

Keywords: endothelial cells, flow-induced vasodilation, shear stress, vascular myogenic response, vascular smooth muscle cells
Abstract
This review aims to survey the current state of mechanotransduction in vascular smooth muscle cells (VSMCs) and endothelial cells (ECs), including their sensing of mechanical stimuli and transduction of mechanical signals that result in the acute functional modulation and longer-term transcriptomic and epigenetic regulation of blood vessels. The mechanosensors discussed include ion channels, plasma membrane-associated structures and receptors, and junction proteins. The mechanosignaling pathways presented include the cytoskeleton, integrins, extracellular matrix, and intracellular signaling molecules. These are followed by discussions on mechanical regulation of transcriptome and epigenetics, relevance of mechanotransduction to health and disease, and interactions between VSMCs and ECs. Throughout this review, we offer suggestions for specific topics that require further understanding. In the closing section on conclusions and perspectives, we summarize what is known and point out the need to treat the vasculature as a system, including not only VSMCs and ECs but also the extracellular matrix and other types of cells such as resident macrophages and pericytes, so that we can fully understand the physiology and pathophysiology of the blood vessel as a whole, thus enhancing the comprehension, diagnosis, treatment, and prevention of vascular diseases.
CLINICAL HIGHLIGHTS.
-
1)
Atherosclerotic cardiovascular diseases occur preferentially at the branch points and curved regions of the arterial tree because of mechanotransduction events elicited by the oscillatory shear stress associated with disturbed, instead of laminar, flow that acts on the vascular endothelial cells in those regions.
-
2)
Vascular smooth muscle cells respond to pressure-induced distension of the vascular wall, resulting in acute vasoconstriction that, if sustained, progresses to vascular remodeling and hypertension.
-
3)
The vascular endothelium exerts both acute and chronic vasodilatory effects on vascular smooth muscle cells to counteract pressure-induced constriction; loss of this interaction between the two cell layers promotes the progression of hypertension and atherosclerosis.
-
4)
Understanding of the mechanisms of mechanotransduction in vascular endothelial cells and smooth muscle cells and their interactions is essential for the elucidation of the pathophysiological basis of important cardiovascular diseases and the development of effective treatments.
1. INTRODUCTION
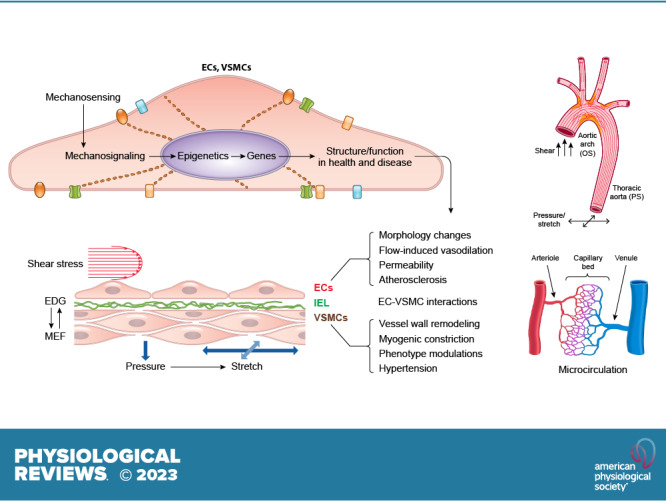
Every cell can detect and respond to changes in its external environment, whether the stimulus involves mechanical, electrical, chemical, magnetic, light, and/or other forms of energy. Although some cells serve as specialized sensors (e.g., cochlear hair cells, photoreceptors), most cells appear to share some basic mechanisms of mechanosensing and downstream signal transduction pathways. Cells in the cardiovascular system are constantly subjected to hemodynamic forces resulting from the pressure generated by the repetitive cardiac contraction and the consequent flow of blood through the systemic and pulmonary circulations. This review addresses the mechanisms of pressure and shear stress detection (i.e., mechanosensing) by vascular smooth muscle cells (VSMCs) and endothelial cells (ECs), as well as the subsequent downstream signaling mechanisms (i.e., mechanosignaling) that are activated to produce single-cell or multicell responses. These processes are grouped together under the term “mechanotransduction.” Other cell types in the cardiovascular system also sense and transduce mechanical forces, including cardiomyocytes (1, 2), red blood cells (3, 4), white blood cells (5), platelets (6), and baroreceptor nerve endings (7, 8). This review focuses on the responses of VSMCs and ECs to hemodynamic forces that lead to regulatory or adaptive mechanisms in the circulatory system, including the vascular myogenic response and flow-induced dilation. These processes are complex and involve not only multiple signaling pathways but also multiple temporal components: from rapid events (milliseconds to seconds) such as changes in membrane potential and intracellular Ca2+ concentration ([Ca2+]), to intermediate events (minutes to hours) such as changes in cell alignment, and to long-term events (days to months) such as vascular remodeling or plaque formation.
Hemodynamic forces are characterized by different modalities (pressure, shear), different vectors (circumferential, radial, axial; FIGURE 1), both static and dynamic components, and a wide range of magnitudes and temporal characteristics, each of which may vary among blood vessels in different regions of the body. For example, as arterial pressure increases during each cardiac cycle, arteries experience a distending stress. This distending stress acts on the longitudinally oriented ECs in the radial direction of the vessel (σr) and on the circumferentially oriented VSMCs in their longitudinal direction (σθ). The magnitudes of σr and σθ vary within each cardiac cycle, with the prevailing pressure level (e.g., artery vs. vein), within a given vascular bed (e.g., as the pressure profile changes from artery to vein), and across regional circulations (e.g., systemic vs. pulmonary). Additional hydrostatic loads may be imposed, or reduced, by gravitational forces acting on standing columns of blood, especially in bipeds. During each cardiac cycle, ECs directly experience a force resulting from the frictional drag of blood (τw, wall shear stress) on their surface; this effect is greatly attenuated in the medial layer where VSMCs reside, because of the presence of the internal elastic lamina. Wall shear stress varies with the location of an EC within a vascular network (whether it is located in a straight segment, bifurcation, or curvature, in arteries, microvessels, or veins, in a cardiac or venous valve leaflet, or in a valve sinus) and depends on the flow rate, vessel size, and location. The direction and magnitude of shear stress not only change in synchrony with the cardiac cycle but also oscillate at lower frequencies within vascular networks, e.g., in arcading vessels during vasomotion. ECs, which align parallel to the flow stream, have responses to σr and τw (9, 10) different from VSMCs, which are physically separated from the flow stream but aligned perpendicularly to it.
FIGURE 1.

Mechanical forces acting on the arterial wall. Modified from Ref. 9, with permission from the American Physiological Society.
For additional perspectives, the reader is referred to previous reviews addressing the topic of vascular mechanotransduction, some of which focus on vascular smooth muscle (12–16) or endothelium (9, 11, 17), others on selected mechanisms such as shear-stress sensing by EC ion channels (18, 19) or pressure sensing by G protein-coupled receptors (GPCRs) in VSMCs (20–23), or EC-VSMC interactions (24, 25), as well as other topics that overlap with vascular mechanotransduction, such as vascular transient receptor potential (TRP) channel function (26–28). In addition to reviewing the primary literature, we aim to integrate relevant concepts to provide an updated, comprehensive review that addresses mechanotransduction by vascular smooth muscle and endothelium in health and disease.
1.1. Mechanoresponses of ECs
1.1.1. Mechanical forces on ECs in the vascular system.
Vascular ECs serve important homeostatic functions in response to chemical and mechanical stimuli (9), including the modulation of vascular remodeling, inflammatory responses, hemostasis/thrombosis, VSMC contraction, and macromolecular permeability. Endothelial dysfunction can lead to pathophysiological changes and vascular disorders (29–31). As indicated above, ECs experience both a radial distending stress (σr) due to transmural pressure and the tangential wall shear stress (τw) due to blood flow (FIGURE 1). Both σr and τw vary with the location of the EC in various segments of the vascular tree because of the different levels of pressure and flow, as well as geometry. τw is a function of (vη/r), where v is mean linear velocity, η is blood viscosity and r is vessel radius. Based on this function, the mean τw in different longitudinal segments of the vascular tree under resting conditions can be estimated as shown in FIGURE 2 (32). Blood is a non-Newtonian fluid, with its viscosity rising at low shear rates (33); however, as a first approximation the Newtonian value of 4 centipoise (cP) is used in this estimation. τw is ∼12 dyn/cm2 in large arteries, but it is >10-fold higher in capillaries, where the high shear stress facilitates the deformation of erythrocytes in traversing the narrow channels. The τw is lowest in postcapillary venules, where the low shear stress is conducive to blood cell aggregation and adhesion. These values apply to systemic vessels in general, but there are regional variations, e.g., in the hepatic and renal circulations, where there are two networks of resistance vessels in series. In the pulmonary circulation, low pressure is accompanied by a low σr, but its greater values of v/r than in the systemic circulation in comparable segments lead to higher τw. Because of the pulsatile nature of the flow dynamics, the values of σr and τw change periodically with each cardiac cycle. ECs respond in both similar and different ways to changes in σr and τw.
FIGURE 2.

Estimated mean wall shear stress (τw, log scale) in different longitudinal segments of the vascular tree under resting condition using the formula 4vη/r, where v is the mean linear velocity, η is blood viscosity, and r is vessel radius. The abscissa scale for the microcirculation [arterioles, capillaries (Caps.), venules] is lengthened in comparison to the large vessels (arteries and veins). From Ref. 32, with permission from Academic Press.
1.1.2. Flow patterns in the arterial tree in relation to the focal nature of atherosclerosis.
Studies by the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group (34) have shown the preferential localization of atherosclerotic lesions at arterial branch points and regions of curvature. The flow patterns exhibit marked variations in the arterial system. The flow in the straight part of the arterial tree (e.g., most of the thoracic aorta) is mainly laminar, whereas the flows at the inner curvature of the aortic arch and arterial branch points are disturbed (FIGURE 3) (35–37). The correlation between the regions of flow disturbance and proneness to atherosclerosis suggests that disturbed flow is an important factor in atherogenesis. The focal nature of atherosclerosis has led to the hypothesis that disturbed flow patterns cause an accelerated EC turnover and an increase in EC permeability to large molecules such as LDL for their entry into the subendothelial layers to cause atherosclerosis (38). Experimental studies on the rabbit aorta (39) have shown that focal regions of disturbed flow (as reflected by nuclear orientation) colocalize with those of increased mitosis and enhanced macromolecular permeability.
FIGURE 3.

Schematic drawing of streamlined antiatherogenic flow (e.g., thoracic aorta) and disturbed atherogenic flow (aortic arch and branch points) in the arterial tree. The figure is based on work from Refs. 35, 36 and adapted from Ref. 37, with permission from the authors.
It is well established that, in the straight part of the arterial tree, the pulsatile blood flow is laminar with a clear direction; the pulsatile shear stress (PS) is antiproliferative, anti-inflammatory, and antioxidative, and hence protective against atherogenesis. In contrast, blood flow at branch points and curvatures (e.g., the inner aspect of aortic arch) is disturbed and oscillatory without a clear direction; such oscillatory shear stress (OS) is proproliferative, proinflammatory, and prooxidative, and hence atherogenic. TransWSS is the multidirectional flow with the average magnitude of wall shear stress (WSS) component acting transversely to the mean vector. The simulation of flow patterns based on micro-computer tomography (CT) imaging of rabbit aorta has demonstrated that high transWSS may serve as an important pathogenic factor for lesion formation, in comparison to shear flow without transWSS such as PS (40). The transWSS provides a three-dimensional (3-D) version of the disturbed flow. Although transWSS is different mechanically from OS, they share a lack of forward direction characteristic of PS and laminar shear stress (LS) and have similar biological effects. In vitro experiments have demonstrated that the application of shear or stretch in directions perpendicular to the aligned ECs leads to stress signaling (41, 42), which may contribute to pathogenesis and cell death in the vessel wall. Furthermore, the high temporal gradients of shear stress may also contribute to EC dysfunctions (43, 44). In general, appropriate hemodynamic forces are essential for physiological functions of ECs, whereas their abnormalities, e.g., due to flow disturbance (9, 40, 45–48), in concert with systemic risk factors (49), lead to atherogenesis. The low and oscillatory shear stress in arterial branches and curvatures causes the sustained activation of atherogenic genes. In contrast, the straight parts of the arterial tree exposed to PS with a definite direction are generally spared from atherosclerotic lesions, and ECs in these regions show downregulation of atherogenic genes and upregulation of antioxidant and growth-arrest genes (9). Thus, the disturbed and laminar flow patterns induce differential molecular signaling in ECs to result in the preferential occurrence of atherosclerotic lesions at arterial branches and curvatures and the sparing of the straight parts.
Disturbed flow also occurs on the aortic side of aortic valvular leaflets to contribute to preferential lesion development (50–52). In the venous system, the disturbed flow due to reflux through dysfunctional or incompetent valves, outflow obstruction, or stasis may cause venous hypertension to induce venous EC dysfunction and inflammation, and hence the development and progression of chronic venous diseases (53).
Understanding of the effects of disturbed flow on EC signaling, gene expression, structure, and function will help to define the molecular and mechanical bases for the role of complex flow patterns in the development of vascular pathologies and clinical consequences. Such information may also lead to the discovery of novel disease-related genes and the development of new therapeutic strategies.
1.1.3. Methods to study the effects of shear stress and stretch.
1.1.3.1. DEVICES FOR IN VITRO STUDIES.
The study of mechanosignaling in native vessels is potentially difficult because of the complexity of the in vivo mechanical environment and limitations in cellular, molecular, and genetic investigations, including the difficulties in obtaining sufficient target cells for analyses and implementing molecular manipulations for mechanistic investigations. To understand the detailed mechanisms of regulation of endothelial mechanosignaling and vascular function, in vitro systems have been established with well-controlled applications of shear stress and stretch. Such in vitro experiments using cultured ECs and VSMCs subjected to mechanical stresses in flow devices with precise control of the pulsatility, frequency, amplitude, and duration allow deciphering of the cellular and molecular mechanisms for mechanosensing, signal transduction, transcriptomic and epigenetic regulations, and functional modulations in relation to vascular homeostasis and pathology.
1.1.3.1.1. Flow devices.
Many types of flow devices have been developed to study the effects of the magnitude and pattern of flow on vascular cells, as described in a previous review (9). These devices are used to investigate the responses of ECs to the atheroprotective PS or laminar shear (LS) versus the atheroprone OS and disturbed flow. OS has oscillation without a clear direction, i.e., the net flow is close to zero. PS and LS have clear directions of net flow, with pulsatility present in PS but not LS. In general, the responses of ECs to PS and LS are similar, and the results are often discussed interchangeably in this review, in contrast to OS. Although these devices have been used primarily to study the effects of flow on ECs, they have also been used to study VSMCs.
There are mainly two types of flow devices: the parallel-plate flow chamber (FIGURE 4) (54–56) and the cone-plate viscometer (57–59). Both systems have been used to study the effects of laminar shear (60, 61); with modifications, they allow the applications of other flow patterns such as PS and OS. The modification of the parallel-plate flow chamber with a computed continuous increase of the width of the channel over its length (FIGURE 4A, tapered chamber) leads to a linear decrease of the shear stress along the channel length from the entrance, thus allowing the study of cellular responses to different magnitudes of shear stress within the same system (62). The incorporation of a step at the entrance of the flow channel to create a reduction of channel height followed by its step increase can mimic the vessel branch point with the creation of flow disturbance immediately beyond the step, thus allowing the study of cellular responses to PS and OS in a single chamber (FIGURE 4A) (63, 64). In the cone-plate viscometer, different levels of LS, disturbed flow, as well as flows with various waveforms, can be generated with modifications of the cone angle and velocity, placement of surface obstacle strips, as well as addition of computerized controllers (65, 66).
FIGURE 4.
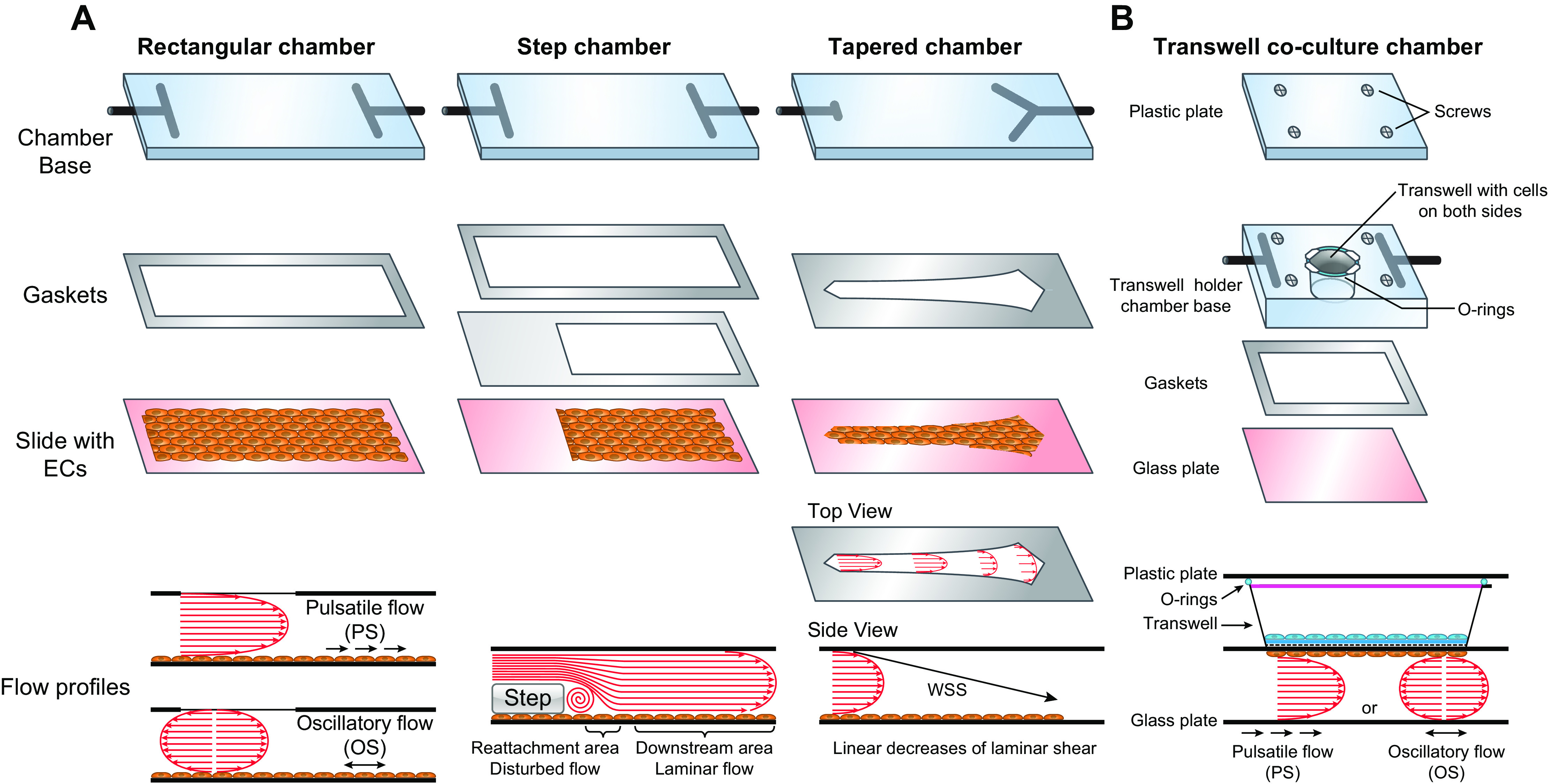
Flow chambers. A: monoculture flow chambers with various geometries for the creation of different flow patterns. B: coculture flow chamber with Transwell for the investigations of cell-cell interactions under different flow patterns. WSS, wall shear stress. See glossary for other abbreviations.
The incorporation of multiple-layered VSMC cultures, or VSMC-embedded ECM gel block with an EC culture on top, into the flow chamber system allows the investigation of the interactions between these two types of vascular cells under controlled flow patterns to reflect the in vivo vascular conditions (67, 68). The porous Transwell has been incorporated into the flow chamber to separate two types of cells (e.g., VSMCs and ECs) to investigate their interactions in responding to flow conditions applied to the EC (FIGURE 4B) (69, 70). This allows the clean separation, visualization, and collection of each cell type, thus enabling the investigation of interactions among different types of vascular cells and providing information reflecting the in vivo vascular conditions.
1.1.3.1.2. Stretch devices.
Stretch devices have been employed for mechanosignaling studies on cultured layers of VSMCs and ECs. Here (FIGURE 5) we only outline some examples of stretch devices used in studies on mechanosignaling in cells cultured on an elastic membrane. In general, a chamber with cells seeded on an elastic membrane coated with extracellular matrix is mounted in the device with various stretching mechanisms. In the mobile chamber system (FIGURE 5A), the elastic membrane (blue color) is stretched by pushing the chamber against the indenter. Biaxial stretch is generated by using an indenter with a geometry that matches with the chamber shape to produce even stretching along all sides (FIGURE 5A, left top view, pink lines), whereas uniaxial stretch is generated by using an indenter with a rectangular geometry (FIGURE 5A, right top view, pink lines) for pulling preferentially along the long axis, with only limited pulling along the short axis (FIGURE 5A). A similar principle is applied for the circular chamber using vacuum stretch systems (FIGURE 5B) with different geometries: Biaxial stretch is applied to a circular chamber well by even pulling in all directions against a loading post with cylindrical geometry (FIGURE 5B, left top view, pink lines), whereas uniaxial stretch is generated by using a loading post with an arctangular shape (FIGURE 5B, right top view, pink lines) to apply membrane pulling only along the short axis. The magnitude of stretch is governed by the position of the indenter or the vacuum strength. Another stretch device format is the unidirectional lateral pulling of an elastic membrane mounted on a fixed post at one end and on a mobile arm at the other end. The mobile arm moves cyclically in a lateral direction to generate uniaxial stretch (FIGURE 5C). A microfluid channel system has been constructed with cells seeded onto an elastic polydimethylsiloxane (PDMS) membrane on top of the channel that can be deformed by pressure variation underneath in response to microfluid effects (FIGURE 5D). Such a device generates circumferential stress with uniaxial stretch as well as bending stress. All these devices can be used to effectively test the stretch regulation of cell function. However, the induction of stretch by the displacement of an elastic membrane can cause the movement of cell growth medium over the surface of cells in the chamber, and hence some level of undefined shear stress. The effects of shear stresses induced in this manner remain to be determined. Single-VSMC and -EC studies for ion channels mainly involve membrane stretch or compression with micropipettes, bead twisting, or osmotic stimuli during patch-clamp recording or calcium imaging, as discussed in sect. 2.1.2.
FIGURE 5.
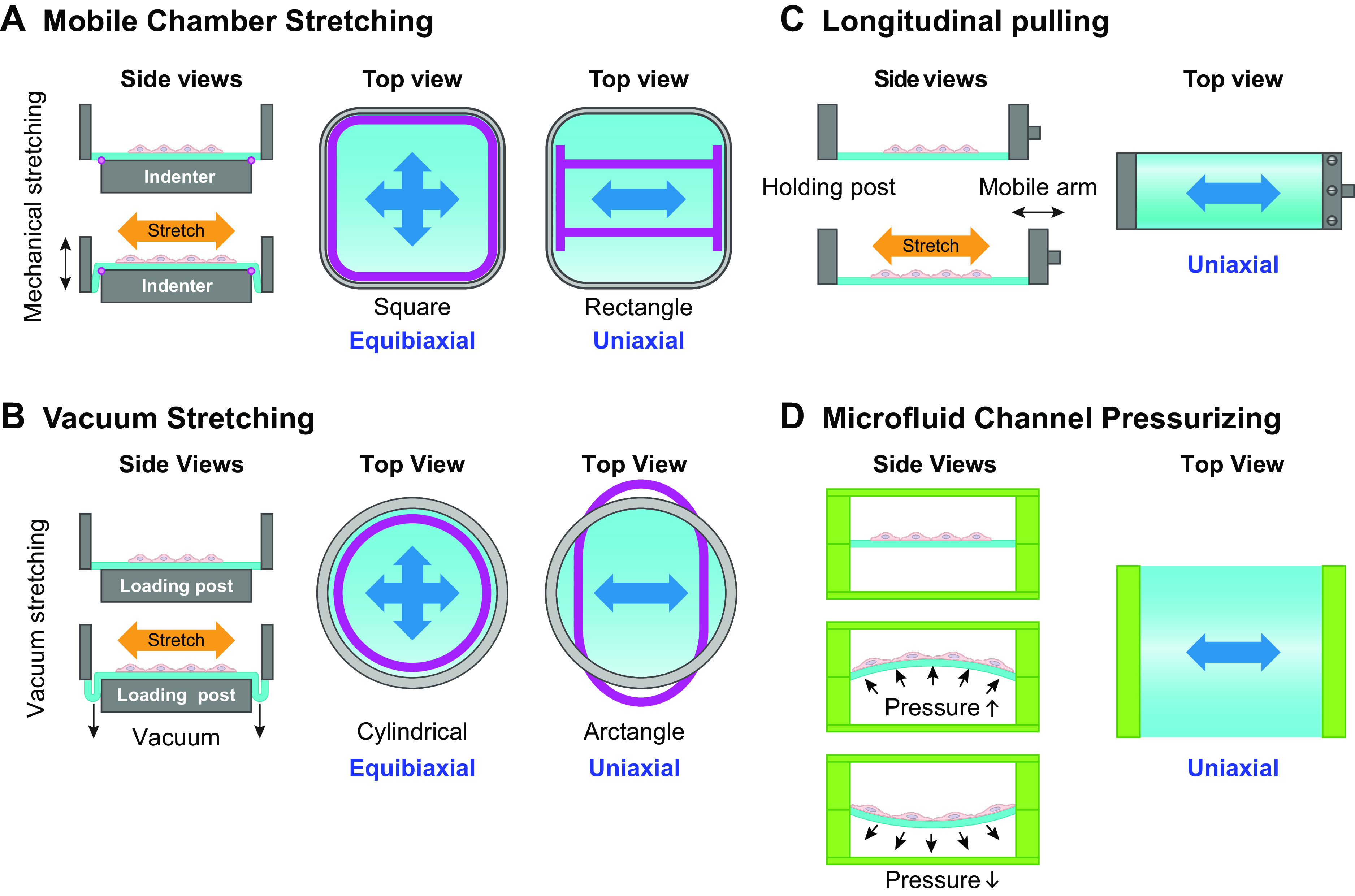
Stretch chambers. A: vertical mobile stretch chambers against indenters. B: stretch chambers with vacuum pulling against loading posts. C: longitudinal mobile stretch chamber for uniaxial stretch. D: microfluid channel-based stretch devices.
1.1.4. Shear stress (or flow)-induced vasodilation.
An increase in shear stress in vivo can result from a number of different factors, e.g., as a consequence of arterial pressure elevation or upstream arterial dilation. Increased shear stress triggers multiple intracellular signaling processes in the endothelium, resulting in EC hyperpolarization and/or production of EC-derived metabolites such as nitric oxide (NO) and inhibition of vascular myogenic tone (71). This process, termed shear stress-induced dilation (or more often flow-induced dilation or flow-mediated dilation in human studies), has been extensively investigated in arteries by pressure myography, with which the effects of flow and pressure can be differentiated by keeping the intravascular pressure constant through adjustment of inflow and outflow pressures (72). Flow-induced dilation (or dilatation) often synergizes with other local blood flow control mechanisms, e.g., metabolic hyperemia, to recruit parallel and/or upstream vessels and produce larger network responses (73).
The mechanisms underlying flow-induced dilation and NO production have been studied extensively in intact arteries as well as in cultured ECs (FIGURE 6). Flow-induced dilation can be mediated in part by Ca2+-dependent production of NO and/or prostanoids from the endothelium (74–76), which diffuse to the VSMC layer and inhibit tone (71). After the onset of flow, arterial dilation is reported to have ∼6- to 15-s delay (72, 77, 78), consistent with the activation of intracellular processes that lead to enhancement of endothelial NO production. However, shear stress also induces endothelium-derived hyperpolarization (EDH), which conducts directly to the VSMC layer via myoendothelial gap junctions in most arteries/arterioles such that blocking gap junctions can inhibit flow-induced dilation in some arteries (79). However, the EDH mechanism may be more prevalent under pathological than normal conditions (80).
FIGURE 6.

Hypothesized relative time course of events contributing to flow-induced dilation. Vm: initial hyperpolarization through Kir2 channels followed by secondary depolarization (dotted red line) due to the activation of a Ca2+-activated Cl− channel. [Ca2+]: initial Ca2+ release from endoplasmic reticulum (ER) stores, followed by sustained Ca2+ entry through various Ca2+-permeable ion channels. These events occur in parallel with or are followed by enhanced endothelial nitric oxide synthase (eNOS) phosphorylation. All these mechanisms combine to increase arterial diameter through inhibition of myogenic tone. See glossary for other abbreviations.
The initial event triggered by increased shear stress is thought to be the activation of one or more K+ conductances, possibly by activation of a mechanosensitive K+ channel, as discussed in sect. 2.1. Hyperpolarization increases the “passive” movement of Ca2+ down its electrochemical gradient into the cytosol (81, 82) of “nonexcitable” ECs, which do not normally express voltage-gated Ca2+ channels (VGCCs) (83–85). The EC isoform of nitric oxide synthase, eNOS, is a Ca2+/calmodulin-regulated enzyme (86–88). A large number of studies have demonstrated that shear stress induces a rise in EC cytosolic Ca2+ levels, both in cultured ECs (for review see Ref. 18) and in intact arteries/arterioles (89, 90). Resting membrane potentials in ECs generally average approximately −30 mV, although a wider range of potentials has been reported in populations of cultured ECs, whose membrane potentials are not as synchronized by gap junction coupling as they are in intact arteries (91). The hyperpolarization triggered by shear stress (as well as by EC-dependent vasodilators) has an upper limit of approximately −90 mV, as determined by the K+ equilibrium potential (EK). Although only an approximate twofold increase in the driving force for Ca2+ entry can potentially occur through this mechanism, shear stress (and most agonists) often induces much greater increases in EC global Ca2+ levels, implying that membrane Ca2+ conductance also changes (92). EC Ca2+ conductance can increase through the opening of non-voltage-gated Ca2+-permeable ion channels, including Piezo1 (93), TRPV4 (94), polycystins (95), and P2X4 (96), as discussed in sect. 2.2. In parallel with EC hyperpolarization, shear stress induces the activation of phospholipase C (PLC), with consequent production of diacylglycerol (DAG) and inositol trisphosphate (IP3) (97, 98). IP3 binds to IP3 receptors and induces rapid Ca2+ release from endoplasmic reticulum (ER) stores. Multiple ion channels are potentially activated by these signaling pathways, including transient receptor potential (TRP) cation channels and store-operated Ca2+ entry channels such as Orai (99). The effects of NO are further enhanced through the Ca2+ sensitization of eNOS by phosphorylation at specific COOH-terminal residues (Refs. 100–103; for review see Ref. 104). Prostanoid production may require the activation of additional downstream events (105).
Although one or more ion channels have been hypothesized to be the EC mechanosensor (for review see Ref. 18), other membrane elements may be upstream of ion channels, including receptor tyrosine kinases (102, 106), mechanosensitive GPCRs (107–111), and junctional proteins (112). Each of these mechanisms is discussed in subsequent sections.
1.2. Mechanoresponses of VSMCs
1.2.1. Intravascular pressure exerts a mechanical force on VSMCs.
An increase in intraluminal pressure leads to expansion of a blood vessel (FIGURE 7A), subjecting VSMCs to varying degrees of stretch, depending on their orientation (FIGURE 7B). In the medial layers of most arteries, VSMCs are oriented circumferentially so that the primary force, σθ, is distributed along the long axis of the spindle-shaped cell (FIGURE 7Ba). The relationship between σθ and pressure is dictated by the law of Laplace, such that stress σθ = T/h, where T = tension and h = wall thickness. In turn, T = (Pint − Pext)·r, where r denotes vessel radius. Therefore, circumferential stress is determined by the vessel radius and the transmural pressure difference. Because Pext (external or tissue hydrostatic pressure) is usually low and nearly constant in the systemic circulation, a change in Pint (intraluminal pressure) is the most physiologically relevant force that determines the acute change in circumferential stress acting on VSMCs. Arterioles and many small arteries may have a significant fraction of VSMCs oriented with their axis differing significantly from 90° to the flow direction (FIGURE 7Bb; Ref. 113), and some vessels even have axially or helically oriented VSMCs (e.g., the outer layer of some arteries and portal vein; FIGURE 7Bc). The orientation of a VSMC will determine the extent to which σθ is transmitted to the mechanosensing (and force bearing) elements of that cell.
FIGURE 7.

Relationships between transmural pressure, σθ, and VSMC orientation. Cross-sectional (A) and axial (B) views of a blood vessel. See glossary for abbreviations.
1.2.2. The vascular myogenic response.
1.2.2.1. DEFINITION.
One of the more significant vascular responses to an increase in Pint (hereafter referred to simply as an increase in pressure) is the acute contractile response of VSMCs to circumferential stretch (FIGURE 8A). This phenomenon was initially described by Bayliss (114). When pressure is elevated within a certain range (e.g., in arteries after an increase in systemic arterial pressure or in arterioles as a result of constriction of the terminal arterioles), the vessel transiently distends and then constricts, reaching a final diameter (df) that is smaller than the initial diameter (di), even as pressure remains elevated. The constriction occurs over a time course of fractions of seconds to minutes and reflects active force development by VSMCs within the vessel wall in response to the increase in σθ. The active constriction to pressure elevation is intrinsic to VSMCs and does not require an intact endothelium or innervation (12). Most small arteries and arterioles exhibit this response, and it is detectable in some veins (115, 116) and lymphatic vessels (117). When pressure is lowered, a “myogenically reactive” vessel typically shows the opposite behavior, dilating and remaining dilated even at the new (lower) pressure (FIGURE 8A). The dilation is typically slower and less often studied than the constriction (12), but in the discussions that follow it is assumed that the processes are the same (but see counterargument in Ref. 118). Collectively, the constriction to elevated pressure and the dilation to reduced pressure are referred to as the “myogenic response,” or more properly the “vascular myogenic response.” The magnitude and speed of myogenic constriction, as well as the sensitivity to the initial stretch (determined in part by the relative distensibility of the vessel), may be interrelated and vary widely among different branching orders of arteries/arterioles and between vessels of similar sizes from different vascular beds. Myogenic constrictions occur in most vessels on the arterial side of the systemic circulation and are strongest at the level of small arterioles (119). Arteries/arterioles from the renal and cerebral circulations have the most pronounced myogenic responses (12); e.g., renal afferent arterioles begin to constrict within 300 ms after an initial pressure-induced distension and can completely close within seconds (118). Coronary and skeletal muscle arteries show myogenic constrictions of intermediate magnitude, whereas mesenteric arteries typically have weaker responses (120) and pulmonary arteries often fail to show any constriction (121). The relative gain of the myogenic response in arteries/arterioles of these various vascular beds is proportional to the degree of blood flow autoregulation in those same beds, pointing to one of its presumed physiological roles (discussed below). The activation of the myogenic response is independent of extrinsic factors related to the endothelium, circulating agonists, and components of blood and perivascular nerves, and yet it is modulated by most of these. In vivo assessment of the myogenic response can be complicated by the influences of all these factors.
FIGURE 8.
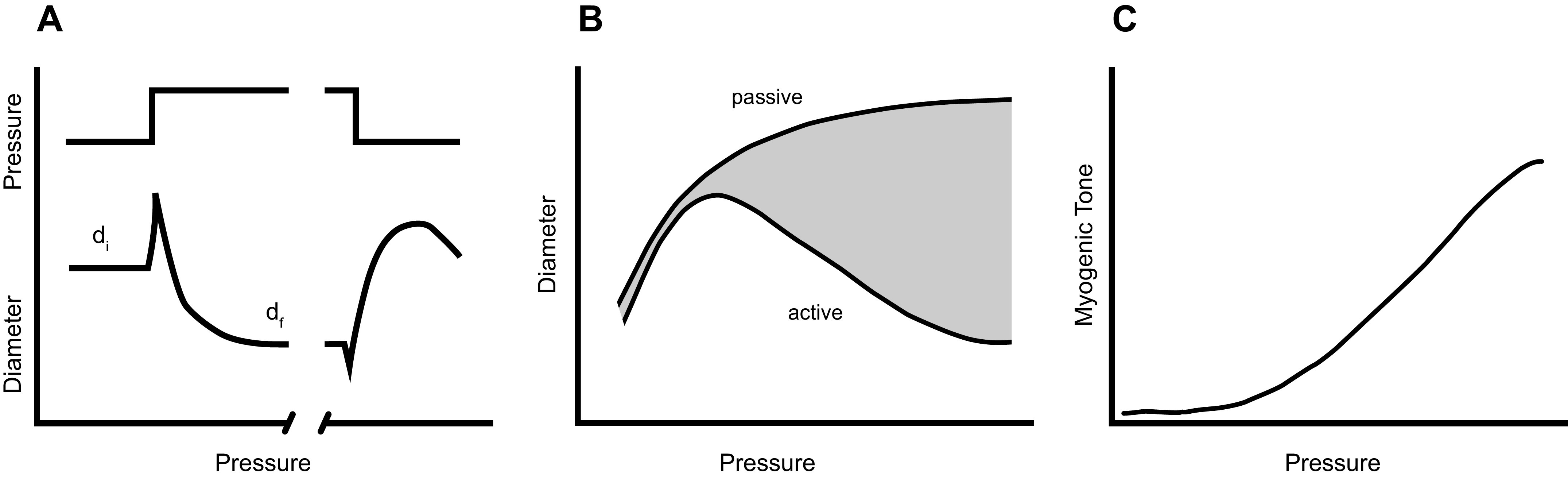
Various representations of the vascular myogenic response. A: time course of arterial myogenic constriction to a step increase in internal pressure. B: plot of arterial diameter as a function of pressure showing progressive constriction in physiological saline (active) vs. dilation in calcium-free physiological saline (passive). C: myogenic tone (passive-active) curves in B plotted as a function of pressure.
1.2.2.2. METHODS FOR STUDYING THE MYOGENIC RESPONSE.
Multiple methods have classically been used to study the myogenic response. Whole organ studies originally suggested that both metabolic and myogenic components contributed to the calculated vascular resistance changes evoked by manipulation of perfusion pressure and/or venous pressure (122, 123). Clever modifications to these whole organ methods were employed in attempts to understand the underlying segmental resistance changes (124, 125). Multiple laboratories extended this line of investigation to studies of the microcirculation using intravital microscopy, where the diameters of individual small arteries and arterioles could be observed while manipulating pressure (126–131). These studies have been summarized in review articles and chapters (132, 133). However, even with the use of these methods, the pressure change at the level of a particular vessel was usually unknown and could not be precisely controlled. Investigations of mechanisms underlying the myogenic response were not advanced until the development of methods to study single small arteries or arterioles under controlled conditions, either under isometric conditions with a wire myograph (134, 135) or under isobaric conditions with “pressure myography” (136, 137). In wire myograph experiments, myogenic tone development is represented by the secondary development of force after an initial stretch (138–141); however, myogenic responses under these conditions are highly variable and labile (115, 141). Myogenic responses are now most often studied with pressure myography of cannulated arteries/arterioles, where it is possible to control Pint and/or Pext in the absence of flow, exchange luminal and extraluminal solutions, and regulate temperature while accurately measuring internal diameter. An additional advantage of pressure myography is that the VSMC layer is maintained in its normal geometry (135, 142). The experimentally measured variable is usually the time course of constriction/dilation, and the data are analyzed and presented in comparison to the responses of the same vessels to equivalent pressure steps after elimination of all active tone in calcium-free solution. The active and passive steady-state diameters are plotted against pressure (FIGURE 8B), with the difference between the two curves representing the myogenic responsiveness of the vessel (shaded area in FIGURE 8B). Active myogenic tone developed at every measured pressure is often described by the relationship shown in FIGURE 8C.
1.2.2.3. PHYSIOLOGICAL ROLE OF THE MYOGENIC RESPONSE.
The myogenic response is proposed to subserve several physiological roles, including 1) the autoregulation of blood flow (the ability of a vascular network to maintain flow at different levels of perfusion pressure); 2) the establishment of a basal level of vascular tone on which vasodilators and vasoconstrictors can bidirectionally regulate flow; 3) the partial regulation of capillary hydrostatic pressure (Pc) if perfusion pressure to an organ falls; 4) protection against excessive increases in Pc and subsequent net fluid movement from capillaries during various conditions associated with elevated vascular pressure, including venous occlusion, increased gravitational hydrostatic loads, and systemic hypertension; and 5) contribution to the initial phase of reactive hyperemia (the transient increase in blood flow to an organ that occurs after a period of ischemia) (143). Metabolic, neural, and shear stress control mechanisms of blood flow can attenuate or potentiate the vascular myogenic response (71, 144). The physiological relevance of the myogenic response is the focus of detailed discussions in previous reviews (71, 143, 145, 146).
1.2.2.4. CONCEPTUALIZING THE UNDERLYING MYOGENIC MECHANISM.
Conceptual arguments for how the myogenic response might be initiated and sustained were advanced by Johnson in his Handbook of Physiology chapter (146). These and earlier discussions (147–149) served as initial attempts to elucidate the underlying mechanisms. Johnson asked how a system with vessel radius as the sensed variable could explain myogenic constriction to a radius lower than control in response to sustained, elevated pressure, because the error signal would have been eliminated once the radius reaches its initial level. Indeed, elimination of the error signal is a long-standing question addressed by subsequent reviews on mechanotransduction in general (150–153). Johnson, and Burton before him (149, 154, 155), proposed that if wall tension, rather than radius, were a sensed variable, tension would increase in response to pressure elevation and return toward, but not completely back to, or below, control as the vessel constricted. In this way, an error signal for regulating wall tension would persist after steady-state constriction of the vessel to a smaller radius. The concept of myogenic control of arteriolar diameter through regulation of wall tension, although not proven, is generally consistent with experimental data (131, 156–158), even though the supporting data are only correlative in nature. Similar, widely accepted arguments have been advanced in favor of acute regulation of wall shear stress (78, 159) and long-term regulation of wall tension during vascular remodeling (160, 161). Although these are useful for discussing homeostasis, additional insights are needed into the underlying cellular mechanisms.
Myogenic constriction is thought to involve stretch activation of one or more VSMC mechanosensors, followed by activation of downstream signaling pathways that results in phosphorylation of a critical myosin light chain serine residue (MLC20) (162–164), leading to increased cross-bridge cycling and actomyosin force development. Multiple signaling pathways in addition to the myogenic response regulate MLC20 phosphorylation via activation of myosin light chain kinase (MLCK) and/or inhibition of myosin light chain phosphatase (MLCP) (165, 166). It is often assumed that the mechanism underlying vascular myogenic constriction is common to all types of arterial vessels, but a few studies in which similar pathways were blocked in arteries from different tissues suggest possible differences in the underlying mechanism(s) (167). Force-sensing elements are considered to be the most upstream components of the myogenic response signaling pathway, whereas phosphorylation of MLC20 is considered the most downstream component. Potential force-sensing elements include ion channels, integrins, focal adhesion complexes, cell junction molecules, cytoskeletal complexes, caveolae, and G protein-coupled receptors (GPCRs). The sections below discuss the evidence for and against these mechanisms.
1.2.2.5. PRESSURE-INDUCED DEPOLARIZATION PRECEDES MYOGENIC CONSTRICTION AND IMPLIES THE INVOLVEMENT OF ION CHANNELS.
A hallmark of the arterial response to pressure elevation is rapid VSMC depolarization (168), which triggers a global VSMC calcium increase (169, 170) through activation of voltage-gated calcium channels (VGCCs) (12). Ca2+ influx down its electrochemical gradient through VGCCs produces a global rise in cytosolic [Ca2+], leading to the activation of MLCK, MLC20 phosphorylation, and vasoconstriction (162, 171). It is well documented that myogenic tone- and pressure-induced constriction are suppressed by L-type Ca2+ channel (Cav1.2) inhibitors (12). Additionally, smooth muscle (SM)-specific deletion of Cav1.2 eliminates the arterial constriction to pressure elevation and lowers peripheral resistance and blood pressure (172), which are effects that would be predicted with the widespread loss of arterial myogenic tone. This Ca2+-sensitive pathway has been the most widely studied mechanism underlying the arterial myogenic response and is the primary reason for extensive discussions of mechanosensitive VSMC ionic mechanisms in sect. 2. However, two additional pressure-sensitive pathways regulate myogenic tone through MLCP and actin polymerization; these are discussed in sects. 5.1 and 5.2, respectively.
Pressure-induced vascular smooth muscle (VSM) depolarization appears to be the key initiating event in the vascular myogenic response. The membrane potential (Vm) of VSMCs is highly pressure dependent, with resting values ranging from approximately −60 mV at 10 mmHg to approximately −30 mV at 160 mmHg (168, 170, 173) (see Figures 1 and 8 in Ref. 173 for data from cerebral and skeletal muscle arteries, respectively). Data are typically compiled from steady-state Vm measurements made at multiple pressures after repeated impalements because measuring the time course of depolarization in a single VSMC before, during, and after a given pressure step usually is precluded by vessel wall movement that can easily dislodge a sharp microelectrode. It has not been possible to selectively prevent pressure-induced VSM depolarization to test whether constriction is blocked by such a maneuver (174). Elevating bath K+ concentration ([K+]) above 15 mM leads to VSMC depolarization and generally results in a blunting of myogenic responsiveness (175), i.e., reducing the slope of the curve in FIGURE 8C, but this intervention directly and indirectly affects the activity of multiple ion channels. Certain agonists also blunt myogenic responsiveness (176, 177), presumably with little or no VSM depolarization (178, 179). The similarity of the KCl and agonist effects suggests that wall stiffening per se may explain the decreased myogenic responsiveness under these conditions, although the possibility of attenuation of the initiating “error signal” cannot be ruled out.
The well-supported observation of pressure-induced depolarization naturally suggests a role for a mechanosensitive ion channel that would permit cation entry or anion efflux, but another possibility would be pressure-induced inhibition of a constitutively active K+ current (180). Initial studies on the ion currents activated by stretch in isolated single smooth muscle cells (SMCs) pointed to a nonselective cation channel with a relative ionic permeability Na+, Ca2+ > K+ ≫ Cl− and a conductance of ∼30 pS (181–185) that could produce depolarization when activated. Although these data were obtained before the molecular identification of TRP and Piezo channels, the measurements are consistent with the ionic permeabilities of several TRP family members, including TRPC6, TRPC3, TRPC7, and TRPM4, and other cation channels such as Piezo1 and ENaC. Subsequent functional studies on arteries have provided evidence for a component of myogenic constriction mediated by ENaC and ASIC channels or specific TRP isoforms. Evidence for these and other ion channels is discussed in sect. 2.2.
2. MECHANOSENSITIVE ION CHANNELS
Before providing detailed analyses of specific channels involved in shear stress-induced EC responses and pressure-induced VSMC responses, we first address general concepts about mechanosensitive channels because the experimental conditions under which mechanosensitivity is determined and the forces required for gating, particularly in biophysical protocols, have important implications for the physiological relevance of a given ion channel.
2.1. Background
2.1.1. General concepts.
What determines whether an ion channel functions as a legitimate, physiological mechanosensor? Many ion channels are polymodal, i.e., they can be gated by different types of stimuli, including electrical, chemical, and mechanical forces. A given channel may have a primary gating modality, but it may also respond to a lesser degree to other stimuli. Examples include channels such as TRPV1 that are sensitive to temperature, plant-derived chemical compounds, pH, and changes in osmolarity (186, 187) and Kv channels that exhibit both voltage sensitivity and mechanosensitivity (188, 189). Even photoreceptors have been found to require a mechanical component to gating under certain conditions (190). Posttranslational modification (phosphorylation, methylation, palmitoylation, etc.) of critical channel residues could potentially lead to switching between primary and secondary gating modalities. Because multiple ion channels have been implicated in VSMC and EC mechanosensitivity, polymodal gating is an important concept to clarify. Thus, although multiple ion channels can potentially be activated by circumferential stretch or shear stress under in vitro conditions, their primary sensitivity may be to nonmechanical factors and thus their contributions to pressure-induced myogenic constriction or flow-induced dilation might be minimal or undetectable in vivo.
2.1.2. Experimental methods for determining single-cell mechanosensitivity and their limitations.
Ion channel mechanosensitivity most often is assessed with patch-clamp electrophysiology methods. Patch clamping is a single-cell voltage-clamp method that enables ionic current contributed by a specific type of ion channel to be measured. For stability, the most commonly used recording mode is the cell-attached recording mode, in which the outside of the cell membrane is exposed to the solution in the patch pipette and the inside of the membrane to the normal cytoplasmic contents; an alternative method is the excised, inside-out recording mode, in which the patch is pulled off after gigaseal formation and the inside surface is exposed to the bath solution. Some studies employ the conventional whole cell recording mode, in which the patch is purposely ruptured and the cell is dialyzed with the pipette solution. Other studies use the perforated-patch recording mode, in which the membrane patch is not ruptured but exposed to a permeabilizing compound added to the patch pipette solution that makes the patch permeable to monovalent cations (and to a lesser extent anions) but not calcium (see details in Ref. 191); this method allows intracellular Ca2+ levels to remain undisturbed during the recording. The other method commonly used in studies of mechanosensitive ion channels involves the insertion of recombinant channels into a lipid bilayer (192). The experiments are performed in a chamber with cis- and trans-compartments connected through a small aperture. After a lipid solution is applied to the aperture, a planar bilayer membrane forms in the hole. Recombinant ion channels can then be inserted into the membrane from a micellar solution or after fusion with liposomes. The ion current associated with channel gating is recorded from the two sides of the chamber. Many electrophysiology studies have been made with recombinant channels to isolate the behavior of a specific ion channel, because studies performed in native cells can be complicated by lower channel density and the presence of multiple mechanosensitive currents.
Conclusions about whether a particular ion channel is mechanosensitive may depend critically on how a mechanical stimulus is applied in an experimental protocol (193, 194). In bilayer studies of recombinant channels, asymmetric lipids are often used to create a hydrophobic mismatch and thereby increase the membrane curvature and tension (see examples below). In experiments with native cells, the types of mechanical stimuli vary widely (FIGURE 9). The approach used most commonly to assess mechanosensitivity of selected ion currents is to apply pressure or suction through a patch-clamp micropipette in the cell-attached or excised patch recording mode. Negative and positive pipette pressure may be equally effective in elevating membrane tension in cell-attached patches (196). Mechanical perturbation of whole cells has been accomplished in multiple ways, including indentation with blunt probes, substrate deformation, and the use of micropipettes or matrix-coated beads to pull on, or twist, the cell membrane (FIGURE 9). However, in some cases, macroscopic currents cannot be evoked by these means even if single-channel mechanosensitive currents are elicited in the same cells (197), raising the issue of possible artifacts arising from glass-lipid interactions in the latter recording mode (189, 198). Stretch activation of some channels becomes easier with repeated testing; this also raises concerns of artifacts (199) due to changes in the structural integrity of the patch (200) or the cortical actin cytoskeleton (CSK) (201). Both ECs and VSMCs have been subjected to LS or OS, acutely or chronically, by flow streams directed from micropipettes or small tubes or by controlling flow in open or closed chambers (FIGURE 4). Osmotic swelling is a widely used mechanical stimulus that requires only a solution change, but swelling can trigger chemical as well as mechanical responses because it dilutes cytoplasmic contents, decreases ionic strength (which in itself alters ion channel conductance), elicits production of metabolites such as arachidonic acid (202), and alters the cytoskeletal composition (200). For these reasons the response of an ion channel to an osmotic stimulus alone is generally not considered to be conclusive evidence of mechanosensitivity (195).
FIGURE 9.
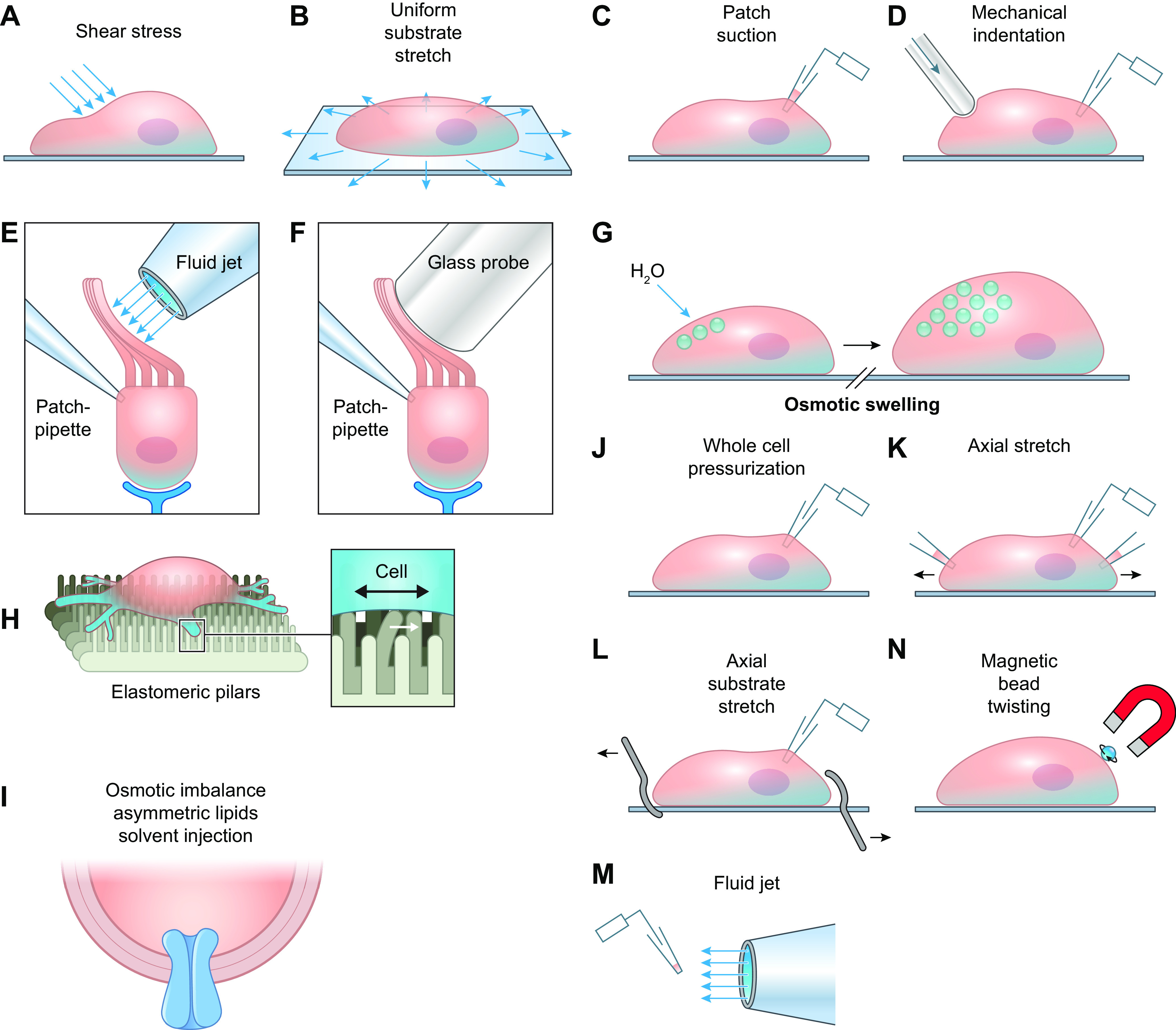
Commonly used mechanotransduction assays for ion channels. A: application of shear stress to a single cell. B: stretch of a cell by substrate deformation. C: deformation of local membrane by patch pipette suction while recording single-channel currents. D: focal indentation of cell with mechanical probe while recording whole-cell current. E: deformation of cell (in this case inner ear hair cell) using fluid jet from micropipette while recording whole-cell current. F: deformation of inner ear hair cell with blunt probe while recording whole-cell current. G: osmotic swelling of cell. H: deformation of cell seeded onto elastomeric pillars. I: deformation of channel incorporated into bilayer by injection of asymmetric lipids. J: pressurization of a single cell through a whole-cell recording pipette. K: stretch of a single cell using two patch pipettes while recording whole-cell current. L: stretch of a single cell on flexible substrate using two blunt pipettes while recording whole-cell current. M: recording of current from patch of an excised, inside-out cell membrane while moving it into a flow stream from a pipette. N: localized deformation of a cell membrane by twisting of a magnetic bead attached to the cell surface. Modified from Ref. 195, with permission from Neuron.
Many studies addressing the mechanosensitivity of ion channels employ supraphysiological forces, such as unusually large osmotic gradients (e.g., 500 mM mannitol) (203) or extreme membrane distortion generated by lipid-glass interactions in cell-attached patches (188, 189, 194, 198, 204). Cell dialysis with certain solutions, including KCl and KBr, can dissolve certain cytoskeletal components (205). Mechanosensitive studies of reconstituted channels suffer from additional problems, as discussed elsewhere (206, 207). It is therefore important to consider whether the mode or magnitude of the mechanical stimulus required to activate a channel in a bilayer, isolated cell, or membrane patch is consistent with the physiologically relevant force the channel would experience in vivo.
2.1.3. Criteria for true mechanosensitivity.
Chalfie and coworkers and Patapoutian et al. propose that a bona fide mechanosensitive ion channel should fulfill strict criteria to ensure that it is a direct, rather than indirect, mechanosensor (203, 208). The proposed criteria include the following:
-
1)
The channel should contain a pore-forming subunit permitting rapid ion conduction.
-
2)
When reconstituted into an artificial, cell-free lipid bilayer, the purified channel should gate when tension is applied to the bilayer.
-
3)
Site-directed mutagenesis of critical channel domains that affect pore selectivity or conductance should alter mechanosensitivity.
-
4)
Forced expression of the channel in a nonmechanosensitive cell should confer mechanosensitivity.
-
5)
Both gene and protein of the channel must be expressed in the purported mechanosensitive cell.
-
6)
Genetic deletion of the channel should abolish mechanosensitivity in a way that rules out the channel having only a developmental role or being a downstream signaling component of another mechanosensor. [However, genetic deletion can disrupt normal signaling complexes, leading to off-target effects, so that the expression of a dominant-negative (dead) channel construct may be an even better approach.]
Whether these criteria are sufficient to distinguish true mechanosensitive channels from those that are indirect mechanosensors is a matter of debate, and this issue is revisited in the summary sections for VSMC and EC ion channels.
Although multiple ion channels have been implicated in mechanosensing, the criteria above are met by only a few select ion channels, including the bacterial channels MscS and MscL and the mammalian channels Piezo1–2, TREK-1/2, TRAAK, and possibly ENaC and ASIC (195, 208). The first truly mechanosensitive ion channels, MscS and MscL, with small and large conductances, respectively, were identified in bacteria (209, 210). When reconstituted into liposomes, the channels retained mechanosensitivity without the need to include other proteins (e.g., CSK components). Under these conditions, membrane tension was altered by incorporation of asymmetric lipids into the two leaflets, and the relationship between channel open probability (Po) and tension was described by a Boltzmann function (211). Mutations in critical domains of MscS and MscL modulated their mechanosensitivity (212), and their combined genetic deletion abolished the response to osmotic swelling (208). Although MscS and MscL exhibit “high-threshold” mechanosensitivity and respond only to near-lytic membrane tensions (188, 196), their properties fulfill many of the requirements for mechanosensitivity proposed above. Mammalian channels such as Piezo1, TREK-1/2, and TRAAK exhibit lower thresholds for mechanosensitivity.
Not all investigators agree that such strict criteria should be applied to eukaryotic mechanosensitive ion channels (152, 198, 208, 213). In eukaryotes, mechanosensitivity may be inherent to not only the pore-forming subunit but also the auxiliary channel subunits, specialized lipid domains such as rafts, and/or CSK-tethering proteins (214, 215). A focus on forces that act only in the plasmalemmal bilayer may oversimplify how stress is distributed to many other elements of the cell, including the CSK, attachment sites to the extracellular matrix (ECM), and cell-cell junctions. Thus, although a mechanosensitive ion channel may change its conformation under stress and become activated (or inactivated) in response to increases in bilayer tension, those changes are also transmitted to the CSK (198), which is composed of multiple elements that bear and distribute a variable fraction of the imposed stress. Complicating this issue, force generated internally (e.g., by VSMC contraction) can be transmitted through the CSK network to the plasmalemma and associated scaffolding proteins, which could serve as a potential feedback mechanism for mechanosensitive ion channel inactivation. Because of this complex distribution of force, a stimulus such as osmotic stress can have widespread effects; for example, although osmotic swelling is predicted to increase tension in the cortical CSK, atomic force microscopy measurements show that the cortical CSK actually softens as the cell osmotically expands, presumably because of the redistribution of stress through internal CSK elements (216).
2.1.4. Basic mechanisms of ion channel mechanosensitivity.
In line with the preceding discussion, two major hypotheses have been proposed for the molecular mechanism of mechanosensitive ion channel gating (FIGURE 10) (217–219): One involves forces intrinsic to the lipid bilayer, and the other involves tethering of the channel (or bilayer) to the CSK or other components. It is well established that some channels [e.g., MscS, MscL, Piezo1, two-pore domain K+ channels (K2P)] retain mechanosensitivity when purified and reconstituted into lipid bilayers in the absence of accessory subunits, CSK, and ECM components. The mechanosensitive gating of a channel resulting from increased bilayer tension is termed the “force-from-lipid (FFL) hypothesis,” which describes how force is transmitted from lipids to proteins (218). Proteins embedded in the membrane are subject to relatively large, anisotropic push-pull forces created by interactions of phospholipid polar head groups and interior acyl tails. At the lipid-water interface of each bilayer a large surface tension pulls outward on an embedded membrane protein, whereas at the bilayer midplane lateral compressive forces push inward (196, 220–222). The crystal structures of MscL and K2P channels provide insight into how changes in membrane tension might alter channel gating (223, 224): tension applied to the bilayer causes it to “thin” (FIGURE 10A), increasing the surface area (ΔA) of the channel and opening the pore (225, 226). Structural analyses also reveal the intimate association of lipid subpopulations with certain channel domains, facilitated for example by interactions between negatively charged phosphatidyl groups and positively charged arginine residues, as in the voltage sensor of Kv channels (188, 227–229). Thus, changes in tension may alter interactions of the channel protein with annular lipids to alter gating (196, 230). The maintenance of mechanosensitivity by the mammalian channels K2P and Piezo in isolated bilayers (203, 231) suggests that FFL is a general principle that applies to both prokaryotic and eukaryotic channels (218). FFL explains the requirement of Kir channels for an interaction with cholesterol (sect. 2.5.3) and the influence of fatty acids on K2P channel gating (sect. 2.5.5).
FIGURE 10.
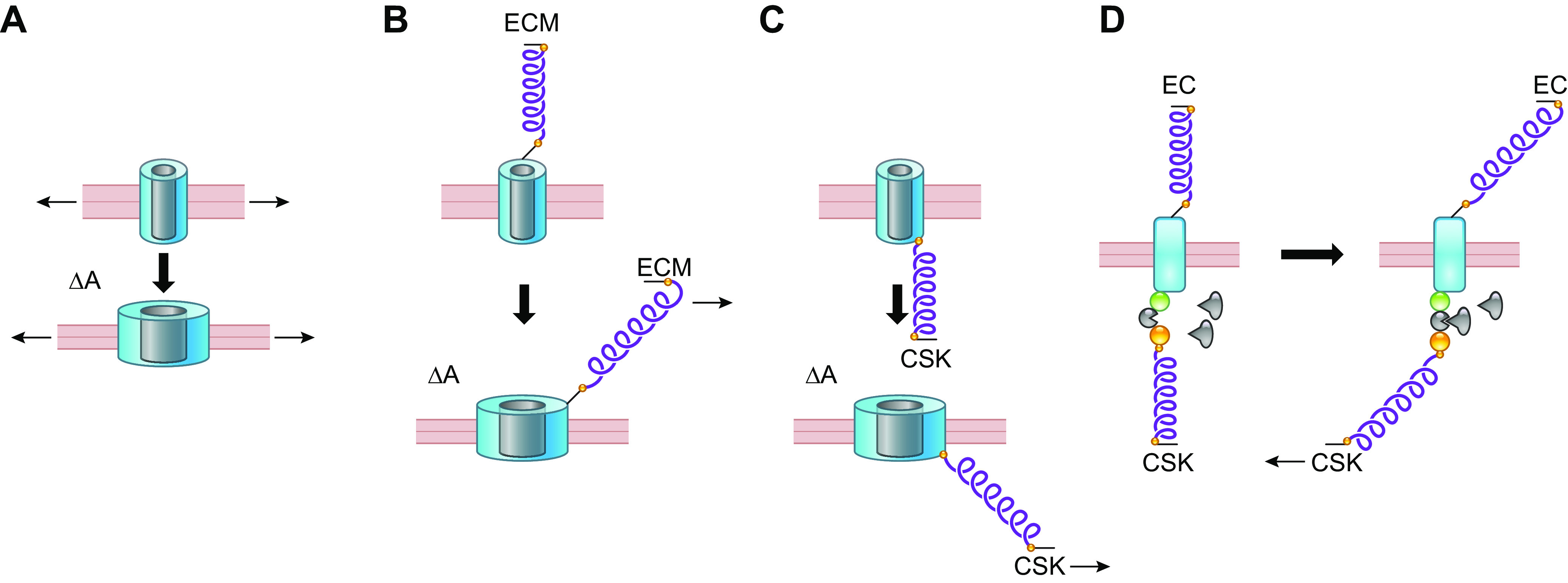
Possible mechanisms for gating a mechanosensitive ion channel. A: changes in bilayer tension alone. B: tension applied to channel through ECM tether. C: tension applied to channel through CSK tether. D: tension on 1 or more tethers exposes an intracellular binding domain. See glossary for abbreviations. Modified from Refs. 217–219, with permission from Developmental Cell, Pflügers Archiv, and Nature, respectively.
Despite evidence in support of FFL, many channels behave differently in native cells than they do after reconstitution into lipid bilayers. For example, Piezo1 channels lose their normal property of rapid inactivation when studied in bilayers (203), possibly because of disruption of their normal association with STOML3, an integral membrane scaffolding protein. Piezo1 sensitivity in sensory neurons to a standardized mechanical stimulus is reduced fivefold after genetic deletion of STOML3 (214). Although the cortical CSK provides stability to the membrane bilayer (218), proteins such as β-spectrin and ankyrin that link the plasma membrane to the cortical CSK, as well as actin, which links the cortical and internal CSK elements, have been demonstrated to be critical for force transmission to mechanosensitive channels in other systems (215, 232, 233). These and many other observations suggest that tethers between channel proteins and ECM proteins (FIGURE 10B), or between channels and CSK components (FIGURE 10C), may be critical to confer normal mechanosensitivity to an ion channel. Ideally, evidence for mechanosensitivity should be supported by studies in intact systems. For example, Caenorhabditis elegans touch receptors are known to be tethered to complexes of both CSK and ECM proteins (208, 219), as discussed in sect. 2.2.2. Tethering may serve as a force multiplier, acting as a lever through which mechanosensitive channels with poor sensitivity can respond to weak but physiologically relevant forces (234). This is sometimes referred to as the “force from filament” mechanism (235). Indeed, there are many precedents at the cellular and multicellular levels for the advantage of a tethered force transmission system, including the tip link connectors in cochlear hair cell stereocilia (236) and the lanceolate endings in skin hair cells, both of which amplify the displacement associated with a given force. Another mechanism by which mechanical forces could gate an ion channel is through CSK and/or ECM tethers that result in the exposure of cryptic intracellular domains upon force application, allowing intracellular domains of the channel to interact with cytoplasmic proteins such as kinases to alter channel activity by phosphorylating critical residues (FIGURE 10D). Although the latter mechanism has not yet been described for a mechanosensitive ion channel, its relevance to other proteins is mentioned in sect. 5.2.
2.1.5. Mechanoprotection of ion channels?
Morris has argued (189, 237) that, because of the susceptibility of ion channels and other transmembrane proteins to mechanical deformation of the lipid bilayer, almost any large protein can potentially be mechanosensitive and that “mechanoprotection” measures, such as the cortical CSK (201), may be needed to delineate and/or protect specific mechanotransduction mechanisms. An example of delineation is the tethering protein system that enables vestibular hair cell ion channels to respond exclusively to mechanical deflection along the tip link axis (238, 239). Examples of protection include the regulation of TREK-1 by the actin CSK (240) and the inhibitory effects that the cortical actin cross-linking protein FlnA (241) normally exerts on the activity of Piezo1 (242) and on the interactions of TPPP1/TRPP2 (243) in VSMCs, such that SM-specific FlnA deletion enhances the stretch sensitivity of both Piezo1 and TRPP1. TRPP1/2 have also been shown to regulate MLC20 phosphorylation and cellular stiffness (244), implying that the function of one class of ion channel can indirectly alter the mechanosensitivity of other classes of channels. The preservation of ion channel interactions with their native lipid microdomains, auxiliary subunits, membrane scaffolding proteins, and other components that may tonically inhibit channel activity is critical to proper interpretation of data regarding mechanosensitive VSMC ion channels. These mechanoprotection mechanisms are likely to be reinforced at the multicellular/tissue level by the ECM and cell-cell junction proteins.
2.1.6. Use of inhibitors to study mechanosensitive ion channels.
In native cell systems including VSMCs, assessing the role of mechanosensitive ion channels is a complicated issue because there may be multiple types of mechanosensitive channels and they are likely to be present at much lower densities than those studied in heterologous expression systems. In addition, ion flux through a mechanosensitive channel can potentially gate a nonmechanosensitive channel (245). Multiple studies of VSMC mechanosensitivity have based their conclusions on the use of inhibitors such as the trivalent lanthanide Gd3+ (184, 245–247), ruthenium red (248, 249), or the tarantula toxin GsMTx4 (250–252). Although some studies have found that Gd3+ blocks or attenuates the vascular myogenic response (253), Gd3+ blocks almost every calcium entry pathway and is a well-known inhibitor of voltage-gated sodium channels (254), voltage-gated calcium channels (255–258), and Kv channels (259). Ruthenium red is an ion channel pore blocker (260) but it also blocks ryanodine receptors (261) and inhibits mitochondrial Ca2+ uptake (262). GsMTx4 was originally screened as a selective inhibitor of cationic mechanosensitive channels (263, 264), and multiple studies have found that GsMTx4 inhibits the myogenic response to various degrees (167, 265). However, GsMTx4 is now known to block other classes of ion channels and nonchannel targets (266–271), and it may directly alter the properties of the lipid bilayer (272). Because L-type calcium channels and MLC20 phosphorylation are required for VSMC contraction and are presumably downstream from mechanosensitive elements, the use of inhibitors with off-target effects on these proteins may yield misleading conclusions. The molecular identification of specific ion channel families and screening of more specific inhibitors, as well as the use of RNA-knockdown approaches and genetically modified mice, have subsequently led to a better understanding of the mechanosensitive ion channels underlying pressure-induced arterial constriction and flow-mediated dilation.
Mechanosensitive ion channels are a likely explanation for both the rapid depolarization of VSMCs in response to elevated pressure and the rapid hyperpolarization of ECs in response to elevated shear stress. The ion channels that could potentially account for these processes are depicted in FIGURE 11. In the following sections we examine the evidence for each of these channels in vascular mechanotransduction, starting with nonselective cation channels (Piezo, ENaC, ASIC, TRPs, P2X), then chloride channels, voltage-gated cation channels (Cav1.2, Cav3, Nav, HCN), and K+ channels (Kv, KCa, Kir, KATP, K2P). We conclude the section with integrative summaries and tables to assess the most relevant ion channels involved in EC and VSMC mechanotransduction.
FIGURE 11.
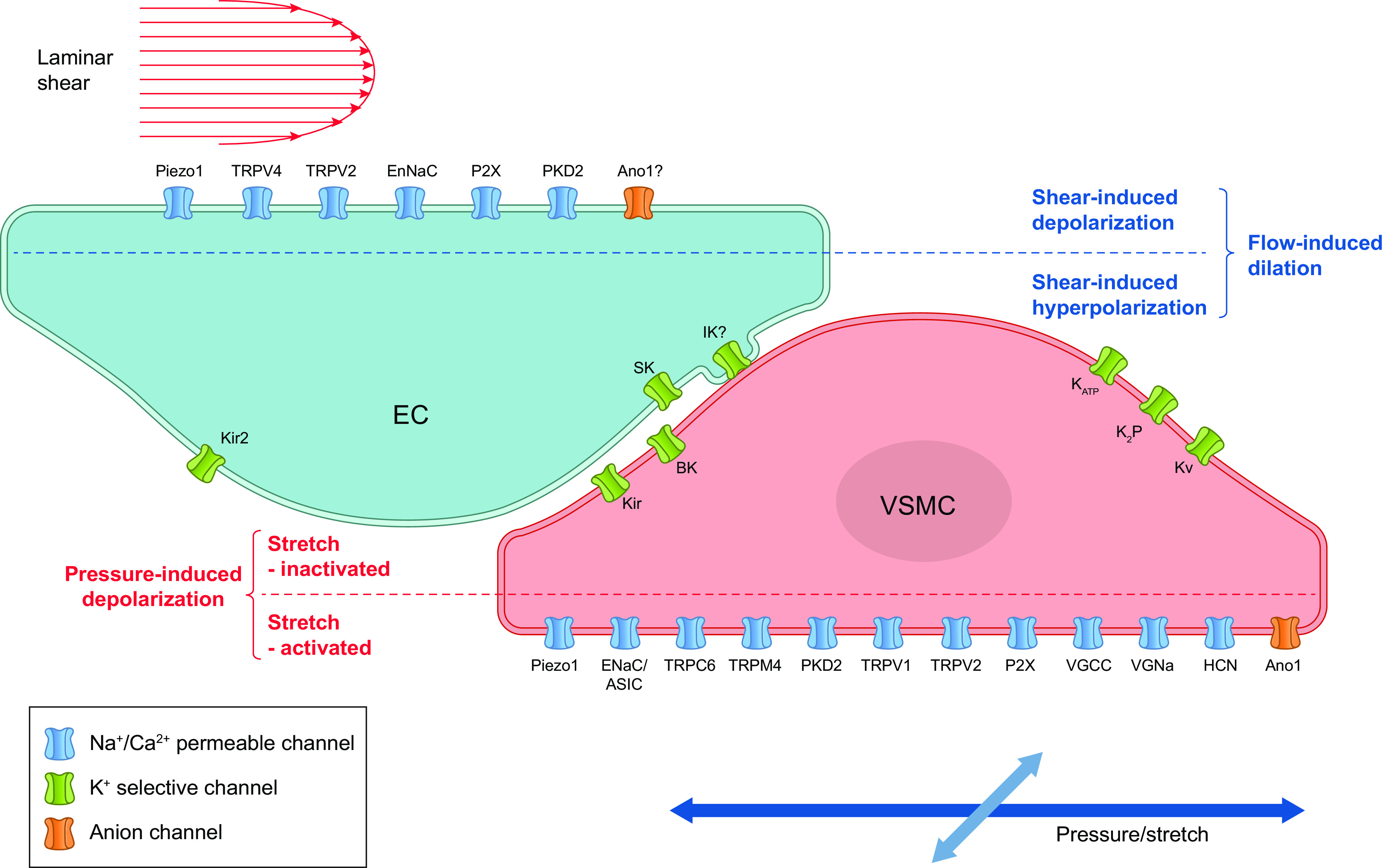
Ion channels in ECs and VSMCs that could potentially account for, or contribute to, flow-induced dilation and pressure-induced depolarization/constriction, respectively. Shear stress-induced activation of cation and Cl− channels in ECs can produce depolarization, but the EC response is normally dominated by the activation of K+ channels, leading to a net hyperpolarization. See glossary for abbreviations.
2.2. Nonselective Cation Channels
2.2.1. Piezo channels.
Piezo1 and Piezo2 are perhaps the best-characterized eukaryotic mechanosensitive channels. Piezo1 was first identified by the Patapoutian laboratory in 2010 (273). It is the largest known ion channel, with a molecular mass of ∼1 million daltons and an estimated 18–38 transmembrane domains. Functional Piezo1 channels are thought to form a trimeric complex of identical subunits (203, 274), in contrast to the more common four-subunit structures of TRP channels and voltage-gated Na+, K+, and Ca2+ channels. The Piezo1 structure is also unique in that it contains a large extracellular COOH-terminal domain, which forms an inverted dome that indents into the plasma membrane and may be critical for sensing membrane tension (275–277). Piezo1 and Piezo2 are nonselective cation channels that are slightly more permeable to Ca2+ than to monovalent cations (273); the relative permeability of Piezo1 to monovalent cations is PK > PCs > PNa > PLi (1.0:0.9:0.8:0.7). Piezo1 has a single-channel conductance of ∼115 pS in equimolar K+ (203) but a lower conductance of ∼30 pS if Ca2+ is present (278), as would occur under physiological conditions. Piezo2 has a lower single-channel conductance. Both Piezo1 and Piezo2 rapidly activate and inactivate in response to mechanical force (279), with Piezo2 exhibiting faster inactivation than Piezo1 (278). Selective pharmacology for Piezo channels is limited. Piezo channels are blocked by “classical” chemical antagonists of mechanosensitive channels, including Gd3+, ruthenium red (278), and GsMTx4 (280). Small-molecule screens identified Yoda1 as a Piezo1 activator (EC50 ∼20 μM) (281) and Dooku1 as a reversible antagonist of Yoda1 that lacks agonist activity (282). Specifically, Yoda1 prolongs activation but does not activate the channel in the absence of force. Yoda1 also has effects on cellular processes that are independent of Piezo1 (283). Although these agonists and antagonists have been widely used in vivo, the functional effects they may produce are not definitive evidence for Piezo1 involvement.
Piezo1 meets most or all of the six criteria listed for inherent mechanosensitivity in sect. 2.1.3. When recombinant Piezo1 channels are incorporated into droplet bilayers with asymmetric lipids, the channels are constitutively active. In contrast, channel activity is absent when incorporated into symmetric lipids, but interventions that increase membrane tension, including hyperosmotic solutions or solvent injection into the cis (intracellular) side (203), lead to channel activation (e.g., see Figure 2 in Ref. 203). In the absence of these stimuli, application of Yoda1 can also stimulate the channel (281). These same interventions do not activate KcsA channels (non-mechano-gated bacterial channels) incorporated into similar droplet bilayers. Heterologous expression of Piezo1 in N2A or HEK cells results in rapidly activating and inactivating inward currents upon application of patch pipette suction that exhibit sigmoidal (Boltzmann) activation as a function of pressure (e.g., see Figure 1 in Ref. 273). Finally, substitution of E2133A mutant Piezo1 channels, with altered pore properties (284), results in ∼50% decrease in single-channel conductance (203). Although these observations fulfill most of the criteria for true mechanosensitivity, the behavior of Piezo1 channels in a bilayer do not recapitulate all its characteristics in native cell systems, such as rapid inactivation. Additional proteins appear to be involved in modulating Piezo mechanosensitivity, including scaffolding proteins such as STOML3 (214), CSK proteins such as FlnA (242), and small molecules such as phosphoinositides (285).
Many lines of evidence support functional roles for Piezo channels in a number of physiological processes. Both gain-of-function and loss-of-function mutations in Piezo1 and Piezo2 have been linked to multiple human diseases (286), including dehydrated hereditary xerocytosis (287, 288), arthrogyropsis (289), and congenital lymphatic dysplasia (290, 291).
Piezo1 is highly expressed in a number of cells of the cardiovascular system, including VSMCs (242), ECs (292), and red blood cells (RBCs) (3, 195). Piezo2 channels appear to be expressed chiefly in peripheral neurons, where they are critical for the normal functioning of nearly all types of low-threshold (i.e., light touch) mechanoreceptors, including lanceolate endings and circumferential endings in hair follicles, as well as proprioception by Merkel cells and Meissner’s corpuscles (293–296). Combined genetic deletion of Piezo1 and Piezo2 from endothelium leads to developmental lethality in mice, indicating their requirement for the normal development of both blood (297) and lymphatic (298) vascular systems. The activation of Piezo1 by OS in developing lymphatic vessels could facilitate the Ca2+ entry required for nuclear factor of activated T cells (NFAT) activation and subsequent induction of Foxc2, a transcription factor critical for valve development (292, 299). Conditional deletion of Piezo1 and Piezo2 (but neither alone) from sensory ganglia containing the baroreceptor cell bodies results in loss or attenuation of the baroreceptor-mediated heart rate reduction in response to blood pressure elevation and increased variability in 24-h blood pressure regulation (8), indicating that both channels are critical for baroreflex control of blood pressure in mice. Piezo1 is required for normal osmoregulation by RBCs, and its deletion leads to RBC dehydration and membrane fragility (3). Piezo1 is also critical for mechanosensitive release of ATP from RBCs (3, 4, 300), which is an important regulator of tone in the microcirculation, as RBCs constantly undergo large-scale deformation while passing through small arterioles, capillaries, and venules (301–304).
2.2.1.1. PIEZO CHANNELS IN VSMC MECHANOTRANSDUCTION.
Little is known about the roles of Piezo channels in VSMCs. The application of suction to cell-attached patches of VSMCs isolated from caudal arteries activated a stretch-activated, nonselective cation current that was nearly abolished in SM-specific Piezo1-knockout mice (242), suggesting that Piezo1 is expressed in VSMCs and can be activated by membrane deformation. Activation of a Piezo1-like current as a function of patch pipette suction in VSMCs (see Figure 2 in Ref. 242) followed a sigmoidal relationship similar to that of heterologously expressed Piezo1 channels. Interestingly, VSMCs from renal arteries and aorta exhibited much lower levels of stretch-activated current. Somewhat surprisingly, both caudal arteries and rostral cerebellar arteries from SM-specific Piezo-knockout mice, which express relatively high levels of Piezo1 compared with other arteries in wild-type (WT) mice, showed no significant defects in the development of myogenic tone, pressure-dependent vasomotion, or response to vasoconstrictors (242). Watts and colleagues (305) reported that the contractile properties of multiple arteries from rat were insensitive to the Piezo1 channel modulators Yoda1 and Dooku1. However, another study found that Piezo1 deletion from VSMCs substantially attenuated the remodeling of caudal arteries when mice were made hypertensive (242), suggesting that Piezo1 plays a role in long-term, but not acute, adaptive responses to elevated pressure (sect. 7.2). It is reasonable to expect that Piezo1 channels may be important for other aspects of VSMC function, but those roles remain to be elucidated.
In summary, Piezo channels meet all the criteria for bona fide mechanosensitive ion channels, can function as true mammalian mechanosensors, and are critical for multiple aspects of cardiovascular function, but they do not appear to play a significant role in acute, pressure-induced depolarization and contraction of VSMCs.
2.2.1.2. PIEZO CHANNELS IN EC MECHANOTRANSDUCTION.
Piezo1 is widely expressed in ECs and is essential for vascular development (93, 282, 297, 306). Only two studies have provided evidence that Piezo2 is expressed in ECs (307, 308), including one in which Piezo2 knockdown suppressed tumor growth via a reduction in angiogenesis (308). It remains to be determined whether Piezo2 is widely expressed in ECs and whether it is regulated by shear stress.
Piezo1 is an important component of a number of shear stress-induced processes in ECs. Evidence first presented by the Beech and Patapoutian laboratories demonstrated that Piezo1 channels were activated by shear stress and pressure (93, 297). Flow induced increases in intracellular [Ca2+] in several types of ECs [human umbilical vein ECs (HUVECs), mouse embryonic ECs, and adult mouse mesenteric artery ECs] that were attenuated by Gd3+, GsMTx4, or EC-specific deletion of Piezo1 (Piezo1 ecKO) (93, 309). Patch-clamp recordings demonstrated that localized application of flow to whole ECs or to outside-out patches excised from ECs activated a nonselective cation current with a single-channel conductance of ∼25 pS (93, 309), consistent with the published characteristics of Piezo1 (273). These flow-evoked cation currents and Ca2+ increases were substantially impaired in Piezo1-deficient ECs (93, 309). Transfection of Piezo1 into HEK293 cells, which may not endogenously express Piezo1 (but see Refs. 310, 311 for contrary evidence), enabled them to respond to flow with Ca2+ influx (93, 297). A consistent observation in these protocols was a sustained activation of Piezo1 in native ECs as opposed to the activation and rapid inactivation of Piezo1 observed in heterologous expression systems. This finding may indicate the presence of as-yet-unidentified auxiliary proteins or interactions of Piezo1 with CSK/ECM elements in ECs, differing levels of sphingomyelinase activity, which modifies Piezo1 inactivation (312), or secondary activation of other channels/processes that depend on Piezo1-mediated Ca2+ influx, such as TRPV4 and/or ATP-P2Y2R signaling (300). The method of shear stress control in these electrophysiology studies was accomplished with a pressurized perfusion tube positioned near the patch-clamped EC, with shear stress subsequently estimated from the dimensions of the tube tip and/or particle tracking (297, 309). Under these conditions, the half-maximal shear stress for activation of Piezo1 expressed in HEK293 cells was 57 dyn/cm2 (297). In contrast, Piezo1-dependent Ca2+ increases in ECs were activated at shear stresses of 5–20 dyn/cm2 in a microfluidic chamber (93). The activation of Piezo1 channels in native ECs needs further investigation under controlled levels of shear stress to determine their threshold for activation, the extent to which they may be differentially activated by LS, OS, or PS, and the exclusion of other Piezo1-dependent Ca2+ influx mechanisms.
Shear stress activation of Piezo1 by itself would lead to EC depolarization, and this was indeed observed in sheets of mesenteric artery ECs, where flow induced ∼5-mV depolarization that was absent in Piezo1-deficient ECs (309). Piezo1-mediated depolarization appeared to be conducted through myoendothelial gap junctions (MEGJs) to the VSMC layer to activate VGCCs. In isolated mouse mesenteric arteries, luminal flow induced vasoconstriction (rather than vasodilation) and that response was abolished in Piezo1 ecKO mice (309). The authors proposed that flow-induced constriction through Piezo1 activation in exercise would facilitate shunting of blood flow away from the gastrointestinal (GI) tract (although activation of the sympathetic nervous system is known to accomplish this). The observation that flow induced the constriction of mesenteric arteries conflicts with observations by other groups of flow-induced dilation in arteries from the mouse mesentery (300, 313) and other regions (for reviews see Refs. 71, 144), so the differences await explanation. In most cases, the depolarizing effect of EC Piezo1 activation in response to shear stress is presumably masked or overwhelmed by the simultaneous activation of Kir2 channels, which leads to net EC hyperpolarization (313, 314) (see sect. 2.5), although it is possible that this does not occur in all arteries. A hyperpolarized endothelium would increase the driving force for Ca2+ entry into the EC cytosol, implying that the major effect of Piezo1 activation in this context may be to increase EC Ca2+ conductance. Both mechanisms acting in concert would promote increases in EC [Ca2+].
The studies just described predict that Piezo1 ecKO mice should have impaired flow-induced dilation. Surprisingly, this aspect of Piezo1 function has not been thoroughly investigated in intact arteries, although there are a number of documented downstream consequences to EC-specific Piezo1 deletion or Piezo1 haplodeficiency that are consistent with impaired NO production (93). These include Piezo1-dependent eNOS expression (315), eNOS phosphorylation, AKT and ERK1/2 phosphorylation (316), tyrosine phosphorylation of VEGFR2, ATP release, and P2Y2 receptor activation (see Ref. 317 for review). However, the number of studies directly assessing deficits in flow-induced responses of intact arteries from Piezo1 ecKO mice, and/or the NO-mediated component of such, are quite limited. Examples include NO production by pulmonary arteries that is substantially attenuated in Piezo1 ecKO mice (318); inhibition by GsMTx4 of a substantial component of flow-induced dilation in rat uterine arteries and near-maximal relaxation of the same vessels by Yoda1 (319); and Yoda1- and pressure/flow-induced EC Ca2+ increases in perfused retinal capillary networks that are independent of TRPV4 channel activation (260). Besides the example of flow-induced constriction mentioned above (309), only a single study has examined the effects of EC-specific Piezo1 deletion on flow-induced dilation of intact arteries (300). In that study, luminal flow-induced dilation of precontracted mouse mesenteric arteries from WT mice was inhibited by ∼60% in arteries from Piezo1 ecKO mice. One complexity to those findings was that a major component of the flow-induced dilation was mediated by Piezo1-dependent ATP release from ECs (which was attenuated in pannexin-deficient arteries lacking the critical mechanism for ATP export). Piezo1 ecKO mice develop hypertension, implying that similar Piezo1-dependent mechanisms are operative in other systemic arteries (300). Clearly, additional studies are needed to assess the degree of impairment of flow-induced dilation in arteries from multiple vascular beds in Piezo1 ecKO mice.
Other EC channels/receptors may depend on Piezo1 for their contributions to shear stress-induced responses, including TRPV4 activation (320) and P2Y2 receptor activation secondary to Piezo1-mediated ATP release (300). Channels known to interact and possibly interfere with Piezo1 activation include TRPV1 (285) (which may not be expressed in ECs) and PKD2 (321). The secondary activation of other Ca2+ conductances could complicate conclusions regarding the direct effects of shear stress on Piezo1-mediated Ca2+ entry. Thus, functional deficits associated with EC-specific deletion of Piezo may not be mediated by Piezo1 channels per se.
In addition to their potential role in flow-mediated dilation, Piezo1 channels are critical for the activation of multiple downstream signaling events in ECs in response to shear stress, including the regulation of PECAM expression (322) and the activation of KLF2 and YAP pathways (for review see Refs. 317, 323). Piezo1 is required for LS-induced EC alignment (297) and for the inflammatory EC phenotype characterized by p65 and VCAM-1 expressions and monocyte adhesion that is induced by OS (324). Piezo1 mediates pressure-induced increases in lung EC permeability through the disruption of adherens junctions (325). Yoda1 enhances the LS-induced ATP release via pannexin1 and regulates P2Y2R-Gq/11 signaling to increase the phosphorylation of AKT, eNOS, Src, PECAM1, and VEGFR2/Flk-1 in ECs (300, 320). However, Yoda-1 can induce Akt and ERK1/2 phosphorylation without the involvement of Piezo1 (283). Both LS and OS can activate Piezo1-P2Y2R-Gq/11 signaling, but only LS leads to α5 integrin-mediated focal adhesion kinase (FAK) phosphorylation, NO production, and atheroprotective responses, whereas NF-κB activation leads to inflammatory responses (324). Piezo1 ecKO mice and Gq/11-knockout mice exhibit reduced integrin activation, inflammation, and lesion formation in atheroprone areas and partially ligated carotid arteries of Ldl−/− mice (324), suggesting that Piezo1 activation has atherogenic effects. These studies indicate that Piezo 1 mediates both anti-inflammatory and inflammatory responses in ECs under different experimental conditions.
2.2.2. DEG/ENaC/ASIC channels.
DEG (degenerins), ENaC (epithelial sodium channel), and ASIC (acid-sensing ion channel) are a closely related family of Na+-permeable ion channels with varying degrees of mechanosensitivity. If pressure-induced depolarization is mediated by a cation channel, the most likely cation is Na+, because of its high out-to-in concentration difference.
2.2.2.1. DEG CHANNELS.
DEG channels in the nematode C. elegans represent the first identified molecular mechanotransduction complex. DEG proteins are encoded by 28 genes in C. elegans and by 9 homologous genes in humans (326). Gentle touch of C. elegans with an eyebrow hair evokes movement away from the stimulus site. The afferent arm of this response is sensed by six touch receptor neurons that express a Na+-permeable, amiloride-sensitive ion channel composed of the pore-forming subunits MEC-4 and MEC-10 (MEC for mechanosensory abnormal) (327). The normal function of the MEC-4/10 channel requires several closely associated intracellular and extracellular subunits, which bind cholesterol and other lipids or ECM proteins or interact with protofilament microtubules (208). Although DEG channels per se are not expressed in mammalian VSMCs or ECs, they are worth mentioning here because they fulfill key criteria for a true mechanosensitive ion channel (sect. 2.1.3) and form the core of a mechanotransduction protein complex involving a cation channel with ECM and CSK tethering elements that is analogous to several mammalian mechanosensitive ion channel complexes.
2.2.2.2. ENAC CHANNELS.
The mammalian equivalent of DEG channels is the epithelial Na+ channel, ENaC, which was first identified in 1986 (328) and named for its role in epithelial Na+ transport in the cortical collecting duct of the kidney. The canonical ENaC channel is composed of three different subunits: α, β, and γENaC (329, 330), encoded by three different genes (331). A δ-subunit subsequently identified in humans can form a functional channel alone or in combination with the other three subunits (333). Each ENaC subunit is composed of two transmembrane segments with an intermediate 50-kDa extracellular loop and intracellular NH2 and COOH termini (334); this loop is critical for proton sensing by ASIC channels and for Na+ sensing by ENaC channels (335). Based on their homology to DEG/ASIC channels and the crystal structure of ASIC1 (336), ENaC subunits are thought to assemble as trimers into a large ion channel complex in association with multiple CSK, ECM, and scaffolding proteins (331). αβγENaC forms a constitutively open channel with 5-pS conductance in physiological Na+ concentration ([Na+]), with an average open probability of 0.5 (337), 100-fold selectivity for Na+ over K+, and low Cl− permeability (338). Incorporation of αENaC into a lipid bilayer produces a channel with 40-pS conductance (in equimolar Na+) and voltage-independent gating (339). The β- and γ-subunits alone are capable of forming an amiloride-sensitive, Na+-permeable channel, although with reduced open probability (340), but the α-subunit of ENaC imparts higher constitutive activity (338). ENaC is regulated by changes in both extracellular and intracellular sodium; increases in extracellular Na+ inhibit ENaC activity (338), and this effect is abolished by treatment with extracellular proteases, suggesting there is a Na+-responsive site on the extracellular loop (341). ENaC channels are blocked by submicromolar concentrations of amiloride, or its more potent analog benzamil, through a well-characterized binding site that is conserved among subunits (331). ENaC activation by proteolytic cleavage may be important in a number of clinical conditions (335). ENaC gain-of-function mutations lead to hypertension (Liddle’s syndrome), and ENaC loss-of-function mutations lead to pseudohypoaldosteronism type 1 with hypotension (335).
Evidence for the mechanosensitivity of ENaC is extensive (342). In bilayer studies, αENaC can be activated by a slight pressure gradient to induce bilayer deformation (339) or inhibited in a voltage-dependent manner by Ca2+ (from either side). In the absence of Ca2+ the channel loses its mechanosensitivity, suggesting that mechanical activation may reflect a release from Ca2+ inhibition (343). Whether this same mechanism explains the mechanosensitivity of the heteromultimeric ENaC channel complex in native cells is not known (344). Heterologous ENaC channels expressed in mammalian cells or oocytes can be activated in a graded manner by patch pipette suction (345) or by a directed flow stream. In oocytes, osmotic swelling leads to αENaC activation (346), although the results of some oocyte studies may be complicated by concomitant ATP production (347). Numerous studies have documented that both native and heterologously expressed ENaC channels are activated by flow/shear stress (348–350), and this response is thought to be mediated by specific amino acid residues either in the second transmembrane domain of ENaC (351) or through residues in the palm and knuckle domain that act as a tether to the glycocalyx (352). In total, these studies show that most of the criteria required for a mechanosensitive ion channel are met by ENaC.
2.2.2.2.1. ENaC channels in VSMC mechanotransduction.
Drummond and colleagues have accumulated a substantial body of evidence in support of the idea that ENaC channels in VSMCs mediate pressure-induced myogenic constriction. VSMCs enzymatically dispersed from mouse renal interlobar arteries were found to express transcripts for all three ENaC subunits, with positive immunostaining in or near the plasma membrane for β and γ, but not α, ENaC (353). Amiloride and benzamil (at 5 µM and 1 µM, respectively) inhibited pressure-induced constrictions of isolated interlobar arteries by 75–80% (353). These inhibitory effects of amiloride on myogenic constriction are consistent with prior and subsequent findings by other laboratories (115, 354). Comparable inhibition of pressure-induced constriction of rat renal afferent arterioles by amiloride and benzamil were observed in vivo with the blood-perfused juxtamedullary nephron technique (355), although another group experienced in that technique found no effect of amiloride (3 µM) or benzamil (1 µM) on pressure-induced afferent arteriole constriction but rather potentiation of constriction by a higher concentration of benzamil (10 µM) (356) through its known effect on the Na+/Ca2+ exchanger (Ref. 357; for detailed discussion see Ref. 358). Although interpretation of such studies is complicated by concerns about other targets of amiloride and benzamil (331, 359–363), subsequent short-term organ culture studies in which mouse interlobar arteries were transfected with siRNA against γENaC or βENaC found ∼40% or ∼60% reduction, respectively, in the degree of pressure-induced constriction (364–366). Furthermore, overexpression of dominant-negative γENaC or βENaC constructs inhibited renal myogenic constriction by ∼60% or 80%, respectively (364–366). Drummond and colleagues also tested the consequences of βENaC deficiency using the βENaCm/m mouse model (367), in which βENaC transcript is reduced but not abolished in most tissues. βENaC-null mice die postnatally (368), whereas βENaCm/m mice are healthy until placed on a low-Na+ diet (367).
Electrophysiological evidence for mechanosensitive ENaC channels in VSMCs was partially supported by the following experiment. After VSMCs were isolated from renal arteries and allowed to adhere to a flexible, collagen-coated substrate, blunt probes were used to deform the substrate (see Figure 1 in Ref. 369) and induce graded stretch of individual patch-clamped cells from βENaC+/+ mice; this resulted in graded, inward currents carried largely by Na+. Such stretch-induced currents were not consistently repeatable for a given cell, possibly because of partial cell detachment from the substrate, but similar currents were nearly absent in VSMCs isolated from βENaCm/m mice (369).
Additional work by the Drummond laboratory using a perfused arteriole-glomerulus preparation showed that myogenic constrictions of afferent arterioles were nearly abrogated in βENaCm/m mice compared with their βENaC+/+ littermates; likewise, myogenic constrictions of isolated interlobar arteries were attenuated and myogenic regulation of renal blood flow and renal vascular resistance was blunted in βENaCm/m mice (370). A subsequent study found that βENaCm/m mice showed evidence of ischemic damage to the kidney (371). Curiously, arterial pressure was elevated by 15 mmHg in those mice (reduced arterial pressure would otherwise be expected from loss of ENaC-mediated Na+ and water reabsorption), which Drummond et al. speculated could be secondary to the ischemic kidney damage and loss of renal myogenic regulation (366). Some of these results concerning the roles of βENaC and γENaC in the myogenic response are supported by studies from another group (372–374).
Drummond and colleagues conclude that pressure-mediated regulation of an ENaC channel complex in VSMCs plays a critical role in myogenic regulation of renal arterial resistance and constitutes a protective mechanism to prevent overperfusion of the kidney (365, 375). They propose that this channel is composed of β and γ, but not α, ENaC subunits, based on the precedent that βγENaC forms a functional, Na+-permeable channel (340) and the finding that VSMCs in mouse renal arteries fail to show positive immunostaining for αENaC [although rat renal afferent arterioles stain positively for all three subunits (355)]. The absence of the α-subunit in VSMCs is predicted to result in low channel activity in the absence of a mechanical stimulus, such as vessel distension, as opposed to the constitutive activity conferred to the epithelial ENaC channel by the α-subunit (338). The third part of the ENaC trimer presumably is formed by an additional β- or γ-subunit [the δ-subunit is not expressed in rat or mouse (333)] or by an ASIC2 subunit (376), as discussed in sect. 2.2.2.3. It follows that a similar mechanism would operate in any artery that expresses βγENaC in VSMCs. Indeed, two studies from the same group provide evidence that ENaC mediates much of the myogenic responsiveness of cerebral arteries (377, 378). In support of this point, Drummond and coworkers (379) have recently shown that overexpression of βENaC in mouse cerebral arteries results in approximately twofold increase in peak myogenic tone.
Although compelling, these studies raise a number of unanswered questions. What would be the consequence of SM-specific βENaC knockout, e.g., using a βENaC floxed mouse, one in which little or no compensation in Na+ reabsorption and subsequent changes in blood volume/pressure or aldosterone would be expected? Is there a role for ENaC in pressure-induced myogenic regulation of other vascular beds? Does ENaC deletion abolish pressure-induced VSMC depolarization? How can near-complete abolition of pressure-induced constriction in the renal arterial vasculature in the studies by Drummond and colleagues be reconciled with the findings that GPCR and/or TRP channels mediate most/all of the myogenic response in other studies (23, 380, 381), including the kidney (see sects. 2.2.3 and 4.1.2)? In summary, although the evidence is highly supportive of a major role for ENaC in myogenic regulation of renal vascular resistance, it is not clear whether ENaC is an exclusive VSMC mechanosensor underlying pressure-induced constriction and/or if its role is specific to the renal (and possibly cerebral) vasculature.
2.2.2.2.2. ENaC channels in EC mechanotransduction.
ENaC is chiefly known as a regulator of epithelial Na+ transport (382). Increased expression of ENaC in the apical membrane of principal cells, under the control of aldosterone (338, 383), leads to increased salt and water reabsorption (as Cl− and H2O movement are coupled to Na+ flux) and consequently increased blood volume and arterial pressure (328). ENaC channels are expressed in ECs of arteries from multiple regions and species (384), although at lower levels than in epithelia (385); the endothelial ENaC channel is often referred to as “EnNaC.” EnNaC can be activated by mechanical forces in addition to the regulation of its channel density by mineralocorticoids and glucocorticoids. However, the evidence for flow- or shear-stress-induced activation of EnNaC is much less convincing than that for ENaC (discussed above). In the conventional whole cell patch-clamp recording mode, a benzamil-sensitive inward Na+ current from HUVECs can be activated by micropipette application of flow (at an undefined shear stress level) or by patch pipette pressurization (386), consistent with an EnNaC current. The possibility of a direct effect on the channel is complicated by shear stress-induced insertion of new ENaC channels into the plasma membrane (387). Whether enhanced EnNaC activity results in, or contributes to, the depolarization associated with shear stress activation of Ca2+-activated Cl− channels (sect. 2.3) is not known. Enhanced EnNaC activity is linked to the impairment of EC-derived NO production through at least two mechanisms (384, 388). EnNaC colocalizes with heme oxygenases 1 and 2 (HO-1/2) in caveolae at the cell surface. Inflammatory agents such as phorbol 12-myristate 13-acetate (TPA) and TNF-α activate HO-1/2 to degrade heme and produce carbon monoxide (CO), which enhances EnNaC activation through an unknown mechanism (386), resulting in inhibition of l-arginine transport and impaired NO production. Increased EnNaC activity leads to increases in local [Na+], which stabilizes (389, 390) and stiffens the cortical actin CSK (391), thus limiting the ability of eNOS to convert l-arginine to NO and impairing NO production (392). Part of this effect is due to impaired eNOS phosphorylation, because EnNaC inhibition in rat mesenteric arteries leads to higher NO production and increased eNOS phosphorylation at Ser1177 (393). Thus, mice with endothelium-specific αENaC knockout have a blunted aldosterone-induced rise in aortic pulse wave velocity compared with control mice, consistent with the contribution of EnNaC to vascular stiffening (394). The levels of shear stress required to activate EnNaC are not fully defined (but see Ref. 387), and it will be important to determine the threshold and saturating levels of shear stress and whether LS, PS, and/or OS preferentially activate EnNaC channels, because enhanced activity of this channel appears to contribute to EC dysfunction in multiple disease states (394–396).
2.2.2.3. ASIC CHANNELS.
ASIC channels are voltage-insensitive sodium channels that are activated by external protons and share substantial homology to DEG and ENaC channels. Like ENaC channels, ASIC channels form large trimeric ion channel complexes, in both homo- and heterotrimeric configurations, with intracellular NH2 and COOH termini and a large extracellular domain (331). To date, six ASIC isoforms have been identified. The crystal structure of ASIC1 predicts that the extracellular domain is critical for H+ sensing (336). ASIC currents inactivate more rapidly and have a lower Na+-to-K+ permeability ratio (3–13, depending on the isoform) than ENaC currents. Some ASIC isoforms, e.g., ASIC1a and ASIC1a/2b, have a low permeability to Ca2+, with a Na+-to-Ca2+ permeability ratio between 1.8 and 18.5 (397). ASICs are permeable to protons, but proton influx is negligible because the extracellular H+ concentration is orders of magnitude lower than the extracellular Na+ concentration (331). The single-channel conductances are 6 pS for ASIC2 and 4 pS for ASIC1 in 2 mM extracellular Ca2+ (estimated from Ref. 398). In general, ASIC channels are less sensitive than ENaC channels to amiloride, with IC50 values of 10–100 mM (331). In mammalian cells, ASIC channels interact with a number of other proteins, including STOML1, STOML3 and CSK and ECM proteins, as well as several PDZ domain proteins that may act as membrane scaffolds (331).
ASIC channels respond to both chemical and mechanical stimuli. ASIC1 channels are considered to be important chemoreceptors in the central nervous system (CNS). Asic1a−/− mice show impaired detection of CO and H+ by neurons in the amygdala (399). ASIC3 channels are implicated in the function of c-type mechanoreceptors, myelinated mechanoreceptors, and proprioceptors (400–403) and are considered to be the most important chemosensor in the peripheral nervous system (PNS), sensing parameters associated with tissue acidosis (400, 404–406). They are implicated in pressure-induced vasodilation, a reflex that delays the decrease in cutaneous blood flow produced by local application of low pressure to the skin, because this reflex is abolished in Asic3−/− mice (407). However, the supraphysiological nature of the mechanical stimuli used in some studies (see sect. 2.1.1) raises the possibility that ASICs may not be mechanosensitive per se but instead modulate the mechanosensitivity of other channels in native cell systems (408, 409). For these reasons, and for the failure to fulfill all the criteria listed above (sect. 2.1.3), such as exhibiting mechanosensitivity after reconstitution into bilayers, the bona fide mechanosensitivity of ASIC channels has been questioned (410, 411).
ASIC2 channels are of particular importance in the cardiovascular system. Although ASIC2 channels are expressed in several CNS regions and in mechanoreceptors of the somatosensory system, including lanceolate fibers (412), Pacinian corpuscles, and Meissner’s corpuscles (402, 413), they are also expressed in the aortic arch and nodose ganglia (NG). Asic2−/− mice show impaired mechanotransduction by aortic arch nerve endings (7): NG neurons isolated from Asic2−/− mice show attenuated depolarization in response to a puff of fluid from a nearby micropipette compared with neurons from WT mice, whereas NG neurons from mice with transgenic ASIC2 overexpression show enhanced depolarization to the same stimulus (7). The attenuated depolarization in NG neurons from Asic2−/− mice results in an impaired baroreceptor reflex (7), i.e., an attenuated pressor response to bilateral carotid occlusion (reduced baroreceptor gain), while the mice exhibit no chemosensory defects (414). However, Asic2−/− mice still show a residual level of baroreceptor sensitivity (7). Whether this remaining component is mediated by other ASIC channel isoforms [e.g., ASIC2a vs. ASIC2b channels (415)], or by other mechanosensitive channels such as Piezo1 [whose ablation also disrupts the baroreflex and more extensively than ASIC2 ablation (8)], has yet to be determined. Collectively, these findings suggest that ASIC channels may not be required for mechanosensitivity per se but modulate the mechanosensitivity of cells in which they are expressed, possibly via interaction with other channels such as Piezo or ENaC.
2.2.2.3.1. ASIC channels in VSMC mechanosensitivity.
ASIC2 channels are expressed in VSMCs of both middle cerebral and renal interlobar arteries (416, 417), and these arteries from Asic2−/− mice show impaired pressure-mediated constrictions ex vivo (416, 417). Furthermore, myogenic regulation of renal blood flow is blunted in Asic2−/− mice, which also show evidence of mild renal ischemia (417). An interesting finding from these studies is that there is an increase in arterial pressure and greater impairment of pressure-induced constriction in Asic2+/− mice than in Asic2−/− mice, which the authors believe is consistent with the existence of a heteromultimeric complex of βγENaC and ASIC2 channels in VSM (417); this idea might explain why deficiency of either βENaC or ASIC2 nearly abolishes myogenic constriction (370, 417) rather than only partial attenuation. Pressure-induced constriction has not been examined in Asic1−/− or Asic2−/− mice or in mice with deletion of multiple ASIC isoforms. An ENaC/ASIC heteromultimeric channel is conceptually possible, as verified by electrophysiology, fluorescence resonance energy transfer (FRET), and coimmunoprecipitation protocols in a forced expression system (376), but its existence in native cells such as VSMCs remains to be demonstrated. This idea might be tested further in SM-specific βENaC-knockout mice in WT and Asic2−/− backgrounds, providing those mice would be viable.
2.2.2.3.2. ASIC channels in EC mechanosensitivity.
No studies to date indicate that ASIC channels are expressed in ECs. A study examining the localization of ASIC1 channels in the cerebral vasculature by immunofluorescence found that ASIC1 colocalized with SM α-actin, but not eNOS, in four different cerebral arteries in both rat and human (418).
2.2.3. Transient receptor potential channels.
Since their initial identification in Drosophila (419), TRP channels have long been speculated to be involved in VSMC mechanotransduction. Several TRP family members exhibit characteristics consistent with the initial characterization of stretch-activated ion channels in isolated VSMCs (181–185).
The TRP superfamily of cation channels is organized into six subfamilies based on sequence homology. The subfamilies are canonical (TRPC), vanilloid (TRPV), melastatin (TRPM), ankyrin (TRPA), mucolipin (TRPML), and polycystin (TRPP). All TRP channels are expressed as six-transmembrane domain polypeptides, and functional ion channels are formed by the assembly of four of these subunits. Most mammalian genomes encode 28 independent TRP subunit genes, but Trpc2, important for the sensing of pheromones by rodents (420), is a nonfunctional pseudogene in humans (421). TRP channels are broadly expressed throughout the body, and all cells express multiple types. At the cellular level, the TRP channels act as fundamental sensors of a multitude of stimuli. TRP channel activators include many external and cytoplasmic ligands, pH, Ca2+, membrane lipids, temperature, osmolarity, reactive oxygen species (ROS), and mechanical stress. TRP channels are notoriously polymodal, as exemplified by TRPV1. TRPV1 acts as the receptor for capsaicin, a substance that is abundant in hot peppers and is also activated by warm temperatures greater than 42°C (186). Thus, peppers rich in capsaicin generate a sensation of heat when eaten.
Collectively, the TRP channels are characterized as “nonselective” cation channels, meaning that under physiological conditions they conduct currents composed of a mixture of cations. The exceptions are TRPV5 and TRPV6, which are highly selective for Ca2+ ions (relative Ca2+-to-Na+ permeability ratio, or PCa/PNa, = ∼100) (422), and TRPM4 and TRPM5, which are selective for monovalent cations and are not permeable to Ca2+ or other divalent ions (423–425). The composition of the mixed cationic currents conducted by specific TRP channels determines their cellular function. For example, TRPV1, TRPV3, TRPV4, and TRPA1 conduct currents with a large Ca2+ fraction compared with other TRP channels, and single-channel activity creates transient, high-amplitude Ca2+ signals within subcellular microdomains that can be detected by total internal reflection fluorescent (TIRF) microscopy or high-speed spinning disk confocal microscopy (426–434). These signals, known as “sparklets,” have been linked to the activation of Ca2+-activated signaling pathways in VSMCs and ECs. Other members of the TRP superfamily, such as the TRPCs, likely conduct currents with a higher Na+ fraction at negative resting membrane potentials (435). The Ca2+ signals resulting from TRPC channel activity are more nuanced compared with TRPV1, TRPV3, TRPV4, and TRPA1 channels and cannot be detected at the single-channel level by optical methods. The primary effect of TRPM4 and TRPM5 channel activity at negative resting membrane potentials is Na+ influx and membrane depolarization. Many of the patch-clamp electrophysiological studies investigating TRP channels have used symmetric cation gradients. Although this approach has been useful for describing the biophysical properties of the channels, it provides limited information about the composition of the currents conducted under native conditions. Comprehensive studies on the ionic composition of currents under physiological conditions would broaden our understanding of the physiologically relevant functions of TRP channels.
This section discusses the current literature concerning the TRP channel superfamily members that are present on (or in) VSMCs and have been implicated in the vascular myogenic response and shear activation in ECs. These studies are based on the concept that increases in intraluminal pressure or shear stress deform the plasma membrane, thereby activating mechanosensitive TRP channels directly or indirectly to produce cation influx that depolarizes the membrane. Membrane depolarization stimulates Ca2+ influx through voltage-dependent Ca2+ channels to cause vasoconstriction (170). It is also conceivable that Ca2+ influx through TRP channels stimulates vasoconstriction directly without the involvement of voltage-gated Ca2+ channels, but this has not been demonstrated experimentally.
2.2.3.1. TRPC6.
TRPC6 was among the first of the TRP channels to be successfully cloned and functionally expressed (436). The initial study describing the channel demonstrated that stimulation of Gq protein-coupled receptors (GqPCRs) induced Ca2+ influx and evoked nonselective cation currents in COS and HEK cells expressing cloned mouse TRPC6 channels. TRPC6 channels were found to be insensitive to Ca2+ store depletion. Hofmann and colleagues (437) subsequently showed that cloned human TRPC6 channels were directly activated by diacylglycerol (DAG), an intracellular second messenger generated by the activity of the enzyme phospholipase C (PLC) acting downstream of GqPCRs. This study also reported that the unitary single-channel amplitude of TRPC6 channels was ∼35 pS and the PCa/PNa was ∼5. However, the Ca2+ fraction of the mixed cation currents conducted by TRPC6 is thought to be lower than that of TRPV4 channels, which have a comparable PCa/PNa, possibly because of a lower permeability to K+ ions (435). TRPC6 subunits are capable of forming heteromultimeric channels with closely related TRPC3 and TRPC7 subunits (438, 439). Interestingly, the biophysical characteristics of TRPC heteromultimeric channels are distinct from those of the homomeric channels (439, 440). The high-resolution (3.8 Å) cryo-EM structure of the cytoplasmic domain of TRPC6 has been reported, providing important insight into intracellular regulation of channel activity (441).
2.2.3.1.1. TRPC6 in VSMC mechanotransduction.
TRPC6 is broadly expressed in rodents and humans, and its mRNA and/or protein have been detected in the heart, lung, kidney, muscle, adipose tissue, GI tract, several brain regions, and other tissues (442). TRPC6 mRNA has been detected in native VSMCs isolated from several vascular beds, including rat cerebral artery (381, 440). In addition, currents that are activated by DAG analogs with single-channel conductance consistent with TRPC6 have been recorded from VSMCs isolated from arteries of multiple vascular beds, including VSMCs from rabbit portal vein, ear artery, and mesenteric artery (443–445) and cultured aortic VSMCs (446). Thus, it appears likely that TRPC6 channels are present and functional on VSMCs of arteries in most, if not all, organs.
Several lines of evidence have implicated the involvement of TRPC6 channels in stretch-activated cation current activity. Brayden and coworkers (447) identified a cation current in VSMCs from rat cerebellar and basilar arteries that was elicited by hypotonic swelling and blocked by Gd3+ as well as the Cl− channel blockers DIDS, tamoxifen and IAA-94. A subsequent study from this group demonstrated that antisense oligonucleotides directed against TRPC6 diminished swelling-activated cation currents in rat cerebral artery VSMCs (381). Park et al. (246) reported that single-channel cation current activity could be activated by application of suction to stretch the plasma membrane in native VSMCs isolated from rabbit pulmonary arteries. These stretch-evoked currents had properties that are consistent with those of TRPC6 channels: unitary conductance of 30 pS, a reversal potential near 0 mV when symmetric cationic solutions were used, and inhibition by Gd3+. In addition, channel activity was stimulated by the DAG analog 1,2-dioctanoyl-sn-glycerol (DOG) and essentially abolished by inhibition of PLC activity (246). Collectively, these findings suggest that in native VSMCs stretch of the plasma membrane rapidly stimulates PLC activity and DAG generation to activate a cation channel with biophysical properties matching TRPC6. This work was extended by Gudermann and coworkers (23), who demonstrated that TRPC6 channels expressed in HEK cells could be activated by administration of OAG but were not directly activated by hypotonic swelling or by application of negative pressure to the plasma membrane. Instead, this report demonstrated that cation currents could be activated by hypotonic swelling of HEK cells coexpressing TRPC6 channels and angiotensin II type 1 receptors (AT1Rs) or other GqPCRs. Importantly, swelling-induced current activation was inhibited in patch-clamp experiments by blocking PLC activity or by adding the nonhydrolyzable GTP analog GDP-β-S to the intracellular solution. Contrasting findings were reported by Spassova et al. (252), who reported that stretch-induced activation of TRPC6 channel currents in HEK cells and membrane patches from CHO cells was independent of PLC activity, suggesting a model of direct channel activation. However, another contradictory study found that the magnitude of stretch-activated currents in COS cells expressing human TRPC6 did not differ from those recorded from transfected control cells (448). Furthermore, Nikolaev et al. (449) reported that application of negative pressure to the plasma membrane of HEK cells transfected with TRPC6 and the membrane patches from lipid bilayers containing reconstituted TRPC6 channels failed to evoke cation currents, indicating that TRPC6 channels lack direct mechanosensitivity. Currently, the weight of evidence favors a model of indirect activation of TRPC6 channels in response to stretch of the plasma membrane that is initiated by GqPCRs and includes the PLC-generated second messengers DAG and IP3. TRPC6 channel activation by DAG may work in conjunction with TRPM4 channel activation by IP3 to produce depolarization in response to pressure elevation (381, 450).
The effects of TRPC6 deficiency on myogenic tone are not entirely consistent. Welsh et al. (381) demonstrated that antisense oligonucleotides directed against TRPC6 attenuated pressure-induced constriction of rat cerebral arteries. In contrast, Schleifenbaum et al. (451) found that myogenic tone in mesenteric arteries from global TRPC6-knockout (Trpc6−/−) mice was not different from that of control mice. However, in a different study, cerebral arteries from Trpc6−/− mice exhibited enhanced (rather than attenuated) myogenic tone, and the mice were hypertensive (452). This phenotype may be related to the compensatory upregulation of DAG-sensitive TRPC3 channels in the Trpc6−/− mice, which could be reversed by TRPC3 silencing (452). Further studies using conditional TRPC6 knockout in VSMCs would be helpful to resolve these issues and fully clarify the channel’s role in pressure-induced vasoconstriction.
2.2.3.1.2. TRPC6 in EC mechanotransduction.
Several studies suggest that TRPC6 channels are expressed in ECs and are important for various aspects of EC function. Mehta and colleagues (453) reported that TRPC6 channels mediated the increased permeability of pulmonary endothelium in response to the activation of TLR4 receptors by endotoxin, and at least two additional studies have implicated TRPC6 channels in control of EC permeability (454, 455). TRPC6 channels also mediate the EC Ca2+ entry required to permit leukocyte transmigration, as demonstrated by Weber et al. (456) using shRNA knockdown strategies. Fleming and colleagues (457) found that the translocation of TRPC6 channels, and the possible Ca2+ influx through them, mediated the subsequent activation of EC KCa channels by 11,12-EET. These studies indicate that TRPC6 channels are important components of EC function, but there is no evidence that TRPC6 is activated directly or indirectly by shear stress in ECs.
2.2.3.2. TRPM4.
Two variations of TRPM4 have been described, a short form that was discovered first and is now known as TRPM4a (458), and a longer form that was initially designated as TRPM4b (424). TRPM4a is a splice variant of TRPM4b and lacks a 174-amino acid domain in the amino terminus (424). TRPM4b is now recognized as the form that is most commonly detected in tissue samples and is described hereafter as “TRPM4.” Patch-clamp analysis of cloned human TRPM4 expressed in HEK cells showed that the channel’s properties are unusual compared with other TRP channels. TRPM4 is activated by intracellular Ca2+, is highly selective for monovalent cations, and is nearly impermeant to Ca2+ ions (423, 424). The unitary conductance of the channel is ∼25 pS (424). Under physiological conditions at negative resting potentials, the mixed monovalent cation current conducted by TRPM4 is largely composed of an inward movement of Na+ ions that has a net depolarizing effect on the plasma membrane (424). The channel displays strong outward rectification in conventional whole cell patch-clamp studies using symmetric cation solutions, likely because of rapid deactivation at negative membrane potentials. The ubiquitous plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) is required for TRPM4 activity (459), and PIP2 depletion has been linked with rapid desensitization of channel activity under standard patch-clamp conditions (460). Protein kinase C (PKC) potentiates channel activation (380, 461, 462) by enhancing the channel’s sensitivity to Ca2+ and by stimulating the trafficking of channel protein to the plasma membrane (463, 464). High-resolution cryo-EM structures of TRPM4 channels have been described (465, 466).
Selective pharmacological activators of TRPM4 channels were not available as of the date of this review. TRPM4 channels are nonselectively inhibited by flufenamic acid (467) and glibenclamide (468), which have effects on other channels that limit their usefulness. The most widely used TRPM4 inhibitor is the tricyclic compound 9-phenanthrol, which inhibits TRPM4 with selectivity if used at an appropriate concentration range (∼10–100 µM) (469). Within this range, 9-phenanthrol does not inhibit closely related TRPM5 channels (469) and does not affect the activity of recombinant TRPC3 or TRPC6 channels expressed in HEK cells (470) or BK, Kv, KIR, or CaV1.2 channel activity in native VSMCs from rat cerebral pial arteries (470). However, 9-phenanthrol has been shown in other studies to activate intermediate-conductance Ca2+-activated K+ channels (471), inhibit the Ca2+-activated Cl− channel TMEM16A/ANO1 (472), and inhibit both delayed- and inward-rectifying K+ channels (473). The halogenated anthranilic amide compound 4-chloro-2-[[2-(2-chlorophenoxy)acetyl]amino]benzoic acid (CBA, also known as compound 5) was recently developed as a selective small-molecule gating inhibitor of human TRPM4, with an IC50 of ∼1.8 µM (474). At a concentration of 10 µM, CBA reportedly has no significant effects against TRPM5, TRPM7, TRPM8, TRPV1, TRPV3, and TRPV6 channels or other Ca2+ and K+ channels. However, CBA does not inhibit mouse TRPM4 and cannot be used to study the function of the channel in rodents (475).
TRPM4 channels are broadly expressed and are found in the GI system, prostate, kidney, testis, heart, lung, brain, skeletal muscle, urinary bladder, and other tissues (423, 424, 476, 477). TRPM4 channels are present on VSMCs from rat aorta (478, 479), as well as cerebral (380), pulmonary (478), and mesenteric (479) arteries. The expression of TRPM4 in cultured mouse aortic ECs has been reported (480), but TRPM4 channels were not detected on the endothelium of rat cerebral arteries (380). TRPM4 is also present in the sinoatrial node and Purkinje fibers of the heart (468). Interestingly, a gain-of-function mutation in TRPM4 is associated with progressive familial heart block type I, an autosomal-dominantly inherited disease characterized by conduction block at the atrioventricular node. A glutamic acid-to-lysine substitution at amino acid position 7 of TRPM4 was found to impair endocytosis of the channel in HEK293 cells, leading to elevated levels of channel protein at the plasma membrane and increased current density in patch-clamp electrophysiology experiments (481).
Several studies have investigated the mechanosensitivity of TRPM4. Purified TRPM4 channels incorporated into liposomes (482) and expressed in HEK cells (449, 450) were not activated when suction was applied to the plasma membrane, suggesting that the channel is not inherently sensitive to mechanical stimuli. However, inward TRPM4 cation current activity was increased in response to suction applied to the plasma membrane in native mouse and rat cerebral pial artery (450, 470, 483–486) and rat parenchymal arteriole VSMCs (487). These studies used the amphotericin B perforated patch-clamp configuration or other methods to minimize disruption of the intracellular environment on the premise that TRPM4 channels are regulated by endogenous intracellular Ca2+ signaling pathways. In agreement with this concept, stretch-induced activation of inward cation currents through TRPM4 in VSMCs isolated from rat cerebral pial arteries was shown to be indirectly activated by a force-sensitive signal transduction cascade initiated by GPCRs that includes PLCγ1 and Ca2+ release from inositol triphosphate receptors (IP3Rs) (450). Rapid trafficking of intracellular TRPM4 channels to the plasma membrane in response to increased activity of PKCδ also contributes to stretch-induced current activity in VSMCs from cerebral pial arteries (462–464). In cerebral pial artery VSMCs from rats and mice, stretch-induced activation of TRPM4 activity is inhibited by the AT1R blocker losartan or by downregulation of the AT1Rb subtype with AT1Rb-targeted morpholinos (450, 484). These findings are consistent with indirect regulation of TRPM4 channels by IP3-mediated sarcoplasmic (SR) Ca2+ release, downstream from a mechanosensitive GPCR such as AT1R (FIGURE 12; sect. 4.1.2). The involvement of mechanosensitive purinergic receptors has also been linked to activation of TRPM4 currents in VSMCs from rat cerebral parenchymal arterioles (487).
FIGURE 12.
