

Abstract
Cyclin D1 is a regulatory subunit of cyclin-Dependent Kinases 4 and 6 (CDK4/6) and regulates progression from G1 to S phase of the cell cycle. Dysregulated cyclin D1-CDK4/6 contributes to abnormal cell proliferation and tumor development. Phosphorylation of threonine 286 of cyclin D1 is necessary for ubiquitin-dependent degradation. Non-phosphorylatable cyclin D1 mutants are stabilized and concentrated in the nucleus, contributing to genomic instability and tumor development. Studies investigating the tumor-promoting functions of cyclin D1 mutants have focused on the use of artificial promoters to drive the expression which unfortunately may not accurately reflect tumorigenic functions of mutant cyclin D1 in cancer development. We have generated a conditional knock-in mouse model where cyclin D1T286A is expressed under the control of its endogenous promoter following Cre-dependent excision of a lox-stop-lox sequence. Acute expression of cyclin D1T286A following tamoxifen-inducible Cre recombinase triggers inflammation, lymphocyte abnormality and ultimately mesenteric tumors in the intestine. Tissue-specific expression of cyclin D1T286A in the uterus and endometrium cooperates with Pten loss to drive endometrial hyperplasia and cancer. Mechanistically, cyclin D1T286A mutant activates NF-κB signaling, augments inflammation, and contributes to tumor development. These results indicate that mutation of cyclin D1 at threonine 286 has a critical role in regulating inflammation and tumor development.
Keywords: cyclin D1, Pten, cyclin D1T286A, inflammation, NF-κB signaling, uterine carcinoma
Introduction
D-type cyclins play a pivotal role in regulating the G1-S phase transition of the cell cycle as regulatory subunits of Cyclin-Dependent Kinases 4 and 6 (CDK4/6) which phosphorylate tumor suppressor retinoblastoma protein (Rb) (1–3). Phosphorylated Rb dissociates from the E2f transcription factor permitting expression of genes necessary for S phase progression. CCND1 transcription depends on Ras-MAP kinase signaling (4, 5) and likewise protein accumulation is also regulated via Ras-dependent mechanisms that control the rapid, ubiquitin-mediated protein degradation at the G1/S phase boundary (6). Glycogen Synthase Kinase 3β (GSK3β) phosphorylates cyclin D1 on threonine 286 (Thr286), resulting in CRM1-mediated nuclear export and subsequent recognition by SCFFbxo4 E3 ligase which directs its degradation by the 26S proteasome (6–8). Substitution of Thr286 to alanine on cyclin D1, cyclin D1T286A, stabilizes cyclin D1 in the nucleus; stabilized, nuclear cyclin D1T286A/CDK4 triggers DNA re-replication, induces genomic instability and promotes tumor initiation and progression (9–13). Consistently, mutation of Thr286 is observed in multiple human cancers (cBioportal.org). While the tumor-promoting function of cyclin D1T286A has been demonstrated using transgenic mouse models (9), a major limitation of the current models is that overexpression of cyclin D1T286A is driven by an exogenous promoter, which may not accurately reflect studying the function of cyclin D1T286A mutant in human cancers. We have generated a novel mouse model that permits the acute expression of cyclin D1T286A from its endogenous promoter. This system will faithfully models human cancers harboring the cyclin D1T286A mutation without affecting the cyclin D1T286A expression in embryogenesis or development. We demonstrate that the acute expression of cyclin D1T286A triggers tumorigenesis in several tissues. Furthermore, we show when cyclin D1T286A mutation occurs concurrently with Pten loss in the uterus, cyclin D1T286A accelerates endometrial hyperplasia and subsequently tumor development.
Results
The characterization of a novel cyclin D1T286A mouse model.
We generated a cyclin D1T286A conditional, knock-in mouse (cyclin D1Fl/WT; Fig. 1A). Exon 5 of cyclin D1 that includes threonine-286 residue was engineered such that it is flanked by a lox-stop-lox sequence. Cyclin D1Fl/WT mice were intercrossed with transgenic CreERT2 mice (CreER) to create cyclin D1Fl/WT CreER mice. We used a single cyclin D1 flox allele to accurately investigate the role of cyclin D1 mutation found in human cancers. We treated 6–8 weeks old cyclin D1Fl/WT CreER mice with tamoxifen for 5 days to activate Cre and induce cyclin D1T286A expression in mice (cyclin D1TA/WT). Excision of WT exon 5 and recombination of mutant exon 5 was confirmed by genomic PCR (Fig. 1B); no recombination was observed in the absence of tamoxifen (Fig. 1B). We confirmed widespread recombination of cyclin D1T286A allele (Fig. 1C) and cyclin D1 protein accumulation following Cre activation by tamoxifen treatment (Fig. 1D). The presence of the cyclin D1T286A mutation was also confirmed by direct sequencing of genomic DNA (Fig. S1A).
Figure 1. Characterization of cyclin D1 knock-in mouse model.
