

Abstract
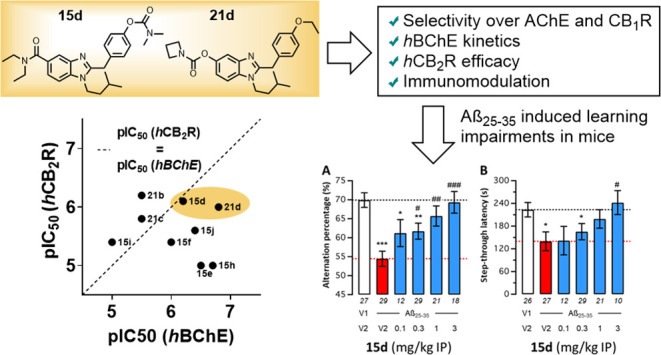
We present the synthesis and characterization of merged human butyrylcholinesterase (hBChE) inhibitor/cannabinoid receptor 2 (hCB2R) ligands for the treatment of neurodegeneration. In total, 15 benzimidazole carbamates were synthesized and tested for their inhibition of human cholinesterases, also with regard to their pseudoirreversible binding mode and affinity toward both cannabinoid receptors in radioligand binding studies. After evaluation in a calcium mobilization assay as well as a β-arrestin 2 (βarr2) recruitment assay, two compounds with balanced activities on both targets were tested for their immunomodulatory effect on microglia activation and regarding their pharmacokinetic properties and blood–brain barrier penetration. Compound 15d, containing a dimethyl carbamate motif, was further evaluated in vivo, showing prevention of Aβ25–35-induced learning impairments in a pharmacological mouse model of Alzheimer’s disease for both short- and long-term memory responses. Additional combination studies proved a synergic effect of BChE inhibition and CB2R activation in vivo.
Introduction
Alzheimer’s disease (AD) has first been described by German neuropathologist and psychiatrist Alois Alzheimer in 1906, following his contact with patient Auguste Deter starting in 1901. He described Mrs. Deter’s symptoms with a “rapidly worsening memory weakness” and “pronounced psychosocial impairment” while being disorientated and confused. After her death, Alzheimer examined her brain and identified the histological features that are still associated with the disease today, e.g., the presence of amyloid plaques and neurofibrillary tangles. While his first lecture discussing the pathological finding in 1906 was barely noticed, the term “Alzheimer’s disease” has nowadays become the term for the majority of dementia cases associated with loss of neurons and synapses in the cerebral cortex.1,2
Over a century later, several drugs have been approved by the U.S. Food and Drug Administration to improve the life and symptoms of AD patients. Unfortunately, none of these drugs can slow down the degradation of neurons as these drugs only enable symptomatic medication. Factors that have been identified in disease progression are extracellular deposits of amyloid beta (Aβ) in senile plaques, hyperphosphorylation of tau proteins, and oxidative stress.3−7 However, memory loss and a significant interference with the patient’s ability to perform everyday activities are primarily caused by substantial deficits of the neurotransmitter acetylcholine (ACh) due to a loss of cholinergic neurons.4,8,9 Therefore, inhibition of ACh esterase (AChE), the enzyme responsible for ACh hydrolysis, is focused on to treat AD symptomatically. Unfortunately, substantial side effects, ranging from convulsion to syncope and pneumonia, limit the use of current AD medication.10 Furthermore, with disease progression, levels of AChE seem to decrease as much as 90%, undermining an effective treatment in the later stages of the disease.11,12
Different to AChE, levels of butyrylcholinesterase (BChE) seem to increase during disease progression, marking it as a promising target for mid- and late-stage treatment of AD.22 Several studies by us and others could already underline beneficial effects of BChE inhibition in vivo regarding neuroprotective properties.14,18,19,21,23−26
Besides targeting of cholinesterases (ChEs), the endocannabinoid system, especially activation of the cannabinoid receptor type 2 (CB2R), has shown to have beneficial effects on neuroinflammation for the treatment of AD.18,19,27−30 The CB2R is mainly expressed on immune cells, and its expression correlates with the aggregation of Aβ plaques, Aβ1–42 concentration, and levels of hyperphosphorylated tau.31,32 The beneficial influence of CB2R agonists on AD progression is assumed to derive from their ability to inhibit synaptic transmission and therefore regulate neurogenic inflammation by reduction of inflammatory peptide release and afferent firing.33 This can be described by the effects of CB2R agonists on microglia, which are active propagators of neuroinflammation. Microglia can be divided into two phenotypes, the pro-inflammatory M1 state and the anti-inflammatory M2 state. As CB2 receptor activation mediates the immunosuppressive, anti-inflammatory effects of the M2 phenotype and attenuates the expression of pro-inflammatory M1 state biomarkers, agonists are expected to have beneficial effects on neurodegenerative diseases, such as AD.34,35
However, as AD is a multifactorial disorder, a multitarget medication may deliver the best results for both symptomatic and causal therapy, resulting in a decelerated disease progression and improved cognitive abilities of patients suffering from AD.36,37 To reach these goals, a combination of both a selective hBChE inhibitor and a hCB2R agonist was aimed for. Although both targets and potential hybrids have been evaluated in the context of AD treatment, a synergistic effect has not yet been proven in vivo, which we wanted to address within the context of this work.38,39
For the design of such hybrids, careful selection of utilized pharmacophores is vital to obtain active hybrid molecules with “balanced” activities. One of the most potent classes of BChE inhibitors (BChEIs) is represented by carbamates, which act in a pseudoirreversible manner, whereas a part of the molecule is transferred onto the ChE.40 Rivastigmine (1) and several tri- and tetracyclic BChEIs, exemplified by compounds 2 and 3, exhibit submicromolar inhibition of the enzyme with rivastigmine being approved for the treatment of AD (Figure 1).13,18,20 To address another disease-related target, also acting in a neuroprotective manner based on a different mechanism, CB2R is regarded as a highly promising target.28,31,41 Therefore, several highly potent CB2R agonists have been reported in the last decades, with compound 4 showing weak inhibition of eqBChE, creating the starting point for our investigations into BChE/CB2R hybridization.42,43 Besides hybridization by introducing a linker between two pharmacophores, as described for compound 5, merged approaches by using a single entity, yielding interesting dual-acting compounds acting on CB2R and BChE, have been reported for compounds 6–8 (Figure 1).14−16,44−46 Unfortunately, these approaches suffer from either low-affinity, non-agonistic behavior on CB2R or an imbalanced profile on their respective targets.
Figure 1.
Key structures targeting BChE and/or CB2R. (a) Carbamate-based ChEIs; (b) benzimidazole CB2R agonist developed by AstraZeneca; and (c) selection of recently reported CB2R/BChE hybrid molecules.13−21
To address this issue, using the core structure of CB2R agonist 4, Dolles et al. designed several benzimidazole-based compounds, wherein docking and binding studies on hBChE and hCB2R revealed moderate inhibition of hBChE while retaining hCB2R affinity, proving that the 2-benzylbenzimidazole core structure is able to interact with hBChE also.14,42 To efficiently introduce the inhibitory effects of pseudoirreversible hBChEIs to the dual-acting core structure of compounds 4 and 6, we utilize small carbamate moieties to obtain hybrid molecules with low molecular weight and high effectiveness on both targets.26,47
Respective molecules consist of the 2-benzylbenzimidazole core, modified on one of its phenolic positions by small, lipophilic carbamates to fit into the orthosteric pocket of cannabinoid receptors, as well as interacting with the active center of hBChE (Figure 2). We explored this scaffold first in 2022, designing and obtaining BChE-selective inhibitors but lacking CB2R activity.21 For the construction of BChE/CB2R hybrids, we utilized smaller carbamate units in different positions of the benzimidazole core, mimicking benzimidazole-based agonists, described by Pagé et al., to introduce CB2R affinity to the benzimidazole-carbamate scaffold of compound 3.17
Figure 2.

Target structures representing benzimidazole-carbamates 15a–j and 21a–e.
Results and Discussion
Chemistry
To allow carbamate introduction at the last synthetic step, it was important to obtain a phenolic group close to the end of synthesis. As a phenol is considered a good nucleophile under basic conditions that can cause undesired side reactions, especially during coupling reaction (iv) and cyclization (v), the introduction of a protection group cleavable with high yields is key to the reaction sequence.
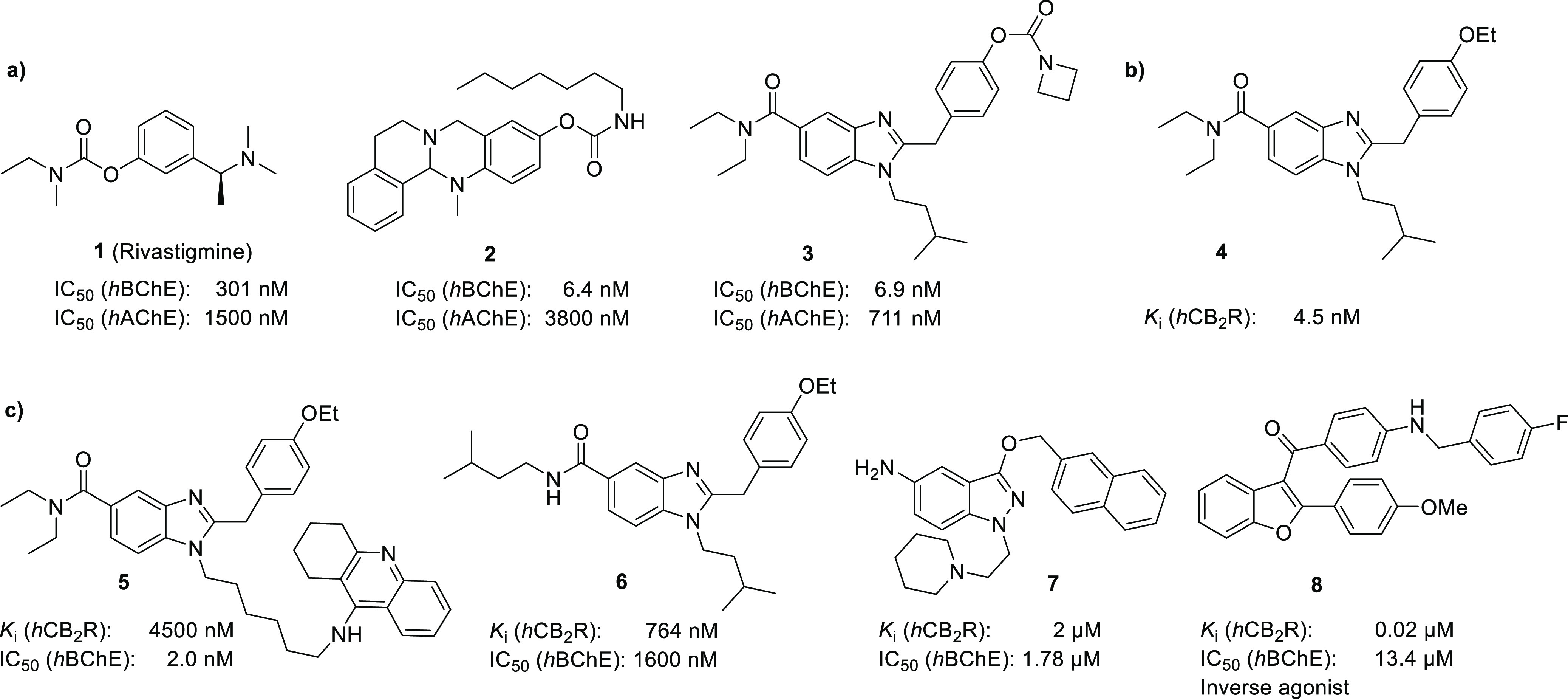
For synthesis of compounds 15a–j, the benzyl protection group was considered optimal as it is robust to both basic and acidic conditions used for workup and reaction methods applied. Therefore, a seven-step synthesis from commercially available chemicals was developed (Scheme 1).
Scheme 1. Synthesis of Hybrid Set 1 (15a–j).
Reagents and conditions: (i) (a) oxalyl chloride, DMF, 0 °C, 1 h; (b) diethylamine, TEA, 0 °C to rt, 4 h; (ii) isopentylamine, TEA, 55 °C, 3 h; (iii) hydrogen, Pd/C, EtOH, rt, overnight; (iv) 2-(4-(benzyloxy)phenyl)acetic acid, HBTU, TEA, DMF, rt, 3 h; (v) acetic acid, 130 °C, 3 h; (vi) hydrogen, Pd/C, MeOH, rt, overnight; (vii) (a) isocyanates, TEA, DCM, rt, 12 h or (b) carbamyl chlorides, NaH, THF, rt, 12 h or (c) 4-nitrophenylbenzyl(methyl)carbamate, tert-butoxide, THF, 55 °C, 12 h.
First, 4-fluoro-3-nitrobenzoic acid was converted to the corresponding acid chloride and reacted with dimethylamine, forming N,N-diethyl-4-fluoro-3-nitrobenzamide 9. Following nucleophile aromatic substitution with isopentylamine yielded N,N-diethyl-4-(isopentylamino)-3-nitrobenzamide 10 in high yields. Reduction of the nitro group was carried out using hydrogen and palladium on carbon as the catalyst, making any further purification besides simple filtration unnecessary. The corresponding aniline 11 was then coupled with benzyl-protected 4-hydroxyphenylacetic acid. In this reaction step, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and triethylamine (TEA) were used to form the active ester that was reacted with aniline 11 to obtain diamide 12. Cyclization to the benzimidazole 13 was carried out by reflux in acetic acid. Then, deprotection of the benzyl ether using hydrogen and palladium on carbon yielded the key intermediate 14 holding the phenol ready for subsequent functionalization. To obtain the desired carbamates 15a–j, three different procedures were used. For aryl-OCO-NHalkyl carbamates 15a–c, as well as benzyl derivative 15h, phenol 14 was treated with TEA and reacted with the respected isocyanates in yields from 45 to 88%, applying mild conditions which are sufficient due to high reactivity of the isocyanates. Aryl-OCO-N(alkyl)2 carbamates 15d–g, as well as derivative 15i, were synthesized by treating phenol 14 with sodium hydride and addition of the respective carbamoyl chlorides, enabling yields from 77 to 92%. As the carbamoyl chlorides are less reactive than the isocyanates used for the synthesis of aryl-OCO-NHalkyl carbamates, a stronger, non-nucleophilic base was needed to accelerate conversion. Due to a lack of chemicals commercially available for direct conversion, N-benzylmethyl was first treated with 4-nitrophenyl chloroformate to yield the corresponding 4-nitrophenyl dialkylcarbamate which was subsequently treated with phenol 14. Using KOtBu and elevated temperatures, a yield of 38% for the N-benzylmethyl derivative 15j was obtained. As elevated temperatures were vital for conversion, KOtBu proved to be sufficient and led to higher yields when compared to sodium hydride which promoted partial hydrolysis of the target molecules under the conditions applied (Scheme 1).
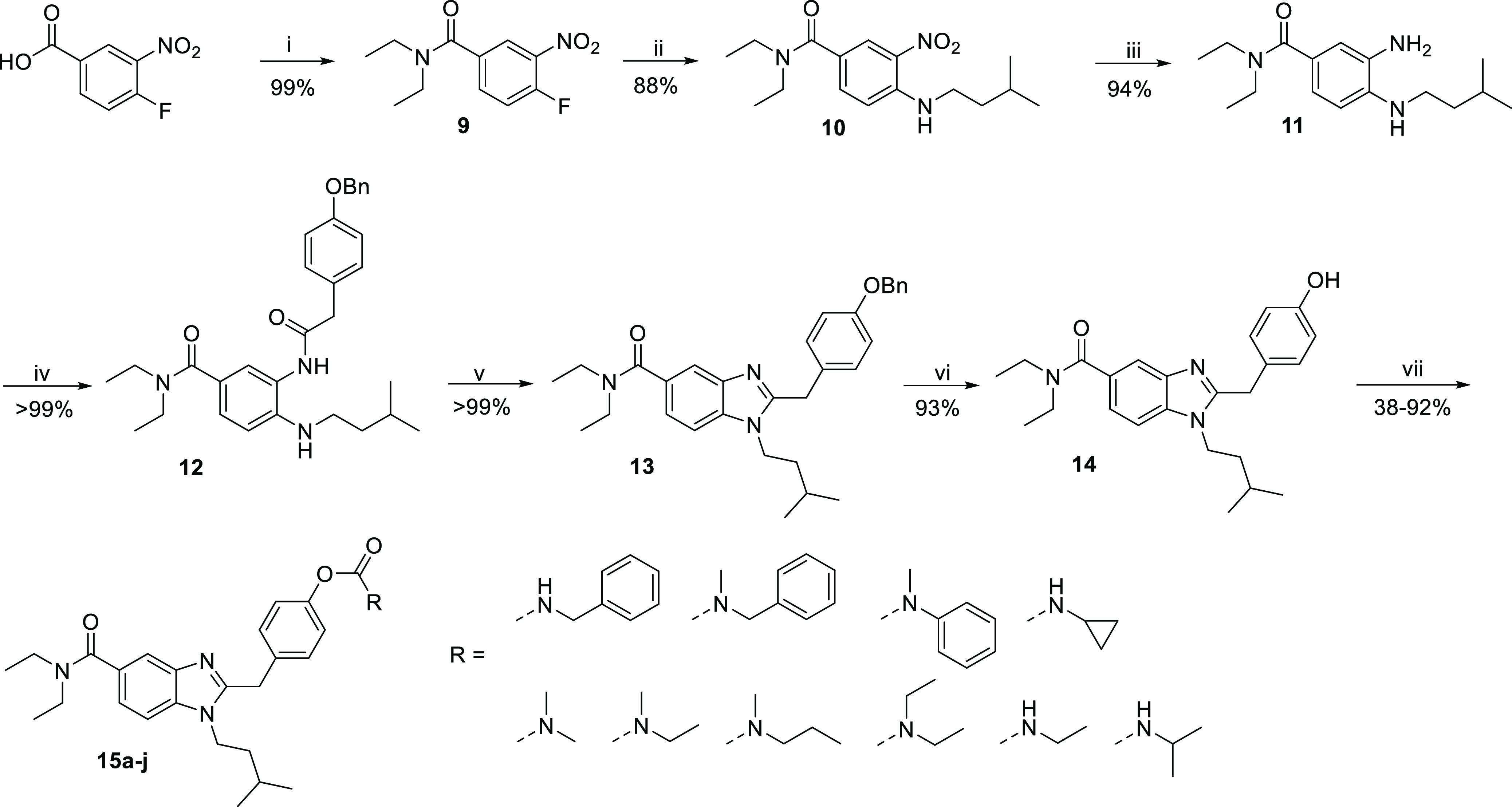
In a related fashion, a carbamate was introduced at the benzimidazole replacing the amide. Instead of the benzyl group, a MOM group was introduced in the first step to protect the phenol as it could simultaneously be cleaved off during cyclization. Subsequent introduction of isopentyl amine via nucleophilic aromatic substitution led to compound 17. Reduction of the nitro group using hydrogen and palladium on carbon as catalyst, making no further purification besides filtration necessary, led to compound 18. The obtained dianiline 18 was then coupled with 2-(4-ethoxyphenyl)acetic acid using HBTU and TEA, followed by cyclization of refluxing amide 19 in acetic acid. As the crude product still contained the MOM protection group, trifluoroacetic acid (TFA) was added to obtain phenol 20. Aryl-OCO-N(alkyl)2 carbamates (21a–c) were synthesized in yields of 44–76% using the respective carbamoyl chlorides and sodium hydride. Azetidine carbamate 21d was synthesized in a yield of 41% using 4-nitrophenyl azetidine carboxylate and KOtBu at 55 °C. Cyclopropyl carbamate (21e) was obtained using cyclopropyl isocyanate and TEA, yielding 43% of compound 21e (Scheme 2).