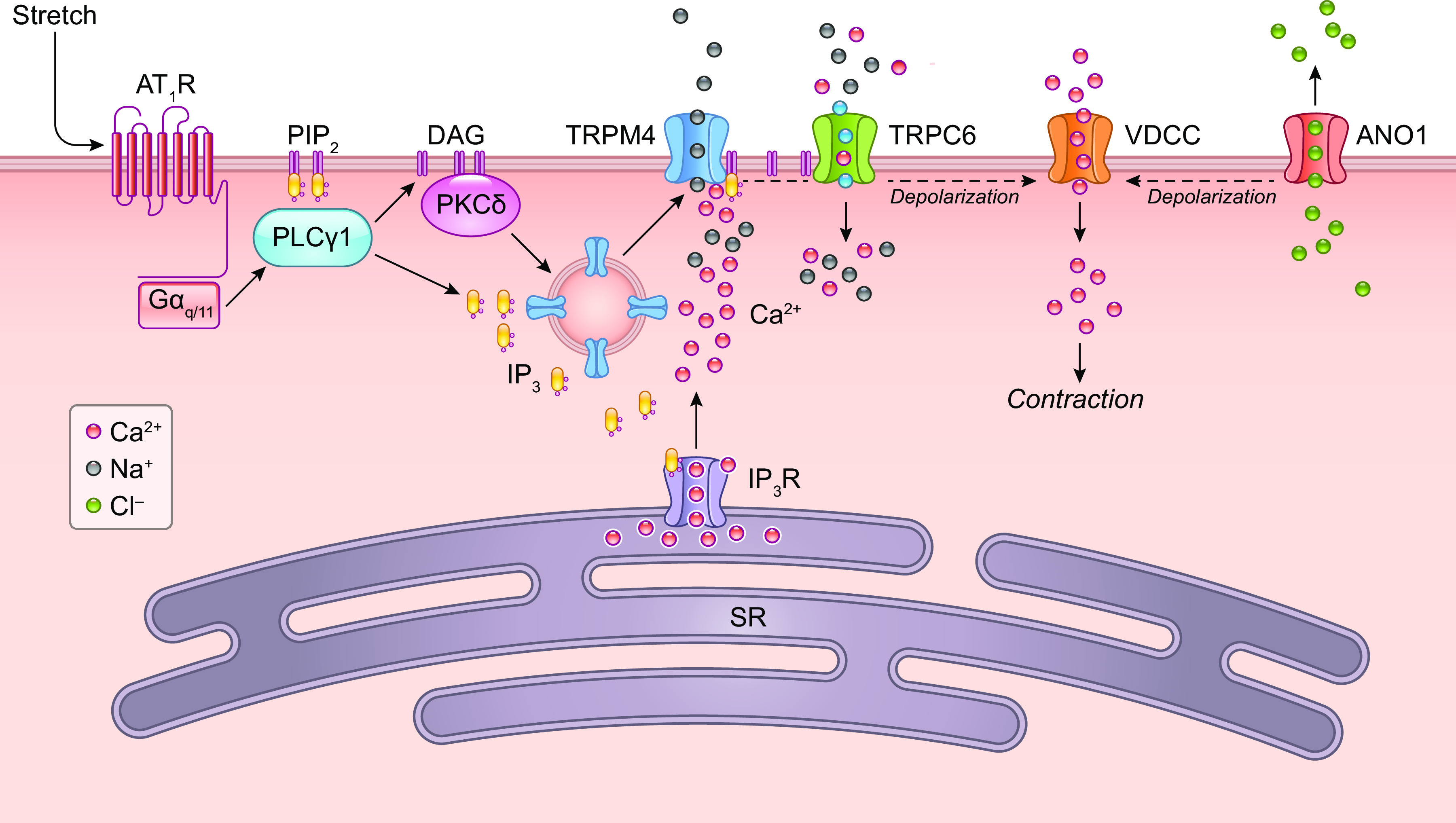
Postulated sequence of TRP, Ano1, K+, and voltage-dependent Ca2+ channel (VDCC) channel activation in VSMCs following a pressure step. The calcium source for Ano1 is not yet defined, but it may be activated by IP3R-mediated Ca2+ release along with TRPM4. SR, sarcoplasmic reticulum. See glossary for other abbreviations.
TRPM4 channel activity is necessary for myogenic constriction of cerebral pial arteries and parenchymal arterioles. Antisense-mediated downregulation of TRPM4 expression in intact rat cerebral pial arteries significantly diminished pressure-induced VSMC depolarization and myogenic tone development in ex vivo pressure myography experiments (380). These findings were later confirmed in experiments that used siRNA to knock down TRPM4 expression in rat cerebral pial arteries (483). Acute administration of the TRPM4 blocker 9-phenanthrol hyperpolarized the membrane potential of VSMCs in intact, pressurized rat cerebral pial arteries to nearly the K+ equilibrium potential and caused a corresponding loss of myogenic tone (470). Reading et al. administered TRPM4 antisense oligonucleotides to the cerebrospinal fluid of rats in vivo and demonstrated delivery of fluorescently tagged oligonucleotides to VSMCs in cerebral arteries and a corresponding decrease in TRPM4 expression (1846). An ex vivo study of pial arteries from rats treated with TRPM4 antisense demonstrated significantly diminished levels of myogenic tone. More importantly, autoregulation of cerebral blood flow in response to changes in mean arterial pressure was impaired in TRPM4 antisense-treated rats. Using an antisense knockdown approach in conjunction with the selective TRPM4 blocker 9-phenanthrol, Li et al. (487) demonstrated an important role for TRPM4 channels in the development of myogenic tone of cerebral parenchymal arterioles from rats. Collectively, these data show that TRPM4 channel activity is necessary for the development of myogenic tone of cerebral pial arteries and parenchymal arterioles and that activity of the channel contributes to the autoregulation of blood flow in the brain. Blockade of TRPM4 with 9-phenanthrol has also been shown to inhibit myogenic tone of isolated skeletal muscle arterioles (488).
Mathar et al. investigated the cardiovascular phenotype of global TRPM4-knockout (Trpm4−/−) mice (480). Interestingly, these mice displayed elevated systolic and diastolic blood pressures and tachycardia compared with wild-type (WT) control animals. Pressure-dependent changes in vascular resistance in perfused hindlimbs did not differ between Trpm4−/− and WT mice, suggesting that myogenic tone was independent of TRPM4 expression in the vasculature of skeletal muscles. However, myogenic tone was only estimated from hindlimb vascular resistance changes rather than pressure myograph analyses. Mean arterial pressure in Trpm4−/− mice was normalized to WT mouse values after intraperitoneal injection of the ganglionic blocker hexamethonium or the α1-adrenergic receptor blocker prazosin (480), suggesting that the hypertension seen in the Trpm4−/− mice was neurogenic in origin. Consistent with this possibility, this study also found that circulating levels of epinephrine and urinary excretion of the corresponding metabolites metanephrine and vanillylmandelic acid were significantly elevated in Trpm4−/− mice compared with control animals (480). These authors also reported that the production and release of catecholamines were increased in adrenal chromaffin cells from Trpm4−/− mice compared with control. Thus, the most apparent consequence of global TRPM4 knockout was the increased production and release of catecholamines, leading to elevated sympathetic tone and neurogenic hypertension. It is possible that the elevated catecholamine levels in Trpm4−/− mice could mask the impaired myogenic response/tone. Further in vivo investigation into the importance of TRPM4 channels on VSMCs in vascular function and the myogenic response in arteries from multiple regions will require the development of smooth muscle-specific, conditional TRPM4-knockout animals.
2.2.3.3. TRPP1 (PKD2).
The polycystins are a gene family (Pkd1, Pkd2, Pkd2L1, and Pkd2L2) that were first identified by their association with polycystic kidney disease. Pkd2, Pkd2L1, and Pkd2L2 encode TRP channels, but Pkd1 does not encode an ion channel. The past nomenclature used to describe the three cation channels that make up the TRPP subfamily is confusing. Pkd1, which was for a time erroneously assigned to the TRP superfamily as TRPP1, encodes a membrane protein (PKD1) that is not an ion channel. There is no convincing evidence that PKD1 forms functional complexes with other PKDs, and the role of the protein is unknown. The products of Pkd2 and Pkd2L1 were initially called TRPP2 and TRPP3, respectively, and, for obscure reasons, PKD2L2 was originally called TRPP5. The International Union of Basic and Clinical Pharmacology (IUPHAR) database now uses TRPP1 for the product of PKD2 and TRPP2 and TRPP3 for PKD2L1 and PKD2L2, respectively. We follow this convention here and specify gene names for clarity when necessary. The older literature (and some current studies) still uses the former terminology for the TRPP family.
TRPP1 (the product of PKD2) was originally identified as a six-transmembrane domain protein that shared regions of homology with PKD1 (489, 490). Less is known about the electrophysiological properties of TRPP channels compared with other members of the TRP superfamily, in part because of the unique subcellular localization of the channel. Several studies have reported that TRPP1 channels localize to the primary cilium, a singular nonmotile structure of uncertain function that is found on nearly every mammalian cell. Ion channels in the primary cilium have proven to be difficult (but not impossible) to study by patch-clamp electrophysiology. These difficulties are amplified by the lack of selective pharmacological tools for the study of TRPP1 channels. Although one report suggests that channel activity can be nonselectively blocked by Cd2+, Ni2+, and SKF96365, these agents have extensive off-target effects (491). An early study showed that the single-channel conductance of human TRPP1 channels transcribed in vitro and reconstituted into artificial lipid bilayers channels was 157 pS when conducting K+ ions (492). This study also demonstrated that the channel was voltage dependent, displaying a lower open probability at positive membrane potentials (492). Cloned mouse Pdk2 expressed in cultured mouse inner medullar collecting duct (mIMCD) cells and Madin–Darby canine kidney (MDCK) cells resulted in the localization of TRPP1 primarily to the endoplasmic reticulum (ER), with a smaller fraction on the plasma membrane (493). Single-channel currents recorded from MDCK cells with the cell-attached patch-clamp technique displayed a unitary conductance of 116 pS and a higher permeability for K+ versus Na+ ions (PNa/PK = 0.19) (493). Liu et al. (494) provided the most complete description of the biophysical properties of both native and heterologously expressed TRPP1 in mIMCD cells and the primary cilia of HEK cells. This study elegantly used specialized methods to patch clamp the primary cilium and found that, contrary to prior reports, expression of PKD1 is not required for the formation of functional TRPP1 cation channels. TRPP1 channels preferentially conducted K+ compared with Na+ (PK/PNa = 2.4) and had very low permeability to Ca2+ (PCa/PNa = 0.06). This study also found that intracellular Ca2+ potentiated channel activity. High-resolution structures of TRPP1 channels have been reported (495, 496), including the structure of a PKD1-TRPP1 heteromeric complex (497).
TRPP1 is broadly distributed, and its expression has been reported in kidney, testis, ovary, GI system, lung, brain, heart, liver, and skeletal muscle (489, 490). Several studies claim that TRPP1 is not abundantly expressed on the plasma membrane but is instead only present on the primary cilium (491, 494) and/or intracellular membranes (493). Other studies have demonstrated the expression of TRPP1 channels on the plasma membrane of VSMCs (498, 499). Jaggar and coworkers (500) reported that posttranslational modification of TRPP1 by small ubiquitin-like modifier 1 (SUMO1) targets the protein for recycling, whereas nonsumoylated TRPP1 protein is resident at the plasma membrane of VSMCs in mouse mesenteric arteries. It is not clear why different laboratories report differences in the subcellular localization of the channel, but it may relate to differences in posttranslational modification and/or trafficking mechanisms in different types of cells. The biophysical properties of TRPP1 channels in the primary cilium have been described in detail (494), but the characteristics of TRPP1 channels on the plasma membrane have not yet been extensively investigated.
2.2.3.3.1. TRPP1/2 in VSMC mechanotransduction.
A few studies have investigated the mechanosensitivity of polycystins localized to the plasma membranes of VSMCs. Honoré and coworkers (243) demonstrated that cation currents stimulated by the application of negative pressure to plasma membrane of VSMCs from mesenteric arteries of mice were diminished by SM-specific knockout of Pkd1. Jaggar and coworkers (499) reported that swelling-induced cation currents were inhibited by Gd3+ and diminished by downregulation of Pkd2 using shRNA. A study from the same group demonstrated that outwardly rectifying, Gd3+-sensitive cation currents could be activated by cell swelling in VSMCs from the hindlimb arteries of mice and that these currents were absent from cells from SM-specific Pkd2-knockout (Pkd2 smKO) mice (498). However, in the absence of selective pharmacology and a complete characterization of the currents produced by TRPP1 channels on the plasma membrane, these Gd3+-sensitive currents cannot be unequivocally assigned to TRPP1.
Conflicting evidence has been reported regarding the importance of Pkd family members in the regulation of myogenic vasoconstriction. Sharif-Naeini et al. (243) found that the myogenic tone of third-order mesenteric resistance arteries from SM-specific Pkd1-knockout (Pkd1 smKO) mice was attenuated by 40–50% compared with vessels from control mice, suggesting that PKD1 contributed to pressure-induced constriction in these vessels by an unknown mechanism. This study also reported that the siRNA-mediated knockdown of Pkd2 (TRPP1) restored stretch-activated cation currents and myogenic tone in mesenteric arteries from smKO mice (243). These data suggest that the primary effect of TRPP1 channels in mesenteric arteries from mice is the suppression of myogenic constriction by an unknown process. However, more recent data from this research group show that Pkd1 deletion or Pkd2 overexpression inhibits endogenous Piezo1 channels, suggesting that the stretch-activated currents in VSMCs originally attributed to Pkd1 may actually have been conducted by Piezo channels (321). Significant clarification is needed as to whether PKD1 actually forms an ion channel or influences the activity of Piezo1 or other ion channels.
A study by Narayanan et al. (499) reported that downregulation of Pkd2 (TRPP1) expression with shRNA decreased pressure-induced VSMC depolarization and myogenic tone in rat cerebral pial arteries. In agreement with these observations, Bulley et al. (498) showed that the pressure-induced vasoconstriction is blunted in hindlimb arteries isolated from Pkd2 smKO mice compared with controls, suggesting that expression of TRPP1 channels was necessary for the development of myogenic tone in this vascular bed. Interestingly, myogenic tone of third-order mesenteric arteries from Pkd2 smKO mice did not differ from controls, but vasoconstriction in response to the adrenergic receptor agonist phenylephrine was blunted in Pkd2 smKO mice. Collectively, these data suggest that TRPP1 channels on the plasma membranes of VSMCs respond to stimuli differently depending on the anatomical location of the vascular bed. Notably, the study from Bulley et al. (498) did not report the effects of SM-specific deletion of Pkd2 on the myogenic tone of cerebral arteries.
Global Pkd2-knockout mice are not viable (501). An analysis of mice that are hemizygous for Pkd2 expression (Pkd2+/−) showed that aortic rings display enhanced contractility in response to phenylephrine compared with wild-type controls that were linked to elevated levels of contractile proteins (502). This study also showed that pressurized (60 mmHg) fourth-order mesenteric arteries from Pkd2+/− mice had greater phenylephrine-induced vasoconstriction than controls but significantly lower increases in vessel wall Ca2+ (502). Elevated contractility of aortic VSMCs from Pkd2+/− mice has been linked to elevated levels of SM α-actin and enhanced phenylephrine-induced increases in RhoA activity and filamentous-to-globular actin ratios (503). However, it is unclear from these studies how TRPP1 expression influences the RhoA pathway at a mechanistic level. A comprehensive study from Bulley et al. (498) reported that inducible SM-specific knockout of TRPP1 rendered the mice hypotensive. Cardiac output, plasma angiotensin II (ANG II), aldosterone, and ANP, and plasma and urine electrolytes were similar between Pkd2 smKO mice and control animals, supporting the authors’ claims that the hypotensive phenotype resulted from diminished contractility of skeletal muscle and mesenteric arteries and decreased vascular resistance (498). This study also showed that Pkd2 smKO mice were resistant to ANG II-induced hypertension. The authors conclude that TRPP1 channels on VSMCs are a major factor in the regulation of total peripheral resistance.
2.2.3.3.2. TRPP1/TRPP2 in EC mechanotransduction.
The primary cilia of both epithelial and endothelial cells are subjected to luminal flow, albeit at different levels, and potentially serve as mechanosensors of shear stress (for review see Refs. 504, 505), but this concept has been disputed (506). Praetorius and Spring (507) first demonstrated that bending the primary cilia of MDCK cells, with either a micropipette or fluid flow over the cell surface, led to both Ca2+ entry and Ca2+ release from intracellular stores, with the local Ca2+ increase spreading as a wave through the cell and to neighboring cells via gap junctions. Deflection of the cilium resulted in a delayed hyperpolarization mediated by IK channels (508). Nauli and colleagues (509) found that Pkd1 and Pkd2 (TRPP1) form heteromeric complexes in the base of the primary cilium in mouse kidney epithelium that are necessary for shear (0.75 dyn/cm2)-induced Ca2+ influx. Ca2+ influx in response to fluid flow was dependent on the expression of PKD1 and was inhibited by blocking antibodies directed against an extracellular epitope of TRPP1 (509). High-resolution imaging of zebrafish embryos revealed that the deflection angle of the EC primary cilia correlated with the cell calcium level and that deletion of PKD2 impaired both the calcium response and vascular morphogenesis (510). Ca2+ influx and NO production induced by higher levels of LS (7.5 dyn/cm2) were abolished in embryonic aortic ECs isolated from Pkd1- and Tg737-deficient/mutant mice (95). Although primary cilia of adult animals may be confined to vascular segments under low flow, these studies suggest that PKD1 and PKD2 (TRPP1) form a mechanosensitive signaling complex in primary cilia that can be activated by shear stress to enhance Ca2+ influx (509). This conclusion has been challenged by recent studies (506). The permeability of Ca2+ through TRPP1 channels is reported to be very low (494), and Clapham and coworkers (506) recently used high-resolution imaging of genetically encoded Ca2+ indicator proteins targeted to the primary cilium to show that stimulation of the primary cilium by flow does not induce an increase in intracellular Ca2+. The authors concluded that the earlier observations may have resulted from technical issues related to insufficient time resolution of the Ca2+-imaging studies and/or motion- and light path-dependent artifacts (506). Thus, if TRPP1 in the primary cilium is mechanosensitive, it likely does not act as a Ca2+ signaling pathway. Work from Nikolaev et al. (449) showed that application of negative pressure failed to evoke single-channel currents in HEK cells expressing human TRPP2 (Pkd2L1), a channel that is closely related to TRPP1. A recent study by Jaggar and colleagues (511) shows that shear stress activates EC Ca2+ influx through TRPP1 (PKD2) channels to induce hyperpolarization through SK/IK channels and eNOS phosphorylation that together evoke vasodilation of mouse mesenteric arteries. EC-specific deletion of Pkd2 impairs these responses by ∼50% and leads to an ∼11% elevation in arterial pressure (511). Pkd2 deletion was not associated with changes in expression of other putative shear stress-activated ion channels, including Piezo1 and TRPV4 or the mechanosensitive protein GPR68 (511). There was no mention in that study of whether PKD2 channels were localized to primary cilia, nor were the possible contributions of PKD1 examined. Collectively, these findings support the idea that PKD2 channels are a component of the shear stress transduction mechanism in ECs, with secondary activation of SK and/or IK channels mediating EC hyperpolarization, but that this mechanism may not fully account for the vasodilation observed in response to shear stress.
2.2.3.4. TRPV1.
Fate-mapping transgenic mice were used to show that TRPV1 channels are present on VSMCs in small arteries and arterioles of skeletal muscles, the heart, and adipose tissue (512–514). Activation of these channels in isolated arterioles with capsaicin caused an increase in VSMC Ca2+ levels (513, 514) and constriction of small arteries in skeletal muscle and the heart. Blockade of TRPV1 channels reversibly dilated isolated skeletal muscle arteries with predeveloped myogenic tone but had no effect on similar-sized mesenteric arteries (488). Myogenic tone of skeletal muscle arterioles was blunted by the TRPV1 blocker BCTC but did not differ between global TRPV1 knockout and control. However, TRPV1 knockout lengthened the time required for recovery of myogenic tone in response to removal and restoration of extracellular Ca2+ (488). Combined blockade of TRPM4 and TRPV1 channels suggested that both are necessary for pressure-induced constriction of skeletal muscle arterioles and that TRPM4 channels are solely responsible for the development of myogenic tone in vessels from TRPV1-knockout mice (488). In agreement with these data, chronic administration of capsaicin to globally desensitize TRPV1 channels did not impair the myogenic constriction of isolated, cannulated skeletal muscle arterioles, suggesting that the channel is not required for this response (512, 515). Stretching of the plasma membrane activates TRPV1 indirectly by a pathway that requires GqPCRs, PLC, and PKC (488). These data indicate that TRPV1 and TRPM4 channels work together to generate myogenic tone in skeletal muscle arteries but that TRPV1 is not involved in this process in mesenteric arteries.
Studies from reporter mice (512, 513) strongly suggest that TRPV1 is not expressed in the endothelium.
2.2.3.5. TRPV2.
TRPV2 was initially characterized as a homolog of TRPV1 that was activated by heat but not by capsaicin or acidic pH (516). Detailed analysis showed that TRPV2 is activated at a higher temperature range (50–54°C) compared with TRPV1 (42–52°C) (516). TRPV2 has also been distinguished from its counterpart by the observation that current amplitude increases with successive activation. Ion substitution experiments demonstrated that the channel is somewhat more permeable to Ca2+ compared with Na+ ions (PCa/PNa ∼3) (516). The channel exhibits dual (inward and outward) rectification through 0 mV in whole cell patch-clamp experiments employing symmetric cationic solutions. High-resolution structures of TRPV2 channels have been reported (517, 518).
No selective pharmacological activators or inhibitors of TRPV2 have been reported as of the date of this review. 2-Aminoethoxydiphenylborane (2-APB) reliably stimulates channel activity, but this compound reportedly also activates TRPV1 and TRPV3 channels, blocks TRPC1, TRPC3, TRPC5, TRPC6, TRPV6, TRPM3, TRPM7, TRPM8, and TRPP2 channels, and influences other signaling pathways. Tranilast (N-[3′,4′-dimethoxycinnamoyl]-anthranilic acid) acts as a pore blocker of TRPV2 channels with an IC50 of ∼10 µM (519, 520), but evidence of significant nonspecificity has been reported (521, 522).
TRPV2 channels are expressed in a subset of sensory neurons, as well as the spleen, heart, kidney, lung, spinal cord, several brain regions, GI tract, and urinary bladder (516, 523). Cells from the immune system, including mast cells from humans and rodents, human and mouse B cells, and mouse monocytes and macrophages, express TRPV2 (524). A few studies have reported the expression of TRPV2 in native VSMCs (362, 525).
Several studies suggest that TRPV2 channels can respond to mechanical stretch. For example, Fois et al. (526) showed that mechanical strain induced by rapid stretch of rat primary alveolar type II cells grown on elastic membranes induced a rapid (within 30 ms) increase in intracellular Ca2+ that was reduced by knockdown of TRPV2 expression and by the nonselective TRPV channel blocker ruthenium red. Both HEK cells expressing recombinant TRPV2 channels and embryonic dorsal root ganglion (DRG) neurons grown on elastic membranes that were stretched to 102.8% of cell length for 15 s demonstrated global increases in intracellular Ca2+ that were blocked by ruthenium red and diminished by the expression of a dominant-negative TRPV2 construct (527). Stretch-induced activation of TRPV2 channels on VSMCs has also been demonstrated. The expression of TRPV2 mRNA and protein has been reported in VSMCs from mouse aorta as well as mesenteric and basilar arteries (525). Downregulation of TRPV2 expression in mouse aortic VSMCs diminished the amplitude of cation currents that were induced by hypotonic swelling (525). A study by McGahon et al. (362) showed that swelling of cells by administration of a hypotonic bathing solution induced Ca2+ influx and stimulated whole cell cation currents in native VSMCs obtained from rat retinal arteries and that these responses were diminished by tranilast. This study also showed that cation currents evoked by suction applied to the plasma membranes of VSMCs from rat retinal arteries were diminished by a blocking antibody targeting an extracellular domain of TRPV2 channels (362).
McGahon et al. (362) provided the only evidence for the involvement of TRPV2 channels in the regulation of myogenic tone. This study showed that the administration of tranilast to rat retinal arteries with established myogenic tone caused vasodilation and that preincubation with a TRPV2-blocking antibody prevented the development of myogenic tone. A study that examined the cardiovascular phenotype of global TRPV2-knockout mice reported diminished stroke volume, ejection fraction, cardiac output, and heart rate compared with control animals (528). The authors also found that TRPV2 knockout did not alter the contractile responses of aortic rings to phenylephrine or the relaxation to probenecid compared with controls. Furthermore, the authors reported no differences in cerebral blood flow, mean arterial pressure, and total peripheral resistance between groups. These authors concluded that TRPV2 channels do not have a substantial role in the regulation of vascular tone (528). This study suggests that any contribution of TRPV2 channels to the development of myogenic tone is limited to vascular segments that do not significantly contribute to total peripheral resistance, but specific studies of pressure-induced constriction in TRPV2-deficient mice need to be conducted.
2.2.3.6. TRPV4.
TRPV4 was originally identified as an osmolarity-sensitive cation channel that is activated by cell swelling induced by hypoosmotic bathing solution (529). TRPV4 channels exhibit dual rectification, high Ca2+ permeability (PCa/PNa = 6–10), and a unitary conductance of ∼60 pS at −60 mV and ∼90–100 pS at +60 mV. The channel is also activated by temperature in the range of 24–32°C (202). TRPV4 is activated by endogenously produced anandamide and epoxyeicosatrienoic acids (EETs), a family of signaling molecules that are generated from arachidonic acid by cytochrome P-450 epoxygenase enzymes (530). A high-resolution structure of TRPV4 has been reported (531).
Excellent pharmacological tools for investigating the function of TRPV4 channels are available. GSK1016790A is a selective small-molecular activator of the channel that is effective in the nanomolar range (EC50 = ∼2–20 nM) (532). Several potent and selective blockers of TRPV4 have been developed, including RN1734 (IC50 < 10 nM) (533), HC 067047 (IC50 = ∼20 nM) (534), and GSK2193874 (IC50 = 50 nM) (535).
2.2.3.6.1. TRPV4 in VSMC mechanotransduction.
TRPV4 channels are broadly expressed and are found in the kidney, liver, lung, spleen, skin, brain, testis, and adipose tissues (536, 537) TRPV4 channels are present on VSMCs in mesenteric and cerebral pial arteries and parenchymal arterioles isolated from rats and mice (430, 432, 538, 539) and in the preglomerular arterioles of neonatal pigs (540). Earley et al. (539) reported that activation of TRPV4 channels in VSMCs of rat cerebral pial arteries caused vasodilation. This study showed that Ca2+ influx through TRPV4 channels stimulated by 11,12-EET activated Ca2+-induced Ca2+ release (CICR) from SR stores in the form of Ca2+ sparks. Ca2+ sparks evoked by 11,12-EET activated BK channels to hyperpolarize the plasma membrane, decrease voltage-dependent Ca2+ entry, and cause vasodilation. A similar role for VSMC TRPV4 channels in regulating the contractile activity of mesenteric arteries in vitro and in vivo has been described (538). A study by Mercado et al. (432) demonstrated that a signaling pathway initiated by the binding of angiotensin II to the AT1R increases TRPV4 activity in isolated cerebral pial artery VSMCs. TRPV4 activity in this context activates the Ca2+ spark/BK channel pathway to oppose the vasoconstrictor effects of ANG II through the Ca2+ spark-dependent mechanism described above.
The “osmosensitive” property of the TRPV4 channel has been intensely investigated (536, 537) under the premise that the channel has an important mechanosensory property in many types of cells (for review, see Refs. 435, 541, 542). This property is not well characterized in VSMCs, but a study by Gebremedhin et al. (543) reported that single-channel cation currents that were sensitive to the selective TRPV4 blocker HC 067047 could be evoked by membrane stretch in cerebral pial artery VSMCs from fawn-hooded hypertensive rats. In addition, TRPV4 channels expressed in HEK cells are not activated by suction applied to the plasma membrane, suggesting an indirect mechanism of mechanical activation (449).
A study by Soni et al. (540) showed that selective blockade of TRPV4 channels with HC 067047 blunted pressure-induced VSMC depolarization and myogenic vasoconstriction of renal preglomerular arteries isolated from neonatal pigs. However, other studies reported that myogenic tone of cerebral and mesenteric arteries from adult global TRPV4-knockout mice did not differ from controls, suggesting that the channel is not involved in pressure sensing in these vascular beds (432, 538). Soni et al. (540) reported that intrarenal infusion of the selective TRPV4 blockers HC 067047 or RN 1734 inhibited autoregulation of blood flow within the kidney in vivo. However, other in vivo studies using adult rodents showed little involvement of TRPV4 channels in cardiovascular control. Basal mean arterial pressure (MAP) does not differ between wild-type and global TRPV4-knockout (Trpv4−/−) mice (544). The only reported cardiovascular phenotype of Trpv4−/− mice is a mildly enhanced sensitivity to hypertensive stimuli (538, 545). Furthermore, acute or chronic (8 days) administration of the selective TRPV4 blocking compounds GSK2193874 and GSK2263095 to rats did not alter MAP or heart rate, indicating that inhibition of the channel has no significant effect on basal cardiovascular regulation (535). Together, these in vivo studies suggest that TRPV4 channels have little effect on total peripheral vascular resistance in adult animals and that the impact of TRPV4 activity on myogenic tone may be developmentally regulated and/or restricted to certain vascular beds.
2.2.3.6.2. TRPV4 in EC mechanotransduction.
TRPV4 channels are widely expressed in ECs and participate in multiple aspects of EC signaling (433, 546, 547). Studies by Sonkusare et al. (433) firmly established the importance of TRPV4-mediated Ca2+ entry in ECs and demonstrated that TRPV4 channels can generate Ca2+ sparklets in ECs, similar to the elementary events mediated by Cav1.2 channels in VSMCs (548).
Multiple laboratories have provided evidence that TRPV4 channels are involved in the EC Ca2+ signaling mechanisms that drive the production of multiple EC-derived factors underlying flow-induced dilation. Initial work investigating the role of TRPV4 in flow-induced dilation utilized soluble TRPV4 antagonists. For example, Gd3+ blocked shear stress-induced Ca2+ mobilization from internal stores in cultured ECs (549), and the nonspecific TRPV4 blocker ruthenium red attenuated shear stress-induced dilation of rat carotid arteries (541) and mouse carotid arteries (550). At least three independent studies have demonstrated some component of flow-induced dilation that is blocked in arteries from Trpv4−/− mice, but the degree of blockade was quite variable among those studies. Mendoza et al. (94) found that flow-induced dilation of mesenteric arteries was attenuated by 50–70% in Trpv4−/− mice compared with WT control animals. Loot et al. (550) found essentially no impairment in flow-induced dilation of carotid arteries from Trpv4−/− mice but ∼40% inhibition of flow-induced dilation when TRPV4 deletion was combined with NO and COX inhibition. In contrast, Kohler and colleagues (551) found that shear stress-induced dilation was completely eliminated in carotid arteries of Trpv4−/− mice. The differences among these findings appear to depend on the various components of the shear stress response operating in the specific type of artery (i.e., NO or prostanoid production or hyperpolarization).
TRPV4 channels were initially thought to be mechanosensitive because of their activation by osmotic stimuli (529), but a recent study demonstrated convincingly that TRPV4 channels (and other TRP channels) are not directly activated by mechanical forces [Nikolaev et al. (449)]. TRPV4 current activation in ECs by hypoosmotic swelling is mediated by arachidonic acid (AA) and its metabolites (202, 552), and TRPV4 channels can be activated downstream from both PLC and phospholipase A2 (PLA2) signaling (431, 553) and by IP3 binding (554), IP3-mediated Ca2+ release (84), and membrane lipids (see Ref. 546 for review). An elegant study by Harraz et al. (1001) demonstrated that TRPV4 channels in brain capillary ECs are activated by PIP2 depletion (PIP2 is a known inhibitor of TRPV4) downstream from GPCR activation; because PIP2 is an activator of Kir channels, GPCR activation also led to the inactivation of Kir channels. ATP was found to dilate pulmonary arteries through the activation of EC TRPV4 channels and Ca2+-dependent regulation of NO production (555). Although the exact mechanism is not known, it is possible that ATP binding to P2Y receptors leads to TRPV4 activation through depletion of membrane PIP2. Alternatively, the mechanism may involve Piezo1 channels, as described in sect. 2.2.1.
TRPV4 channel activation in ECs is linked to Piezo1 activity. Ingber and colleagues (556) demonstrated that, in principle, TRPV4 channels in cultured ECs could be activated within 4 ms by twisting forces applied to β1 integrins, but the extent of whole cell TRPV4 activation and the implications for downstream signaling were not shown. More recent studies show that TRPV4 activation enhances and sustains the Ca2+ influx initiated by Piezo1 channels. In HUVECs, LS (4–12 dyn/cm2) evoked a sustained Ca2+ transient (320). Endogenous Piezo1 channels mediated only the initial transient component of this response, whereas the sustained component required the activation of TRPV4 channels, which have higher Ca2+ conductance and slower inactivation than Piezo1 channels. The transient and sustained components of the shear stress-induced response could be recapitulated in an HEK293T cell system. In an HEK293T cell line with endogenous Piezo1 deleted (310), transfection with Piezo1 exhibited transient Ca2+ responses, whereas cells expressing both Piezo1 and TRPV4 exhibited sustained Ca2+ responses to shear stress (320). Ca2+ entry through both channels may be a general principle common to many cell types because similar responses to shear stress were also characteristic of pancreatic acinar cells (557), where TRPV4 expression was needed to amplify and prolong the otherwise transient Ca2+ increases mediated by Piezo1 channels. TRPV4 activation in these studies appeared to be mediated by the Piezo1-dependent Ca2+ entry that activates PLA2 to form 5′,6′-EET (530), which in turn activates TRPV4. In pancreatic acinar cells, the sustained Ca2+ response was blocked both by a TRPV4 antagonist and by tissue-specific KO of TRPV4; it will be important to confirm this mechanism in ECs with EC-specific Trpv4-knockout mice.
In summary, TRPV4 activation by shear stress may involve a complicated interplay of Piezo1 channel activation, ATP release, and P2Y receptor activation that leads to both PIP2 depletion and the production of AA metabolites downstream from PLA2 activation. Together, these mechanisms drive the Ca2+ influx needed for NO and prostanoid productions that contribute to flow-mediated dilation. At some point, the sustained Ca2+ influx from the opening of the two channels in tandem can lead to a cascade of deleterious effects (557), including the breakdown of adherens junctions and hyperpermeability in ECs (320). The factors that determine this progression to pathological Ca2+ influx are not yet known.
There are additional effects of shear stress on ECs that are mediated by TRPV4 channels. LS enhances the translocation of TRPV4 to colocalize with KCa2.3 (SK3) in caveolae for LS-induced SK3 signaling, which is attenuated by blocking TRPV4. These results suggest that TRPV4 mediates the LS-induced activation of SK3 (558). Stochastic optical reconstruction microscopy (dSTORM) and diffraction-limited total internal reflection fluorescence microscopy (TIRFM) demonstrated that LS causes the rapid disruption of the clustering of TRPV4 with β-catenin and their interactions at basolateral junctions and membrane on the basal side, as well as the redistribution of TRPV4 to outside the clusters at the cell membrane on the basal side; such redistribution can be blocked by the inhibition of FAK and integrin α5β1 (559). These results demonstrate that the spatial distribution of TRPV4 may play a role in its shear regulation and interactions with other mechanosensing molecules.
Microvascular ECs subjected to 15% static uniaxial stretch exhibit activation of TRPV4 and a rapid Ca2+ influx (556, 560), along with PI3K activation, to lead to integrin β1 activation and relay to downstream signaling (e.g., AKT and ERK activations). These effects of stretch-induced signaling can be abolished by siTRPV4 and RuR (560). In microvascular ECs, 10% cyclic uniaxial stretch induces the perpendicular alignments of stress fibers and focal adhesion assembly (e.g., integrin, vinculin, FAK). Such stretch-induced alignments require TRPV2/4 and integrin β1 activation and are abolished by the inhibition of TRPV4 or PI3K (560). In vivo studies of overventilation-induced mouse lung injury demonstrate that TRPV4 is activated by overventilation and serves as a major determinant for the increased pulmonary permeability (561, 562). The overstretch-induced TRPV4 activation is coupled with serum glucocorticoid-regulated kinase-1 (SGK1) such that inhibition of TRPV4 or SKG1 abolishes pulmonary inflammation (e.g., IL-1β, MCP-1, and RANTES, etc.) to prevent injury (562). Collectively, these studies demonstrate that TRPV4 plays an important role in regulating vascular homeostasis in ECs.
2.2.3.7. TRPML1.
The members of the TRPML subfamily are nonselective cation channels that are primarily localized to endosomes and lysosomes (563). The biophysical properties of these intracellular ion channels have been studied with patch-clamp electrophysiology by expressing channels with point mutations that cause the channel to localize to the plasma membrane (564). Another approach used vacuolin-1 to swell endosomes and lysosomes to a size sufficiently large for patch-clamping (565). Vacuolin-1 does not increase the size of these intracellular organelles in primary cells, and the compound’s utility appears to be restricted to the study of cultured cells (566, 567). TRPML1 channel activity is potentiated by acidic pH and is dependent on phosphatidylinositol 3,5-bisphosphate [PtdIns(3,5)P2], a specific phosphoinositide species that is abundant in the membranes of late endosomes but rare within early endosomes and the plasma membrane (568). The channel has a unitary conductance on the order of 30–40 pS and is strongly inwardly rectifying (i.e., cations flow out from the vesicle lumen into the cytosol). Activation of TRPML1 channels on the membranes of late endosomes and lysosomes causes intracellular Ca2+ release because of the steep concentration gradient between the late endosomal lumen ([Ca2+] ≈ 2 mM) and the cytosol ([Ca2+] ≈ 100 nM). The high-resolution structure of TRPML1 has been reported (569). The endogenous regulation of TRPML1 activity has not been extensively characterized. One report suggested that the mammalian target of rapamycin (mTOR) directly phosphorylates and inactivates TRPML1 (570), whereas another study indicates that TRPML1 activity is stimulated by exogenous oxidants and endogenously generated ROS in a manner that is independent of mTOR. A surface-expressing mutant form of TRPML1 was not activated by stretch applied to the plasma membrane, suggesting that the channel lacks inherent sensitivity to mechanical stimuli (449).
Selective inhibitors of TRPML1 are not currently available. Two compounds, ML-SA1 (571, 572) and MK6-83 (573), have been identified as selective activators of all three TRPML channels.
TRPML1 is universally expressed (574), and its channel activity contributes to the maturation, fusion, and trafficking of endosomes and lysosomes (575). Point mutations in TRPML1 cause the rare but severe inherited neurodegenerative disease mucolipidosis type IV (574, 576). A few studies have investigated TRPML1 channel function in proliferative (cultured) VSMCs isolated from coronary arteries and provide evidence that the second messenger nicotinic acid adenine dinucleotide phosphate (NAADP) can stimulate Ca2+ release from lysosomes, a response that is diminished by downregulation of TRPML1 expression (577, 578), but this finding is controversial (579). TRPML1 has also been linked to apoptosis associated with activation of the Fas ligand receptor in proliferative VSMCs (577, 580). Expression of TRPML1, but not TRPML2 or TRPML3, is reported in native VSMCs isolated from mouse cerebral and mesenteric arteries (567).
Trpml1−/− mice recapitulate aspects of the pathology of mucolipidosis type IV in human patients. Eight-month-old Trpml1−/− mice display dense inclusion bodies in all cell types in brain, elevated plasma gastrin levels, vacuolization in parietal cells, retinal degeneration, drastically distended bladders, and neurological defects (581). Thakore et al. (567) showed that 5- to 6-mo-old Trpml1−/− mice exhibit spontaneous systolic hypertension, which the authors attributed to elevated vascular resistance associated with arterial VSMC hypercontractility. The responses of cerebral and mesenteric arteries from global TRPML1-knockout (Trpml1−/−) mice to increases in intraluminal pressure are highly exaggerated compared with wild type, suggesting the involvement of TRPML1 in the myogenic response. This study found that the arterial hypercontractility results from the inactivation of a critical Ca2+ signaling pathway that regulates VSMC contractility. In VSMCs, ryanodine receptors are functionally coupled with large-conductance Ca2+-activated K+ (BK) channels on the plasma membrane (582). Ca2+ released from the SR through clusters of ryanodine receptors generates transient large-amplitude Ca2+ signals (Ca2+ sparks) at the plasma membrane that activate clusters of BK channels to produce large transient outward K+ currents that hyperpolarize the membrane and relax VSMCs (582). This pathway provides essential negative feedback to limit the magnitude and duration of pressure-induced vasoconstriction (583). Thakore et al. showed that Ca2+ sparks and the corresponding K+ currents are absent in VSMCs from Trpml1−/− mice. This study also used superresolution microscopy to show that TRPML1 channels on late endosomes form nanoscale Ca2+ signaling complexes with ryanodine receptors on the SR. The authors put forth the concept that Ca2+ released from late endosomes through TRPML1 channels is required for the formation of Ca2+ sparks by stimulating Ca2+-induced Ca2+ release from ryanodine receptors. This scheme implies that the activity of TRPML1 channels is stimulated by increases in intraluminal pressure to activate this negative feedback pathway. However, it is not clear how intracellular TRPML1 channels are activated by increases in intraluminal pressure. One possibility is pressure-induced ROS generation. TRPML1 channels are sensitized or activated by exogenous oxidants and endogenously generated ROS (584). Prosser et al. (585) showed that physiological stretch of cardiomyocytes rapidly activates the generation of intracellular ROS by NADPH oxidase 2 via a process that is dependent on microtubules. A similar mechanism, if present in VSMCs, could account for the activation of TRPML1 channels in response to increases in intraluminal pressure.
The following TRP channels are present on VSMCs and/or previously implicated in mechanotransduction but do not appear to be directly involved in the development of myogenic tone.
2.2.3.8. TRPC1.
Store-operated calcium entry (SOCE) is a process common to most cells, whereby agonist-evoked depletion of calcium stores activates a calcium entry mechanism that refills the stores (586, 587). In noncontractile VSMCs, SOCE is primarily involved in proliferation and migration, but it may also activate Ca2+/CaM-dependent enzymes and control the expression of multiple ion channels (588). SOCE is not present in native contractile SMCs (589). For immune cells and many nonexcitable cells, the archetypal SOCE channel is the so-called CRAC channel (590), shown subsequently to be composed of Orai channels (591) and regulated by STIM proteins (592) after store depletion (593, 594). However, the characteristics of Orai/STIM channels often do not match those of SOCEs in VSMCs or ECs (595, 596). TRPC1 has been implicated as a major Ca2+ entry pathway in SOCE (28, 597, 598), and TRPC1 has been proposed to contribute to SOCE in VSMCs (595, 599), but other evidence suggests that TRPC1 channels only indirectly contribute to SOCE by their activation by Ca2+ influx through Orai/STIM channels (600).
TRPC1 is particularly relevant to this review because a major publication identified it as the stretch-activated cation channel in vertebrate cells (601). Maroto et al. (601) isolated a membrane protein fraction from frog oocytes that reconstituted high MscCa (a Ca2+-permeable cation channel) activity; this protein was identified as TRPC1. Heterologous expression of human TRPC1 in oocytes resulted in >100-fold increase in MscCa channel density in cell-attached patches, whereas injection of TRPC1 siRNA abolished endogenous MscCa activity (601). However, a subsequent study, which included the same authors, found that transiently expressing TRPC1 in COS or CHO cells did not significantly alter the amplitude of mechanosensitive currents in those cells (448), thus questioning the original findings. A more recent study also confirms that TRPC1, along with other TRP channels, is not directly gated by membrane stretch (449).
There are only limited functional studies of TRPC1 in intact arteries. Baudel et al. (595) provide some evidence that TRPC1 mediates SOCE in response to norepinephrine. Dietrich et al. (602) reported that pressure-induced vasoconstriction of cerebral arteries was not impaired in global TRPC1-knockout (Trpc1−/−) mice and that swelling-induced cation currents in cerebral pial artery VSMCs did not differ between Trpc1−/− mice and wild-type control mice. These data suggest that TRPC1 channels are not directly involved in smooth muscle mechanosensing. Notably, there is no evidence that TRPC1 forms homomeric ion channels in native VSMCs.
2.2.3.9. TRPC3.
TRPC3 channels are closely related to TRPC6 and share many of the same basic biophysical properties, including sensitivity to DAG and its analogs (437). TRPC3 is upregulated in Trpc6−/− mice (603) and may compensate for TRPC6 deficiency (452). Whether the reverse occurs in VSMCs is unknown, but acute knockdown of TRPC3 channel expression (over a time course in which TRPC6 was not likely to be upregulated) did not affect the development of myogenic tone of rat cerebral pial arteries (604), suggesting that TRPC3 is not directly involved in pressure-induced vasoconstriction under normal conditions.
2.2.3.10. VSMC TRP CHANNEL SUMMARY.
The studies discussed above provide evidence that multiple types of TRP channels can be involved in pressure-induced vasoconstriction. However, there is little convincing evidence that any of the TRP channels present on VSMCs meet the criteria outlined in sect. 2.1.3 for a true mechanosensitive ion channel. In contrast, there is strong evidence supporting a model of indirect activation of TRPC6, TRPM4, and possibly TRPV1 by signaling pathways that are initiated by other mechanosensitive elements, e.g., GPCRs (sect. 4.1.2). Furthermore, acute knockdown approaches provide evidence for the involvement of TRPC6 and TRPM4, perhaps working in series, for the initiation and maintenance of the myogenic response of arteries and arterioles in the brain (FIGURE 11). TRPM4 also appears to be important for myogenic tone of skeletal muscle arterioles. Unfortunately, compensatory mechanisms associated with global knockout of these channels have obscured a fuller understanding of their influence on vascular regulation in vivo. Further investigations using conditional knockout animals and next-generation selective pharmacology are needed. The work from Bulley et al. using conditional knockout animals makes a compelling case that VSMC TRPP1 channels are critically involved in the regulation of total peripheral resistance, but critical issues remain to be resolved. For example, TRPP1 was shown to be important for the development of myogenic tone in skeletal muscle and not mesenteric arteries, but the mechanisms of the underlying differences have not been examined. Nor have the consequences of TRPP1 deletion been tested in cerebral and renal circulations, where myogenic responses are strongest. It is not clear how stretch activates TRPP1 channels, or why some laboratories report that expression of the channel is restricted to the primary cilium. Further investigations into the respective signaling mechanisms should be insightful. The current data suggest that the involvement of TRPV2 and TRPV4 in the development of myogenic tone may be restricted to specific vascular beds that do not have a large impact on total peripheral resistance. TRPML1 channels appear to be fundamental for the initiation of a signaling pathway that provides negative feedback to balance myogenic vasoconstriction. Together, the current evidence suggests that multiple TRP channel family members may contribute to the myogenic response, with varying importance in different regional vascular beds. Future studies investigating the relative expression and impact of particular TRP channels throughout the vasculature with a combination of fate-mapping reporters and conditional knockouts will add considerably to our understanding.
2.2.3.11. EC TRP CHANNEL SUMMARY.
The most important TRP channels involved in flow-induced EC Ca2+ entry appear to be TRPV4, TRPV2, and PKD2. However, none of these channels is directly mechanosensitive; the only bona fide mechanosensitive channel demonstrated to date in ECs is Piezo1. Shear stress activation of Piezo1 appears to produce transient Ca2+ influx that initiates a cascade of events leading to activation of these TRP channels through a number of intracellular signaling pathways. Of these TRP channels, only TRPV4 and PKD2 have been shown to mediate a portion of flow-induced dilation in studies using knockout mice rather than soluble inhibitors with possible off-target effects. The relative contributions of Ca2+ entry through these TRP channels probably vary with the type of artery and with the mixture of NO, prostanoid, and hyperpolarization mechanisms that are activated in a given artery to initiate flow-induced dilation. However, assessing the contributions of these channels per se to flow-induced dilation is complicated by the simultaneous activation of other mechanisms, such as ATP release and subsequent P2Y receptor activation, that may play equal or more important roles in many arteries.
TRP channels contribute to other aspects of EC signaling besides flow-induced dilation. TRPA1 is expressed in cerebral vascular endothelium (19), and its activation by ROS leads to vasodilation (428). TRPA1 activation provides a neuroprotective role following ischemic stroke (426) and contributes to neurovascular coupling (1847). Endothelial TRPV4 channels are activated in overventilation-induced mouse lung injury and contribute substantially to the increased pulmonary permeability and inflammation in that model, which are abolished by TRPV4 inhibition. LS enhances the interaction between TRPV4 and KCa2.3 (SK3) in caveolae for LS-induced SK3 signaling. LS also causes disruption of the clustering of TRPV4 with β-catenin at basolateral junctions and basal membrane via an integrin α5β1- and FAK-dependent pathway. OS induces the expression of EC TRPC6 and TRPV1 mRNA in conjunction with the increases of GATA1/4 and TNF-α that are seen in human atherosclerotic lesions, suggesting a potential role of these TRPs in EC inflammation. Moreover, TRPV2/4-dependent pathways in ECs mediate the activation of integrin β1, AKT, and ERK, in addition to the perpendicular alignment of stress fibers and focal adhesion assembly. These results demonstrate that the expression level, spatial distribution, and heteromerization status of TRP channels may play a role in EC mechanosensing. Further studies using conditional knockout animals and next-generation selective pharmacology are needed to elucidate the roles of all TRP channel family members in their molecular and functional roles in EC mechanoregulation.
2.2.4. P2X channels.
The purine nucleotide ATP is released by many types of cells in response to mechanical stresses (605, 606). ATP is metabolized by ectonucleotidases into ADP, AMP, and adenosine, and all four compounds have effects on vascular tone that are mediated by two types of nucleotide-binding receptors, P1 and P2 (607). P1 receptors (4 subtypes) are GPCRs that signal through Gs or Gi proteins. P2 receptors are traditionally classified as ionotropic (P2X) or metabotropic (P2Y) receptors. P2Y receptors (8 subtypes) are GPCRs and are discussed in sect. 4.1; P2X receptors are ligand-gated ion channels.
2.2.4.1. P2X CHANNELS IN VSMC MECHANOTRANSDUCTION.
The seven subtypes of P2X receptors are nonselective cation channels, permeable to both Na+ and Ca2+; all are gated by ATP and related nucleotides that, upon binding, induce a conformational change in the inner pore of the channel (608). P2X channels are expressed in a wide variety of cell types, including VSMCs and ECs. VSMCs are exposed to ATP from the abluminal surface, whereas ECs are exposed to luminal ATP. Shear stress induces ATP release from ECs (609, 610), and possibly RBCs (611, 612), but not VSMCs (609). Burnstock and colleagues (613, 614) showed that corelease of ATP with NE from perivascular nerve terminals induces vasoconstriction of arteries via P2X1 receptors, independent of α-adrenoreceptors on VSMCs. Inscho et al. (615) reported a role for P2X1 receptors in the renal myogenic response. Using the juxtamedullary nephron technique with tubuloglomerular feedback-dependent influences minimized, blockade of P2X1 receptors inhibited pressure-mediated vasoconstriction of rat renal afferent arterioles. In addition, the autoregulatory responses to increased renal perfusion pressure were significantly blunted in P2x1−/− mice (615). Although P2X channels have been proposed to be intrinsically mechanosensitive, recent studies suggest that ATP release rather than channel gating is mechanosensitive, as discussed in sect. 2.2.4.2.
2.2.4.2. P2X CHANNELS IN EC MECHANOTRANSDUCTION.
The release and subsequent action of extracellular ATP is an important component of flow-induced dilation, which acts through both P2X and P2Y receptors on ECs (616). Initial studies of cultured ECs showed shear stress-dependent Ca2+ influx when exposed to LS in the presence of extracellular ATP (617–619); subsequent work revealed the involvement of P2X4 channels in this response. P2X channels do not appear to be direct mechanosensors but instead mediate responses to the mechanosensitive release of ATP (620). Shear stress-induced activation of calcium influx and inward current carried by Na+/Ca2+ in the presence of ATP can be replicated in oocytes or HEK293 cells expressing heterologous P2X4 channels (96, 620). The first application of 100 µM ATP elicits an inward current, and a second application produces a current that is attenuated by ∼50%, consistent with desensitization of P2X4 receptors. However, this desensitization does not occur under LS (5 dyn/cm2), suggesting that LS may stabilize the open state of the channel (620).
Mechanistic insights into the mechanosensitivity of ATP release come from studies of podocytes, but the underlying mechanisms in ECs may be similar. Podocytes respond to hypoosmotic swelling or ATP application with an inward current (measured under whole cell voltage clamp) and a transient intracellular Ca2+ increase (621). These responses are absent in the presence of the P2X receptor blocker trinitrophenyl-ATP and in podocytes from P2rx4−/− mice. Likewise, HEK293 cells expressing P2X4 channels are responsive to the same hypoosmotic stimulus only in the presence of ATP (621). In WT podocytes, the inward current evoked by cell swelling is blocked by apyrase (to degrade ATP) and impaired by stabilization of the actin cytoskeleton, cholesterol depletion, or blockade of SNARE proteins, suggesting that it is mediated by ATP release from prepackaged vesicles. These studies confirm that P2X channels are not intrinsically mechanosensitive and suggest that ATP release occurs via mechanosensitive reorganization of the CSK (621).
ATP release occurs through a number of mechanisms, including vesicular exocytosis, ABC transporters, undocked connexin hemichannels, and pannexin channels (for review see Ref. 622). In RBCs, ATP release in response to hypoxia or membrane deformation is mediated primarily through pannexin1 (611, 612, 623). Studies of mice with EC-specific knockout of Panx1 by Isakson and colleagues (624) provide evidence for a critical role for pannexin1 in ATP-P2YR-TRPV4 signaling in pulmonary artery endothelium.
P2RX4 activation in ECs also triggers a negative feedback mechanism to downregulate P2RX4 expression under LS by reducing the binding of transcription factor SP1 to P2RX4 promotor via the ROS pathways (625). These results may reflect the roles of P2RX4 in the short-term regulation of vascular tone and the long-term maintenance of vascular homeostasis. With a chemiluminescence imaging technique, LS has been demonstrated to induce localized ATP release and a consequent focal Ca2+ wave at the caveolin-1-coated membrane raft. This focal ATP/Ca2+ response is abolished by caveolin knockdown. These findings suggest a role for caveolae as mechanosensitive domains in coordinating cellular events (626), as well as the complicated interactions among multiple cell membrane compartments in mechanotransduction. P2RX4-knockout (P2rx4−/−) mice exhibit higher arterial pressure with impaired Ca2+ influx in ECs and NO production in response to step increases of LS (3, 8, and 15 dyn/cm2), which result in impaired ATP-dependent vasodilation without affecting acetylcholine-induced vasodilation (627). These findings demonstrate that P2RX4 is an important player in hemodynamic regulation of vascular homeostasis.
2.2.5. TMC-1/2 channels.
The detection of sound by cochlear hair cells has long been considered a model of mechanotransduction. Sound vibration deflects hair bundles, composed of F-actin-filled microvilli, which exert a force on extracellular filaments (tip links) that interconnect hair cell stereocilia (FIGURE 9, E AND F); force is detected by mechanosensitive ion channels in the tips of the stereocilia (628). Studies over the past decade show that the central components of the hair cell mechanotransduction system are two mechanosensitive ion channels, TMC-1 and TMC-2 (transmembrane channel-like proteins 1 and 2), which are members of a family of eight Tmc genes conserved from C. elegans to humans. TMC-1 is a nonselective cation channel with a single-channel conductance between 35 and 40 pS (629, 630) and a relatively high permeability to Ca2+ (631). TMC-2 has properties similar to TMC-1 (630). Mutations in the putative channel pore of TMC-1 alter its single-channel conductance and ion selectivity (629). TMC-1 and TMC-2 each contain 10 transmembrane domains with intercellular NH2 and COOH termini, but no high-resolution crystal structures have yet been resolved. Structural models predict that their closest homology is to the TMEM16 family of ion channels (239). Hair cell mechanotransduction is not significantly altered in Tmc-1−/− mice or in Tmc-2−/− mice but is abolished in Tmc-1−/−;Tmc-2−/− double-knockout mice, suggesting redundancy in the roles of the two channels. Loss of function in Tmc-1−/−;Tmc-2−/− mice can be rescued by reexpression of TMC-1 or TMC-2 in hair cells with viral vectors (629). Thus, TMC-1 fulfills essentially all the criteria described in sect. 2.1.3 for a true mechanosensitive ion channel, but its possible interactions with TMC-2 and other channel subunits or channel-associated proteins remain to be determined.
The TMC-1/2 channel complex requires a number of other proteins for normal functioning. These include a possible accessory channel protein (TMIE) (632), a cadherin (CDH23), a protocadherin (PCDH15), a tetraspanin (LHFPL5), and a Ca2+- and integrin-binding protein (CIB2) that may be critical for linking the TMC-1/2 channel to the ankyrin/spectrin CSK of the cell cortex (239). Deletion of any one of these components disrupts the detection of hair cell mechanosensitive current (632–634) and/or impairs hair cell mechanotransduction (635, 636). Collectively, evidence suggests that this group of proteins comprises a multiprotein complex containing an ion channel tethered to the ECM and to the CSK and that integrity of the entire complex is required for normal hair cell mechanotransduction. Many of these studies have been published over the past 3 years, and it is likely that other critical components of the complex are yet to be identified.
Although the hair cell is a highly specialized structure, the properties of TMC-1/2 suggest that these channels could potentially be involved in other aspects of mechanotransduction. Searches of published gene databases from RNA sequencing (RNA-seq) analyses of ECs and VSMCs in short-term culture indicate that TMC-1 is not highly expressed in VSMCs (637, 638), but one study supports TMC-1 expression in ECs (637–639). TMC-2 is not expressed in either VSMCs or ECs (639). Neither TMC-1 nor TMC-2 is expressed in visceral smooth muscle (640). No aspect of arterial function has been examined in Tmc-1−/− or Tmc-2−/− mice. The expression and potential function of these channels in EC mechanotransduction may merit future investigation.
2.2.6. TMEM63 channels.
In plants, members of the OSCA family of ion channels mediate Ca2+ influx in response to osmotic stimuli (641, 642). Recent studies by Patapoutian and colleagues (643) show that OSCA1.1 and OSCA1.2 channels are nonselective, high-threshold cation channels, conserved across eukaryotes, that can be activated both by hypoosmotic solutions and by patch pipette suction. OSCA1.2 channels exhibit mechanosensitivity when incorporated into bilayers, indicating that they can be directly gated by changes in bilayer tension without the need for other proteins (643). The closest mammalian homologs to OSCA channels are the TMEM63 family of proteins. Several members, including TMEM63A and TMEM63B, appear to be functional ion channels. After expression of TMEM63A or TMEM63B in HEK cells deficient in Piezo1, either patch pipette suction or mechanical indentation evokes a nonselective, Gd3+-sensitive cation current with slow activation/inactivation kinetics (643). TMEM63 channels have a relatively high threshold for mechanosensitivity (643, 644). The other properties of these channels, e.g., ionic conductance and possible modulation by auxiliary subunits, remain to be determined.
TMEM63 proteins are expressed in many tissues, including the nervous system, heart, skeletal muscle, and stomach (643). Searches of published gene databases from RNA-seq analyses of ECs and VSMCs in short-term culture indicate that TMEM63a and TMEM63b, but not TMEM63c, are expressed both in VSMCs and ECs (637–639). Their potential roles in vascular mechanosensitivity remain unknown.
2.3. Chloride Channels
The intracellular Cl− concentration ([Cl−]in) varies greatly among different cell types, depending on the activity and expression of the various Cl− exchangers and cotransporters. Assuming a constant extracellular Cl− concentration ([Cl−]out), the value of the Cl− equilibrium potential () is directly determined by [Cl−]in, and the difference between and Vm at any point in time determines whether net Cl− movement will be inward or outward. Activation of a Cl− channel can thus produce depolarization or hyperpolarization.
In VSM, is estimated to be between −18 and −35 mV (645–647) because of active Cl− accumulation through the Cl−/ exchanger and the Na+-K+-2Cl− cotransporter NKCC (647). Because the resting Vm in VSM ranges from −40 to −65 mV (648–650), pressure-induced activation of a Cl− channel would lead to net Cl− movement out of the cell, potentially contributing to depolarization. In ECs the Cl− equilibrium potential is regulated by multiple ion exchangers, particularly NKCC. Although has not been definitively determined in ECs, Vm is typically approximately −30 mV and Cl− channel activation appears to consistently produce depolarization (see below).
2.3.1. Cl− channels in VSM mechanotransduction.
Studies in the 1970s and 1980s suggested that Cl− efflux from VSMCs was associated with norepinephrine (NE)-induced arterial constriction (651–653). Multiple reports provided evidence for a NE-activated Cl− conductance in venous SMCs (654–656); subsequent patch-clamp studies identified a Ca2+-activated Cl− current in both vascular and nonvascular SM, as summarized by Large and Wang (648). In arterial myocytes, this current could be activated both by Ca2+ release from internal stores and by Ca2+ entry (648). Whole cell Cl− currents associated with Ca2+ release are often evident as STICs (spontaneous transient inward currents) (657). Although the majority of such studies point to a prominent role for a Ca2+-activated Cl− channel, VSMCs also express several other Cl− channels that could potentially be involved, including volume-regulated anion channels (658–660), CFTR (661–663), CIC family channels (664), bestrophin channels (665), and cGMP-regulated Cl− channels (666).
Hypoosmotic cell swelling activates Cl− currents in isolated VSMCs from many types of arteries (cerebral, renal and pulmonary) as well as in portal vein myocytes (667–669). The evidence supporting a Ca2+-activated Cl− channel in these studies derives largely from the use of pharmacological inhibitors and ion substitution protocols. Specifically, the reversal potential of the swelling-evoked Cl− current shifts with changes in , and the current is largely blocked by Cl− channel inhibitors and/or chelation of Ca2+ in bath or pipette solutions (648). Whether the degree of membrane deformation induced by the hypoosmotic stimulus in these studies corresponds to the physiological forces experienced by VSM in vivo is subject to the limitations discussed in sect. 2.1.2. Cation channels can also be simultaneously activated with Cl− channels by cell swelling, and high concentrations (≥100 μM) of “classical” Cl− channel inhibitors used in many VSM studies, including DIDS, niflumic acid, flufenamic acid, tamoxifen, glibenclamide, NPPB, 9-AC, and IAA-94, are known to have inhibitory effects on both monovalent cation and calcium channels, nor do they discriminate well between the different families of Cl− channels (670–672). For example, the Cl− channel inhibitors tamoxifen, DIDS, and IAA-94 all blocked swelling-activated current- and pressure-induced constriction of cerebral arteries, but subsequent ionic substitution protocols revealed a reversal potential for swelling-activated current consistent with a cation channel rather than a Cl− channel (447); however, the use of Ca2+-free intracellular and extracellular solutions in that study likely minimized contributions from Ca2+-activated Cl− channels.
The molecular identity of the Ca2+-activated Cl− channel in VSM was unknown until the discovery of the anoctamin family of transmembrane proteins in 2008 (673–675). TMEM16A (encoded by ANO1) has nine paralogs ranging from TMEM16B (ANO2) to TMEM16K (ANO10). TMEM16A and TMEM16B are the only bona fide ion channels (673–675). The other paralogs have diverse cellular functions (676) but do not appear to be ion channels activated by physiological Ca2+ concentrations (677); thus most studies of Ca2+ activated Cl− channels have focused on TMEM16A and TMEM16B. Ano1 encodes a protein of 960 amino acids that is 91% homologous with the human channel, which has 123 additional amino acid residues at its NH2 terminus (675); TMEM16A and B have little homology with other families of ion channels. From this point on, we refer to TMEM16A as ANO1. The anion permeability sequence for ANO1 is SCN− > - > I− > Br− > Cl− > F− (674), with a single-channel conductance for Cl− of ∼8 pS; these characteristics are consistent with those reported for native Ca2+-activated Cl− currents (675, 678). Channel gating depends on both Ca2+ and voltage, with ANO1 current exhibiting outward rectification at nanomolar intracellular Ca2+ levels that linearizes at millimolar Ca2+ concentrations. ANO1 channels are thought to be relatively insensitive to Ca2+(675, 679), but evidence from neurons, epithelial cells, and interstitial cells of Cajal (ICCs) suggests that the channels may be localized near IP3Rs and respond to IP3R-mediated Ca2+ release (for review see Ref. 680). However, definitive evidence that ANO1 is activated by IP3R-mediated Ca2+ release in VSMCs is lacking. The Jaggar laboratory reports that swelling-induced activation of ANO1 Cl− currents is unaffected by the depletion of SR Ca2+ stores with thapsigargin or block of voltage-dependent Ca2+ channels (VDCCs) (245). They propose that ANO1 channels are closely coupled with a stretch-activated Ca2+-permeable cation channel that is blocked by Gd3+ and SKF-9665, likely TRPC6, and that activation of the stretch-dependent companion channel provides the Ca2+ signal necessary for ANO1 activity (245). The Ca2+ source for ANO1 activation in VSMCs, and the channel’s overall importance in vascular homeostasis, might best be resolved with additional studies of SM-specific Ano1-knockout mice, which will be less reliant on soluble inhibitors of Ca2+-activated Cl− channels with possible off-target effects.
ANO1 is also regulated by membrane lipids and fatty acids in a complex way. ANO1 activity may be strongly influenced by the lipid environment of the plasma membrane (681–686), and relief from PIP2 inhibition [e.g., by agonist-mediated PIP2 depletion (see sect. 4.1.2) (1164)] may be a physiological mechanism for activation of ANO1 current in VSM. All these properties of ANO1 are consistent with the characteristics of the unidentified Ca2+-activated Cl− current in earlier studies on VSMCs (648).
ANO1 is widely expressed in VSMCs (687–692), and evidence from multiple laboratories suggests that it is critical for a large fraction of agonist-induced constriction in many arteries (693–696). In contrast, TMEM16B appears to be expressed only at low levels in the few vascular beds in which it has been examined (691, 692), and its functional role in VSM remains largely undefined. A role for ANO1 in vascular tone development is consistent with reports that the channel is overexpressed in systemic (697) and pulmonary (698) hypertension and may contribute to the chronically elevated vascular resistance in those animal models. After its identification as a Ca2+-activated Cl− channel, improved methods have been developed for inhibiting ANO1, including high-throughput screens to find selective pharmacological inhibitors (699, 700), siRNA-mediated knockdown (245, 687), and the engineering of mice with tissue-specific genetic deletion of Ano1 (695). Specific inhibitors of ANO1, such as T16A(inh)-A01, CaCC(inh)-A01, and MONNA, block or attenuate responses to contractile agonists in mouse portal vein, thoracic aorta, and carotid artery (699), rat cerebral arteries (694), and rat coronary arteries (701), but even those compounds may partially act through off-target effects, including the inhibition of VGCCs (702). More recently identified selective ANO1 inhibitors, such as benzbromarone and Ani9, which are efficient blockers of ANO1-mediated slow wave generation in interstitial cells of Cajal (ICCs) (700, 703, 704), have not yet been tested for their effects on the arterial myogenic response (701). Constitutive expression of siRNA against Ano1 in SM resulted in complex effects on agonist-mediated constriction: it attenuated the responses of mouse aorta and tail arteries to NE and arginine vasopressin, but the responsiveness of mesenteric and femoral arteries of the siRNA-treated animals showed no difference from controls and there was no effect on arterial pressure (705); however, it is to be noted that this approach only reduced ANO1 protein expression by at most 30% and it also reduced the expression of L-type Ca2+ channel protein.
The specific role of ANO1 in pressure-induced VSM depolarization and constriction has not been investigated to the same degree as its role in agonist-induced responses. An early study found that myogenic constriction of rat cerebral arteries was associated with Cl− efflux that was blocked by tamoxifen (706). Results from another study suggest that pressure-induced constrictions of renal arterioles are substantially attenuated by Cl− channel inhibitors (707). Two studies to date have tested the consequences of selective ANO1 inhibition on pressure-induced arterial constriction. Bulley et al. (245) demonstrated the inhibition of a swelling-activated Ca2+-activated Cl− current in VSMCs isolated from rat isolated cerebral arteries by an ANO1 antibody and the inhibition of a similar current in VSMCs from arteries maintained in organ culture with siRNA to Ano1. siRNA treatment blunted pressure-induced depolarization of denuded cerebral arteries and pressure-induced myogenic tone development by ∼50%. An important finding of that study was that the cation channel blockers Gd3+ and SKF96365 blocked the swelling-activated ANO1 current in cerebral VSMCs but did not block the currents activated by patch pipette suction from recombinant ANO1 channels in HEK cells, suggesting that ANO1 currents may not be activated directly by cell swelling but rather secondary to the activation of nonselective cation channels (245). A subsequent study by the same group found evidence that ANO1 channels colocalize with TRPC6 (but not TRPC3, TRPM4, or IP3R1) channels in the same macromolecular complex. Furthermore, the TRPC6 activator Hyp9 stimulated Cl− currents that were blocked by ANO1 inhibitors or by knockdown with siRNA (708). These findings suggest that local Ca2+ signals generated from TRPC6 activation are the trigger for ANO1 activation (rather than stretch directly) and that the activities of those two channels together amplify the depolarization underlying pressure-induced vasoconstriction. Limitations of these studies include the use of nonselective TRPC6 inhibitors and the use of two very different mechanical stimuli (cell swelling and patch pipette suction, both of which may have exaggerated the magnitude of the mechanostimulus in intact arteries) in the protocols that compared the respective contributions of ANO1 and TRPC6 (FIGURE 11). Nevertheless, this is an interesting idea that could be further explored by testing the effects of genetic deletion of ANO1 and TRPC6 channels, alone and in combination, on pressure-induced vasoconstriction.
Only a few studies to date have examined the consequences of genetic deletion of Ano1 on VSMC contractile function. Global deletion of Ano1 is embryonically lethal (674), thus precluding studies of the VSM response to agonists or pressure. Haplodeletion of Ano1 resulted in mice with reduced current density of Ca2+-activated Cl− channels in tail artery VSMCs and reduced contractile responses of the aorta to NE, vasopressin, and K+, but the mice had normal arterial pressure and enhanced contractile responses of tail and saphenous arteries to the same agonists (709). These paradoxical responses were associated with enhanced Ca2+ influx and increases in expressions of Piezo1 and plasma membrane Ca2+ ATPase but not L-type Ca2+ channels (assessed by tandem mass spectrometry). In another study, inducible deletion of Ano1 from VSMCs (Ano1 smKO) resulted in mice with chronically lower systemic arterial pressure 4 wk after beginning tamoxifen induction (695). Ca2+-activated Cl− currents were essentially absent in VSMCs from aorta and tail and retinal arteries from Ano1 smKO mice; constrictor responses to angiotensin II and to U46619 were blunted in aorta and retinal arteries but not in mesenteric arteries. Elevations in hindlimb perfusion pressure in response to boli of the agonist U46619 were blunted in Ano1 smKO mice (695). Collectively, those results suggest that ANO1 plays a predominant role in controlling agonist-mediated tone in many types of small peripheral arteries that would contribute to total vascular resistance. Unfortunately, neither of those studies (695, 709) assessed pressure-induced constriction per se in small arteries or arterioles, which account for the largest fraction of vascular resistance. Additionally, changes in the expression of ion channels and proteins controlling Ca2+ influx in studies of haplodeficient mice or with systemic siRNA knockdown (687, 705, 709) raise concerns about possible compensatory effects of Ano1 deletion. In summary, the effect of Ano1 deletion in VSM supports the idea that ANO1 mediates agonist-induced constriction in some, but not all, arteries. The role of ANO1 in pressure-induced constriction of arteries/arterioles remains to be more widely and systematically studied with genetic deletion strategies that selectively target the channel.
There are precedents for ANO1 also playing a role in controlling spontaneous rhythmicity in VSMCs and closely related systems. For example, a Cl− current with the characteristics of ANO1 is thought to be critical for the generation of action potentials (APs) underlying arterial vasomotion (710, 711). Likewise, lymphatic vessels from Ano1 smKO mice have a threefold reduction in the frequency of spontaneous action potentials and associated contractions and do not exhibit normal pressure-induced chronotropy; however, ANO1 deletion does not completely abolish spontaneous contractions (692), suggesting that one or more additional ion channels are involved in lymphatic pacemaking. In the gastrointestinal (GI) tract, ANO1 is not expressed in longitudinal or circular SMCs, but it is highly expressed in the cells that control GI pacemaking, interstitial cells of Cajal (ICCs), where it has been shown convincingly to be the ion channel that initiates slow waves (700, 712–714). Despite a large number of studies on the role of ANO1 in GI pacemaking, its role in stretch-induced slow-wave chronotropy, which is mediated by PGE2 production from ICCs (715), is not yet known.
2.3.2. Cl− channels in EC mechanotransduction.
ECs express both Ca2+-activated Cl− channels, which participate in various aspects of Ca2+ signaling, and volume-regulated Cl− (VRAC) channels, which function as a transport pathway for amino acids and organic osmolytes (597). VRAC channels are insensitive to shear stress per se, but shear stress modifies their response to osmotic stimuli (716).
Barakat and colleagues found that LS induced the activation of a Cl− current in cultured bovine aortic ECs (BAECs), as first demonstrated with membrane-potential sensitive dyes (717) and then by whole cell patch-clamp experiments. A reduction in extracellular Cl− enhances EC depolarization in response to LS, consistent with an important role for Cl− efflux in promoting depolarization. Cl− channel activation produced depolarization, which was preceded by a faster phase of hyperpolarization mediated by K+ channels (717) (see sect. 2.5). Shear stress-mediated depolarization occurred only in some cells, whereas the initial hyperpolarization phase occurred in all cells tested, suggesting that the Cl− channel is not expressed in all ECs (717). The threshold for Cl− channel activation was 0.3 dyn/cm2, and the current saturated at 3.5 dyn/cm2 (718). A detailed study demonstrated that LS (0.3–14 dyn/cm2) increased Cl− current over 2–6.5 min, and this became slowly desensitized over 10 min. Both LS (3.5 dyn/cm2) and PS (3.5 ± 3.5 dyn/cm2, 1 Hz) induce Cl− current in ECs (718). It is of interest that the regulation of Cl− current by OS at 0 ± 3.5 dyn/cm2 is dependent on oscillation frequency: Cl− current is significantly induced at 0.04 Hz, less at 0.2 Hz, and not at 1 Hz. By adjusting the perfusate viscosity, it was shown that the induction of Cl− current is dependent on shear stress but not shear rate (718). Activation of this Cl− channel has been shown to contribute to Akt phosphorylation (718), suggesting a functional role in the eventual phosphorylation of eNOS (104). Collectively, these characteristics of the channel are consistent with its contribution to an anti-inflammatory EC phenotype. The identity of the EC Cl− channel(s) mediating these responses is not known. Studies examining ANO1 expression in arteries have generally failed to show evidence for ANO1 expression in the EC layer (695, 719). Until the channel is identified molecularly, genetic deletion strategies cannot be used to rigorously test its role in ECs or endothelium-dependent aspects of vascular function.
2.4. Voltage-Gated Cation Channels
Essentially all major classes of voltage-gated ion channels exhibit voltage- and time-dependent conductances that are inherently mechanosensitive (270). The pore-forming α-subunits of NaV, Cav, and Kv share similar structures: α1 is the pore-forming subunit, containing 24 transmembrane segments separated into four homologous repeat domains joined by intracellular loops. Each domain contains six transmembrane segments (S1–S6), with segments S1–S4 forming a voltage-sensing domain and segments S5 and S6 forming the pore domain (720, 721). The conceptual basis for mechanosensitivity of these channels is that a large, tetrameric protein complex with moving voltage-sensor domains embedded in a lipid bilayer will have a sufficiently different lateral pressure profile at the protein-lipid interface that mechanical perturbation (e.g., thinning) of the bilayer will alter the channel conformation (199, 271, 722, 723). However, this line of reasoning implies that almost any protein in a bilayer could be mechanosensitive, thus undermining the first criterion listed in sect. 2.1.3 to assess ion channel mechanosensitivity. Mechanosensitivity has been most extensively studied for Kv channels (see discussion below) (188, 237, 724) and, to a lesser extent, for NaV and Cav channels. Membrane stretch may be a specific stimulus in this context because it modifies channel activation and/or inactivation differently for these three classes of channels (196, 269, 270).
This section applies only to VSMCs because ECs do not express voltage-gated channels (for review and exceptions see Ref. 597). Nav channels have been proposed to be important for conducted electrical responses along the endothelium (725), but this idea is controversial (726) and lacks convincing experimental support.
2.4.1. Voltage-gated sodium channels.
Human cells express nine closely related voltage-gated Na+ (NaV) channel subtypes, NaV1.1–1.9, along with a tenth family member, NaX, whose function is not as well understood. Both neurons and VSMCs express multiple NaV isoforms (727, 728). NaV1.5 is the dominant NaV isoform in cardiac pacemaking cells and lymphatic VSMCs (727).
Morris and Juranka (196) demonstrated that Nav1.5 α-subunits exhibit intrinsic mechanosensitivity when recombinantly expressed in oocytes. Mechanosensitivity was tested by recording macroscopic Nav1.5 currents during the application of suction (approximately −30 mmHg) to cell-attached patches through the recording pipette. Stretch reversibly accelerated both activation and inactivation of the channel by ∼1.4-fold. At holding potentials near the activation threshold for APs, peak Nav1 current was enhanced ∼1.5 times by stretch, an effect the authors speculated would modulate cardiac pacemaking, possibly in a feedforward fashion, to trigger arrythmias (196, 270, 729). Strege et al. (730, 731) identified a voltage-gated sodium current that was likely to be Nav1.5 (SCN5A) in native ICCs from the human intestinal tract. Voltage-activated sodium currents were enhanced 25–30% by perfusion with bath solution at a high flow rate (10 mL/min), presumably reflecting a shear stress-sensitive response (the level of SS was not quantified). These sodium currents were blocked by disruption of the actin cytoskeleton but not by disruption of microtubules or intermediate filaments (731). The authors subsequently identified an interaction between the COOH terminus of Nav1.5 and the PDZ domain of syntrophin γ2, a cortical actin CSK protein: disruption of their interaction by cell dialysis with decoy peptides to the last 10 aa of SCN5A or to a 98-aa sequence corresponding to the syntrophin γ2 PDZ domain eliminated the perfusion-induced enhancement of sodium current. The authors postulated that mechanosensitive modulation of Nav1.5 may account for the stretch-sensitive responses of ICCs (732).
Although multiple reports indicate that Nav channels are expressed in VSM (727, 733, 734), their roles and the specific isoforms expressed are largely unknown, and voltage-gated sodium channels (VGSCs) are ignored in most reviews of VSM contractile function (735–739). Although most arteries are not rhythmically contractile and their VSMCs do not spontaneously fire APs (649), many small arterioles exhibit vasomotion that is driven by VSMC APs (173, 740–742), and rhythmic contractions can be induced in some quiescent arteries under appropriate conditions (743–745), but whether the emergent AP activity in these cases involves VGSCs is not known. Nevertheless, rhythmic contractile activity and AP firing are not required for pressure-induced constriction, suggesting that VGSCs do not play an essential role in the myogenic response.
2.4.2. Voltage-gated calcium channels.
Early electrophysiological studies identified several distinct voltage-dependent Ca2+ conductances in various cell types. Molecular cloning subsequently identified three subfamilies of mammalian CaV channels: CaV1 containing four members (L-type currents), CaV2 containing three members (P/Q-, N-, and R-type currents), and CaV3 containing three members (T-type currents). CaV α1-subunits typically contain a long COOH-terminal tail with multiple serine and tyrosine phosphorylation sites (746), enabling posttranslational modification of channel properties by intracellular kinases. The α1-subunits of CaV1 and CaV2 associate with four auxiliary β- and α2δ-subunits that confer differing properties to the channels. CaV2 channels are expressed primarily in presynaptic nerve terminals (747). Cav1 and Cav3 channels are expressed in many excitable cells, including skeletal, cardiac, and smooth muscle. VSMCs express both L (Cav1.2)- and T (Cav3.1, Cav3.2)-type voltage-gated calcium channels (VGCCs).
The most definitive evidence for intrinsic mechanosensitivity of VGCCs has been obtained for N-type (Cav2) channels. Calabrese et al. (748) expressed the N-type α1B channel subunit in HEK cells, with or without auxiliary subunits, and found evidence for channel mechanosensitivity in both cell-attached and whole cell configurations. Slight pressurization of the cells (by <10 mmHg) did not alter whole cell capacitance but increased peak current by ∼1.5 times with an increase in cell size. Stretch increased both open- and closed-state inactivation and shifted inactivation to more negative potentials without affecting activation. Interestingly, recombinant T-type channels (Cav3.3, α1I) were insensitive to similar mechanical stimuli (748). Although these experiments establish a precedent for mechanosensitive modulation of Cav channels, only two reports suggest that N-type VGCCs might be expressed in VSMCs (749, 750); it is not clear whether they are widely expressed in arteries, nor have any studies tested their possible role in myogenic responsiveness. As mentioned above (sect. 2.3.2), VGCCs are not expressed in native ECs.
2.4.2.1. L-TYPE VGCCS.
Cav1.2 channels are generally expressed at a low density in VSMCs, but even small currents can substantially change Vm, because of the high input resistance of these cells (751). Cav1.2 channels are strongly voltage dependent, with an activation threshold approximately −60 mV. Po increases steeply around −55 mV and reaches a plateau at approximately −40 mV (752), which closely matches the values of measured Vm in VSMCs of arteries over the range of pressures that produce myogenic constriction (FIGURE 4). Cav1.2 plays a critical role in basal vascular tone development and myogenic responsiveness. Cav1.2 channels have a dihydropyridine binding sequence conserved among cardiac, neuronal, and smooth muscle isoforms; hence, myogenic tone is quite sensitive to dihydropyridines and other Cav1.2 blockers (with exceptions mentioned in the next section) (12). SM-specific deletion of Cav1.2 eliminates pressure-induced myogenic tone, attenuates arterial responses to vasoconstrictors, decreases vascular resistance, and lowers arterial pressure (172).
A small body of literature suggests that Cav1.2 channels have some degree of intrinsic mechanosensitivity. Whole cell Cav1.2 currents in isolated rabbit cerebral artery myocytes were increased after exposure to a 16% hypotonic solution or in response to elevation of the bath perfusion rate (at an unknown shear stress level) (753). Studies by the Farrugia laboratory confirmed the mechanosensitivity of Cav1.2 channels cloned from human jejunum. When recombinant Cav1.2 channels were expressed in HEK 293 cells along with the β2 channel subunit, whole cell currents were enhanced 23% by the application of shear stress (of unknown magnitude, achieved by rapid bath perfusion). A 10% increase in peak current was observed by the application of 5 mmHg pressure to inflate the cell through the whole cell patch pipette (754). These mechanical stimuli produced modest increases in peak current (755) and accelerated both activation and inactivation kinetics (754) but did not shift the activation threshold, ruling down the secondary involvement of kinases such as PKA. Subsequent studies by the same laboratory showed that mechanosensitivity to shear stress was retained after truncation of the Cav1.2 COOH terminus, also ruling out possible mechanosensitive regulation by PKA-mediated phosphorylation at its canonical serine reside. The preservation of mechanosensitivity in truncated Cav1.2 channels also eliminates critical roles for the autoinhibitory domain and the proline-rich, protein-interaction domain (754). Collectively, these results further support the conclusion that mechanosensitive regulation of Cav1.2 is kinase independent.
A study by Honoré and coworkers provided further insight into another possible mechanism of Cav1.2 mechanosensitivity (745). Ba2+ currents recorded in cell-attached patches from heterologously expressed Cav1.2 channels (along with accessory subunits) were enhanced at depolarized potentials by patch pipette suction, consistent with mechanosensitive Cav1.2 channels (756). Determination of the current-voltage (I-V) relationship during maintained pipette suction led to the conclusion (745) that membrane stretch shifted the I-V curve to the left by 10 mV [in contrast to the findings of Lyford et al. (754)]. This leftward shift in the I-V curve for Cav1.2 channel activation is similar to that observed when the channel is phosphorylated by PKA (757). These results were confirmed in native VSMCs from SM-specific Piezo1-knockout mice, to eliminate the possible influence of stretch-activated Piezo1 channels. SM-specific deletion of FlnA, a cytoskeletal protein known to associate with multiple ion channels, receptors, and the actin cytoskeletal network (756), essentially eliminated pressure-induced myogenic tone of caudal arteries and third-order renal arteries while having minimal effect on KCl-mediated vasoconstriction (745). FlnA deletion also resulted in blunting of pressure-induced Ca2+ increases in VSMCs of the caudal artery, and FlnA deletion in a Piezo1-deficient background reduced stretch activation of Cav1.2 by ∼50% (745). Whether the stretch-induced shift in Cav1.2 activation was mediated by FlnA was not determined, and the mechanism by which FlnA mediates force transduction to Cav1.2 remains unknown. One possibility is that FlnA regulates the association of Cav1.2 channels with cholesterol and/or Cav-1, both of which are known to influence myogenic responsiveness (758, 759). These provocative findings point to CSK-based mechanosensitive regulation of Cav1.2 as a significant contributor to pressure-induced arterial constriction. They do not, however, account for the pressure-induced depolarization of VSMCs that precedes myogenic constriction. If pressure-induced Cav1.2 channel activation would be sufficient to produce arterial constriction in the absence of VSMC depolarization (which is not yet known), then that finding would contradict many of the assumptions about the signaling mechanisms underlying myogenic constriction.
2.4.2.2. T-TYPE VGCCS.
Initial studies on the expression of different VGCC isoforms in arteries provided evidence for T- as well as L-type VGCCs (760–762); however, early studies relied on the use of antagonists such as mibefradil and Ni2+ that were thought at the time to be selective for T-type VGCCs but later found to also inhibit L-type channels (763, 764). Subsequent studies using patch clamp and RT-PCR methods revealed that VSMCs from some arteries do indeed express two of the three isoforms (Cav.3.1 and Cav3.2) of T-type VGCCs (763, 765–767). A few in vivo and ex vivo studies of arteries or arterioles have suggested that components of the myogenic response, particularly in smaller vessels, were calcium dependent but nifedipine insensitive (767–770), possibly implicating Cav3 channels. Other studies, again relying primarily on the use of inhibitors such as mibefradil, suggested that Cav3 channels played a substantial role in arterial myogenic constriction and/or blood flow autoregulation (771, 772). However, more recent studies, with judicious use of inhibitors and/or employing Cav3 knockout mice, have concluded that T-type channels play no role in pressure-induced constriction (773) or only a subtle role at very low pressures, where VSMCs would be hyperpolarized to potentials within the window current of Cav3 channels (774).
There are no published reports that Cav3 channels are intrinsically mechanosensitive. Rather, under the same conditions in which N-type channels Ca2+ exhibit mechanosensitivity in a heterologous expression system, Cav3.3(α1I) channels are not (748). However, at least one study suggests that expression of Cav3 channels may be required to enable the normal mechanosensitivity of D-hair receptors (775), although in this case it is likely that Cav3 channels are activated secondary to another mechanosensory protein. Studies by Welsh and colleagues (776, 777) show that a major role of Cav3.2 channels in VSMCs is to couple RYR2-mediated Ca2+ release to the activation of BK channels, which is a mechanism that counteracts myogenic tone. Indeed, myogenic constriction is enhanced in Cav3.2−/− arteries because of a reduction in STOCs (776). These latter observations indicate that Cav3 channels do not play a direct role in pressure-induced constriction.
2.4.2.3. HCN CHANNELS.
These hyperpolarization-activated, cyclic-nucleotide-gated cation (HCN) channels are a class of voltage-gated cation channels activated by hyperpolarization rather than by depolarization. HCN channels share substantial homology to Nav, Cav, and Kv channels, but the selectivity pore of HCN channels allows permeation of both K+ and Na+ ions. Single-channel HCN conductance is <10 pS, with PNa/PK ∼0.4. HCN channels are also permeable to Ca2+ ions (778), and they may be regulated by intracellular Ca2+ as well (779, 780). HCN channels are most closely related to Kv10 and Kv11 channels, with the addition of a cytosolic cyclic nucleotide binding domain (781). Cyclic nucleotides are not required for HCN channel opening, but cAMP differentially modulates the open probability of the four differentially expressed HCN isoforms.
HCN channels are not distributed as widely as Cav and Kv channels, but they are expressed in many mechanosensitive cells (see Table 1 in Ref. 722 and Table 1 in Ref. 782), including cochlear inner hair cells (783, 784) and sensory neurons (781, 785). HCN currents have been extensively studied in the context of pacemaking by sinoatrial node (SAN) cells, where transcripts for all four isoforms are expressed in the mouse (786). Substantial evidence suggests that HCN channels are involved in controlling the pacemaking rate of SAN cells and mechanosensory neurons, where they contribute to IF (or Ih), the “funny” current (782, 787). They are activated after the cardiac action potential and contribute to depolarization during the diastolic depolarization phase of the pacemaking cycle (788). Classic blockers of HCN channels include external Cs+ (1–5 mM), ivabradine (10 µM), and the reputedly specific If inhibitor ZD7288 (1 µM) (789), although ivabradine, and to a lesser extent Cs+, have off-target effects on Nav, Kv, and/or other K+ channels (782, 790). HCN-knockout mouse models are available and have been tested in a number of studies investigating SAN pacemaking, but the results are complicated by the expression of multiple HCN isoforms, including their incorporation into heteromeric channels, as well as by the compensatory upregulation of some isoforms upon deletion of others (787).
Although numerous studies link HCN channels to mechanosensitive control of cardiac pacemaking (787, 791), only a single study has directly tested the mechanosensitivity of HCN channels. Lin et al. (722) expressed human HCN2 channels in oocytes and recorded currents in cell-attached macropatches before, during, and after the application of pipette suction in combination with various voltage protocols. The mechanosensitivity of HCN was more complicated than that of Cav, Nav, and Kv channels because HCN channels exhibit mode switching (792), which is evident as hysteresis in the I-V relationship when assessed by sawtooth ramp voltage protocols at different rates. The balance of stretch inactivation versus stretch activation varied with the frequency of applied rhythmic voltage waveforms and led to the conclusion that rhythmic waveforms (mimicking repetitive action potentials) would keep HCN channels “disequilibrated” at a high Po, but with a net effect that stretch inactivation would dominate at lower frequencies (722). The influence of cAMP and auxiliary HCN channel subunits (e.g., HCNE) on this mechanosensitive behavior remains unknown.
Only a few studies have examined the role of HCN channels in VSMCs. The VSMCs in most arteries have stable resting membrane potentials and do not typically show spontaneous depolarization leading to AP firing (649), except for those in portal vein (793) and small arterioles (173, 740, 794). HCN channels have not been shown to have a functional role in arterial tone development, but one study reported that they play an indirect role in tone development of airway SM (795). There is molecular and functional evidence for the roles of HCN channels in pacemaking for urinary bladder SM (796–798), uterine SM (799), and ICCs (800). Greenwood and Prestwich (793) recorded an inwardly rectifying cation current in rat and rabbit isolated portal vein cells that had the characteristics of an HCN channel: activated by hyperpolarization, permeable to Na+ and K+, and blockable by Cs+, ZD7288, and ivabradine. Cs+ produced a modest slowing of spontaneous contractions of isometric portal vein segments, but ZD7288 had complicated effects, including an increase in basal force production. Portal vein expresses mRNA for HCN isoforms 2–4 (793). McCloskey et al. (801) recorded currents with similar characteristics from isolated sheep lymphatic VSMCs: slowly activating, hyperpolarizing currents blocked by Cs+ and ZD7288 but not by Ba2+. In isolated lymphatic segments, both compounds lowered spontaneous contraction frequency by ∼50%. Negrini et al. (802) found evidence for expression of both the message and protein of all four HCN isoforms in rat diaphragm lymphatic smooth muscle. Although electrophysiological evidence for functional HCN channels was not provided, the spontaneous pumping of in situ lymphatic segments was found to be significantly inhibited by Cs+, ivabradine, and ZD7288, albeit at much higher concentrations than typically used in other studies. At least one other study has also implicated HCN channels in the spontaneous pacemaking of lymphatic VSMCs (803). Collectively, these studies suggest that HCN channels may play a role in VSMCs of vessels with spontaneous pacemaking. A background mechanosensitive HCN conductance in arteries that do not spontaneously fire APs could potentially contribute to pressure-induced constriction, but this idea remains to be tested.
2.5. Potassium Channels
Potassium channels potentially may be involved in both shear stress-induced hyperpolarization of ECs and pressure-induced depolarization of VSMCs. Mechanosensitive activation of a K+ channel would lead to hyperpolarization because EK is more negative than the resting membrane potential of either cell type. Therefore, mechanosensitive activation of an EC K+ channel is consistent with the rapid hyperpolarization observed upon initiation of shear stress that is often a component of flow-induced dilation. In contrast, pressure-induced depolarization of VSMCs is more consistent with activation of a cation channel leading to inward cation flux or activation of a chloride channel leading to outward Cl− flux. A mechanosensitive K+ current could produce VSMC depolarization if two criteria are met: 1) a background (basal) K+ channel is active at rest (e.g., in a relaxed artery) and 2) mechanical stimulation by stretch results in inactivation of that channel. We discuss the potential roles of four major types of K+ channels, Kv, KCa, Kir, and K2P, with a view toward their potential activation by shear stress in ECs or inactivation by pressure in VSMCs.
2.5.1. Kv channels.
Kv channels share a structure similar to the other voltage-gated channels discussed above (721). For Kv channels, both the NH2 and COOH termini are cytoplasmic. The α-subunits of Kv channels usually form complexes with auxiliary subunits that modulate channel trafficking and/or gating (804). Kv channels are widely expressed among many types of excitable cells and include the classic delayed-rectifier K+ channel (KDR) in squid axon and the Shaker K+ channel from Drosophila. Kv channels can be classified into 12 subfamilies. Kv family members showing delayed-rectifier activity include Kv1.2, Kv2.1, and Kv3.1; these channels are also characterized by slow inactivation and susceptibility to blockade by millimolar levels of tetraethylammonium (TEA). Kv family members Kv1.4, Kv3.4, and Kv4.2 (A-type channels) exhibit fast inactivation and are preferentially blocked by 4-aminopyridine (4-AP). Some Kv channel subfamilies are selectively blocked by peptide toxins (805). Kv7 channels, also called KCNQ channels, activate at more negative potentials than other Kv channel family members (806, 807) and are regulated by G protein signaling (808, 809). This subfamily includes Kv7.2 and Kv7.3 channels, which mediate the so-called M currents implicated in the development of pacemaker potentials in some neurons (810–812). Kv7 channels are blocked by XE991 and linopirdine or related compounds (813). Kv10–12 channels [also called ether-à-go-go (Kv10), ether-à-go-go related (Kv11), and ether-à-go-go like (Kv12) K+ channels] are delayed-rectifier Kv channels expressed in the heart, various neurons, vascular and visceral SMCs, and endocrine cells (814). Of these, Kv11.1 is distinguished by its rapid voltage-dependent inactivation and is the most widely studied because of its role in the cardiac action potential (815).
Multiple Kv family members, including Kv1, 2, 3, 4, 7, and 11, are expressed in VSMCs. Evidence for consistent expression of functional Kv channels in ECs is lacking (597, 816). The half-maximal activation voltage varies widely among Kv subfamilies. Because the Vm of VSMCs in most arteries and arterioles is between −60 and −40 mV and depolarizes further with vessel distension, substantial activation of Kv channels occurs at almost all intravascular pressure levels. It is not surprising that Kv channel inhibitors such as 4-AP and TEA are widely documented to be potent vasoconstrictors (817) and that multiple agonists, e.g., pyrimidines, cause VSM dilation primarily or in part through inhibition of Kv (818). Chen et al. (819) found that the expression of dominant-negative Kv1 channels enhanced the myogenic gain of isolated cerebral arteries in organ culture. Likewise, the Kv7 inhibitor linopirdine enhanced myogenic responses, and a Kv7 activator inhibited myogenic responses (806). These and many other studies suggest that depolarization-induced activation of multiple Kv family members contributes to negative feedback regulation of pressure-induced tone in peripheral arteries and arterioles (12, 650).
Kv channels exhibit both stretch activation and inactivation under appropriate conditions (237, 820), similar to the mechanosensitivity of heterologously expressed Nav channels (196). The mechanosensitive behavior of a Shaker Kv channel with a mutated inactivation domain that permitted simplified analysis of activation/inactivation was studied extensively by Morris and colleagues (237). Stretch activation was observed when Po was low (i.e., at more negative voltages), whereas stretch inactivation was observed when Po was high. Subsequent studies (189, 724, 821) suggested that the voltage-dependent transition of the channel was inherently mechanosensitive. The MacKinnon laboratory compared the gating properties of Kv1.2 and other Kv channels in oocytes using patch-clamp versus two-electrode voltage-clamp recording modes (188) and concluded that differences in channel behavior in the two recording modes could be explained by increased lateral tension on the membrane patch due to adhesion of the lipid to glass in cell-attached patches. The authors estimated a 50% increase in Po at a physiological Vm if membrane tension increased by only 1/10 the value required to activate the bacterial mechanosensitive channel MscL (211, 822), implying that mechanosensitivity may be present under physiological conditions. Morris et al. (189) pointed out that the outside-out recording mode used in those studies would result in a higher resting bilayer tension and hence an overestimation of the inherent channel mechanosensitivity. These studies support the inherent mechanosensitivity of Kv channels but highlight the difficulties in translating this property to physiological conditions, as discussed in sect. 2.1.2.
Stretch activation of Kv channels would produce membrane hyperpolarization, which runs counter to the idea that Kv channels mediate pressure-induced VSM depolarization; however, acceleration of Kv channel inactivation by mechanical forces (189, 237) could potentially promote depolarization. This, of course, would depend on the balance of mechanical forces acting on Kv channel activation versus inactivation (724). Simulations by the Morris laboratory suggest that mechanosensitive Kv gating inhibits overall cell excitability (189); this prediction is corroborated by evidence from other studies of native cells and intact tissues, suggesting that mechanosensitive activation of Kv channels shortens the duration of the atrial action potential (823) and acts as a mechanosensitive brake on the excitability of neurons involved in touch and pain sensation (824). Unfortunately, very little information is available to date regarding mechanosensitive inhibition of Kv channels in VSMCs, and whether the magnitudes of mechanical forces influencing Kv channel gating in VSMCs are within physiologically relevant ranges to promote pressure-induced depolarization and constriction is a critical issue that remains unstudied. Retailleau et al. (745) found no significant effect of pipette pressure on K+ channel current in cell-attached patches of caudal artery VSMCs from mice deficient in Piezo1. Hypoosmotic swelling did not alter KDR current under the same conditions in which it inactivated Kir channels (180), suggesting that KDR channels are not mechanosensitive. However, Gollasch and colleagues (451) found that activation of AT1R GPCRs by hypoosmotic swelling (see sect. 4.1.2) led to suppression of an XE991-sensitive Kv current in patch-clamped VSMCs and that XE991 enhanced the myogenic tone of isolated mesenteric and renal arteries. Although these observations suggest that inhibition of a Kv7 channel might contribute to the development of myogenic tone, XE991-sensitive K+ current and myogenic contractions persist in mesenteric arteries from mice lacking functional Kv7.3, Kv7.4, or Kv7.5 channels (451), indicating that XE991 may be acting on other VSMC channels. Kv channels also are subject to inhibitory regulation by PKC and Ca2+ downstream from Gαq/11 signaling, such that indirect regulation of these channels might contribute to pressure-induced VSMC depolarization. These findings, particularly the potential pressure-induced inactivation of Kv7 channels, need further confirmation and investigation.
2.5.2. KCa channels.
KCa channels are comprised of three subfamilies: large-conductance (200–300 pS) BK channels, intermediate-conductance (32–39 pS) IK channels, and small-conductance (4–14 pS) SK channels. KCa α-subunits assemble as tetramers of four channel subunits but differ from Kv channels in that each subunit has an extracellular amino terminus and an intracellular carboxy terminus. Although KCa channels are generally activated by both membrane depolarization and increases in intracellular Ca2+, the subfamilies exhibit different electrophysiological properties, calcium sensitivities, and tissue expression profiles.
2.5.2.1. KCA CHANNELS IN VSMCS.
2.5.2.1.1. BK channels.
The pore-forming α-subunits of BK channels (KCa1.x) may or may not associate with auxiliary β- and γ-subunits. Four mammalian β-subunits and four smaller, leucine-rich repeat-containing γ-subunits (LRRC) have been identified (825, 826). Iberiotoxin (IbTX) and penitrem A are somewhat specific BK inhibitors; paxilline is widely used, but it also inhibits Ca2+-activated Cl− channels (827). BK channels are activated by both membrane depolarization and elevated cytosolic [Ca2+] but are less sensitive to Ca2+ than IK or SK channels. At physiological membrane potentials of arteriolar VSMCs, BK channels have a cytosolic Ca2+ set point of >3 μM (828), compared with cytosolic [Ca2+] levels of 300–400 nM in arterioles with myogenic tone (169, 829). The Ca2+ and/or voltage sensitivity of the channel can be enhanced by phosphorylation of COOH terminus domains on the α-subunit (830, 831) and by association with the β1 (832, 833) and γ auxiliary subunits (834, 835). BK channel function is thought to be modulated by NO, arachidonic acid, and COOH-terminal phosphorylation by multiple protein kinases (836), although at least one recent study suggests that NO does not directly affect BK channel activity under physiological conditions (837).
BK channels are expressed in a wide variety of VSMCs and have been established as important regulators of vascular tone (838). Like Kv channels, their activation provides a negative feedback mechanism to counteract VSM depolarization and vasoconstriction (12, 650); unlike Kv channels, BK channels are normally silent at the resting Vm of VSM and become activated only upon depolarization or increases in intracellular Ca2+. BK channel activation does not require a global (cytoplasmic) Ca2+ increase but only localized Ca2+ events associated with the ryanodine receptors (RYRs), specifically RYR3 (839) and RYR2 (840, 841). Ca2+ sparks are rapid elementary Ca2+ events through RYRs (840, 842, 843), and the functional coupling of BK channels and RYRs has been demonstrated in VSMCs from multiple types of arteries (844), although it is absent in others (Refs. 845, 846; for recent reviews see Refs. 847, 848). The synchronized openings of BK channels generate spontaneous transient outward currents (STOCs) that result in membrane hyperpolarization and vasodilation (848, 849). This negative-feedback regulation of Vm is facilitated by the functional coupling of BK channels with L-type Ca2+ (Cav1.2) channels, as demonstrated first by Leblanc and colleagues (850) and in subsequent studies (828), showing that the caveolin protein Cav-1 (851) and scaffolding proteins such as AKAP150 (852) and junctophilin (853) promote the interaction of BK and Cav1.2 in a complex that likely includes multiple other components (854). A similar functional interaction between RYR and BK channels has been demonstrated for Cav3.2 channels (855, 856). A cytoplasmic linker to the BK channel α-subunit had been initially thought to be mechanoprotective, but this was subsequently shown not to be the case (857).
Several lines of evidence suggest that BK channels are activated by membrane stretch. Dopico et al. (858) found that a K+ channel with conductance of ∼260 pS was activated by pipette suction in excised inside-out patches of mesenteric artery VSMCs. Unlike the behavior of Kv channels (237), stretch did not change voltage-dependent gating of the BK channel. Channel activation was independent of Ca2+, making it unlikely that activation occurred secondary to Ca2+ entry through another pathway (858). In contrast, Wu and Davis (181) observed that a large-conductance Ca2+-activated K+ channel was activated in response to longitudinal stretch of single VSMCs from coronary arteries, but in that case channel activation was secondary to activation of a smaller-conductance Ca2+-permeable cation channel. Similar evidence for the mechanosensitive activation of BK channels has been confirmed in other cell types (859, 860).
Stretch activation of BK channels, either direct or indirect, would promote hyperpolarization. Consistent with this action, almost all studies examining the effects of BK channel inhibitors on myogenic constriction have shown that BK channel activation opposes myogenic constriction (861). Studies of genetically modified mice reinforce this conclusion in that disruption of the gene encoding the BK β1 accessory subunit shifts the coupling between Ca2+ sparks and STOCs to more depolarized potentials and results in elevated systemic vascular tone and higher arterial pressure (833). Although arterial myogenic responses per se were not tested in that study, a subsequent study found that BK β1 disruption also increases basal arterial tone (862). These findings confirm that BK channel activation opposes myogenic constriction and are consistent with several other studies (649, 833, 839, 843). However, there are no published reports that BK channels in VSM are inactivated by stretch; this implies that they do not play a direct role in pressure-induced vasoconstriction. BK channels are inhibited by PKC and other kinases (for review see Ref. 836), so that their indirect regulation through these mechanisms could theoretically contribute to pressure-induced VSMC depolarization (for review see Ref. 847).
2.5.2.1.2. IK Ca channels.
In contrast to BK channels, IK channels (KCa3.1, SK4) exhibit variable expression in VSMCs. The majority of evidence suggests that this channel is expressed at relatively low levels in quiescent (contractile) VSMCs and thus may contribute little to resting Vm and tone. However, IK channels are upregulated in proliferating VSMCs (863, 864), and their expression may underlie that phenotypic switch. The IK inhibitor TRAM-34, which is potent at nanomolar concentrations and specific for IK over BK and SK, prevents the angioplasty-induced phenotypic switch of coronary VSMCs, thereby limiting the restenosis process.
The ability to assess the possibility of a direct contribution of IK channels to pressure-induced constriction, although improbable (865), is limited by the off-target effects of TRAM-34 on several other types of ion channels, notably Cav1.2 (866, 867). Studies of arterial myogenic constriction in IK-knockout mice do not appear to have been performed.
2.5.2.1.3. SK Ca channels.
SK channels (KCa2.1–3) are widely expressed in excitable cells, where they play important roles in pacemaking and AP bursting (868–870). They are blocked with reasonable selectivity by the neurotoxin apamin (871). The activation of SK channels in detrusor muscle silences activity of the urinary bladder during the filling phase (872–874), and SK channel activity in PDGF receptor (PDGFR)α+ interstitial cells is critical in mediating purinergic input to that organ (875, 876). There are no studies to date suggesting that SK channels in VSMCs are inactivated by stretch to produce or facilitate pressure-induced arterial constriction.
2.5.2.2. KCA CHANNELS IN ECS.
2.5.2.2.1. BK channels.
The expression of functional BK channels in ECs is a long-standing controversy. Multiple studies have reported evidence for BK channels in cultured ECs (see Ref. 877 for review), but this finding may represent phenotypic drift in passaged cells. ECs freshly isolated from certain arteries/arterioles express message and/or protein for BK channels (878), but ECs from other arteries do not (879, 880). Multiple electrophysiological studies of native arterial ECs exhibiting SK and IK currents have failed to detect functional BK currents (879, 881, 882). One explanation for these discrepancies may relate to the finding that endothelial BK channels may localize to caveolae, where their interaction with caveolin inhibits their function (883); thus, different pathological conditions that alter membrane lipid domains, such as hypoxia (884) or cholesterol depletion (885), may potentially relieve EC BK channels from normal inhibition by caveolin. This idea remains to be tested, but most evidence to date suggests that IK and SK channels account for all the EC-mediated arterial hyperpolarization (881), with little role for BK channels.
2.5.2.2.2. IK channels.
IK channels are expressed at relatively high levels in ECs and are widely implicated in mediating EDH (886). ECs from a variety of arteries/arterioles express IK channels, which colocalize with IP3Rs in EC projections to VSMCs through holes in the internal elastic lamina. EC agonists such as ACh bind their respective GPCRs and signal downstream to produce spatially localized release of Ca2+ from IP3Rs (“Ca2+ pulsars”) in these projections, thus activating IK channels (887). A number of other proteins also colocalize to this signaling complex, including eNOS (888), protein kinases (431), TRPA1 (889), and TRPV4 channels (431, 433), with their interactions facilitated by scaffolding proteins such as AKAP150 (431) (for review see Ref. 890). Agonists that activate this signaling complex elicit EDH, which is transmitted to the VSM layer through myoendothelial gap junctions, producing vasodilation by inhibiting myogenic tone (891). The IP3R/Ca2+ pulsar/IK channel axis in ECs appears to be functionally similar to the IP3R/Ca2+ spark/BK channel axis in VSMCs.
The importance of this signaling complex in ECs is illustrated by the phenotype of IK channel-knockout mice. Global deletion of the KCa3.1 gene abolished TRAM-34-sensitive currents in aortic ECs and attenuated ACh-induced hyperpolarization and dilation in carotid arteries. The systemic consequences of IK channel deletion were a significant increase in arterial pressure and mild hypertrophy of the left ventricle (552).
2.5.2.2.3. SK channels.
SK channels are widely expressed in ECs (891–893); both SK and IK channels are needed to account for the entirety of EDH and ACh-mediated dilation. A subpopulation of SK channels colocalizes with IK channels in EC projections, but SK channels are also distributed around the cell surface (894) in caveolar and lipid rafts (895). In most vascular beds, SK channel activation appears to be the more important component of EDH (896). Nelson and colleagues (897) used a transgenic mouse in which SK3 (KCa2.3) gene expression could be experimentally controlled by dietary doxycycline. In the absence of doxycycline, SK3 channel expression in the transgenic mouse was threefold higher than in WT mice, resulting in a reduction in myogenic tone of mesenteric arteries. Conversely, SK3 deletion by doxycycline treatment led to pronounced increases in myogenic tone and arterial pressure (897). These effects on EDH and EC-mediated dilation were even more pronounced in mice with combined KCa1/SK3 deficiency (881). Importantly, the deletion of the SK3 channel reduced the carotid artery dilation induced by increased shear stress (from 3 to 7 dyn/cm2, accomplished by increasing the viscosity of the luminal solution with 5% dextran) from 2% to 14%, whereas deletion of IK1 channels did not affect the response to increased shear stress (881). These results suggest that SK, but not IK, channels play a role in flow-induced carotid artery dilation.
With respect to longer-term responses to elevated shear stress, KCa2.3 and KCa3.1 can be upregulated by LS via a CaMKK-Akt-p300 pathway, whereas OS only elevates KCa3.1 but not KCa2.3 (898). KCa2.3 and KCa3.1 may modulate vascular tone through distinct mechanisms; KCa2.3, but not KCa3.1, colocalizes with TRVP4 at caveolae (899) under static conditions. LS enhances TRPV4 translocation to caveolae to interact with KCa2.3 (558) as well as KCa3.1 (899). Cav-1 knockout leads to the impairment of NO-mediated vasodilation, consistent with the idea that these KCa channels coordinate with TRVP4 to regulate that process (899), although Cav-1 interactions with other EC channels could also explain this observation.
2.5.3. Kir channels.
Like Kv channels, Kir channels are composed of tetramers of individual Kir channel subunits. Each subunit contains two transmembrane domains with cytoplasmic NH2 and COOH termini connected by an extracellular loop that folds inward to form part of the selectivity filter. At least seven Kir channel subfamilies have been identified and can be classified into four groups: “classical” (Kir2.x, Kir5.x), “K+ transport” (Kir1.x, Kir4.x, Kir7.x), G protein-regulated (Kir3.x), and ATP-sensitive (Kir6.x) channels (900). Kir channels share ∼40% amino acid identity across subfamilies and ∼60% identity within subfamilies (901).
Kir channels are characterized by notable inward rectification in their current-voltage (I-V) relationship, which is conferred by voltage-dependent block of the channel pore by Mg2+ and polyamines (901). The degree of rectification varies among the subfamilies, from strong to weak (Kir2 and Kir3 > Kir4 > Kir1 and Kir6). In addition to regulation by intracellular Mg2+ and polyamines, Kir channels are also sensitive to PIP2, membrane lipid composition, intracellular and extracellular pH, extracellular K+, intracellular Na+, and phosphorylation by protein kinases. Two Kir families are defined by their regulatory interactions with other membrane proteins: Kir3 with βγ G protein subunits and Kir6 with sulfonylurea subunits (900, 902). Ba2+ and Cs+ are the most commonly used Kir channel blockers (903). Ba2+ is inhibitory for most Kir channel isoforms at concentrations between 10 and 100 µM and is more effective at hyperpolarized membrane potentials. Over that concentration range, Ba2+ does not substantially inhibit Kv or KCa channels, and for that reason it is commonly used to assess the roles of Kir channels in native tissues.
Kir3 channels have been demonstrated to exhibit mechanosensitivity. Ji et al. (904) found that the G protein-gated muscarinic K+ current in rabbit atrial myocytes was inactivated within 500 ms by the application of positive pressure to the patch pipette, a response that was reversible and preventable by 5 mM Ba2+. Because these are characteristics of a G protein-regulated (Kir3.x) channel, the authors retested the mechanosensitivity of Kir3.1 and Kir3.4 channels after their heterologous expression in oocytes. Oocyte swelling produced by a 50% reduction in osmotic strength of the bath solution led to reversible decreases in Kir3.1/3.4 current by 18% (904) and decreases in Kir3.4 current by 27%. Noninjected oocytes showed no such responses to a similar hypoosmotic challenge, nor did cells expressing Kir1.1 or Kir2.1 channels. Importantly, the swelling-induced inhibition of Kir3.x channels was a relatively slow response that required 10 min to fully develop, implying the involvement of second messengers and/or multistep signaling cascades or CSK disruption. It is interesting that another laboratory studying the mechanosensitivity of these channels in cardiac myocytes reported them to be activated, rather than inactivated, by membrane stretch (905).
Of relevance to vascular mechanotransduction, Kir2.1 and Kir2.2 are strongly rectifying K+ channels that are widely expressed in VSM (650, 861, 906–908) and ECs (313, 314, 909). Their large inward conductances at negative voltages contribute to the maintenance of a stable VSMC resting potential, whereas their reduced conductances at potentials positive to Vrest permit depolarization and AP generation in VSMCs that fire APs. Close inspection of the relatively flat part of the I-V curve from EK to 0 mV for an idealized Kir channel reveals a small, outward Ba2+-sensitive current with a region of negative-slope conductance (see Figure 1 in Ref. 910) between EK and Vrest (650, 906). Modest elevations in extracellular K+ (5–15 mM) shift the I-V curve to the right, leading to hyperpolarization upon Kir activation. Conductance plots confirm that outward Kir current increases at any given Vm as extracellular K+ increases (910). Because of the high input resistance of most VSMCs, Kir currents as small as 2 pA/cell can produce a significant change in Vm (650, 910). This is thought to be the mechanism by which Kir channels mediate hyperpolarization of ECs and/or VSMCs in response to elevated extracellular K+ produced by metabolically active parenchymal cells in tissues such as brain and skeletal muscle (910–914). This property may also enable Kir channels to amplify responses initiated by other K+ conductances (906), including those originating in the endothelium and conducted to the VSM layer through myoendothelial gap junctions (915).
2.5.3.1. KIR CHANNELS IN VSMC MECHANOTRANSDUCTION.
Stretch activation of Kir current in VSMCs is the opposite of what is needed to produce pressure-induced arterial depolarization/constriction. However, the possibility that Kir channels in VSMCs, and other tissues, might be inactivated by stretch gains support from at least two studies. Welsh and colleagues (180) found that hypoosmotic swelling (reduction in bath osmolarity from 305 to 250 mosM) of isolated rat cerebral artery myocytes, which express Kir2.1 and Kir2.2 channels, led to a relatively rapid, ∼50% reduction in whole cell Kir current at −120 mV holding potential. This effect was blocked by the PKC inhibitor calphostin C, with a high degree of Ca2+ buffering in the whole cell patch pipette, suggesting that current inactivation might be mediated by a Ca2+-independent isoform of PKC (180). Subsequent work by the same laboratory significantly extended these observations. Sancho et al. (902) studied Ba2+-sensitive (30 µM) Kir current in both VSMCs and ECs isolated from rat cerebral arteries. Kir currents in ECs were activated by low levels of fluid shear stress (0.1 dyn/cm2) in a PIP2-dependent manner. Preventing PIP2-Kir channel interactions with neomycin, a known disruptor of such interactions (916), blocked the shear stress-induced activation. In contrast, Kir currents in VSMCs were inactivated by hypoosmotic swelling (300 to 205 mosM) or by patch pipette pressurization, but neither of those effects was affected by PIP2 depletion. Instead, cholesterol-Kir channel interactions were critical such that depleting cholesterol content with methyl-β-cyclodextrin (MβCD) blocked swelling-induced suppression of VSM Kir current. These results suggest that an interaction between cholesterol and Kir channels is required for mechanosensitive inactivation of Kir current. Although Kir2.1 and Kir2.2 channels are both expressed in cerebral artery ECs and VSMCs, the identity of the mechanosensitive Kir isoform in either cell type was not established because the individual Kir isoforms could not be selectively blocked. One limitation of this study is that neomycin also blocks PLC (917), RYR receptors (918), and several types of ion channels either directly or through their interactions with PIP2 (919–922). Although Sancho et al. (902) did not definitively identify the Kir channel(s) mediating these mechanosensitive responses, Kir2.1 and Kir2.2 channels were highly expressed and Kir2.4 channels were not expressed. It will be important to perform similar protocols on VSMCs and ECs isolated from mice selectively deficient in Kir2.1 and/or Kir2.2 channels (907), as well as using the same methods to test the mechanosensitivity of heterologously expressed Kir2.1/2.2 channels (909). Nevertheless, these studies collectively suggest that Kir is more than simply a background current and that the interaction of Kir channels with the appropriate membrane lipid in their respective cell types (PIP2 for ECs and cholesterol for VSMCs) may be required for the sensitivity of Kir channels to mechanical stimuli.
A limitation of these two studies (180, 902) is that inhibition of VSMC Kir current has not been demonstrated in response to a physiologically relevant force. As discussed in sect. 2.1.2, the cell swelling method to expand the plasma membrane can produce many indirect effects, including alterations in the concentrations of numerous intracellular components. Thus, a critical question remains: To what degree are the pressure-induced depolarization and vasoconstriction that occur in intact arteries explained by inhibition of Kir current? In support of their patch-clamp data, Sancho et al. (902) and Wu et al. (180) found that 30 µM Ba2+ elicited more constriction of cerebral arteries held at low pressure than at high pressure, consistent with the hypothesis that Kir channels are more inactivated at higher levels of pressure/stretch. Furthermore, Ba2+ produced a substantial degree of arterial constriction in the presence of MβCD, which is consistent with a dependence of VSM Kir channels on interaction with cholesterol for their mechanosensitivity (902). However, it should be noted that other laboratories have found the effects of Ba2+ on tone and Vm to differ substantially among different types of arteries/arterioles in vivo (861, 923), although in those studies the amount of spontaneous vascular tone varied widely and the pressures under which the effects of Ba2+ were tested were in most cases unknown. An argument against a strong or sole role for Kir channels in transducing pressure is that 100 µM Ba2+ constricts skeletal muscle (cremaster or cheek pouch) arterioles by only 15–20% (861, 923), whereas the same vessels can exhibit pressure-induced constriction up to 50% (924–926). These comparisons imply that Kir channel inhibition contributes at most to only a fraction of the pressure-induced constriction. Indeed, in vessels with some of the strongest myogenic responsiveness (e.g., renal afferent arterioles) Ba2+ increases tone and attenuates, but does not block, myogenic constriction (927, 928). Surprisingly, there are very few studies in which Ba2+, at a concentration selective for Kir channels, has been tested on arterial myogenic responsiveness across a range of pressures (929), so this issue remains to be resolved. Additionally, it is not known whether pressure-induced constrictions are preserved in arteries from mice deficient in Kir2.1 or Kir2.2 (907), although in the case of Kir2.1, deletion would need to be SM specific.
2.5.3.2. KIR CHANNELS IN EC MECHANOTRANSDUCTION.
Kir channels were one of the first mammalian ion channels found to be regulated by physiological levels of mechanical force, as reported by Davies and coworkers (930) in 1988. They described a shear stress-activated K+ channel in ECs (930) in which whole cell K+ currents with inward rectification were activated in ECs seeded into the open ends of capillary tubes perfused with physiological solution. The currents activated within seconds (half-maximal activation at 0.7 dyn/cm2) and rapidly inactivated upon cessation of flow. This initial observation has since been confirmed by other groups (313, 909), with Kir2.1 identified as the shear stress-sensitive Kir isoform (314). Activation of the channel by physiologically relevant levels of shear stress leads to EC hyperpolarization and enhanced passive Ca2+ entry (due to the absence of VGCCs in ECs) to promote NO production from eNOS (909). Subsequent studies by the Hoger and Levitan laboratories show that Kir2.1 deficiency or inhibition results in the reduction of shear-induced K+ current, loss of NO production, and impaired flow-induced vasodilation in mouse mesenteric arteries and human microvessels (313, 314). Kir2.1 deficiency also results in the loss of flow-mediated AKT and eNOS phosphorylation, contributing to impaired shear stress-mediated NO production (313, 931). Other shear stress-induced EC processes that require activation of EC K+ channels include cGMP formation (932), upregulation of transforming growth factor beta (TGF-β) (933) and thrombomodulin (934), and downregulation of endothelin-1 (ET-1) protein expression (935).
How does shear stress activate Kir channels? In patch-clamp studies of flow-activated Kir channels, the channel in the membrane patch is physically protected by the recording pipette from shear stress. Modeling studies by Barakat (936) predict that the thermal energy required for direct activation of the channel is 10- to 100-fold greater than the energy generated by physiological levels of shear stress. These findings suggest that Kir channels are not directly activated by shear stress. However, a number of groups have shown that Kir2.1 channel gating is regulated by cholesterol and other membrane lipids (902, 937–939). For example, the Ba2+-sensitive (30 µM) Kir current in ECs isolated from rat cerebral arteries is activated by low levels of fluid shear stress (0.1 dyn/cm2) in a PIP2-dependent manner (902) such that preventing PIP2-Kir channel interactions with neomycin (916) blocked shear stress-induced channel activation. Levitan and coworkers (940) have identified specific binding sites for cholesterol in the transmembrane domain of Kir2.1 between adjacent subunits of the channel. Because shear stress has been shown to increase membrane fluidity and reorder membrane lipids on a relatively fast timescale (941–943), it has the potential to directly activate Kir channels. Increases in membrane cholesterol decrease membrane fluidity and inhibit Kir2.1 channel gating (944), but whether this mechanism can produce the rapid, graded regulation of Kir2.1 gating that occurs in response to shear stress remains to be determined. Interactions of Kir2 channels with cholesterol explain the suppression of Kir2 channel activation by exposure to VLDL lipoproteins (945). The reduction of Kir2.1 current in Kir2.1+/−;Apoe−/− mice leads to impaired flow-induced NO production and vasodilation, as well as an increase in lesion formation in the normally atheroresistant descending aorta. Such atheroprone responses can be reversed by EC-specific overexpression of Kir2.1, supporting a role for Kir2.1 channels in atheroprotection (946).
2.5.4. KATP channels.
Kir6.1 and Kir6.2 channels assemble with SUR1 or SUR2 subunits to form heterotetrameric ATP-sensitive K+ “KATP” channels. KATP channels are weak rectifiers, with nearly linear I-V relationships, but are strongly regulated by the intracellular ADP-to-ATP ratio, which is detected by the SUR subunit. Kir6.2/SUR1 channels are expressed in heart, Kir6.2/SUR2 channels in pancreatic islets, and Kir6.1/SUR2B channels in arterial and lymphatic SM (947, 948). KATP channels are thought to act as metabolic sensors, detecting increases in the ADP-to-ATP ratio if the tissue becomes ischemic (949), although in VSM the evidence is often conflicting and there are examples of arteries in which hypoxia/ischemia-induced vasodilation does not involve VSMC KATP channel activation (950, 951).
In arterial SM, KATP channel-mediated hyperpolarization leads to inactivation of VGCCs, reducing Ca2+ entry and causing dilation (908). However, KATP channels have little activity in many types of arteries under normoxic conditions, and KATP channel activators, such as pinacidil, cromakalim, and levcromakalim are powerful vasodilators that antagonize pressure-induced constriction (768, 952, 953). The KATP channel inhibitor glibenclamide produces variable effects on resting Vm and tone of arteries, with constriction in some cases (952) and no effect in others (929, 954–956). Glibenclamide sensitivity also depends on the prevailing Po2 level, with a greater effect in vivo when the tissues are equilibrated with low Po2 that simulates normal tissue levels (957) and a lesser effect on ex vivo arteries that often are equilibrated with unphysiologically high Po2. Glibenclamide generally has little or no effect on myogenic or pressure-induced responsiveness of arteries, including rat skeletal muscle small arteries (958), rat mesenteric small arteries (929, 958), and rat cerebral arteries (672). Importantly, there are no reports to date suggesting that KATP channels might be inactivated by pressure and thereby contribute to myogenic constriction.