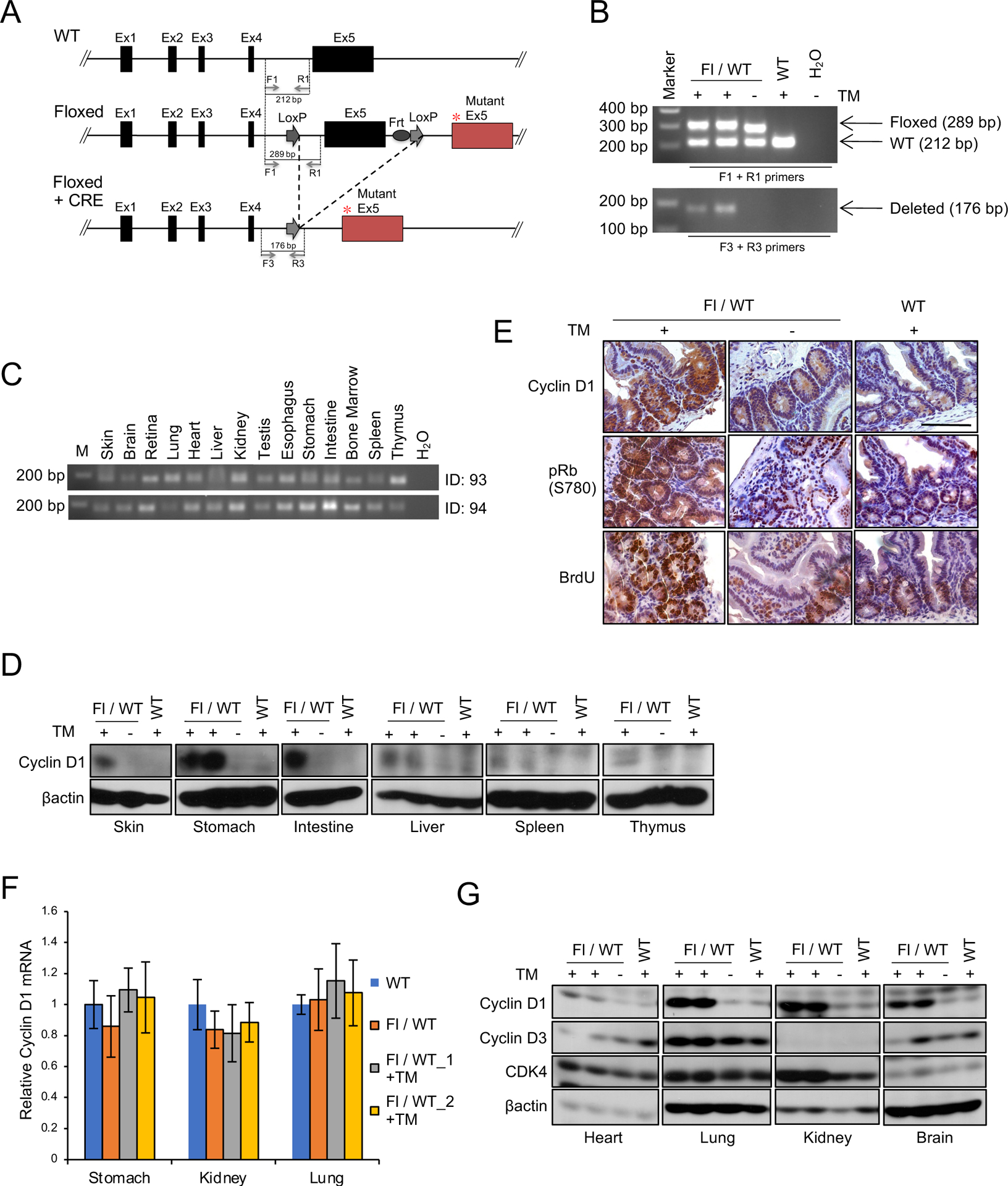
(A) The scheme of cyclin D1 knock-in allele. * indicates T286A mutation in exon 5 (Ex5). (B) PCR analysis of genomic DNA from cyclin D1 Flox/WT or WT/WT mice with or without oral administration of tamoxifen (TM) for 5 days using primers F1+R1 or F3+R3 indicated in (A). (C) PCR analysis of genomic DNA for tissues indicated from two independent cyclin D1 Flox/WT mice (ID: 93 and 94) following administration of TM for 5 days using primers F3+R3. (D) Western blot analysis of lysates for tissues indicated from cyclin D1 Flox/WT or WT/WT mice with or without oral administration of TM using antibodies to cyclin D1 and βactin. (E) Representative images for immunohistochemistry analysis of intestine from Flox/WT or WT/WT mice with or without oral administration of TM using antibodies to cyclin D1, phosphorylation of Rb at serine 780 (pRb S780), and BrdU. BrdU was administered by intraperitoneal injection two hours prior to the analysis of mice. (F) QPCR analysis of cyclin D1 mRNA expression in stomach, kidney and lung from Flox/WT and WT/WT mice with or without oral administration of TM. Two independent cyclin D1 Flox/WT mice (Fl/WT_1 and Fl/WT_2) were analyzed. (G) Western blot analysis of lysates from tissues indicated from cyclin D1 Flox/WT or WT/WT mice with or without oral administration of TM using antibodies to cyclin D1, cyclin D3, CDK4 and βactin.
Expression of cyclin D1T286A was associated with increased Rb phosphorylation at serine 780 and BrdU positive cells (Fig. 1E). Furthermore, increased cyclin D1 protein accumulation reflects reduced protein degradation, but not increased CCND1 expression (Fig. 1F). The expression of cyclin D3 and CDK4 is unchanged following treatment of tamoxifen in Cyclin D1Fl/WT CreER mice, demonstrating specific induction of the cyclin D1T286A mutant (Fig. 1G). Hematopoietic cells, where cyclins D2 and D3 are primary D-type cyclins, exhibited no abnormality in D1Fl/WT CreER mice treated with tamoxifen consistent with the normal regulation of cyclin D2 and cyclin D3 (14) (Fig. S1B).
Cyclin D1T286A mice develop mesenteric tumors.
To ascertain the impact of acute cyclin D1T286A expression, 6–8-week-old mice were exposed to tamoxifen and monitored for 20 months. Cyclin D1TA/WT mice developed multiple disorders with increased age and overall survival for cyclin D1TA/WT mice (D1TA_All) was poor compared with WT mice (Fig. 2A). Neoplastic events included a papillary carcinoma that maintained high expression of cyclin D1 and exhibited increased Rb phosphorylation at serine 780 (Fig. S1C). The major finding in cyclin D1TA/WT mice was mesenteric tumors in the intestine with approximately 50% penetrance in cyclin D1TA/WT mice (Fig. 2B). When compared to WT mice, the survival of the cyclin D1TA/WT mice that have only mesenteric tumors (D1TA_Mes) was similar to the survival of all cyclin D1TA/WT mice (D1TA_All) (Fig. 2A). Hematoxylin and eosin (H&E) staining revealed that mesenteric tumors include massive inflammation (Fig. 2C). Immunohistochemical (IHC) staining for CD3, a marker for T cells, and CD45R, a marker for B cells, revealed a mixed population of T cells and B cells infiltrate these tumors, which ultimately induces lymphoma (Fig. 2C). Further analyses revealed that mesenteric tumors have increased p-Rb (S780) and BrdU incorporation (Fig. 2D). We previously demonstrated that cyclin D1 overexpression causes DNA damage, leading to genomic instability, which in turn, develops tumors (10, 12). Consistently, we detected increased p-Chk1 (S345) and p-Chk2 (T68), both of which are responsible for initial DNA damage responses (Fig. 2D).
Figure 2. Endogenous expression of cyclin D1T286A induces mesenteric tumors in mice.