Scheme 2. Synthesis of Hybrid Set 2 (20a–e).
Reagents and conditions: (i) MOMCl, potassium carbonate, DIPEA, acetone, 0 °C to rt, 4 h; (ii) isopentylamine, TEA, EtOH, 55 °C, overnight; (iii) hydrogen, Pd/C, EtOH, rt, overnight; (iv) 2-(4-ethoxyphenyl)acetic acid, HBTU, TEA, DMF, rt, 2 h; (v) (a) acetic acid, 130 °C, 2 h; (b) trifluoro acetic acid, rt, 30 min; (vi) (a) cyclopropyl isocyanate, TEA, rt, overnight or (b) carbamoyl chlorides, NaH, THF, rt, 12 h or (c) 4-nitrophenylazetidine carboxylate, tert-butoxide, THF, 55 °C, 12 h.
Pharmacological Profile
Inhibition of ChEs and Radioligand Binding Studies on CBRs
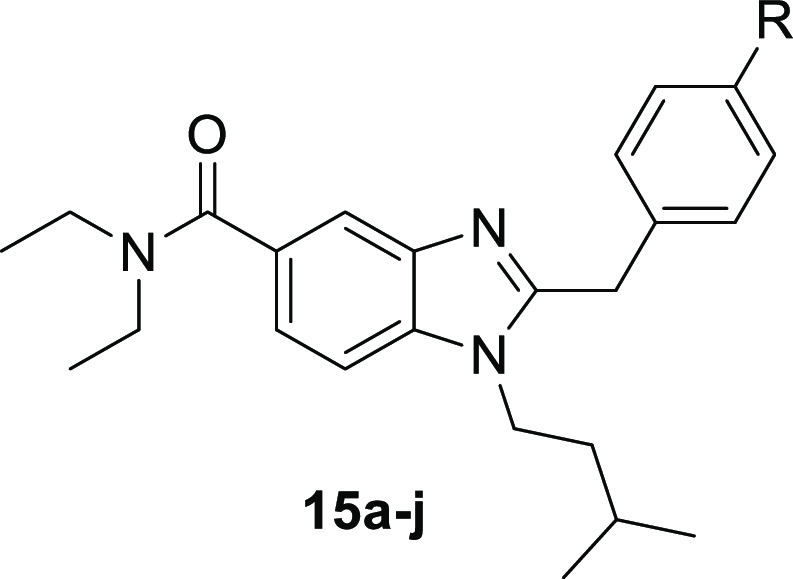
All target compounds were tested for their ability to inhibit hBChE, as well as hAChE to evaluate influence of various small carbamate units introduced at the respective phenol moiety. Therefore, the established Ellman’s assay using hChEs and either butyrylthiocholine (BTC) or acetylthiocholine (ATC) as the substrate was used to calculate IC50 values on both enzymes. The use of human enzyme for both ChEs is vital due to a different activity profile when compared to eqBChE or eeAChE, reported by Dolles et al.14,42 The introduction of various small carbamate units led to an interesting pharmacological profile for this novel class of benzimidazole-based carbamates, ranging from two-digit micromolar to two-digit nanomolar inhibition of hBChE. Dialkylated, aliphatic, acyclic carbamates (15d–g) exhibit better inhibition for smaller groups, making dimethylated compound (15d) significantly more potent than diethylated compound (15g). This trend is then reversed for sterically less demanding, monoalkylated compounds (15a–c). The tremendous loss in activity between the isopropyl carbamate (15b) and cyclopropyl carbamate (15c) could be explained by a steric clash of the α-methyl unit which is significantly reduced in its cyclic form. While clear trends are observed for aliphatic compounds, carbamates containing an aromatic unit (15h–j) exhibit a more ambiguous profile. While benzylic compound 15h has an IC50 value of 186 nM, N-methylbenzyl carbamate 15j is about two-fold weaker active while keeping nearly the same selectivity for hBChE. This shows a resemblance to N-methylethyl carbamate 15e which has a similar IC50 value but a higher selectivity for hBChE, suggesting that aromatic units close to the carbamate influence hAChE selectivity. This is further underlined by N-methylphenyl carbamate (15i), the only ligand with a higher selectivity for hAChE than that for hBChE (Table 1).
Table 1. In Vitro Results of the Inhibition of hBChE, hAChE, and Radioligand Binding to hCB2R, rCB1R of Compounds 14, 15a–ja.
n.d. not determined.
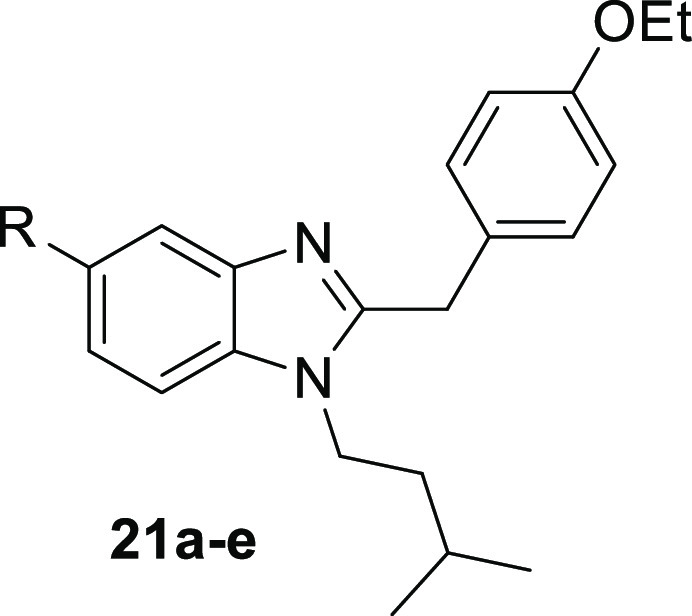
Introduction of the carbamate at the benzimidazole led to a loss of affinity, exemplified by dimethylcarbamate 21a which is more than 100-fold less active than its counterpart 15d. Affinity is partially regained by the introduction of cyclic structures, wherein azetidine 21d is more beneficial for binding to hBChE than pyrrolidine 21c. N-Methylethyl carbamate 21b and cyclopropyl carbamate 21e also showed micromolar inhibition, underlining that carbamate introduction on the benzimidazole is less advantageous than at the phenyl moiety (Table 2).
Table 2. In Vitro Results of the Inhibition of hBChE, hAChE, and Radioligand Binding of hCB2R, rCB1R of Compounds 20, 21a–ea.
n.d. not determined.
For the CB2R binding assay, hCB2R expressed in human embryonic kidney (HEK) cells were used to prepare membrane homogenates. [3H]CP55,940 was used as a radioligand, and compound 4 was used as a positive control. According to the affinity profile of compounds 15a–j, the tested ligands can be divided into two groups, monoalkyl carbamates and dialkyl carbamates. All monoalkyl carbamates except benzyl derivative 15h have no affinity toward hCB2R. However, all dialkyl carbamates except 15g show micromolar to submicromolar affinity toward the receptor. This could be explained by the substitution pattern at the nitrogen as 15g is the only compound that is substituted with two ethyl groups, while all other dialkyl carbamates carry at least one methyl group. Hydroxy derivative 14 shows an IC50 of 2.2 μM (Table 1).
Compound set 2, carrying the carbamate moiety at the benzimidazole, shows an overall good affinity toward hCB2R. Dialkyl carbamates 21a and 21b exhibit an IC50 of 1.2 μM and 578 nM against [3H]CP55,940. Cyclic carbamates 21c and 21d show an IC50 of 1.5 and 1.0 μM, respectively, making compound 21d, which is the only compound modified at the benzimidazole site with a submicromolar inhibition of hBChE, a promising hybrid candidate. Additionally, affinity of cyclopropyl carbamate 21e is comparable to compounds 21a–d with an IC50 value of 1.0 μM, while hydroxy derivative 20 exhibits a five-fold lower affinity (Table 2).
As CB1R activity is connected to several adverse side effects, additional radioligand binding assays for rCB1R were performed to investigate CB2R selectivity.48 CB1R membrane homogenates were freshly prepared from brains of adult female rats, and [3H]CP55,940 was used as the radioligand. None of the investigated compounds showed affinity toward CB1R, confirming our design approach. The only exception is compound 21b, which was not further investigated due to its weak inhibition of hBChE also.
Kinetic Characterization at hBChE, Exclusion of MOR Off-Target Binding, and Functional Activity on CB2R
The binding mode of compounds 15d and 21d regarding hBChE was further characterized kinetically (Figure 3). For the most part, carbamate-based BChE inhibitors act in a pseudoirreversible fashion, whereas the process of inhibition can be described by a three-step mechanism. First, an enzyme–inhibitor complex is formed, followed by transfer of the carbamate moiety onto the enzyme (EC). In the last step of pseudoirreversible inhibition, decarbamylation of enzyme takes place, resulting in reactivation of the enzyme activity. Formation of the EC complex can be described by Michaelis–Menten kinetics and therefore expressed by Michaelis constant KM and maximum reaction rate νmax, while the decarbamylation process can be described by rate constant k4 (Figure 3B).26,47,49,50
Figure 3.
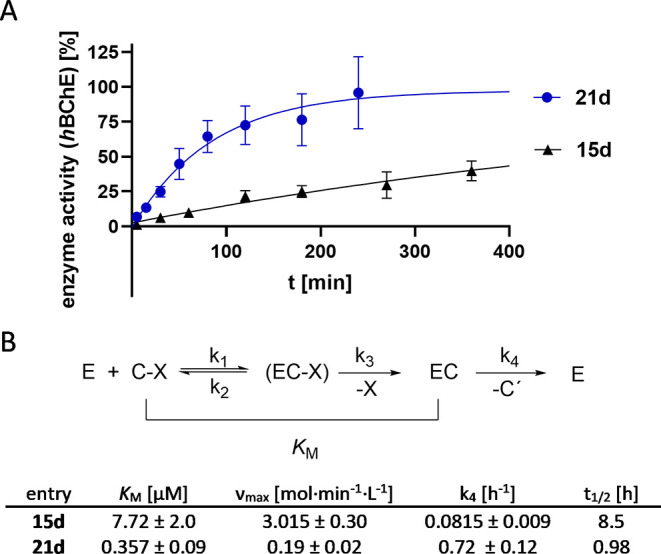
Kinetic investigations into compounds 15d and 21d. (A) Time-dependent enzyme regeneration from carbamylated state [EC]. (B) Kinetic characterization of compounds 15d and 21d on hBChE.
Dimethyl carbamate 15d shows a KM of 7.7 μM, a factor of 20 higher than compound 21d, while νmax of compound 15d is 15 times higher (Figures 3 and S2). Interestingly, dimethyl carbamate 15d exhibits a prolonged half-life of 8.5 h, when compared to compound 21d with a half-life of about 1 h, meaning that reconstitution of hBChE takes significantly longer. Both results confirm pseudoirreversible mode of action.
Subsequent to radioligand binding studies, compounds 15a,c,d,e,h,j and 21d, respectively, which displayed submicromolar inhibition of hBChE, and therefore represent possible hybrid compounds, were screened for their functional activity using a calcium mobilization assay utilizing Chinese hamster ovarian (CHO-K1) cells stably expressing Gαq16 with hCB2R. Calcium flux upon interaction with an agonist was monitored using Fura-2AM and an automated plate reader (Figure 4A). In addition, activation of the hCB2R-βarr2 pathway by compounds 3, 15a,c,d,e,h,j, and 21d was assessed using NanoBiT bioassays (Figures 4B and S3). Additionally, potential off-target activity at the human μ-opioid receptor (hMOR) of compounds 3, 15a,c,d,e,h,j, and 21d, due to their structural similarity to etonitazene, as observed in our previous approaches, was investigated using a similar MOR-βarr2 assay.14,19
Figure 4.

Functional activity screening of compounds 15a,c,d,e,h,j and 21d. (A) Calcium mobilization assay at hCB2R. Calcium flux was measured at 10 μM using Fura-2AM and normalized to the maximum response caused by compound 4; n ≥ 3; (B) NanoBiT hCB2R βarr2 recruitment assay. Receptor activation was monitored at 10 μM and normalized to the maximum response caused by compound 4; n = 3.
The calcium-screening assay revealed a heterogeneous activation profile, wherein both sets of carbamates are represented (Figure 4A). Compounds with an efficacy >20% at 10 μM were tested at various concentrations to determine their potency (Figure 5A). Using a hCB2R βarr2 recruitment screening assay, we observed a rather similar profile (Figures 4B, and S3).
Figure 5.
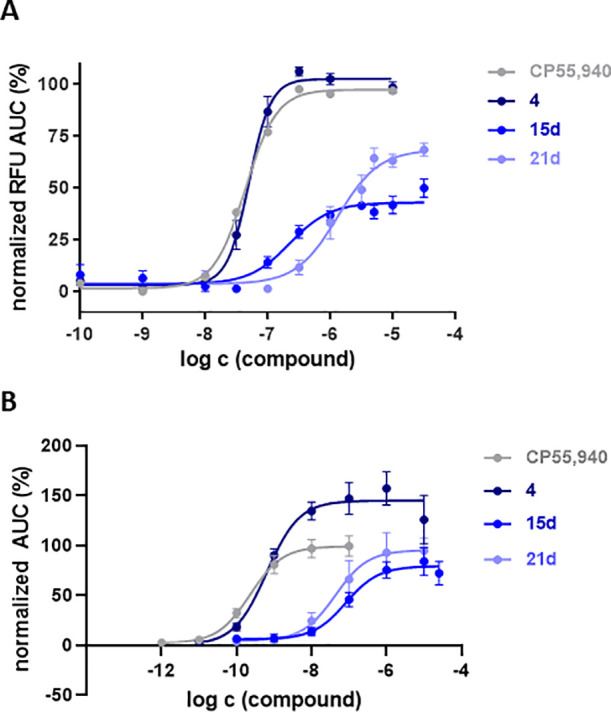
Functional activity at hCB2R of 15d and 21d, measured using a calcium mobilization assay, normalized to compound 4 (A) and a NanoBiT βarr2 recruitment assay (B), normalized to CP55,940.
Analyzing the dose–response curves for calcium mobilization at CB2R, three compounds can be pointed out. Dimethylcarbamate 15d exhibits an EC50 of 244 nM with a maximal activation (Emax) of 42%, while N-benzylmethyl derivative 15h exhibits an EC50 value of 7.71 μM with an Emax of 22% (data not shown). Benzimidazole-modified carbamate 21d showed the highest efficacy of all tested compounds (Emax = 68%) and an EC50 value of 1.3 μM. Positive control 4 (EC50 = 158 nM) was characterized as a full agonist with respect to CP55,940 (Figure 5A).
Due to the advantageous BChE inhibition profile of compounds 15d and 21d, both compounds were further characterized after initial screening, and various concentration ranges were tested in the βarr2 recruitment assay, alongside CP55,940 and compound 4 (Figure 5B). Here, we found an EC50 value of 0.63 nM for positive control 4 with an Emax value of 149%, relative to CP55,940. Compound 21d had a potency of 42.6 nM and a (relative) efficacy of 95%. For 15d, we observed a similar potency and (relative) efficacy, at 82.0 nM and 79%, respectively.
None of the compounds showed any sign of hMOR activity in a NanoBit βarr2 recruitment assay, indicating that we were able to “design-out” undesired MOR affinity when compared to previously developed hybrids based on the scaffold of compound 4 (data not shown).14,19
Microglia Activation
After characterization of respective benzimidazole-carbamates as partial agonists on CB2R, we wanted to evaluate the activity of compounds 15d and 21d in a biologically more complex environment. As the immunomodulatory effect of CB2R agonists is microglia-mediated, compounds 15d and 21d were tested for the ability to suppress the production of neurotoxic factors. Induction of the pro-inflammatory M1 state was triggered by exposure of murine microglial N9 cells to lipopolysaccharide (LPS) (100 ng/mL) for 24 h within or without the respective compounds 15d and 21d in increasing concentrations. While nitrite accumulation was measured using the Griess test, interleukin-1 beta (IL1β) release, cytokine-inducible nitric oxide synthases (iNOSs), triggering receptor expressed on myeloid cells 2 (TREM2) and transforming growth factor-beta 2 (TGFβ2) expression was detected by Western blot. While IL1β and iNOS are markers of the pro-inflammatory M1 state, TREM2 and TGFβ2 are the markers of the anti-inflammatory M2 state.
Both compounds significantly decrease the expression of NOS at 1.0 and 2.5 μM for compounds 15d and 21d, respectively. Additionally, IL1β release after treatment with LPS was significantly reduced by compounds 15d and 21d at 5.0 and 1.0 μM, respectively. A comparable effect could be observed for nitrite accumulation. No parallel change in TREM2 and TGFβ2 expression was detected (Figure 6). This indicates that both molecules have an immunomodulatory effect as they reduce the pro-inflammatory LPS-mediated activation of microglia, but no phenotypic switch from the pro-inflammatory M1 to the myeloid-phagocytic, and therefore, anti-inflammatory M2 phenotype was observed. This data is in accordance with previous experiments regarding BChE/CB2R hybridization, whereby the work of Scheiner et al., who reported a comparable effect on the M1/M2 states, deserves special attention.19
Figure 6.

Effects of compounds 15d and 21d on microglia activation in N9 cells after LPS (100 ng/mL) treatment in the presence of increasing concentrations (1, 2.5, and 5 μM) of compounds 15d and 21d. (A) Expression of iNOS, TREM2, and TGFβ2 was evaluated by Western blot analysis (left) and quantified through densitometry; (B) IL1β release was measured by Western blot analysis and quantified through densitometry; and (C) nitrite release was quantified using the Griess test. All quantitative data are presented as means ± SEM from at least three independent experiments. Statistical significance between different treatments was calculated by using one-way analysis of variance (ANOVA) followed by post hoc comparison through Bonferroni’s test. #p < 0.05, ##p < 0.01, ###p < 0.001, compared to LPS-activated microglia; *p < 0.05, **p < 0.01, and ***p < 0.001, compared to control; two-way ANOVA (Dunnett’s post hoc comparison test).
