

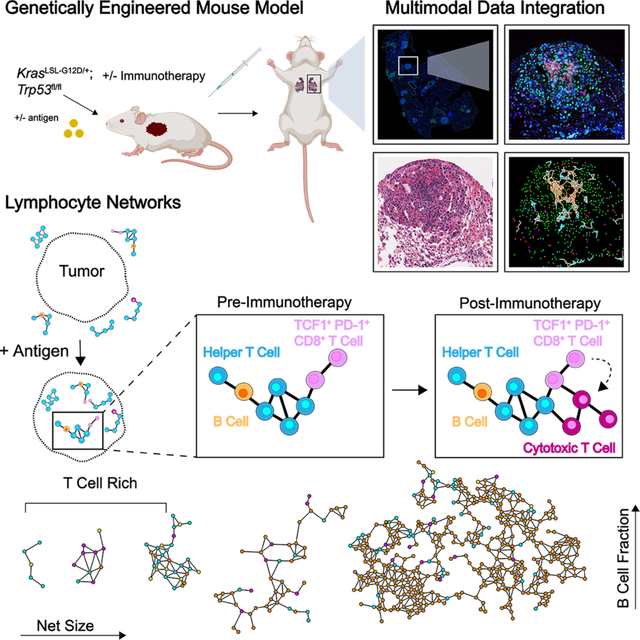
SUMMARY
Lymphocytes are key for immune surveillance of tumors, but our understanding of the spatial organization and physical interactions that facilitate lymphocyte anti-cancer functions is limited. We used multiplexed imaging, quantitative spatial analysis, and machine learning to create high-definition maps of lung tumors from a Kras/Trp53-mutant mouse model and human resections. Networks of interacting lymphocytes (‘lymphonets’) emerged as a distinctive feature of the anti-cancer immune response. Lymphonets nucleated from small T cell clusters and incorporated B cells with increasing size. CXCR3-mediated trafficking modulated lymphonet size and number, but T cell antigen expression directed intratumoral localization. Lymphonets preferentially harbored TCF1+ PD-1+ progenitor CD8+ T cells involved in responses to immune checkpoint blockade (ICB) therapy. Upon treatment of mice with ICB or an antigen-targeted vaccine, lymphonets retained progenitor and gained cytotoxic CD8+ T cell populations, likely via progenitor differentiation. These data show that lymphonets create a spatial environment supportive of CD8+ T cell anti-tumor responses.
eTOC Blurb
Gaglia et al. find striking changes in the spatial arrangement of immune cells in response to tumor antigens. T and B cells are recruited in lymphocyte networks (‘lymphonets’), which contain progenitor T cells. After immunotherapy, lymphonets gain cytotoxic T cells, likely due to progenitor cell differentiation and activation in this distinct immune environment.
Graphical Abstract
INTRODUCTION
During cancer progression, immune cells proliferate, migrate, and adapt in an attempt to impede tumor spread1,2. Tumor cells respond by inducing programs that suppress immune cell function3. Detailed characterization of the functional states of immune cells and their spatial organization relative to tumor cells is therefore needed to identify the features of anti-tumor immunity4,5. One way to accomplish this is using highly multiplexed spatial profiling, a set of analytical methods and computational approaches that provide quantitative descriptions of the i) identities and molecular characteristics of immune, tumor, and stromal cells, ii) physical and chemical factors that influence the spatial organization of these cell types, and iii) changes in spatial features of over time and space that constitute tumor responses to therapy6–8.
The Kras/Trp53-mutant (KP) lung adenocarcinoma model, which includes several variants, is prototypical of genetically engineered mouse models (GEMMs) of cancer having many of the features of human tumors. In this model, tumorigenesis is synchronously initiated in multiple cells by intratracheal delivery of lentivirus-encoded Cre recombinase into KrasLSL-G12D/+; Trp53fl/fl animals9,10. This gives rise to ~10–15 tumor nodules per 2-dimensional lung cross-section and progression from hyperplasia to adenocarcinoma occurs over the course of 1–5 months. Because these tumors have low rates of somatic mutations, they are not highly immunogenic11. To overcome this, T cell antigens are introduced by way of the tumor-initiating lentiviruses. In the LucOS variant of the KP model, two model CD8+ T cell antigens, the SIINFEKL (SIIN) epitope from chicken ovalbumin and the synthetic peptide SIYRYYGL (SIY), are expressed as a fusion to luciferase in tumor cells12. Conventional single-marker immunohistochemistry (IHC) analysis of tumor-bearing lung tissue from KP-LucOS versus control (KP-Cre) mice has shown that expression of tumor-specific LucOS antigens substantially increases the number of CD8+ T cells infiltrating tumors. However, despite this engagement of immunosurveillance mechanisms, tumor growth rebounds within weeks with a concomitant decline in the CD8+ T cell response12.
While dissociative single-cell methods like scRNA-seq, CYTOF, and FACS can provide deep insight into tumorigenesis and immunosurveillance in GEMMs13, these methods lack information on cell-cell interactions and locations of cell populations. Conventional histology and IHC provide positional information, however, they do not supply the detailed molecular information needed to identify and phenotype cells precisely.
Here, we used multiplexed tissue imaging to characterize spatial features of tumor-immune interactions in KP-LucOS lung tumors, including when chemokine-mediated trafficking was modulated, and after treatment with antigen-targeted vaccine or immune checkpoint blockade (ICB). This study establishes generally useful methods for spatial analysis of GEMMs and identifies lymphocyte networks (lymphonets) which harbor stem-like progenitor CD8 T cells as components of functional T cell responses in early tumor lesions and following immunotherapy.
RESULTS
Spatial analysis of KP GEMM tumor-immune microenvironment by multimodal data integration
To generate high-content spatial maps of tumor and immune-cell interactions in KP-lung tumors under multiple biologically informative conditions, KP mice were exposed to different tumor-initiating lentiviruses via intratracheal delivery and treated with immune therapies (Fig. 1A). Six to nine weeks after tumor initiation, H&E staining, mRNA in situ hybridization (ISH) and 24-plex CyCIF14 (Table S1) were performed on serial whole-slide sections (~1cm2) of formalin-fixed, paraffin-embedded (FFPE) tissue containing 2 or 3 lung lobes. Histopathological annotation of H&E images provided data on the position of tumor nodules and normal anatomic structures, including medium-large airways and blood vessels (Fig. S1A). RNA in situ hybridization (ISH) provided information on critical chemokines (e.g., Cxcl9, Cxcl10) that are difficult to image in tissues using antibodies. For CyCIF, a 24-plex antibody panel was developed that included lineage-specific transcription factors such as NKX2–1 (TTF-1) and intermediate filament protein pan-cytokeratin (Pan-CK), both markers of epithelial/tumor cells, and vimentin (VIM), a marker of mesenchymal cells, as well as markers expressed on specific lymphoid and myeloid cells (CD45, CD3e, B220, NKp46, CD11b, CD11c, Ly6G, CD103) (Figs. 1B–1D, S1B). These immune markers made it possible to delineate cell types with increasing depth, separating lymphoid and myeloid lineages, and subdividing them into T cell, B cell, natural killer (NK) cell, neutrophil, CD103+ dendritic cell (DC), alveolar macrophage, and tumor-associated macrophage (TAM) populations (Fig. 1D; see Fig. S1C for cell-type classification dendrogram). Additional markers (CD4, CD8, FOXP3) made it possible to distinguish T helper (Th), T cytotoxic (Tc), and T regulatory (Treg) cell populations, and functional markers were used to define the states of these cells with respect to Ki67 positivity (proliferation), cytotoxicity markers (granzyme B, GZMB; perforin, PRF), the presence of immune inhibitory receptors PD-1 and TIM-3, and expression of the T cell transcription factor (TCF1), a key regulator of T cell function and differentiation (Figs. 1C–1D, S1C).
Figure 1. Spatial analysis of KP GEMM tumor-immune microenvironment by multimodal data integration.
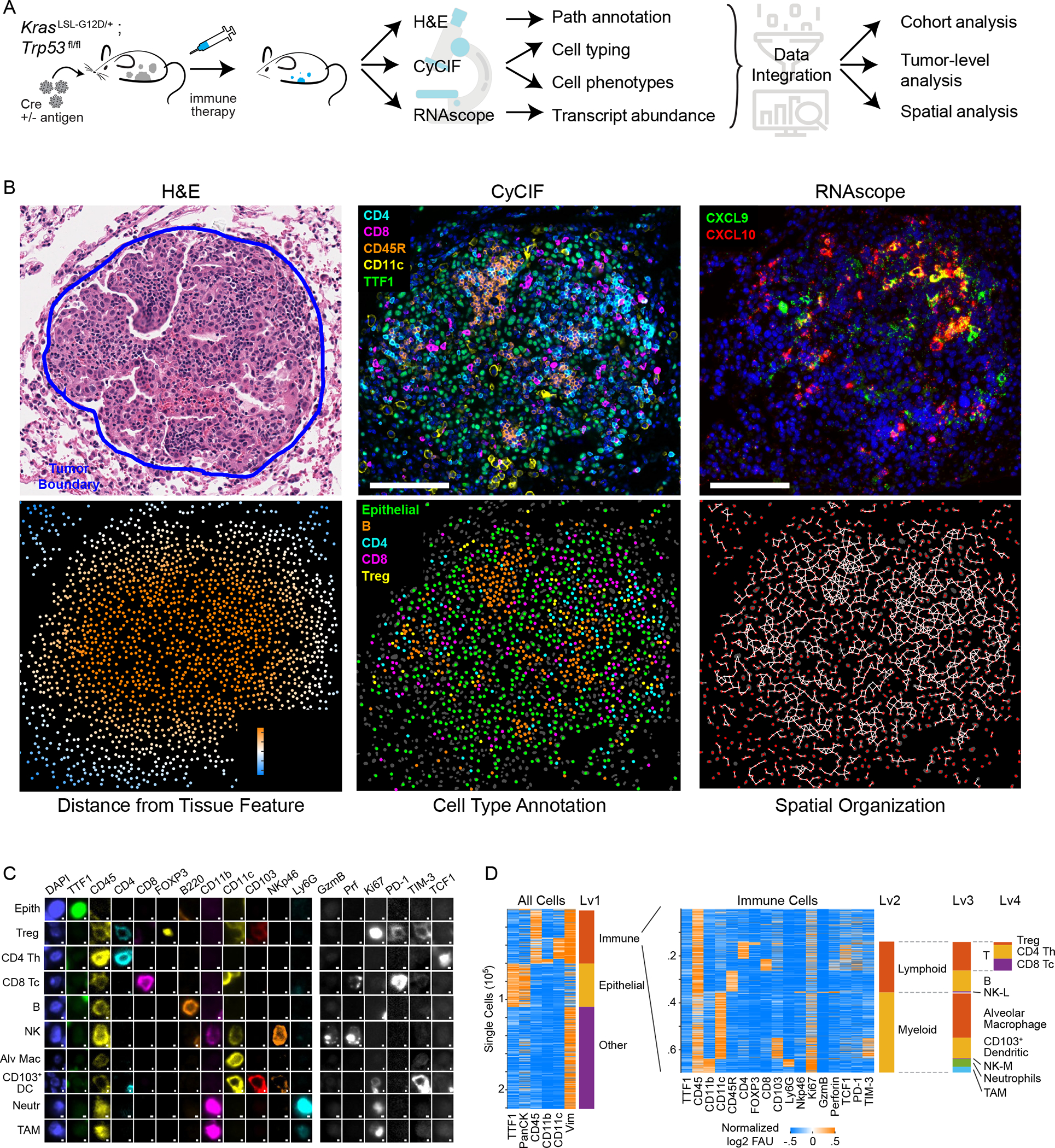
(A) Schematic: KP lung cancer GEMM, treatments, and multi-modality data integration. (B) Images acquired from KP-LucOS GEMM tumor nodule (expressing CD8+ T cell antigens): H&E, multiplexed CyCIF image of immune/tumor markers (DNA, blue), Cxcl9, Cxcl10 RNAScope™ (DNA, blue) (serial sections), map showing distance of cells from tumor edge, cell-type annotation map, and ‘graph’ map of physically interacting cells (Delaunay Triangulation). (C) Gallery of lineage, cell-state, and functional markers from CyCIF images of KP LucOS. Scalebar: 1μm. (D) Sequential clustering of CyCIF data using marker combinations in Figure S1C for immune, epithelial/tumor, stromal populations (rows=individual cells). See also Figure S1 and Table S1.