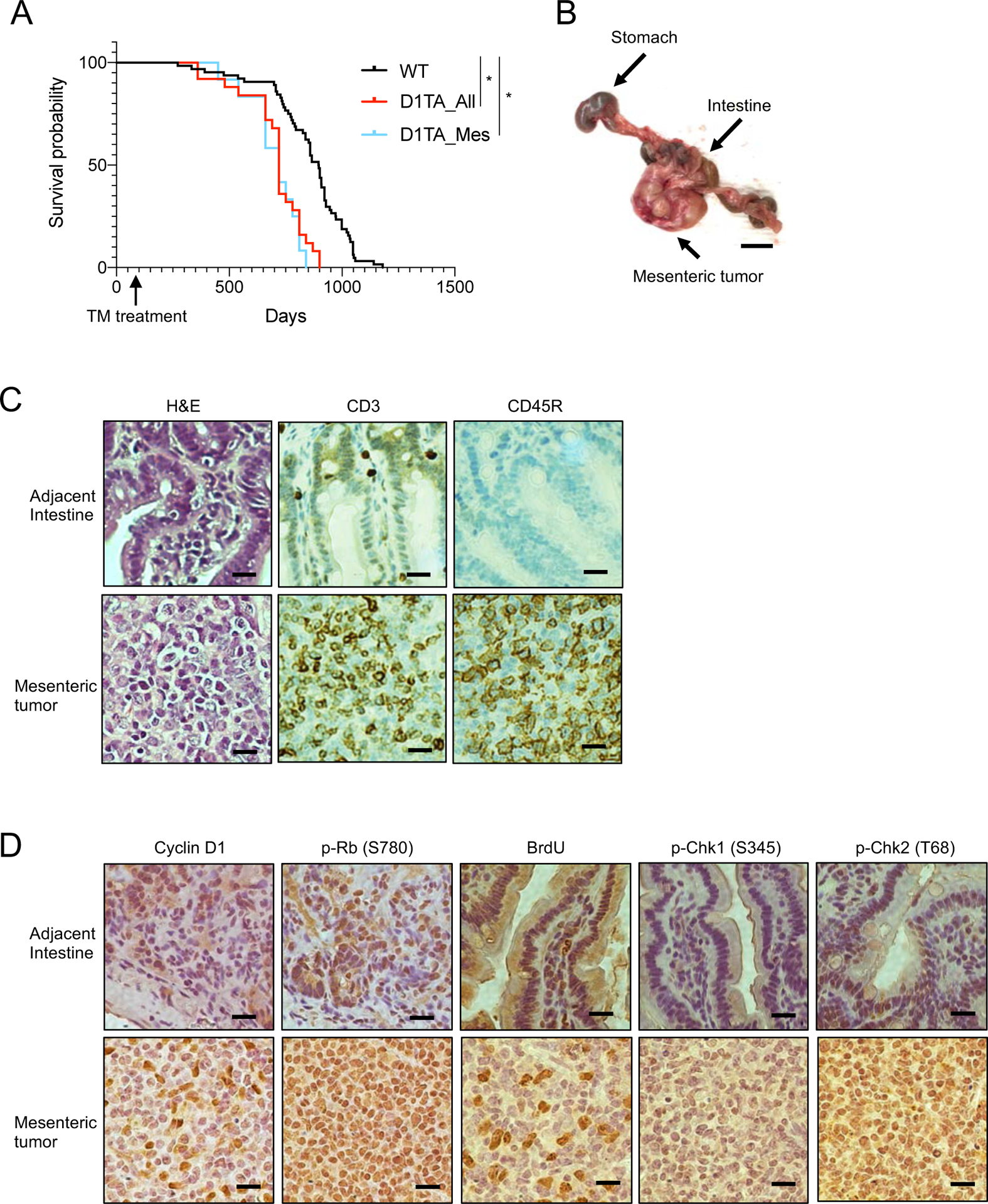
(A) Tumor free survival curves of cyclin D1 Fl/WT treated with tamoxifen (TM) at the age of 8–12 weeks old and WT mice. D1TA_All indicates all cyclin D1 T286A mice and D1TA_Mes indicates cyclin D1 T286A mice only with mesenteric tumors. *p < 0.01 (Log-rank test; n=64 for B6, n=26 for D1TA_All and n=13 for D1TA_Mes). (B) Representative image of mesenteric tumor from cyclin D1T286A mouse. Scale bar indicates 10 mm. (C-D) Representative images for HE staining and immunohistochemistry analysis of mesenteric tumor and adjacent intestine from cyclin D1 T286A mouse using antibodies to CD3, CD45R, cyclin D1, phosphorylation of Rb (S780), BrdU, phosphorylation of Chk1 (S345) and phosphorylation of Chk2 (T68). BrdU was administered by intraperitoneal injection two hours prior to the analysis of mice. Scale bar indicates 20μm.
Endogenous cyclin D1T286A expression accelerates Pten+/− mediated endometrial cancer development.
CCND1 mutations are observed in endometrial cancers (~7%; cBioportal) and the most common mutation of cyclin D1 is at T286 (Fig. 3A). In endometrial carcinomas, deletion or mutation of the Phosphatase and tensin homolog deleted on Chromosome 10 (PTEN) frequently co-occurs with CCND1 mutations (Fig. S1D; cBioportal. org). Deletion or inactivating mutations of PTEN in the uterus can trigger endometrial hyperplasia or overt carcinoma in mice (15, 16). Multiple groups reported that deletion of mutation of Pten in endometrial cells causes endometrial hyperplasia within 5 months (15); we reasoned that expression of cyclin D1T286A would cooperate with Pten loss to trigger endometrial carcinoma development and accelerate presentation of the phenotype. To test this hypothesis, we utilized Sprr2f-Cre transgene for conditional gene targeting in the endometrial epithelium (15, 17). We intercrossed mice harboring cyclin D1Fl/WT and/or PtenFl/WT with CreSprr2f, generating mice expressing cyclin D1T286A (cyclin D1TA/WT), Pten+/− (Pten+/−) and cyclin D1 TA/Pten+/− (cyclin D1TA/WT Pten+/−) only in endometrial cells.
Figure 3. Cyclin D1T286A accelerates Pten+/− induced endometrial hyperplasia development and progression.
