify liver injury in NASH (Ibrahim et al., 2018).
Caspase-2 is induced by endoplasmic reticulum (ER) stress and TNF-α in experimental NASH, which promotes proteolytic activation of site 1 protease (S1P) (Kim et al., 2018). Genetic or pharmacologic ablation of caspase-2 attenuates NASH by abrogating SREBP activation, enhancing triglyceride secretion, increasing energy expenditure, increasing AMPK activity (Kim et al., 2018), and reducing lipo-apoptosis and hedgehog pathway activation (Machado et al., 2015b). CFLAR directly targets ASK-1 to block its dimerization, which attenuates steatohepatitis in an animal model (Wang et al., 2017).
Necroptosis is a distinct type of cell death attributed to the necrosome, a protein kinase complex whose identified components include kinase receptor-interacting serine/threonine-protein kinase 1 (RIPK1), RIPK3, and the mixed-lineage kinase domain-like protein (MLK1), a pseudokinase (Schwabe and Luedde, 2018). Apoptosis and necroptosis can regulate each other reciprocally but have distinct morphologic features; apoptosis involves pyknosis, or reduction of cell and nuclear volume, and chromatin condensation, which has a modest collateral effect, and necroptosis involves cell swelling and then membrane rupture, which elicits a wide range of pericellular inflammation and damage. Among other pathways downstream of necroptosis, RIPK3 may activate the NLRP3 inflammasome (Lawlor et al., 2015). Moreover, inhibition of RIPK3 attenuates injury and fibrosis in the MCD diet model of NAFLD (Gautheron et al., 2014).
Pyroptosis is a specialized form of cell death in NASH that is similar to the action of inflammatory caspases and, like necroptosis, induces cell swelling and release of inflammatory signals (Beier and Banales, 2018; Jorgensen and Miao, 2015). A key mediator of pyroptosis is gasdermin-D, and its genetic depletion protects against experimental NASH by reducing several inflammatory cytokines and reducing steatosis (Xu et al., 2018). Moreover, the N-terminal domain of gasdermin has been proposed as a biomarker of steatohepatitis (Xu et al., 2018).
Dysregulation of autophagy, a highly conserved lysosomal pathway that maintains energy homeostasis, is increasingly implicated in NASH pathogenesis (Czaja, 2016). This link was sparked by the discovery that autophagy in hepatocytes not only degrades protein substrates but also lipids (lipophagy) (Singh et al., 2009). Autophagy is downregulated in NASH (Fukuo et al., 2014) and obesity (Yang et al., 2010), which may contribute to lipid accumulation in liver and adipose tissue. Moreover, impaired autophagy in macrophages (Ilyas et al., 2019) contributes to alcohol-induced liver injury; thus, altered macrophage autophagy may also be relevant to NAFLD, given the close similarity between the two diseases. Among several soluble signals that regulate autophagy, omega-3 and omega-6 polyunsaturated fatty acids, which are degraded by soluble epoxide hydrolase (Kooner et al., 2011), promote autophagy and M2-mediated macrophage polarization and attenuate ER stress in murine liver and adipose tissue (López-Vicario et al., 2015). Thus, inhibitors of sEH stabilize the concentrations of these beneficial fatty acids and could have therapeutic potential. These findings also reinforce an increasing focus on lipid mediators of injury and resolution, as reviewed above.
Fibrosis in NASH
Fibrosis is the sole histologic feature of NASH that predicts clinical outcomes (Dulai et al., 2017; Sanyal et al., 2019); therefore, it is critical to clarify the mechanisms of fibrogenesis and fibrosis regression in this disease. Recent articles have reviewed general pathways of fibrosis and its degradation in the liver (Koyama and Brenner, 2017; Tsuchida and Friedman, 2017), and here we focus primarily on NASH-specific mechanisms.
The cellular source of fibrosis in NASH is the hepatic stellate cell. This peri-sinusoidal resident cell type undergoes a specialized trans-differentiation or “activation,” which comprises a transition from a quiescent vitamin A-rich cell to one that is fibrogenic, proliferative, and proinflammatory (Tsuchida and Friedman, 2017). NASH-specific drivers may derive from dysregulated hepatocytes that elicit paracrine signals, from autocrine and paracrine cytokines and chemokines released by inflammatory cells, following a number of cell signaling and intracellular events that promote an activated phenotype (Figure 5). In addition, several circulating molecules derived from other tissues can promote fibrotic responses in the liver through direct or indirect actions on hepatic stellate cells (see below). Informaticsbased approaches are increasingly utilized to uncover molecular signatures from tissue and stellate cells that can identify targets and predict outcomes (Hicks et al., 2017; Marcher et al., 2019; Ramnath et al., 2018; Wooden et al., 2017). From such studies, transcriptional (Marcher et al., 2019) and epigenetic (Moran-Salvador et al., 2019) drivers can be inferred and are remarkably conserved across different models of cellular activation, suggesting that stellate cell activation is a common pathway downstream of multiple fibrogenic signals in liver injury.
Figure 5. Cellular and signaling events in NASH.
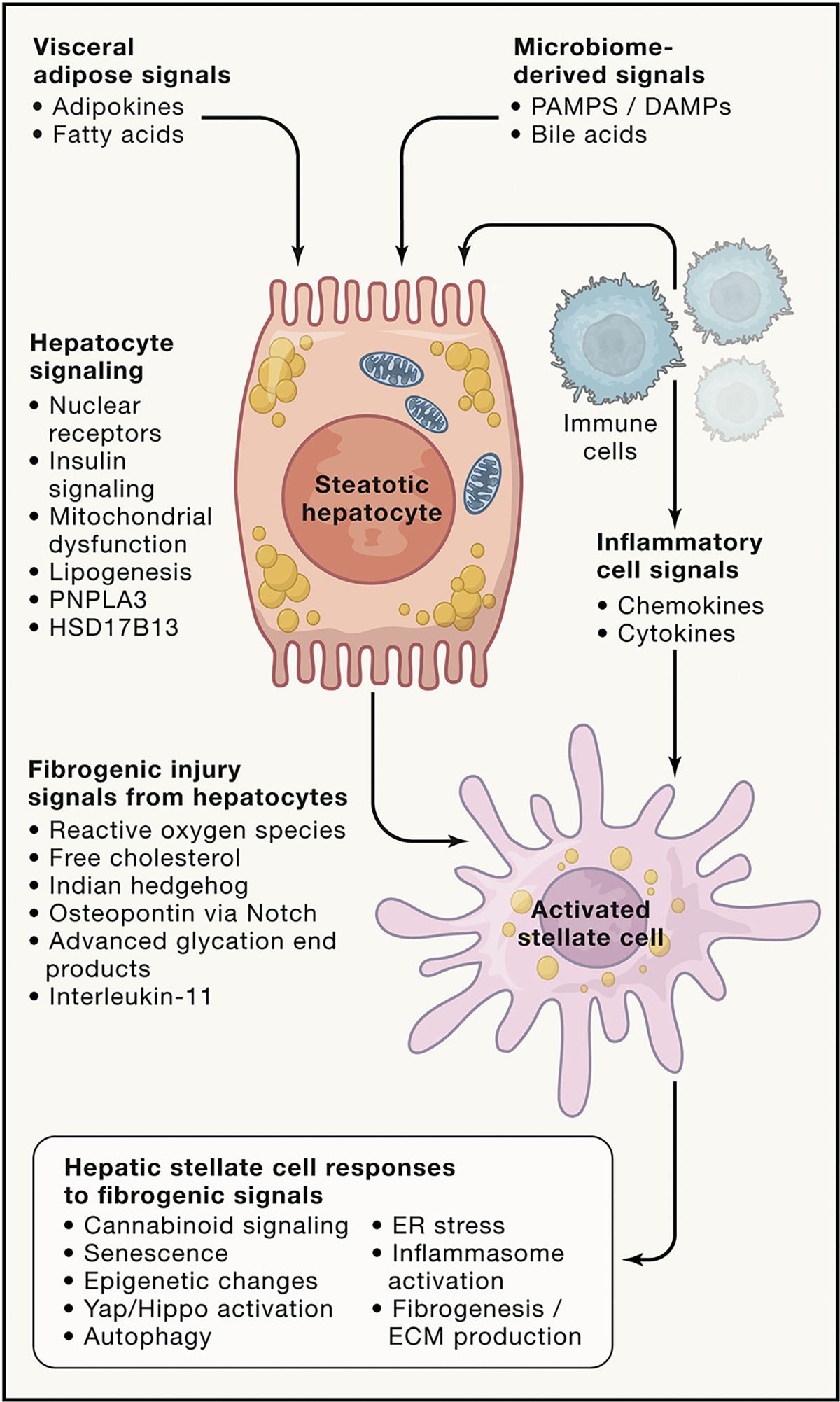
NASH results from a cascade of cellular and signaling events that begin with external stimuli derived from visceral adipose tissue and the gut microbiome as well as inflammatory and immune cells. These inputs converge on hepatocytes to generate a range of intracellular responses, including activation of nuclear receptors (FXR, LXR, PXR, and vitamin D receptor), altered insulin signaling, mitochondrial dysfunction, and lipogenesis. Genetic variants in PNPLA3, HSD17B13, and others may influence the propensity for cell injury and liberation of proinflammatory and fibrogenic signals. Hepatic stellate cells are activated by these signals and undergo a series of intracellular responses that culminate in enhanced extracellular matrix (e.g., collagen) production. It is not certain whether these events characterize every individual with NASH and fibrosis or whether there are endophenotypes of disease that engage different mediators and cells to yield a similar histologic picture. It is also not known which events or signals represent the most critical points of therapeutic vulnerability.
Paracrine signals in the liver that drive fibrosis
Hepatocytes, as the main target of cell injury and fat accumulation, are a rich source of soluble signals that can drive stellate cell activation. The lipotoxic response to hepatocellular injury leads to release of reactive oxygen species (ROS), which have long been recognized as fibrogenic stimuli of stellate cells. Oxidant stress can be generated by NADPH oxidase 4 (NOX4), among others, which has been linked to the concentration of advanced glycation end products and their cognate receptor AGER1, which is downregulated in the liver of individuals with NASH (Dehnad et al., 2020).
Lipotoxic drivers of fibrosis derived from hepatocytes are not well characterized, but strong candidates are oxidized phospholipids, and their neutralization in experimental NASH using a single-chain antibody attenuates disease (Sun et al., 2020). Other sources of ROS and toxic lipids (Chu et al., 2018b) include macrophages (Leroux et al., 2012; Ye et al., 2012) and LSECs (Hammoutene and Rautou, 2019). A key feature of the lipotoxic response is enhanced free cholesterol, which is directly fibrogenic toward stellate cells (Tomita et al., 2014). Apoptosis of hepatocytes, in part through lipotoxicity, is a critical feature of NASH histopathology, and apoptotic bodies are fibrogenic toward hepatic stellate cells (Canbay et al., 2003). It is likely that other lipotoxic mediators underlie the transition from NAFLD to NASH, including mitochondrion-derived danger signals (An et al., 2020) and oxidized phospholipids (Sun et al., 2020). Further identification of these lipotoxic species could unearth therapeutic targets by seeking their neutralization.
Other hepatocyte-derived signals include Ihh (see above; Wang et al., 2016), osteopontin downstream of c-jun and leptin-mediated signaling (Coombes et al., 2016; Schulien et al., 2019), and advanced glycation end product signaling downstream of high-mobility group box-1 (Ge et al., 2018; Sakasai-Sakai et al., 2019), among others. In addition, paracrine and endocrine leptin, which are elevated in obesity and NASH, are fibrogenic (Coombes et al., 2016; Saxena et al., 2004), whereas adiponectin, which is decreased in NASH, is antifibrotic (Handy et al., 2011). Bone morphogenic protein 8 (BMP8) is a member of the transforming growth factor β1 (TGF-β1) family that drives hepatic stellate cells into a more inflammatory and fibrogenic phenotype (Vacca et al., 2020).
Paracrine inflammatory cell signals that drive fibrosis
Several chemokines and their receptors have been implicated in NASH fibrosis, including CCR2 (Seki et al., 2009b), CCL5/CCR5 (Li et al., 2017; Seki et al., 2009a), CXCL10 (Tomita et al., 2016), and CCL20 (Chu et al., 2018b; Hanson et al., 2019); CXCL9 is antifibrotic (Wasmuth et al., 2009). There are several chemokines that contribute to other pathways of NASH pathogenesis apart from fibrosis (Roh and Seki, 2018).