The resulting data were analyzed using several computational approaches. For CyCIF, images were stitched and registered and then segmented to identify single cells (typically ~1–5×105 cells per sample/mouse) and staining intensities quantified at a single-cell level; for mRNA ISH, foci were identified, their densities quantified, and data registered to CyCIF images from serial sections. Distance metrics were used to characterize cell positions relative to boundaries between tumor nodules and non-neoplastic lung tissue (‘tumor edge’) and blood vessels (Fig. 1B). Single cell positions were used to identify interacting cells in physical proximity and to create ‘graphs’ of interacting cell “networks” (Fig. 1B).
Tumor-antigen expression reorganizes the immune landscape in KP-lung cancer
We first profiled immune responses triggered by the LucOS CD8+ T cell antigens 8 weeks after lentiviral infection, a timepoint that represents a transition between a functional and dysfunctional CD8+ T cell response12,15,16. The tumor burden in LucOS mice was significantly lower than Cre mice (Fig. 2A, Table S2), however, the presence in LucOS mice of immunogenic SIIN and SIY CD8+ T cell antigens resulted in only modest differences in immune cell composition when lung tissue was examined as a whole (both tumor and non-tumor compartments together). For example, the numbers of neutrophils and B cells were slightly higher in LucOS whole lungs as compared to Cre lungs, and Treg cells and CD103+ DCs were slightly lower, but these differences did not reach statistical significance (Figs. 2B–2D, S2A–S2C).
Figure 2. Tumor antigen expression reorganizes KP lung cancer immune landscape.
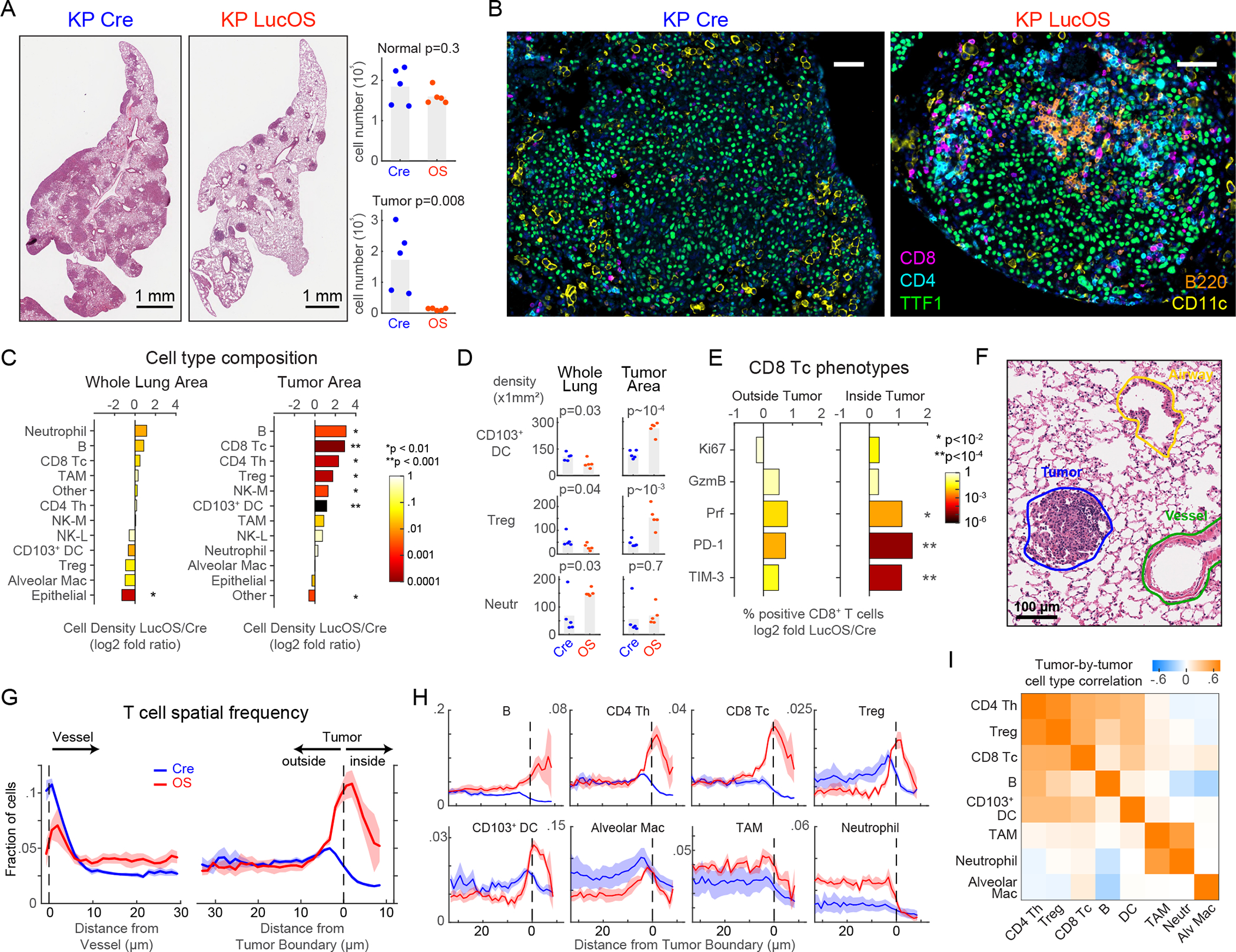
(A-B) H&E, CyCIF images (taken from whole-slide images) of KP-Cre versus KP-LucOS (antigen-expressing) tumors and quantification of normal and tumor-cell number (n=5 mice/group, bar=mean). (C) Log2 fold ratio of cell-type densities between LucOS and Cre in whole-lung and tumor areas (n=5 mice/group, color: p-value). (D) Cell-density measurements for indicated immune cell types in whole-lung and tumor areas (n=5 mice/group, bar=mean). (E) Log2 ratio between LucOS and Cre density of CD8+ T cells positive for indicated single phenotypic markers (right, inside tumor; left, outside tumor, n=5 mice/group). (F) Representative pathology annotation of H&E. (G-H) T cell spatial frequency relative to vessels and tumor boundaries (G); (H) frequency of indicated cell types from tumor boundaries (Cre and LucOS, n=5 mice/group, mean±SEM). (I) Tumor-by-tumor correlation values within LucOS-tumor nodules for indicated cell types (n=29 tumors). In all mouse experiments in this manuscript, all tumor nodules were analyzed from 2–3 lung lobes/mouse for each experiment. p-values, two-tailed t-test on mean of n=5 mice/group. See also Figure S1 and S2, and Table S2.
By contrast, when tumor areas were examined separately from non-neoplastic areas, the density of all lymphocyte subsets (Tc, Th, Treg, B cells) was significantly higher in LucOS tumors as compared to Cre tumors, increasing 3.3 to 8-fold (Figs. 2B–2D, S2A–S2C). Increased infiltration in LucOS tumors was observed even for Treg cells that were less abundant in LucOS as compared to Cre lung as a whole (>3-fold higher in LucOS versus Cre tumors) (Fig. 2D). Both NK (myeloid lineage marker-defined, see Fig. S1C) and CD103+ DCs were also significantly increased within LucOS tumors but not in whole-lung tissues (Figs. 2C–2D,S2D). Notably, the ratio of Tc cells to Treg cells was significantly increased in LucOS tumors (5.8-fold; this was also true, to a lesser extent in non-tumor tissue) (Fig. S2E); a higher ratio is a hallmark of a more immune-permissive tumor microenvironment (TME)17. Additionally, Tc cells inside tumors were enriched for expression of the cytotoxicity-associated marker Prf and the inhibitory receptors PD-1 and TIM-3, suggestive of a greater functional antitumor response moving toward T cell exhaustion (Fig. 2E). Flow cytometry analysis of T cell populations from dissociated tumor-bearing lung lobes from the same mice was consistent with the whole-lung area analysis rather than the tumor-area analysis; no significant changes in Tc, Th or Treg populations were observed, though trends toward increased Tc cells and decreased Treg cells resulted in an increased Tc/Treg ratio (Fig. S2F). Thus, enumeration of T cell populations by dissociative techniques does not fully capture the tumor-specific changes evident by high-plex tissue imaging.
To investigate the effects of LucOS-antigen expression on the spatial distribution of immune cells relative to blood vessels and the tumor margin, we combined CyCIF with anatomical annotations from H&E images (Figs. 2F, S1A). In both Cre and LucOS samples, we observed immune cell accumulation near blood vessels. LucOS mice had a greater accumulation of B cells, Tc cells and neutrophils whereas Cre tumors had more Treg cells, CD103+ DCs and alveolar macrophages (Figs. 2G, S2G). Lymphocytes in Cre animals were excluded from tumors, whereas in LucOS animals, the lymphocytes breached the tumor boundary and infiltrated into the tumor (Figs. 2B, 2G–2H). Moreover, the degree of infiltration by different types of lymphocytes (B, CD4+ Th, CD8+ Tc, Treg cells) was highly positively correlated in individual tumor nodules (Fig. 2I), suggesting coordinated infiltration into tumors. By contrast, most types of myeloid cells were evenly distributed in the normal lung tissue, without evidence of perivascular accumulation. Myeloid cells were more abundant at the tumor margin but did not infiltrate into tumors in either Cre or LucOS mice with the exception of dendritic cells, which readily infiltrated the tumor in the LucOS model with spatial patterns similar to those of lymphocytes (Figs. 2D, 2H–2I). Tumor exclusion was particularly evident in the case of neutrophils, which were substantially more abundant in LucOS than Cre lungs (Fig. 2H).