Neurotoxicity and Neuroprotection
Due to its balanced profile on both targets, hBChE and hCB2R, its selectivity over both hAChE and rCB1R, and its submicromolar potency and immunomodulatory effect, compound 15d was chosen for further investigations. To examine potential neurotoxic potential of 15d prior to potential in vivo studies, murine hippocampal neuronal (HT22) cells were treated with dimethylcarbamate 15d and its expected metabolite 14 in a MTT assay. To evaluate the potential neuroprotection of compound 15d, a glutamate-assay, as previously described, was performed.19,52−55 The glutamate-sensitive cell HT22 cell line lacks ionotropic glutamate receptors, for which reason high concentrations of glutamate lead to reactive oxygen species (ROS) accumulation.56
Both compounds, 15d and 14, show a negligible decrease in cell viability but no neurotoxicity at tested doses (Figure S1A). Compound 15d could not protect against glutamate-induced neurotoxicity (Figure S1B). As used mouse hippocampal HT22 cells do not express the cannabinoid receptors, these results indicate that the immunomodulatory effect observed for microglia activation of compound 15d is CB2R-mediated.57,58
In Vivo Studies
Behavioral Studies
In vivo neuroprotection of compound 15d was then analyzed in the pharmacological model of AD induced in mice after injection of oligomerized Aβ25–35 peptide.59 The amyloid peptide was administered intracerebroventricularly (ICV) at day 1. Compound 15d was administered intraperitoneally (IP) o.d. in the 0.1–3 mg/kg dose range from day 1 to 7. This model in the given application scheme allows detection of neuroprotective effects upon repeated administration.26,60 The learning abilities of mice were then tested using two complementary behavioral tests: spontaneous alternation in the Y-maze was analyzed on day 8, thus 24 h after the last drug injection, to monitor spatial working (thus short-term) memory deficits, and passive avoidance response was analyzed on day 9 (training) and 10 (retention) to monitor long-term, non-spatial memory. Compound 15d dose-dependently prevented Aβ25–35-induced learning deficits in both tests, with an active dose range of 0.3–3.0 mg/kg IP for spontaneous alternation (Figure 7A) and 3 mg/kg IP for passive avoidance (Figure 7B). In both tests, the highest dose led to a complete blockade of Aβ25–35-induced learning deficits, showing drug efficacy of compound 15d. The compound therefore showed a strong effect, especially compared to IC50 values measured. This efficacy likely relies on a synergy of the BChE/CB2R target combination. Additionally, very favorable bioavailability or a conjectural tertiary target could also be possible.
Figure 7.
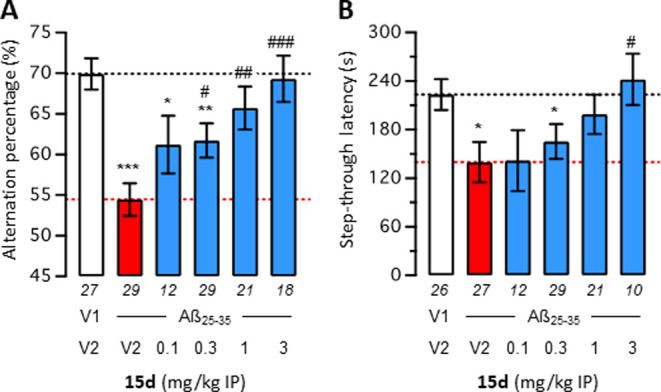
Effect of compound 15d on Aβ25–35-induced learning impairments in mice: spontaneous alternation performance (A) and retention latency in the passive avoidance test (B). Mice were treated with Aβ25–35 (9 nmol, 3 μL ICV) or vehicle (3 μL of ddH2O, V1) on day 1, then received either vehicle (DMSO 60% in saline, V2) or compound 15d (0.1–3.0 mg/kg) IP between day 1 and 7. Mice were then tested for spontaneous alternation on day 8 and passive avoidance on day 9 (training) and day 10 (retention). Data show mean ± SEM of the number of animals indicated below each bar graph. ANOVA: F(5,130) = 6788; p < 0.0001 in (A); Kruskal–Wallis ANOVA: H = 11.75; p = 0.0038 in (B). *p < 0.05, **p < 0.01, and ***p < 0.001 vs V1/V2-treated group; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs Aβ25–35/V2-treated group; Dunnett’s test in (A); Dunn’s test in (B).
To verify that a synergic effect of the compound 15d on its primary targets is the most likely explanation, a drug combination was analyzed using two compounds selective for each of the targets of compound 15d: compound 2 is a selective BChE inhibitor with a IC50(hBChE) of 6.4 nM, and compound 6 is a CB2R agonist with a Ki of 764 nM.14,18 Both compounds have separately been evaluated before and therefore considered reasonable candidates for combination studies.14,26 The CB2R affinity of compound 6 corresponds to the one of compound 15d, while the higher affinity of compound 2 was used to mask the weak residual hBChE affinity of compound 6 in the mixture.
A combination study was performed, with calculation of the combination index (CI) based on the isobologram representation of drug combination.60−62 The effects of compounds 2 and 6 alone were tested in the 0.1–1 mg/kg dose range (Figure 8A,C), and then, a combination of the low 0.1 mg/kg doses was tested. Both drugs alone and the combination showed significant protection (Figure 8A) or attenuation (Figure 8C) of Aβ25–35-induced learning deficits. Calculation of the CI (Figure 8B,D; Table S1) showed that both effects were synergic, with CIs ≪ 1.0. This experiment confirmed the potential of BChE and CB2R target combination and strengthened the interest in compound 15d, attenuating Aβ25–35-induced learning deficits in low doses.
Figure 8.
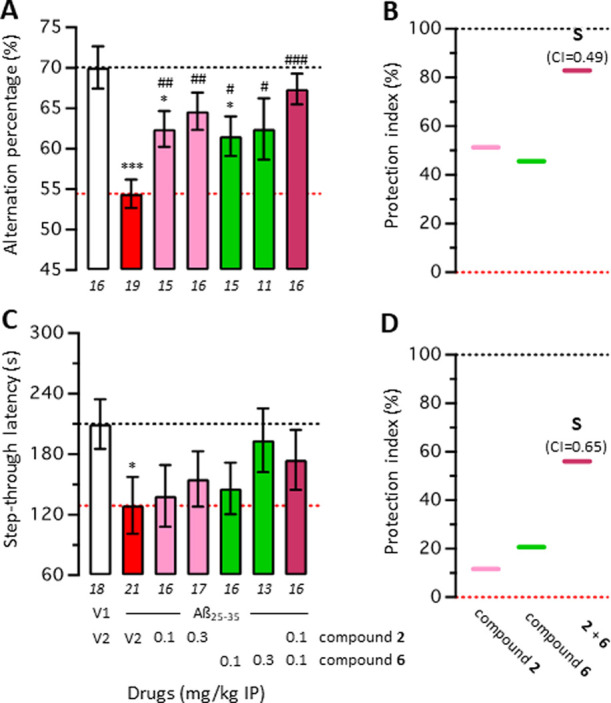
Combination studies between the BChE inhibitor 2 and the CB2R agonist 6 in Aβ25–35-treated mice: spontaneous alternation in the Y-maze (A) and step-through passive avoidance (C). Mice were treated with Aβ25–35 (9 nmol, 3 μL ICV) or vehicle (3 μL of ddH2O, V1) on day 1, then received either vehicle (DMSO 60% in saline, V2) or the drugs (0.1 and 0.3 mg/kg) IP between day 1 and 7. Mice were then tested for spontaneous alternation on day 8 and passive avoidance on day 9 (training) and day 10 (retention). Data show mean ± SEM of the number of animals indicated below each bar graph. (B,D) Protection is shown using a cursor-on-scale representation, with data from (V1 + V2)-treated group as 100% and (Aβ25–35 + V2)-treated group as 0%. ANOVA: F(6,101) = 4.979; p = 0.0002 in (A); Kruskal–Wallis ANOVA: H = 7.959; p = 0.2411 in (C). *p < 0.05 and ***p < 0.001 vs V1/V2-treated group; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs Aβ25–35/V2-treated group; Dunnett’s test in (A); Dunn’s test in (C). S: synergic effect with CI < 1.
Pharmacokinetic Studies
Following the successful application of compound 15d as neuroprotectant against Aβ25–35-induced learning impairments, initial pharmacokinetic (PK) studies were performed with compounds 15d and 21d. Expected metabolites 14 and 20 were additionally quantified in plasma and brain samples. Therefore, a single dose of 2 mg/kg of the respective compounds 15d and 21d was applied through intravenous (IV) administration, followed by blood and brain sampling at five time points. Sample analysis was conducted not only for the concentration of the injected compounds but also for their respective metabolites resulting from the expected transfer of the carbamate unit.
As shown in Figure 9A,C, dimethylamine 15d was significantly higher concentrated in plasma, as well as in the brain, when compared to azetidine derivative 21d, reaching both the highest plasma and brain concentrations 5 min after injection. Apparently, compound 21d outperforms compound 15d in terms of terminal elimination half-life t1/2 while the absolute concentration of compound 15d at 5 and 15 min is still high in plasma, as well as in the brain (Tables S2–S7). Even though elimination kinetics appear fast, pseudoirreversible BChE inhibitors, depending on their kinetic characteristics, will have already transferred their carbamate unit onto the enzyme (at least in part). This leads to a prolonged activity at the respective target, surpassing elimination half-life of the respective inhibitor.63 Additionally, comparable drugs against AD, such as rivastigmine, are also applied using a transdermal patch, enabling a constant and controlled release into the bloodstream of the drug, reducing a possible high application frequency.64 In this regard, the encouraging behavioral studies of compound 15d have to be mentioned, as the drug was applied once daily IP, leading to complete prevention of Aβ25–35-induced learning deficits in both the short-term and long-term memory tests.
Figure 9.

Initial PK characterization of compounds 15d and 21d, as well as their metabolites 14 and 20 after IV injection of compounds 15d and 21d, respectively. (A) Plasma and brain concentration–time curve of compound 15d; (B) plasma and brain concentration–time curve of compound 14; (C) plasma and brain concentration–time curve of compound 21d; (D) plasma and brain concentration–time curve of compound 20; (E) selected PK parameters of compound 15d; (F) selected PK parameters of compound 14; (G) selected PK parameters of compound 21d; and (H) selected PK parameters of compound 20.
Interestingly, the extent of phenolic metabolite formation, as result of, e.g., hydrolysis by BChE, of compound 15d to phenolic compound 14, is significantly lower than the phenolic metabolite formation of compound 21d compared to compound 20 (Figure 9B,D). This indicates a higher carbamate stability of dimethylamine derivative 15d, when compared to azetidine derivative 21d, similar to previously examined (de-)carbamoylation kinetics.
Both investigated compounds 15d and 21d were able to cross the blood–brain barrier, reaching Cmax values of 194 and 38 ng·mL–1 for dimethylamine derivative 15d and azetidine derivative 21d, respectively, underlining the beneficial structural properties of the investigated benzimidazole-carbamate hybrids (Figure 9E,G). Relative brain-to-plasma ratios are higher for compound 21d (0.323) when compared to compound 15d (0.057). Especially, metabolite 20 concentrates in the brain, with a 2.14 times higher area under the curve (AUC) value for brain compared to plasma, when injected IV. Nevertheless, compounds injected IP may exhibit a different profile, as the primary route of absorption after IP injection is into mesenteric vessels, that drain into the portal vein and pass through the liver, therefore potentially resulting in hepatic metabolism.65 While both routes of administration can be considered parenteral, IP injections lead to a PK profile with higher similarity to oral administration, wherefore the successful behavioral studies may suggest beneficial properties regarding (oral) bioavailability.65
A more detailed examination of central nervous system (CNS) penetration of both the target compounds and their metabolites seems necessary for elucidating the pro-cognitive effects of such irreversibly acting compounds, and in related studies, e.g., the same route of administration is desirable.
Conclusions
In summary, low-molecular-weight molecules (<550 g/mol), maintaining beneficial, drug-like properties, based on a 2-benzylbenzimidazole core structure, have been obtained, employing a six- to seven-step synthesis. Evaluation on ChEs and CBRs revealed a multifaceted set of compounds, able to address both BChE and CB2R with selectivity over AChE, CB1R, and MOR. Furthermore, several ligands could be characterized as partial agonists of CB2R in a calcium mobilization assay. As CB2R agonism can suppress pro-inflammatory microglia activation, compounds 15d and 21d were tested for their effect on microglia activation, showing a pronounced immunomodulation. A similar effect could not be observed on CBR-deficient HT22 cells, suggesting that the immunomodulatory effect of compound 15d is indeed CB2R-mediated. After no neurotoxic effect of compound 15d could be observed, the compound was examined in vivo, using an established AD mouse model. Herein, compound 15d dose-dependently attenuated Aβ25–35-induced learning impairments in spontaneous alternation at low doses of 0.3 mg/kg and passive avoidance at 3 mg/kg, indicating a synergic effect of BChE/CB2R target combination. To confirm the proposed over-additive (synergic) effect, a combination study was performed, proving a synergic effect of BChE inhibition and CB2R activation in vivo, further underlining the approach of BChE/CB2R hybridization for the treatment of neurodegeneration. Nevertheless, an additional beneficial contribution of a conjectural tertiary target cannot be excluded until a complete pharmacological profiling of the molecule has been carried out.
Subsequent initial PK characterization of compounds 15d and 21d, also in the context of their respective phenolic metabolites, may path a way for the therapeutic application of CB2R/BChE hybrids against neurodegenerative diseases.
Experimental Section
General Information
All reagents were purchased from Sigma-Aldrich (St. Louis, Missouri), ABCR (Karlsruhe, Germany), and Fluorochem (Hadfield, United Kingdom) and used without further purification. THF was dried by refluxing over sodium under an argon atmosphere, and DCM was dried over magnesium sulfate. Thin-layer chromatography was performed on silica gel 60 [alumina foils with a fluorescent indicator (254 nm)]. For detection, staining by potassium permanganate, ninhydrin or UV light (254 and 366 nm) was used. For column chromatography, silica gel 60 (particle size 0.040–0.063 mm) was used. Nuclear magnetic resonance spectra were recorded with a Bruker AV-400 NMR instrument (Bruker, Karlsruhe, Germany) in deuterated solvents, and chemical shifts are expressed in parts per million relative to the solvent residues used for NMR. Purity was determined by HPLC (Shimadzu Products), containing a DGU-20A3R degassing unit, a LC20AB solvent delivery unit, and a SPD-20A UV/VIS detector. UV detection was measured at 254 nm. Mass spectra were obtained by a LCMS 2020 (Shimadzu Products). As a stationary phase, a Synergi 4U fusion-RP (150 mm × 4.6 mm) column was used, and as a mobile phase, a gradient of MeOH/water with 0.1% formic acid was used. Parameters: A = water, B = MeOH, V(B)/[V(A) + V(B)] = from 5 to 90% over 10 min, V(B)/[V(A) + V(B)] = 90% for 5 min, and V(B)/[V(A) + V(B)] = from 90 to 5% over 3 min. The method was performed with a flow rate of 1.0 mL/min. Compounds were only used for biological evaluation if the purity was ≥95% and were dried under high vacuum (<0.1 mbar) beforehand.
Chemistry
General Procedures for the Synthesis of Target Compounds (15a–j and 21a–e)
General Procedure for Carbamate Formation Using Isocyanates (GP1)
The respective phenol (1.0 equiv) was dissolved in anhydrous DCM before TEA (1.5–1.7 equiv) was added. After addition of the isocyanate (1.4–1.5 equiv), the solution was stirred for 12 h at rt. Then, the solution was diluted with DCM and washed with ammonium chloride (two times). The combined organic layers were washed with brine and dried over sodium sulfate. Subsequent column chromatography (DCM/methanol) yielded the corresponding carbamate.
General Procedure for Carbamate Formation Using Carbamoyl Chlorides (GP2)
The respective phenol (1.0 equiv) was dissolved in anhydrous THF before NaH in paraffin oil (1.2 equiv) was added. After addition of the carbamoyl chloride (1.2 equiv), the solution was stirred for 12 h at rt. Then, the solution was diluted with DCM and washed with ammonium chloride (two times). The combined organic layers were washed with brine and dried over sodium sulfate. Subsequent column chromatography (DCM/methanol) yielded the corresponding carbamate.
General Procedure for Carbamate Formation Using 4-Nitrophenyl Dialkylcarbamates (GP3)
The respective phenol (1.0 equiv) was dissolved in anhydrous THF before KOtBu (1.3 equiv) was added. After addition of the 4-nitrophenyl dialkylcarbamates (1.2 equiv), the solution was stirred for 12 h at 55 °C. Then, the solution was diluted with DCM and washed with ammonium chloride (two times). The combined organic layers were washed with brine and dried over sodium sulfate. Subsequent column chromatography (DCM/methanol) yielded the corresponding carbamate.
N,N-Diethyl-4-fluoro-3-nitrobenzamide (9)
4-Fluoro-3-nitrobenzoic acid (5.00 g, 27.0 mmol, 1.0 equiv) was dissolved in DCM and treated with catalytic amounts of DMF before oxalyl chloride (2.55 mL, 3.77 g, 1.2 equiv) was added at 0 °C. After stirring the solution for 1 h at 0 °C, a mixture of diethylamine (3.09 mL, 2.17 g, 29.7 mmol, 1.1 eq) and TEA was added slowly at 0 °C and stirred for 4 h at rt. The combined organic layers were washed with 1 M hydrochloric acid (2×) and 1 M NaOHaq. Drying over sodium sulfate and evaporation of the solvent under reduced pressure yielded N,N-diethyl-4-fluoro-3-nitrobenzamide (9) (6.44 g, 26.8 mmol, 99%) as a light, brown oil. 1H-NMR (400 MHz, CDCl3): δ = 8.11 (dd, J = 7.0, 2.1 Hz, 1H), 7.79–7.53 (m, 1H), 7.34 (dd, J = 10.3, 8.7 Hz, 1H), 3.36 (dq, J = 64.0, 7.1 Hz, 4H), 1.26–1.13 (m, 6H) ppm; 13C-NMR (101 MHz, CDCl3): δ = 167.78, 157.19, 154.52, 134.13, 133.99, 133.90, 124.70, 124.67, 119.11, 118.90, 77.16, 43.67, 39.91, 14.41, 14.12 ppm; ESI-MS: m/z = 241.15 [M + H]+, calcd 241.09.
N,N-Diethyl-4-(isopentylamino)-3-nitrobenzamide (10)
N,N-Diethyl-4-fluoro-3-nitrobenzamide (9) (2.50 g, 10.4 mmol, 1.0 equiv) was dissolved in ethanol (20 mL), before isoamylamine (1.44 mL, 994 mg, 11.4 mmol, 1.1 equiv) and TEA (2.16 mL, 1.58 g, 15.6 mmol, 1.5 equiv) dissolved in 35 mL of ethanol were added. The solution was stirred for 3 h at 55 °C, resulting in an orange solution that was then evaporated under reduced pressure. The residue was taken up in diethyl ether (150 mL) and washed with citric acid (2.5% in water, 100 mL) and water (100 mL). The combined aqueous phases were extracted with diethyl ether (100 mL). The combined organic layers were then dried over magnesium sulfate, and the solvent was evaporated under reduced pressure to yield N,N-diethyl-4-(isopentylamino)-3-nitrobenzamide (10) (2.82 g, 9.19 mmol, 88%) as an orange oil. 1H-NMR (400 MHz, CDCl3): δ = 8.27 (d, J = 2.0 Hz, 1H), 8.14 (br s, 1H), 7.56 (dd, J = 8.9, 2.1 Hz, 1H, arom.), 6.88 (d, J = 8.9 Hz, 1H), 3.45 (dt, J = 7.7, 4.0 Hz, 4H), 3.34 (td, J = 7.4, 5.0 Hz, 2H), 1.78 (dq, J = 13.3, 6.7 Hz, 1H), 1.64 (q, J = 7.1 Hz, 2H), 1.22 (t, J = 7.1 Hz, 6H), 0.99 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 169.60, 146.14, 135.36, 130.75, 125.78, 123.55, 114.09, 41.53, 37.88, 26.11, 22.60 ppm; ESI-MS: m/z = 308.25 [M + H]+, 615.45 [2M + H]+, calcd 308.19.