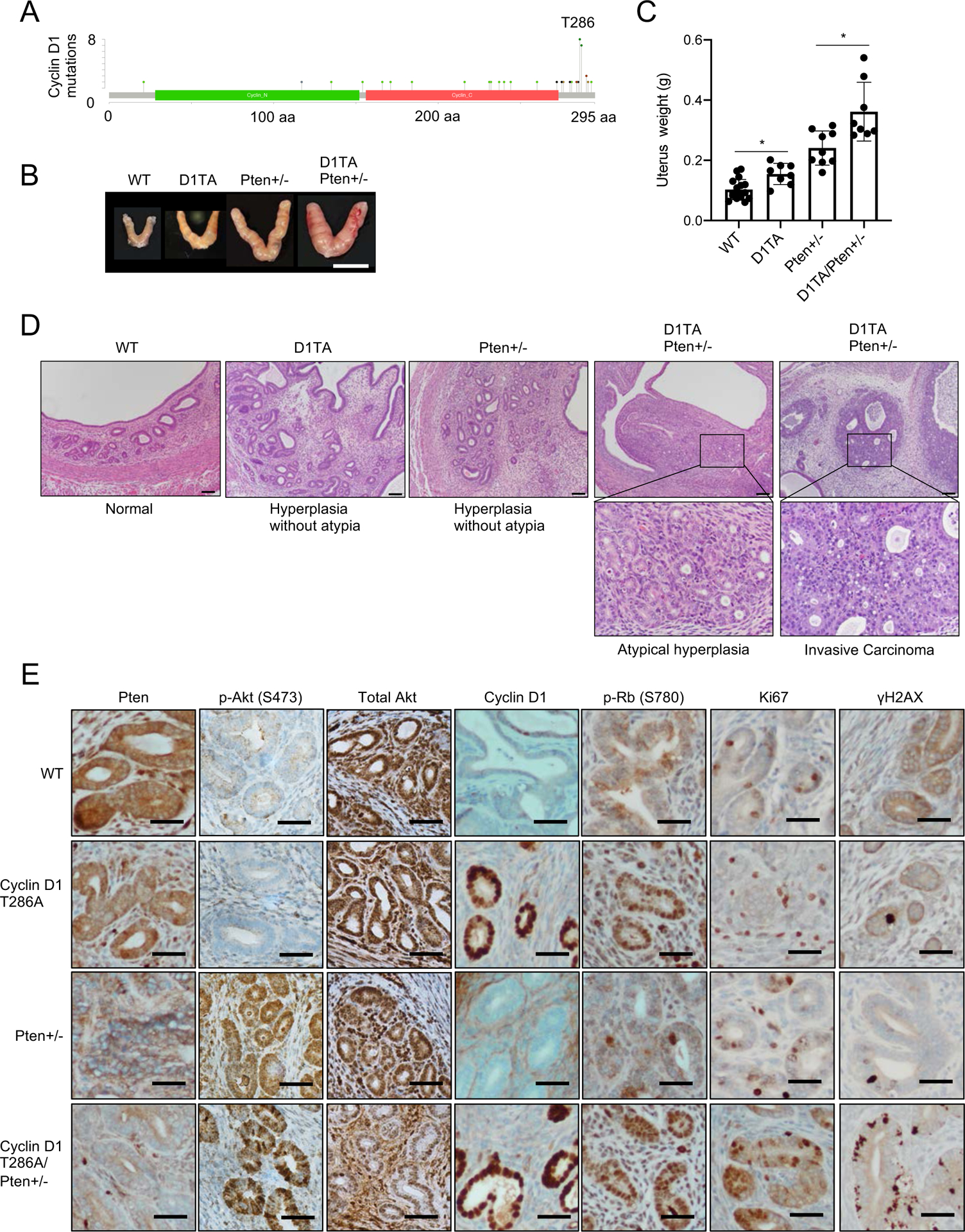
(A) The mutations of cyclin D1 observed in endometrial carcinoma (cBioportal.org). (B) Representative images of the uterus from 10-month-old WT, cyclin D1T286A (D1TA), Pten+/− and cyclin D1T286A (D1TA)/Pten+/−. Scale bar indicates 10 mm. (C) Average weights of the uterus from (B). Data represents mean ± SD, *p < 0.01 (two-tailed Student’s t-test; n=15 for WT, n=7 for D1TA, n=8 for Pten+/− and n=7 for D1TA/Pten+/−). (D) Representative images of H&E staining for uterus from (B). WT shows normal endometrial histology, while D1TA and Pten+/− mice exhibit endometrial hyperplasia (without atypia). Only D1TA/Pten+/− mice show endometrial neoplasia ranging from non-invasive endometrial intraepithelial neoplasia/atypical endometrial hyperplasia to invasive endometrial carcinoma. Scale bar indicates 100μm. Images of H&E staining from 7 tumors and sections of each genotype were observed. (E) Representative images for immunohistochemistry analysis of uteri from (B) using antibodies to Pten, phosphorylation of Akt (S473), total Akt, cyclin D1, phosphorylation of Rb (S780), Ki67 and γH2AX. Scale bar indicates 50μm.
By 10 months of age, cyclin D1TA/WT or Pten+/− mice exhibited a modest increase in uterine weight, while mice expressing cyclin D1TA/WT Pten+/− exhibited markedly increased uterine weight relative to wild type (Fig. 3B, C). Histopathological analysis revealed endometrial hyperplasia without atypia in cyclin D1TA/WT and Pten+/− mice while neoplastic lesions comprising atypical endometrial hyperplasia or carcinoma were observed only in cyclin D1TA/WT Pten+/− mice (Fig. 3D). Further histological analysis revealed that only cyclin D1TA/WT Pten+/− mice developed neoplastic endometrial lesions, equivalent to the spectrum of human endometrial neoplasia ranging from atypical endometrial hyperplasia/endometrial intraepithelial neoplasia to invasive endometrial carcinoma (Fig. 3D). At 10 months, cyclin D1TA/WT Pten+/− mice exhibited invasive carcinomas limited to the endometrium without evidence of myometrial invasion. In contrast, cyclin D1TA/WT or Pten+/− mice developed endometrial hyperplasia (without atypia) but no neoplasia (Fig. 3D). Together these data demonstrate endogenous cyclin D1TA expression accelerates Pten+/− induced endometrial hyperplasia and/or neoplasia.
Consistent with attenuation of Pten in Pten+/− and cyclin D1TA/WT Pten+/− mice, phosphorylated Akt (S473), a downstream target of PI3K signaling, is increased in Pten+/− uterus (Fig. 3E). IHC revealed accumulation of cyclin D1 in cyclin D1TA/WT and cyclin D1TA/WT Pten+/− mice along with increased phosphorylation of Rb (S780) and increased Ki67 positive staining (Fig. 3E). γH2AX positive cells in cyclin D1TA/WT Pten+/− mice was observed (Fig. 3E), analogous to the mesenteric tumors that developed in cyclin D1TA/WT mice. In contrast, phosphorylated ERK (Thr202/Tyr204) and total ERK were unchanged (Fig. S1E), indicating the observed phenotype of endometrial tumor development occurred through Pten loss and cyclin D1 accumulation. Taken together these results indicate that endogenous expression of cyclin D1T286A mutant promotes Pten+/− mediated endometrial hyperplasia and subsequently neoplasia.
NF-κB pathway is activated upon cyclin D1T286A expression.
To dissect the mechanism whereby cyclin D1TA/WT mice develop tumors we generated cyclin D1Fl/WT CreERT2 mouse embryonic fibroblasts (MEFs). This system permits us to induce cyclin D1T286A expression acutely upon 4-hydroxytamoxifen (4OHT) treatment (Fig. S2A, B). We knocked down pten in cyclin D1Fl/WT CreERT2 MEFs and treated cells with 4OHT to induce cyclin D1T286A expression (Fig. 4A). Knockdown of Pten had no significant impact on MAPK signaling in the cyclin D1T286A background (Fig. S2C). RNA sequencing revealed substantial changes in gene expression with distinct clusters among each group (Fig. 4B; Fig. S3A). Gene Set Enrichment Analysis (GSEA) and heat maps revealed that the p53 pathway is activated upon cyclin D1T286A expression, consistent with a DNA damage response in cyclin D1TA/WT mice and in cyclin D1TA/WT Pten+/− mice. Further analysis revealed that interferon alpha/beta and gamma pathways are activated after cyclin D1T286A induction compared with WT (Fig. 4C, Fig. S3B). Given that NFκB signaling produces cytokine production, induces an inflammatory response, and regulates interferon pathways (18, 19), we focused on the NFκB signaling pathway. GSEA and heat map analysis revealed that targets of NFκB pathway are upregulated following induction of cyclin D1T286A compared with WT regardless of the Pten status (Fig. 4D; Fig. S3B); in contrast, TNFα signaling was not differentially regulated (Fig. S3C).