KATP channels are also expressed in ECs (Refs. 959, 960; for review see Ref. 961). The expression of functional KATP channels is upregulated in pulmonary ECs subjected to elevated shear stress (from 1 to 10 dyn/cm2) for 24 h, as measured by patch-clamp protocols, and cessation of flow results in EC depolarization (962), as measured with a voltage-sensitive dye. Depolarization is blocked by a high concentration of the KATP channel activator cromakalim (962) and is absent in ECs deficient in Kir6.2, which is the KATP channel isoform expressed in pulmonary ECs (963, 964). These findings suggest that the depolarization is due to the inactivation of EC KATP channels following flow cessation. The depolarization triggers the generation of ROS and activation of NADPH oxidase 2, which are thought to drive signaling pathways for revascularization of the ischemic lung (964, 965). Although it is not known how widespread this mechanism is, it is important to note that flow-adapted cultured ECs may better represent the in vivo situation than many studies of ECs under static conditions.
2.5.5. K2P channels.
There are 15 identified K2P channel members comprising six subfamilies. These are categorized in large part by their regulation by stretch, pH, polyunsaturated fatty acids (PUFAs), temperature, second messengers, and/or volatile anesthetics (966–968). K2P channels are composed of two pore domains in tandem, forming dimeric channels. Many K2P family members lack strong voltage dependence and are insensitive to the conventional K+ channel inhibitors tetraethylammonium, 4-aminopyridine, glibenclamide, Ba2+, charybdotoxin, and apamin (969, 970). Unfortunately, there are no high-affinity selective K2P channel inhibitors, but some members are blocked by millimolar concentrations of methionine and other sulfur-containing amino acids that do not significantly affect other K+ channels (971, 972). In many of the tissues in which they are expressed, including neurons, VSMCs, and cardiomyocytes, K2P channels are active when cells are at their resting membrane potential; therefore, they are thought to comprise a significant fraction of the background K+ leak current contributing to the resting potential (973–976). Of most relevance to this review and to VSM are the K2P channels of the TREK family, TREK-1, TREK-2, and TRAAK.
K2P channels exhibit several characteristics of true mechanosensitivity (sect. 2.1.3). Both TREK and TRAAK channels exhibit sigmoidal increases in Po in response to increasing suction pressure applied to the membrane through a patch pipette (969). In the whole cell recording mode, however, both TREK and TRAAK are more sensitive to negative than positive patch pipette pressure (969), and TREK-1 currents are inhibited by cell shrinkage in hyperosmotic solutions (977). These observations raise the possibility that TREK-1 may actually be inhibited by membrane stretch. The mechanosensitivity of TREK-1 and TREK-2 is lost with deletion of the COOH termini (978, 979). Like Piezo channels, the mechanosensitivity of TREK and TRAAK channels is retained in bilayers (224) and can be altered by adjusting the cylindrical versus conical lipids that control membrane curvature in the bilayer (970, 977, 980). TREK-1 channels become constitutively active in excised patches in which CSK connections are minimal or disrupted (975, 981), suggesting that even if the channels directly sense bilayer tension they might normally be inhibited by their interaction with CSK elements (233).
Although TREK and TRAAK channels fulfill many of the criteria listed in sect. 2.1.3 for true mechanosensitive channels, their primary regulatory modalities appear to be heat, intracellular H+ (982), and/or polyunsaturated fatty acids (PUFAs) instead of mechanical stimuli. Regulation by PUFAs has been more extensively characterized in TRAAK than TREK-1 channels, but the sensitivity of both depends critically on the carbonyl chain length, with long-chain PUFAs such as arachidonic acid (AA) being the most effective activators (983, 984). TREK-1 channels are inhibited by polycationic molecules such as poly-l-lysine and spermine, but that inhibition is reversed by PIP2 (985). Thus, PIP2 hydrolysis as a result of GqPCR stimulation could lead to TREK-1 inhibition.
What functional roles might the mechanosensitive properties of TREK channels confer on the cells/tissues in which they are expressed? Several early studies provided evidence for K+-selective currents that were regulated by membrane stretch and/or fatty acids. The critical role of fatty acids in mediating stretch activation of K+ channels in some SMCs (986, 987), described before the molecular identification of K2P channels, is likely mediated by one or more K2P channels. In neonatal cardiomyocytes, Kim and Clapham (988) found that two types of K+ channels, with conductances of 160 and 69 pS, were activated by intracellular H+, arachidonic acid (AA), and other free fatty acids but were insensitive to adenosine triphosphate (ATP) or calcium; these observations are consistent with the behavior of a K2P channel. Koh and Sanders (989) described a K+ channel with a conductance of 95 pS in murine colonic SMCs that was activated by patch pipette suction or cell elongation (989); this channel was subsequently termed the “SDK” channel. The SDK channel and a K+ channel with similar characteristics in bladder and uterine SM are strongly regulated by AA and by activators of soluble guanylate cyclase including nitric oxide (989–991). Many of these properties are now known to match those of TREK-1 (KCNK2), which was identified in the same tissues (992) and has a single-channel conductance of ∼100 pS in symmetric K+ solutions. TREK-1 is widely expressed in visceral and vascular smooth muscle and endothelium, as well as in other tissues. In murine myometrium TREK1 is upregulated during pregnancy, and its blockade by methionine leads to an increase in uterine excitability and contractility, suggesting that it is important for stabilizing membrane potential until its downregulation before parturition (991). Likewise, bladder SM expresses TREK-1, and its inhibition by methionine (993, 994) or deletion in Trek-1−/− mice (995) results in elevated basal tone and enhanced spontaneous activity of the detrusor muscle. In cells where mechanosensitive cation channels such as Piezo1 or TRPP1 are also expressed, the activation of TREK and/or TRAAK channels can counteract the contributions of cation channel activation (224, 996). Collectively, these observations suggest that TREK-1 channels are important for stabilizing membrane potential and lowering excitability in bladder, myometrium, and some portions of the GI tract, when reduced excitability is physiologically advantageous (e.g., receptive relaxation).
The idea that TREK-1 channels might be important for stabilizing membrane potential makes their activation unlikely to play a critical role in the vascular myogenic response. TREK-1 current activation by increased stretch would produce hyperpolarization and is not compatible with the established observation that pressure elevation leads to VSMC depolarization and vasoconstriction. Only a few studies to date have addressed the role of TREK channels in the regulation of arterial tone and/or myogenic responses. Garry et al. (997) and Blondeau et al. (998) found that TREK-1 is expressed in basilar and mesenteric artery endothelium and that EC-dependent vasodilation to ACh and BK are impaired in Trek-1−/− mice. Namiranian et al. (999) found that TREK-1 was expressed in cerebral arteries but that Trek-1−/− mice had normal arterial pressure, normal blood pressure (BP) regulation and normal cardiac function; furthermore, their responsiveness to phenylephrine (PE) and a nitric oxide donor was normal in middle cerebral and basilar arteries. These studies suggest that TREK-1 is important for EC-dependent vasodilation (and imply that its activation would antagonize myogenic tone) but not for the direct induction of myogenic tone or the mediation of myogenic constriction in response to pressure elevation.
2.6. Summary for Mechanosensitive Ion Channels
2.6.1. VSM ion channel summary.
Electrophysiological evidence exists for a large number of potentially mechanosensitive ion channels in VSM. However, only a few of these (Piezo1, TREK-1, and possibly ENaC) meet most of the criteria listed in sect. 2.1.3 for a true mechanosensitive channel with a critical role in pressure-induced constriction. The channels with most relevance to pressure-induced constriction are listed in TABLE 1. It is somewhat surprising that Piezo1, the best-characterized mechanosensitive cation channel, with clear roles in other mechanotransduction processes, does not appear to be involved in arterial myogenic constriction. However, its role has only been examined in two arteries (caudal and superior cerebellar) that are not typically used for myogenic studies, and additional testing in other myogenically active arteries (cerebral, renal, skeletal muscle) from SM-specific Piezo1 KO mice is needed. Other cation channels such as Nav and HCN are only expressed in certain specialized blood vessels and therefore cannot explain the myogenic constriction common to most arteries and arterioles. Channels such as TREK-1 and Kv have some degree of intrinsic mechanosensitivity, but their activation by pressure elevation (e.g., BK channels) would produce hyperpolarization rather than the ubiquitously observed depolarization. Cav1.2 exhibits intrinsic mechanosensitivity but possibly only under nonphysiological conditions. Assessment of its intrinsic mechanosensitivity in pressure-induced constriction is complicated by the fact that Cav1.2 is the primary pathway for Ca2+ entry downstream from other mechanosensitive proteins. Its potential role in VSM mechanotransduction is further complicated by its regulation by CSK proteins (sect. 2.4.2.1) and ECM-integrin interactions (sect. 5.2). Thus, Cav1.2 could potentially mediate pressure-induced constriction without being gated by depolarization from other channels (implying that depolarization is an epiphenomenon), but this idea has not been tested; however, at the present time, the weight of the evidence favors Cav1.2 regulation by voltage-dependent activation.
Table 1.
VSM ion channels potentially contributing to pressure-induced constriction
| Channel | Ionic Permeability (physiological conditions) | Mechanical Activation | Proposed Role in VSMC Pressure-Induced Constriction (when activated) | References* |
|---|---|---|---|---|
| Piezo1/2 | Inward cations (nonselective) | Inherent mechanosensitivity | No significant function reported | (242, 305) |
| ENaC | Inward Na+ | Possible inherent mechanosensitivity | Pressure-induced depolarization (renal and cerebral arteries) | (369–371, 379) |
| ASIC | Inward Na+ | Unknown | Pressure-induced depolarization | (416, 417) |
| TRPC6 | Inward cations (nonselective) | GqPCRs | Pressure-induced depolarization | (246, 381, 452) |
| TRPM4 | Inward Na+ | GqPCRs | Pressure-induced depolarization | (380, 450, 470, 480, 483–488) |
| TRPV1 | Inward cations (nonselective) | GqPCRs | Pressure-induced depolarization (specific vascular bed) | (488, 512, 515) |
| TRPP1 (PKD2) | Inward cations (nonselective) | Unknown | Pressure-induced depolarization | (243, 498, 499, 502) |
| TRPV2 | Inward cations (nonselective) | Unknown | Pressure-induced depolarization (retinal arteries only) | (362, 525, 528) |
| TRPV4 | Inward cations (nonselective) | Unknown | Activation of Ca2+ sparks and BK channels to oppose depolarization | (430, 432, 535, 538–540, 543, 545) |
| TRPML1 | Intracellular Ca2+ release | Unknown | Activation of Ca2+ sparks and BK channels to oppose depolarization | (567) |
| ANO1 | Outward Cl− | Ca2+ influx via TRPC6; possible activation from Ca2+ stores | Pressure-induced depolarization | (245, 693–696, 699, 701, 705, 708, 709) |
| Kv | Outward K+ | Secondary to membrane depolarization | Oppose depolarization | (188, 237, 806, 819, 820) |
| BK | Outward K+ | Secondary to membrane depolarization and Ca2+ influx | Oppose depolarization | (181, 858, 862) |
| Kir | Outward K+ | Possible inherent mechanosensitivity through lipid bilayer | Pressure-induced inhibition would promote depolarization. | (180, 902, 910–914) |
| KATP | Outward K+ | Unknown | Oppose depolarization | (672, 929, 952, 954–956, 958) |
| K2P | Outward K+ | Unknown | Oppose depolarization | (997–999) |
| P2X | Inward cations (nonselective) | Stretch-activated ATP release? | Pressure-induced depolarization | (615) |
| TMEM63 | Inward cations? | Possible inherent mechanosensitivity | Unknown | (637–639) |
| VGSC | Na+ | Secondary to membrane depolarization | Unknown | (196, 730, 731) |
| VGCC | Ca2+ | Secondary to membrane depolarization | Primary Ca2+ entry mechanism | (748, 753–756) |
| HCN | Inward cations (nonselective) | Secondary to membrane hyperpolarization | Unknown | (722, 793, 801, 802, 1000) |
*References are primarily to papers that address the potential mechanosensitivity of the channel. BK, large-conductance Ca2+-activated K+; HCN, hyperpolarization-activated, cyclic nucleotide-gated cation; K2P, 2-pore domain K+. See glossary for other abbreviations.
Evidence to date indicates that the primary mechanism of pressure-induced depolarization occurs via activation of a second messenger-gated channel (or channels) downstream from the activation of another membrane mechanosensor, probably one or more GPCRs (see FIGURE 13). The ion channels most likely involved in this scheme, in order of the best supporting evidence, are TRPM4, TRPC6, ANO1, and possibly TRPV1 in certain vascular beds. TRPM4 is activated by IP3-mediated Ca2+ release from the SR following PLC activation. TRPC6 is activated by PLC-mediated DAG production. TRPV1 is activated by heat and H+, but its sensitivity to heat may be enhanced by PKC-mediated phosphorylation, thus contributing to myogenic tone. ANO1 is activated by Ca2+ release from stores in other cell types, but evidence for that mechanism in VSM is lacking at present; instead, evidence from a single laboratory suggests that it may be activated by Ca2+ entry through TRPC6. The lower arterial pressures in Trpc6−/− and Trpm4−/− and SM-specific Pkd2-knockout mice are consistent with, but do not prove, impaired myogenic tone. Studies of global knockout mice may be complicated by compensation by other mechanisms (upregulation of TRPC3 in Trpc6−/− mice and upregulation of adrenergic signaling in Trpm4−/− mice). There are no studies to date assessing myogenic responses in Trpm4−/− mice or in SM-specific Trpm4- or Trpc6-knockout mice; such studies are critically needed to resolve these discrepancies.
FIGURE 13.

Postulated sequence of TRP, Ano1, K+, and voltage-dependent Ca2+ channel (VDCC) channel activation in VSMCs following a pressure step. Em, membrane potential. See glossary for other abbreviations.
How ENaC, ASIC, and Kir channels fit into the schemes shown in FIGURES 12 AND 13 is not clear. ENaC fulfills many of the criteria for a true mechanosensitive ion channel, but its best-characterized role is in epithelial Na+ transport, where its activity is regulated primarily by the relatively slow process of insertion of new channels into the plasma membrane. ENaC is expressed in the SM layer of myogenically active arteries, its deletion impairs myogenic constriction, and its overexpression enhances myogenic constriction. However, nearly all the evidence for its role in these processes comes mostly from a single laboratory, and further confirmation from multiple laboratories is needed. The best evidence for the inherent mechanosensitivity of ENaC channels comes not from studies of stretch on VSMCs but from the effects of flow or shear stress on epithelia. Mechanosensitive ENaC activation in VSMCs is apparently not linked to GPCR signaling, and the published evidence for the roles of TRP and ENaC channels appears to be mutually exclusive, so it is difficult to reconcile a major role for ENaC with the extensive evidence for mechanosensitive GPCRs and downstream second messenger-gated channels. With respect to ASIC channels, their primary gating modality appears to be detection of chemical rather than mechanical stimuli. The potential role of ASIC2 in the myogenic response could be to modify ENaC sensitivity in some, as yet undefined, way; again, additional confirmation by multiple laboratories is needed. Likewise, the possible contribution of pressure-induced inactivation of Kir channels to myogenic constriction needs to be more definitively established. The evidence for their role in pressure-induced constriction again comes from a single laboratory, and their activation to date has only been demonstrated by nonphysiological stimuli such as osmotic swelling and patch pipette suction. Their physiological role in these processes has not been rigorously assessed either by genetic deletion strategies that are now the standard for testing essentially all other putative mechanosensitive ion channels.
In summary, to claim a role for direct mechanosensitivity of a particular ion channel, it is necessary to define the sensing mechanism according to the criteria listed in sect. 2.1.3. This approach has the greatest potential to drive the field forward. At the present time, the schemes shown in FIGURES 12 AND 13 have the strongest experimental support to date for explaining the roles of ion channels in pressure-induced myogenic constriction.
An impediment to progress on mechanisms of VSMC mechanotransduction is that high-throughput screens for potential mechanosensitive proteins are more difficult to perform in VSMCs than ECs because of the problems associated with rapid VSMC dedifferentiation in culture. An example is the elegant high-throughput screen for flow-sensitive mechanosensitive proteins in cultured ECs devised by Patapoutian and coworkers (107), which uncovered a previously unappreciated mechanosensitive role for GPR68. Such approaches could potentially reveal additional stretch-sensitive proteins in VSMCs, especially if they could be conducted in cultured VSMCs with a preserved contractile phenotype.
2.6.2. EC ion channel summary.
The most immediate EC response to shear stress is the activation of ion channels that lead to flow-induced dilation. The channels with most relevance to pressure-induced constriction are listed in TABLE 2. Critical roles for TRPV4 and PKD2 channels are supported by the observations that the genetic deletion of either Trpv4 or Pkd2 impairs or abolishes flow-induced dilation; the degree of impairment appears to vary among different arteries. Neither of these two channels is a direct mechanosensor. Piezo1 is the only known truly mechanosensitive ion channel in ECs, and its well-documented activation by shear stress triggers the opening of TRPV4 through PLA2-mediated production of AA metabolites to enhance and sustain the Ca2+ influx initiated by Piezo1; whether PKD2 channels are activated downstream from Piezo1 or by another mechanism is not yet known, but, regardless, they do not appear to be localized to primary cilia in ECs. TRPV2 channels may be activated by similar mechanisms downstream from Piezo1, but this has not yet been tested in ECs. Unfortunately, the effects of EC-specific Piezo1 knockout have only been studied by two groups in mesenteric arteries: in one case flow induced vasoconstriction (309), and in the other case flow induced vasodilation through Piezo1-mediated Ca2+ entry, ATP release, and activation of P2Y receptors (300). The contribution of Piezo1 channels to flow-induced dilation of other arteries needs to be critically evaluated, as well as components of flow-induced dilation possibly not mediated by Piezo1 or Piezo1-induced ATP release. The combination of Ca2+ entry through Piezo1, TRPV4, and PKD2 (and perhaps TRPV2) channels appears to be a major mechanism driving NO and prostanoid production, but this likely depends on the type of artery involved.
Table 2.
EC ion channels potentially involved in shear stress-mediated Ca2+ signaling and flow-induced dilation
| Channel | Ionic Permeability (physiological conditions) | Mechanical Activation | Proposed Role in EC Shear Stress-Induced Ca2+ Influx | References |
|---|---|---|---|---|
| Piezo1/2 | Inward cations (nonselective) | Inherent mechanosensitivity | Ca2+ influx, depolarization | (93, 260, 297, 300, 309, 318–320) |
| ENaC | Inward Na+ | Possible inherent mechanosensitivity | SS-induced depolarization | (384, 386, 388–391, 394) |
| ASIC | Inward Na+ | Unknown | Unknown; not expressed? | |
| TRPC6 | Inward cations (nonselective) | GqPCRs? | Ca2+ influx | (453–457) |
| TRPM4 | Inward Na+ | GqPCRs? | Unknown; disputed expression | (380, 480) |
| TRPV1 | Inward cations (nonselective) | GqPCRs? | Unknown; not expressed? | (512, 513) |
| TRPP1 (PKD2) | Inward cations (nonselective) | Unknown | Disputed | (506, 509–511) |
| TRPV4 | Inward cations (nonselective) | Unknown | Ca2+ influx | (94, 202, 320, 433, 546, 547, 550, 552, 556, 560, 1001) |
| Ca2+-activated Cl− | Outward Cl− | Unknown; secondary to Ca2+ influx/release? | Depolarization | (717, 718) |
| SK | Outward K+ | Secondary to Ca2+ influx/release | Hyperpolarization | (881, 891–896) |
| IK | Outward K+ | Secondary to Ca2+ influx/release | Hyperpolarization | (552, 886) |
| Kir | Outward K+ | Possible inherent mechanosensitivity through lipid bilayer | Hyperpolarization | (313, 314, 902, 909, 930, 937–940) |
| KATP | Outward K+ | Unknown | Inactivation causes depolarization | (959, 960, 963, 964) |
| P2X | Inward cations (nonselective) | Stretch-activated ATP release? | Ca2+ influx? | (615, 617–619) |
| TMEM63 | Inward cations? | Possible inherent mechanosensitivity | Unknown | (637–639) |
Shear stress activates K+ channels in parallel with its activation of these Ca2+ entry mechanisms. SK and IK channels in ECs are activated through both Ca2+ entry and Ca2+ release mechanisms. There is some disagreement about the relative contributions of IK and SK channels to flow-induced dilation (80). Kir channels are activated by shear stress through an as-yet-undefined mechanism, perhaps involving dynamic shifts in membrane lipids, but their activation is critical because the genetic deletion of Kir2 channels impairs flow-induced dilation (313). K+ efflux through SK, IK, and Kir channels combines to produce an EC hyperpolarization that enhances the driving force for additional Ca2+ entry into ECs and is also conducted to the VSMC layer through MEGJs, reinforcing the vasodilatory action of NO and prostanoids. A Ca2+-activated Cl− channel in ECs is also activated downstream from shear stress, probably via TRPV4-mediated Ca2+ influx (because its activation is delayed relative to Kir2 activation), and the resulting Cl− efflux attenuates or reverses the K+ channel-mediated hyperpolarization.
EnNaC and possibly P2X4 channels may be directly activated by shear stress in ECs, but whether they contribute to flow-induced dilation is not clear. The effects of EnNaC activation may primarily be longer term, leading to changes in EC ionic composition and CSK stiffening.
Chronically elevated levels of shear stress also lead to changes in the expression of some ion channels, e.g., KATP channels. These changes may amplify or counteract some of the processes described above.
Shear stress-mediated Ca2+ influx can result in physiologically beneficial effects (e.g., flow-induced dilation), but it may also lead to deleterious pathological effects on ECs, including disruption of the integrity of the permeability barrier (320) or a switch from an atheroprotective to atheroprone EC layer. The mechanisms that determine how sustained EC Ca2+ levels are fine-tuned to control this switch remain to be determined.
3. OTHER PLASMA MEMBRANE-ASSOCIATED MECHANOSENSORS
3.1. Plasma Membrane-Associated Mechanosensors in VSMCs
Mechanical stretch rapidly activates multiple signaling pathways (1002), including plasma membrane-bound enzymes that have been proposed to be mechanosensors (1003–1005). Relevant enzymes in VSMCs whose activation could potentially mediate arterial myogenic constriction include phospholipase C (PLC), phospholipase D (PLD), and phospholipase A2 (PLA2), all of which are involved in the metabolism of membrane-associated lipids. Although the direct mechanosensitivity of phospholipases may be in question, there is abundant evidence for the relevance of these enzyme-linked signaling cascades in both short- and long-term responses of VSMCs to stretch/pressure.
3.1.1. PLC and PLD.
PLC activation leads to cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into the potent second messenger molecules DAG and IP3, each of which has multiple downstream targets in VSMCs (FIGURE 14) (12). DAG and IP3 regulate multiple ion channels, some of which are discussed in the context of TRP channel regulation (see sect. 2.2). IP3 may also activate some channels (e.g., TRPC3) independent of IP3R-mediated Ca2+ release (1006). DAG can also be produced from the action of PLD on phosphatidylcholine, a process demonstrated to occur in arteries in response to agonists such as NE (1007) and vasopressin (1008), and is produced by ECs after exposure to cyclic strain (1009). PLD has been shown to be a source for DAG-mediated activation of TRPC channels in VSMCs (1010), but its role in vascular myogenic signaling has not been studied to date. In contrast, PLC activation has been shown in many studies to play a central role in pressure-induced constrictions of arteries and arterioles.
FIGURE 14.

Signaling pathways downstream from PLC, PLD, and PLA2. Based on Ref. 12, with permission from the American Physiological Society. DHETE, dihydroxyeicosatetraenoic acid; HETE, hydroxyeicosatetraenoic acid; Pchol, phosphatidylcholine. See glossary for other abbreviations.
Mammals express six families of PLC, including some linked to G proteins, tyrosine kinases, or small G proteins and phospholipids (1011). Mechanical stimulation of the plasma membrane is known to activate PLC in multiple cell types, including photoreceptors (190), osteoblasts (1012), lymphocytes (1013), neurons (1014), ECs (1015), and SMCs (1016). Early studies on the mechanism of the vascular myogenic response offered support for the idea that PLC activity is essential for the vascular myogenic response. Osol and coworkers (1017) first showed that predeveloped myogenic tone of rat posterior cerebral arteries was reversed by the presumably selective PLC inhibitor U-73122. Pressure-induced constriction was subsequently shown to be blocked by PLC inhibition in many arteries, including renal afferent arterioles (1018), posterior cerebral arteries (1019), and mesenteric arteries (1020), but not in all arteries (167). Likewise, pressure-induced VSM depolarization was reversed by PLC inhibition (1019). Biochemical analyses provided direct evidence that elevation in transmural pressure increases PLC activity in VSMCs (1021), although by necessity those measurements were made at later time points (90 s and 15 min) than pressure-induced constrictions in the same arteries. The results of some of these studies can be questioned on the basis of off-target effects of PLC inhibitors such as U-73122, 2-nitro-4 carboxyphenylcarboxyphenyl-N,N-diphenylcarbamate, and neomycin (1022, 1023), but they are supported by more recent findings such as those of Gonzales et al. (450), who found that siRNA-mediated downregulation of the PLCγ1 isoform had effects similar to pharmacological inhibition of PLC in abolishing the development of myogenic tone and pressure-induced depolarization of VSMCs in rat cerebral pial arteries.
A primary target of DAG production downstream from PLC/PLD is PKC, which is a family of ubiquitous serine and threonine protein kinases implicated in the regulation of a wide variety of VSM functions. The PKC family is composed of 10 isozymes classified as conventional, novel, or atypical according to the mechanisms of their regulation. Conventional isoforms (α and γ) bind to DAG in the presence of Ca2+, novel isoforms (δ, ε, θ, and η) bind to DAG in the absence of Ca2+, and atypical isoforms (ζ and ι/λ) bind to anionic phospholipids rather than DAG or Ca2+ (1024). Early evidence pointing to PKC involvement in the mechanosensitive response of VSMCs was reported by Hill et al. (1025), who found that the PKC inhibitors H7 and staurosporine caused a concentration-dependent decrease in the myogenic tone of skeletal muscle arterioles and that the PKC activator indolactam increased myogenic tone. Although H7 and staurosporine target ATP binding sites and therefore lack specificity, several studies from other laboratories confirmed the effects of pharmacological manipulation of PKC activity on the myogenic tone and myogenic responsiveness of arteries from several organs, including rat cerebral arteries (174, 1026), rat renal afferent arterioles (1027), human coronary arteries (1028), and human subcutaneous arteries (1029). However, PKC inhibition does not block myogenic constriction in all arterial vessels (1025, 1030), and it is interesting that the negative results were found primarily in arterioles (167), which typically exhibit stronger myogenic constrictions than arteries (1031). More recently, Hong et al. (1032) found that the selective AT1R blocker candesartan partially inhibited the myogenic tone of isolated first-order rat cremaster muscle arterioles, that the synthetic DAG analog 1-oleoyl-2-acetyl-sn-glycerol (OAG) reversed this loss of myogenic tone, and that this rescue was prevented by pharmacological block of PKC. These data provide evidence that PKC activity is stimulated by the increase of intraluminal pressure downstream of AT1R, likely by DAG generated through PLC activity.
What downstream targets of PKC activation are relevant to pressure-induced arterial constriction? Earley et al. (462) reported that the degree of VSMC depolarization or cerebral artery vasoconstriction in response to a PKC activator was significantly decreased after acute knockdown of TRPM4 expression, suggesting that PKC-dependent regulation of TRPM4 activity contributes to the control of cerebral artery myogenic tone. Subsequent studies using siRNA-mediated knockdown techniques implicated PKCδ in the translocation of TRPM4 protein to the plasma membrane (463, 464). Collectively, these data suggest that intraluminal pressure elevation stimulates the activity of PKCδ in VSMCs to increase the amount of TRPM4 channel protein at the plasma membrane, thereby enhancing the inward cation current conducted by TRPM4 channels. Other VSM ion channels besides TRPM4 are known to be regulated by PKC, including L-type VGCCs (746), other TRP isoforms (1033), and ANO1 (1034); however, the link between PKC-mediated regulation of these channels and pressure-induced constriction is not as well established as for TRPM4. PKC activation could also enhance pressure-dependent constriction via nonchannel mechanisms, e.g., by enhancement of the Ca2+ sensitivity of contractile proteins (1035–1037) and/or regulation of MLCP through phosphorylation of CPI-17 (1038, 1039).
3.1.2. PLA2.
Mammalian PLA2 isoforms can be classified into three large groups: Ca2+-dependent secretory sPLA2 and cytosolic cPLA2 and Ca2+-independent PLA2, categorized according to their enzymatic properties, structures, and regulatory functions (1040). Plasma membrane expansion induced by hypoosmotic solutions reduces the lateral packing of membrane lipids, which has been demonstrated to strongly modulate the catalytic activity of PLA2 (1003). Although this effect on PLA2 was demonstrated in unilamellar vesicles (1003), it may, in principle, apply to other systems. There is considerable evidence for the involvement of downstream metabolites of PLA2 in pressure-induced constriction of arteries. PLA2 activation leads to the production of AA (FIGURE 14), which in turn is metabolized by lipoxygenases, cyclooxygenases, and cytochrome P-450 family enzymes (1041) to produce a number of secondary products that may be critical for pressure-induced arterial constriction. Studies by the Harder laboratory provided multiple lines of evidence that the P-450 metabolite and potent vasoconstrictor 20-hydroxyeicosatetraenoic acid (20-HETE) contributes to myogenic regulation of cerebral blood flow. For example, pressure elevation of isolated cerebral arteries increases the production of 20-HETE by sixfold; inhibitors of 20-HETE production, inactive analogs of 20-HETE, or competitors of 20-HETE block pressure-induced cerebral artery constriction in vitro; and inhibitors of 20-HETE production attenuate autoregulation of cerebral blood flow in vivo (1042, 1043). 20-HETE appears to act both by enhancing L-type calcium channel current (1044), which promotes myogenic constriction, and by inhibiting large-conductance, Ca2+-activated K+ current (1045), which opposes myogenic constriction. Evidence for a similar role for 20-HETE in pressure-induced constriction of renal and skeletal muscle arterioles is summarized by Roman (1046). Precedents for stretch-mediated activation of PLA2 can also be found in the GI tract, where stretch-induced increases in the slow-wave frequency of intramuscular interstitial cells of Cajal (732) appear to be mediated by PGE2 production downstream from EP3 receptors, because chronotropic responses to stretch are inhibited by indomethacin and absent in cyclooxygenase II-deficient mice (715).
Collectively, there is strong evidence that the membrane-bound enzymes PLC and PLA2 are activated after stretch/pressure in VSMCs and participate in, or mediate, myogenic constriction; however, there is no direct evidence that these enzymes are intrinsically mechanosensitive. Instead, nearly all evidence cited above can be explained by the intrinsic mechanosensitivity of GPCRs, as discussed in sect. 4.1. PLC activity is highly regulated by Gαq/11-coupled GPCRs (1011). Evidence for the regulation of PLA2 by GPCR signaling, although supported by a few studies (1041, 1047–1050), is not as extensive.
3.1.3. Caveola-mediated VSMC responses.
Caveolae are flask-shaped plasma membrane invaginations enriched in cholesterol. They are abundant in both VSMCs and ECs (1051). Multiple studies have suggested that various plasma membrane proteins, including enzymes, GPCRs, and ion channels, localize specifically to, or are concentrated in, caveolae (1052). Caveolin proteins are critical structural components of caveolae and also inhibitory binding partners for multiple proteins, including NOS, PKC, and adenylate cyclase (1053). Global Cav-1-knockout mice exhibited complete loss of caveolae in VSMCs and multiple alterations in vascular function (759). Aortic rings with phenylephrine-induced tone from Cav-1−/− mice showed enhanced relaxation to ACh that was mediated by increased NO production. Small arteries from Cav-1−/− mice showed blunted vasoconstriction to ANG II and ET-1 (759). Subsequent studies by other groups confirmed reduced agonist-mediated constrictions in various types of arteries with Cav1 deletion or cholesterol depletion (1054–1056). Adebiyi et al. (758) examined the effects of Cav1 deletion on myogenic tone using pressure myography of cerebral arteries. Pressure elevation led to blunted myogenic constrictions in Cav-1−/− arteries compared with WT arteries, accompanied by reductions in pressure-induced depolarization and global calcium increases in cerebral artery VSMCs. Inhibition of NO did not restore myogenic tone, suggesting that the impairment was not due to excess NO production. However, Ca2+ spark, STOC, and BK channel activity were elevated in cerebral artery VSMCs of Cav-1−/− mice (758, 1057), suggesting that attenuated myogenic responses could be explained by increased activity of that pathway, possibly through enhanced activation of RYR channels in the absence of Cav-1.
3.2. Plasma Membrane and Associated Structures as EC Mechanosensors
In vitro investigations have demonstrated that the application of shear stress to ECs can activate multiple mechanosensors (FIGURE 15). These include integrins (1058, 1059), tyrosine kinase receptors (e.g., Flk-1) (1059, 1060), G proteins and G protein-coupled receptors (1061), ion channels (627), and intercellular junction proteins (112, 1062). Other possible mechanosensors are membrane components and structures (FIGURE 15) such as membrane lipid rafts (1063), caveolae (1064), and glycocalyx (1065). The mechanosensing of shear stress is mediated through adaptor molecules (e.g., SH2) to trigger a cascade of signaling pathways that modulate the expression of functional genes, i.e., those concerned with proliferation or growth arrest, inflammation or anti-inflammation, and many other physiological and pathophysiological processes.
FIGURE 15.
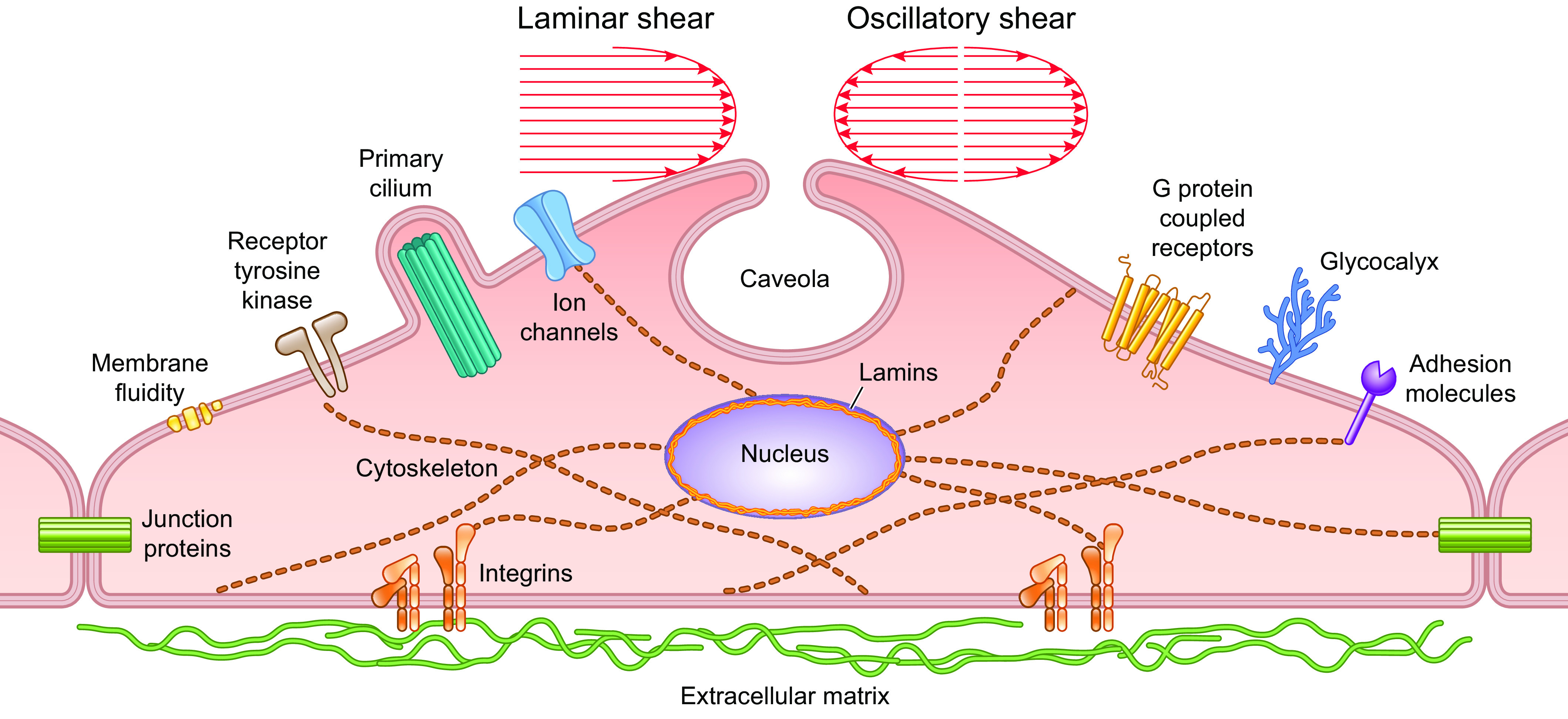
Schematic drawing showing mechanosensing molecules in endothelial cells.
3.2.1. Mechanoregulation of membrane fluidity in EC.
The EC membrane is composed of continuous liquidlike phospholipid bilayers, which can relay the outside-in signaling in response to mechanical stimuli, thus serving as a first-line mechanism for mechanotransduction (for review see Refs. 1066, 1067). To determine the role of EC membrane fluidity in endothelial mechanosensing, fluorescent probes such as 9-(dicyanovinyl)-julolidine (DCVJ) (941) and 1,1′-dihexadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate [DiIC16(13)] (942, 1068) have been incorporated into membrane lipids before subjecting the ECs to shear stress. The results show that LS causes rapid and transient increases of membrane fluidity as a function of the magnitude of shear stress (941). The confocal microscopy fluorescence recovery after photobleaching (FRAP) method was used to determine the DiI diffusion coefficient (D). The results indicate that the membrane fluidity undergoes transient changes with spatial features that vary with the magnitude of LS: Modest shear (10 dyn/cm2) causes a rapid (5 s) increase at the upstream side of the cell with a decrease at the downstream side, followed by a secondary increase upstream (peaks at 7 min) without changes downstream. High LS (20 dyn/cm2) leads to increases both upstream and downstream of the cell at 5 s; by 30 s the upstream D becomes higher than that at downstream (1068). Interestingly, if the application of shear is ramped over time (e.g., 20 dyn/cm2 over 1 min of time) D decreases at both upstream and downstream, and such decreases in D are accompanied by an attenuation of the shear-induced MAPK activation (1063). An increase of D by benzyl alcohol treatment enhances the shear-induced MAPK activation (1063). A decrease of D by cholesterol treatment leads to the suppression of shear-induced ATP release (942). These findings indicate that the membrane fluidity (and its inverse: microviscosity) can play an important role in mediating the spatiotemporal modulation of mechanotransduction in ECs.
Pulmonary arterial ECs respond to 10% step stretch and hypoosmolarity with increased lipid order and decreased membrane fluidity, in contrast to the decrease in membrane lipid order and increase in membrane fluidity induced by LS (943, 1069). Such opposite responses may be due to the differential effects on the membrane cholesterol content, which is decreased by LS but increased by stretch (943). A decrease in fluidity by addition of membrane cholesterol has been found to attenuate the shear stress-induced VEGFR phosphorylation, and an increase in fluidity with methyl-β-cyclodextrin (MβCD) treatment mitigates the stretch-induced PDGFR phosphorylation (943). Although it is unclear how and why stretch and shear modulate membrane cholesterol in opposite ways, it is clear that fluidity plays an important role in modulating transmembrane translocations of molecules for their interactions and activations.
3.2.2. Caveola-mediated EC responses (lipid rafts).
Among the membrane microdomains in ECs, the abundant caveolae (caveolin-coated lipid rafts) can be regulated by shear stress. It has been demonstrated that LS upregulates caveolin expression at the EC plasma membrane and changes caveola distribution (1070). Multiple signaling molecules have been shown to localize in caveolae (for review see Ref. 1071), and disruption of caveolae (by manipulation of membrane lipid content or knockdown of Cav-1) markedly attenuates shear-induced endothelial responses (1072). The results on flow modulation of caveola distribution and on membrane lipid and caveolin-dependent EC regulation suggest that the caveolin-containing membrane lipid rafts may play an important role in mechanotransduction. LS induces the expression and phosphorylation of Cav-1, eNOS, and ERKs in ECs and enhances their colocalization with the luminal surface of caveolae to allow rapid mechanotransduction (1070, 1073). Preconditioning ECs with LS enhances their sensitivities to subsequent challenges of step-flow increases, leading to a sensitization of tyrosine phosphorylation pathways but a desensitization of the MAPK pathway (1070). The shear-induced Cav-1 phosphorylation leads to its dissociation from eNOS, thus allowing Ca2+/calmodulin to activate eNOS/NO production (1074). Cav1−/− mice demonstrate elevated eNOS activity and NO production, with impaired aorta ring contractile tone and augmented relaxation in response to acetylcholine, as well as thickening of the artery wall with EC hyperproliferation and no change in lumen size (759, 1075). In the left external carotid artery (LCA) ligation model, the LCA exhibits an inward remodeling with a decrease in lumen size in wild-type (WT) mice but an outward arterial remodeling (wall thickening without decreases of lumen size) in Cav1−/− mice (1076). Flow (but not acetylcholine)-induced, NO-dependent vasodilation is impaired in isolated, preconstricted carotid arteries of Cav1−/− mice, and this impairment can be reversed by caveolin expression in ECs (1076), suggesting that Cav-1 is necessary for flow-induced eNOS activation. Despite the increase of eNOS activity and NO production, both pressure-induced myogenic tone and flow-induced dilation were found to be decreased in mesenteric resistance arteries of Cav1−/− mice, and these decreases could be restored toward WT levels by inhibiting eNOS (1077). These results suggest a complicated role of caveolae in mechanotransduction. The caveola structures are completely abolished in these Cav1−/− mice, but the knockout is not lethal, suggesting that there may be redundant/compensatory mechanisms (perhaps involving other lipid rafts). In addition, Cav-1 may appear outside the caveola structure functioning as a lipid sensor to modulate lipid transport (among plasma membrane, Golgi, ER, mitochondria, and endolysosome) and cellular cholesterol metabolism (for review see Ref. 1078). Both the mechanical environment and the lipid content are critical factors in regulating vascular function, but the role of Cav-1 in the interplay between mechanosensing and lipid sensing remains to be determined. With the chemiluminescence imaging technique, it has been demonstrated that shear stress induces a localized ATP release and subsequent Ca2+ wave at the caveolae (626). With the use of a FRET-based ATP biosensor, the shear-induced EC mitochondrial ATP production has been shown to be Cav-1/caveolae dependent (1079), suggesting the multiple roles of caveolae in shear regulation of ATP/Ca2+ signaling. It has been demonstrated that, in contrast to LS, OS causes a decrease of membrane fluidity, an increase of membrane cholesterol content, and the activation and translocation of integrin α5 to the lipid raft, which in turn activates the NOD-, LRR- and Pyrin domain-containing protein 3 (NLRP3) pathway for endothelial inflammation (1080). Such OS-induced α5-integrin translocation and activation are mediated by the association of actin with the lipid raft (1080).
It has been reported that LS (6 h) increases the density of caveolin protein and caveola structure on the luminal plasma membrane of rat lung EC, suggesting an increase of cellular sensitivity for mechanotransduction (1070). Fifteen to twenty percent uniaxial stretch (3 min) and hypoosmolarity (5 min) cause a rapid disassembly of caveolae in mouse lung ECs and HeLa cells in an actin- and ATP-independent manner. The disassembly of caveolae is believed to release more membrane surface area to maintain the membrane tension (1081). Indeed, ECs isolated from the lungs of Cav1−/− mice without caveolae are not able to maintain membrane integrity and undergo a sudden increase of tension upon hypoosmotic shock (1081). The impact of caveolae dynamics on the regulation of cell signaling and functions under different types of mechanical stresses remains to be determined.
3.2.3. Glycocalyx-mediated EC responses.
The EC membrane surface has the carbohydrate coating of endothelial surface glycocalyx (ESG). ESG contains many glycoproteins such as sialic acid (SA), heparan sulfate (HS), chondroitin sulfate (CS), hyaluronic acid (HA), etc. and plays a role in transmitting mechanosignals in ECs (for detailed review see Refs. 1082, 1083). Abnormalities of the glycocalyx have been linked to multiple diseases, including atherosclerosis (1084–1086). ESG exhibits robust structures in straight vessels with LS but not in vascular locations with flow disturbance (1087). It has been proposed that the robust ESG in endothelium functions as a protective barrier to maintain homeostasis of the vessel wall (1088), e.g., normal permeability (1089). The glycocalyx has the potential to influence shear stress-induced regulation of EC ion channels through force-tether interactions of glycosylated amino acid residues in the extracellular domains of the channels (352). It is well accepted that cultured ECs do have a glycocalyx structure at the surface but with lower density and thickness in comparison with the in vivo condition (1090–1092); furthermore, in vitro studies generally cannot recapitulate the multitude of factors (such as blood cells and vascular locations) that affect the ESG in vivo. Because of the difficulty in studying the details of glycocalyx function in vivo, however, the roles for glycocalyx in EC mechanotransduction under different forms of shear stress have been studied mainly in vitro (for review see Ref. 1093). These in vitro studies mainly investigate the impacts of enzymatic manipulation of glycoproteins on EC responses under shear, but the integrity and levels of ESG structures under these conditions remain unclear. Heparinase III treatment, which causes the depletion of ESG (mainly HA), abolishes NO production in response to LS and high OS but has no effect on NO production induced by bradykinin (1065). A detailed study has shown that depletions of HS, HA, and SA, but not CS, block the LS induction of NO; in contrast, such ESG depletions do not affect the LS-induced PGI2 production, suggesting the involvement of different mechanosensing/transduction pathways (1094). An ex vivo study has shown that HS and SA regulate the LS-induced NO production through ROS-dependent pathways, whereas HA regulation is ROS independent (1095). These findings highlight the intricacies of mechanotransduction mediated by various glycoproteins. A study using FACS analyses for direct detection of EC surface ESGs has demonstrated the expression and presence of HS that is linked to syndecans and glypican as the functional mechanosensing structure, but the expressions of CS and HA are weak (1096). The removal of glycocalyx abolishes the EC alignment in response to LS and decreases the speed of EC migration (1097). ESG has been shown to be involved in shear-regulated signaling events by mediating the expression of EC junction proteins (e.g., PECAM1) (1098), glypican localization in lipid rafts (1099), Cav-1 expression and distribution (1100), PECAM-1-G protein interactions (1101), integrin signaling (1102), as well as actin remodeling (1103, 1104). A study on inside-out signaling using FACS and immunostaining in a cone-plate shear system has shown that HA and HS, but not SA, are induced and evenly distributed on the EC apical surface in response to atheroprotective flow and that they are suppressed and irregularly distributed under atheroprone flow. A recent study demonstrates that LS induces, whereas OS reduces, the expression of HA and its cell surface localization. Such LS-induced HA is mediated by the upregulation of KLF2 and the consequent HAS2 expression at the EC surface to modulate EC glycolysis (1105). The multifaceted functional roles of ESG in flow regulation of EC functions, especially in vivo, remain to be elucidated.
3.2.4. Primary cilia-mediated EC responses.
Mechanosensing by the primary cilia is a controversial topic (1106). EC primary cilia are the 9 + 0 bundle core of the microtubule-based structures that extend from the basal body of the cell, connect to the CSK, and protrude through the apical membrane. Some data suggest that primary cilia act as EC flow sensors to regulate vascular function (95, 504, 1107–1111). Studies on zebrafish have demonstrated that ECs display cilia in aorta and vein at 24–28 hours postfertilization (hpf), when the blood flow is very low; the cilia are significantly reduced at 48 hpf, when the flow increases with heart maturation. The disruption of heart muscle development results in the retention of cilia in ECs (510). In addition, the angles (degrees) of cilia bending are correlated to the heart rate and blood viscosity as well as Ca2+ signals; therefore, cilia may serve as mechanosensing structures in this context (510). The potential roles of TRPP1/2 channels in acute mechanosensing by primary cilia are discussed in sect. 2.2.3.3. Another mechanosensing/mechanotransduction mechanism by which primary cilia might regulate EC function involves changes in the CSK and/or ciliary structure. An in vivo study demonstrated that the distributions of endothelial primary cilia are elevated in the inner curvature of the aorta arch and internal carotid sinus of wild-type C57BL/6 mice, where flow is disturbed, as well as in/around atherosclerotic lesions and cuff-induced disturbed flow areas in Apoe−/− mice (1110). In vitro studies reported that high LS (15 dyn/cm2) leads to the disassembly of primary cilia in ECs (1112, 1113), whereas low LS (1 dyn/cm2) promotes ciliogenesis (1113). LS induces endothelial-to-mesenchymal transition (EndoMT) in Tg373-mutant and Ift88-deficient nonciliated ECs, as evidenced by the decreased expression of EC marker genes and increased expression of mesenchymal marker genes, in a TGF-β signaling-dependent manner (1114–1116). These results suggest that cilia are involved in maintaining the EC differentiation state to contribute to homeostasis. However, findings that high shear causes cilia to disassemble, whereas low shear has the opposite effects, and the correlation of the presence of primary cilia with areas of disturbed flow, suggest that cilia may also be involved in regulating EC dysfunction. Further investigations are needed to determine the functional roles of cilia in EC mechanotransduction.
4. RECEPTORS AND JUNCTION PROTEINS AS MECHANOSENSORS
4.1. GPCRs as Mechanosensors
A growing body of literature demonstrates that mechanical forces can activate specific G protein-coupled receptors (GPCRs) independent of their chemical ligands. GPCRs comprise the largest family of signaling proteins, including >800 members, enabling cells to detect chemical, electric, magnetic, and mechanical stimuli and light. GPCRs consist of seven alpha-helical transmembrane domains containing long, multiloop extracellular NH2 and COOH termini. The binding of a ligand to a GPCR causes a conformational change, allowing the GPCR to act as a guanine nucleotide-exchange factor; it can then activate an associated heterotrimeric G protein by exchanging the GDP bound to the G protein for a GTP. The α-subunit of the G protein, together with the bound GTP, can then dissociate from the β- and γ-subunits to further affect intracellular signaling proteins, depending on the α-subunit type (Gs, Gi/o, Gq/11, G12/13) with which those signaling proteins associate (1117). GPCRs exhibit “conformational flexibility” in that their active conformations can be influenced by interactions with other GPCRs, ligands, and intracellular signaling proteins, as well as being subject to posttranslational modification. GPCRs can undergo allosteric modulation and oligomerization, exhibit signaling bias, and participate in compartmental or constitutive signaling (1118). Constitutive signaling (an active receptor conformation in the absence of an agonist) potentially enables GPCR mechanosensitivity.
4.1.1. Direct evidence for GPCR mechanosensitivity.
Frangos and colleagues (1061, 1119) first suggested that GPCRs might be sensors of shear stress. Evidence for stretch activation of GPCRs was subsequently provided by Komuro and colleagues (1120), who demonstrated that the type 1 angiotensin receptor, AT1R, a member of the GqPCR family, could be activated by mechanical stress through a mechanism independent of its ligand, angiotensin II. In cultured cardiomyocytes adhered to a deformable silicone substrate, the application of 20% stretch induced the activation of multiple ERK family kinases, which was significantly inhibited either by an ANG II receptor antagonist or by the inverse agonist candesartan (1121). AT1R activation was not mediated by endogenous release or production of ANG II, because mechanosensitive ERK activation persisted in the presence of an angiotensin-converting enzyme (ACE) inhibitor or ANG II neutralizing antibody; it also persisted in cells containing a mutated ANG II binding site and in cells with genetic deletion of ANG II (1120). Not all GPCRs in this system were found to be mechanosensitive, e.g., ET1AR was not (1122). Subsequent studies have produced an extensive list of mechanosensitive GPCRs with potential relevance to cardiovascular biology, including AT1R, the bradykinin receptor B2R, the sphingosine-1-phosphate receptor S1PR, the dopamine receptor D5R, the muscarinic receptor M5R, the leukotriene 1 receptor CysLT1R, and the vasopressin receptor V1AR, almost all of which are expressed in VSM (1123–1129).
4.1.2. GPCR-mediated mechanotransduction in VSMCs.
4.1.2.1. EVIDENCE FOR GPCR-MEDIATED MECHANOTRANSDUCTION.
Data reported by several laboratories support the concept that AT1R and other GPCRs in the plasma membrane of VSMCs transduce increases in intraluminal pressure to initiate signal transduction cascades that cause membrane depolarization and contraction. Studies by Osol and coworkers (1017) showed that myogenic tone of rat posterior cerebral arteries was reversed by administration of pertussis toxin, an inhibitor of Gi/o GPCRs. In line with these results, nonspecific G protein activation with AlF-4 or NaF significantly increased the myogenic tone of arteries (1017) and arterioles (1130). However, interpretation of many of these early studies is limited by their use of nonspecific GPCR inhibitors and/or activators.
4.1.2.1.1. P2YR and S1PR.
A small body of literature suggests that myogenic tone and/or the arterial response to pressure elevation requires signaling through P2Y receptors and/or sphingosine 1-phosphate receptors (S1PRs). Activation of these GPCRs is not thought to be mediated through their intrinsic mechanosensitivity but by binding of their endogenous ligands, autocoids released from VSMCs in response to stretch. The mechanisms of pressure-induced autocoid release from VSMCs are still undefined.
Kauffenstein et al. presented evidence that signaling through P2YR GPCRs on VSMCs is required for normal myogenic responsiveness. Ectonucleotidase inhibition enhances myogenic tone in mouse mesenteric arteries (1131), and myogenic tone was potentiated in Entpd1−/− arteries (1132). In a subsequent study, P2Y6 receptor blockade blunted myogenic tone development in mouse mesenteric arteries and human subcutaneous arteries, and myogenic constriction was substantially attenuated in mesenteric arteries of P2ry6−/− mice (1131). In addition, the enhanced myogenic tone observed in mesenteric arteries after the development of chronic heart failure was prevented in P2ry6−/− mice (1131). These results suggest that mechanosensitive release of UTP (the preferred agonist for P2Y6 receptors) may be required for the development of pressure-induced myogenic tone. The downstream signaling pathway appears to involve Rho kinase activation and MLCP (1131). These findings are in part consistent with earlier work by Brayden and coworkers (1133), who found that pharmacological inhibition of P2Y4 and P2Y6 receptors almost completely inhibited myogenic tone of rat parenchymal arterioles and that partial knockdown of either receptor using antisense oligonucleotides reduced myogenic tone by ∼45%. The signaling pathway downstream from P2Y receptors involved activation of TRPM4 channels (487). However, in that study inhibition of ectonucleotidase activity had no effect on myogenic tone (1133), suggesting that the response was not mediated by release of nucleotides but perhaps by mechanical activation of P2Y receptors. The discrepancies between these studies remain to be resolved.
Bolz et al. (1134) observed that an S1P receptor (S1PR) antagonist potently inhibited myogenic tone of hamster skeletal muscle arterioles. S1P is a bioactive lipid and vasoconstrictor, produced by the enzyme sphingosine kinase (Sphk1), which may translocate from the cytosol to the plasma membrane in response to stretch. Overexpression of a dominant-negative Sphk1 mutant or coexpression of dominant-negative mutants of RhoA/Rho kinase together with Sphk1 completely inhibited development of myogenic tone of arteries in short-term organ culture (1135). It was concluded that S1P contributes to the development of myogenic constriction through the activation of RhoA/Rho kinase. The mechanism of pressure-induced S1P production remains to be resolved. A subsequent study by this group demonstrated that TNF-α exerts its effects on myogenic tone through S1PR (1136).
4.1.2.1.2. AT1R and Gαq/11 GPCRs.
In a landmark paper, Gudermann and coworkers (23) performed extensive studies using patch-clamped HEK293 cells coexpressing AT1R and TRPC6 channels, with TRPC6 current used as a downstream readout of agonist activation or mechanoactivation of the GPCR. The membrane-permeable DAG analog OAG activated whole cell currents with an I-V relationship and permeability signature consistent with TRPC6, but the mechanical stimulus (hypoosmotic solution at 250 mosM) did not trigger the currents unless the cells coexpressed AT1R (23). Consistent with a requirement for AT1R in mechanosensitivity, swelling-induced TRPC6 currents were largely prevented by the inverse AT1R agonists losartan and candesartan. The cells also exhibited mechanosensitivity when AT1R was replaced by other GqPCRs, including H1R, M5R, and V1aR, or when TRPC6 was replaced by other DAG-sensitive TRP channels (TRPC3 or TRPC7); in each case, swelling-induced TRP currents were nearly completely prevented by an appropriate inverse agonist (23). Mechanosensitivity was not observed when the cells were transfected with Gs-coupled GPCRs or treated with pertussis toxin to inhibit Gi/o-coupled GPCRs; therefore mechanosensitivity appeared to be specific to Gq-coupled GPCRs, although this conclusion is at odds with the findings of a more recent study by Erdogmus et al.(1137) showing evidence for the mechanosensitivity of Gs- and Gi/o-coupled GPCRs in addition to Gq-coupled GPCRs.
The compelling results of the study by Mederos y Schnitzler et al. (23) support the hypothesis that mechanosensitivity resides in GPCRs and suggest that downstream regulation of PLC/PKC drives production of the second messenger(s) DAG and IP3, which directly or indirectly activate cation/chloride channels to produce depolarization. Additional observations in support of this scheme included the following: 1) swelling-induced activation of TRPC6 current in H1R-expressing cells was blocked by a PLC inhibitor or a nonhydrolyzable GTP analog and 2) IP3 was generated in a graded manner by exposure of M5R-expressing COS cells to solutions containing graded reductions in osmolarity (23). Although this study predominantly used TRPC6 current as a readout for mechanoactivation, at least one additional downstream target of stretch-mediated GPCR activation must be present in VSM because myogenic constriction is preserved in TRPC6-deficient arteries (452). When this scheme was tested in various VSM systems, the authors found that the AT1R inverse agonist losartan had the following effects: it blocked swelling-induced increases in intracellular Ca2+ in isolated renal VSMCs expressing a high density of AT1Rs, attenuated constriction to pressure elevation in isolated arteries, and decreased the rise in perfusion pressure that otherwise occurred in response to increased flow in isolated, perfused kidneys (23). Mechanosensitivity could even be conferred to non-myogenically active arteries, such as the aorta, after forced expression of AT1R (23). Also consistent with a role for mechanosensitive GPCRs was the observation that genetic deletion of RGS2, a negative regulator of Gq/11-coupled GPCRs, enhanced pressure-induced responses in the context of renal autoregulation (23, 1138). Similar observations have been made in Rgs5−/− arteries (1139, 1140). Transactivation of the EGF receptor (EGFR) secondary to mechanoactivation of AT1R (1141) could explain the observation that EGFR-mediated signaling is required for myogenic responsiveness (1142, 1143).
Subsequent studies, using selective pharmacological inhibitors of AT1R, acute AT1R knockdown, and/or mice deficient in AT1R or other GPCRs (487), have confirmed the basic principle that pressure-induced constriction of arteries is transduced largely, or primarily, by Gq/11-coupled GPCRs. Convincing evidence has been provided that AT1R activity is necessary for myogenic vasoconstriction in a number of arteries/arterioles, including mouse mesenteric arteries (451, 1020, 1144), first-order rat cremaster muscle arterioles (1032), mouse and rat cerebral pial arteries (450, 484), and mouse parenchymal arterioles (1145). A contrasting study on rat cerebral pial arteries reported that agonists of Gq/11-coupled GPCRs failed to enhance stretch‐induced TRPC currents and did not augment myogenic constriction (249), leading to the conclusion that TRPC currents and GPCRs do not interact in the production of myogenic tone. Apart from that study, the collective data support a model in which PLC in VSMCs is activated downstream of Gq/11-coupled GPCRs in response to increases in intraluminal pressure, leading to increased production of DAG and IP3. DAG and IP3 stimulate signaling pathways that trigger TRPM4 and TRPC6 cation channels (and possibly other channels) to depolarize the plasma membrane of VSMCs and increase the open probability of L-type voltage-gated Ca2+ channels (FIGURE 12), thus promoting global Ca2+ influx to drive myosin light chain kinase activation and contraction. DAG-mediated activation of PKC may also lead to the modulation of other ion channels, such as TRPV1 (488), that contribute to the development of pressure-induced constriction.
4.1.2.2. UNRESOLVED ISSUES REGARDING THE ROLE OF GPCRS IN MYOGENIC CONSTRICTION.
The roles of GPCRs in myogenic constriction are the subject of several excellent reviews (see Refs. (21, 23, 1146, 1147). Despite the compelling evidence for GPCR-mediated arterial constriction to pressure elevation, several unresolved issues remain:
-
1)
Does hypoosmotic swelling of HEK293 cells expressing TRPC6 and a GPCR (23) replicate the actual forces experienced by VSMCs in the arterial wall in response to pressure elevation (see sect. 2.1.2)? As a check against nonspecific effects of cell swelling, Mederos y Schnitzler et al. (23) found that application of positive patch pipette pressure also activated cation current in HEK293 cells coexpressing AT1R and TRPC6, and this was prevented by the AT1R inverse agonist losartan; however, as discussed in sect. 2.1.2, patch pipette suction/pressurization may not reproduce a physiological stimulus at the cell membrane. It is also possible that the forced expression of GPCRs and/or TRPC6 channels in studies of heterologous cell systems may artificially sensitize those signaling pathways to a given mechanical stimulus, i.e., the normal mechanosensitivity of Gq/11-coupled GPCRs may be enhanced and/or the density of TRPC6 channels used for readout may be raised to a sufficiently high level that they respond to what would otherwise be subphysiological levels of PLC-produced second messengers.
-
2)
What accounts for the fraction of pressure-sensitive myogenic tone that is not explained by mechanosensitive Gq/11-coupled GPCRs? In some systems AT1R was found to account for nearly all myogenic constriction (451), whereas in others it accounts for a lower fraction, from 64% to <50% (23, 1128, 1144, 1148), or none (167). In these other cases, were there unidentified GPCRs that were not blocked or were there contributions from other mechanosensitive proteins, such as ion channels (621, 1128)?
-
3)
How would the multiple GqPCRs expressed in native cells (including VSMCs) integrate a given mechanical stimulus and then feed into downstream effectors, e.g., to modulate DAG or IP3 production? Do different GqPCRs have different degrees of mechanosensitivity [as suggested in some studies (1144)]? For example, Mederos y Schnitzler et al. (23) found this hierarchy: H1R > AT1R M5R > V1AR, but to what extent is that an actual difference or merely reflective of the relative expressions of the respective GPCRs in the HEK cell assay? To what extent might differential GPCR expression in various vascular beds and different branching orders of vessels (1149) explain the well-documented regional differences in myogenic responsiveness (12)?
-
4)
Does AT1R play a central role in acute pressure transduction by most/all arteries? Of the various GPCRs (including AT1R, B2R, H1R, CysLT1R, and V1AR) expressed in VSMCs and shown to exhibit mechanosensitivity in response to cell swelling in heterologous expression systems (23), only AT1R has been shown by genetic deletion studies to be required for myogenic responsiveness in vivo. The identity of the particular AT1R isotype necessary for the generation of myogenic tone in rats and mice is controversial (451, 484, 1144, 1145), but it appears that relative expression levels dictate the comparative importance of AT1aR and AT1bR for the development of myogenic tone in a given artery and that the expression levels of these receptors vary considerably among different segments of the same vascular bed. The findings of one study suggest that AT1aR might detect pressures in the 120–160 mmHg range whereas AT1bR might detect pressures in the 60–120 mmHg range (1144), but other GPCRs contributing to pressure sensing could also be differentially expressed or differentially upregulated in these two different knockout mice. This observation is, however, not applicable to human arteries, which express only a single AT1R isoform. Another study demonstrated that myogenic constrictions in mesenteric and cerebral arteries, and in the perfused renal vasculature, were attenuated in Agtr1−/− mice and SM-specific Agtr1a−/− mice but not Agtr1b−/− mice (1150). In contrast, myogenic tone of cerebral and mesenteric arteries was not affected in Agtr1a−/− mice (484).
-
5)
Finally, a recent study is contradictory to the developing consensus that mechanosensitive Gq/11-coupled GPCRs mediate pressure-induced constriction. Offermanns and coworkers (1151) reported that inducible, SM-specific knockout of Gq/11 proteins did not significantly impair the development of pressure-induced myogenic tone of mesenteric arteries and posterior cerebral arteries from mice. Pressure-induced constriction, however, was attenuated by ∼80% in mesenteric arteries and ∼50% in posterior cerebral arteries from SM-specific G12/13-knockout mice and from mice with SM-specific deletion of LARG, suggesting that myogenic vasoconstriction depends critically on G12/13-mediated Rho/Rho kinase signaling but not on Gq/11 signaling (1151); this implies that myogenic responses are not transduced through Gq/11-coupled GPCRs to regulate PLC and downstream ion channels. Curiously, pressure-induced increases in VSMC intracellular Ca2+ concentration ([Ca2+]i) were also preserved in mesenteric arteries of both SM-specific G12/13- and SM-specific Gq/11-knockout mice. Although the myogenic responsiveness of arteries from other regions were not tested in that study, measurements of hindlimb and renal vascular resistance and perfusion were consistent with systemwide impairment in myogenic responses in G12/13-knockout mice. The reasons for the discrepancies between this study and the others cited above are not clear. Further work, including systematic studies of myogenic responsiveness of arteries from different vascular beds in both SM-specific Gq/11- and G12/13-knockout mice, is needed to resolve this issue, particularly in arteries shown in other studies to be regulated by mechanosignaling through AT1R.
4.1.3. GPCR- and G protein-mediated mechanotransduction in ECs.
Studies by Frangos and colleagues (1119, 1152) in ECs loaded with azidoanilido α-32P-GTP (AAGTP) suggested that Gαq/11 and Gαi3/o are G proteins that can be activated by LS to bind to the EC membrane. In γ-32P-GTP-loaded phospholipid vesicles constructed with purified G proteins, LS caused G protein activation in a dose-dependent manner that was positively correlated with EC membrane fluidity (1119, 1152). Exposure of ECs to GDP-β-S, a pan-inhibitor for G proteins, attenuated the LS-induced early release of cAMP and initial burst of NO production in a dose-dependent manner but had less effect on long-term responses (1061). The Gi/o inhibitor pertussis toxin (PTx) had little effect on shear-induced responses (1061), suggesting that Gi/o-coupled receptors are not involved in the shear-regulated EC mechanotransduction. Furthermore, PTx had little effect on LS-induced Ras activation, but overexpression of Gαq and Gβγ subunits enhanced the shear responses (1153). The use of a FRET-based biosensor to study the conformational changes of B2 bradykinin receptor (BDKRB2, a receptor coupled to both Gq/11 and Gi proteins) showed that LS and osmotic stress changed its dynamics similar to that after ligand binding, suggesting that BDKRB2 can sense mechanical stimuli and mediate mechanotransduction (110). These results suggest that Gαq/11, but not Gi/o, proteins are coupled to mechanosensitive GPCRs in ECs; these findings both support and contradict the findings of Erdogmus et al. (1137) described in sect. 4.1.4.
A recent study by Patapoutian and coworkers (107) using a high-throughput screening system with siRNA libraries identified GPR68, a Gαq/11 coupled receptor, as the GPCR most sensitive to physiological levels of shear stress in ECs. Transfection of GPR68 into HEK293T cells enabled them to respond to shear stress (20 dyn/cm2) with robust intracellular Ca2+ increases, primarily through IP3R-mediated Ca2+ release, whereas HEK cells transfected with other GPCRs, including the angiotensin receptor AGTR1, the endothelin receptor A (EDNRA), BDKRB2, the muscarinic cholinergic receptor (CHRM5), the histamine H1 receptor (H1R), the parathyroid hormone 1 receptor (PTH1R), and GPR132, were unresponsive to the equivalent shear stress. GPR68-expressing HEK293T cells responded to LS, PS, and disturbed shear. GPR68 expression was shown to be required for LS-induced Ca2+ signaling in mouse microvascular ECs (107). The expression of GPR68 was especially high in the EC layer of small arteries but not in arteries or venules. Mesenteric arteries from Gpr68−/− mice showed ∼50% impairment of flow-induced dilation compared with vessels from WT mice but no impairment in ACh-mediated dilation, suggesting that GPR68 is specifically involved in EC mechanotransduction. The lack of response of HEK293T cells expressing H1R to shear stress (107) conflicts with the findings of Erdogmus et al. (1137), who not only demonstrated a critical role for H1R in shear stress-induced Ca2+ responses in ECs but also found that expressing H1R in HEK293T cells conferred flow-induced Ca2+ signaling to them. The same study (1137) also found that other Gq/11-coupled GPCRs (including AGTR1), as well as Gs-and Gi/o-coupled GPCRs containing the COOH-terminal helix 8 domain, were mechanosensitive (see sect. 4.1.4); these differences remain to be resolved. Some of these issues are addressed in a recent review on GPCRs in EC mechanosensing (1154).
There are reports showing that OS (0 to 14 dyn/cm2 oscillation at 1 Hz) activates Gαq/11, causing it to dissociate from PECAM-1, whereas the ramped steady laminar shear (14 dyn/cm2) does not affect such association (1155). The Gαq/11-PECAM-1 association is mediated by heparan sulfate-dependent (1101), but Piezo1-independent (322), mechanisms to activate the Akt signaling pathway (1101). In vivo investigation in mouse aorta demonstrated the association between Gαq/11 and PECAM-1 in the straight part of the descending aorta and its absence in atheroprone areas in the arterial tree such as the renal branch (1155, 1156). The detailed mechanisms of interactions among GPCRs (e.g., GPR68, AGTR1, BDKRB2, and H1R), G proteins (e.g., Gαq/11), and other membrane proteins (e.g., PECAM1 and RTKs) in orchestrating EC mechanotransduction remain to be elucidated.
4.1.4. Molecular mechanisms of GPCR mechanosensitivity.
Only a few studies have investigated the molecular mechanism of GPCR mechanosensitivity. Komuro and colleagues (1157) used cysteine accessibility mapping strategies to demonstrate that stretch-induced activation of AT1R expressed heterologously in HEK293 cells may involve an anticlockwise rotation and shift of the seventh transmembrane domain (TM7) into the ligand-binding pocket of the receptor. Carboxyl residues of inverse agonists such as candesartan appear to block the movement of TM7 by binding to specific residues in that pocket, thereby stabilizing the receptor in its inactive conformation (1157). This stretch-induced active conformation of AT1R is distinct from that induced by agonist binding, with the two states also inducing different β-arrestin conformations (23, 1147, 1158). Other studies have suggested that TM6 might be mechanosensitive (1159) or that mechanical stretch acts by stabilizing β-arrestin-activated conformations of AT1R (1160, 1161). The possibility that the effects of agonists and mechanical forces may converge at the level of GPCRs suggests an alternate explanation for the well-established synergism between pressure-induced responses and agonist responses, which was previously explained by convergence of downstream signaling pathways (176, 177, 1162).
A recent study by Mederos y Schnitzler and colleagues (1137) has provided the most detailed insights into the mechanosensitivity of GPCRs. These authors studied H1R, a Gq/11-coupled GPCR, which they found to be expressed at a higher level than any other GPCR in HUVECs. Shear stress (4 and 20 dyn/cm2) or osmotic swelling induced HUVEC Ca2+ transients that were partially inhibited by the inverse H1R agonist mepyramine. Consistent with a key role for H1R in the EC Ca2+ response to shear stress, the authors observed an ∼50% reduction in flow-induced dilation and nitrate production in mesenteric arteries from H1R−/− mice or in mesenteric arteries from C57BL/6J mice in the presence of the inverse H1R agonists mepyramine and desloratadine. Flow-induced dilation in mesenteric arteries from WT mice and from H1/2/3/4R−/− mice was also completely blocked by the Gq/11 protein inhibitor YM254890 (1137), suggesting mechanosensitive contributions by other histamine receptors. HEK293 cells transfected with H1R also exhibited Ca2+ transients induced by hypoosmotic swelling that were almost completely blocked by mepyramine. Removal of the helix 8 domain of H1R eliminates the hypo-osmotic swelling-induced Ca2+ transient. FRET reporter studies pointed to the unfolding/refolding of the helix 8 domain (H8) on the COOH terminus of H1R as the mechanosensitive mechanism and confirmed that mechanical activation of H1R was independent of agonist binding (1137). H8 is a common feature of Gq/11-coupled GPCRs, which were previously shown to be mechanosensitive in the context of pressure-induced VSMC depolarization (23). Deleting or disrupting the structural integrity of H8 on H1R or on AT1R (the most-studied Gq/11 protein in VSMCs but not highly expressed in HUVECs) abolished the mechanosensitivity of both receptors to hypoosmotic swelling. Transfer of the H8 domain to otherwise nonmechanosensitive GPCRs, such as GnRH and CXCR4, conferred mechanosensitivity on them (1137). Importantly, transfection of HEK293 cells with D2R, a Gi/o-coupled GPCR that also contains H8, together with a Kir3 channel, which is normally regulated by D2R, enabled them to respond to hypoosmotic swelling with increases in Kir current. Similarly, the Gs-coupled adenosine A2A receptor, which possesses the H8 domain, is also mechanosensitive (1137). This very compelling set of data strongly suggests that GPCRs containing a complete H8 domain on their COOH termini are enabled to transduce mechanical force. Some of the HEK studies may need to be confirmed with shear stress rather than osmotic swelling as the mechanical stimulus, since it has been shown that the two stimuli exert different effects on membrane lipid order and membrane fluidity (943).
4.1.5. PIP2 availability and GPCR mechanoactivation.
An additional factor related to GPCR activation is the availability of PIP2. This negatively charged phospholipid resides primarily in the inner leaflet of the plasma membrane and is a substrate for PLC. PIP2 levels are dynamic, reflecting the net activity of GqPCR-stimulated hydrolysis by phospholipases and that of lipid kinases and phosphatases (1164). Since PIP2 is a known regulator of nearly 100 different ion channels (1165, 1166), changes in PIP2 levels alter the function of many PIP2-regulated proteins. Specifically, depletion of PIP2 by increases in GqPCR activity exerts an inhibitory effect on the ion channels activated by PIP2 but a disinhibitory effect on the ion channels tonically inhibited by PIP2 (1164). Nelson and coworkers (1001) have elegantly demonstrated the reciprocal regulation of Kir2.1 and TRPV4 channels by PIP2 in endothelium in response to GqPCR agonists; this could equally apply to mechanoactivation of GqPCRs since PIP2 availability depends on not only the activation state of the GPCR but also the downstream activity of PLC (1167). PIP2 conversion to DAG also results in a change in tension of the plasma membrane bilayer. In photoreceptors, the resulting compression of the inner leaflet causes a tiny change in the diameter of the rhabdomere, which is amplified by the stacking of rhodopsin molecules in rhabdomere microvilli to a degree sufficient to mechanically gate TRP channels (190). In principle, a similar change in bilayer tension (see sect. 2.1.4) could regulate mechanosensitive channels in VSMCs and ECs, but the highly specialized structure of the photoreceptor is likely required to produce changes in mechanical forces that are physiologically relevant.
4.2. Membrane Receptors and Junction Proteins as Mechanosensors
4.2.1. Membrane receptors.
LS activates receptor tyrosine kinases such as vascular endothelial growth factor receptor 2 (Flk-1) to result in Flk-1 clustering, phosphorylation, and association with the Src homology 2 (SH2) domain-containing adaptor proteins Shc and Grb2 to cause MAPK activation and MCP1 expression (1059). LS causes the recruitment of casitas B-lineage lymphoma protein (Cbl) to Flk-1 and subsequent activation of ERK but not JNK; VEGF treatment causes Flk-1 to recruit Nckβ, but not Cbl, to lead to ERK activation (1168). These findings suggest a difference in the chemical and mechanical regulatory mechanisms for the cell surface receptors. The LS-induced Flk-1/Cbl association leads to the activation of phosphatidylinositol-3-kinase (PI3K), Akt, and IκB kinase (IKK). Treatment with U1498 (a Flk-1 inhibitor) or mutation of Cbl attenuates the shear-induced PI3K/Akt activation and NF-κB nuclear translocation (1168–1170), leading to consequent modulations of gene transcription and phenotype. LS has also been shown to induce a magnitude- and time-dependent Tie2 phosphorylation that is positively correlated to the activations of PI3K and Akt (1171), but the causality and functional consequences are not reported in the study. It has been shown that Tie2 knockdown in endothelial progenitor cells abolishes the flow-induced PI3K/Akt/eNOS activation and the consequential reendothelialization in the denuded carotid artery (106), suggesting Tie2 as an upstream regulator for the shear induction of PI3K/Akt/eNOS. Tie1 is predominantly expressed in the atheroprone areas of the arterial tree in mice, and atherosclerotic lesion formation on high-fat diet has been found to be reduced in Tie1+/−;Apoe−/− mice compared with Tie1+/+;Apoe−/− mice (1172). An in vitro study has shown that LS attenuates Tie1 expression in mouse ECs and that deletion of Tie1 causes an augmentation of the shear-induced phosphorylation of Tie2 and expressions of eNOS and IκBα, as well as a further reduction of the shear-suppressed ICAM-1 expression (1172).
A recent study has identified plexinD1 as a mechanosensitive receptor to regulate EC functions under PS and OS (1173). PlexinD1 is a member of the semaphorin (SEMA) family of cell-guidance signaling proteins, which is composed of an extracellular SEMA binding domain and a transmembrane domain, with a cytoplasmic tail that contains GTPase-activating protein (GAP) and Rho binding domains (RBDs). PlexinD1 plays important roles in cardiovascular development and disease progression (for review see Refs. 1174, 1175). Knockdown or mutation of plexinD1 significantly attenuates the atheroprotective flow-induced EC alignment, phosphorylation of Flk1, ERKs, Akt, and eNOS, as well as KLF expression. Mechanistic investigation demonstrated that upon shear application plexinD1 forms a mechanosensing complex with Flk1, VE-cadherin (VE-cad), NRP1, PI3K, and Scr in a ligand-independent manner for mechanotransduction. EC-specific inducible knockout enhances lesion formation in the descending aorta (1173). It is interesting that plexinD1 knockdown also attenuates the atheroprone flow-induced MCP1 and VCAM1 expressions (1173). These findings add a new family of receptors that can coordinate mechanosensing for vascular health. The detailed mechanisms of the upstream shear regulation of plexinD1 expression/activation and the downstream mechanotransduction remain to be determined.
Cyclic stretch activates multiple RTKs, including Flk-1 and Tie2 (1176, 1177). A 15% uniaxial stretch of rat coronary microvascular ECs enhances their response to VEGF-induced proliferation and increases the expression of ANG I/II (1177), suggesting potential vascular dysfunction. A 50% area increase of HUVECs by biaxial stretch causes a rapid Flk-1 activation, secretions of von Willebrand factor (vWF) and IL-8, as well as the translocation of P-selectin to the EC surface to induce leukocyte adhesion (1176). Such vWF release is attenuated by knockdown of Flk-1 or PLCg and treatment with BAPTA [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, a Ca2+ chelator] (1176), demonstrating that Flk-1 mediates the stretch-induced vWF release. It is interesting that knockdown of Flk-1 attenuates the stretch-induced Akt/eNOS activation, indicating that Flk-1 also mediates the stretch-induced Akt/eNOS activation to decrease vWF release (1176). An in vivo study in a partial portal vein ligation (PPVL) hypertension mouse model demonstrates that stretch causes the translocation of P-selectin to the EC surface; this effect is attenuated by the inhibition of Flk-1 (1176). However, eNOS knockout enhances the PPVL-induced P-selectin membrane localization. It is likely that Flk-1 mediates both inflammation (increases of IL-8/P-selectin) and homeostasis (eNOS activation) pathways to orchestrate the stretch regulation of vascular function.
4.2.2. Junction proteins.
Cells interconnect through a number of different transmembrane proteins, including adherens junctions, tight junctions, connexins, and desmosomes (1178). Each of these classes of junctional proteins has distinct mechanical properties and components that link to subcellular signaling pathways, and all have been implicated in mechanosensitivity in various contexts. Adherens junctions and connexins are widely expressed in almost all cell types. Tight junctions are found mostly in epithelium and endothelium. Desmosomes form strong cell-cell adhesions by virtue of their extensive coupling to the subcellular intermediate filament network. They are enriched in tissues subjected to repeated mechanical stress, e.g., cardiomyocytes and VSMCs (1179). The role of the desmosome in mechanotransduction is not well studied, but new FRET-based desmosome tension sensors have recently been developed that promise to advance that field (1180–1182). Here, we briefly discuss the role of adherens junctions, both because they are the principal cell-cell junctions and because evidence exists for their role in VSMC and EC mechanotransduction.
Adherens junctions are formed by the homotypic interactions of the cadherin family of proteins. E-cadherins are expressed primarily in epithelium, VE-cadherin (VE-cad) is expressed primarily in endothelium, and N-cadherin is expressed in other cell types, including neurons and VSMCs (1178). VSMCs also express R-cadherin, T-cadherin, and cadherin-11 (1183). Cadherins are composed of an NH2-terminal extracellular domain, a transmembrane domain, and a cytoplasmic COOH terminus that binds to the actin CSK via interactions with the catenin family of proteins (1184). The catenin family includes p120 catenin and α-, β-, and γ-catenin. The cytoplasmic domains of cadherins serve as a protein scaffold to facilitate interactions with α-catenin, β-catenin, and plakoglobin to form a “cadherin-catenin complex.” α-Catenin and β-catenin are primarily responsible for interacting with actin and associated proteins, where α-catenin has a role analogous to that of talin in the focal adhesion. α-Catenin is autoinhibited in the absence of applied tension, but under tension it partially unfolds to unmask a cryptic binding site for vinculin (1185, 1186). As mentioned in sect. 5.2, vinculin has three major domains: a head domain that binds to α-catenin, β-catenin, and talin, a proline-rich domain that binds to VASP and Arp2/3, and a tail domain that binds to paxillin, F-actin, and PIP2 (1190). Vinculin exists in an autoinhibited state in the cytoplasm, but when bound to α-catenin it partially unfolds and is released from autoinhibition; subsequent phosphorylation by kinases such as Abl (1187, 1188) enables its binding to actin, where it exhibits catch-bond behavior (1189). Changede and Sheetz (1190) have shown that this property of both α-catenin and vinculin confers potential mechanosensitivity to the cadherin junction. In addition, the link between cadherins and intracellular proteins involved in actin polymerization, such as paxillin, VASP, and Arp2/3, suggests the possibility that cadherin-mediated mechanotransduction could regulate dynamic actin assembly in VSMCs.
4.2.2.1. JUNCTION PROTEINS IN SMCS.
Cadherins are involved in multiple aspects of VSMC function (1183), particularly in phenotype switching, but only a few studies have implicated them in VSMC mechanotransduction. ACh-induced activation of airway SM contraction increases the recruitment of β-catenin to N-cadherin, and blocking that recruitment inhibits ∼50% of the force development (1191, 1192). The reduction in force is not associated with a reduction in MLC20 phosphorylation or ACh-induced actin polymerization, but actin polymerization is required for the recruitment of β-catenin to N-cadherin (1191). One study provides evidence that cadherins might be involved in myogenic tone and pressure-induced arterial constriction. Meininger and coworkers (1193) found that pressure-induced constriction of isolated skeletal muscle arterioles was attenuated by an inhibitory peptide containing the HAV sequence that binds to the ectodomain of N-cadherin; likewise, pressure-induced constriction was attenuated by a function-blocking antibody to N-cadherin. Collectively, these three studies (1191–1193) suggest a critical role for N-cadherin in the contraction response of vascular and visceral smooth muscle in response to agonists and pressure, but they leave unresolved the issue of whether this is simply a structural requirement for force transmission to neighboring VSMCs through existing cadherin junctions or whether there exists an underlying signaling mechanism involving the assembly of an intracellular cadherin-catenin complex that regulates one or more of the three pathways shown in FIGURE 16. This is a potential area for future research.
FIGURE 16.
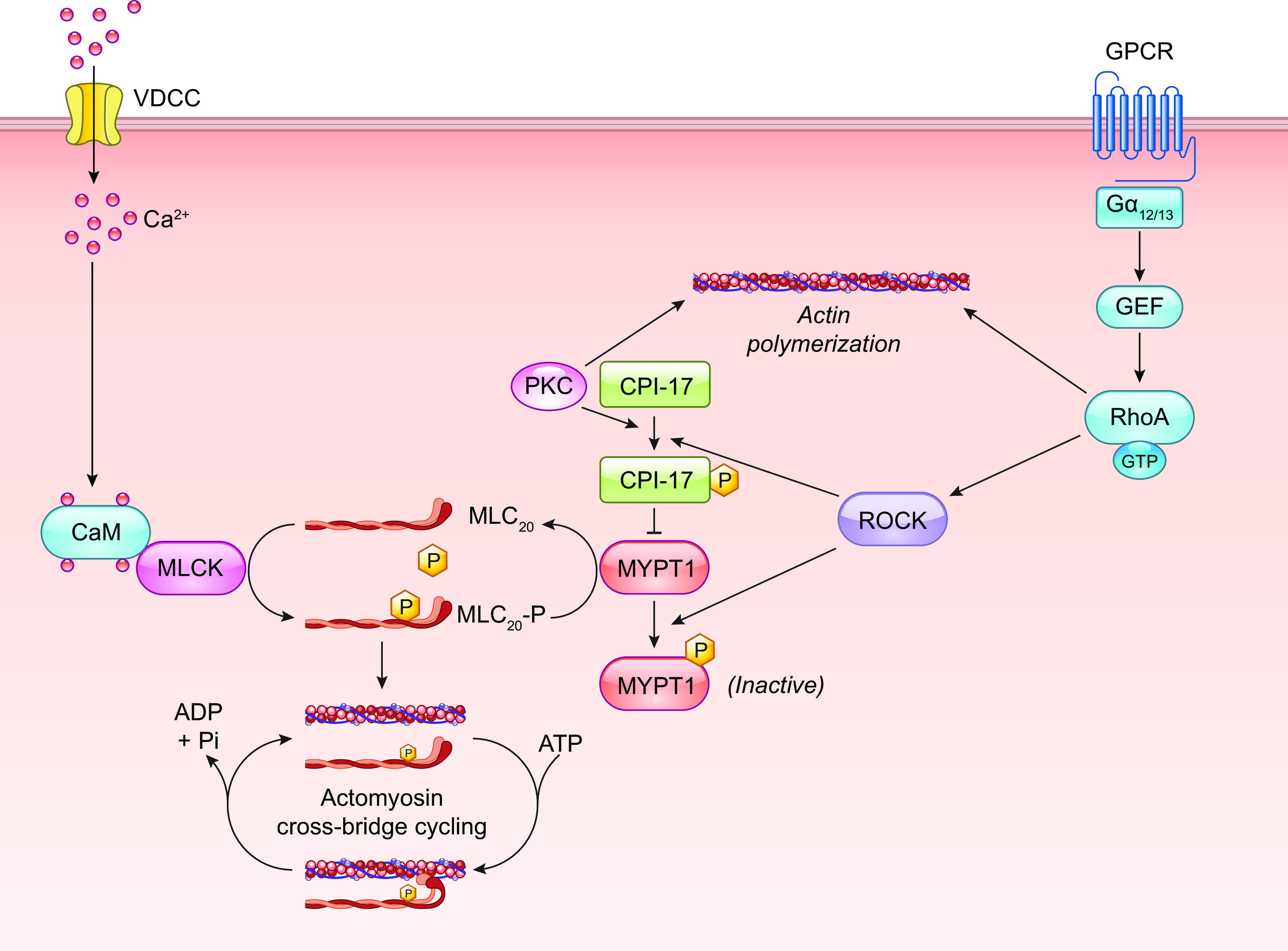
Ca2+-independent regulation of MLCP and constriction through Rho-ROCK signaling and control of actin polymerization. See glossary for abbreviations.
4.2.2.2. JUNCTION PROTEINS IN ECS.
The application of external shear to an EC monolayer transmits forces throughout the EC, including the apical, basal, and intercellular spaces (for review see Ref. 1194). The interactions between neighboring ECs are mainly controlled by the junction proteins, including gap junction proteins [e.g., connexins (Cxs)], adherens junction proteins (e.g., PECAM1 and VE-cad), and tight junction proteins (e.g., claudins). Early studies have shown that the EC gap junction protein Cx43, but not Cx37 or 40, is preferentially increased under disturbed flow in vivo and in vitro (1062, 1195). In a recent study on the role of connexin 37 in regulating atherosclerosis plaque formation in vivo, a restrictor casting was used to modify shear flow in Apoe−/−;Cx37−/− mouse carotid arteries with the creation of areas with high versus low shear stresses as well as OS (1196). It was found that the depletion of Cx37 increases the plaque size and lipid content while decreasing the stability at oscillatory shear flow areas. Studies on EC-specific Cx40-knockout Apoe−/− mice have also shown that the depletion of Cx40 exaggerates lesion formation in OS areas (1197). These results indicate that connexins play important roles in atherogenesis at the flow-disturbed areas because of their contributions to intercellular communication and junction integrity. EC-specific Cx43 knockout has been shown to lead to hypotension with elevated NO and ANG I/II production in mice (1198), suggesting a significant role of Cx43 in regulating vascular dilation and homeostasis. In contrast, global Cx40 knockout leads to 25% higher arterial pressure and attenuates acetylcholine-induced vasodilation (1199). These results indicate the multiple roles of connexins in the regulation of mechanical responses in the vascular system. Several studies have shown the activation of adherent junction proteins PECAM-1 and VE-cad by LS (1200–1204). PECAM-1/VE-cad/Flk-1 have been found to form a mechanosensory complex that mediates LS-induced EC alignment and AKT/PI3K/NF-κB signaling (112). The application of LS rapidly induces the interaction between VE-cad and Flk-1 (1200), with the transactivation of Flk-1 via the transmembrane domain of VE-cad, leading to downstream PI3K signaling (1205). Using FRET-based tension sensors for VE-cad and PECAM-1, Schwartz and colleagues (1204) showed that LS causes the junctional tension to increase rapidly across PECAM-1 but decrease across VE-cad; these findings suggest that the flow regulation of junction proteins is an active process rather than a passive resultant of cytoskeleton remodeling. Studies on Apoe−/− mice have shown that PECAM-1 depletion reduces lesion formation at areas of the aorta with disturbed flow (1206, 1207). A study on Ldlr−/− mice has shown that Pecam-1 knockout attenuates the atherosclerotic lesion formation in the inner curvature of the aortic arch but enhances it in the aortic sinus, arterial branches, and descending aorta (1208), suggesting that PECAM-1 may play different roles in the response to different flow patterns.
LS (10 dyn/cm2 for 3 h) to cultured ECs decreases the expression of the tight junction protein occludin but increases its phosphorylation to enhance shear-induced hydraulic conductivity (1209). Such positive correlation between occludin phosphorylation and hydraulic conductivity found under steady LS at 10 dyn/cm2 does not occur under PS at 10 ± 5, 10 ± 10, and 10 ± 15 dyn/cm2 or OS at 0 ± 20 dyn/cm2 (1210). It has been shown that steady LS (10–20 dyn/cm2) enhances microvascular endothelial barrier function by increasing ZO1 and occludin expressions and their junctional localization, whereas pathological levels of high steady LS (40 dyn/cm2) and PS at 10 ± 10, 20 ± 20, and 40 ± 40 dyn/cm2 do not (1211). It has also been found that 24-h LS (10 dyn/cm2) causes an increase of junctional localization of occludin and ZO1, a reduction of occludin phosphorylation, and an enhancement of endothelial integrity; such flow regulation of tight junction remodeling is dependent on VE-cad-Rac1-Tiam1 signaling, suggesting the signal is relayed among junctional proteins (1212). These studies demonstrate that the regulations of junction proteins are flow pattern specific; however, the interplay among junction proteins in regulating endothelial functions under different flow conditions remains to be elucidated.
Cyclic stretch modulates EC junctions (for review see Refs. 1213, 1214), and Piezo1 has been identified to play a key role in mediating this vascular junction remodeling (1215, 1216). Biaxial stretch of 20% leads to transient (at 5–30 min) EC junction (VE-cad staining) disruption by actomyosin contraction. Time course phosphoproteomics studies reveal that Rho and Rac are the potential regulatory pathways correlated with the Piezo1-mediated Ca2+ influx and junction disruption (1215). Stretch with a duration of 1–3 h leads to downregulation of Piezo1, attenuation of Ca2+ influx, phosphorylation of filamin, increased EC stiffness, and restoration of cadherin junctions (1215). These biphasic changes (i.e., transient junction disruption at 5–30 min and restoration at 1–3 h) provide the mechanisms for the dynamic stretch-regulated junction remodeling. An in vivo study demonstrated that EC-specific Piezo1 depletion enhances hyperventilation-induced pulmonary leakage by decreasing VE-cad and activating Src and paxillin, suggesting that Piezo1 is also involved in long-term (4–72 h) stretch-induced disruption of vascular junctions. This mechanistic study demonstrated that stretch activates Piezo1 and calpain (a Ca2+-dependent cysteine protease downstream of Piezo1) to cleave Src, reduce VE-cad phosphorylation, and destabilize EC junctions. Activation of Piezo1 or calpain mitigates the junction disruption induced by a high level of cyclic stretch (18% elongation) (1216). These results indicate that mechanical stretch induces diverse dynamics of multiple mechanotransduction pathways for EC regulation.
5. MECHANOSIGNALING PATHWAYS
5.1. Rho/ROCK Pathway
5.1.1. Rho/ROCK pathway in VSMC and myogenic tone.
A signaling pathway centered on Ras homology family A (RhoA) and Rho-associated protein kinases (ROCKs) influences the contractility of VSMCs (for review, see Ref. 1217). RhoA is a small GTPase downstream of GPCRs activated by guanine nucleotide-exchange factors (GEFs) that exchange bound GDP for GTP, and it is inactivated by GTPase-activating proteins (GAPs), which enhance the intrinsic GTP hydrolysis activity of RhoA. RhoA controls the actions of several downstream effector proteins that impact on VSMC proliferation, migration, differentiation, and contractility. ROCK1 and ROCK2, the two most intensely investigated RhoA effectors, are closely related serine-threonine protein kinases activated by interactions with GTP-bound RhoA. Both are present in VSMCs. Activated ROCK2 alters contractility by phosphorylating myosin phosphatase target subunit 1 (MYPT1), a regulatory subunit of myosin light chain phosphatase (MLCP), at threonine residue 855 of the rat protein (1218). Phosphorylation by ROCK decreases MLCP activity, thereby increasing the net phosphorylation level of the myosin regulatory light chain (MLC20) by MLCK and enhancing VSMC contractility (FIGURE 16). MLCP activity is also regulated by CPI-17, a small cytosolic protein that, when phosphorylated at Thr-38 by ROCK or PKC, binds to and inhibits the catalytic subunit of MLCP (1219). Loss of ROCK activity decreases MLCP phosphorylation, increasing its phosphatase activity and lowering MLC phosphorylation levels, thereby promoting muscle relaxation. Importantly, modulation of ROCK activity can control the diameter of blood vessels in a manner that is independent of changes in cytosolic Ca2+ levels. This contractile process is referred to as “Ca2+ sensitization” and has been reviewed extensively (1039, 1220, 1221). The Rho/ROCK/MLCP signaling pathway likely explains the observations by multiple laboratories that arterial myogenic tone/constriction does not always depend on VGCC-mediated Ca2+ influx and that pressure-induced [Ca2+]i changes do not always correlate with the degree of arterial constriction (174, 1151, 1222, 1223) (for review see Ref. 1224).
Many studies have investigated the involvement of the RhoA/ROCK pathway in the development of vascular myogenic tone. VanBavel et al. (1225) demonstrated that the ROCK inhibitor Y-27632 caused a concentration-dependent inhibition of myogenic tone in isolated rat mesenteric arteries without affecting pressure-induced changes in intracellular [Ca2+]. Other studies report similar effects of pharmacological blockade of ROCK in several types of blood vessels, including rat tail (1226), cerebral (174, 1227, 1228), ophthalmic (1030), and skeletal muscle (166) arteries. These findings obtained by ex vivo pressure myography suggest that tonic activity of ROCK is necessary for the development of basal myogenic vasoconstriction. However, the available in vivo data are less convincing. Global knockout of Rock1 or Rock2 on the C57BL/6 background is lethal during embryonic development (1229). Studies using Rock1+/− and Rock2+/− heterozygous mice have shown that, although levels of targeted proteins are lower in vascular tissues, systolic and diastolic blood pressures and cardiac output do not differ, suggesting that decreases in neither Rock1 nor Rock2 expression alter total vascular resistance (1230–1232). In addition, it has been reported that twofold overexpression of Rock2 in the vasculature did not change resting systolic blood pressure (1230). Thus, it appears that altering ROCK expression levels does not affect the resistance arteries. The reasons for these discrepancies are not apparent. Notably, the effects of conditional SM-specific knockout of Rock1 or Rock2 on vasoconstriction have not been reported.
Evidence that increases in intraluminal pressure directly activate the RhoA/ROCK pathway in VSMCs is scant. Investigating the possible involvement of sphingosine kinase 1 (SPHK1) as a modulator of the RhoA/ROCK pathway in the myogenic response, Bolz and coworkers (1134) reported that heterologous overexpression of SPHK1 increased the myogenic tone of hamster gracilis arteries. This response was blunted by heterologous coexpression of dominant-negative mutant forms of RhoA or ROCK together with SPHK1 (1134). However, that study did not report the effects of expression of dominant-negative forms of RhoA and ROCK independent of SPHK1 overexpression, and no mechanistic link between elevated intraluminal pressure and increased SPHK1 activity was identified. G12/G13 proteins activate Rho/ROCK signaling through direct interaction of their α-subunits with RhoGEFs, which include p115-RhoGEF, PDZ-RhoGEF, and ARHGEF12 (also referred to as leukemia-associated RhoGEF, LARG) (1233). ARHGEF12 is expressed in arteries and is the predominant RhoGEF in mouse aorta (1163). Offermanns and coworkers (1151) showed that inducible SM-specific knockout of the ARHGEF12 causes the loss of pressure-dependent RhoA activation and myogenic constriction of mouse mesenteric arteries. These results are in apparent contradiction to a previous finding that conditional SM-specific knockout of ARHGEF12 or G12/13 did not affect resting arterial pressure (1163), implying that this pathway does not significantly affect vascular resistance. However, SM-specific knockout of G12/G13 was found to blunt the myogenic constriction of mesenteric arteries (1151). These data support a pathway in which pressure elevation is sensed by a G12/13-coupled GPCR to activate RhoA/ROCK through the ARHGEF12 (FIGURE 16).
In summary, the RhoA/ROCK pathway appears to be critical for Ca2+ sensitization of VSM (1039, 1220, 1224), but there is conflicting evidence on whether this pathway is a critical mediator of pressure-induced arterial constriction.
5.1.2. Rho/ROCK pathway in EC mechanosignaling.
Rho family GTPases have been shown to be important for the modulation of shear-induced signaling and CSK remodeling (1234, 1235). It has been demonstrated that LS activates all three Rho family GTPases: RhoA, Cdc42, and Rac1. Thus, LS causes a transient nuclear translocation of Cdc42 to activate the JNK/AP1 pathway and a sustained nuclear translocation of RhoA to result in P160ROCK activation, stress fiber formation, and cell alignment (1236–1238). Using a FRET-based biosensor, Schwartz and colleagues (1239) have shown that LS induces a directional Rac1 activation downstream to the flow-induced new integrin-ECM binding sites, leading to NF-κB activation and cell alignment. LS induces a polarized Cdc42 activation in the direction of flow to modulate the localization of microtubule organizing center (MTOC) to the downstream direction of flow (1240). Although RhoA/ROCK and Rac are involved in shear-induced EC polarization (elongation, alignment, and directional migration), the inhibition of Cdc42 does not affect EC polarization (1241). These studies demonstrated the differential roles of RhoGTPases in regulating LS-induced EC polarization, CSK remodeling, and cell alignment. It has been reported that inhibition of RhoGTPases with C3 isoenzyme retards EC migration into denuded surfaces to delay wound healing under both LS and disturbed flow (1242). However, the inhibition of ROCK enhances EC migration to accelerate reendothelization in the areas denuded by the stent but does not affect the migration of ECs without stenting (1243), suggesting that the different mechanical environments introduced by stents have significant effects on ROCK-mediated EC migration. These results suggest that all RhoGTPases (Rho/Rock, Rac, and cdc42) are involved in signal transduction under different flow patterns, but they have differential effects on EC migration. The detailed roles of RhoGTPases and their spatiotemporal differences in regulating EC functions require further investigation.
RhoA has been shown to mediate the perpendicular alignment of BAECs induced by 10% uniaxial stretch, as evidenced by the reorientation of cell alignment by inhibition of the Rho/ROCK/mDia pathway (1244, 1245). The same level of stretch-induced alignment can be achieved with only 3% uniaxial stretch after overexpression of the constitutive active form of Rho (RhoV14) (1244), indicating a cooperative effect of mechanical and biochemical stimuli for the regulation of EC alignment.
5.2. Cytoskeleton, Integrins, and Extracellular Matrix in Mechanosignaling
5.2.1. Cytoskeleton, integrins, and extracellular matrix in VSMC mechanosignaling.
Mechanosensing and mechanotransduction potentially can be mediated by elements of the CSK itself, by interactions between the CSK and the ECM surrounding the cell, and/or by interactions between adjacent cells through junctional proteins. These mechanisms are discussed in the context of pressure-induced constriction of VSM.
5.2.1.1. CYTOSKELETON.
The CSK is an intricate structure composed of actin fibers, microtubules, intermediate filaments, and dozens of associated proteins. A mechanical analogy describes CSK components as being organized into an interconnected network of struts and coils embedded in a cytoplasmic gel (1246, 1247). Experiments by Ingber (153) and colleagues have documented the “tensegrity” properties of the CSK, which align with established physicochemical principles governing mechanical structures. Tensegrity in the context of a cell refers to the force balance of CSK elements, some under tension and others under compression, to enable the maintenance of a stable, three-dimensional structure that can resist perturbations by relatively large external forces. This concept has profound implications for explaining how forces are transduced and redistributed within the CSK; how ECM proteins resist cell tension and promote structural CSK rearrangement; and how internally generated actomyosin forces can result in the unloading of tension-bearing elements of the CSK to provide a potential mechanism for feedback inhibition of the response to external force application.
The individual elements of the CSK are organized into multiple levels or domains. The cortical CSK is a thin, interwoven meshwork composed largely of actin, spectrin, and ankyrin filaments. Being closely associated with the plasmalemma, it determines and/or redistributes stress on the membrane bilayer and associated proteins. In VSMCs, the submembranous CSK is populated with dense bodies, enriched in vinculin and α-actinin, through which internal tension development is transmitted to the ECM and neighboring cells (1248–1250). The deeper CSK is composed of a lattice network of actin and myosin filaments that are somewhat organized but not into defined sarcomeres as in striated muscle (1251). Intermediate filaments, enriched in vimentin and desmin, interconnect all these structures (1252). Perinuclear microtubules are the stiffest CSK components, whose resistance to compression opposes the axial shortening of the cell. Agents that disrupt microtubules, such as colchicine and nocodazole, increase the basal tone of arteries/arterioles independent of the endothelium (1253) and enhance the constriction in response to agonists (1254–1256). Agents that stabilize microtubules inhibit, or have little effect on, basal vascular tone (1253, 1257). The microtubule network in differentiated VSMCs is thought to be relatively stable as opposed to the cortical actin network, which can undergo dynamic changes under physiological conditions.
5.2.1.2. DYNAMIC ACTIN ASSEMBLY.
The actin CSK is dynamic, and its assembly/disassembly has important implications for tone development in VSM. Actin exists in the cell as both soluble 42-kDa monomers (G-actin) and insoluble filamentous actin (F-actin) polymers of variable size. Each G-actin monomer is polarized, with plus and minus ends that mediate head-to-tail interactions with other G-actin monomers that are oriented in the same direction when polymerized into filaments. F-actin ends grow at different rates, with monomers added to the plus end at 5–10 times the rate of their removal at the minus end (1258). F-actin fibers are cross linked by the 100-kDa protein α-actinin. F-actin assembly/disassembly is facilitated by the actin-binding proteins cofilin and profilin (1259). Cofilin preferentially binds to ADP-actin and sequesters actin monomers in the ADP-bound form, preventing their reincorporation into filaments and producing G-actin monomers with barbed ends that provide nucleation sites for the formation of new F-actin polymers. Profilin can reverse this effect by stimulating the exchange of bound ADP for ATP, resulting in the formation of ATP-actin monomers, which dissociate from cofilin and are then available for assembly into filaments (1258). Cofilin activity is regulated by phosphorylation at serine 3 (1260), which facilitates its binding to scaffolding and focal adhesion proteins (1261) and abolishes its severing function (1262, 1263). In VSMCs, cofilin activity appears to be restricted to the cortical CSK and it does not interfere with the stability of cycling actomyosin cross bridges. Evidence that actin filaments themselves can sense and transduce mechanical tension (1264) comes from in vitro assays using purified proteins, where tension applied to actin filaments 1) lowers the activity of cofilin (1265), 2) increases the binding of cofilin to actin (1266), and 3) increases the affinity of actin for myosin II motor domains (1267). The extent to which this level of regulation occurs in intact, native VSMCs is not known.
G- and F-actins are typically measured by cell fractionation assays or fluorescence imaging methods. VSM contraction is associated with a decrease in the G-actin fraction and an increase in the F-actin fraction, resulting in an increase in the F- to G-actin ratio. A typical finding for VSMCs is 20–30% G-actin and 70–80% F-actin in the absence of vasoconstrictor agonist (or at low distending pressure), with agonist stimulation leading to a 30–40% decrease in G-actin and ∼10% increase in F-actin (1250). More subtle stimuli, such as pressure elevation, would be expected to produce more modest changes. Cerebral artery VSMCs contain a substantial pool of G-actin that is significantly reduced upon pressure elevation, with concomitant elevation in F-actin. Intact arteries lose tone in response to global actin cytoskeleton disruptors such as cytochalasin (a relatively crude approach). Conversely, compounds that promote F-actin polymerization and stabilize preexisting actin filaments cause a decrease in G-actin content and arterial constriction (1268). The process of pressure-induced F-actin polymerization in VSMCs has been visualized by confocal microscopy in mouse tail arteries (743). Actin depolymerization has also been shown to be an important component of agonist-mediated vasodilation of skeletal muscle arterioles (1269). It is important to note that inhibition of only a relatively small fraction of actin polymerization is sufficient to alter force development, as confirmed by confocal imaging (1270, 1271) and atomic force microscopy (AFM) studies (1272–1274). In AFM studies, the stiffness of the cortical CSK (1275) is altered by cytochalasin but not by ML-7, an inhibitor of MLCK and subsequent actomyosin-mediated force development. Likewise, ML-7 does not inhibit actin polymerization (1272, 1273). These observations point to the possibility that at least two different pools of actin, possibly composed of different actin isoforms, are distributed within the VSMC, one in the cortical CSK and another involved in actomyosin force development (1274). Reinforcement of the cortical actin CSK is thought to aid in the transmission of tension generated by internal cross-bridge cycling (1273, 1276).
Actin polymerization is controlled by a complex sequence of steps first elucidated by Pollard et al. (1277) in studies of migrating nonmuscle cells, but the underlying principles also appear to apply to differentiated smooth muscle. Gunst and colleagues have performed the most relevant mechanistic studies of this process using airway smooth muscle. Their data suggest a key role for another actin binding protein, Arp2/3 (1278, 1279), in creating nucleation sites for the formation of new cortical F-actin filaments (FIGURE 17). The Arp2/3 complex requires activation by the nuclear protein factor N-WASP (neuronal Wiskott–Aldrich syndrome protein), whose recruitment is catalyzed by the small GTPase cdc42 (1280). F-actin assembly under these conditions also depends on RhoA-mediated assembly and phosphorylation of a number of focal adhesion proteins and kinases (1281), including the adaptor proteins paxillin and CrkII (1282, 1283), PAK (p21-activated kinase), Rho kinase (1284, 1285), and GIT (G protein interacting target) proteins (1273). Cofilin phosphorylation/dephosphorylation is critical to this process (1260). α-Actinin is not required for actin polymerization but is recruited to dense plaques and dense bodies during this process and required for force transmission (1286). The roles for these proteins in airway smooth muscle were elegantly elucidated by Gunst and colleagues in tissue extracts, freshly dissociated SM cells, and isometric preparations of airway smooth muscle, in which dominant-negative peptides, shRNA constructs, or kinase-inactive mutants of critical proteins could be delivered intracellularly by transient permeabilization. Studies by other laboratories provided evidence that these same pathways are involved in dynamic F-actin assembly in agonist-stimulated VSMCs (1287, 1288) and length-dependent VSMC force development (1289), although whether they are also involved in myogenic constriction to pressure elevation is not yet known. The time course over which new actin polymerization occurs is unlikely to account for the very rapid phase (<300 ms) of myogenic constriction in some arterioles (118), but it could explain components of the slower phase(s) of the response (1290).
FIGURE 17.
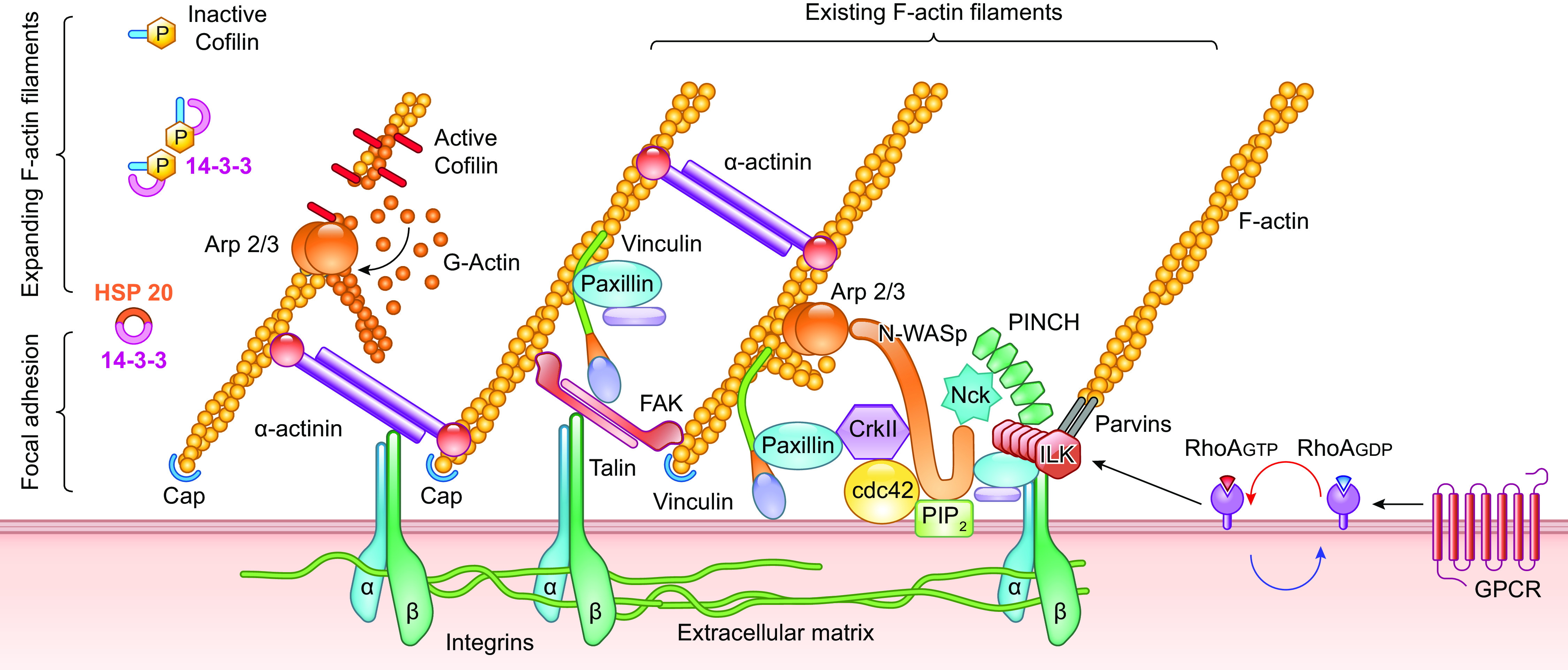
Regulation of actin polymerization in VSMCs by GPCR and integrin signaling at the focal adhesion. See glossary for abbreviations.
5.2.1.3. ACTIN ASSEMBLY AS AN ALTERNATIVE MECHANISM OF SMOOTH MUSCLE FORCE DEVELOPMENT.
How does the dynamic assembly of F-actin in response to applied force integrate with the two pathways for regulating MLC20 phosphorylation discussed above (sect. 5.1.1)? As in striated muscle, actomyosin cross-bridge cycling in VSMCs is the fundamental mechanism for tension development. A mechanical stimulus or the binding of a contractile agonist to its appropriate GPCR results in Ca2+/CaM-mediated activation of MLCK and subsequent phosphorylation of MLC20 at serine residue 19 (1291), enabling the cyclical activation of heavy chain myosin heads (smooth muscle type II), via their ATPase activity, to produce force by ratcheting along a central lattice of actin filaments (1292). This pathway is counterbalanced by the RhoA-ROCK-MYPT pathway that regulates MLC20 phosphorylation through MLCP to control Ca2+ sensitivity. A well-known property of smooth muscle is that force development can be maintained during a sustained contraction at a low rate of ATP hydrolysis by the actomyosin ATPase, i.e., the so-called “latch state” (163). The molecular basis of the latch state has been attributed to a subpopulation of slowly cycling, dephosphorylated cross bridges, but an alternative possibility is that dynamic changes in actin polymerization underlie sustained force development (1289, 1293). In line with this idea, Saito et al. (1294) found that cytochalasin interfered with tension development of rat aorta without concomitant changes in global Ca2+ levels, MLC20 phosphorylation, or myosin ATPase activity. In studies on cerebral arteries, cytochalasin D abolished the constriction to pressure elevation while actually enhancing the pressure-induced global [Ca2+] increase (1295). Indeed, a number of studies in vascular and nonvascular smooth muscle have confirmed the importance of dynamic actin assembly (1271, 1273, 1294, 1296, 1297), including studies of myogenic responses of rat cerebral arteries (1298), mouse mesenteric arteries (1299), and skeletal muscle arterioles (1032). Collectively, these studies point to actin reinforcement of the CSK as a third mechanism controlling force development in vascular (and nonvascular) smooth muscle that is distinct from the two canonical pathways regulating MLCK and MLCP (FIGURE 16).
How might actin polymerization be initiated through the N-WASP-Arp2/3 pathway in response to the application of mechanical force such as pressure elevation? A number of contractile agonists in VSM work through Gα12/13-, Gαq/11-, and Gi-coupled GPCRs (1300, 1301) to activate guanine nucleotide-exchange factors, including LARG and multiple RhoGEFs (1220), which in turn activate RhoA (1302, 1303). Mechanical activation of these GPCRs might initiate this process (see sect. 4.1), but the downstream signaling pathways are also linked to integrin-dependent signaling cascades through RhoGEF12 (1304). Gunst and colleagues (1273, 1281) provide evidence that existing focal adhesions and/or their expansion are required upstream of N-WASP, because they serve as a scaffolding to anchor new, cortical F-actin polymers that are generated. The specific focal adhesion proteins involved are discussed in the following section, but a few are mentioned here in the immediate context of N-WASP-Arp2/3 signaling. A potentially critical protein in the initial steps of this pathway is integrin-linked kinase (ILK), a 59-kDa protein that binds to the cytoplasmic domain of β-integrins and serves as a scaffolding protein in focal adhesions (FIGURE 17). ILK, like ROCK, was initially thought to be a Ca2+-independent kinase capable of MLC20 diphosphorylation at threonine 18 (in addition to monophosphorylation at serine 19), which increases actomyosin MgATPase activity at subsaturating actin concentrations (1291, 1305, 1306), but more recent studies call into question its kinase activity (1307, 1308). ILK, PINCH (particularly interesting Cys-His-rich protein), and α-parvin form an ILK-PINCH-parvin (IPP) complex that is recruited to focal adhesions from the cytoplasm through their interactions with paxillin and vinculin (1309, 1310). A proline-rich domain in vinculin binds to VASP and Arp2/3; a tail domain binds to paxillin and F-actin (1178). Gunst and colleagues used recombinant GFP-tagged ILK and PINCH constructs to demonstrate the recruitment of these proteins to focal adhesions in tracheal smooth muscle in response to ACh stimulation. The importance of this pathway was confirmed when expression of a peptide fragment of the NH2-terminal LIM1 domain of PINCH, which competes with endogenous PINCH, inhibited assembly of the IPP complex and consequent activation of N-WASP, actin polymerization, and tension development (1311). Additional studies by other laboratories indicate that other scaffolding proteins and kinases in focal adhesions, including CAS (Crk-associated substrate) and kinases Src, Abl, and PYK2, are involved in an alternate pathway to stimulate actin polymerization through N-WASP (1187, 1188, 1287, 1312, 1313). Collectively, these studies point to integrins coupled to focal adhesion proteins and kinases as upstream regulators of actin polymerization under conditions in which MLC20 phosphorylation is not altered.
There are at least four additional pathways that could potentially mediate force augmentation through F-actin polymerization. The first involves 1) signaling through PAK/PYK2-p38 MAPK-MK2 to regulate HSP27 (1314, 1315), by which phosphorylated HSP27 directly promotes actin polymerization (1316). The participation of MAP kinases in this pathway is perhaps an explanation for the finding that MAPK inhibition interferes with the arteriolar myogenic response (1317–1319). In addition to this pathway, there are others that regulate the balance of cofilin and profilin phosphorylation, thereby determining the extent to which these actin-binding proteins are either sequestered by focal adhesion proteins or free to interact with actin, including 2) RhoA-ROCK-LIMK-14/3/3-cofilin (1261, 1320, 1321); 3) p130 Cas-profilin (1322); and 4) PKA/PKG-VASP-profilin (1323). The control of actin polymerization through pathways involving ROCK and PKC (1298, 1324, 1325) has the potential for cross talk with pathways regulating MLC20 phosphorylation through MLCK/MLCP. The ways in which these pathways might converge to control actin dynamics in SM are the subject of multiple reviews (1038, 1250, 1288, 1316). It is not possible to provide a detailed discussion, as the evidence for their involvement in myogenic constriction is confined to a single study (1298); however, this is a potentially important area for further research.
5.2.1.4. INTEGRINS AND ECM.
A vast body of evidence demonstrates that mechanical signals can be transduced from outside to inside the cell through the interactions of cell surface receptors with ECM proteins. VSMCs, like most cells, express multiple cell surface receptors, including integrins, syndecans, and glypicans. Here we focus on integrins. Integrins are a class of membrane-spanning glycoproteins, composed of α- and β-heterodimers with extracellular domains that interact with ECM proteins and short cytoplasmic tails that interact with focal adhesion proteins (1326). VSMCs express at least 11 of the >24 identified integrins, with α1β1, α3β1, α5β1, αvβ1, and αvβ3 being the most widely expressed (1327). ECM proteins expressed in the vascular wall include collagens I, II, IV, V, and VI, fibronectin, vitronectin, osteopontin, elastin, and several types of laminins (1327). Integrin α-subunits specify the ECM proteins with which they interact, e.g., α5β1 integrin binds to fibronectin and α1β1 binds to collagen and laminin (1328). Integrin β-subunits mediate interactions with specific focal adhesion proteins, e.g., talin, which links β-integrin tails to the actin CSK (1329). Integrin engagement with an ECM protein results in so-called “outside-in” signaling, initiating the recruitment of numerous proteins to the underlying focal adhesion and subsequent activation of phosphorylation cascades involving both receptor and nonreceptor tyrosine kinases (1330, 1331). Integrins can also transmit signals “inside-out,” in which signals from the focal adhesion modulate the conformation of the integrin extracellular domain to change its affinity and/or avidity for ECM proteins (1332, 1333). Outside-in signaling is the focus of the following discussion since it is most relevant to the role of integrins and the ECM in mechanotransduction by VSMCs.
Focal adhesions begin as nascent adhesions that contain only a few integrins (e.g., during cell migration), become focal complexes as intracellular adaptor proteins are recruited, and mature into focal adhesions once those adaptor proteins link to the actin CSK (1334). A mature focal adhesion is organized into at least three distinct layers, as originally predicted by Geiger and Yamada (1335) and confirmed by superresolution microscopy (1336): 1) a signaling layer most closely associated with integrins in the plasma membrane that includes paxillin, focal adhesion kinase (FAK), Src kinase, and ILK; 2) a force transduction layer, ∼30 nm below the plasma membrane, that includes the adaptor proteins talin and vinculin; and 3) a layer of actin regulating proteins, ∼60 nm below the plasma membrane, that include paxillin, zyxin, α-actinin, VASP, and p130 Cas (1249). Focal adhesions are dynamic structures undergoing constant turnover of their individual elements at different rates. Many studies investigating this topic have been conducted on the lamellipodia of migrating cells; however, in differentiated VSM it is likely that the majority of focal adhesions in VSMCs exist as stable, membrane-associated dense plaques (1250).
Integrin binding to ECM triggers a series of subcellular events that result in the activation of multiple signaling pathways. For example, the application of small (1–5 µm) beads coated with an integrin ligand, e.g., ECM protein, integrin Ab, or Arg-Gly-Asp (RGD)-containing peptide (a binding sequence found in multiple integrins), results in the clustering of surface integrins under the beads (1190) and the accumulation of various focal adhesion proteins. This method was used in conjunction with an ultrasensitive magnetometer probe by Ingber and colleagues to examine how mechanical forces applied through integrin-ECM bonds are transduced. Ferromagnetic beads coated with integrin ligand were attached to cells, followed by application of a twisting force while the cellular response to that force (angular strain) was recorded (1337). The application of force through integrin-ECM bonds resulted in a linear increase in CSK stiffness as predicted by tensegrity models (153). Integrin clustering under these conditions is followed by the recruitment of focal adhesion proteins to the vicinity of the bead, resulting in an enhanced (>6-fold) and sustained adhesion force (1338). Integrin clustering catalyzes the phosphorylation of multiple focal adhesion proteins and the activation of associated kinases, but this only occurs if the ligand is cross linked (e.g., insoluble ECM protein) with multiple integrin binding sites; soluble ECM ligands typically do not produce this complex response or even may compete with insoluble ligands for binding (1339).
Integrin-ECM binding and focal adhesion expansion initiate signaling cascades in which a key upstream event is the activation of FAK. For example, α5β1 integrin engagement with fibronectin (FN) results in talin and vinculin recruitment to the focal adhesion (1340). Talin uncoils under mechanical load to unmask cryptic binding sites for vinculin (1341, 1342). These interactions catalyze FAK phosphorylation at Tyr397, creating a high-affinity binding site for Src (1343, 1344). Src is then autophosphorylated at Tyr419 (1345–1347), leading to phosphorylation of the docking proteins paxillin and p130 Cas (1348, 1349) and subsequent activation of downstream signaling pathways. In VSMCs, Src activation leads to activation of Rho-GTPases and PLCγ, resulting in the production of IP3 (1350). Integrins may also connect to the actin CSK via the ILK-PINCH-parvin complex (1307). The time courses over which these events occur vary widely: talin associates rapidly (<2 s) with β-integrin tails (1342, 1351), whereas vinculin is recruited to focal adhesions over the course of several seconds (1352) and the expansion of a focal adhesion in the direction of applied force requires a few minutes (1353). Multiple steps in this process are potentially mechanosensitive. Mechanical loading enhances the phosphorylation status of some CSK proteins (1354) and exposes cryptic binding sites in others, e.g., myosin (1355) and talin (1342), to facilitate interactions with other focal adhesion proteins.
Studies by Gunst and coworkers suggest that these processes operate in differentiated SMCs in response to applied mechanical force. In response to contractile stimulation, FAK and Src in tracheal SMCs are phosphorylated at their respective residues mentioned above (1356), catalyzing the assembly of macromolecular adhesion complexes at the CSK-matrix junction, including the recruitment of paxillin and vinculin to the focal adhesion (1310) as well as paxillin phosphorylation (1281, 1357). Contractile force development under these conditions also depends on the interaction of β1 integrin and α-actinin (1286). Similar responses have been noted in response to localized mechanical stress of cultured airway SMCs by another group (1358). Studies using atomic force microscopy have shown that pulling on FN-coated beads attached to cultured VSMCs induces a counteracting force generated by the cell, in essence a myogenic-like response at the molecular level (1359).