2-(4-(Benzyloxy)phenyl)acetic Acid
4-Hydroxyphenylacetic acid (2.5 g, 16.4 mmol, 1.0 equiv), benzyl bromide (2.04 mL, 2.94 g, 17.2 mmol, 1.05 equiv), potassium hydroxide (2.30 g, 41.0 mmol, 2.5 equiv), and sodium iodide (49.2 mg, 328 μmol, 0.02 equiv) were dissolved in ethanol (75 mL) and refluxed for 20 h. The solution was then cooled to rt, before hydrochloric acid (3.0 M, 75 mL) was added. The resulting precipitate was filtered, washed with water (50 mL), and dried in vacuo, yielding 2-(4-(benzyloxy)phenyl)acetic acid (1.84 g, 7.60 mmol, 46%) as a white solid (mp 121.9 °C). 1H-NMR (400 MHz, CDCl3): δ = 7.44–7.36 (m, 4H), 7.36–7.29 (m, 1H), 7.22–7.18 (m, 2H), 6.97–6.92 (m, 2H), 5.05 (s, 2H), 3.59 (s, 2H); 13C-NMR (101 MHz, CDCl3): δ = 176.88, 158.27, 137.10, 130.58, 128.74, 128.12, 127.60, 125.75, 115.19, 70.20, 40.10 ppm; ESI-MS: m/z = 243.10 [M + H]+, 265.00 [M + Na]+, calcd 243.10.
3-Amino-N,N-diethyl-4-(isopentylamino)benzamide (11)
N,N-Diethyl-4-(isopentylamino)-3-nitrobenzamide (10) (1.50 g, 4.89 mmol, 1.0 equiv) was dissolved in ethanol (18 mL), before palladium on carbon (10 wt %, 150 mg) was added. The solvent was purged with hydrogen, before the solution was stirred overnight under a hydrogen atmosphere. After reaction control indicated consumption of the starting material, the suspension was filtrated over Celite. Evaporation of the starting material under reduced pressure yielded 3-amino-N,N-diethyl-4-(isopentylamino)benzamide (11) (1.28 g, 4.62 mmol, 94%) as a purple oil. 1H-NMR (400 MHz, CDCl3): δ = 6.86 (dd, J = 8.1, 1.9 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 3.42 (q, J = 7.3 Hz, 4H), 3.13 (t, J = 7.4 Hz, 2H), 1.79–1.69 (m, 1H), 1.60–1.52 (m, 2H), 1.16 (t, J = 7.0 Hz, 6H), 0.96 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 172.02, 139.26, 133.40, 126.23, 119.40, 115.36, 109.94, 42.22, 38.48, 26.09, 22.63 ppm; ESI-MS: m/z = 278.15 [M + H]+, 555.40 [2M + H]+, calcd 278.22.
3-(2-(4-(Benzyloxy)phenyl)acetamido)-N,N-diethyl-4-(isopentylamino)benzamide (12)
2-(4-(Benzyloxy)phenyl)acetic acid (11) (770 mg, 3.18 mmol, 1.1 equiv), HBTU (1.21 g, 3.18 mmol, 1.1 equiv), and TEA (600 μL, 438 mg, 4.34 mmol, 1.5 equiv) were dissolved in DMF (12 mL), before 3-amino-N,N-diethyl-4-(isopentylamino)benzamide (800 mg, 2.89 mmol, 1.0 equiv) in DMF (8 mL) was added. The solution was then stirred for 3 h at rt, and the solvent was evaporated under reduced pressure. Subsequent column chromatography (ethyl acetate/petroleum ether, 4:1) yielded 3-(2-(4-(benzyloxy)phenyl)acetamido)-N,N-diethyl-4-(isopentylamino)benzamide (12) (993 mg, 1.98 mmol, 69%) as a purple oil. 1H-NMR (400 MHz, CDCl3): δ = 9.02 (s, 1H), 7.46–7.26 (m, 8H), 7.04–6.90 (m, 4H), 6.39 (d, J = 8.4 Hz, 1H), 5.02 (s, 2H), 3.61 (s, 2H), 3.48–3.32 (m, 4H), 2.91 (t, J = 7.3 Hz, 2H), 2.01 (s, 1H), 1.68–1.55 (m, 1H), 1.37 (td, J = 7.2 Hz, 2H), 1.18–1.09 (m, 6H), 0.92 (d, J = 6.8 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 172.03, 171.14, 157.63, 143.90, 136.87, 130.00, 128.34, 128.19, 127.70, 127.22, 125.22, 125.08, 123.23, 122.23, 114.78, 109.92, 69.76, 42.59, 41.43, 38.03, 25.71, 22.46 ppm; ESI-MS: m/z = 502.35 [M + H]+, 1003.45 [2M + H]+, calcd 502.30.
2-(4-(Benzyloxy)benzyl)-N,N-diethyl-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (13)
3-(2-(4-(Benzyloxy)phenyl)acetamido)-N,N-diethyl-4-(isopentylamino)benzamide (12) (970 mg, 1.93 mmol, 1.0 equiv) was dissolved in acetic acid (15 mL) and stirred for 3 h at 130 °C. The reaction mixture was cooled to rt before ammonia (25% in water) was added until a pH > 10. Then, the aqueous phase was extracted with DCM (3 × 75 mL), the combined organic layers were dried over magnesium sulfate, and the solvent was extracted under reduced pressure, yielding 2-(4-(benzyloxy)benzyl)-N,N-diethyl-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (13) (665 mg, 1.37 mmol, 71%) as a brown oil. 1H-NMR (400 MHz, CDCl3): δ = 7.76 (br s, 1H), 7.42–7.28 (m, 7H), 7.16 (d, J = 8.6 Hz, 2H), 6.94–6.89 (m, 2H), 5.03 (s, 2H), 4.27 (s, 2H), 4.04–3.91 (m, 2H), 3.65–3.27 (m, 4H), 1.62–1.51 (m, 1H), 1.43–1.34 (m, 2H), 1.23–1.19 (m, 6H), 0.90 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.70, 157.91, 154.20, 136.88, 135.66, 131.20, 129.55, 128.57, 128.26, 127.97, 127.39, 121.47, 117.46, 115.27, 109.50, 70.07, 42.67, 38.12, 33.74, 26.14, 22.36 ppm; ESI-MS: m/z = 484.35 [M + H]+, 967.70 [2M + H]+, calcd 484.29.
N,N-Diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14)
2-(4-(Benzyloxy)benzyl)-N,N-diethyl-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (13) (290 mg, 599 μmol, 1.0 equiv) was dissolved in methanol (10 mL), before palladium on carbon (10 wt %, 29.0 mg) was added. The solvent was purged with hydrogen, before the solution was stirred overnight under a hydrogen atmosphere. After reaction control indicated consumption of the starting material, the suspension was filtrated over Celite. Evaporation of the solvent under reduced pressure yielded N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (214 mg, 545 μmol, 91%) as a brown solid (mp 166.6 °C). 1H-NMR (400 MHz, CDCl3): δ = 7.71 (br s, 1H), 7.31–7.24 (m, 2H), 6.94–6.88 (m, 2H), 6.74–6.66 (m, 2H), 4.16 (s, 2H), 3.97 (dd, J = 9.7, 6.8 Hz, 2H), 3.57–3.25 (m, 5H), 1.63–1.48 (m, 1H), 1.40–1.32 (m, 2H), 1.26–1.03 (m, 6H), 0.88 (d, J = 6.6, 1.4 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.99, 156.71, 154.90, 140.93, 135.51, 131.15, 129.44, 125.90, 121.54, 116.99, 116.23, 109.88, 42.80, 38.12, 33.49, 26.21, 22.43 ppm; ESI-MS: m/z = 394.25 [M + H]+, 787.45 [2M + H]+, calcd 394.24.
4-Nitrophenyl-benzyl(methyl)carbamate
N-Methyl benzylamine (319 μL, 300 mg, 2.48 mmol, 1.0 equiv) was dissolved in anhydrous DCM (18 mL), before TEA (1.03 mL, 751 mg, 7.44 mmol, 3.0 equiv) and 4-nitrophenyl chloroformate (551 mg, 2.73 mmol, 1.1 equiv) were added subsequently. The reaction mixture was stirred for 2 h at rt before it was diluted with DCM (12 mL) and washed with hydrochloric acid (2 × 25 mL) and dried over magnesium sulfate. Subsequent column chromatography (DCM) yielded the product 4-nitrophenyl benzyl(methyl)carbamate (643 mg, 2.25 mmol, 91%) as an off-white oil. 1H-NMR (400 MHz, CDCl3): δ = 8.26 (dd, J = 8.9, 6.1 Hz, 2H), 7.58–7.21 (m, 7H), 4.61 (d, J = 34.8 Hz, 2H), 3.04 (d, J = 10.6 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ = 156.49, 129.03, 128.92, 128.21, 127.99, 127.32, 125.22, 125.19, 122.39, 53.18, 35.11, 34.37 ppm; ESI-MS: m/z = 287.10 [M + H]+, calcd 287.10.
4-Nitrophenyl azetidine-1-carboxylate
Azetidine (150 μL, 127 mg, 2.22 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (15 mL), before TEA (922 μL, 673 mg, 6.66 mmol, 3.0 equiv) and 4-nitrophenyl chloroformate (537 mg, 2.66 mmol, 1.2 equiv) were added. The reaction mixture was stirred for 2 h at rt before its dilution with dichloromethane and (25 mL), then washed with citric acid (2 × 25 mL) and brine (25 mL). After drying over sodium sulfate, subsequent column chromatography (petroleum ether/ethyl acetate, 5:2) yielded the product 4-nitrophenyl azetidine-1-carboxylate (443 mg, 2.00 mmol, 90%) as an off-white solid (mp 70.7–71.5 °C). 1H-NMR (400 MHz, CDCl3): δ = 8.22 (d, J = 9.2 Hz, 2H, arom.), 7.30 (d, J = 9.1 Hz, 2H, arom.), 4.19 (dt, J = 41.9, 7.7 Hz, 4H, CH2N), 2.36 (p, 2H, CH2CH2CH2); 13C-NMR (101 MHz, CDCl3): δ = 156.23, 152.60, 144.78, 125.17, 122.07, 55.03, 49.39, 15.89 ppm; ESI-MS: m/z = 223.05 [M + H]+, calcd 223.07.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl Ethylcarbamate (15a)
Following GP1, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (95.0 mg, 241 μmol, 1.0 equiv), ethyl isocyanate (28.2 μL, 25.7 mg, 362 μmol, 1.5 equiv), and TEA (50.1 μL, 36.6 mg, 362 μmol, 1.5 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl ethylcarbamate (15a) (50.0 mg, 108 μmol, 45%) as a purple oil after column chromatography (DCM/MeOH, 19:1). 1H-NMR (400 MHz, CDCl3): δ = 7.73 (br s, 1H), 7.30–7.24 (m, 2H), 7.19 (d, J = 8.2 Hz, 2H), 7.03 (d, J = 8.1 Hz, 2H), 5.30 (t, J = 5.8 Hz, 1H), 4.28 (s, 2H), 4.01–3.90 (m, 2H), 3.62–3.21 (m, 6H), 1.63–1.48 (m, 1H), 1.40 (dt, J = 12.0, 6.7 Hz, 2H), 1.26–1.11 (m, 9H), 0.89 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.83, 154.42, 153.91, 150.27, 141.80, 135.69, 132.79, 131.19, 129.29, 122.10, 121.50, 117.52, 109.62, 42.71, 38.25, 36.13, 33.86, 26.15, 22.40, 15.13 ppm; ESI-MS: m/z = 465.25 [M + H]+, calcd 465.28.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl Isopropylcarbamate (15b)
According to GP1, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (55.0 mg, 140 μmol, 1.0 equiv), isopropyl isocyanate (19.3 μL, 16.7 mg, 196 μmol, 1.4 equiv), and TEA (32.9 μL, 24.0 mg, 238 μmol, 1.7 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl isopropylcarbamate (15b) (51.0 mg, 106 μmol, 76%) as an off-white solid (mp 58.3 °C) after column chromatography (DCM/methanol, 20:1). 1H-NMR (400 MHz, CDCl3): δ = 7.73 (br s, 1H), 7.31–7.24 (m, 2H), 7.20 (d, J = 8.1 Hz, 2H), 7.04 (d, J = 7.7 Hz, 2H), 5.02 (t, J = 8.8 Hz, 1H), 4.28 (s, 2H), 4.04–3.89 (m, 2H), 3.91–3.77 (m, 1H), 3.65–3.26 (m, 4H), 1.61–1.50 (m, 1H), 1.45–1.37 (m, 2H), 1.27–1.13 (m, 12H), 0.89 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.84, 153.90, 153.63, 150.25, 142.00, 135.79, 132.87, 131.20, 129.32, 122.13, 121.51, 117.63, 109.61, 43.52, 42.74, 38.31, 34.00, 26.19, 22.94, 22.44 ppm; ESI-MS: m/z = 479.30 [M + H]+, calcd 479.30.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl Cyclopropylcarbamate (15c)
According to GP1, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (40.0 mg, 102 μmol, 1.0 equiv), cyclopropyl isocyanate (11.9 mg, 143 μmol, 1.4 equiv), and TEA (24.0 μL, 17.5 mg, 143 μmol, 1.7 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl cyclopropylcarbamate (15c) (34.0 mg, 71.3 μmol, 70%) as an off-white oil after column chromatography (DCM/methanol, 24:1). 1H-NMR (400 MHz, CDCl3): δ = 7.72 (br s, 1H), 7.29–7.22 (m, 2H), 7.18 (d, J = 8.1 Hz, 2H), 7.01 (d, J = 8.1 Hz, 2H), 5.53 (s, 1H), 4.26 (s, 2H), 3.94 (t, 2H), 3.59–3.26 (m, 4H), 2.61 (s, 1H), 1.59–1.49 (m, 1H), 1.44–1.35 (m, 2H), 1.24–1.10 (m, 6H), 0.87 (d, J = 6.6 Hz, 6H), 0.73–0.67 (m, 2H), 0.59–0.52 (m, 2H); 13C-NMR (101 MHz, CDCl3): δ = 171.84, 155.16, 153.87, 150.16, 141.97, 135.78, 132.94, 131.20, 129.31, 122.08, 121.50, 117.61, 109.61, 42.73, 38.29, 33.97, 26.17, 23.32, 22.43, 7.27, 6.80 ppm; ESI-MS: m/z = 477.30 [M + H]+, calcd 477.28.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl dimethylcarbamate (15d)
Following GP2, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (100 mg, 254 μmol, 1.0 equiv), dimethylcarbamyl chloride (28.1 μL, 32.9 mg, 305 μmol, 1.2 equiv), and NaH (60% in mineral oil, 12.2 mg, 305 μmol, 1.2 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl dimethylcarbamate (15d) (98.0 mg, 211 μmol, 83%) as an off-white solid (mp 123.8–124.0 °C) after column chromatography (DCM/MeOH, 32:1). 1H-NMR (400 MHz, CDCl3): δ = 7.79 (br s, 1H), 7.38–7.29 (m, 2H), 7.27–7.22 (m, 2H), 7.10–7.05 (m, 2H), 4.34 (s, 2H), 4.07–3.96 (m, 2H), 3.68–3.30 (m, 4H), 3.09 (s, 3H), 3.01 (s, 3H), 1.60 (dq, J = 13.1, 6.6, 2.1 Hz, 2H), 1.47 (td, J = 10.5, 6.4 Hz, 4H), 1.31–1.15 (m, 6H), 0.94 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.81, 154.86, 153.90, 150.78, 135.70, 132.79, 131.48, 131.41, 129.36, 122.32, 121.71, 117.55, 109.74, 42.82, 38.34, 36.83, 36.57, 33.92, 26.25, 22.48 ppm; ESI-MS: m/z = 465.25 [M + H]+, calcd 465.28.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl ethyl(methyl)carbamate (15e)
According to GP2, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (45.0 mg, 114 μmol, 1.0 equiv), methyl(ethyl)carbamic chloride (16.7 mg, 137 μmol, 1.2 equiv), and sodium hydride (60% in paraffin oil, 5.48 mg, 137 μmol, 1.2 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl methyl(propyl)carbamate 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl ethyl(methyl)carbamate (15e) (42.0 mg, 87.7 μmol, 77%) as an off-white oil after column chromatography (DCM/methanol, 20:1). 1H-NMR (400 MHz, CDCl3): δ = 7.75 (br s, 1H), 7.32–7.25 (m, 3H), 7.21 (d, J = 8.3 Hz, 3H), 7.04 (d, J = 8.0 Hz, 3H), 4.30 (s, 3H), 4.03–3.94 (m, 3H), 3.58–3.30 (m, 6H), 2.99 (d, J = 29.5 Hz, 5H), 1.62–1.52 (m, 1H), 1.49–1.40 (m, 2H), 1.25–1.14 (m, 9H), 0.91 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.82, 154.36, 153.92, 150.75, 141.86, 135.75, 132.79, 131.30, 129.30, 122.27, 121.59, 117.59, 109.65, 44.17, 42.75, 38.33, 34.33, 33.97, 33.91, 26.21, 22.46, 13.30, 12.53 ppm; ESI-MS: m/z = 479.30 [M + H]+, calcd 479.30.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl methyl(propyl)carbamate (15f)
According to GP2, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (30.0 mg, 76.1 μmol, 1.0 equiv), methyl(propyl)carbamic chloride (12.4 mg, 91.3 μmol, 1.2 equiv), and sodium hydride (60% in paraffin oil, 3.65 mg, 91.3 μmol, 1.2 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl methyl(propyl)carbamate (15f) (34.4 mg, 69.4 μmol, 92%) as an off-white oil after column chromatography (DCM/methanol, 30:1). 1H-NMR (400 MHz, CDCl3): δ = 7.74 (br s, 1H), 7.32–7.25 (m, 2H), 7.21 (d, J = 8.3 Hz, 2H), 7.04 (t, J = 7.2 Hz, 2H), 4.29 (s, 2H), 4.00–3.92 (m, 2H), 3.61–3.26 (m, 6H), 2.99 (d, J = 30.1 Hz, 3H), 1.67–1.39 (m, 5H), 1.25–1.12 (m, 6H), 0.95–0.87 (m, 9H); 13C-NMR (101 MHz, CDCl3): δ = 171.84, 154.76, 153.92, 150.75, 142.00, 135.81, 132.84, 131.25, 129.31, 122.24, 121.53, 117.64, 109.62, 51.06, 50.98, 42.73, 38.34, 34.89, 34.52, 34.04, 26.21, 22.46, 21.33, 20.69, 11.19 ppm; ESI-MS: m/z = 493.35 [M + H]+, calcd 493.31.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl Diethylcarbamate (15g)
According to GP2, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (55.0 mg, 140 μmol, 1.0 equiv), diethylcarbamic chloride (19.5 μL, 22.8 mg, 168 μmol, 1.2 equiv), and sodium hydride (60% in paraffin oil, 7.72 mg, 168 μmol, 1.2 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl diethylcarbamate (15g) (56.9 mg, 115 μmol, 82%) as an off-white oil after column chromatography (DCM/methanol, 26:1). 1H-NMR (400 MHz, CDCl3): δ = 7.75 (s, 1H), 7.32–7.25 (m, 2H), 7.22 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.5 Hz, 2H), 4.30 (s, 2H), 4.04–3.90 (m, 2H), 3.56–3.31 (m, 8H), 1.61–1.53 (m, 1H), 1.48–1.41 (m, 2H), 1.24–1.16 (m, 12H), 0.91 (d, J = 6.5 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.84, 154.16, 153.93, 150.76, 141.96, 135.80, 132.75, 131.29, 129.30, 122.28, 121.57, 117.63, 109.64, 42.75, 42.35, 42.00, 38.36, 34.04, 26.22, 22.47, 14.33, 13.48 ppm; ESI-MS: m/z = 493.35 [M + H]+, calcd 493.31.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl Benzylcarbamate (15h)
According to GP1, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (60.0 mg, 152 μmol, 1.0 equiv), benzyl isocyanate (26.3 μL, 28.3 mg, 213 μmol, 1.4 equiv), and TEA (31.5 μL, 23.0 mg, 228 μmol, 1.5 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl benzylcarbamate (15h) (70.1 mg, 133 μmol, 88%) after column chromatography (DCM/methanol, 15:1) as a white solid (mp 65.1–67.3 °C) . 1H-NMR (400 MHz, CDCl3): δ = 7.75 (s, 1H), 7.35–7.24 (m, 7H), 7.22 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.2 Hz, 2H), 5.64 (t, J = 6.0 Hz, 1H), 4.42 (d, J = 6.0 Hz, 2H), 4.29 (s, 2H), 4.02–3.91 (m, 4H), 3.50 (d, J = 7.1 Hz, 4H), 1.65–1.51 (m, 1H), 1.47–1.39 (m, 2H), 1.20 (t, 6H), 0.92 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.84, 154.65, 153.87, 150.25, 141.96, 138.13, 135.78, 133.04, 131.25, 129.37, 128.82, 127.77, 127.74, 122.11, 121.55, 117.62, 109.64, 45.37, 42.76, 38.33, 33.99, 26.20, 22.46 ppm; ESI-MS: m/z = 527.30 [M + H]+, calcd 527.30.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl methyl(phenyl)carbamate (15i)
According to GP2, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (62.0 mg, 157 μmol, 1.0 equiv), methyl(phenyl)carbamic chloride (32.0 mg, 188 μmol, 1.2 equiv), and sodium hydride (60% in paraffin oil, 7.52 mg, 188 μmol, 1.2 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl methyl(phenyl)carbamate (15i) (72.6 mg, 138 μmol, 88%) as an off-white oil after column chromatography (DCM/methanol, 26:1). 1H-NMR (400 MHz, CDCl3): δ = 7.69 (br s, 1H, arom.), 7.34–7.13 (m, 9H, arom.), 7.00 (d, J = 8.1 Hz, 2H), 4.23 (s, 2H), 3.96–3.85 (m, 2H), 3.63–3.08 (m, 7H), 1.57–1.45 (m, 1H), 1.40–1.33 (m, 2H), 1.24–1.04 (m, 6H), 0.85 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.77, 153.80, 153.78, 150.43, 142.88, 142.08, 135.78, 133.08, 131.12, 129.23, 129.03, 126.58, 125.86, 122.02, 121.39, 117.60, 109.52, 42.62, 38.24, 38.20, 33.97, 26.11, 22.37 ppm; ESI-MS: m/z = 527.35 [M + H]+, calcd 527.30.