Figure 4. NFkB signaling is activated upon cyclin D1 T286A expression.
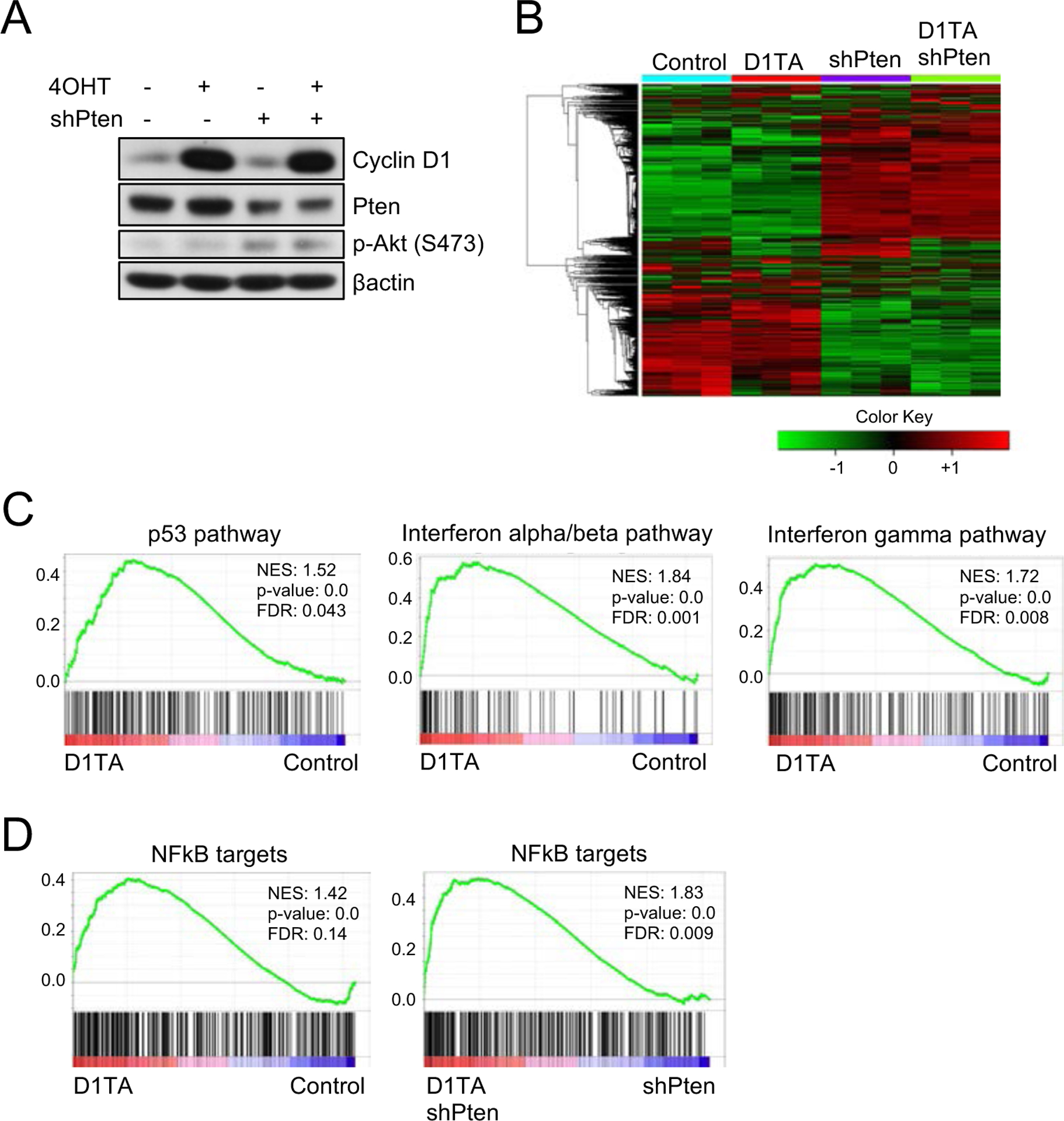
(A) Western blot analysis of lysates from primary cyclin D1 Flox/WT mouse embryonic fibroblasts (MEFs) with the treatment of 4-hydroxytamoxifen (4OHT), knockdown of Pten, or both using antibodies to cyclin D1, Pten, phosphorylation of Akt (S473) and βactin. (B) Heatmap with hierarchical clustering of samples from (A). (C-D) Gene Set Enrichment Analysis (GSEA) of primary MEFs expressing cyclin D1 T286A compared to WT MEFs for E2F targets, p53 pathway, Interferon alpha/beta pathway, Interferon-gamma pathway and NFkB targets.
Cyclin D1T286A regulates phosphorylation of p65 (RelA).
Given that p65 (RelA) is the main transcription factor that plays a central role in regulating inflammation, cell proliferation, and cell survival through cytokine production, chemokine production, and transcription (18), we investigated p65. Phosphorylated p65 (S536) was increased after cyclin D1T286A induction while the expression of total p65 and IKB was unchanged (Fig. 5A). Phosphorylated p53 (S15) was slightly increased (Fig. 5A). We next assessed whether increased phosphorylation of p65 is dependent on CDK4/6 kinase activity using the CDK4/6 inhibitor palbociclib. Palbociclib treatment reduced phosphorylation of p65 (Fig. 5B lane 2). Because palbociclib-mediated CDK4/6 inhibition is reversible, we removed the CDK4/6 inhibitor from the medium and monitored the status of p65 phosphorylation. Phosphorylation of p65 recovered at 24 hours after palbociclib withdrawal (Fig. 5B). Co-immunoprecipitation revealed the weak endogenous interaction of p65 and cyclin D1, suggesting the potential regulation of p65 by cyclin D1 (Fig. 5C). To assess whether p65 is a direct substrate for cyclin D1-CDK4/6 complex, we performed an in vitro kinase assay. Phosphorylation of p65 by cyclin D1T286A-CDK4/6 was not detected while a known substrate, Rb was strongly phosphorylated (Fig. S4) suggesting that p65 regulation by cyclin D1T286A is indirect.
Figure 5. p65 (RelA) is regulated by cyclin D1-CDK4/6 complex.