Antigen expression is associated with intratumoral localization of lymphocyte networks (‘lymphonets’)
The co-occurrence of different types of lymphocytes in LucOS tumors (Fig. 2I) prompted us to look for evidence of cell-cell interactions among lymphocytes. We applied the Visinity method recently developed by our group18 to interactively identify and quantify spatial arrangements among cells in whole-slide tissue images (see STAR Methods). This method organizes cells into a 2-dimensional embedding based on the cell types within a neighborhood of defined diameter (50μm); cells close to each other in this representation are surrounded by similar cell types (Fig. 3A). When applied to the ~2.6 million cells in the combined datasets from Cre and LucOS mouse lungs, the shared embedding space revealed a clear separation of neighborhood composition in both normal lung and tumor (Figs. S3A–S3C). The lymphoid population accumulated in two areas of the plot (clusters), at the intersection of normal and tumor neighborhoods and encompassed both B and T cells (Figs. 3B, S3A–S3C), quantitatively demonstrating the spatial coordination of lymphocytes within cellular neighborhoods.
Figure 3. Antigen expression is associated with intratumoral localization of lymphonets.
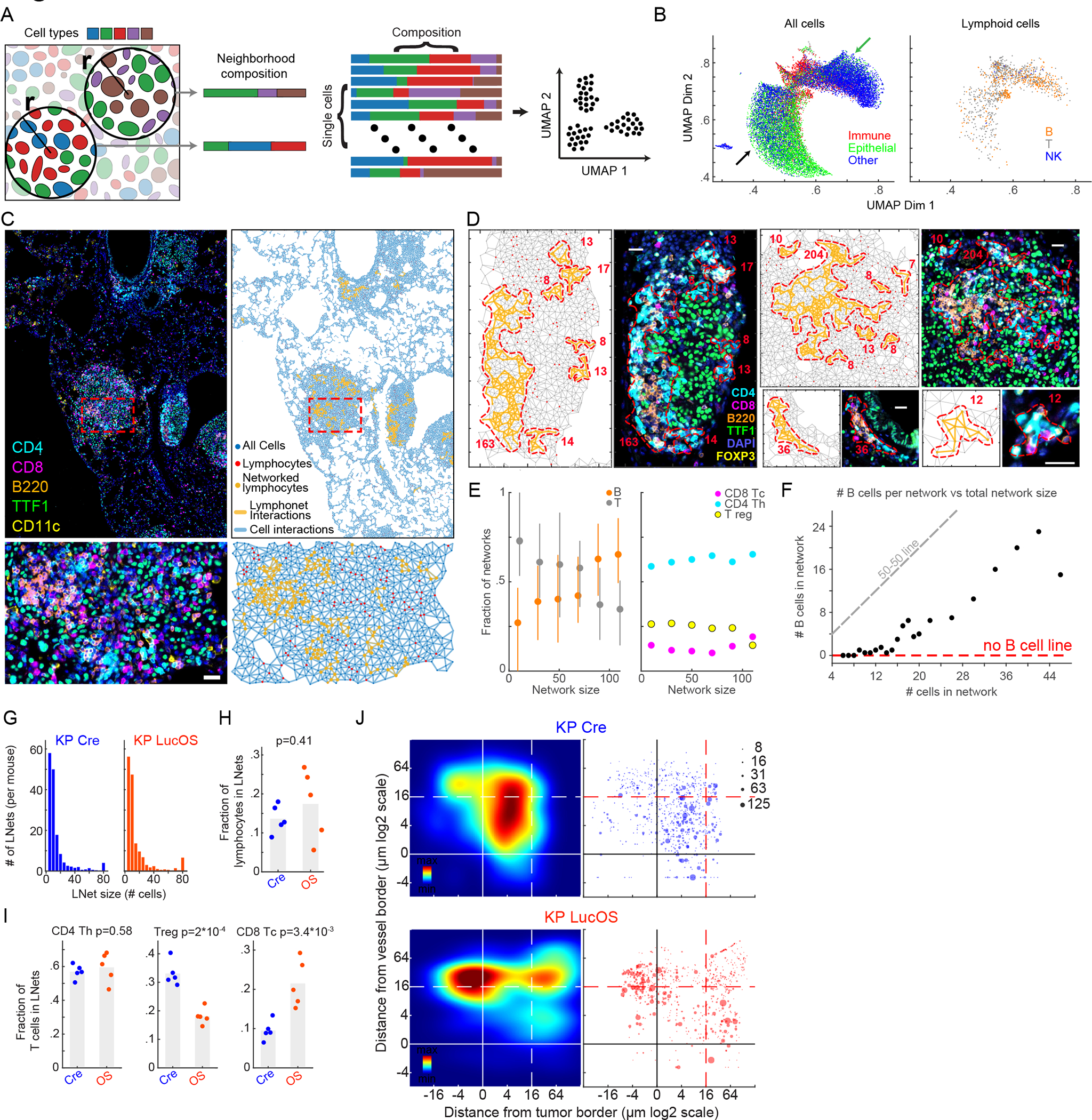
(A) Schematic of Visinity neighborhood quantification. Each cell is assigned to a unique neighborhood (all cells within a specified radius to the reference cell). Feature vectors are calculated representing weighted presence of each cell type within a neighborhood. Similar neighborhood vectors correspond to spatial patterns. (B) Visinity embedding of Cre and LucOS; arrows indicate immune neighborhoods enriched in normal (green) and tumor areas (black). (C) CyCIF images and corresponding graphic maps of interacting cell populations (Delaunay Triangulation) in LucOS. (D) Example lymphonets. (E) Lymphonet composition across network sizes. Left, B, T cells; right, T cell subtypes (mean±25th percentile). (F) Number of B cells/network versus lymphonet size (mean). (G) Number of lymphonets identified/mouse of indicated size in Cre- and LucOS-lung tissue. (H) Fraction of B and T lymphocytes and (I) T cell subsets in lymphonets in Cre versus LucOS (n=5 mice/group, bar=mean, two-tailed t-test). (J) Left, density plots of lymphonets by distance from closest blood vessel (y-axis) and tumor (x-axis) in Cre and LucOS. Dot size represents lymphonet size (n=5 mice/group). See also Figure S3 and Table S2.
To characterize these T and B cell clusters, we generated graphs of cell-cell interactions by performing Delaunay Triangulation19,20 on each specimen individually (Figs. 3C–3D); Delaunay Triangulation identifies networks of cells that directly contact each other. We identified lymphocyte cell-cell networks that ranged from small clusters of <10 lymphocytes to >100 lymphocytes that were in direct contact (Figs. 3C–3D; Fig. 3D shows examples of lymphonets ranging in size from 8 to 204 cells). Across Cre and LucOS mice, a minority of lymphocytes were organized into lymphonets using as a cutoff ≥6 lymphocytes connected by direct cell-cell contacts (mean 15.5±6.8%sd of total lymphocytes present in lymphonets, Fig. S3D). We detected an average of ~77 lymphonets per mouse lung lobe with an average of 17 cells/network. Analysis of lymphonet composition showed that Th and B cells were the most common structural elements; >50% of individual lymphonets had a majority of either Th or B cells (31 and 23%, respectively) in contrast to 2% of lymphonets comprised of a majority of Tc cells or 8% of majority Treg cells (Fig. S3E). The fraction of B and T cells was strongly correlated with lymphonet size; small lymphonets were enriched in T cells and large lymphonets in B cells (Fig. 3E). Notably, lymphonets having <16 cells were almost exclusively composed of T cells and the frequency of B cells increased linearly after this threshold (Fig. 3F). This relationship between network size and cell composition suggests that lymphonets nucleate from a core of T cells and subsequently grow by recruiting B cells.
The overall number and size of lymphonets increased in a tumor-dependent manner (P Cre/LucOS vs KP Cre), but did not change substantially with LucOS antigen expression (KP Cre vs KP LucOS) across the lung tissues (Figs. 3G–3H, S3F–S3G). Lymphonet number (but not size) was correlated with tumor burden in Cre mice, but not LucOS mice (Figs. S3H–S3I). This suggests that tumors and lymphonets develop in concert in the absence of tumor-antigen expression. The composition of lymphonets in LucOS versus Cre mice differed substantially, with LucOS lymphonets containing significantly more Tc cells and significantly fewer Tregs as compared to lymphonets in Cre lungs (Fig. 3I). Analysis of myeloid populations showed that CD103+ DCs were more proximal to lymphonets and interacted more frequently with lymphonets than other myeloid subtypes in both LucOS and Cre mice (Figs. S3J–S3K). Thus, CD103+ DCs may play a role in lymphonet formation or maintenance, likely through their function as antigen presenting cells. In addition, LucOS antigen expression dramatically relocalized lymphonets relative to histopathological features (Fig. 3J): in LucOS lungs, the majority of lymphonets were located inside tumors whereas in Cre mice most lymphonets were located outside of tumors, with a substantial fraction residing within 20 μm of a major blood vessel (Fig. 3J). These findings reveal a strong correlation between T cell antigen expression and lymphonet formation inside tumors.
To investigate temporal control over lymphonet composition, we compared lymphonets from 6-week and 9-week LucOS mice. We found that lymphonet size increased significantly over time and there was also a trend toward increased number (Fig. S3L). Interestingly, the composition of lymphonets was largely unchanged, with similar proportions of Th, Treg, and B cells at both timepoints (Figs. S3M–S3N). As observed previously (Figs. 3E–3F) small lymphonets were predominantly composed of Th cells and large lymphonets were predominantly composed of B cells (Fig. S3N). Notably, the proportion of Tc cells decreased significantly from 6 to 9 weeks (Figs. S3M–S3N), which may reflect contraction of the Tc compartment that occurs between these timepoints12,15,16. Altogether, our findings suggest lymphonet composition in LucOS mice is determined by lymphonet size rather than tumor size or tumor age.
CXCR3 ligands modulate lymphonet formation and size but not intratumoral localization
The recruitment of activated Th and Tc cells to the TME is mediated in part by binding of the CXCL9 and CXCL10 chemokines (and also CXCL11 in human) to CXCR3 receptors on T cells21,22. Given that small lymphonets predominantly contained T cells (Figs. 3E–3F), we hypothesized that CXCR3-mediated recruitment of T cells might contribute to lymphonet nucleation. Because Cxcl9 and Cxcl10 levels are tightly controlled at a transcriptional level23, and antibodies suitable for imaging these cytokines in tissue are unavailable, we measured cytokine distribution using RNA ISH (Figs. 1A–1B, 4A). In total, the levels of Cxcl9 and Cxcl10 mRNA in lung tissue were modestly increased in LucOS compared with Cre mice (changes were not statistically significant; Fig. 4B). Cxcl9 and Cxcl10 were expressed across multiple cell types in both LucOS and Cre samples and were expressed in a higher proportion of T cells, CD103+ DCs, TAMs and epithelial cells in LucOS mice (Figs. S4A–S4B). In LucOS (but not Cre) mice, Cxcl9 expression was strongly localized within tumors (Fig. S4C–S4D). Compared with Cre mice, B and T cells were localized closer to the Cxcl9 and Cxcl10-positive cells in LucOS mice (Fig. 4C) and these immune populations were predominantly present within lymphonets (Fig. 4D). Overall, the likelihood of lymphocytes belonging to lymphonets was negatively correlated with the distance to the closest Cxcl9 or Cxcl10-positive cell (i.e., lymphocytes are more likely to be networked when they are closer to Cxcl9/10-expressing cells) (Fig. 4E). Thus, lymphonets are spatially correlated with chemokine expression in LucOS mice.