4-((5-(Diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl benzyl(methyl)carbamate (15j)
According to GP3, N,N-diethyl-2-(4-hydroxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-carboxamide (14) (20.0 mg, 50.8 μmol, 1.0 equiv), 4-nitrophenyl benzyl(methyl)carbamate (17.4 mg, 61.0 μmol, 1.2 equiv), and potassium tert-butoxide (7.39 mg, 66.0 μmol, 1.3 equiv) were used to obtain 4-((5-(diethylcarbamoyl)-1-isopentyl-1H-benzo[d]imidazole-2-yl)methyl)phenyl benzyl(methyl)carbamate (15j) (10.4 mg, 19.2 μmol, 38%) as an off-white oil after column chromatography (DCM/methanol, 30:1). 1H-NMR (400 MHz, CDCl3): δ = 7.78 (s, 1H), 7.39–7.23 (m, 9H), 7.08 (dd, J = 19.9, 8.1 Hz, 2H), 4.57 (d, J = 34.7 Hz, 2H), 4.34 (s, 2H), 4.03–3.96 (m, 2H), 3.60–3.32 (m, 4H), 2.99 (d, J = 12.5 Hz, 3H), 1.66–1.54 (m, 1H), 1.51–1.41 (m, 2H, CH2), 1.25–1.16 (m, 6H), 0.93 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 171.70, 153.78, 150.76, 137.14, 135.57, 132.80, 131.67, 129.45, 128.89, 128.82, 128.21, 127.77, 127.47, 122.37, 121.90, 117.44, 109.83, 53.04, 42.89, 38.36, 34.55 (d, 1C), 33.88, 26.27, 22.50 ppm; ESI-MS: m/z = 541.35 [M + H]+, calcd 541.31.
Fluoro-4-(methoxymethoxy)-2-nitrobenzene (16)
4-Fluoro-3-nitrophenol (250 mg, 1.59 mmol, 1.0 equiv) was dissolved in anhydrous acetone (4 mL), and potassium carbonate (878 mg, 6.36 mmol, 4.0 equiv) was added. The suspension was cooled to 0 °C before MOMCl (242 μL, 256 mg, 3.18 mmol, 2.0 equiv) was added dropwise. The reaction mixture was stirred for 4 h at rt while the pH was adjusted to 8–9 with DIPEA. After the solvent was evaporated under reduced pressure, the residue was taken up with citric acid and extracted with DCM (3 × 50 mL). The combined organic phases were washed with sodium hydroxide (1.0 M in water) and brine and dried over sodium sulfate. Evaporation of the solvent yielded 1-fluoro-4-(methoxymethoxy)-2-nitrobenzene (16) (338 mg, quant.) as a light orange oil. 1H-NMR (400 MHz, CDCl3): δ = 7.70 (dd, J = 6.0, 3.0 Hz, 1H), 7.34–7.25 (m, 1H), 7.18 (t, J = 9.7 Hz, 1H), 5.17 (s, 2H), 3.47 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 155.12–147.14 (m, 2C), 137.51 (d, J = 12.3 Hz), 123.78 (d, J = 7.7 Hz), 119.07 (d, J = 22.5 Hz), 113.10 (d, J = 2.9 Hz), 95.13, 56.43 ppm; ESI-MS: m/z = 202.10 [M + H]+, calcd 202.05.
N-Isopentyl-4-(methoxymethoxy)-2-nitroaniline (17)
1-Fluoro-4-(methoxymethoxy)-2-nitrobenzene (16) (315 mg, 1.57 mmol, 1.0 equiv) was dissolved in ethanol (7 mL), before isopentyl amine (201 μL, 151 mg, 1.73 mmol, 1.1 equiv) and TEA (326 μL, 238 mg, 2.36 mmol, 1.5 equiv) were added. The solution was stirred at 55 °C overnight. Then, ethanol was evaporated under reduced pressure, and the residue was taken up with citric acid and extracted with ethyl acetate (3 × 50 mL). The combined organic phases were washed with brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. Subsequent column chromatography (DCM/methanol; 9:1) yielded N-isopentyl-4-(methoxymethoxy)-2-nitroaniline (17) (279 mg, 1.04 mmol, 66%) as an orange oil. 1H-NMR (400 MHz, CDCl3): δ = 7.87 (d, J = 2.9 Hz, 1H), 7.33–7.22 (m, 1H), 6.86 (d, J = 9.3 Hz, 1H), 5.14 (s, 2H), 3.51 (s, 3H), 3.37–3.26 (m, 2H), 1.91–1.71 (m, 1H), 1.64 (td, J = 7.1 Hz, 2H), 1.00 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 146.81, 141.95, 128.23, 115.18, 112.63, 95.59, 56.22, 41.76, 38.03, 26.10, 22.61 ppm; ESI-MS: m/z = 269.10 [M + H]+, calcd 269.15.
N1-Isopentyl-4-(methoxymethoxy)benzene-1,2-diamine (18)
N-Isopentyl-4-(methoxymethoxy)-2-nitroaniline (17) (257 mg, 959 μmol, 1.0 equiv) was dissolved in ethanol (20 mL), before palladium on carbon (10 wt % 25.7 mg) was added. The solvent was purged with hydrogen, before the solution was stirred overnight under a hydrogen atmosphere. After reaction control indicated consumption of the starting material, the suspension was filtrated over Celite. Evaporation of the solvent under reduced pressure yielded N1-isopentyl-4-(methoxymethoxy)benzene-1,2-diamine (18) (227 mg, 954 μmol, 99%) as a brown oil. 1H-NMR (400 MHz, CDCl3): δ = 6.63 (dd, J = 9.2, 1.5 Hz, 1H), 6.49 (dd, J = 6.3, 2.6 Hz, 2H), 5.08 (s, 2H), 3.47 (s, 3H), 3.17–2.94 (m, 2H), 1.86–1.67 (m, 1H), 1.60–1.48 (m, 2H), 0.95 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 136.86, 131.74, 114.38, 107.56, 105.92, 105.63, 95.43, 55.93, 43.71, 38.84, 26.26, 22.79 (2C) ppm; ESI-MS: m/z = 239.20 [M + H]+, calcd 239.17.
2-(4-Ethoxyphenyl)-N-(2-(isopentylamino)-5-(methoxymethoxy)phenyl)acetamide (19)
2-(4-Ethoxyphenyl)acetic acid (189 mg, 1.05 mmol, 1.1 equiv) was dissolved in DMF (4 mL), before HBTU (398 mg, 1.05 mmol, 1.1 equiv) and TEA (197 μL, 144 mg, 1.43 mmol, 1.5 equiv) were added. Then, N1-isopentyl-4-(methoxymethoxy)benzene-1,2-diamine (18) (227 mg, 954 μmol, 1.0 equiv) was added, and the solution was stirred for 2 h at rt. After evaporation of the solvent, the residue was taken up with ammonium chloride (saturated solution in water) and extracted with ethyl acetate (2 × 50 mL). The combined organic phases were washed sodium bicarbonate (saturated solution in water, 2 × 70 mL) and brine (70 mL), dried over sodium sulfate, and the solvent was evaporated under reduced pressure, yielding 2-(4-ethoxyphenyl)-N-(2-(isopentylamino)-5-(methoxymethoxy)phenyl)acetamide (19) (224 mg, 559 μmol, 59%) as a blue oil. The product was used without further purification. 1H-NMR (400 MHz, CDCl3): δ = 7.41 (br s, 1H), 7.28–7.24 (m, 2H), 6.91 (d, J = 8.5 Hz, 2H), 6.77–6.76 (m, 2H), 5.08 (s, 2H), 4.02–3.95 (m, 2H), 3.70 (s, 2H), 3.45 (s, 3H), 2.86 (t, J = 7.5 Hz, 2H), 1.63–1.50 (m, 1H), 1.41–1.31 (m, 2H), 0.92–0.83 (m, 9H); 13C-NMR (101 MHz, CDCl3): δ = 170.22, 144.38, 130.87, 130.75, 130.38, 126.61, 115.31, 114.42, 114.15, 112.30, 106.60, 103.55, 95.32, 63.60, 56.08, 43.96, 40.28, 36.66, 26.11, 22.67, 14.95 ppm; ESI-MS: m/z = 401.25 [M + H]+, calcd 401.24.
2-(4-Ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20)
2-(4-Ethoxyphenyl)-N-(2-(isopentylamino)-5-(methoxymethoxy)phenyl)acetamide (19) (224 mg, 559 μmol, 1.0 equiv) was dissolved in acetic acid (4 mL) and stirred at 130 °C for 2 h. Afterward the pH was adjusted to 8 by 25% NH3 in water and extracted with DCM (3 × 25 mL). The crude product was treated with TFA for 30 min. TFA was evaporated under nitrogen flow, before subsequent column chromatography (DCM/methanol; 18:1) yielded 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20) (136 mg, 401 μmol, 72%) as an off-white oil. 1H-NMR (400 MHz, CDCl3): δ = 8.31 (s, 1H), 7.43 (d, J = 2.2 Hz, 1H), 7.14–7.10 (m, 2H), 7.04 (d, J = 8.7 Hz, 1H), 6.89 (dd, J = 8.7, 2.2 Hz, 1H), 6.82–6.75 (m, 2H), 4.21 (s, 2H), 3.99–3.84 (m, 4H), 1.60–1.48 (m, 1H), 1.44–1.25 (m, 4H), 0.87 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 158.00, 153.66, 152.99, 142.18, 129.64, 129.06, 128.16, 114.91, 113.30, 109.75, 104.87, 63.56, 42.63, 38.12, 33.42, 26.26, 22.46, 14.89 ppm; ESI-MS: m/z = 339.20 [M + H]+, calcd 339.20.
2-(4-Ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl dimethylcarbamate (21a)
According to GP2, 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20) (32.0 mg, 94.4 μmol, 1.0 equiv), dimethylcarbamyl chloride (10.4 μL, 12.2 mg, 113 μmol, 1.2 equiv), and sodium hydride (60% in mineral oil, 5.00 mg, 123 μmol, 1.3 equiv) were used to obtain 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl dimethylcarbamate (21a) (28.0 mg, 68.3 μmol, 72%) as an off-white oil after column chromatography (DCM/methanol, 20:1). 1H-NMR (400 MHz, CDCl3): δ = 7.45 (d, J = 2.1 Hz, 1H), 7.18 (d, J = 8.7 Hz, 1H), 7.14–7.09 (m, 2H), 7.00 (dd, J = 8.7, 2.2 Hz, 1H), 6.83–6.78 (m, 2H), 4.22 (s, 2H), 4.02–3.86 (m, 4H), 3.07 (d, J = 44.7 Hz, 6H), 1.60–1.47 (m, 1H), 1.40–1.31 (m, 5H), 0.87 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 158.06, 155.72, 154.16, 147.00, 142.68, 132.94, 129.56, 128.12, 117.11, 114.91, 112.39, 109.24, 63.57, 42.70, 38.16, 36.84, 36.59, 33.84, 26.21, 22.44, 14.89 ppm; ESI-MS: m/z = 410.20 [M + H]+, calcd 410.24.
2-(4-Ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl ethyl(methyl)carbamate (21b)
According to GP2, 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20) (22.0 mg, 64.9 μmol, 1.0 equiv), methyl(ethyl)carbamic chloride (9.50 mg, 77.9 μmol, 1.2 equiv), and sodium hydride (60% in paraffin oil, 3.38 mg, 84.4 μmol, 1.3 equiv) were used to obtain 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl ethyl(methyl)carbamate (21b) (12.1 mg, 28.5 μmol, 44%) as an off-white oil after column chromatography (ethyl acetate/petroleum ether, 20:1). 1H-NMR (400 MHz, CDCl3): δ = 7.47 (br s, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.13 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.8 Hz, 1H), 6.84–6.79 (m, 2H), 4.25 (s, 2H), 4.04–3.90 (m, 4H), 3.60–3.35 (m, 2H), 3.05 (d, J = 39.7 Hz, 3H), 1.62–1.48 (m, 1H), 1.41–1.18 (m, 9H), 0.88 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 158.03, 155.27, 153.91, 147.10, 141.99, 132.63, 129.49, 127.82, 117.31, 114.86, 112.15, 109.24, 63.49, 44.09, 42.67, 38.05, 33.61, 26.12, 22.35, 14.79 ppm; ESI-MS: m/z = 424.25 [M + H]+, calcd 424.26.
2-(4-Ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl Pyrrolidine-1-carboxylate (21c)
According to GP2, 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20) (32.0 mg, 94.4 μmol, 1.0 equiv), pyrrolidine-1-carbonyl chloride (12.5 μL, 15.1 mg, 113 μmol, 1.2 equiv), and sodium hydride (60% in mineral oil, 5.00 mg, 123 μmol, 1.3 equiv) were used to obtain 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl pyrrolidine-1-carboxylate (21c) (31.1 mg, 71.3 μmol, 76%) as an off-white oil after column chromatography (DCM/methanol, 24:1). 1H-NMR (400 MHz, CDCl3): δ = 7.47 (d, J = 2.1 Hz, 1H), 7.18 (d, J = 8.7 Hz, 1H), 7.14–7.10 (m, 2H), 7.04 (dd, J = 8.6, 2.2 Hz, 1H), 6.84–6.78 (m, 2H), 4.23 (s, 2H), 4.08–3.86 (m, 4H), 3.55 (dt, J = 44.7, 6.4 Hz, 4H), 2.02–1.86 (m, 4H), 1.59–1.48 (m, 1H), 1.41–1.32 (m, 5H), 0.88 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 158.08, 154.10, 153.99, 146.93, 132.90, 129.57, 128.14, 117.21, 114.92, 112.42, 109.22, 63.58, 46.59, 46.49, 42.72, 38.18, 33.87, 26.23, 25.96, 25.12, 22.46, 14.90 ppm; ESI-MS: m/z = 436.30 [M + H]+, calcd 436.26.
2-(4-Ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl Azetidine-1-carboxylate (21d)
According to GP3, 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20) (30.0 mg, 88.5 μmol, 1.0 equiv), 4-nitrophenyl azetidine-1-carboxylate (23.5 mg, 106 μmol, 1.2 equiv), and potassium tert-butoxide (12.9 mg, 115 μmol, 1.3 equiv) were used to obtain 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl azetidine-1-carboxylate (21d) (15.2 mg, 36.0 μmol, 41%) as an off-white oil after column chromatography (ethyl acetate). 1H-NMR (400 MHz, CDCl3): δ = 7.47 (d, J = 2.1 Hz, 1H), 7.19 (d, J = 8.7 Hz, 1H), 7.15–7.10 (m, 2H), 7.02 (ddd, J = 8.6, 2.3, 0.9 Hz, 1H), 6.84–6.79 (m, 2H), 4.32–4.07 (m, 6H), 4.02–3.90 (m, 4H), 2.39–2.29 (m, 2H), 1.60–1.49 (m, 1H), 1.40–1.34 (m, 5H), 0.88 (d, J = 6.6 Hz, 6H); 13C-NMR (101 MHz, CDCl3): δ = 158.12, 155.12, 154.16, 146.65, 142.42, 132.90, 129.61, 128.02, 117.12, 114.96, 112.30, 109.34, 63.61, 50.36, 49.43, 42.77, 38.18, 33.82, 26.25, 22.47, 15.93, 14.92 ppm; ESI-MS: m/z = 422.25 [M + H]+, calcd 422.24.