Macrophages can promote or attenuate NASH fibrosis based on their polarization between proinflammatory and reparative phenotypes (Kazankov et al., 2019). Their most critical role in affecting fibrosis is through proreparative, fibrolytic macrophages, which, in the mouse, are CD11b+, F4/80+, and Ly6Clow (Ramachandran et al., 2012). Conversely, TLR4 signaling in proinflammatory macrophages in response to endotoxin generates more TGF-β1, a key fibrogenic cytokine (Rivera et al., 2007).
Mediators of stellate cell responses in NASH
Many convergent signals affect stellate cell behavior in NASH; some are profibrotic, and others are antifibrotic. Among extracellular signals, Notch activation, cannabinoids, and IL-11 are especially relevant to NASH fibrosis. Notch activation in hepatocytes activates the SOX9 transcription factor to stimulate osteopontin secretion, which, in turn, activates stellate cells (Zhu et al., 2018). The CB1 receptor is expressed on stellate cells and drives fibrogenesis in addition to pleotropic effects on other liver cell types (Mallat and Lotersztajn, 2008). In contrast, CB2 signaling has the opposite effect on stellate cells and can also antagonize profibrotic signaling by TH17 lymphocytes (Guillot et al., 2014). Antagonism of CB1 receptors is an appealing therapeutic approach for NASH, provided that the compound does not cross the blood-brain barrier, where it can induce severe depression in some people. IL-11, whose expression by hepatocytes is induced by the fibrogenic cytokine TGF-β1, has been identified as a potent inflammatory and fibrotic signal toward stellate cells in experimental NASH models, and its antagonism greatly attenuates disease in these models (Widjaja et al., 2019).
Intracellular events underlying stellate cell activation include autophagy and ER stress (Hernández-Gea et al., 2012, 2013), responses elicited in part by liver injury and, possibly, hypoxemia, leading to HIF-1 activation (Asrani et al., 2014). Other metabolic pathways may also be relevant to NASH. For example, generation of α-keto-glutarate from glutamine via glutaminolysis is required for stellate cell activation, which is under the control of the YAP transcription factor (Du et al., 2018). YAP also drives stellate cell activation (Martin et al., 2016), which can be antagonized by omega-3 polyunsaturated fatty acids (Zhang et al., 2016b). A growing list of epigenetic modifications regulating stellate cell activation has been uncovered (Barcena-Varela et al., 2021; Eslam et al., 2018; Schuppan et al., 2018). Many miRNAs affect stellate cell responses; miR-590–5p has been implicated specifically in NASH (Hanson et al., 2019). Global changes in methylation have also been reported during stellate cell activation (Page et al., 2016), and these epigenetic determinants of fibrosis may be heritable (Zeybel et al., 2012). Finally, senescence of hepatic stellate cells may contribute to an altered immune microenvironment that can affect tumorigenesis associated with obesity (Lujambio et al., 2013; Yoshimoto et al., 2013).
Systemic regulators of fibrogenesis in NASH
NASH is one component of the multisystem metabolic syndrome that affects adipose tissue, gut, and muscle, among other organs. Thus, hepatokines (Lebensztejn et al., 2016) and systemic signals derived from non-hepatic tissues can influence hepatic fibrosis, although the relative contribution of liver-derived and systemic molecules to fibrosis is uncertain. Adipose tissue is a rich source of TNF-α, IL-6, and the adipokines leptin, adiponectin, resistin, and retinol-binding protein 4 (Saxena and Anania, 2015), and circulating levels of leptin are correlated with the severity of NAFLD (Polyzos et al., 2016). Visceral fat is a particularly rich source of IL-6 and TNF-α (van der Poorten et al., 2008). Systemic insulin resistance, which is largely regulated by skeletal muscle (Shulman, 2000), may also promote NASH fibrosis (Bugianesi et al., 2004; Khan et al., 2019) by increasing connective tissue growth factor, which is fibrogenic (Paradis et al., 2001).
The gut has emerged as a major driver or NASH through the effect of the microbiome and its interaction with gut-liver hormonal signaling. In particular, endotoxin and gut-derived PAMPs and DAMPs are directly fibrogenic by signaling through TLR4 and other innate immune receptors on stellate cells. Conversely, intestinal FXR signaling induces FGF15 in mice and FGF19 in humans, which are potently antifibrotic (Schumacher et al., 2019), and FXR agonists are being evaluated as potential therapeutic agents for NASH.
NASH and HCC
Clinical considerations.
The NASH epidemic has provoked a dramatic increase in HCC (Berkan-Kawińska and Piekarska, 2020; Mittal et al., 2016; Negro, 2020). As noted above, NASH is the fastest-rising cause of HCC in the United States (Younossi et al., 2018) and the most rapidly increasing indication for liver transplantation among individuals with HCC (Younossi et al., 2019a). Despite some progress in clarifying HCC pathogenesis in NASH, there are significant unmet challenges, and there is inadequate knowledge of the natural history, key pathogenic drivers, and optimal animal models that mimic the disease to establish therapies.
HCC in NASH is more likely to be diagnosed at later stages, when tumors are larger and outcomes are poorer, for two key reasons. First, NASH is often unrecognized and may present as “cryptogenic cirrhosis,” where the first manifestation is HCC. Second, in individuals with NASH, a larger fraction (~35%–50%) of tumors arises before the individuals are cirrhotic compared with other liver diseases (e.g., chronic HBV or HCV), where cirrhosis is already present in ~90% of people (Dyson et al., 2014; Mittal et al., 2016; Paradis et al., 2009; Piscaglia et al., 2016). Thus, many people are unaware that they have liver disease, and even those with recognized NASH are not routinely screened for HCC until cirrhosis is present, which may be too late because of the higher propensity for tumors to arise prior to cirrhosis. Thus, the poorer outcomes for HCC in the NASH population may reflect the later stage at the time of diagnosis rather than inherent differences in tumor biology.
The unique clinical features of HCC in NASH underscore the critical need to better identify high-risk individuals through improved surveillance strategies to detect underlying NASH and improved screening methods for HCC that do not rely on establishing a diagnosis of cirrhosis, otherwise early HCCs will continue to be missed (Berkan-Kawińska and Piekarska, 2020; Reig et al., 2019). Efforts to develop more sensitive screening strategies that detect circulating tumor cells or their DNA or specific epigenetic changes using liquid biopsy or other non-invasive technologies are advancing but have not yet been validated sufficiently to be implemented in clinical practice (Chalasani et al., 2020; Villanueva, 2019; von Felden et al., 2020).
The larger fraction of individuals with NASH who develop HCC before cirrhosis is present compared with other etiologies underscores the contribution of systemic or metabolic risk factors unique to NASH. Moreover, NASH not only confers risk of HCC as the primary etiology but is often overlooked as a co-factor in driving HCC in people with other etiologies, especially hepatitis C (Chen et al., 2008), hepatitis B (Chan et al., 2017; Chen et al., 2008), and alcohol-associated liver disease (Nair et al., 2002).
Genetic risk factors for HCC in individuals with NAFLD have not yet been explored adequately. To date, the best validated genetic risk is the PNPLA3 variant, which, as noted above, also confers an increased risk of NASH, especially in obese people (PNPLA3 rs738409, c.444 C > G p.I148M) (Liu et al., 2014; Singal et al., 2014; Trépo et al., 2014). In the United States, the PNPLA3 GG genotype (rs738409: C > G) is associated with increased risk of underlying cirrhosis in individuals with HCC (odds ratio [OR] = 2.48; 95% confidence interval [CI], 1.05–5.87), and the adjusted OR of HCC in individuals with NASH with a homozygous GG genotype is 3.21 (95% CI, 1.68–6.41) compared with those with CC or CG genotypes (Hassan et al., 2013). Single studies have also identified the rs641738 variant of MBOAT7 as enhancing the risk of HCC in non-cirrhotic Italian people with NASH (Donati et al., 2017), and a variant of TM6SF2 (rs58542926) that promotes NASH has an independent effect on HCC risk that is additive with the risk allele of PNPLA3 (Yang et al., 2019). As disease awareness expands and cohort sizes increase, additional variants are likely to emerge, and such studies will need to distinguish between variants that heighten the risk of NASH and those that specifically increase the risk of HCC beyond their effect on disease severity.
Although specific pathogenic factors unique to NASH are not fully elucidated, it is clear that obesity and metabolic syndrome associated with NASH impose specific risks apart from those linked to liver disease severity. Indeed, obesity increases the risk of all cancers, with the most heightened added risk for HCC compared with other tumors (Bhaskaran et al., 2014; Calle et al., 2003). Along with obesity, type 2 diabetes imposes a significant risk of HCC among individuals with NASH cirrhosis (Doycheva et al., 2020; Hassan et al., 2013; Yang et al., 2020). Concurrently, metabolic dysregulation associated with obesity and metabolic syndrome create a milieu that promotes oxidant stress, DNA damage, and other permissive conditions, as reviewed below.
NASH-related mechanisms of HCC
NAFLD and NASH are characterized by enhanced lipid accumulation in hepatocytes that can generate pathogenic drivers of carcinogenesis, in particular oxidative DNA damage (Masarone et al., 2018; Tanaka et al., 2013). As a substrate for oxidant generation, hepatic steatosis can also increase STAT-1 and STAT-3 stability and activity through oxidation of a specific phosphatase that normally inactivates these molecules, concurrently promoting NASH, fibrosis, and HCC (Grohmann et al., 2018). Oxidative damage also stimulates the DNA damage response, whose consequences may include increased DNA-dependent protein kinase (DNA-PK), which leads to error-prone DNA repair that may confer resistance to conventional chemotherapy (Cornell et al., 2015; Li et al., 2020). DNA damage induced by the unconventional prefoldin RPB5 interactor (URI) may also stimulate IL-17A-mediated inflammation, which is blocked in an experimental model by antibodies to this cytokine (Gomes et al., 2016); a similar mechanism may also underlie HCC associated with alcohol-associated liver disease (Ma et al., 2020).
Hepatic steatosis is amplified by increased dietary cholesterol, which enhances HCC growth in a rodent model and promotes oncogenic signaling and tumor-promoting gene expression, mimicking similar changes that have been documented in human NASH-HCC (Liang et al., 2018). Human NASH and NASH-related HCC are also characterized by enhanced expression of the hepatocyte cholesterol biosynthetic enzyme squalene epoxidase, which generates increased NADP+ and cholesterol ester to epigenetically inactivate the PTEN tumor suppressor; these events promote cell growth and oxidative stress (Liu et al., 2018). Inhibition of squalene epoxidase with the clinically approved antifungal compound terbinafine attenuates HCC in cultured HCC cells and animal models (Liu et al., 2018). Remarkably, hepatic carcinogenesis in NASH may also be regulated by the circadian clock, which is controlled by opposing activities of FXR and CAR (Kettner et al., 2016). Studies have additionally implicated dysregulation of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) in NASH-HCC pathogenesis, which can modulate several metabolic pathways, including gluconeogenesis, mitochondrial biogenesis, fatty acid oxidation, the antioxidant response, and DNL (Piccinin et al., 2019). Evidence that tumorigenesis requires DNL is based on studies demonstrating that inhibition of acetyl-CoA carboxylase, which promotes DNL, is anti-tumorigenic (Lally et al., 2019). Steatotic hepatocytes are also sensitized to cell death through apoptosis, which may promote tumorigenesis in experimental NASH (Hirsova et al., 2020). On the other hand, the apoptosis-antagonizing transcription factor (AATF) promotes tumorigenesis in animal models (Kumar et al., 2019), so the ultimate contribution of apoptosis to HCC is unsettled.
Autophagy, a lysosomal degradative pathway described above, is especially important to catabolize lipids and proteins in maintaining cellular health in healthy and steatotic hepatocytes (Czaja, 2016; Lee et al., 2018; Singh et al., 2009). Impaired autophagy associated with lipid accumulation can activate mammalian target of rapamycin (mTOR), which may increase hepatic oxidative stress (Kim et al., 2011). Impaired autophagy in NAFLD and fat accumulation are also associated with increased ER stress, which may further promote inflammation mediated by TNF-α (Maurel et al., 2014; Nakagawa et al., 2014). In response to autophagy, which is stimulated by oxidative stress, transcriptional activity of nuclear factor erythroid 2-related factor (NRF2) may be increased to generate an antioxidant response, which can promote survival not only of steatotic hepatocytes but also of tumor cells (Ichimura et al., 2013; Xu et al., 2019).