Integrin-ECM interactions are known to initiate or enhance Ca2+ signaling in VSMCs. Davis and coworkers examined the effects of integrin-ECM interactions using microapplication of coated beads to VSMCs freshly isolated from rat skeletal muscle arterioles. The attachment of FN- or α5 integrin antibody-coated beads (with minimal or no associated force) led to enhancement of L-type Ca2+ channel (Cav1.2) current, as recorded with whole cell patch-clamp techniques (1360). The enhancement occurred only upon exposure to cross-linked ligands (e.g., ECM on beads) and not to soluble ligands. In contrast, attachment of beads coated with vitronectin, β3 antibody, or ligands for αvβ3 integrin led to a reduction in Cav1.2 current, suggesting that α5β1 and αvβ3 integrins reciprocally regulate Cav1.2 channels (1361). A subsequent study found that the enhancement in Cav1.2 current in response to α5β1 integrin engagement was blocked by dialyzing the cell with selective inhibitors of FAK or Src (1362). Current enhancement under these conditions was mediated both by Cav1.2 phosphorylation at the canonical PKA phosphorylation site Ser1901 and by tyrosine phosphorylation at Tyr2122 near the intracellular COOH terminus of Cav1.2 (1363). These results are consistent with reports that the activation of tyrosine phosphorylation cascades is involved in myogenic responses of arteries/arterioles (Refs. 1364, 1365; for review see Ref. 1366). Although these studies establish a precedent for the regulation of a VSM ion channel by integrin-ECM interactions, they stopped short of showing that the subsequent application of force through established integrin-ECM bonds results in enhancement of the same pathways or activation of other pathways, perhaps involving other ion channels. However, subsequent studies by the same laboratory found that VSMC BK channels were also activated by the application of α5β1 integrin ligands on beads (1367, 1368) and that these channels may have been activated by the integrin-mediated increase in calcium spark activity (707, 1369). These results align with those from multiple other laboratories showing that the Ca2+ increases in VSMCs are stimulated by integrin-ECM interactions (1370–1372) as well as evidence that other ion channels in other cell types are regulated by the activation of integrin signaling (556, 1373), as reviewed in Refs. 1361, 1374. It should be noted, however, that the roles for integrins in the vascular myogenic response may be limited to compartmental (707, 1369), rather than global, Ca2+ signaling in VSMCs (1193, 1375).
Several laboratories have translated these findings to studies of intact arteries/arterioles. Initial studies had suggested a role for tyrosine kinase activation in the arterial myogenic response. For example, the nonselective tyrosine kinase inhibitor herbimycin A attenuated pressure-induced constriction of rat cerebral arteries (1376, 1377), and pervanadate (a tyrosine phosphatase inhibitor) or PP1 (a more selective Src inhibitor) inhibited myogenic tone of rat skeletal muscle arterioles (1365, 1366, 1378). Meininger and coworkers (1379) found that RGD-containing peptides cause a transient constriction, followed by dilation, of isolated skeletal muscle arterioles, with the dilation phase being attenuated by pretreatment with β3 integrin antibody. Both α5β1 and αvβ3 integrins contain RGD-binding sequences (1380), and these two integrins are known to have reciprocal or antagonistic actions in other contexts via their activation of Rho- and Rac-GTPases (1381), which could explain their effects on arteriolar tone. Although RGD peptides also have effects on endothelium-derived NO (1373, 1382), it is to be noted that the RGD-evoked dilation of skeletal muscle arterioles occurred under conditions in which NO production was blocked. A biphasic effect on arteriolar tone is consistent with the involvement of multiple integrins, at least one linked to constriction and another to dilation, as in the studies described above implicating α5β1 and αvβ3 integrins in the regulation of Cav1.2 channels in VSMCs (1360), although at least a component of the constriction might be mediated by endothelin production from the endothelium (1383). Two subsequent studies showed that not just tone but also myogenic constriction to pressure elevation were impaired by blockade of α5β1 or β3 integrins (1193, 1384). Other integrin-specific peptides are also vasoactive in arterioles. For example, a Leu-Asp-Val (LDV)-containing peptide, which interacts with α4β1 integrin, causes arteriolar vasoconstriction associated with the enhancement of Cav1.2 current in VSMCs (1385). Collectively, these studies strongly implicate integrin-linked signaling in VSMCs as at least one component of pressure-induced myogenic constriction.
5.2.2. Cytoskeleton, integrins, and extracellular matrix in EC mechanosignaling.
Many of the fundamentals presented in sect. 5.2.1 on cytoskeleton, integrins, and extracellular matrix in VSMC mechanosignaling are also applicable to corresponding sections for ECs below.
5.2.2.1. CYTOSKELETON.
The stress fiber alignment induced by LS/PS along the shear axis determines the role of the CSK in regulating the mechanical properties of ECs and their mechanotransduction. Early studies demonstrated that the application of low LS (5 dyn/cm2) caused rapid increases of G-actin from 54 ± 0.8% to 87 ± 4.2%, concurrent with increases of endothelin-1 (ET-1) expression, in porcine aortic ECs (1386). Such low shear-induced actin depolymerization and ET-1 expression was Ca2+ and PKC dependent (1387). With a UV-cross linking technique, it has been shown that the 51-kDa G-actin binds to the 3′-untranslated region (UTR) of eNOS mRNA to reduce eNOS expression (1388), suggesting a potential role of direct regulation of gene expression by the dynamics of actin remodeling. Multiple studies have demonstrated that disruption of actin, tubulin, or microfilaments markedly attenuates or abolishes LS-induced EC signaling, gene expression, and modulation of function (1237, 1386, 1389–1391). Moreover, it has been established that the direction of shear flow plays an important role in regulating EC signaling and function (9). In ECs prealigned on a micropatterned surface (15-µm strips), the restriction-induced cell death is modulated by directional shear flow, being rescued by parallel shear and enhanced by perpendicular shear; these processes are found to be dependent on Rho and actin (42). Parallel shear induces atheroprotective signals (e.g., Akt and eNOS), whereas perpendicular shear induces inflammatory signals (e.g., p-p65) (1392). These results demonstrate the importance of the direction of shear force on the CSK in regulating EC functions. The actin CSK is associated with focal adhesion sites (FASs), which are loaded with signaling molecules (such as integrins, FAK, and Src) and interact with the EC and extracellular environment to modulate cell-substrate interactions.
In addition to anchoring the FAS for signaling events, the CSK is linked to the nucleoskeleton to transduce the tension for nuclear mechanics and epigenetic chromatin regulation through the linkers of nucleoskeleton and cytoskeleton (LINC) complex (for review see Refs. 1393, 1394). With the use of magnetic beads coated with Arg-Gly-Asp (RGD) to directly engage integrins and actin, it has been shown that stretch leads to nuclear deformation, chromatin stretching, and gene expression in CHO cells; disruption of actin abolished force-induced chromatin remodeling and gene expression (1395). It has also been demonstrated that uniaxial stretch or compression of the CHO cells is translated into the stretching or compression of chromatins, respectively, to modulate gene expression (1396). The LINC complex includes Nesprins, Sun domain proteins (Sun), Emerin, and Lamins. It has been reported that Nesprin3 expression and localization are not directly regulated by shear but necessary for LINC assembly in ECs to direct LS-induced MTOC polarization and modulate cell migration direction (1397). Moreover, it has been reported that PS, but not OS, induces Nesprin1/2 localization in the nucleoenvelope to increase β-catenin association and induce EC tight junction protein expression for enhanced integrity of the endothelium. Inhibition of β-catenin leads to a decrease in permeability of mouse aorta (1398). Disruption of LINC with a dominant-negative mutant for the Kash domain of Nesprin leads to EC detachment, attenuates EC migration and angiogenesis, and abolishes laminar shear-induced EC alignment but does not affect EC proliferation (1399). EC Lamin A/C expression is passage dependent, with the level decreasing over time. LS downregulates Lamin A/C in ECs with lower passage [population doubling level (PDL) of 13 or less] compared with those of high passage (PDL 60 ± 5) (1400). LS downregulates Lamin A/C in ECs with lower passages compared with those of high passage (1400). Lamin regulation of mechanotransduction and gene expression involves the recruitment of histone modifiers (e.g., methyltransferases and HDACs) to serve as anchor points for chromatin remodeling (1401–1403) and epigenetic regulation of cell functions. The role of Lamins in mechanical regulation of EC epigenetics remains to be elucidated.
5.2.2.2. INTEGRINS.
It has been well established that integrins are major mechanosensing molecules for EC responses under flow (1058, 1404, 1405). Early studies include the analyses of adaptor Shc2 recruitment to activated integrins and the uses of specific antibodies against active integrin. The results have shown that LS activates integrins (including αvβ3, α2β1, α5β1, and α6β1) in an ECM protein-specific manner to regulate specific EC signaling pathways such as Ras-MAPK, PKA-PI3K, PKC-PI3K, Src-Akt-eNOS, and NF-κB (1059, 1236, 1406–1408). It has been demonstrated that OS causes the activation and translocation of integrin α5 to lipid rafts, with the consequent activation of the NLRP3 pathway for EC inflammation (1080). It has also been demonstrated that the application of OS to ECs on fibronectin causes a sustained activation of integrin β1 to lead to the sustained association of β1 with TGF-β receptors (TbRs), activation of SMAD2, and induction of EC inflammatory phenotype, whereas the application of OS to ECs on laminin causes transient activations of integrin β1- and β3-TβRs, as well as Smad2, to attenuate inflammatory responses (1409).
In vitro (18% elongation) and in vivo (hyperventilation) studies on pulmonary ECs have shown that cyclic stretch induces tyrosine phosphorylation of integrin β4, together with the expression of inflammatory genes (IL-6, IL-8, MCP-1, and RANTES); overexpression of β4 mutant constructs (tyrosine-to-phenylalanine mutations or cytosolic signaling domain deletion) in ECs attenuates the stretch-induced inflammatory responses and prevents hyperventilation-induced lung injury (1410). Integrin α5β1 has been reported to mediate the stretch activation of adaptor protein p66Shc and JNK in human aortic ECs (HAECs) to increase and decrease NO bioactivity, with cell damage. Inhibition of integrin α5β1 or knockdown of p66Shc and JNK reverses these effects (1411). Integrins have also been reported to mediate stretch-induced HUVEC alignment by regulating the CSK and focal adhesions (1412). The detailed mechanisms of stretch regulation of EC function by integrin activation remain to be elucidated.
5.2.2.3. EXTRACELLULAR MATRIX.
The interactions between ECs and ECM trigger integrin activation and consequent signaling events (1405). The major ECM proteins in the vessel wall include fibronectin, collagen, and vitronectin. Earlier studies have demonstrated that the activation of EC integrins by LS is ECM protein specific (1406, 1407). A recent study has shown that OS causes sustained integrin β1-TβRI/II and integrin β1/β3-TβRI association, Smad2 phosphorylation, and NF-κB expression in ECs cultured on fibronectin and that these reactions are transient in ECs cultured on laminin. Atherosclerotic lesions in patients exhibit an augmentation of Smad2 phosphorylation and a markedly higher level of fibronectin than laminin in the EC layer and neointimal region, suggesting a role of fibronectin-integrin-TβR1-pSmad2-NF-κB pathway in mediating the OS-induced vascular inflammation (1409).
In addition to ECM composition, the rigidity of the matrix also affects EC mechanoresponses. EC dysfunctions have been reported in hypertensive and aged arteries, which are stiffer than healthy young ones (1413–1415). With the use of polyacrylamide (PA) gel to control the substrate stiffness in studying EC responses to flow, it has been found that ECs on a substrate of 2.5 kPa (relevant to young healthy vessels) show a more elongated morphology, more tight junctions, lesser Rho activation, faster responses in ERK and eNOS phosphorylation, and augmented NO production under LS, in comparison to the ECs on the more rigid 10-kPa substrate (relevant to aged, stiffer vessels) or glass slides (1416). Jo, Brewster, and colleagues (1417) showed that, in in vitro experiments on HAECs, OS induces higher thromospondin 1 (TSP1) and collagen1 expressions in comparison to static conditions or LS and that inhibition of TSP1 activation attenuates the OS-induced collagen1 expression. In vivo experiments using the carotid artery partial ligation model to create flow disturbances in young and old mice demonstrated that disturbed flow increases arterial stiffness and induces the expression of genes related to CSK remodeling, including collagen 1 and TSP1. TSP1-knockout animals exhibit a marked reduction of the arterial stiffness induced by disturbed flow (1417). These studies demonstrate the cooperative effects of substrate compliance and flow dynamics in regulating EC functions.
5.2.2.4. MECHANOSENSITIVE EC REMODELING.
With multifluorophore labeling of EC structural proteins, it has been found that LS induces the remodeling of CSK, FAs, and EC lamellipodia dynamics (1418–1420). ECs respond to LS by enhancing actin polarization with increases of lamellipodia and actin-anchored FA formation (with vinculin and paxillin) downstream of the shear direction (1418, 1419, 1421). Such changes of CSK dynamics modulate EC-ECM interactions to lead to EC alignment and directional migration. It is well established that LS induces the formation of actin stress fibers in ECs and their elongation and alignment along the direction of flow (1422). This remodeling is shear pattern dependent: ECs are elongated and aligned rapidly under high shear without flow reversal at 40 ± 20 dyn/cm2, are less elongated and aligned slowly under medium shear with flow reversal at 20 ± 40 and 10 ± 15 dyn/cm2, and do not undergo elongation or alignment under zero net shear with flow reversal at 0 ± 40 and 0 ± 20 dyn/cm2 (1423). It has been demonstrated that such remodeling processes are dependent on Ca2+, tyrosine kinases, and microtubules (1389). LS has been shown to cause changes in the intercellular junction VE-cad complex, with the phosphorylated β-catenin bridging between VE-cad and actin CSK (1424, 1425). Furthermore, LS has been shown to increase EC stiffness as a result of stress fiber formation (1426). With an intracellular particle tracking technique, it has been shown that the changes in EC mechanical properties under LS are anisotropic in that the EC is more “liquidlike” in the direction along the shear axis and more “solidlike” in the perpendicular direction (1427), which may contribute to EC alignment.
Shear stress and stretch regulate EC mechanotransduction in convergent (e.g., signaling pathways) as well as divergent (e.g., Ca2+ influx) ways. Shear and cyclic stretch have been shown to have synergistic effects in regulating BAEC morphology and actin alignment (1428). With the use of an elastic tube (Sylgard silicone) to apply 1.9–6.4 dyn/cm2 LS in conjunction with 2–7.3% (diameter changes) strain to pulmonary ECs (1429), it has been shown that the combination of shear stress > 2 dyn/cm2 with 2% stretch significantly decreases the shape index and actin angles, indicating their synergistic effect for greater elongation and alignment (1428). Further investigation of LS (80 dyn/cm2) or OS (20 ± 10 dyn/cm2) in conjunction with 2% cyclic stretch has demonstrated that OS, but not steady LS, is synergistic with stretch in regulating HAEC morphological responses (1430). It has also been shown that 4% stretch causes greater enhancement of Cx43 expression induced by OS (0.3 ± 3 dyn/cm2) than that induced by PS (6 ± 3 dyn/cm2) (1431). Application of cyclic stretch (4%) with different stress phase angle (SPA) together with shear stress (10 ± 10 dyn/cm2) has demonstrated that tight junction occludens-1 expression is significantly higher at atheroprotective SPA (0°) compared with atheroprone SPA (−180°) (1432). These results suggest critical roles for the interplay of multiple mechanical factors in regulating endothelial integrity.
5.3. YAP/TAZ Pathway
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) have been identified as novel sensors that can respond to signals resulting from cell shape and ECM rigidity (1433) and relay these mechanical cues to the nucleus. In investigating the roles of YAP/TAZ in shear stress-induced signaling in ECs, S. Chien, Y.-S. Li, and others have demonstrated that YAP and TAZ serve as mechanoresponsive molecules to regulate EC growth and inflammatory responses (1434, 1435). In response to LS and PS, YAP is phosphorylated at serine 127 through the activation of β3 integrin, Gα13 (1434), RhoA (1435), and LATS1/2 (1436) pathways. The phosphorylated YAP is retained in the cytosol as an inactive form without nuclear translocation (1433). Depletion of YAP causes decreases in CCNA1 and MCP-1 (1436), as well as ICAM-1 and VCAM-1 (1437) in ECs, thus leading to antiproliferation and anti-inflammation responses (1435). Overexpression of the active form of YAP reverses the PS-induced suppressions of EC proliferation and inflammation (1435). OS causes phosphorylation of YAP at Y357, but not at S127 and S397, and a sustained YAP nuclear localization in an integrin α5β1-dependent manner; fibronectin was found to increase YAP phosphorylation at only Y357 and not S127 (1438). Another study reported that OS causes YAP S127 phosphorylation in ECs on Matrigel but not on fibronectin (FN). Additional experiments demonstrated that FN suppresses YAP S127 phosphorylation via an α5β1/PDE4D B55α/PP2A pathway (1439). A negative mutant of YAPY357 attenuates the YAPY357 phosphorylation, VCAM-1 expression, and monocyte adhesion induced by OS (1438). In vivo studies using Apoe−/− mice have demonstrated the presence of YAPS127p in atheroprotective areas and YAPY357p in atheroprone areas. Decreases of the phosphorylation status by YAP knockdown, CRISPR-based EC-specific YAP knockout, or inhibition of YAP upstream molecules can mitigate the disturbed flow-mediated atherogenesis (1438). It has been demonstrated that LS causes a transient activation of YAP in human pulmonary arterial ECs at 10 min, followed by subsidence at 6 h and beyond (time points between 10 min and 6 h were not reported). Under static conditions, inactive cytosolic YAP binds to angiomotins (AMOTs); the interaction of AMOTs with actin in response to LS causes the release of YAP for transient nuclear translocation to regulate gene expression (1440). This transient YAP nuclear translocation is dependent on actin remodeling but not LAST1/2. These results suggest an additional potential mechanism by which long-term PS/LS induces stress fiber formation to reduce actin-AMOT interaction and YAP nuclear translocation and hence the deactivation of YAP for vascular homeostasis. A study on ECs of the dorsal aorta of zebrafish during embryogenesis demonstrated that the onset of blood flow upon the establishment of perfusion leads to YAP nuclear translocation and transcription activation and that such YAP nuclear localization can be reversed by blood flow cessation and rescued by restarting blood flow (1440). Lumen stenosis and vessel retraction are observed in the dorsal vessels in YAP-mutant larvae or in wild-type larvae after flow cessation (1440), suggesting that YAP plays an important role in vessel maintenance. These findings indicate that YAP mediates temporal modulation in mechanotransduction and that it plays critical roles in the mechanoregulation of EC functions and vascular remodeling and disease progression (for review see Ref. 1441).
Stretch-induced HUVEC proliferation has been shown to be mediated by the YAP/TAZ pathway; inhibition of YAP, but not TAZ, causes significant attenuation of the 15% uniaxial stretch-induced EC proliferation (1442). In vivo experiments demonstrated that EC-specific knockout of YAP/TAZ leads to abnormalities in vascular networks, which may be caused by the EC remodeling (proliferation and migration) to impair junctional integrity (1442). Studies of vascular ECM stiffening induced by pulmonary arterial hypertension (PAH) revealed that YAP/TAZ mediates the stiffness-induced miR103/301 expressions, which in turn increase the proliferations of pulmonary ECs, SMCs, and arterial adventitial fibroblasts, as well as collagen deposition, to further promote ECM remodeling. Inhibition of YAP/TAZ-miR103/301 ameliorates the pulmonary matrix stiffening (1443). Further investigations revealed that YAP/TAZ mediates the ECM stiffness-induced expression of metabolic enzymes in pulmonary vascular cells to coordinate glutaminolysis and glycolysis to reprogram pulmonary EC proliferation and migration to drive pulmonary hypertension (1444). These findings show that YAP and TAZ serve as critical mechanoresponsive modulators for vascular development and pathogenesis.
5.4. AMPK, Akt, PKA, and eNOS
Multiple signaling pathways have been shown to mediate EC responses to shear stress (for review see Refs. 9, 1766), as summarized in FIGURE 18.
FIGURE 18.
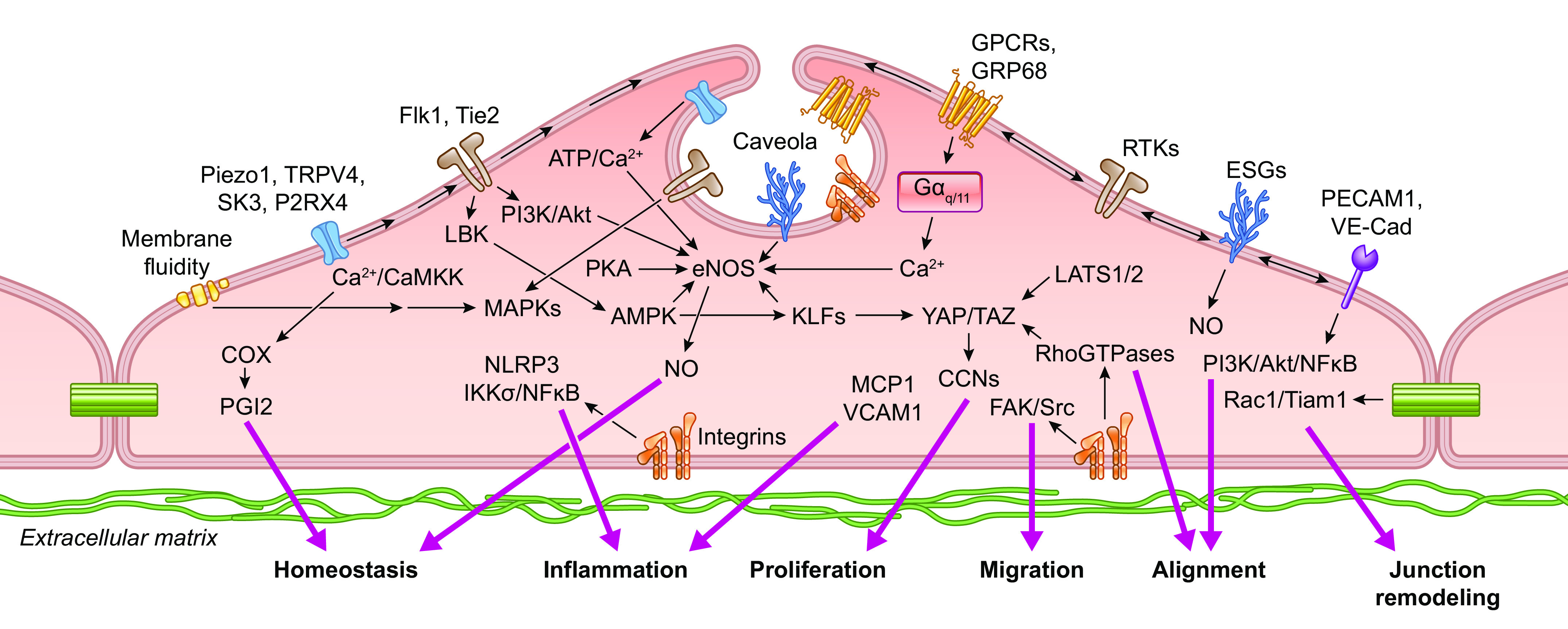
Schematic drawing showing mechanotransduction pathways in endothelial cells. ESG, endothelial surface glycocalyx; CNN, cyclins; LBK, liver kinase B1 (also called LKB1). See glossary for other abbreviations.
Earlier studies on EC mechanotransduction focused on the immediate signaling events in response to LS, such as those mediated by MAPKs (1446–1450) and Akt/PI3K (1451, 1452). Recent studies have placed emphasis on the differential regulation by different flow patterns. NO production plays critical roles in regulating vascular tone. Among the mechanotransduction pathways, AMPK, Akt, and PKA are kinases that modulate human eNOS activity through phosphorylation (Ser114,615,635,1177 and Thr495) (for review see Ref. 104). It has been reported that AMPK is specifically activated by LS but not by OS (1453) or pathologically low shear stress (1454). AMPK, which is a master switch that senses cellular energy (ATP) and stresses for metabolic regulation, plays important roles in modulating vascular functions (for review see Ref. 1455). LS induces AMPK phosphorylation, which in turn leads to eNOS activation by phosphorylation at S1179 (the bovine analog of human eNOSs1177), to produce NO (1456, 1457). In addition, LS-induced AMPK-eNOS activation is required for the increase of NO production following eNOS deacetylation in response to the association of eNOS with the NAD-dependent deacetylase sirtuin 1 (Sirt1) (1458). Furthermore, LS enhances a junction-stabilizing protein [membrane-associated guanylate kinase with inverted domain structure 1 (MAGI1)] that colocalizes with VE-cad at cell borders to induce eNOS activation. This MAGI1-mediated process involves AMPK and KLF4 pathways (1459). In addition to eNOS activation, the LS-induced AMPK activation also downregulates the transcription factor forkhead box O (FoxO) to reduce ANG II and hydroxymethylglutaryl-coenzyme A reductase (HCR), resulting in a decrease of inflammation and an increase of vasodilation (1460, 1461). Pathological low LS has been shown to cause the nuclear translocation of liver kinase B1 (LKB1) to decrease AMPK, which in turn attenuates NADH oxidase (p47phox) and enhances Na+/H+ exchanger 1 (NHE1) activity to activate hyaluronidase 2 (Hyal2) to degrade the glycocalyx. Such low LS-induced glycocalyx dysfunction mediated by AMPK has been validated in the low-flow regions of the mouse carotid artery subjected to partial ligation (1454, 1462). These results suggest a major role of AMPK in regulating EC homeostasis under atheroprotective flow via the activation of eNOS, reductions of free radicals, and inhibition of inflammatory pathways. Although it has been reported that Akt activation is not flow pattern specific (1453), Melchior and Frangos (1156) demonstrated that phospho-Akt is preferentially distributed with PECAM in cell junctions in the straight part of mouse aorta but is diffusely distributed at perinuclear locations at artery bifurcations. In vitro experiments showed that Akt can be activated by PS, but not OS, in a Gαq/11-dependent manner (1156). The PS/LS-induced Akt-eNOS activation is PECAM dependent, whereas that of AMPK-eNOS is not (1457), suggesting divergent routes in shear modulation of AMPK and Akt pathways for NO regulation. AMPK and Akt phosphorylate eNOSS1179 in a Ca2+-dependent manner to modulate EC NO activity under LS, but the phosphorylation of eNOSS116 and eNOST497 is not affected by LS (1463). LS induces phosphorylation of eNOSS635 (bovine analog of human S633) in a PKA-dependent, but PI3K/Akt/Ca2+-independent, pathway (1463, 1464). Although PI3K/Akt and PKA have been shown to modulate NO production for EC homeostasis, there are also reports suggesting that the same pathways lead to EC dysfunctions under low shear (2 dyn/cm2) (1465, 1466). These findings suggest the diverse roles of these pathways in EC regulation. The details of the spatiotemporal dynamics for the mechanotransduction for pathophysiological regulation of vascular functions remain to be determined.
5.5. Integration of Acute Mechanosensing Processes by VSMCs and ECs
5.5.1. Pressure-induced vasoconstriction.
The VSMC sections of this review have focused on various components of pressure-induced myogenic constriction as the primary example of acute VSMC mechanotransduction. A recent review article provides an excellent summary of most of the known mechanisms shown experimentally to be involved in, or interfere with, myogenic constriction and presents a complicated flow diagram (see Figure 2 in Ref. 16). That scheme shows mechanisms we have discussed in the present review and a few that we have not covered [e.g., NCX (1467–1469), EGFR (1143), MAP kinases (1317–1319) and ROS (1470)], in part because they are not yet supported by data from genetic knockout animals. Although comprehensive, the flow diagram does not provide information about which of the mechanisms have the strongest experimental support, which ones are more important for myogenic constriction, or which ones predominate at various time points. The following discussion and summary figure (FIGURE 19) attempt to address these points.
FIGURE 19.
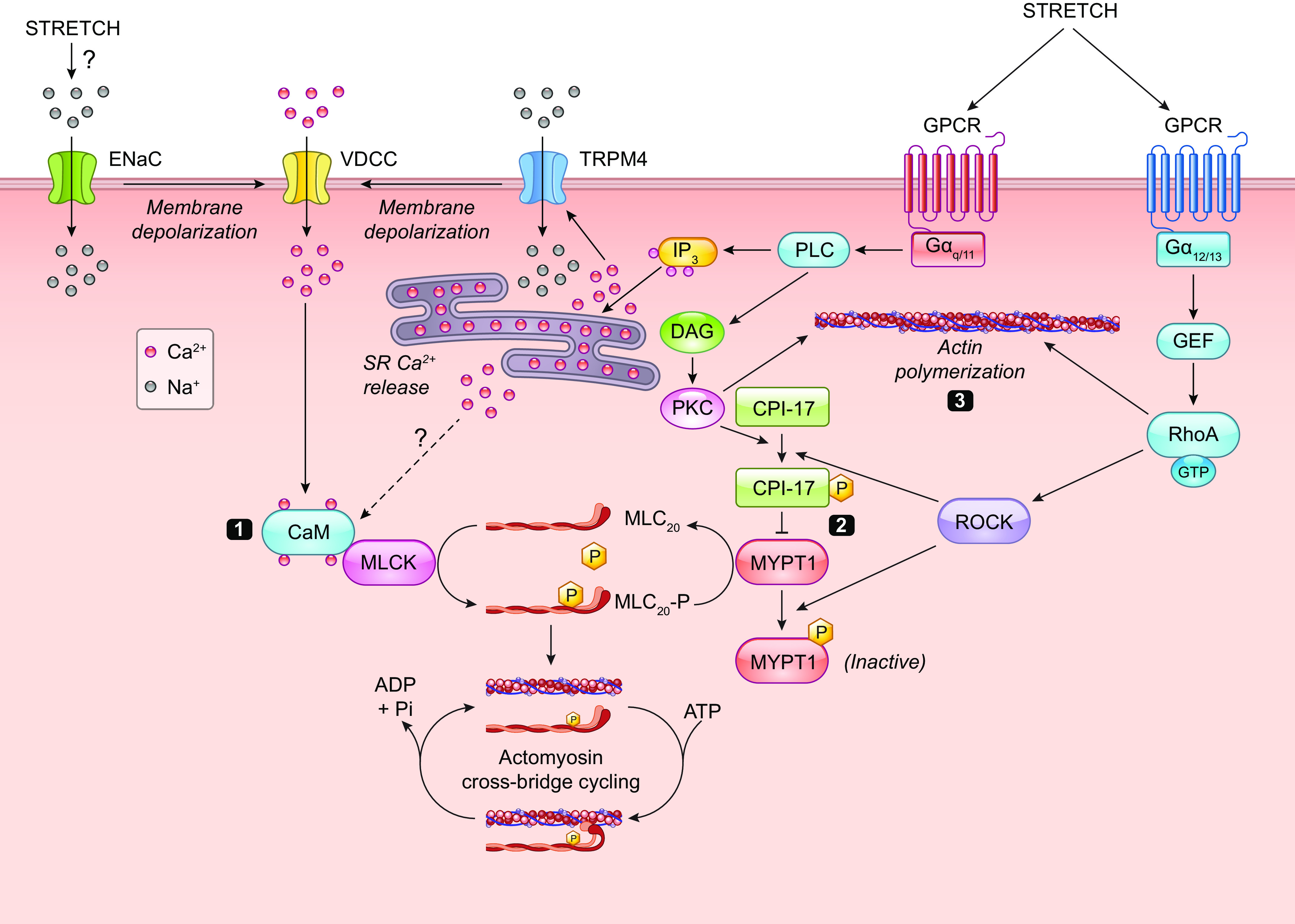
Three major signaling pathways for myogenic constriction: 1) Ca2+-dependent regulation of MLCK downstream from VGCC gating by mechanosensitive GPCRs and second messenger-gated ion channels. TRPM4 is shown here as a representative but not exclusive ion channel target downstream from Gq/11 GPCRs. 2) Ca2+-independent regulation of MLPC through RhoK-ROCK signaling. 3) Rho regulation of actin polymerization. See glossary for abbreviations.
The rapid initiation of the myogenic response (within a few hundred milliseconds), along with the membrane depolarization that precedes it, almost certainly implicate one or more ion channels in the underlying mechanism. Of the large number of potential ion channels expressed by VSMCs, only a very select few can be considered true mechanosensors. Piezo1 is the most prominent and best-characterized mechanosensitive ion channel, yet it does not appear to be involved in the acute phase of pressure-induced constriction (although it is involved in vascular wall remodeling). The weight of experimental evidence, integrated from multiple laboratories, suggests that multiple second messenger-gated VSMC ion channels are responsible for pressure-induced depolarization; none of these channels meets the rigorous criteria for a mechanosensitive channel, but instead they are activated secondary to an upstream mechanosensor. To date, the best evidence suggests that these mechanosensors are Gαq/11-coupled GPCRs. More than one GPCR may be involved, but evidence from several different arteries implicates AT1R. The intrinsic mechanosensitivity of the GPCR results in the activation of membrane-bound PLC and catalysis of DAG and IP3 from PIP2. DAG activates TRPC6 channels, and IP3 elicits Ca2+ release from IP3R-sensitive stores to activate TRPM4 and possibly TRPV1 channels. PKD2 and ANO1 channels are potentially activated by IP3-mediated Ca2+ release or by Ca2+ influx through TRPC6 channels. The permeabilities of these VSMC ion channels (represented by TRPM4 in FIGURE 19) are consistent with cation influx or anion efflux to produce depolarization. Depolarization increases the open probability of Cav1.2 calcium channels, which conduct sufficient Ca2+ ions to induce the global Ca2+ increases associated with MLCK activation (pathway 1 in FIGURE 19) and vasoconstriction. The sensitivities of the channels in this Ca2+-dependent pathway could potentially be enhanced by metabolites of arachidonic acid (1043) and/or kinase-dependent phosphorylation downstream from other mechanosensitive processes such as CSK activation (745) or integrin activation (1471).
This scheme does not reconcile all the existing data, notably omitting a role for ENaC channels in VSMCs. Is an ENaC or an ENaC/ASIC heteromeric channel working in parallel with the GPCR-TRP channel mechanism, as illustrated in FIGURE 19? ENaC is proposed to be a direct mechanosensor or part of a multiprotein mechanosensitive complex. ENaC activation, even if independent of a mechanosensitive GPCR, would allow increased Na+ influx and contribute to the depolarization that regulates VGCCs. A recent review (1472) attempts to resolve the issue of parallel mechanisms by arguing for a mechanosensitive complex composed of a βγENaC/ASIC2 heterotrimeric channel, allowing Na+ influx and depolarization, with channel mechanosensitivity being modulated by both ECM-integrin interactions and internal tethering to CSK proteins (see Figure 5 in Ref. 1472). An analogy is made to the DEG mechanosensitive complex comprising the touch receptor in C. elegans (sect. 2.2.2), whose function is compromised to varying degrees by the loss of individual components (326). It is argued that such a fundamentally important mechanism would be evolutionarily conserved. However, this could also be an argument for the need of redundant mechanisms, and it conflicts with experimental observations that different mechanisms appear to operate in arteries from different vascular beds. In mammals, a mechanotransduction complex is perhaps better represented by mammalian touch receptors such as Meissner’s corpuscles or baroreceptor nerve endings, which include specialized structures for amplifying force, rather than a generalized mechanotransduction mechanism with relatively low sensitivity and specificity. Piezo channels play central roles in both of those systems, and yet they apparently are not needed for acute, pressure-induced constriction of arteries (242). Perhaps a better analogy would be the cochlear hair cell in mammals, where DEG and/or TRP channels were once thought to be central components (1473) but recent evidence now favors a complex consisting of the nonselective, Ca2+-permeable mechanosensitive cation channels TMC-1/2 (sect. 2.2), with extracellular cadherins transmitting force by the ECM-rich tip links to those channels, and an integrin-binding protein mediating intracellular tethering to the ankyrin-spectrin CSK (239). However, TMC-1 and TMC-2 do not appear to be expressed in VSMCs. An argument against parallel VSMC mechanotransduction mechanisms involving ENaC and GPCR-TRP signaling is that the evidence for the role of each in pressure-induced constriction appears to be mutually exclusive. TRP (1474) and VGCC channel-mediated Ca2+ entry (1475) downstream from the DEG complex (208) has been demonstrated in C. elegans, but no data exist supporting a comparable scheme in mammalian blood vessels.
How do the RhoA-MLCP and actin polymerization mechanisms (pathways 2 and 3, respectively, in FIGURE 19) integrate with the mechanosensitive regulation of pathway 1? The activation of RhoGEFs through their interactions with the α-subunits of G12/13-coupled GPCRs and subsequent activation of ROCK and inhibition of MLCP is potentially a relatively fast process that could operate concomitantly with ion channel-mediated Ca2+ influx; alternatively, production of some degree of basal tone through this pathway may be required for subsequent Ca2+-dependent regulation of that tone by mechanosensitive GPCRs and ion channels. Thus, both pathways 1 and 2 would work together to enhance MLC20 phosphorylation and force development following the application of force to VSMCs. In contrast, the regulation of actin polymerization through Arp2/3-mediated mechanisms (pathway 3) is predicted to be a relatively slow process, requiring multiple steps of protein phosphorylation as well as the recruitment and molecular assembly of cytoskeletal proteins, and it is likely that this process would be involved in sustained force production, possibly when signaling through the other pathways wanes (e.g., because of Ca2+- or voltage-dependent inactivation of Cav1.2).
5.5.2. Flow-induced vasodilation.
The EC sections of this review have discussed the various components of flow-induced dilation as the primary example of acute EC mechanotransduction.
Multiple laboratories have provided evidence that flow-induced dilation can be blocked or impaired by global or EC-specific knockout of Piezo1, TRPV4, Kir2.1, H1R, Pkd2, GPR68, Gq/11, or P2YR, all of which are involved in various aspects of shear stress-mediated Ca2+ signaling in ECs. These genetic approaches are generally considered to provide the most compelling evidence for a critical role of a particular gene or protein. The observation that loss of a single component results in complete or nearly complete abolition of flow-induced dilation would normally suggest that these pathways are sequential; however, it is difficult to explain how the three GPCRs on this list (H1R, GPR68, and P2YR) could be activated sequentially. One possible explanation is that different mechanisms (GPCRs, G proteins, and/or ion channels) are operating in different arteries, similar to the way in which various endothelium-derived mechanisms—NO, prostanoids, EDH—contribute to the vasodilation of arteries in different regions of the body, as well as to the responses of a given type of artery during different stages of vascular disease.
We propose the general scheme in FIGURE 20, which attempts to integrate the contributions of several “mechanosensitive” EC ion channels with GPCR signaling mechanisms that underlie flow-induced dilation.
FIGURE 20.
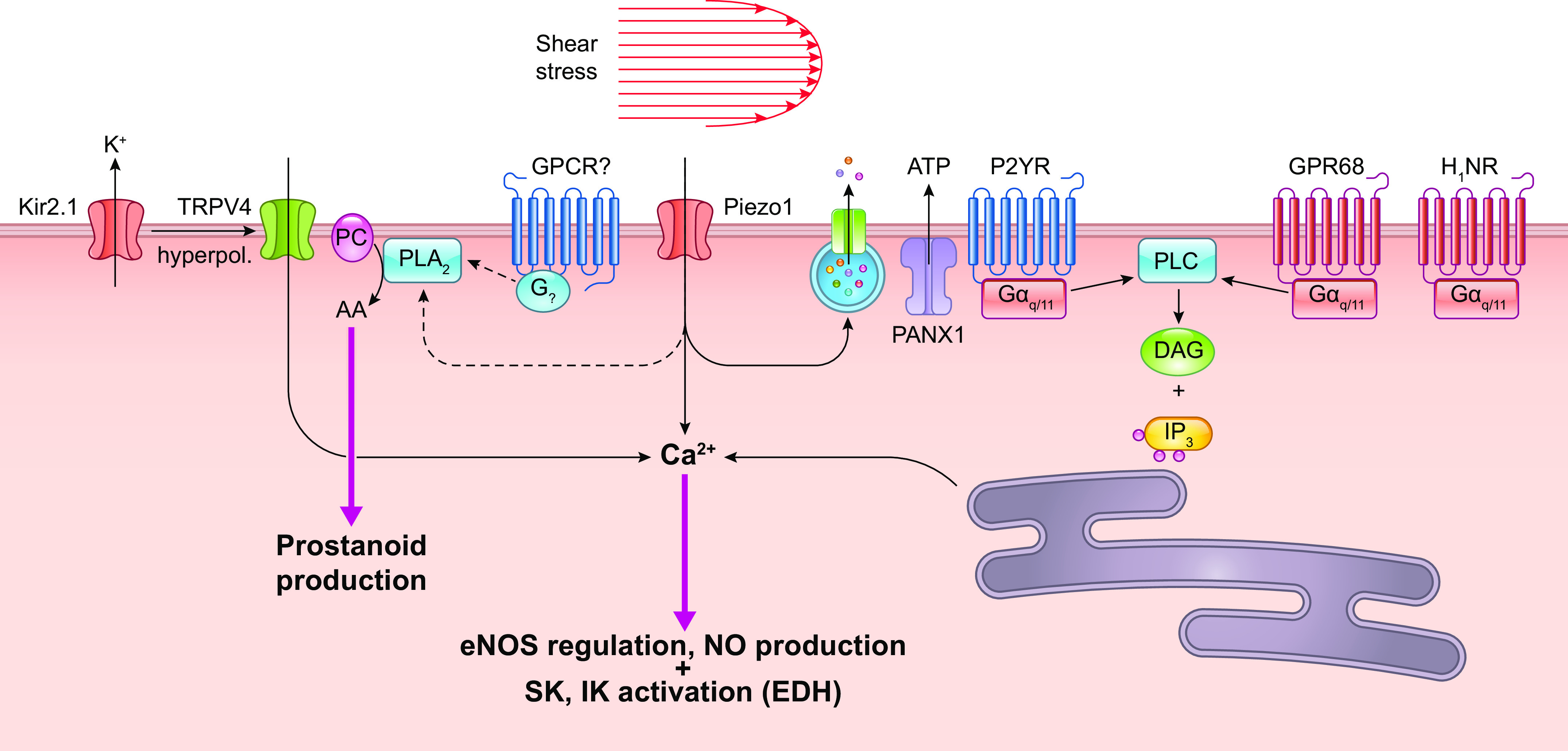
Components of the Ca2+ entry and release mechanisms regulated by shear stress in ECs. Shear stress-activated proteins are colored in red. See glossary for abbreviations.
To date, evidence supports Piezo1 channels as the only bona fide mechanosensitive channel in ECs (see TABLE 2). (Exceptions might be Kir2 and EnNaC, as discussed below.) Shear stress-induced activation of Piezo1 results in rapid Ca2+ entry that leads to the activation of TRPV4 channels and triggers ATP release. TRPV4 activation depends on the activity of PLA2 to produce undefined intermediate metabolite(s); PLA2 may be activated by Piezo1-mediated Ca2+ influx, but another possibility is that PLA2 is activated through coupling to an undefined mechanosensitive GPCR (1041). Ca2+ influx through TRPV4 channels is needed to amplify and sustain the influx through Piezo1. PKD2 channels (not shown) also contribute to this Ca2+ influx, but it is not clear how they are activated. Ca2+ influx triggers ATP efflux from intracellular vesicles and/or pannexin hemichannels. ATP binds to surface P2Y receptors that couple to PLC through Gαq/11 proteins. P2X channels may contribute to EC Ca2+ influx in response to ATP release; their relative contributions in this context are not known. GPR68 and H1N are mechanosensitive GPCRs that also couple to PLC through Gαq/11 proteins. These signals converge to activate PLC, resulting in DAG and IP3 production, with IP3 triggering IP3R-mediated Ca2+ release from ER Ca2+ stores.
In parallel with Piezo channel activation, shear stress activates Kir2 channels. The activation mechanism is as yet undefined, but it may involve shear stress-induced changes in membrane fluidity and/or interactions with membrane phospholipids such as PIP2. Kir2 channel activation results in a rapid EC hyperpolarization that increases the passive driving force for Ca2+ entry, thus enhancing influx through Piezo and TRPV4 channels. Ca2+ release from stores activates SK and IK channels, which further enhance and sustain the hyperpolarization. At the same time, Cl− channels may be activated in some ECs (possibly via Ca2+ entry or release) and the resulting Cl− efflux will counteract the hyperpolarization to some degree.
The combined changes in intracellular Ca2+ and production of AA-derived metabolites from PLA2 activation trigger phosphorylation cascades that promote the production of NO and various prostanoids, which diffuse from the EC layer to the VSMCs to promote vasodilation. Concomitantly, K+ channel-mediated EC hyperpolarization is conducted through MEGJs to the VSMC layer (EDH) to effect vasodilation by inhibition of VSMC L-type Ca2+ channels. How EnNaC channels, which may be intrinsically mechanosensitive but not Ca2+ permeable, fit into this scheme is not understood; their activity may be related to the regulation of longer-term events in ECs.
The activities of the proteins in this scheme are subject to regulation by multiple additional factors. Flow-induced dilation is impaired in Cav-1-deficient arteries, which could be explained if some of the ion channels shown in FIGURE 20 were localized to caveolae or Cav-1 lipid rafts. Likewise, the glycocalyx might influence the transmission of shear stress to Piezo1 and Kir2 channels through connections to various ECM or CSK proteins, but those processes are not currently understood. Other EC mechanosensors such as the PECAM/VeCad/VEGFR2 complex appear to regulate longer-term EC processes such as EC alignment, gene expression, protein phosphorylation, and vascular wall remodeling.
6. MECHANICAL REGULATION OF TRANSCRIPTOME AND EPIGENOME
The cellular responses to changes of mechanical environments are mediated by mechanotransduction, in which the mechanical signaling modulates the expression profiles of genes and proteins to result in phenotypical and functional responses. The expressions of functional gene sets are based on the epigenomic and transcriptomic regulations, and such regulations are integral components of the comprehensive mechanotransduction processes.
6.1. Mechanoregulation of VSMC Epigenome
Epigenomic studies for mechanotransduction generally require cultured cells, and it is not easy to harvest a large number of VSMCs. VSMCs dedifferentiate rapidly in culture, switching from a contractile to a synthetic phenotype and losing the expression of certain contractile proteins and certain ion channels (83) while increasing the expression of proproliferative and proinflammatory markers and ECM proteins (1476–1478). For example, the expression levels of the L-type channel decline rapidly in VSMCs in culture (1479, 1480), with consequent changes in the expression of many other genes (1481). This process can be ameliorated to some extent by the continuous application of mechanical force (1482, 1483) or the use of culture medium supplement, but the supplemental factors may alter the expression of many (including unknown) genes to complicate the interpretation of the results. Thus, studies on epigenome of VSMCs present more challenge than ECs, and hence there are less definitive studies on the epigenome of VSMCs.
Histone modifications in VSMCs modulate the hyperplasia and neointimal restenosis induced by vascular injury and vein graft. Inhibition of HDAC 1, 2, or 3 prevents mitogen-induced VSMC proliferation via growth arrest in the G1 phase due to inhibition of retinoblastoma protein phosphorylation (1484–1488). Local administration of the HDAC inhibitor trichostatin A (TSA) prevents the proliferation of VSMCs and neointimal hyperplasia induced by balloon injury in rat carotid artery via activation of the KLF4/p21/p27 signaling pathway (1485). Enhancer of zeste homolog 2 (EZH2), the H3K27 methyltransferase component of polycomb repressive complex 2, is upregulated in the neointimal hyperplasia induced by wire injury of the rat carotid artery and by treatment of VSMCs with PDGF-BB; such EZH2 upregulations result in an increased level of H3K27me3 in VSMCs (1486). PDGF-BB causes the enrichment of H3K27me3 at the promotor regions of a tumor suppressor gene p16INK4A (cyclin-dependent kinase inhibitor) to lead to VSMC proliferation (1486). Inhibition of EZH2/1 by the chemical inhibitor UNC1999 suppresses the PDGF-BB-induced VSMC proliferation and injury-induced neointima formation by reducing H3K27me3 at p16INK4A (1486). JMJD2A, a histone demethylase, is upregulated by high glucose and arterial balloon injury to result in the reduction of H3K9me3 (1487). The inhibition of demethylase JMJD2A by the chemical inhibitor 2,4-pyridinedicarboxylic acid (2,4-PDCA) or siRNA attenuates neointimal formation in balloon-injured diabetic rats via the suppression of VSMC proliferation, migration, and inflammation by restoring H3K9me3 (1487). Histone-lysine N-methyltransferase Suv39h1 is significantly upregulated by ANG II and carotid balloon injury to lead to VSMC proliferation (1488). The downregulation of Suv39h1 promotes p21 and p27Kip1 expression to inhibit ANG II-induced VSMC proliferation and injury-induced neointimal hyperplasia, whereas the overexpression of SuV39h1 has opposite effects (1488). Although there is evidence that histone modifications in VSMCs exert significant effects on the responses of VSMCs to injury, their roles and the underlying mechanisms remain to be elucidated, especially by studies on the responses of cultured VSMCs to cyclic stretch.
In addition to histone modifications, epigenetic regulation by noncoding RNAs also plays key roles in regulating VSMC functions in vascular diseases (for review see Refs. 1489, 1490) and modulating VSMC phenotype switch (for review see Refs. 1489, 1491, 1492). Targeting VSMC miRNAs has potential therapeutic efficacy for vascular diseases (for review see Ref. 1493). It has been shown that stretch-dependent VSMC differentiation involves miRs and that Dicer knockout abolishes the stretch-induced VSMC contractile marker expression (for review see Ref. 1494). In vivo studies have shown that miR-145 is downregulated in VSMCs of atherosclerotic lesions (1495) and arteries in pulmonary hypertension (1496) to enhance disease progression and that miR-33 downregulation in grafted veins leads to increased Smad2/5 phosphorylation, BMP3 expression, and neointimal hyperplasia (1497). It has also been demonstrated in vein grafts in vivo and cyclic stretch in vitro that miR-29a is significantly upregulated to reduce DNA demethylase TET1 and hence VSMC contractile gene expression (1498). These findings suggest the roles of miRs in the mechanosensitive regulation of VSMC function. In vitro studies have demonstrated that cyclic stretch causes downregulation of miR-19b-3p to increase connective tissue growth factor (CTGF) and induce rat VSMC proliferation (1499), upregulation of miR-124-3p to decrease laminin A/C and cause VSMC apoptosis (1500), as well as upregulation of AP-1 to increase miR-21 with ensuing VSMC apoptosis (1501). However, the impact of cyclic stretch in regulating VSMC miRs is still not well understood. Recent transcriptomic studies demonstrate that long noncoding (lnc)RNAs play significant roles in mechanosensitive regulation of VSMC function. With a lncRNA microarray, it has been shown that 20% cyclic stretch leads to the downregulation of 281 lncRNAs and upregulation of 199 lncRNAs (1502) in human aortic VSMCs. Cross-examination of these 580 lncRNAs and the stretch-regulated mRNAs has led to the identification of 25 potential mRNA-lncRNA interactions (1502). Gene Ontology (GO) analyses of the differentially expressed mRNAs suggest that cyclic stretch regulates cytokine activation, cell differentiation, stress responses, and ECM remodeling in VSMCs, with implications for hypertension development (1502). The detailed mechanisms by which cyclic stretch regulates noncoding RNAs in modulating VSMC function remain to be determined.
6.2. Transcriptomic and Epigenomic Regulation in ECs
Conventional gene expression studies provide important but limited information on the snapshot of a transcript at one time point. With the advancement of the next-generation sequencing (NGS) technologies, the profiles of transcripts, epigenetic modifications, as well as chromatin remodeling can be revealed. Using systems biology approaches to analyze the multi-omic profiles allows the comprehension and prediction of the regulations of the networks, interactions, dynamics, and mechanisms of the genomic responses that govern cell functions and fates.
6.2.1. Transcription factors.
With conventional studies, the key transcription factors (TFs) for regulating EC homeostasis and dysfunctions have been identified. These TFs have been investigated by using NGS and systems biology approaches to reveal the interactions and networks in regulating EC gene expression under PS and OS.
6.2.1.1. TFS FOR EC HOMEOSTASIS: KRüPPEL-LIKE FACTORS.
A group of key TFs for the regulation of EC homeostasis is the Krüppel-like factor (KLF) family of zinc finger-containing transcription factors. In particular, KLF2/4 are highly induced in ECs under PS in comparison to OS (1503). The PS-induced KLF2/4 expression is mediated through signaling pathways involving ERK5, MEF2, and AMPK to positively regulate eNOS expression (1504) and consequently EC homeostasis. In addition to their conventional AMPK/MEF2/ERK5-mediated induction, KLFs are also modulated by epigenetic regulation involving microRNAs in response to different flow patterns. Disturbed flow causes the methylation of KLF4 promoter regions by DNA methyltransferase 3A (DNMT3A) to suppress KLF4 expression (1505). PS decreases miR-92a with an increase of KLF2 expression, and overexpression of miR-92a attenuates the flow induction of KLF2 (1506). Inhibition of miR-92a in Ldlr−/− or Apoe−/− mice increases KLF2 and KLF4 and attenuates atherosclerotic lesion formation (1507, 1508). Reduced EC dicer expression promotes EC inflammation and atherosclerosis in Apoe−/− mice under high-fat diet, and this is in part due to the suppression of miR103, which targets KLF4 and contributes to atherosclerosis (1509). Klf2+/−;Apoe−/− mice show significant increases of LDL uptake (under atherogenic diet) and atherosclerotic lesion formation in comparison to Klf2+/+;Apoe−/− mice (1510). Atherosclerosis is enhanced by EC-specific KLF4 knockdown and reduced by EC-specific KLF4 overexpression (1511). Further analysis has demonstrated that KLF4 binds to p300 to activate the transcription of the atheroprotective gene thrombomodulin (TM), while attenuating transcription of the atheroprone gene VCAM-1 by preventing p300-NF-κB interaction at the VCAM1 promoter region (1511). In addition, siRNA of KLF2 attenuates the PS-induced activation (phosphorylation and nuclear localization) of ATF2 and increases the expressions of proinflammatory genes (1512). It has also been demonstrated that preconditioning ECs with LS protects ECs from TNF-α- and simvastatin-induced EC inflammation (1513, 1514). The protective status induced by LS remains for hours after flow cessation; thus, after the initial suppression of transient MCP-1 induction, ECs are also protected against the second phase of flow induction of MCP-1 (1515), indicating an important atheroprotective role of LS. It is possible that the LS-induced KLFs compete with NF-κB for p300 recruitment and block the NF-κB/ATF2/cJun signaling for inflammatory and oxidative gene expressions (1516) (see sect. 6.2.1.3).
6.2.1.2. TFS FOR EC DYSFUNCTION (NF-κb, ap-1, ETC.).
Earlier studies demonstrated that the activation of the proinflammatory transcription factor NF-κB is shear sensitive (1517–1519) and mediates the inflammatory gene expression induced by low shear or high shear gradient (44, 1520, 1521). NF-κB is a complex composed of cRel proteins (including Rel A p65) with transactivation domains and p50/p52 subunits that direct NF-κB specificities. Upon activation, NF-κB is released from its inhibiting proteins, IκBs, and translocates to the nucleus for transcriptional activation (1522). NF-κB activation has a complicated time course with an oscillatory feature (biphasic and beyond) (1523, 1524). The biphasic/oscillatory activations of NF-κB in ECs under shear flow (1518) are IκBα dependent (1525). The NF-κB activation is significantly greater under steady low LS and OS (2 and 2 ± 2 dyn/cm2, respectively) than under high LS (16 dyn/cm2); furthermore, it is sustained under low shear but transient under high shear (1517, 1526). Flow-induced NF-κB activation is dependent on fibronectin (integrin αvβ3) but not collagen (integrin β1) (1406, 1519, 1527). With the use of a flow constriction cuff on the mouse carotid artery, it has been shown that the NF-κB/inflammation induced by flow disturbance is dependent on JNK (AP-1) (1528), suggesting a cross talk between MAPK and NF-κB pathways. Besides its direct interaction with the promoter regions of proinflammatory genes to regulate their transcriptions, NF-κB also induces hypoxia-induced factor-1alpha (HIF-1α) under low LS (4–5 dyn/cm2) to promote glycolysis and hence the enhancement of EC proliferation and dysfunction in vitro and in vivo (1529).
6.2.1.3. NGS/SYSTEMS BIOLOGY ANALYSES OF THE TFS.
Beyond the aforementioned conventional studies of the key TFs for EC homeostasis and dysfunction, a systems biology analysis has been performed on the time course of dynamic changes of the EC transcriptome under PS and OS (1530, 1531). Analyses of TF profiles showed that OS increases E2F1, early growth response 1 (EGR1), and HIF-1α, with the potential of cross-interactions with other TFs such as Fos protooncogene (FOS), serum response factor (SRF), and forkhead box O1 (FoxO1), to regulate the gene networks related to EC oxidative stress, inflammation, and cell cycle progression. In contrast, the PS-induced KLFs may interact with CCAAT enhancer binding protein beta (CEBPB) to regulate EC homeostasis. Furthermore, the analyses of histone modification of TF genes showed that PS caused the H3K27ac enrichment of SP1 and KLFs (1532), whereas OS caused the H3K27ac enrichment of FOX, CPEG, YAP, and TAZ (1532, 1533), which may in turn upregulate these TFs and the downstream gene expressions to modulate EC fates. These shear-dependent TFs, identified from studies on cultured ECs under shear conditions in vitro, were validated in vivo by single-cell RNA sequencing (scRNA-seq) and ATAC-seq studies using the mouse partial carotid artery ligation model (1534). Analysis of the scATAC-seq results revealed numerous TF binding motifs that are differentially regulated by disturbed flow in proatherogenic ECs (E8 cluster) in the partially ligated left carotid artery (LCA) in comparison to the healthy ECs (E2 cluster) under pulsatile flow in the right carotid artery (RCA). The TF binding motifs for KLF4 were enriched in the healthy ECs, whereas those for TEF, ETV3, RELA, FOS/JUN, TEAD1, and STAT1 were enriched in ECs exposed to disturbed flow. These results validate KLF2/4 as a predominant master TF under stable flow or PS conditions. In contrast, it is confirmed that RELA, FOS/JUN, and TEAD1 are TFs activated under disturbed flow conditions. Additionally, TEF, ETV3, and STAT1 were identified as potential novel TFs induced by disturbed flow. These TFs may play critical roles in regulating vascular functions and may serve as potential therapeutic targets for disease mitigation (for review see Ref. 1535). Such network modeling can contribute significantly to TF-targeted therapeutic development.
6.2.2. Transcriptional regulation of gene expression.
With the NGS/systems biology approaches, the transcriptional regulation of EC gene expression under different flow patterns has been shown to play an important role in modulating EC functions and vascular cell interactions. High-throughput approaches using microarrays and RNA-seq have demonstrated that, in general, LS and PS induce atheroprotective genes such as Klf2 and Enos, whereas disturbed flow and OS increase inflammatory genes such as VCAM-1 and MCP-1 (1531, 1536, 1537). However, some atheroprotective and atheroprone genes increase their expressions under both LS/PS and disturbed flow/OS conditions. In an in vivo study, Davies (1538) and colleagues have shown the coexistence of atheroprotective and atheroprone gene expressions in areas of disturbed flow in the swine aorta. These results indicate the complex hemodynamic regulation of EC gene expressions and hence vascular function with heterogeneous responses. The involvement of multiple ECs with high heterogeneity in the development of atherosclerotic diseases highlights the insufficiency of the bulk approach, which implies a “uniform” single cell type and would mask important information on the heterogeneity of cells in their responses to shear.
The advancement of single-cell (sc) sequencing technology provides the tools to gain information on the heterogeneous responses of individual cells under various pathophysiological conditions. Single-cell analysis of normal mouse aorta has established three distinct subpopulations of ECs (1539): 1) ECs that express canonical EC markers (e.g., VE-cad) with the Reactome features of ECM organization and integrin signaling, 2) ECs that express genes involved in lipid transport and angiogenesis with Reactome features of plasma lipoprotein assembly and clearance pathways, and 3) ECs that express genes specific to lymphatic ECs. Flow cytometry and in situ hybridization have validated these distinct EC subgroups. Interestingly, the EC subgroups in arteries from mice fed with western diet are similar to those from normal mouse arteries. Another scRNA-seq study of sorted ECs from the mouse aorta has led to the identification of three distinct clusters (plus a potential VSMC group): 1) mature differentiated ECs, 2) mesenchymal-like ECs, and 3) differentiated ECs with an inflammatory phenotype. Further analysis of the different genes in clusters 1 and 2 indicates that they are at different stages of EC differentiation, and pseudotime trajectory analysis indicates that clusters 1 and 3 are rooted at cluster 2, suggesting the differentiation of the mesenchymal-like population in cluster 2 into the other two types of ECs (1540). scRNA-seq analysis of the macrophage populations in atherosclerosis have been conducted using the CD45-sorted leukocyte populations isolated from Ldlr−/− mice on normal chow or western diet. Among 13 distinct aortic cell clusters identified, 3 are found predominantly in atherosclerotic vessels, including inflammatory macrophages (M1-like), resident-like macrophages (M2-like), and macrophages with “triggered receptor expressed on myeloid cells 2” (trem2). Whereas the first two clusters have more overlapping macrophage functions, the third cluster expresses genes that are related to lipid metabolism, cholesterol efflux, and oxidative stress. These three populations of macrophages can be detected in atherosclerotic vessels in Apoe−/− mice as well as human atherosclerotic lesions (1541). A recent study profiling ECs in the mouse partial ligation model with scRNA-seq and assay for transposase-accessible chromatin sequencing (ATAC-seq) showed that arterial ECs are highly heterogeneous, dynamic, and reprogrammed from healthy atheroprotective phenotype to proatherogenic phenotype (1534). This partial carotid ligation model study using single-cell analyses identified eight EC subpopulations along with other types of cells such as VSMCs, immune cells, and fibroblasts (1534). Disturbed flow increases the EC populations expressing atheroprone pathways, whereas steady LS increases the EC populations expressing atheroprotective pathways. Pseudotime trajectory and chromatin accessibility profile analyses demonstrate that disturbed flow promotes Endo-MT and endothelial-to-immunelike cell transition (Endo-ICLT). Although the effect of flow on Endo-MT and its role in atherosclerosis are established (1542–1547), the pathophysiological consequence of Endo-ICLT remains to be determined. As discussed above, the scATAC-seq study further demonstrated that KLF2/4 and NRF1 are known TFs for the regulation of EC homeostasis under steady shear, whereas RELA, TEAD, STAT1, and FOS/JUN are TFs that may play important roles in the regulation of EC heterogeneity and plasticity under disturbed flow (1534). Another study using mice with the same partial carotid ligation protocol also demonstrated the heterogeneity of vascular cells with the identification of 10 distinct disturbed flow-induced cell subpopulations, including ECs, VSMCs, macrophages, dendritic cells, lymphocytes, and granulocytes (1548). Among the EC subpopulations, four EC subgroups express high Cd36, Dickkopf WNT signaling pathway inhibitor 2 (Dkk2), Kallikrein Related Peptidase 8 (Klk8), or LDL receptor related protein 1 (Lpr1). The detections of Dkk2- and Cd36-expressing cells in the lesser curvature, but not the greater curvature, of the aorta arch support the notion that those cell types are induced by disturbed flow. However, the functional roles of those cells remain to be determined (1548). For macrophages, which interact with ECs, four subgroups have been identified to be enriched by disturbed flow; analyses of the top enriched genes suggest a potential increase in macrophage proliferation/apoptosis, which may contribute to macrophage accumulation in the vessel wall in atherogenesis. Furthermore, analysis of VSMC subpopulations demonstrated that disturbed flow enriches VSMCs expressing the osteoblastic marker Secreted phosphoprotein 1 (Spp1), suggesting a potential contribution to vascular calcification (1548). These findings provide valuable insights into the complexity of EC dynamics and transdifferentiation, as well as their interactions with multiple cell types in the vessel wall and the recruitment of blood cells. The information gained will allow deciphering the roles of the subpopulations of cells in controlling disease progression.
Although in vivo studies at the tissue and organ levels provide important information on the vascular responses to mechanical stimuli, it has been difficult to pinpoint the exact in situ mechanotransduction processes at the cellular/molecular/genomic levels. Single-cell profiling, by determining the differential responses at the transcriptomic/epigenomic levels of individual cells, is a major step to identify the critical targets for further investigations. Genomewide single-cell studies (e.g., scRNA-seq, sc-ATAC-seg) have shown that each cell type is not homogeneous, that the EC populations vary at different vasculature locations, and that these populations change over the time course of the applications of stimuli or treatments (1534, 1539, 1541). The trajectory of the population shifts suggests that the endothelium can be directed to various differentiation/dedifferentiation routes and to recruit different blood cells over time. Such vascular population dynamics and genomewide heterogeneity can be investigated with single-cell sequencing and spatial analysis technologies, such as multiplex imaging technology, multifluorophore FRET pairs (1549), 10X Visium (1550), and NanoString GeoMx (1551), which provide powerful tools to define the role of cellular heterogeneity in modulating homeostasis and pathogenesis. In addition, by relating the knowledge of cellular heterogeneity along the vascular tree to their local mechanical environments, the bioinformatic data analyses will allow the prediction of the mechanotransduction pathways and interactions in the native states of the cells. Furthermore, such information will allow the incorporation of guide RNA-based locus-targeting labels and gene modulation to alter the cell populations and mitigate disease progression. The use of these novel interdisciplinary approaches will allow the understanding of the vascular wall as a system. Investigations along these lines will lead to the development of new strategies for the treatment and prevention of atherosclerotic diseases.
6.2.3. Noncoding RNAs.
With the NGS/systems biology approaches, it has been shown that shear regulation of noncoding RNAs plays an important role in modulating EC functions and vascular cell interactions (TABLE 3).
Table 3.
Shear regulation of noncoding RNAs
| Condition | Targets | Pathways | Affected Genes | Functions | References | |
|---|---|---|---|---|---|---|
| EC miRs | ||||||
| miR-10a | PS | GATA6 ↓ | RARa-RANRE | VAMC1 ↓ | Inflammation ↓ | (1552) |
| miR-19a | LS | Cyclin D1↓ | Cyclin D1 ↓ | Proliferation ↓ | (1553) | |
| miR-21* | LS | PTEN ↓ | PI3K/Akt pathway↑ | Apoptosis ↓, eNOS ↑ | Homeostasis ↑ | (1554) |
| miR-23b | PS | CCNH ↓ | CAK ↓, E2F1 ↓, pRB ↓ | CAK complex ↓ | Proliferation ↓ | (1555, 1556) |
| miR-27b | PS | E2F1 ↓ | (1555) | |||
| miR-27a/b | LS | SEMAs ↓ | SEMAs ↓ | Pericyte recruitment ↑ | (1557) | |
| miR-101 | LS | mTOR ↓ | mTOR pathway ↓ | mTOR ↓ | Proliferation ↓ | (1558) |
| miR-126 | LS | SDC4 ↑, CECR4 ↑, SDF-1 ↓, VCAM1 ↓ | Vascular remodeling ↑ | (1559) | ||
| miR-21* | OS | PPARa ↓ | AP1 pathway ↑ | VCAM1 ↑, MCP1 ↑, miR-21 ↑ | Inflammation ↑ | (1560) |
| miR-92a | OS | KLF2 ↓ | eNOS pathway ↓ | KLF2 ↓, eNOS ↓ | Vasodilation ↓ | (1506) |
| miR-663 | OS | Predicted inflammatory gene ↑ | Inflammation ↑ | (1561) | ||
| EC lncRNAs | ||||||
| LINC00341 | PS | PRC2 recruitment | VCAM1 ↓ | Inflammation ↓ | (1562) | |
| SENCR | LS | CKAP4 | CDH5 internalization | Junction stability ↑ | (1563) | |
| LEENE | PS | PolII/KLF4/MED1 interaction | eNOS ↑ | Inflammation ↓ | (1564) | |
| STEEL | LS | PARP1 recruitment | KLF2/eNOS ↑ | Angiogenesis ↑ | (1565) | |
| MANTIS | LS | BGR1 recruitment | ICAM ↓ | Inflammation ↓ | (1566) | |
| AF131217.1 | LS | miR-128 | KLF2 ↑, ICAM ↓, VCAM1 ↓ | Inflammation ↓ | (1567, 1568) | |
| HOTAIR | LS (Rotation) | Predicted miR-185, 23b, 21 ↓ | (1569) | |||
| Extracellular miRs | ||||||
| miR-92a | OS | Mediated by VE | Macrophage: KLF2 ↑ | Inflammation ↑ | (1570) | |
| miR-126 | OS | Mediated by Ago2 | SMC: FOXO3, Bcl2, IRS1 ↑ | SMC turnover ↑ | (1571) | |
eNOS, endothelial nitric oxide synthase; LS, steady laminar shear. See glossary for other abbreviations.
6.2.3.1. MICRORNAS.
In addition to the conventional protein-coding RNAs, noncoding RNAs play critical roles in regulating the pathogenesis and progression of various human disorders, including cardiovascular diseases. MicroRNAs (miRs) are abundant noncoding RNAs consisting of ∼22 nucleotides; they can be detected in various cell types in the vessel wall as well as the circulating blood (1572). miRs have been identified as important players in the regulation of endothelial proliferation and inflammation under shear stress (see Ref. 46 for review) and used as potential biomarkers for many diseases, including atherosclerotic disorders (1573). S. Chien, Y.-S. Li, and others have shown that the miR-23b induced by PS causes a decrease in E2F1 and hypophosphorylation of Rb (1555), thus targeting cyclin H to reduce the activities of “cyclin-dependent kinase activating kinase” (CAK) and RNA polymerase II (PolII) (1556). Knocking down miR-23b results in a decrease of the PS-induced EC growth arrest (1555, 1556). miR19a reduces cyclin D1 (1553), and miR101 suppresses the mTOR pathway (1558) to cause cell growth arrest. Interestingly, miR-21 has been reported to be induced by both PS and OS. Under PS, miR-21 is increased to reduce the target gene PTEN, thus leading to an increase of eNOS phosphorylation and decrease of apoptosis (1554). However, OS has also been shown to induce miR-21, which directly targets PPARα to increase AP1 activity and downstream inflammatory gene expressions (1560). The details of the differential miR-21 regulations under different flow patterns remain to be elucidated. miR-92a is downregulated by PS to increase KLF2 expression and activate the downstream NO pathway to enhance EC homeostasis (1506). miR-663 expression is induced by OS, and knocking down miR-663 attenuates the OS-induced monocyte adhesion, suggesting its proinflammatory role in ECs (1561). miR-10a has a higher expression level in ECs under PS than under OS (1574), and it decreases transcription factor GATA6 to suppress inflammatory gene expression. Hence the decrease of miR-10a in ECs under PS leads to the increases of GATA6 and its downstream VCAM-1 expression to enhance the EC inflammatory phenotype (1574). It has also been reported that atheroprotective flow induces miR-126 expression to decrease VCAM-1 and increase syndecan-4 levels (1559), suggesting a role of miR-126 in reducing leukocyte adhesion. In general, one miR can target multiple mRNAs, and multiple miRs can target the same mRNA in a coordinated fashion, to cause their degradation or inhibit their translation. In addition, the miRs are transcribed with their host genes and manifest their functions in a cell- and stimulus-specific manner.
6.2.3.2. LONG NONCODING RNAS.
Long noncoding RNAs (lncRNAs), which are noncoding RNAs >200 nucleotides in length, have been identified as important regulators for EC functions in addition to miRs. lncRNAs can interact directly with DNA and chromatin to regulate gene transcription (1575) or interact with mRNA or miR to modulate gene levels (1576). Chien and coworkers, and others, have investigated the roles of lncRNAs in regulating EC functions in response to different flow patterns. Analysis of RNA-seq data on HUVECs subjected to PS and OS for 24 h has led to the identification of lncRNAs that function through different modes. For example, LINC00341 is an atheroprotective flow-induced lncRNA that interacts with polycomb repressive complex 2 and VCAM-1 promoter to suppress VCAM-1 expression and monocyte adhesion to ECs (1562). LINC00520, an enhancer-derived lncRNA, is also highly induced in ECs by atheroprotective flow. Predominantly associated with chromatins, LINC00520 serves as a guide to facilitate RNA polymerase II binding to the eNOS promoter to induce eNOS expression, thus termed “lncRNA that enhances eNOS expression” (LEENE) (1564). These studies demonstrate the emerging essential role of lncRNAs in promoting EC homeostasis (1564).
PS, but not OS, has been shown to induce several lncRNAs: SENCR targets cytoskeletal association protein 4 (CKAP4) to decrease cadherin 5 (CDH5/VE-Cad) internalization, stabilize EC-EC junctions, and reduce endothelium permeability (1563); STEEL guides PARP1 to the promotor regions of KLF2 and eNOS to enhance angiogenesis (1565); MANTIS decreases monocyte-EC interaction and attenuates ICAM-1 expression by reducing the binding of transcription factor BGR1 to the ICAM-1 promoter (1566); and AF131217.1 targets miR-128-3p to increase KLF2 and decrease ICAM-1 and VCAM-1 expressions to inhibit EC inflammatory responses (1567). With the use of a rotating shaker, it has been demonstrated that HOTAIR expression increases as a function of shear stress (reflected in the rotation speed in an orbital shaker), with a negative correlation with several miRs, including miR-23b, suggesting that it acts as a RNA sponge to inhibit miR functions (1569). This finding, obtained in an orbital shaker, in which the shear pattern is not well defined, seems to be contradictory to the results under flow that LS induces miR-23b to cause EC growth arrest (1555, 1556). Hence, the functional role of the rotation-induced HOTAIR remains to be ascertained. Other lncRNAs, such as LINC00323 (1577), MALAT1 (1578–1580), H19 (1581), MEG3 (1582–1585), and LISPR1 (1586), have been reported to regulate EC transcription, epigenetics, proliferation, apoptosis, aging, inflammation, migration, and angiogenesis. However, their mechano-responsiveness remains to be determined. These recent studies reveal several novel pathways for hemodynamic regulation of EC functions.
An NGS study has identified 199 lncRNAs and 97 mRNAs to be differentially expressed (DE) in pulmonary microvascular ECs subjected to 20% cyclic stretch for 2 h (1587). Analyses of the lncRNA/mRNA coexpression networks reveal the enrichment of EC responses to hypoxia, oxidative stress, and inflammation. Four potential lncRNAs (n335470, n406639, n333984, and n337322) have been identified to act in cis with inflammatory genes (1587), and they may play important roles in regulating the stress responses in these ECs, but the functions of these lncRNAs in EC responses to stretch in vitro and in vivo remain to be determined. A study on HUVECs subjected to 18% uniaxial cyclic stretch shows that there are 960 mRNAs, 74 miRNAs, and 155 lncRNAs among the DE transcripts. A lncRNA-associated competing endogenous RNA (CeRNA) network has been constructed based on the public databases miRcode and miRTarBase, with the prediction of miRNA-mRNA and lncRNA-miR interactions. Analyses of the top 20 nodes for these interactions have identified lncRNA NEAT1 as a potential major regulator for the HUVEC transcriptome (1588). NEAT1 has been reported to function as a miR sponge to modulate a variety of human diseases, including cancer and metabolic and cardiovascular diseases (1589). Expression studies and function validation reveal that NEAT1 is a functional lncRNA downregulated by cyclic stretch, which can lead to increases of EC apoptosis (1588). The comprehensive data sets for stretch regulation of EC transcriptome and epigenome remain to be established.
6.2.3.3. EXTRACELLULAR MIRS AND INTERCELLULAR COMMUNICATION OF MIRS.
Circulating miRs can serve as biomarkers for cardiovascular diseases (see Ref. 1590 for review). As mentioned above, miR-21 (1554, 1560), miR-27b (1557), miR-92a (1506), and miR-126 (1559, 1571) have been shown to be shear-regulated miRs in ECs. Some of these EC-derived miRs (such as miR-21) also function in VSMCs (1591, 1592) and macrophages (1593) to promote proliferation and migration, respectively, thus playing a role in integrating the vascular function among these cell types. Overexpression of miR-27b in ECs increases EC-pericyte interactions (1557). miR-126 and miR-92a have been shown to be transported from ECs to VSMCs and macrophages, respectively, to regulate their functions (1570, 1571). Coculturing with ECs causes an increase of miR-126 in VSMCs without enhancing miR biosynthesis; this increase can be abolished by the application of laminar shear to the EC layer. The miR-126 secreted by ECs is associated with Ago2 but not the cell membrane. The detection of exogenous biotinylated miR-126 (b-miR-126) in VSMCs incubated with the conditioned media of EC transfected with b-miR-126 indicates that miR-126 can be transferred from ECs to VSMCs. Knockdown and overexpression of EC miR-126 decreased and increased, respectively, the miR-126 levels in VSMCs incubated with the EC-conditioned media, thus confirming that ECs are the source of VSMC miR-126. The increase of miR-126 in VSMCs leads to an increase in VSMC turnover. The finding of a significant reduction of the arterial ligation-induced atherosclerotic lesions in miR-126−/− mice suggests a proatherogenic role for miR-126 (1570, 1571). It has also been demonstrated that OS enhances the secretion of miR-126 and miR-200a through the activation of vesicle-associated membrane protein 3 (VAMP3) and synaptosomal-associated protein 23 (SNAP23). Inhibition of VAMP3 and SNAP23 ameliorates the VSMC hyperplasia induced by partial carotid artery ligation, indicating the critical role of EC-VSMC interaction in atherogenesis (1594). Macrophage-EC interactions also play critical roles in vascular homeostasis. A recent study shows that macrophages cocultured with ECs subjected to OS or TNF-α treatment exhibit high expression levels of inflammatory genes compared with macrophages cocultured with ECs subjected to PS. These responses can be modulated by alterations in EC miR-92a level, suggesting miR-92a as a mediator for EC-macrophage interactions (1570). The detection of Dy547 in macrophages cocultured with ECs transfected with Dy547-miR-92a indicates the transmission of miR-92a from ECs to macrophages. Further analysis has demonstrated that, unlike miR-126 secretion, the EC secretion of miR-92a is predominantly via extracellular vesicles (EVs). The EC-derived EV miR-92a reduces macrophage KLF4, increases inflammatory gene expression and LDL uptake, and reduces macrophage migration, thus leading to an atheroprone phenotype (1570). The roles of extracellular miR-21 and -27 b as mediators for intercellular communications in the vascular system remain to be determined.
6.2.4. Epigenetic modifications.
Epigenetic regulation, including the modifications of the epigenome (mainly methylation and acetylation of DNA and histone) and epitranscriptome (mainly methylation of RNA), plays important roles in modulating endothelial gene expression and functions (1595).
6.2.4.1. DNA AND RNA MODIFICATIONS.
Epigenetic modulations of DNA and RNA, mainly through their methylation/demethylation, have been demonstrated to play important roles in regulating the transcript profiles of cells in response to chemical and mechanical stimuli (for review see Ref. 1596). DNA modifications are mainly involved in transcriptional regulation through the chromatin structural changes that modulate the accessibility of the transcription machinery in its interaction with DNA. RNA modifications are involved in the posttranscriptional regulation of transcripts (including coding and noncoding RNAs) for the recruitment of modified RNA binding proteins to modulate RNA splicing, translocation, stability, degradation, and translation (for review see Ref. 1597). It is of importance to understand the epigenomic modifications as components of mechanotransduction that mediate, at least in part, the mechanoregulated functional gene profiles for vascular health and disease.
DNA methylation of the 5th carbon of cytosine (5mC) typically acts as a transcription suppressor by preventing the recruitment of transcription factors in the formation of transcription machinery. DNA methyltransferases (DNMTs) are the enzymes that mediate DNA methylation. Jo’s (1598) and Chien’s (1445) groups have shown that OS increases DNMT1 expression and DNA methylation in vitro and in vivo. Inhibition of DNMTs by 5′-Aza-2-deoxycytidine (5Aza) mitigates the disturbed flow-induced atherogenesis (1445, 1598). A genomewide study has shown differential DNA methylation in atheroprone versus atheroprotective regions in mouse arteries, with the identification of 11 mechanosensitive genes, including the transcription factors HOX5A and KLF3, whose downregulation may be involved in generating the EC inflammatory phenotype under OS (1598). Davies and colleagues (1505) showed in human aortic ECs and swine aorta that the sites for DNA methylation in response to OS are the KLF4 promoter and that this methylation is mediated by DNMT3A. Inhibition of DNMTs by RG108 or 5Aza reduces the OS-induced methylation of KLF4 promoter and increases KLF4 expression (1505). It has been shown that OS causes an increase of DNMT1 binding to cyclin A promoter to induce its hypermethylation and that inhibition of DNMT1 abolishes the OS induction of cyclin A (1599). Interestingly, OS decreases DNMT1 binding to the CTGF promoter to induce its hypomethylation, but the manipulation of DNMT1 has little effect on the induction of CTGF by OS (1599). These results suggest the involvement of multiple cofactors in the flow regulation of gene expression by DNA methylation. A recent study has demonstrated that the level of N6-methyladenine (m6A) DNA modification in leukocytes is negatively correlated to the severity of atherosclerotic lesions in patients, whereas 5mC does not show such correlation (1600). With the atherosclerotic mouse model, it has been found that western diet (WD) reduces the DNA m6A level of leukocytes, with no effect on N6A-DNA methyltransferase (N6AMT-1), but it induces the expression of demethylase AlkB homolog1 (ALKBH1) (1600). In addition, WD induces the expression of lncRNA MIAT in leukocytes and at the aortic root. MIAT has been identified as a potential target for ALKBH1 (1600) and is implicated to play a faciliatory role in inducing atherosclerosis in mice and humans (1601, 1602). In vitro experiments have demonstrated that oxidized-LDL induces m6A modifications and MIAT expression in THP1 and ECs (1600). m6A chromatin immunoprecipitation (ChIP) has shown that ox-LDL increases m6A modification at the MIAT promoter region and that knockdown of ALKBH1 attenuates the ox-LDL-induced MIAT expression (1600). However, the effects of depletion of ALKBH1 and MIAT on vascular functions have not been tested. It is of great interest to investigate the roles of multiple forms of DNA modifications in the hemodynamic regulation of vascular homeostasis and atherosclerotic disease.
In addition to the role of DNA modification in transcriptional regulation, investigations on RNA modifications have generated emerging mechanisms in posttranscriptional regulation of gene expression. Distinct types of RNA modification, such as methylation and acetylation, have been identified to control RNA fate through modulations of translocation, stability, splicing, and translation (1603). N6-methyladenosine (m6A) RNA methylation is the most abundant RNA modification observed in mammalian cells; it is regulated by methyltransferases (writers), m6A binding proteins (readers), and demethylases (erasers) (1597). Multiple types of RNAs, including mRNAs, lncRNAs, microRNAs, rRNAs, and tRNAs, can be modified with m6A modification (1604). Dysregulation of RNA modifications has been linked to cancer development, disruption of stem cell differentiation, and, more recently, cardiovascular dysfunctions such as VSMC differentiation, heart failure, abdominal aortic aneurysm, vascular calcification, and hypertension (for review see Ref. 1605). The progression of an atherosclerotic lesion is associated with the dynamic changes of m6A levels and modifiers (decrease in early lesion and increase in late stage) (1606), suggesting functional roles of m6A in atherogenesis. Only recently have the roles of m6A in the flow regulation of EC biology begun to be unraveled. Chien and colleagues (1607) have recently identified the m6A methyltransferase METTL3 (writer) as a major regulator for the flow-modulated epitranscriptome. Compared with PS, OS upregulates METTL3 expression in ECs, which is accompanied by augmented RNA m6A hypermethylation that leads to EC inflammatory responses. Knockdown of METTL3 abrogates this OS-induced m6A hypermethylation and EC inflammation, whereas METTL3 overexpression in ECs under PS leads to changes resembling the effects of OS. RNA-sequencing and m6A enhanced cross linking and immunoprecipitation (eCLIP) sequencing experiments have revealed that the OS-enriched epitranscriptome mediated by METTL3/m6A involves EC stress, inflammation, and metabolic functions (1607). Importantly, knockdown of METTL3 via lentiviral-shRNA administration prevents the atherogenic process in mice (1607). These findings have shown that epitranscriptomic regulation of RNA methylation is a novel aspect of mechanotransduction that plays an important role in the mechanoregulation of vascular function in health and disease.
6.2.4.2. HISTONE MODIFICATIONS.
In addition to DNA modifications, histone and nonhistone protein modifications are also dynamic processes that regulate gene expression in a complex manner. In general, histone acetylation leads to increases of euchromatin accessibility and hence transcription activation, whereas histone methylation leads to increases of heterochromatin to have the opposite effects (1608). However, the sites and levels of modifications at different histones may lead to diverse outcomes. The main regulators for histone acetylation and deacetylation are histone acetyltransferases (HATs) and deacetylases (HDACs), respectively, and those for histone methylation and demethylation are histone methyltransferases (HMTs) and demethylases (HDMs), respectively. The flow regulation of HATs is not clearly understood. An early study showed that atheroprotective flow increases p300/HAT activity and the acetylation of NF-κB p65 at eNOS promoter regions and that inhibition of p300/HAT activation abolishes the shear induction of eNOS expression (1609). It has been proposed that NF-κB at promoter regions may act as the mediator that balances the recruitments of p300/HAT and HDACs to modulate gene expression during angiogenesis (1610).
Multiple studies show that shear stresses modify histone and nonhistone proteins to change gene expression in ECs (TABLE 4). LS induces the phosphorylation and nuclear export of HDAC5, leading to the release of the associated MEF2 to induce KLF2 transcription. Mutations of HDAC5 phosphorylation sites S259/S498 abolish the LS-induced HDAC5 nuclear export and KLF2 expression (1611). In contrast, OS induces the expression and nuclear localization of HDAC1/2/3 and 5/7 to lead to their associations with NF-E2-related factor 2 (Nrf2) and MEF2, respectively (1612). Such associations cause the deacetylations of these factors to reduce the expressions of NADPH quinone oxidoreductase1 (NQO1) and KLF2, thus exerting adverse effects on EC homeostasis. LS elevates the phosphorylation and cytosolic retention of HDAC5/7 to prevent Nrf2/MEF2 deacetylation and increase the expressions of NQO1 and KLF2 for EC homeostasis. In the same study, it has also been found that inhibition of HDAC3 can attenuate the increase of cyclin A, decrease of p21 expression, and EC proliferation induced by OS (1612). In addition, it has been reported that the OS-induced HDAC3/5/7 interact with retinoic acid receptor-α to repress miR-10a expression and increase GATA6-mediated VCAM-1 expression (1574). In addition to HDAC regulation, it has been shown that the deacetylase Sirt1 is expressed at higher levels in the atheroprotective regions of the mouse arteries (1458). An in vitro mechanistic study for flow regulation of Sirt1 shows that atheroprotective flow increases AMPK activation to phosphorylate eNOS, thus priming the Sirt1 binding and deacetylation of the phosphorylated eNOS, and consequently NO production (1458). These studies have demonstrated the involvement of histone methylation and acetylation in the mechanoregulation of EC functions.
Table 4.
Shear regulation of epigenetic modifications
| Condition | Enzymes | Affected Genes | Functions | References | |
|---|---|---|---|---|---|
| DNA modification | |||||
| Methyltransferases | |||||
| 5mC | OS | DNMT1 | HOX5A ↓, KLF3 ↓, etc. | Inflammation ↑ | (1598) |
| OS | DNMT1 | Cyclin A ↑, CTGF ↑ | Dysfunction ↑ | (1445, 1599) | |
| OS | DNMT3A | KLF4 ↓ | (1505) | ||
| Demethylases | |||||
| m6A | Neg corr. with lesion | ALKBH1 | MIAT↑ | Lesion ↓ | (1600) |
| RNA modification | |||||
| Methyltransferase | |||||
| m6A | OS | METTL3 | KLF4 ↓, NLRP1↑ | Inflammation ↑ | (1607) |
| Protein acetylation | |||||
| Acetylase | |||||
| eNOS | LS | HAT | eNOS ↑ | Homeostasis ↑ | (1609) |
| Deacetylases | |||||
| MEF2 | LS | HDAC5 (cytosolic) | KLF2 ↑ | Homeostasis ↑ | (1611) |
| MEF2, Nrf2 | OS | HDAC1/2/3, 5/7 (nucleic) | NQO1, KLF2 ↓ | Dysfunction ↑ | (1612) |
| PARa | OS | HDAC 3/5/7 | miR-10a ↑, VCAM1 ↑ | Dysfunction ↑ | (1574) |
| eNOS | PS | SIRT1 | eNOS ↑ | Vasodilation ↑ | (1458) |
| Histone acetylation | |||||
| Acetylase | |||||
| H3K27 | PS | ChIP-seq: e.g., IRPR3 ↑, KLF4 ↑ | Homeostasis ↑ | (1532) | |
| H3K27 | OS | ChIP-seq: e.g., ETS ↑ | Dysfunction ↑ | (1533) | |
ChIP-seq, chromatin immunoprecipitation-sequencing; eNOS, endothelial nitric oxide synthase. See GLOSSARY for other abbreviations.
High-throughput experiments with various modified histone ChIP-seq have been performed to elucidate the genomewide histone modifications in gene expression under different flow patterns. Shyy and colleagues (1532) focused on the PS-enriched H3K27ac and KLF pathway in ECs, and Schnittler and colleagues (1533) focused on the OS-enriched H3K27ac and YAP pathway in ECs. These studies have demonstrated that H3K27ac is differentially regulated by PS versus OS and that the H3K27ac enrichment is mainly located at the promoter and enhancer regions of the genes. Analyses of TF binding regions with H3K27Kac (1532) have shown that SP1/KLF and FOX/CPEB motifs are among the top-ranked regions under PS and OS, respectively. Integrated analyses of H3K27ac/H3K4me-ChIP-seq and KLF4 ATAC-seq have led to the identification of 18 genes that are upregulated by both PS and KLF4 (1532). Among those, inositol 1,4,5-trisphosphate receptor 3 (ITPR3) is found to regulate calcium influx and NO production in ECs under PS (1532). Bondareva et al. (1533), using the GO term and KEGG pathway analyses, have identified the genes with OS-enriched H3K27ac and shown their involvement in signaling cascades relevant to atherogenesis (including cell cycle progression, metabolism, survival, and apoptosis). With TF binding analysis, ETS/KLF/AP1 and YAP/TAZ are found to be the top-ranked motifs enriched in ECs under OS; this is in agreement with the findings that OS induces YAP/TAZ activation, nuclear translocation, and binding to target gene promoters (1533). Nakato et al. (1613) performed analyses of 131 data sets of active histone ChIP-seq and RNA-seq of nine different sources of ECs to reveal that all isolated ECs express EC markers but with variations in H3K27ac/H3K4me3 marks and enhancers across cell types. Interestingly, principal component analysis (PCA) shows that the ECs can be clustered into two groups, ECs from upper body (e.g., aorta, coronary, carotid, and pulmonary arteries) and ECs from lower body (e.g., renal artery, umbilical artery, umbilical vein, and saphenous vein), suggesting developmental diversity.
In conclusion, ECs respond to shear flows to modulate the landscape of histone methylations and acetylations, which in turn control the accessibility for recruitment and activation of transcription factors. Although the activation marks for flow regulation of histone modifications (e.g., H3K27ac) have been investigated in detail, there is a relative lack of information on histone repressive marks (e.g., H3K9me2/3), probably because of the low signal outputs for profiling studies. It has been demonstrated that histone modifications affect chromatin states, i.e., euchromatin versus heterochromatin, which in turn modulate nuclear rigidity independent of the lamin contribution. The increase in euchromatin due to histone modifications (e.g., acetylation) leads to reductions of nuclear rigidity and DNA damage; these effects are reversed by the inhibition of HDACs (for review see Ref. 1614). Although it has been well established that nuclear skeleton/LINC-mediated mechanotransduction is important for the regulation of gene expression, there is a paucity of information on the mechanisms by which the chromatin-dependent nuclear rigidity affects mechanotransduction and the consequential gene expression.
7. VASCULAR MECHANOTRANSDUCTION IN HEALTH AND DISEASE
7.1. Mechanotransduction in Hypertension
Systemic hypertension (HT) is a major risk factor for many human diseases, including stroke, myocardial infarction, renovascular disease, and vascular dementia. HT is associated with increased vascular resistance, enhanced VSMC contractility, rarefaction of small blood vessels, and vascular wall remodeling. This section focuses on the role of vascular mechanotransduction in the development of HT.
Many different animal models of HT provide evidence for enhanced arterial myogenic responsiveness, either preceding or in association with the development of HT. Thus, at a given pressure small arteries and arterioles from hypertensive animals may exhibit a higher basal myogenic tone, show an exaggerated myogenic constriction to a standardized step pressure increase (1615, 1616), and/or maintain a constriction over an extended pressure range (Refs. 924, 1617; see FIGURE 8). Evidence in support of enhanced myogenic responsiveness in HT comes from studies of large arteries, small arteries, and arterioles from multiple vascular beds, including the renal (1618–1621), cerebral (1622, 1623), skeletal muscle (924), and mesenteric (1624) circulations. Other studies, however, find that arterial responsiveness to pressure in HT is unchanged (1625–1628) or impaired (1617, 1629, 1630). It is likely that multiple factors underlie these disparate findings, including the model of hypertension used (see below), the size and location of the artery studied, and the time point at which measurements were made during the development of HT. For example, enhanced myogenic gain is commonly observed in the SHR (spontaneous hypertensive rat) model of HT, whereas impaired myogenic responsiveness is often observed in high-salt models of HT (1629–1631). Furthermore, increased myogenic gain is most often observed during the developmental phase of HT and less consistently observed after it has been established (1624, 1632). Myogenic gain varies between large and small arteries within the same hypertensive vascular bed, and changes in HT may reflect the partial normalization of pressure in the downstream segments of a vascular network (1632). When these factors are considered, the preponderance of evidence suggests that some degree of enhanced myogenic responsiveness of small arteries and/or arterioles is associated with, or contributes to, the development of HT. Indeed, this is the premise of many reviews on HT (1633–1635).
Whether vasoconstriction is an initiating factor in the development of HT or a consequence of HT is a long-standing issue addressed by many investigators (1616, 1636–1638). Regardless, the enhanced constriction of small arteries and arterioles in HT is generally considered to be a protective mechanism to prevent elevated systemic pressure from being transmitted downstream to capillaries in the respective organ. For example, enhanced myogenic constriction of renal interlobular and afferent arterioles in HT confers protection on the glomerulus by attenuating the fraction of elevated systemic arterial pressure transmitted to glomerular capillaries (127, 365, 1639–1642). Impairment of this mechanism leads to subclinical edema that can compromise functions of the target organ (1643). Insufficient myogenic autoregulation in the hypertensive cerebral circulation promotes focal cerebral ischemia, breakdown of the blood-brain barrier, and infiltration of immune cell-generated cytokines (1644). Impaired myogenic responses in the retinal vasculature accelerate the development of diabetic retinopathy (1645). Even in the mesenteric circulation, arterial lesions can result from the elevated pressure associated with HT (1646). When HT progresses to include rarefaction of the smallest arterioles and/or capillaries (1647–1650), perfusion of the entire capillary network is compromised. The renal circulation is particularly important because myogenic and tubular-glomerular feedback mechanisms interact to determine the degree of renal pressure/flow autoregulation (1651), which in turn affects the production of renin; the conversion of renin to angiotensin II has systemic effects on arterial tone and promotes VSMC growth and dedifferentiation (1642, 1652).
The literature suggests that the mechanisms underlying enhanced myogenic responsiveness in HT involve many of the VSM mechanotransduction mechanisms discussed in previous sections of this review. One of the earliest mechanistic observations was an enhanced depolarization of cerebral artery VSMCs from the SHR compared with normotensive rats (1622), suggesting an increase in cation permeability. Subsequent studies of hypertensive arteries support this idea and provide evidence for the enhanced activities of multiple ion channels, including VGCCs (1653–1656), TRP channels (1657), ENaC channels (372), and Ca2+-activated Cl− channels (693). Augmented myogenic responses in HT are also associated with increased PKC activity (1658), enhanced production of cytochrome P-450 metabolites such as 20-HETE (1657, 1659), phenotypic switching of IP3Rs (1660), and decreased signaling through the RYR2-BK channel axis that would otherwise oppose myogenic constriction (841). Evidence also points to enhanced Ca2+ sensitivity of the contractile machinery (1661) and to other pathways, including increases in production of ROS (1621, 1662) and endothelin in ECs (1663, 1664) and Ca2+ entry through the STIM/Orai mechanism in VSMCs (1665).
The development of HT also involves alterations in EC mechanotransduction (15, 1666–1668). In response to physicochemical stimuli in HT, ECs release endothelium-derived contracting factors (EDCFs), including endothelins and angiotensin II, as well as cyclooxygenase-derived prostanoids and superoxide anions, which have been identified as being responsible for the impairment of endothelium-dependent vasodilation in patients with essential HT (1669). The vasodilation response to acetylcholine is increased by indomethacin, a cyclooxygenase inhibitor, and vitamin C, an antioxidant, by restoring NO availability. Thus, HT involves a decline in EC function with a decrease in NO bioavailability and an increase in the production of EDCFs.
Studying the role of alterations of vasomotor responses of microvessels in HT, Koller (15) concluded that ECs have a central role in the early functional adaptations to HT. Pressure-induced myogenic constriction of arterioles is enhanced in HT because of the augmented release of EC-derived constrictor factors that modulate arteriolar Ca2+- and K+-permeable ion channels in VSMCs to regulate VSMC membrane potential, whereas flow/shear stress-induced arteriolar dilation is reduced in HT because of the impaired NO component of the response (for review see Ref. 1670). The alterations in synthesis and/or action of EC-derived mediators may be initiated by the elevated hemodynamic forces in HT, probably via the enhancement of ROS release and the ensuing interference with synthesis and/or action of EC-derived mediators.
Harrison and colleagues (1668) studied the effect of hyperstretch on confluent human aortic ECs cocultured with human monocytes (MCs). An increase in stretch from the baseline of 5% to 10% enhanced the conversion of the MCs to CD14+/CD16+ intermediate MCs and MCs bearing the CD209 marker, increased their mRNA expressions of IL-6, IL-1β, IL-23, chemokine (C-C motif) ligand 4, and TNF-α, and activated STAT3. Inhibition of STAT3 or NO, neutralization of IL-6, and scavenging of H2O2 prevented the formation of intermediate MCs in response to increased stretch. The authors showed increased infiltration of MCs with activated STAT3 and MC-derived cells in aortas and kidneys of mice with angiotensin II-induced HT; they also showed that hypertensive patients have increased intermediate and nonclassical MCs, with STAT3 activation in their intermediate MCs. These are likely the results, in part, of a loss of NO signaling and increased release of IL-6 and H2O2 by the dysfunctional EC and a parallel increase in STAT activation in adjacent MCs.
Both innate and adaptive immune responses regulate EC function under HT (1671, 1672). There is evidence that the HT-associated vascular EC dysfunction is related to systemic and local vascular inflammation (1673, 1674).
Data from the Framingham offspring cohort suggest that the severity of HT is positively associated with the degree of impairment of EC function (1675). Whether EC dysfunction is a cause or an effect of HT remains controversial; there seems to be a complex and potentially bidirectional relationship.
7.2. VSM Mechanotransduction in Vascular Wall Remodeling
Increased myogenic gain facilitates chronic arterial constriction, which when combined with elevated systemic pressure eventually leads to remodeling of the arterial wall (1676). Remodeling has been defined as an active structural change that involves cell growth, death, and migration as well as synthesis and/or degradation of ECM proteins (1677, 1678). Vascular wall remodeling can be viewed as a progression of mechanistic steps that include:
-
1)
rapid changes in VSMC signaling that lead to Ca2+-dependent and -independent regulation of MLC20 phosphorylation, occurring over a fraction of a second to several seconds;
-
2)
changes in myofilament organization, including actin polymerization, occurring within seconds to minutes;
-
3)
changes in VSMC attachment and orientation, occurring within minutes to hours;
-
4)
phenotypic changes in VSMCs and other vascular wall cells, requiring nuclear transcription and gene expression, occurring over hours to days;
-
5)
overt structural changes in ECM composition, occurring over days to weeks.
7.2.1. Vascular wall remodeling in hypertension.
Historically, in vivo studies describing HT-associated changes in luminal diameter or altered medial wall thickness of arteries and arterioles often yielded conflicting and confusing results. These resulted from studying vessels with varying levels of active tone or different or unknown levels of intraluminal pressure, from different regional circulations, and/or of different branching orders (1634, 1637, 1638, 1679). Consistency was improved by normalizing wall cross-sectional area to vessel diameter and further improved when several major laboratories proposed a scheme to classify the types of arterial remodeling in terms of both diameter and wall cross-sectional area (1680). This scheme has now become the de facto standard of remodeling nomenclature. According to this system, inward and outward remodeling are defined as a decreased or increased luminal diameter, respectively, and hypertrophic and hypotrophic remodeling are defined as an increased or decreased wall cross-sectional area, respectively. Eutrophic remodeling is defined as parallel changes in inner and outer diameter without a change in wall cross-sectional area.
The type of remodeling that occurs in HT is determined both by the underlying cause and by the extent of the chronic change in pressure and/or flow (1634). Thus, spontaneous HT, renovascular HT, reduced pressure, reduced flow, sympathetic stimulation, elevated circulating levels of vasoconstrictors, and impaired EC production of vasodilators produce different types of arterial wall remodeling (see Ref. 1681 for references]. However, the two most common types of remodeling associated with many forms of HT are inward, hypertrophic remodeling (1682–1684) and inward, eutrophic remodeling (1685–1687). Inward, eutrophic remodeling can also be induced by chronic low blood flow (1688–1690). Outward, hypertrophic remodeling occurs with chronic high blood flow (1690–1692). Outward, hypotrophic remodeling occurs with the chronic use of vasodilators (1693). The observation that flow inhibits inward remodeling initiated by flow or agonists (1694) implies that significant interactions between VSMCs and ECs result from the chronic effects of pressure and flow on these respective cell types.
It is well established that prolonged vasoconstriction induces wall remodeling, with the degree of tone determining the direction (inward vs. outward) of remodeling (1618, 1695–1697). This link between vascular tone and the induction of arterial wall remodeling suggests a critical role for VSMC mechanosensing. Thus, the degree of myogenic tone appears to determine the degree of remodeling such that low myogenic tone in large arteries is associated with more subtle remodeling than what occurs in small arteries/arterioles with high myogenic tone (1698, 1699). Even blood flow-dependent arterial remodeling is thought to be directed by the level of vascular tone (1700). vanBavel and coworkers (1698, 1701, 1702) found that the optimal diameter for active tension development of small mesenteric arteries shifts along with that for passive tension such that the relationship between the active and passive biomechanics is maintained during inward remodeling. As a result, the reduced diameter and increased medial thickness result in the normalization of wall stress (1698), according to σθ = [(Pint − Pext)·r]/h, a finding confirmed by other laboratories (1625, 1703, 1704). A mechanistic model of remodeling has been proposed in which the level of VSMC activation is the controlled variable (1697). This model predicts that the acute changes in VSMC activation leading to constriction are followed by a slow, compensatory phase of remodeling that eventually results in structural changes in the arterial wall. The predictions of this model are in qualitative agreement with the experimental findings of many laboratories. It should be noted that the behavior of the model does not depend on myogenic responsiveness per se (i.e., this behavior is independent of whether the active, steady-state pressure-diameter relationship has a positive or negative slope) but simply on the level of VSMC activation, regardless of the stimulus. This is consistent with the finding that prolonged agonist-mediated constriction can induce inward modeling in the absence of a pressure change (1618, 1705, 1706). The model is limited in that it is based only on the behavior of a single segment of artery. As vanBavel and coworkers (1698) pointed out, normalization of wall stress may be adaptive for the VSMC or the vessel but can be maladaptive for the arterial network or the cardiovascular system as a whole. More complex models of remodeling that incorporate entire networks, as well as the effects of both pressure and flow (1707, 1708), have been valuable in understanding what variables are being controlled during arterial wall remodeling.
7.2.2. Plasticity in vascular wall remodeling.
What factors determine how VSMCs switch from acute to chronic adaptation in response to elevated pressure, i.e., from normal to enhanced myogenic responses, with eventual progression to remodeling? Both visceral and vascular SMCs exhibit a remarkable degree of inherent “plasticity” that is manifested under both normal and pathological conditions. In general, SMCs have a much wider active length-tension (L-T) relationship than skeletal muscle cells, allowing the development of substantial active tension over the wide range of lengths experienced by these cells surrounding the blood vessel (1709, 1710). Force development of SM decreases as the muscle shortens to lesser lengths (175, 1711), reflecting the phenomenon of shortening-dependent cross-bridge inactivation, which limits the degree of constriction that can occur. Studies of airway smooth muscle (ASM) by Gunst and coworkers (1712–1714) suggest that SMCs “remember” their contraction history in that the length of the contractile element established during isometric contraction affects the force developed during subsequent stretch of the muscle. This degree of plasticity seems to be a general property of SM, as much of the behavior observed in ASM (1715, 1716) is common to VSM (175, 1717, 1718), gastrointestinal SM (1719), and bladder SM (1720, 1721). The plasticity of ASM is further illustrated by studies in which unstimulated ASM is maintained at various suboptimal lengths for 24 h followed by the determination of active and passive L-T relationships (1716, 1722). The new length for optimal force development adapts to the previously imposed length, whether it is lower or higher than the original optimal length. It is easy to envision how this process could be extended to produce the results described in arteries by vanBavel and coworkers (1698, 1701, 1702) in which inward remodeling results in a leftward shift in the active L-T relationship.
Martinez-Lemus and coworkers provide evidence for another aspect of VSMC plasticity. When resistance arteries are exposed to a vasoconstrictor for a period of ∼4 h, visible rearrangement of VSMCs is observed, with cells adjusting their length to increase the amount of overlap along their longitudinal axis. This reduces the ability of the vessel to return to its original (pretreatment) diameter in the absence of the vasoconstrictor; such responses are not observed after exposure to the vasoconstrictor for only 5 min (1705). The process by which the VSMCs lengthen and reorient under these conditions depends on signaling through tyrosine phosphorylation pathways, suggesting the involvement of integrin-ECM interactions (1723, 1724); this is consistent with the findings of other laboratories (1695, 1725). There are other pathways involved in this particular aspect of arterial wall remodeling that include actin polymerization and the activation of MMPs and transglutaminases (TGs), which are discussed below. These dynamic changes in VSMC positioning appear to precede the longer-term changes in vessel mechanical properties and wall structure observed by other investigators studying remodeling of small arteries in animal models of HT (1676, 1678).
Given the highly plastic nature of VSMCs, it is not surprising that the active mechanical properties of arteries change during vascular remodeling (1698, 1701). The underlying mechanisms appear to involve changes in both myosin and actin filaments, as well as intermediate filaments. Myosin filaments themselves are known to be highly plastic (1726). Sustained contractile activation leads to increases in the number of myosin filaments (1727) and their organization (1728–1730) so that a higher myosin density is associated with longer filament lengths. New sarcomeres are added in response to chronic lengthening of ASM, which allows the maintenance of a broad L-T relationship (1731). Intermediate filaments, which are required for normal SM force development (1252, 1732–1734), also undergo extensive reorganization in response to increased mechanical load (1735, 1736). Their reorganization is mediated in part by the disassembly of vimentin, which is facilitated by its contraction-induced phosphorylation (1735–1738). Because intermediate filaments anchor the CSK at dense plaques, they are likely to serve as templates for the formation of new F-actin polymers (1252, 1358, 1739). Thus, it is not surprising that actin polymerization in VSMCs is also involved in arterial wall remodeling, because this process contributes to force development in response to acute mechanical stress, as discussed in sect. 7.2.1. For example, the recovery of force in rabbit femoral arteries after maintenance at a shorter length involves actin polymerization (1740), and severing actin filaments largely prevents the remodeling of the passive diameter of skeletal muscle arterioles induced by prolonged vasoconstrictor exposure (1741). At least one study of VSMCs suggests that such changes in actin dynamics require myosin phosphorylation (1742), indicating an additional level of interaction between myosin and actin filaments in this process.
The most well-documented aspect of arterial wall remodeling is a change in the passive L-T relationship (1678, 1683). Inward remodeling consistently produces leftward shifts in both the passive L-T curve and the passive stress-strain relationship (1743), indicating that the artery can no longer relax to its original diameter at any pressure. Studies on the mechanisms of VSMC rearrangement after 4 h of prolonged constriction show that 75% of the reduction in passive diameter is mediated by actin polymerization in VSMCs (1741). Indeed, even short-term exposure (10 min) to the vasoconstrictor serotonin, in combination with NOS inhibition, leads to detectable actin remodeling and a reduction in the maximal passive vessel diameter (1706). The shape of the passive L-T curve is thought to depend largely on the contributions of collagen and elastin (1710, 1744), and there is minimal contribution of the endothelium, as shown by studies in which the EC layer is removed (1710). Longer-term remodeling eventually leads to overt changes in ECM composition, including the loss of elastin, altered fibronectin and collagen deposition, and/or collagen cross linking. Such changes occur primarily in the medial layer and the internal elastic lamina but also in the adventitia, depending on the size of the artery (1745). The loss of elastin leads to increased arterial stiffness, which is known to precede the development of overt HT (1746, 1747).
7.2.3. Mechanisms of vascular wall remodeling.
The activation of both MMPs and TGs has been shown to participate in wall remodeling. For example, the inward remodeling of skeletal muscle arterioles after 4-h exposure to vasoconstrictors is associated with an increase in enzymatic activity of MMP-2 that can be prevented by blocking MMP activation (1748). Although MMP activity is typically associated with ECM degradation (1749), MMPs can influence remodeling in other ways, e.g., by generating vasoactive fragments of matrix proteins (1339) and by cleaving big ET-1 to produce a peptide with vasoconstrictor properties (1750). TGs, which are calcium-dependent enzymes that catalyze stable cross linking of ECM proteins through amine incorporation or deamidation (1751), also mediate various aspects of vascular wall remodeling. They can act on many of the ECM proteins found in the arterial wall, including fibronectin, osteopontin, and collagen, with effects that are normally reversed only by natural turnover of the cross-linked proteins. vanBavel and colleagues first showed that the inward remodeling of rat skeletal muscle arteries induced by low flow or by prolonged exposure to endothelin could be blocked by inhibitors of TG2 (tissue-type TG); in addition, stimulation of TG2 with retinoic acid or exogenous administration of TG2 induced inward modeling in the same vessels (1689). Similar findings have since been confirmed by multiple laboratories (242, 1706, 1743, 1752–1754).
How do TGs become activated in the process of arterial remodeling? Because TGs associate with integrins and can function as G proteins, they have the potential to stimulate integrin clustering and RhoA activation, thereby regulating actin polymerization (1755, 1756). For example, TG activation by dithiothreitol in isolated skeletal muscle arterioles induces constriction and inward remodeling via actin polymerization (1743). This process may also involve ROS production (1757, 1758), as multiple studies have shown that inward hypertrophic remodeling is attenuated in the presence of ROS inhibitors (see summary Table 1 in Ref. 1757). An important study by Retailleau, Honoré, and coworkers (242) provides compelling evidence that Piezo1 cation channels are critical for promoting the VSMC Ca2+ entry required for TG activation. Using a Piezo1 reporter mouse, those authors identified the caudal artery and the rostral cerebellar artery as representative arteries expressing high levels of Piezo1. SM-specific deletion of Piezo1 did not alter acute myogenic responsiveness in either of these arteries in normotensive animals (Figures S3 and S4 in Ref. 242) but attenuated the increases in wall thickness and cross-sectional area (CSA) associated with arterial remodeling in caudal arteries induced by a chronic angiotensin II infusion model of HT. Piezo1 deletion also reduced CSA in a DOCA/salt/uninephrectomy model of HT (242). In addition, the authors showed that enhancing endogenous Piezo1 activity by SM-specific deletion of filamin A (FlnA) could trigger remodeling in normotensive arteries. FlnA is an actin cross-linking protein that stabilizes the cortical actin CSK and appears to constitutively inhibit the activity of Piezo1 and other mechanosensitive ion channels (243, 1759). SM-specific deletion of FlnA induces outward hypertrophic remodeling of the aorta and carotid arteries (1760). FlnA deletion from caudal arteries induces inward remodeling in the absence of hypertension (242) and is associated with a global rise in VSMC [Ca2+] and activation of TG that are both blocked by Piezo1 deletion (242). These results strongly suggest that Piezo1 activation mediates a global VSMC Ca2+ increase that activates TGs and leads to arterial remodeling. However, there are several unexplained aspects of that study that warrant further investigation. For example, Jaggar and coworkers showed that Pkd2 smKO mice were resistant to ANG II-induced hypertension (498). The remodeling associated with the HT resulting from either angiotensin II infusion or DOCA/salt/uninephrectomy in Retailleau et al. (242) did not lead to reductions in luminal diameters or left shifts in the passive pressure-diameter curves of either caudal or cerebellar Piezo1+/+ arteries, and yet, paradoxically, diameters were reduced in hypertensive Piezo1-deficient arteries. The remodeling in normotensive arteries with FlnA-deficient VSMCs and enhanced Piezo1 activity led to a reduced diameter, a left-shifted passive pressure-diameter curve, and wall thickening without a change in cross-sectional area, i.e., inward eutrophic remodeling (242), yet only certain aspects of this remodeling were prevented by simultaneous Piezo1 deletion. The differences in the various aspects of remodeling among control, Piezo1-deficient, and Piezo1-overactive arteries remain incompletely explained, as do several other important questions: Is increased wall stress the signal for Piezo1 activation in the initial process of remodeling? Does Piezo1 activity decline after wall stress has been normalized? Is Piezo1 channel activation also critical for other types of arterial remodeling? Caudal and rostral cerebellar arteries were chosen for the study by Retailleau et al. (242) because they expressed the highest levels of Piezo1; is Piezo1 as important for remodeling of arteries with more modest expression? It will be important for other groups to confirm these findings as well as extend them to other models of remodeling.
Finally, vascular wall remodeling may be accompanied by VSM hypertrophy and hyperplasia, which require phenotypic changes and nuclear signaling. VSMCs are normally quiescent, highly differentiated cells that express proteins consistent with a contractile phenotype, including SM α-actin, SM myosin heavy chain, calponin, caldesmon, SM-22α, and myocardin (1761–1763). The HT-associated, inward remodeling discussed above does not result in VSMC hypertrophy or phenotype switching, which is more typically associated with atherosclerosis and vascular injury. However, under some conditions chronic mechanical stress can trigger VSMCs to reenter their cell cycle and dedifferentiate, acquiring a proliferative and migrating phenotype associated with a decline in the expression of contractile proteins. This usually occurs in combination with the infiltration of immune cells into the wall and/or activation of resident macrophages (1764), as discussed in sect. 7.3. The accumulation of these dedifferentiated VSMCs in the vessel wall leads to neointimal hyperplasia, medial thickening, and increased wall stiffness. The phenotypic switch of VSMCs is promoted by enhanced ROS production and may involve noncoding RNAs (1635, 1765). Detailed discussions of these processes are beyond the scope of this review, as the role of VSMC mechanotransduction in these processes is unlikely to be one of the most important determinants.
In summary, HT is often associated with exaggerated levels of myogenic tone and constriction, which become major drivers of vascular wall inward remodeling, ultimately increasing systemic vascular resistance. Existing evidence indicates roles for Piezo1 channels, enhanced ROS production, and activation of MMPs and TGs in this process, but the interactions of these mechanisms and the sequences of events linking them together need further investigation.
7.3. EC Mechanotransduction in Health and Disease
A prevalent hypothesis for the focal distribution of atherosclerosis is that the distinct flow patterns at various regions of the arteries exert differential effects on ECs to lead to their development of atheroprotective versus atheroprone phenotypes. Thus, the pulsatile laminar shear in straight segments of the arterial tree is atheroprotective, whereas the disturbed flow at the inner curvature of the aortic arch and bifurcation points is atheroprone. This hypothesis has been the basis of the studies on mechanotransduction in many laboratories, including ours, and has led to the discovery of an array of mechanosensing mechanisms and molecular pathways that mediate endothelial responses to atheroprotective versus atheroprone flow patterns (9, 47, 1766–1768).
The “Pathobiological Determinants of Atherosclerosis in Youth” (PDAY) Research Group showed the preferential localization of fatty streaks and atherosclerotic lesions in regions of the arterial tree with low shear stress and flow disturbance (1769), i.e., mainly at arterial branch points and regions of curvature (1770, 1771). Simulations of flow patterns in patient arteries with/without lesions also demonstrate atheroprotective flow at the lesion-free distal internal carotid artery but atheroprone reversal flow at the lesion-prone carotid sinus (1772). There are also similar findings in the outer (atheroprotective) versus inner (atheroprone) curvature of human and rodent aortic arch (for review see Refs. 9, 1773). The formation of atheroma in the artery wall involves the imbibement of LDL by macrophages. Although the entry of monocytes into the vessel wall and their transformation into macrophages is a major atherogenic factor (for review see Refs. 1774–1776), a complementary factor is the entry of LDL (for review see Refs. 1777, 1778). Thus, the accelerated EC turnover and the ensuing increase in EC permeability to LDL to enter the subendothelial space contribute to the focal nature of atherosclerosis (for review see Ref. 9). Experimental studies on the rabbit aorta have shown the colocalization of focal regions of disturbed flow (as reflected by nuclear orientation) with increased mitosis and enhanced macromolecular permeability (39).
7.3.1. Animal models for in vivo studies.
7.3.1.1. FOCAL DISTRIBUTION OF LESIONS (ROLE OF ARTERIAL GEOMETRY).
As a result of the variations in geometric features, the blood flow patterns and wall shear stress profiles vary along the arterial tree to result in the focal nature of atherosclerotic lesion formation. In the aortic arch, the wall shear stress at the inner curvature, where the lesions are commonly observed, is low with flow disturbance compared with the outer curvature (FIGURE 21A). In the cranial part of the descending aorta with only minor branching points, shear stress is high and flow is laminar; it is generally spared of lesions. Lesions occur preferentially at the arterial branch points (such as those at the carotid, renal, and femoral arterial branches), where flow is disturbed and shear stress profiles are complex (for review see Ref. 9). Although the flow patterns in large animals (e.g., pigs and dogs) are more relevant for human disease, rodents have generally been used as animal models because of the cost-effectiveness and applicability of genetic manipulations. Two strains of mice, Apoe−/− and Ldlr−/−, in conjunction with modulations of lipid metabolism, have been most commonly used to study the effects of blood flows on the development, progression, and mitigation of atherosclerosis. With the feeding of high-fat/high-cholesterol diet, these mice develop fatty streaks and lesions in ∼8 wk (for review see Ref. 1779). The resulting lesions present a focal nature with flow pattern implications. Many of the proinflammatory and proliferative genes induced by disturbed flow in vitro are detected in the lesion area, whereas the laminar flow-induced anti-inflammatory/antiproliferative genes are present in the straight part of the aorta, which is generally spared from lesion formation. These experimental animals provide valuable models for the correlation of flow patterns and lesion development/progression.
FIGURE 21.
Modulation of flow patterns in vivo. A: a schematic drawing of the flow patterns in native arteries. B: vessel constriction with cuffs to generate high shear at the cuff-narrowed neck and flow disturbance at the outlet of the neck. C: total ligation of left carotid artery (LCA) to create the flow cessation and back flow. D: ligations of external carotid (ECA)/internal carotid (ICA)/ophthalmic (OA) arteries on LCA to create low and disturbed flows. RCA, right carotid artery; STA, superficial temporal artery.
7.3.1.2. CUFFING (STENOSIS FOR FLOW REDUCTION AND DISTURBANCE).
Application of perivascular flow constrictors with various geometries can create a stenosis to generate flow disturbance (FIGURE 21B), thus enabling the studies of gene expression, cell phenotype, and lesion formation in a controlled manner. The use of a straight perivascular cuff (FIGURE 21B, left) to reduce the diameter of the common carotid artery by 30% causes an accelerated lesion formation in both Apoe−/− and Ldlr−/− mice (with lesion formation evident at 3 and 6 wk, respectively). This procedure leads to increases of ICAM-1 and VCAM-1 and a decrease of eNOS expression in regions proximal and distal to the cuff (1780), where the flow patterns are disturbed because of the change of vessel diameter along the flow path. The application of a tapered perivascular cuff (FIGURE 21B, right) leads to a low-shear stress region at the wider proximal region, a region with increasing shear stress along the tapered channel, followed by a region of flow disturbance with vortices and oscillations at the distal widened region beyond the cuff (1781). Lesion formations are mainly observed at the proximal (low shear) and distal (disturbed flow) regions, whereas the vessel segments with high shear are spared from lesion formation (1782). Analyses of the compositions of the lesions at the proximal low-shear region show vulnerable soft lesions with lower calcium, less VSMCs and collagen, and more lipids, inflammatory mediators, and MMP compared with lesions at the distal disturbed flow region (1782, 1783). These studies provide evidence of the impact of flow modulation on vascular disease progression.
7.3.1.3. ARTERIAL LIGATION (COMPLETE AND PARTIAL LIGATION).
Complete ligation of the left common carotid artery (FIGURE 21C), which causes blood flow cessation without alterations of systemic arterial pressure and pulsation, leads to EC inflammation, VSMC proliferation, leukocyte recruitment, and neointima formation in the artery proximal to the ligation (1571, 1784, 1785), with a significant increase of MMP-9 and vascular remodeling (1786). In a rabbit model, the ligation of left carotid artery (LCA) causes the doubling of flow in the right carotid artery (RCA) and vascular remodeling in both carotid arteries, with the fenestrae area being reduced in LCA and increased in RCA (1787). There is a lack of flow characterization of this complete ligation model, and the resulting flow cessation does not allow the study of disturbed flow. A model with flow disturbance that is more relevant to in vivo pathophysiological conditions has been achieved by the partial constriction of one of the carotid arteries of the rat to reduce the flow, with the other remaining patent and having an increase in flow (1788). The flow reduction in the constricted artery causes significant endothelial proliferation and increases in PDGF-a/b and PDGFR expressions (1789). Lesion formation was not investigated in these two studies. Jo and colleagues (1790) have modified the partial ligation method for Apoe−/− mice with ligations of the left external/internal carotid arteries and occipital artery (FIGURE 21D), leaving the superior thyroid artery open, thus significantly reducing the flow in the LCA with little effect on the RCA. This procedure decreases endothelial KLF2 and eNOS expressions and increases ICAM-1 and VCAM-1 expressions in the partially ligated LCA, with the consequential development of advanced lesion formation (1790). This model has been used to elucidate the changes in mechanotransduction, gene expression, and EC phenotypes induced by flow disturbance in vivo and provides an excellent experimental platform for studying pathophysiological responses and testing potential targets for disease treatments.
7.3.2. Mechanotransduction in atherosclerotic cardiovascular diseases.
As presented in previous parts of this review, atherosclerosis is mainly an inflammatory disease that involves EC dysfunction, VSMC dedifferentiation and hyperplasia, and monocyte/macrophage recruitment. The atherosclerotic lesions are found primarily in regions of the cardiovascular system with disturbed flow. To understand the pathogenesis of atherosclerotic cardiovascular diseases and develop therapeutical strategies, it is essential to elucidate the mechanisms of mechanotransduction in response to disturbed flow. This section summarizes the effects of disturbed flow in vivo or OS in vitro on mechanosensing, signal transduction, and transcriptomic and epigenomic regulation, as presented in the previous sections of this review.
A major pathophysiological consequence of disturbed flow is vascular inflammation and the ensuing activation and adhesion of monocytes to ECs. OS activates Piezo1 to induce integrin-mediated FAK phosphorylation and the consequent NF-κB activation for inflammatory responses (324). Disturbed flow/OS acts on ECs to increase the expression of inflammatory genes such as VCAM-1 and MCP-1 (1531, 1536) and the inflammation marker TNF-α (1791). NF-κB activation is greater and more sustained under OS than under LS (1517, 1528). Flow-regulated NF-κB activation is dependent on the ECM protein fibronectin (integrin αvβ3) but not collagen (integrin β1) (1406, 1519, 1527). NF-κB also induces hypoxia-induced factor-1alpha (HIF-1α) under low LS to promote glycolysis and hence the enhancement of EC proliferation and dysfunction (1529).
OS causes the activation and translocation of integrin α5 to the lipid raft to activate the NLRP3 pathway (1080). OS also causes sustained integrin β1-TβRI/II and integrin β1/β3-TβRI association, Smad2 phosphorylation, and NF-κB expression in ECs on fibronectin (1409). Atherosclerotic lesions in patients exhibit augmentations of Smad2 phosphorylation and fibronectin level, suggesting a role of the fibronectin-integrin-TβR1-pSmad2-NF-κB pathway in mediating the OS-induced vascular inflammation (1409). Disturbed flow and OS increase arterial stiffness and induce the expressions of genes related to vascular remodeling such as collagen1 (1417).
OS causes YAP phosphorylation at tyrosine 357, but not at serine 127, and a sustained YAP nuclear localization (1438). A negative mutant of YAPY357 attenuates the VCAM-1 expression and monocyte adhesion induced by OS (1438). Studies on Apoe−/− mice showed YAPY357p in atheroprone areas but YAPS127p in atheroprotective areas. YAP knockdown, EC-specific YAP knockout, or inhibition of YAP upstream molecules can mitigate the disturbed flow-induced atherogenesis (1438).
Single-cell transcriptomic studies in the mouse partial ligation model showed that disturbed flow increases the EC populations expressing atheroprone pathways, promotes Endo-MT and Endo-ICL (1534), and enriches the VSMCs expressing Spp1 (1548). Three clusters of macrophages can be detected in atherosclerotic vessels in Apoe−/− mice as well as human atherosclerotic lesions (1541).
Regarding TFs, OS increases E2F1, EGR1, and HIF1a, with the potential of cross-interactions with other TFs such as FOS, SRF, and FoxO1 to regulate the gene networks related to EC oxidative stress, inflammation, and cell cycle progression. OS causes H3K27ac enrichment of FOX, CPEG, YAP, and TAZ (1532, 1533), which may upregulate these TFs and the downstream gene expressions to modulate EC fates.