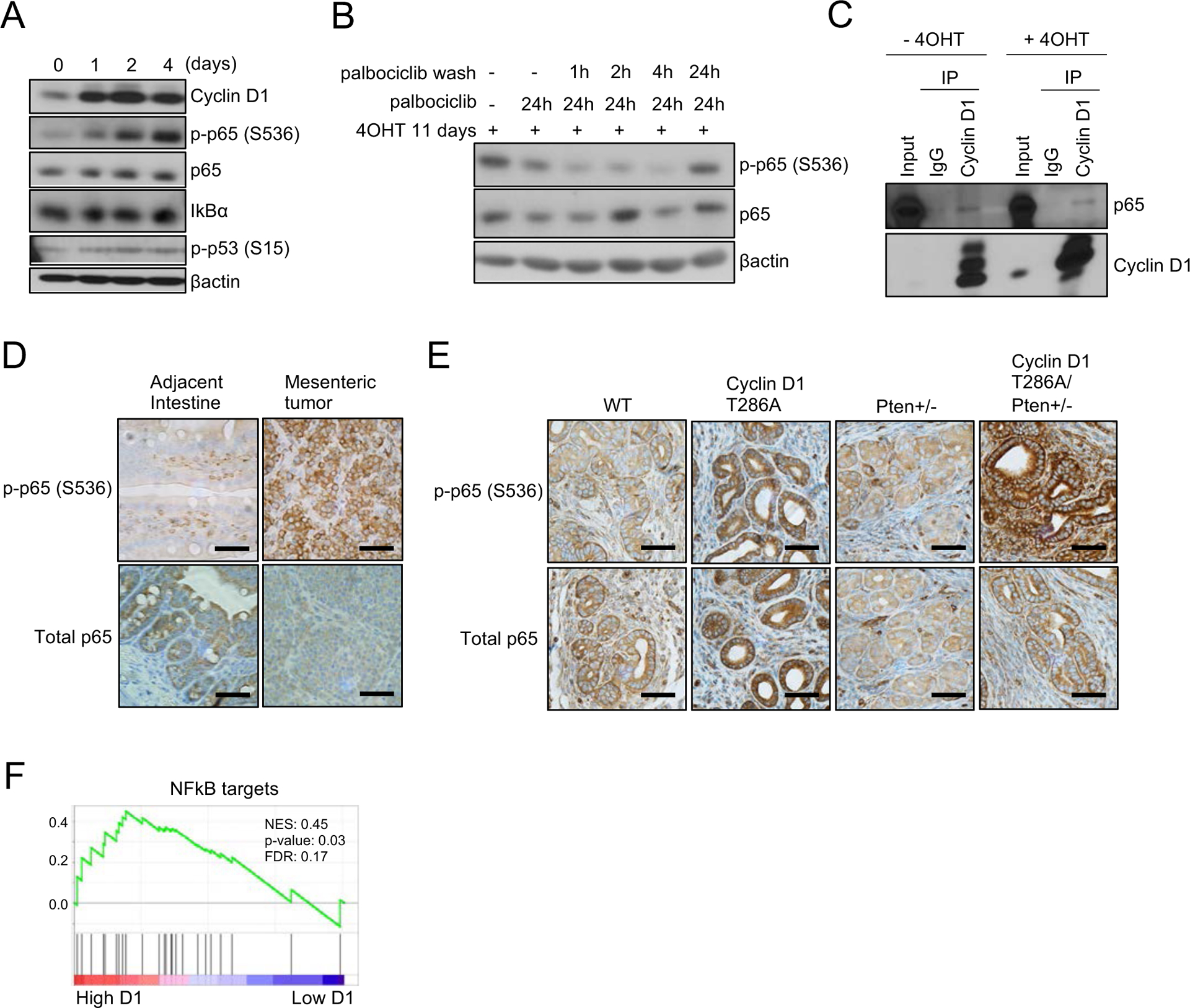
(A) Western blot analysis of lysates from primary cyclin D1 Flox/WT MEFs treated with 4-hydroxytamoxifen (4OHT) for 1 day, 2 days and 4 days using antibodies to cyclin D1, phosphorylation of p53 (S15), phosphorylation of p65 (S536), p65, IKB and βactin. (B) Primary cyclin D1 Flox/WT MEFs were treated with 4OHT for 11 days followed by palbociclib treatment (1 μM) for 24 hours, and then palbociclib was removed by washing cells. Lysates were harvested at 1, 2, 4, and 24 hours after palbociclib removal and subjected for western blot analysis using antibodies to phosphorylation of p65 (S536), p65 and βactin. (C) Lysates from primary cyclin D1 Flox/WT MEFs treated with or without 4OHT were subjected to immunoprecipitation with normal IgG or anti-cyclin D1 antibody. Immune complexes were analyzed by western blot for p65 and cyclin D1. (D) Representative images for immunohistochemistry analysis of mesenteric tumor and adjacent intestine using antibodies to phosphorylation of p65 (S536) and p65. Scale bar indicates 50μm. (E) Representative images for immunohistochemistry analysis of uterus from 10-month-old WT, cyclin D1T286A (D1TA), Pten+/− and cyclin D1 T286A (D1TA)/Pten+/− mice using antibodies to phosphorylation of p65 (S536) and p65. Scale bar indicates 50μm. (F) Gene Set Enrichment Analysis (GSEA) of human endometrial carcinoma with high expression of cyclin D1 compared to that with low expression of cyclinD1 for NFkB targets. Expression data for cyclin D1 in endometrial carcinoma were obtained from The Cancer Genome Atlas Program (TCGA) database.
We subsequently evaluated the relevance of increased p65 phosphorylation in two of our mouse models. IHC staining revealed that phosphorylation of p65 is upregulated in mesenteric tumors from cyclin D1TA/WT mice, and endometrial epithelial cells from cyclin D1TA/WT and cyclin D1TA/WT Pten+/− mice (Fig. 5D–E). We further noticed that endometrial epithelial cells from cyclin D1TA/WT Pten+/− mice have a stronger signal of phosphorylated p65 compared with endometrial epithelial cells from Pten+/− mice (Fig. 5E). This is consistent with a previous report that the PI3K-Akt signaling pathway regulates phosphorylation of p65 (20). Finally, we assessed whether there is increased activation of NFκB signaling in human endometrial tumors with high expression of cyclin D1 compared to tumors with low expression of cyclin D1 using publicly available data. GSEA analysis revealed that cyclin D1 expression is positively correlated with NFκB targets in endometrial cancers (Fig. 5F), consistent with the observation in our mouse models. Together, these results indicate that the expression of cyclin D1T286A activates NFκB signaling, which in turn, induces inflammation and lymphocytes hyperplasia in cyclin D1TA/WT mice or endometrial hyperplasia in cyclin D1TA/WT CreSprr2f mice.
Discussion
Mutant cyclin D1T286A, a non-phosphorylatable mutant accumulates in the nucleus, triggers DNA re-replication, induces genomic instability, and in so doing initiates tumorigenesis in model systems. A shortcoming of the experimental systems used to date reflects the use of overexpression systems such as transgenic mice. Use of such an overexpression system can be limited given expression of mutant cyclin D1 in human cancer is driven by its own promoter. Here we describe a novel knock-in mouse model where expression of cyclin D1T286A is regulated by its endogenous promoter upon Cre recombinase activation permitting investigation of its expression under conditions more accurately reflecting human disease. Consistent with cyclin D1T286A functioning as a driver oncogene in human cancer, knock-in mice develop overt malignancy by 9 months post-activation of cyclin D1T28A expression with a majority of the mice eventually developing mesenteric tumors within 24 months. Mesenteric tumors contain a population mixed with T cells and B cells including massive inflammation in mice. While O-GlcNAcylation is reported to regulate cyclin D1 stability (21), we have not observed changes in O-GlcNAcylation. Modification of T286 is not linked with any other modifications besides ubiquitin in our studies that include mas-spec analysis.