As discussed above, the gut microbiome is increasingly implicated in NASH pathogenesis, and more recent observations extend its role to the pathogenesis of HCC (Chu et al., 2018a; Henao-Mejia et al., 2012; Kanda et al., 2020; Sydor et al., 2020). A distinct gut microbiome has been linked to HCC pathogenesis in NASH through production of bacterially derived deoxycholate, which enhances a senescence-associated secretome that promotes inflammation in the liver (Yoshimoto et al., 2013). The gut’s conversion of primary to secondary bile acids may also regulate the liver’s antitumor response mediated by NK T cells (Ma et al., 2018) and by increasing FGF19 production by the gut, which promotes hepatocyte proliferation (Sydor et al., 2020). These effects may also be amplified by activation of TLR4 and TLR9 signaling in the liver (Henao-Mejia et al., 2012).
Although most studies of HCC pathogenesis associated with NASH focus on dysregulated signaling by hepatocytes, other resident and infiltrating cells in the liver also promote tumorigenesis. TLR4 signaling in macrophages and stellate cells, for example, provokes inflammasome activation in both cell types, which is proinflammatory and fibrogenic (Anstee et al., 2019; Dong et al., 2020; Nguyen et al., 2018; Sharifnia et al., 2015). In turn, there are a number of potential mechanisms by which stellate cell activation and fibrogenesis may promote cancer, including generation of factors that promote hepatocyte growth and survival, inflammation, and tumor evasion and involving Yap/Taz, Notch, and hedgehog, among others (Barry et al., 2020; Schwabe et al., 2020; Zhang et al., 2016a).
Finally, extensive characterization of transcriptomic, genetic, and epigenetic abnormalities in human HCC in individuals with hepatitis B or C has led to detailed classification systems (Nakagawa et al., 2016; Sia et al., 2017; Villanueva, 2019; Zucman-Rossi et al., 2015). Although none has yet been compiled for NASH-HCC, similar efforts are underway and are likely to uncover novel, actionable targets for chemopreventive or treatment strategies in individuals at high risk or those with HCC. Recent mouse models and clinical data indicate that NASH-HCC is less responsive to emerging immunotherapies because of reduced infiltration of CD4+ T cells, impaired immune surveillance, and NASH-related aberrant T cell activation, which induces tissue damage (Heinrich et al., 2021; Pfister et al., 2021).
Conclusions
There are significant challenges when tackling the global epidemic of NAFLD and its related metabolic and hepatic complications, but there is a solid and mechanistic foundation on which therapeutic progress can be based. NAFLD is almost universally present before overt onset of NASH, liver fibrosis, and type 2 diabetes. Obstacles remain in developing better tools to predict who is at risk for NAFLD, especially for non-obese individuals, and in advancing non-invasive modalities to detect and stage NAFLD/NASH/liver fibrosis. Drug approval for NASH currently relies on liver biopsy, which has slowed progress in evaluating the efficacy of candidate therapies. Importantly, however, preclinical studies have demonstrated that, if hepatic steatosis can be reversed through metabolic interventions, then liver inflammation, liver fibrosis, and diabetes can resolve, providing a rationale and roadmap for development of new strategies to address metabolic dysregulation in NAFLD and NASH. Although these metabolic abnormalities are not the sole defects in this multifaceted disease, they nonetheless are at the nexus of convergent drivers of disease progression, and their targeting is likely to be a foundational element of any successful therapeutic regimen. Therefore, we expect that treatment response can be enhanced by strategically combining multiple agents targeting distinct metabolic pathways to not only achieve resolution of NASH and improvement in fibrosis but also a reduction in cardiovascular disease risk and future risk of cancer.
ACKNOWLEDGMENTS
The authors received funding support from the Department of Health and Human Services (NIH/NIDDK/NHLBI/NIEHS/NCATS): R01 DK56621 and DK128289 (to S.L.F.); R01 DK113984, R01 DK114793, R01 DK116774, R01 DK119968, RC2 DK120534, and P30 DK045735 (to G.I.S.); and P42 ES01033, UL1 TR001442, U01 DK061734, R01 DK106419, R01 DK121378, R01 DK124318, P30 DK120515, and P01 HL147835 (to R.L.).
Footnotes
DECLARATION OF INTERESTS
R.L. serves as a consultant for Anylam/Regeneron, Amgen, Arrowhead Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, CohBar, Eli Lilly, Galmed, Gilead, Glympse bio, Inipharm, Intercept, Ionis, Janssen, Madrigal, Metacrine, NGM Biopharmaceuticals, Novartis, Novo Nordisk, Pfizer, Sagimet, 89 bio, and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Astrazeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, Genfit, Gilead, Intercept, Inventiva, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, Pfizer, and Siemens. He is also co-founder of Liponexus; S.L.F. is a consultant to 89 Bio, Amgen, Axcella Health, Blade Therapeutics, Bristol Myers Squibb, Can-Fite Biopharma, Casma Therapeutics, ChemomAb, Escient Pharmaceuticals, Forbion, Galmed, Gordian Biotechnology, Glycotest, Glympse Bio, In sitro, Morphic Therapeutics, North Sea Therapeutics, Novartis, Ono Pharmaceuticals, Pfizer Pharmaceuticals, Scholar Rock, and Surrozen and has stock options (all less than 1% of company value) in Blade Therapeutics, Escient, Galectin, Galmed, Genfit, Glympse, Hepgene, Lifemax, Metacrine, Morphic Therapeutics, Nimbus, North Sea Therapeutics, Scholar Rock, and Surrozen. G.I.S. serves on the scientific advisory boards for Merck, Novo Nordisk, AstraZeneca, Gilead Sciences, Esperion, Generian, Levels, 89bio, and Janseen Research and Development. G.I.S. receives investigator-initiated support from AstraZeneca, Gilead Sciences, and Merck. G.I.S. is an inventor on Yale patents for liver-targeted mitochondrial uncoupling agents and controlled-release mitochondrial uncoupling agents for the treatment of NAFLD, NASH, T2D, and related metabolic disorders and is a scientific-cofounder of TLC.
REFERENCES
- Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, Kozlitina J, Stender S, Wood GC, et al. (2018). A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 378, 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulizi A, Vatner DF, Ye Z, Wang Y, Camporez JP, Zhang D, Kahn M, Lyu K, Sirwi A, Cline GW, et al. (2020). Membrane-bound sn-1,2-diacylglycerols explain the dissociation of hepatic insulin resistance from hepatic steatosis in MTTP knockout mice. J. Lipid Res. 61, 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, and Angulo P (2005). The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129, 113–121. [DOI] [PubMed] [Google Scholar]
- Adorini L, Pruzanski M, and Shapiro D (2012). Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov. Today 17, 988–997. [DOI] [PubMed] [Google Scholar]
- Alegre F, Pelegrin P, and Feldstein AE (2017). Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 37, 119–127. [DOI] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, and Cohen P (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 7, 261–269. [DOI] [PubMed] [Google Scholar]
- An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, Kuramitsu K, Lai M, and Popov YV (2020). Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 11, 2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DP, Eriksson-Hogling D, Bäckdahl J, Thorell A, Löfgren P, Rydén M, Arner P, and Hoffstedt J (2017). Omentectomy in Addition to Bariatric Surgery-a 5-Year Follow-up. Obes. Surg. 27, 1115–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstee QM, Seth D, and Day CP (2016). Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 150, 1728–1744.e7. [DOI] [PubMed] [Google Scholar]
- Anstee QM, Reeves HL, Kotsiliti E, Govaere O, and Heikenwalder M (2019). From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16, 411–428. [DOI] [PubMed] [Google Scholar]
- Arab JP, Karpen SJ, Dawson PA, Arrese M, and Trauner M (2017). Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 65, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrani SK, Talwalkar JA, Kamath PS, Shah VH, Saracino G, Jennings L, Gross JB, Venkatesh S, and Ehman RL (2014). Role of magnetic resonance elastography in compensated and decompensated liver disease. J. Hepatol. 60, 934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcena-Varela M, Paish H, Alvarez L, Uriarte I, Latasa MU, Santamaria E, Recalde M, Garate M, Claveria A, Colyn L, et al. (2021). Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 70, 388–400. [DOI] [PubMed] [Google Scholar]
- Barry AE, Baldeosingh R, Lamm R, Patel K, Zhang K, Dominguez DA, Kirton KJ, Shah AP, and Dang H (2020). Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 8, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, Yang K, Kumari M, Gross RW, et al. (2011). Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 52, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BasuRay S, Smagris E, Cohen JC, and Hobbs HH (2017). The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 66, 1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayol SA, Simbi BH, Fowkes RC, and Stickland NC (2010). A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic Fatty liver disease in rat offspring. Endocrinology 151, 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayoumi A, Grønbæk H, George J, and Eslam M (2020). The Epigenetic Drug Discovery Landscape for Metabolic-associated Fatty Liver Disease. Trends Genet. 36, 429–441. [DOI] [PubMed] [Google Scholar]
- Beaven SW, and Tontonoz P (2006). Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu. Rev. Med. 57, 313–329. [DOI] [PubMed] [Google Scholar]
- Beaven SW, Matveyenko A, Wroblewski K, Chao L, Wilpitz D, Hsu TW, Lentz J, Drew B, Hevener AL, and Tontonoz P (2013). Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 18, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier JI, and Banales JM (2018). Pyroptosis: An inflammatory link between NAFLD and NASH with potential therapeutic implications. J. Hepatol. 68, 643–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamouche-Trouillet S, and Postic C (2016). Emerging role of miR-21 in non-alcoholic fatty liver disease. Gut 65, 1781–1783. [DOI] [PubMed] [Google Scholar]
- Berkan-Kawinska A, and Piekarska A (2020). Hepatocellular carcinoma in non-alcohol fatty liver disease - changing trends and specific challenges. Curr. Med. Res. Opin. 36, 235–243. [DOI] [PubMed] [Google Scholar]
- Betancourt-Garcia MM, Arguelles A, Montes J, Hernandez A, Singh M, and Forse RA (2017). Pediatric Nonalcoholic Fatty Liver Disease: the Rise of a Lethal Disease Among Mexican American Hispanic Children. Obes. Surg. 27, 236–244. [DOI] [PubMed] [Google Scholar]
- Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, and Smeeth L (2014). Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5$24 million UK adults. Lancet 384, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindesbøll C, Fan Q, Nørgaard RC, MacPherson L, Ruan H-B, Wu J, Pedersen TÅ, Steffensen KR, Yang X, Matthews J, et al. (2015). Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 56, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borén J, Adiels M, Björnson E, Matikainen N, Söderlund S, Rämö J, Ståhlman M, Ripatti P, Ripatti S, Palotie A, et al. (2020). Effects of TM6SF2 E167K on hepatic lipid and very low-density lipoprotein metabolism in humans. JCI Insight 5, e144079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borji M, Nourbakhsh M, Shafiee SM, Owji AA, Abdolvahabi Z, Hesari Z, Ilbeigi D, Seiri P, and Yousefi Z (2019). Down-Regulation of SIRT1 Expression by mir-23b Contributes to Lipid Accumulation in HepG2 Cells. Biochem Genet. 57, 507–521. [DOI] [PubMed] [Google Scholar]
- Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, et al. (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady GF, Kwan R, Ulintz PJ, Nguyen P, Bassirian S, Basrur V, Nesvizhskii AI, Loomba R, and Omary MB (2018). Nuclear lamina genetic variants, including a truncated LAP2, in twins and siblings with nonalcoholic fatty liver disease. Hepatology 67, 1710–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenachot X, Ramadori G, Ioris RM, Veyrat-Durebex C, Altirriba J, Aras E, Ljubicic S, Kohno D, Fabbiano S, Clement S, et al. (2017). Hepatic protein tyrosine phosphatase receptor gamma links obesity-induced inflammation to insulin resistance. Nat. Commun. 8, 1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (2008). Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96. [DOI] [PubMed] [Google Scholar]
- Browning JD, and Horton JD (2004). Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumbaugh DE, Tearse P, Cree-Green M, Fenton LZ, Brown M, Scherzinger A, Reynolds R, Alston M, Hoffman C, Pan Z, et al. (2013). Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J. Pediatr. 162, 930–936.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruschi FV, Claudel T, Tardelli M, Caligiuri A, Stulnig TM, Marra F, and Trauner M (2017). The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 65, 1875–1890. [DOI] [PubMed] [Google Scholar]
- Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD, Brosch M, Rosendahl J, Berg T, Ridinger M, et al. (2015). A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 47, 1443–1448. [DOI] [PubMed] [Google Scholar]
- Bugianesi E, Manzini P, D’Antico S, Vanni E, Longo F, Leone N, Massarenti P, Piga A, Marchesini G, and Rizzetto M (2004). Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 39, 179–187. [DOI] [PubMed] [Google Scholar]
- Calle EE, Rodriguez C, Walker-Thurmond K, and Thun MJ (2003). Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 348, 1625–1638. [DOI] [PubMed] [Google Scholar]
- Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N, De Mollerat Du Jeu X, Llorente C, Boyer J, and Feldstein AE. (2019). Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J. Clin. Invest. 129, 4091–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camporez JP, Wang Y, Faarkrog K, Chukijrungroat N, Petersen KF, and Shulman GI (2017). Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl. Acad. Sci. USA 114, E11285–E11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, and Gores GJ (2003). Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Invest. 83, 655–663. [DOI] [PubMed] [Google Scholar]
- Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, Guebre-Egziabher F, Jurczak MJ, Kahn M, Guigni BA, et al. (2013). CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 110, 1869–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carino A, Cipriani S, Marchian S, Biagioli M, Santorelli C, Donini A, Zampella A, Monti MC, and Fiorucci S (2017). BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci. Rep. 7, 42801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussy C, Soni M, Cui J, Bettencourt R, Schork N, Chen CH, Ikhwan MA, Bassirian S, Cepin S, Gonzalez MP, et al. ; Familial NAFLD Cirrhosis Research Consortium (2017). Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Invest. 127, 2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C, Bettencourt R, Rizo E, Richards L, Xu ZZ, et al. (2019). A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 10, 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, and Sanyal AJ (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357. [DOI] [PubMed] [Google Scholar]
- Chalasani NP, Ramasubramanian TS, Bhattacharya A, Olson MC, Edwards VD, Roberts LR, Kisiel JB, Reddy KR, Lidgard GP, Johnson SC, et al. (2020). A Novel Blood-based Panel of Methylated DNA and Protein Markers for Detection of Early-Stage Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. Published online September 2, 2020. 10.1016/j.cgh.2020.08.065. [DOI] [PubMed] [Google Scholar]
- Chan AW, Wong GL, Chan HY, Tong JH, Yu YH, Choi PC, Chan HL, To KF, and Wong VW (2017). Concurrent fatty liver increases risk of hepatocellular carcinoma among patients with chronic hepatitis B. J. Gastroenterol. Hepatol. 32, 667–676. [DOI] [PubMed] [Google Scholar]
- Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, and Dierkhising RA (2011). Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 141, 1249–1253. [DOI] [PubMed] [Google Scholar]
- Chavez-Talavera O, Tailleux A, Lefebvre P, and Staels B (2017). Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 152, 1679–1694.e3. [DOI] [PubMed] [Google Scholar]
- Chen CL, Yang HI, Yang WS, Liu CJ, Chen PJ, You SL, Wang LY, Sun CA, Lu SN, Chen DS, and Chen CJ (2008). Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology 135, 111–121. [DOI] [PubMed] [Google Scholar]
- Chen W, Chang B, Li L, and Chan L (2010). Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 52, 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Thomsen M, and Vitetta L (2019). Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J. Cell. Biochem. 120, 2713–2720. [DOI] [PubMed] [Google Scholar]
- Chennamsetty I, Coronado M, Contrepois K, Keller MP, Carcamo-Orive I, Sandin J, Fajardo G, Whittle AJ, Fathzadeh M, Snyder M, et al. (2016). Nat1 Deficiency Is Associated with Mitochondrial Dysfunction and Exercise Intolerance in Mice. Cell Rep. 17, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, Kellum JM, Min H, Luketic VA, and Sanyal AJ (2008). Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 48, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY (2013). Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YC, Lim TE, Fu Y, Shin EM, Tergaonkar V, and Han W (2019). Indian Hedgehog links obesity to development of hepatocellular carcinoma. Oncogene 38, 2206–2222. [DOI] [PubMed] [Google Scholar]
- Chu H, Williams B, and Schnabl B (2018a). Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res. 2, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X, Jin Q, Chen H, Wood GC, Petrick A, Strodel W, Gabrielsen J, Benotti P, Mirshahi T, Carey DJ, et al. (2018b). CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J. Transl. Med. 16, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Boerwinkle E, Mosley TH Jr., and Hobbs HH (2006). Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272. [DOI] [PubMed] [Google Scholar]
- Collin de l’Hortet A, Takeishi K, Guzman-Lepe J, Morita K, Achreja A, Popovic B, Wang Y, Handa K, Mittal A, Meurs N, et al. (2019). Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 30, 385–401.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes JD, Choi SS, Swiderska-Syn M, Manka P, Reid DT, Palma E, Briones-Orta MA, Xie G, Younis R, Kitamura N, et al. (2016). Osteopontin is a proximal effector of leptin-mediated non-alcoholic steatohepatitis (NASH) fibrosis. Biochim. Biophys. Acta 1862, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell L, Munck JM, Alsinet C, Villanueva A, Ogle L, Willoughby CE, Televantou D, Thomas HD, Jackson J, Burt AD, et al. (2015). DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. Cancer Res. 21, 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillard C, Bergeron N, Prud’homme D, Bergeron J, Tremblay A, Bouchard C, Mauriège P, and Després JP (1999). Gender difference in postprandial lipemia : importance of visceral adipose tissue accumulation. Arterioscler. Thromb. Vasc. Biol. 19, 2448–2455. [DOI] [PubMed] [Google Scholar]
- Csak T, Pillai A, Ganz M, Lippai D, Petrasek J, Park JK, Kodys K, Dolganiuc A, Kurt-Jones EA, and Szabo G (2014). Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 34, 1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Chen CH, Lo MT, Schork N, Bettencourt R, Gonzalez MP, Bhatt A, Hooker J, Shaffer K, Nelson KE, et al. ; For The Genetics Of Nafld In Twins Consortium (2016). Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study. Hepatology 64, 1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson DJ, Steele T, Wilding JP, Halford JC, Harrold JA, Hamer M, and Karpe F (2017). What have human experimental overfeeding studies taught us about adipose tissue expansion and susceptibility to obesity and metabolic complications? Int. J. Obes. 41, 853–865. [DOI] [PubMed] [Google Scholar]
- Czaja MJ (2016). Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 61, 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva HE, Teterina A, Comelli EM, Taibi A, Arendt BM, Fischer SE, Lou W, and Allard JP (2018). Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 8, 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Weiner A, and Amit I (2020). The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181, 1207–1217. [DOI] [PubMed] [Google Scholar]
- Dehnad A, Fan W, Jiang JX, Fish SR, Li Y, Das S, Mozes G, Wong KA, Olson KA, Charville GW, et al. (2020). AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J. Clin. Invest. 130, 4320–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Després JP, Moorjani S, Lupien PJ, Tremblay A, Nadeau A, and Bouchard C (1990). Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 10, 497–511. [DOI] [PubMed] [Google Scholar]
- Ding J, Li M, Wan X, Jin X, Chen S, Yu C, and Li Y (2015). Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 5, 13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, Petta S, Maier S, Rosso C, De Luca L, et al. (2017). MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 7, 4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Zhuang Q, Ning M, Wu S, Lu L, and Wan X (2020). Palmitic acid stimulates NLRP3 inflammasome activation through TLR4-NF-κB signal pathway in hepatic stellate cells. Ann. Transl. Med. 8, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doycheva I, Zhang T, Amjad W, and Thuluvath PJ (2020). Diabetes and Hepatocellular Carcinoma: Incidence Trends and Impact of Liver Disease Etiology. J. Clin. Exp. Hepatol. 10, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, and Diehl AM (2018). Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 154, 1465–1479.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Lin Z, Wang Z, Liu D, Tian D, and Xia L (2020). SPOCK1 overexpression induced by platelet-derived growth factor-BB promotes hepatic stellate cell activation and liver fibrosis through the integrin α5β1/PI3K/Akt signaling pathway. Lab. Invest. 100, 1042–1056. [DOI] [PubMed] [Google Scholar]
- du Plessis J, van Pelt J, Korf H, Mathieu C, van der Schueren B, Lannoo M, Oyen T, Topal B, Fetter G, Nayler S, et al. (2015). Association of Adipose Tissue Inflammation With Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 149, 635, 48.e14. [DOI] [PubMed] [Google Scholar]
- Dudley KJ, Sloboda DM, Connor KL, Beltrand J, and Vickers MH (2011). Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 6, e21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, Sebastiani G, Ekstedt M, Hagstrom H, Nasr P, et al. (2017). Increased risk of mortality by fibrosis stage in non-alcoholic fatty liver disease: Systematic Review and Meta-analysis. Hepatology 65, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, Aslam T, Patanwala I, Gaggar S, Cole M, et al. (2014). Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 60, 110–117. [DOI] [PubMed] [Google Scholar]
- Elbadawy M, Yamanaka M, Goto Y, Hayashi K, Tsunedomi R, Hazama S, Nagano H, Yoshida T, Shibutani M, Ichikawa R, et al. (2020). Efficacy of primary liver organoid culture from different stages of non-alcoholic steatohepatitis (NASH) mouse model. Biomaterials 237, 119823. [DOI] [PubMed] [Google Scholar]
- Erion DM, Popov V, Hsiao JJ, Vatner D, Mitchell K, Yonemitsu S, Nagai Y, Kahn M, Gillum MP, Dong J, et al. (2013). The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology 154, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslam M, and George J (2019). Genetic Insights for Drug Development in NAFLD. Trends Pharmacol. Sci. 40, 506–516. [DOI] [PubMed] [Google Scholar]
- Eslam M, Valenti L, and Romeo S (2018). Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 68, 268–279. [DOI] [PubMed] [Google Scholar]
- Estes C, Razavi H, Loomba R, Younossi Z, and Sanyal AJ (2018). Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A, and Klein S (2009). Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 106, 15430–15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Tamboli RA, Magkos F, Marks-Shulman PA, Eckhauser AW, Richards WO, Klein S, and Abumrad NN (2010). Surgical removal of omental fat does not improve insulin sensitivity and cardiovascular risk factors in obese adults. Gastroenterology 139, 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Lu H, Guo Y, Zhu T, Garcia-Barrio MT, Jiang Z, Willer CJ, Zhang J, and Chen YE (2016). Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 150, 1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al. (2015). Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 21, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang QH, Shen QL, Li JJ, Yang Y, Guo JJ, Cheng Y, Zhou HC, Niu WY, Chen LM, Li CJ, and Sun B (2019). Inhibition of microRNA-124a attenuates non-alcoholic fatty liver disease through upregulation of adipose triglyceride lipase and the effect of liraglutide intervention. Hepatol. Res. 49, 743–757. [DOI] [PubMed] [Google Scholar]
- Feaver RE, Cole BK, Lawson MJ, Hoang SA, Marukian S, Blackman BR, Figler RA, Sanyal AJ, Wamhoff BR, and Dash A (2016). Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 1, e90954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery C, Dufour S, Rabøl R, Shulman GI, and Petersen KF (2012). Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 61, 2711–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondevila MF, Fernandez U, Gonzalez-Rellan MJ, Da Silva Lima N, Buque X, Gonzalez-Rodriguez A, Alonso C, Iruarrizaga-Lejarreta M, Delgado TC, Varela-Rey M, et al. (2021). The L-α-Lysophosphatidylinositol/GProtein-Coupled Receptor 55 System Induces the Development of Nonalcoholic Steatosis and Steatohepatitis. Hepatology 73, 606–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs CD, Traussnigg SA, and Trauner M (2016). Nuclear Receptor Modulation for the Treatment of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis.36, 69–86. [DOI] [PubMed] [Google Scholar]
- Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, Uchiyama A, Kon K, Ikejima K, and Watanabe S (2014). Abnormality of autophagic function and cathepsin expression in the liver from patients with nonalcoholic fatty liver disease. Hepatol. Res. 44, 1026–1036. [DOI] [PubMed] [Google Scholar]
- Gai Z, Visentin M, Gui T, Zhao L, Thasler WE, Häusler S, Hartling I, Cremonesi A, Hiller C, and Kullak-Ublick GA (2018). Effects of Farnesoid X Receptor Activation on Arachidonic Acid Metabolism, NF-kB Signaling, and Hepatic Inflammation. Mol. Pharmacol. 94, 802–811. [DOI] [PubMed] [Google Scholar]
- Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, et al. (2014). A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 6, 1062–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Arriazu E, Magdaleno F, Antoine DJ, Dela Cruz R, Theise N, and Nieto N (2018). High Mobility Group Box-1 Drives Fibrosis Progression Signaling via the Receptor for Advanced Glycation End Products in Mice. Hepatology 68, 2380–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, Blüher M, Czech MP, and Tabas I (2018). Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijón MA, Riekhof WR, Zarini S, Murphy RC, and Voelker DR (2008). Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J. Biol. Chem. 283, 30235–30245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AL, Teijeiro A, Burén S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, Perna C, and Djouder N (2016). Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 30, 161–175. [DOI] [PubMed] [Google Scholar]
- González-Terán B, Matesanz N, Nikolic I, Verdugo MA, Sreeramkumar V, Hernández-Cosido L, Mora A, Crainiciuc G, Sáiz ML, Bernardo E, et al. (2016). p38γ and p38δ reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 35, 536–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, Rasmiena AA, Kaur S, Gulati T, Goh PK, et al. (2018). Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 175, 1289–1306.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, and Sabatini DM (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22. [DOI] [PubMed] [Google Scholar]
- Guillot A, Hamdaoui N, Bizy A, Zoltani K, Souktani R, Zafrani ES, Mallat A, Lotersztajn S, and Lafdil F (2014). Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology 59, 296–306. [DOI] [PubMed] [Google Scholar]