2-(4-Ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl Cyclopropylcarbamate (21e)
According to GP1, 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-ol (20) (22.0 mg, 64.9 μmol, 1.0 equiv), cyclopropyl isocyanate (7.55 mg, 90.9 μmol, 1.4 equiv), and TEA (15.2 μL, 11.1 mg, 110 μmol, 1.7 equiv) were used to obtain 2-(4-ethoxybenzyl)-1-isopentyl-1H-benzo[d]imidazole-5-yl cyclopropylcarbamate (21e) (11.8 mg, 28.0 μmol, 43%) as an off-white oil after column chromatography (DCM/methanol, 40:1). 1H-NMR (400 MHz, CDCl3): δ = 7.49 (s, 1H), 7.20 (d, J = 8.7 Hz, 1H), 7.13 (d, J = 8.5 Hz, 2H), 7.08–7.04 (m, 1H), 6.85–6.79 (m, 2H), 5.30 (s, 1H), 4.25 (s, 2H), 4.02–3.88 (m, 4H), 2.80–2.65 (m, 1H), 1.63–1.49 (m, 1H), 1.40–1.33 (m, 5H), 0.89 (d, J = 6.6 Hz, 6H), 0.83–0.74 (m, 2H), 0.68–0.61 (m, 2H); 13C-NMR (101 MHz, CDCl3): δ = 158.18, 155.88, 154.21, 146.62, 142.20, 132.86, 129.64, 127.90, 117.21, 115.01, 112.18, 109.44, 99.39, 63.63, 42.82, 38.20, 33.76, 26.27, 22.48, 14.93, 7.01 ppm; ESI-MS: m/z = 422.20 [M + H]+, calcd 422.24.
In Vitro Studies on ChEs and CBRs
ChE Inhibition Studies
hBChE (E.C. 3.1.1.8, from humans) was kindly provided by Oksana Lockridge from the University of Nebraska Medical Center. For determination of IC50 values, KM, and νmax, the enzyme stock solution (2.2 mg/mL) was diluted with Dulbecco’s phosphate-buffered saline (1:1250) and stored at 4 °C. For determination of k4, the stock solution was used without previous dilution. hAChE, ATC, and BTC iodines, as well as DTNB, were obtained from Sigma-Aldrich (Steinheim, Germany). A phosphate solution was prepared by dissolving NaH2PO4 in water (55 mM) and adjusting a pH 8.0 by adding 0.1 M NaOH (buffer A). A 10 mM DNTB stock solution was diluted with buffer to 0.3 mM (buffer B). All experiments were conducted at 25 °C.
IC50 Determination
The stock solutions of the test compounds were prepared in DMSO (4 mM) and diluted with buffer to a starting concentration of 10 or 100 μM, depending on the activity of the compound. Further desired dilutions were prepared on a 96-well plate (>1% DMSO). Then, 130 μL of 12 different concentrations of compound dilution and 10 μL of the respective enzyme (≈2.5 units/mL) were incubated for 20 min (irreversible). Afterward, 3 μL of either ATC or BTC iodide solution (45 mM in water) was added, and enzyme activity was immediately observed via UV (λ = 412 nm) for 3 min at seven points with an interval of 30 s. Each concentration was repeated as a triplicate.
Determination of KM and νmax
To determine KM and νmax, a similar setup, as for the calculation of IC50 values, was used. At least seven different concentrations were measured at six different time points. The obtained enzyme activities (in percent) were plotted against time and fitted to eq 1 to determine the rate constant kobs using GraphPad Prism 9.
| 1 |
A is the enzyme activity at time t, A0 is the enzyme activity at time t = 0, and A∞ is the enzyme activity at infinite time. The obtained kobs values were plotted against the concentration using GraphPad Prism 9 to obtain KM and νmax.
Decarbamylation
For the measurement of decarbamylation kinetics, a high amount of enzyme was incubated with a suitable amount of the inhibitor for 1 h. The concentration of the inhibitor was chosen so that the enzyme was inhibited to >90%. After incubation, the solution was diluted 1000 fold so that no enzyme was carbamylated anymore, and enzyme activity was measured at several (at least six) time points as described above. To determine full enzyme activity, a batch of the enzyme was treated with buffer instead of inhibitor solution. The enzyme activity in percent was plotted against the time after dilution to give the first-order rate constant k4 using software GraphPad Prism 9.
All procedures used to determine the binding properties of the inhibitors on ChEs have been established before.14,18,19,26,42,47,66−73
Radioligand Binding Studies for CBRs
Radioligand binding studies were performed as previously described by our group.14,19,42,51,74,75hCB2R-HEK cells were a kind gift from AbbVie Laboratories (Chicago, U.S.). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose supplemented with 10% fetal calve serum, 1% penicillin/streptomycin, and 25 μg/mL zeocin in a 37 °C incubator in the presence of 5% CO2. Cells were passaged three times a week. The respective hCB2R membranes were prepared as described in the literature.74 The rCB1R membrane homogenate was prepared from brains of adult, female rats, supplied by Prof. Dr. Kristina Lorenz from the University of Würzburg.76,77rCB1R membrane homogenates were shock-frozen in liquid nitrogen and stored at −80 °C. The respective rCB1R membranes were freshly prepared according to the protocol described by Catani and Rinaldi-Carmona for preparation of membrane homogenates.77,78
Saturation and competition binding assays were carried out according to the protocol previously established in M. Decker’s research group.74 Radioactive [3H]CP55,940 was bought from PerkinElmer LAS (Rodgau, Germany). Rimonabant (CB1R reference compound) and compound 4 were synthesized in-house. Competition binding assays were carried out in 96-well multiscreen filter plates (Millipore) with seven concentrations (0.01 nM to 0.324 mM) of the target compound and 0.63 nM [3H]CP55,940. The positive controls were the selective ligands rimonabant and compound 4 for the assays over CB1R and CB2R, respectively. The stock solutions were prepared by dissolving in DMSO in a concentration of 5 mM. The dilution series of all stock solutions was prepared in binding buffer (50 mM Tris–HCl; 5 mM MgCl2·6H2O; 2.5 mM EDTA; 2 mg/mL BSA; pH = 7.4). Reactions were started by adding the membrane homogenate (12.5 μg/well for rCB1R or 8 μg/well for hCB2R) diluted in binding buffer to the wells. After 3 h incubation at room temperature, the reaction was stopped by vacuum filtration, and each well was washed with cold assay buffer (4 × 200 μL). The filter plate was dried at 45 °C. Then, IRGA Safe plus-scintillation cocktail (PerkinElmer) was added (20 μL/well). The activity was counted in a Micro Beta Trilux counter (Turku, Wallac). The positive controls for CB1R and CB2R were used for normalization of the obtained values for the test compounds. IC50 values were determined from sigmoidal dose–response curves, applying nonlinear regression and one-side fit logIC50 as curve fitting functions, using GraphPad Prism 9.
Calcium Mobilization Assay for CB2R
Chinese hamster ovarian (CHO-K1) cells stably expressing Gαq16 with either hCB1 or hCB2 receptor were cultivated in Ham’s nutrient mixture F12 (Merck) supplemented in 10% fetal calf serum (FCS) and penicillin/streptomycin (90 U/mL) and plated onto 96-well plates (30,000 cells/well) to a final volume of 100 μL/well and incubated at 37 °C for 24 h. Cells were washed with freshly prepared assay buffer made up from 10× Hanks’ balanced salt solution (HBSS, containing final concentrations of 137.0 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 0.4 mM MgSO4, 0.5 mM MgCl2, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 4.2 mM NaHCO3, and 5.6 mM d-glucose), 1.0 M HEPES (final concentration 20 mM), bovine serum albumin (1%), and Probenecid (dissolved in 1 M NaOH, final concentration 2.7 mM) adjusted to pH 7.4. Cells were loaded with 100 μL of Fura-2AM (4 μM) in assay buffer at 37 °C for 1 h. The dye was removed, and the wells were washed with assay buffer. Then, 60 μL of assay buffer (100 μL for controls) was added to the dye-loaded cells. Calcium flux was monitored using an automated plate reader (FlexStation 3, molecular devices) at an excitation wavelength of 340 and 380 nm and an emission wavelength of 510 nm. After measuring the baseline for 30 s, test compounds in assay buffer (<0.5% DMSO) with various concentrations, compounds 4, and CP55,940 (positive control) or DMSO (vehicle control) were added automatically, and fluorescence was monitored for 5 min. All measurements were performed in duplicate in at least three independent experiments. EC50 values were analyzed from the respective area under curve with sigmoidal dose–response curve fitting using GraphPad Prism 9 software.
β-Arrestin 2 Recruitment Assays
Activity-based bioassays monitoring the recruitment of the intracellular protein βarr2 to the ligand-activated receptor were used to assess activity at hCB2R and off-target activity at hMOR. The development and application of these NanoBiT assays have been described previously.75,79−82 HEK293T cells stably expressing the hCB2R-βarr2 or hMOR-βarr2 system were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich, Darmstadt, Germany), 100 IU/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (all from Thermo Fisher Scientific), under a humidified atmosphere at 37 °C and 5% CO2. The day prior to the assays, cells were seeded in poly-d-lysine (Sigma-Aldrich)-coated 96-well plates (5 × 104 cells/well) and incubated overnight. The next day, cells were rinsed with Opti-MEM I Reduced Serum (Thermo Fisher Scientific). For the evaluation of CB2-βarr2 association, 100 μL of Opti-MEM was then added. Next, 25 μL of the 20-fold diluted Nano-Glo Live Cell Reagent (which contains the substrate) (Promega, Madison, WI, USA) was added to each well, and luminescence was monitored for 10–15 min using a TriStar2 LB 942 Multimode Microplate Reader (Berthold Technologies GmbH & Co., Germany) until the signal stabilized. Subsequently, 10 μL of 13.5× concentrated test solutions was added to the wells, and luminescence was recorded during 2 h. For screening at hCB2-βarr2R, compounds 4, 15a,c,d,e,h, 14j, and 21d were tested at a concentration of 10 μM. At a later stage, concentration ranges of compounds 3, 15d, and 21d were tested at both receptor systems. Test solutions were prepared by serial dilution in Opti-MEM. Here, CP55,940 (Sigma-Aldrich) served as the reference standard for normalization of the obtained data. Appropriate vehicle controls were present on each plate. For the assessment of hMOR activity, the same protocol was applied; however, here, 90 μL of Opti-MEM was used after rinsing, and 20 μL of a 6.75× concentrated test solution was added after equilibration. Again, compounds 4, 15a,c,d,e,h, j, and 21d were screened for MOR activity at a concentration of 10 μM. Hydromorphone was used as a positive control, and solvent controls were taken along on each plate. Raw data were corrected for inter-well variability using Microsoft Excel 2019. Mean AUC values were calculated and blank-corrected. All values were normalized to the Emax of the reference CP55,940 (set at 100%). Potency (EC50) and efficacy (Emax) of compounds 4, 15d, and 21d were calculated by fitting the generated concentration–response curves to a nonlinear regression (three-parametric logistic fit) model, using GraphPad Prism 9 software (San Diego, CA, USA). Results are represented as the AUC ± standard error of the mean (SEM) derived from a minimum of three independent experiments, run in duplicate. Data points were excluded for the highest concentrations in case of a signal reduction of minimally 20% compared to the closed lower dilution. The Grubbs test was used to detect potential outliers (p value <0.05); however, no outliers were found (212 data points). Raw luminescence values from screening experiments were processed in a similar way. Data are represented as % CB2-βarr2 activation (normalized to activation by 10 μM of compound 4), depicting the normalized AUC ± SEM.
Effects on Microglia
Cell Cultures
Mouse N9 microglial cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, and 2 mM glutamine (all cell cultures’ reagents were purchased from Aurogene Srl, Rome, Italy). At confluence, after a short wash with sterile PBS, microglia was trypsinized for 5 min at 37° C, and trypsin was inactivated with a complete DMEM. Detached cells were then collected, centrifuged for 5 min at 300g, and resuspended to be counted. For experiments, microglial cells were plated at the density of 2.5 × 105 in a 35 mmØ dish and exposed to 100 ng/mL LPS, in the presence or absence of increasing concentrations of the compound to be tested. After 24 h of treatment, microglial conditioned media were collected and partly used for the nitrite measurement, partly filtered through 0.22 μm filters, concentrated using Microcon YM-3 (Millipore, Billerica, MA), and resuspended in 12 μL of 4× loading buffer (0.2 M Tris-HCl, pH 6.8; 8% sodium dodecyl sulfate; 40% glycerol; 0.4% bromophenol blue, and 0.4 M dithiothreitol; Sigma-Aldrich) for Western blot analysis. In parallel, microglial cells were collected in 2× loading buffer (LB; 50 μL per dish) for Western blot analysis.
Western Blotting
Concentrated microglial conditioned media and cell samples were briefly sonicated and loaded into 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad). After electrophoresis and transfer onto nitrocellulose membranes (GE Healthcare, Milano, Italy), membranes were blocked for 1 h in blocking solution PBS—0.1% Tween-20 (Sigma-Aldrich) and 4% nonfat dry milk (Bio-Rad)—and incubated overnight at 4 °C with primary antibodies in PBS—0.1% Tween-20. Primary antibodies used were rabbit anti-iNOS, rabbit anti-IL1β, rabbit anti-TREM2, rabbit anti-TGFβ2, and mouse anti-GAPDH (all 1:1000 dilution except for anti-GAPDH used 1:20,000 dilution, all obtained from Santa Cruz Biotechnology). Membranes were then incubated with specific secondary antibodies conjugated to horseradish peroxidase (goat anti-rabbit and goat anti-mouse, both at 1:2000 dilution and from Santa Cruz) for 90 min at room temperature in PBS—0.1% Tween-20. Labeled proteins were visualized by using the Clarity Western ECL substrate (Bio-Rad) and detected using Bio-Rad Image Lab software with a ChemiDoc MP imaging system (Bio-Rad).
Nitrite Assay
Accumulation of nitrite in microglial conditioned media was measured by a colorimetric assay based on the Griess reaction. A nitrate standard curve was performed with NaNO2 at known concentrations. Sulfanilamide (5 mM; Sigma-Aldrich) was added to the culture medium, and the standard curve was obtained. Sulfanilamide reacts with nitrite under acidic conditions to form a diazonium cation, which subsequently couples to N-naphthyl-ethylenediamine dihydrochloride (40 mM; Sigma-Aldrich) to produce a colored azo dye. After 15 min of incubation at room temperature in the dark, absorbance was read at 540 nm with a multiplate spectrophotometric reader (BioRad Laboratories Srl, Segrate, Milano, Italy).
Statistical Analysis
All quantitative data are presented as means ± SE from at least three independent experiments. Statistical significance between different treatments was calculated using GraphPad Prism 6 software applying one-way analysis of variance (ANOVA) followed by post hoc comparison through Bonferroni’s test. A value of p < 0.05 was considered statistically significant.
Neurotoxicity and Neuroprotection (Oxytosis)
Cell Culture
HT22 cells were grown in DMEM (Sigma-Aldrich, Munich Germany) supplemented with 10% (v/v) FCS and 1% (v/v) penicillin–streptomycin. Cells were subcultured every 2 days and incubated at 37 °C with 5% CO2 in a humidified incubator. Compounds were dissolved in DMSO (Sigma-Aldrich, Munich, Germany) as stock solutions and diluted further into culture medium. To determine cell viability, a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich, Munich, Germany) assay was used. MTT solution (4 mg/mL in PBS) was diluted 1:10 with medium and added to the wells after removal of the old medium. Cells were incubated for 3 h, and then, lysis buffer (10% sodium dodecyl sulfide) was applied. The next day, the absorbance at 560 nm was determined with a multiwell plate photometer (Tecan, SpectraMax 250).
Toxicity and Oxytosis Assay
5 × 103 cells per well were seeded into sterile 96-well plates and incubated overnight. For the neurotoxicity assay, medium was removed, and increasing concentrations of the compound diluted with medium from a 0.1 M stock solution were added to the wells. DMSO (0.05%) in DMEM served as control. Cells were incubated for 24 h if neurotoxicity was determined by using a colorimetric MTT assay. For the oxytosis assay, 5 mM glutamate (monosodium l-glutamate, Sigma-Aldrich, Munich, Germany) together with increasing concentrations of the respective compounds was added to the cells and incubated for 24 h. As a positive control, a mixture of 25 mM quercetin (Sigma-Aldrich, Munich, Germany) and 5 mM glutamate was used. After 24 h incubation, cell viability was determined by using a colorimetric MTT assay, as described above. Results are presented as percentage of untreated control cells. Data are expressed as means ± SEM of three independent experiments. Analysis was accomplished by using GraphPad Prism 9 software by applying one-way ANOVA followed by Dunnett’s multiple comparison post-test. Levels of significance: *p < 0.05; **p < 0.01; and ***p < 0.001.
In Vivo Studies
Protocols for behavioral experiments were established before.14,19,26,47,52,53,59,83,84 In this work, compound 15d was tested for their neuroprotective properties in the in vivo pharmacological model of AD induced by intracerebroventricular (ICV) injection of the oligomerized Aβ25–35 peptide in the mouse. Parent compounds 2, 3, and 5, as well as 6, were already tested under the same conditions but further evaluated in this work.14,26 Compounds were injected IP between days 1 and 7. The oligomerized Aβ25–35 peptide was injected ICV on the first day of the study, and behavioral evaluation was performed between days 8 and 10.
Animals
Male swiss mice, 6 weeks old, weighing 32 ± 2 g, from JANVIER labs (Saint Berthevin, France) were kept in for housing, and experiments took place within the animal facility building of the University of Montpellier (CECEMA, Office of Veterinary Services agreement # B-34-172-23). Upon arrival, animals were divided into groups and housed with access to food and water ad libitum, except during behavioral experiments. Mice were kept in a temperature- and humidity-controlled animal facility on a 12 h/12 h light/dark cycle (lights off at 07:00 PM). Behavioral experiments were conducted between 9:00 and 17:00 in a sound-attenuated and air-regulated experimental room. All animal procedures were conducted in strict adherence to the European Union directive of September 22, 2010 (2010/63/UE) and the ARRIVE guidelines. The project was authorized by the French National Ethic Committee (APAFIS no. 148515034).85
Drugs and Injections
All experiments followed previously described protocols.19,26,47,52,55,59 The Aβ25–35 peptide was solubilized at 3 μM in water (vehicle solution V1) and oligomerized by incubation at 37 °C for 4 days. Mice were anesthetized with 2.5% isoflurane and injected ICV with Aβ25–35 (9 nmol in 3 μL). Compounds were dissolved in 100% DMSO at a concentration of 1 mg/mL and then diluted with physiological saline (0.9% NaCl) to a final percentage of 60% DMSO (vehicle solution V2). After compound injections, the behavior of the mice in their home cage was checked visually. Weight was examined daily, and the treatments did not affect their daily weight gain.
Spontaneous Alternation in the Y-Maze
Animals were tested for spontaneous alternation performance in the Y-maze, an index of spatial working memory.86,87 Made of gray polyvinylchloride, the Y-maze has arms of dimensions 40 cm × 13 cm; 3 cm at bottom, 10 cm at top, converging at an equal angle. Each mouse was placed at the end of one arm and allowed to move freely for 8 min. Arm entries, including possible returns into the same arm, were recorded. An alternation was defined as successive entry in the three different arms. The percentage of alternation was calculated as the number of maximum alternations = total arm entries minus 2 and percentage of alternation = (actual alternations/maximum alternations) × 100. Animals showing less than eight entries in 8 min or alternation percentages >90% or <20% were discarded from the calculations. Attrition was routinely <5%.