In ECs, OS induces miR-21, which targets PPARα to increase AP1 activity and inflammatory gene expressions (1560), and miR-663, which mediates the OS-induced monocyte adhesion (1561). The increase in inflammatory genes in macrophages cocultured with ECs in response to OS is mediated by miR-92a (1570). Atheroprotective lncRNAs, such as LEENE, are induced by PS but not OS (1562–1567).
OS increases DNMT1 expression and DNA methylation in vitro and in vivo (1445, 1598) as well as the binding of DNMT1 to cyclin A promoter to induce its hypermethylation (1599). Inhibition of DNMTs by 5Aza mitigates the disturbed flow-induced atherogenesis (1445, 1598, 1599). A genomewide study has shown differential DNA methylation in atheroprone versus atheroprotective regions in mouse arteries, with the identification of 11 mechanosensitive genes whose downregulation may be involved in generating the EC inflammatory phenotype under OS (1792). In human aortic ECs and swine aorta, the sites for DNA methylation in response to OS have been identified on the KLF4 promoter (1505). OS increases METTL3 expression and RNA m6A hypermethylation to lead to EC inflammatory responses and metabolic changes (1607).
OS induces the expression and nuclear localization of HDAC1/2/3 and 5/7 to deacetylate Nrf2 and MEF2, respectively (1612), thus reducing the expressions of NQO1 and KLF2 and exerting detrimental effects on EC homeostasis (1612). The OS-induced HDAC3/5/7 interact with retinoic acid receptor-α to repress miR-10a expression and increase the GATA6-mediated VCAM-1 expression that recruits macrophages to ECs (1574). OS enriches H3K27ac at the FOX/CPEB motifs, which may lead to the regulation of signaling cascades relevant to atherogenesis (1533). With TF binding analysis, ETS/KLF/AP1 and YAP/TAZ are found to be the top-ranked motifs enriched in ECs under OS; this is in agreement with the findings that OS induces YAP/TAZ activation, nuclear translocation, and binding to target gene promoters (1533).
EC-VSMC interactions are critical for flow regulation of vascular functions. OS enhances EC secretion of miR-126 to lead to VSMC dysfunction, which can be abolished by knocking down EC Drosha, miR-126, or Ago2 as well as by LS application (1571). An in vivo study showed that inhibition of vesicle trafficking proteins VAMP3 and SNAP23 can attenuate the disturbed flow-enhanced miR-126 to reduce VSMC hyperplasia and lesion formation (1594). OS induces FGF-4, VEGF-A, and PDGF-BB in ECs to enhance the migration and proliferation of the cocultured VSMCs; these effects are dependent on EC Cav-1 or ERK activation (1793). The proliferative genes induced by disturbed flow in vitro are detected in areas of atherosclerotic lesion of the aorta (1793). These findings suggest a role for EC-VSMC paracrine interaction in atherogenesis.
7.3.3. Therapeutic strategies.
In vitro and in vivo studies have established multiple signaling pathways, transcription factors, epigenetic modulations, and target genes that mediate the gene expression and functional outcome in response to disturbed flow and OS. These findings serve to identify the potential therapeutic targets as candidates for treatments of atherosclerotic cardiovascular diseases, including mechanosensors (e.g., Piezo1), signaling pathways (e.g., NF-κB), transcription regulators (e.g., YAP/TAZ), noncoding RNAs (e.g., miR-10a, miR-126), as well as epigenetic regulators (e.g., DNMTs, METTL3).
The efficacy of such treatments has been demonstrated by various interventional experiments in vivo, as exemplified by the following: EC-specific knockout of Piezo1 mitigates lesion formation in the artery tree of Ldlr−/− mice (324). The lesion formation induced by partial ligation in Apoe−/− mice is reduced by inhibition or EC-specific knockout of YAP (1434, 1435) and by inhibition of miR-10a (1574), miR-126 (1445, 1792), DNMT1 (1445, 1598), or METTL3 (1607), These in vivo studies provide strong support for further investigations and development of these therapeutic options for atherosclerotic cardiovascular diseases. There are also experimental studies that use biochemical reagents to counter the actions of atherogenic flow or mimic the effects of atheroprotective flow to mitigate atherosclerotic disease progression (for review see Ref. 1794). Examples include the Piezo1 activator Yoda1, AMPK activators (metformin, AICAR), KLF2 activation by statins, YAP/TAZ inhibitors (statins, verteporfin), and DNMT inhibitor (5Aza). Among these reagents, statins, metformin, and verteporfin are FDA-approved drugs for treating cardiovascular lipid disorders, type 2 diabetes, and retinal blood vessel diseases. It would be of interest to assess the therapeutic efficacy of these “atheroprotective flow mimicking” drugs on mechanotransduction in atherosclerosis patients.
A drug delivery system with homing mechanisms to recognize the injured endothelium in the vasculature is ideal for effective targeted therapy. Recently, nanomedicine has been developed for vascular disease detection, diagnosis, and targeted treatment. Nanoparticles (NPs) are used as carriers loaded with various agents for imaging, homing, and therapeutics (for review see Refs. 1795–1797). The biomimetic NPs combine the synthetic nanomaterials and cellular components with camouflage for the biological system to avoid immunoresponse, thus allowing for prolonged circulation time and targeted delivery to specific sites of interest (for review see Refs. 1798, 1799). An example is the recently developed platelet membrane-cloaked NPs (PNPs), which present platelet surface functional molecules such as immunomodulators and integrins (1800). This approach has the advantages of minimizing phagocytosis by macrophages and achieving selective adherence to injured vasculature for therapeutic delivery (1800, 1801). Loading PNPs with the antiproliferation drug docetaxel significantly reduces the neointima thickening in denuded rat carotid arteries (1800). Incorporation of imaging agents, such as lipid-chelated gadolinium, in PNPs allows magnetic resonance image (MRI)-based tracking of PNP delivery and targeting in living tissues (1801), thus providing beneficial therapeutic effects. Nanomedicine-based approaches can be used to deliver various therapeutics, including drugs, proteins, antibodies, nucleic acids (e.g., siRNAs, miRs), and viruses for the effective treatment of cardiovascular diseases (for review see Refs. 1802–1804).
8. INTERACTIONS BETWEEN ECs AND VSMCs
ECs and VSMCs in the arterial wall are separated by the internal elastic lamina (IEL), which forms a fenestrated sheet between these cells in the intima and media, respectively. The size, number, and density of the pores in the IEL vary with the type and location of the vessel, as well as age and other factors (1805, 1806). The fenestrae provide a conduit for the passage of water and small molecules but serve as a major barrier for the transport of large molecules and the transmission of shear stress. The IEL fenestrations have been shown to be altered in pathophysiological states, e.g., decreasing in size in hypertension (25, 1807, 1808) and increasing in size in hypercholesterolemia (1809), suggesting their potential pathophysiological role in disease progression. There are excellent recent reviews on EC-VSMC interactions, such as Refs. 24 and 1810.
The IEL fenestrae allow the protrusion of ECs through the pores to establish contact with VSMCs, thus forming a myoendothelial junction (MEJ). The density of MEJs is generally low in large arteries and higher in small arteries and arterioles (1811), implying that direct connections between the two cell layers are more important for the functioning of resistance than conduit vessels. Electrical continuity between ECs and VSMCs at the MEJ occurs via Cx43 and/or Cx40 gap junctions (892, 1812); this is supported by the observation that EC and VSMC membrane potentials oscillate together in vessels with spontaneous vasomotion (1813, 1814). Both Cx43 and Cx40 are permeable to cations, enabling the transfer of charge as well as the diffusion of small molecules such as Ca2+ and IP3 (1815). Connexin permeability can, in principle, be regulated by connexin phosphorylation, but it is not known whether this process dynamically regulates MEJ function in vivo.
MEJs permit bidirectional signaling between ECs and VSMCs, facilitated by signaling microdomains in the two respective cell types at their points of contact (1816–1818). Endothelium-derived hyperpolarization (EDH) is the well-described phenomenon whereby hyperpolarization originating in the EC layer is conducted to the VSMC layer through MEJs (891, 1819). EDH is preserved with blockade of NO and prostanoid production, indicating that it does not depend on humoral factors. Most studies agree that EDH is blocked by combined inhibition of SK and IK channels in ECs (882, 896, 1820, 1821). Myoendothelial feedback (MEF) is the process in the reverse direction whereby depolarizing signals in the VSMC layer spread to ECs through MEJs. MEF was first suggested by Dora et al. (1822) and confirmed by many subsequent studies, e.g., Ref. 85 (see Table 1 in Ref. 1817 for references). Although depolarization can potentially be transmitted from VSMCs to ECs, there is evidence that MEF is mediated by Ca2+ and/or IP3 movement from VSMCs to ECs through MEJs. Modeling studies by Welsh and colleagues (915, 1823) using experimentally determined cell-cell resistances for EC-EC, VSMC-VSMC, and EC-VSMC gap junctions help to explain why hyperpolarizing signals originating in ECs can be readily transmitted to VSMCs but depolarizing signals in VSMCs are much more limited in their spread. MEF is thought to facilitate the production of EC-derived vasodilators to limit vasoconstriction (1817). For most small arteries and arterioles, tone is generally fixed around an intermediate set point so that it can be acutely modulated in either direction by the interactions between ECs and VSMCs. Complete constriction leads to underperfusion of downstream capillary beds, whereas maximal dilation limits any further increase in blood flow.
Shear stress regulates the interactions between ECs and VSMCs. PS and OS activate multiple EC mechanosensors (FIGURE 15), including ion channels (sects. 2.2.2–2.2.4), GPCRs (sect. 4.1), and RTKs (sect. 4.2) to trigger mechanotransduction cascades (FIGURE 18), e.g., AMPK and PI3K/Akt (sect. 5.4), resulting in the release of endothelium-derived factors (including NO, ATP, and PGI2) for VSMC-mediated vasodilation (1824, 1825). Furthermore, high LS (6–25 dyn/cm2) suppresses, whereas low shear (1.8 dyn/cm2) increases, vasoconstrictor ET-1 expression and secretion in ECs, and such shear regulation of ET-1 is mediated by PKC and the cGMP/NO pathway (1826).
To investigate the mechanisms of EC-VSMC interactions, in vitro coculture perfusion systems are often employed (e.g., Refs. 67, 69, 1827) with ECs and VSMCs cultured on the two sides of the ECM-coated porous membrane to mimic the IEL, followed by subjection of ECs to mechanical stimuli for experimental studies. However, because VSMCs rapidly dedifferentiate to a noncontractile, proliferative phenotype in culture, these types of studies provide very limited information about contractile regulation.
Under static conditions, coculture with VSMCs (proliferative phenotype) causes ECs to exhibit a proliferative and inflammatory phenotype, as evidenced by the reduction of eNOS (70), activation of NF-κB (1828), induction of adhesion molecule expression (e.g., ICAM-1, VCAM-1, and E-selectin) (70, 1829), enhancement of platelet adhesion (1830), and increases of proliferation (1831) and migration (1832). Such coculture-induced EC dysfunctions can be mitigated by LS. It has been shown that the E-selectin expression in ECs induced by coculture with VSMCs is mediated by the production of IL1 and IL6b in ECs and the consequential activation of MAPKs and NF-κB that bind to the E-selectin promoter for its transcriptional activation (1829). Another study shows that VSMC coculture induces transient increases of anti-inflammatory miRs (miR-146a, -708, -451, and -98) for 6–24 h in ECs, followed by their significant decreases below basal levels. Application of LS restores the levels of these EC miRs in a Nfr2-dependent manner and reduces the expression of their target inflammatory genes (IRAK, IKKg, IL6R, CHUK) (1833), thus resulting in EC anti-inflammatory responses. The anti-inflammatory role of miR-146a has been validated in mouse carotid artery by its prevention of injury-induced neointima formation (1833). The increase of EC proliferation when cocultured with VSMCs (demonstrated by BrdU incorporation assays) is attenuated by LS through the induction of Sirt1 and downregulation of Cx40 (1831), thus exerting protective effects on ECs. The EC migration induced by coculture with VSMCs has been correlated with an increase in HDAC6 and decrease in tubulin acetylation; causal studies demonstrate that inhibition of HDAC6 abolishes the VSMC coculture-induced EC migration. In EC monoculture, LS also induces HDAC6 and migration and attenuates proliferation but to lesser extents compared with VSMC coculture (1832).
In an EC-VSMC coculture system, Redmond and colleagues (1834) have shown that EC NO production can be significantly increased by high LS (15 dyn/cm2) but not low LS (0.5 dyn/cm2), whereas ET-1 expression is not affected by shear. However, the regulation of ET-1 binding to VSMCs in an EC-VSMC coculture system is significantly increased by high LS but not low PS. Such high LS-induced ET-1 binding to VSMCs is due to increases in the expression of ET-1 receptors (ET-A and ET-B). The LS-induced VSMC ET-A/B expression is EC dependent and regulated, at least in part, by NO activity (1834). These results suggest a complicated feedback regulatory mechanism that balances the NO production by ECs and ET-1 binding in VSMCs to regulate vascular tone. Moreover, pathological low shear stress increases EC production of ROS through an AT1R-dependent pathway (1835), which may lead to vasoconstriction. Thus, mechanical forces modulate EC-VSMC interactions to set vascular tone and play roles in vascular diseases such as hypertension (for review see Refs. 1836–1839).
In vitro coculture with ECs under static conditions causes VSMCs to assume a migratory and proliferative phenotype (1571, 1840–1842). An earlier study on EC-VSMC coculture showed differential regulations of EC and VSMC G proteins by LS: The application of high LS (15 dyn/cm2) to ECs causes an increase of Giα3 expression in ECs but a decrease in Giα1/2 in VSMCs compared with the results under low LS (0.5 dyn/cm2). This EC-dependent VSMC Gα1/2 reduction is mediated by 6-keto-PGF1α and can be abolished by the inhibition of EC cyclooxygenase (1843), suggesting that flow regulates vascular tone via, at least in part, a paracrine mechanism. In coculture, ECs activate VSMCs, as indicated by the increase of VSMC migration through an Akt-dependent pathway (1842), and such EC-induced VSMC migration can be inhibited by subjecting the ECs to LS (15 dyn/cm2) (1842). In contrast, low LS and OS (4 and 0 ± 4 dyn/cm2, respectively) induce fibroblast growth factor-4 (FGF-4), vascular endothelial growth factor A (VEGF-A), and platelet-derived growth factor-BB (PDGF-BB) in ECs to induce migration and proliferation of cocultured VSMCs; these effects are dependent on Cav-1 or ERK activation in ECs (1793). Furthermore, the uremic toxin-induced VSMC proliferation and migration in the coculture system can be blocked by subjecting ECs to normal and high LS (12 and 20 dyn/cm2, respectively) but not by low shear and OS (1793). It has been further demonstrated that LS (12 dyn/cm2) promotes the phenotypic change of VSMCs from synthetic to contractile, as evidenced by the increases of SM α-actin, SM-MHC, calponin, h-caldesmon, and SM22a, decreases of MCP-1 and IL8, as well as reduction of proliferation, in the coculture system. The effects of shearing on VSMC phenotype are specific to ECs, but not fibroblasts, in coculture conditions (1841). Mechanistically, LS activates the peroxisome proliferator-activated receptor α/δ (PPARα/δ) pathway to increase EC PGI2 expression and secretion, which in turn induce VSMC SM22α transcription. Importantly, knockdown of EC PGI2 production abolishes the LS-promoted VSMC contractile phenotype switch (1841), supporting the significant role of this EC-derived metabolite in regulating VSMC tone.
Recent studies have demonstrated that extracellular miRs can mediate EC-VSMC interactions (1571, 1594, 1844). EC miR-143/145 have been identified to be induced by LS or KLF2 overexpression and transmitted to VSMCs through the secretion of extracellular vesicles (EVs), as evidenced by the sensitivity of this process to Triton X-100 treatment and detection of extracellular miR-143/145 in the EV fraction of conditioned medium. The transmitted miR-143/145 reduce target gene expressions [e.g., KLF4, Ca2+/calmodulin-dependent protein kinase 2d (CAMK2d), and metalloproteinases 3 (MMP3)] in VSMCs. Knockdown of the EC miR biosynthesis key enzyme class 2 ribonuclease III (Drosha) abolishes the EC-VSMC interaction. The formation of atherosclerotic lesions is significantly reduced in Apoe−/− mice treated with miR-143/145-containing EVs derived from ECs with KLF2 overexpression (1844). Other studies have demonstrated that miR-126-3p and miR-200a-3p mediate EC-VSMC interaction via EV-free mechanisms, as evidenced by the sensitivity of the transmission to RNase and protein kinase treatments and the detection of miR-126 in the EV-excluded fraction of the conditioned medium (1571, 1594). Extracellular miR-126 is associated with the RNA binding protein Argonaute 2 (Ago2) to protect and facilitate its transmission. EC miR-126 transmission to VSMCs has been further validated with the transfection of labeled miR-126 and tagged Ago2 into EC before coculture and detection of the exogeneous labels and tags in VSMCs in coculture; these experiments exclude the potential issue that EC coculture may induce endogenous VSMC miR-126 biosynthesis. The transmitted miR-126 targets and represses VSMC forkhead box O3 (FOXO3), B cell lymphoma 2 (Bcl2), and insulin receptor substrate 1 (IRS1) mRNAs to increase VSMC proliferation and apoptosis. The secretion of miR-126 from ECs and the subsequent VSMC dysfunction can be abolished by knocking down EC Drosha, miR-126, and Ago2, as well as by the application of LS, whereas OS leads to opposite effects (1571). Further investigations on EC exocytosis SNARE proteins have identified that OS, but not PS, induces the SNARE family members vesicle-associated membrane protein 3 (VAMP3) and synaptosomal-associated protein 23 (SNAP23) through a mTOR-dependent pathway. Knocking down VAMP3 or SNAP23, or inhibition of mTOR by rapamycin treatment, attenuates the OS-induced miR-126 secretion and the consequent VSMC dysfunctions. Elevated levels of VAMP3 and SNAP23 can be detected in mouse arteries in response to disturbed flow, and rapamycin treatment decreases VAMP3 and SNAP23 levels to mitigate the partial ligation-induced lesion formation, whereas overexpression of VAMP3 and SNAP23 enhance lesion formation (1594). These results demonstrate the multitude of regulatory mechanisms that mediate the paracrine interactions between ECs and VSMCs.
The studies outlined above have established that EC-VSMC coculture under static conditions promotes the activation of both ECs and VSMCs to lead to vascular cell dysfunctions (e.g., inflammation, migration, and proliferation). These dysfunctions are mitigated by atheroprotective shear and aggravated by atheroprone shear. Such interactions involve a paracrine mechanism in which EC secretions such as NO, prostanoids, ATP, growth factors, and miRs act on VSMCs. Although ECs are also activated by coculture with VSMCs, the mechanisms by which VSMCs act on ECs are less well understood. Circulating leukocytes (monocytes, neutrophils, and lymphocytes) can interact with ECs, and these interactions are modulated by different types of shear stress (1845). LS causes leukocyte pseudopod retraction, decreased activities of GPCRs [e.g., surface formyl peptide receptors (FPRs)], and reduced integrin CD18 expression in circulating leukocytes (for review see Ref. 5); these changes may reduce their interactions with ECs, and thereby also affect VSMCs, to modulate vascular homeostasis and dysfunction (FIGURE 22).
FIGURE 22.
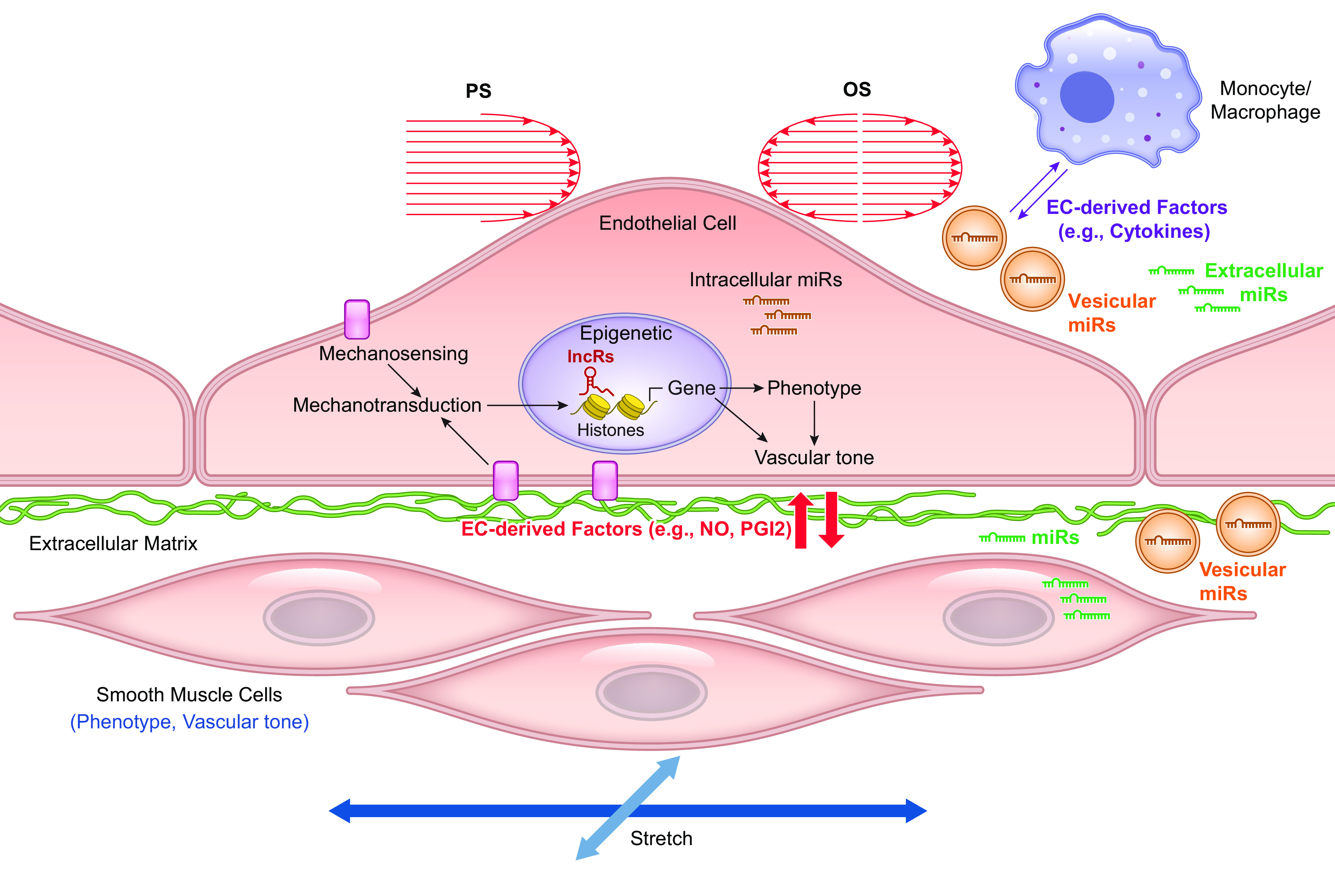
Shear flow regulation of vascular cell interactions. Shear stress modulates endothelial function through EC mechanosensing and mechanotransduction processes to regulate the epigenome, transcriptome, and phenotypes, as well as the interactions (indicated by colored arrows) between ECs and the neighboring cells. Such regulation leads to homeostasis in health and pathophysiological changes in disease. lncR, long noncoding RNA. See glossary for other abbreviations.
9. CONCLUSIONS AND PERSPECTIVES
The circulation of blood generates mechanical stresses on blood vessels, including the flow-induced shear stress acting on ECs and the pressure-induced distending stresses acting on VSMCs. This review surveys the current state of these mechanotransduction processes, including the sensing of mechanical stimuli, their intracellular signaling, transduction into transcriptomic and epigenetic regulation, and the impact of mechanotransduction on the functional modulation of blood vessels in health and disease.
Although remarkable progress has been made in this rapidly advancing field, there are still a lot of unknowns. For example, among the large number of mechanosensors reported, very few are inherently mechanosensitive according to the criteria presented in sect. 2.1.3. Some of the experimental procedures used to study mechanosensing, e.g., hypoosmotic swelling, may also evoke nonmechanical responses, and many of the pharmacological agents used are nonselective. Culture conditions change cellular phenotype (particularly for VSMCs) and may be confounding. The use of tissue-specific inducible knockout animals and local delivery of next-generation selective pharmacology will add considerably to our understanding of mechanosensing. There is a need to elucidate the interactions among various types of mechanosensors and their hierarchical relationships. Cells in different vasculatures may have different reactivities to mechanical stimuli; hence studies on mechanotransduction need to be performed on vascular cells in various regional circulations and in different vascular segments with appropriate stimuli.
The interactions between ECs and VSMCs play an important role in regulating vascular functions. There is a need to further elucidate the mechanisms of these interactions in health and disease, as well as the interactions of ECs and VSMCs with other types of cells, including macrophages, other types of leukocytes, and pericytes.
Most of the mechanosensitive mechanisms have been determined from in vitro experiments; it is important to assess their roles in hemodynamic regulation in vivo, including flow-induced vasodilation and pressure-induced vasoconstriction as well as their relevance to disease states such as hypertension. Studies with new tools (e.g., 3-D bioprinting and organoids) that can create environments that more closely mimic the in vivo setting will also help to close the gaps.
Mechanotransduction involves the modulation of expression profiles of genes and proteins to result in phenotypical and functional responses. The expressions of functional gene sets are based on the epigenomic and transcriptomic regulations. There is a great need to elucidate the epigenomic and transcriptomic bases of hemodynamic regulation of vascular homeostasis in health and disease. There is also a need to elucidate the mechanisms by which the chromatin-dependent nuclear rigidity affects mechanotransduction and the consequential gene expressions. Investigations on nuclear biomechanics in mechanotransduction would be highly desirable.
It is important to determine the details of spatiotemporal dynamics of mechanotransduction in regulating vascular functions, with the aid of various NGS approaches. Such quantitative, spatiotemporal information can be used to perform systems analysis to gain novel insights into the localized regulations of mechanotransduction in modulating vascular functions in health and disease.
Ultimately, we should aim to apply the knowledge gained in mechanotransduction research to clinical conditions for the diagnosis, treatment, and prevention of disease in patients.
GRANTS
The research studies by the authors are supported by grants from NHLBI of the National Institutes of Health: HL106579, HL108735, HL121365, HL122578, R35HL155008, RF1NS110044, R33NS115132, P20GM130459.
DISCLOSURES
S. Earley is an editor of Physiological Reviews and was not involved and did not have access to information regarding the peer-review process or final disposition of this article. An alternate editor oversaw the peer-review and decision-making process for this article. No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.J.D., S.E., Y.-S.L., and S.C. conceived and designed research; M.J.D., Y.-S.L., S.E., and S.C. prepared figures; M.J.D., S.E., Y.-S.L., and S.C. drafted manuscript; M.J.D., S.E., Y.-S.L., and S.C. edited and revised manuscript; M.J.D., S.E., Y.-S.L., and S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors appreciate the help of Drs. Zhen Bowman Chen, Juan-Carlos del Alamo, Michael Hill, Han-Joong Jo, Luis Martinez-Lemus, John Y.J. Shyy, and Shankar Subramaniam for valuable suggestions in certain sections. Drs. Min Li and Alexei Stuckel assisted with analysis of RNA-seq data for VSMC and EC ion channels.
Some of the figures were prepared in BioRender, for which appropriate publication licenses have been obtained.
GLOSSARY
- 2-APB
2-aminoethoxydiphenylborane
- 2,4-PDCA
2,4-pyridinedicarboxylic acid
- 4-AP
4-aminopyridine
- 5Aza
5′-Aza-2-deoxycytidine
- A2AR
adenosine receptor
- AA
arachidonic acid
- AAGTP
azidoanilido α-32P-GTP
- Abl
Tyrosine-protein kinase ABL1
- ACE
angiotensin converting enzyme
- ADP
adenosine diphosphate
- AFM
atomic force microscopy
- AGTR1
angiotensin II receptor
- AKAP(150)
A-kinase anchoring protein
- AKT
Protein kinase B
- AlF
aluminum fluoride
- ALKBH1
AlkB homolog1
- AMOT
angiomotin
- AMP
adenosine monophosphate
- Ang II
angiotensin II
- ANO1 (or TMEM16A)
member of TMEM16 family of calcium-activated Cl− channels
- ANP
atrial natriuretic peptide
- Apr2/3
actin related protein 2/3 complex
- ASIC
family of acid-sensing ion channels
- ASM
airway smooth muscle
- AT1R
angiotensin II type 1 receptor
- ATAC-seq
Assay for Transposase-Accessible Chromatin sequencing
- BAEC
bovine aortic endothelial cell
- BAPTA
1,2-biso-aminophenoxy:ethane-N,N,N′,N′-tetraacetic acid
- BDKRB2 (B2R)
gene encoding B2 bradykinin GPCR
- BK
large(big)-conductance, calcium-activated K+ channel
- BTP2
N-[4-[3,5-bistrifluoromethyl:pyrazol-1-yl]phenyl]-4-methylthiadiazole-5-carboxamide
- cAMP
cyclic AMP, or 3′,5′-cyclic adenosine monophosphate
- CAK
cyclin-dependent kinase activating kinase
- CaM
calmodulin
- CAS
Crk-associated substrate
- Cav1
subfamily of VGCCs including L-type channels
- Cav1.2
L-type VGCC
- Cav2
subfamily of VGCCs including P/Q-, N-, and R-type channels
- Cav3
subfamily of VGCCs including T-type channels
- Cav-1
caveolin-1
- Cbl
casitas B-lineage lymphoma protein
- Cdc42
cell division control protein 42 of Rho subfamily of Ras-related GTP-binding proteins
- CDH23
gene encoding cadherin 23 protein
- CEBPB
CCAAT enhancer binding protein beta
- CeRNA
competing endogenous RNA
- ChIP
chromatin immunoprecipitation
- CHO
Chinese hamster ovary (cell line)
- CIB2
gene encoding calcium and integrin binding family member 2 protein
- CICR
calcium-induced calcium release
- CKAP4
cytoskeletal association protein 4
- CNS
central nervous system
- COOH
carboxy (terminus)
- COS
fibroblast-like cell line derived from monkey kidney
- COX
cyclooxygenase
- cPLA2
Cytosolic PLA2
- Crk11
Cysteine-rich receptor-like protein kinase 11
- CS
chondroitin sulfate
- CSA
cross-sectional area
- CSK
cytoskeleton
- CTGF
connective tissue growth factor
- Cx
connexin
- CXCR4
gene encoding C-X-C motif chemokine receptor 4
- CysLT1R
leukotriene receptor
- D5R
dopamine D5 receptor
- D
diffusion coefficient
- DAG
diacylglycerol
- DCVJ
9-(dicyanovinyl)-julolidine
- DE
differentially expressed
- DEG
degenerins family of sodium channels
- df
final diameter
- di
initial diameter
- DIDS
4,4′-Diisothiocyanato-2,2′-stilbenedisulfonic acid disodium salt, chloride channel inhibitor
- DiIC
1,1′-dihexadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- Dkk2
Dickkopf WNT signaling pathway inhibitor 2
- DNMT
DNA methyltransferase
- DOCA
deoxycorticosterone acetate
- DOG
1,2-dioctanoyl-sn-glycerol (DAG analog)
- Dooku1
reversible antagonist of Yoda1
- dSTORM
stochastic optical reconstruction microscopy
- EC
endothelial cell
- ecKO
endothelial cell-specific knock out
- eCLIP
enhanced crosslinking and immunoprecipitation
- ECM
extracellular matrix
- EDH
endothelium dependent hyperpolarization
- EDCF
endothelium-derived constricting factor
- EDHF
endothelium-derived hyperpolarizing factor
- EETs
epoxyeicosatrienoic acids
- EGFR
epidermal growth factor receptor
- ECl
Cl− equilibrium potential
- EK
K+ equilibrium potential
- Em
membrane potential
- ENaC
epithelial sodium channel
- EnNaC
endothelial sodium channel (possible EC variant of ENaC)
- Endo-ICLT
endothelial to immune-like cell transition
- EndoMT
endothelial-to-mesenchymal transition
- eNOS
endothelial isoform of nitric oxide synthase
- ER
endoplasmic reticulum
- ERK(1/2)
members of family of extracellular signal-regulated protein kinases
- ESG
endothelial surface glycocalyx
- ET-1
endothelin-1
- EV
extracellular vesicle
- EZH2
Enhancer of zeste homologue 2
- FACS
fluorescence activated cell sorting
- F-actin
filamentous actin
- FAK
focal adhesion kinase
- FAS
focal adhesion site
- FFL
force-from-lipids
- FLK-1
vascular endothelial growth factor receptor
- FlnA
filamin A
- FN
fibronectin
- FoxO
forkhead box O
- FPR
surface formyl peptide receptor
- FRAP
fluorescence recovery after photobleaching
- FRET
Forster (or fluorescence) resonance energy transfer
- G-actin
monomeric actin
- GAP
GTPase-activating protein
- GATA
family of transcription factors that bind to the DNA sequence “GATA”
- GDP
guanosine diphosphate
- GEF
guanine nucleotide-exchange factor
- GI
gastrointestinal
- GIT
G-protein interacting target proteins
- GnRH
gonadotropin-releasing hormone
- GO
Gene Ontology
- GPCR
G-protein coupled receptor
- GqPCR
Gq family of G-protein-coupled receptors
- GPR68
G protein-coupled receptor 68
- Gs
alpha subunit of heterotrimeric G proteins that activate adenylyl cyclase
- Gi
alpha subunit of heterotrimeric G proteins that inhibit adenylyl cyclase
- G(α)q/11
alpha subunit of heterotrimeric G proteins coupling GPCRs to phospholipase C
- G(α)12/13
alpha subunit of heterotrimeric G proteins coupling GPCRs to Rho small GTPases
- GsMTx4
spider venom peptide that inhibits cationic mechanosensitive channels
- GTP
guanosine triphosphate
- H1R
histamine H1 receptor
- HA
hyaluronic acid
- HAT
histone acetyltransferase
- HCN
Hyperpolarization-activated cyclic nucleotide–gated (cation channel)
- HCR
hydroxy-methylglutaryl coenzyme A reductase
- HDAC
histone deacetylase
- HDM
histone demethylase
- HEK(293)
immortalized human embryonic kidney cell line; T variant with SV40 large T antigen
- HETE
family of hydroxyeicosatetraenoic acids formed from AA metabolism
- 20-HETE
20-hydroxyeicosatetraenoic acid
- HIF-1α
hypoxia-induced factor-1 alpha
- HMT
histone methyltransferase
- HS
heparan sulfate
- HSP27
heat shock protein 27
- HT
hypertension
- Hyal2
hyaluronidase 2
- HUVEC
human umbilical vein endothelial cell
- IAA-94
indanyloxyacetic acid inhibitor of chloride channels
- IbTX
iberiotoxin (BK channel inhibitor)
- ICCs
Interstitial Cells of Cajal
- IK
intermediate-conductance, calcium-activated K+ channel
- IKK
IκB kinase
- ILK
integrin-linked kinase
- IP3
inositol 1,4,5-trisphosphate
- IP3R
inositol triphosphate receptor
- ITPR3
inositol 1,4,5-trisphosphate receptor3
- I-V
current-voltage
- JMID2A
JmiC histone demethylase
- JNK
c-Jun N-terminal kinase
- K2P
family of K+ channels with two pore domains
- KATP
ATP-sensitive K+ channels
- KCa
calcium-activated K+ channels
- KDR
delayed rectifier K+
- Kir
inward-rectifying K+ channel
- Kv
voltage-gated family of K+ channels
- KLF
Krüppel-like factor
- Klk8
Kallikrein Related Peptidase 8
- L-T
length-tension
- LARG (ARHGEF12)
leukemia-associated RhoGEF
- LCA
left carotid artery
- LDV
Leu-Asp-Val peptide sequence
- LEENE
LncRNA that enhances eNOS expression. Gene name LINC00520
- LHFPL5
LHFPL tetraspan subfamily member 5 protein
- LIMK
Lim domain kinase
- LINC
linkers of nucleoskeleton and cytoskeleton
- LKB1
liver kinase B1
- LncRNA
Long noncoding RNA
- Lpr1
LDL receptor related protein 1
- LRRC
leucine-rich repeat-containing (γ-subunit of BK channel)
- LS
laminar shear
- M5R
muscarinic M5 receptor
- m6A
N6-methyladenine
- MAGI1
membrane-associated guanylate kinase with inverted domain structure 1
- MβCD
methyl-β-cyclodextrin
- MDCK
Madin-Darby canine kidney (cell line)
- MEC(4,10)
member(s) of degenerins family of sodium channels
- METTL3
methyltransferase 3
- mIMCD
mouse inner medullar collecting duct
- miR
MicroRNA
- MK2
MAPK activated protein kinase 2
- ML-7
Hexahydro-1-[(5-iodo-1-naphthalenyl)sulfonyl]-1H-1,4-diazepine hydrochloride, MLCK inhibitor
- MLC20
20-kDa regulatory myosin light chain subunit
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- MMP
matrix metalloproteinase
- MscS
family of small conductance mechanosensitive ion channels
- MscL
family of large conductance mechanosensitive ion channels
- MscCa
family of calcium-permeable mechanosensitive ion channels
- MTOC
microtubule organizing center
- MYPT
myosin phosphatase targeting protein
- MβCD
methyl-β-cyclodextrin
- N2A
mouse neural crest-derived cell line
- N-[3′,4′-dimethoxycinnamoyl]-anthranilic acid
Tranilast
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NaF
sodium fluoride
- NE
norepinephrine
- NEAT
nuclear paraspeckle assembly transcript
- NFAT
Nuclear factor of activated T-cells
- NCX
Na+-Ca2+ exchanger
- NG
nodose ganglia
- NHE1
Na+-H+ exchanger 1
- NLRP3
NOD-, LRR- and Pyrin domain-containing protein 3
- NO
nitric oxide
- NOS
nitric oxide synthase (any or all isoforms)
- NP
nanoparticle
- NQO1
NADPH quinone oxidoreductase1
- Nrf2
NF-E2-related factor 2
- N-WASP
neuronal Wiskott-Aldrich syndrome protein
- OAG
1-oleoyl-2-acetyl-sn-glycerol (DAG analog)
- Orai
family of calcium release-activated calcium channel proteins
- OS
oscillatory shear stress
- OSCA
family of osmo-sensitive ion channels
- p130Cas
docking protein
- P2RX4
P2X purinoreceptor 4
- P450
cytochrome P450 superfamily of enzymes
- p47phox
NADH oxidase
- PA
polyacrylamide
- PAH
pulmonary arterial hypertension
- PAK
p21 (Rac) activated kinase
- Pc
capillary hydrostatic pressure
- PCa
Ca2+ permeability
- PK
K+ permeability
- PNa
Na+ permeability
- 2,4-PDCA
2,4-pyridinedicarboxylic acid
- Pc
capillary (hydrostatic) pressure
- PCDH15
protocadherin related 15 protein
- PDAY
Pathobiological Determinants of Atherosclerosis in Youth
- PDE4D
phosphodiesterase 4D
- PDGF
platelet-derived growth factor
- PDL
population doubling level
- PDMS
polydimethylsiloxane
- PE
phenylephrine
- PECAM-1
platelet and endothelial cell adhesion molecule 1
- Pext
external or tissue hydrostatic pressure
- PGI2
prostacyclin
- PI3K
phosphatidylinositol-3-kinase
- PINCH
particularly interesting Cys-His-rich protein
- Pint
intraluminal pressure
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKA
protein kinase A
- PKC
protein kinase C
- PLA2
phospholipase A2
- PLC
phospholipase C
- PLD
phospholipase D
- PNa
Na+ permeability
- PNP
platelet-membrane-cloaked NP
- PNS
peripheral nervous system
- Po
channel open probability
- PP1
1-(1,1-Dimethylethyl)-1-(4-methylphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine, Src inhibitor
- PP2
3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine, Src inhibitor
- PP2A
protein phosphatase 2A
- PPARα/δ
peroxisome proliferator-activated receptor α/δ
- PPVL
partial portal vein ligation
- PS
pulsatile shear stress
- PTx
pertussis toxin
- PUFA
polyunsaturated fatty acid
- PYK2
protein tyrosine kinase 2
- r
vessel radius
- Rac1
Rac family small GTPase 1
- RANTES
C-C motif chemokine ligand 5
- RBC
red blood cell
- RCA
right carotid artery
- RCAEC
rat carotid artery endothelium
- AGTR1
angiotensin II receptor type 1
- RGD
Arg-Gly-Asp (peptide sequence)
- RGS(2,5)
regulator of G-protein signaling
- RhoA
Ras homology family A
- RhoV14
constitutive active form of Rho
- RMAECs
rat mesenteric artery ECs
- RNA
ribonucleic acid
- RNA-seq
RNA sequencing technique
- ROCK
Rho-associated protein kinase
- ROS
reactive oxygen species
- RuR
ruthenium red
- RYR
ryanodine receptor
- S1PR1
sphingosine-1-phosphate receptor
- SA
sialic acid
- SAN
sino-atrial node
- sc
single-cell
- SDK
stretch-dependent K+ channel
- SH2
Src homology 2 (domain)
- SHR
spontaneous hypertensive rat
- SIRT1
sirtuin 1
- SK
small-conductance, calcium-activated K+ channel
- SK3
KCa2.3
- SKF96365
1-[2-(4-Methoxyphenyl)-2-[3-(4-methoxyphenyl)propoxy]ethyl-1H-imidazole hydrochloride, SOCE inhibitor
- SKG1
serum glucocorticoid-regulated kinase-1
- SM
smooth muscle
- SMC
smooth muscle cell
- smKO
smooth muscle cell-specific knock out
- SNAP23
synaptosomal-associated protein 23
- SNARE
family of proteins mediating vesicle fusion
- SOCE
Store-operated calcium entry
- SPA
stress phase angle
- SPHK1
sphingosine kinase 1
- sPLA2
secretory PLA2
- Spp1
marker secreted phosphoprotein 1
- SR
sarcoplasmic reticulum
- Src
member of family of non-receptor tyrosine kinases
- SRF
serum response factor
- STIM
stromal interaction molecule, family of dynamic sensors of Ca2+ in the ER
- STIC
spontaneous transient inward current
- STOC
spontaneous transient outward current
- STOML(1,3)
stomatin-like family of proteins
- SUMO1
small ubiquitin-like modifier 1
- Sun
Sun domain proteins
- SuV39h1
SUV39H1 histone lysine methyltransferase
- TAZ
transcriptional coactivator with PDZ-binding motif
- TEA
tetraethylammonium
- TET1
Tet methylcytosine dioxgenase 1
- TG
transglutaminase
- TbRs
TGF-β receptors
- TIRF
total internal reflection fluorescent microscopy
- TIRFM
diffraction-limited total internal reflection fluorescence microscopy
- TM
thrombomodulin
- TMC(-1,-2)
transmembrane channel-like protein
- TMEM
transmembrane protein family of calcium-activated ion channels and phospholipid scramblases
- TMEM16A (or ANO1)
member of TMEM16 family of calcium-activated Cl− channels
- TMIE
gene encoding transmembrane inner ear protein
- TNFa
tumor necrosis factor alpha (inflammatory cytokine)
- TPA
phorbol 12-myristate 13-acetate (PKC activator)
- TRAAK
(membrane tension-activated K+ channel), member of two-pore-domain potassium channel family
- TREK (1,2)
(Twik-related K+ channel 1), members of two-pore-domain potassium channel family
- TRAM-34
(Triarylmethane-34) 1-[(2-Chlorophenyl)diphenylmethyl]-1H-pyrazole; IK channel blocker
- trem2
myeloid cells 2
- TRP
transient receptor potential channel
- TRPA
ankyrin subfamily of TRP channels
- TRPC
canonical subfamily of TRP channels
- Trpc6 −/−
TRPC6 knockout mice
- Trpm4 −/−
TRPM4 knockout mice
- TRPM
melastatin subfamily of TRP channels
- TRPML
mucolipin subfamily of TRP channels
- TRPP
polycystin subfamily of TRP channels
- TRPV
vanilloid subfamily of TRP channels
- TSA
trichostatin A
- TSP1
thromospondin 1
- V
velocity
- V1AR
vasopressin 1A receptor
- VAMP3
vesicle-associated membrane protein 3
- VASP
vasodilator stimulated phosphoprotein
- VDCC
voltage-dependent calcium channel (same as VGCC)
- VE-cad
VE cadherin
- VEGFR
vascular endothelial growth factor receptor
- VGCC
voltage-gated calcium channel
- Vm
membrane potential
- VRAC
volume-regulated anion channel
- VSM
vascular smooth muscle
- VSMC
vascular smooth muscle cells
- WD
Western Diet
- WT
wild-type
- XE991
10,10-bis(4-Pyridinylmethyl)-9(10H)-anthracenone dihydrochloride; Kv7 inhibitor
- YAP
Yes-associated protein
- Yoda1
2-[5-[[(2,6-Dichlorophenyl)methyl]thio]-1,3,4-thiadiazol-2-yl]pyrazine, chemical activator of Piezo1 channels
- ▵A
change in surface area
- η
viscosity
- σϴ
longitudinal distending stress (acting on VSMCs)
- σr
radial distending stress (acting on ECs)
- τw
wall shear stress
REFERENCES
- 1. Schönleitner P, Schotten U, Antoons G. Mechanosensitivity of microdomain calcium signalling in the heart. Prog Biophys Mol Biol 130: 288–301, 2017. doi: 10.1016/j.pbiomolbio.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 2. Chen-Izu Y, Izu LT. Mechano-chemo-transduction in cardiac myocytes. J Physiol 595: 3949–3958, 2017. doi: 10.1113/JP273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M, Patapoutian A. Piezo1 links mechanical forces to red blood cell volume. Elife 4: e07370, 2015. doi: 10.7554/eLife.07370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cinar E, Zhou S, DeCourcey J, Wang Y, Waugh RE, Wan J. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci USA 112: 11783–11788, 2015. doi: 10.1073/pnas.1507309112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shin HY, Fukuda S, Schmid-Schönbein GW. Fluid shear stress-mediated mechanotransduction in circulating leukocytes and its defect in microvascular dysfunction. J Biomech 120: 110394, 2021. doi: 10.1016/j.jbiomech.2021.110394. [DOI] [PubMed] [Google Scholar]
- 6. Hansen CE, Qiu Y, McCarty OJ, Lam WA. Platelet mechanotransduction. Annu Rev Biomed Eng 20: 253–275, 2018. doi: 10.1146/annurev-bioeng-062117-121215. [DOI] [PubMed] [Google Scholar]
- 7. Lu Y, Ma X, Sabharwal R, Snitsarev V, Morgan D, Rahmouni K, Drummond HA, Whiteis CA, Costa V, Price M, Benson C, Welsh MJ, Chapleau MW, Abboud FM. The ion channel ASIC2 is required for baroreceptor and autonomic control of the circulation. Neuron 64: 885–897, 2009. doi: 10.1016/j.neuron.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zeng WZ, Marshall KL, Min S, Daou I, Chapleau MW, Abboud FM, Liberles SD, Patapoutian A. PIEZOs mediate neuronal sensing of blood pressure and the baroreceptor reflex. Science 362: 464–467, 2018. doi: 10.1126/science.aau6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327–387, 2011. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thoumine O, Ziegler T, Girard PR, Nerem RM. Elongation of confluent endothelial cells in culture: the importance of fields of force in the associated alterations of their cytoskeletal structure. Exp Cell Res 219: 427–441, 1995. doi: 10.1006/excr.1995.1249. [DOI] [PubMed] [Google Scholar]
- 11. Fang Y, Wu D, Birukov KG. Mechanosensing and mechanoregulation of endothelial cell functions. Compr Physiol 9: 873–904, 2019. doi: 10.1002/cphy.c180020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79: 387–423, 1999. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 13. Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol (1985) 91: 973–983, 2001. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- 14. Osol G. Mechanotransduction by vascular smooth muscle. J Vasc Res 32: 275–292, 1995. doi: 10.1159/000159102. [DOI] [PubMed] [Google Scholar]
- 15. Koller A. Signaling pathways of mechanotransduction in arteriolar endothelium and smooth muscle cells in hypertension. Microcirculation 9: 277–294, 2002. doi: 10.1038/sj.mn.7800142. [DOI] [PubMed] [Google Scholar]
- 16. Jackson WF. Myogenic tone in peripheral resistance arteries and arterioles: the pressure is on. Front Physiol 12: 699517, 2021. doi: 10.3389/fphys.2021.699517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev 75: 519–560, 1995. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerhold KA, Schwartz MA. Ion channels in endothelial responses to fluid shear stress. Physiology (Bethesda) 31: 359–369, 2016. doi: 10.1152/physiol.00007.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thakore P, Earley S. Transient receptor potential channels and endothelial cell calcium signaling. Compr Physiol 9: 1249–1277, 2019. doi: 10.1002/cphy.c180034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nilius B, Honoré E. Sensing pressure with ion channels. Trends Neurosci 35: 477–486, 2012. doi: 10.1016/j.tins.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 21. Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Delmas P, Patel A, Honoré E. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol 48: 83–89, 2010. doi: 10.1016/j.yjmcc.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 22. Folgering JH, Sharif-Naeini R, Dedman A, Patel A, Delmas P, Honoré E. Molecular basis of the mammalian pressure-sensitive ion channels: focus on vascular mechanotransduction. Prog Biophys Mol Biol 97: 180–195, 2008. doi: 10.1016/j.pbiomolbio.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 23. Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J 27: 3092–3103, 2008. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M, Qian M, Kyler K, Xu J. Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Front Cardiovasc Med 5: 151, 2018. doi: 10.3389/fcvm.2018.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Méndez-Barbero N, Gutiérrez-Muñoz C, Blanco-Colio LM. Cellular crosstalk between endothelial and smooth muscle cells in vascular wall remodeling. Int J Mol Sci 22: 7284, 2021. doi: 10.3390/ijms22147284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 95: 645–690, 2015. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedersen SF, Nilius B. Transient receptor potential channels in mechanosensing and cell volume regulation. Methods Enzymol 428: 183–207, 2007. doi: 10.1016/S0076-6879(07)28010-3. [DOI] [PubMed] [Google Scholar]
- 28. Sharif-Naeini R, Dedman A, Folgering JH, Duprat F, Patel A, Nilius B, Honoré E. TRP channels and mechanosensory transduction: insights into the arterial myogenic response. Pflugers Arch 456: 529–540, 2008. doi: 10.1007/s00424-007-0432-y. [DOI] [PubMed] [Google Scholar]
- 29. Dimmeler S, Haendeler J, Zeiher AM. Regulation of endothelial cell apoptosis in atherothrombosis. Curr Opin Lipidol 13: 531–536, 2002. doi: 10.1097/00041433-200210000-00009. [DOI] [PubMed] [Google Scholar]
- 30. Garin G, Berk BC. Flow-mediated signaling modulates endothelial cell phenotype. Endothelium 13: 375–384, 2006. doi: 10.1080/10623320601061599. [DOI] [PubMed] [Google Scholar]
- 31. Gimbrone MA Jr. Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis. Am J Cardiol 75: 67B–70B, 1995. doi: 10.1016/0002-9149(95)80016-l. [DOI] [PubMed] [Google Scholar]
- 32. Chien S. Biophysical behavior of red cells in suspensions. In: The Red Blood Cell (2nd ed.). New York: Academic Press, 1975, p. 1031–1133. [Google Scholar]
- 33. Baskurt OK, Meiselman HJ. Blood rheology and hemodynamics. Semin Thromb Hemost 29: 435–450, 2003. doi: 10.1055/s-2003-44551. [DOI] [PubMed] [Google Scholar]
- 34.Anonymous. Natural history of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY Study. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb 13: 1291–1298, 1993. doi: 10.1161/01.ATV.13.9.1291. [DOI] [PubMed] [Google Scholar]
- 35. Goldsmith HL, Karino T. Flow patterns and the localization pf vascular disease in the circulation. In: Endovascular Interventional Neuroradiology, edited by Holtzmann RN, Stein BM, Winston H.. New York: Springer, 1985, p. 25–66. [Google Scholar]
- 36. Karino T. Microscopic structure of disturbed flows in the arterial and venous systems, and its implication in the localization of vascular diseases. Int Angiol 5: 297–313, 1986. [PubMed] [Google Scholar]
- 37. Chien S. Shu Chien: an Autobiography and Tributes at his 80th Birthday and Beyond, edited by Li YS, Flores L.. Singapore: World Scientific, 2018, p. 61. [Google Scholar]
- 38. Weinbaum S, Tzeghai G, Ganatos P, Pfeffer R, Chien S. Effect of cell turnover and leaky junctions on arterial macromolecular transport. Am J Physiol Heart Circ Physiol 248: H945–H960, 1985. doi: 10.1152/ajpheart.1985.248.6.H945. [DOI] [PubMed] [Google Scholar]
- 39. Chien S. Molecular and mechanical bases of focal lipid accumulation in arterial wall. Prog Biophys Mol Biol 83: 131–151, 2003. doi: 10.1016/s0079-6107(03)00053-1. [DOI] [PubMed] [Google Scholar]
- 40. Mohamied Y, Rowland EM, Bailey EL, Sherwin SJ, Schwartz MA, Weinberg PD. Change of direction in the biomechanics of atherosclerosis. Ann Biomed Eng 43: 16–25, 2015. doi: 10.1007/s10439-014-1095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaunas R, Usami S, Chien S. Regulation of stretch-induced JNK activation by stress fiber orientation. Cell Signal 18: 1924–1931, 2006. doi: 10.1016/j.cellsig.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 42. Wu CC, Li YS, Haga JH, Kaunas R, Chiu JJ, Su FC, Usami S, Chien S. Directional shear flow and Rho activation prevent the endothelial cell apoptosis induced by micropatterned anisotropic geometry. Proc Natl Acad Sci USA 104: 1254–1259, 2007. doi: 10.1073/pnas.0609806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bao X, Lu C, Frangos JA. Mechanism of temporal gradients in shear-induced ERK1/2 activation and proliferation in endothelial cells. Am J Physiol Heart Circ Physiol 281: H22–H29, 2001. doi: 10.1152/ajpheart.2001.281.1.H22. [DOI] [PubMed] [Google Scholar]
- 44. Bao X, Lu C, Frangos JA. Temporal gradient in shear but not steady shear stress induces PDGF-A and MCP-1 expression in endothelial cells: role of NO, NF kappa B, and egr-1. Arterioscler Thromb Vasc Biol 19: 996–1003, 1999. doi: 10.1161/01.ATV.19.4.996. [DOI] [PubMed] [Google Scholar]
- 45. Gimbrone MA Jr, Topper JN, Nagel T, Anderson KR, Garcia CG. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci 902: 230–239, 2000. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 46. Lee DY, Chiu JJ. Atherosclerosis and flow: roles of epigenetic modulation in vascular endothelium. J Biomed Sci 26: 56, 2019. doi: 10.1186/s12929-019-0551-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech 38: 1949–1971, 2005. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 48. Nerem RM. Vascular fluid mechanics, the arterial wall, and atherosclerosis. J Biomech Eng 114: 274–282, 1992. doi: 10.1115/1.2891384. [DOI] [PubMed] [Google Scholar]
- 49. Wissler RW. An overview of the quantitative influence of several risk factors on progression of atherosclerosis in young people in the United States. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Am J Med Sci 310, Suppl 1: S29–S36, 1995. doi: 10.1097/00000441-199512000-00006. [DOI] [PubMed] [Google Scholar]
- 50. Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc Lond B Biol Sci 362: 1445–1457, 2007. doi: 10.1098/rstb.2007.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Davies PH. Endothelial mechanotransduction. In: Cellular Mechanotransduction, edited by Mofrad MR, Kamm RD.. Cambridge, UK: Cambridge University Press, 2009. [Google Scholar]
- 52. Yoganathan AP, He Z, Casey Jones S. Fluid mechanics of heart valves. Annu Rev Biomed Eng 6: 331–362, 2004. doi: 10.1146/annurev.bioeng.6.040803.140111. [DOI] [PubMed] [Google Scholar]
- 53. Bergan JJ, Schmid-Schönbein GW, Smith PD, Nicolaides AN, Boisseau MR, Eklof B. Chronic venous disease. N Engl J Med 355: 488–498, 2006. doi: 10.1056/NEJMra055289. [DOI] [PubMed] [Google Scholar]
- 54. Lawrence MB, McIntire LV, Eskin SG. Effect of flow on polymorphonuclear leukocyte/endothelial cell adhesion. Blood 70: 1284–1290, 1987. [PubMed] [Google Scholar]
- 55. Koslow AR, Stromberg RR, Friedman LI, Lutz RJ, Hilbert SL, Schuster P. A flow system for the study of shear forces upon cultured endothelial cells. J Biomech Eng 108: 338–341, 1986. doi: 10.1115/1.3138625. [DOI] [PubMed] [Google Scholar]
- 56. Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science 227: 1477–1479, 1985. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 57. Malek AM, Ahlquist R, Gibbons GH, Dzau VJ, Izumo S. A cone-plate apparatus for the in vitro biochemical and molecular analysis of the effect of shear stress on adherent cells. Methods Cell Sci 17: 165–176, 1995. doi: 10.1007/BF00996123. [DOI] [Google Scholar]
- 58. McIntire LV, Eskin SG. Mechanical and biochemical aspects of leukocyte interactions with model vessel walls. Kroc Found Ser 16: 209–219, 1984. [PubMed] [Google Scholar]
- 59. Davies PF, Dewey CF Jr, Bussolari SR, Gordon EJ, Gimbrone MA Jr.. Influence of hemodynamic forces on vascular endothelial function. In vitro studies of shear stress and pinocytosis in bovine aortic cells. J Clin Invest 73: 1121–1129, 1984. doi: 10.1172/JCI111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wong AK, LLanos P, Boroda N, Rosenberg SR, Rabbany SY. A parallel-plate flow chamber for mechanical characterization of endothelial cells exposed to laminar shear stress. Cell Mol Bioeng 9: 127–138, 2016. doi: 10.1007/s12195-015-0424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shankaran H, Neelamegham S. Effect of secondary flow on biological experiments in the cone-plate viscometer: methods for estimating collision frequency, wall shear stress and inter-particle interactions in non-linear flow. Biorheology 38: 275–304, 2001. [PubMed] [Google Scholar]
- 62. Usami S, Chen HH, Zhao Y, Chien S, Skalak R. Design and construction of a linear shear stress flow chamber. Ann Biomed Eng 21: 77–83, 1993. doi: 10.1007/BF02368167. [DOI] [PubMed] [Google Scholar]
- 63. Chiu JJ, Wang DL, Chien S, Skalak R, Usami S. Effects of disturbed flow on endothelial cells. J Biomech Eng 120: 2–8, 1998. doi: 10.1115/1.2834303. [DOI] [PubMed] [Google Scholar]
- 64. Truskey GA, Barber KM, Robey TC, Olivier LA, Combs MP. Characterization of a sudden expansion flow chamber to study the response of endothelium to flow recirculation. J Biomech Eng 117: 203–210, 1995. doi: 10.1115/1.2796002. [DOI] [PubMed] [Google Scholar]
- 65. Franzoni M, Cattaneo I, Ene-Iordache B, Oldani A, Righettini P, Remuzzi A. Design of a cone-and-plate device for controlled realistic shear stress stimulation on endothelial cell monolayers. Cytotechnology 68: 1885–1896, 2016. doi: 10.1007/s10616-015-9941-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DePaola N, Gimbrone MA Jr, Davies PF, Dewey CF Jr.. Vascular endothelium responds to fluid shear stress gradients. Arterioscler Thromb 12: 1254–1257, 1992. doi: 10.1161/01.atv.12.11.1254. [DOI] [PubMed] [Google Scholar]
- 67. Ziegler T, Alexander RW, Nerem RM. An endothelial cell-smooth muscle cell co-culture model for use in the investigation of flow effects on vascular biology. Ann Biomed Eng 23: 216–225, 1995. doi: 10.1007/BF02584424. [DOI] [PubMed] [Google Scholar]
- 68. Heydarkhan-Hagvall S, Chien S, Nelander S, Li YC, Yuan S, Lao J, Haga JH, Lian I, Nguyen P, Risberg B, Li YS. DNA microarray study on gene expression profiles in co-cultured endothelial and smooth muscle cells in response to 4- and 24-h shear stress. Mol Cell Biochem 281: 1–15, 2006. doi: 10.1007/s11010-006-0168-6. [DOI] [PubMed] [Google Scholar]
- 69. Nackman GB, Fillinger MF, Shafritz R, Wei T, Graham AM. Flow modulates endothelial regulation of smooth muscle cell proliferation: a new model. Surgery 124: 353–360, 1998. [PubMed] [Google Scholar]
- 70. Chiu JJ, Chen LJ, Lee PL, Lee CI, Lo LW, Usami S, Chien S. Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood 101: 2667–2674, 2003. doi: 10.1182/blood-2002-08-2560. [DOI] [PubMed] [Google Scholar]
- 71. Laughlin MH, Davis MJ, Secher NH, van Lieshout JJ, Arce-Esquivel AA, Simmons GH, Bender SB, Padilla J, Bache RJ, Merkus D, Duncker DJ. Peripheral circulation. Compr Physiol 2: 321–447, 2012. doi: 10.1002/cphy.c100048. [DOI] [PubMed] [Google Scholar]
- 72. Kuo L, Davis MJ, Chilian WM. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol Heart Circ Physiol 259: H1063–H1070, 1990. doi: 10.1152/ajpheart.1990.259.4.H1063. [DOI] [PubMed] [Google Scholar]
- 73. Carlson BE, Arciero JC, Secomb TW. Theoretical model of blood flow autoregulation: roles of myogenic, shear-dependent, and metabolic responses. Am J Physiol Heart Circ Physiol 295: H1572–H1579, 2008. doi: 10.1152/ajpheart.00262.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sun D, Liu H, Yan C, Jacobson A, Ojaimi C, Huang A, Kaley G. COX-2 contributes to the maintenance of flow-induced dilation in arterioles of eNOS-knockout mice. Am J Physiol Heart Circ Physiol 291: H1429–H1435, 2006. doi: 10.1152/ajpheart.01130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am J Physiol Cell Physiol 266: C628–C636, 1994. doi: 10.1152/ajpcell.1994.266.3.C628. [DOI] [PubMed] [Google Scholar]
- 76. Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 459: 793–806, 2010. doi: 10.1007/s00424-009-0767-7. [DOI] [PubMed] [Google Scholar]
- 77. Rubanyi GM, Romero JC, Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. Am J Physiol Heart Circ Physiol 250: H1145–H1149, 1986. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- 78. Koller A, Kaley G. Endothelial regulation of wall shear stress and blood flow in skeletal muscle microcirculation. Am J Physiol Heart Circ Physiol 260: H862–H868, 1991. doi: 10.1152/ajpheart.1991.260.3.H862. [DOI] [PubMed] [Google Scholar]
- 79. Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol Heart Circ Physiol 280: H2462–H2469, 2001. doi: 10.1152/ajpheart.2001.280.6.H2462. [DOI] [PubMed] [Google Scholar]
- 80. Schmidt K, de Wit C. Endothelium-derived hyperpolarizing factor and myoendothelial coupling: the in vivo perspective. Front Physiol 11: 602930, 2020. doi: 10.3389/fphys.2020.602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schilling WP. Effect of membrane potential on cytosolic calcium of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 257: H778–H784, 1989. doi: 10.1152/ajpheart.1989.257.3.H778. [DOI] [PubMed] [Google Scholar]
- 82. Cannell MB, Sage SO. Bradykinin-evoked changes in cytosolic calcium and membrane currents in cultured bovine pulmonary artery endothelial cells. J Physiol 419: 555–568, 1989. doi: 10.1113/jphysiol.1989.sp017886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Adams DJ, Barakeh J, Laskey R, Van Breemen C. Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J 3: 2389–2400, 1989. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- 84. Hong K, Cope EL, DeLalio LJ, Marziano C, Isakson BE, Sonkusare SK. TRPV4 (transient receptor potential vanilloid 4) channel-dependent negative feedback mechanism regulates Gq protein-coupled receptor-induced vasoconstriction. Arterioscler Thromb Vasc Biol 38: 542–554, 2018. doi: 10.1161/ATVBAHA.117.310038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Garland CJ, Bagher P, Powell C, Ye X, Lemmey HA, Borysova L, Dora KA. Voltage-dependent Ca2+ entry into smooth muscle during contraction promotes endothelium-mediated feedback vasodilation in arterioles. Sci Signal 10: eaal3806, 2017. doi: 10.1126/scisignal.aal3806. [DOI] [PubMed] [Google Scholar]
- 86. Fleming I, Busse R. Signal transduction of eNOS activation. Cardiovasc Res 43: 532–541, 1999. doi: 10.1016/s0008-6363(99)00094-2. [DOI] [PubMed] [Google Scholar]
- 87. Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 299: 818–824, 2001. [PubMed] [Google Scholar]
- 88. Michel T, Vanhoutte PM. Cellular signaling and NO production. Pflugers Arch 459: 807–816, 2010. doi: 10.1007/s00424-009-0765-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Falcone JC, Kuo L, Meininger GA. Endothelial cell calcium increases during flow-induced dilation in isolated arterioles. Am J Physiol Heart Circ Physiol 264: H653–H659, 1993. doi: 10.1152/ajpheart.1993.264.2.H653. [DOI] [PubMed] [Google Scholar]
- 90. Muller JM, Davis MJ, Kuo L, Chilian WM. Changes in coronary endothelial cell Ca2+ concentration during shear stress- and agonist-induced vasodilation. Am J Physiol Heart Circ Physiol 276: H1706–H1714, 1999. doi: 10.1152/ajpheart.1999.276.5.H1706. [DOI] [PubMed] [Google Scholar]
- 91. Daut J, Mehrke G, Nees S, Newman WH. Passive electrical properties and electrogenic sodium transport of cultured guinea-pig coronary endothelial cells. J Physiol 402: 237–254, 1988. doi: 10.1113/jphysiol.1988.sp017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Himmel HM, Whorton AR, Strauss HC. Intracellular calcium, currents, and stimulus-response coupling in endothelial cells. Hypertension 21: 112–127, 1993. doi: 10.1161/01.hyp.21.1.112. [DOI] [PubMed] [Google Scholar]
- 93. Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad RK, Evans PC, Ainscough JF, Beech DJ. Piezo1 integration of vascular architecture with physiological force. Nature 515: 279–282, 2014. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M, Zhang DX. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298: H466–H476, 2010. doi: 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 117: 1161–1171, 2008. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yamamoto K, Korenaga R, Kamiya A, Ando J. Fluid shear stress activates Ca2+ influx into human endothelial cells via P2X4 purinoceptors. Circ Res 87: 385–391, 2000. doi: 10.1161/01.res.87.5.385. [DOI] [PubMed] [Google Scholar]
- 97. Bhagyalakshmi A, Berthiaume F, Reich KM, Frangos JA. Fluid shear stress stimulates membrane phospholipid metabolism in cultured human endothelial cells. J Vasc Res 29: 443–449, 1992. doi: 10.1159/000158963. [DOI] [PubMed] [Google Scholar]
- 98. Nollert MU, Eskin SG, McIntire LV. Shear stress increases inositol trisphosphate levels in human endothelial cells. Biochem Biophys Res Commun 170: 281–287, 1990. doi: 10.1016/0006-291x(90)91271-s. [DOI] [PubMed] [Google Scholar]
- 99. Kwan HY, Leung PC, Huang Y, Yao X. Depletion of intracellular Ca2+ stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells. Circ Res 92: 286–292, 2003. doi: 10.1161/01.res.0000054625.24468.08. [DOI] [PubMed] [Google Scholar]
- 100. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399: 597–601, 1999. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res 90: 904–910, 2002. doi: 10.1161/01.RES.0000016506.04193.96. [DOI] [PubMed] [Google Scholar]
- 102. Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 93: 354–363, 2003. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 103. Jin ZG, Wong C, Wu J, Berk BC. Flow shear stress stimulates Gab1 tyrosine phosphorylation to mediate protein kinase B and endothelial nitric-oxide synthase activation in endothelial cells. J Biol Chem 280: 12305–12309, 2005. doi: 10.1074/jbc.M500294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol 42: 271–279, 2007. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 105. Russell-Puleri S, Dela Paz NG, Adams D, Chattopadhyay M, Cancel L, Ebong E, Orr AW, Frangos JA, Tarbell JM. Fluid shear stress induces upregulation of COX-2 and PGI2 release in endothelial cells via a pathway involving PECAM-1, PI3K, FAK, and p38. Am J Physiol Heart Circ Physiol 312: H485–H500, 2017. doi: 10.1152/ajpheart.00035.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Yang Z, Xia WH, Zhang YY, Xu SY, Liu X, Zhang XY, Yu BB, Qiu YX, Tao J. Shear stress-induced activation of Tie2-dependent signaling pathway enhances reendothelialization capacity of early endothelial progenitor cells. J Mol Cell Cardiol 52: 1155–1163, 2012. doi: 10.1016/j.yjmcc.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 107. Xu J, Mathur J, Vessières E, Hammack S, Nonomura K, Favre J, Grimaud L, Petrus M, Francisco A, Li J, Lee V, Xiang FL, Mainquist JK, Cahalan SM, Orth AP, Walker JR, Ma S, Lukacs V, Bordone L, Bandell M, Laffitte B, Xu Y, Chien S, Henrion D, Patapoutian A. GPR68 senses flow and is essential for vascular physiology. Cell 173: 762–775.e16, 2018. doi: 10.1016/j.cell.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Melchior B, Frangos JA. Galphaq/11-mediated intracellular calcium responses to retrograde flow in endothelial cells. Am J Physiol Cell Physiol 303: C467–C473, 2012. doi: 10.1152/ajpcell.00117.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Barauna VG, Magalhaes FC, Campos LC, Reis RI, Kunapuli SP, Costa-Neto CM, Miyakawa AA, Krieger JE. Shear stress-induced Ang II AT1 receptor activation: G-protein dependent and independent mechanisms. Biochem Biophys Res Commun 434: 647–652, 2013. doi: 10.1016/j.bbrc.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 110. Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci USA 103: 15463–15468, 2006. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dela Paz NG, Melchior B, Frangos JA. Shear stress induces Galphaq/11 activation independently of G protein-coupled receptor activation in endothelial cells. Am J Physiol Cell Physiol 312: C428–C437, 2017. doi: 10.1152/ajpcell.00148.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431, 2005. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 113. Walmsley JG, Gore RW, Dacey RG Jr, Damon DN, Duling BR. Quantitative morphology of arterioles from the hamster cheek pouch related to mechanical analysis. Microvasc Res 24: 249–271, 1982. doi: 10.1016/0026-2862(82)90016-4. [DOI] [PubMed] [Google Scholar]
- 114. Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol 28: 220–231, 1902. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Henrion D, Laher I, Klaasen A, Bevan JA. Myogenic tone of rabbit facial vein and posterior cerebral artery is influenced by changes in extracellular sodium. Am J Physiol Heart Circ Physiol 266: H377–H383, 1994. doi: 10.1152/ajpheart.1994.266.2.H377. [DOI] [PubMed] [Google Scholar]
- 116. Dubroca C, You D, Lévy BI, Loufrani L, Henrion D. Involvement of RhoA/Rho kinase pathway in myogenic tone in the rabbit facial vein. Hypertension 45: 974–979, 2005. doi: 10.1161/01.HYP.0000164582.63421.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Davis MJ, Davis AM, Ku CW, Gashev AA. Myogenic constriction and dilation of isolated lymphatic vessels. Am J Physiol Heart Circ Physiol 296: H293–H302, 2009. doi: 10.1152/ajpheart.01040.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Loutzenhiser R, Bidani A, Chilton L. Renal myogenic response: kinetic attributes and physiological role. Circ Res 90: 1316–1324, 2002. doi: 10.1161/01.res.0000024262.11534.18. [DOI] [PubMed] [Google Scholar]
- 119. Davis MJ. Myogenic response gradient in an arteriolar network. Am J Physiol Heart Circ Physiol 264: H2168–H2179, 1993. doi: 10.1152/ajpheart.1993.264.6.H2168. [DOI] [PubMed] [Google Scholar]
- 120. Halpern W, Osol G. Resistance vessels in hypertension. Prog Clin Biol Res 219: 211–223, 1986. [PubMed] [Google Scholar]
- 121. Davis MJ, Gilmore JP, Joyner WL. Responses of pulmonary allograft and cheek pouch arterioles in the hamster to alterations in extravascular pressure in different oxygen environments. Circ Res 49: 133–140, 1981. doi: 10.1161/01.res.49.1.133. [DOI] [PubMed] [Google Scholar]
- 122. Johnson PC. Myogenic nature of increase in intestinal vascular resistance with venous pressure elevation. Circ Res 7: 992–999, 1959. doi: 10.1161/01.res.7.6.992. [DOI] [PubMed] [Google Scholar]
- 123. Johnson PC, Hanson KM. Effect of arterial pressure on arterial and venous resistance of intestine. J Appl Physiol 17: 503–508, 1962. doi: 10.1152/jappl.1962.17.3.503. [DOI] [PubMed] [Google Scholar]
- 124. Grände PO, Mellander S. Characteristics of static and dynamic regulatory mechanisms in myogenic microvascular control. Acta Physiol Scand 102: 231–245, 1978. doi: 10.1111/j.1748-1716.1978.tb06067.x. [DOI] [PubMed] [Google Scholar]
- 125. Mellander S. On the control of capillary fluid transfer by precapillary and postcapillary vascular adjustments. Microvasc Res 15: 319–330, 1978. doi: 10.1016/0026-2862(78)90032-8. [DOI] [PubMed] [Google Scholar]
- 126. Ping P, Johnson PC. Role of myogenic response in enhancing autoregulation of flow during sympathetic nerve stimulation. Am J Physiol Heart Circ Physiol 263: H1177–H1184, 1992. doi: 10.1152/ajpheart.1992.263.4.H1177. [DOI] [PubMed] [Google Scholar]
- 127. Loutzenhiser R, Bidani AK, Wang X. Systolic pressure and the myogenic response of the renal afferent arteriole. Acta Physiol Scand 181: 407–413, 2004. doi: 10.1111/j.1365-201X.2004.01312.x. [DOI] [PubMed] [Google Scholar]
- 128. Davis MJ. Control of bat wing capillary pressure and blood flow during reduced perfusion pressure. Am J Physiol Heart Circ Physiol 255: H1114–H1129, 1988. doi: 10.1152/ajpheart.1988.255.5.H1114. [DOI] [PubMed] [Google Scholar]
- 129. Bouskela E, Wiederhielm CA. Microvascular myogenic reaction in the wing of the intact unanesthetized bat. Am J Physiol Heart Circ Physiol 237: H59–H65, 1979. doi: 10.1152/ajpheart.1979.237.1.H59. [DOI] [PubMed] [Google Scholar]
- 130. Slaaf DW, Reneman RS, Wiederhielm CA. Pressure regulation in muscle of unanesthetized bats. Microvasc Res 33: 315–326, 1987. doi: 10.1016/0026-2862(87)90026-4. [DOI] [PubMed] [Google Scholar]
- 131. Burrows ME, Johnson PC. Arteriolar responses to elevation of venous and arterial pressures in cat mesentery. Am J Physiol Heart Circ Physiol 245: H796–H807, 1983. doi: 10.1152/ajpheart.1983.245.5.H796. [DOI] [PubMed] [Google Scholar]
- 132. Renkin EM. Control of microcirculation and blood-tissue exchange. In: Handbook of Physiology. The Cardiovascular System. Microcirculation, edited by Renkin EM, Michel CC, Geiger SR.. Bethesda, MD: American Physiological Society, 1984, p. 627–687. [Google Scholar]
- 133. Johnson PC. The myogenic response. In: Handbook of Physiology. The Cardiovascular System. Vascular Smooth Muscle, edited by Bohr DF, Somlyo AP, Sparks HV Jr.. Bethesda, MD: American Physiological Society, 1980, p. 409–442. [Google Scholar]
- 134. Mulvany MJ, Aalkjaer C. Structure and function of small arteries. Physiol Rev 70: 921–961, 1990. doi: 10.1152/physrev.1990.70.4.921. [DOI] [PubMed] [Google Scholar]
- 135. Buus NH, VanBavel E, Mulvany MJ. Differences in sensitivity of rat mesenteric small arteries to agonists when studied as ring preparations or as cannulated preparations. Br J Pharmacol 112: 579–587, 1994. doi: 10.1111/j.1476-5381.1994.tb13114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Duling BR, Gore RW, Dacey RG Jr, Damon DN. Methods for isolation, cannulation, and in vitro study of single microvessels. Am J Physiol Heart Circ Physiol 241: H108–H116, 1981. doi: 10.1152/ajpheart.1981.241.1.H108. [DOI] [PubMed] [Google Scholar]
- 137. Halpern W, Osol G, Coy GS. Mechanical behavior of pressurized in vitro prearteriolar vessels determined with a video system. Ann Biomed Eng 12: 463–479, 1984. doi: 10.1007/BF02363917. [DOI] [PubMed] [Google Scholar]
- 138. Sparks HV Jr, Bohr DF. Effect of stretch on passive tension and contractility of isolated vascular smooth muscle. Am J Physiol Heart Circ Physiol 202: 835–840, 1962. doi: 10.1152/ajplegacy.1962.202.5.835. [DOI] [PubMed] [Google Scholar]
- 139. Sparks HV Jr. Effect of quick stretch on isolated vascular smooth muscle. Circ Res 15, Suppl: 254–260, 1964. [PubMed] [Google Scholar]
- 140. Nakayama K. Calcium-dependent contractile activation of cerebral artery produced by quick stretch. Am J Physiol Heart Circ Physiol 242: H760–H768, 1982. doi: 10.1152/ajpheart.1982.242.5.H760. [DOI] [PubMed] [Google Scholar]
- 141. Cobine CA, Callaghan BP, Keef KD. Role of L-type calcium channels and PKC in active tone development in rabbit coronary artery. Am J Physiol Heart Circ Physiol 292: H3079–H3088, 2007. doi: 10.1152/ajpheart.01261.2006. [DOI] [PubMed] [Google Scholar]
- 142. Wenceslau CF, McCarthy CG, Earley S, England SK, Filosa JA, Goulopoulou S, Gutterman DD, Isakson BE, Kanagy NL, Martinez-Lemus LA, Sonkusare SK, Thakore P, Trask AJ, Watts SW, Webb RC. Guidelines for the measurement of vascular function and structure in isolated arteries and veins. Am J Physiol Heart Circ Physiol 321: H77–H111, 2021. doi: 10.1152/ajpheart.01021.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Davis MJ. Physiological role(s) of the vascular myogenic response. Microcirculation 19: 99–114, 2012. doi: 10.1111/j.1549-8719.2011.00131.x. [DOI] [PubMed] [Google Scholar]
- 144. Davis MJ, Hill MA, Kuo L. Local regulation of microvascular perfusion. In: Handbook of Physiology: Microcirculation II, Regulation of Microvascular Blood Flow, edited by Tuma R, Duran W, Ley K.. New York: Elsevier, 2008, p. 1–127. [Google Scholar]
- 145. Davis MJ, Meininger GA. The myogenic response in microvascular networks. In: Mechanotransduction by the Vascular Wall, edited by Rubanyi G. Mt. Kisco, NY: Futura Publishing Company, 1993, p. 37–60. [Google Scholar]
- 146. Johnson PC. The myogenic response. In: Handbook of Physiology. The Cardiovascular System. Vascular Smooth Muscle. Bethesda, MD: American Physiological Society, 1981, p. 409–422. [Google Scholar]
- 147. Folkow B. Description of the myogenic hypothesis. Circ Res 15, Suppl: 279–287, 1964. [PubMed] [Google Scholar]
- 148. Baez S. Bayliss response in the microcirculation. Fed Proc 27: 1410–1415, 1968. [PubMed] [Google Scholar]
- 149. Burton AC. Relation of structure to function of the tissues of the wall of blood vessels. Physiol Rev 34: 619–642, 1954. doi: 10.1152/physrev.1954.34.4.619. [DOI] [PubMed] [Google Scholar]
- 150. Sachs F. Mechanical transduction in biological systems. Crit Rev Biomed Eng 16: 141–169, 1988. [PubMed] [Google Scholar]
- 151. Sachs R. Biophysics of mechanoreception. Membr Biochem 6: 173–195, 1986. doi: 10.3109/09687688609065448. [DOI] [PubMed] [Google Scholar]
- 152. Ingber DE. Mechanosensation through integrins: cells act locally but think globally. Proc Natl Acad Sci USA 100: 1472–1474, 2003. doi: 10.1073/pnas.0530201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol 59: 575–599, 1997. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- 154. Burton AC. Physical principles of circulatory phenomena: the physical equilibria of the heart and blood vessels. In: Handbook of Physiology, edited by Hamilton WF. Washington, DC.: American Physiological Society, 1962, p. 85–106. [Google Scholar]
- 155. Burton AC. On the physical equilibrium of small blood vessels. Am J Physiol 164: 319–329, 1951. doi: 10.1152/ajplegacy.1951.164.2.319. [DOI] [PubMed] [Google Scholar]
- 156. Burrows ME, Johnson PC. Diameter, wall tension, and flow in mesenteric arterioles during autoregulation. Am J Physiol Heart Circ Physiol 241: H829–H837, 1981. doi: 10.1152/ajpheart.1981.241.6.H829. [DOI] [PubMed] [Google Scholar]
- 157. VanBavel E, Mulvany MJ. Role of wall tension in the vasoconstrictor response of cannulated rat mesenteric small arteries. J Physiol 477: 103–115, 1994. doi: 10.1113/jphysiol.1994.sp020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Davis MJ, Davidson J. Force-velocity relationship of myogenically active arterioles. Am J Physiol Heart Circ Physiol 282: H165–H174, 2002. doi: 10.1152/ajpheart.2002.282.1.H165. [DOI] [PubMed] [Google Scholar]
- 159. Koller A, Sun D, Kaley G. Role of shear stress and endothelial prostaglandins in flow- and viscosity-induced dilation of arterioles in vitro. Circ Res 72: 1276–1284, 1993. doi: 10.1161/01.res.72.6.1276. [DOI] [PubMed] [Google Scholar]
- 160. Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res 116: 1448–1461, 2015. doi: 10.1161/CIRCRESAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Pries AR, Secomb TW. Modeling structural adaptation of microcirculation. Microcirculation 15: 753–764, 2008. doi: 10.1080/10739680802229076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zou H, Ratz PH, Hill MA. Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am J Physiol Heart Circ Physiol 269: H1590–H1596, 1995. doi: 10.1152/ajpheart.1995.269.5.H1590. [DOI] [PubMed] [Google Scholar]
- 163. Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science 211: 495–497, 1981. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- 164. Obara K, Uchino M, Koide M, Yamanaka A, Nakayama K. Stretch-induced triphosphorylation of myosin light chain and myogenic tone in canine basilar artery. Eur J Pharmacol 534: 141–151, 2006. doi: 10.1016/j.ejphar.2005.12.086. [DOI] [PubMed] [Google Scholar]
- 165. Kitazawa T, Masuo M, Somlyo AP. G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc Natl Acad Sci USA 88: 9307–9310, 1991. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Moreno-Domínguez A, Colinas O, El-Yazbi A, Walsh EJ, Hill MA, Walsh MP, Cole WC. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol 591: 1235–1250, 2013. doi: 10.1113/jphysiol.2012.243576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Jackson WF, Boerman EM. Regional heterogeneity in the mechanisms of myogenic tone in hamster arterioles. Am J Physiol Heart Circ Physiol 313: H667–H675, 2017. doi: 10.1152/ajpheart.00183.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Harder DR. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ Res 55: 197–202, 1984. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- 169. Meininger GA, Zawieja DC, Falcone JC, Hill MA, Davey JP. Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am J Physiol Heart Circ Physiol 261: H950–H959, 1991. doi: 10.1152/ajpheart.1991.261.3.H950. [DOI] [PubMed] [Google Scholar]
- 170. Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508: 199–209, 1998. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Zou H, Ratz PH, Hill MA. Temporal aspects of Ca2+ and myosin phosphorylation during myogenic and norepinephrine-induced arteriolar constriction. J Vasc Res 37: 556–567, 2000. doi: 10.1159/000054089. [DOI] [PubMed] [Google Scholar]
- 172. Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J 22: 6027–6034, 2003. doi: 10.1093/emboj/cdg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kotecha N, Hill MA. Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am J Physiol Heart Circ Physiol 289: H1326–H1334, 2005. doi: 10.1152/ajpheart.00323.2005. [DOI] [PubMed] [Google Scholar]
- 174. Lagaud G, Gaudreault N, Moore ED, Van Breemen C, Laher I. Pressure-dependent myogenic constriction of cerebral arteries occurs independently of voltage-dependent activation. Am J Physiol Heart Circ Physiol 283: H2187–H2195, 2002. doi: 10.1152/ajpheart.00554.2002. [DOI] [PubMed] [Google Scholar]
- 175. Jackson PA, Duling BR. Myogenic response and wall mechanics of arterioles. Am J Physiol Heart Circ Physiol 257: H1147–H1155, 1989. doi: 10.1152/ajpheart.1989.257.4.H1147. [DOI] [PubMed] [Google Scholar]
- 176. Faber JE, Meininger GA. Selective interaction of α-adrenoceptors with myogenic regulation of microvascular smooth muscle. Am J Physiol Heart Circ Physiol 259: H1126–H1133, 1990. doi: 10.1152/ajpheart.1990.259.4.H1126. [DOI] [PubMed] [Google Scholar]
- 177. Meininger GA, Faber JE. Adrenergic facilitation of myogenic response in skeletal muscle arterioles. Am J Physiol Heart Circ Physiol 260: H1424–H1432, 1991. doi: 10.1152/ajpheart.1991.260.5.H1424. [DOI] [PubMed] [Google Scholar]
- 178. Van Helden DF. Electrophysiology of neuromuscular transmission in guinea-pig mesenteric veins. J Physiol 401: 469–488, 1988. doi: 10.1113/jphysiol.1988.sp017173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hermsmeyer K. Excitation of vascular muscle by norepinephrine. Ann Biomed Eng 11: 567–577, 1983. doi: 10.1007/BF02364086. [DOI] [PubMed] [Google Scholar]
- 180. Wu BN, Luykenaar KD, Brayden JE, Giles WR, Corteling RL, Wiehler WB, Welsh DG. Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 292: H1085–H1094, 2007. doi: 10.1152/ajpheart.00926.2006. [DOI] [PubMed] [Google Scholar]
- 181. Wu X, Davis MJ. Characterization of stretch-activated cation current in coronary smooth muscle cells. Am J Physiol Heart Circ Physiol 280: H1751–H1761, 2001. doi: 10.1152/ajpheart.2001.280.4.H1751. [DOI] [PubMed] [Google Scholar]
- 182. Davis MJ, Donovitz JA, Hood JD. Stretch-activated single-channel and whole-cell currents in vascular smooth muscle cells. Am J Physiol Cell Physiol 262: C1083–C1088, 1992. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- 183. Ohya Y, Adachi N, Nakamura Y, Setoguchi M, Abe I, Fujishima M. Stretch-activated channels in arterial smooth muscle of genetic hypertensive rats. Hypertension 31: 254–258, 1998. doi: 10.1161/01.hyp.31.1.254. [DOI] [PubMed] [Google Scholar]
- 184. Setoguchi M, Ohya Y, Abe I, Fujishima M. Stretch-activated whole-cell currents in smooth muscle cells from mesenteric resistance artery of guinea-pig. J Physiol 501: 343–353, 1997. doi: 10.1111/j.1469-7793.1997.343bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kirber MT, Walsh JV Jr, Singer JJ. Stretch-activated ion channels in smooth muscle: a mechanism for the initiation of stretch-induced contraction. Pflugers Arch 412: 339–345, 1988. doi: 10.1007/BF01907549. [DOI] [PubMed] [Google Scholar]
- 186. Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824, 1997. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 187. Sharif-Naeini R, Witty MF, Séguéla P, Bourque CW. An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci 9: 93–98, 2006. doi: 10.1038/nn1614. [DOI] [PubMed] [Google Scholar]
- 188. Schmidt D, del Mármol J, MacKinnon R. Mechanistic basis for low threshold mechanosensitivity in voltage-dependent K+ channels. Proc Natl Acad Sci USA 109: 10352–10357, 2012. doi: 10.1073/pnas.1204700109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Morris CE, Prikryl EA, Joós B. Mechanosensitive gating of Kv channels. PLoS One 10: e0118335, 2015. doi: 10.1371/journal.pone.0118335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Hardie RC, Franze K. Photomechanical responses in Drosophila photoreceptors. Science 338: 260–263, 2012. doi: 10.1126/science.1222376. [DOI] [PubMed] [Google Scholar]
- 191. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391: 85–100, 1981. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 192. Zakharian E. Recording of ion channel activity in planar lipid bilayer experiments. Methods Mol Biol 998: 109–118, 2013. doi: 10.1007/978-1-62703-351-0_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Morris CE. Mechanosensitive ion channels. J Membr Biol 113: 93–107, 1990. doi: 10.1007/BF01872883. [DOI] [PubMed] [Google Scholar]
- 194. Sachs F. Mechanical transduction by ion channels: how forces reach the channel. Soc Gen Physiol Ser 52: 209–218, 1997. [PubMed] [Google Scholar]
- 195. Ranade SS, Syeda R, Patapoutian A. Mechanically activated ion channels. Neuron 87: 1162–1179, 2015. doi: 10.1016/j.neuron.2015.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Morris CE, Juranka PF. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J 93: 822–833, 2007. doi: 10.1529/biophysj.106.101246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Morris CE, Horn R. Failure to elicit neuronal macroscopic mechanosensitive currents anticipated by single-channel studies. Science 251: 1246–1249, 1991. doi: 10.1126/science.1706535. [DOI] [PubMed] [Google Scholar]
- 198. Sachs F. Mechanical transduction and the dark energy of biology. Biophys J 114: 3–9, 2018. doi: 10.1016/j.bpj.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Morris CE. Why are so many ion channels mechanosensitive? In: Cell Physiology Source Book, edited by Sperelakis N. Amsterdam: Elsevier, 2011. [Google Scholar]
- 200. Sachs F. Stretch-activated ion channels: what are they? Physiology (Bethesda) 25: 50–56, 2010. doi: 10.1152/physiol.00042.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Morris CE. Mechanoprotection of the plasma membrane in neurons and other non-erythroid cells by the spectrin-based membrane skeleton. Cell Mol Biol Lett 6: 703–720, 2001. [PubMed] [Google Scholar]
- 202. Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA 101: 396–401, 2004. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, Martinac B, Patapoutian A. Piezo1 channels are inherently mechanosensitive. Cell Rep 17: 1739–1746, 2016. doi: 10.1016/j.celrep.2016.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ruknudin A, Song MJ, Sachs F. The ultrastructure of patch-clamped membranes: a study using high voltage electron microscopy. J Cell Biol 112: 125–134, 1991. doi: 10.1083/jcb.112.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Terakawa S, Nakayama T. Are axoplasmic microtubules necessary for membrane excitation? J Membr Biol 85: 65–77, 1985. doi: 10.1007/BF01872006. [DOI] [PubMed] [Google Scholar]
- 206. Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev 81: 685–740, 2001. doi: 10.1152/physrev.2001.81.2.685. [DOI] [PubMed] [Google Scholar]
- 207. Rossier BC. Mechanosensitivity of the epithelial sodium channel (ENaC): controversy or pseudocontroversy? J Gen Physiol 112: 95–96, 1998. doi: 10.1085/jgp.112.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Arnadóttir J, Chalfie M. Eukaryotic mechanosensitive channels. Annu Rev Biophys 39: 111–137, 2010. doi: 10.1146/annurev.biophys.37.032807.125836. [DOI] [PubMed] [Google Scholar]
- 209. Sukharev SI, Blount P, Martinac B, Blattner FR, Kung C. A large-conductance mechanosensitive channel in E. coli encoded by MscL alone. Nature 368: 265–268, 1994. doi: 10.1038/368265a0. [DOI] [PubMed] [Google Scholar]
- 210. Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science 282: 2220–2226, 1998. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- 211. Sukharev SI, Sigurdson WJ, Kung C, Sachs F. Energetic and spatial parameters for gating of the bacterial large conductance mechanosensitive channel, MscL. J Gen Physiol 113: 525–540, 1999. doi: 10.1085/jgp.113.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Okada K, Moe PC, Blount P. Functional design of bacterial mechanosensitive channels. Comparisons and contrasts illuminated by random mutagenesis. J Biol Chem 277: 27682–27688, 2002. doi: 10.1074/jbc.M202497200. [DOI] [PubMed] [Google Scholar]
- 213. Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci STKE 2002: PE6, 2002., doi: 10.1126/stke.2002.119.pe6. [DOI] [PubMed] [Google Scholar]
- 214. Poole K, Herget R, Lapatsina L, Ngo HD, Lewin GR. Tuning Piezo ion channels to detect molecular-scale movements relevant for fine touch. Nat Commun 5: 3520, 2014. doi: 10.1038/ncomms4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Zhang W, Cheng LE, Kittelmann M, Li J, Petkovic M, Cheng T, Jin P, Guo Z, Göpfert MC, Jan LY, Jan YN. Ankyrin repeats convey force to gate the NOMPC mechanotransduction channel. Cell 162: 1391–1403, 2015. doi: 10.1016/j.cell.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Spagnoli C, Beyder A, Besch S, Sachs F. Atomic force microscopy analysis of cell volume regulation. Phys Rev E Stat Nonlin Soft Matter Phys 78: 031916, 2008. doi: 10.1103/PhysRevE.78.031916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Orr AW, Helmke BP, Blackman BR, Schwartz MA. Mechanisms of mechanotransduction. Dev Cell 10: 11–20, 2006. doi: 10.1016/j.devcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 218. Teng J, Loukin S, Anishkin A, Kung C. The force-from-lipid (FFL) principle of mechanosensitivity, at large and in elements. Pflugers Arch 467: 27–37, 2015. doi: 10.1007/s00424-014-1530-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Gillespie PG, Walker RG. Molecular basis of mechanosensory transduction. Nature 413: 194–202, 2001. doi: 10.1038/35093011. [DOI] [PubMed] [Google Scholar]
- 220. Gullingsrud J, Schulten K. Lipid bilayer pressure profiles and mechanosensitive channel gating. Biophys J 86: 3496–3509, 2004. doi: 10.1529/biophysj.103.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Wiggins P, Phillips R. Membrane-protein interactions in mechanosensitive channels. Biophys J 88: 880–902, 2005. doi: 10.1529/biophysj.104.047431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Cox CD, Bavi N, Martinac B. Biophysical principles of ion-channel-mediated mechanosensory transduction. Cell Rep 29: 1–12, 2019. doi: 10.1016/j.celrep.2019.08.075. [DOI] [PubMed] [Google Scholar]
- 223. Vásquez V, Sotomayor M, Cordero-Morales J, Schulten K, Perozo E. A structural mechanism for MscS gating in lipid bilayers. Science 321: 1210–1214, 2008. doi: 10.1126/science.1159674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Brohawn SG, Su Z, MacKinnon R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci USA 111: 3614–3619, 2014. doi: 10.1073/pnas.1320768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Perozo E, Cortes DM, Sompornpisut P, Kloda A, Martinac B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature 418: 942–948, 2002. doi: 10.1038/nature00992. [DOI] [PubMed] [Google Scholar]
- 226. Perozo E, Kloda A, Cortes DM, Martinac B. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat Struct Biol 9: 696–703, 2002. doi: 10.1038/nsb827. [DOI] [PubMed] [Google Scholar]
- 227. Schmidt D, Jiang QX, MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444: 775–779, 2006. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 228. Li Q, Wanderling S, Sompornpisut P, Perozo E. Structural basis of lipid-driven conformational transitions in the KvAP voltage-sensing domain. Nat Struct Mol Biol 21: 160–166, 2014. doi: 10.1038/nsmb.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Anishkin A, Loukin SH, Teng J, Kung C. Feeling the hidden mechanical forces in lipid bilayer is an original sense. Proc Natl Acad Sci USA 111: 7898–7905, 2014. doi: 10.1073/pnas.1313364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Flegler VJ, Rasmussen A, Borbil K, Boten L, Chen HA, Deinlein H, Halang J, Hellmanzik K, Löffler J, Schmidt V, Makbul C, Kraft C, Hedrich R, Rasmussen T, Böttcher B. Mechanosensitive channel gating by delipidation. Proc Natl Acad Sci USA 118: e2107095118, 2021. doi: 10.1073/pnas.2107095118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Brohawn SG, Campbell EB, MacKinnon R. Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 516: 126–130, 2014. doi: 10.1038/nature14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Hayakawa K, Tatsumi H, Sokabe M. Actin stress fibers transmit and focus force to activate mechanosensitive channels. J Cell Sci 121: 496–503, 2008. doi: 10.1242/jcs.022053. [DOI] [PubMed] [Google Scholar]
- 233. Li Fraine S, Patel A, Duprat F, Sharif-Naeini R. Dynamic regulation of TREK1 gating by Polycystin 2 via a Filamin A-mediated cytoskeletal mechanism. Sci Rep 7: 17403, 2017. doi: 10.1038/s41598-017-16540-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Poole K, Moroni M, Lewin GR. Sensory mechanotransduction at membrane-matrix interfaces. Pflugers Arch 467: 121–132, 2015. doi: 10.1007/s00424-014-1563-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Chuang YC, Chen CC. Force from filaments: the role of the cytoskeleton and extracellular matrix in the gating of mechanosensitive channels. Front Cell Dev Biol 10: 886048, 2022. doi: 10.3389/fcell.2022.886048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Denk W, Holt JR, Shepherd GM, Corey DP. Calcium imaging of single stereocilia in hair cells: localization of transduction channels at both ends of tip links. Neuron 15: 1311–1321, 1995. doi: 10.1016/0896-6273(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 237. Gu CX, Juranka PF, Morris CE. Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys J 80: 2678–2693, 2001. doi: 10.1016/S0006-3495(01)76237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Hudspeth AJ. How hearing happens. Neuron 19: 947–950, 1997. doi: 10.1016/S0896-6273(00)80385-2. [DOI] [PubMed] [Google Scholar]
- 239. Zheng W, Holt JR. The mechanosensory transduction machinery in inner ear hair cells. Annu Rev Biophys 50: 31–51, 2021. doi: 10.1146/annurev-biophys-062420-081842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lauritzen I, Chemin J, Honoré E, Jodar M, Guy N, Lazdunski M, Jane Patel A. Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cytoskeleton. EMBO Rep 6: 642–648, 2005. doi: 10.1038/sj.embor.7400449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2: 138–145, 2001. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 242. Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, Jodar M, Moro C, Offermanns S, Feng Y, Demolombe S, Patel A, Honoré E. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep 13: 1161–1171, 2015. doi: 10.1016/j.celrep.2015.09.072. [DOI] [PubMed] [Google Scholar]
- 243. Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJ, Honoré E. Polycystin-1 and -2 dosage regulates pressure sensing. Cell 139: 587–596, 2009. doi: 10.1016/j.cell.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 244. Nigro EA, Distefano G, Chiaravalli M, Matafora V, Castelli M, Pesenti Gritti A, Bachi A, Boletta A. Polycystin-1 regulates actomyosin contraction and the cellular response to extracellular stiffness. Sci Rep 9: 16640, 2019. doi: 10.1038/s41598-019-53061-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W, Jaggar JH. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res 111: 1027–1036, 2012. doi: 10.1161/CIRCRESAHA.112.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Park KS, Kim Y, Lee YH, Earm YE, Ho WK. Mechanosensitive cation channels in arterial smooth muscle cells are activated by diacylglycerol and inhibited by phospholipase C inhibitor. Circ Res 93: 557–564, 2003. doi: 10.1161/01.RES.0000093204.25499.83. [DOI] [PubMed] [Google Scholar]
- 247. Wellner MC, Isenberg G. Stretch effects on whole-cell currents of guinea-pig urinary bladder myocytes. J Physiol 480: 439–448, 1994. doi: 10.1113/jphysiol.1994.sp020373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Scotland RS, Chauhan S, Davis C, De Felipe C, Hunt S, Kabir J, Kotsonis P, Oh U, Ahluwalia A. Vanilloid receptor TRPV1, sensory C-fibers, and vascular autoregulation: a novel mechanism involved in myogenic constriction. Circ Res 95: 1027–1034, 2004. doi: 10.1161/01.RES.0000148633.93110.24. [DOI] [PubMed] [Google Scholar]
- 249. Anfinogenova Y, Brett SE, Walsh MP, Harraz OF, Welsh DG. Do TRPC-like currents and G protein-coupled receptors interact to facilitate myogenic tone development? Am J Physiol Heart Circ Physiol 301: H1378–H1388, 2011. doi: 10.1152/ajpheart.00460.2011. [DOI] [PubMed] [Google Scholar]
- 250. Fanchaouy M, Bychkov R, Meister JJ, Beny JL. Stretch-elicited calcium responses in the intact mouse thoracic aorta. Cell Calcium 41: 41–50, 2007. doi: 10.1016/j.ceca.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 251. Ducret T, El Arrouchi J, Courtois A, Quignard JF, Marthan R, Savineau JP. Stretch-activated channels in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Cell Calcium 48: 251–259, 2010. doi: 10.1016/j.ceca.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 252. Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA 103: 16586–16591, 2006. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Takenaka T, Suzuki H, Okada H, Hayashi K, Kanno Y, Saruta T. Mechanosensitive cation channels mediate afferent arteriolar myogenic constriction in the isolated rat kidney. J Physiol 511: 245–253, 1998. doi: 10.1111/j.1469-7793.1998.245bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Li GR, Baumgarten CM. Modulation of cardiac Na+ current by gadolinium, a blocker of stretch-induced arrhythmias. Am J Physiol Heart Circ Physiol 280: H272–H279, 2001. doi: 10.1152/ajpheart.2001.280.1.H272. [DOI] [PubMed] [Google Scholar]
- 255. Shim AL, Kamkin AG, Kamkina OV, Kazanskii VE, Mitrokhin VM, Bilichenko AS, Filatova TS, Abramochkin DV. Gadolinium as an inhibitor of ionic currents in isolated rat ventricular cardiomyocytes. Bull Exp Biol Med 168: 187–192, 2019. doi: 10.1007/s10517-019-04672-0. [DOI] [PubMed] [Google Scholar]
- 256. Lacampagne A, Gannier F, Argibay J, Garnier D, Le Guennec JY. The stretch-activated ion channel blocker gadolinium also blocks L-type calcium channels in isolated ventricular myocytes of the guinea-pig. Biochim Biophys Acta 1191: 205–208, 1994. doi: 10.1016/0005-2736(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 257. Biagi BA, Enyeart JJ. Gadolinium blocks low- and high-threshold calcium currents in pituitary cells. Am J Physiol Cell Physiol 259: C515–C520, 1990. doi: 10.1152/ajpcell.1990.259.3.C515. [DOI] [PubMed] [Google Scholar]
- 258. Beedle AM, Hamid J, Zamponi GW. Inhibition of transiently expressed low- and high-voltage-activated calcium channels by trivalent metal cations. J Membr Biol 187: 225–238, 2002. doi: 10.1007/s00232-001-0166-2. [DOI] [PubMed] [Google Scholar]
- 259. Elinder F, Århem P. The modulatory site for the action of gadolinium on surface charges and channel gating. Biophys J 67: 84–90, 1994. doi: 10.1016/S0006-3495(94)80457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Harraz OF, Klug NR, Senatore AJ, Hill-Eubanks DC, Nelson MT. Piezo1 is a mechanosensor channel in central nervous system capillaries. Circ Res 130: 1531–1546, 2022. doi: 10.1161/CIRCRESAHA.122.320827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Imagawa T, Smith JS, Coronado R, Campbell KP. Purified ryanodine receptor from skeletal muscle sarcoplasmic reticulum is the Ca2+-permeable pore of the calcium release channel. J Biol Chem 262: 16636–16643, 1987. doi: 10.1016/S0021-9258(18)49303-9. [DOI] [PubMed] [Google Scholar]
- 262. Dedkova EN, Ji X, Lipsius SL, Blatter LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am J Physiol Cell Physiol 286: C406–C415, 2004. doi: 10.1152/ajpcell.00155.2003. [DOI] [PubMed] [Google Scholar]
- 263. Suchyna TM, Johnson JH, Hamer K, Leykam JF, Gage DA, Clemo HF, Baumgarten CM, Sachs F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J Gen Physiol 115: 583–598, 2000. doi: 10.1085/jgp.115.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Oswald RE, Suchyna TM, McFeeters R, Gottlieb P, Sachs F. Solution structure of peptide toxins that block mechanosensitive ion channels. J Biol Chem 277: 34443–34450, 2002. doi: 10.1074/jbc.M202715200. [DOI] [PubMed] [Google Scholar]
- 265. Lee HA, Baek EB, Park KS, Jung HJ, Kim JI, Kim SJ, Earm YE. Mechanosensitive nonselective cation channel facilitation by endothelin-1 is regulated by protein kinase C in arterial myocytes. Cardiovasc Res 76: 224–235, 2007. doi: 10.1016/j.cardiores.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 266. Bowman CL, Gottlieb PA, Suchyna TM, Murphy YK, Sachs F. Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: history, properties, mechanisms and pharmacology. Toxicon 49: 249–270, 2007. doi: 10.1016/j.toxicon.2006.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Louis M, Zanou N, Van Schoor M, Gailly P. TRPC1 regulates skeletal myoblast migration and differentiation. J Cell Sci 121: 3951–3959, 2008. doi: 10.1242/jcs.037218. [DOI] [PubMed] [Google Scholar]
- 268. Pan NC, Zhang T, Hu S, Liu C, Wang Y. Fast desensitization of acetylcholine receptors induced by a spider toxin. Channels (Austin) 15: 507–515, 2021. doi: 10.1080/19336950.2021.1961459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Morris CE. Why are so many ion channels mechanosensitive? In: Cell Physiology Sourcebook, edited by Sperelakis N Amsterdam: Elsevier, 2011, p. 493–505. [Google Scholar]
- 270. Morris CE. Voltage-gated channel mechanosensitivity: fact or friction? Front Physiol 2: 25, 2011. doi: 10.3389/fphys.2011.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Morris CE. Pacemaker, potassium, calcium, sodium: stretch modulation of voltage-gated channels. In: Cardiac Mechano-Electric Feedback and Arrhythmias, edited by Kohl P, Sachs F, Franz M.. Philadelphia, PA: Elsevier Saunders, 2011, p. 42–49 [Google Scholar]
- 272. Suchyna TM, Tape SE, Koeppe RE 2nd, Andersen OS, Sachs F, Gottlieb PA. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 430: 235–240, 2004. doi: 10.1038/nature02743. [DOI] [PubMed] [Google Scholar]
- 273. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330: 55–60, 2010. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, Li R, Gao N, Xiao B, Yang M. Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 527: 64–69, 2015. doi: 10.1038/nature15247. [DOI] [PubMed] [Google Scholar]
- 275. Guo YR, MacKinnon R. Structure-based membrane dome mechanism for Piezo mechanosensitivity. Elife 6: e33660, 2017. doi: 10.7554/eLife.33660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Haselwandter CA, MacKinnon R. Piezo’s membrane footprint and its contribution to mechanosensitivity. Elife 7: e41968, 2018. doi: 10.7554/eLife.41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Yang X, Lin C, Chen X, Li S, Li X, Xiao B. Structure deformation and curvature sensing of PIEZO1 in lipid membranes. Nature 604: 377–383, 2022. doi: 10.1038/s41586-022-04574-8. [DOI] [PubMed] [Google Scholar]
- 278. Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, Montal M, Patapoutian A. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483: 176–181, 2012. doi: 10.1038/nature10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Gnanasambandam R, Gottlieb PA, Sachs F. The kinetics and the permeation properties of Piezo channels. Curr Top Membr 79: 275–307, 2017. doi: 10.1016/bs.ctm.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 280. Bae C, Sachs F, Gottlieb PA. The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry 50: 6295–6300, 2011. doi: 10.1021/bi200770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Syeda R, Xu J, Dubin AE, Coste B, Mathur J, Huynh T, Matzen J, Lao J, Tully DC, Engels IH, Petrassi HM, Schumacher AM, Montal M, Bandell M, Patapoutian A. Chemical activation of the mechanotransduction channel Piezo1. Elife 4: e07369, 2015. doi: 10.7554/eLife.07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Evans EL, Cuthbertson K, Endesh N, Rode B, Blythe NM, Hyman AJ, Hall SJ, Gaunt HJ, Ludlow MJ, Foster R, Beech DJ. Yoda1 analogue (Dooku1) which antagonizes Yoda1-evoked activation of Piezo1 and aortic relaxation. Br J Pharmacol 175: 1744–1759, 2018. doi: 10.1111/bph.14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Dela Paz NG, Frangos JA. Yoda1-induced phosphorylation of Akt and ERK1/2 does not require Piezo1 activation. Biochem Biophys Res Commun 497: 220–225, 2018. doi: 10.1016/j.bbrc.2018.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Coste B, Murthy SE, Mathur J, Schmidt M, Mechioukhi Y, Delmas P, Patapoutian A. Piezo1 ion channel pore properties are dictated by C-terminal region. Nat Commun 6: 7223, 2015. doi: 10.1038/ncomms8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Borbiro I, Badheka D, Rohacs T. Activation of TRPV1 channels inhibits mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Sci Signal 8: ra15, 2015. doi: 10.1126/scisignal.2005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Wu J, Lewis AH, Grandl J. Touch, tension, and transduction—the function and regulation of Piezo ion channels. Trends Biochem Sci 42: 57–71, 2017. doi: 10.1016/j.tibs.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Bae C, Gnanasambandam R, Nicolai C, Sachs F, Gottlieb PA. Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc Natl Acad Sci USA 110: E1162–E1168, 2013. doi: 10.1073/pnas.1219777110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Albuisson J, Murthy SE, Bandell M, Coste B, Louis-Dit-Picard H, Mathur J, Fénéant-Thibault M, Tertian G, de Jaureguiberry JP, Syfuss PY, Cahalan S, Garçon L, Toutain F, Simon Rohrlich P, Delaunay J, Picard V, Jeunemaitre X, Patapoutian A. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat Commun 4: 1884, 2013. doi: 10.1038/ncomms2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. McMillin MJ, Beck AE, Chong JX, Shively KM, Buckingham KJ, Gildersleeve HI, et al. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am J Hum Genet 94: 734–744, 2014. doi: 10.1016/j.ajhg.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Lukacs V, Mathur J, Mao R, Bayrak-Toydemir P, Procter M, Cahalan SM, Kim HJ, Bandell M, Longo N, Day RW, Stevenson DA, Patapoutian A, Krock BL. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat Commun 6: 8329, 2015. doi: 10.1038/ncomms9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Fotiou E, Martin-Almedina S, Simpson MA, Lin S, Gordon K, Brice G, Atton G, Jeffery I, Rees DC, Mignot C, Vogt J, Homfray T, Snyder MP, Rockson SG, Jeffery S, Mortimer PS, Mansour S, Ostergaard P. Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat Commun 6: 8085, 2015. doi: 10.1038/ncomms9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Choi D, Park E, Jung E, Cha B, Lee S, Yu J, Kim PM, Lee S, Hong YJ, Koh CJ, Cho CW, Wu Y, Jeon NL, Wong AK, Shin L, Kumar SR, Bermejo-Moreno I, Srinivasan RS, Cho IT, Hong YK. Piezo1 incorporates mechanical force signals to genetic program that governs lymphatic valve development and maintenance. JCI Insight 4: e125068, 2019. doi: 10.1172/jci.insight.125068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Woo SH, Lukacs V, de Nooij JC, Zaytseva D, Criddle CR, Francisco A, Jessell TM, Wilkinson KA, Patapoutian A. Piezo2 is the principal mechanotransduction channel for proprioception. Nat Neurosci 18: 1756–1762, 2015. doi: 10.1038/nn.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Woo SH, Ranade S, Weyer AD, Dubin AE, Baba Y, Qiu Z, Petrus M, Miyamoto T, Reddy K, Lumpkin EA, Stucky CL, Patapoutian A. Piezo2 is required for Merkel-cell mechanotransduction. Nature 509: 622–626, 2014. doi: 10.1038/nature13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Schrenk-Siemens K, Wende H, Prato V, Song K, Rostock C, Loewer A, Utikal J, Lewin GR, Lechner SG, Siemens J. PIEZO2 is required for mechanotransduction in human stem cell-derived touch receptors. Nat Neurosci 18: 10–16, 2015. doi: 10.1038/nn.3894. [DOI] [PubMed] [Google Scholar]
- 296. Nonomura K, Woo SH, Chang RB, Gillich A, Qiu Z, Francisco AG, Ranade SS, Liberles SD, Patapoutian A. Piezo2 senses airway stretch and mediates lung inflation-induced apnoea. Nature 541: 176–181, 2017. doi: 10.1038/nature20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA 111: 10347–10352, 2014. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Nonomura K, Lukacs V, Sweet DT, Goddard LM, Kanie A, Whitwam T, Ranade SS, Fujimori T, Kahn ML, Patapoutian A. Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc Natl Acad Sci USA 115: 12817–12822, 2018. doi: 10.1073/pnas.1817070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hägerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, Calmes JM, Adams RH, Mäkinen T, Kiefer F, Kwak BR, Petrova TV. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell 22: 430–445, 2012. doi: 10.1016/j.devcel.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 300. Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest 126: 4527–4536, 2016. doi: 10.1172/JCI87343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol Heart Circ Physiol 269: H2155–H2161, 1995. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- 302. Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. ATP: the red blood cell link to NO and local control of the pulmonary circulation. Am J Physiol Heart Physiol 271: H2717–H2722, 1996. doi: 10.1152/ajpheart.1996.271.6.H2717. [DOI] [PubMed] [Google Scholar]
- 303. Skalak R, Branemark PI. Deformation of red blood cells in capillaries. Science 164: 717–719, 1969. doi: 10.1126/science.164.3880.717. [DOI] [PubMed] [Google Scholar]
- 304. Chien S. Red cell deformability and its relevance to blood flow. Annu Rev Physiol 49: 177–192, 1987. doi: 10.1146/annurev.ph.49.030187.001141. [DOI] [PubMed] [Google Scholar]
- 305. Miron TR, Flood ED, Tykocki NR, Thompson JM, Watts SW. Identification of Piezo1 channels in perivascular adipose tissue (PVAT) and their potential role in vascular function. Pharmacol Res 175: 105995, 2022. doi: 10.1016/j.phrs.2021.105995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Morley LC, Shi J, Gaunt HJ, Hyman AJ, Webster PJ, Williams C, Forbes K, Walker JJ, Simpson NA, Beech DJ. Piezo1 channels are mechanosensors in human fetoplacental endothelial cells. Mol Hum Reprod 24: 510–520, 2018. doi: 10.1093/molehr/gay033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Ferrari LF, Bogen O, Green P, Levine JD. Contribution of Piezo2 to endothelium-dependent pain. Mol Pain 11: 65, 2015. doi: 10.1186/s12990-015-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Yang H, Liu C, Zhou RM, Yao J, Li XM, Shen Y, Cheng H, Yuan J, Yan B, Jiang Q. Piezo2 protein: a novel regulator of tumor angiogenesis and hyperpermeability. Oncotarget 7: 44630–44643, 2016. doi: 10.18632/oncotarget.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Rode B, Shi J, Endesh N, Drinkhill MJ, Webster PJ, Lotteau SJ, Bailey MA, Yuldasheva NY, Ludlow MJ, Cubbon RM, Li J, Futers TS, Morley L, Gaunt HJ, Marszalek K, Viswambharan H, Cuthbertson K, Baxter PD, Foster R, Sukumar P, Weightman A, Calaghan SC, Wheatcroft SB, Kearney MT, Beech DJ. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat Commun 8: 350, 2017. doi: 10.1038/s41467-017-00429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Dubin AE, Murthy S, Lewis AH, Brosse L, Cahalan SM, Grandl J, Coste B, Patapoutian A. Endogenous Piezo1 can confound mechanically activated channel identification and characterization. Neuron 94: 266–270.e3, 2017. doi: 10.1016/j.neuron.2017.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Zhang J, Yuan H, Yao X, Chen S. Endogenous ion channels expressed in human embryonic kidney (HEK-293) cells. Pflugers Arch 474: 665–680, 2022. doi: 10.1007/s00424-022-02700-z. [DOI] [PubMed] [Google Scholar]
- 312. Shi J, Hyman AJ, De Vecchis D, Chong J, Lichtenstein L, Futers TS, Rouahi M, Salvayre AN, Auge N, Kalli AC, Beech DJ. Sphingomyelinase disables inactivation in endogenous PIEZO1 channels. Cell Rep 33: 108225, 2020. doi: 10.1016/j.celrep.2020.108225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, Phillips SA, Levitan I. Inwardly rectifying K+ channels are major contributors to flow-induced vasodilatation in resistance arteries. J Physiol 595: 2339–2364, 2017. doi: 10.1113/JP273255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Hoger JH, Ilyin VI, Forsyth S, Hoger A. Shear stress regulates the endothelial Kir2.1 ion channel. Proc Natl Acad Sci USA 99: 7780–7785, 2002. doi: 10.1073/pnas.102184999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Bartoli F, Debant M, Chuntharpursat-Bon E, Evans EL, Musialowski KE, Parsonage G, Morley LC, Futers TS, Sukumar P, Bowen TS, Kearney MT, Lichtenstein L, Roberts LD, Beech DJ. Endothelial Piezo1 sustains muscle capillary density and contributes to physical activity. J Clin Invest 132: e141775, 2022. doi: 10.1172/JCI141775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Iring A, Jin YJ, Albarrán-Juárez J, Siragusa M, Wang S, Dancs PT, Nakayama A, Tonack S, Chen M, Künne C, Sokol AM, Günther S, Martínez A, Fleming I, Wettschureck N, Graumann J, Weinstein LS, Offermanns S. Shear stress-induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. J Clin Invest 129: 2775–2791, 2019. doi: 10.1172/JCI123825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Beech DJ, Kalli AC. Force sensing by Piezo channels in cardiovascular health and disease. Arterioscler Thromb Vasc Biol 39: 2228–2239, 2019. doi: 10.1161/ATVBAHA.119.313348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Lhomme A, Gilbert G, Pele T, Deweirdt J, Henrion D, Baudrimont I, Campagnac M, Marthan R, Guibert C, Ducret T, Savineau JP, Quignard JF. Stretch-activated Piezo1 channel in endothelial cells relaxes mouse intrapulmonary arteries. Am J Respir Cell Mol Biol 60: 650–658, 2019. doi: 10.1165/rcmb.2018-0197OC. [DOI] [PubMed] [Google Scholar]
- 319. John L, Ko NL, Gokin A, Gokina N, Mandalà M, Osol G. The Piezo1 cation channel mediates uterine artery shear stress mechanotransduction and vasodilation during rat pregnancy. Am J Physiol Heart Circ Physiol 315: H1019–H1026, 2018. doi: 10.1152/ajpheart.00103.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Swain SM, Liddle RA. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J Biol Chem 296: 100171, 2021. doi: 10.1074/jbc.RA120.015059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Peyronnet R, Martins JR, Duprat F, Demolombe S, Arhatte M, Jodar M, Tauc M, Duranton C, Paulais M, Teulon J, Honoré E, Patel A. Piezo1-dependent stretch-activated channels are inhibited by Polycystin-2 in renal tubular epithelial cells. EMBO Rep 14: 1143–1148, 2013. doi: 10.1038/embor.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Dela Paz NG, Frangos JA. Rapid flow-induced activation of Galphaq/11 is independent of Piezo1 activation. Am J Physiol Cell Physiol 316: C741–C752, 2019. doi: 10.1152/ajpcell.00215.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Douguet D, Patel A, Xu A, Vanhoutte PM, Honoré E. Piezo ion channels in cardiovascular mechanobiology. Trends Pharmacol Sci 40: 956–970, 2019. doi: 10.1016/j.tips.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 324. Albarrán-Juárez J, Iring A, Wang S, Joseph S, Grimm M, Strilic B, Wettschureck N, Althoff TF, Offermanns S. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J Exp Med 215: 2655–2672, 2018. doi: 10.1084/jem.20180483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Friedrich EE, Hong Z, Xiong S, Zhong M, Di A, Rehman J, Komarova YA, Malik AB. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc Natl Acad Sci USA 116: 12980–12985, 2019. doi: 10.1073/pnas.1902165116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Bounoutas A, Chalfie M. Touch sensitivity in Caenorhabditis elegans. Pflugers Arch 454: 691–702, 2007. doi: 10.1007/s00424-006-0187-x. [DOI] [PubMed] [Google Scholar]
- 327. Syntichaki P, Tavernarakis N. Genetic models of mechanotransduction: the nematode Caenorhabditis elegans. Physiol Rev 84: 1097–1153, 2004. doi: 10.1152/physrev.00043.2003. [DOI] [PubMed] [Google Scholar]
- 328. Palmer LG, Frindt G. Amiloride-sensitive Na channels from the apical membrane of the rat cortical collecting tubule. Proc Natl Acad Sci USA 83: 2767–2770, 1986. doi: 10.1073/pnas.83.8.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature 361: 467–470, 1993. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 330. Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P. Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett 318: 95–99, 1993. doi: 10.1016/0014-5793(93)81336-X. [DOI] [PubMed] [Google Scholar]
- 331. Kellenberger S, Schild L. International Union of Basic and Clinical Pharmacology. XCI. structure, function, and pharmacology of acid-sensing ion channels and the epithelial Na+ channel. Pharmacol Rev 67: 1–35, 2015. doi: 10.1124/pr.114.009225. [DOI] [PubMed] [Google Scholar]
- 332. Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem 270: 27411–27414, 1995. doi: 10.1074/jbc.270.46.27411. [DOI] [PubMed] [Google Scholar]
- 333. Giraldez T, Rojas P, Jou J, Flores C, Alvarez de la Rosa D. The epithelial sodium channel delta-subunit: new notes for an old song. Am J Physiol Renal Physiol 303: F328–F338, 2012. doi: 10.1152/ajprenal.00116.2012. [DOI] [PubMed] [Google Scholar]
- 334. Canessa CM, Merillat AM, Rossier BC. Membrane topology of the epithelial sodium channel in intact cells. Am J Physiol Cell Physiol 267: C1682–C1690, 1994. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 335. Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na+ Channel regulation by extracellular and intracellular factors. Annu Rev Physiol 80: 263–281, 2018. doi: 10.1146/annurev-physiol-021317-121143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 337. Palmer LG, Frindt G. Gating of Na channels in the rat cortical collecting tubule: effects of voltage and membrane stretch. J Gen Physiol 107: 35–45, 1996. doi: 10.1085/jgp.107.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82: 735–767, 2002. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 339. Awayda MS, Ismailov II, Berdiev BK, Benos DJ. A cloned renal epithelial Na+ channel protein displays stretch activation in planar lipid bilayers. Am J Physiol Cell Physiol 268: C1450–1459, 1995. doi: 10.1152/ajpcell.1995.268.6.C1450. [DOI] [PubMed] [Google Scholar]
- 340. Bonny O, Chraibi A, Loffing J, Jaeger NF, Gründer S, Horisberger JD, Rossier BC. Functional expression of a pseudohypoaldosteronism type I mutated epithelial Na+ channel lacking the pore-forming region of its alpha subunit. J Clin Invest 104: 967–974, 1999. doi: 10.1172/JCI6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Chraïbi A, Horisberger JD. Na self inhibition of human epithelial Na channel: temperature dependence and effect of extracellular proteases. J Gen Physiol 120: 133–145, 2002. doi: 10.1085/jgp.20028612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Fronius M, Clauss WG. Mechano-sensitivity of ENaC: may the (shear) force be with you. Pflugers Arch 455: 775–785, 2008. doi: 10.1007/s00424-007-0332-1. [DOI] [PubMed] [Google Scholar]
- 343. Ismailov II, Berdiev BK, Shlyonsky VG, Benos DJ. Mechanosensitivity of an epithelial Na+ channel in planar lipid bilayers: release from Ca2+ block. Biophys J 72: 1182–1192, 1997. doi: 10.1016/S0006-3495(97)78766-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Benos DJ. Sensing tension: recognizing ENaC as a stretch sensor. Hypertension 44: 616–617, 2004. doi: 10.1161/01.HYP.0000144467.43205.ed. [DOI] [PubMed] [Google Scholar]
- 345. Kizer N, Guo XL, Hruska K. Reconstitution of stretch-activated cation channels by expression of the alpha-subunit of the epithelial sodium channel cloned from osteoblasts. Proc Natl Acad Sci USA 94: 1013–1018, 1997. doi: 10.1073/pnas.94.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Ji HL, Fuller CM, Benos DJ. Osmotic pressure regulates alpha beta gamma-rENaC expressed in Xenopus oocytes. Am J Physiol Cell Physiol 275: C1182–C1190, 1998. doi: 10.1152/ajpcell.1998.275.5.C1182. [DOI] [PubMed] [Google Scholar]
- 347. Ma HP, Li L, Zhou ZH, Eaton DC, Warnock DG. ATP masks stretch activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 282: F501–F505, 2002. doi: 10.1152/ajprenal.00147.2001. [DOI] [PubMed] [Google Scholar]
- 348. Satlin LM, Sheng S, Woda CB, Kleyman TR. Epithelial Na+ channels are regulated by flow. Am J Physiol Renal Physiol 280: F1010–F1018, 2001. doi: 10.1152/ajprenal.2001.280.6.F1010. [DOI] [PubMed] [Google Scholar]
- 349. Carattino MD, Sheng S, Kleyman TR. Epithelial Na+ channels are activated by laminar shear stress. J Biol Chem 279: 4120–4126, 2004. doi: 10.1074/jbc.M311783200. [DOI] [PubMed] [Google Scholar]
- 350. Althaus M, Bogdan R, Clauss WG, Fronius M. Mechano-sensitivity of epithelial sodium channels (ENaCs): laminar shear stress increases ion channel open probability. FASEB J 21: 2389–2399, 2007. doi: 10.1096/fj.06-7694com. [DOI] [PubMed] [Google Scholar]
- 351. Abi-Antoun T, Shi S, Tolino LA, Kleyman TR, Carattino MD. Second transmembrane domain modulates epithelial sodium channel gating in response to shear stress. Am J Physiol Renal Physiol 300: F1089–F1095, 2011. doi: 10.1152/ajprenal.00610.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Knoepp F, Ashley Z, Barth D, Baldin JP, Jennings M, Kazantseva M, Saw EL, Katare R, Alvarez de la Rosa D, Weissmann N, Fronius M. Shear force sensing of epithelial Na+ channel (ENaC) relies on N-glycosylated asparagines in the palm and knuckle domains of alphaENaC. Proc Natl Acad Sci USA 117: 717–726, 2020. doi: 10.1073/pnas.1911243117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Jernigan NL, Drummond HA. Vascular ENaC proteins are required for renal myogenic constriction. Am J Physiol Renal Physiol 289: F891–F901, 2005. doi: 10.1152/ajprenal.00019.2005. [DOI] [PubMed] [Google Scholar]
- 354. Oyabe A, Masumoto N, Ueta K, Nakayama K. Amiloride-sensitive pressure-induced myogenic contraction in rat cerebral artery. Fundam Clin Pharmacol 14: 369–377, 2000. doi: 10.1111/j.1472-8206.2000.tb00418.x. [DOI] [PubMed] [Google Scholar]
- 355. Guan Z, Pollock JS, Cook AK, Hobbs JL, Inscho EW. Effect of epithelial sodium channel blockade on the myogenic response of rat juxtamedullary afferent arterioles. Hypertension 54: 1062–1069, 2009. doi: 10.1161/HYPERTENSIONAHA.109.137992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Wang X, Breaks J, Loutzenhiser K, Loutzenhiser R. Effects of inhibition of the Na+/K+/2Cl− cotransporter on myogenic and angiotensin II responses of the rat afferent arteriole. Am J Physiol Renal Physiol 292: F999–F1006, 2007. doi: 10.1152/ajprenal.00343.2006. [DOI] [PubMed] [Google Scholar]