- Guo L, Zhang P, Chen Z, Xia H, Li S, Zhang Y, Kobberup S, Zou W, and Lin JD (2017). Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J. Clin. Invest. 127, 4449–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A, Kabashima A, Pavelko KD, Madden B, Alhuwaish H, et al. (2019). Integrin β1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 71, 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, Hirschey M, Vaitheesvaran B, Farese RV Jr., Kurland IJ, et al. (2012). Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 15, 873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoutene A, and Rautou PE (2019). Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 70, 1278–1291. [DOI] [PubMed] [Google Scholar]
- Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, Koo SH, and Lee MO (2017). RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep. 20, 124–135. [DOI] [PubMed] [Google Scholar]
- Han CY, Rho HS, Kim A, Kim TH, Jang K, Jun DW, Kim JW, Kim B, and Kim SG (2018). FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 24, 2985–2999. [DOI] [PubMed] [Google Scholar]
- Han YH, Shin KO, Kim JY, Khadka DB, Kim HJ, Lee YM, Cho WJ, Cha JY, Lee BJ, and Lee MO (2019). A maresin 1/RORα/12-lipoxygenase autoregulatory circuit prevents inflammation and progression of nonalcoholic steatohepatitis. J. Clin. Invest. 129, 1684–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy JA, Fu PP, Kumar P, Mells JE, Sharma S, Saxena NK, and Anania FA (2011). Adiponectin inhibits leptin signalling via multiple mechanisms to exert protective effects against hepatic fibrosis. Biochem. J. 440, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson A, Piras IS, Wilhelmsen D, Still CD, Chu X, Petrick A, Gerhard GS, and DiStefano JK (2019). Chemokine ligand 20 (CCL20) expression increases with NAFLD stage and hepatic stellate cell activation and is regulated by miR-590–5p. Cytokine 123, 154789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan MM, Kaseb A, Etzel CJ, El-Serag H, Spitz MR, Chang P, Hale KS, Liu M, Rashid A, Shama M, et al. (2013). Genetic variation in the PNPLA3 gene and hepatocellular carcinoma in USA: risk and prognosis prediction. Mol. Carcinog. 52 (Suppl 1), E139–E147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich B, Brown ZJ, Diggs LP, Vormehr M, Ma C, Subramanyam V, Rosato U, Ruf B, Walz JS, McVey JC, et al. (2021). Steatohepatitis Impairs T-cell-Directed Immunotherapies Against Liver Tumors in Mice. Gastroenterology 160, 331–345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsley RN, Varadharajan V, Brown AL, Gromovsky AD, Schugar RC, Ramachandiran I, Fung K, Kabbany MN, Banerjee R, Neumann CK, et al. (2019). Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 8, e49882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, and Samuel VT (2016). The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 27, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, and Friedman SL (2012). Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, Devi LA, and Friedman SL (2013). Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 59, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks DF, Goossens N, Blas-García A, Tsuchida T, Wooden B, Wallace MC, Nieto N, Lade A, Redhead B, Cederbaum AI, et al. (2017). Transcriptome-based repurposing of apigenin as a potential anti-fibrotic agent targeting hepatic stellate cells. Sci. Rep. 7, 42563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsova P, and Gores GJ (2015). Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 1, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, Charlton MR, Shah VH, Malhi H, and Gores GJ (2016). Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 150, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsova P, Weng P, Salim W, Bronk SF, Griffith TS, Ibrahim SH, and Gores GJ (2017). TRAIL Deletion Prevents Liver, but Not Adipose Tissue, Inflammation during Murine Diet-Induced Obesity. Hepatol. Commun. 1, 648–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsova P, Bohm F, Dohnalkova E, Nozickova B, Heikenwalder M, Gores GJ, and Weber A (2020). Hepatocyte apoptosis is tumor promoting in murine nonalcoholic steatohepatitis. Cell Death Dis. 11, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland ML, Lowe R, Caton PW, Gemma C, Carbajosa G, Danson AF, Carpenter AA, Loche E, Ozanne SE, and Rakyan VK (2016). Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science 353, 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwath JA, Hurr C, Butler SD, Guruju M, Cassell MD, Mark AL, Davisson RL, and Young CN (2017). Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI Insight 2, e90170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyles L, Fernández-Real JM, Federici M, Serino M, Abbott J, Charpentier J, Heymes C, Luque JL, Anthony E, Barton RH, et al. (2018). Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 24, 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, et al. (2007). A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology 46, 297–306. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Hayashi T, Fujimoto WY, Kahn SE, Leonetti DL, McNeely MJ, and Boyko EJ (2015). Visceral abdominal fat accumulation predicts the conversion of metabolically healthy obese subjects to an unhealthy phenotype. Int. J. Obes. 39, 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SH, Hirsova P, and Gores GJ (2018). Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. (2013). Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631. [DOI] [PubMed] [Google Scholar]
- Ilyas G, Cingolani F, Zhao E, Tanaka K, and Czaja MJ (2019). Decreased Macrophage Autophagy Promotes Liver Injury and Inflammation from Alcohol. Alcohol. Clin. Exp. Res. 43, 1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J, Jahangir Z, and Al-Qarni AA (2020). Microsomal Triglyceride Transfer Protein: From Lipid Metabolism to Metabolic Diseases. Adv. Exp. Med. Biol. 1276, 37–52. [DOI] [PubMed] [Google Scholar]
- Irimia JM, Meyer CM, Segvich DM, Surendran S, DePaoli-Roach AA, Morral N, and Roach PJ (2017). Lack of liver glycogen causes hepatic insulin resistance and steatosis in mice. J. Biol. Chem. 292, 10455–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MD (2008). Role of body fat distribution and the metabolic complications of obesity. J. Clin. Endocrinol. Metab. 93 (11, Suppl 1), S57–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, and Miao EA (2015). Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki T, Sekikawa A, Murata K, Maegawa H, Takamiya T, Okamura T, El-Saed A, Miyamatsu N, Edmundowicz D, Kita Y, et al. (2006). Japanese men have larger areas of visceral adipose tissue than Caucasian men in the same levels of waist circumference in a population-based study. Int. J. Obes. 30, 1163–1165. [DOI] [PubMed] [Google Scholar]
- Kanda T, Goto T, Hirotsu Y, Masuzaki R, Moriyama M, and Omata M (2020). Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 21, 1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, and Grønbæk H (2019). The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 16, 145–159. [DOI] [PubMed] [Google Scholar]
- Keitel V, and Häussinger D (2018). Role of TGR5 (GPBAR1) in Liver Disease. Semin. Liver Dis. 38, 333–339. [DOI] [PubMed] [Google Scholar]
- Kelley DE, McKolanis TM, Hegazi RA, Kuller LH, and Kalhan SC (2003). Fatty liver in type 2 diabetes mellitus: relation to regional adiposity, fatty acids, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 285, E906–E916. [DOI] [PubMed] [Google Scholar]
- Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, Katchy CA, Lee C, Moore DD, and Fu L (2016). Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 30, 909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RS, Bril F, Cusi K, and Newsome PN (2019). Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 70, 711–724. [DOI] [PubMed] [Google Scholar]
- Kim D, and Kim WR (2017). Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 15, 474–485. [DOI] [PubMed] [Google Scholar]
- Kim JK, Gavrilova O, Chen Y, Reitman ML, and Shulman GI (2000). Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J. Biol. Chem. 275, 8456–8460. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, and Karin M (2018). ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 175, 133–145.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. ; Nonalcoholic Steatohepatitis Clinical Research Network (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia KS, Dimas AS, Hassanali N, et al. ; DIAGRAM; MuTHER (2011). Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 43, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec AK, Abrahams SR, Thornton S, Palumbo JS, Mullins ES, Divanovic S, Weiler H, Owens AP 3rd, Mackman N, Goss A, et al. (2017). Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J. Clin. Invest. 127, 3152–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, and Brenner DA (2017). Liver inflammation and fibrosis. J. Clin. Invest. 127, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel O, and Tacke F (2017). Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 17, 306–321. [DOI] [PubMed] [Google Scholar]
- Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, Liepelt A, Lefebvre E, Luedde T, Hellerbrand C, et al. (2018). Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 67, 1270–1283. [DOI] [PubMed] [Google Scholar]
- Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, Branco DPP, Mossanen JC, Luedde T, Trautwein C, et al. (2020). Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 69, 551–563. [DOI] [PubMed] [Google Scholar]
- Kumar DP, Santhekadur PK, Seneshaw M, Mirshahi F, Uram-Tuculescu C, and Sanyal AJ (2019). A Regulatory Role of Apoptosis Antagonizing Transcription Factor in the Pathogenesis of Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma. Hepatology 69, 1520–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA, et al. (2012). Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 15, 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, Still CD, Gerhard GS, Han X, Dziura J, et al. (2011). Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 108, 16381–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumashiro N, Yoshimura T, Cantley JL, Majumdar SK, Guebre-Egziabher F, Kursawe R, Vatner DF, Fat I, Kahn M, Erion DM, et al. (2013). Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 57, 1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafoz E, Ruart M, Anton A, Oncins A, and Hernández-Gea V (2020). The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 9, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, et al. (2019). Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 29, 174–182.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor DA, Relton C, Sattar N, and Nelson SM (2012). Maternal adiposity–a determinant of perinatal and offspring outcomes? Nat. Rev. Endocrinol. 8, 679–688. [DOI] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensztejn DM, Flisiak-Jackiewicz M, Bia1okoz-Kalinowska I, Bobrus-Chociej A, and Kowalska I. (2016). Hepatokines and non-alcoholic fatty liver disease. Acta Biochim. Pol. 63, 459–467. [DOI] [PubMed] [Google Scholar]
- Lee H-Y, Birkenfeld AL, Jornayvaz FR, Jurczak MJ, Kanda S, Popov V, Frederick DW, Zhang D, Guigni B, Bharadwaj KG, et al. (2011). Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54, 1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, et al. (2010). Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 12, 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim Y, Friso S, and Choi SW (2017). Epigenetics in non-alcoholic fatty liver disease. Mol. Aspects Med. 54, 78–88. [DOI] [PubMed] [Google Scholar]
- Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML, Fujiwara N, Goossens N, Chou HI, Parvin-Nejad FP, et al. (2018). Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat. Commun. 9, 4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, Naveau S, Prévot S, Makhzami S, Perlemuter G, and Cassard-Doulcier AM (2012). Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 57, 141–149. [DOI] [PubMed] [Google Scholar]
- Levine R, and Fritz IB (1956). The relation of insulin to liver metabolism. Diabetes 5, 209–219, discussion, 219–222. [DOI] [PubMed] [Google Scholar]
- Lewis GF, Zinman B, Groenewoud Y, Vranic M, and Giacca A (1996). Hepatic glucose production is regulated both by direct hepatic and extrahepatic effects of insulin in humans. Diabetes 45, 454–462. [DOI] [PubMed] [Google Scholar]
- Li S, Brown MS, and Goldstein JL (2010). Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 107, 3441–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, and Hobbs HH (2012). Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Invest. 122, 4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. ; MetaHIT Consortium; MetaHIT Consortium (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. [DOI] [PubMed] [Google Scholar]
- Li BH, He FP, Yang X, Chen YW, and Fan JG (2017). Steatosis induced CCL5 contributes to early-stage liver fibrosis in nonalcoholic fatty liver disease progress. Transl. Res. 180, 103–117.e4. [DOI] [PubMed] [Google Scholar]
- Li YH, Zhong M, Zang HL, and Tian XF (2020). MTA1 Promotes Hepatocellular Carcinoma Progression by Downregulation of DNA-PK-Mediated H1.2T146 Phosphorylation. Front. Oncol. 10, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, Chiu J, Dong L, Arfianti E, Haigh WG, et al. (2018). Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 9, 4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang N, Damdimopoulos A, Goñi S, Huang Z, Vedin LL, Jakobsson T, Giudici M, Ahmed O, Pedrelli M, Barilla S, et al. (2019). Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARα. Nat. Commun. 10, 1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Chang PF, Lin HF, Liu K, Chang MH, and Ni YH (2016). Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J. Hepatol. 65, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, Day CP, Daly AK, Reeves HL, and Anstee QM (2014). Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 61, 75–81. [DOI] [PubMed] [Google Scholar]
- Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, Zhou Y, Zhang Y, Xu W, Yang Y, et al. (2018). Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 10, eaap9840. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhang Y, Graham S, Wang X, Cai D, Huang M, Pique-Regi R, Dong XC, Chen YE, Willer C, and Liu W (2020). Causal relationships between NAFLD, T2D and obesity have implications for disease subphenotyping. J. Hepatol. 73, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, and Adams LA (2019). The 20% Rule of NASH Progression: The Natural History of Advanced Fibrosis and Cirrhosis due to NASH. Hepatology 70, 1885–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, and Sanyal AJ (2013). The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 10, 686–690. [DOI] [PubMed] [Google Scholar]
- Loomba R, Quehenberger O, Armando A, and Dennis EA (2015a). Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for non-invasive diagnosis of nonalcoholic steatohepatitis. J. Lipid Res. 56, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, Nguyen P, Hernandez C, Richards L, Salotti J, et al. ; Genetics of NAFLD in Twins Consortium (2015b). Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 149, 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, Dulai PS, Caussy C, Bettencourt R, Highlander SK, et al. (2017). Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 25, 1054–1062.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Gindin Y, Jiang Z, Lawitz E, Caldwell S, Djedjos CS, Xu R, Chung C, Myers RP, Subramanian GM, et al. (2018). DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 3, e96685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Lim JK, Patton H, and El-Serag HB (2020). AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 158, 1822–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Rius B, Hwang SH, Titos E, Lopategi A, Hammock BD, Arroyo V, and Clària J (2015). Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 112, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, et al. ; EPIC-InterAct Consortium; Cambridge FPLD1 Consortium (2017). Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 49, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, and Lowe SW (2013). Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, Pei Y, Knight LR, Deveau M, Chen Y, et al. (2018). Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 155, 1971–1984.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Hyötyläinen T, Leivonen M, Arola J, Orho-Melander M, Orešič M, and Yki-Järvinen H (2016a). The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of nonalcoholic fatty liver disease in humans. J. Hepatol. 65, 1263–1265. [DOI] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Sädevirta S, Leivonen M, Arola J, Orešič M, Hyötyläinen T, and Yki-Järvinen H (2016b). Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 64, 1167–1175. [DOI] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Nidhina Haridas PA, Dwivedi OP, Hyötyläinen T, Ali A, Juuti A, Leivonen M, Tukiainen T, Ahonen L, et al. (2017). Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 67, 128–136. [DOI] [PubMed] [Google Scholar]
- Lyu K, Zhang Y, Zhang D, Kahn M, Ter Horst KW, Rodrigues MRS, Gaspar RC, Hirabara SM, Luukkonen PK, Lee S, et al. (2020). A Membrane-Bound Diacylglycerol Species Induces PKCε-Mediated Hepatic Insulin Resistance. Cell Metab. 32, 654–664.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu K, Zhang D, Song J, Li X, Perry RJ, Samuel VT, and Shulman GI (2021). Short-term overnutrition induces white adipose tissue insulin resistance through sn-1,2-diacylglycerol/PKCε/insulin receptor Thr1160 phosphorylation. JCI Insight 6, e139946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, et al. (2018). Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, de Boer YS, Koh C, Chen Y, Du X, et al. ; for the Nonalcoholic Steatohepatitis Clinical Research Network (2019). 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 69, 1504–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY, Alexandrov LB, Koyama Y, Nishio T, Benner C, et al. (2020). IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 72, 946–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, and Diehl AM (2015a). Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol. 63, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado MV, Michelotti GA, Pereira Tde.A., Boursier J, Kruger L, Swiderska-Syn M, Karaca G, Xie G, Guy CD, Bohinc B, et al. (2015b). Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 64, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magkos F, Fabbrini E, Mohammed BS, Patterson BW, and Klein S (2010). Increased whole-body adiposity without a concomitant increase in liver fat is not associated with augmented metabolic dysfunction. Obesity (Silver Spring) 18, 1510–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, Varela JE, Brunt EM, Patterson BW, and Klein S (2012). Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142, 1444, 6.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, Eriksson P, and van’t Hooft F (2014). TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 111, 8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M, Surewaard BGJ, Rath D, et al. (2019). Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 25, 641–655. [DOI] [PubMed] [Google Scholar]
- Mallat A, and Lotersztajn S (2008). Cannabinoid receptors as novel therapeutic targets for the management of non-alcoholic steatohepatitis. Diabetes Metab. 34, 680–684. [DOI] [PubMed] [Google Scholar]
- Mancina RM, Matikainen N, Maglio C, Söderlund S, Lundbom N, Hakkarainen A, Rametta R, Mozzi E, Fargion S, Valenti L, et al. (2015). Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J. Clin. Endocrinol. Metab. 100, E821–E825. [DOI] [PubMed] [Google Scholar]
- Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, Borén J, Montalcini T, Pujia A, Wiklund O, et al. (2016). The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 150, 1219–1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcher AB, Bendixen SM, Terkelsen MK, Hohmann SS, Hansen MH, Larsen BD, Mandrup S, Dimke H, Detlefsen S, and Ravnskjaer K (2019). Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci. Rep. 9, 2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Pritchett J, Llewellyn J, Mullan AF, Athwal VS, Dobie R, Harvey E, Zeef L, Farrow S, Streuli C, et al. (2016). PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 7, 12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, Federico A, and Persico M (2018). Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 9547613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel M, Samali A, and Chevet E (2014). Endoplasmic reticulum stress: at the crossroads of inflammation and metabolism in hepatocellular carcinoma development. Cancer Cell 26, 301–303. [DOI] [PubMed] [Google Scholar]
- McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, and Grove KL (2009). Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 119, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlvride S, Nikolova V, Fan HM, McDonald JAK, Wahlström A, Bellafante E, Jansen E, Adorini L, Shapiro D, Jones P, et al. (2019). Obeticholic acid ameliorates dyslipidemia but not glucose tolerance in mouse model of gestational diabetes. Am. J. Physiol. Endocrinol. Metab. 317, E399–E410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metwally M, Bayoumi A, Romero-Gomez M, Thabet K, John M, Adams LA, Huo X, Aller R, García-Monzón C, Teresa Arias-Loste M, et al. (2019). A polymorphism in the Irisin-encoding gene (FNDC5) associates with hepatic steatosis by differential miRNA binding to the 3’UTR. J. Hepatol. 70, 494–500. [DOI] [PubMed] [Google Scholar]
- Mirea AM, Tack CJ, Chavakis T, Joosten LAB, and Toonen EJM (2018). IL-1 Family Cytokine Pathways Underlying NAFLD: Towards New Treatment Strategies. Trends Mol. Med. 24, 458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsche MA, Hobbs HH, and Cohen JC (2018). Patatin-like phospholipase domain–containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J. Biol. Chem. 293, 6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, May SB, Kramer JR, Richardson PA, and Davila JA (2016). Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 14, 124–131.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Salvador E, Garcia-Macia M, Sivaharan A, Sabater L, Zaki MYW, Oakley F, Knox A, Page A, Luli S, Mann J, and Mann DA (2019). Fibrogenic Activity of MECP2 Is Regulated by Phosphorylation in Hepatic Stellate Cells. Gastroenterology 157, 1398–1412.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou GN, et al. (2017). NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 66, 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Gambino R, Cassader M, Paschetta E, and Sircana A (2018). Specialized Proresolving Mediators: Enhancing Nonalcoholic Steatohepatitis and Fibrosis Resolution. Trends Pharmacol. Sci. 39, 387–401. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, Dong J, Zhang D, Jurczak MJ, Löffler MG, et al. (2009). The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 9, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Mason A, Eason J, Loss G, and Perrillo RP (2002). Is obesity an independent risk factor for hepatocellular carcinoma in cirrhosis? Hepatology 36, 150–155. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, and Accili D (2001). The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest. 108, 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress co-operates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Wei L, Song WM, Higashi T, Ghoshal S, Kim RS, Bian CB, Yamada S, Sun X, Venkatesh A, et al. ; Precision Liver Cancer Prevention Consortium (2016). Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 30, 879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nati M, Haddad D, Birkenfeld AL, Koch CA, Chavakis T, and Chatzigeorgiou A (2016). The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH). Rev. Endocr. Metab. Disord. 17, 29–39. [DOI] [PubMed] [Google Scholar]
- Nazare JA, Smith JD, Borel AL, Haffner SM, Balkau B, Ross R, Massien C, Alméras N, and Després JP (2012). Ethnic influences on the relations between abdominal subcutaneous and visceral adiposity, liver fat, and cardiometabolic risk profile: the International Study of Prediction of IntraAbdominal Adiposity and Its Relationship With Cardiometabolic Risk/IntraAbdominal Adiposity. Am. J. Clin. Nutr. 96, 714–726. [DOI] [PubMed] [Google Scholar]
- Negro F (2020). Natural history of NASH and HCC. Liver Int. 40 (Suppl 1), 72–76. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA, Clark JM, Bass NM, Van Natta ML, Unalp-Arida A, Tonascia J, Zein CO, Brunt EM, Kleiner DE, McCullough AJ, et al. ; NASH Clinical Research Network (2010). Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology 52, 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, et al. ; NASH Clinical Research Network (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J, Jiao J, Smoot K, Watt GP, Zhao C, Song X, Stevenson HL, McCormick JB, Fisher-Hoch SP, Zhang J, et al. (2018). Toll-like receptor 4: a target for chemoprevention of hepatocellular carcinoma in obesity and steatohepatitis. Oncotarget 9, 29495–29507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Guo Z, Johnson CM, Hensrud DD, and Jensen MD (2004). Splanchnic lipolysis in human obesity. J. Clin. Invest. 113, 1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki Y, Petersen MC, Zhang D, Vatner DF, Perry RJ, Abulizi A, Haedersdal S, Zhang XM, Butrico GM, Samuel VT, et al. (2020). Metabolic control analysis of hepatic glycogen synthesis in vivo. Proc. Natl. Acad. Sci. USA 117, 8166–8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, Singh S, Madamba EV, Bettencourt R, Richards L, et al. (2020). A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell Metab. 32, 878–888.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H, Thompson W, Karns RA, Mayhew CN, McGrath PS, et al. (2019). Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 30, 374–384.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Han SN, Zhang JY, Dioletis E, Nemeth BT, Pacher P, Feng D, Bataller R, Cabezas J, Stärkel P, et al. (2018). Digoxin Suppresses Pyruvate Kinase M2-Promoted HIF-1α Transactivation in Steatohepatitis. Cell Metab. 27, 339–350.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page A, Paoli P, Moran Salvador E, White S, French J, and Mann J (2016). Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 64, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AK, and Kirkland JL (2016). Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp. Gerontol. 86, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, Kahali B, Carr JJ, Harris TB, Jhun MA, et al. (2013). Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 58, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis V, Perlemuter G, Bonvoust F, Dargere D, Parfait B, Vidaud M, Conti M, Huet S, Ba N, Buffet C, and Bedossa P (2001). High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 34, 738–744. [DOI] [PubMed] [Google Scholar]
- Paradis V, Zalinski S, Chelbi E, Guedj N, Degos F, Vilgrain V, Bedossa P, and Belghiti J (2009). Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 49, 851–859. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, and Shulman GI (2014). Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat. Med. 20, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. (2015). Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160, 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perseghin G, Price TB, Petersen KF, Roden M, Cline GW, Gerow K, Rothman DL, and Shulman GI (1996). Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N. Engl. J. Med. 335, 1357–1362. [DOI] [PubMed] [Google Scholar]
- Peter A, Kantartzis K, Machicao F, Machann J, Wagner S, Templin S, Königsrainer I, Königsrainer A, Schick F, Fritsche A, et al. (2012). Visceral obesity modulates the impact of apolipoprotein C3 gene variants on liver fat content. Int. J. Obes. 36, 774–782. [DOI] [PubMed] [Google Scholar]