Figure 4. CXCR3 ligands modulate lymphonet formation and size but not intratumoral localization.
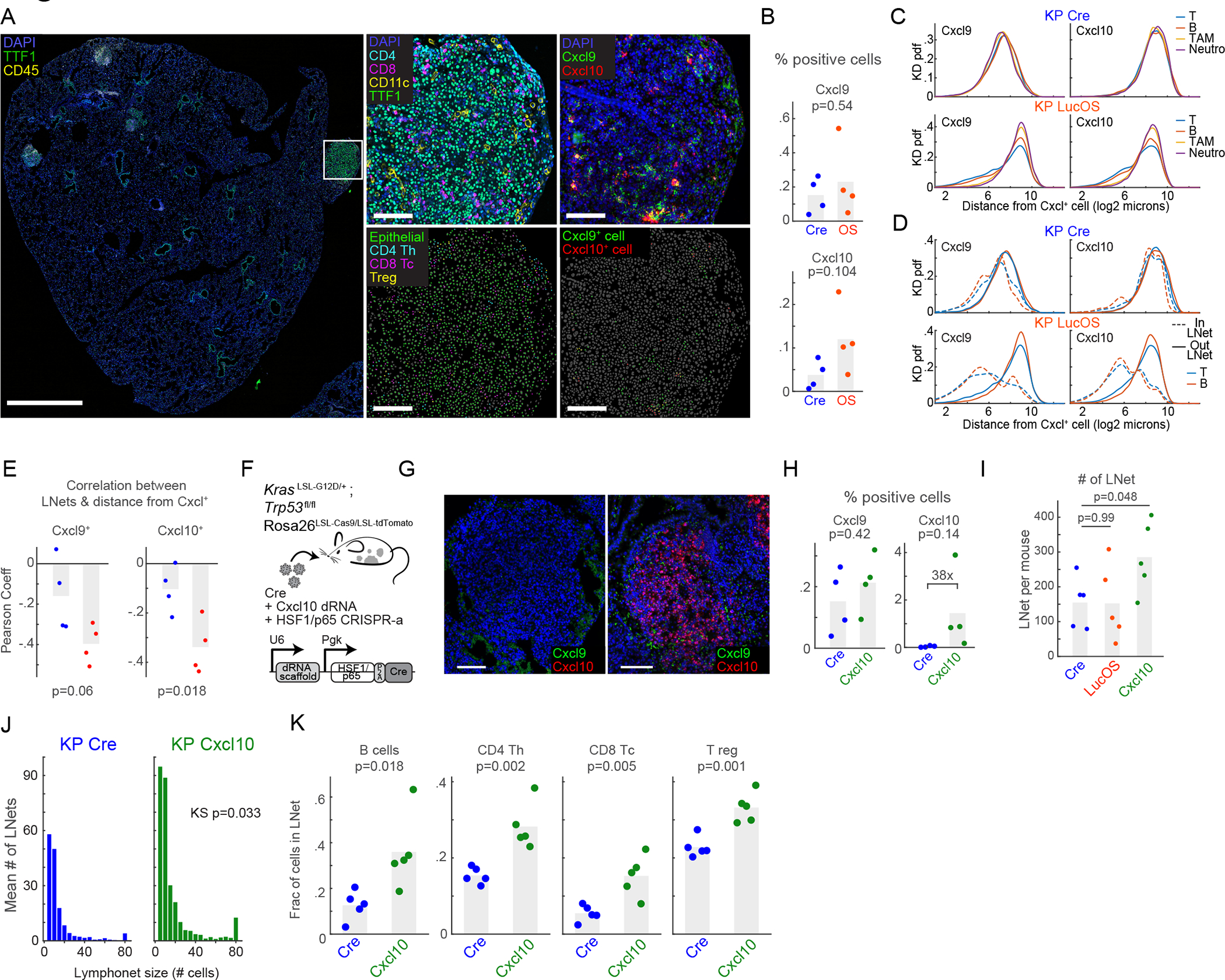
(A) CyCIF and RNAScope™ images from LucOS tumor (serial sections); cell type/state calls indicated. (B) %total cells expressing Cxcl9 and Cxcl10 mRNA in Cre- versus LucOS-lung tissue (n=4 mice/group, bar=mean). (C-D) Probability density functions of distance of (C) indicated immune-cell populations or (D) T and B cells in or out of lymphonets from Cxcl9 and Cxcl10 mRNA-expressing cells in Cre and LucOS. (E) Correlation between likelihood of lymphocytes belonging to lymphonets and their distance to the closest Cxcl9 or Cxcl10 mRNA-expressing cells in Cre (blue) and LucOS (red) (n=4 mice/group, bar=mean). (F) Schematic: lentiviral system to deliver dRNAs and HSF1/p65 activation complex for CRISPR-a Cxcl10 in KP Cas9 mice. (G) Images of Cxcl9 and Cxcl10 mRNAs using RNAscope™ in KP-Cre versus KP Cxcl10-activated tumor nodules. (H) %total cells expressing Cxcl9 and Cxcl10 mRNA in KP-Cre versus KP-Cxcl10 (n=4 mice/group, bar=mean). (I) Number of lymphonets/mouse in KP-Cre, KP-LucOS, and KP-Cxcl10 (n=5 mice/group, bar=mean). (J) Histogram of mean number of lymphonets/mouse of indicated size in KP-Cre and KP-Cxcl10 (n=5 mice/group, two-tailed KS test). (K) Plots of fraction of lymphocyte populations within lymphonets in KP-Cre and KP-Cxcl10 (n=5 mice/group, bar=mean,). All p-value are from two-tailed t-test unless specified. See also Figure S4 and Table S2.
To test whether CXCR3 ligands promote lymphonet formation, we used CRISPR-activation to ectopically express Cxcl10 in KP-Cre tumors (Fig. 4F), resulting in a 38-fold induction of Cxcl10 mRNA levels (Figs. 4G–4H). Concomitantly lymphonet number and size increased significantly (Figs. 4I–4J) and involved recruitment of B cells and all T cell subsets (Fig. 4K). Lymphonets were more proximal to blood vessels in mice over-expressing Cxcl10 compared to control mice but remained excluded from the inside of tumors (Fig. S4E). These data show that expression of Cxcl10 in the TME can promote formation and growth of lymphonets but that additional antigen-dependent mechanisms are required for lymphonet localization to tumors.
Spatial analysis reveals dynamic shifts in Tc cell states with immunotherapy treatments
To investigate the role of lymphonets in anti-tumor Tc responses, we first assayed Tc differentiation states and functional potential using markers associated with cytotoxicity (GZMB, PRF) and proliferation (Ki67), inhibitory receptors (PD-1, TIM-3), and the transcription factor TCF1. LucOS mice were exposed to one of two immunotherapy regimens previously shown to improve the anti-tumor functionality of the Tc response15: (i) therapeutic vaccination (Vax) against SIIN and SIY antigens, and (ii) antibody-mediated PD-1/CTLA-4 immune checkpoint blockade (ICB) (Fig. S5A). For vaccination, LucOS mice were injected subcutaneously with SIIN and SIY 30-mer peptides and cyclic-di-GMP as an adjuvant 6 weeks post-tumor initiation followed by a booster at 8 weeks; mice were sacrificed at 9 weeks for analysis. Vax treatment resulted in a significant reduction of tumor burden (Fig. S5B;15). For ICB therapy, a mixture of anti-PD-1 and anti-CTLA-4 antibodies or isotype controls were administered by intraperitoneal injection starting 8-weeks post-tumor initiation (three doses spaced 3-days apart: day 0, 3, 6) and mice were then sacrificed, also 9 weeks after tumor initiation. Anti-PD-1 and anti-CTLA-4 ICB treatment is not known to result in a significant reduction in tumor burden in this model (Fig. S5C), but has been shown to increase tumor-specific effector Tc activity and synergize with chemotherapy15,24.
The resulting data were analyzed using Palantir, an algorithm that uses multidimensional expression data to align single cells along differentiation trajectories, thereby capturing continuity in cell states and stochasticity in cell-fate determination25. Three predominant CD8 T cell states (S1 to S3, Figs. 5A, S5D) were identified in both Vax and ICB mice and gated using a supervised approach typical of FACS data analysis (see STAR Methods). Phenotypic markers used here do not empirically demonstrate cell functionality but are suggestive of differentiation state and potential activity of Tc cell subsets. State S1 had high levels of TCF1 expression and no expression of markers of activation/exhaustion (PD-1, TIM-3) or cytotoxicity (GZMB, PRF) and therefore corresponded to a naïve T cell state (Figs. 5B, S5E). S2 had high expression of GZMB and/or PRF and the proliferation marker Ki67, indicative of a proliferative, cytotoxic T cell state. S3 had low expression of GZMB, PRF, and Ki67 and high expression of inhibitory receptors PD-1 and TIM-3, denoting an exhausted T cell state. The three discrete states we identified were interconnected by cells – about one-third of the total – having transitional phenotypes (T1, T2, T3) in which the expression of multiple markers was graded and mixed (Figs. 5A–5B, S5D–S5E).
Figure 5. Spatial analysis reveals dynamic shifts in Tc cell states and localization with immunotherapy.