- 357. Kleyman TR, Cragoe EJ Jr.. Amiloride and its analogs as tools in the study of ion transport. J Membr Biol 105: 1–21, 1988. doi: 10.1007/BF01871102. [DOI] [PubMed] [Google Scholar]
- 358. Loutzenhiser R, Aaronson PI. Role of epithelial sodium channels in the renal myogenic response? Hypertension 55: e6, 2010. doi: 10.1161/HYPERTENSIONAHA.109.147132. [DOI] [PubMed] [Google Scholar]
- 359. Lopez-Charcas O, Rivera M, Gomora JC. Block of human CaV3 channels by the diuretic amiloride. Mol Pharmacol 82: 658–667, 2012. doi: 10.1124/mol.112.078923. [DOI] [PubMed] [Google Scholar]
- 360. Rüsch A, Kros CJ, Richardson GP. Block by amiloride and its derivatives of mechano-electrical transduction in outer hair cells of mouse cochlear cultures. J Physiol 474: 75–86, 1994. doi: 10.1113/jphysiol.1994.sp020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Lane JW, McBride DW, Hamill OP. Ionic effects on amiloride block of the mechanosensitive channel in Xenopus oocytes. Br J Pharmacol 108: 116–119, 1993. doi: 10.1111/j.1476-5381.1993.tb13449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. McGahon MK, Fernández JA, Dash DP, McKee J, Simpson DA, Zholos AV, McGeown JG, Curtis TM. TRPV2 channels contribute to stretch-activated cation currents and myogenic constriction in retinal arterioles. Invest Ophthalmol Vis Sci 57: 5637–5647, 2016. doi: 10.1167/iovs.16-20279. [DOI] [PubMed] [Google Scholar]
- 363. Castañeda MS, Tonini R, Richards CD, Stocker M, Pedarzani P. Benzamil inhibits neuronal and heterologously expressed small conductance Ca2+-activated K+ channels. Neuropharmacology 158: 107738, 2019. doi: 10.1016/j.neuropharm.2019.107738. [DOI] [PubMed] [Google Scholar]
- 364. Jernigan NL, Drummond HA. Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous beta and gammaENaC. Am J Physiol Renal Physiol 291: F1184–F1191, 2006. doi: 10.1152/ajprenal.00177.2006. [DOI] [PubMed] [Google Scholar]
- 365. Drummond HA, Stec DE. betaENaC acts as a mechanosensor in renal vascular smooth muscle cells that contributes to renal myogenic blood flow regulation, protection from renal injury and hypertension. J Nephrol Res 1: 1–9, 2015. doi: 10.17554/j.issn.2410-0579.2015.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Drummond HA. betaENaC is a molecular component of a VSMC mechanotransducer that contributes to renal blood flow regulation, protection from renal injury, and hypertension. Front Physiol 3: 341, 2012. doi: 10.3389/fphys.2012.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Pradervand S, Barker PM, Wang Q, Ernst SA, Beermann F, Grubb BR, Burnier M, Schmidt A, Bindels RJ, Gatzy JT, Rossier BC, Hummler E. Salt restriction induces pseudohypoaldosteronism type 1 in mice expressing low levels of the beta-subunit of the amiloride-sensitive epithelial sodium channel. Proc Natl Acad Sci USA 96: 1732–1737, 1999. doi: 10.1073/pnas.96.4.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB Jr, Stokes JB, Welsh MJ, Williamson RA. Disruption of the beta subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci USA 96: 1727–1731, 1999. doi: 10.1073/pnas.96.4.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Chung WS, Weissman JL, Farley J, Drummond HA. betaENaC is required for whole cell mechanically gated currents in renal vascular smooth muscle cells. Am J Physiol Renal Physiol 304: F1428–F1437, 2013. doi: 10.1152/ajprenal.00444.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Ge Y, Gannon K, Gousset M, Liu R, Murphey B, Drummond HA. Impaired myogenic constriction of the renal afferent arteriole in a mouse model of reduced betaENaC expression. Am J Physiol Renal Physiol 302: F1486–F1493, 2012. doi: 10.1152/ajprenal.00638.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Drummond HA, Grifoni SC, Abu-Zaid A, Gousset M, Chiposi R, Barnard JM, Murphey B, Stec DE. Renal inflammation and elevated blood pressure in a mouse model of reduced beta-ENaC. Am J Physiol Renal Physiol 301: F443–F449, 2011. doi: 10.1152/ajprenal.00694.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Choi SK, Yeon SI, Kwon Y, Byeon S, Lee YH. Involvement of epithelial Na+ channel in the elevated myogenic response in posterior cerebral arteries from spontaneously hypertensive rats. Sci Rep 7: 45996, 2017. doi: 10.1038/srep45996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Kim EC, Choi SK, Lim M, Yeon SI, Lee YH. Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS One 8: e84194, 2013. doi: 10.1371/journal.pone.0084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Kim EC, Ahn DS, Yeon SI, Lim M, Lee YH. Epithelial Na+ channel proteins are mechanotransducers of myogenic constriction in rat posterior cerebral arteries. Exp Physiol 97: 544–555, 2012. doi: 10.1113/expphysiol.2011.062232. [DOI] [PubMed] [Google Scholar]
- 375. Drummond HA, Grifoni SC, Jernigan NL. A new trick for an old dogma: ENaC proteins as mechanotransducers in vascular smooth muscle. Physiology (Bethesda) 23: 23–31, 2008. doi: 10.1152/physiol.00034.2007. [DOI] [PubMed] [Google Scholar]
- 376. Meltzer RH, Kapoor N, Qadri YJ, Anderson SJ, Fuller CM, Benos DJ. Heteromeric assembly of acid-sensitive ion channel and epithelial sodium channel subunits. J Biol Chem 282: 25548–25559, 2007. doi: 10.1074/jbc.M703825200. [DOI] [PubMed] [Google Scholar]
- 377. Drummond HA, Gebremedhin D, Harder DR. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension 44: 643–648, 2004. doi: 10.1161/01.HYP.0000144465.56360.ad. [DOI] [PubMed] [Google Scholar]
- 378. VanLandingham LG, Gannon KP, Drummond HA. Pressure-induced constriction is inhibited in a mouse model of reduced betaENaC. Am J Physiol Regul Integr Comp Physiol 297: R723–R728, 2009. doi: 10.1152/ajpregu.00212.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Nemeth Z, Ryan MJ, Granger JP, Drummond HA. Expression of exogenous epithelial sodium channel beta subunit in the mouse middle cerebral artery increases pressure-induced constriction. Am J Hypertens 34: 1227–1235, 2021. doi: 10.1093/ajh/hpab098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res 95: 922–929, 2004. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- 381. Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90: 248–250, 2002. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 382. Goralski JL, Boucher RC, Button B. Osmolytes and ion transport modulators: new strategies for airway surface rehydration. Curr Opin Pharmacol 10: 294–299, 2010. doi: 10.1016/j.coph.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Chambers L, Dorrance AM. Regulation of ion channels in the microcirculation by mineralocorticoid receptor activation. Curr Top Membr 85: 151–185, 2020. doi: 10.1016/bs.ctm.2020.02.001. [DOI] [PubMed] [Google Scholar]
- 384. Wang S, Meng F, Mohan S, Champaneri B, Gu Y. Functional ENaC channels expressed in endothelial cells: a new candidate for mediating shear force. Microcirculation 16: 276–287, 2009. doi: 10.1080/10739680802653150. [DOI] [PubMed] [Google Scholar]
- 385. Zhang J, Yuan HK, Chen S, Zhang ZR. Detrimental or beneficial: role of endothelial ENaC in vascular function. J Cell Physiol 237: 29–48, 2022. doi: 10.1002/jcp.30505. [DOI] [PubMed] [Google Scholar]
- 386. Guo D, Liang S, Wang S, Tang C, Yao B, Wan W, Zhang H, Jiang H, Ahmed A, Zhang Z, Gu Y. Role of epithelial Na+ channels in endothelial function. J Cell Sci 129: 290–297, 2016. doi: 10.1242/jcs.168831. [DOI] [PubMed] [Google Scholar]
- 387. Cosgun ZC, Sternak M, Fels B, Bar A, Kwiatkowski G, Pacia MZ, Herrnböck L, Lindemann M, Stegbauer J, Höges S, Chlopicki S, Kusche-Vihrog K. Rapid shear stress-dependent ENaC membrane insertion is mediated by the endothelial glycocalyx and the mineralocorticoid receptor. Cell Mol Life Sci 79: 235, 2022. doi: 10.1007/s00018-022-04260-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Tarjus A, Maase M, Jeggle P, Martinez-Martinez E, Fassot C, Loufrani L, Henrion D, Hansen PBL, Kusche-Vihrog K, Jaisser F. The endothelial alphaENaC contributes to vascular endothelial function in vivo. PLoS One 12: e0185319, 2017. doi: 10.1371/journal.pone.0185319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Mazzochi C, Benos DJ, Smith PR. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am J Physiol Renal Physiol 291: F1113–F1122, 2006. doi: 10.1152/ajprenal.00195.2006. [DOI] [PubMed] [Google Scholar]
- 390. Oda T, Makino K, Yamashita I, Namba K, Maéda Y. Distinct structural changes detected by X-ray fiber diffraction in stabilization of F-actin by lowering pH and increasing ionic strength. Biophys J 80: 841–851, 2001. doi: 10.1016/S0006-3495(01)76063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Jeggle P, Callies C, Tarjus A, Fassot C, Fels J, Oberleithner H, Jaisser F, Kusche-Vihrog K. Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 61: 1053–1059, 2013. doi: 10.1161/HYPERTENSIONAHA.111.199455. [DOI] [PubMed] [Google Scholar]
- 392. Kusche-Vihrog K, Tarjus A, Fels J, Jaisser F. The epithelial Na+ channel: a new player in the vasculature. Curr Opin Nephrol Hypertens 23: 143–148, 2014. doi: 10.1097/01.mnh.0000441054.88962.2c. [DOI] [PubMed] [Google Scholar]
- 393. Su Y, Edwards-Bennett S, Bubb MR, Block ER. Regulation of endothelial nitric oxide synthase by the actin cytoskeleton. Am J Physiol Cell Physiol 284: C1542–C1549, 2003. doi: 10.1152/ajpcell.00248.2002. [DOI] [PubMed] [Google Scholar]
- 394. Jia G, Habibi J, Aroor AR, Hill MA, Yang Y, Whaley-Connell A, Jaisser F, Sowers JR. Epithelial sodium channel in aldosterone-induced endothelium stiffness and aortic dysfunction. Hypertension 72: 731–738, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Paar M, Pavenstädt H, Kusche-Vihrog K, Drüppel V, Oberleithner H, Kliche K. Endothelial sodium channels trigger endothelial salt sensitivity with aging. Hypertension 64: 391–396, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03348. [DOI] [PubMed] [Google Scholar]
- 396. Warnock DG, Kusche-Vihrog K, Tarjus A, Sheng S, Oberleithner H, Kleyman TR, Jaisser F. Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat Rev Nephrol 10: 146–157, 2014. doi: 10.1038/nrneph.2013.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Paukert M, Babini E, Pusch M, Gründer S. Identification of the Ca2+ blocking site of acid-sensing ion channel (ASIC) 1: implications for channel gating. J Gen Physiol 124: 383–394, 2004. doi: 10.1085/jgp.200308973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. de Weille J, Bassilana F. Dependence of the acid-sensitive ion channel, ASIC1a, on extracellular Ca2+ ions. Brain Res 900: 277–281, 2001. doi: 10.1016/s0006-8993(01)02345-9. [DOI] [PubMed] [Google Scholar]
- 399. Ziemann AE, Allen JE, Dahdaleh NS, Drebot II, Coryell MW, Wunsch AM, Lynch CM, Faraci FM, Howard MA 3rd, Welsh MJ, Wemmie JA. The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell 139: 1012–1021, 2009. doi: 10.1016/j.cell.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Wu WL, Cheng CF, Sun WH, Wong CW, Chen CC. Targeting ASIC3 for pain, anxiety, and insulin resistance. Pharmacol Ther 134: 127–138, 2012. doi: 10.1016/j.pharmthera.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 401. Lin SH, Sun WH, Chen CC. Genetic exploration of the role of acid-sensing ion channels. Neuropharmacology 94: 99–118, 2015. doi: 10.1016/j.neuropharm.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 402. Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32: 1071–1083, 2001. doi: 10.1016/S0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 403. Molliver DC, Immke DC, Fierro L, Paré M, Rice FL, McCleskey EW. ASIC3, an acid-sensing ion channel, is expressed in metaboreceptive sensory neurons. Mol Pain 1: 35, 2005. [ doi: 10.1186/1744-8069-1-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Chen WN, Chen CC. Acid mediates a prolonged antinociception via substance P signaling in acid-induced chronic widespread pain. Mol Pain 10: 30, 2014. doi: 10.1186/1744-8069-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Sluka KA, Price MP, Breese NM, Stucky CL, Wemmie JA, Welsh MJ. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 106: 229–239, 2003. doi: 10.1016/S0304-3959(03)00269-0. [DOI] [PubMed] [Google Scholar]
- 406. Lin CC, Chen WN, Chen CJ, Lin YW, Zimmer A, Chen CC. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc Natl Acad Sci USA 109: E76–E83, 2012. doi: 10.1073/pnas.1108903108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Fromy B, Lingueglia E, Sigaudo-Roussel D, Saumet JL, Lazdunski M. Asic3 is a neuronal mechanosensor for pressure-induced vasodilation that protects against pressure ulcers. Nat Med 18: 1205–1207, 2012. doi: 10.1038/nm.2844. [DOI] [PubMed] [Google Scholar]
- 408. Petroff EY, Price MP, Snitsarev V, Gong H, Korovkina V, Abboud FM, Welsh MJ. Acid-sensing ion channels interact with and inhibit BK K+ channels. Proc Natl Acad Sci USA 105: 3140–3144, 2008. doi: 10.1073/pnas.0712280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Omerbašić D, Schuhmacher LN, Bernal Sierra YA, Smith ES, Lewin GR. ASICs and mammalian mechanoreceptor function. Neuropharmacology 94: 80–86, 2015. doi: 10.1016/j.neuropharm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 410. Drew LJ, Rohrer DK, Price MP, Blaver KE, Cockayne DA, Cesare P, Wood JN. Acid-sensing ion channels ASIC2 and ASIC3 do not contribute to mechanically activated currents in mammalian sensory neurones. J Physiol 556: 691–710, 2004. doi: 10.1113/jphysiol.2003.058693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Roza C, Puel JL, Kress M, Baron A, Diochot S, Lazdunski M, Waldmann R. Knockout of the ASIC2 channel in mice does not impair cutaneous mechanosensation, visceral mechanonociception and hearing. J Physiol 558: 659–669, 2004. doi: 10.1113/jphysiol.2004.066001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. García-Añoveros J, Samad TA, Zuvela-Jelaska L, Woolf CJ, Corey DP. Transport and localization of the DEG/ENaC ion channel BNaC1alpha to peripheral mechanosensory terminals of dorsal root ganglia neurons. J Neurosci 21: 2678–2686, 2001. doi: 10.1523/JNEUROSCI.21-08-02678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Wu WL, Lin YW, Min MY, Chen CC. Mice lacking Asic3 show reduced anxiety-like behavior on the elevated plus maze and reduced aggression. Genes Brain Behav 9: 603–614, 2010. doi: 10.1111/j.1601-183X.2010.00591.x. [DOI] [PubMed] [Google Scholar]
- 414. Price MP, Lewin GR, McIlwrath SL, Cheng C, Xie J, Heppenstall PA, Stucky CL, Mannsfeldt AG, Brennan TJ, Drummond HA, Qiao J, Benson CJ, Tarr DE, Hrstka RF, Yang B, Williamson RA, Welsh MJ. The mammalian sodium channel BNC1 is required for normal touch sensation. Nature 407: 1007–1011, 2000. doi: 10.1038/35039512. [DOI] [PubMed] [Google Scholar]
- 415. Cheng YR, Jiang BY, Chen CC. Acid-sensing ion channels: dual function proteins for chemo-sensing and mechano-sensing. J Biomed Sci 25: 46, 2018. doi: 10.1186/s12929-018-0448-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Gannon KP, Vanlandingham LG, Jernigan NL, Grifoni SC, Hamilton G, Drummond HA. Impaired pressure-induced constriction in mouse middle cerebral arteries of ASIC2 knockout mice. Am J Physiol Heart Circ Physiol 294: H1793–H1803, 2008. doi: 10.1152/ajpheart.01380.2007. [DOI] [PubMed] [Google Scholar]
- 417. Gannon KP, McKey SE, Stec DE, Drummond HA. Altered myogenic vasoconstriction and regulation of whole kidney blood flow in the ASIC2 knockout mouse. Am J Physiol Renal Physiol 308: F339–F348, 2015. doi: 10.1152/ajprenal.00572.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Lin LH, Jin J, Nashelsky MB, Talman WT. Acid-sensing ion channel 1 and nitric oxide synthase are in adjacent layers in the wall of rat and human cerebral arteries. J Chem Neuroanat 61-62: 161–168, 2014. doi: 10.1016/j.jchemneu.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2: 1313–1323, 1989. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 420. Lucas P, Ukhanov K, Leinders-Zufall T, Zufall F. A diacylglycerol-gated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: mechanism of pheromone transduction. Neuron 40: 551–561, 2003. doi: 10.1016/s0896-6273(03)00675-5. [DOI] [PubMed] [Google Scholar]
- 421. Yildirim E, Dietrich A, Birnbaumer L. The mouse C-type transient receptor potential 2 (TRPC2) channel: alternative splicing and calmodulin binding to its N terminus. Proc Natl Acad Sci USA 100: 2220–2225, 2003. doi: 10.1073/pnas.0438036100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Müller D, Hoenderop JG, Meij IC, van den Heuvel LP, Knoers NV, den Hollander AI, Eggert P, García-Nieto V, Claverie-Martín F, Bindels RJ. Molecular cloning, tissue distribution, and chromosomal mapping of the human epithelial Ca2+ channel (ECAC1). Genomics 67: 48–53, 2000. doi: 10.1006/geno.2000.6203. [DOI] [PubMed] [Google Scholar]
- 423. Nilius B, Prenen J, Droogmans G, Voets T, Vennekens R, Freichel M, Wissenbach U, Flockerzi V. Voltage dependence of the Ca2+-activated cation channel TRPM4. J Biol Chem 278: 30813–30820, 2003. doi: 10.1074/jbc.M305127200. [DOI] [PubMed] [Google Scholar]
- 424. Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109: 397–407, 2002. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 425. Hofmann T, Chubanov V, Gudermann T, Montell C. TRPM5 is a voltage-modulated and Ca2+-activated monovalent selective cation channel. Curr Biol 13: 1153–1158, 2003. doi: 10.1016/s0960-9822(03)00431-7. [DOI] [PubMed] [Google Scholar]
- 426. Pires PW, Earley S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 7: e35316, 2018. doi: 10.7554/eLife.35316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Pires PW, Sullivan MN, Pritchard HA, Robinson JJ, Earley S. Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am J Physiol Heart Circ Physiol 309: H2031–H2041, 2015. doi: 10.1152/ajpheart.00140.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG, Earley S. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal 8: ra2, 2015. doi: 10.1126/scisignal.2005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Sullivan MN, Francis M, Pitts NL, Taylor MS, Earley S. Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Mol Pharmacol 82: 464–472, 2012. doi: 10.1124/mol.112.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Tajada S, Moreno CM, O’Dwyer S, Woods S, Sato D, Navedo MF, Santana LF. Distance constraints on activation of TRPV4 channels by AKAP150-bound PKCalpha in arterial myocytes. J Gen Physiol 149: 639–659, 2017. doi: 10.1085/jgp.201611709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, Santana LF, Nelson MT. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci Signal 7: ra66, 2014. doi: 10.1126/scisignal.2005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, Brayden JE, Santana LF. Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J Gen Physiol 143: 559–575, 2014. doi: 10.1085/jgp.201311050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601, 2012. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Senning EN, Gordon SE. Activity and Ca2+ regulate the mobility of TRPV1 channels in the plasma membrane of sensory neurons. Elife 4: e03819, 2015. doi: 10.7554/eLife.03819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Hill-Eubanks DC, Gonzales AL, Sonkusare SK, Nelson MT. Vascular TRP channels: performing under pressure and going with the flow. Physiology (Bethesda) 29: 343–360, 2014. doi: 10.1152/physiol.00009.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem 272: 29672–29680, 1997. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- 437. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 438. Dietrich A, Kalwa H, Rost BR, Gudermann T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflugers Arch 451: 72–80, 2005. doi: 10.1007/s00424-005-1460-0. [DOI] [PubMed] [Google Scholar]
- 439. Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99: 7461–7466, 2002. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Maruyama Y, Nakanishi Y, Walsh EJ, Wilson DP, Welsh DG, Cole WC. Heteromultimeric TRPC6-TRPC7 channels contribute to arginine vasopressin-induced cation current of A7r5 vascular smooth muscle cells. Circ Res 98: 1520–1527, 2006. doi: 10.1161/01.RES.0000226495.34949.28. [DOI] [PubMed] [Google Scholar]
- 441. Azumaya CM, Sierra-Valdez F, Cordero-Morales JF, Nakagawa T. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). J Biol Chem 293: 10381–10391, 2018. doi: 10.1074/jbc.RA118.003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Riccio A, Medhurst AD, Mattei C, Kelsell RE, Calver AR, Randall AD, Benham CD, Pangalos MN. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res Mol Brain Res 109: 95–104, 2002. doi: 10.1016/s0169-328x(02)00527-2. [DOI] [PubMed] [Google Scholar]
- 443. Albert AP, Large WA. Synergism between inositol phosphates and diacylglycerol on native TRPC6-like channels in rabbit portal vein myocytes. J Physiol 552: 789–795, 2003. doi: 10.1113/jphysiol.2003.052977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Albert AP, Piper AS, Large WA. of a constitutively active Ca2+-permeable non-selective cation channel in rabbit ear artery myocytes. J Physiol 549: 143–156, 2003. doi: 10.1113/jphysiol.2002.038190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Saleh SN, Albert AP, Peppiatt CM, Large WA. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol 577: 479–495, 2006. doi: 10.1113/jphysiol.2006.119305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res 88: 325–332, 2001. doi: 10.1161/01.RES.88.3.325. [DOI] [PubMed] [Google Scholar]
- 447. Welsh DG, Nelson MT, Eckman DM, Brayden JE. Swelling-activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. J Physiol 527: 139–148, 2000. doi: 10.1111/j.1469-7793.2000.t01-1-00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honoré E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455: 1097–1103, 2008. doi: 10.1007/s00424-007-0359-3. [DOI] [PubMed] [Google Scholar]
- 449. Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, Vásquez V, Laver DR, Martinac B. Mammalian TRP ion channels are insensitive to membrane stretch. J Cell Sci 132: jcs238360, 2019. doi: 10.1242/jcs.238360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, Hill-Eubanks DC, Nelson MT, Earley S. A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal 7: ra49, 2014. doi: 10.1126/scisignal.2004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Schleifenbaum J, Kassmann M, Szijarto I, Hercule H, Tano JY, Weinert S, Heidenreich M, Pathan A, Anistan Y, Alenina N, Rusch N, Jentsch T, Bader M, Gollasch M. Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res 115: 263–272, 2014. doi: 10.1161/CIRCRESAHA.115.302882. [DOI] [PubMed] [Google Scholar]
- 452. Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol 25: 6980–6989, 2005. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, Birnbaumer L, Malik AB, Mehta D. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med 209: 1953–1968, 2012. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem 282: 7833–7843, 2007. doi: 10.1074/jbc.M608288200. [DOI] [PubMed] [Google Scholar]
- 455. Samapati R, Yang Y, Yin J, Stoerger C, Arenz C, Dietrich A, Gudermann T, Adam D, Wu S, Freichel M, Flockerzi V, Uhlig S, Kuebler WM. Lung endothelial Ca2+ and permeability response to platelet-activating factor is mediated by acid sphingomyelinase and transient receptor potential classical 6. Am J Respir Crit Care Med 185: 160–170, 2012. doi: 10.1164/rccm.201104-0717OC. [DOI] [PubMed] [Google Scholar]
- 456. Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med 212: 1883–1899, 2015. doi: 10.1084/jem.20150353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R. Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol 27: 2612–2618, 2007. doi: 10.1161/ATVBAHA.107.152074. [DOI] [PubMed] [Google Scholar]
- 458. Xu XZ, Moebius F, Gill DL, Montell C. Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc Natl Acad Sci USA 98: 10692–10697, 2001. doi: 10.1073/pnas.191360198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, Voets T. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J 25: 467–478, 2006. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Zhang Z, Okawa H, Wang Y, Liman ER. Phosphatidylinositol 4,5-bisphosphate rescues TRPM4 channels from desensitization. J Biol Chem 280: 39185–39192, 2005. doi: 10.1074/jbc.M506965200. [DOI] [PubMed] [Google Scholar]
- 461. Nilius B, Prenen J, Tang J, Wang C, Owsianik G, Janssens A, Voets T, Zhu MX. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J Biol Chem 280: 6423–6433, 2005. doi: 10.1074/jbc.M411089200. [DOI] [PubMed] [Google Scholar]
- 462. Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol 292: H2613–H2622, 2007. doi: 10.1152/ajpheart.01286.2006. [DOI] [PubMed] [Google Scholar]
- 463. Crnich R, Amberg GC, Leo MD, Gonzales AL, Tamkun MM, Jaggar JH, Earley S. Vasoconstriction resulting from dynamic membrane trafficking of TRPM4 in vascular smooth muscle cells. Am J Physiol Cell Physiol 299: C682–C694, 2010. doi: 10.1152/ajpcell.00101.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Garcia ZI, Bruhl A, Gonzales AL, Earley S. Basal protein kinase Cdelta activity is required for membrane localization and activity of TRPM4 channels in cerebral artery smooth muscle cells. Channels (Austin) 5: 210–214, 2011. doi: 10.4161/chan.5.3.15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Winkler PA, Huang Y, Sun W, Du J, Lü W. Electron cryo-microscopy structure of a human TRPM4 channel. Nature 552: 200–204, 2017. doi: 10.1038/nature24674. [DOI] [PubMed] [Google Scholar]
- 466. Guo J, She J, Zeng W, Chen Q, Bai XC, Jiang Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 552: 205–209, 2017. doi: 10.1038/nature24997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G, Nilius B. Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium 37: 267–278, 2005. doi: 10.1016/j.ceca.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 468. Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc Res 73: 531–538, 2007. doi: 10.1016/j.cardiores.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 469. Grand T, Demion M, Norez C, Mettey Y, Launay P, Becq F, Bois P, Guinamard R. 9-Phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br J Pharmacol 153: 1697–1705, 2008. doi: 10.1038/bjp.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Gonzales AL, Garcia ZI, Amberg GC, Earley S. Pharmacological inhibition of TRPM4 hyperpolarizes vascular smooth muscle. Am J Physiol Cell Physiol 299: C1195–C1202, 2010. doi: 10.1152/ajpcell.00269.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Garland CJ, Smirnov SV, Bagher P, Lim CS, Huang CY, Mitchell R, Stanley C, Pinkney A, Dora KA. TRPM4 inhibitor 9-phenanthrol activates endothelial cell intermediate conductance calcium-activated potassium channels in rat isolated mesenteric artery. Br J Pharmacol 172: 1114–1123, 2015. doi: 10.1111/bph.12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Burris SK, Wang Q, Bulley S, Neeb ZP, Jaggar JH. 9-Phenanthrol inhibits recombinant and arterial myocyte TMEM16A channels. Br J Pharmacol 172: 2459–2468, 2015. doi: 10.1111/bph.13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Veress R, Baranyai D, Hegyi B, Kistamás K, Dienes C, Magyar J, Bányász T, Nánási PP, Szentandrássy N, Horváth B. Transient receptor potential melastatin 4 channel inhibitor 9-phenanthrol inhibits K+ but not Ca2+ currents in canine ventricular myocytes. Can J Physiol Pharmacol 96: 1022–1029, 2018. doi: 10.1139/cjpp-2018-0049. [DOI] [PubMed] [Google Scholar]
- 474. Ozhathil LC, Delalande C, Bianchi B, Nemeth G, Kappel S, Thomet U, Ross-Kaschitza D, Simonin C, Rubin M, Gertsch J, Lochner M, Peinelt C, Reymond JL, Abriel H. Identification of potent and selective small molecule inhibitors of the cation channel TRPM4. Br J Pharmacol 175: 2504–2519, 2018. doi: 10.1111/bph.14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. Arullampalam P, Preti B, Ross-Kaschitza D, Lochner M, Rougier JS, Abriel H. Species-specific effects of cation channel TRPM4 small-molecule inhibitors. Front Pharmacol 12: 712354, 2021. doi: 10.3389/fphar.2021.712354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Parajuli SP, Hristov KL, Sullivan MN, Xin W, Smith AC, Earley S, Malysz J, Petkov GV. Control of urinary bladder smooth muscle excitability by the TRPM4 channel modulator 9-phenanthrol. Channels (Austin) 7: 537–540, 2013. doi: 10.4161/chan.26289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Smith AC, Parajuli SP, Hristov KL, Cheng Q, Soder RP, Afeli SA, Earley S, Xin W, Malysz J, Petkov GV. TRPM4 channel: a new player in urinary bladder smooth muscle function in rats. Am J Physiol Renal Physiol 304: F918–F929, 2013. doi: 10.1152/ajprenal.00417.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Yang XR, Lin MJ, McIntosh LS, Sham JS. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol 290: L1267–L1276, 2006. doi: 10.1152/ajplung.00515.2005. [DOI] [PubMed] [Google Scholar]
- 479. Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circ Res 99: 119–131, 2006. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- 480. Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londoño JE, Uhl S, Voets T, Hummel B, van den Bergh A, Herijgers P, Nilius B, Flockerzi V, Schweda F, Freichel M. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J Clin Invest 120: 3267–3279, 2010. doi: 10.1172/JCI41348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Schulze-Bahr E, Brink P, Pongs O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest 119: 2737–2744, 2009. doi: 10.1172/JCI38292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Constantine M, Liew CK, Lo V, Macmillan A, Cranfield CG, Sunde M, Whan R, Graham RM, Martinac B. Heterologously-expressed and liposome-reconstituted human transient receptor potential melastatin 4 channel (TRPM4) is a functional tetramer. Sci Rep 6: 19352, 2016. doi: 10.1038/srep19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483. Gonzales AL, Amberg GC, Earley S. Ca2+ release from the sarcoplasmic reticulum is required for sustained TRPM4 activity in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 299: C279–C288, 2010. doi: 10.1152/ajpcell.00550.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Pires PW, Ko EA, Pritchard HA, Rudokas M, Yamasaki E, Earley S. The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J Physiol 595: 4735–4753, 2017. doi: 10.1113/JP274310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Morita H, Honda A, Inoue R, Ito Y, Abe K, Nelson MT, Brayden JE. Membrane stretch-induced activation of a TRPM4-like nonselective cation channel in cerebral artery myocytes. J Pharmacol Sci 103: 417–426, 2007. doi: 10.1254/jphs.fp0061332. [DOI] [PubMed] [Google Scholar]
- 486. Gonzales AL, Earley S. Endogenous cytosolic Ca2+ buffering is necessary for TRPM4 activity in cerebral artery smooth muscle cells. Cell Calcium 51: 82–93, 2012. doi: 10.1016/j.ceca.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Li Y, Baylie RL, Tavares MJ, Brayden JE. TRPM4 channels couple purinergic receptor mechanoactivation and myogenic tone development in cerebral parenchymal arterioles. J Cereb Blood Flow Metab 34: 1706–1714, 2014. doi: 10.1038/jcbfm.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Phan TX, Ton HT, Gulyás H, Pórszász R, Tóth A, Russo R, Kay MW, Sahibzada N, Ahern GP. TRPV1 in arteries enables a rapid myogenic tone. J Physiol 600: 1651–1666, 2022. doi: 10.1113/JP281873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272: 1339–1342, 1996. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 490. Wu G, Mochizuki T, Le TC, Cai Y, Hayashi T, Reynolds DM, Somlo S. Molecular cloning, cDNA sequence analysis, and chromosomal localization of mouse Pkd2. Genomics 45: 220–223, 1997. doi: 10.1006/geno.1997.4920. [DOI] [PubMed] [Google Scholar]
- 491. Pelucchi B, Aguiari G, Pignatelli A, Manzati E, Witzgall R, Del Senno L, Belluzzi O. Nonspecific cation current associated with native polycystin-2 in HEK-293 cells. J Am Soc Nephrol 17: 388–397, 2006. doi: 10.1681/ASN.2004121146. [DOI] [PubMed] [Google Scholar]
- 492. Gonzalez-Perrett S, Batelli M, Kim K, Essafi M, Timpanaro G, Moltabetti N, Reisin IL, Arnaout MA, Cantiello HF. Voltage dependence and pH regulation of human polycystin-2-mediated cation channel activity. J Biol Chem 277: 24959–24966, 2002. doi: 10.1074/jbc.M105084200. [DOI] [PubMed] [Google Scholar]
- 493. Luo Y, Vassilev PM, Li X, Kawanabe Y, Zhou J. Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol Cell Biol 23: 2600–2607, 2003. doi: 10.1128/MCB.23.7.2600-2607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Liu X, Vien T, Duan J, Sheu SH, DeCaen PG, Clapham DE. Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. Elife 7: e33183, 2018. doi: 10.7554/eLife.33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Grieben M, Pike AC, Shintre CA, Venturi E, El-Ajouz S, Tessitore A, Shrestha L, Mukhopadhyay S, Mahajan P, Chalk R, Burgess-Brown NA, Sitsapesan R, Huiskonen JT, Carpenter EP. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat Struct Mol Biol 24: 114–122, 2017. doi: 10.1038/nsmb.3343. [DOI] [PubMed] [Google Scholar]
- 496. Shen PS, Yang X, DeCaen PG, Liu X, Bulkley D, Clapham DE, Cao E. The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell 167: 763–773.e11, 2016. doi: 10.1016/j.cell.2016.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, Zhou Q, Mei C, Shi Y. Structure of the human PKD1-PKD2 complex. Science 361: eaat9819, 2018. doi: 10.1126/science.aat9819. [DOI] [PubMed] [Google Scholar]
- 498. Bulley S, Fernández-Peña C, Hasan R, Leo MD, Muralidharan P, Mackay CE, Evanson KW, Moreira-Junior L, Mata-Daboin A, Burris SK, Wang Q, Kuruvilla KP, Jaggar JH. Arterial smooth muscle cell PKD2 (TRPP1) channels regulate systemic blood pressure. Elife 7: e42628, 2018. doi: 10.7554/eLife.42628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Narayanan D, Bulley S, Leo MD, Burris SK, Gabrick KS, Boop FA, Jaggar JH. Smooth muscle cell transient receptor potential polycystin-2 (TRPP2) channels contribute to the myogenic response in cerebral arteries. J Physiol 591: 5031–5046, 2013. doi: 10.1113/jphysiol.2013.258319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Hasan R, Leo MD, Muralidharan P, Mata-Daboin A, Yin W, Bulley S, Fernandez-Peña C, MacKay CE, Jaggar JH. SUMO1 modification of PKD2 channels regulates arterial contractility. Proc Natl Acad Sci USA 116: 27095–27104, 2019. doi: 10.1073/pnas.1917264116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H Jr, Kucherlapati R, Edelmann W, Somlo S. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93: 177–188, 1998. doi: 10.1016/S0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 502. Qian Q, Hunter LW, Du H, Ren Q, Han Y, Sieck GC. Pkd2+/− vascular smooth muscles develop exaggerated vasocontraction in response to phenylephrine stimulation. J Am Soc Nephrol 18: 485–493, 2007. doi: 10.1681/ASN.2006050501. [DOI] [PubMed] [Google Scholar]
- 503. Du H, Wang X, Wu J, Qian Q. Phenylephrine induces elevated RhoA activation and smooth muscle alpha-actin expression in Pkd2+/- vascular smooth muscle cells. Hypertens Res 33: 37–42, 2010. doi: 10.1038/hr.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Van der Heiden K, Egorova AD, Poelmann RE, Wentzel JJ, Hierck BP. Role for primary cilia as flow detectors in the cardiovascular system. Int Rev Cell Mol Biol 290: 87–119, 2011. doi: 10.1016/B978-0-12-386037-8.00004-1. [DOI] [PubMed] [Google Scholar]
- 505. Ando J, Yamamoto K. Flow detection and calcium signalling in vascular endothelial cells. Cardiovasc Res 99: 260–268, 2013. doi: 10.1093/cvr/cvt084. [DOI] [PubMed] [Google Scholar]
- 506. Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, Clapham DE. Primary cilia are not calcium-responsive mechanosensors. Nature 531: 656–660, 2016. doi: 10.1038/nature17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 184: 71–79, 2001. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 508. Praetorius HA, Frokiaer J, Nielsen S, Spring KR. Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol 191: 193–200, 2003. doi: 10.1007/s00232-002-1055-z. [DOI] [PubMed] [Google Scholar]
- 509. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 510. Goetz JG, Steed E, Ferreira RR, Roth S, Ramspacher C, Boselli F, Charvin G, Liebling M, Wyart C, Schwab Y, Vermot J. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep 6: 799–808, 2014. doi: 10.1016/j.celrep.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 511. MacKay CE, Leo MD, Fernández-Peña C, Hasan R, Yin W, Mata-Daboin A, Bulley S, Gammons J, Mancarella S, Jaggar JH. Intravascular flow stimulates PKD2 (polycystin-2) channels in endothelial cells to reduce blood pressure. Elife 9: e60401, 2020. doi: 10.7554/eLife.60401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Phan TX, Ton HT, Gulyás H, Pórszász R, Tóth A, Russo R, Kay MW, Sahibzada N, Ahern GP. TRPV1 expressed throughout the arterial circulation regulates vasoconstriction and blood pressure. J Physiol 598: 5639–5659, 2020. doi: 10.1113/JP279909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O’Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 31: 5067–5077, 2011. doi: 10.1523/JNEUROSCI.6451-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514. Phan TX, Ton HT, Chen Y, Basha ME, Ahern GP. Sex-dependent expression of TRPV1 in bladder arterioles. Am J Physiol Renal Physiol 311: F1063–F1073, 2016. doi: 10.1152/ajprenal.00234.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Tóth A, Czikora A, Pasztor ET, Dienes B, Bai P, Csernoch L, Rutkai I, Csató V, Manyine IS, Pórszász R, Edes I, Papp Z, Boczan J. Vanilloid receptor-1 (TRPV1) expression and function in the vasculature of the rat. J Histochem Cytochem 62: 129–144, 2014. doi: 10.1369/0022155413513589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398: 436–441, 1999. doi: 10.1038/18906. [DOI] [PubMed] [Google Scholar]
- 517. Zubcevic L, Herzik MA Jr, Chung BC, Liu Z, Lander GC, Lee SY. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat Struct Mol Biol 23: 180–186, 2016. doi: 10.1038/nsmb.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Huynh KW, Cohen MR, Jiang J, Samanta A, Lodowski DT, Zhou ZH, Moiseenkova-Bell VY. Structure of the full-length TRPV2 channel by cryo-EM. Nat Commun 7: 11130, 2016. doi: 10.1038/ncomms11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Hisanaga E, Nagasawa M, Ueki K, Kulkarni RN, Mori M, Kojima I. Regulation of calcium-permeable TRPV2 channel by insulin in pancreatic beta-cells. Diabetes 58: 174–184, 2009. doi: 10.2337/db08-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Nie L, Oishi Y, Doi I, Shibata H, Kojima I. Inhibition of proliferation of MCF-7 breast cancer cells by a blocker of Ca2+-permeable channel. Cell Calcium 22: 75–82, 1997. doi: 10.1016/s0143-4160(97)90107-x. [DOI] [PubMed] [Google Scholar]
- 521. Koch SE, Nieman ML, Robbins N, Slone S, Worley M, Green LC, Chen Y, Barlow A, Tranter M, Wang H, Lorenz JN, Rubinstein J. Tranilast blunts the hypertrophic and fibrotic response to increased afterload independent of cardiomyocyte transient receptor potential vanilloid 2 channels. J Cardiovasc Pharmacol 72: 40–48, 2018. doi: 10.1097/FJC.0000000000000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522. Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res 91: 15–28, 2015. doi: 10.1016/j.phrs.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 523. Nedungadi TP, Dutta M, Bathina CS, Caterina MJ, Cunningham JT. Expression and distribution of TRPV2 in rat brain. Exp Neurol 237: 223–237, 2012. doi: 10.1016/j.expneurol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Parenti A, De Logu F, Geppetti P, Benemei S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br J Pharmacol 173: 953–969, 2016. doi: 10.1111/bph.13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Muraki K, Iwata Y, Katanosaka Y, Ito T, Ohya S, Shigekawa M, Imaizumi Y. TRPV2 is a component of osmotically sensitive cation channels in murine aortic myocytes. Circ Res 93: 829–838, 2003. doi: 10.1161/01.RES.0000097263.10220.0C. [DOI] [PubMed] [Google Scholar]
- 526. Fois G, Weimer M, Busch T, Felder ET, Oswald F, von Wichert G, Seufferlein T, Dietl P, Felder E. Effects of keratin phosphorylation on the mechanical properties of keratin filaments in living cells. FASEB J 27: 1322–1329, 2013. doi: 10.1096/fj.12-215632. [DOI] [PubMed] [Google Scholar]
- 527. Shibasaki K, Murayama N, Ono K, Ishizaki Y, Tominaga M. TRPV2 enhances axon outgrowth through its activation by membrane stretch in developing sensory and motor neurons. J Neurosci 30: 4601–4612, 2010. doi: 10.1523/JNEUROSCI.5830-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Rubinstein J, Lasko VM, Koch SE, Singh VP, Carreira V, Robbins N, Patel AR, Jiang M, Bidwell P, Kranias EG, Jones WK, Lorenz JN. Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am J Physiol Heart Circ Physiol 306: H574–H584, 2014. doi: 10.1152/ajpheart.00854.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2: 695–702, 2000. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 530. Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424: 434–438, 2003. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 531. Deng Z, Paknejad N, Maksaev G, Sala-Rabanal M, Nichols CG, Hite RK, Yuan P. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat Struct Mol Biol 25: 252–260, 2018. doi: 10.1038/s41594-018-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, Chendrimada TP, Lashinger ES, Gordon E, Evans L, Misajet BA, Demarini DJ, Nation JH, Casillas LN, Marquis RW, Votta BJ, Sheardown SA, Xu X, Brooks DP, Laping NJ, Westfall TD. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther 326: 432–442, 2008. doi: 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- 533. Vincent F, Acevedo A, Nguyen MT, Dourado M, DeFalco J, Gustafson A, Spiro P, Emerling DE, Kelly MG, Duncton MA. Identification and characterization of novel TRPV4 modulators. Biochem Biophys Res Commun 389: 490–494, 2009. doi: 10.1016/j.bbrc.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 534. Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, Voets T. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci USA 107: 19084–19089, 2010. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4: 159ra148, 2012. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- 536. Delany NS, Hurle M, Facer P, Alnadaf T, Plumpton C, Kinghorn I, See CG, Costigan M, Anand P, Woolf CJ, Crowther D, Sanseau P, Tate SN. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol Genomics 4: 165–174, 2001. doi: 10.1152/physiolgenomics.2001.4.3.165. [DOI] [PubMed] [Google Scholar]
- 537. Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525–535, 2000. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol 297: H1096–H1102, 2009. doi: 10.1152/ajpheart.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279, 2005. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- 540. Soni H, Peixoto-Neves D, Matthews AT, Adebiyi A. TRPV4 channels contribute to renal myogenic autoregulation in neonatal pigs. Am J Physiol Renal Physiol 313: F1136–F1148, 2017. doi: 10.1152/ajprenal.00300.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Kohler R, Hoyer J. Role of TRPV4 in the mechanotransduction of shear stress in endothelial cells. In: TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades, edited by Liedtke WB, Heller S.. Boca Raton, FL: CRC, 2007. [PubMed] [Google Scholar]
- 542. Liedtke WB. TRPV Channels’ function in osmo- and mechanotransduction. In: TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades, edited by Liedtke WB, Heller S. Boca Raton, FL: CRC, 2007. [PubMed] [Google Scholar]
- 543. Gebremedhin D, Zhang DX, Weihrauch D, Uche NN, Harder DR. Detection of TRPV4 channel current-like activity in Fawn Hooded hypertensive (FHH) rat cerebral arterial muscle cells. PLoS One 12: e0176796, 2017. doi: 10.1371/journal.pone.0176796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278: 22664–22668, 2003. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- 545. Nishijima Y, Zheng X, Lund H, Suzuki M, Mattson DL, Zhang DX. Characterization of blood pressure and endothelial function in TRPV4-deficient mice with l-NAME- and angiotensin II-induced hypertension. Physiol Rep 2: e00199, 2014. doi: 10.1002/phy2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Chen YL, Sonkusare SK. Endothelial TRPV4 channels and vasodilator reactivity. Curr Top Membr 85: 89–117, 2020. doi: 10.1016/bs.ctm.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem 277: 13569–13577, 2002. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 548. Amberg GC, Navedo MF, Nieves-Cintrón M, Molkentin JD, Santana L. Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J Physiol 579: 187–201, 2007. doi: 10.1113/jphysiol.2006.124420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Hoyer J, Köhler R, Distler A. Mechanosensitive Ca2+ oscillations and STOC activation in endothelial cells. FASEB J 12: 359–366, 1998. doi: 10.1096/fasebj.12.3.359. [DOI] [PubMed] [Google Scholar]
- 550. Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, Fleming I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc Res 80: 445–452, 2008. doi: 10.1093/cvr/cvn207. [DOI] [PubMed] [Google Scholar]
- 551. Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J, Köhler R. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One 2: e827, 2007. doi: 10.1371/journal.pone.0000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Si H, Heyken WT, Wölfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C, Hoyer J, Köhler R. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res 99: 537–544, 2006. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- 553. Berna-Erro A, Izquierdo-Serra M, Sepúlveda RV, Rubio-Moscardo F, Doñate-Macián P, Serra SA, Carrillo-Garcia J, Peralvarez-Marin A, Gonzalez-Nilo F, Fernandez-Fernandez JM, Valverde MA. Structural determinants of 5',6'-epoxyeicosatrienoic acid binding to and activation of TRPV4 channel. Sci Rep 7: 10522, 2017. doi: 10.1038/s41598-017-11274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Takahashi N, Hamada-Nakahara S, Itoh Y, Takemura K, Shimada A, Ueda Y, Kitamata M, Matsuoka R, Hanawa-Suetsugu K, Senju Y, Mori MX, Kiyonaka S, Kohda D, Kitao A, Mori Y, Suetsugu S. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P(2). Nat Commun 5: 4994, 2014. doi: 10.1038/ncomms5994. [DOI] [PubMed] [Google Scholar]
- 555. Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE, Sonkusare SK. Nitric oxide-dependent feedback loop regulates transient receptor potential vanilloid 4 (TRPV4) channel cooperativity and endothelial function in small pulmonary arteries. J Am Heart Assoc 6: 007157, 2017. doi: 10.1161/JAHA.117.007157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556. Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2: 435–442, 2010. doi: 10.1039/c0ib00034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Swain SM, Romac JM, Shahid RA, Pandol SJ, Liedtke W, Vigna SR, Liddle RA. TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest 130: 2527–2541, 2020. doi: 10.1172/JCI134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Lu T, Wang XL, Chai Q, Sun X, Sieck GC, Katusic ZS, Lee HC. Role of the endothelial caveolae microdomain in shear stress-mediated coronary vasorelaxation. J Biol Chem 292: 19013–19023, 2017. doi: 10.1074/jbc.M117.786152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Baratchi S, Knoerzer M, Khoshmanesh K, Mitchell A, McIntyre P. Shear stress regulates TRPV4 channel clustering and translocation from adherens junctions to the basal membrane. Sci Rep 7: 15942, 2017. doi: 10.1038/s41598-017-16276-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, Ingber DE. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res 104: 1123–1130, 2009. doi: 10.1161/CIRCRESAHA.108.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol 293: L923–L932, 2007. doi: 10.1152/ajplung.00221.2007. [DOI] [PubMed] [Google Scholar]
- 562. Michalick L, Erfinanda L, Weichelt U, van der Giet M, Liedtke W, Kuebler WM. Transient receptor potential vanilloid 4 and serum glucocorticoid-regulated kinase 1 are critical mediators of lung injury in overventilated mice in vivo. Anesthesiology 126: 300–311, 2017. doi: 10.1097/ALN.0000000000001443. [DOI] [PubMed] [Google Scholar]
- 563. Wang W, Zhang X, Gao Q, Xu H. TRPML1: an ion channel in the lysosome. Handb Exp Pharmacol 222: 631–645, 2014. doi: 10.1007/978-3-642-54215-2_24. [DOI] [PubMed] [Google Scholar]
- 564. Xu H, Delling M, Li L, Dong X, Clapham DE. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc Natl Acad Sci USA 104: 18321–18326, 2007. doi: 10.1073/pnas.0709096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565. Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, Ferrer M, Xu H. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci USA 112: E1373–E1381, 2015. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Shang S, Zhu F, Liu B, Chai Z, Wu Q, Hu M, Wang Y, Huang R, Zhang X, Wu X, Sun L, Wang Y, Wang L, Xu H, Teng S, Liu B, Zheng L, Zhang C, Zhang F, Feng X, Zhu D, Wang C, Liu T, Zhu MX, Zhou Z. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J Cell Biol 215: 369–381, 2016. doi: 10.1083/jcb.201603081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Thakore P, Pritchard HA, Griffin CS, Yamasaki E, Drumm BT, Lane C, Sanders KM, Feng Earley Y, Earley S. TRPML1 channels initiate Ca2+ sparks in vascular smooth muscle cells. Sci Signal 13: eaba1015, 2020. doi: 10.1126/scisignal.aba1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat Commun 1: 38, 2010. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569. Li M, Zhang WK, Benvin NM, Zhou X, Su D, Li H, Wang S, Michailidis IE, Tong L, Li X, Yang J. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat Struct Mol Biol 24: 205–213, 2017. doi: 10.1038/nsmb.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Onyenwoke RU, Sexton JZ, Yan F, Díaz MC, Forsberg LJ, Major MB, Brenman JE. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem J 470: 331–342, 2015. doi: 10.1042/BJ20150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Grimm C, Jörs S, Saldanha SA, Obukhov AG, Pan B, Oshima K, Cuajungco MP, Chase P, Hodder P, Heller S. Small molecule activators of TRPML3. Chem Biol 17: 135–148, 2010. doi: 10.1016/j.chembiol.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3: 731, 2012. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Chen CC, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, Bracher F, Biel M, Wahl-Schott C, Grimm C. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun 5: 4681, 2014. doi: 10.1038/ncomms5681. [DOI] [PubMed] [Google Scholar]
- 574. Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G. Identification of the gene causing mucolipidosis type IV. Nat Genet 26: 118–123, 2000. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 575. Venkatachalam K, Wong CO, Zhu MX. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58: 48–56, 2015. doi: 10.1016/j.ceca.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576. Berman ER, Livni N, Shapira E, Merin S, Levij IS. Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. J Pediatr 84: 519–526, 1974. doi: 10.1016/s0022-3476(74)80671-2. [DOI] [PubMed] [Google Scholar]
- 577. Zhang F, Jin S, Yi F, Li PL. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J Cell Mol Med 13: 3174–3185, 2009. doi: 10.1111/j.1582-4934.2008.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Zhang F, Xu M, Han WQ, Li PL. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1-/- cells. Am J Physiol Cell Physiol 301: C421–C430, 2011. doi: 10.1152/ajpcell.00393.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459: 596–600, 2009. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580. Xu M, Li X, Walsh SW, Zhang Y, Abais JM, Boini KM, Li PL. Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes. Am J Physiol Cell Physiol 304: C458–C466, 2013. doi: 10.1152/ajpcell.00342.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Venugopal B, Browning MF, Curcio-Morelli C, Varro A, Michaud N, Nanthakumar N, Walkley SU, Pickel J, Slaugenhaupt SA. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. Am J Hum Genet 81: 1070–1083, 2007. doi: 10.1086/521954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 270: 633–637, 1995. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 583. Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol 508: 211–221, 1998. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, Delling M, Marugan J, Ferrer M, Xu H. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7: 12109, 2016. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333: 1440–1445, 2011. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 586. Putney JW Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS. Mechanisms of capacitative calcium entry. J Cell Sci 114: 2223–2229, 2001. doi: 10.1242/jcs.114.12.2223. [DOI] [PubMed] [Google Scholar]
- 587. Parekh AB. On the activation mechanism of store-operated calcium channels. Pflugers Arch 453: 303–311, 2006. doi: 10.1007/s00424-006-0089-y. [DOI] [PubMed] [Google Scholar]
- 588. Spinelli AM, Trebak M. Orai channel-mediated Ca2+ signals in vascular and airway smooth muscle. Am J Physiol Cell Physiol 310: C402–C413, 2016. doi: 10.1152/ajpcell.00355.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Krishnan V, Ali S, Gonzales AL, Thakore P, Griffin CS, Yamasaki E, Alvarado MG, Johnson MT, Trebak M, Earley S. STIM1-dependent peripheral coupling governs the contractility of vascular smooth muscle cells. Elife 11: e70278, 2022. doi: 10.7554/eLife.70278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590. Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355: 353–356, 1992. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 591. Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature 443: 230–233, 2006. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 592. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169: 435–445, 2005. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem 281: 20661–20665, 2006. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 594. Hogan PG, Rao A. Store-operated calcium entry: mechanisms and modulation. Biochem Biophys Res Commun 460: 40–49, 2015. doi: 10.1016/j.bbrc.2015.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Baudel M, Shi J, Large WA, Albert AP. Insights into activation mechanisms of store-operated TRPC1 channels in vascular smooth muscle. Cells 9: 179, 2020. doi: 10.3390/cells9010179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol 559: 685–706, 2004. doi: 10.1113/jphysiol.2004.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev 81: 1415–1459, 2001. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 598. Gees M, Owsianik G, Nilius B, Voets T. TRP channels. Compr Physiol 2: 563–608, 2012. doi: 10.1002/cphy.c110026. [DOI] [PubMed] [Google Scholar]
- 599. Lopez JJ, Jardin I, Sanchez-Collado J, Salido GM, Smani T, Rosado JA. TRPC channels in the SOCE scenario. Cells 9: 126, 2020. doi: 10.3390/cells9010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600. Lopez JJ, Jardin I, Albarrán L, Sanchez-Collado J, Cantonero C, Salido GM, Smani T, Rosado JA. Molecular basis and regulation of store-operated calcium entry. Adv Exp Med Biol 1131: 445–469, 2020. doi: 10.1007/978-3-030-12457-1_17. [DOI] [PubMed] [Google Scholar]
- 601. Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol 7: 179–185, 2005. doi: 10.1038/ncb1218. [DOI] [PubMed] [Google Scholar]
- 602. Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch 455: 465–477, 2007. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- 603. Quick K, Zhao J, Eijkelkamp N, Linley JE, Rugiero F, Cox JJ, Raouf R, Gringhuis M, Sexton JE, Abramowitz J, Taylor R, Forge A, Ashmore J, Kirkwood N, Kros CJ, Richardson GP, Freichel M, Flockerzi V, Birnbaumer L, Wood JN. TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biol 2: 120068, 2012. doi: 10.1098/rsob.120068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3 mediates pyrimidine receptor-induced depolarization of cerebral arteries. Am J Physiol Heart Circ Physiol 288: H2055–H2061, 2005. doi: 10.1152/ajpheart.00861.2004. [DOI] [PubMed] [Google Scholar]
- 605. Woo SH, Trinh TN. P2 receptors in cardiac myocyte pathophysiology and mechanotransduction. Int J Mol Sci 22: 251, 2020. doi: 10.3390/ijms22010251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Praetorius HA, Leipziger J. ATP release from non-excitable cells. Purinergic Signal 5: 433–446, 2009. doi: 10.1007/s11302-009-9146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev 50: 413–492, 1998. [PubMed] [Google Scholar]
- 608. Li M, Kawate T, Silberberg SD, Swartz KJ. Pore-opening mechanism in trimeric P2X receptor channels. Nat Commun 1: 44, 2010. doi: 10.1038/ncomms1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Bodin P, Burnstock G. Synergistic effect of acute hypoxia on flow-induced release of ATP from cultured endothelial cells. Experientia 51: 256–259, 1995. doi: 10.1007/BF01931108. [DOI] [PubMed] [Google Scholar]
- 610. Burnstock G. Release of vasoactive substances from endothelial cells by shear stress and purinergic mechanosensory transduction. J Anat 194: 335–342, 1999. doi: 10.1046/j.1469-7580.1999.19430335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611. Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol 299: H1146–H1152, 2010. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612. Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP release. Am J Physiol Cell Physiol 281: C1158–C1164, 2001. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- 613. Sneddon P, Burnstock G. ATP as a co-transmitter in rat tail artery. Eur J Pharmacol 106: 149–152, 1984. doi: 10.1016/0014-2999(84)90688-5. [DOI] [PubMed] [Google Scholar]
- 614. Hopwood AM, Burnstock G. ATP mediates coronary vasoconstriction via P2x-purinoceptors and coronary vasodilatation via P2y-purinoceptors in the isolated perfused rat heart. Eur J Pharmacol 136: 49–54, 1987. doi: 10.1016/0014-2999(87)90777-1. [DOI] [PubMed] [Google Scholar]
- 615. Inscho EW, Cook AK, Imig JD, Vial C, Evans RJ. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J Clin Invest 112: 1895–1905, 2003. doi: 10.1172/JCI18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Liu C, Mather S, Huang Y, Garland CJ, Yao X. Extracellular ATP facilitates flow-induced vasodilatation in rat small mesenteric arteries. Am J Physiol Heart Circ Physiol 286: H1688–H1695, 2004. doi: 10.1152/ajpheart.00576.2003. [DOI] [PubMed] [Google Scholar]
- 617. Ando J, Komatsuda T, Kamiya A. Cytoplasmic calcium response to fluid shear stress in cultured vascular endothelial cells. In Vitro Cell Dev Biol 24: 871–877, 1988. doi: 10.1007/BF02623896. [DOI] [PubMed] [Google Scholar]
- 618. Mo M, Eskin SG, Schilling WP. Flow-induced changes in Ca2+ signaling of vascular endothelial cells: effect of shear stress and ATP. Am J Physiol Heart Circ Physiol 260: H1698–H1707, 1991. doi: 10.1152/ajpheart.1991.260.5.H1698. [DOI] [PubMed] [Google Scholar]
- 619. Dull RO, Davies PF. Flow modulation of agonist (ATP)-response (Ca2+) coupling in vascular endothelial cells. Am J Physiol Heart Circ Physiol 261: H149–H154, 1991. doi: 10.1152/ajpheart.1991.261.1.H149. [DOI] [PubMed] [Google Scholar]
- 620. Kessler S, Clauss WG, Fronius M. Laminar shear stress modulates the activity of heterologously expressed P2X4 receptors. Biochim Biophys Acta 1808: 2488–2495, 2011. doi: 10.1016/j.bbamem.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 621. Forst AL, Olteanu VS, Mollet G, Wlodkowski T, Schaefer F, Dietrich A, Reiser J, Gudermann T, Mederos y Schnitzler M, Storch U. Podocyte purinergic P2X4 channels are mechanotransducers that mediate cytoskeletal disorganization. J Am Soc Nephrol 27: 848–862, 2016. doi: 10.1681/ASN.2014111144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622. Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res 95: 269–280, 2012. doi: 10.1093/cvr/cvs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. El-Lakany MA, Kowalewska PM, Sandow SL, Ellis CG, Welsh DG. Autocrine P2X4 receptor activation in RBCs drives oxygen-dependent hyperemic responses in mouse skeletal muscle capillaries (Abstract). FASEB J 36: R3060, 2022. doi: 10.1096/fasebj.2022.36.S1.R3060. [DOI] [Google Scholar]
- 624. Daneva Z, Ottolini M, Chen YL, Klimentova E, Kuppusamy M, Shah SA, Minshall RD, Seye CI, Laubach VE, Isakson BE, Sonkusare SK. Endothelial pannexin 1-TRPV4 channel signaling lowers pulmonary arterial pressure in mice. Elife 10: e67777, 2021. doi: 10.7554/eLife.67777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625. Korenaga R, Yamamoto K, Ohura N, Sokabe T, Kamiya A, Ando J. Sp1-mediated downregulation of P2X4 receptor gene transcription in endothelial cells exposed to shear stress. Am J Physiol Heart Circ Physiol 280: H2214–H2221, 2001. doi: 10.1152/ajpheart.2001.280.5.H2214. [DOI] [PubMed] [Google Scholar]
- 626. Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M, Ando J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci 124: 3477–3483, 2011. doi: 10.1242/jcs.087221. [DOI] [PubMed] [Google Scholar]
- 627. Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, Fukuda T, Sato T, Sekine K, Kato S, Isshiki M, Fujita T, Kobayashi M, Kawamura K, Masuda H, Kamiya A, Ando J. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat Med 12: 133–137, 2006. doi: 10.1038/nm1338. [DOI] [PubMed] [Google Scholar]
- 628. Kachar B, Parakkal M, Kurc M, Zhao Y, Gillespie PG. High-resolution structure of hair-cell tip links. Proc Natl Acad Sci USA 97: 13336–13341, 2000. doi: 10.1073/pnas.97.24.13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Pan B, Akyuz N, Liu XP, Asai Y, Nist-Lund C, Kurima K, Derfler BH, György B, Limapichat W, Walujkar S, Wimalasena LN, Sotomayor M, Corey DP, Holt JR. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron 99: 736–753.e6, 2018. doi: 10.1016/j.neuron.2018.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630. Jia Y, Zhao Y, Kusakizako T, Wang Y, Pan C, Zhang Y, Nureki O, Hattori M, Yan Z. TMC1 and TMC2 proteins are pore-forming subunits of mechanosensitive ion channels. Neuron 105: 310–321.e3, 2020. doi: 10.1016/j.neuron.2019.10.017. [DOI] [PubMed] [Google Scholar]
- 631. Ohmori H. Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. J Physiol 359: 189–217, 1985. doi: 10.1113/jphysiol.1985.sp015581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Cunningham CL, Qiu X, Wu Z, Zhao B, Peng G, Kim YH, Lauer A, Müller U. TMIE defines pore and gating properties of the mechanotransduction channel of mammalian cochlear hair cells. Neuron 107: 126–143.e8, 2020. doi: 10.1016/j.neuron.2020.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633. Zhao B, Wu Z, Grillet N, Yan L, Xiong W, Harkins-Perry S, Muller U. TMIE is an essential component of the mechanotransduction machinery of cochlear hair cells. Neuron 84: 954–967, 2014. doi: 10.1016/j.neuron.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634. Giese AP, Tang YQ, Sinha GP, Bowl MR, Goldring AC, Parker A, Freeman MJ, Brown SD, Riazuddin S, Fettiplace R, Schafer WR, Frolenkov GI, Ahmed ZM. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat Commun 8: 43, 2017. doi: 10.1038/s41467-017-00061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635. Xiong W, Grillet N, Elledge HM, Wagner TF, Zhao B, Johnson KR, Kazmierczak P, Müller U. TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell 151: 1283–1295, 2012. doi: 10.1016/j.cell.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636. Alagramam KN, Goodyear RJ, Geng R, Furness DN, van Aken AF, Marcotti W, Kros CJ, Richardson GP. Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One 6: e19183, 2011. doi: 10.1371/journal.pone.0019183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637. Lötzer K, Döpping S, Connert S, Gräbner R, Spanbroek R, Lemser B, Beer M, Hildner M, Hehlgans T, van der Wall M, Mebius RE, Lovas A, Randolph GJ, Weih F, Habenicht AJ. Mouse aorta smooth muscle cells differentiate into lymphoid tissue organizer-like cells on combined tumor necrosis factor receptor-1/lymphotoxin beta-receptor NF-kappaB signaling. Arterioscler Thromb Vasc Biol 30: 395–402, 2010. doi: 10.1161/ATVBAHA.109.191395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638. Shirvani SM, Mookanamparambil L, Ramoni MF, Chin MT. Transcription factor CHF1/Hey2 regulates the global transcriptional response to platelet-derived growth factor in vascular smooth muscle cells. Physiol Genomics 30: 61–68, 2007. doi: 10.1152/physiolgenomics.00277.2006. [DOI] [PubMed] [Google Scholar]
- 639. Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res 103: 654–661, 2008. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Project SMTS. Smooth Muscle Transcriptome Sequencing Project (Online). Reno, NV: University of Nevada, Reno. https://med.unr.edu/physio/transcriptome.
- 641. Yuan F, Yang H, Xue Y, Kong D, Ye R, Li C, Zhang J, Theprungsirikul L, Shrift T, Krichilsky B, Johnson DM, Swift GB, He Y, Siedow JN, Pei ZM. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosensing in Arabidopsis. Nature 514: 367–371, 2014. doi: 10.1038/nature13593. [DOI] [PubMed] [Google Scholar]
- 642. Liu X, Wang J, Sun L. Structure of the hyperosmolality-gated calcium-permeable channel OSCA1.2. Nat Commun 9: 5060, 2018. doi: 10.1038/s41467-018-07564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643. Murthy SE, Dubin AE, Whitwam T, Jojoa-Cruz S, Cahalan SM, Mousavi SAR, Ward AB, Patapoutian A. OSCA/TMEM63 are an evolutionarily conserved family of mechanically activated ion channels. Elife 7: e41844, 2018. doi: 10.7554/eLife.41844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644. Zhao X, Yan X, Liu Y, Zhang P, Ni X. Co-expression of mouse TMEM63A, TMEM63B and TMEM63C confers hyperosmolarity activated ion currents in HEK293 cells. Cell Biochem Funct 34: 238–241, 2016. doi: 10.1002/cbf.3185. [DOI] [PubMed] [Google Scholar]
- 645. Kitamura K, Yamazaki J. Chloride channels and their functional roles in smooth muscle tone in the vasculature. Jpn J Pharmacol 85: 351–357, 2001. doi: 10.1254/jjp.85.351. [DOI] [PubMed] [Google Scholar]
- 646. Bulley S, Jaggar JH. Cl− channels in smooth muscle cells. Pflugers Arch 466: 861–872, 2014. doi: 10.1007/s00424-013-1357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647. Hübner CA, Schroeder BC, Ehmke H. Regulation of vascular tone and arterial blood pressure: role of chloride transport in vascular smooth muscle. Pflugers Arch 467: 605–614, 2015. doi: 10.1007/s00424-014-1684-y. [DOI] [PubMed] [Google Scholar]
- 648. Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl- conductance in smooth muscle. Am J Physiol Cell Physiol 271: C435–C454, 1996. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- 649. Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol 269: H348–H355, 1995. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 650. Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 268: C799–C822, 1995. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 651. Smith JM, Jones AW. Calcium-dependent fluxes of potassium-42 and chloride-36 during norepinephrine activation of rat aorta. Circ Res 56: 507–516, 1985. doi: 10.1161/01.RES.56.4.507. [DOI] [PubMed] [Google Scholar]
- 652. Casteels R, Kitamura K, Kuriyama H, Suzuki H. The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol 271: 41–61, 1977. doi: 10.1113/jphysiol.1977.sp011989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653. Wahlström BA, Svennerholm B. Potentiation and inhibition of noradrenaline induced contractions of the rat portal vein in anion substituted solutions. Acta Physiol Scand 92: 404–411, 1974. doi: 10.1111/j.1748-1716.1974.tb05758.x. [DOI] [PubMed] [Google Scholar]
- 654. Pacaud P, Loirand G, Mironneau C, Mironneau J. Noradrenaline activates a calcium-activated chloride conductance and increases the voltage-dependent calcium current in cultured single cells of rat portal vein. Br J Pharmacol 97: 139–146, 1989. doi: 10.1111/j.1476-5381.1989.tb11934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655. Van Helden DF. An alpha-adrenoceptor-mediated chloride conductance in mesenteric veins of the guinea-pig. J Physiol 401: 489–501, 1988. doi: 10.1113/jphysiol.1988.sp017174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656. Byrne NG, Large WA. Mechanism of action of alpha-adrenoceptor activation in single cells freshly dissociated from the rabbit portal vein. Br J Pharmacol 94: 475–482, 1988. doi: 10.1111/j.1476-5381.1988.tb11550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657. Hogg RC, Wang Q, Helliwell RM, Large WA. Properties of spontaneous inward currents in rabbit pulmonary artery smooth muscle cells. Pflugers Arch 425: 233–240, 1993. doi: 10.1007/BF00374172. [DOI] [PubMed] [Google Scholar]
- 658. Liu GX, Vepa S, Artman M, Coetzee WA. Modulation of human cardiovascular outward rectifying chloride channel by intra- and extracellular ATP. Am J Physiol Heart Circ Physiol 293: H3471–H3479, 2007. doi: 10.1152/ajpheart.00357.2007. [DOI] [PubMed] [Google Scholar]
- 659. Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157: 447–458, 2014. doi: 10.1016/j.cell.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660. Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344: 634–638, 2014. doi: 10.1126/science.1252826. [DOI] [PubMed] [Google Scholar]
- 661. Robert R, Norez C, Becq F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl− transport of mouse aortic smooth muscle cells. J Physiol 568: 483–495, 2005. doi: 10.1113/jphysiol.2005.085019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662. Robert R, Savineau JP, Norez C, Becq F, Guibert C. Expression and function of cystic fibrosis transmembrane conductance regulator in rat intrapulmonary arteries. Eur Respir J 30: 857–864, 2007. doi: 10.1183/09031936.00060007. [DOI] [PubMed] [Google Scholar]
- 663. Meissner A, Yang J, Kroetsch JT, Sauvé M, Dax H, Momen A, Noyan-Ashraf MH, Heximer S, Husain M, Lidington D, Bolz SS. Tumor necrosis factor-alpha-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation 125: 2739–2750, 2012. doi: 10.1161/CIRCULATIONAHA.111.047316. [DOI] [PubMed] [Google Scholar]
- 664. Matchkov VV, Secher Dam V, Bødtkjer DM, Aalkjær C. Transport and function of chloride in vascular smooth muscles. J Vasc Res 50: 69–87, 2013. doi: 10.1159/000345242. [DOI] [PubMed] [Google Scholar]
- 665. Dam VS, Boedtkjer DM, Aalkjaer C, Matchkov V. The bestrophin- and TMEM16A-associated Ca2+-activated Cl− channels in vascular smooth muscles. Channels (Austin) 8: 361–369, 2014. doi: 10.4161/chan.29531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666. Matchkov VV, Aalkjaer C, Nilsson H. Distribution of cGMP-dependent and cGMP-independent Ca2+-activated Cl- conductances in smooth muscle cells from different vascular beds and colon. Pflugers Arch 451: 371–379, 2005. doi: 10.1007/s00424-005-1472-9. [DOI] [PubMed] [Google Scholar]
- 667. Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume regulated chloride channels in canine vascular smooth muscle cells. J Physiol 507: 729–736, 1998. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668. Greenwood IA, Large WA. Properties of a Cl- current activated by cell swelling in rabbit portal vein vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 275: H1524–H1532, 1998. doi: 10.1152/ajpheart.1998.275.5.H1524. [DOI] [PubMed] [Google Scholar]
- 669. Yano S, Ishikawa T, Tsuda H, Obara K, Nakayama K. Ionic mechanism for contractile response to hyposmotic challenge in canine basilar arteries. Am J Physiol Cell Physiol 288: C702–C709, 2005. doi: 10.1152/ajpcell.00367.2003. [DOI] [PubMed] [Google Scholar]
- 670. Greenwood IA, Leblanc N. Overlapping pharmacology of Ca2+-activated Cl- and K+ channels. Trends Pharmacol Sci 28: 1–5, 2007. doi: 10.1016/j.tips.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 671. Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov 8: 153–171, 2009. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672. Doughty JM, Miller AL, Langton PD. Non-specificity of chloride channel blockers in rat cerebral arteries: block of the L-type calcium channel. J Physiol 507: 433–439, 1998. doi: 10.1111/j.1469-7793.1998.433bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673. Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322: 590–594, 2008. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 674. Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134: 1019–1029, 2008. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675. Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455: 1210–1215, 2008. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 676. Oh U, Jung J. Cellular functions of TMEM16/anoctamin. Pflugers Arch 468: 443–453, 2016. doi: 10.1007/s00424-016-1790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677. Duran C, Qu Z, Osunkoya AO, Cui Y, Hartzell HC. ANOs 3-7 in the anoctamin/Tmem16 Cl- channel family are intracellular proteins. Am J Physiol Cell Physiol 302: C482–C493, 2012. doi: 10.1152/ajpcell.00140.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678. Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol 67: 719–758, 2005. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 679. Xiao Q, Yu K, Perez-Cornejo P, Cui Y, Arreola J, Hartzell HC. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc Natl Acad Sci USA 108: 8891–8896, 2011. doi: 10.1073/pnas.1102147108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680. Jin X, Shah S, Du X, Zhang H, Gamper N. Activation of Ca2+-activated Cl- channel ANO1 by localized Ca2+ signals. J Physiol 594: 19–30, 2016. doi: 10.1113/jphysiol.2014.275107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681. Pritchard HA, Leblanc N, Albert AP, Greenwood IA. Inhibitory role of phosphatidylinositol 4,5-bisphosphate on TMEM16A-encoded calcium-activated chloride channels in rat pulmonary artery. Br J Pharmacol 171: 4311–4321, 2014. doi: 10.1111/bph.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682. Tembo M, Wozniak KL, Bainbridge RE, Carlson AE. Phosphatidylinositol 4,5-bisphosphate (PIP2) and Ca2+ are both required to open the Cl- channel TMEM16A. J Biol Chem 294: 12556–12564, 2019. doi: 10.1074/jbc.RA118.007128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683. De Jesús-Pérez JJ, Cruz-Rangel S, Espino-Saldaña AE, Martinez-Torres A, Qu Z, Hartzell HC, Corral-Fernandez NE, Perez-Cornejo P, Arreola J. Phosphatidylinositol 4,5-bisphosphate, cholesterol, and fatty acids modulate the calcium-activated chloride channel TMEM16A (ANO1). Biochim Biophys Acta Mol Cell Biol Lipids 1863: 299–312, 2018. doi: 10.1016/j.bbalip.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684. Le SC, Jia Z, Chen J, Yang H. Molecular basis of PIP2-dependent regulation of the Ca2+-activated chloride channel TMEM16A. Nat Commun 10: 3769, 2019. doi: 10.1038/s41467-019-11784-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685. Arreola J, Hartzell HC. Wasted TMEM16A channels are rescued by phosphatidylinositol 4,5-bisphosphate. Cell Calcium 84: 102103, 2019. doi: 10.1016/j.ceca.2019.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 686. Yu K, Jiang T, Cui Y, Tajkhorshid E, Hartzell HC. A network of phosphatidylinositol 4,5-bisphosphate binding sites regulates gating of the Ca2+-activated Cl- channel ANO1 (TMEM16A). Proc Natl Acad Sci USA 116: 19952–19962, 2019. doi: 10.1073/pnas.1904012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687. Dam VS, Boedtkjer DM, Nyvad J, Aalkjaer C, Matchkov V. TMEM16A knockdown abrogates two different Ca2+-activated Cl- currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch 466: 1391–1409, 2014. doi: 10.1007/s00424-013-1382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688. Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol 588: 2305–2314, 2010. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689. Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, Jaggar JH. TMEM16A channels generate Ca2+-activated Cl- currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol 301: H1819–H1827, 2011. doi: 10.1152/ajpheart.00404.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690. Wang M, Yang H, Zheng LY, Zhang Z, Tang YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG, Guan YY. Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation 125: 697–707, 2012. doi: 10.1161/CIRCULATIONAHA.111.041806. [DOI] [PubMed] [Google Scholar]
- 691. Davis AJ, Forrest AS, Jepps TA, Valencik ML, Wiwchar M, Singer CA, Sones WR, Greenwood IA, Leblanc N. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol 299: C948–C959, 2010. doi: 10.1152/ajpcell.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692. Zawieja SD, Castorena JA, Gui P, Li M, Bulley SA, Jaggar JH, Rock JR, Davis MJ. Ano1 mediates pressure-sensitive contraction frequency changes in mouse lymphatic collecting vessels. J Gen Physiol 151: 532–554, 2019. doi: 10.1085/jgp.201812294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693. Matchkov VV, Boedtkjer DM, Aalkjaer C. The role of Ca2+ activated Cl- channels in blood pressure control. Curr Opin Pharmacol 21: 127–137, 2015. doi: 10.1016/j.coph.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 694. Li RS, Wang Y, Chen HS, Jiang FY, Tu Q, Li WJ, Yin RX. TMEM16A contributes to angiotensin II-induced cerebral vasoconstriction via the RhoA/ROCK signaling pathway. Mol Med Rep 13: 3691–3699, 2016. doi: 10.3892/mmr.2016.4979. [DOI] [PubMed] [Google Scholar]
- 695. Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijártó IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, Schroeder BC, Hübner CA. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest 124: 675–686, 2014. doi: 10.1172/JCI70025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696. Mohanakumar S, Majgaard J, Telinius N, Katballe N, Pahle E, Hjortdal V, Boedtkjer D. Spontaneous and alpha-adrenoceptor-induced contractility in human collecting lymphatic vessels require chloride. Am J Physiol Heart Circ Physiol 315: H389–H401, 2018. doi: 10.1152/ajpheart.00551.2017. [DOI] [PubMed] [Google Scholar]
- 697. Wang B, Li C, Huai R, Qu Z. Overexpression of ANO1/TMEM16A, an arterial Ca2+-activated Cl- channel, contributes to spontaneous hypertension. J Mol Cell Cardiol 82: 22–32, 2015. doi: 10.1016/j.yjmcc.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 698. Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, Singer CA, Valencik ML, Greenwood IA, Leblanc N. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 303: C1229–C1243, 2012. doi: 10.1152/ajpcell.00044.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699. Davis AJ, Shi J, Pritchard HA, Chadha PS, Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP, Greenwood IA. Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh)-A01. Br J Pharmacol 168: 773–784, 2013. doi: 10.1111/j.1476-5381.2012.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700. Hwang SJ, Basma N, Sanders KM, Ward SM. Effects of new-generation inhibitors of the calcium-activated chloride channel anoctamin 1 on slow waves in the gastrointestinal tract. Br J Pharmacol 173: 1339–1349, 2016. doi: 10.1111/bph.13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701. Askew Page HR, Dalsgaard T, Baldwin SN, Jepps TA, Povstyan O, Olesen SP, Greenwood IA. TMEM16A is implicated in the regulation of coronary flow and is altered in hypertension. Br J Pharmacol 176: 1635–1648, 2019. doi: 10.1111/bph.14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702. Boedtkjer DM, Kim S, Jensen AB, Matchkov VM, Andersson KE. New selective inhibitors of calcium-activated chloride channels—T16A(inh)-A01, CaCC(inh)-A01 and MONNA—what do they inhibit? Br J Pharmacol 172: 4158–4172, 2015. doi: 10.1111/bph.13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703. Seo Y, Kim J, Chang J, Kim SS, Namkung W, Kim I. Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur J Med Chem 160: 245–255, 2018. doi: 10.1016/j.ejmech.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 704. Drumm BT, Rembetski BE, Baker SA, Sanders KM. Tonic inhibition of murine proximal colon is due to nitrergic suppression of Ca2+ signaling in interstitial cells of Cajal. Sci Rep 9: 4402, 2019. doi: 10.1038/s41598-019-39729-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705. Jensen AB, Joergensen HB, Dam VS, Kamaev D, Boedtkjer D, Füchtbauer EM, Aalkjaer C, Matchkov VV. Variable contribution of TMEM16A to tone in murine arterial vasculature. Basic Clin Pharmacol Toxicol 123: 30–41, 2018. doi: 10.1111/bcpt.12984. [DOI] [PubMed] [Google Scholar]
- 706. Doughty JM, Langton PD. Measurement of chloride flux associated with the myogenic response in rat cerebral arteries. J Physiol 534: 753–761, 2001. doi: 10.1111/j.1469-7793.2001.t01-1-00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707. Yip KP, Balasubramanian L, Kan C, Wang L, Liu R, Ribeiro-Silva L, Sham JS. Intraluminal pressure triggers myogenic response via activation of calcium spark and calcium-activated chloride channel in rat renal afferent arteriole. Am J Physiol Renal Physiol 315: F1592–F1600, 2018. doi: 10.1152/ajprenal.00239.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708. Wang Q, Leo MD, Narayanan D, Kuruvilla KP, Jaggar JH. Local coupling of TRPC6 to ANO1/TMEM16A channels in smooth muscle cells amplifies vasoconstriction in cerebral arteries. Am J Physiol Cell Physiol 310: C1001–C1009, 2016. doi: 10.1152/ajpcell.00092.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709. Matchkov VV, Black Joergensen H, Kamaev D, Hoegh Jensen A, Beck HC, Skryabin BV, Aalkjaer C. A paradoxical increase of force development in saphenous and tail arteries from heterozygous ANO1 knockout mice. Physiol Rep 8: e14645, 2020. doi: 10.14814/phy2.14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710. Boedtkjer DM, Matchkov VV, Boedtkjer E, Nilsson H, Aalkjaer C. Vasomotion has chloride-dependency in rat mesenteric small arteries. Pflugers Arch 457: 389–404, 2008. doi: 10.1007/s00424-008-0532-3. [DOI] [PubMed] [Google Scholar]
- 711. Matchkov VV. Mechanisms of cellular synchronization in the vascular wall. Mechanisms of vasomotion. Dan Med Bull 57: B4191, 2010. [PubMed] [Google Scholar]
- 712. Hwang SJ, Blair PJ, Britton FC, O’Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM, Ward SM. Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol 587: 4887–4904, 2009. doi: 10.1113/jphysiol.2009.176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713. Choi S, Kang HG, Wu MJ, Jiao HY, Shin DH, Hong C, Jun JY. Effects of Ca2+-activated Cl- channel ANO1 inhibitors on pacemaker activity in interstitial cells of Cajal. Cell Physiol Biochem 51: 2887–2899, 2018. doi: 10.1159/000496041. [DOI] [PubMed] [Google Scholar]
- 714. Malysz J, Gibbons SJ, Saravanaperumal SA, Du P, Eisenman ST, Cao C, Oh U, Saur D, Klein S, Ordog T, Farrugia G. Conditional genetic deletion of Ano1 in interstitial cells of Cajal impairs Ca2+ transients and slow waves in adult mouse small intestine. Am J Physiol Gastrointest Liver Physiol 312: G228–G245, 2017. doi: 10.1152/ajpgi.00363.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715. Forrest AS, Hennig GW, Jokela-Willis S, Park CD, Sanders KM. Prostaglandin regulation of gastric slow waves and peristalsis. Am J Physiol Gastrointest Liver Physiol 296: G1180–G1190, 2009. doi: 10.1152/ajpgi.90724.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716. Romanenko VG, Davies PF, Levitan I. Dual effect of fluid shear stress on volume-regulated anion current in bovine aortic endothelial cells. Am J Physiol Cell Physiol 282: C708–C718, 2002. doi: 10.1152/ajpcell.00247.2001. [DOI] [PubMed] [Google Scholar]
- 717. Barakat AI, Leaver EV, Pappone PA, Davies PF. A flow-activated chloride-selective membrane current in vascular endothelial cells. Circ Res 85: 820–828, 1999. doi: 10.1161/01.res.85.9.820. [DOI] [PubMed] [Google Scholar]
- 718. Gautam M, Shen Y, Thirkill TL, Douglas GC, Barakat AI. Flow-activated chloride channels in vascular endothelium. Shear stress sensitivity, desensitization dynamics, and physiological implications. J Biol Chem 281: 36492–36500, 2006. doi: 10.1074/jbc.M605866200. [DOI] [PubMed] [Google Scholar]
- 719. Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY. Studies on expression and function of the TMEM16a calcium-activated chloride channel. Proc Natl Acad Sci USA 106: 21413–21418, 2009. doi: 10.1073/pnas.0911935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720. Noreng S, Li T, Payandeh J. Structural pharmacology of voltage-gated sodium channels. J Mol Biol 433: 166967, 2021. doi: 10.1016/j.jmb.2021.166967. [DOI] [PubMed] [Google Scholar]
- 721. Hille B, Schwarz W. Potassium channels as multi-ion single-file pores. J Gen Physiol 72: 409–442, 1978. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722. Lin W, Laitko U, Juranka PF, Morris CE. Dual stretch responses of mHCN2 pacemaker channels: accelerated activation, accelerated deactivation. Biophys J 92: 1559–1572, 2007. doi: 10.1529/biophysj.106.092478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723. Morris CE, Laitko U. The mechanosensitivity of voltage-gated channels may contribute to cardiac mechanoelectric feedback. In: Cardiac Mechano-Electric Feedback and Arrhythmias, edited by Sachs F, Franz M, Kohl P. Philadelphia, PA: Elsevier Saunders, 2005, p. 33–41. [Google Scholar]
- 724. Laitko U, Morris CE. Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J Gen Physiol 123: 135–154, 2004. doi: 10.1085/jgp.200308965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725. Figueroa XF, Chen CC, Campbell KP, Damon DN, Day KH, Ramos S, Duling BR. Are voltage-dependent ion channels involved in the endothelial cell control of vasomotor tone? Am J Physiol Heart Circ Physiol 293: H1371–H1383, 2007. doi: 10.1152/ajpheart.01368.2006. [DOI] [PubMed] [Google Scholar]
- 726. Welsh DG, Tran CH, Plane F, Sandow S. Letter to the editor: “Are voltage-dependent ion channels involved in the endothelial cell control of vasomotor tone?” Am J Physiol Heart Circ Physiol 293: H2007, 2007. doi: 10.1152/ajpheart.00722.2007. [DOI] [PubMed] [Google Scholar]
- 727. Telinius N, Majgaard J, Kim S, Katballe N, Pahle E, Nielsen J, Hjortdal V, Aalkjaer C, Boedtkjer DB. Voltage-gated sodium channels contribute to action potentials and spontaneous contractility in isolated human lymphatic vessels. J Physiol 593: 3109–3122, 2015. doi: 10.1113/JP270166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728. Ho WS, Davis AJ, Chadha PS, Greenwood IA. Effective contractile response to voltage-gated Na+ channels revealed by a channel activator. Am J Physiol Cell Physiol 304: C739–C747, 2013. doi: 10.1152/ajpcell.00164.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729. Banderali U, Juranka PF, Clark RB, Giles WR, Morris CE. Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin) 4: 12–21, 2010. doi: 10.4161/chan.4.1.10260. [DOI] [PubMed] [Google Scholar]
- 730. Strege PR, Ou Y, Sha L, Rich A, Gibbons SJ, Szurszewski JH, Sarr MG, Farrugia G. Sodium current in human intestinal interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol 285: G1111–G1121, 2003. doi: 10.1152/ajpgi.00152.2003. [DOI] [PubMed] [Google Scholar]
- 731. Strege PR, Holm AN, Rich A, Miller SM, Ou Y, Sarr MG, Farrugia G. Cytoskeletal modulation of sodium current in human jejunal circular smooth muscle cells. Am J Physiol Cell Physiol 284: C60–C66, 2003. doi: 10.1152/ajpcell.00532.2001. [DOI] [PubMed] [Google Scholar]
- 732. Won KJ, Sanders KM, Ward SM. Interstitial cells of Cajal mediate mechanosensitive responses in the stomach. Proc Natl Acad Sci USA 102: 14913–14918, 2005. doi: 10.1073/pnas.0503628102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733. Sturek M, Hermsmeyer K. Calcium and sodium channels in spontaneously contracting vascular muscle cells. Science 233: 475–478, 1986. doi: 10.1126/science.2425434. [DOI] [PubMed] [Google Scholar]
- 734. Hollywood MA, Cotton KD, Thornbury KD, McHale NG. Tetrodotoxin-sensitive sodium current in sheep lymphatic smooth muscle. J Physiol 503: 13–20, 1997. doi: 10.1111/j.1469-7793.1997.013bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735. D’Angelo G, Davis MJ, Meininger GA. Calcium and mechanotransduction of the myogenic response. Am J Physiol Heart Circ Physiol 273: H175–H182, 1997. doi: 10.1152/ajpheart.1997.273.1.H175. [DOI] [PubMed] [Google Scholar]
- 736. Somlyo AP, Himpens B. Cell calcium and its regulation in smooth muscle. FASEB J 3: 2266–2276, 1989. doi: 10.1096/fasebj.3.11.2506092. [DOI] [PubMed] [Google Scholar]
- 737. Somlyo AP, Kitazawa T, Kobayashi S, Gong MC, Somlyo AV. Pharmacomechanical coupling: the membranes talk to the crossbridges. Adv Exp Med Biol 304: 185–208, 1991. doi: 10.1007/978-1-4684-6003-2_17. [DOI] [PubMed] [Google Scholar]
- 738. Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature 372: 231–236, 1994. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 739. Somlyo AP, Wu X, Walker LA, Somlyo AV. Pharmacomechanical coupling: the role of calcium, G-proteins, kinases and phosphatases. Rev Physiol Biochem Pharmacol 440: 183–187, 1999. doi: 10.1111/j.1469-7793.1997.013bi.x. [DOI] [PubMed] [Google Scholar]
- 740. Emerson GG, Segal SS. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 86: 94–100, 2000. doi: 10.1161/01.res.86.1.94. [DOI] [PubMed] [Google Scholar]
- 741. Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H. Hypothesis for the initiation of vasomotion. Circ Res 88: 810–815, 2001. doi: 10.1161/hh0801.089603. [DOI] [PubMed] [Google Scholar]
- 742. Aalkjær C, Boedtkjer D, Matchkov V. Vasomotion—what is currently thought? Acta Physiol (Oxf) 202: 253–269, 2011. doi: 10.1111/j.1748-1716.2011.02320.x. [DOI] [PubMed] [Google Scholar]
- 743. Flavahan NA, Bailey SR, Flavahan WA, Mitra S, Flavahan S. Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol 288: H660–H669, 2005. doi: 10.1152/ajpheart.00608.2004. [DOI] [PubMed] [Google Scholar]
- 744. Shabir S, Borisova L, Wray S, Burdyga T. Rho-kinase inhibition and electromechanical coupling in rat and guinea-pig ureter smooth muscle: Ca2+-dependent and -independent mechanisms. J Physiol 560: 839–855, 2004. doi: 10.1113/jphysiol.2004.070615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745. Retailleau K, Arhatte M, Demolombe S, Peyronnet R, Baudrie V, Jodar M, Bourreau J, Henrion D, Offermanns S, Nakamura F, Feng Y, Patel A, Duprat F, Honoré E. Arterial myogenic activation through smooth muscle filamin A. Cell Rep 14: 2050–2058, 2016. doi: 10.1016/j.celrep.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 746. Keef KD, Hume JR, Zhong J. Regulation of cardiac and smooth muscle Ca2+ channels (CaV1.2a,b) by protein kinases. Am J Physiol Cell Physiol 281: C1743–C1756, 2001. doi: 10.1152/ajpcell.2001.281.6.C1743. [DOI] [PubMed] [Google Scholar]
- 747. Dolphin AC. Functions of presynaptic voltage-gated calcium channels. Function (Oxf) 2: zqaa027, 2021. doi: 10.1093/function/zqaa027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748. Calabrese B, Tabarean IV, Juranka P, Morris CE. Mechanosensitivity of N-type calcium channel currents. Biophys J 83: 2560–2574, 2002. doi: 10.1016/S0006-3495(02)75267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749. Andreasen D, Friis UG, Uhrenholt TR, Jensen BL, Skøtt O, Hansen PB. Coexpression of voltage-dependent calcium channels Cav1.2, 2.1a, and 2.1b in vascular myocytes. Hypertension 47: 735–741, 2006. doi: 10.1161/01.HYP.0000203160.80972.47. [DOI] [PubMed] [Google Scholar]
- 750. Hansen PB. New role of P/Q-type voltage-gated calcium channels: from transmitter release to contraction of renal vasculature. J Cardiovasc Pharmacol 65: 406–411, 2015. doi: 10.1097/FJC.0000000000000184. [DOI] [PubMed] [Google Scholar]
- 751. Jackson WF. Ion channels and vascular tone. Hypertension 35: 173–178, 2000. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752. Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol 259: C3–C18, 1990. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 753. Amano S, Ishikawa T, Nakayama K. Facilitation of L-type Ca2+ currents by fluid flow in rabbit cerebral artery myocytes. J Pharmacol Sci 98: 425–429, 2005. doi: 10.1254/jphs.fp0050387. [DOI] [PubMed] [Google Scholar]
- 754. Lyford GL, Strege PR, Shepard A, Ou Y, Ermilov L, Miller SM, Gibbons SJ, Rae JL, Szurszewski JH, Farrugia G. a1C (CaV1.2) L-type calcium channel mediates mechanosensitive calcium regulation. Am J Physiol Cell Physiol 283: C1001–C1008, 2002. doi: 10.1152/ajpcell.00140.2002. [DOI] [PubMed] [Google Scholar]
- 755. Farrugia G, Holm AN, Rich A, Sarr MG, Szurszewski JH, Rae JL. A mechanosensitive calcium channel in human intestinal smooth muscle cells. Gastroenterology 117: 900–905, 1999. doi: 10.1016/s0016-5085(99)70349-5. [DOI] [PubMed] [Google Scholar]
- 756. Razinia Z, Mäkelä T, Ylänne J, Calderwood DA. Filamins in mechanosensing and signaling. Annu Rev Biophys 41: 227–246, 2012. doi: 10.1146/annurev-biophys-050511-102252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757. Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc Natl Acad Sci USA 103: 16574–16579, 2006. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758. Adebiyi A, Zhao G, Cheranov SY, Ahmed A, Jaggar JH. Caveolin-1 abolishment attenuates the myogenic response in murine cerebral arteries. Am J Physiol Heart Circ Physiol 292: H1584–H1592, 2007. doi: 10.1152/ajpheart.00584.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759. Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293: 2449–2452, 2001. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 760. Kuga T, Sadoshima J, Tomoike H, Kanaide H, Akaike N, Nakamura M. Actions of Ca2+ antagonists on two types of Ca2+ channels in rat aorta smooth muscle cells in primary culture. Circ Res 67: 469–480, 1990. doi: 10.1161/01.res.67.2.469. [DOI] [PubMed] [Google Scholar]
- 761. Hermsmeyer K, Mishra S, Miyagawa K, Minshall R. Physiologic and pathophysiologic relevance of T-type calcium-ion channels: potential indications for T-type calcium antagonists. Clin Ther 19, Suppl: 18–26, 1997. doi: 10.1016/S0149-2918(97)80034-3. [DOI] [PubMed] [Google Scholar]
- 762. Hansen PB, Jensen BL, Andreasen D, Skøtt O. Differential expression of T- and L-type voltage-dependent calcium channels in renal resistance vessels. Circ Res 89: 630–638, 2001. doi: 10.1161/hh1901.097126. [DOI] [PubMed] [Google Scholar]
- 763. Harraz OF, Welsh DG. T-type Ca2+ channels in cerebral arteries: approaches, hypotheses, and speculation. Microcirculation 20: 299–306, 2013. doi: 10.1111/micc.12038. [DOI] [PubMed] [Google Scholar]
- 764. To KH, Gui P, Li M, Zawieja SD, Castorena-Gonzalez JA, Davis MJ. T-type, but not L-type, voltage-gated calcium channels are dispensable for lymphatic pacemaking and spontaneous contractions. Sci Rep 10: 70, 2020. doi: 10.1038/s41598-019-56953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765. Ganitkevich VY, Isenberg G. Contribution of two types of calcium channels to membrane conductance of single myocytes from guinea-pig coronary artery. J Physiol 426: 19–42, 1990. doi: 10.1113/jphysiol.1990.sp018125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766. Morita H, Shi J, Ito Y, Inoue R. T-channel-like pharmacological properties of high voltage-activated, nifedipine-insensitive Ca2+ currents in the rat terminal mesenteric artery. Br J Pharmacol 137: 467–476, 2002. doi: 10.1038/sj.bjp.0704892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767. Kuo IY, Ellis A, Seymour VA, Sandow SL, Hill CE. Dihydropyridine-insensitive calcium currents contribute to function of small cerebral arteries. J Cereb Blood Flow Metab 30: 1226–1239, 2010. doi: 10.1038/jcbfm.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768. Hill MA, Meininger GA. Calcium entry and myogenic phenomena in skeletal muscle arterioles. Am J Physiol Heart Circ Physiol 267: H1085–H1092, 1994. doi: 10.1152/ajpheart.1994.267.3.H1085. [DOI] [PubMed] [Google Scholar]
- 769. Welsh DG, Jackson WF, Segal SS. Oxygen-induces electromechanical coupling in arteriolar smooth muscle cells: a role for L-type Ca2+ channels. Am J Physiol Heart Circ Physiol 274: H2018–H2024, 1998. doi: 10.1152/ajpheart.1998.274.6.H2018. [DOI] [PubMed] [Google Scholar]
- 770. Kuo IY, Wölfle SE, Hill CE. T-type calcium channels and vascular function: the new kid on the block? J Physiol 589: 783–795, 2011. doi: 10.1113/jphysiol.2010.199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 771. Feng MG, Li M, Navar LG. T-type calcium channels in the regulation of afferent and efferent arterioles in rats. Am J Physiol Renal Physiol 286: F331–F337, 2004. doi: 10.1152/ajprenal.00251.2003. [DOI] [PubMed] [Google Scholar]
- 772. Griffin KA, Hacioglu R, Abu-Amarah I, Loutzenhiser R, Williamson GA, Bidani AK. Effects of calcium channel blockers on “dynamic” and “steady-state step” renal autoregulation. Am J Physiol Renal Physiol 286: F1136–F1143, 2004. doi: 10.1152/ajprenal.00401.2003. [DOI] [PubMed] [Google Scholar]
- 773. Frandsen RH, Salomonsson M, Hansen PB, Jensen LJ, Braunstein TH, Holstein-Rathlou NH, Sorensen CM. No apparent role for T-type Ca2+ channels in renal autoregulation. Pflugers Arch 468: 541–550, 2016. doi: 10.1007/s00424-015-1770-9. [DOI] [PubMed] [Google Scholar]
- 774. Abd El-Rahman RR, Harraz OF, Brett SE, Anfinogenova Y, Mufti RE, Goldman D, Welsh DG. Identification of L- and T-type Ca2+ channels in rat cerebral arteries: role in myogenic tone development. Am J Physiol Heart Circ Physiol 304: H58–H71, 2013. doi: 10.1152/ajpheart.00476.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 775. Shin JB, Martinez-Salgado C, Heppenstall PA, Lewin GR. A T-type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci 6: 724–730, 2003. doi: 10.1038/nn1076. [DOI] [PubMed] [Google Scholar]
- 776. Harraz OF, Brett SE, Zechariah A, Romero M, Puglisi JL, Wilson SM, Welsh DG. Genetic ablation of CaV3.2 channels enhances the arterial myogenic response by modulating the RyR-BKCa axis. Arterioscler Thromb Vasc Biol 35: 1843–1851, 2015. doi: 10.1161/ATVBAHA.115.305736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777. Harraz OF, Abd El-Rahman RR, Bigdely-Shamloo K, Wilson SM, Brett SE, Romero M, Gonzales AL, Earley S, Vigmond EJ, Nygren A, Menon BK, Mufti RE, Watson T, Starreveld Y, Furstenhaupt T, Muellerleile PR, Kurjiaka DT, Kyle BD, Braun AP, Welsh DG. CaV3.2 channels and the induction of negative feedback in cerebral arteries. Circ Res 115: 650–661, 2014. doi: 10.1161/CIRCRESAHA.114.304056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778. Yu X, Duan KL, Shang CF, Yu HG, Zhou Z. Calcium influx through hyperpolarization-activated cation channels (Ih channels) contributes to activity-evoked neuronal secretion. Proc Natl Acad Sci USA 101: 1051–1056, 2004. doi: 10.1073/pnas.0305167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 779. Hagiwara N, Irisawa H. Modulation by intracellular Ca+2 of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J Physiol 409: 121–141, 1989. doi: 10.1113/jphysiol.1989.sp017488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780. Zaza A, Maccaferri G, Mangoni M, DiFrancesco D. Intracellular calcium does not directly modulate cardiac pacemaker (if) channels. Pflugers Arch 419: 662–664, 1991. doi: 10.1007/BF00370312. [DOI] [PubMed] [Google Scholar]
- 781. Combe CL, Gasparini S. Ih from synapses to networks: HCN channel functions and modulation in neurons. Prog Biophys Mol Biol 166: 119–132, 2021. doi: 10.1016/j.pbiomolbio.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 782. Benzoni P, Bertoli G, Giannetti F, Piantoni C, Milanesi R, Pecchiari M, Barbuti A, Baruscotti M, Bucchi A. The funny current: even funnier than 40 years ago. Uncanonical expression and roles of HCN/f channels all over the body. Prog Biophys Mol Biol 166: 189–204, 2021. doi: 10.1016/j.pbiomolbio.2021.08.007. [DOI] [PubMed] [Google Scholar]
- 783. Horwitz GC, Risner-Janiczek JR, Jones SM, Holt JR. HCN channels expressed in the inner ear are necessary for normal balance function. J Neurosci 31: 16814–16825, 2011. doi: 10.1523/JNEUROSCI.3064-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784. Bal R, Oertel D. Hyperpolarization-activated, mixed-cation current (Ih) in octopus cells of the mammalian cochlear nucleus. J Neurophysiol 84: 806–817, 2000. doi: 10.1152/jn.2000.84.2.806. [DOI] [PubMed] [Google Scholar]
- 785. Doan TN, Stephans K, Ramirez AN, Glazebrook PA, Andresen MC, Kunze DL. Differential distribution and function of hyperpolarization-activated channels in sensory neurons and mechanosensitive fibers. J Neurosci 24: 3335–3343, 2004. doi: 10.1523/JNEUROSCI.5156-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 786. Boyett MR, Yanni J, Tellez J, Bucchi A, Mesirca P, Cai X, Logantha S, Wilson C, Anderson C, Ariyaratnam J, Stuart L, Nakao S, Abd Allah E, Jones S, Lancaster M, Stephenson R, Chandler N, Smith M, Bussey C, Monfredi O, Morris G, Billeter R, Mangoni ME, Zhang H, Hart G, D’Souza A. Regulation of sinus node pacemaking and atrioventricular node conduction by HCN channels in health and disease. Prog Biophys Mol Biol 166: 61–85, 2021. doi: 10.1016/j.pbiomolbio.2021.06.008. [DOI] [PubMed] [Google Scholar]
- 787. DiFrancesco ML, Mesirca P, Bidaud I, Isbrandt D, Mangoni ME. The funny current in genetically modified mice. Prog Biophys Mol Biol 166: 39–50, 2021. doi: 10.1016/j.pbiomolbio.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 788. Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol 58: 299–327, 1996. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 789. BoSmith RE, Briggs I, Sturgess NC. Inhibitory actions of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br J Pharmacol 110: 343–349, 1993. doi: 10.1111/j.1476-5381.1993.tb13815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790. Novella Romanelli M, Sartiani L, Masi A, Mannaioni G, Manetti D, Mugelli A, Cerbai E. HCN channels modulators: the need for selectivity. Curr Top Med Chem 16: 1764–1791, 2016. doi: 10.2174/1568026616999160315130832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791. Accili EA, Proenza C, Baruscotti M, DiFrancesco D. From funny current to HCN channels: 20 years of excitation. News Physiol Sci 17: 32–37, 2002. [DOI] [PubMed] [Google Scholar]
- 792. Männikkö R, Pandey S, Larsson HP, Elinder F. Hysteresis in the voltage dependence of HCN channels: conversion between two modes affects pacemaker properties. J Gen Physiol 125: 305–326, 2005. doi: 10.1085/jgp.200409130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793. Greenwood IA, Prestwich SA. Characteristics of hyperpolarization-activated cation currents in portal vein smooth muscle cells. Am J Physiol Cell Physiol 282: C744–C753, 2002. doi: 10.1152/ajpcell.00393.2001. [DOI] [PubMed] [Google Scholar]
- 794. Haddock RE, Hill CE. Rhythmicity in arterial smooth muscle. J Physiol 566: 645–656, 2005. doi: 10.1113/jphysiol.2005.086405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795. McGovern AE, Robusto J, Rakoczy J, Simmons DG, Phipps S, Mazzone SB. The effect of hyperpolarization-activated cyclic nucleotide-gated ion channel inhibitors on the vagal control of guinea pig airway smooth muscle tone. Br J Pharmacol 171: 3633–3650, 2014. doi: 10.1111/bph.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796. Kashyap M, Singh N, Yoshimura N, Chermansky C, Tyagi P. Constitutively active HCN channels constrain detrusor excitability and modulate evoked contractions of human bladder. Am J Clin Exp Urol 8: 163–176, 2020. [PMC free article] [PubMed] [Google Scholar]
- 797. Mader F, Müller S, Krause L, Springer A, Kernig K, Protzel C, Porath K, Rackow S, Wittstock T, Frank M, Hakenberg OW, Köhling R, Kirschstein T. Hyperpolarization-activated cyclic nucleotide-gated non-selective (HCN) ion channels regulate human and murine urinary bladder contractility. Front Physiol 9: 753, 2018. doi: 10.3389/fphys.2018.00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798. He P, Deng J, Zhong X, Zhou Z, Song B, Li L. Identification of a hyperpolarization-activated cyclic nucleotide-gated channel and its subtypes in the urinary bladder of the rat. Urology 79: 1411.e7–1411.e13, 2012. doi: 10.1016/j.urology.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 799. Alotaibi M, Kahlat K, Nedjadi T, Djouhri L. Effects of ZD7288, a hyperpolarization-activated cyclic nucleotide-gated (HCN) channel blocker, on term-pregnant rat uterine contractility in vitro. Theriogenology 90: 141–146, 2017. doi: 10.1016/j.theriogenology.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 800. Si X, Huang L, Gong Y, Lu J, Lin L. Role of calcium in activation of hyperpolarization-activated cyclic nucleotide-gated channels caused by cholecystokinin octapeptide in interstitial cells of Cajal. Digestion 85: 266–275, 2012. doi: 10.1159/000337077. [DOI] [PubMed] [Google Scholar]
- 801. McCloskey KD, Toland HM, Hollywood MA, Thornbury KD, McHale NG. Hyperpolarization-activated inward current in isolated sheep mesenteric lymphatic smooth muscle. J Physiol 521: 201–211, 1999. doi: 10.1111/j.1469-7793.1999.00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802. Negrini D, Marcozzi C, Solari E, Bossi E, Cinquetti R, Reguzzoni M, Moriondo A. Hyperpolarization-activated cyclic nucleotide-gated channels in peripheral diaphragmatic lymphatics. Am J Physiol Heart Circ Physiol 311: H892–H903, 2016. doi: 10.1152/ajpheart.00193.2016. [DOI] [PubMed] [Google Scholar]
- 803. Majgaard J, Skov FG, Kim S, Hjortdal VE, Boedtkjer DM. Positive chronotropic action of HCN channel antagonism in human collecting lymphatic vessels. Physiol Rep 10: e15401, 2022. doi: 10.14814/phy2.15401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804. Choe S. Potassium channel structures. Nat Rev Neurosci 3: 115–121, 2002. doi: 10.1038/nrn727. [DOI] [PubMed] [Google Scholar]
- 805. Matsumura K, Yokogawa M, Osawa M. Peptide toxins targeting KV channels. Handb Exp Pharmacol 267: 471–505, 2021. doi: 10.1007/164_2021_500. [DOI] [PubMed] [Google Scholar]
- 806. Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, Greenwood IA. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol 588: 3277–3293, 2010. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807. Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, Byron KL. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther 325: 475–483, 2008. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808. Zaika O, Lara LS, Gamper N, Hilgemann DW, Jaffe DB, Shapiro MS. Angiotensin II regulates neuronal excitability via phosphatidylinositol 4,5-bisphosphate-dependent modulation of Kv7 (M-type) K+ channels. J Physiol 575: 49–67, 2006. doi: 10.1113/jphysiol.2006.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809. Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314: 1454–1457, 2006. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 810. Verneuil J, Brocard C, Trouplin V, Villard L, Peyronnet-Roux J, Brocard F. The M-current works in tandem with the persistent sodium current to set the speed of locomotion. PLoS Biol 18: e3000738, 2020. doi: 10.1371/journal.pbio.3000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811. Drion G, Bonjean M, Waroux O, Scuvée-Moreau J, Liégeois JF, Sejnowski TJ, Sepulchre R, Seutin V. M-type channels selectively control bursting in rat dopaminergic neurons. Eur J Neurosci 31: 827–835, 2010. doi: 10.1111/j.1460-9568.2010.07107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812. Forti L, Cesana E, Mapelli J, D’Angelo E. Ionic mechanisms of autorhythmic firing in rat cerebellar Golgi cells. J Physiol 574: 711–729, 2006. doi: 10.1113/jphysiol.2006.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813. Chow LW, Leung YM. The versatile Kv channels in the nervous system: actions beyond action potentials. Cell Mol Life Sci 77: 2473–2482, 2020. doi: 10.1007/s00018-019-03415-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814. Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ channels: structure, function and clinical significance. Physiol Rev 92: 1393–1478, 2012. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- 815. Shibasaki T. Conductance and kinetics of delayed rectifier potassium channels in nodal cells of the rabbit heart. J Physiol 387: 227–250, 1987. doi: 10.1113/jphysiol.1987.sp016571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816. Adams DJ. Ionic channels in vascular endothelial cells. Trends Cardiovasc Med 4: 18–26, 1994. doi: 10.1016/1050-1738(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 817. Nelson MT, Brayden JE. Regulation of arterial tone by calcium-dependent K+ channels and ATP-sensitive K+ channels. Cardiovasc Drug Ther 7, Suppl: 605–610, 1993. doi: 10.1007/BF00877627. [DOI] [PubMed] [Google Scholar]
- 818. Luykenaar KD, Brett SE, Wu BN, Wiehler WB, Welsh DG. Pyrimidine nucleotides suppress KDR currents and depolarize rat cerebral arteries by activating Rho kinase. Am J Physiol Heart Circ Physiol 286: H1088–H1100, 2004. doi: 10.1152/ajpheart.00903.2003. [DOI] [PubMed] [Google Scholar]
- 819. Chen TT, Luykenaar KD, Walsh EJ, Walsh MP, Cole WC. Key role of Kv1 channels in vasoregulation. Circ Res 99: 53–60, 2006. doi: 10.1161/01.RES.0000229654.45090.57. [DOI] [PubMed] [Google Scholar]
- 820. Morris CE, Sigurdson WJ. Stretch-inactivated ion channels coexist with stretch-activated ion channels. Science 243: 807–809, 1989. doi: 10.1126/science.2536958. [DOI] [PubMed] [Google Scholar]
- 821. Tabarean IV, Morris CE. Membrane stretch accelerates activation and slow inactivation in Shaker channels with S3-S4 linker deletions. Biophys J 82: 2982–2994, 2002. doi: 10.1016/S0006-3495(02)75639-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 822. Moe P, Blount P. Assessment of potential stimuli for mechano-dependent gating of MscL: effects of pressure, tension, and lipid headgroups. Biochemistry 44: 12239–12244, 2005. doi: 10.1021/bi0509649. [DOI] [PubMed] [Google Scholar]
- 823. Otway R, Vandenberg JI, Guo G, Varghese A, Castro ML, Liu J, Zhao J, Bursill JA, Wyse KR, Crotty H, Baddeley O, Walker B, Kuchar D, Thorburn C, Fatkin D. Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol 49: 578–586, 2007. doi: 10.1016/j.jacc.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 824. Hao J, Padilla F, Dandonneau M, Lavebratt C, Lesage F, Noël J, Delmas P. Kv1.1 channels act as mechanical brake in the senses of touch and pain. Neuron 77: 899–914, 2013. doi: 10.1016/j.neuron.2012.12.035. [DOI] [PubMed] [Google Scholar]
- 825. Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci USA 109: 7917–7922, 2012. doi: 10.1073/pnas.1205435109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 826. Evanson KW, Bannister JP, Leo MD, Jaggar JH. LRRC26 is a functional BK channel auxiliary gamma subunit in arterial smooth muscle cells. Circ Res 115: 423–431, 2014. doi: 10.1161/CIRCRESAHA.115.303407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827. Sones WR, Leblanc N, Greenwood IA. Inhibition of vascular calcium-gated chloride currents by blockers of KCa1.1, but not by modulators of KCa2.1 or KCa2.3 channels. Br J Pharmacol 158: 521–531, 2009. doi: 10.1111/j.1476-5381.2009.00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 828. Jackson WF, Blair KL. Characterization and function of Ca2+-activated K+ channels in arteriolar muscle cells. Am J Physiol Heart Circ Physiol 274: H27–H34, 1998. doi: 10.1152/ajpheart.1998.274.1.H27. [DOI] [PubMed] [Google Scholar]
- 829. Brekke JF, Jackson WF, Segal SS. Arteriolar smooth muscle Ca2+ dynamics during blood flow control in hamster cheek pouch. J Appl Physiol (1985) 101: 307–315, 2006. doi: 10.1152/japplphysiol.01634.2005. [DOI] [PubMed] [Google Scholar]
- 830. Braun AP. Structural insights into the cytoplasmic domain of a human BK channel. Channels (Austin) 5: 1–3, 2011. doi: 10.4161/chan.5.1.14818. [DOI] [PubMed] [Google Scholar]
- 831. Braun AF, Sy L. Contribution of potential EF hand motifs to the calcium-dependent gating of a mouse brain large conductance, calcium-sensitive K+ channel. J Physiol 533: 681–695, 2001. doi: 10.1111/j.1469-7793.2001.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 832. Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 833. Plüger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel β1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ Res 87: E53–E60, 2000. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- 834. Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature 466: 513–516, 2010. doi: 10.1038/nature09162. [DOI] [PubMed] [Google Scholar]
- 835. Gonzalez-Perez V, Xia XM, Lingle CJ. Functional regulation of BK potassium channels by gamma1 auxiliary subunits. Proc Natl Acad Sci USA 111: 4868–4873, 2014. doi: 10.1073/pnas.1322123111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836. Kyle BD, Braun AP. The regulation of BK channel activity by pre- and post-translational modifications. Front Physiol 5: 316, 2014. doi: 10.3389/fphys.2014.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 837. Ali S, Solano AS, Gonzales AL, Thakore P, Krishnan V, Yamasaki E, Earley S. Nitric oxide signals through IRAG to inhibit TRPM4 channels and dilate cerebral arteries. Function (Oxf) 2: zqab051, 2021. doi: 10.1093/function/zqab051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 838. Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256: 532–535, 1992. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 839. Löhn M, Jessner W, Fürstenau M, Wellner M, Sorrentino V, Haller H, Luft FC, Gollasch M. Regulation of calcium sparks and spontaneous transient outward currents by RyR3 in arterial vascular smooth muscle cells. Circ Res 89: 1051–1057, 2001. doi: 10.1161/hh2301.100250. [DOI] [PubMed] [Google Scholar]
- 840. Ji G, Feldman ME, Greene KS, Sorrentino V, Xin HB, Kotlikoff MI. RYR2 proteins contribute to the formation of Ca2+ sparks in smooth muscle. J Gen Physiol 123: 377–386, 2004. doi: 10.1085/jgp.200308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841. Kaßmann M, Szijártó IA, García-Prieto CF, Fan G, Schleifenbaum J, Anistan YM, Tabeling C, Shi Y, Le Noble F, Witzenrath M, Huang Y, Markó L, Nelson MT, Gollasch M. Role of ryanodine type 2 receptors in elementary Ca2+ signaling in arteries and vascular adaptive responses. J Am Heart Assoc 8: e010090, 2019. doi: 10.1161/JAHA.118.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842. Gollasch M, Wellman GC, Knot HJ, Jaggar JH, Damon DH, Bonev AD, Nelson MT. Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circ Res 83: 1104–1114, 1998. doi: 10.1161/01.RES.83.11.1104. [DOI] [PubMed] [Google Scholar]
- 843. Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, Rubart M, Stevenson AS, Lederer WJ, Knot HJ, Bonev AD, Nelson MT. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand 164: 577–587, 1998. doi: 10.1046/j.1365-201X.1998.00462.x. [DOI] [PubMed] [Google Scholar]
- 844. Pérez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol 113: 229–238, 1999. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845. Westcott EB, Jackson WF. Heterogeneous function of ryanodine receptors, but not IP3 receptors, in hamster cremaster muscle feed arteries and arterioles. Am J Physiol Heart Circ Physiol 300: H1616–H1630, 2011. doi: 10.1152/ajpheart.00728.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846. Westcott EB, Goodwin EL, Segal SS, Jackson WF. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol 590: 1849–1869, 2012. doi: 10.1113/jphysiol.2011.222083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847. Jackson WF. Calcium-dependent ion channel and the regulation of arteriolar myogenic tone. Front Physiol 12: 770450, 2021. doi: 10.3389/fphys.2021.770450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848. Fan G, Cui Y, Gollasch M, Kassmann M. Elementary calcium signaling in arterial smooth muscle. Channels (Austin) 13: 505–519, 2019. doi: 10.1080/19336950.2019.1688910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 849. Wellman GC, Santana LF, Bonev AD, Nelson MT. Role of phospholamban in the modulation of arterial Ca2+ sparks and Ca2+-activated K+ channels by cAMP. Am J Physiol Cell Physiol 281: C1029–C1037, 2001. doi: 10.1152/ajpcell.2001.281.3.C1029. [DOI] [PubMed] [Google Scholar]
- 850. Guia A, Wan X, Courtemanche M, Leblanc N. Local Ca2+ entry through L-type Ca2+ channels activates Ca2+-dependent K+ channels in rabbit coronary myocytes. Circ Res 84: 1032–1042, 1999. doi: 10.1161/01.res.84.9.1032. [DOI] [PubMed] [Google Scholar]
- 851. Suzuki Y, Yamamura H, Ohya S, Imaizumi Y. Caveolin-1 facilitates the direct coupling between large conductance Ca2+-activated K+ (BKCa) and Cav1.2 Ca2+ channels and their clustering to regulate membrane excitability in vascular myocytes. J Biol Chem 288: 36750–36761, 2013. doi: 10.1074/jbc.M113.511485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 852. Nystoriak MA, Nieves-Cintrón M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell’acqua ML, Scott JD, Santana LF, Navedo MF. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res 114: 607–615, 2014. doi: 10.1161/CIRCRESAHA.114.302168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 853. Pritchard HA, Griffin CS, Yamasaki E, Thakore P, Lane C, Greenstein AS, Earley S. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc Natl Acad Sci USA 116: 21874–21881, 2019. doi: 10.1073/pnas.1911304116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 854. Liu G, Shi J, Yang L, Cao L, Park SM, Cui J, Marx SO. Assembly of a Ca2+-dependent BK channel signaling complex by binding to beta2 adrenergic receptor. EMBO J 23: 2196–2205, 2004. doi: 10.1038/sj.emboj.7600228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 855. Hashad AM, Harraz OF, Brett SE, Romero M, Kassmann M, Puglisi JL, Wilson SM, Gollasch M, Welsh DG. Caveolae link CaV3.2 channels to BKCa-mediated feedback in vascular smooth muscle. Arterioscler Thromb Vasc Biol 38: 2371–2381, 2018. doi: 10.1161/ATVBAHA.118.311394. [DOI] [PubMed] [Google Scholar]
- 856. Fan G, Kaßmann M, Hashad AM, Welsh DG, Gollasch M. Differential targeting and signalling of voltage-gated T-type Cav 3.2 and L-type Cav 1.2 channels to ryanodine receptors in mesenteric arteries. J Physiol 596: 4863–4877, 2018. doi: 10.1113/JP276923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857. Wang W, Huang H, Hou D, Liu P, Wei H, Fu X, Niu W. Mechanosensitivity of STREX-lacking BKCa channels in the colonic smooth muscle of the mouse. Am J Physiol Gastrointest Liver Physiol 299: G1231–G1240, 2010. doi: 10.1152/ajpgi.00268.2010. [DOI] [PubMed] [Google Scholar]
- 858. Dopico AM, Kirber MT, Singer JJ, Walsh JV Jr.. Membrane stretch directly activates large conductance Ca2+-activated K+ channels in mesenteric artery smooth muscle cells. Am J Hypertens 7: 82–89, 1994. doi: 10.1093/ajh/7.1.82. [DOI] [PubMed] [Google Scholar]
- 859. Taniguchi J, Guggino WB. Membrane stretch: a physiological stimulator of Ca2+-activated K+ channels in thick ascending limb. Am J Physiol Renal Physiol 257: F347–F352, 1989. doi: 10.1152/ajprenal.1989.257.3.F347. [DOI] [PubMed] [Google Scholar]
- 860. Mienville J, Barker JL, Lange GD. Mechanosensitive properties of BK channels from embryonic rat neuroepithelium. J Membr Biol 153: 211–216, 1996. doi: 10.1007/s002329900124. [DOI] [PubMed] [Google Scholar]
- 861. Jackson WF. Ion channels and the regulation of myogenic tone in peripheral arterioles. Curr Top Membr 85: 19–58, 2020. doi: 10.1016/bs.ctm.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 862. Löhn M, Lauterbach B, Haller H, Pongs O, Luft FC, Gollasch M. β1-Subunit of BK channels regulates arterial wall [Ca2+] and diameter in mouse cerebral arteries. J Appl Physiol (1985) 91: 1350–1354, 2001. doi: 10.1152/jappl.2001.91.3.1350. [DOI] [PubMed] [Google Scholar]
- 863. Tharp DL, Wamhoff BR, Turk JR, Bowles DK. Upregulation of intermediate-conductance Ca2+-activated K+ channel (IKCa1) mediates phenotypic modulation of coronary smooth muscle. Am J Physiol Heart Circ Physiol 291: H2493–H2503, 2006. doi: 10.1152/ajpheart.01254.2005. [DOI] [PubMed] [Google Scholar]