- Petersen MC, and Shulman GI (2018). Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 98, 2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Laurent D, Rothman DL, Cline GW, and Shulman GI (1998). Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J. Clin. Invest. 101, 1203–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Laurent D, Yu C, Cline GW, and Shulman GI (2001). Stimulating effects of low-dose fructose on insulin-stimulated hepatic glycogen synthesis in humans. Diabetes 50, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, De-Paoli AM, Taylor SI, Gorden P, and Shulman GI (2002). Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Invest. 109, 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, Di-Pietro L, Cline GW, and Shulman GI (2003). Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300, 1140–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, Cobelli C, and Shulman GI (2006). Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc. Natl. Acad. Sci. USA 103, 18273–18277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, et al. (2007). The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 104, 12587–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Hariri A, Nelson-Williams C, Foo JN, Zhang XM, Dziura J, Lifton RP, and Shulman GI (2010). Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N. Engl. J. Med. 362, 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang X-M, et al. (2016). Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest. 126, 4361–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, and Sabatini DM (2011). mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta S, Valenti L, Tuttolomondo A, Dongiovanni P, Pipitone RM, Cammà C, Cabibi D, Di Marco V, Fracanzani AL, Badiali S, et al. (2017). Interferon lambda 4 rs368234815 TT>δG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. Hepatology 66, 1885–1893. [DOI] [PubMed] [Google Scholar]
- Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, Gupta R, Qiu M, Deczkowska A, Weiner A, et al. (2021). NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. Published online March 24, 2021. 10.1038/s41586-021-03362-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinin E, Villani G, and Moschetta A (2019). Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 16, 160–174. [DOI] [PubMed] [Google Scholar]
- Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, Montalcini T, Hedfalk K, and Romeo S (2014). Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 1841, 574–580. [DOI] [PubMed] [Google Scholar]
- Pingitore P, Sasidharan K, Ekstrand M, Prill S, Lindén D, and Romeo S (2019). Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 20, 1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, Burza MA, Indiveri C, Ferro Y, Montalcini T, et al. (2014). PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 23, 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscaglia F, Svegliati-Baroni G, Barchetti A, Pecorelli A, Marinelli S, Tiribelli C, and Bellentani S; HCC-NAFLD Italian Study Group (2016). Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 63, 827–838. [DOI] [PubMed] [Google Scholar]
- Planas-Paz L, Sun T, Pikiolek M, Cochran NR, Bergling S, Orsini V, Yang Z, Sigoillot F, Jetzer J, Syed M, et al. (2019). YAP, but Not RSPO-LGR4/5, Signaling in Biliary Epithelial Cells Promotes a Ductular Reaction in Response to Liver Injury. Cell Stem Cell 25, 39–53.e10. [DOI] [PubMed] [Google Scholar]
- Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, and Mantzoros CS (2016). Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia 59, 30–43. [DOI] [PubMed] [Google Scholar]
- Previs SF, Cline GW, and Shulman GI (1999). A critical evaluation of mass isotopomer distribution analysis of gluconeogenesis in vivo. Am. J. Physiol. 277, E154–E160. [DOI] [PubMed] [Google Scholar]
- Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, Contos MJ, Sterling RK, Fuchs M, Zhou H, et al. (2009). The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 50, 1827–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Seale JP, and Donnelly R (1999). Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats - effects of feeding. J. Endocrinol. 162, 207–214. [DOI] [PubMed] [Google Scholar]
- Rabøl R, Petersen KF, Dufour S, Flannery C, and Shulman GI (2011). Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc. Natl. Acad. Sci. USA 108, 13705–13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, Cantley J, Wilks D, Preston E, Hegarty BD, Leitges M, et al. (2011). Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 54, 1447–1456. [DOI] [PubMed] [Google Scholar]
- Rafiq N, Bai C, Fang Y, Srishord M, McCullough A, Gramlich T, and Younossi ZM (2009). Long-term follow-up of patients with nonalcoholic fatty liver. Clin. Gastroenterol. Hepatol. 7, 234–238. [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, et al. (2012). Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 109, E3186–E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, Greenwood R, Sikaroodi M, Lam V, Crotty P, et al. (2013). Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 11, 868–875. [DOI] [PubMed] [Google Scholar]
- Ramli MNB, Lim YS, Koe CT, Demircioglu D, Tng W, Gonzales KAU, Tan CP, Szczerbinska I, Liang H, Soe EL, et al. (2020). Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 159, 1471–1486.e12. [DOI] [PubMed] [Google Scholar]
- Ramnath D, Irvine KM, Lukowski SW, Horsfall LU, Loh Z, Clouston AD, Patel PJ, Fagan KJ, Iyer A, Lampe G, et al. (2018). Hepatic expression profiling identifies steatosis-independent and steatosis-driven advanced fibrosis genes. JCI Insight 3, e120274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, Chen W, and Diehl AM (2011). Increased production of sonic hedgehog by ballooned hepatocytes. J. Pathol. 224, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, Kudlich T, Hermanns HM, Bantel H, Beyersdorf N, and Geier A (2016). Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 196, 97–105. [DOI] [PubMed] [Google Scholar]
- Rebrin K, Steil GM, Mittelman SD, and Bergman RN (1996). Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J. Clin. Invest. 98, 741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebuffé-Scrive M, Anderson B, Olbe L, and Björntorp P (1990). Metabolism of adipose tissue in intraabdominal depots in severely obese men and women. Metabolism 39, 1021–1025. [DOI] [PubMed] [Google Scholar]
- Reddy JK (2001). Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPAR alpha, and steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G1333–G1339. [DOI] [PubMed] [Google Scholar]
- Reig M, Gambato M, Man NK, Roberts JP, Victor D, Orci LA, and Toso C (2019). Should Patients With NAFLD/NASH Be Surveyed for HCC? Transplantation 103, 39–44. [DOI] [PubMed] [Google Scholar]
- Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, and Wallace M (2007). Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 47, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh YS, and Seki E (2018). Chemokines and Chemokine Receptors in the Development of NAFLD. Adv. Exp. Med. Biol. 1061, 45–53. [DOI] [PubMed] [Google Scholar]
- Rókusz A, Veres D, Szücs A, Bugyik E, Mózes M, Paku S, Nagy P, and Dezso} K. (2017). Ductular reaction correlates with fibrogenesis but does not contribute to liver regeneration in experimental fibrosis models. PLoS ONE 12, e0176518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, and Hobbs HH (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman DL, Magnusson I, Cline G, Gerard D, Kahn CR, Shulman RG, and Shulman GI (1995). Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA 92, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusu V, Hoch E, Mercader JM, Tenen DE, Gymrek M, Hartigan CR, DeRan M, von Grotthuss M, Fontanillas P, Spooner A, et al. ; MEDIA Consortium; SIGMA T2D Consortium (2017). Type 2 Diabetes Variants Disrupt Function of SLC16A11 through Two Distinct Mechanisms. Cell 170, 199–212.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakasai-Sakai A, Takata T, Takino JI, and Takeuchi M (2019). The Relevance of Toxic AGEs (TAGE) Cytotoxicity to NASH Pathogenesis: A Mini-Review. Nutrients 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2012). Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2018). Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 27, 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, and Shulman GI (2004). Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, and Shulman GI (2007). Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest. 117, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B, et al. (2012). Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 55, 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal AJ, and Pacana T (2015). A Lipidomic Readout of Disease Progression in A Diet-Induced Mouse Model of Nonalcoholic Fatty Liver Disease. Trans. Am. Clin. Climatol. Assoc. 126, 271–288. [PMC free article] [PubMed] [Google Scholar]
- Sanyal AJ, Harrison SA, Ratziu V, Abdelmalek MF, Diehl AM, Caldwell S, Shiffman ML, Schall RA, Jia C, McColgan B, et al. (2019). The Natural History of Advanced Fibrosis due to Nonalcoholic Steatohepatitis: Data from the Simtuzumab Trials. Hepatology 70, 1913–1927. [DOI] [PubMed] [Google Scholar]
- Sato K, Marzioni M, Meng F, Francis H, Glaser S, and Alpini G (2019). Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 69, 420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NK, and Anania FA (2015). Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 26, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NK, Titus MA, Ding X, Floyd J, Srinivasan S, Sitaraman SV, and Anania FA (2004). Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 18, 1612–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulien I, Hockenjos B, Schmitt-Graeff A, Perdekamp MG, Follo M, Thimme R, and Hasselblatt P (2019). The transcription factor c-Jun/AP-1 promotes liver fibrosis during non-alcoholic steatohepatitis by regulating Osteopontin expression. Cell Death Differ. 26, 1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher JD, Kong B, Wu J, Rizzolo D, Armstrong LE, Chow MD, Goedken M, Lee YH, and Guo GL (2019). Direct and Indirect Effects of FGF15 and FGF19 on Liver Fibrosis Development. Hepatology 71, 670–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan D, Surabattula R, and Wang XY (2018). Determinants of fibrosis progression and regression in NASH. J. Hepatol. 68, 238–250. [DOI] [PubMed] [Google Scholar]
- Schuster S, Cabrera D, Arrese M, and Feldstein AE (2018). Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 15, 349–364. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, and Luedde T (2018). Apoptosis and necroptosis in the liver: a matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 15, 738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Tabas I, and Pajvani UB (2020). Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 158, 1913–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M, Lass A, Zimmermann R, Eichmann TO, and Zechner R (2009). Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 297, E289–E296. [DOI] [PubMed] [Google Scholar]
- Schwimmer JB, Johnson JS, Angeles JE, Behling C, Belt PH, Borecki I, Bross C, Durelle J, Goyal NP, Hamilton G, et al. (2019). Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 157, 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, and Schwabe RF (2009a). CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 119, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF, and Brenner DA (2009b). CCR2 promotes hepatic fibrosis in mice. Hepatology 50, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2017). Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. 31, 1273–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifnia T, Antoun J, Verriere TG, Suarez G, Wattacheril J, Wilson KT, Peek RM Jr., Abumrad NN, and Flynn CR (2015). Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G270–G278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpton SR, Ajmera V, and Loomba R (2019). Emerging Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease: From Composition to Function. Clin. Gastroenterol. Hepatol. 17, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI (2000). Cellular mechanisms of insulin resistance. J. Clin. Invest. 106, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, Putra J, Camprecios G, Bassaganyas L, Akers N, et al. (2017). Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 153, 812–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, and Waljee AK (2014). The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am. J. Gastroenterol. 109, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, and Loomba R (2015). Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 13, 643–654.e1–9, quiz e639–e640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagris E, Gilyard S, BasuRay S, Cohen JC, and Hobbs HH (2016). Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J. Biol. Chem. 291, 10659–10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith U (2015). Abdominal obesity: a marker of ectopic fat accumulation. J. Clin. Invest. 125, 1790–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somm E, Henry H, Bruce SJ, Aeby S, Rosikiewicz M, Sykiotis GP, Asrih M, Jornayvaz FR, Denechaud PD, Albrecht U, et al. (2017). β-Klotho deficiency protects against obesity through a crosstalk between liver, microbiota, and brown adipose tissue. JCI Insight 2, e91809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JD, Alves TC, Befroy DE, Perry RJ, Mason GF, Zhang XM, Munk A, Zhang Y, Zhang D, Cline GW, et al. (2020a). Dissociation of Muscle Insulin Resistance from Alterations in Mitochondrial Substrate Preference. Cell Metab. 32, 726–735.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen W, Athavale D, Ge X, Desert R, Das S, Han H, and Nieto N (2020b). Osteopontin takes center stage in chronic liver disease. Hepatology 73, 1594–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookoian S, and Pirola CJ (2019). Genetics of Nonalcoholic Fatty Liver Disease: From Pathogenesis to Therapeutics. Semin. Liver Dis. 39, 124–140. [DOI] [PubMed] [Google Scholar]