CCND1 mutations co-occur with PTEN loss in uterine cancers suggesting cooperativity in this malignancy (cBioportal.org). Indeed, mice harboring cyclin D1T286A or Pten+/− develop endometrial hyperplasia without atypia, while mice both mutant D1 and loss of one pten allele develop invasive carcinoma. Given the strong DNA damage response, we considered whether p53 mutation might be selected for; however, only one p53 mutant tumor was confirmed while others retained wild type p53. In addition, the remaining allele of Pten is not mutated or deleted in cyclin D1TA Pten+/− mice. The retention of wild type p53 likely reflects the ability of cyclin D1T286A to activate PRMT5 which can inactivate p53 by direct arginine methylation (12).
We discovered that the NFκB pathway is activated upon cyclin D1T286A expression. Although the phosphorylation of p65, a transcription factor that plays a central role in regulating NFκB signaling, is increased after acute expression of cyclin D1T286A mutant, we did not observe the direct phosphorylation of p65 by cyclin D1 and CDK4/6. Although we did observe an interaction between p65 and cyclin D1 (Fig. 5C), the molecular mechanisms whereby p65 is regulated by cyclin D1 and CDK4/6 remains to be elucidated. One possible mode of p65 regulation could be through PRMT5. PRMT5 dimethylates arginine 30 of the p65 to drive the gene expression (22). Further analysis is needed to investigate the cooperation among cyclin D1, PRMT5 and p65 in regulating inflammation and tumorigenesis.
Cyclin D1T286A expression accelerates Pten+/− induced endometrial hyperplasia in mice. These observations are consistent with previous studies that loss of Pten induces NFκB signaling through Akt, which results in resistance to chemotherapy, lung fibrosis and prostate cancer development (23–26). Since an inflammatory signature can promote both tumor development and metastasis, acute expression of cyclin D1T286A might accelerate cancer invasion through NFκB signaling-mediated inflammatory signature. In conclusion, endogenous expression of cyclin D1T286A mutant induces NFκB signaling mediated inflammation and promotes tumor development. This study strongly supports the utilization of a CDK4/6 inhibitor in a clinical setting to suppress cell cycle progression, cell proliferation, and inflammation in cancers.
Materials and Methods
Animal husbandry
Mouse experiments were conducted in accordance with Institutional Animal Care and Committee (IACUC) protocols and Animal Resource Center (ARC) at Case Western Reserve University. Cre-ERT2 and Pten knockout mice were obtained from Jackson Laboratory. Sprr2f-Cre mice were provided by Dr. Leone. Cyclin D1Flox/WT mice were generated in Cyagen Biosciences Inc. Tamoxifen (Sigma) was administered orally to Cyclin D1Flox/WT Cre-ERT2 once a day for 5 days at a dose of 200mg/kg of body weight to generate Cyclin D1TA/WT mice. 1–2 mg of BrdU was administered by intraperitoneal injection two hours prior to analysis. The genotypes and DNA recombination were verified by genomic PCR. PCR primers are as follows: cyclin D1Flox/WT F1: 5’-GTGGAAGGCTTCCAGGTGTGAC-3’, R1: 5’-CCTAGTATGGACCCACAGACACCC-3’, F3: 5’-GCCCCATTCCTACATATCTCA-3’ and R3: 5’-TGGATCCACCTAATAACTTCGTA-3’.
Cell culture
Authenticated human embryonic kidney (HEC) 293T cells were purchased from ATCC and cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. For viral production, viral expression plasmids were transfected into 293T cells with lipofectamine together with packaging plasmid. Virus supernatants harvested 48 to 72 hours after transfection were used to infect melanoma cells with polybrene (10μg/mL). Mouse Embryonic Fibroblasts (MEFs) harboring flox allele of cyclin D1T286A were generated using embryos at day 13.5 according to the standard procedure and cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, 2mM L-Glutamine and MEM Non-Essential Amino Acids Solution.
Immunohistochemistry staining
Tumors from mice were fixed with 4% PFA, dehydrated, and embedded in paraffin followed by sectioned with a microtome. Sections were blocked with 10% goat serum, incubated with the primary antibodies, and subsequently incubated with biotinylated antibodies. Signal was developed with ABC substrate kit (Vector) followed by DAB reaction (Vector), and counterstained with Hematoxylin (Thermo Scientific). Antibodies for p-Rb (Ser780), p-Chk1 (Ser345), Pten, p-Akt (Ser473), Akt, and γH2AX were obtained from Cell Signaling Technology. Antibodies for Ki-67 (ab15580), p-Chk2 (Thr68), CD3 and CD45R were purchased from Abcam. BrdU antibody was obtained from BD Biosciences. Cyclin D1 (72–13G) was generated in our laboratory.
Flow cytometry
For the BrdU incorporation assay, cells were cultured in a medium containing 10 μM BrdU for 40 min and stained with an anti-BrdU mouse monoclonal antibody followed by 7-AAD staining according to the manufacturer’s instructions (BD Biosciences). BrdU positive cells were measured with BD LSRFortessa (BD Biosciences).
qRT-PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen), and reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. qRT-PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and the data were normalized to GAPDH.
Protein analysis
Cells were lysed in EBC buffer (50 mM Tris pH 8.0, 120 mM NaCl, 1 mM EDTA, 0.5% NP40) containing 1mM phenylmethylsulfonyl fluoride, 20 U/ml aprotinin, 1μM leupeptin, 1mM dithiothreitol, 0.1mM NaF, 0.1mM Sodium orthovanadate, and 10mM β-glycerophosphate. Proteins were resolved by SDS-PAGE, transferred to a membrane, and immunoblotted with the indicated antibodies as follows; CDK4 (H-303) was obtained from Santa Cruz. Cyclin D3 (DCS22), p-p53 (Ser15), p-p65 (Ser536), p65 (D14E12) and IkBα were purchased from Cell Signaling Technology. Raptor was purchased from Bethyl laboratories. β actin (AC-15) was purchased from Sigma. Cyclin D1 (72–13G) was generated in our laboratory. Membranes were incubated with horseradish peroxidase-conjugated anti-mouse or rabbit antibodies and signals were developed with the ECL system (PerkinElmer) according to the manufacturer’s instructions. For kinase assay, Recombinant GST-RB was purified as described previously (27, 28) and recombinant p65 was purchased from Active Motif. Purified cyclin D1/CDK4 or cyclin D1T286A/CDK4, 1 μg of protein substrate, 2.5mM EGTA, 10 mM β-glycerophosphate, 0.1mM sodium orthovandadate, 1.0 mM NaF and 10 μCi [Γ-P32] ATP were mixed in kinase buffer (50 mM Tris-HCl pH7.5, 10 mM MgCl2, and 1 mM DTT). The Samples were incubated in 1.5 ml microfuge tubes for 30 minutes. The reaction was terminated by the addition of 10 μl of SDS sample buffer, mixed, and then heated to 90° C for 10 minutes. Subsequently, samples were applied to an SDS gel for electrophoresis. The destained gel is then dried onto filter paper and the dried gel is then subjected to autoradiography to visualize any [P32] transferred to the substrate protein.