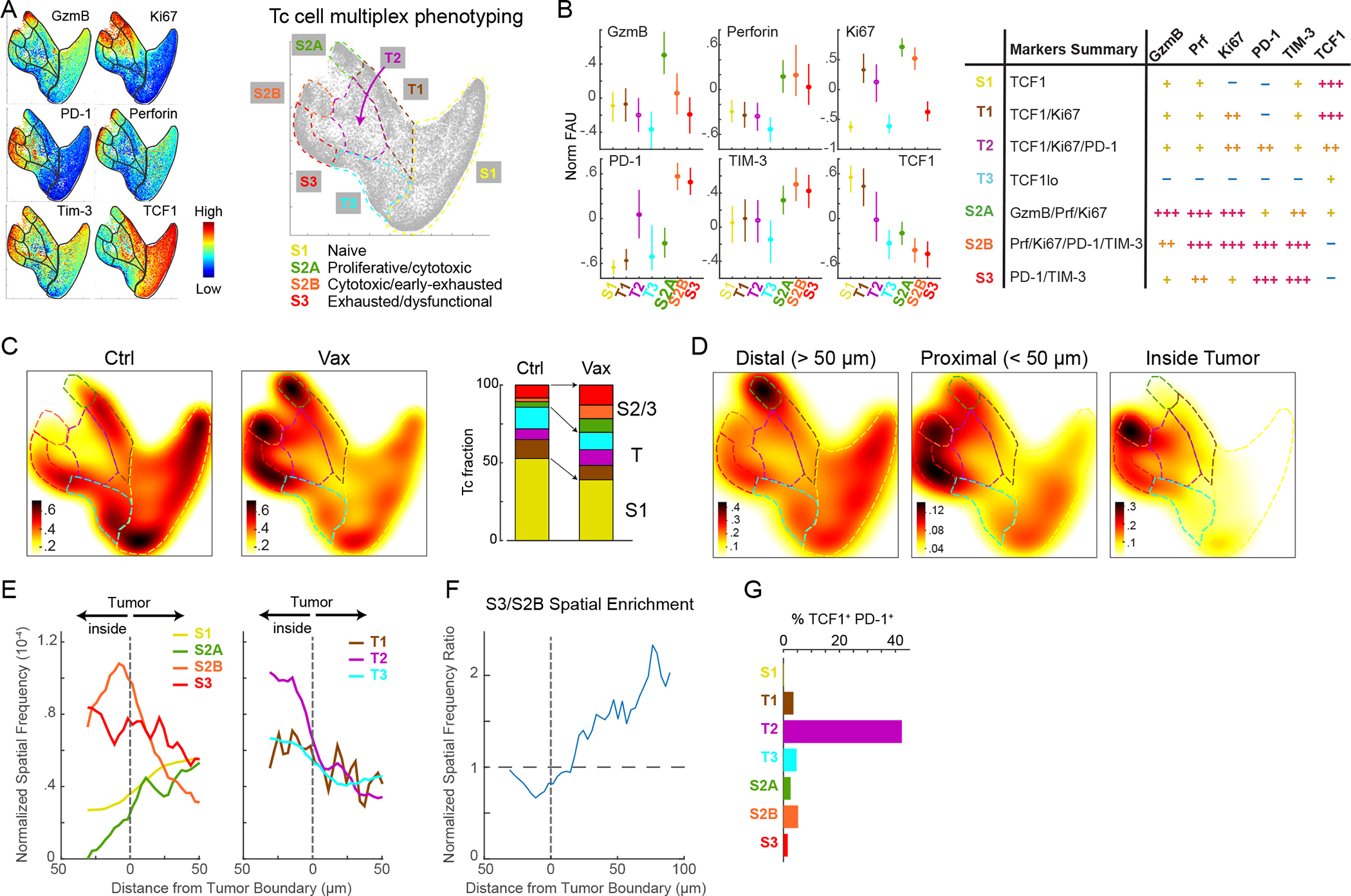
(A) Palantir projection of CD8+ Tc populations in KP-LucOS mice treated with SIINFEKL (SIIN) and SIYRYYGL (SIY) long-peptide vaccine (Vax) or PBS/Ctrl (n=104 cells sampled from n=8 and 7 mice/treatment). Expression levels of indicated markers are color mapped (normalized between 0.1 and 99thpercentile). Tc states (S1, S2A, S2B, S3) defined by multiparameter measurements indicated at extremes of representation, connected by transitional phenotypes (T1-T3); schematic, right. (B) Normalized fluorescence units for markers in indicated Tc cell states and transitions (mean±25th percentile); summary of Tc states and transitions; table, right. (C) Heat map of Tc cell densities in Palantir projections for Ctrl and Vax groups (n=104 cells/treatment). Right, stacked-bar graph of Tc cell fractions in each state and transition. (D) Heat map of Tc densities in Palantir projections for LucOS following Vax by indicated distance from tumor boundary and (E) their spatial frequency from tumor boundary (Vax). (F) Enrichment of S3 versus S2B relative to boundary. (G) %Tc cells that are TCF1+ PD-1+ in Tc cell states/transitions. See also Figure S5 and Table S2.
Using this division of cell types and states, we examined shifts in Tc phenotype induced by the two immunotherapy regimens. In the untreated LucOS cohorts, the majority of Tc cells were naïve (S1), but Vax and ICB protocols shifted cells into cytotoxic (S2) and exhausted (S3) states (Figs. 5C, S5F). In the Vax cohort, the cytotoxic (S2) population split into two groups distinguished by levels of PD-1 and TIM-3 expression (S2A and S2B in Figs. 5A–5C): the S2A state had low PD-1/TIM-3 expression and appeared to have greater cytotoxic potential, expressing high levels of both GZMB and PRF whereas cells in the S2B state expressed high levels of PD-1/TIM-3 cells and lower levels of GZMB. In the phenotypic landscape, cells in the S2B state were adjacent to the exhausted (S3) population, suggesting S2B may represent a cell state directly preceding exhaustion/dysfunction. In the ICB cohort, the S2 state did not split and resembled the PD-1/TIM-3high GZMBlow state of S2B Vax cells (Figs. S5D–S5F). These data suggest that Vax is substantially more effective than ICB in generating cytotoxic and proliferative effector T cell states.
Functionally distinct Tc cell states are spatially segregated in the tumor microenvironment
To characterize the spatial distribution of Tc states relative to tumor cells, we split the Palantir phenotypic landscape depending on whether immune cells (i) resided inside tumors, (ii) were proximal to the edge of tumors (<50μm of an edge), or (iii) were distal to tumors (>50μm away from edges) (Figs. 5D–5E, S5G–S5I). Strikingly, we found that the proliferative/cytotoxic S2A state, which was unique to Vax mice, was found distal to tumors (Figs. 5D–5E), whereas the cytotoxic/early-exhausted S2B (Vax) and S2 (ICB) states were enriched inside tumors (Figs. 5D–5E, S5G–S5H). We therefore posit that cells in the distal S2A state are poised to enter tumors at which point they differentiate to an S2B state. The exhausted S3 population in both Vax and ICB mice was found proximal to tumor edges and more frequently outside of tumors compared to S2B suggesting the S3 cell state is associated with progressive exclusion from tumors (Figs. 5D–5F, S5H–S5I). This finding suggests that Tc cells exit tumors upon upregulating suppressive inhibitory receptors and downregulating cytotoxic activity.
Neither the Vax nor ICB protocols significantly changed the fraction (~30%) of CD8+ T cells that displayed transitional phenotypes (T1-T3; Figs. 5C, S5F). This may be due to flux through transitional states as Tc cells differentiate from naïve S1 to effector S2 and exhausted S3 states. In Vax, T2 cells were spatially enriched inside tumors (Fig. 5E) and were substantially enriched for cells co-expressing TCF1 and PD-1 (an 8- and 200- fold increase was observed relative to other T cell states, Fig. 5G); this enrichment was also observed in T1 and T2 states in ICB (Fig. S5J). TCF1+ PD-1+ CD8+ T cells have recently been shown to play a critical role in driving therapeutic responses to ICB in both mice and humans26–29. Such cells are thought to be in a progenitor-like state and induced to differentiate into cells with cytotoxic function in response to treatment28,29. Our data suggest that TCF1+ PD-1+ progenitor CD8+ T cells are enriched in specific transitional states that efficiently traffic into tumors and can establish residence within the tumor bed.
One limitation of multiplexed imaging methods in characterizing T cell phenotypes is the inability to detect tumor antigen-specific T cell populations in fixed tissue. Thus, to determine whether the Tc phenotypes we observed in tumors were tumor-antigen specific, we performed a flow cytometric analysis of SIIN and SIY-specific Tc cells in dissociated lung samples from the Vax- and ICB-treated mice using peptide-MHC tetramers. We found that the majority of TIM-3+ and PD-1+ TIM-3+ cells, which resembled the exhausted S3 state, were specific to LucOS antigens in control mice and this association increased with ICB and Vax to >70% of this population (Figs. S5K–S5L). In contrast, the majority of TCF1+ cells resembling the naïve S1 state were not tumor-antigen specific; however, TCF1+ PD-1+ (T2) cells were more enriched for Tetramer+ cells. Most Tc cells with a proliferative (Ki67+) and/or cytotoxic (GZMB+) phenotype resembling the S2 state were also not tumor antigen-specific in control mice but both treatments significantly expanded the Tetramer+ cell proportions, with Vax increasing the proportion of Tetramer+ cells to >50% (Figs. S5K–S5L). Interestingly, increases in T cell populations induced by Vax were restricted to the Tetramer+ cell fraction whereas ICB increased both Tetramer+ and Tetramer− cells. These findings indicate that vaccination against SIIN and SIY specifically targets SIIN- and SIY-specific T cells whereas ICB additionally acts on other Tc populations. These additional Tc populations may be responding to tumor-associated antigens in the model30 or may be “bystander” T cells specific to non-tumor antigens31.
TCF1+ PD-1+ progenitor CD8+ T cells reside within intratumoral lymphonets
We next used data from Vax-treated LucOS mice to investigate how changes in lymphonets are related with changes in Tc cell phenotypes. While Vax did not substantially change the overall size, number, or localization of lymphonets (Figs. S6A–S6B), it did increase association of Tc cells but not other T cell subsets across most lymphonets (Figs. 6A–6B, S6C–S6E). Remarkably, the TCF1+ PD-1+ progenitor Tc state was the most highly and significantly enriched state in lymphonets (Fig. 6C, KS p-value=10−3). Moreover, across the Vax cohort the total number of Tc cells in lymphonets was linearly correlated with the number of TCF1+ PD-1+ cells (Fig. 6D). The Tc compartment of lymphonets was also predominantly comprised of the transitional T2 state containing TCF1+ PD-1+ progenitor cells (Figs. 6E–6F, S6F); this was true of lymphonets both inside and outside of tumors, however, cells in the T2 state were mostly found within tumors (Fig. 5E). Lymphonets were similarly enriched for transitional phenotypes containing TCF1+ PD-1+ cells in the ICB cohort (i.e., T1 and T2, Figs. 6G–6H, S6G). Notably, the only Tc state that increased in lymphonets following Vax or ICB treatment was the cytotoxic S2 state (S2B for Vax and S2 for ICB, Figs. 6E–6H). Thus, after either Vax or ICB treatment, cells with cytotoxic potential colocalized with TCF1+ PD-1+ progenitor cells in lymphonets. Given that TCF1+ PD-1+ progenitor cells give rise to cytotoxic Tc cells in tumors29, these data suggest that lymphonets are the site of differentiation of progenitor cells into cytotoxic cells in response to immunotherapy.
Figure 6. TCF1+ PD-1+ progenitor CD8+ T cells reside within intratumoral lymphonets.