- Spengler EK, and Loomba R (2015). Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin. Proc. 90, 1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender S, and Loomba R (2020). PNPLA3 Genotype and Risk of Liver and All-Cause Mortality. Hepatology 71, 777–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender S, Kozlitina J, Nordestgaard BG, Tybjærg-Hansen A, Hobbs HH, and Cohen JC (2017). Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 49, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PRJ, Reese CB, McCormick F, Tempst P, et al. (1998). Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279, 710–714. [DOI] [PubMed] [Google Scholar]
- Stols-Gonçalves D, Tristão LS, Henneman P, and Nieuwdorp M. (2019). Epigenetic Markers and Microbiota/Metabolite-Induced Epigenetic Modifications in the Pathogenesis of Obesity, Metabolic Syndrome, Type 2 Diabetes, and Non-alcoholic Fatty Liver Disease. Curr. Diab. Rep. 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Seidman JS, Zhao P, Troutman TD, Spann NJ, Que X, Zhou F, Liao Z, Pasillas M, Yang X, et al. (2020). Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 31, 189–206.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydor S, Best J, Messerschmidt I, Manka P, Vilchez-Vargas R, Brodesser S, Lucas C, Wegehaupt A, Wenning C, Aßmuth S, et al. (2020). Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin. Transl. Gastroenterol. 11, e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, et al. (2010). Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51, 1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, and Iracheta-Vellve A (2015). Inflammasome activation in the liver: Focus on alcoholic and non-alcoholic steatohepatitis. Clin. Res. Hepatol. Gastroenterol. 39 (Suppl 1), S18–S23. [DOI] [PubMed] [Google Scholar]
- Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, and Dobbins RL (2005). Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 288, E462–E468. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Miyanishi K, Kobune M, Kawano Y, Hoki T, Kubo T, Hayashi T, Sato T, Sato Y, Takimoto R, and Kato J (2013). Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 48, 1249–1258. [DOI] [PubMed] [Google Scholar]
- ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, Romijn JA, Nieuwdorp M, Zhang D, Samuel VT, et al. (2017). Hepatic Diacylglycerol-Associated Protein Kinase Cε Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep. 19, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Horst KW, Vatner DF, Zhang D, Cline GW, Ackermans MT, Nederveen AJ, Verheij J, Demirkiran A, van Wagensveld BA, Dallinga-Thie GM, et al. (2020). Hepatic Insulin Resistance Is Not Pathway Selective in Humans With Nonalcoholic Fatty Liver Disease. Diabetes care 44, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thabet K, Asimakopoulos A, Shojaei M, Romero-Gomez M, Mangia A, Irving WL, Berg T, Dore GJ, Grønbæk H, Sheridan D, et al. ; International Liver Disease Genetics Consortium (2016). MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat. Commun. 7, 12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangapandi VR, Knittelfelder O, Brosch M, Patsenker E, Vvedenskaya O, Buch S, Hinz S, Hendricks A, Nati M, Herrmann A, et al. (2021). Loss of hepatic Mboat7 leads to liver fibrosis. Gut 70, 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. (2009). TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Stokowski RP, Kershenobich D, Ballinger DG, and Hinds DA (2010). Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet. 42, 21–23. [DOI] [PubMed] [Google Scholar]
- Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H, et al. (2014). Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 59, 154–169. [DOI] [PubMed] [Google Scholar]
- Tomita K, Freeman BL, Bronk SF, LeBrasseur NK, White TA, Hirsova P, and Ibrahim SH (2016). CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Sci. Rep. 6, 28786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres DM, Williams CD, and Harrison SA (2012). Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 10, 837–858. [DOI] [PubMed] [Google Scholar]
- Trépo E, and Valenti L (2020). Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 72, 1196–1209. [DOI] [PubMed] [Google Scholar]
- Trépo E, Nahon P, Bontempi G, Valenti L, Falleti E, Nischalke HD, Hamza S, Corradini SG, Burza MA, Guyot E, et al. (2014). Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 59, 2170–2177. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, and Friedman SL (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411. [DOI] [PubMed] [Google Scholar]
- Uyeda K, and Repa JJ (2006). Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4, 107–110. [DOI] [PubMed] [Google Scholar]
- Vacca M, Leslie J, Virtue S, Lam BYH, Govaere O, Tiniakos D, Snow S, Davies S, Petkevicius K, Tong Z, et al. (2020). Bone morphogenetic protein 8B promotes the progression of non-alcoholic steatohepatitis. Nat. Metab. 2, 514–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valladolid-Acebes I, Åvall K, Recio-López P, Moruzzi N, Bryzgalova G, Björnholm M, Krook A, Alonso EF, Ericsson M, Landfors F, et al. (2021). Lowering apolipoprotein CIII protects against high-fat diet-induced metabolic derangements. Sci. Adv. 7, eabc2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, London R, Peduto T, Chisholm DJ, and George J (2008). Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology 48, 449–457. [DOI] [PubMed] [Google Scholar]
- Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez JP, Cline GW, Jurczak MJ, et al. (2015). Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 112, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A (2019). Hepatocellular Carcinoma. N. Engl. J. Med. 380, 1450–1462. [DOI] [PubMed] [Google Scholar]
- von Felden J, Garcia-Lezana T, Schulze K, Losic B, and Villanueva A (2020). Liquid biopsy in the clinical management of hepatocellular carcinoma. Gut 69, 2025–2034. [DOI] [PubMed] [Google Scholar]
- Wang X, Lopategi A, Ge X, Lu Y, Kitamura N, Urtasun R, Leung TM, Fiel MI, and Nieto N (2014). Osteopontin induces ductular reaction contributing to liver fibrosis. Gut 63, 1805–1818. [DOI] [PubMed] [Google Scholar]
- Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, and Tabas I (2016). Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 24, 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PX, Ji YX, Zhang XJ, Zhao LP, Yan ZZ, Zhang P, Shen LJ, Yang X, Fang J, Tian S, et al. (2017). Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 23, 439–449. [DOI] [PubMed] [Google Scholar]
- Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, et al. (2020). Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 31, 969–986.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmuth HE, Lammert F, Zaldivar MM, Weiskirchen R, Hellerbrand C, Scholten D, Berres ML, Zimmermann H, Streetz KL, Tacke F, et al. (2009). Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology 137, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA, and Mehal WZ (2009). Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1248–G1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattacheril J, Lavine JE, Chalasani NP, Guo X, Kwon S, Schwimmer J, Molleston JP, Loomba R, Brunt EM, Chen YI, et al. (2017). Genome-Wide Associations Related to Hepatic Histology in Nonalcoholic Fatty Liver Disease in Hispanic Boys. J. Pediatr. 190, 100–107.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehmeyer MH, Zyriax BC, Jagemann B, Roth E, Windler E, Schulze Zur Wiesch J, Lohse AW, and Kluwe J (2016). Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine (Baltimore) 95, e3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein G, Zelber-Sagi S, Preis SR, Beiser AS, DeCarli C, Speliotes EK, Satizabal CL, Vasan RS, and Seshadri S (2018). Association of Nonalcoholic Fatty Liver Disease With Lower Brain Volume in Healthy Middle-aged Adults in the Framingham Study. JAMA Neurol. 75, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski SR, Kasmi KC, Jonscher KR, and Friedman JE (2017). Developmental origins of NAFLD: a womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 14, 81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, D’Agostino GA, Ng B, Lim WW, Tan J, Paleja BS, et al. (2019). Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 157, 777–792.e14. [DOI] [PubMed] [Google Scholar]
- Williams AL, Jacobs SB, Moreno-Macías H, Huerta-Chagoya A, Churchhouse C, Márquez-Luna C, García-Ortíz H, Gómez-Vázquez MJ, Burtt NP, Aguilar-Salinas CA, et al. ; SIGMA Type 2 Diabetes Consortium (2014a). Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MJ, Clouston AD, and Forbes SJ (2014b). Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 146, 349–356. [DOI] [PubMed] [Google Scholar]
- Wooden B, Goossens N, Hoshida Y, and Friedman SL (2017). Using Big Data to Discover Diagnostics and Therapeutics for Gastrointestinal and Liver Diseases. Gastroenterology 152, 53–67.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TJ, Wang YC, Wu TH, Lee CF, Chan KM, and Lee WC (2012). Inhibition of allogenic T-cell cytotoxicity by hepatic stellate cell via CD4+ CD25+ Foxp3+ regulatory T cells in vitro. Transplant. Proc. 44, 1055–1059. [DOI] [PubMed] [Google Scholar]
- Wueest S, Item F, Lucchini FC, Challa TD, Müller W, Blüher M, and Konrad D (2016). Mesenteric Fat Lipolysis Mediates Obesity-Associated Hepatic Steatosis and Insulin Resistance. Diabetes 65, 140–148. [DOI] [PubMed] [Google Scholar]
- Xie G, Karaca G, Swiderska-Syn M, Michelotti GA, Krüger L, Chen Y, Premont RT, Choi SS, and Diehl AM (2013). Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 58, 1801–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, et al. (2019). Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 75, 644–660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, Zhang Z, Zhang D, Fan D, Nie Y, et al. (2018). Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 68, 773–782. [DOI] [PubMed] [Google Scholar]
- Xu D, Xu M, Jeong S, Qian Y, Wu H, Xia Q, and Kong X (2019). The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 9, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, and Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Miura K, Zhang B, Matsushita H, Yang YM, Liang S, Song J, Roh YS, and Seki E (2017). TRIF Differentially Regulates Hepatic Steatosis and Inflammation/Fibrosis in Mice. Cell. Mol. Gastroenterol. Hepatol. 3, 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Trépo E, Nahon P, Cao Q, Moreno C, Letouzé E, Imbeaud S, Gustot T, Deviere J, Debette S, et al. (2019). PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int. J. Cancer 144, 533–544. [DOI] [PubMed] [Google Scholar]
- Yang JD, Ahmed F, Mara KC, Addissie BD, Allen AM, Gores GJ, and Roberts LR (2020). Diabetes Is Associated With Increased Risk of Hepatocellular Carcinoma in Patients With Cirrhosis From Nonalcoholic Fatty Liver Disease. Hepatology 71, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y, Man K, Lo CM, Li X, and Xu A (2012). Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 61, 1058–1067. [DOI] [PubMed] [Google Scholar]
- Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, Zhang J, Shi J, Xu A, Lin S, and Wang Y (2016). Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 65, 988–997. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Yoshitaka H, Hamaguchi M, Kojima T, Fukuda T, Ohbora A, and Fukui M (2017). Nonoverweight nonalcoholic fatty liver disease and incident cardiovascular disease: A post hoc analysis of a cohort study. Medicine (Baltimore) 96, e6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, and Wymer M (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, and Bugianesi E (2018). Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20. [DOI] [PubMed] [Google Scholar]
- Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, Duseja A, Eguchi Y, Wong VW, Negro F, Yilmaz Y, et al. ; Global Nonalcoholic Steatohepatitis Council (2019a). Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 17, 748–755.e3. [DOI] [PubMed] [Google Scholar]
- Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, Wai-Sun Wong V, Yilmaz Y, George J, Fan J, and Vos MB (2019b). Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 69, 2672–2682. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Gupta S, Maurya MR, Subramaniam S, and Loomba R (2016). Serum microRNAs explain discordance of non-alcoholic fatty liver disease in monozygotic and dizygotic twins: a prospective study. Gut 65, 1546–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng N, Huang R, Li N, Jiang H, Li R, Wang F, Chen W, Xia M, and Wang Q (2018). MiR-451a attenuates free fatty acids-mediated hepatocyte steatosis by targeting the thyroid hormone responsive spot 14 gene. Mol. Cell. Endocrinol. 474, 260–271. [DOI] [PubMed] [Google Scholar]
- Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, Oakley F, Burt AD, Wilson CL, Anstee QM, et al. (2012). Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat. Med. 18, 1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Goossens N, Guo J, Tsai MC, Chou HI, Altunkaynak C, Sangiovanni A, Iavarone M, Colombo M, Kobayashi M, et al. (2016a). A hepatic stellate cell gene expression signature associated with outcomes in hepatitis C cirrhosis and hepatocellular carcinoma after curative resection. Gut 65, 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Chang Y, Shi Z, Han X, Han Y, Yao Q, Hu Z, Cui H, Zheng L, Han T, and Hong W (2016b). ω-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci. Rep. 6, 30029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RN, Zheng RD, Mi YQ, Zhou D, Shen F, Chen GY, Zhu CY, Pan Q, and Fan JG (2016c). APOC3 rs2070666 Is Associated with the Hepatic Steatosis Independently of PNPLA3 rs738409 in Chinese Han Patients with Nonalcoholic Fatty Liver Diseases. Dig. Dis. Sci. 61, 2284–2293. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, Fang C, Lu YX, Yang X, Gao MM, et al. (2018). The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 24, 84–94. [DOI] [PubMed] [Google Scholar]
- Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, et al. (2018). Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 10, eaat0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucman-Rossi J, Villanueva A, Nault JC, and Llovet JM (2015). Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149, 1226–1239.e4. [DOI] [PubMed] [Google Scholar]