Step-through Passive Avoidance
The test measures non-spatial/contextual long-term memory.19,88 The apparatus was a grid-floor, two-compartment box separated by a guillotine door: (1) illuminated (60 W lamp, 40 cm above) with white polyvinylchloride walls and transparent cover (15 × 20 × 15 cm) and (2) black polyvinylchloride walls and cover (15 × 20 × 15 cm). Scrambled foot shocks (0.3 mA, 3 s) were delivered to the grid floor via a shock generator scrambler (Lafayette Instruments, Lafayette, MA). During training, the guillotine door was initially closed. Each mouse was placed into the white compartment. After 5 s, the door was raised. When the mouse entered the darkened compartment with all paws on the grid floor, the door was gently closed. A scrambled foot shock was delivered for 3 s. Step-through latency (i.e., the latency spent to enter the dark compartment) and level of sensitivity to the shock (0 = no sign; 1 = flinching reactions; 2 = flinching and vocalization reactions) were recorded. 24 h after training, retention tests were performed by placing each mouse into the white compartment for 5 s, raising the door, and recording step-through latency, up to 300 s. Animals showing latencies during training and retention <10 s and low shock sensitivity (0 or 1) were discarded from the calculations. Attrition was routinely <5%.
Statistical Analyses
Data were presented as mean ± SEM and analyzed using ANOVA (F value) followed by Dunnett’s test or a Kruskal–Wallis non-parametric ANOVA (H value) followed by Dunn’s multiple comparison test. The level of statistical significance considered was p < 0.05.
CI Calculations
Isobologram analyses were used to evaluate the nature of the interaction of two drugs at a given effect level. As expected dose–response curves followed a bi-phasic effect, only the ascending part of the curve was considered in calculations.62 Isobologram representation followed the concept of Fraser,61 and we previously published such analyses.62,84 The concentration required to produce a given effect (e.g., where ICx = IC50) is determined for drug A (ICx,A) and drug B (ICx,B) and indicated on the x and y axes of a two-coordinate plot, forming the two points, (ICx,A, 0) and (0, ICx,B). The line connecting these two points is the line of additivity. In a mix of drugs A + B, the concentrations of A and B contained in the combination that provide the same effect are represented by the coordinates (CA,x, CB,x). In the plot, synergy/additivity/antagonism is indicated when (CA,x, CB,x) is located below/on/above the line, respectively. Operationally, a CI is calculated as: CI = CA,x/ICx,A + CB,x/ICx,B, where CA,x and CB,x are the concentrations of drug A and B used in a combination that generates x % of the maximal combination effect. CI is the combination index. ICx,A and ICx,B are the concentrations of drugs A and B needed alone to produce x % of the maximal effect. A CI less than/equal to/more than 1 indicates synergy/additivity/antagonism, respectively. To calculate CI based on isobologram representation, alternation percentages and passive avoidance latencies were expressed as percentage of protection (PP) for each treatment group with PP(V1/V2) set as 100% and PP(Aβ25–35/V2) as 0% [Maurice, 2016; Martin et al., 2020].60,62 All calculations are presented in Table S1.
PK Measurements
Initial PK characterization was carried out by service provider Bienta/Enamine Ltd. (Kyiv, Ukraine) under direction of Yuliia Holota. Study design, animal selection, handling, and treatment were all in accordance with the Enamine PK study protocols and Institutional Animal Care and Use Guidelines. Animal treatment and samples preparation were conducted by the Animal Laboratory personnel at Enamine/Bienta. Male CD-1 mice [9 weeks old, body weight ranged from 29.8 to 35.8 g and average body weight across all groups 32.3 g, standard deviation (SD) = 1.7 g] were used in this study. The animals were randomly assigned to the treatment groups before the PK study; all animals were fasted for 4 h before dosing. Five time points (5, 15, 60, 120, and 240 min) and IV route of administration were set for this PK study. Each of the time point treatment group included three animals. There was also a control group of one animal. Target dosing of 2 mg/kg was applied using the respective compounds 15d and 21d solubilized in DMSO—saline (2:8). Mice were injected IP with 2,2,2-tribromoethanol from Sigma-Aldrich (St. Louis, Missouri) at the dose of 150 mg/kg prior to drawing the blood. Blood collection was performed from the orbital sinus in microtainers containing KF + Na2EDTA from Vacutest Kima (Arzergrande, Italy). Animals were sacrificed by cervical dislocation after the blood sample collection. Each blood sample was centrifuged for 10 min at 3000 rpm. Brain samples (right lobe) were collected and weighed. The samples were immediately processed, flash-frozen, and stored at −70 °C until subsequent analysis.
Plasma samples (40 μL) were mixed with 200 μL of internal standard [IS(90)] solution. After mixing by pipetting and centrifuging for 4 min at 6000 rpm, 2 μL of each supernatant was injected into the LC–MS/MS system.
The mixture of two compounds (verapamil and camptothecin) (50 and 300 ng/mL, respectively, in the water–methanol mixture 1:9, v/v) was used as internal standard [IS(90)] for quantification of compounds 15d and 21d and its metabolites 14 and 20 in plasma samples.
Brain samples were homogenized with 5 volumes of IS(80) solution (1 w + 5 v) using zirconium oxide beads in The Bullet Blender homogenizer for 30 s at speed 8. After this, the samples were centrifuged for 4 min at 14,000 rpm, and 2 μL of each supernatant was injected into the LC–MS/MS system.
The mixture of two compounds (verapamil and camptothecin) (50 and 300 ng/mL, respectively, in the water–methanol mixture, 1:4, v/v) was used as an internal standard [IS(80)] for quantification of compounds 15d and 21d and its metabolites 14 and 20 in the brain samples.
Analyses of plasma and brain samples were conducted by the Bioanalytical Laboratory personnel at Enamine/Bienta. The concentrations of all analyzed compounds in samples were determined using the high-performance liquid chromatography/tandem mass spectrometry (HPLC-MS/MS) method. Shimadzu HPLC system comprised two isocratic pumps LC-10ADvp, an autosampler SIL-20AC, a sub-controller FCV-14AH, and a degasser DGU-14A. Mass spectrometric analysis was performed using an API 3000 (triple-quadrupole) instrument from AB Sciex (Canada) with an electro-spray (ESI) interface. The data acquisition and system control were performed using Analyst 1.6.3 software from AB Sciex.
Calibration standards for quantification of the analytes in plasma samples: compounds were dissolved in DMSO, and resulting solutions with a concentration of 2 mg/mL were used for calibration standard preparation (stock solutions). The stock solution of compounds was consecutively diluted with IS(90) to get a series of calibration solutions with final concentrations of 10,000, 4000, 2000, 1000, 400, 200, 100, 40, 20, 10, 4, 2, 1, 0.4, 0.2, and 0.1 ng/mL for each analyte. The calibration curves were constructed using blank mouse plasma samples. To obtain calibration standards, blank plasma samples (40 μL) were mixed with 200 μL of the corresponding calibration solution. After mixing by pipetting and centrifuging for 4 min at 6000 rpm, 2 μL of each supernatant was injected into the LC–MS/MS system.
Calibration standards for quantification of compounds in the brain samples: the stock solutions of compounds 15d and 21d were consecutively diluted with IS(80) to get a series of calibration solutions with final concentrations of 10,000, 4000, 2000, 1000, 400, 200, 100, 40, 20, 10, 4, 2, 1, 0.4, 0.2, and 0.1 ng/mL; UW-MD 283 stock was dissolved at 10,000, 4000, 2000, 1000, 400, 200, 100, 40, 20, 10, 4, 2, 1, 0.4, and 0.2 ng/mL. The calibration curves were constructed using blank mouse brain samples. To obtain calibration standards, blank brain samples (weight 100 ± 1 mg) were homogenized in 500 μL of corresponding calibration solution using zirconium oxide beads (115 ± 5 mg) in The Bullet Blender homogenizer for 30 s at speed 8. After this, the samples were centrifuged for 4 min at 14 000 rpm, and 2 μL of each supernatant was injected into the LC–MS/MS system.
Acknowledgments
hBChE was kindly donated by Dr. O. Lockridge (University of Nebraska Medical Center). We thank the CECEMA-UM animal facility for animal housing and experiments.
Glossary
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- Aβ
amyloid β
- AD
Alzheimer’s disease
- ATC
acetylthiocholine
- ANOVA
analysis of variance
- BChE
butyrylcholinesterase
- BChEIs
butyrylcholinesterase inhibitors
- BTC
butyrylthiocholine
- CBR
cannabinoid receptor
- CHO
Chinese hamster ovarian
- cns
central nervous system
- DALY
disability adjusted life years
- DCM
dichloromethane
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- eq
equivalent
- DIPEA
diisopropyl ethylamine
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- EDTA
2,2′,2″,2‴-(ethane-1,2diylbis(azanetriyl))tetraacetic acid
- FDA
Food and Drug Administration
- HBTU
(2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium-hexafluorophosphate)
- HEK
human embryonic kidney
- HPLC
high-performance liquid chromatography
- ICV
intracerebroventricular
- IL1β
interleukin-1 beta
- iNOS
cytokine-inducible nitric oxide synthase
- IP
intraperitoneal
- IV
intravenous
- KOtBu
potassium tert-butoxide
- MOM
methoxymethyl
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide
- NMR
nuclear magnetic resonance
- PK
pharmacokinetic
- ROS
reactive oxygen species
- SI
selectivity index
- SDS
sodium dodecyl sulfate
- ST-PA
step-through passive avoidance test vehicle
- TEA
triethylamine
- TGFβ2
transforming growth factor-beta 2
- TREM2
triggering receptor expressed on myeloid cells 2
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- YMT
spontaneous alternation test in the Y-maze
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00541.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. P.S. performed synthesis, in vitro testing on ChEs, and animal experiments under supervision of T.M.; A.T., S.S., and M.H.D. (the latter under supervision of C.P.S.) performed in vitro testing on CBRs and MOR; E.P. (under supervision of B.M.) performed testing on microglial cells; A.M. and A.C. performed additional animal experiments; J.H. performed neuroprotectivity and neurotoxicity experiments; T.M. was responsible for supervision of behavioral animal experiments, provided support on their execution, and conducted additional animal testing; M.D. was accountable for the oversight and development of the whole project. All authors have given approval to the final version of the manuscript.
M.D. acknowledges funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG DE1546/10-1 and 12-1) and by the International Doctoral Program “Receptor Dynamics” within the Elite Network of Bavaria (Elitenetzwerk Bayern) for awarding Ph.D. positions to P.S. and A.T. The BayFrance (Franco-Bavarian University cooperation, FK03-2020) is gratefully acknowledged for funding of travel costs. M.H.D. acknowledges funding from the Research Foundation Flanders (FWO; grant 1S54521N).
The authors declare no competing financial interest.
Supplementary Material
References
- Cipriani G.; Dolciotti C.; Picchi L.; Bonuccelli U. Alzheimer and his disease: a brief history. Neurol. Sci. 2011, 32, 275–279. 10.1007/s10072-010-0454-7. [DOI] [PubMed] [Google Scholar]
- Niikura T.; Tajima H.; Kita Y. Neuronal Cell Death in Alzheimer’s Disease and a Neuroprotective Factor, Humanin. Curr. Neuropharmacol. 2006, 4, 139–147. 10.2174/157015906776359577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621–629. 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- Francis P. T.; Palmer A. M.; Snape M.; Wilcock G. K. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol., Neurosurg. Psychiatry 1999, 66, 137–147. 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K. A.; Alafuzoff I.; Attems J.; Beach T. G.; Cairns N. J.; Crary J. F.; Dickson D. W.; Hof P. R.; Hyman B. T.; Jack C. R. Jr.; Jicha G. A.; Knopman D. S.; Kovacs G. G.; Mackenzie I. R.; Masliah E.; Montine T. J.; Nelson P. T.; Schmitt F.; Schneider J. A.; Serrano-Pozo A.; Thal D. R.; Toledo J. B.; Trojanowski J. Q.; Troncoso J. C.; Vonsattel J. P.; Wisniewski T. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015, 129, 757–762. 10.1007/s00401-015-1407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery W. R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free Radicals Biol. 1997, 23, 134–147. 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Pohanka M. Alzheimer’s Disease and Oxidative Stress: A Review. Curr. Med. Chem. 2013, 21, 356–364. 10.2174/09298673113206660258. [DOI] [PubMed] [Google Scholar]
- H Ferreira-Vieira T.; M Guimaraes I.; R Silva F.; M Ribeiro F. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. 10.2174/1570159x13666150716165726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry A. V. Jr.; Buccafusco J. J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- Ali T. B.; Schleret T. R.; Reilly B. M.; Chen W. Y.; Abagyan R. Adverse Effects of Cholinesterase Inhibitors in Dementia, According to the Pharmacovigilance Databases of the United-States and Canada. PLoS One 2015, 10, 01443377. 10.1371/journal.pone.0144337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T.; Brückner M. K.; Lange M.; Bigl V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development—A study of molecular forms. Neurochem. Int. 1992, 21, 381–396. 10.1016/0197-0186(92)90189-x. [DOI] [PubMed] [Google Scholar]
- Giacobini E. Cholinergic function and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, 1–5. 10.1002/gps.935. [DOI] [PubMed] [Google Scholar]
- Bolognesi M. L.; Bartolini M.; Cavalli A.; Andrisano V.; Rosini M.; Minarini A.; Melchiorre C. Design, Synthesis, and Biological Evaluation of Conformationally Restricted Rivastigmine Analogues. J. Med. Chem. 2004, 47, 5945–5952. 10.1021/jm049782n. [DOI] [PubMed] [Google Scholar]
- Dolles D.; Hoffmann M.; Gunesch S.; Marinelli O.; Moller J.; Santoni G.; Chatonnet A.; Lohse M. J.; Wittmann H. J.; Strasser A.; Nabissi M.; Maurice T.; Decker M. Structure-Activity Relationships and Computational Investigations into the Development of Potent and Balanced Dual-Acting Butyrylcholinesterase Inhibitors and Human Cannabinoid Receptor 2 Ligands with Pro-Cognitive in Vivo Profiles. J. Med. Chem. 2018, 61, 1646–1663. 10.1021/acs.jmedchem.7b01760. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Naranjo P.; Perez-Macias N.; Campillo N. E.; Perez C.; Aran V. J.; Giron R.; Sanchez-Robles E.; Martin M. I.; Gomez-Canas M.; Garcia-Arencibia M.; Fernandez-Ruiz J.; Paez J. A. Cannabinoid agonists showing BuChE inhibition as potential therapeutic agents for Alzheimer’s disease. Eur. J. Med. Chem. 2014, 73, 56–72. 10.1016/j.ejmech.2013.11.026. [DOI] [PubMed] [Google Scholar]
- Montanari S.; Mahmoud A. M.; Pruccoli L.; Rabbito A.; Naldi M.; Petralla S.; Moraleda I.; Bartolini M.; Monti B.; Iriepa I.; Belluti F.; Gobbi S.; Di Marzo V.; Bisi A.; Tarozzi A.; Ligresti A.; Rampa A. Discovery of novel benzofuran-based compounds with neuroprotective and immunomodulatory properties for Alzheimer’s disease treatment. Eur. J. Med. Chem. 2019, 178, 243–258. 10.1016/j.ejmech.2019.05.080. [DOI] [PubMed] [Google Scholar]
- Pagé D.; Balaux E.; Boisvert L.; Liu Z.; Milburn C.; Tremblay M.; Wei Z.; Woo S.; Luo X.; Cheng Y. X.; Yang H.; Srivastava S.; Zhou F.; Brown W.; Tomaszewski M.; Walpole C.; Hodzic L.; St-Onge S.; Godbout C.; Salois D.; Payza K. Novel benzimidazole derivatives as selective CB2 agonists. Bioorg. Med. Chem. Lett. 2008, 18, 3695–3700. 10.1016/j.bmcl.2008.05.073. [DOI] [PubMed] [Google Scholar]
- Sawatzky E.; Al-Momani E.; Kobayashi R.; Higuchi T.; Samnick S.; Decker M. A Novel Way To Radiolabel Human Butyrylcholinesterase for Positron Emission Tomography through Irreversible Transfer of the Radiolabeled Moiety. ChemMedChem 2016, 11, 1540–1550. 10.1002/cmdc.201600223. [DOI] [PubMed] [Google Scholar]
- Scheiner M.; Dolles D.; Gunesch S.; Hoffmann M.; Nabissi M.; Marinelli O.; Naldi M.; Bartolini M.; Petralla S.; Poeta E.; Monti B.; Falkeis C.; Vieth M.; Hubner H.; Gmeiner P.; Maitra R.; Maurice T.; Decker M. Dual-Acting Cholinesterase-Human Cannabinoid Receptor 2 Ligands Show Pronounced Neuroprotection in Vitro and Overadditive and Disease-Modifying Neuroprotective Effects in Vivo. J. Med. Chem. 2019, 62, 9078–9102. 10.1021/acs.jmedchem.9b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J.; Hijikuro I.; Kihara T.; Murugesh M. G.; Fuse S.; Tsumura Y.; Akaike A.; Niidome T.; Takahashi T.; Sugimoto H. Design, synthesis and evaluation of carbamate-modified (-)-N(1)-phenethylnorphysostigmine derivatives as selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1721–1723. 10.1016/j.bmcl.2010.01.035. [DOI] [PubMed] [Google Scholar]
- Spatz P.; Zimmermann T.; Steinmüller S.; Hofmann J.; Maurice T.; Decker M. Novel benzimidazole-based pseudo-irreversible butyrylcholinesterase inhibitors with neuroprotective activity in an Alzheimer’s disease mouse model. RSC Med. Chem. 2022, 13, 944–954. 10.1039/d2md00087c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig N. H.; Utsuki T.; Yu Q.; Zhu X.; Holloway H. W.; Perry T.; Lee B.; Ingram D. K.; Lahiri D. K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. 10.1185/03007990152673800. [DOI] [PubMed] [Google Scholar]
- Du C.; Wang L.; Guan Q.; Yang H.; Chen T.; Liu Y.; Li Q.; Lyu W.; Lu X.; Chen Y.; Liu Y.; Liu H.; Feng F.; Liu W.; Liu Z.; Li W.; Chen Y.; Sun H. N-Benzyl Benzamide Derivatives as Selective Sub-Nanomolar Butyrylcholinesterase Inhibitors for Possible Treatment in Advanced Alzheimer’s Disease. J. Med. Chem. 2022, 65, 11365–11387. 10.1021/acs.jmedchem.2c00944. [DOI] [PubMed] [Google Scholar]