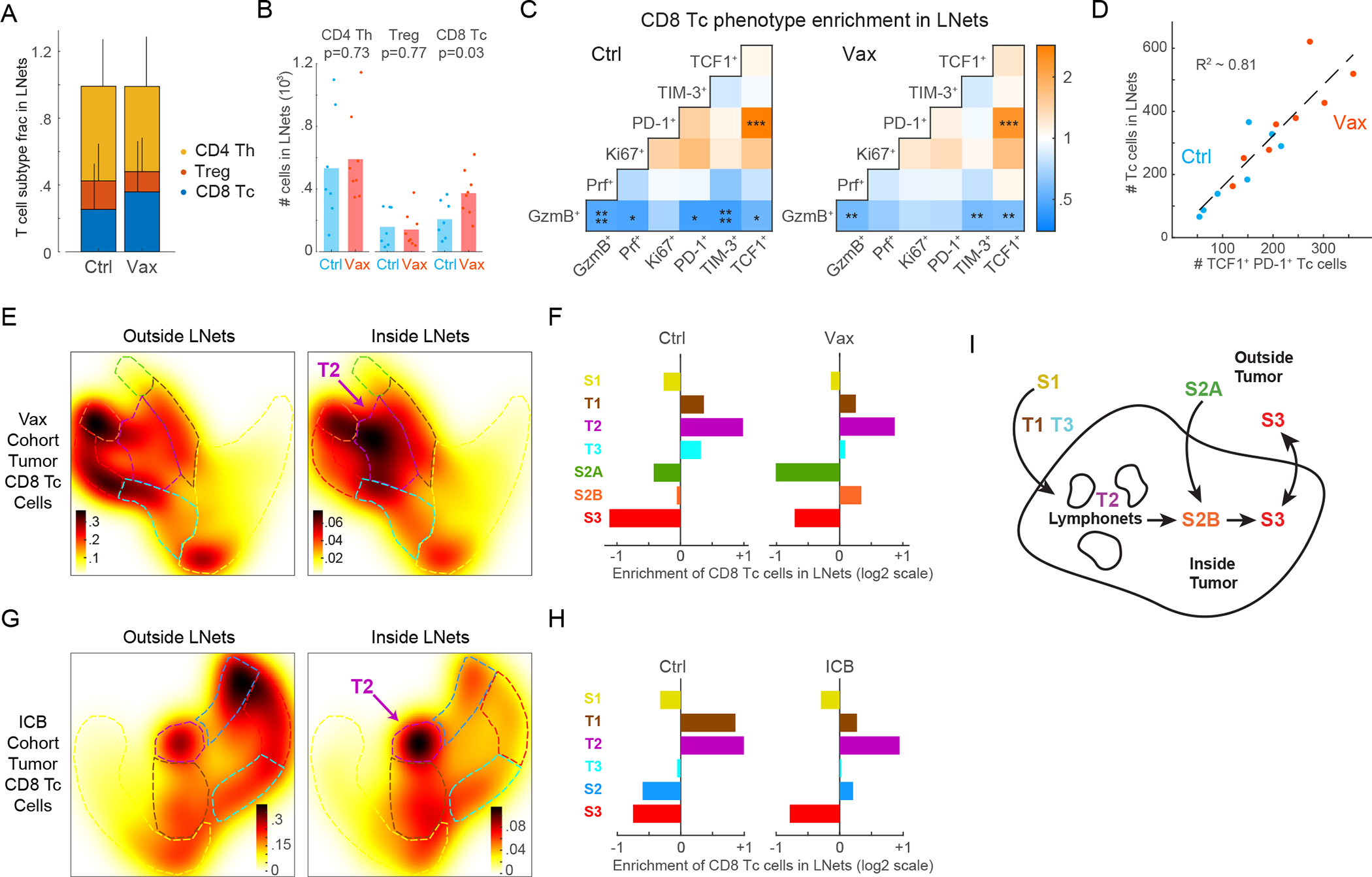
(A) Proportion of T cell subtypes in lymphonets (Ctrl n=7, Vax n=8 mice, mean+SD, same LucOS cohort in Figure 5). (B) Number of T cell subtypes present in lymphonets (bar=mean, two-tailed t-test). (C) Pairwise enrichment analysis of marker co-expression in Tc cells in Ctrl and Vax groups (KS p-value *p<0.05,**p<0.01,***p<10−3,****p<10−4). (D) Plot of Tc cells present in lymphonets versus TCF1+ PD-1+ cells in Ctrl and Vax per mouse (dotted line, linear regression, R2=0.81). (E) Heat map of cell densities of tumor-localized Tc cells present outside and inside lymphonets in Palantir projections for Vax-treated cohort (n=3,736 and 806 cells, respectively). (F) Enrichment of tumor-localized Tc cells in lymphonets for Ctrl and Vax mice. (G) Heat map of cell densities of tumor-localized Tc cells present outside and inside lymphonets in Palantir projections for anti-PD-1 and anti-CTLA-4 treated (ICB) cohort (n=6 mice/group, n=4,276 and 1,041 cells, respectively). (H) Enrichment of tumor-localized Tc cells in lymphonets for Ctrl and ICB mice (n=6 mice/group). (I) Schematic of data interpretation from Figures 5 and 6. See also Figure S6 and Table S2.
Taken together, our data support a model wherein Tc cells migrate into intratumoral lymphonets upon differentiation from an TCF1+ S1 state into a T2 TCF1+ PD-1+ state (Fig. 6I). ICB and Vax immunotherapies promote differentiation of TCF1+ PD-1+ cells to a cytotoxic S2B state within tumors, and these cells then progress to a tumor-excluded exhausted S3 state upon upregulation of inhibitory receptors and downregulation of cytotoxic activity.
Lymphonets enriched for TCF1+ PD-1+ progenitor CD8+ T cells are abundant in early-stage human lung adenocarcinoma
To begin to investigate the relevance of these findings to human disease we used a panel of CyCIF-qualified antibodies to characterize the features of lymphonets in whole slide sections of early-stage human lung adenocarcinoma from 14 patients (Table S3); these early-stage human tumors are likely analogous to the early-stage tumors we studied in the KP-LucOS GEMM. We performed sequential clustering of ~7.8 million cells from these images and identified tumor and stromal cells (Fig. 7A, Lv1) and immune cells (~3.4 million cells) for further cell-type calling (Fig. 7A, Lv2-Lv4). Human specimens had highly variable fractions of tumor, stromal, and lymphocyte subtypes (Fig. 7B). In histopathologically-annotated tumor areas, we identified many lymphonets per sample, and they varied substantially in size. Similar to lymphonets in mice, the vast majority of these networks in human tumors were small (Figs. 7C–7D, S7A–S7C), and the fraction of B cells was positively correlated to lymphonet size (Fig. 7E). We found that the number of lymphonets with >500 cells matched the number of TLS as scored by pathology review (linear regression coefficient=0.99, R2=0.74, Figs. S7A–S7B, Table S4). These findings suggest that anti-cancer immune responses in both early-stage human and mouse lung cancer is characterized by a preponderance of small lymphocyte networks.
Figure 7. Lymphonets enriched for TCF1+ PD-1+ progenitor CD8 T cells are abundant in early-stage human lung adenocarcinoma.
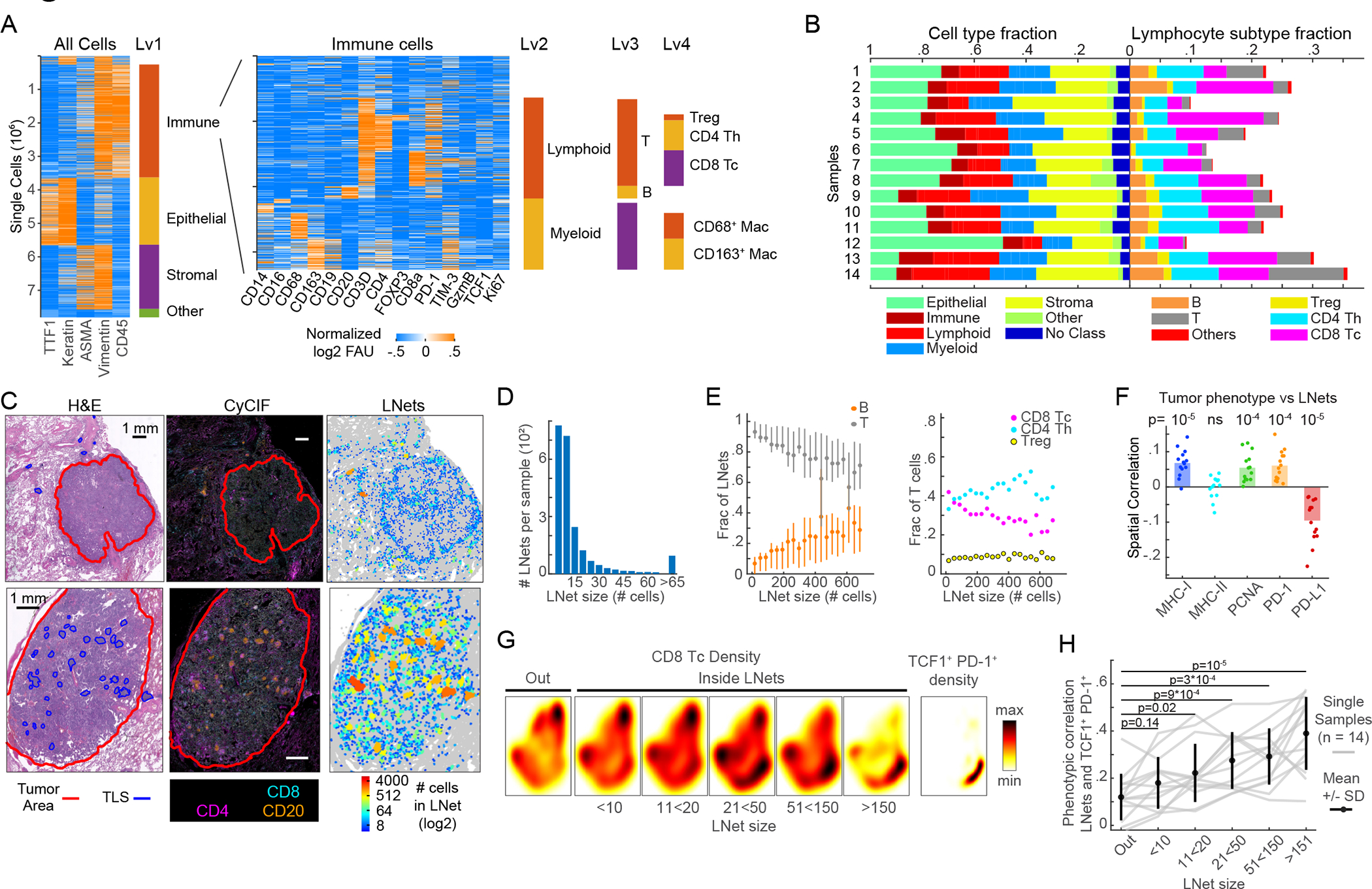
(A) Sequential clustering of immune, epithelial/tumor, stromal and ‘other’ cell populations (Lv1); immune cells were further clustered into lymphoid and myeloid (Lv2) and immune subsets (Lv3, Lv4). Rows=individual cells. 7.8 × 106 cells plotted from n=14 human lung adenocarcinomas. Immune clusters shown in heat map (right). (B) Horizontal-stacked bar graphs of cell-type fractions (Lv1–2) and lymphocyte-subtype fractions (Lv3-Lv4). (C) H&E, CyCIF representative images; map indicates lymphonet size. Top: tumor with small lymphonets (n<64 cells). Bottom: tumor with large lymphonets (n>64 cells). Scalebar: 1mm. (D) Histogram: average number of lymphonets/sample (n=14) by lymphonet size. (E) Composition of lymphonets by lymphocyte type across different network sizes (mean±25th percentile). (F) Spatial correlation of lymphocytes’ likelihood of belonging to a lymphonet and the likelihood of non-lymphoid cells expressing the indicated markers (n=14 samples, bar=mean, Pearson correlation and p-values). (G) Heat map of density of total Tc in and out of lymphonets of different sizes; density of TCF1+ PD-1+ CD8+ T cells in Palantir projection from 14 human lung adenocarcinomas (n=21*103 cells sampled from n=14 samples). (H) Phenotypic correlation of Palantir distributions of TCF1+ PD-1+ CD8+ Tc cells and lymphonets binned by lymphonet size (correlation of likelihood of CD8+ Tc belonging to a lymphonet (binned by size) and the likelihood of CD8+ Tc being TCF1+ PD-1+); gray lines represent data from individual tumors (n=14, n=3000 cells/sample); black line=mean±SD; Pearson correlation and two-tailed t-test. See also Figure S7, Tables S3 and S4.