RNA-seq and data analyses
Quality control of total RNA samples is executed using Qubit Fluorometer (Invitrogen) for RNA quantification and Fragment Analyzer (Agilent) to assess RNA quality using a cut-off of RIN > 7.0 to select specimens for further analysis. For library preparation, the New England BioLabs NEBNext Ultra II Directional RNA Library Prep Kit for Illumina with NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) is used. This protocol starts by using the rRNA Depletion kit to remove ribosomal RNA (rRNA) from 250ng of total RNA using probes targeting all rRNAs. The resulting purified mRNA is used as input for the Ultra II Directional kit in which libraries are tagged with unique adapter-indexes. Final libraries are validated on the Fragment Analyzer, quantified via qPCR, and pooled at equimolar ratios. Pooled libraries are diluted, denatured, and loaded onto the Illumina NextSeq system, following the NextSeq User Guide for a single read 75 cycle run. Sequencing reads generated from the Illumina platform were assessed for quality and trimmed for adapter sequences using TrimGalore! v0.4.2 (Babraham Bioinformatics), a wrapper script for FastQC and cutadapt. Reads that passed quality control were then aligned to the mouse reference genome (mm10) using the STAR aligner v2.5.3 (29). The alignment for the sequences was guided using the GENCODE annotation for mm10. The aligned reads were analyzed for differential expression using Cufflinks v2.2.1 (30), and the RNASeq analysis package that reports the fragments per kilobase of exon per million fragments mapped (FPKM) for each gene. Differential analysis report was generated using the cuffdiff command performed in a pairwise manner for each group. Differential genes were identified using a significance cutoff of q-value < 0.05 (B&H corrected for multiple testing). The genes were then subjected to gene set enrichment analysis (GenePattern, Broad Institute) to determine any relevant processes that may be differentially overrepresented for the conditions tested. Additional pathway analysis was performed using iPathwayGuide (Advaita Bio). The datasets were deposited in the GEO database (GEO accession: GSE188299).
Statistical analysis
The normality and equal variance of data were evaluated by the Shapiro-Wilk test and F-test, respectively. When two groups were normally distributed, the parametric test was applied such as Student’s t-test for equal variance and Welch’s t-test for unequal variance. When the group was not normally distributed, a non-parametric test such as the Mann-Whitney U test was applied. For the xenograft experiment, an unpaired two-tailed student’s t-test was applied after data were transformed to log values. The error bars represent standard deviation (SD) of the mean and significance was defined as a p-value less than 0.05 with two-tailed unless otherwise specified. Details and significant values are described in the figure legends.
Supplementary Material
Significance statement:
Non-phosphorylatable cyclin D1T286A, induces inflammation-mediated lymphocyte disorder and mesenteric tumor formation. Uterine-specific expression of cyclin D1T286A accelerates Pten loss driven endometrial hyperplasia to promote uterine cancer.
Acknowledgments
We thank assistance from Dr. E. Ricky Chan in the Institute for Computational Biology for RNA sequencing analysis, Simone Edelheit in the Genomics Core for RNA sequencing sample preparation, and the Animal Resource Center at Case Western Reserve University. We thank assistance from Melodie Parrish in the Translational Science Resource at The Medical University of South Carolina for Hematoxylin and Eosin staining and immunohistochemistry staining. This work was supported by grants from National Cancer Institute: R01 CA093237 and P01 CA098101 (J.A.D.); R01 CA121275 and Advancing a Healthier Wisconsin Endowment Award (G.L.).
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79(4):551–5. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672–7. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73(6):1059–65. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. [DOI] [PubMed] [Google Scholar]
- 5.Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–39. [DOI] [PubMed] [Google Scholar]
- 6.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24(3):355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes & development. 1997;11(8):957–72. [DOI] [PubMed] [Google Scholar]
- 8.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes & development. 1998;12(22):3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gladden AB, Woolery R, Aggarwal P, Wasik MA, Diehl JA. Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene. 2006;25(7):998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes & development. 2007;21(22):2908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pontano LL, Aggarwal P, Barbash O, Brown EJ, Bassing CH, Diehl JA. Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability. Mol Cell Biol. 2008;28(23):7245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18(4):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Chitnis N, Nakagawa H, Kita Y, Natsugoe S, Yang Y, et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015;5(3):288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ando K, Ajchenbaum-Cymbalista F, Griffin JD. Regulation of G1/S transition by cyclins D2 and D3 in hematopoietic cells. Proceedings of the National Academy of Sciences. 1993;90(20):9571–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caserta E, Egriboz O, Wang H, Martin C, Koivisto C, Pecót T, et al. Noncatalytic PTEN missense mutation predisposes to organ-selective cancer development in vivo. Genes & development. 2015;29(16):1707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer research. 2008;68(14):5619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, et al. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Disease models & mechanisms. 2010;3(3–4):181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal transduction and targeted therapy. 2017;2:17023-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Molecular cancer. 2013;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon HJ, Choi GE, Ryu S, Kwon SJ, Kim SC, Booth C, et al. Stepwise phosphorylation of p65 promotes NF-κB activation and NK cell responses during target cell recognition. Nature communications. 2016;7:11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masclef L, Dehennaut V, Mortuaire M, Schulz C, Leturcq M, Lefebvre T, et al. Cyclin D1 Stability Is Partly Controlled by O-GlcNAcylation. Front Endocrinol (Lausanne). 2019;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(33):13516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes & development. 2008;22(11):1490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustin JA, Maehama T, Dixon JE, Donner DB. The PTEN tumor suppressor protein inhibits tumor necrosis factor-induced nuclear factor kappa B activity. The Journal of biological chemistry. 2001;276(29):27740–4. [DOI] [PubMed] [Google Scholar]
- 25.Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, et al. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell. 2019;18(1):e12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu L, Zhu N, Findley HW, Zhou M. Loss of PTEN Expression Induces NF-kB Via PI3K/Akt Pathway Involving Resistance to Chemotherapy in Acute Lymphoblastic Leukemia Cell Lines. Blood. 2004;104(11):4438-. [Google Scholar]
- 27.Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71(2):323–34. [DOI] [PubMed] [Google Scholar]
- 28.Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14(3):2066–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England). 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.