- 864. Bi D, Toyama K, Lemaître V, Takai J, Fan F, Jenkins DP, Wulff H, Gutterman DD, Park F, Miura H. The intermediate conductance calcium-activated potassium channel KCa3.1 regulates vascular smooth muscle cell proliferation via controlling calcium-dependent signaling. J Biol Chem 288: 15843–15853, 2013. doi: 10.1074/jbc.M112.427187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 865. Mishra RC, Wulff H, Hill MA, Braun AP. Inhibition of myogenic tone in rat cremaster and cerebral arteries by SKA-31, an activator of endothelial KCa2.3 and KCa3.1 channels. J Cardiovasc Pharmacol 66: 118–127, 2015. doi: 10.1097/FJC.0000000000000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 866. Schilling T, Eder C. TRAM-34 inhibits nonselective cation channels. Pflugers Arch 454: 559–563, 2007. doi: 10.1007/s00424-007-0232-4. [DOI] [PubMed] [Google Scholar]
- 867. McNeish AJ, Jimenez Altayo F, Garland CJ. Evidence both L-type and non-L-type voltage-dependent calcium channels contribute to cerebral artery vasospasm following loss of NO in the rat. Vascul Pharmacol 53: 151–159, 2010. doi: 10.1016/j.vph.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 868. Autuori E, Sedlak P, Xu L, C Ridder M, Tedoldi A, Sah P. rSK1 in rat neurons: a controller of membrane rSK2? Front Neural Circuits 13: 21, 2019. doi: 10.3389/fncir.2019.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869. Matschke LA, Rinné S, Snutch TP, Oertel WH, Dolga AM, Decher N. Calcium-activated SK potassium channels are key modulators of the pacemaker frequency in locus coeruleus neurons. Mol Cell Neurosci 88: 330–341, 2018. doi: 10.1016/j.mcn.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 870. Zhang Y, Huang H. SK Channels regulate resting properties and signaling reliability of a developing fast-spiking neuron. J Neurosci 37: 10738–10747, 2017. doi: 10.1523/JNEUROSCI.1243-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 871. Wulff H, Köhler R. Endothelial small-conductance and intermediate-conductance KCa channels: an update on their pharmacology and usefulness as cardiovascular targets. J Cardiovasc Pharmacol 61: 102–112, 2013. doi: 10.1097/FJC.0b013e318279ba20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 872. Ro S, Hatton WJ, Koh SD, Horowitz B. Molecular properties of small-conductance Ca2+-activated K+ channels expressed in murine colonic smooth muscle. Am J Physiol Gastrointest Liver Physiol 281: G964–G973, 2001. doi: 10.1152/ajpgi.2001.281.4.G964. [DOI] [PubMed] [Google Scholar]
- 873. Li N, Ding H, He X, Li Z, Liu Y. Expression and function of the small-conductance Ca2+-activated K+ channel is decreased in urinary bladder smooth muscle cells from female guinea pig with partial bladder outlet obstruction. Int Urol Nephrol 49: 1147–1155, 2017. doi: 10.1007/s11255-017-1592-0. [DOI] [PubMed] [Google Scholar]
- 874. Lee K, Isogai A, Antoh M, Kajioka S, Eto M, Hashitani H. Role of K+ channels in regulating spontaneous activity in the muscularis mucosae of guinea pig bladder. Eur J Pharmacol 818: 30–37, 2018. doi: 10.1016/j.ejphar.2017.10.024. [DOI] [PubMed] [Google Scholar]
- 875. Lee H, Koh BH, Peri LE, Sanders KM, Koh SD. Functional expression of SK channels in murine detrusor PDGFR+ cells. J Physiol 591: 503–513, 2013. doi: 10.1113/jphysiol.2012.241505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876. Lee H, Koh BH, Peri LE, Sanders KM, Koh SD. Purinergic inhibitory regulation of murine detrusor muscles mediated by PDGFRalpha+ interstitial cells. J Physiol 592: 1283–1293, 2014. doi: 10.1113/jphysiol.2013.267989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 877. Sandow SL, Grayson TH. Limits of isolation and culture: intact vascular endothelium and BKCa. Am J Physiol Heart Circ Physiol 297: H1–H7, 2009. doi: 10.1152/ajpheart.00042.2009. [DOI] [PubMed] [Google Scholar]
- 878. Ungvari Z, Csiszar A, Koller A. Increases in endothelial Ca2+ activate KCa channels and elicit EDHF-type arteriolar dilation via gap junctions. Am J Physiol Heart Circ Physiol 282: H1760–H1767, 2002. doi: 10.1152/ajpheart.00676.2001. [DOI] [PubMed] [Google Scholar]
- 879. Gauthier KM, Liu C, Popovic A, Albarwani S, Rusch NJ. Freshly isolated bovine coronary endothelial cells do not express the BK Ca channel gene. J Physiol 545: 829–836, 2002. doi: 10.1113/jphysiol.2002.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 880. Jackson WF. Endothelial cell ion channel expression and function in arterioles and resistance arteries. In: Vascular Ion Channels in Physiology and Disease, edited by Levitan I, Dopico AM.. Cham, Switzerland: Springer International Publishing, 2016, p. 3–36. [Google Scholar]
- 881. Brähler S, Kaistha A, Schmidt VJ, Wölfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J, Köhler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119: 2323–2332, 2009. doi: 10.1161/CIRCULATIONAHA.108.846634. [DOI] [PubMed] [Google Scholar]
- 882. Hannah RM, Dunn KM, Bonev AD, Nelson MT. Endothelial SKCa and IKCa channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 31: 1175–1186, 2011. doi: 10.1038/jcbfm.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883. Wang XL, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, Sieck GC, Lee HC. Caveolae targeting and regulation of large conductance Ca2+-activated K+ channels in vascular endothelial cells. J Biol Chem 280: 11656–11664, 2005. doi: 10.1074/jbc.M410987200. [DOI] [PubMed] [Google Scholar]
- 884. Hughes JM, Riddle MA, Paffett ML, Gonzalez Bosc LV, Walker BR. Novel role of endothelial BKCa channels in altered vasoreactivity following hypoxia. Am J Physiol Heart Circ Physiol 299: H1439–H1450, 2010. doi: 10.1152/ajpheart.00124.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 885. Riddle MA, Hughes JM, Walker BR. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am J Physiol Cell Physiol 301: C1404–C1414, 2011. doi: 10.1152/ajpcell.00013.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886. Köhler R, Ruth P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflugers Arch 459: 969–976, 2010. doi: 10.1007/s00424-010-0819-z. [DOI] [PubMed] [Google Scholar]
- 887. Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci USA 105: 9627–9632, 2008. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 888. Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, Dwyer ST, Looft-Wilson R, Lysiak JJ, Gaston B, Palmer L, Isakson BE. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler Thromb Vasc Biol 31: 399–407, 2011. doi: 10.1161/ATVBAHA.110.215939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 889. Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ Res 104: 987–994, 2009. doi: 10.1161/CIRCRESAHA.108.189530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890. Wolpe AG, Ruddiman CA, Hall PJ, Isakson BE. Polarized proteins in endothelium and their contribution to function. J Vasc Res 58: 65–91, 2021. doi: 10.1159/000512618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 891. Garland CJ, Dora KA. EDH: endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol (Oxf) 219: 152–161, 2017. doi: 10.1111/apha.12649. [DOI] [PubMed] [Google Scholar]
- 892. Brasen JC, de Wit C, Sorensen CM. Myoendothelial coupling through Cx40 contributes to EDH-induced vasodilation in murine renal arteries: evidence from experiments and modelling. Acta Physiol 222: e12906, 2018. doi: 10.1111/apha.12906. [DOI] [PubMed] [Google Scholar]
- 893. Wei R, Lunn SE, Tam R, Gust SL, Classen B, Kerr PM, Plane F. Vasoconstrictor stimulus determines the functional contribution of myoendothelial feedback to mesenteric arterial tone. J Physiol 596: 1181–1197, 2018. doi: 10.1113/JP274797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894. Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (KCa) and connexins: possible relationship to vasodilator function? J Anat 209: 689–698, 2006. doi: 10.1111/j.1469-7580.2006.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895. Absi M, Burnham MP, Weston AH, Harno E, Rogers M, Edwards G. Effects of methyl beta-cyclodextrin on EDHF responses in pig and rat arteries; association between SKCa channels and caveolin-rich domains. Br J Pharmacol 151: 332–340, 2007. doi: 10.1038/sj.bjp.0707222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 896. Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol 553: 183–189, 2003. doi: 10.1113/jphysiol.2003.051896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897. Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res 93: 124–131, 2003. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- 898. Takai J, Santu A, Zheng H, Koh SD, Ohta M, Filimban LM, Lemaître V, Teraoka R, Jo H, Miura H. Laminar shear stress upregulates endothelial Ca2+-activated K+ channels KCa2.3 and KCa3.1 via a Ca2+/calmodulin-dependent protein kinase kinase/Akt/p300 cascade. Am J Physiol Heart Circ Physiol 305: H484–H493, 2013. doi: 10.1152/ajpheart.00642.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899. Goedicke-Fritz S, Kaistha A, Kacik M, Markert S, Hofmeister A, Busch C, Bänfer S, Jacob R, Grgic I, Hoyer J. Evidence for functional and dynamic microcompartmentation of Cav-1/TRPV4/KCa in caveolae of endothelial cells. Eur J Cell Biol 94: 391–400, 2015. doi: 10.1016/j.ejcb.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 900. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 901. Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol 59: 171–191, 1997. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 902. Sancho M, Fabris S, Hald BO, Brett SE, Sandow SL, Poepping TL, Welsh DG. Membrane lipid-KIR2.x channel interactions enable hemodynamic sensing in cerebral arteries. Arterioscler Thromb Vasc Biol 39: 1072–1087, 2019. doi: 10.1161/ATVBAHA.119.312493. [DOI] [PubMed] [Google Scholar]
- 903. French RJ, Shoukimas JJ. An ion’s view of the potassium channel. The structure of the permeation pathway as sensed by a variety of blocking ions. J Gen Physiol 85: 669–698, 1985. doi: 10.1085/jgp.85.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 904. Ji S, John SA, Lu Y, Weiss JN. Mechanosensitivity of the cardiac muscarinic potassium channel. A novel property conferred by Kir3.4 subunit. J Biol Chem 273: 1324–1328, 1998. doi: 10.1074/jbc.273.3.1324. [DOI] [PubMed] [Google Scholar]
- 905. Pleumsamran A, Kim D. Membrane stretch augments the cardiac muscarinic K+ channel activity. J Membr Biol 148: 287–297, 1995. doi: 10.1007/BF00235046. [DOI] [PubMed] [Google Scholar]
- 906. Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ, Welsh DG. Kir channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol 586: 1147–1160, 2008. doi: 10.1113/jphysiol.2007.145474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 907. Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res 87: 160–166, 2000. doi: 10.1161/01.RES.87.2.160. [DOI] [PubMed] [Google Scholar]
- 908. Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev 77: 1165–1232, 1997. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 909. Fancher IS, Levitan I. Endothelial inwardly-rectifying K+ channels as a key component of shear stress-induced mechanotransduction. Curr Top Membr 85: 59–88, 2020. doi: 10.1016/bs.ctm.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 910. Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9: 1397–1403, 2006. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 911. McCarron JG, Halpern W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am J Physiol Heart Circ Physiol 259: H902–H908, 1990. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- 912. Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20: 717–726, 2017. doi: 10.1038/nn.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 913. Rivers RJ, Hein TW, Zhang C, Kuo L. Activation of barium-sensitive inward rectifier potassium channels mediates remote dilation of coronary arterioles. Circulation 104: 1749–1753, 2001. doi: 10.1161/hc4001.098053. [DOI] [PubMed] [Google Scholar]
- 914. Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396: 269–272, 1998. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 915. Welsh DG, Tran CHT, Hald BO, Sancho M. The conducted vasomotor response: function, biophysical basis, and pharmacological control. Annu Rev Pharmacol Toxicol 58: 391–410, 2018. doi: 10.1146/annurev-pharmtox-010617-052623. [DOI] [PubMed] [Google Scholar]
- 916. Arbuzova A, Martushova K, Hangyás-Mihályné G, Morris AJ, Ozaki S, Prestwich GD, McLaughlin S. Fluorescently labeled neomycin as a probe of phosphatidylinositol-4,5-bisphosphate in membranes. Biochim Biophys Acta 1464: 35–48, 2000. doi: 10.1016/s0005-2736(99)00243-6. [DOI] [PubMed] [Google Scholar]
- 917. Haruna T, Yoshida H, Nakamura TY, Xie LH, Otani H, Ninomiya T, Takano M, Coetzee WA, Horie M. Alpha1-adrenoceptor-mediated breakdown of phosphatidylinositol 4,5-bisphosphate inhibits pinacidil-activated ATP-sensitive K+ currents in rat ventricular myocytes. Circ Res 91: 232–239, 2002. doi: 10.1161/01.res.0000029971.60214.49. [DOI] [PubMed] [Google Scholar]
- 918. Laver DR, Hamada T, Fessenden JD, Ikemoto N. The ryanodine receptor pore blocker neomycin also inhibits channel activity via a previously undescribed high-affinity Ca2+ binding site. J Membr Biol 220: 11–20, 2007. doi: 10.1007/s00232-007-9067-3. [DOI] [PubMed] [Google Scholar]
- 919. Huang K, Wang X, Liu Y, Zhao Y. CRAC channel is inhibited by neomycin in a Ptdlns(4,5)P2-independent manner. Cell Biochem Funct 33: 97–100, 2015. doi: 10.1002/cbf.3088. [DOI] [PubMed] [Google Scholar]
- 920. Mann SE, Johnson M, Meredith FL, Rennie KJ. Inhibition of K+ currents in type I vestibular hair cells by gentamicin and neomycin. Audiol Neurootol 18: 317–326, 2013. doi: 10.1159/000354056. [DOI] [PubMed] [Google Scholar]
- 921. Karashima Y, Prenen J, Meseguer V, Owsianik G, Voets T, Nilius B. Modulation of the transient receptor potential channel TRPA1 by phosphatidylinositol 4,5-biphosphate manipulators. Pflugers Arch 457: 77–89, 2008. doi: 10.1007/s00424-008-0493-6. [DOI] [PubMed] [Google Scholar]
- 922. Török TL, Rácz D, Sáska Z, David AZ, Tabi T, Zillikens S, Nada SA, Klebovich I, Gyires K, Magyar K. Reverse Na+/Ca2+-exchange mediated Ca2+-entry and noradrenaline release in Na+-loaded peripheral sympathetic nerves. Neurochem Int 53: 338–345, 2008. doi: 10.1016/j.neuint.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 923. Hayoz S, Pettis J, Bradley V, Segal SS, Jackson WF. Increased amplitude of inward rectifier K+ currents with advanced age in smooth muscle cells of murine superior epigastric arteries. Am J Physiol Heart Circ Physiol 312: H1203–H1214, 2017. doi: 10.1152/ajpheart.00679.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 924. Falcone JC, Granger HJ, Meininger GA. Enhanced myogenic activation in skeletal muscle arterioles from spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 265: H1847–H1855, 1993. doi: 10.1152/ajpheart.1993.265.6.H1847. [DOI] [PubMed] [Google Scholar]
- 925. Hill MA, Davis MJ, Meininger GA. Cyclooxygenase inhibition potentiates myogenic activity in skeletal muscle arterioles. Am J Physiol Heart Circ Physiol 258: H127–H133, 1990. doi: 10.1152/ajpheart.1990.258.1.H127. [DOI] [PubMed] [Google Scholar]
- 926. Davis MJ, Gilmore JP, Joyner WL. Autoregulatory responses of arterioles in hamster pulmonary allograft and cheek pouch tissue to changes in transmural pressure. Microvasc Res 20: 105, 1980. [Google Scholar]
- 927. Chilton L, Smirnov SV, Loutzenhiser K, Wang X, Loutzenhiser R. Segment-specific differences in the inward rectifier K+ current along the renal interlobular artery. Cardiovasc Res 92: 169–177, 2011. doi: 10.1093/cvr/cvr179. [DOI] [PubMed] [Google Scholar]
- 928. Chilton L, Loutzenhiser R. Functional evidence for an inward rectifier potassium current in rat renal afferent arterioles. Circ Res 88: 152–158, 2001. doi: 10.1161/01.res.88.2.152. [DOI] [PubMed] [Google Scholar]
- 929. Chlopicki S, Nilsson H, Mulvany MJ. Initial and sustained phases of myogenic response of rat mesenteric small arteries. Am J Physiol Heart Circ Physiol 281: H2176–H2183, 2001. doi: 10.1152/ajpheart.2001.281.5.H2176. [DOI] [PubMed] [Google Scholar]
- 930. Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature 331: 168–170, 1988. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 931. Cooke JP, Rossitch E Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest 88: 1663–1671, 1991. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 932. Ohno M, Gibbons GH, Dzau VJ, Cooke JP. Shear stress elevates endothelial cGMP: role of a potassium channel and G protein coupling. Circulation 88: 193–197, 1993. doi: 10.1161/01.CIR.88.1.193. [DOI] [PubMed] [Google Scholar]
- 933. Ohno M, Cooke JP, Dzau VJ, Gibbons GH. Fluid shear stress induces endothelial transforming growth factor beta-1 transcription and production. Modulation by potassium channel blockade. J Clin Invest 95: 1363–1369, 1995. doi: 10.1172/JCI117787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 934. Malek AM, Jackman R, Rosenberg RD, Izumo S. Endothelial expression of thrombomodulin is reversibly regulated by fluid shear stress. Circ Res 74: 852–860, 1994. doi: 10.1161/01.res.74.5.852. [DOI] [PubMed] [Google Scholar]
- 935. Malek AM, Izumo S. Molecular aspects of signal transduction of shear stress in the endothelial cell. J Hypertens 12: 989–999, 1994. [PubMed] [Google Scholar]
- 936. Barakat AI. A model for shear stress-induced deformation of a flow sensor on the surface of vascular endothelial cells. J Theor Biol 210: 221–236, 2001. doi: 10.1006/jtbi.2001.2290. [DOI] [PubMed] [Google Scholar]
- 937. D’Avanzo N, Cheng WW, Doyle DA, Nichols CG. Direct and specific activation of human inward rectifier K+ channels by membrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem 285: 37129–37132, 2010. doi: 10.1074/jbc.C110.186692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 938. Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J 83: 3211–3222, 2002. doi: 10.1016/S0006-3495(02)75323-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 939. Singh DK, Rosenhouse-Dantsker A, Nichols CG, Enkvetchakul D, Levitan I. Direct regulation of prokaryotic Kir channel by cholesterol. J Biol Chem 284: 30727–30736, 2009. doi: 10.1074/jbc.M109.011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 940. Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem 288: 31154–31164, 2013. doi: 10.1074/jbc.M113.496117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 941. Haidekker MA, L’Heureux N, Frangos JA. Fluid shear stress increases membrane fluidity in endothelial cells: a study with DCVJ fluorescence. Am J Physiol Heart Circ Physiol 278: H1401–H1406, 2000. doi: 10.1152/ajpheart.2000.278.4.H1401. [DOI] [PubMed] [Google Scholar]
- 942. Yamamoto K, Ando J. Endothelial cell and model membranes respond to shear stress by rapidly decreasing the order of their lipid phases. J Cell Sci 126: 1227–1234, 2013. doi: 10.1242/jcs.119628. [DOI] [PubMed] [Google Scholar]
- 943. Yamamoto K, Ando J. Vascular endothelial cell membranes differentiate between stretch and shear stress through transitions in their lipid phases. Am J Physiol Heart Circ Physiol 309: H1178–H1185, 2015. doi: 10.1152/ajpheart.00241.2015. [DOI] [PubMed] [Google Scholar]
- 944. Sancho M, Welsh DG. KIR channels in the microvasculature: regulatory properties and the lipid-hemodynamic environment. Curr Top Membr 85: 227–259, 2020. doi: 10.1016/bs.ctm.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 945. Fang Y, Mohler ER 3rd, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL, Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res 98: 1064–1071, 2006. doi: 10.1161/01.RES.0000218776.87842.43. [DOI] [PubMed] [Google Scholar]
- 946. Fancher IS, Ahn SJ, Adamos C, Osborn C, Oh MJ, Fang Y, Reardon CA, Getz GS, Phillips SA, Levitan I. Hypercholesterolemia-induced loss of flow-induced vasodilation and lesion formation in apolipoprotein E-deficient mice critically depend on inwardly rectifying K+ channels. J Am Heart Assoc 7: e007430, 2018. doi: 10.1161/JAHA.117.007430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 947. Foster MN, Coetzee WA. KATP channels in the cardiovascular system. Physiol Rev 96: 177–252, 2016. doi: 10.1152/physrev.00003.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 948. Davis MJ, Kim HJ, Zawieja SD, Castorena‐Gonzalez JA, Gui P, Li M, Saunders BT, Zinselmeyer BH, Randolph GJ, Remedi MS, Nichols CG. Kir6.1-dependent KATP channels in lymphatic smooth muscle and vessel dysfunction in mice with Kir6.1 gain-of-function. J Physiol 598: 3107–3127, 2020. doi: 10.1113/JP279612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 949. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 440: 470–476, 2006. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 950. Lynch FM, Austin C, Heagerty AM, Izzard AS. Adenosine and hypoxic dilation of rat coronary small arteries: roles of the ATP-sensitive potassium channel, endothelium, and nitric oxide. Am J Physiol Heart Circ Physiol 290: H1145–H1150, 2006. doi: 10.1152/ajpheart.00314.2005. [DOI] [PubMed] [Google Scholar]
- 951. Jackson WF. Arteriolar oxygen reactivity: where is the sensor and what is the mechanism of action? J Physiol 594: 5055–5077, 2016. doi: 10.1113/JP270192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 952. Jackson WF. Arteriolar tone is determined by activity of ATP-sensitive potassium channels. Am J Physiol Heart Circ Physiol 265: H1797–H1803, 1993. doi: 10.1152/ajpheart.1993.265.5.H1797. [DOI] [PubMed] [Google Scholar]
- 953. Reslerova M, Loutzenhiser R. Renal microvascular actions of calcitonin gene-related peptide. Am J Physiol Renal Physiol 274: F1078–F1085, 1998. doi: 10.1152/ajprenal.1998.274.6.F1078. [DOI] [PubMed] [Google Scholar]
- 954. Burns WR, Cohen KD, Jackson WF. K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation 11: 279–293, 2004. doi: 10.1080/10739680490425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 955. Asano M, Masuzawa-Ito K, Matsuda T. Charybdotoxin-sensitive K+ channels regulate the myogenic tone in the resting state of arteries from spontaneously hypertensive rats. Br J Pharmacol 108: 214–222, 1993. doi: 10.1111/j.1476-5381.1993.tb13465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 956. Omote M, Mizusawa H. Interaction between the L-type Ca2+ channel and the Ca2+-activated K+ channel in the fluctuating myogenic contraction in the rabbit basilar artery. Jpn J Physiol 46: 353–356, 1996. doi: 10.2170/jjphysiol.46.353. [DOI] [PubMed] [Google Scholar]
- 957. Jackson WF. Potassium channels and regulation of the microcirculation. Microcirculation 5: 85–90, 1998. [PubMed] [Google Scholar]
- 958. Watanabe J, Horiguchi S, Keitoku M, Karibe A, Takeuchi M, Suzuki S, Satoh S, Shirato K. The role of extracellular cations in the development of myogenic contraction in isolated rat small arteries. Jpn Circ J 60: 239–246, 1996. doi: 10.1253/jcj.60.239. [DOI] [PubMed] [Google Scholar]
- 959. Li Y, Aziz Q, Anderson N, Ojake L, Tinker A. Endothelial ATP-sensitive potassium channel protects against the development of hypertension and atherosclerosis. Hypertension 76: 776–784, 2020. doi: 10.1161/HYPERTENSIONAHA.120.15355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 960. Aziz Q, Li Y, Anderson N, Ojake L, Tsisanova E, Tinker A. Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J Biol Chem 292: 17587–17597, 2017. doi: 10.1074/jbc.M117.810325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 961. Aziz Q, Li Y, Tinker A. Endothelial biology and ATP-sensitive potassium channels. Channels (Austin) 12: 45–46, 2018. doi: 10.1080/19336950.2017.1412151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 962. Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol 285: C959–C967, 2003. doi: 10.1152/ajpcell.00511.2002. [DOI] [PubMed] [Google Scholar]
- 963. Chatterjee S, Levitan I, Wei Z, Fisher AB. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 13: 633–644, 2006. doi: 10.1080/10739680600930255. [DOI] [PubMed] [Google Scholar]
- 964. Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, Birnbaum MJ, Fisher AB. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol 302: H105–H114, 2012. doi: 10.1152/ajpheart.00298.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965. Browning E, Wang H, Hong N, Yu K, Buerk DG, DeBolt K, Gonder D, Sorokina EM, Patel P, De Leon DD, Feinstein SI, Fisher AB, Chatterjee S. Mechanotransduction drives post ischemic revascularization through KATP channel closure and production of reactive oxygen species. Antioxid Redox Signal 20: 872–886, 2014. doi: 10.1089/ars.2012.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966. Goonetilleke L, Quayle J. TREK-1 K+ channels in the cardiovascular system: their significance and potential as a therapeutic target. Cardiovasc Ther 30: e23–e29, 2012. doi: 10.1111/j.1755-5922.2010.00227.x. [DOI] [PubMed] [Google Scholar]
- 967. Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J 38: 305–318, 2009. doi: 10.1007/s00249-008-0326-8. [DOI] [PubMed] [Google Scholar]
- 968. Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 24: 339–346, 2001. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- 969. Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 17: 4283–4290, 1998. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 970. Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. Lysophospholipids open the two-pore domain mechano-gated K+ channels TREK-1 and TRAAK. J Biol Chem 275: 10128–10133, 2000. doi: 10.1074/jbc.275.14.10128. [DOI] [PubMed] [Google Scholar]
- 971. Park KJ, Baker SA, Cho SY, Sanders KM, Koh SD. Sulfur-containing amino acids block stretch-dependent K+ channels and nitrergic responses in the murine colon. Br J Pharmacol 144: 1126–1137, 2005. doi: 10.1038/sj.bjp.0706154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 972. Baker SA, Hennig GW, Han J, Britton FC, Smith TK, Koh SD. Methionine and its derivatives increase bladder excitability by inhibiting stretch-dependent K+ channels. Br J Pharmacol 153: 1259–1271, 2008. doi: 10.1038/sj.bjp.0707690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 973. Millar JA, Barratt L, Southan AP, Page KM, Fyffe RE, Robertson B, Mathie A. A functional role for the two-pore domain potassium channel TASK-1 in cerebellar granule neurons. Proc Natl Acad Sci USA 97: 3614–3618, 2000. doi: 10.1073/pnas.97.7.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 974. Lauritzen I, Zanzouri M, Honoré E, Duprat F, Ehrengruber MU, Lazdunski M, Patel AJ. K+-dependent cerebellar granule neuron apoptosis. Role of task leak K+ channels. J Biol Chem 278: 32068–32076, 2003. doi: 10.1074/jbc.M302631200. [DOI] [PubMed] [Google Scholar]
- 975. Sanders KM, Koh SD. Two-pore-domain potassium channels in smooth muscles: new components of myogenic regulation. J Physiol 570: 37–43, 2006. doi: 10.1113/jphysiol.2005.098897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 976. Dedman A, Sharif-Naeini R, Folgering JH, Duprat F, Patel A, Honoré E. The mechano-gated K2P channel TREK-1. Eur Biophys J 38: 293–303, 2009. doi: 10.1007/s00249-008-0318-8. [DOI] [PubMed] [Google Scholar]
- 977. Patel AJ, Lazdunski M, Honoré E. Lipid and mechano-gated 2P domain K+ channels. Curr Opin Cell Biol 13: 422–428, 2001. doi: 10.1016/s0955-0674(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 978. Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem 275: 17412–17419, 2000. doi: 10.1074/jbc.M000445200. [DOI] [PubMed] [Google Scholar]
- 979. Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 274: 26691–26696, 1999. doi: 10.1074/jbc.274.38.26691. [DOI] [PubMed] [Google Scholar]
- 980. Lundbaek JA, Andersen OS. Lysophospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J Gen Physiol 104: 645–673, 1994. doi: 10.1085/jgp.104.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 981. Maingret F, Fosset M, Lesage F, Lazdunski M, Honore E. TRAAK is a mammalian neuronal mechano-gated K+ channel. J Biol Chem 274: 1381–1387, 1999. doi: 10.1074/jbc.274.3.1381. [DOI] [PubMed] [Google Scholar]
- 982. Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honoré E. TREK-1 is a heat-activated background K+ channel. EMBO J 19: 2483–2491, 2000. doi: 10.1093/emboj/19.11.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 983. Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 17: 3297–3308, 1998. doi: 10.1093/emboj/17.12.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 984. Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J 24: 44–53, 2005. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985. Lopes CM, Rohács T, Czirják G, Balla T, Enyedi P, Logothetis DE. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol 564: 117–129, 2005. doi: 10.1113/jphysiol.2004.081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 986. Ordway RW, Petrou S, Kirber MT, Walsh JV Jr, Singer JJ. Stretch activation of a toad smooth muscle K+ channel may be mediated by fatty acids. J Physiol 484: 331–337, 1995. doi: 10.1113/jphysiol.1995.sp020668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 987. Petrou S, Ordway RW, Kirber MT, Dopico AM, Hamilton JA, Walsh JV Jr, Singer JJ. Direct effects of fatty acids and other charged lipids on ion channel activity in smooth muscle cells. Prostaglandins Leukot Essent Fatty Acids 52: 173–178, 1995. doi: 10.1016/0952-3278(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 988. Kim D, Clapham DE. Potassium channels in cardiac cells activated by arachidonic acid and phospholipids. Science 244: 1174–1176, 1989. doi: 10.1126/science.2727703. [DOI] [PubMed] [Google Scholar]
- 989. Koh SD, Sanders KM. Stretch-dependent potassium channels in murine colonic smooth muscle cells. J Physiol 533: 155–163, 2001. doi: 10.1111/j.1469-7793.2001.0155b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 990. Koh SD, Campbell JD, Carl A, Sanders KM. Nitric oxide activates multiple potassium channels in canine colonic smooth muscle. J Physiol 489: 735–743, 1995. doi: 10.1113/jphysiol.1995.sp021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 991. Monaghan K, Baker SA, Dwyer L, Hatton WC, Sik Park K, Sanders KM, Koh SD. The stretch-dependent potassium channel TREK-1 and its function in murine myometrium. J Physiol 589: 1221–1233, 2011. doi: 10.1113/jphysiol.2010.203869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992. Koh SD, Monaghan K, Sergeant GP, Ro S, Walker RL, Sanders KM, Horowitz B. TREK-1 regulation by nitric oxide and cGMP-dependent protein kinase. An essential role in smooth muscle inhibitory neurotransmission. J Biol Chem 276: 44338–44346, 2001. doi: 10.1074/jbc.M108125200. [DOI] [PubMed] [Google Scholar]
- 993. Lei Q, Pan XQ, Chang S, Malkowicz SB, Guzzo TJ, Malykhina AP. Response of the human detrusor to stretch is regulated by TREK-1, a two-pore-domain (K2P) mechano-gated potassium channel. J Physiol 592: 3013–3030, 2014. doi: 10.1113/jphysiol.2014.271718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 994. Baker SA, Hatton WJ, Han J, Hennig GW, Britton FC, Koh SD. Role of TREK-1 potassium channel in bladder overactivity after partial bladder outlet obstruction in mouse. J Urol 183: 793–800, 2010. doi: 10.1016/j.juro.2009.09.079. [DOI] [PubMed] [Google Scholar]
- 995. Pineda RH, Hypolite J, Lee S, Carrasco A Jr, Iguchi N, Meacham RB, Malykhina AP. Altered detrusor contractility and voiding patterns in mice lacking the mechanosensitive TREK-1 channel. BMC Urol 19: 40, 2019. doi: 10.1186/s12894-019-0475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 996. Peyronnet R, Sharif-Naeini R, Folgering JH, Arhatte M, Jodar M, El Boustany C, Gallian C, Tauc M, Duranton C, Rubera I, Lesage F, Pei Y, Peters DJ, Somlo S, Sachs F, Patel A, Honoré E, Duprat F. Mechanoprotection by polycystins against apoptosis is mediated through the opening of stretch-activated K2P channels. Cell Rep 1: 241–250, 2012. doi: 10.1016/j.celrep.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 997. Garry A, Fromy B, Blondeau N, Henrion D, Brau F, Gounon P, Guy N, Heurteaux C, Lazdunski M, Saumet JL. Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep 8: 354–359, 2007. doi: 10.1038/sj.embor.7400916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 998. Blondeau N, Pétrault O, Manta S, Giordanengo V, Gounon P, Bordet R, Lazdunski M, Heurteaux C. Polyunsaturated fatty acids are cerebral vasodilators via the TREK-1 potassium channel. Circ Res 101: 176–184, 2007. doi: 10.1161/CIRCRESAHA.107.154443. [DOI] [PubMed] [Google Scholar]
- 999. Namiranian K, Lloyd EE, Crossland RF, Marrelli SP, Taffet GE, Reddy AK, Hartley CJ, Bryan RM Jr.. Cerebrovascular responses in mice deficient in the potassium channel, TREK-1. Am J Physiol Regul Integr Comp Physiol 299: R461–R469, 2010. doi: 10.1152/ajpregu.00057.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000. Beckett EA, Hollywood MA, Thornbury KD, McHale NG. Spontaneous electrical activity in sheep mesenteric lymphatics. Lymphat Res Biol 5: 29–43, 2007. doi: 10.1089/lrb.2007.5104. [DOI] [PubMed] [Google Scholar]
- 1001. Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 7: e38689, 2018. doi: 10.7554/eLife.38689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1002. Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J 12: 1681–1692, 1993. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1003. Lehtonen JY, Kinnunen PK. Phospholipase A2 as a mechanosensor. Biophys J 68: 1888–1894, 1995. doi: 10.1016/S0006-3495(95)80366-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1004. Vandenburgh HH, Shansky J, Karlisch P, Solerssi RL. Mechanical stimulation of skeletal muscle generates lipid-related second messengers by phospholipase activation. J Cell Physiol 155: 63–71, 1993. doi: 10.1002/jcp.1041550109. [DOI] [PubMed] [Google Scholar]
- 1005. Matsumoto H, Baron CB, Coburn RF. Smooth muscle stretch-activated phospholipase C activity. Am J Physiol Cell Physiol 268: C458–C465, 1995. doi: 10.1152/ajpcell.1995.268.2.C458. [DOI] [PubMed] [Google Scholar]
- 1006. Xi Q, Adebiyi A, Zhao G, Chapman KE, Waters CM, Hassid A, Jaggar JH. IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ Res 102: 1118–1126, 2008. doi: 10.1161/CIRCRESAHA.108.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1007. Jones AW, Shukla SD, Geisbuhler BB. Stimulation of phospholipase D activity and phosphatidic acid production by norepinephrine in rat aorta. Am J Physiol Cell Physiol 264: C609–C616, 1993. doi: 10.1152/ajpcell.1993.264.3.C609. [DOI] [PubMed] [Google Scholar]
- 1008. Kamishima T, McCarron JG. Regulation of the cytosolic Ca2+ concentration by Ca2+ stores in single smooth muscle cells from rat cerebral arteries. J Physiol 501: 497–508, 1997. doi: 10.1111/j.1469-7793.1997.497bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1009. Evans L, Frenkel L, Brophy CM, Rosales O, Sudhaker CB, Li G, Du W, Sumpio BE. Activation of diacylglycerol in cultured endothelial cells exposed to cyclic strain. Am J Physiol Cell Physiol 272: C650–C656, 1997. doi: 10.1152/ajpcell.1997.272.2.C650. [DOI] [PubMed] [Google Scholar]
- 1010. Albert AP, Piper AS, Large WA. Role of phospholipase D and diacylglycerol in activating constitutive TRPC-like cation channels in rabbit ear artery myocytes. J Physiol 566: 769–780, 2005. doi: 10.1113/jphysiol.2005.090852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1011. Kadamur G, Ross EM. Mammalian phospholipase C. Annu Rev Physiol 75: 127–154, 2013. doi: 10.1146/annurev-physiol-030212-183750. [DOI] [PubMed] [Google Scholar]
- 1012. Hoberg M, Gratz HH, Noll M, Jones DB. Mechanosensitivity of human osteosarcoma cells and phospholipase C beta2 expression. Biochem Biophys Res Commun 333: 142–149, 2005. doi: 10.1016/j.bbrc.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 1013. Nam JH, Lee HS, Nguyen YH, Kang TM, Lee SW, Kim HY, Kim SJ, Earm YE, Kim SJ. Mechanosensitive activation of K+ channel via phospholipase C-induced depletion of phosphatidylinositol 4,5-bisphosphate in B lymphocytes. J Physiol 582: 977–990, 2007. doi: 10.1113/jphysiol.2007.128413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1014. Shah L, Bansal V, Rye PL, Mumtaz N, Taherian A, Fisher TE. Osmotic activation of phospholipase C triggers structural adaptation in osmosensitive rat supraoptic neurons. J Physiol 592: 4165–4175, 2014. doi: 10.1113/jphysiol.2014.273813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1015. Berk BC, Corson MA, Peterson TE, Tseng H. Protein kinases as mediators of fluid shear stress stimulated signal transduction in endothelial cells: a hypothesis for calcium-dependent and calcium-independent events activated by flow. J Biomech 28: 1439–1450, 1995. doi: 10.1016/0021-9290(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 1016. Nahorski SR, Wilcox RA, Mackrill JJ, Challiss RA. Phosphoinositide-derived second messengers and the regulation of Ca2+ in vascular smooth muscle. J Hypertens Suppl 12: S133–S143, 1994. [PubMed] [Google Scholar]
- 1017. Osol G, Laher I, Kelley M. Myogenic tone is coupled to phospholipase C and G protein activation in small cerebral arteries. Am J Physiol Heart Circ Physiol 265: H415–H420, 1993. doi: 10.1152/ajpheart.1993.265.1.H415. [DOI] [PubMed] [Google Scholar]
- 1018. Inscho EW, Cook AK, Mui V, Imig JD. Calcium mobilization contributes to pressure-mediated afferent arteriolar vasoconstriction. Hypertension 31: 421–428, 1998. doi: 10.1161/01.hyp.31.1.421. [DOI] [PubMed] [Google Scholar]
- 1019. Jarajapu YP, Knot HJ. Role of phospholipase C in development of myogenic tone in rat posterior cerebral arteries. Am J Physiol Heart Circ Physiol 283: H2234–H2238, 2002. doi: 10.1152/ajpheart.00624.2002. [DOI] [PubMed] [Google Scholar]
- 1020. Björling K, Joseph PD, Egebjerg K, Salomonsson M, Hansen JL, Ludvigsen TP, Jensen LJ. Role of age, Rho-kinase 2 expression, and G protein-mediated signaling in the myogenic response in mouse small mesenteric arteries. Physiol Rep 6: e13863, 2018. doi: 10.14814/phy2.13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1021. Narayanan J, Imig M, Roman RJ, Harder DR. Pressurization of isolated renal arteries increases inositol trisphosphate and diacylglycerol. Am J Physiol Heart Circ Physiol 266: H1840–H1845, 1994. doi: 10.1152/ajpheart.1994.266.5.H1840. [DOI] [PubMed] [Google Scholar]
- 1022. Wang JP. U-73122, an aminosteroid phospholipase C inhibitor, may also block Ca2+ influx through phospholipase C-independent mechanism in neutrophil activation. Naunyn Schmiedebergs Arch Pharmacol 353: 599–605, 1996. doi: 10.1007/BF00167177. [DOI] [PubMed] [Google Scholar]
- 1023. Langton PD, Farley R, Everitt DE. Neomycin inhibits K+-induced force and Ca2+ channel current in rat arterial smooth muscle. Pflugers Arch 433: 188–193, 1996. doi: 10.1007/s004240050266. [DOI] [PubMed] [Google Scholar]
- 1024. Wu-Zhang AX, Newton AC. Protein kinase C pharmacology: refining the toolbox. Biochem J 452: 195–209, 2013. doi: 10.1042/BJ20130220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1025. Hill MA, Falcone JC, Meininger GA. Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am J Physiol Heart Circ Physiol 259: H1586–H1594, 1990. doi: 10.1152/ajpheart.1990.259.5.H1586. [DOI] [PubMed] [Google Scholar]
- 1026. Osol G, Laher I, Cipolla M. Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ Res 68: 359–367, 1991. doi: 10.1161/01.res.68.2.359. [DOI] [PubMed] [Google Scholar]
- 1027. Kirton CA, Loutzenhiser R. Alterations in basal protein kinase C activity modulate renal afferent arteriolar myogenic reactivity. Am J Physiol Heart Circ Physiol 275: H467–H475, 1998. doi: 10.1152/ajpheart.1998.275.2.H467. [DOI] [PubMed] [Google Scholar]
- 1028. Miller FJ Jr, Dellsperger KC, Gutterman DD. Myogenic constriction of human coronary arterioles. Am J Physiol Heart Circ Physiol 273: H257–H264, 1997. doi: 10.1152/ajpheart.1997.273.1.H257. [DOI] [PubMed] [Google Scholar]
- 1029. Coats P, Johnston F, MacDonald J, McMurray JJ, Hillier C. Signalling mechanisms underlying the myogenic response in human subcutaneous resistance arteries. Cardiovasc Res 49: 828–837, 2001. doi: 10.1016/s0008-6363(00)00314-x. [DOI] [PubMed] [Google Scholar]
- 1030. Ito I, Jarajapu YP, Grant MB, Knot HJ. Characteristics of myogenic tone in the rat ophthalmic artery. Am J Physiol Heart Circ Physiol 292: H360–H368, 2007. doi: 10.1152/ajpheart.00630.2006. [DOI] [PubMed] [Google Scholar]
- 1031. Davis MJ. Myogenic response gradient in an arteriolar network. Am J Physiol Heart Circ Physiol 264: H2168–H2179, 1993. doi: 10.1152/ajpheart.1993.264.6.H2168. [DOI] [PubMed] [Google Scholar]
- 1032. Hong K, Zhao G, Hong Z, Sun Z, Yang Y, Clifford PS, Davis MJ, Meininger GA, Hill MA. Mechanical activation of angiotensin II type 1 receptors causes actin remodelling and myogenic responsiveness in skeletal muscle arterioles. J Physiol 594: 7027–7047, 2016. doi: 10.1113/JP272834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1033. Vennekens R, Nilius B. Insights into TRPM4 function, regulation and physiological role. Handb Exp Pharmacol 179: 269–285, 2007. doi: 10.1007/978-3-540-34891-7_16. [DOI] [PubMed] [Google Scholar]
- 1034. Dutta AK, Khimji AK, Liu S, Karamysheva Z, Fujita A, Kresge C, Rockey DC, Feranchak AP. PKCalpha regulates TMEM16A-mediated Cl− secretion in human biliary cells. Am J Physiol Gastrointest Liver Physiol 310: G34–G42, 2016. doi: 10.1152/ajpgi.00146.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1035. Hill MA, Davis MJ, Song J, Zou H. Calcium dependence of indolactam-mediated contractions in resistance vessels. J Pharmacol Exp Ther 276: 867–874, 1996. [PubMed] [Google Scholar]
- 1036. Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem 266: 1708–1715, 1991. doi: 10.1016/S0021-9258(18)52353-X. [DOI] [PubMed] [Google Scholar]
- 1037. Kitazawa T, Somlyo AP. Modulation of Ca2+ sensitivity by agonists in smooth muscle. Adv Exp Med Biol 304: 97–109, 1991. doi: 10.1007/978-1-4684-6003-2_10. [DOI] [PubMed] [Google Scholar]
- 1038. Walsh MP, Cole WC. The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 33: 1–12, 2013. doi: 10.1038/jcbfm.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1039. Szasz T, Webb RC. Rho-mancing to sensitize calcium signaling for contraction in the vasculature: role of rho kinase. Adv Pharmacol 78: 303–322, 2017. doi: 10.1016/bs.apha.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 1040. Murakami M, Nakatani Y, Atsumi GI, Inoue K, Kudo I. Regulatory functions of phospholipase A2. Crit Rev Immunol 37: 127–195, 2017. doi: 10.1615/CritRevImmunol.v37.i2-6.20. [DOI] [PubMed] [Google Scholar]
- 1041. Exton JH. Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta 1212: 26–42, 1994. doi: 10.1016/0005-2760(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 1042. Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, Narayanan J, Falck JR, Okamoto H, Roman RJ, Nithipatikom K, Campbell WB, Harder DR. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res 87: 60–65, 2000. doi: 10.1161/01.res.87.1.60. [DOI] [PubMed] [Google Scholar]
- 1043. Harder DR, Narayanan J, Gebremedhin D. Pressure-induced myogenic tone and role of 20-HETE in mediating autoregulation of cerebral blood flow. Am J Physiol Heart Circ Physiol 300: H1557–H1565, 2011. doi: 10.1152/ajpheart.01097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1044. Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER, Harder DR. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J Physiol 507: 771–781, 1998. doi: 10.1111/j.1469-7793.1998.771bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1045. Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. 20-HETE is an endogenous inhibitor of the large-conductance Ca2+-activated K+ channel in renal arterioles. Am J Physiol Regul Integr Comp Physiol 270: R228–R237, 1996. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- 1046. Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 1047. Burch RM, Luini A, Axelrod J. Phospholipase A2 and phospholipase C are activated by distinct GTP-binding proteins in response to alpha 1-adrenergic stimulation in FRTL5 thyroid cells. Proc Natl Acad Sci USA 83: 7201–7205, 1986. doi: 10.1073/pnas.83.19.7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1048. Hussain T, Lokhandwala MF. Dopamine-1 receptor G-protein coupling and the involvement of phospholipase A2 in dopamine-1 receptor mediated cellular signaling mechanisms in the proximal tubules of SHR. Clin Exp Hypertens 19: 131–140, 1997. doi: 10.3109/10641969709080810. [DOI] [PubMed] [Google Scholar]
- 1049. Rosenstock M, Danon A, Rimon G. Dual regulation of PLA2 and PGI2 production by G proteins in bovine aortic endothelial cells. Am J Physiol Cell Physiol 271: C555–C562, 1996. doi: 10.1152/ajpcell.1996.271.2.C555. [DOI] [PubMed] [Google Scholar]
- 1050. Denson DD, Li J, Wang X, Eaton DC. Activation of BK channels in GH3 cells by a c-PLA2-dependent G-protein signaling pathway. J Neurophysiol 93: 3146–3156, 2005. doi: 10.1152/jn.00865.2004. [DOI] [PubMed] [Google Scholar]
- 1051. Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev 84: 1341–1379, 2004. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 1052. Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA. Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Mol Pharmacol 62: 983–992, 2002. doi: 10.1124/mol.62.5.983. [DOI] [PubMed] [Google Scholar]
- 1053. Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev 54: 431–467, 2002. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 1054. Dreja K, Voldstedlund M, Vinten J, Tranum-Jensen J, Hellstrand P, Swärd K. Cholesterol depletion disrupts caveolae and differentially impairs agonist-induced arterial contraction. Arterioscler Thromb Vasc Biol 22: 1267–1272, 2002. doi: 10.1161/01.atv.0000023438.32585.a1. [DOI] [PubMed] [Google Scholar]
- 1055. Je HD, Gallant C, Leavis PC, Morgan KG. Caveolin-1 regulates contractility in differentiated vascular smooth muscle. Am J Physiol Heart Circ Physiol 286: H91–H98, 2004. doi: 10.1152/ajpheart.00472.2003. [DOI] [PubMed] [Google Scholar]
- 1056. Shaw L, Sweeney MA, O’Neill SC, Jones CJ, Austin C, Taggart MJ. Caveolae and sarcoplasmic reticular coupling in smooth muscle cells of pressurised arteries: the relevance for Ca2+ oscillations and tone. Cardiovasc Res 69: 825–835, 2006. doi: 10.1016/j.cardiores.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 1057. Cheng X, Jaggar JH. Genetic ablation of caveolin-1 modifies Ca2+ spark coupling in murine arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 290: H2309–H2319, 2006. doi: 10.1152/ajpheart.01226.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1058. Schwartz MA. Integrin signaling revisited. Trends Cell Biol 11: 466–470, 2001. doi: 10.1016/s0962-8924(01)02152-3. [DOI] [PubMed] [Google Scholar]
- 1059. Chen KD, Li YS, Kim M, Li S, Yuan S, Chien S, Shyy JY. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J Biol Chem 274: 18393–18400, 1999. doi: 10.1074/jbc.274.26.18393. [DOI] [PubMed] [Google Scholar]
- 1060. Wang Y, Miao H, Li S, Chen KD, Li YS, Yuan S, Shyy JY, Chien S. Interplay between integrins and FLK-1 in shear stress-induced signaling. Am J Physiol Cell Physiol 283: C1540–C1547, 2002. doi: 10.1152/ajpcell.00222.2002. [DOI] [PubMed] [Google Scholar]
- 1061. Kuchan MJ, Jo H, Frangos JA. Role of G proteins in shear stress-mediated nitric oxide production by endothelial cells. Am J Physiol Cell Physiol 267: C753–C758, 1994. doi: 10.1152/ajpcell.1994.267.3.C753. [DOI] [PubMed] [Google Scholar]
- 1062. DePaola N, Davies PF, Pritchard WF Jr, Florez L, Harbeck N, Polacek DC. Spatial and temporal regulation of gap junction connexin43 in vascular endothelial cells exposed to controlled disturbed flows in vitro. Proc Natl Acad Sci USA 96: 3154–3159, 1999. doi: 10.1073/pnas.96.6.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1063. Butler PJ, Tsou TC, Li JY, Usami S, Chien S. Rate sensitivity of shear-induced changes in the lateral diffusion of endothelial cell membrane lipids: a role for membrane perturbation in shear-induced MAPK activation. FASEB J 16: 216–218, 2002. doi: 10.1096/fj.01-0434fje. [DOI] [PubMed] [Google Scholar]
- 1064. Park H, Go YM, Darji R, Choi JW, Lisanti MP, Maland MC, Jo H. Caveolin-1 regulates shear stress-dependent activation of extracellular signal-regulated kinase. Am J Physiol Heart Circ Physiol 278: H1285–H1293, 2000. doi: 10.1152/ajpheart.2000.278.4.H1285. [DOI] [PubMed] [Google Scholar]
- 1065. Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res 93: e136–e142, 2003. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 1066. Ando J, Yamamoto K. Hemodynamic forces, endothelial mechanotransduction, and vascular diseases. Magn Reson Med Sci 21: 258–266, 2022. doi: 10.2463/mrms.rev.2021-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1067. Yamamoto K, Ando J. Emerging role of plasma membranes in vascular endothelial mechanosensing. Circ J 82: 2691–2698, 2018. doi: 10.1253/circj.CJ-18-0052. [DOI] [PubMed] [Google Scholar]
- 1068. Butler PJ, Norwich G, Weinbaum S, Chien S. Shear stress induces a time- and position-dependent increase in endothelial cell membrane fluidity. Am J Physiol Cell Physiol 280: C962–C969, 2001. doi: 10.1152/ajpcell.2001.280.4.C962. [DOI] [PubMed] [Google Scholar]
- 1069. Yamamoto K, Nogimori Y, Imamura H, Ando J. Shear stress activates mitochondrial oxidative phosphorylation by reducing plasma membrane cholesterol in vascular endothelial cells. Proc Natl Acad Sci USA 117: 33660–33667, 2020. doi: 10.1073/pnas.2014029117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1070. Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am J Physiol Heart Circ Physiol 285: H1720–H1729, 2003. doi: 10.1152/ajpheart.00344.2002. [DOI] [PubMed] [Google Scholar]
- 1071. Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol 23: 1161–1168, 2003. doi: 10.1161/01.ATV.0000070546.16946.3A. [DOI] [PubMed] [Google Scholar]
- 1072. van der Meer AD, Kamphuis MM, Poot AA, Feijen J, Vermes I. Lowering caveolin-1 expression in human vascular endothelial cells inhibits signal transduction in response to shear stress. Int J Cell Biol 2009: 532432, 2009. doi: 10.1155/2009/532432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1073. Rizzo V, Sung A, Oh P, Schnitzer JE. Rapid mechanotransduction in situ at the luminal cell surface of vascular endothelium and its caveolae. J Biol Chem 273: 26323–26329, 1998. doi: 10.1074/jbc.273.41.26323. [DOI] [PubMed] [Google Scholar]
- 1074. Rizzo V, McIntosh DP, Oh P, Schnitzer JE. In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association. J Biol Chem 273: 34724–34729, 1998. doi: 10.1074/jbc.273.52.34724. [DOI] [PubMed] [Google Scholar]
- 1075. Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138, 2001. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 1076. Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116: 1284–1291, 2006. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1077. Albinsson S, Shakirova Y, Rippe A, Baumgarten M, Rosengren BI, Rippe C, Hallmann R, Hellstrand P, Rippe B, Swärd K. Arterial remodeling and plasma volume expansion in caveolin-1-deficient mice. Am J Physiol Regul Integr Comp Physiol 293: R1222–R1231, 2007. doi: 10.1152/ajpregu.00092.2007. [DOI] [PubMed] [Google Scholar]
- 1078. Pol A, Morales-Paytuví F, Bosch M, Parton RG. Non-caveolar caveolins—duties outside the caves. J Cell Sci 133: jcs241562, 2020. doi: 10.1242/jcs.241562. [DOI] [PubMed] [Google Scholar]
- 1079. Yamamoto K, Imamura H, Ando J. Shear stress augments mitochondrial ATP generation that triggers ATP release and Ca2+ signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol 315: H1477–H1485, 2018. doi: 10.1152/ajpheart.00204.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1080. Sun X, Fu Y, Gu M, Zhang L, Li D, Li H, Chien S, Shyy JY, Zhu Y. Activation of integrin alpha5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc Natl Acad Sci USA 113: 769–774, 2016. doi: 10.1073/pnas.1524523113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1081. Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, Morone N, Parton RG, Raposo G, Sens P, Lamaze C, Nassoy P. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144: 402–413, 2011. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1082. Weinbaum S, Cancel LM, Fu BM, Tarbell JM. The glycocalyx and its role in vascular physiology and vascular related diseases. Cardiovasc Eng Technol 12: 37–71, 2021. doi: 10.1007/s13239-020-00485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1083. Zeng Y, Zhang XF, Fu BM, Tarbell JM. The role of endothelial surface glycocalyx in mechanosensing and transduction. Adv Exp Med Biol 1097: 1–27, 2018. doi: 10.1007/978-3-319-96445-4_1. [DOI] [PubMed] [Google Scholar]
- 1084. Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis 252: 136–146, 2016. doi: 10.1016/j.atherosclerosis.2016.07.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1085. Dogné S, Flamion B. Endothelial glycocalyx impairment in disease: focus on hyaluronan shedding. Am J Pathol 190: 768–780, 2020. doi: 10.1016/j.ajpath.2019.11.016. [DOI] [PubMed] [Google Scholar]
- 1086. Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J Intern Med 280: 97–113, 2016. doi: 10.1111/joim.12465. [DOI] [PubMed] [Google Scholar]
- 1087. Mitra R, O’Neil GL, Harding IC, Cheng MJ, Mensah SA, Ebong EE. Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr Atheroscler Rep 19: 63, 2017. doi: 10.1007/s11883-017-0691-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1088. Harding IC, Mitra R, Mensah SA, Nersesyan A, Bal NN, Ebong EE. Endothelial barrier reinforcement relies on flow-regulated glycocalyx, a potential therapeutic target. Biorheology 56: 131–149, 2019. doi: 10.3233/BIR-180205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1089. Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res 87: 198–210, 2010. doi: 10.1093/cvr/cvq062. [DOI] [PubMed] [Google Scholar]
- 1090. Chappell D, Jacob M, Paul O, Rehm M, Welsch U, Stoeckelhuber M, Conzen P, Becker BF. The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture. Circ Res 104: 1313–1317, 2009. doi: 10.1161/CIRCRESAHA.108.187831. [DOI] [PubMed] [Google Scholar]
- 1091. Ebong EE, Macaluso FP, Spray DC, Tarbell JM. Imaging the endothelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy. Arterioscler Thromb Vasc Biol 31: 1908–1915, 2011. doi: 10.1161/ATVBAHA.111.225268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1092. Potter DR, Damiano ER. The hydrodynamically relevant endothelial cell glycocalyx observed in vivo is absent in vitro. Circ Res 102: 770–776, 2008. doi: 10.1161/CIRCRESAHA.107.160226. [DOI] [PubMed] [Google Scholar]
- 1093. Haymet AB, Bartnikowski N, Wood ES, Vallely MP, McBride A, Yacoub S, Biering SB, Harris E, Suen JY, Fraser JF. Studying the endothelial glycocalyx in vitro: what is missing? Front Cardiovasc Med 8: 647086, 2021. doi: 10.3389/fcvm.2021.647086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1094. Pahakis MY, Kosky JR, Dull RO, Tarbell JM. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun 355: 228–233, 2007. doi: 10.1016/j.bbrc.2007.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1095. Kumagai R, Lu X, Kassab GS. Role of glycocalyx in flow-induced production of nitric oxide and reactive oxygen species. Free Radic Biol Med 47: 600–607, 2009. doi: 10.1016/j.freeradbiomed.2009.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096. Koo A, Dewey CF Jr, Garcia-Cardena G. Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am J Physiol Cell Physiol 304: C137–C146, 2013. doi: 10.1152/ajpcell.00187.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1097. Yao Y, Rabodzey A, Dewey CF Jr.. Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol 293: H1023–H1030, 2007. doi: 10.1152/ajpheart.00162.2007. [DOI] [PubMed] [Google Scholar]
- 1098. Nikmanesh M, Shi ZD, Tarbell JM. Heparan sulfate proteoglycan mediates shear stress-induced endothelial gene expression in mouse embryonic stem cell-derived endothelial cells. Biotechnol Bioeng 109: 583–594, 2012. doi: 10.1002/bit.23302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1099. Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong EE, Rizzo V, Tarbell JM. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am J Physiol Heart Circ Physiol 305: H811–H820, 2013. doi: 10.1152/ajpheart.00764.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1100. Harding IC, Mitra R, Mensah SA, Herman IM, Ebong EE. Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J Transl Med 16: 364, 2018. doi: 10.1186/s12967-018-1721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1101. Dela Paz NG, Melchior B, Shayo FY, Frangos JA. Heparan sulfates mediate the interaction between platelet endothelial cell adhesion molecule-1 (PECAM-1) and the Galphaq/11 subunits of heterotrimeric G proteins. J Biol Chem 289: 7413–7424, 2014. doi: 10.1074/jbc.M113.542514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1102. Barnes JM, Kaushik S, Bainer RO, Sa JK, Woods EC, Kai F, Przybyla L, Lee M, Lee HW, Tung JC, Maller O, Barrett AS, Lu KV, Lakins JN, Hansen KC, Obernier K, Alvarez-Buylla A, Bergers G, Phillips JJ, Nam DH, Bertozzi CR, Weaver VM. A tension-mediated glycocalyx-integrin feedback loop promotes mesenchymal-like glioblastoma. Nat Cell Biol 20: 1203–1214, 2018. doi: 10.1038/s41556-018-0183-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1103. Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc Natl Acad Sci USA 101: 16483–16488, 2004. doi: 10.1073/pnas.0407474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1104. Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol (Camb) 6: 338–347, 2014. doi: 10.1039/c3ib40199e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1105. Wang G, Kostidis S, Tiemeier GL, Sol W, de Vries MR, Giera M, Carmeliet P, van den Berg BM, Rabelink TJ. Shear stress regulation of endothelial glycocalyx structure is determined by glucobiosynthesis. Arterioscler Thromb Vasc Biol 40: 350–364, 2020. doi: 10.1161/ATVBAHA.119.313399. [DOI] [PubMed] [Google Scholar]
- 1106. Ferreira R, Fukui H, Chow R, Vilfan A, Vermot J. The cilium as a force sensor-myth versus reality. J Cell Sci 132: jcs213496, 2019. doi: 10.1242/jcs.213496. [DOI] [PubMed] [Google Scholar]
- 1107. Egorova AD, van der Heiden K, Poelmann RE, Hierck BP. Primary cilia as biomechanical sensors in regulating endothelial function. Differentiation 83: S56–S61, 2012. doi: 10.1016/j.diff.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 1108. Zaragoza C, Márquez S, Saura M. Endothelial mechanosensors of shear stress as regulators of atherogenesis. Curr Opin Lipidol 23: 446–452, 2012. doi: 10.1097/MOL.0b013e328357e837. [DOI] [PubMed] [Google Scholar]
- 1109. Nauli SM, Jin X, Hierck BP. The mechanosensory role of primary cilia in vascular hypertension. Int J Vasc Med 2011: 376281, 2011. doi: 10.1155/2011/376281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1110. Van der Heiden K, Hierck BP, Krams R, de Crom R, Cheng C, Baiker M, Pourquie MJ, Alkemade FE, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 196: 542–550, 2008. doi: 10.1016/j.atherosclerosis.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 1111. Dinsmore C, Reiter JF. Endothelial primary cilia inhibit atherosclerosis. EMBO Rep 17: 156–166, 2016. doi: 10.15252/embr.201541019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1112. Iomini C, Tejada K, Mo W, Vaananen H, Piperno G. Primary cilia of human endothelial cells disassemble under laminar shear stress. J Cell Biol 164: 811–817, 2004. doi: 10.1083/jcb.200312133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1113. Sheng X, Sheng Y, Gao S, Fan F, Wang J. Low fluid shear stress promoted ciliogenesis via Dvl2 in hUVECs. Histochem Cell Biol 154: 639–654, 2020. doi: 10.1007/s00418-020-01908-3. [DOI] [PubMed] [Google Scholar]
- 1114. Ten Dijke P, Egorova AD, Goumans MJ, Poelmann RE, Hierck BP. TGF-beta signaling in endothelial-to-mesenchymal transition: the role of shear stress and primary cilia. Sci Signal 5: pt2, 2012. doi: 10.1126/scisignal.2002722. [DOI] [PubMed] [Google Scholar]
- 1115. Singh S, Adam M, Matkar PN, Bugyei-Twum A, Desjardins JF, Chen HH, Nguyen H, Bazinet H, Michels D, Liu Z, Mebrahtu E, Esene L, Joseph J, Ehsan M, Qadura M, Connelly KA, Leong-Poi H, Singh KK. Endothelial-specific loss of IFT88 promotes endothelial-to-mesenchymal transition and exacerbates bleomycin-induced pulmonary fibrosis. Sci Rep 10: 4466, 20 20. doi: 10.1038/s41598-020-61292-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1116. Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, Poelmann RE, Hierck BP. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res 108: 1093–1101, 2011. doi: 10.1161/CIRCRESAHA.110.231860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1117. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev 85: 1159–1204, 2005. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 1118. Geppetti P, Veldhuis NA, Lieu T, Bunnett NW. G protein-coupled receptors: dynamic machines for signaling pain and itch. Neuron 88: 635–649, 2015. doi: 10.1016/j.neuron.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 1119. Gudi SR, Clark CB, Frangos JA. Fluid flow rapidly activates G proteins in human endothelial cells—involvement of G proteins in mechanochemical signal transduction. Circ Res 79: 834–839, 1996. doi: 10.1161/01.RES.79.4.834. [DOI] [PubMed] [Google Scholar]
- 1120. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6: 499–506, 2004. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 1121. Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Mizuno T, Takano H, Hiroi Y, Ueki K, Tobe K. Mechanical stress activates protein kinase cascade of phosphorylation in neonatal rat cardiac myocytes. J Clin Invest 96: 438–446, 1995. doi: 10.1172/JCI118054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1122. Yasuda N, Akazawa H, Qin Y, Zou Y, Komuro I. A novel mechanism of mechanical stress-induced angiotensin II type 1-receptor activation without the involvement of angiotensin II. Naunyn Schmiedebergs Arch Pharmacol 377: 393–399, 2008. doi: 10.1007/s00210-007-0215-1. [DOI] [PubMed] [Google Scholar]
- 1123. Moran CS, Rush CM, Dougan T, Jose RJ, Biros E, Norman PE, Gera L, Golledge J. Modulation of kinin B2 receptor signaling controls aortic dilatation and rupture in the angiotensin II-infused apolipoprotein E-deficient mouse. Arterioscler Thromb Vasc Biol 36: 898–907, 2016. doi: 10.1161/ATVBAHA.115.306945. [DOI] [PubMed] [Google Scholar]
- 1124. Wendt TS, Li YJ, Gonzales RJ. Ozanimod, an S1PR1 ligand, attenuates hypoxia plus glucose deprivation-induced autophagic flux and phenotypic switching in human brain VSM cells. Am J Physiol Cell Physiol 320: C1055–C1073, 2021. doi: 10.1152/ajpcell.00044.2021. [DOI] [PubMed] [Google Scholar]
- 1125. Duru EA, Fu Y, Davies MG. Role of S-1-P receptors and human vascular smooth muscle cell migration in diabetes and metabolic syndrome. J Surg Res 177: e75–e82, 2012. doi: 10.1016/j.jss.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1126. Natarajan A, Han G, Chen SY, Yu P, White R, Jose P. The D5 dopamine receptor mediates large-conductance, calcium- and voltage-activated potassium channel activation in human coronary artery smooth muscle cells. J Pharmacol Exp Ther 332: 640–649, 2010. doi: 10.1124/jpet.109.159871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1127. Mazzetti L, Franchi-Micheli S, Nistri S, Quattrone S, Simone R, Ciuffi M, Zilletti L, Failli P. The ACh-induced contraction in rat aortas is mediated by the Cys Lt1 receptor via intracellular calcium mobilization in smooth muscle cells. Br J Pharmacol 138: 707–715, 2003. doi: 10.1038/sj.bjp.0705087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1128. Storch U, Blodow S, Gudermann T, Mederos y Schnitzler M. Cysteinyl leukotriene 1 receptors as novel mechanosensors mediating myogenic tone together with angiotensin II type 1 receptors-brief report. Arterioscler Thromb Vasc Biol 35: 121–126, 2015. doi: 10.1161/ATVBAHA.114.304844. [DOI] [PubMed] [Google Scholar]
- 1129. Wisniewski K, Galyean R, Tariga H, Alagarsamy S, Croston G, Heitzmann J, Kohan A, Wisniewska H, Laporte R, Rivière PJ, Schteingart CD. New, potent, selective, and short-acting peptidic V1a receptor agonists. J Med Chem 54: 4388–4398, 2011. doi: 10.1021/jm200278m. [DOI] [PubMed] [Google Scholar]
- 1130. Lui J, Hill MA, Meininger GA. G protein involvement in arteriolar myogenic reactivity (Abstract). FASEB J 7: A893, 1993. [Google Scholar]
- 1131. Kauffenstein G, Tamareille S, Prunier F, Roy C, Ayer A, Toutain B, Billaud M, Isakson BE, Grimaud L, Loufrani L, Rousseau P, Abraham P, Procaccio V, Monyer H, de Wit C, Boeynaems JM, Robaye B, Kwak BR, Henrion D. Central role of P2Y6 UDP receptor in arteriolar myogenic tone. Arterioscler Thromb Vasc Biol 36: 1598–1606, 2016. doi: 10.1161/ATVBAHA.116.307739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1132. Kauffenstein G, Drouin A, Thorin-Trescases N, Bachelard H, Robaye B, D’Orleans-Juste P, Marceau F, Thorin E, Sévigny J. NTPDase1 (CD39) controls nucleotide-dependent vasoconstriction in mouse. Cardiovasc Res 85: 204–213, 2010. doi: 10.1093/cvr/cvp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1133. Brayden JE, Li Y, Tavares MJ. Purinergic receptors regulate myogenic tone in cerebral parenchymal arterioles. J Cereb Blood Flow Metab 33: 293–299, 2013. doi: 10.1038/jcbfm.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1134. Bolz SS, Vogel L, Sollinger D, Derwand R, Boer C, Pitson SM, Spiegel S, Pohl U. Sphingosine kinase modulates microvascular tone and myogenic responses through activation of RhoA/Rho kinase. Circulation 108: 342–347, 2003. doi: 10.1161/01.CIR.0000080324.12530.0D. [DOI] [PubMed] [Google Scholar]
- 1135. Peter BF, Lidington D, Harada A, Bolz HJ, Vogel L, Heximer S, Spiegel S, Pohl U, Bolz SS. Role of sphingosine-1-phosphate phosphohydrolase 1 in the regulation of resistance artery tone. Circ Res 103: 315–324, 2008. doi: 10.1161/CIRCRESAHA.108.173575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1136. Yang J, Noyan-Ashraf MH, Meissner A, Voigtlaender-Bolz J, Kroetsch JT, Foltz W, Jaffray D, Kapoor A, Momen A, Heximer SP, Zhang H, van Eede M, Henkelman RM, Matthews SG, Lidington D, Husain M, Bolz SS. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-phosphate signaling. Circulation 126: 196–206, 2012. doi: 10.1161/CIRCULATIONAHA.111.039644. [DOI] [PubMed] [Google Scholar]
- 1137. Erdogmus S, Storch U, Danner L, Becker J, Winter M, Ziegler N, Wirth A, Offermanns S, Hoffmann C, Gudermann T, Mederos YSM. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat Commun 10: 5784, 2019. doi: 10.1038/s41467-019-13722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1138. Hercule HC, Tank J, Plehm R, Wellner M, da Costa Goncalves AC, Gollasch M, Diedrich A, Jordan J, Luft FC, Gross V. Regulator of G protein signalling 2 ameliorates angiotensin II-induced hypertension in mice. Exp Physiol 92: 1014–1022, 2007. doi: 10.1113/expphysiol.2007.038240. [DOI] [PubMed] [Google Scholar]
- 1139. Hong K, Li M, Nourian Z, Meininger GA, Hill MA. Angiotensin II type 1 receptor mechanoactivation involves RGS5 (Regulator of G Protein Signaling 5) in skeletal muscle arteries: impaired trafficking of RGS5 in hypertension. Hypertension 70: 1264–1272, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1140. Arnold C, Demirel E, Feldner A, Genové G, Zhang H, Sticht C, Wieland T, Hecker M, Heximer S, Korff T. Hypertension-evoked RhoA activity in vascular smooth muscle cells requires RGS5. FASEB J 32: 2021–2035, 2018. doi: 10.1096/fj.201700384RR. [DOI] [PubMed] [Google Scholar]
- 1141. Forrester SJ, Kawai T, O'Brien S, Thomas W, Harris RC, Eguchi S. Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu Rev Pharmacol Toxicol 56: 627–653, 2016. doi: 10.1146/annurev-pharmtox-070115-095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1142. Amin AH, Abd Elmageed ZY, Partyka M, Matrougui K. Mechanisms of myogenic tone of coronary arteriole: role of down stream signaling of the EGFR tyrosine kinase. Microvasc Res 81: 135–142, 2011. doi: 10.1016/j.mvr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1143. Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation 110: 3587–3593, 2004. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 1144. Blodow S, Schneider H, Storch U, Wizemann R, Forst AL, Gudermann T, Mederos y Schnitzler M. Novel role of mechanosensitive AT1B receptors in myogenic vasoconstriction. Pflugers Arch 466: 1343–1353, 2014. doi: 10.1007/s00424-013-1372-3. [DOI] [PubMed] [Google Scholar]
- 1145. Yamasaki E, Thakore P, Krishnan V, Earley S. Differential expression of angiotensin II type 1 receptor subtypes within the cerebral microvasculature. Am J Physiol Heart Circ Physiol 318: H461–H469, 2020. doi: 10.1152/ajpheart.00582.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1146. Kauffenstein G, Laher I, Matrougui K, Guérineau NC, Henrion D. Emerging role of G protein-coupled receptors in microvascular myogenic tone. Cardiovasc Res 95: 223–232, 2012. doi: 10.1093/cvr/cvs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1147. Mederos y Schnitzler M, Storch U, Gudermann T. AT1 receptors as mechanosensors. Curr Opin Pharmacol 11: 112–116, 2011. doi: 10.1016/j.coph.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 1148. Storch U, Mederos y Schnitzler M, Gudermann T. G protein-mediated stretch reception. Am J Physiol Heart Circ Physiol 302: H1241–H1249, 2012. doi: 10.1152/ajpheart.00818.2011. [DOI] [PubMed] [Google Scholar]
- 1149. Davis MJ. Myogenic response gradient in an arteriolar network. In: Resistance Arteries, Structure and Function: Proceedings of the Third International Symposium on Resistance Arteries, edited by Mulvany MJ, Aalkjaer C, Heagerty AM, Nyborg NC, Strandgaard S.. Amsterdam: Elsevier Science Publishers, 1991, p. 51–55. [Google Scholar]
- 1150. Cui Y, Kassmann M, Nickel S, Zhang C, Alenina N, Anistan YM, Schleifenbaum J, Bader M, Welsh DG, Huang Y, Gollasch M. Myogenic vasoconstriction requires canonical Gq/11 signaling of the angiotensin II type 1 receptor. J Am Heart Assoc 11: e022070, 2022. doi: 10.1161/JAHA.121.022070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1151. Chennupati R, Wirth A, Favre J, Li R, Bonnavion R, Jin YJ, Wietelmann A, Schweda F, Wettschureck N, Henrion D, Offermanns S. Myogenic vasoconstriction requires G12/G13 and LARG to maintain local and systemic vascular resistance. Elife 8: e49374, 2019. doi: 10.7554/eLife.49374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1152. Gudi S, Nolan JP, Frangos JA. Modulation of GTPase activity of G proteins by fluid shear stress and phospholipid composition. Proc Natl Acad Sci USA 95: 2515–2519, 1998. doi: 10.1073/pnas.95.5.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1153. Gudi S, Huvar I, White CR, McKnight NL, Dusserre N, Boss GR, Frangos JA. Rapid activation of Ras by fluid flow is mediated by Galphaq and Gbetagamma subunits of heterotrimeric G proteins in human endothelial cells. Arterioscler Thromb Vasc Biol 23: 994–1000, 2003. doi: 10.1161/01.ATV.0000073314.51987.84. [DOI] [PubMed] [Google Scholar]
- 1154. Hu Y, Chen M, Wang M, Li X. Flow-mediated vasodilation through mechanosensitive G protein-coupled receptors in endothelial cells. Trends Cardiovasc Med 32: 61–70, 2022. doi: 10.1016/j.tcm.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 1155. Otte LA, Bell KS, Loufrani L, Yeh JC, Melchior B, Dao DN, Stevens HY, White CR, Frangos JA. Rapid changes in shear stress induce dissociation of a G alphaq/11-platelet endothelial cell adhesion molecule-1 complex. J Physiol 587: 2365–2373, 2009. doi: 10.1113/jphysiol.2009.172643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1156. Melchior B, Frangos JA. Distinctive subcellular Akt-1 responses to shear stress in endothelial cells. J Cell Biochem 115: 121–129, 2014. doi: 10.1002/jcb.24639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1157. Yasuda N, Miura S, Akazawa H, Tanaka T, Qin Y, Kiya Y, Imaizumi S, Fujino M, Ito K, Zou Y, Fukuhara S, Kunimoto S, Fukuzaki K, Sato T, Ge J, Mochizuki N, Nakaya H, Saku K, Komuro I. Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep 9: 179–186, 2008. doi: 10.1038/sj.embor.7401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1158. Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal 3: ra46, 2010. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159. Audet M, Bouvier M. Restructuring G-protein- coupled receptor activation. Cell 151: 14–23, 2012. doi: 10.1016/j.cell.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 1160. Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. Allosteric modulation of beta-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem 289: 28271–28283, 2014. doi: 10.1074/jbc.M114.585067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1161. Deupi X. Relevance of rhodopsin studies for GPCR activation. Biochim Biophys Acta 1837: 674–682, 2014. doi: 10.1016/j.bbabio.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 1162. Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res 104: 1399–1409, 2009. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 1163. Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 1164. Harraz OF, Hill-Eubanks D, Nelson MT. PIP2: a critical regulator of vascular ion channels hiding in plain sight. Proc Natl Acad Sci USA 117: 20378–20389, 2020. doi: 10.1073/pnas.2006737117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1165. Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC. Phosphoinositides regulate ion channels. Biochim Biophys Acta 1851: 844–856, 2015. doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1166. Hilgemann DW, Dai G, Collins A, Lariccia V, Magi S, Deisl C, Fine M. Lipid signaling to membrane proteins: from second messengers to membrane domains and adapter-free endocytosis. J Gen Physiol 150: 211–224, 2018. doi: 10.1085/jgp.201711875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1167. Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507–520, 2002. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- 1168. Wang Y, Chang J, Chen KD, Li S, Li JY, Wu C, Chien S. Selective adapter recruitment and differential signaling networks by VEGF vs. shear stress. Proc Natl Acad Sci USA 104: 8875–8879, 2007. doi: 10.1073/pnas.0703088104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1169. Wang Y, Flores L, Lu S, Miao H, Li YS, Chien S. Shear stress regulates the Flk-1/Cbl/PI3K/NF-kappaB pathway via actin and tyrosine kinases. Cell Mol Bioeng 2: 341–350, 2009. doi: 10.1007/s12195-009-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1170. Wang Y, Chang J, Li YC, Li YS, Shyy JY, Chien S. Shear stress and VEGF activate IKK via the Flk-1/Cbl/Akt signaling pathway. Am J Physiol Heart Circ Physiol 286: H685–H692, 2004. doi: 10.1152/ajpheart.00237.2003. [DOI] [PubMed] [Google Scholar]
- 1171. Lee HJ, Koh GY. Shear stress activates Tie2 receptor tyrosine kinase in human endothelial cells. Biochem Biophys Res Commun 304: 399–404, 2003. doi: 10.1016/s0006-291x(03)00592-8. [DOI] [PubMed] [Google Scholar]
- 1172. Woo KV, Qu X, Babaev VR, Linton MF, Guzman RJ, Fazio S, Baldwin HS. Tie1 attenuation reduces murine atherosclerosis in a dose-dependent and shear stress-specific manner. J Clin Invest 121: 1624–1635, 2011. doi: 10.1172/JCI42040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1173. Mehta V, Pang KL, Rozbesky D, Nather K, Keen A, Lachowski D, Kong Y, Karia D, Ameismeier M, Huang J, Fang Y, Del Rio Hernandez A, Reader JS, Jones EY, Tzima E. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature 578: 290–295, 2020. doi: 10.1038/s41586-020-1979-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1174. Zhang YF, Zhang Y, Jia DD, Yang HY, Cheng MD, Zhu WX, Xin H, Li PF, Zhang YF. Insights into the regulatory role of Plexin D1 signalling in cardiovascular development and diseases. J Cell Mol Med 25: 4183–4194, 2021. doi: 10.1111/jcmm.16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1175. Cavallero S, Blázquez-Medela AM, Satta S, Hsiai TK. Endothelial mechanotransduction in cardiovascular development and regeneration: emerging approaches and animal models. Curr Top Membr 87: 131–151, 2021. doi: 10.1016/bs.ctm.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1176. Xiong Y, Hu Z, Han X, Jiang B, Zhang R, Zhang X, Lu Y, Geng C, Li W, He Y, Huo Y, Shibuya M, Luo J. Hypertensive stretch regulates endothelial exocytosis of Weibel-Palade bodies through VEGF receptor 2 signaling pathways. Cell Res 23: 820–834, 2013. doi: 10.1038/cr.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1177. Zheng W, Christensen LP, Tomanek RJ. Stretch induces upregulation of key tyrosine kinase receptors in microvascular endothelial cells. Am J Physiol Heart Circ Physiol 287: H2739–H2745, 2004. doi: 10.1152/ajpheart.00410.2004. [DOI] [PubMed] [Google Scholar]
- 1178. Angulo-Urarte A, van der Wal T, Huveneers S. Cell-cell junctions as sensors and transducers of mechanical forces. Biochim Biophys Acta Biomembr 1862: 183316, 2020. doi: 10.1016/j.bbamem.2020.183316. [DOI] [PubMed] [Google Scholar]
- 1179. Nekrasova O, Green KJ. Desmosome assembly and dynamics. Trends Cell Biol 23: 537–546, 2013. doi: 10.1016/j.tcb.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1180. Baddam SR, Arsenovic PT, Narayanan V, Duggan NR, Mayer CR, Newman ST, Abutaleb DA, Mohan A, Kowalczyk AP, Conway DE. The desmosomal cadherin Desmoglein-2 experiences mechanical tension as demonstrated by a FRET-based tension biosensor expressed in living cells. Cells 7: 66, 2018. doi: 10.3390/cells7070066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1181. Daday C, Kolšek K, Gräter F. The mechano-sensing role of the unique SH3 insertion in plakin domains revealed by molecular dynamics simulations. Sci Rep 7: 11669, 2017. doi: 10.1038/s41598-017-11017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1182. Price AJ, Cost AL, Ungewiß H, Waschke J, Dunn AR, Grashoff C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat Commun 9: 5284, 2018. doi: 10.1038/s41467-018-07523-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1183. Frismantiene A, Philippova M, Erne P, Resink TJ. Cadherins in vascular smooth muscle cell (patho)biology: quid nos scimus? Cell Signal 45: 23–42, 2018. doi: 10.1016/j.cellsig.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 1184. Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem 61: 514–523, 1996. doi: 10.1002/(SICI)1097-4644(19960616)61:4<514::AID-JCB4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 1185. Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ, Dunn AR. Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science 346: 1254211, 2014. doi: 10.1126/science.1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1186. Charras G, Yap AS. Tensile forces and mechanotransduction at cell-cell junctions. Curr Biol 28: R445–R457, 2018. doi: 10.1016/j.cub.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 1187. Chen S, Wang R, Li QF, Tang DD. Abl knockout differentially affects p130 Crk-associated substrate, vinculin, and paxillin in blood vessels of mice. Am J Physiol Heart Circ Physiol 297: H533–H539, 2009. doi: 10.1152/ajpheart.00237.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1188. Jia L, Tang DD. Abl activation regulates the dissociation of CAS from cytoskeletal vimentin by modulating CAS phosphorylation in smooth muscle. Am J Physiol Cell Physiol 299: C630–C637, 2010. doi: 10.1152/ajpcell.00095.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1189. Huang DL, Bax NA, Buckley CD, Weis WI, Dunn AR. Vinculin forms a directionally asymmetric catch bond with F-actin. Science 357: 703–706, 2017. doi: 10.1126/science.aan2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1190. Changede R, Sheetz M. Integrin and cadherin clusters: a robust way to organize adhesions for cell mechanics. Bioessays 39: 1–12, 2017. doi: 10.1002/bies.201600123. [DOI] [PubMed] [Google Scholar]
- 1191. Wang T, Wang R, Cleary RA, Gannon OJ, Tang DD. Recruitment of beta-catenin to N-cadherin is necessary for smooth muscle contraction. J Biol Chem 290: 8913–8924, 2015. doi: 10.1074/jbc.M114.621003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1192. Jansen SR, Van Ziel AM, Baarsma HA, Gosens R. beta-Catenin regulates airway smooth muscle contraction. Am J Physiol Lung Cell Mol Physiol 299: L204–L214, 2010. doi: 10.1152/ajplung.00020.2010. [DOI] [PubMed] [Google Scholar]
- 1193. Jackson TY, Sun Z, Martinez-Lemus LA, Hill MA, Meininger GA. N-cadherin and integrin blockade inhibit arteriolar myogenic reactivity but not pressure-induced increases in intracellular Ca. Front Physiol 1: 165, 2010. doi: 10.3389/fphys.2010.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1194. Davies PF, Tripathi SC. Mechanical stress mechanisms and the cell: an endothelial paradigm. Circ Res 72: 239–245, 1993. doi: 10.1161/01.res.72.2.239. [DOI] [PubMed] [Google Scholar]
- 1195. Gabriels JE, Paul DL. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ Res 83: 636–643, 1998. doi: 10.1161/01.res.83.6.636. [DOI] [PubMed] [Google Scholar]
- 1196. Pfenniger A, Meens MJ, Pedrigi RM, Foglia B, Sutter E, Pelli G, Rochemont V, Petrova TV, Krams R, Kwak BR. Shear stress-induced atherosclerotic plaque composition in ApoE-/- mice is modulated by connexin37. Atherosclerosis 243: 1–10, 2015. doi: 10.1016/j.atherosclerosis.2015.08.029. [DOI] [PubMed] [Google Scholar]
- 1197. Denis JF, Scheckenbach KE, Pfenniger A, Meens MJ, Krams R, Miquerol L, Taffet S, Chanson M, Delmar M, Kwak BR. Connexin40 controls endothelial activation by dampening NFkappaB activation. Oncotarget 8: 50972–50986, 2017. doi: 10.18632/oncotarget.16438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1198. Liao Y, Day KH, Damon DN, Duling BR. Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci USA 98: 9989–9994, 2001. doi: 10.1073/pnas.171305298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1199. de Wit C, Roos F, Bolz SS, Kirchhoff S, Krüger O, Willecke K, Pohl U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res 86: 649–655, 2000. doi: 10.1161/01.res.86.6.649. [DOI] [PubMed] [Google Scholar]
- 1200. Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci USA 99: 9462–9467, 2002. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1201. Kaufman DA, Albelda SM, Sun J, Davies PF. Role of lateral cell-cell border location and extracellular/transmembrane domains in PECAM/CD31 mechanosensation. Biochem Biophys Res Commun 320: 1076–1081, 2004. doi: 10.1016/j.bbrc.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 1202. Fujiwara K, Masuda M, Osawa M, Kano Y, Katoh K. Is PECAM-1 a mechanoresponsive molecule? Cell Struct Funct 26: 11–17, 2001. doi: 10.1247/csf.26.11. [DOI] [PubMed] [Google Scholar]
- 1203. Conway DE, Coon BG, Budatha M, Arsenovic PT, Orsenigo F, Wessel F, Zhang J, Zhuang Z, Dejana E, Vestweber D, Schwartz MA. VE-cadherin phosphorylation regulates endothelial fluid shear stress responses through the polarity protein LGN. Curr Biol 27: 2219–2225.e5, 2017. doi: 10.1016/j.cub.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1204. Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol 23: 1024–1030, 2013. doi: 10.1016/j.cub.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1205. Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, Yun S, Thomas JL, Schwartz MA. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol 208: 975–986, 2015. doi: 10.1083/jcb.201408103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1206. Stevens HY, Melchior B, Bell KS, Yun S, Yeh JC, Frangos JA. PECAM-1 is a critical mediator of atherosclerosis. Dis Model Mech 1: 175–181, 2008. doi: 10.1242/dmm.000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1207. Harry BL, Sanders JM, Feaver RE, Lansey M, Deem TL, Zarbock A, Bruce AC, Pryor AW, Gelfand BD, Blackman BR, Schwartz MA, Ley K. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 28: 2003–2008, 2008, 2008. doi: 10.1161/ATVBAHA.108.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1208. Goel R, Schrank BR, Arora S, Boylan B, Fleming B, Miura H, Newman PJ, Molthen RC, Newman DK. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol 28: 1996–2002, 2008. doi: 10.1161/ATVBAHA.108.172270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1209. DeMaio L, Chang YS, Gardner TW, Tarbell JM, Antonetti DA. Shear stress regulates occludin content and phosphorylation. Am J Physiol Heart Circ Physiol 281: H105–H113, 2001. doi: 10.1152/ajpheart.2001.281.1.H105. [DOI] [PubMed] [Google Scholar]
- 1210. Pang Z, Antonetti DA, Tarbell JM. Shear stress regulates HUVEC hydraulic conductivity by occludin phosphorylation. Ann Biomed Eng 33: 1536–1545, 2005. doi: 10.1007/s10439-005-7786-0. [DOI] [PubMed] [Google Scholar]
- 1211. Garcia-Polite F, Martorell J, Del Rey-Puech P, Melgar-Lesmes P, O’Brien CC, Roquer J, Ois A, Principe A, Edelman ER, Balcells M. Pulsatility and high shear stress deteriorate barrier phenotype in brain microvascular endothelium. J Cereb Blood Flow Metab 37: 2614–2625, 2017. doi: 10.1177/0271678X16672482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1212. Walsh TG, Murphy RP, Fitzpatrick P, Rochfort KD, Guinan AF, Murphy A, Cummins PM. Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J Cell Physiol 226: 3053–3063, 2011. doi: 10.1002/jcp.22655. [DOI] [PubMed] [Google Scholar]
- 1213. Salameh A, Dhein S. Effects of mechanical forces and stretch on intercellular gap junction coupling. Biochim Biophys Acta 1828: 147–156, 2013. doi: 10.1016/j.bbamem.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 1214. Yamashiro Y, Yanagisawa H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin Sci (Lond) 134: 2399–2418, 2020. doi: 10.1042/CS20190488. [DOI] [PubMed] [Google Scholar]
- 1215. Miroshnikova YA, Manet S, Li X, Wickström SA, Faurobert E, Albiges-Rizo C. Calcium signaling mediates a biphasic mechanoadaptive response of endothelial cells to cyclic mechanical stretch. Mol Biol Cell 32: 1724–1736, 2021. doi: 10.1091/mbc.E21-03-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1216. Zhong M, Wu W, Kang H, Hong Z, Xiong S, Gao X, Rehman J, Komarova YA, Malik AB. Alveolar stretch activation of endothelial Piezo1 protects adherens junctions and lung vascular barrier. Am J Respir Cell Mol Biol 62: 168–177, 2020. doi: 10.1165/rcmb.2019-0024OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1217. de Godoy MA, Rattan S. Role of rho kinase in the functional and dysfunctional tonic smooth muscles. Trends Pharmacol Sci 32: 384–393, 2011. doi: 10.1016/j.tips.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1218. Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol 587: 2537–2553, 2009. doi: 10.1113/jphysiol.2008.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1219. Eto M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem 284: 35273–35277, 2009. doi: 10.1074/jbc.R109.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1220. Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 1221. Budzyn K, Marley PD, Sobey CG. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci 27: 97–104, 2006. doi: 10.1016/j.tips.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 1222. Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol 283: H2260–H2267, 2002. doi: 10.1152/ajpheart.00634.2002. [DOI] [PubMed] [Google Scholar]
- 1223. Gokina NI, Knot HJ, Nelson MT, Osol G. Increased Ca2+ sensitivity as a key mechanism of PKC-induced constriction in pressurized cerebral arteries. Am J Physiol Heart Circ Physiol 277: H1178–H1188, 1999. doi: 10.1152/ajpheart.1999.277.3.H1178. [DOI] [PubMed] [Google Scholar]
- 1224. Schubert R, Lidington D, Bolz SS. The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc Res 77: 8–18, 2008. doi: 10.1016/j.cardiores.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 1225. VanBavel E, van der Meulen ET, Spaan JA. Role of Rho-associated protein kinase in tone and calcium sensitivity of cannulated rat mesenteric small arteries. Exp Physiol 86: 585–592, 2001. doi: 10.1113/eph8602217. [DOI] [PubMed] [Google Scholar]
- 1226. Schubert R, Kalentchuk VU, Krien U. Rho kinase inhibition partly weakens myogenic reactivity in rat small arteries by changing calcium sensitivity. Am J Physiol Heart Circ Physiol 283: H2288–H2295, 2002. doi: 10.1152/ajpheart.00549.2002. [DOI] [PubMed] [Google Scholar]
- 1227. Gokina NI, Park KM, McElroy-Yaggy K, Osol G. Effects of Rho kinase inhibition on cerebral artery myogenic tone and reactivity. J Appl Physiol (1985) 98: 1940–1948, 2005. doi: 10.1152/japplphysiol.01104.2004. [DOI] [PubMed] [Google Scholar]
- 1228. Li Y, Brayden JE. Rho kinase activity governs arteriolar myogenic depolarization. J Cereb Blood Flow Metab 37: 140–152, 2017. doi: 10.1177/0271678X15621069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1229. Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, Oshima H, Oshima M, Taketo MM, Narumiya S. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol Cell Biol 23: 5043–5055, 2003. doi: 10.1128/MCB.23.14.5043-5055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1230. Shimizu T, Fukumoto Y, Tanaka S, Satoh K, Ikeda S, Shimokawa H. Crucial role of ROCK2 in vascular smooth muscle cells for hypoxia-induced pulmonary hypertension in mice. Arterioscler Thromb Vasc Biol 33: 2780–2791, 2013. doi: 10.1161/ATVBAHA.113.301357. [DOI] [PubMed] [Google Scholar]
- 1231. Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest 118: 1632–1644, 2008. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1232. Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation 112: 2959–2965, 2005. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1233. Kozasa T, Hajicek N, Chow CR, Suzuki N. Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J Biochem 150: 357–369, 2011. doi: 10.1093/jb/mvr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1234. Langille BL. Morphologic responses of endothelium to shear stress: reorganization of the adherens junction. Microcirculation 8: 195–206, 2001. doi: 10.1038/sj/mn/7800085. [DOI] [PubMed] [Google Scholar]
- 1235. Ishida T, Takahashi M, Corson MA, Berk BC. Fluid shear stress-mediated signal transduction: how do endothelial cells transduce mechanical force into biological responses? Ann NY Acad Sci 811: 12–23, 1997. doi: 10.1111/j.1749-6632.1997.tb51984.x. [DOI] [PubMed] [Google Scholar]
- 1236. Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J 20: 4639–4647, 2001. doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1237. Shiu YT, Li S, Yuan S, Wang Y, Nguyen P, Chien S. Shear stress-induced c-fos activation is mediated by Rho in a calcium-dependent manner. Biochem Biophys Res Commun 303: 548–555, 2003. doi: 10.1016/S0006-291X(03)00388-7. [DOI] [PubMed] [Google Scholar]
- 1238. Li S, Chen BP, Azuma N, Hu YL, Wu SZ, Sumpio BE, Shyy JY, Chien S. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J Clin Invest 103: 1141–1150, 1999. doi: 10.1172/JCI5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1239. Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, Schwartz MA. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J 21: 6791–6800, 2002. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1240. Tzima E, Kiosses WB, del Pozo MA, Schwartz MA. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J Biol Chem 278: 31020–31023, 2003. doi: 10.1074/jbc.M301179200. [DOI] [PubMed] [Google Scholar]
- 1241. Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol 161: 429–439, 2003. doi: 10.1083/jcb.200210135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1242. Hsu PP, Li S, Li YS, Usami S, Ratcliffe A, Wang X, Chien S. Effects of flow patterns on endothelial cell migration into a zone of mechanical denudation. Biochem Biophys Res Commun 285: 751–759, 2001. doi: 10.1006/bbrc.2001.5221. [DOI] [PubMed] [Google Scholar]
- 1243. Hsiao ST, Spencer T, Boldock L, Prosseda SD, Xanthis I, Tovar-Lopez FJ, Van Beusekom HM, Khamis RY, Foin N, Bowden N, Hussain A, Rothman A, Ridger V, Halliday I, Perrault C, Gunn J, Evans PC. Endothelial repair in stented arteries is accelerated by inhibition of Rho-associated protein kinase. Cardiovasc Res 112: 689–701, 2016. doi: 10.1093/cvr/cvw210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1244. Kaunas R, Nguyen P, Usami S, Chien S. Cooperative effects of Rho and mechanical stretch on stress fiber organization. Proc Natl Acad Sci USA 102: 15895–15900, 2005. doi: 10.1073/pnas.0506041102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1245. Huang L, Helmke BP. Polarized actin structural dynamics in response to cyclic uniaxial stretch. Cell Mol Bioeng 8: 160–177, 2015. doi: 10.1007/s12195-014-0370-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1246. Ingber DE. Cellular tensegrity: defining new rules of biological design that govern the cytoskeleton. J Cell Sci 104: 613–627, 1993. doi: 10.1242/jcs.104.3.613. [DOI] [PubMed] [Google Scholar]
- 1247. Pollack GH. The role of aqueous interfaces in the cell. Adv Colloid Interface Sci 103: 173–196, 2003. doi: 10.1016/S0001-8686(02)00095-7. [DOI] [PubMed] [Google Scholar]
- 1248. Small JV, Gimona M. The cytoskeleton of the vertebrate smooth muscle cell. Acta Physiol Scand 164: 341–348, 1998. doi: 10.1046/j.1365-201X.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- 1249. Martino F, Perestrelo AR, Vinarský V, Pagliari S, Forte G. Cellular mechanotransduction: from tension to function. Front Physiol 9: 824, 2018. doi: 10.3389/fphys.2018.00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1250. Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295: C576–C587, 2008. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1251. Herrera AM, McParland BE, Bienkowska A, Tait R, Paré PD, Seow CY. ‘Sarcomeres’ of smooth muscle: functional characteristics and ultrastructural evidence. J Cell Sci 118: 2381–2392, 2005. doi: 10.1242/jcs.02368. [DOI] [PubMed] [Google Scholar]
- 1252. Tang DD. Intermediate filaments in smooth muscle. Am J Physiol Cell Physiol 294: C869–C878, 2008. doi: 10.1152/ajpcell.00154.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1253. Platts SH, Falcone JC, Holton WT, Hill MA, Meininger GA. Alteration of microtubule polymerization modulates arteriolar vasomotor tone. Am J Physiol Heart Circ Physiol 277: H100–H106, 1999. doi: 10.1152/ajpheart.1999.277.1.H100. [DOI] [PubMed] [Google Scholar]
- 1254. Leite R, Webb RC. Microtubule disruption potentiates phenylephrine-induced vasoconstriction in rat mesenteric arterial bed. Eur J Pharmacol 351: R1–R3, 1998. doi: 10.1016/S0014-2999(98)00358-6. [DOI] [PubMed] [Google Scholar]
- 1255. Paul RJ, Bowman PS, Kolodney MS. Effects of microtubule disruption on force, velocity, stiffness and [Ca2+]i in porcine coronary arteries. Am J Physiol Heart Circ Physiol 279: H2493–H2501, 2000. doi: 10.1152/ajpheart.2000.279.5.H2493. [DOI] [PubMed] [Google Scholar]
- 1256. Sheridan BC, McIntyre RC Jr, Meldrum DR, Cleveland JC Jr, Agrafojo J, Banerjee A, Harken AH, Fullerton DA. Microtubules regulate pulmonary vascular smooth muscle contraction. J Surg Res 62: 284–287, 1996. doi: 10.1006/jsre.1996.0209. [DOI] [PubMed] [Google Scholar]
- 1257. Battistella-Patterson AS, Wang S, Wright GL. Effect of disruption of the cytoskeleton on smooth muscle contraction. Can J Physiol Pharmacol 75: 1287–1299, 1997. [PubMed] [Google Scholar]
- 1258. Cooper GM. The Cell: a Molecular Approach. Sunderland, MA: Sinauer Associates, 2000. [Google Scholar]
- 1259. Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol 9: 364–370, 1999. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- 1260. Zhao R, Du L, Huang Y, Wu Y, Gunst SJ. Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J Biol Chem 283: 36522–36531, 2008. doi: 10.1074/jbc.M805294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1261. Gohla A, Bokoch GM. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr Biol 12: 1704–1710, 2002. doi: 10.1016/s0960-9822(02)01184-3. [DOI] [PubMed] [Google Scholar]
- 1262. Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem 270: 17582–17587, 1995. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- 1263. Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1: 73–86, 1996. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- 1264. Galkin VE, Orlova A, Egelman EH. Actin filaments as tension sensors. Curr Biol 22: R96–R101, 2012. doi: 10.1016/j.cub.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1265. Hayakawa K, Tatsumi H, Sokabe M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J Cell Biol 195: 721–727, 2011. doi: 10.1083/jcb.201102039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1266. Hayakawa K, Sakakibara S, Sokabe M, Tatsumi H. Single-molecule imaging and kinetic analysis of cooperative cofilin-actin filament interactions. Proc Natl Acad Sci USA 111: 9810–9815, 2014. doi: 10.1073/pnas.1321451111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1267. Uyeda TQ, Iwadate Y, Umeki N, Nagasaki A, Yumura S. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS One 6: e26200, 2011. doi: 10.1371/journal.pone.0026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1268. Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J 16: 72–76, 2002. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 1269. Clifford PS, Ferguson BS, Jasperse JL, Hill MA. Arteriolar vasodilation involves actin depolymerization. Am J Physiol Heart Circ Physiol 315: H423–H428, 2018. doi: 10.1152/ajpheart.00723.2017. [DOI] [PubMed] [Google Scholar]
- 1270. Adler KB, Krill J, Alberghini TV, Evans JN. Effect of cytochalasin D on smooth muscle contraction. Cell Motil 3: 545–551, 1983. doi: 10.1002/cm.970030521. [DOI] [PubMed] [Google Scholar]
- 1271. Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol 519: 829–840, 1999. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1272. An SS, Laudadio RE, Lai J, Rogers RA, Fredberg JJ. Stiffness changes in cultured airway smooth muscle cells. Am J Physiol Cell Physiol 283: C792–C801, 2002. doi: 10.1152/ajpcell.00425.2001. [DOI] [PubMed] [Google Scholar]
- 1273. Zhang W, Huang Y, Wu Y, Gunst SJ. A novel role for RhoA GTPase in the regulation of airway smooth muscle contraction. Can J Physiol Pharmacol 93: 129–136, 2015. doi: 10.1139/cjpp-2014-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1274. North AJ, Gimona M, Lando Z, Small JV. Actin isoform compartments in chicken gizzard smooth muscle cells. J Cell Sci 107: 445–455, 1994. doi: 10.1242/jcs.107.3.445. [DOI] [PubMed] [Google Scholar]
- 1275. Bitar KN, Kaminski MS, Hailat N, Cease KB, Strahler JR. Hsp27 is a mediator of sustained smooth muscle contraction in response to bombesin. Biochem Biophys Res Commun 181: 1192–1200, 1991. doi: 10.1016/0006-291x(91)92065-r. [DOI] [PubMed] [Google Scholar]
- 1276. Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc 5: 32–39, 2008. doi: 10.1513/pats.200704-048VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1277. Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct 29: 545–576, 2000. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 1278. May RC. The Arp2/3 complex: a central regulator of the actin cytoskeleton. Cell Mol Life Sci 58: 1607–1626, 2001. doi: 10.1007/PL00000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1279. Higgs HN, Pollard TD. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu Rev Biochem 70: 649–676, 2001. doi: 10.1146/annurev.biochem.70.1.649. [DOI] [PubMed] [Google Scholar]
- 1280. Zhang W, Wu Y, Du L, Tang DD, Gunst SJ. Activation of the Arp2/3 complex by N-WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol 288: C1145–C1160, 2005. doi: 10.1152/ajpcell.00387.2004. [DOI] [PubMed] [Google Scholar]
- 1281. Zhang W, Huang Y, Gunst SJ. The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J Biol Chem 287: 33996–34008, 2012. doi: 10.1074/jbc.M112.369603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1282. Tang DD, Zhang W, Gunst SJ. The adapter protein CrkII regulates neuronal Wiskott-Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J Biol Chem 280: 23380–23389, 2005. doi: 10.1074/jbc.M413390200. [DOI] [PubMed] [Google Scholar]
- 1283. Carlier MF, Ducruix A, Pantaloni D. Signalling to actin: the Cdc42-N-WASP-Arp2/3 connection. Chem Biol 6: R235–R240, 1999. doi: 10.1016/s1074-5521(99)80107-0. [DOI] [PubMed] [Google Scholar]
- 1284. Zhang W, Bhetwal BP, Gunst SJ. Rho kinase collaborates with p21-activated kinase to regulate actin polymerization and contraction in airway smooth muscle. J Physiol 596: 3617–3635, 2018. doi: 10.1113/JP275751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1285. Zhang W, Huang Y, Gunst SJ. p21-Activated kinase (Pak) regulates airway smooth muscle contraction by regulating paxillin complexes that mediate actin polymerization. J Physiol 594: 4879–4900, 2016. doi: 10.1113/JP272132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1286. Zhang W, Gunst SJ. Dynamic association between alpha-actinin and beta-integrin regulates contraction of canine tracheal smooth muscle. J Physiol 572: 659–676, 2006. doi: 10.1113/jphysiol.2006.106518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1287. Anfinogenova Y, Wang R, Li QF, Spinelli AM, Tang DD. Abl silencing inhibits CAS-mediated process and constriction in resistance arteries. Circ Res 101: 420–428, 2007. doi: 10.1161/CIRCRESAHA.107.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1288. Tang DD, Anfinogenova Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther 13: 130–140, 2008. doi: 10.1177/1074248407313737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1289. Tejani AD, Walsh MP, Rembold CM. Tissue length modulates “stimulated actin polymerization,” force augmentation, and the rate of swine carotid arterial contraction. Am J Physiol Cell Physiol 301: C1470–C478, 2011. doi: 10.1152/ajpcell.00149.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1290. Gunst SJ, Fredberg JJ. The first three minutes: smooth muscle contraction, cytoskeletal events, and soft glasses. J Appl Physiol (1985) 95: 413–425, 2003. doi: 10.1152/japplphysiol.00277.2003. [DOI] [PubMed] [Google Scholar]
- 1291. Takeya K, Wang X, Sutherland C, Kathol I, Loutzenhiser K, Loutzenhiser RD, Walsh MP. Involvement of myosin regulatory light chain diphosphorylation in sustained vasoconstriction under pathophysiological conditions. J Smooth Muscle Res 50: 18–28, 2014. doi: 10.1540/jsmr.50.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1292. Driska SP, Aksoy MO, Murphy RA. Myosin light chain phosphorylation associated with contraction in arterial smooth muscle. Am J Physiol Cell Physiol 240: C222–C233, 1981. doi: 10.1152/ajpcell.1981.240.5.C222. [DOI] [PubMed] [Google Scholar]
- 1293. Jones KA, Perkins WJ, Lorenz RR, Prakash YS, Sieck GC, Warner DO. F-actin stabilization increases tension cost during contraction of permeabilized airway smooth muscle in dogs. J Physiol 519: 527–538, 1999. doi: 10.1111/j.1469-7793.1999.0527m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1294. Saito SY, Hori M, Ozaki H, Karaki H. Cytochalasin D inhibits smooth muscle contraction by directly inhibiting contractile apparatus. J Smooth Muscle Res 32: 51–60, 1996. doi: 10.1540/jsmr.32.51. [DOI] [PubMed] [Google Scholar]
- 1295. Gokina NI, Osol G. Actin cytoskeletal modulation of pressure-induced depolarization and Ca2+ influx in cerebral arteries. Am J Physiol Heart Circ Physiol 282: H1410–H1420, 2002. doi: 10.1152/ajpheart.00441.2001. [DOI] [PubMed] [Google Scholar]
- 1296. Obara K, Yabu H. Effect of cytochalasin B on intestinal smooth muscle cells. Eur J Pharmacol 255: 139–147, 1994. doi: 10.1016/0014-2999(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 1297. Rembold CM, Tejani AD, Ripley ML, Han S. Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol 293: C993–C1002, 2007. doi: 10.1152/ajpcell.00090.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1298. Moreno-Domínguez A, El-Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, Cole DM, Kargacin GJ, Walsh MP, Cole WC. Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem 289: 20939–20952, 2014. doi: 10.1074/jbc.M114.553743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1299. Schubert KM, Qiu J, Blodow S, Wiedenmann M, Lubomirov LT, Pfitzer G, Pohl U, Schneider H. The AMP-related kinase (AMPK) induces Ca2+-independent dilation of resistance arteries by interfering with actin filament formation. Circ Res 121: 149–161, 2017. doi: 10.1161/CIRCRESAHA.116.309962. [DOI] [PubMed] [Google Scholar]
- 1300. Fukuhara S, Chikumi H, Gutkind JS. RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene 20: 1661–1668, 2001. doi: 10.1038/sj.onc.1204182. [DOI] [PubMed] [Google Scholar]
- 1301. Tanabe S, Kreutz B, Suzuki N, Kozasa T. Regulation of RGS-RhoGEFs by Galpha12 and Galpha13 proteins. Methods Enzymol 390: 285–294, 2004. doi: 10.1016/S0076-6879(04)90018-3. [DOI] [PubMed] [Google Scholar]
- 1302. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167–180, 2005. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 1303. Togashi H, Emala CW, Hall IP, Hirshman CA. Carbachol-induced actin reorganization involves Gi activation of Rho in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 274: L803–L809, 1998. doi: 10.1152/ajplung.1998.274.5.L803. [DOI] [PubMed] [Google Scholar]
- 1304. Takefuji M, Krüger M, Sivaraj KK, Kaibuchi K, Offermanns S, Wettschureck N. RhoGEF12 controls cardiac remodeling by integrating G protein- and integrin-dependent signaling cascades. J Exp Med 210: 665–673, 2013. doi: 10.1084/jem.20122126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1305. Walsh MP. Vascular smooth muscle myosin light chain diphosphorylation: mechanism, function, and pathological implications. IUBMB Life 63: 987–1000, 2011. doi: 10.1002/iub.527. [DOI] [PubMed] [Google Scholar]
- 1306. Deng JT, Van Lierop JE, Sutherland C, Walsh MP. Ca2+-independent smooth muscle contraction. a novel function for integrin-linked kinase. J Biol Chem 276: 16365–16373, 2001. doi: 10.1074/jbc.M011634200. [DOI] [PubMed] [Google Scholar]
- 1307. Wickström SA, Lange A, Montanez E, Fässler R. The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J 29: 281–291, 2010. doi: 10.1038/emboj.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1308. Fukuda K, Knight JD, Piszczek G, Kothary R, Qin J. Biochemical, proteomic, structural, and thermodynamic characterizations of integrin-linked kinase (ILK): cross-validation of the pseudokinase. J Biol Chem 286: 21886–21895, 2011. doi: 10.1074/jbc.M111.240093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1309. Legate KR, Montañez E, Kudlacek O, Fässler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol 7: 20–31, 2006. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- 1310. Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol 286: C433–C447, 2004. doi: 10.1152/ajpcell.00030.2003. [DOI] [PubMed] [Google Scholar]
- 1311. Zhang W, Wu Y, Wu C, Gunst SJ. Integrin-linked kinase regulates N-WASp-mediated actin polymerization and tension development in tracheal smooth muscle. J Biol Chem 282: 34568–34580, 2007. doi: 10.1074/jbc.M704966200. [DOI] [PubMed] [Google Scholar]
- 1312. Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol 140: 961–972, 1998. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1313. Tang DD, Tan J. Role of Crk-associated substrate in the regulation of vascular smooth muscle contraction. Hypertension 42: 858–863, 2003. doi: 10.1161/01.HYP.0000085333.76141.33. [DOI] [PubMed] [Google Scholar]
- 1314. Srinivasan R, Forman S, Quinlan RA, Ohanian J, Ohanian V. Regulation of contractility by Hsp27 and Hic-5 in rat mesenteric small arteries. Am J Physiol Heart Circ Physiol 294: H961–H969, 2008. doi: 10.1152/ajpheart.00939.2007. [DOI] [PubMed] [Google Scholar]
- 1315. Larsen JK, Yamboliev IA, Weber LA, Gerthoffer WT. Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am J Physiol Lung Cell Mol Physiol 273: L930–L940, 1997. doi: 10.1152/ajplung.1997.273.5.L930. [DOI] [PubMed] [Google Scholar]
- 1316. Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol (1985) 91: 963–972, 2001. doi: 10.1152/jappl.2001.91.2.963. [DOI] [PubMed] [Google Scholar]
- 1317. Massett MP, Ungvari Z, Csiszar A, Kaley G, Koller A. Different roles of PKC and MAP kinases in arteriolar constrictions to pressure and agonists. Am J Physiol Heart Circ Physiol 283: H2282–H2287, 2002. doi: 10.1152/ajpheart.00544.2002. [DOI] [PubMed] [Google Scholar]
- 1318. Spurrell BE, Murphy TV, Hill MA. Intraluminal pressure stimulates MAPK phosphorylation in arterioles: temporal dissociation from myogenic contractile response. Am J Physiol Heart Circ Physiol 285: H1764–H1773, 2003. doi: 10.1152/ajpheart.00468.2003. [DOI] [PubMed] [Google Scholar]
- 1319. Loufrani L, Lehoux S, Tedgui A, Lévy BI, Henrion D. Stretch induces mitogen-activated protein kinase activation and myogenic tone through 2 distinct pathways. Arterioscler Thromb Vasc Biol 19: 2878–2883, 1999. doi: 10.1161/01.ATV.19.12.2878. [DOI] [PubMed] [Google Scholar]
- 1320. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70: 389–399, 1992. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 1321. Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol 39: 1071–1076, 2007. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 1322. Gutsche-Perelroizen I, Lepault J, Ott A, Carlier MF. Filament assembly from profilin-actin. J Biol Chem 274: 6234–6243, 1999. doi: 10.1074/jbc.274.10.6234. [DOI] [PubMed] [Google Scholar]
- 1323. Wu Y, Gunst SJ. Vasodilator-stimulated phosphoprotein (VASP) regulates actin polymerization and contraction in airway smooth muscle by a vinculin-dependent mechanism. J Biol Chem 290: 11403–11416, 2015. doi: 10.1074/jbc.M115.645788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1324. El-Yazbi AF, Abd-Elrahman KS, Moreno-Dominguez A. PKC-mediated cerebral vasoconstriction: role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem Pharmacol 95: 263–278, 2015. doi: 10.1016/j.bcp.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 1325. Löhn M, Kämpf D, Gui-Xuan C, Haller H, Luft FC, Gollasch M. Regulation of arterial tone by smooth muscle myosin type II. Am J Physiol Cell Physiol 283: C1383–C1389, 2002. doi: 10.1152/ajpcell.01369.2000. [DOI] [PubMed] [Google Scholar]
- 1326. Vuori K. Integrin signaling: Tyrosine phosphorylation events in focal adhesions. J Membr Biol 165: 191–199, 1998. doi: 10.1007/s002329900433. [DOI] [PubMed] [Google Scholar]
- 1327. Glukhova MA, Koteliansky VE. Integrins, cytoskeletal and extracellular matrix proteins in developing smooth muscle cells of human aorta. In: The Vascular Smooth Muscle Cell: Molecular and Biological Responses to the Extracellular Matrix, edited by Schwartz SM, Mecham RP.. San Diego, CA: Academic Press, 1995. [Google Scholar]
- 1328. van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res 305: 285–298, 2001. doi: 10.1007/s004410100417. [DOI] [PubMed] [Google Scholar]
- 1329. Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 69: 11–25, 1992. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 1330. Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science 268: 233–239, 1995. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 1331. Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: molecular hierarchies of cytoskeletal, and signaling molecules. J Cell Biol 131: 791–805, 1995. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1332. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 1333. Wrighton KH. Cell adhesion: the ‘ins’ and ‘outs’ of integrin signalling. Nat Rev Mol Cell Biol 14: 752, 2013. doi: 10.1038/nrm3708. [DOI] [PubMed] [Google Scholar]
- 1334. Zuidema A, Wang W, Sonnenberg A. Crosstalk between cell adhesion complexes in regulation of mechanotransduction. Bioessays 42: e2000119, 2020. doi: 10.1002/bies.202000119. [DOI] [PubMed] [Google Scholar]
- 1335. Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol 3: a005033, 2011. doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1336. Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol 9: 858–867, 2007. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1337. Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science 260: 1124–1127, 1993. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 1338. Roca-Cusachs P, Gauthier NC, Del Rio A, Sheetz MP. Clustering of alpha5beta1 integrins determines adhesion strength whereas alphavbeta3 and talin enable mechanotransduction. Proc Natl Acad Sci USA 106: 16245–16250, 2009. doi: 10.1073/pnas.0902818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1339. Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol 156: 1489–1498, 2000. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1340. Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol 179: 1043–1057, 2007. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1341. Lee SE, Kamm RD, Mofrad MR. Force-induced activation of talin and its possible role in focal adhesion mechanotransduction. J Biomech 40: 2096–2106, 2007. doi: 10.1016/j.jbiomech.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 1342. del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science 323: 638–641, 2009. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1343. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol 14: 1680–1688, 1994. doi: 10.1128/mcb.14.3.1680-1688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1344. Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein tyrosine kinase, pp125FAK. Mol Cell Biol 14: 147–155, 1994. doi: 10.1128/mcb.14.1.147-155.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1345. Berk BC, Corson MA. Angiotensin II signal transduction in vascular smooth muscle—Role of tyrosine kinases. Circ Res 80: 607–616, 1997. doi: 10.1161/01.res.80.5.607. [DOI] [PubMed] [Google Scholar]
- 1346. Ishida M, Marrero MB, Schieffer B, Ishida T, Bernstein KE, Berk BC. Angiotensin II activates pp60c-src in vascular smooth muscle cells. Circ Res 77: 1053–1059, 1995. doi: 10.1161/01.res.77.6.1053. [DOI] [PubMed] [Google Scholar]
- 1347. Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest 103: 789–797, 1999. doi: 10.1172/JCI4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1348. Turner CE, Pietras KM, Taylor DS, Molloy CJ. Angiotensin II stimulation of rapid paxillin tyrosine phosphorylation correlates with the formation of focal adhesions in rat aortic smooth muscle cells. J Cell Sci 108 : 333–342, 1995. doi: 10.1242/jcs.108.1.333. [DOI] [PubMed] [Google Scholar]
- 1349. Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays 19: 137–145, 1997. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 1350. Marrero MB, Schieffer B, Bernstein KE, Ling BN. Angiotensin II-induced tyrosine phosphorylation in mesangial and vascular smooth muscle cells. Clin Exp Pharmacol Physiol 23: 83–88, 1996. [PubMed] [Google Scholar]
- 1351. Jiang G, Giannone G, Critchley DR, Fukumoto E, Sheetz MP. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature 424: 334–337, 2003. doi: 10.1038/nature01805. [DOI] [PubMed] [Google Scholar]
- 1352. Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol 159: 695–705, 2002. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1353. Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol 153: 1175–1186, 2001. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1354. Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 127: 1015–1026, 2006. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1355. Veigel C, Molloy JE, Schmitz S, Kendrick-Jones J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat Cell Biol 5: 980–986, 2003. doi: 10.1038/ncb1060. [DOI] [PubMed] [Google Scholar]
- 1356. Tang DD, Gunst SJ. Roles of focal adhesion kinase and paxillin in the mechanosensitive regulation of myosin phosphorylation in smooth muscle. J Appl Physiol (1985) 91: 1452–1459, 2001. doi: 10.1152/jappl.2001.91.3.1452. [DOI] [PubMed] [Google Scholar]
- 1357. Tang D, Mehta D, Gunst SJ. Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. Am J Physiol Cell Physiol 276: C250–C258, 1999. doi: 10.1152/ajpcell.1999.276.1.C250. [DOI] [PubMed] [Google Scholar]
- 1358. Deng L, Fairbank NJ, Fabry B, Smith PG, Maksym GN. Localized mechanical stress induces time-dependent actin cytoskeletal remodeling and stiffening in cultured airway smooth muscle cells. Am J Physiol Cell Physiol 287: C440–448, 2004. doi: 10.1152/ajpcell.00374.2003. [DOI] [PubMed] [Google Scholar]
- 1359. Sun Z, Martinez-Lemus LA, Hill MA, Meininger GA. Extracellular matrix-specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. Am J Physiol Cell Physiol 295: C268–C278, 2008. doi: 10.1152/ajpcell.00516.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1360. Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium current in arteriolar smooth muscle by αvβ3 and α5β1 integrin ligands. J Cell Biol 143: 241–252, 1998. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1361. Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, Wilson E. Regulation of ion channels by integrins. Cell Biochem Biophys 36: 41–66, 2002. doi: 10.1385/CBB:36:1:41. [DOI] [PubMed] [Google Scholar]
- 1362. Wu X, Davis GE, Meininger GA, Wilson E, Davis MJ. Regulation of the L-type calcium channel by α5β1 integrin requires signaling between focal adhesion proteins. J Biol Chem 276: 30285–30292, 2001. doi: 10.1074/jbc.M102436200. [DOI] [PubMed] [Google Scholar]
- 1363. Gui P, Wu X, Ling S, Stotz SC, Winkfein RJ, Wilson E, Davis GE, Braun AP, Zamponi GW, Davis MJ. Integrin receptor activation triggers converging regulation of Cav1.2 calcium channels by c-Src and protein kinase A pathways. J Biol Chem 281: 14015–14025, 2006. doi: 10.1074/jbc.M600433200. [DOI] [PubMed] [Google Scholar]
- 1364. Murphy TV, Spurrell BE, Hill MA. Tyrosine phosphorylation following alterations in arteriolar intraluminal pressure and wall tension. Am J Physiol Heart Circ Physiol 281: H1047–H1056, 2001. doi: 10.1152/ajpheart.2001.281.3.H1047. [DOI] [PubMed] [Google Scholar]
- 1365. Murphy TV, Spurrell BE, Hill MA. Mechanisms underlying pervanadate-induced contraction of rat cremaster muscle arterioles. Eur J Pharmacol 442: 107–114, 2002. doi: 10.1016/s0014-2999(02)01498-x. [DOI] [PubMed] [Google Scholar]
- 1366. Murphy TV, Spurrell BE, Hill MA. Cellular signalling in arteriolar myogenic constriction: involvement of tyrosine phosphorylation pathways. Clin Exp Pharmacol Physiol 29: 612–619, 2002. doi: 10.1046/j.1440-1681.2002.03698.x. [DOI] [PubMed] [Google Scholar]
- 1367. Wu X, Yang Y, Gui P, Sohma Y, Meininger GA, Davis GE, Braun AP, Davis MJ. Potentiation of large conductance, calcium-activated K+ channels by α5β1 integrin activation in arteriolar smooth muscle. J Physiol 586: 1699–1713, 2008. doi: 10.1113/jphysiol.2007.149500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1368. Yang Y, Wu X, Gui P, Wu J, Sheng JZ, Ling S, Braun AP, Davis GE, Davis MJ. α5β1 integrin engagement increases large conductance Ca2+-activated K+ channel current and Ca2+ sensitivity through c-Src-mediated channel phosphorylation. J Biol Chem 285: 131–141, 2010. doi: 10.1074/jbc.M109.033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1369. Mufti RE, Zechariah A, Sancho M, Mazumdar N, Brett SE, Welsh DG. Implications of alphavbeta3 integrin signaling in the regulation of Ca2+ waves and myogenic tone in cerebral arteries. Arterioscler Thromb Vasc Biol 35: 2571–2578, 2015. doi: 10.1161/ATVBAHA.115.305619. [DOI] [PubMed] [Google Scholar]
- 1370. Chan WL, Holstein-Rathlou NH, Yip KP. Integrin mobilizes intracellular Ca2+ in renal vascular smooth muscle cells. Am J Physiol Cell Physiol 280: C593–C603, 2001. doi: 10.1152/ajpcell.2001.280.3.C593. [DOI] [PubMed] [Google Scholar]
- 1371. Umesh A, Thompson MA, Chini EN, Yip KP, Sham JS. Integrin ligands mobilize Ca2+ from ryanodine receptor-gated stores and lysosome-related acidic organelles in pulmonary arterial smooth muscle cells. J Biol Chem 281: 34312–34323, 2006. doi: 10.1074/jbc.M606765200. [DOI] [PubMed] [Google Scholar]