As in KP-mouse tumors, smaller lymphonets in human tumors were composed of T cells, with the B cell fraction increasing with lymphonet size (Fig. 7E). Uniquely to human samples, the CD8 T cell fraction decreased as lymphonets increased in size, being replaced by CD4 Th cells (Fig. 7E). A positive spatial correlation (i.e., increased probability of spatial proximity) between major histocompatibility class I (MHC I) expression on non-lymphoid cells in tumors and lymphonets was observed (Fig. 7F), suggesting lymphonet organization in early-stage human lung cancer may be regulated by CD8+ T cell antigen presentation. A negative spatial correlation (i.e., increased probability of being spatially distant) was observed between non-lymphoid cells in tumors expressing PD-L1 and lymphonets (Fig. 7F), which implies that PD-L1 may promote their distancing from lymphonets. Subsets of myeloid cells exhibited similar negative spatial correlation (Fig. S7D) perhaps due to high expression of PD-L1 on myeloid cells such as TAMs (Fig. S7E). MHC II was expressed in many cell types, including B cells, TAMs, and epithelial cells (Fig. S7E), as previously reported for lung tissue32; however, no correlation was observed between lymphonet formation and MHC II expression on non-lymphoid cells in tumors or myeloid cells (Figs. 7F, S7D). When we profiled Tc cells with markers of functional potential and used Palantir to identify the TCF1 and PD-1 co-expressing population of progenitor CD8+ T cells we found that Tc cells were present both outside and inside of lymphonets, but TCF1+ PD-1+ progenitor cells were largely restricted to lymphonets (Fig. 7G) and became increasingly enriched as lymphonet size increased (Fig. 7G–H). Altogether, these findings reveal that lymphonets as identified in the KP-GEMM model are found in abundance in human lung adenocarcinomas where they may have a similar function in supporting progenitor CD8+ T cell maturation.
DISCUSSION
Multiplexed imaging of the KP GEMM of lung cancer revealed striking changes in the spatial arrangements of lymphocytes and dendritic cells following expression of tumor antigens (in the KP-LucOS model) and consequent induction of T cell mediated anti-tumor immunity. Both T and B cells were recruited to tumors when tumor antigens were expressed with lymphocytes forming networks of cells that directly contacted each other. We termed these networks of 6 to several hundred interacting cells ‘lymphonets.’ The smallest primarily contained T cells, but the proportion of B cells increased as networks enlarged. A key feature of lymphonets is that they contain TCF1+ PD-1+ CD8+ T cell progenitors and gain cytotoxic CD8+ T cells following treatment with ICB or antigen-targeted vaccines, most likely due to differentiation and activation of the progenitor cells. We speculate that paracrine and juxtacrine signaling among cells in lymphonets promotes or coordinates this critical aspect of induced anti-tumor immunity.
Compartmentalized and structured rather than mixed organization of lymphocytes with respect to tumors has previously been correlated with tumor control33, particularly with respect to TLS formation across multiple cancer types34,35. TLS are aggregates of immune cells with cellular composition and organization resembling secondary lymphoid organs. Fully mature TLS generally contain B and T cell zones and germinal centers, containing follicular dendritic cells. The presence of TLS is predictive of better patient survival and response to ICB and vaccine immunotherapies across multiple cancer types36,37. However, it remains unclear whether TLS directly facilitate anti-tumor immune responses or are merely evidence of a prior immune response with potential for reinvigoration by immunotherapy. Characterization of dynamic changes within TLS over time or with therapy is difficult to investigate in humans and studies in mice have been limited due to the absence of TLS formation in most transplantable tumor models38.
In the KP-LucOS model, we previously described the formation of mature TLS peritumorally around 20 weeks post-tumor initiation39, a time-point correlated with loss of functional anti-tumor CD8+ T cell immunity and lack of response to anti-PD-1/anti-CTLA-4 ICB therapy12,15,16. In comparison to TLS, the lymphonets we describe here (at 9-weeks post-tumor initiation) are coincident across conditions with functional Tc responses in tumors and are less structured, lacking distinct T and B cell zones; however, we did find a significant association between lymphonets and cross-presenting CD103+ dendritic cells. It is possible that some lymphonets represent precursors to the TLS observed later during tumor progression. Additional spatial profiling of the TME longitudinally between 9- and 20-weeks post-tumor initiation is needed to investigate the connection between and TLS and lymphonets, to identify factors that support anti-tumor Tc immunity in lymphonets, and to distinguish bystander and immunosuppressive functions.
Multiparametric analysis of key functional Tc cell markers in LucOS tumors defined three major Tc cell states, naïve (S1), cytotoxic (S2), and dysfunctional/exhausted (S3), and characterized the flux through these states and connecting transitional phenotypes (T1-T3) in response to immunotherapies. Tumor antigen-targeted vaccination (Vax) and anti-PD-1/anti-CTLA-4 ICB shifted Tc cells from the naïve S1 state to the S2 and S3 states. Parallel flow cytometry analysis of SIIN and SIY antigen-specific Tc cells in dissociated lung tissue showed that the majority of S3 cells were tumor-specific as were many S2 cells, especially post-treatment. These differentiated functional states were phenotypically related to cells exhibiting intermediate transitional phenotypes (T1-T3). TCF1+ PD-1+ cells that have been described as giving rise to cytotoxic and exhausted CD8+ T cell populations in response to ICB therapy40 occupied intratumoral transition states and were tightly associated with lymphonets both before and after immunotherapy treatment. After therapy, S2 cells colocalized with TCF1+ PD-1+ cells in lymphonets, consistent with progenitor cells seeding the S2 population. Notably, vaccination resulted in two S2 populations (cytotoxic S2A T cells marked by Ki67 and high expression of GZMB, and cytotoxic/early-exhausted S2B T cells marked by low expression of inhibitory receptors) that were spatially segregated; only the S2B population localized to tumors and lymphonets while the S2A population was present outside of tumors. The exhausted/dysfunctional T cells (S3) were largely excluded to just outside of the tumor margin. We hypothesize that in contrast to the S2B (and ICB S2 populations), S2A cells are not derived from intratumoral TCF1+ PD-1+ cells and instead seed directly from the periphery. Upon entering tumors, S2A cells may pass through the S2B state before they become terminally exhausted (S3). Consistent with this, we previously reported that vaccination acutely promotes substantial peripheral Tc expansion rather than expanding the existing Tc populations in the lung by flow cytometric analysis15. In contrast to Vax, ICB induced only the intratumoral S2B-like S2 state associated with TCF1+ PD-1+ progenitor cells, and this may help to explain the central role of progenitor cells in driving ICB response in mice and humans.
Consistent with our observation in mice that intratumoral lymphonets harbor TCF1+ PD-1+ progenitor CD8+ T cells, we found that TCF1+ PD-1+ cells were also localized to lymphonets in human lung cancer resections. Localization of stem-like cells (defined as CXCR5+ TCF1+) to intratumoral lymphocyte ‘niches’ has been previously reported in human renal cell carcinoma, where the ‘niches’ were proposed to support generation of cytotoxic T cells41. These niches were not mature TLS and instead were defined by lymphocyte aggregation around MHC II-expressing cells, presumably marking regions rich in antigen presenting cells. Interestingly, we did not find a correlation between MHC II expression and lymphonets of any size in human lung cancer, but we did observe a significant association between CD103+ DCs (also expressing MHC II) and lymphonets in mice. The expression of MHC II on multiple cell populations and the lack of DC-specific markers in the human antibody panel prevented us from validating this CD103+ DC phenotype in human lung tumors. However, MHC I expression level was correlated with lymphonets in human tumors, and this may suggest that antigen presentation to CD8+ T cells is necessary for lymphonet formation and/or that lymphonets promote MHC I upregulation (perhaps through T cell secretion of IFNγ). Consistent with this observation, lymphonets in the mouse were found intratumorally only following expression of LucOS T cell antigens and these lymphonets were significantly associated with cells expressing IFNγv-induced chemokines (Cxcl9, Cxcl10). We also observed that ectopic expression of Cxcl10 was able to increase the size and number of lymphonets in Cre mice lacking LucOS antigen expression. Pelka et al.4 recently reported a significant association between formation of “immune hubs” enriched in T lymphocytes (similar to the lymphonets reported here) and expression of CXCR3 ligands during a productive anti-tumor immune response to mismatch repair deficient (MMRd) human colorectal cancer. Our findings provide mechanistic evidence that CXCR3 ligands such as CXCL9 and CXCL10 actively promote the formation of lymphocyte niches correlated with productive anti-tumor immunity; however, localization of these cell networks inside tumors depends on antigen expression or associated factors.
Lymphonets in KP-LucOS mice were predominantly composed of Th and B cells, with the B cell fraction increasing with lymphonet size in both mouse and human. An association of B cell gene signatures with better patient survival and response to ICB therapy has been found across many cancer types38. Interestingly, however, B cells in cancer have been demonstrated to have both pro- and anti-tumorigenic functions. For example, B regulatory cells contribute to tumor-promoting inflammation and suppression of anti-tumor T cell responses, while antibody-producing plasma cells (frequently associated with TLS) are more commonly associated with tumor control38. Future imaging studies with additional markers of B cell states paired with spatial transcriptomics in KP GEMM could clarify the function of B cells and Th cells in lymphonets and how they might support TCF1+ PD-1+ progenitor CD8+ T cell function. Given that antigen is necessary for nucleation of lymphonets inside KP-lung tumors and MHC I expression is associated with lymphonets in human lung cancer, one hypothesis is that B cells regulate CD8+ T cells and support Th cell function through their role as antigen presenting cells42.
Limitations of the Study
Antibody panels focused on effector T cell states; additional antibodies are required to fully characterize other T cell populations and myeloid cells43. Analysis of dendritic cells (DCs) was also limited to cross-presenting CD103+ DCs analyzed in mouse studies and MHC II-expressing cells in human. Immunogenic model antigens were expressed throughout tumors, which may not represent situations of limited antigen availability. Multiparametric measurements used here permit inference of dynamic properties, but not direct visualization of transitions over time.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sandro Santagata (ssantagata@bics.bwh.harvard.edu).