- Li Q.; Chen Y.; Xing S.; Liao Q.; Xiong B.; Wang Y.; Lu W.; He S.; Feng F.; Liu W.; Chen Y.; Sun H. Highly Potent and Selective Butyrylcholinesterase Inhibitors for Cognitive Improvement and Neuroprotection. J. Med. Chem. 2021, 64, 6856–6876. 10.1021/acs.jmedchem.1c00167. [DOI] [PubMed] [Google Scholar]
- Viayna E.; Coquelle N.; Cieslikiewicz-Bouet M.; Cisternas P.; Oliva C. A.; Sanchez-Lopez E.; Ettcheto M.; Bartolini M.; De Simone A.; Ricchini M.; Rendina M.; Pons M.; Firuzi O.; Perez B.; Saso L.; Andrisano V.; Nachon F.; Brazzolotto X.; Garcia M. L.; Camins A.; Silman I.; Jean L.; Inestrosa N. C.; Colletier J. P.; Renard P. Y.; Munoz-Torrero D. Discovery of a Potent Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase with Antioxidant Activity that Alleviates Alzheimer-like Pathology in Old APP/PS1 Mice. J. Med. Chem. 2020, 64, 812–839. 10.1021/acs.jmedchem.0c01775. [DOI] [PubMed] [Google Scholar]
- Hoffmann M.; Stiller C.; Endres E.; Scheiner M.; Gunesch S.; Sotriffer C.; Maurice T.; Decker M. Highly Selective Butyrylcholinesterase Inhibitors with Tunable Duration of Action by Chemical Modification of Transferable Carbamate Units Exhibit Pronounced Neuroprotective Effect in an Alzheimer’s Disease Mouse Model. J. Med. Chem. 2019, 62, 9116–9140. 10.1021/acs.jmedchem.9b01012. [DOI] [PubMed] [Google Scholar]
- Ashton J. C.; Glass M. The Cannabinoid CB2 Receptor as a Target for Inflammation-Dependent Neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso E.; Ferrer I. Cannabinoids for treatment of Alzheimer’s disease: moving toward the clinic. Front. Pharmacol. 2014, 5, 37. 10.3389/fphar.2014.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contino M.; Capparelli E.; Colabufo N. A.; Bush A. I.. The CB2 Cannabinoid System: A New Strategy in Neurodegenerative Disorder and Neuroinflammation; Frontiers Media SA, 2017; Vol. 11, p 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte C.; Blanchet M. R.; Laviolette M.; Flamand N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. 10.1007/s00018-016-2300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A. J.; Zubko O.; Reeves S. J.; Howard R. J. Endocannabinoid system alterations in Alzheimer’s disease: A systematic review of human studies. Brain Res. 2020, 1749, 147135. 10.1016/j.brainres.2020.147135. [DOI] [PubMed] [Google Scholar]
- Galiègue S.; Mary S.; Marchand J.; Dussossoy D.; Carriere D.; Carayon P.; Bouaboula M.; Shire D.; Fur G.; Casellas P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- McKenna M.; McDougall J. J. Cannabinoid control of neurogenic inflammation. Br. J. Pharmacol. 2020, 177, 4386–4399. 10.1111/bph.15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A. P.; Denovan-Wright E. M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 806417. 10.3389/fphar.2021.806417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiesh H. M.; Sharma C.; Goyal S. N.; Jha N. K.; Ojha S. Pharmacological Properties, Therapeutic Potential and Molecular Mechanisms of JWH133, a CB2 Receptor-Selective Agonist. Front. Pharmacol. 2021, 12, 702675. 10.3389/fphar.2021.702675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K.; Grundke-Iqbal I. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers. Dement. 2010, 6, 420–424. 10.1016/j.jalz.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proschak E.; Stark H.; Merk D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- Mangiatordi G. F.; Intranuovo F.; Delre P.; Abatematteo F. S.; Abate C.; Niso M.; Creanza T. M.; Ancona N.; Stefanachi A.; Contino M. Cannabinoid Receptor Subtype 2 (CB2R) in a Multitarget Approach: Perspective of an Innovative Strategy in Cancer and Neurodegeneration. J. Med. Chem. 2020, 63, 14448–14469. 10.1021/acs.jmedchem.0c01357. [DOI] [PubMed] [Google Scholar]
- Seghetti F.; Gobbi S.; Belluti F.; Rampa A.; Bisi A. Interplay Between Endocannabinoid System and Neurodegeneration: Focus on Polypharmacology. Curr. Med. Chem. 2022, 29, 4796–4830. 10.2174/0929867328666211115124639. [DOI] [PubMed] [Google Scholar]
- Xing S.; Li Q.; Xiong B.; Chen Y.; Feng F.; Liu W.; Sun H. Structure and therapeutic uses of butyrylcholinesterase: Application in detoxification, Alzheimer’s disease, and fat metabolism. Med. Res. Rev. 2020, 41, 858–901. 10.1002/med.21745. [DOI] [PubMed] [Google Scholar]
- Abate G.; Uberti D.; Tambaro S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology 2021, 10, 542. 10.3390/biology10060542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolles D.; Nimczick M.; Scheiner M.; Ramler J.; Stadtmüller P.; Sawatzky E.; Drakopoulos A.; Sotriffer C.; Wittmann H.-J.; Strasser A.; Decker M. Aminobenzimidazoles and Structural Isomers as Templates for Dual-Acting Butyrylcholinesterase Inhibitors andhCB2R Ligands To Combat Neurodegenerative Disorders. ChemMedChem 2016, 11, 1270–1283. 10.1002/cmdc.201500418. [DOI] [PubMed] [Google Scholar]
- Pagé D.; Brochu M.-C.; Yang H.; Brown W.; St-Onge S.; Martin E.; Salois D. Novel Benzimidazole Derivatives as Selective CB2 Inverse Agonists. Lett. Drug Des. Discovery 2006, 3, 298–303. 10.2174/157018006777574177. [DOI] [Google Scholar]
- Jiang X.; Zhang Z.; Zuo J.; Wu C.; Zha L.; Xu Y.; Wang S.; Shi J.; Liu X. H.; Zhang J.; Tang W. Novel cannabidiol-carbamate hybrids as selective BuChE inhibitors: Docking-based fragment reassembly for the development of potential therapeutic agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 223, 113735. 10.1016/j.ejmech.2021.113735. [DOI] [PubMed] [Google Scholar]
- Montanari S.; Allara M.; Scalvini L.; Kostrzewa M.; Belluti F.; Gobbi S.; Naldi M.; Rivara S.; Bartolini M.; Ligresti A.; Bisi A.; Rampa A. New Coumarin Derivatives as Cholinergic and Cannabinoid System Modulators. Molecules 2021, 26, 3254. 10.3390/molecules26113254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo S.; Tarozzi A.; Bartolini M.; Da Costa G.; Bisi A.; Gobbi S.; Belluti F.; Ligresti A.; Allara M.; Monti J. P.; Andrisano V.; Di Marzo V.; Hrelia P.; Rampa A. 2-Arylbenzofuran-based molecules as multipotent Alzheimer’s disease modifying agents. Eur. J. Med. Chem. 2012, 58, 519–532. 10.1016/j.ejmech.2012.10.045. [DOI] [PubMed] [Google Scholar]
- Scheiner M.; Hoffmann M.; He F.; Poeta E.; Chatonnet A.; Monti B.; Maurice T.; Decker M. Selective Pseudo-irreversible Butyrylcholinesterase Inhibitors Transferring Antioxidant Moieties to the Enzyme Show Pronounced Neuroprotective Efficacy In Vitro and In Vivo in an Alzheimer’s Disease Mouse Model. J. Med. Chem. 2021, 64, 9302–9320. 10.1021/acs.jmedchem.1c00534. [DOI] [PubMed] [Google Scholar]
- Abate C.; Riganti C.; Pati M. L.; Ghigo D.; Berardi F.; Mavlyutov T.; Guo L. W.; Ruoho A. Development of sigma-1 (σ 1) receptor fluorescent ligands as versatile tools to study σ 1 receptors. Eur. J. Med. Chem. 2016, 108, 577–585. 10.1016/j.ejmech.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnetta L.; Bermudez M.; Riefolo F.; Matera C.; Claro E.; Messerer R.; Littmann T.; Wolber G.; Holzgrabe U.; Decker M. Fluorination of Photoswitchable Muscarinic Agonists Tunes Receptor Pharmacology and Photochromic Properties. J. Med. Chem. 2019, 62, 3009–3020. 10.1021/acs.jmedchem.8b01822. [DOI] [PubMed] [Google Scholar]
- Scheiner M.; Sink A.; Hoffmann M.; Vrigneau C.; Endres E.; Carles A.; Sotriffer C.; Maurice T.; Decker M. Photoswitchable Pseudoirreversible Butyrylcholinesterase Inhibitors Allow Optical Control of Inhibition in Vitro and Enable Restoration of Cognition in an Alzheimer’s Disease Mouse Model upon Irradiation. J. Am. Chem. Soc. 2022, 144, 3279–3284. 10.1021/jacs.1c13492. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Soacha D. A.; Fender J.; Ramirez Y. A.; Collado J. A.; Munoz E.; Maitra R.; Sotriffer C.; Lorenz K.; Decker M. Photo-Rimonabant: Synthesis and Biological Evaluation of Novel Photoswitchable Molecules Derived from Rimonabant Lead to a Highly Selective and Nanomolar “Cis-On” CB1R Antagonist. ACS Chem. Neurosci. 2021, 12, 1632–1647. 10.1021/acschemneuro.1c00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunesch S.; Hoffmann M.; Kiermeier C.; Fischer W.; Pinto A. F. M.; Maurice T.; Maher P.; Decker M. 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol. 2020, 29, 101378. 10.1016/j.redox.2019.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J.; Fayez S.; Scheiner M.; Hoffmann M.; Oerter S.; Appelt-Menzel A.; Maher P.; Maurice T.; Bringmann G.; Decker M. Sterubin: Enantioresolution and Configurational Stability, Enantiomeric Purity in Nature, and Neuroprotective Activity in Vitro and in Vivo. Chem.—Eur. J. 2020, 26, 7299–7308. 10.1002/chem.202001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J.; Ginex T.; Espargaro A.; Scheiner M.; Gunesch S.; Arago M.; Stigloher C.; Sabate R.; Luque F. J.; Decker M. Azobioisosteres of Curcumin with Pronounced Activity against Amyloid Aggregation, Intracellular Oxidative Stress, and Neuroinflammation. Chem.—Eur. J. 2021, 27, 6015–6027. 10.1002/chem.202005263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J.; Spatz P.; Walther R.; Gutmann M.; Maurice T.; Decker M. Synthesis and Biological Evaluation of Flavonoid-Cinnamic Acid Amide Hybrids with Distinct Activity against Neurodegeneration in Vitro and in Vivo. Chem.—Eur. J. 2022, 28, e202200786 10.1002/chem.202200786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S.; Wood M.; Maher P. Oxidative Stress Induces a Form of Programmed Cell Death with Characteristics of Both Apoptosis and Necrosis in Neuronal Cells. J. Neurochem. 2002, 71, 95–105. 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Soriano-Castell D.; Kepchia D.; Duggan B. M.; Currais A.; Schubert D.; Maher P. Cannabinol inhibits oxytosis/ferroptosis by directly targeting mitochondria independently of cannabinoid receptors. Free Radical Biol. Med. 2022, 180, 33–51. 10.1016/j.freeradbiomed.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D.; Kepchia D.; Liang Z.; Dargusch R.; Goldberg J.; Maher P. Efficacy of Cannabinoids in a Pre-Clinical Drug-Screening Platform for Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7719–7730. 10.1007/s12035-019-1637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T.; Lockhart B. P.; Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996, 706, 181–193. 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- Maurice T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 2016, 296, 270–278. 10.1016/j.bbr.2015.09.020. [DOI] [PubMed] [Google Scholar]
- Fraser T. R. Lecture on the Antagonism between the Actions of Active Substances. Br. Med. J. 1872, 2, 485–487. 10.1136/bmj.2.618.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P.; de Witte P. A. M.; Maurice T.; Gammaitoni A.; Farfel G.; Galer B. Fenfluramine acts as a positive modulator of sigma-1 receptors. Epilepsy Behav. 2020, 105, 106989. 10.1016/j.yebeh.2020.106989. [DOI] [PubMed] [Google Scholar]
- Guay D. R. Rivastigmine Transdermal Patch: Role in the Management of Alzheimer’s Disease. Consult. Pharm. 2008, 23, 598–609. 10.4140/tcp.n.2008.598. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Rivastigmine Transdermal Patch. Adis Drug Evaluation 2011, 71, 1209–1231. 10.2165/11206380-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Turner P. V.; Brabb T.; Pekow C.; Vasbinder M. A. Administration of Substances to Laboratory Animals: Routes of Administration and Factors to Consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Tikhonova I. G.; Decker M. Probing the mid-gorge of cholinesterases with spacer-modified bivalent quinazolinimines leads to highly potent and selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2011, 19, 1222–1235. 10.1016/j.bmc.2010.12.034. [DOI] [PubMed] [Google Scholar]
- Chen X.; Wehle S.; Kuzmanovic N.; Merget B.; Holzgrabe U.; Konig B.; Sotriffer C. A.; Decker M. Acetylcholinesterase inhibitors with photoswitchable inhibition of beta-amyloid aggregation. ACS Chem. Neurosci. 2014, 5, 377–389. 10.1021/cn500016p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras F. H.; Kling B.; Heilmann J.; Decker M. Neuroprotective Tri- and Tetracyclic BChE Inhibitors Releasing Reversible Inhibitors upon Carbamate Transfer. ACS Med. Chem. Lett. 2012, 3, 914–919. 10.1021/ml3001825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras F. H.; Kling B.; Sawatzky E.; Heilmann J.; Decker M. Cyclic acyl guanidines bearing carbamate moieties allow potent and dirigible cholinesterase inhibition of either acetyl- or butyrylcholinesterase. Bioorg. Med. Chem. 2014, 22, 5020–5034. 10.1016/j.bmc.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Gentzsch C.; Chen X.; Spatz P.; Kosak U.; Knez D.; Nose N.; Gobec S.; Higuchi T.; Decker M. Synthesis and Initial Characterization of a Reversible, Selective 18F-Labeled Radiotracer for Human Butyrylcholinesterase. Mol. Imaging Biol. 2021, 23, 505–515. 10.1007/s11307-021-01584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch C.; Hoffmann M.; Ohshima Y.; Nose N.; Chen X.; Higuchi T.; Decker M. Synthesis and Initial Characterization of a Selective, Pseudo-irreversible Inhibitor of Human Butyrylcholinesterase as PET Tracer. ChemMedChem 2021, 16, 1427–1437. 10.1002/cmdc.202000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatzky E.; Wehle S.; Kling B.; Wendrich J.; Bringmann G.; Sotriffer C. A.; Heilmann J.; Decker M. Discovery of Highly Selective and Nanomolar Carbamate-Based Butyrylcholinesterase Inhibitors by Rational Investigation into Their Inhibition Mode. J. Med. Chem. 2016, 59, 2067–2082. 10.1021/acs.jmedchem.5b01674. [DOI] [PubMed] [Google Scholar]
- Scheiner M.; Sink A.; Spatz P.; Endres E.; Decker M. Photopharmacology on Acetylcholinesterase: Novel Photoswitchable Inhibitors with Improved Pharmacological Profiles. ChemPhotoChem 2020, 5, 149–159. 10.1002/cptc.202000119. [DOI] [Google Scholar]
- Dolles D.; Strasser A.; Wittmann H.-J.; Marinelli O.; Nabissi M.; Pertwee R. G.; Decker M. The First Photochromic Affinity Switch for the Human Cannabinoid Receptor 2. Adv. Ther. 2018, 1, 1700032. 10.1002/adtp.201700032. [DOI] [Google Scholar]
- Rodriguez-Soacha D. A.; Steinmuller S. A. M.; Isbilir A.; Fender J.; Deventer M. H.; Ramirez Y. A.; Tutov A.; Sotriffer C.; Stove C. P.; Lorenz K.; Lohse M. J.; Hislop J. N.; Decker M. Development of an Indole-Amide-Based Photoswitchable Cannabinoid Receptor Subtype 1 (CB1R) “Cis-On” Agonist. ACS Chem. Neurosci. 2022, 13, 2410–2435. 10.1021/acschemneuro.2c00160. [DOI] [PubMed] [Google Scholar]
- Devane W. A.; Dysarz F. A. III; Johnson M. R.; Melvin L. S.; Howlett A. C. Determination and Characterization of a Cannabinoid Receptor in Rat Brain. Mol. Pharmacol. 1998, 34, 605–613. [PubMed] [Google Scholar]
- Rinaldi-Carmona M.; Barth F.; Héaulme M.; Shire D.; Calandra B.; Congy C.; Martinez S.; Maruani J.; Neliat G.; Caput D.; Ferrara P.; Soubrie P.; Brelière J. C.; Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- Catani V. M.; Gasperi V. Assay of CB1 Receptor Binding. Methods Mol. Biol. 2016, 1412, 41–55. 10.1007/978-1-4939-3539-0_5. [DOI] [PubMed] [Google Scholar]
- Cannaert A.; Storme J.; Franz F.; Auwarter V.; Stove C. P. Detection and Activity Profiling of Synthetic Cannabinoids and Their Metabolites with a Newly Developed Bioassay. Anal. Chem. 2016, 88, 11476–11485. 10.1021/acs.analchem.6b02600. [DOI] [PubMed] [Google Scholar]
- Cannaert A.; Storme J.; Hess C.; Auwarter V.; Wille S. M. R.; Stove C. P. Activity-Based Detection of Cannabinoids in Serum and Plasma Samples. Clin. Chem. 2018, 64, 918–926. 10.1373/clinchem.2017.285361. [DOI] [PubMed] [Google Scholar]
- Cannaert A.; Vasudevan L.; Friscia M.; Mohr A. L. A.; Wille S. M. R.; Stove C. P. Activity-Based Concept to Screen Biological Matrices for Opiates and (Synthetic) Opioids. Clin. Chem. 2018, 64, 1221–1229. 10.1373/clinchem.2018.289496. [DOI] [PubMed] [Google Scholar]
- Vasudevan L.; Vandeputte M.; Deventer M.; Wouters E.; Cannaert A.; Stove C. P. Assessment of structure-activity relationships and biased agonism at the Mu opioid receptor of novel synthetic opioids using a novel, stable bio-assay platform. Biochem. Pharmacol. 2020, 177, 113910. 10.1016/j.bcp.2020.113910. [DOI] [PubMed] [Google Scholar]
- Lahmy V.; Meunier J.; Malmstrom S.; Naert G.; Givalois L.; Kim S. H.; Villard V.; Vamvakides A.; Maurice T. Blockade of Tau Hyperphosphorylation and Aβ1–42 Generation by the Aminotetrahydrofuran Derivative ANAVEX2-73, a Mixed Muscarinic and σ1 Receptor Agonist, in a Nontransgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacol 2013, 38, 1706–1723. 10.1038/npp.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T.; Strehaiano M.; Simeon N.; Bertrand C.; Chatonnet A. Learning performances and vulnerability to amyloid toxicity in the butyrylcholinesterase knockout mouse. Behav. Brain Res. 2016, 296, 351–360. 10.1016/j.bbr.2015.08.026. [DOI] [PubMed] [Google Scholar]
- Kilkenny C.; Browne W.; Cuthill I. C.; Emerson M.; Altman D. G. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T.; Bayle J.; Privat A. Learning impairment following acute administration of the calcium channel antagonist nimodipine in mice. Behav. Pharmacol. 1995, 6, 167–175. 10.1097/00008877-199503000-00009. [DOI] [PubMed] [Google Scholar]
- Sarter M.; Bodewitz G.; Stephens D. N. Attenuation of scopolamine-induced impairment of spontaneous alternation behaviour by antagonist but not inverse agonist and agonist beta-carbolines. Psychopharmacology 1988, 94, 491–495. 10.1007/bf00212843. [DOI] [PubMed] [Google Scholar]
- Ögren S. O.; Stiedl O.. Encyclopedia of Psychopharmacology; Springer: Springer Berlin, Heidelberg, 2010; Vol. 2, pp 960–967. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.