- 1372. Balasubramanian L, Ahmed A, Lo CM, Sham JS, Yip KP. Integrin-mediated mechanotransduction in renal vascular smooth muscle cells: activation of calcium sparks. Am J Physiol Regul Integr Comp Physiol 293: R1586–R1594, 2007. doi: 10.1152/ajpregu.00025.2007. [DOI] [PubMed] [Google Scholar]
- 1373. Schwartz MA, Denninghoff K. αv Integrins mediate the rise in intracellular calcium in endothelial cells on fibronectin even though they play a minor role in adhesion. J Biol Chem 269: 11133–11137, 1994. doi: 10.1016/S0021-9258(19)78101-0. [DOI] [PubMed] [Google Scholar]
- 1374. Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, Wilson E. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 281: H1835–H1862, 2001. doi: 10.1152/ajpheart.2001.281.5.H1835. [DOI] [PubMed] [Google Scholar]
- 1375. Sun Z, Li Z, Meininger GA. Mechanotransduction through fibronectin-integrin focal adhesion in microvascular smooth muscle cells: is calcium essential? Am J Physiol Heart Circ Physiol 302: H1965–H1973, 2012. doi: 10.1152/ajpheart.00598.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1376. Masumoto N, Nakayama K, Oyabe A, Uchino M, Ishii K, Obara K, Tanabe Y. Specific attenuation of the pressure-induced contraction of rat cerebral artery by herbimycin A. Eur J Pharmacol 330: 55–63, 1997. doi: 10.1016/s0014-2999(97)00166-0. [DOI] [PubMed] [Google Scholar]
- 1377. Masumoto N, Tanabe Y, Saito M, Nakayama K. Attenuation of pressure-induced myogenic contraction and tyrosine phosphorylation by fasudil, a cerebral vasodilator, in rat cerebral artery. Br J Pharmacol 130: 219–230, 2000. doi: 10.1038/sj.bjp.0703292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1378. Spurrell BE, Murphy TV, Hill MA. Tyrosine phosphorylation modulates arteriolar tone but is not fundamental to myogenic response. Am J Physiol Heart Circ Physiol 278: H373–H382, 2000. doi: 10.1152/ajpheart.2000.278.2.H373. [DOI] [PubMed] [Google Scholar]
- 1379. Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle αvβ3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res 79: 821–826, 1996. doi: 10.1161/01.RES.79.4.821. [DOI] [PubMed] [Google Scholar]
- 1380. Burridge K. Focal adhesions: a personal perspective on a half century of progress. FEBS J 284: 3355–3361, 2017. doi: 10.1111/febs.14195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1381. Inman A, Smutny M. Feeling the force: multiscale force sensing and transduction at the cell-cell interface. Semin Cell Dev Biol 120: 53–65, 2021. doi: 10.1016/j.semcdb.2021.06.006. [DOI] [PubMed] [Google Scholar]
- 1382. Muller JM, Chilian WM, Davis MJ. Integrin signaling transduces shear stress-dependent vasodilation of coronary arterioles. Circ Res 80: 320–326, 1997. doi: 10.1161/01.res.80.3.320. [DOI] [PubMed] [Google Scholar]
- 1383. Mogford JE, Davis GE, Meininger GA. RGDN peptide interaction with endothelial α5β1 integrin causes sustained endothelin-dependent vasoconstriction of rat skeletal muscle arterioles. J Clin Invest 100: 1647–1653, 1997. doi: 10.1172/JCI119689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1384. Martinez-Lemus LA, Crow T, Davis MJ, Meininger GA. αvβ3- and α5β1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol Heart Circ Physiol 289: H322–H329, 2005. doi: 10.1152/ajpheart.00923.2003. [DOI] [PubMed] [Google Scholar]
- 1385. Waitkus-Edwards KR, Martinez-Lemus LA, Wu X, Trzeciakowski JP, Davis MJ, Davis GE, Meininger GA. α4β1 integrin activation of L-type calcium channels in vascular smooth muscle causes arteriolar vasoconstriction. Circ Res 90: 473–480, 2002. doi: 10.1161/hh0402.105899. [DOI] [PubMed] [Google Scholar]
- 1386. Morita T, Kurihara H, Maemura K, Yoshizumi M, Yazaki Y. Disruption of cytoskeletal structures mediates shear stress-induced endothelin-1 gene expression in cultured porcine aortic endothelial cells. J Clin Invest 92: 1706–1712, 1993. doi: 10.1172/JCI116757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1387. Morita T, Kurihara H, Maemura K, Yoshizumi M, Nagai R, Yazaki Y. Role of Ca2+ and protein kinase C in shear stress-induced actin depolymerization and endothelin 1 gene expression. Circ Res 75: 630–636, 1994. doi: 10.1161/01.res.75.4.630. [DOI] [PubMed] [Google Scholar]
- 1388. Searles CD, Ide L, Davis ME, Cai H, Weber M. Actin cytoskeleton organization and posttranscriptional regulation of endothelial nitric oxide synthase during cell growth. Circ Res 95: 488–495, 2004. doi: 10.1161/01.RES.0000138953.21377.80. [DOI] [PubMed] [Google Scholar]
- 1389. Malek AM, Izumo S. Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J Cell Sci 109: 713–726, 1996. doi: 10.1242/jcs.109.4.713. [DOI] [PubMed] [Google Scholar]
- 1390. Cheng M, Wu J, Liu X, Li Y, Nie Y, Li L, Chen H. Low shear stress-induced interleukin-8 mRNA expression in endothelial cells is mechanotransduced by integrins and the cytoskeleton. Endothelium 14: 265–273, 2007. doi: 10.1080/10623320701678169. [DOI] [PubMed] [Google Scholar]
- 1391. Liu B, Lu S, Hu YL, Liao X, Ouyang M, Wang Y. RhoA and membrane fluidity mediates the spatially polarized Src/FAK activation in response to shear stress. Sci Rep 4: 7008, 2014. doi: 10.1038/srep07008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1392. Wang C, Baker BM, Chen CS, Schwartz MA. Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol 33: 2130–2136, 2013. doi: 10.1161/ATVBAHA.113.301826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1393. Amar K, Wei F, Chen J, Wang N. Effects of forces on chromatin. APL Bioeng 5: 041503, 2021. doi: 10.1063/5.0065302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1394. Lityagina O, Dobreva G. The LINC between mechanical forces and chromatin. Front Physiol 12: 710809, 2021. doi: 10.3389/fphys.2021.710809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1395. Tajik A, Zhang Y, Wei F, Sun J, Jia Q, Zhou W, Singh R, Khanna N, Belmont AS, Wang N. Transcription upregulation via force-induced direct stretching of chromatin. Nat Mater 15: 1287–1296, 2016. doi: 10.1038/nmat4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1396. Sun J, Chen J, Amar K, Wu Y, Jiang M, Wang N. LAP2beta transmits force to upregulate genes via chromatin domain stretching but not compression. Acta Biomater 2021: S1742-7061(21)00700-5, 2021. doi: 10.1016/j.actbio.2021.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1397. Morgan JT, Pfeiffer ER, Thirkill TL, Kumar P, Peng G, Fridolfsson HN, Douglas GC, Starr DA, Barakat AI. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Mol Biol Cell 22: 4324–4334, 2011. doi: 10.1091/mbc.E11-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1398. Yang F, Zhang Y, Zhu J, Wang J, Jiang Z, Zhao C, Yang Q, Huang Y, Yao W, Pang W, Han L, Zhou J. Laminar flow protects vascular endothelial tight junctions and barrier function via maintaining the expression of long non-coding RNA MALAT1. Front Bioeng Biotechnol 8: 647, 2020. doi: 10.3389/fbioe.2020.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1399. Denis KB, Cabe JI, Danielsson BE, Tieu KV, Mayer CR, Conway DE. The LINC complex is required for endothelial cell adhesion and adaptation to shear stress and cyclic stretch. Mol Biol Cell 32: 1654–1663, 2021. doi: 10.1091/mbc.E20-11-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1400. Jiang Y, Ji JY. Expression of nuclear lamin proteins in endothelial cells is sensitive to cell passage and fluid shear stress. Cell Mol Bioeng 11: 53–64, 2018. doi: 10.1007/s12195-017-0513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1401. Harr JC, Gonzalez-Sandoval A, Gasser SM. Histones and histone modifications in perinuclear chromatin anchoring: from yeast to man. EMBO Rep 17: 139–155, 2016. doi: 10.15252/embr.201541809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1402. Demmerle J, Koch AJ, Holaska JM. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem 287: 22080–22088, 2012. doi: 10.1074/jbc.M111.325308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1403. van Steensel B, Belmont AS. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169: 780–791, 2017. doi: 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1404. Mo FE. Shear-regulated extracellular microenvironments and endothelial cell surface integrin receptors intertwine in atherosclerosis. Front Cell Dev Biol 9: 640781, 2021. doi: 10.3389/fcell.2021.640781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1405. Shyy JY, Chien S. Role of integrins in endothelial mechanosensing of shear stress. Circ Res 91: 769–775, 2002. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 1406. Orr AW, Ginsberg MH, Shattil SJ, Deckmyn H, Schwartz MA. Matrix-specific suppression of integrin activation in shear stress signaling. Mol Biol Cell 17: 4686–4697, 2006. doi: 10.1091/mbc.e06-04-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1407. Jalali S, del Pozo MA, Chen K, Miao H, Li Y, Schwartz MA, Shyy JY, Chien S. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci USA 98: 1042–1046, 2001. doi: 10.1073/pnas.98.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1408. Yang B, Rizzo V. Shear stress activates eNOS at the endothelial apical surface through beta1 containing integrins and caveolae. Cell Mol Bioeng 6: 346–354, 2013. doi: 10.1007/s12195-013-0276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1409. Yang TL, Lee PL, Lee DY, Wang WL, Wei SY, Lee CI, Chiu JJ. Differential regulations of fibronectin and laminin in Smad2 activation in vascular endothelial cells in response to disturbed flow. J Biomed Sci 25: 1, 2018. doi: 10.1186/s12929-017-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1410. Chen W, Epshtein Y, Ni X, Dull RO, Cress AE, Garcia JG, Jacobson JR. Role of integrin beta4 in lung endothelial cell inflammatory responses to mechanical stress. Sci Rep 5: 16529, 2015. doi: 10.1038/srep16529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1411. Spescha RD, Glanzmann M, Simic B, Witassek F, Keller S, Akhmedov A, Tanner FC, Lüscher TF, Camici GG. Adaptor protein p66(Shc) mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension 64: 347–353, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02129. [DOI] [PubMed] [Google Scholar]
- 1412. Huang W, Sakamoto N, Miyazawa R, Sato M. Role of paxillin in the early phase of orientation of the vascular endothelial cells exposed to cyclic stretching. Biochem Biophys Res Commun 418: 708–713, 2012. doi: 10.1016/j.bbrc.2012.01.083. [DOI] [PubMed] [Google Scholar]
- 1413. Tesauro M, Mauriello A, Rovella V, Annicchiarico-Petruzzelli M, Cardillo C, Melino G, Di Daniele N. Arterial ageing: from endothelial dysfunction to vascular calcification. J Intern Med 281: 471–482, 2017. doi: 10.1111/joim.12605. [DOI] [PubMed] [Google Scholar]
- 1414. Rodríguez-Mañas L, El-Assar M, Vallejo S, López-Dóriga P, Solis J, Petidier R, Montes M, Nevado J, Castro M, Gomez-Guerrero C, Peiro C, Sanchez-Ferrer CF. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 8: 226–238, 2009. doi: 10.1111/j.1474-9726.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- 1415. Juonala M, Viikari JS, Rönnemaa T, Helenius H, Taittonen L, Raitakari OT. Elevated blood pressure in adolescent boys predicts endothelial dysfunction: the cardiovascular risk in young Finns study. Hypertension 48: 424–430, 2006. doi: 10.1161/01.HYP.0000237666.78217.47. [DOI] [PubMed] [Google Scholar]
- 1416. Kohn JC, Zhou DW, Bordeleau F, Zhou AL, Mason BN, Mitchell MJ, King MR, Reinhart-King CA. Cooperative effects of matrix stiffness and fluid shear stress on endothelial cell behavior. Biophys J 108: 471–478, 2015. doi: 10.1016/j.bpj.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1417. Kim CW, Pokutta-Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, Dalal S, Chadid T, Kuo KM, Raykin J, Li H, Yanagisawa H, Gleason RL Jr, Jo H, Brewster LP. Disturbed flow promotes arterial stiffening through thrombospondin-1. Circulation 136: 1217–1232, 2017. doi: 10.1161/CIRCULATIONAHA.116.026361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1418. Mott RE, Helmke BP. Mapping the dynamics of shear stress-induced structural changes in endothelial cells. Am J Physiol Cell Physiol 293: C1616–C1626, 2007. doi: 10.1152/ajpcell.00457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1419. Hu YL, Chien S. Dynamic motion of paxillin on actin filaments in living endothelial cells. Biochem Biophys Res Commun 357: 871–876, 2007. doi: 10.1016/j.bbrc.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1420. Hu YL, Li S, Miao H, Tsou TC, del Pozo MA, Chien S. Roles of microtubule dynamics and small GTPase Rac in endothelial cell migration and lamellipodium formation under flow. J Vasc Res 39: 465–476, 2002. doi: 10.1159/000067202. [DOI] [PubMed] [Google Scholar]
- 1421. Li S, Butler P, Wang Y, Hu Y, Han DC, Usami S, Guan JL, Chien S. The role of the dynamics of focal adhesion kinase in the mechanotaxis of endothelial cells. Proc Natl Acad Sci USA 99: 3546–3551, 2002. doi: 10.1073/pnas.052018099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1422. Galbraith CG, Skalak R, Chien S. Shear stress induces spatial reorganization of the endothelial cell cytoskeleton. Cell Motil Cytoskeleton 40: 317–330, 1998. doi: 10.1002/(SICI)1097-0169(1998)40:4<317::AID-CM1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 1423. Helmlinger G, Geiger RV, Schreck S, Nerem RM. Effects of pulsatile flow on cultured vascular endothelial cell morphology. J Biomech Eng 113: 123–131, 1991. doi: 10.1115/1.2891226. [DOI] [PubMed] [Google Scholar]
- 1424. Ukropec JA, Hollinger MK, Woolkalis MJ. Regulation of VE-cadherin linkage to the cytoskeleton in endothelial cells exposed to fluid shear stress. Exp Cell Res 273: 240–247, 2002. doi: 10.1006/excr.2001.5453. [DOI] [PubMed] [Google Scholar]
- 1425. Lertkiatmongkol P, Liao D, Mei H, Hu Y, Newman PJ. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr Opin Hematol 23: 253–259, 2016. doi: 10.1097/MOH.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1426. Sato M, Levesque MJ, Nerem RM. Micropipette aspiration of cultured bovine aortic endothelial cells exposed to shear stress. Arteriosclerosis 7: 276–286, 1987. doi: 10.1161/01.atv.7.3.276. [DOI] [PubMed] [Google Scholar]
- 1427. del Alamo JC, Norwich GN, Li YS, Lasheras JC, Chien S. Anisotropic rheology and directional mechanotransduction in vascular endothelial cells. Proc Natl Acad Sci USA 105: 15411–15416, 2008. doi: 10.1073/pnas.0804573105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1428. Zhao S, Suciu A, Ziegler T, Moore JE Jr, Bürki E, Meister JJ, Brunner HR. Synergistic effects of fluid shear stress and cyclic circumferential stretch on vascular endothelial cell morphology and cytoskeleton. Arterioscler Thromb Vasc Biol 15: 1781–1786, 1995. doi: 10.1161/01.ATV.15.10.1781. [DOI] [PubMed] [Google Scholar]
- 1429. Moore JE Jr, Bürki E, Suciu A, Zhao S, Burnier M, Brunner HR, Meister JJ. A device for subjecting vascular endothelial cells to both fluid shear stress and circumferential cyclic stretch. Ann Biomed Eng 22: 416–422, 1994. doi: 10.1007/BF02368248. [DOI] [PubMed] [Google Scholar]
- 1430. Owatverot TB, Oswald SJ, Chen Y, Wille JJ, Yin FC. Effect of combined cyclic stretch and fluid shear stress on endothelial cell morphological responses. J Biomech Eng 127: 374–382, 2005. doi: 10.1115/1.1894180. [DOI] [PubMed] [Google Scholar]
- 1431. Kwak BR, Silacci P, Stergiopulos N, Hayoz D, Meda P. Shear stress and cyclic circumferential stretch, but not pressure, alter connexin43 expression in endothelial cells. Cell Commun Adhes 12: 261–270, 2005. doi: 10.1080/15419060500514119. [DOI] [PubMed] [Google Scholar]
- 1432. Berardi DE, Tarbell JM. Stretch and shear interactions affect intercellular junction protein expression and turnover in endothelial cells. Cell Mol Bioeng 2: 320–331, 2009. doi: 10.1007/s12195-009-0073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1433. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183, 2011. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 1434. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540: 579–582, 2016. doi: 10.1038/nature20602. [DOI] [PubMed] [Google Scholar]
- 1435. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA 113: 11525–11530, 2016. doi: 10.1073/pnas.1613121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1436. Xu S, Koroleva M, Yin M, Jin ZG. Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl Res 176: 18–28.e2, 2016. doi: 10.1016/j.trsl.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1437. Liu D, Lv H, Liu Q, Sun Y, Hou S, Zhang L, Yang M, Han B, Wang G, Wang X, Du W, Nie H, Zhang R, Huang X, Hou J, Yu B. Atheroprotective effects of methotrexate via the inhibition of YAP/TAZ under disturbed flow. J Transl Med 17: 378, 2019. doi: 10.1186/s12967-019-02135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1438. Li B, He J, Lv H, Liu Y, Lv X, Zhang C, Zhu Y, Ai D. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J Clin Invest 129: 1167–1179, 2019. doi: 10.1172/JCI122440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1439. Yun S, Hu R, Schwaemmle ME, Scherer AN, Zhuang Z, Koleske AJ, Pallas DC, Schwartz MA. Integrin alpha5beta1 regulates PP2A complex assembly through PDE4D in atherosclerosis. J Clin Invest 129: 4863–4874, 2019. doi: 10.1172/JCI127692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1440. Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, Ando K, Miyazaki T, Yokota Y, Schmelzer E, Belting HG, Affolter M, Lecaudey V, Mochizuki N. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell 40: 523–536.e6, 2017. doi: 10.1016/j.devcel.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 1441. He J, Bao Q, Yan M, Liang J, Zhu Y, Wang C, Ai D. The role of Hippo/yes-associated protein signalling in vascular remodelling associated with cardiovascular disease. Br J Pharmacol 175: 1354–1361, 2018. doi: 10.1111/bph.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1442. Neto F, Klaus-Bergmann A, Ong YT, Alt S, Vion AC, Szymborska A, Carvalho JR, Hollfinger I, Bartels-Klein E, Franco CA, Potente M, Gerhardt H. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife 7: e31037, 2018. doi: 10.7554/eLife.31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1443. Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Black SM, Fratz S, Fineman JR, Vargas SO, Haley KJ, Waxman AB, Chau BN, Fredenburgh LE, Chan SY. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep 13: 1016–1032, 2015. doi: 10.1016/j.celrep.2015.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1444. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 126: 3313–3335, 2016. doi: 10.1172/JCI86387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1445. Zhou J, Li YS, Wang KC, Chien S. Epigenetic mechanism in regulation of endothelial function by disturbed flow: induction of DNA hypermethylation by DNMT1. Cell Mol Bioeng 7: 218–224, 2014. doi: 10.1007/s12195-014-0325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1446. Yan C, Takahashi M, Okuda M, Lee JD, Berk BC. Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells. Dependence on tyrosine kinases and intracellular calcium. J Biol Chem 274: 143–150, 1999. doi: 10.1074/jbc.274.1.143. [DOI] [PubMed] [Google Scholar]
- 1447. Li YS, Shyy JY, Li S, Lee J, Su B, Karin M, Chien S. The Ras-JNK pathway is involved in shear-induced gene expression. Mol Cell Biol 16: 5947–5954, 1996. doi: 10.1128/MCB.16.11.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1448. Li S, Kim M, Hu YL, Jalali S, Schlaepfer DD, Hunter T, Chien S, Shyy JY. Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J Biol Chem 272: 30455–30462, 1997. doi: 10.1074/jbc.272.48.30455. [DOI] [PubMed] [Google Scholar]
- 1449. Jo H, Sipos K, Go YM, Law R, Rong J, McDonald JM. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal Jun kinase in endothelial cells. Gi2- and Gbeta/gamma-dependent signaling pathways. J Biol Chem 272: 1395–1401, 1997. doi: 10.1074/jbc.272.2.1395. [DOI] [PubMed] [Google Scholar]
- 1450. Azuma N, Akasaka N, Kito H, Ikeda M, Gahtan V, Sasajima T, Sumpio BE. Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress. Am J Physiol Heart Circ Physiol 280: H189–H197, 2001. doi: 10.1152/ajpheart.2001.280.1.H189. [DOI] [PubMed] [Google Scholar]
- 1451. Go YM, Park H, Maland MC, Darley-Usmar VM, Stoyanov B, Wetzker R, Jo H. Phosphatidylinositol 3-kinase gamma mediates shear stress-dependent activation of JNK in endothelial cells. Am J Physiol 275: H1898–H1904, 1998. doi: 10.1152/ajpheart.1998.275.5.H1898. [DOI] [PubMed] [Google Scholar]
- 1452. Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res 83: 334–341, 1998. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 1453. Guo D, Chien S, Shyy JY. Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ Res 100: 564–571, 2007. doi: 10.1161/01.RES.0000259561.23876.c5. [DOI] [PubMed] [Google Scholar]
- 1454. Yang H, Zhu L, Chao Y, Gu Y, Kong X, Chen M, Ye P, Luo J, Chen S. Hyaluronidase2 (Hyal2) modulates low shear stress-induced glycocalyx impairment via the LKB1/AMPK/NADPH oxidase-dependent pathway. J Cell Physiol 233: 9701–9715, 2018. doi: 10.1002/jcp.26944. [DOI] [PubMed] [Google Scholar]
- 1455. Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res 105: 114–127, 2009. doi: 10.1161/CIRCRESAHA.109.201590. [DOI] [PubMed] [Google Scholar]
- 1456. Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, Kassab GS, Garland T Jr, Shyy JY. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol 26: 1281–1287, 2006. doi: 10.1161/01.ATV.0000221230.08596.98. [DOI] [PubMed] [Google Scholar]
- 1457. Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci 118: 4103–4111, 2005. doi: 10.1242/jcs.02541. [DOI] [PubMed] [Google Scholar]
- 1458. Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY. Shear stress, SIRT1, and vascular homeostasis. Proc Natl Acad Sci USA 107: 10268–10273, 2010. doi: 10.1073/pnas.1003833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1459. Ghimire K, Zaric J, Alday-Parejo B, Seebach J, Bousquenaud M, Stalin J, Bieler G, Schnittler HJ, Rüegg C. MAGI1 mediates eNOS activation and NO production in endothelial cells in response to fluid shear stress. Cells 8: 388, 2019. doi: 10.3390/cells8050388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1460. Fisslthaler B, Fleming I, Keserü B, Walsh K, Busse R. Fluid shear stress and NO decrease the activity of the hydroxy-methylglutaryl coenzyme A reductase in endothelial cells via the AMP-activated protein kinase and FoxO1. Circ Res 100: e12–e21, 2007. doi: 10.1161/01.RES.0000257747.74358.1c. [DOI] [PubMed] [Google Scholar]
- 1461. Dixit M, Bess E, Fisslthaler B, Härtel FV, Noll T, Busse R, Fleming I. Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovasc Res 77: 160–168, 2008. doi: 10.1093/cvr/cvm017. [DOI] [PubMed] [Google Scholar]
- 1462. Zhang J, Kong X, Wang Z, Gao X, Ge Z, Gu Y, Ye P, Chao Y, Zhu L, Li X, Chen S. AMP-activated protein kinase regulates glycocalyx impairment and macrophage recruitment in response to low shear stress. FASEB J 33: 7202–7212, 2019. doi: 10.1096/fj.201801869RRR. [DOI] [PubMed] [Google Scholar]
- 1463. Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. Shear stress stimulates phosphorylation of eNOS at Ser635 by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol 283: H1819–H1828, 2002. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- 1464. Boo YC, Sorescu GP, Bauer PM, Fulton D, Kemp BE, Harrison DG, Sessa WC, Jo H. Endothelial NO synthase phosphorylated at SER635 produces NO without requiring intracellular calcium increase. Free Radic Biol Med 35: 729–741, 2003. doi: 10.1016/S0891-5849(03)00397-6. [DOI] [PubMed] [Google Scholar]
- 1465. Zhu L, Yang H, Chao Y, Gu Y, Zhang J, Wang F, Yu W, Ye P, Chu P, Kong X, Chen S. Akt phosphorylation regulated by IKKϵ in response to low shear stress leads to endothelial inflammation via activating IRF3. Cell Signal 80: 109900, 2021. doi: 10.1016/j.cellsig.2020.109900. [DOI] [PubMed] [Google Scholar]
- 1466. Kong X, Chen L, Ye P, Wang Z, Zhang J, Ye F, Chen S. The role of HYAL2 in LSS-induced glycocalyx impairment and the PKA-mediated decrease in eNOS-Ser-633 phosphorylation and nitric oxide production. Mol Biol Cell 27: 3972–3979, 2016. doi: 10.1091/mbc.E16-04-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1467. Zhang J. New insights into the contribution of arterial NCX to the regulation of myogenic tone and blood pressure. Adv Exp Med Biol 961: 329–343, 2013. doi: 10.1007/978-1-4614-4756-6_28. [DOI] [PubMed] [Google Scholar]
- 1468. Kashihara T, Nakayama K, Matsuda T, Baba A, Ishikawa T. Role of Na+/Ca2+ exchanger-mediated Ca2+ entry in pressure-induced myogenic constriction in rat posterior cerebral arteries. J Pharmacol Sci 110: 218–222, 2009. doi: 10.1254/jphs.09054sc. [DOI] [PubMed] [Google Scholar]
- 1469. Raina H, Ella SR, Hill MA. Decreased activity of the smooth muscle Na+/Ca2+ exchanger impairs arteriolar myogenic reactivity. J Physiol 586: 1669–1681, 2008. doi: 10.1113/jphysiol.2007.150268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1470. Nowicki PT, Flavahan S, Hassanain H, Mitra S, Holland S, Goldschmidt-Clermont PJ, Flavahan NA. Redox signaling of the arteriolar myogenic response. Circ Res 89: 114–116, 2001. doi: 10.1161/hh1401.094367. [DOI] [PubMed] [Google Scholar]
- 1471. Chao JT, Gui P, Zamponi GW, Davis GE, Davis MJ. Spatial association of the Cav1.2 calcium channel with α5β1-integrin. Am J Physiol Cell Physiol 300: C477–C489, 2011. doi: 10.1152/ajpcell.00171.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1472. Drummond HA. What evolutionary evidence implies about the identity of the mechanoelectrical couplers in vascular smooth muscle cells. Physiology (Bethesda) 36: 292–306, 2021. doi: 10.1152/physiol.00008.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1473. Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50: 277–289, 2006. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 1474. Geffeney SL, Cueva JG, Glauser DA, Doll JC, Lee TH, Montoya M, Karania S, Garakani AM, Pruitt BL, Goodman MB. DEG/ENaC but not TRP channels are the major mechanoelectrical transduction channels in a C. elegans nociceptor. Neuron 71: 845–857, 2011. doi: 10.1016/j.neuron.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1475. Suzuki H, Kerr R, Bianchi L, Frøkjaer-Jensen C, Slone D, Xue J, Gerstbrein B, Driscoll M, Schafer WR. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39: 1005–1017, 2003. doi: 10.1016/j.neuron.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 1476. Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol 31: 1495–1505, 2011. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1477. Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev 75: 487–517, 1995. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 1478. Kaur H, Carvalho J, Looso M, Singh P, Chennupati R, Preussner J, Günther S, Albarrán-Juárez J, Tischner D, Classen S, Offermanns S, Wettschureck N. Single-cell profiling reveals heterogeneity and functional patterning of GPCR expression in the vascular system. Nat Commun 8: 15700, 2017. doi: 10.1038/ncomms15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1479. Kuga T, Kobayashi S, Hirakawa Y, Kanaide H, Takeshita A. Cell cycle-dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res 79: 14–19, 1996. doi: 10.1161/01.res.79.1.14. [DOI] [PubMed] [Google Scholar]
- 1480. Richard S, Neveu D, Carnac G, Bodin P, Travo P, Nargeot J. Differential expression of voltage-gated Ca2+-currents in cultivated aortic myocytes. Biochim Biophys Acta 1160: 95–104, 1992. doi: 10.1016/0167-4838(92)90042-c. [DOI] [PubMed] [Google Scholar]
- 1481. Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ Res 95: 406–414, 2004. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 1482. Halka AT, Turner NJ, Carter A, Ghosh J, Murphy MO, Kirton JP, Kielty CM, Walker MG. The effects of stretch on vascular smooth muscle cell phenotype in vitro. Cardiovasc Pathol 17: 98–102, 2008. doi: 10.1016/j.carpath.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 1483. Jensen LF, Bentzon JF, Albarrán-Juarez J. The phenotypic responses of vascular smooth muscle cells exposed to mechanical cues. Cells 10: 2209, 2021. doi: 10.3390/cells10092209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1484. Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol 31: 851–860, 2011. doi: 10.1161/ATVBAHA.110.221952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1485. Kee HJ, Kwon JS, Shin S, Ahn Y, Jeong MH, Kook H. Trichostatin A prevents neointimal hyperplasia via activation of Krüppel like factor 4. Vascul Pharmacol 55: 127–134, 2011. doi: 10.1016/j.vph.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 1486. Liang J, Li Q, Cai W, Zhang X, Yang B, Li X, Jiang S, Tian S, Zhang K, Song H, Ai D, Zhang X, Wang C, Zhu Y. Inhibition of polycomb repressor complex 2 ameliorates neointimal hyperplasia by suppressing trimethylation of H3K27 in vascular smooth muscle cells. Br J Pharmacol 176: 3206–3219, 2019. doi: 10.1111/bph.14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1487. Qi H, Jing Z, Xiaolin W, Changwu X, Xiaorong H, Jian Y, Jing C, Hong J. Histone Demethylase JMJD2A inhibition attenuates neointimal hyperplasia in the carotid arteries of balloon-injured diabetic rats via transcriptional silencing: inflammatory gene expression in vascular smooth muscle cells. Cell Physiol Biochem 37: 719–734, 2015. doi: 10.1159/000430390. [DOI] [PubMed] [Google Scholar]
- 1488. Zhang J, Chen J, Yang J, Xu C, Hu Q, Wu H, Cai W, Guo Q, Gao W, He C, Yang C, Yang J. Suv39h1 downregulation inhibits neointimal hyperplasia after vascular injury. Atherosclerosis 288: 76–84, 2019. doi: 10.1016/j.atherosclerosis.2019.06.909. [DOI] [PubMed] [Google Scholar]
- 1489. Leeper NJ, Maegdefessel L. Non-coding RNAs: key regulators of smooth muscle cell fate in vascular disease. Cardiovasc Res 114: 611–621, 2018. doi: 10.1093/cvr/cvx249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1490. Maguire EM, Xiao Q. Noncoding RNAs in vascular smooth muscle cell function and neointimal hyperplasia. FEBS J 287: 5260–5283, 2020. doi: 10.1111/febs.15357. [DOI] [PubMed] [Google Scholar]
- 1491. Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L. miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 4: 197–205, 2011. doi: 10.1161/CIRCGENETICS.110.958702. [DOI] [PubMed] [Google Scholar]
- 1492. Vacante F, Denby L, Sluimer JC, Baker AH. The function of miR-143, miR-145 and the MiR-143 host gene in cardiovascular development and disease. Vascul Pharmacol 112: 24–30, 2019. doi: 10.1016/j.vph.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1493. Albinsson S, Swärd K. Targeting smooth muscle microRNAs for therapeutic benefit in vascular disease. Pharmacol Res 75: 28–36, 2013. doi: 10.1016/j.phrs.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 1494. Albinsson S, Bhattachariya A, Hellstrand P. Stretch-dependent smooth muscle differentiation in the portal vein-role of actin polymerization, calcium signaling, and microRNAs. Microcirculation 21: 230–238, 2014. doi: 10.1111/micc.12106. [DOI] [PubMed] [Google Scholar]
- 1495. Riches-Suman K. Diverse roles of microRNA-145 in regulating smooth muscle (dys)function in health and disease. Biochem Soc Trans 49: 353–363, 2021. doi: 10.1042/BST20200679. [DOI] [PubMed] [Google Scholar]
- 1496. Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, Upton P, Xin M, van Rooij E, Olson EN, Morrell NW, MacLean MR, Baker AH. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res 111: 290–300, 2012. doi: 10.1161/CIRCRESAHA.112.267591. [DOI] [PubMed] [Google Scholar]
- 1497. Huang K, Bao H, Yan ZQ, Wang L, Zhang P, Yao QP, Shi Q, Chen XH, Wang KX, Shen BR, Qi YX, Jiang ZL. MicroRNA-33 protects against neointimal hyperplasia induced by arterial mechanical stretch in the grafted vein. Cardiovasc Res 113: 488–497, 2017. doi: 10.1093/cvr/cvw257. [DOI] [PubMed] [Google Scholar]
- 1498. Liu JT, Liu Z, Chen Y, Qi YX, Yao QP, Jiang ZL. MicroRNA-29a involvement in phenotypic transformation of venous smooth muscle cells via ten-eleven translocation methylcytosinedioxygenase 1 in response to mechanical cyclic stretch. J Biomech Eng 142: 051009, 2020. doi: 10.1115/1.4044581. [DOI] [PubMed] [Google Scholar]
- 1499. Wang WB, Li HP, Yan J, Zhuang F, Bao M, Liu JT, Qi YX, Han Y. CTGF regulates cyclic stretch-induced vascular smooth muscle cell proliferation via microRNA-19b-3p. Exp Cell Res 376: 77–85, 2019. doi: 10.1016/j.yexcr.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 1500. Bao H, Li HP, Shi Q, Huang K, Chen XH, Chen YX, Han Y, Xiao Q, Yao QP, Qi YX. Lamin A/C negatively regulated by miR-124-3p modulates apoptosis of vascular smooth muscle cells during cyclic stretch application in rats. Acta Physiol (Oxf) 228: e13374, 2020. doi: 10.1111/apha.13374. [DOI] [PubMed] [Google Scholar]
- 1501. Song J, Hu B, Qu H, Bi C, Huang X, Zhang M. Mechanical stretch modulates microRNA 21 expression, participating in proliferation and apoptosis in cultured human aortic smooth muscle cells. PLoS One 7: e47657, 2012. doi: 10.1371/journal.pone.0047657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1502. Mantella LE, Singh KK, Sandhu P, Kantores C, Ramadan A, Khyzha N, Quan A, Al-Omran M, Fish JE, Jankov RP, Verma S. Fingerprint of long non-coding RNA regulated by cyclic mechanical stretch in human aortic smooth muscle cells: implications for hypertension. Mol Cell Biochem 435: 163–173, 2017. doi: 10.1007/s11010-017-3065-2. [DOI] [PubMed] [Google Scholar]
- 1503. Wang N, Miao H, Li YS, Zhang P, Haga JH, Hu Y, Young A, Yuan S, Nguyen P, Wu CC, Chien S. Shear stress regulation of Krüppel-like factor 2 expression is flow pattern-specific. Biochem Biophys Res Commun 341: 1244–1251, 2006. doi: 10.1016/j.bbrc.2006.01.089. [DOI] [PubMed] [Google Scholar]
- 1504. Young A, Wu W, Sun W, Benjamin Larman H, Wang N, Li YS, Shyy JY, Chien S, García-Cardena G. Flow activation of AMP-activated protein kinase in vascular endothelium leads to Krüppel-like factor 2 expression. Arterioscler Thromb Vasc Biol 29: 1902–1908, 2009. doi: 10.1161/ATVBAHA.109.193540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1505. Jiang YZ, Jiménez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res 115: 32–43, 2014. doi: 10.1161/CIRCRESAHA.115.303883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1506. Wu W, Xiao H, Laguna-Fernandez A, Villarreal G Jr, Wang KC, Geary GG, Zhang Y, Wang WC, Huang HD, Zhou J, Li YS, Chien S, Garcia-Cardena G, Shyy JY. Flow-dependent regulation of Kruppel-Like Factor 2 is mediated by microRNA-92a. Circulation 124: 633–641, 2011. doi: 10.1161/CIRCULATIONAHA.110.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1507. Loyer X, Potteaux S, Vion AC, Guérin CL, Boulkroun S, Rautou PE, Ramkhelawon B, Esposito B, Dalloz M, Paul JL, Julia P, Maccario J, Boulanger CM, Mallat Z, Tedgui A. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ Res 114: 434–443, 2014. doi: 10.1161/CIRCRESAHA.114.302213. [DOI] [PubMed] [Google Scholar]
- 1508. Chen Z, Wen L, Martin M, Hsu CY, Fang L, Lin FM, Lin TY, Geary MJ, Geary GG, Zhao Y, Johnson DA, Chen JW, Lin SJ, Chien S, Huang HD, Miller YI, Huang PH, Shyy JY. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation 131: 805–814, 2015. doi: 10.1161/CIRCULATIONAHA.114.013675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1509. Hartmann P, Zhou Z, Natarelli L, Wei Y, Nazari-Jahantigh M, Zhu M, Grommes J, Steffens S, Weber C, Schober A. Endothelial Dicer promotes atherosclerosis and vascular inflammation by miRNA-103-mediated suppression of KLF4. Nat Commun 7: 10521, 2016. doi: 10.1038/ncomms10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1510. Atkins GB, Wang Y, Mahabeleshwar GH, Shi H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI, Jain MK. Hemizygous deficiency of Krüppel-like factor 2 augments experimental atherosclerosis. Circ Res 103: 690–693, 2008. doi: 10.1161/CIRCRESAHA.108.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1511. Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y, Sharma N, Liao X, Hale A, Boerboom L, Feaver RE, Gao H, Desai A, Schmaier A, Gerson SL, Wang Y, Atkins GB, Blackman BR, Simon DI, Jain MK. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J Clin Invest 122: 4727–4731, 2012. doi: 10.1172/JCI66056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1512. Fledderus JO, van Thienen JV, Boon RA, Dekker RJ, Rohlena J, Volger OL, Bijnens AP, Daemen MJ, Kuiper J, van Berkel TJ, Pannekoek H, Horrevoets AJ. Prolonged shear stress and KLF2 suppress constitutive proinflammatory transcription through inhibition of ATF2. Blood 109: 4249–4257, 2007. doi: 10.1182/blood-2006-07-036020. [DOI] [PubMed] [Google Scholar]
- 1513. Rossi J, Rouleau L, Emmott A, Tardif JC, Leask RL. Laminar shear stress prevents simvastatin-induced adhesion molecule expression in cytokine activated endothelial cells. Eur J Pharmacol 649: 268–276, 2010. doi: 10.1016/j.ejphar.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 1514. Berk BC, Abe JI, Min W, Surapisitchat J, Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann NY Acad Sci 947: 93–109, 2001. doi: 10.1111/j.1749-6632.2001.tb03932.x. [DOI] [PubMed] [Google Scholar]
- 1515. Shyy YJ, Hsieh HJ, Usami S, Chien S. Fluid shear stress induces a biphasic response of human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc Natl Acad Sci USA 91: 4678–4682, 1994. doi: 10.1073/pnas.91.11.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1516. Liu M, Kluger MS, D’Alessio A, García-Cardeña G, Pober JS. Regulation of arterial-venous differences in tumor necrosis factor responsiveness of endothelial cells by anatomic context. Am J Pathol 172: 1088–1099, 2008. doi: 10.2353/ajpath.2008.070603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1517. Mohan S, Mohan N, Sprague EA. Differential activation of NF-kappa B in human aortic endothelial cells conditioned to specific flow environments. Am J Physiol Cell Physiol 273: C572–C578, 1997. doi: 10.1152/ajpcell.1997.273.2.C572. [DOI] [PubMed] [Google Scholar]
- 1518. Lan Q, Mercurius KO, Davies PF. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem Biophys Res Commun 201: 950–956, 1994. doi: 10.1006/bbrc.1994.1794. [DOI] [PubMed] [Google Scholar]
- 1519. Bhullar IS, Li YS, Miao H, Zandi E, Kim M, Shyy JY, Chien S. Fluid shear stress activation of IkappaB kinase is integrin-dependent. J Biol Chem 273: 30544–30549, 1998. doi: 10.1074/jbc.273.46.30544. [DOI] [PubMed] [Google Scholar]
- 1520. Weber KS, Draude G, Erl W, de Martin R, Weber C. Monocyte arrest and transmigration on inflamed endothelium in shear flow is inhibited by adenovirus-mediated gene transfer of IkappaB-alpha. Blood 93: 3685–3693, 1999. [PubMed] [Google Scholar]
- 1521. Mohan S, Mohan N, Valente AJ, Sprague EA. Regulation of low shear flow-induced HAEC VCAM-1 expression and monocyte adhesion. Am J Physiol Cell Physiol 276: C1100–C1107, 1999. doi: 10.1152/ajpcell.1999.276.5.C1100. [DOI] [PubMed] [Google Scholar]
- 1522. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168: 37–57, 2017. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1523. Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306: 704–708, 2004. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 1524. Hatanaka N, Seki T, Inoue JI, Tero A, Suzuki T. Critical roles of IkappaBalpha and RelA phosphorylation in transitional oscillation in NF-kappaB signaling module. J Theor Biol 462: 479–489, 2019. doi: 10.1016/j.jtbi.2018.11.023. [DOI] [PubMed] [Google Scholar]
- 1525. Ganguli A, Persson L, Palmer IR, Evans I, Yang L, Smallwood R, Black R, Qwarnstrom EE. Distinct NF-kappaB regulation by shear stress through Ras-dependent IkappaBalpha oscillations: real-time analysis of flow-mediated activation in live cells. Circ Res 96: 626–634, 2005. doi: 10.1161/01.RES.0000160435.83210.95. [DOI] [PubMed] [Google Scholar]
- 1526. Mohan S, Hamuro M, Koyoma K, Sorescu GP, Jo H, Natarajan M. High glucose induced NF-kappaB DNA-binding activity in HAEC is maintained under low shear stress but inhibited under high shear stress: role of nitric oxide. Atherosclerosis 171: 225–234, 2003. doi: 10.1016/j.atherosclerosis.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 1527. Chen J, Green J, Yurdagul A Jr, Albert P, McInnis MC, Orr AW. alphavbeta3 Integrins mediate flow-induced NF-kappaB activation, proinflammatory gene expression, and early atherogenic inflammation. Am J Pathol 185: 2575–2589, 2015. doi: 10.1016/j.ajpath.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1528. Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, Chaudhury H, Luong Le A, Mason JC, Udalova I, Gsell W, Jones H, Haskard DO, Krams R, Evans PC. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-kappaB regulation that promotes arterial inflammation. Circ Res 108: 950–959, 2011. doi: 10.1161/CIRCRESAHA.110.233841. [DOI] [PubMed] [Google Scholar]
- 1529. Feng S, Bowden N, Fragiadaki M, Souilhol C, Hsiao S, Mahmoud M, Allen S, Pirri D, Ayllon BT, Akhtar S, Thompson AA, Jo H, Weber C, Ridger V, Schober A, Evans PC. Mechanical activation of hypoxia-inducible factor 1alpha drives endothelial dysfunction at atheroprone sites. Arterioscler Thromb Vasc Biol 37: 2087–2101, 2017. doi: 10.1161/ATVBAHA.117.309249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1530. Maurya MR, Gupta S, Li JY, Ajami NE, Chen ZB, Shyy JY, Chien S, Subramaniam S. Longitudinal shear stress response in human endothelial cells to atheroprone and atheroprotective conditions. Proc Natl Acad Sci USA 118: e2023236118, 2021. doi: 10.1073/pnas.2023236118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1531. Ajami NE, Gupta S, Maurya MR, Nguyen P, Li JY, Shyy JY, Chen Z, Chien S, Subramaniam S. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc Natl Acad Sci USA 114: 10990–10995, 2017. doi: 10.1073/pnas.1707517114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1532. He M, Huang TS, Li S, Hong HC, Chen Z, Martin M, Zhou X, Huang HY, Su SH, Zhang J, Wang WT, Kang J, Huang HD, Zhang J, Chien S, Shyy JY. Atheroprotective flow upregulates ITPR3 (Inositol 1,4,5-Trisphosphate Receptor 3) in vascular endothelium via KLF4 (Krüppel-Like Factor 4)-mediated histone modifications. Arterioscler Thromb Vasc Biol 39: 902–914, 2019. doi: 10.1161/ATVBAHA.118.312301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1533. Bondareva O, Tsaryk R, Bojovic V, Odenthal-Schnittler M, Siekmann AF, Schnittler HJ. Identification of atheroprone shear stress responsive regulatory elements in endothelial cells. Cardiovasc Res 115: 1487–1499, 2019. doi: 10.1093/cvr/cvz027. [DOI] [PubMed] [Google Scholar]
- 1534. Andueza A, Kumar S, Kim J, Kang DW, Mumme HL, Perez JI, Villa-Roel N, Jo H. Endothelial reprogramming by disturbed flow revealed by single-cell RNA and chromatin accessibility study. Cell Rep 33: 108491, 2020. doi: 10.1016/j.celrep.2020.108491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1535. Niu N, Xu S, Xu Y, Little PJ, Jin ZG. Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol Sci 40: 253–266, 2019. doi: 10.1016/j.tips.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1536. Qiao C, Meng F, Jang I, Jo H, Chen YE, Zhang J. Deep transcriptomic profiling reveals the similarity between endothelial cells cultured under static and oscillatory shear stress conditions. Physiol Genomics 48: 660–666, 2016. doi: 10.1152/physiolgenomics.00025.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1537. Chu TJ, Peters DG. Serial analysis of the vascular endothelial transcriptome under static and shear stress conditions. Physiol Genomics 34: 185–192, 2008. doi: 10.1152/physiolgenomics.90201.2008. [DOI] [PubMed] [Google Scholar]
- 1538. Davies PF. Endothelial transcriptome profiles in vivo in complex arterial flow fields. Ann Biomed Eng 36: 563–570, 2008. doi: 10.1007/s10439-007-9400-0. [DOI] [PubMed] [Google Scholar]
- 1539. Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM. Single-cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell populations. Circulation 140: 147–163, 2019. doi: 10.1161/CIRCULATIONAHA.118.038362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1540. Lukowski SW, Patel J, Andersen SB, Sim SL, Wong HY, Tay J, Winkler I, Powell JE, Khosrotehrani K. Single-cell transcriptional profiling of aortic endothelium identifies a hierarchy from endovascular progenitors to differentiated cells. Cell Rep 27: 2748–2758.e3, 2019. doi: 10.1016/j.celrep.2019.04.102. [DOI] [PubMed] [Google Scholar]
- 1541. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 122: 1661–1674, 2018. doi: 10.1161/CIRCRESAHA.117.312509. [DOI] [PubMed] [Google Scholar]
- 1542. Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, Mammoto A, Chen J, Ariaans M, Francis SE, Van der Heiden K, Ridger V, Evans PC. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep 7: 3375, 2017. doi: 10.1038/s41598-017-03532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1543. Kwak BR, Bäck M, Bochaton-Piallat ML, Caligiuri G, Daemen MJ, Davies PF, Hoefer IE, Holvoet P, Jo H, Krams R, Lehoux S, Monaco C, Steffens S, Virmani R, Weber C, Wentzel JJ, Evans PC. Biomechanical factors in atherosclerosis: mechanisms and clinical implications. Eur Heart J 35: 3013–3020, 2014. doi: 10.1093/eurheartj/ehu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1544. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res 114: 565–577, 2018. doi: 10.1093/cvr/cvx253. [DOI] [PubMed] [Google Scholar]
- 1545. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 7: 11853, 2016. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1546. Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, Baker AH. Endothelial to mesenchymal transition in cardiovascular disease: JACC State-of-the-Art Review. J Am Coll Cardiol 73: 190–209, 2019. doi: 10.1016/j.jacc.2018.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1547. Lai B, Li Z, He M, Wang Y, Chen L, Zhang J, Yang Y, Shyy JY. Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am J Physiol Heart Circ Physiol 315: H1293–H1303, 2018. doi: 10.1152/ajpheart.00213.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1548. Li F, Yan K, Wu L, Zheng Z, Du Y, Liu Z, Zhao L, Li W, Sheng Y, Ren L, Tang C, Zhu L. Single-cell RNA-seq reveals cellular heterogeneity of mouse carotid artery under disturbed flow. Cell Death Discov 7: 180, 2021. doi: 10.1038/s41420-021-00567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1549. Roebroek T, Vandenberg W, Sipieter F, Hugelier S, Stove C, Zhang J, Dedecker P. Simultaneous readout of multiple FRET pairs using photochromism. Nat Commun 12: 2005, 2021. doi: 10.1038/s41467-021-22043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1550. Wang N, Li X, Wang R, Ding Z. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnol J 16: e2100041, 2021. doi: 10.1002/biot.202100041. [DOI] [PubMed] [Google Scholar]
- 1551. Zollinger DR, Lingle SE, Sorg K, Beechem JM, Merritt CR. GeoMx RNA assay: high multiplex, digital, spatial analysis of RNA in FFPE tissue. Methods Mol Biol 2148: 331–345, 2020. doi: 10.1007/978-1-0716-0623-0_21. [DOI] [PubMed] [Google Scholar]
- 1552. Lee DY, Yang TL, Huang YH, Lee CI, Chen LJ, Shih YT, Wei SY, Wang WL, Wu CC, Chiu JJ. Induction of microRNA-10a using retinoic acid receptor-alpha and retinoid x receptor-alpha agonists inhibits atherosclerotic lesion formation. Atherosclerosis 271: 36–44, 2018. doi: 10.1016/j.atherosclerosis.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 1553. Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, Li YS, Chien S, Wang N. MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci USA 107: 3240–3244, 2010. doi: 10.1073/pnas.0914882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1554. Weber M, Baker MB, Moore JP, Searles CD. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun 393: 643–648, 2010. doi: 10.1016/j.bbrc.2010.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1555. Wang KC, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, Wang N, Shyy JY, Li YS, Chien S. Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc Natl Acad Sci USA 107: 3234–3239, 2010. doi: 10.1073/pnas.0914825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1556. Wang KC, Nguyen P, Weiss A, Yeh YT, Chien HS, Lee A, Teng D, Subramaniam S, Li YS, Chien S. MicroRNA-23b regulates cyclin-dependent kinase-activating kinase complex through cyclin H repression to modulate endothelial transcription and growth under flow. Arterioscler Thromb Vasc Biol 34: 1437–1445, 2014. doi: 10.1161/ATVBAHA.114.303473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1557. Demolli S, Doddaballapur A, Devraj K, Stark K, Manavski Y, Eckart A, Zehendner CM, Lucas T, Korff T, Hecker M, Massberg S, Liebner S, Kaluza D, Boon RA, Dimmeler S. Shear stress-regulated miR-27b controls pericyte recruitment by repressing SEMA6A and SEMA6D. Cardiovasc Res 113: 681–691, 2017. doi: 10.1093/cvr/cvx032. [DOI] [PubMed] [Google Scholar]
- 1558. Chen K, Fan W, Wang X, Ke X, Wu G, Hu C. MicroRNA-101 mediates the suppressive effect of laminar shear stress on mTOR expression in vascular endothelial cells. Biochem Biophys Res Commun 427: 138–142, 2012. doi: 10.1016/j.bbrc.2012.09.026. [DOI] [PubMed] [Google Scholar]
- 1559. Mondadori dos Santos A, Metzinger L, Haddad O, M’Baya-Moutoula E, Taïbi F, Charnaux N, Massy ZA, Hlawaty H, Metzinger-Le Meuth V. miR-126 is involved in vascular remodeling under laminar shear stress. Biomed Res Int 2015: 497280, 2015. doi: 10.1155/2015/497280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1560. Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, Li JY, Chien S. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci USA 108: 10355–10360, 2011. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1561. Ni CW, Qiu H, Jo H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am J Physiol Heart Circ Physiol 300: H1762–H1769, 2011. doi: 10.1152/ajpheart.00829.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1562. Huang TS, Wang KC, Quon S, Nguyen P, Chang TY, Chen Z, Li YS, Subramaniam S, Shyy J, Chien S. LINC00341 exerts an anti-inflammatory effect on endothelial cells by repressing VCAM1. Physiol Genomics 49: 339–345, 2017. doi: 10.1152/physiolgenomics.00132.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1563. Lyu Q, Xu S, Lyu Y, Choi M, Christie CK, Slivano OJ, Rahman A, Jin ZG, Long X, Xu Y, Miano JM. SENCR stabilizes vascular endothelial cell adherens junctions through interaction with CKAP4. Proc Natl Acad Sci USA 116: 546–555, 2019. doi: 10.1073/pnas.1810729116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1564. Miao Y, Ajami NE, Huang TS, Lin FM, Lou CH, Wang YT, Li S, Kang J, Munkacsi H, Maurya MR, Gupta S, Chien S, Subramaniam S, Chen Z. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun 9: 292, 2018. doi: 10.1038/s41467-017-02113-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1565. Man HS, Sukumar AN, Lam GC, Turgeon PJ, Yan MS, Ku KH, Dubinsky MK, Ho JJ, Wang JJ, Das S, Mitchell N, Oettgen P, Sefton MV, Marsden PA. Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc Natl Acad Sci USA 115: 2401–2406, 2018. doi: 10.1073/pnas.1715182115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1566. Leisegang MS, Bibli SI, Günther S, Pflüger-Müller B, Oo JA, Höper C, Seredinski S, Yekelchyk M, Schmitz-Rixen T, Schurmann C, Hu J, Looso M, Sigala F, Boon RA, Fleming I, Brandes RP. Pleiotropic effects of laminar flow and statins depend on the Kruppel-like factor-induced lncRNA MANTIS. Eur Heart J 40: 2523–2533, 2019. doi: 10.1093/eurheartj/ehz393. [DOI] [PubMed] [Google Scholar]
- 1567. Lu Q, Meng Q, Qi M, Li F, Liu B. Shear-sensitive lncRNA AF131217.1 inhibits inflammation in HUVECs via regulation of KLF4. Hypertension 73: e25–e34, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12476. [DOI] [PubMed] [Google Scholar]
- 1568. Jiang H, Ge Z, Zhang L, Yang Y, Zhai X, Chen Z, Wei Q. Long noncoding RNA AF131217.1 regulated coronary slow flow-induced inflammation affecting coronary slow flow via KLF4. Braz J Cardiovasc Surg 37: 525–533, 2022. doi: 10.21470/1678-9741-2020-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1569. da Silva RA, Ferreira MR, Gomes AM, Zambuzzi WF. LncRNA HOTAIR is a novel endothelial mechanosensitive gene. J Cell Physiol 235: 4631–4642, 2020. doi: 10.1002/jcp.29340. [DOI] [PubMed] [Google Scholar]
- 1570. Chang YJ, Li YS, Wu CC, Wang KC, Huang TC, Chen Z, Chien S. Extracellular microRNA-92a mediates endothelial cell-macrophage communication. Arterioscler Thromb Vasc Biol 39: 2492–2504, 2019. doi: 10.1161/ATVBAHA.119.312707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1571. Zhou J, Li YS, Nguyen P, Wang KC, Weiss A, Kuo YC, Chiu JJ, Shyy JY, Chien S. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: role of shear stress. Circ Res 113: 40–51, 2013. doi: 10.1161/CIRCRESAHA.113.280883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1572. Sun W, Julie Li YS, Huang HD, Shyy JY, Chien S. microRNA: a master regulator of cellular processes for bioengineering systems. Annu Rev Biomed Eng 12: 1–27, 2010. doi: 10.1146/annurev-bioeng-070909-105314. [DOI] [PubMed] [Google Scholar]
- 1573. Churov A, Summerhill V, Grechko A, Orekhova V, Orekhov A. MicroRNAs as potential biomarkers in atherosclerosis. Int J Mol Sci 20: 5547, 2019. doi: 10.3390/ijms20225547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1574. Lee DY, Lin TE, Lee CI, Zhou J, Huang YH, Lee PL, Shih YT, Chien S, Chiu JJ. MicroRNA-10a is crucial for endothelial response to different flow patterns via interaction of retinoid acid receptors and histone deacetylases. Proc Natl Acad Sci USA 114: 2072–2077, 2017. doi: 10.1073/pnas.1621425114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1575. Roberts TC, Morris KV, Weinberg MS. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 9: 13–20, 2014. doi: 10.4161/epi.26700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1576. Rinn JL, Chang HY. Long noncoding RNAs: molecular modalities to organismal functions. Annu Rev Biochem 89: 283–308, 2020. doi: 10.1146/annurev-biochem-062917-012708. [DOI] [PubMed] [Google Scholar]
- 1577. Fiedler J, Breckwoldt K, Remmele CW, Hartmann D, Dittrich M, Pfanne A, Just A, Xiao K, Kunz M, Müller T, Hansen A, Geffers R, Dandekar T, Eschenhagen T, Thum T. Development of long noncoding RNA-based strategies to modulate tissue vascularization. J Am Coll Cardiol 66: 2005–2015, 2015. doi: 10.1016/j.jacc.2015.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1578. Zhu R, Hu X, Xu W, Wu Z, Zhu Y, Ren Y, Cheng L. LncRNA MALAT1 inhibits hypoxia/reoxygenation-induced human umbilical vein endothelial cell injury via targeting the microRNA-320a/RAC1 axis. Biol Chem 401: 349–360, 2020. doi: 10.1515/hsz-2019-0316. [DOI] [PubMed] [Google Scholar]
- 1579. Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med 19: 1418–1425, 2015. doi: 10.1111/jcmm.12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1580. Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, Boon RA, Dimmeler S. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114: 1389–1397, 2014. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- 1581. Hofmann P, Sommer J, Theodorou K, Kirchhof L, Fischer A, Li Y, Perisic L, Hedin U, Maegdefessel L, Dimmeler S, Boon RA. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res 115: 230–242, 2019. doi: 10.1093/cvr/cvy206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1582. Wu Z, He Y, Li D, Fang X, Shang T, Zhang H, Zheng X. Long noncoding RNA MEG3 suppressed endothelial cell proliferation and migration through regulating miR-21. Am J Transl Res 9: 3326–3335, 2017. [PMC free article] [PubMed] [Google Scholar]
- 1583. Song J, Huang S, Wang K, Li W, Pao L, Chen F, Zhao X. Long non-coding RNA MEG3 attenuates the angiotensin II-induced injury of human umbilical vein endothelial cells by interacting with p53. Front Genet 10: 78, 2019. doi: 10.3389/fgene.2019.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1584. Ruan W, Zhao F, Zhao S, Zhang L, Shi L, Pang T. Knockdown of long noncoding RNA MEG3 impairs VEGF-stimulated endothelial sprouting angiogenesis via modulating VEGFR2 expression in human umbilical vein endothelial cells. Gene 649: 32–39, 2018. doi: 10.1016/j.gene.2018.01.072. [DOI] [PubMed] [Google Scholar]
- 1585. Boon RA, Hofmann P, Michalik KM, Lozano-Vidal N, Berghäuser D, Fischer A, Knau A, Jaé N, Schürmann C, Dimmeler S. Long noncoding RNA Meg3 controls endothelial cell aging and function: implications for regenerative angiogenesis. J Am Coll Cardiol 68: 2589–2591, 2016. doi: 10.1016/j.jacc.2016.09.949. [DOI] [PubMed] [Google Scholar]
- 1586. Josipovic I, Pflüger B, Fork C, Vasconez AE, Oo JA, Hitzel J, Seredinski S, Gamen E, Heringdorf DM, Chen W, Looso M, Pullamsetti SS, Brandes RP, Leisegang MS. Long noncoding RNA LISPR1 is required for S1P signaling and endothelial cell function. J Mol Cell Cardiol 116: 57–68, 2018. doi: 10.1016/j.yjmcc.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 1587. Wang D, Dai C, Zhang X, Gu C, Liu M, Liu H, Yang F, Wu H, Wang Y. Identification and functional analysis of long non-coding RNAs in human pulmonary microvascular endothelial cells subjected to cyclic stretch. Front Physiol 12: 655971, 2021. doi: 10.3389/fphys.2021.655971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1588. Huang Z, Winata WA, Zhang K, Zhao Y, Li Y, Zhou N, Zhou S, Fu W, Qiao B, Li G, Shao Y, Zheng J, Dong R. Reconstruction of a lncRNA-associated ceRNA network in endothelial cells under circumferential stress. Cardiol Res Pract 2020: 1481937, 2020., doi: 10.1155/2020/1481937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1589. Chen Y, Li Z, Chen X, Zhang S. Long non-coding RNAs: from disease code to drug role. Acta Pharm Sin B 11: 340–354, 2021. doi: 10.1016/j.apsb.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1590. Zhang L, Zhang Y, Zhao Y, Wang Y, Ding H, Xue S, Li P. Circulating miRNAs as biomarkers for early diagnosis of coronary artery disease. Expert Opin Ther Pat 28: 591–601, 2018. doi: 10.1080/13543776.2018.1503650. [DOI] [PubMed] [Google Scholar]
- 1591. Sun P, Tang LN, Li GZ, Xu ZL, Xu QH, Wang M, Li L. Effects of MiR-21 on the proliferation and migration of vascular smooth muscle cells in rats with atherosclerosis via the Akt/ERK signaling pathway. Eur Rev Med Pharmacol Sci 23: 2216–2222, 2019. doi: 10.26355/eurrev_201903_17269. [DOI] [PubMed] [Google Scholar]
- 1592. Huang S, Xu T, Huang X, Li S, Qin W, Chen W, Zhang Z. miR-21 regulates vascular smooth muscle cell function in arteriosclerosis obliterans of lower extremities through AKT and ERK1/2 pathways. Arch Med Sci 15: 1490–1497, 2019. doi: 10.5114/aoms.2018.78885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1593. Gao L, Zeng H, Zhang T, Mao C, Wang Y, Han Z, Chen K, Zhang J, Fan Y, Gu J, Wang C. MicroRNA-21 deficiency attenuated atherogenesis and decreased macrophage infiltration by targeting Dusp-8. Atherosclerosis 291: 78–86, 2019. doi: 10.1016/j.atherosclerosis.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 1594. Zhu JJ, Liu YF, Zhang YP, Zhao CR, Yao WJ, Li YS, Wang KC, Huang TS, Pang W, Wang XF, Wang X, Chien S, Zhou J. VAMP3 and SNAP23 mediate the disturbed flow-induced endothelial microRNA secretion and smooth muscle hyperplasia. Proc Natl Acad Sci USA 114: 8271–8276, 2017. doi: 10.1073/pnas.1700561114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1595. Chen Z, Li S, Subramaniam S, Shyy JY, Chien S. Epigenetic regulation: a new frontier for biomedical engineers. Annu Rev Biomed Eng 19: 195–219, 2017. doi: 10.1146/annurev-bioeng-071516-044720. [DOI] [PubMed] [Google Scholar]
- 1596. Zhao LY, Song J, Liu Y, Song CX, Yi C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 11: 792–808, 2020. doi: 10.1007/s13238-020-00733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1597. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20: 608–624, 2019. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 1598. Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, Fan Y, Jordan IK, Jo H. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest 124: 3187–3199, 2014. doi: 10.1172/JCI74792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1599. Zhang YP, Huang YT, Huang TS, Pang W, Zhu JJ, Liu YF, Tang RZ, Zhao CR, Yao WJ, Li YS, Chien S, Zhou J. The mammalian target of rapamycin and DNA methyltransferase 1 axis mediates vascular endothelial dysfunction in response to disturbed flow. Sci Rep 7: 14996, 2017. doi: 10.1038/s41598-017-15387-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1600. Wu L, Pei Y, Zhu Y, Jiang M, Wang C, Cui W, Zhang D. Association of N6-methyladenine DNA with plaque progression in atherosclerosis via myocardial infarction-associated transcripts. Cell Death Dis 10: 909, 2019. doi: 10.1038/s41419-019-2152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1601. Zhong X, Ma X, Zhang L, Li Y, Li Y, He R. MIAT promotes proliferation and hinders apoptosis by modulating miR-181b/STAT3 axis in ox-LDL-induced atherosclerosis cell models. Biomed Pharmacother 97: 1078–1085, 2018. doi: 10.1016/j.biopha.2017.11.052. [DOI] [PubMed] [Google Scholar]
- 1602. Sun G, Li Y, Ji Z. Up-regulation of MIAT aggravates the atherosclerotic damage in atherosclerosis mice through the activation of PI3K/Akt signaling pathway. Drug Deliv 26: 641–649, 2019. doi: 10.1080/10717544.2019.1628116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1603. Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat Rev Genet 22: 119–131, 2021. doi: 10.1038/s41576-020-00295-8. [DOI] [PubMed] [Google Scholar]
- 1604. Lee M, Kim B, Kim VN. Emerging roles of RNA modification: m6A and U-tail. Cell 158: 980–987, 2014. doi: 10.1016/j.cell.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 1605. Qin Y, Li L, Luo E, Hou J, Yan G, Wang D, Qiao Y, Tang C. Role of m6A RNA methylation in cardiovascular disease. Int J Mol Med 46: 1958–1972, 2020. doi: 10.3892/ijmm.2020.4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1606. Quiles-Jiménez A, Gregersen I, Mittelstedt Leal de Sousa M, Abbas A, Kong XY, Alseth I, Holm S, Dahl TB, Skagen K, Skjelland M, Aukrust P, Bjøras M, Halvorsen B. N6-methyladenosine in RNA of atherosclerotic plaques: an epitranscriptomic signature of human carotid atherosclerosis. Biochem Biophys Res Commun 533: 631–637, 2020. doi: 10.1016/j.bbrc.2020.09.057. [DOI] [PubMed] [Google Scholar]
- 1607. Chien CS, Li JY, Chien Y, Wang ML, Yarmishyn AA, Tsai PH, Juan CC, Nguyen P, Cheng HM, Huo TI, Chiou SH, Chien S. METTL3-dependent N6-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci USA 118: e2025070118, 2021.doi: 10.1073/pnas.2025070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1608. Wade PA. Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene 20: 3166–3173, 2001. doi: 10.1038/sj.onc.1204340. [DOI] [PubMed] [Google Scholar]
- 1609. Chen W, Bacanamwo M, Harrison DG. Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J Biol Chem 283: 16293–16298, 2008. doi: 10.1074/jbc.M801803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1610. Aurora AB, Biyashev D, Mirochnik Y, Zaichuk TA, Sánchez-Martinez C, Renault MA, Losordo D, Volpert OV. NF-kappaB balances vascular regression and angiogenesis via chromatin remodeling and NFAT displacement. Blood 116: 475–484, 2010. doi: 10.1182/blood-2009-07-232132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1611. Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115: 2971–2979, 2010. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1612. Lee DY, Lee CI, Lin TE, Lim SH, Zhou J, Tseng YC, Chien S, Chiu JJ. Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc Natl Acad Sci USA 109: 1967–1972, 2012. doi: 10.1073/pnas.1121214109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1613. Nakato R, Wada Y, Nakaki R, Nagae G, Katou Y, Tsutsumi S, et al. Comprehensive epigenome characterization reveals diverse transcriptional regulation across human vascular endothelial cells. Epigenetics Chromatin 12: 77, 2019. doi: 10.1186/s13072-019-0319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1614. Stephens AD. Chromatin rigidity provides mechanical and genome protection. Mutat Res 821: 111712, 2020. doi: 10.1016/j.mrfmmm.2020.111712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1615. Izzard AS, Heagerty AM. Hypertension and the vasculature: arterioles and the myogenic response. J Hypertens 13: 1–4, 1995. [PubMed] [Google Scholar]
- 1616. Heagerty AM, Izzard AS. Small-artery changes in hypertension. J Hypertens 13: 1560–1565, 1995. [PubMed] [Google Scholar]
- 1617. Osol G, Halpern W. Myogenic properties of cerebral blood vessels from normotensive and hypertensive rats. Am J Physiol Heart Circ Physiol 249: H914–H921, 1985. doi: 10.1152/ajpheart.1985.249.5.H914. [DOI] [PubMed] [Google Scholar]
- 1618. Bakker EN, Sorop O, Spaan JA, VanBavel E. Remodeling of resistance arteries in organoid culture is modulated by pressure and pressure pulsation and depends on vasomotion. Am J Physiol Heart Circ Physiol 286: H2052–H2056, 2004. doi: 10.1152/ajpheart.00978.2003. [DOI] [PubMed] [Google Scholar]
- 1619. Hayashi K, Epstein M, Loutzenhiser R. Enhanced myogenic responsiveness of renal interlobular arteries in spontaneously hypertensive rats. Hypertension 19: 153–160, 1992. doi: 10.1161/01.HYP.19.2.153. [DOI] [PubMed] [Google Scholar]
- 1620. Ren Y, D’Ambrosio MA, Liu R, Pagano PJ, Garvin JL, Carretero OA. Enhanced myogenic response in the afferent arteriole of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 298: H1769–1775, 2010. doi: 10.1152/ajpheart.00537.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1621. Xu N, Jiang S, Persson PB, Persson EA, Lai EY, Patzak A. Reactive oxygen species in renal vascular function. Acta Physiol (Oxf) 229: e13477, 2020. doi: 10.1111/apha.13477. [DOI] [PubMed] [Google Scholar]
- 1622. Harder DR, Smeda J, Lombard J. Enhanced myogenic depolarization in hypertensive cerebral arterial muscle. Circ Res 57: 319–322, 1985. doi: 10.1161/01.RES.57.2.319. [DOI] [PubMed] [Google Scholar]
- 1623. Ahn DS, Choi SK, Kim YH, Cho YE, Shin HM, Morgan KG, Lee YH. Enhanced stretch-induced myogenic tone in the basilar artery of spontaneously hypertensive rats. J Vasc Res 44: 182–191, 2007. doi: 10.1159/000100374. [DOI] [PubMed] [Google Scholar]
- 1624. Izzard AS, Bund SJ, Heagerty AM. Myogenic tone in mesenteric arteries from spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 270: H1–H6, 1996. doi: 10.1152/ajpheart.1996.270.1.H1. [DOI] [PubMed] [Google Scholar]
- 1625. Hughes JM, Bund SJ. Influence of experimental reduction of media/lumen ratio on arterial myogenic properties of spontaneously hypertensive and Wistar-Kyoto rats. Clin Sci (Lond) 106: 163–171, 2004. doi: 10.1042/CS20030225. [DOI] [PubMed] [Google Scholar]
- 1626. Hughes JM, Bund SJ. Arterial myogenic properties of the spontaneously hypertensive rat. Exp Physiol 87: 527–534, 2002. doi: 10.1113/eph8702399. [DOI] [PubMed] [Google Scholar]
- 1627. Bund SJ. Contractility of resistance arteries of spontaneously hypertensive rats related to their media: lumen ratio. J Hypertens 18: 1223–1231, 2000. doi: 10.1097/00004872-200018090-00008. [DOI] [PubMed] [Google Scholar]
- 1628. Bund SJ. Spontaneously hypertensive rat resistance artery structure related to myogenic and mechanical properties. Clin Sci (Lond) 101: 385–393, 2001. doi: 10.1042/CS20010036. [DOI] [PubMed] [Google Scholar]
- 1629. Takenaka T, Forster H, De Micheli A, Epstein M. Impaired myogenic responsiveness of renal microvessels in Dahl salt-sensitive rats. Circ Res 71: 471–480, 1992. doi: 10.1161/01.res.71.2.471. [DOI] [PubMed] [Google Scholar]
- 1630. Fan F, Geurts AM, Murphy SR, Pabbidi MR, Jacob HJ, Roman RJ. Impaired myogenic response and autoregulation of cerebral blood flow is rescued in CYP4A1 transgenic Dahl salt-sensitive rat. Am J Physiol Regul Integr Comp Physiol 308: R379–R390, 2015. doi: 10.1152/ajpregu.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1631. Ge Y, Murphy SR, Fan F, Williams JM, Falck JR, Liu R, Roman RJ. Role of 20-HETE in the impaired myogenic and TGF responses of the Af-Art of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 307: F509–F515, 2014. doi: 10.1152/ajprenal.00273.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1632. Bohlen HG. The microcirculation in hypertension. J Hypertens Suppl 7: S117–S124, 1989. [PubMed] [Google Scholar]
- 1633. Schiffrin EL. How structure, mechanics, and function of the vasculature contribute to blood pressure elevation in hypertension. Can J Cardiol 36: 648–658, 2020. doi: 10.1016/j.cjca.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 1634. Feihl F, Liaudet L, Levy BI, Waeber B. Hypertension and microvascular remodelling. Cardiovasc Res 78: 274–285, 2008. doi: 10.1093/cvr/cvn022. [DOI] [PubMed] [Google Scholar]
- 1635. Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, Montezano AC. Vascular smooth muscle contraction in hypertension. Cardiovasc Res 114: 529–539, 2018. doi: 10.1093/cvr/cvy023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1636. Mulvany MJ. Resistance vessel growth and remodelling: cause or consequence in cardiovascular disease. J Hum Hypertens 9: 479–485, 1995. [PubMed] [Google Scholar]
- 1637. Folkow B. Early structural changes in hypertension: pathophysiology and clinical consequences. J Cardiovasc Pharmacol 22, Suppl 1: S1–S6, 1993. [PubMed] [Google Scholar]
- 1638. Folkow B. Hypertensive structural changes in systemic precapillary resistance vessels: how important are they for in vivo haemodynamics? J Hypertens 13: 1546–1559, 1995. [PubMed] [Google Scholar]
- 1639. Fan L, Gao W, Nguyen BV, Jefferson JR, Liu Y, Fan F, Roman RJ. Impaired renal hemodynamics and glomerular hyperfiltration contribute to hypertension-induced renal injury. Am J Physiol Renal Physiol 319: F624–F635, 2020. doi: 10.1152/ajprenal.00239.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1640. Loutzenhiser R, Griffin K, Williamson G, Bidani A. Renal autoregulation: new perspectives regarding the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol 290: R1153–R1167, 2006. doi: 10.1152/ajpregu.00402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1641. Bidani AK, Griffin KA, Williamson G, Wang X, Loutzenhiser R. Protective importance of the myogenic response in the renal circulation. Hypertension 54: 393–398, 2009. doi: 10.1161/HYPERTENSIONAHA.109.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1642. Burke M, Pabbidi MR, Farley J, Roman RJ. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol 12: 845–858, 2014. doi: 10.2174/15701611113116660149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1643. Rossitto G, Mary S, Chen JY, Boder P, Chew KS, Neves KB, Alves RL, Montezano AC, Welsh P, Petrie MC, Graham D, Touyz RM, Delles C. Tissue sodium excess is not hypertonic and reflects extracellular volume expansion. Nat Commun 11: 4222, 2020. doi: 10.1038/s41467-020-17820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1644. Shekhar S, Liu R, Travis OK, Roman RJ, Fan F. Cerebral autoregulation in hypertension and ischemic stroke: a mini review. J Pharm Sci Exp Pharmacol 2017: 21–27, 2017. [PMC free article] [PubMed] [Google Scholar]
- 1645. Tecilazich F, Feke GT, Mazzantini S, Sobrin L, Lorenzi M. Defective myogenic response of retinal vessels is associated with accelerated onset of retinopathy in type 1 diabetic individuals. Invest Ophthalmol Vis Sci 57: 1523–1529, 2016. doi: 10.1167/iovs.15-18356. [DOI] [PubMed] [Google Scholar]
- 1646. Holmberg J, Bhattachariya A, Alajbegovic A, Rippe C, Ekman M, Dahan D, Hien TT, Boettger T, Braun T, Swärd K, Hellstrand P, Albinsson S. Loss of vascular myogenic tone in miR-143/145 knockout mice is associated with hypertension-induced vascular lesions in small mesenteric arteries. Arterioscler Thromb Vasc Biol 38: 414–424, 2018. doi: 10.1161/ATVBAHA.117.310499. [DOI] [PubMed] [Google Scholar]
- 1647. Boudier HA. Arteriolar and capillary remodelling in hypertension. Drugs 58: 37–40, 1999. [PubMed] [Google Scholar]
- 1648. Prewitt RL, Chen II, Dowell RF. Microvascular alterations in the one-kidney, one-clip renal hypertensive rat. Am J Physiol 246: H728–H732, 1984. doi: 10.1152/ajpheart.1984.246.5.H728. [DOI] [PubMed] [Google Scholar]
- 1649. Hashimoto H, Prewitt RL, Efaw CW. Alterations in the microvasculature of one-kidney, one-clip hypertensive rats. Am J Physiol Heart Circ Physiol 253: H933–H940, 1987. doi: 10.1152/ajpheart.1987.253.4.H933. [DOI] [PubMed] [Google Scholar]
- 1650. Hutchins PM. Arteriolar rarefaction in hypertension. Bibl Anat 18: 166–168, 1979. [PubMed] [Google Scholar]
- 1651. Hayashi K, Epstein M, Saruta T. Altered myogenic responsiveness of the renal microvasculature in experimental hypertension. J Hypertens 14: 1387–1401, 1996. doi: 10.1097/00004872-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 1652. Carlström M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev 95: 405–511, 2015. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1653. Shibuya J, Ohyanagi M, Iwasaki T. Enhanced myogenic response in resistance small arteries from spontaneously hypertensive rats: relationship to the voltage-dependent calcium channel. Am J Hypertens 11: 767–773, 1998. doi: 10.1016/S0895-7061(98)00055-7. [DOI] [PubMed] [Google Scholar]
- 1654. Matsuda K, Lozinskaya I, Cox RH. Augmented contributions of voltage-gated Ca2+ channels to contractile responses in spontaneously hypertensive rat mesenteric arteries. Am J Hypertens 10: 1231–1239, 1997. doi: 10.1016/s0895-7061(97)00225-2. [DOI] [PubMed] [Google Scholar]
- 1655. Zhou Y, Fan J, Zhu H, Ji L, Fan W, Kapoor I, Wang Y, Wang Y, Zhu G, Wang J. Aberrant splicing induced by dysregulated Rbfox2 produces enhanced function of CaV1.2 calcium channel and vascular myogenic tone in hypertension. Hypertension 70: 1183–1192, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09301. [DOI] [PubMed] [Google Scholar]
- 1656. Cox RH, Fromme S. Expression of calcium channel subunit variants in small mesenteric arteries of WKY and SHR. Am J Hypertens 28: 1229–1239, 2015. doi: 10.1093/ajh/hpv024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1657. Toth P, Csiszar A, Tucsek Z, Sosnowska D, Gautam T, Koller A, Schwartzman ML, Sonntag WE, Ungvari Z. Role of 20-HETE, TRPC channels, and BKCa in dysregulation of pressure-induced Ca2+ signaling and myogenic constriction of cerebral arteries in aged hypertensive mice. Am J Physiol Heart Circ Physiol 305: H1698–H1708, 2013. doi: 10.1152/ajpheart.00377.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1658. Bazan E, Campbell AK, Rapoport RM. Protein kinase C activity in blood vessels from normotensive and spontaneously hypertensive rats. Eur J Pharmacol 227: 343–348, 1992. doi: 10.1016/0922-4106(92)90014-m. [DOI] [PubMed] [Google Scholar]
- 1659. Szarka N, Amrein K, Horvath P, Ivic I, Czeiter E, Buki A, Koller A, Toth P. Hypertension-induced enhanced myogenic constriction of cerebral arteries is preserved after traumatic brain injury. J Neurotrauma 34: 2315–2319, 2017. doi: 10.1089/neu.2016.4962. [DOI] [PubMed] [Google Scholar]
- 1660. Eid AH, El-Yazbi AF, Zouein F, Arredouani A, Ouhtit A, Rahman MM, Zayed H, Pintus G, Abou-Saleh H. Inositol 1,4,5-trisphosphate receptors in hypertension. Front Physiol 9: 1018, 2018. doi: 10.3389/fphys.2018.01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1661. Matchkov VV, Tarasova OS, Mulvany MJ, Nilsson H. Myogenic response of rat femoral small arteries in relation to wall structure and [Ca2+]. Am J Physiol Heart Circ Physiol 283: H118–H125, 2002. doi: 10.1152/ajpheart.00690.2001. [DOI] [PubMed] [Google Scholar]
- 1662. Vogel PA, Yang X, Moss NG, Arendshorst WJ. Superoxide enhances Ca2+ entry through L-type channels in the renal afferent arteriole. Hypertension 66: 374–381, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1663. Huang A, Koller A. Endothelin and prostaglandin H2 enhance arteriolar myogenic tone in hypertension. Hypertension 30: 1210–1215, 1997. doi: 10.1161/01.hyp.30.5.1210. [DOI] [PubMed] [Google Scholar]
- 1664. Falcone JC, Meininger GA. Endothelin mediates a component of the enhanced myogenic responsiveness of arterioles from hypertensive rats. Microcirculation 6: 305–313, 1999. [PubMed] [Google Scholar]
- 1665. Giachini FR, Lima VV, Filgueira FP, Dorrance AM, Carvalho MH, Fortes ZB, Webb RC, Tostes RC. STIM1/Orai1 contributes to sex differences in vascular responses to calcium in spontaneously hypertensive rats. Clin Sci (Lond) 122: 215–226, 2012. doi: 10.1042/CS20110312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1666. Dharmashankar K, Widlansky ME. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep 12: 448–455, 2010. doi: 10.1007/s11906-010-0150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1667. Konukoglu D, Uzun H. Endothelial dysfunction and hypertension. Adv Exp Med Biol 956: 511–540, 2017. doi: 10.1007/5584_2016_90. [DOI] [PubMed] [Google Scholar]
- 1668. Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, Elijovich F, Laffer CL, Gnecco JS, Noonan J, Maffia P, Jasiewicz-Honkisz B, Czesnikiewicz-Guzik M, Mikolajczyk T, Sliwa T, Dikalov S, Weyand CM, Guzik TJ, Harrison DG. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res 114: 1547–1563, 2018. doi: 10.1093/cvr/cvy112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1669. Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol 157: 527–536, 2009. doi: 10.1111/j.1476-5381.2009.00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1670. Goto K, Ohtsubo T, Kitazono T. Endothelium-dependent hyperpolarization (EDH) in hypertension: the role of endothelial ion channels. Int J Mol Sci 19: 315, 2018. doi: 10.3390/ijms19010315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1671. Magen E, Feldman A, Cohen Z, Alon DB, Linov L, Mishal J, Schlezinger M. Potential link between C3a, C3b and endothelial progenitor cells in resistant hypertension. Am J Med Sci 339: 415–419, 2010. doi: 10.1097/MAJ.0b013e3181d7d496. [DOI] [PubMed] [Google Scholar]
- 1672. Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension 54: 619–624, 2009. doi: 10.1161/HYPERTENSIONAHA.109.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1673. Giannotti G, Doerries C, Mocharla PS, Mueller MF, Bahlmann FH, Horvàth T, Jiang H, Sorrentino SA, Steenken N, Manes C, Marzilli M, Rudolph KL, Lüscher TF, Drexler H, Landmesser U. Impaired endothelial repair capacity of early endothelial progenitor cells in prehypertension: relation to endothelial dysfunction. Hypertension 55: 1389–1397, 2010. doi: 10.1161/HYPERTENSIONAHA.109.141614. [DOI] [PubMed] [Google Scholar]
- 1674. Hage FG, Oparil S, Xing D, Chen YF, McCrory MA, Szalai AJ. C-reactive protein-mediated vascular injury requires complement. Arterioscler Thromb Vasc Biol 30: 1189–1195, 2010. doi: 10.1161/ATVBAHA.110.205377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1675. Benjamin EJ, Larson MG, Keyes MJ, Mitchell GF, Vasan RS, Keaney JF Jr, Lehman BT, Fan S, Osypiuk E, Vita JA. Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study. Circulation 109: 613–619, 2004. doi: 10.1161/01.CIR.0000112565.60887.1E. [DOI] [PubMed] [Google Scholar]
- 1676. Mulvany MJ. Structure and function of small arteries in hypertension. J Hypertens Suppl 8: S225–S232, 1990. [PubMed] [Google Scholar]
- 1677. Renna NF, de Las Heras N, Miatello RM. Pathophysiology of vascular remodeling in hypertension. Int J Hypertens 2013: 808353, 2013. doi: 10.1155/2013/808353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1678. Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension 21: 391–397, 1993. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 1679. Bohr DF, Dominiczak AF, Webb RC. Pathophysiology of the vasculature in hypertension. Hypertension 18: III69–III75, 1991. doi: 10.1161/01.hyp.18.5_suppl.iii69. [DOI] [PubMed] [Google Scholar]
- 1680. Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL, Heistad DD. Vascular remodeling. Hypertension 28: 505–506, 1996. [PubMed] [Google Scholar]
- 1681. Martinez-Lemus LA, Hill MA, Meininger GA. The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology (Bethesda) 24: 45–57, 2009. doi: 10.1152/physiol.00029.2008. [DOI] [PubMed] [Google Scholar]
- 1682. Brown IA, Diederich L, Good ME, DeLalio LJ, Murphy SA, Cortese-Krott MM, Hall JL, Le TH, Isakson BE. Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arterioscler Thromb Vasc Biol 38: 1969–1985, 2018. doi: 10.1161/ATVBAHA.118.311229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1683. Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension 13: 968–972, 1989. doi: 10.1161/01.hyp.13.6.968. [DOI] [PubMed] [Google Scholar]
- 1684. Baumbach GL, Hajdu MA. Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension 21: 816–826, 1993. doi: 10.1161/01.hyp.21.6.816. [DOI] [PubMed] [Google Scholar]
- 1685. Li JS, Schiffrin EL. Effect of calcium channel blockade or angiotensin-converting enzyme inhibition on structure of coronary, renal, and other small arteries in spontaneously hypertensive rats. J Cardiovasc Pharmacol 28: 68–74, 1996. doi: 10.1097/00005344-199607000-00011. [DOI] [PubMed] [Google Scholar]
- 1686. Girardot D, Demeilliers B, deBlois D, Moreau P. ERK1/2-mediated vasoconstriction normalizes wall stress in small mesenteric arteries during NOS inhibition in vivo. J Cardiovasc Pharmacol 42: 339–347, 2003. doi: 10.1097/00005344-200309000-00004. [DOI] [PubMed] [Google Scholar]
- 1687. Pistea A, Bakker EN, Spaan JA, Hardeman MR, van Rooijen N, VanBavel E. Small artery remodeling and erythrocyte deformability in L-NAME-induced hypertension: role of transglutaminases. J Vasc Res 45: 10–18, 2008. doi: 10.1159/000109073. [DOI] [PubMed] [Google Scholar]
- 1688. Langille L, O’Donnell F. Reduction in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 231: 405–407, 1986. doi: 10.1126/science.3941904. [DOI] [PubMed] [Google Scholar]
- 1689. Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res 96: 119–126, 2005. doi: 10.1161/01.RES.0000151333.56089.66. [DOI] [PubMed] [Google Scholar]
- 1690. Buus CL, Pourageaud F, Fazzi GE, Janssen G, Mulvany MJ, De Mey JG. Smooth muscle cell changes during flow-related remodeling of rat mesenteric resistance arteries. Circ Res 89: 180–186, 2001. doi: 10.1161/hh1401.093575. [DOI] [PubMed] [Google Scholar]
- 1691. Ceiler DL, De Mey JG. Chronic NG-nitro-L-arginine methyl ester treatment does not prevent flow-induced remodeling in mesenteric feed arteries and arcading arterioles. Arterioscler Thromb Vasc Biol 20: 2057–2063, 2000. doi: 10.1161/01.ATV.20.9.2057. [DOI] [PubMed] [Google Scholar]
- 1692. Dumont O, Loufrani L, Henrion D. Key role of the NO-pathway and matrix metalloprotease-9 in high blood flow-induced remodeling of rat resistance arteries. Arterioscler Thromb Vasc Biol 27: 317–324, 2007. doi: 10.1161/01.ATV.0000254684.80662.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1693. Ledingham JM, Phelan EL, Cross MA, Laverty R. Prevention of increases in blood pressure and left ventricular mass and remodeling of resistance arteries in young New Zealand genetically hypertensive rats: the effects of chronic treatment with valsartan, enalapril and felodipine. J Vasc Res 37: 134–145, 2000. doi: 10.1159/000025724. [DOI] [PubMed] [Google Scholar]
- 1694. Pistea A, Bakker EN, Spaan JA, VanBavel E. Flow inhibits inward remodeling in cannulated porcine small coronary arteries. Am J Physiol Heart Circ Physiol 289: H2632–H2640, 2005. doi: 10.1152/ajpheart.00205.2005. [DOI] [PubMed] [Google Scholar]
- 1695. Heerkens EH, Izzard AS, Heagerty AM. Integrins, vascular remodeling, and hypertension. Hypertension 49: 1–4, 2007. doi: 10.1161/01.HYP.0000252753.63224.3b. [DOI] [PubMed] [Google Scholar]
- 1696. Bakker EN, van der Meulen ET, van den Berg BM, Everts V, Spaan JA, VanBavel E. Inward remodeling follows chronic vasoconstriction in isolated resistance arteries. J Vasc Res 39: 12–20, 2002. doi: 10.1159/000048989. [DOI] [PubMed] [Google Scholar]
- 1697. Jacobsen JC, Mulvany MJ, Holstein-Rathlou NH. A mechanism for arteriolar remodeling based on maintenance of smooth muscle cell activation. Am J Physiol Regul Integr Comp Physiol 294: R1379–R1389, 2008. doi: 10.1152/ajpregu.00407.2007. [DOI] [PubMed] [Google Scholar]
- 1698. van den Akker J, Schoorl MJ, Bakker EN, Vanbavel E. Small artery remodeling: current concepts and questions. J Vasc Res 47: 183–202, 2010. doi: 10.1159/000255962. [DOI] [PubMed] [Google Scholar]
- 1699. Sonoyama K, Greenstein A, Price A, Khavandi K, Heagerty T. Vascular remodeling: implications for small artery function and target organ damage. Ther Adv Cardiovasc Dis 1: 129–137, 2007. doi: 10.1177/1753944707086358. [DOI] [PubMed] [Google Scholar]
- 1700. Bakker EN, Matlung HL, Bonta P, de Vries CJ, van Rooijen N, Vanbavel E. Blood flow-dependent arterial remodelling is facilitated by inflammation but directed by vascular tone. Cardiovasc Res 78: 341–348, 2008. doi: 10.1093/cvr/cvn050. [DOI] [PubMed] [Google Scholar]
- 1701. Tuna BG, Bakker EN, VanBavel E. Relation between active and passive biomechanics of small mesenteric arteries during remodeling. J Biomech 46: 1420–1426, 2013. doi: 10.1016/j.jbiomech.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 1702. Tuna BG, Bakker EN, VanBavel E. Smooth muscle biomechanics and plasticity: relevance for vascular calibre and remodelling. Basic Clin Pharmacol Toxicol 110: 35–41, 2012. doi: 10.1111/j.1742-7843.2011.00794.x. [DOI] [PubMed] [Google Scholar]
- 1703. Gruionu G, Hoying JB, Pries AR, Secomb TW. Structural remodeling of the mouse gracilis artery: coordinated changes in diameter and medial area maintain circumferential stress. Microcirculation 19: 610–618, 2012. doi: 10.1111/j.1549-8719.2012.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1704. Izzard AS, Rizzoni D, Agabiti-Rosei E, Heagerty AM. Small artery structure and hypertension: adaptive changes and target organ damage. J Hypertens 23: 247–250, 2005. doi: 10.1097/00004872-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 1705. Martinez-Lemus LA, Hill MA, Bolz SS, Pohl U, Meininger GA. Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: implications for functional remodeling. FASEB J 18: 708–710, 2004. doi: 10.1096/fj.03-0634fje. [DOI] [PubMed] [Google Scholar]
- 1706. Foote CA, Castorena-Gonzalez JA, Staiculescu MC, Clifford PS, Hill MA, Meininger GA, Martinez-Lemus LA. Brief serotonin exposure initiates arteriolar inward remodeling processes in vivo that involve transglutaminase activation and actin cytoskeleton reorganization. Am J Physiol Heart Circ Physiol 310: H188–H198, 2016. doi: 10.1152/ajpheart.00666.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1707. Pries AR, Secomb TW. Making microvascular networks work: angiogenesis, remodeling, and pruning. Physiology (Bethesda) 29: 446–455, 2014. doi: 10.1152/physiol.00012.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1708. Pries AR, Reglin B, Secomb TW. Remodeling of blood vessels: responses of diameter and wall thickness to hemodynamic and metabolic stimuli. Hypertension 46: 725–731, 2005. doi: 10.1161/01.HYP.0000184428.16429.be. [DOI] [PubMed] [Google Scholar]
- 1709. Cox RH. Arterial wall mechanics and composition and the effects of smooth muscle activation. Am J Physiol 229: 807–812, 1975. doi: 10.1152/ajplegacy.1975.229.3.807. [DOI] [PubMed] [Google Scholar]
- 1710. Dobrin PB. Mechanical properties of arteries. Physiol Rev 58: 397–460, 1978. doi: 10.1152/physrev.1978.58.2.397. [DOI] [PubMed] [Google Scholar]
- 1711. Gunst SJ, Wu MF, Smith DD. Contraction history modulates isotonic shortening velocity in smooth muscle. Am J Physiol Cell Physiol 265: C467–C476, 1993. doi: 10.1152/ajpcell.1993.265.2.C467. [DOI] [PubMed] [Google Scholar]
- 1712. Gunst SJ. Effect of length history on contractile behavior of canine tracheal smooth muscle. Am J Physiol Cell Physiol 250: C146–C154, 1986. doi: 10.1152/ajpcell.1986.250.1.C146. [DOI] [PubMed] [Google Scholar]
- 1713. Gunst SJ. Contractile force of canine airway smooth muscle during cyclical length changes. J Appl Physiol Respir Environ Exerc Physiol 55: 759–769, 1983. doi: 10.1152/jappl.1983.55.3.759. [DOI] [PubMed] [Google Scholar]
- 1714. Gunst SJ, Russell JA. Contractile force of canine tracheal smooth muscle during continuous stretch. J Appl Physiol Respir Environ Exerc Physiol 52: 655–663, 1982. doi: 10.1152/jappl.1982.52.3.655. [DOI] [PubMed] [Google Scholar]
- 1715. Gunst SJ, Meiss RA, Wu MF, Rowe M. Mechanisms for the mechanical plasticity of tracheal smooth muscle. Am J Physiol Cell Physiol 268: C1267–C1276, 1995. doi: 10.1152/ajpcell.1995.268.5.C1267. [DOI] [PubMed] [Google Scholar]
- 1716. Gunst SJ, Wu MF. Selected contribution: plasticity of airway smooth muscle stiffness and extensibility: role of length-adaptive mechanisms. J Appl Physiol (1985) 90: 741–749, 2001. doi: 10.1152/jappl.2001.90.2.741. [DOI] [PubMed] [Google Scholar]
- 1717. Seow CY. Response of arterial smooth muscle to length perturbation. J Appl Physiol (1985) 89: 2065–2072, 2000. doi: 10.1152/jappl.2000.89.5.2065. [DOI] [PubMed] [Google Scholar]
- 1718. Cox RH. Mechanics of canine iliac artery smooth muscle in vitro. Am J Physiol 230: 462–470, 1976. doi: 10.1152/ajplegacy.1976.230.2.462. [DOI] [PubMed] [Google Scholar]
- 1719. Löfgren M, Fagher K, Wede OK, Arner A. Decreased shortening velocity and altered myosin isoforms in guinea-pig hypertrophic intestinal smooth muscle. J Physiol 544: 707–714, 2002. doi: 10.1113/jphysiol.2002.027060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1720. Almasri AM, Ratz PH, Speich JE. Length adaptation of the passive-to-active tension ratio in rabbit detrusor. Ann Biomed Eng 38: 2594–2605, 2010. doi: 10.1007/s10439-010-0021-7. [DOI] [PubMed] [Google Scholar]
- 1721. Speich JE, Almasri AM, Bhatia H, Klausner AP, Ratz PH. Adaptation of the length-active tension relationship in rabbit detrusor. Am J Physiol Renal Physiol 297: F1119–F1128, 2009. doi: 10.1152/ajprenal.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1722. Wang L, Paré PD, Seow CY. Selected contribution: effect of chronic passive length change on airway smooth muscle length-tension relationship. J Appl Physiol (1985) 90: 734–740, 2001. doi: 10.1152/jappl.2001.90.2.734. [DOI] [PubMed] [Google Scholar]
- 1723. Hill MA, Potocnik SJ, Martinez-Lemus LA, Meininger GA. Delayed arteriolar relaxation after prolonged agonist exposure: functional remodeling involving tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 285: H849–H856, 2003. doi: 10.1152/ajpheart.00986.2002. [DOI] [PubMed] [Google Scholar]
- 1724. Hong Z, Sun Z, Li Z, Mesquitta WT, Trzeciakowski JP, Meininger GA. Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc Res 96: 73–80, 2012. doi: 10.1093/cvr/cvs239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1725. Heerkens EH, Shaw L, Ryding A, Brooker G, Mullins JJ, Austin C, Ohanian V, Heagerty AM. alphaV integrins are necessary for eutrophic inward remodeling of small arteries in hypertension. Hypertension 47: 281–287, 2006. doi: 10.1161/01.HYP.0000198428.45132.02. [DOI] [PubMed] [Google Scholar]
- 1726. Kuo KH, Wang L, Paré PD, Ford LE, Seow CY. Myosin thick filament lability induced by mechanical strain in airway smooth muscle. J Appl Physiol (1985) 90: 1811–1816, 2001. doi: 10.1152/jappl.2001.90.5.1811. [DOI] [PubMed] [Google Scholar]
- 1727. Seow CY. Myosin filament assembly in an ever-changing myofilament lattice of smooth muscle. Am J Physiol Cell Physiol 289: C1363–C1368, 2005. doi: 10.1152/ajpcell.00329.2005. [DOI] [PubMed] [Google Scholar]
- 1728. Kuo KH, Herrera AM, Wang L, Paré PD, Ford LE, Stephens NL, Seow CY. Structure-function correlation in airway smooth muscle adapted to different lengths. Am J Physiol Cell Physiol 285: C384–C390, 2003. doi: 10.1152/ajpcell.00095.2003. [DOI] [PubMed] [Google Scholar]
- 1729. Smolensky AV, Ford LE. The extensive length-force relationship of porcine airway smooth muscle. J Appl Physiol (1985) 102: 1906–1911, 2007. doi: 10.1152/japplphysiol.01169.2006. [DOI] [PubMed] [Google Scholar]
- 1730. Pratusevich VR, Seow CY, Ford LE. Plasticity in canine airway smooth muscle. J Gen Physiol 105: 73–94, 1995. doi: 10.1085/jgp.105.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1731. Bossé Y, Sobieszek A, Paré PD, Seow CY. Length adaptation of airway smooth muscle. Proc Am Thorac Soc 5: 62–67, 2008. doi: 10.1513/pats.200705-056VS. [DOI] [PubMed] [Google Scholar]
- 1732. Wang R, Li Q, Tang DD. Role of vimentin in smooth muscle force development. Am J Physiol Cell Physiol 291: C483–C489, 2006. doi: 10.1152/ajpcell.00097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1733. Wede OK, Löfgren M, Li Z, Paulin D, Arner A. Mechanical function of intermediate filaments in arteries of different size examined using desmin deficient mice. J Physiol 540: 941–949, 2002. doi: 10.1113/jphysiol.2001.014910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1734. Loufrani L, Matrougui K, Li Z, Levy BI, Lacolley P, Paulin D, Henrion D. Selective microvascular dysfunction in mice lacking the gene encoding for desmin. FASEB J 16: 117–119, 2002. doi: 10.1096/fj.01-0505fje. [DOI] [PubMed] [Google Scholar]
- 1735. Tang DD, Bai Y, Gunst SJ. Silencing of p21-activated kinase attenuates vimentin phosphorylation on Ser-56 and reorientation of the vimentin network during stimulation of smooth muscle cells by 5-hydroxytryptamine. Biochem J 388: 773–783, 2005. doi: 10.1042/BJ20050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1736. Li QF, Spinelli AM, Wang R, Anfinogenova Y, Singer HA, Tang DD. Critical role of vimentin phosphorylation at Ser-56 by p21-activated kinase in vimentin cytoskeleton signaling. J Biol Chem 281: 34716–34724, 2006. doi: 10.1074/jbc.M607715200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1737. Wang R, Li QF, Anfinogenova Y, Tang DD. Dissociation of Crk-associated substrate from the vimentin network is regulated by p21-activated kinase on ACh activation of airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 292: L240–L248, 2007. doi: 10.1152/ajplung.00199.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1738. Goto H, Tanabe K, Manser E, Lim L, Yasui Y, Inagaki M. Phosphorylation and reorganization of vimentin by p21-activated kinase (PAK). Genes Cells 7: 91–97, 2002. doi: 10.1046/j.1356-9597.2001.00504.x. [DOI] [PubMed] [Google Scholar]
- 1739. Gunst SJ, Tang DD, Opazo Saez A. Cytoskeletal remodeling of the airway smooth muscle cell: a mechanism for adaptation to mechanical forces in the lung. Respir Physiol Neurobiol 137: 151–168, 2003. doi: 10.1016/s1569-9048(03)00144-7. [DOI] [PubMed] [Google Scholar]
- 1740. Bednarek ML, Speich JE, Miner AS, Ratz PH. Active tension adaptation at a shortened arterial muscle length: inhibition by cytochalasin-D. Am J Physiol Heart Circ Physiol 300: H1166–H1173, 2011. doi: 10.1152/ajpheart.00009.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1741. Staiculescu MC, Galiñanes EL, Zhao G, Ulloa U, Jin M, Beig MI, Meininger GA, Martinez-Lemus LA. Prolonged vasoconstriction of resistance arteries involves vascular smooth muscle actin polymerization leading to inward remodelling. Cardiovasc Res 98: 428–436, 2013. doi: 10.1093/cvr/cvt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1742. Chen X, Pavlish K, Benoit JN. Myosin phosphorylation triggers actin polymerization in vascular smooth muscle. Am J Physiol Heart Circ Physiol 295: H2172–H2177, 2008. doi: 10.1152/ajpheart.91437.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1743. Castorena-Gonzalez JA, Staiculescu MC, Foote CA, Polo-Parada L, Martinez-Lemus LA. The obligatory role of the actin cytoskeleton on inward remodeling induced by dithiothreitol activation of endogenous transglutaminase in isolated arterioles. Am J Physiol Heart Circ Physiol 306: H485–H495, 2014. doi: 10.1152/ajpheart.00557.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1744. Armentano RL, Levenson J, Barra JG, Fischer EI, Breitbart GJ, Pichel RH, Simon A. Assessment of elastin and collagen contribution to aortic elasticity in conscious dogs. Am J Physiol Heart Circ Physiol 260: H1870–H1877, 1991. doi: 10.1152/ajpheart.1991.260.6.H1870. [DOI] [PubMed] [Google Scholar]
- 1745. Clifford PS, Ella SR, Stupica AJ, Nourian Z, Li M, Martinez-Lemus LA, Dora KA, Yang Y, Davis MJ, Pohl U, Meininger GA, Hill MA. Spatial distribution and mechanical function of elastin in resistance arteries: a role in bearing longitudinal stress. Arterioscler Thromb Vasc Biol 31: 2889–2896, 2011. doi: 10.1161/ATVBAHA.111.236570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1746. Oh YS, Berkowitz DE, Cohen RA, Figueroa CA, Harrison DG, Humphrey JD, Larson DF, Leopold JA, Mecham RP, Ruiz-Opazo N, Santhanam L, Seta F, Shyy JY, Sun Z, Tsao PS, Wagenseil JE, Galis ZS. A Special Report on the NHLBI initiative to study cellular and molecular mechanisms of arterial stiffness and its association with hypertension. Circ Res 121: 1216–1218, 2017. doi: 10.1161/CIRCRESAHA.117.311703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1747. Safar ME. Arterial stiffness as a risk factor for clinical hypertension. Nat Rev Cardiol 15: 97–105, 2018. doi: 10.1038/nrcardio.2017.155. [DOI] [PubMed] [Google Scholar]
- 1748. Martinez-Lemus LA, Zhao G, Galiñanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol 300: H2005–H2015, 2011. doi: 10.1152/ajpheart.01066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1749. Martinez-Lemus LA, Galinanes EL. Matrix metalloproteinases and small artery remodeling. Drug Discov Today Dis Models 8: 21–28, 2011. doi: 10.1016/j.ddmod.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1750. Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res 85: 906–911, 1999. doi: 10.1161/01.res.85.10.906. [DOI] [PubMed] [Google Scholar]
- 1751. Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci 27: 534–539, 2002. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- 1752. Huelsz-Prince G, Belkin AM, VanBavel E, Bakker EN. Activation of extracellular transglutaminase 2 by mechanical force in the arterial wall. J Vasc Res 50: 383–395, 2013. doi: 10.1159/000354222. [DOI] [PubMed] [Google Scholar]
- 1753. Engholm M, Pinilla E, Mogensen S, Matchkov V, Hedegaard ER, Chen H, Mulvany MJ, Simonsen U. Involvement of transglutaminase 2 and voltage-gated potassium channels in cystamine vasodilatation in rat mesenteric small arteries. Br J Pharmacol 173: 839–855, 2016. doi: 10.1111/bph.13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1754. van den Akker J, VanBavel E, van Geel R, Matlung HL, Guvenc Tuna B, Janssen GM, van Veelen PA, Boelens WC, De Mey JG, Bakker EN. The redox state of transglutaminase 2 controls arterial remodeling. PLoS One 6: e23067, 2011. doi: 10.1371/journal.pone.0023067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1755. Bakker EN, Pistea A, VanBavel E. Transglutaminases in vascular biology: relevance for vascular remodeling and atherosclerosis. J Vasc Res 45: 271–278, 2008. doi: 10.1159/000113599. [DOI] [PubMed] [Google Scholar]
- 1756. Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell 17: 1606–1619, 2006. doi: 10.1091/mbc.e05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1757. Staiculescu MC, Foote C, Meininger GA, Martinez-Lemus LA. The role of reactive oxygen species in microvascular remodeling. Int J Mol Sci 15: 23792–23835, 2014. doi: 10.3390/ijms151223792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1758. Matlung HL, Bakker EN, VanBavel E. Shear stress, reactive oxygen species, and arterial structure and function. Antioxid Redox Signal 11: 1699–1709, 2009. doi: 10.1089/ars.2008.2408. [DOI] [PubMed] [Google Scholar]
- 1759. Kainulainen T, Pender A, D’Addario M, Feng Y, Lekic P, McCulloch CA. Cell death and mechanoprotection by filamin a in connective tissues after challenge by applied tensile forces. J Biol Chem 277: 21998–22009, 2002. doi: 10.1074/jbc.M200715200. [DOI] [PubMed] [Google Scholar]
- 1760. Retailleau K, Arhatte M, Demolombe S, Jodar M, Baudrie V, Offermanns S, Feng Y, Patel A, Honoré E, Duprat F. Smooth muscle filamin A is a major determinant of conduit artery structure and function at the adult stage. Pflugers Arch 468: 1151–1160, 2016. doi: 10.1007/s00424-016-1813-x. [DOI] [PubMed] [Google Scholar]
- 1761. Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp 283: 174–191, 2007. doi: 10.1002/9780470319413.ch14. [DOI] [PubMed] [Google Scholar]
- 1762. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 118: 692–702, 2016. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1763. Miano JM. Vascular smooth muscle cell differentiation-2010. J Biomed Res 24: 169–180, 2010. doi: 10.1016/S1674-8301(10)60026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1764. Caillon A, Paradis P, Schiffrin EL. Role of immune cells in hypertension. Br J Pharmacol 176: 1818–1828, 2019. doi: 10.1111/bph.14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1765. Miano JM, Long X. The short and long of noncoding sequences in the control of vascular cell phenotypes. Cell Mol Life Sci 72: 3457–3488, 2015. doi: 10.1007/s00018-015-1936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1766. Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol 34: 2191–2198, 2014. doi: 10.1161/ATVBAHA.114.303422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1767. Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol 292: H1209–H1224, 2007. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 1768. Gimbrone MA Jr, García-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118: 620–636, 2016. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1769. Cornhill JF, Herderick EE, Stary HC. Topography of human aortic sudanophilic lesions. Monogr Atheroscler 15: 13–19, 1990. [PubMed] [Google Scholar]
- 1770. Endo S, Goldsmith HL, Karino T. Flow patterns and preferred sites of atherosclerotic lesions in the human aorta—I. Aortic arch. Biorheology 51: 239–255, 2014. doi: 10.3233/BIR-14005. [DOI] [PubMed] [Google Scholar]
- 1771. Endo S, Goldsmith HL, Karino T. Flow patterns and preferred sites of atherosclerotic lesions in the human aorta—II. Abdominal aorta. Biorheology 51: 257–274, 2014. doi: 10.3233/BIR-14006. [DOI] [PubMed] [Google Scholar]
- 1772. Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia-Cardeña G, Gimbrone MA Jr.. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci U S A 101: 14871–14876, 2004. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1773. Davies PF, Civelek M, Fang Y, Fleming I. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res 99: 315–327, 2013. doi: 10.1093/cvr/cvt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1774. Ross R. The pathogenesis of atherosclerosis—an update. N Engl J Med 314: 488–500, 1986. doi: 10.1056/NEJM198602203140806. [DOI] [PubMed] [Google Scholar]
- 1775. Ross R, Agius L. The process of atherogenesis—cellular and molecular interaction: from experimental animal models to humans. Diabetologia 35: S34–S40, 1992. [DOI] [PubMed] [Google Scholar]
- 1776. Kang H, Li X, Xiong K, Song Z, Tian J, Wen Y, Sun A, Deng X. The entry and egress of monocytes in atherosclerosis: a biochemical and biomechanical driven process. Cardiovasc Ther 2021: 6642927, 2021. doi: 10.1155/2021/6642927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1777. Schwartz CJ, Valente AJ, Sprague EA, Kelley JL, Nerem RM. The pathogenesis of atherosclerosis: an overview. Clin Cardiol 14: I1–I16, 1991. doi: 10.1002/clc.4960141302. [DOI] [PubMed] [Google Scholar]
- 1778. Steinberg D, Witztum JL. Lipoproteins and atherogenesis. Current concepts. JAMA 264: 3047–3052, 1990. doi: 10.1001/jama.1990.03450230083034. [DOI] [PubMed] [Google Scholar]
- 1779. Emini Veseli B, Perrotta P, De Meyer GR, Roth L, Van der Donckt C, Martinet W, De Meyer GR. Animal models of atherosclerosis. Eur J Pharmacol 816: 3–13, 2017. doi: 10.1016/j.ejphar.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 1780. von der Thüsen JH, van Berkel TJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation 103: 1164–1170, 2001. doi: 10.1161/01.cir.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 1781. Cheng C, van Haperen R, de Waard M, van Damme LC, Tempel D, Hanemaaijer L, van Cappellen GW, Bos J, Slager CJ, Duncker DJ, van der Steen AF, de Crom R, Krams R. Shear stress affects the intracellular distribution of eNOS: direct demonstration by a novel in vivo technique. Blood 106: 3691–3698, 2005. doi: 10.1182/blood-2005-06-2326. [DOI] [PubMed] [Google Scholar]
- 1782. Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113: 2744–2753, 2006. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 1783. Cheng C, Tempel D, van Haperen R, de Boer HC, Segers D, Huisman M, van Zonneveld AJ, Leenen PJ, van der Steen A, Serruys PW, de Crom R, Krams R. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest 117: 616–626, 2007. doi: 10.1172/JCI28180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1784. Lin WL, Chang CF, Shi CS, Shi GY, Wu HL. Recombinant lectin-like domain of thrombomodulin suppresses vascular inflammation by reducing leukocyte recruitment via interacting with Lewis Y on endothelial cells. Arterioscler Thromb Vasc Biol 33: 2366–2373, 2013. doi: 10.1161/ATVBAHA.113.301221. [DOI] [PubMed] [Google Scholar]
- 1785. Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 17: 2238–2244, 1997. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 1786. Godin D, Ivan E, Johnson C, Magid R, Galis ZS. Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation 102: 2861–2866, 2000. doi: 10.1161/01.CIR.102.23.2861. [DOI] [PubMed] [Google Scholar]
- 1787. Wong LC, Langille BL. Developmental remodeling of the internal elastic lamina of rabbit arteries: effect of blood flow. Circ Res 78: 799–805, 1996. doi: 10.1161/01.RES.78.5.799. [DOI] [PubMed] [Google Scholar]
- 1788. Miyashiro JK, Poppa V, Berk BC. Flow-induced vascular remodeling in the rat carotid artery diminishes with age. Circ Res 81: 311–319, 1997. doi: 10.1161/01.res.81.3.311. [DOI] [PubMed] [Google Scholar]
- 1789. Mondy JS, Lindner V, Miyashiro JK, Berk BC, Dean RH, Geary RL. Platelet-derived growth factor ligand and receptor expression in response to altered blood flow in vivo. Circ Res 81: 320–327, 1997. doi: 10.1161/01.res.81.3.320. [DOI] [PubMed] [Google Scholar]
- 1790. Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 297: H1535–H1543, 2009. doi: 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1791. Thilo F, Vorderwülbecke BJ, Marki A, Krueger K, Liu Y, Baumunk D, Zakrzewicz A, Tepel M. Pulsatile atheroprone shear stress affects the expression of transient receptor potential channels in human endothelial cells. Hypertension 59: 1232–1240, 2012. doi: 10.1161/HYPERTENSIONAHA.111.183608. [DOI] [PubMed] [Google Scholar]
- 1792. Dunn J, Thabet S, Jo H. Flow-dependent epigenetic DNA methylation in endothelial gene expression and atherosclerosis. Arterioscler Thromb Vasc Biol 35: 1562–1569, 2015. doi: 10.1161/ATVBAHA.115.305042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1793. Jia L, Wang L, Wei F, Li C, Wang Z, Yu H, Chen H, Wang B, Jiang A. Effects of Caveolin-1-ERK1/2 pathway on endothelial cells and smooth muscle cells under shear stress. Exp Biol Med (Maywood) 245: 21–33, 2020. doi: 10.1177/1535370219892574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1794. Xu S. Therapeutic potential of blood flow mimetic compounds in preventing endothelial dysfunction and atherosclerosis. Pharmacol Res 155: 104737, 2020. doi: 10.1016/j.phrs.2020.104737. [DOI] [PubMed] [Google Scholar]
- 1795. Schiener M, Hossann M, Viola JR, Ortega-Gomez A, Weber C, Lauber K, Lindner LH, Soehnlein O. Nanomedicine-based strategies for treatment of atherosclerosis. Trends Mol Med 20: 271–281, 2014. doi: 10.1016/j.molmed.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 1796. Gorabi AM, Kiaie N, Reiner Ž, Carbone F, Montecucco F, Sahebkar A. The therapeutic potential of nanoparticles to reduce inflammation in atherosclerosis. Biomolecules 9: 416, 2019. doi: 10.3390/biom9090416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1797. Flores AM, Ye J, Jarr KU, Hosseini-Nassab N, Smith BR, Leeper NJ. Nanoparticle therapy for vascular diseases. Arterioscler Thromb Vasc Biol 39: 635–646, 2019. doi: 10.1161/ATVBAHA.118.311569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1798. Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J Control Release 220: 600–607, 2015. doi: 10.1016/j.jconrel.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1799. Jin K, Luo Z, Zhang B, Pang Z. Biomimetic nanoparticles for inflammation targeting. Acta Pharm Sin B 8: 23–33, 2018. doi: 10.1016/j.apsb.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1800. Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526: 118–121, 2015. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1801. Wei X, Ying M, Dehaini D, Su Y, Kroll AV, Zhou J, Gao W, Fang RH, Chien S, Zhang L. Nanoparticle functionalization with platelet membrane enables multifactored biological targeting and detection of atherosclerosis. ACS Nano 12: 109–116, 2018. doi: 10.1021/acsnano.7b07720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1802. Yan C, Quan XJ, Feng YM. Nanomedicine for gene delivery for the treatment of cardiovascular diseases. Curr Gene Ther 19: 20–30, 2019. doi: 10.2174/1566523218666181003125308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1803. Khodabandehlou K, Masehi-Lano JJ, Poon C, Wang J, Chung EJ. Targeting cell adhesion molecules with nanoparticles using in vivo and flow-based in vitro models of atherosclerosis. Exp Biol Med (Maywood) 242: 799–812, 2017. doi: 10.1177/1535370217693116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1804. Gadde S, Rayner KJ. Nanomedicine Meets microRNA: Current advances in RNA-based nanotherapies for atherosclerosis. Arterioscler Thromb Vasc Biol 36: e73–e79, 2016. doi: 10.1161/ATVBAHA.116.307481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1805. Robertson AM, Watton PN. Mechanobiology of the arterial wall. In: Transport in Biological Media, edited by Becker S, Kuznetsov AV. Amsterdam: Elsevier, 2013, p. 275–347. [Google Scholar]
- 1806. González JM, Briones AM, Starcher B, Conde MV, Somoza B, Daly C, Vila E, McGrath I, González MC, Arribas SM. Influence of elastin on rat small artery mechanical properties. Exp Physiol 90: 463–468, 2005. doi: 10.1113/expphysiol.2005.030056. [DOI] [PubMed] [Google Scholar]
- 1807. Briones AM, González JM, Somoza B, Giraldo J, Daly CJ, Vila E, González MC, McGrath JC, Arribas SM. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J Physiol 552: 185–195, 2003. doi: 10.1113/jphysiol.2003.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1808. Laurent S, Boutouyrie P, Lacolley P. Structural and genetic bases of arterial stiffness. Hypertension 45: 1050–1055, 2005. doi: 10.1161/01.HYP.0000164580.39991.3d. [DOI] [PubMed] [Google Scholar]
- 1809. Kwon HM, Kim D, Hong BK, Byun KH, Oh SH, Kna JS, Kim HS, Schwartz RS, Lerman A. Ultrastructural changes of the internal elastic lamina in experimental hypercholesterolemic porcine coronary arteries. J Korean Med Sci 13: 603–611, 1998. doi: 10.3346/jkms.1998.13.6.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1810. Méndez-Barbero N, Gutiérrez-Muñoz C, Blanco-Colio LM. Cellular crosstalk between endothelial and smooth muscle cells in vascular wall remodeling. Int J Mol Sci 22: 7284, 2021. doi: 10.3390/ijms22147284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1811. Straub AC, Zeigler AC, Isakson BE. The myoendothelial junction: connections that deliver the message. Physiology (Bethesda) 29: 242–249, 2014. doi: 10.1152/physiol.00042.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1812. Isakson BE, Duling BR. Heterocellular contact at the myoendothelial junction influences gap junction organization. Circ Res 97: 44–51, 2005. doi: 10.1161/01.RES.0000173461.36221.2e. [DOI] [PubMed] [Google Scholar]
- 1813. Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87: 474–479, 2000. doi: 10.1161/01.res.87.6.474. [DOI] [PubMed] [Google Scholar]
- 1814. von der Weid PY, Bény JL. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. J Physiol 471: 13–24, 1993. doi: 10.1113/jphysiol.1993.sp019888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1815. Isakson BE, Ramos SI, Duling BR. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ Res 100: 246–254, 2007. doi: 10.1161/01.RES.0000257744.23795.93. [DOI] [PubMed] [Google Scholar]
- 1816. Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol Rev 66: 513–569, 2014. doi: 10.1124/pr.112.007351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1817. Lemmey HAL, Garland CJ, Dora KA. Intrinsic regulation of microvascular tone by myoendothelial feedback circuits. Curr Top Membr 85: 327–355, 2020. doi: 10.1016/bs.ctm.2020.01.004. [DOI] [PubMed] [Google Scholar]
- 1818. Murphy TV, Sandow SL. Agonist-evoked endothelial Ca2+ signalling microdomains. Curr Opin Pharmacol 45: 8–15, 2019. doi: 10.1016/j.coph.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 1819. Nakashima M, Mombouli JV, Taylor AA, Vanhoutte PM. Endothelium-dependent hyperpolarization caused by bradykinin in human coronary arteries. J Clin Invest 92: 2867–2871, 1993. doi: 10.1172/JCI116907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1820. Sheng JZ, Braun AP. Small and intermediate conductance, Ca-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. Am J Physiol Cell Physiol 293: C458–C467, 2007. doi: 10.1152/ajpcell.00036.2007. [DOI] [PubMed] [Google Scholar]
- 1821. Cipolla MJ, Smith J, Kohlmeyer MM, Godfrey JA. SKCa and IKCa channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: effect of ischemia and reperfusion. Stroke 40: 1451–1457, 2009. doi: 10.1161/STROKEAHA.108.535435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1822. Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci USA 94: 6529–6534, 1997. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1823. Hald BO, Welsh DG, Holstein-Rathlou NH, Jacobsen JC. Origins of variation in conducted vasomotor responses. Pflugers Arch 467: 2055–2067, 2015. doi: 10.1007/s00424-014-1649-1. [DOI] [PubMed] [Google Scholar]
- 1824. Furchgott RF. Nitric oxide: from basic research on isolated blood vessels to clinical relevance in diabetes. An R Acad Nac Med (Madr) 115: 317–331, 1998. [PubMed] [Google Scholar]
- 1825. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 1826. Kuchan MJ, Frangos JA. Shear stress regulates endothelin-1 release via protein kinase C and cGMP in cultured endothelial cells. Am J Physiol Heart Circ Physiol 264: H150–H156, 1993. doi: 10.1152/ajpheart.1993.264.1.H150. [DOI] [PubMed] [Google Scholar]
- 1827. Redmond EM, Cahill PA, Sitzmann JV. Perfused transcapillary smooth muscle and endothelial cell co-culture—a novel in vitro model. In Vitro Cell Dev Biol Anim 31: 601–609, 1995. doi: 10.1007/BF02634313. [DOI] [PubMed] [Google Scholar]
- 1828. Chiu JJ, Chen LJ, Chang SF, Lee PL, Lee CI, Tsai MC, Lee DY, Hsieh HP, Usami S, Chien S. Shear stress inhibits smooth muscle cell-induced inflammatory gene expression in endothelial cells: role of NF-kappaB. Arterioscler Thromb Vasc Biol 25: 963–969, 2005. doi: 10.1161/01.ATV.0000159703.43374.19. [DOI] [PubMed] [Google Scholar]
- 1829. Chiu JJ, Chen LJ, Lee CI, Lee PL, Lee DY, Tsai MC, Lin CW, Usami S, Chien S. Mechanisms of induction of endothelial cell E-selectin expression by smooth muscle cells and its inhibition by shear stress. Blood 110: 519–528, 2007. doi: 10.1182/blood-2006-08-040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1830. Tull SP, Anderson SI, Hughan SC, Watson SP, Nash GB, Rainger GE. Cellular pathology of atherosclerosis: smooth muscle cells promote adhesion of platelets to cocultured endothelial cells. Circ Res 98: 98–104, 2006. doi: 10.1161/01.RES.0000198386.69355.87. [DOI] [PubMed] [Google Scholar]
- 1831. Yao QP, Qi YX, Zhang P, Cheng BB, Yan ZQ, Jiang ZL. SIRT1 and Connexin40 Mediate the normal shear stress-induced inhibition of the proliferation of endothelial cells co-cultured with vascular smooth muscle cells. Cell Physiol Biochem 31: 389–399, 2013. doi: 10.1159/000343376. [DOI] [PubMed] [Google Scholar]
- 1832. Wang YH, Yan ZQ, Qi YX, Cheng BB, Wang XD, Zhao D, Shen BR, Jiang ZL. Normal shear stress and vascular smooth muscle cells modulate migration of endothelial cells through histone deacetylase 6 activation and tubulin acetylation. Ann Biomed Eng 38: 729–737, 2010. doi: 10.1007/s10439-009-9896-6. [DOI] [PubMed] [Google Scholar]
- 1833. Chen LJ, Chuang L, Huang YH, Zhou J, Lim SH, Lee CI, Lin WW, Lin TE, Wang WL, Chen L, Chien S, Chiu JJ. MicroRNA mediation of endothelial inflammatory response to smooth muscle cells and its inhibition by atheroprotective shear stress. Circ Res 116: 1157–1169, 2015. doi: 10.1161/CIRCRESAHA.116.305987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1834. Redmond EM, Cahill PA, Sitzmann JV. Flow-mediated regulation of endothelin receptors in cocultured vascular smooth muscle cells: an endothelium-dependent effect. J Vasc Res 34: 425–435, 1997. doi: 10.1159/000159253. [DOI] [PubMed] [Google Scholar]
- 1835. Chao Y, Ye P, Zhu L, Kong X, Qu X, Zhang J, Luo J, Yang H, Chen S. Low shear stress induces endothelial reactive oxygen species via the AT1R/eNOS/NO pathway. J Cell Physiol 233: 1384–1395, 2018. doi: 10.1002/jcp.26016. [DOI] [PubMed] [Google Scholar]
- 1836. Chen M, Li X. Role of TRPV4 channel in vasodilation and neovascularization. Microcirculation 28: e12703, 2021. doi: 10.1111/micc.12703. [DOI] [PubMed] [Google Scholar]
- 1837. Hu Y, Chen M, Wang M, Li X. Flow-mediated vasodilation through mechanosensitive G protein-coupled receptors in endothelial cells. Trends Cardiovasc Med 32: 61–70, 2022. doi: 10.1016/j.tcm.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 1838. Gao Y, Chen T, Raj JU. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am J Respir Cell Mol Biol 54: 451–460, 2016. doi: 10.1165/rcmb.2015-0323TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1839. Marasciulo FL, Montagnani M, Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem 13: 1655–1665, 2006. doi: 10.2174/092986706777441968. [DOI] [PubMed] [Google Scholar]
- 1840. Sakamoto N, Ohashi T, Sato M. Effect of fluid shear stress on migration of vascular smooth muscle cells in cocultured model. Ann Biomed Eng 34: 408–415, 2006. doi: 10.1007/s10439-005-9043-y. [DOI] [PubMed] [Google Scholar]
- 1841. Tsai MC, Chen L, Zhou J, Tang Z, Hsu TF, Wang Y, Shih YT, Peng HH, Wang N, Guan Y, Chien S, Chiu JJ. Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ Res 105: 471–480, 2009. doi: 10.1161/CIRCRESAHA.109.193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1842. Wang HQ, Huang LX, Qu MJ, Yan ZQ, Liu B, Shen BR, Jiang ZL. Shear stress protects against endothelial regulation of vascular smooth muscle cell migration in a coculture system. Endothelium 13: 171–180, 2006. doi: 10.1080/10623320600760282. [DOI] [PubMed] [Google Scholar]
- 1843. Redmond EM, Cahill PA, Sitzmann JV. Flow-mediated regulation of G-protein expression in cocultured vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol 18: 75–83, 1998. doi: 10.1161/01.atv.18.1.75. [DOI] [PubMed] [Google Scholar]
- 1844. Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol 14: 249–256, 2012. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 1845. Chen CN, Chang SF, Lee PL, Chang K, Chen LJ, Usami S, Chien S, Chiu JJ. Neutrophils, lymphocytes, and monocytes exhibit diverse behaviors in transendothelial and subendothelial migrations under coculture with smooth muscle cells in disturbed flow. Blood 107: 1933–1942, 2006. doi: 10.1182/blood-2005-08-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1846. Reading SA, Brayden JE. Central role of TRPM4 channels in cerebral blood flow regulation. Stroke 38: 2322–2328, 2007. doi: 10.1161/STROKEAHA.107.483404. [DOI] [PubMed] [Google Scholar]
- 1847. Thakore P, Alvarado MG, Ali S, Mughal A, Pires PW, Yamasaki E, Pritchard HA, Isakson BE, Tran CHT, Earley S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife 10: e63040, 2021. doi: 10.7554/eLife.63040. [DOI] [PMC free article] [PubMed] [Google Scholar]