Materials Availability
As described in the Key Resources Table, cell lines are available upon request, mouse models are available from Jackson Laboratories or upon request, and plasmids are available from Addgene.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb PD1 | BioXCell | Clone 29F.1A12; Cat# BE0273; RRID: AB_2687796 |
| InVivoMAb CTLA4 | BioXCell | Clone 9H10; Cat# BE0131; RRID: AB_10950184 |
| InVivoMAb rat IgG2a | BioXCell | Clone 2A3; Cat# BE0089; RRID: AB1107769 |
| InVivoMAb polyclonal Syrian Hamster IgG | BioXCell | Cat# BE0087; RRID: AB_1107782 |
| t-CyCIF: anti-human and mouse TTF1 | Abcam | Clone EPR5955(2); Cat# ab206726; RRID: AB_2857980 |
| t-CyCIF: anti-mouse B220 (CD45R) | ThermoFisher Scientific | Clone RA3–6B2; Cat# 41–0452-80; RRID: AB_2573598 |
| t-CyCIF: anti-mouse CD45 | BioLegend | Clone 30-F11; Cat# 103123; RRID: AB_493534 |
| t-CyCIF: anti-mouse FOXP3 | ThermoFisher Scientific | Clone FJK-16s; Cat# 11–5773-82; RRID: AB_465243 |
| t-CyCIF: anti-mouse CD4 | ThermoFisher Scientific | Clone 4SM95; Cat# 41-9766–82; RRID: AB_2573637 |
| t-CyCIF: anti-mouse CD8α | Cell Signaling Technology | Clone D4W2Z; Cat# 98941; RRID: AB_2756376 |
| t-CyCIF: anti-mouse CD103 | R&D Systems | Clone Polyclonal; Cat# AF1990; RRID: AB_2128618 |
| t-CyCIF: anti-mouse CD11c | Cell Signaling Technology | Clone D1V9Y; Cat# 97585; RRID: AB_2800282 |
| t-CyCIF: anti-human and mouse CD11b | Abcam | Clone EPR1344; Cat# ab204471; RRID: AB_2650514 |
| t-CyCIF: anti-mouse Nkp46 | R&D Systems | Clone Polyclonal; Cat# FAB2225F-025; RRID: AB_2149149 |
| t-CyCIF: anti-mouse CD3e | Cell Signaling Technology | Clone D4V8L; Cat# 99940; RRID: AB_2755035 |
| t-CyCIF: anti-human and mouse Ki-67 | Cell Signaling Technology | Clone D3B5; Cat# 12075; RRID: AB_2728830 |
| t-CyCIF: anti-mouse PD-L1 | Cell Signaling Technology | Clone D5V3B; Cat# 64988s; RRID: AB_2799672 |
| t-CyCIF: anti-mouse PD-1 | Cell Signaling Technology | Clone D7D5W; Cat# 61237; RRID: AB_2799604 |
| t-CyCIF: anti-mouse Granzyme B | Cell Signaling Technology | Clone E5V2L; Cat# 44153; RRID: AB_2857976 |
| t-CyCIF: anti-mouse Perforin | Cell Signaling Technology | Clone E3W4I; Cat# 31647; RRID: AB_2857978 |
| t-CyCIF: anti-mouse TIM3 | Cell Signaling Technology | Clone D3M9R; Cat# 83882; RRID: AB_2800033 |
| t-CyCIF: anti-mouse Ly6G | eBioscience | Clone 1A8-Ly6G; Cat#: 12–9668-82; RRID: AB_2572720 |
| t-CyCIF: anti-human and mouse TCF1 | Cell Signaling Technology | Clone C63D9; Cat# 6709; RRID: AB_2797631 |
| t-CyCIF: anti-human and mouse Vimentin | Cell Signaling Technology | Clone D21H3; Cat# 9854; RRID: AB_10829352 |
| t-CyCIF: anti-human and mouse αSMA | Cell Signaling Technology | Clone D4K9N; Cat# 76113; RRID: |
| t-CyCIF: anti-mouse F4/80 | Cell Signaling Technology | Clone D2S9R; Cat# 70076; RRID: AB_2799771 |
| t-CyCIF: anti-human and mouse Pan-Keratin | ThermoFisher Scientific | Clone AE1/AE3; Cat# 53–9003-82; RRID: AB_1834350 |
| t-CyCIF: anti-human and mouse PCNA | Abcam | Clone PC10; Cat# ab201674; RRID: AB_2857977 |
| t-CyCIF: anti-human CD4 | R&D | Clone Polyclonal; Cat# FAB8165G; RRID: AB_2728839 |
| t-CyCIF: anti-human CCR6 | Abcam | Clone EPR22259; Cat# ab243852; RRID: AB_2860033 |
| t-CyCIF: anti-human Granzyme B | Agilent Dako | Clone GrB-7; Cat# M7235; RRID: AB_2114697 |
| t-CyCIF: anti-human and mouse TCF1 | Cell Signaling Technology | Clone C63D9; Cat# 6444; RRID: AB_2797627 |
| t-CyCIF: anti-human FOXP3 | eBioscience | Clone 236A/E7; Cat# 41–4777-82; RRID: AB_2573609 |
| t-CyCIF: anti-human CD8α | eBioscience | Clone AMC908; Cat# 50–0008-82; RRID: AB_2574149 |
| t-CyCIF: anti-human and mouse TTF1 | Abcam | Clone EPR5955(2); Cat# ab206726; RRID: AB_2857980 |
| t-CyCIF: anti-human PD-L1 | Cell Signaling Technology | Clone E1L3N; Cat# 14123; RRID: AB_2798397 |
| t-CyCIF: anti-human CD20 | eBioscience | Clone L26; Cat# 50– 0202-82; RRID: AB_11150959 |
| t-CyCIF: anti-human TIM-3 | Cell Signaling Technology | Clone D5D5R; Cat# 54669; RRID: AB_2799468 |
| t-CyCIF: anti-human CD45 | BioLegend | Clone HI30; Cat# 304008; RRID: AB_314396 |
| t-CyCIF: anti-human PD-1 | Abcam | Clone EPR4877(2); Cat# ab201825; RRID: AB_2728811 |
| t-CyCIF: anti-human CD163 | Abcam | Clone EPR14643–36; Cat# ab218293; RRID: AB_2889155 |
| t-CyCIF: anti-human CD68 | Cell Signaling Technology | Clone D4B9C; Cat# 79594; RRID: AB_2799935 |
| t-CyCIF: anti-human and mouse Ki-67 | Cell Signaling Technology | Clone D3B5; Cat# 12075; RRID: AB_2728830 |
| t-CyCIF: anti-human HLA-DPB1 | Abcam | Clone EPR11226; Cat# ab201527; RRID: AB_2890211 |
| t-CyCIF: anti-human CD3D | Abcam | Clone EP4426; Cat# ab208514; RRID: AB_2728789 |
| t-CyCIF: anti-human HLA A | Abcam | Clone EP1395Y; Cat# ab199837; RRID: AB_2728798 |
| t-CyCIF: anti-human and mouse PCNA | Cell Signaling Technology | Clone PC10; Cat# 8580; RRID: AB_11178664 |
| t-CyCIF: anti-human αSMA | Abcam | Clone EPR5368; Cat# ab202509; RRID: AB_2868435 |
| t-CyCIF: anti-human and mouse Vimentin | Cell Signaling Technology | Clone D21H3; Cat# 9856; RRID: AB_10834530 |
| t-CyCIF: anti-human CD16 | Santa Cruz | Clone DJ130c; Cat# sc-20052 AF488; RRID: AB_2890161 |
| t-CyCIF: anti-human and mouse Pan-Keratin | eBioscience | Clone AE1/AE3; Cat# 41–9003-82; RRID: AB_11218704 |
| t-CyCIF: anti-human CD14 | Abcam | Clone EPR3653; Cat# ab196169; RRID: AB_2890135 |
| t-CyCIF: anti-human CD19 | Abcam | Clone EPR5906; Cat# ab196468; RRID: AB_2889156 |
| t-CyCIF: anti-human CD103 | Abcam | Clone EPR4166(2); Cat# ab225153; RRID: AB_2884945 |
| FC: anti-mouse CD3e | BD Biosciences | Clone 145–2C11; Cat# 565922; RRID: AB_2738278 |
| FC: anti-mouse CD8ɑ | BD Biosciences | Clone 53–6.7; Cat# 563786, 612759; RRID: AB_2732919, AB_2870090 |
| FC: anti-mouse CD4 | ThermoFisher | Clone: RM4–5; Cat# 46–0042-82; RRID: AB_1834431 |
| FC: anti-mouse Foxp3 | ThermoFisher | Clone FJK-16s; Cat# 48–5773-82; RRID AB_1518812 |
| FC: anti-mouse CD44 | BD Biosciences | Clone IM7; Cat# 563736; RRID: AB_2738395 |
| FC: anti-mouse CD45 | ThermoFisher | Clone 30-F11; Cat# 47– 0451-80; RRID: AB_1548790 |
| FC: anti-human Granzyme B | BD Biosciences | Clone GB11; Cat# 515408, 562462; RRID: AB_2562196, AB_2737618 |
| FC: anti-human Ki67 | BD Biosciences | Clone B56; Cat# 561277; RRID: AB_10611571 |
| FC: anti-mouse PD1 (CD279) | BioLegend | Clone RMP1–30; Cat# 109120; RRID: AB_2566641 |
| FC: TCF1/TCF7 | Cell Signaling Technology | Clone C63D9; Cat# 6444; RRID: AB_2797627 |
| FC: TIM3 | BioLegend | Clone RMT3–23; Cat# 119721; RRID: AB_2616907 |
| FC: H-2Kb SIINFEKL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb SIYRYYGL monomer | NIH Tetramer Core Facility | Custom |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human formalin fixed paraffin embedded tissue samples from lung adenocarcinoma cases | Partners Healthcare Institutional Review Board at Brigham Health, Boston, MA, USA | Excess tissue, discarded tissue protocol number 2018P001627 |
| Chemicals, peptides, and recombinant proteins | ||
| SMLVLLPDEVSGLEQLESIINYEKLTEWTS | New England Peptide | Custom |
| SMLVLLPDEVSGLEQLESIINFEKLTEWTS peptide | New England Peptide | Custom |
| Cyclic-di-GMP | Invitrogen | Cat# tlrl-nacdg |
| Mirus TransIT LT1 | Mirus Bio | Cat# MIR 2300 |
| Polybrene Infection Reagent | Millipore | Cat# TR-1003-G |
| Collagenase IV | Worthington Biochemical | Cat# LS004189 |
| DNase I | Sigma-Aldrich | Cat# 10104159001 |
| Streptavidin, allophycocyanin conjugate | Invitrogen | Cat# S32362 |
| Mirus TransIT LT1 | Mirus Bio | Cat# MIR 2300 |
| Zombie Fixable Viability Kit | BioLegend | Cat# 423102 |
| Critical commercial assays | ||
| Mouse CXCL10/IP-10/CRG-2 DuoSet ELISA | R&D Systems | Cat# DY466–05 |
| Fisherbrand Superfrost Plus Microscope Slides | ThermoFisher Scientific | Cat# 12–550-15 |
| Intracellular Fixation & Permeabilization Buffer Set Kit | ThermoFisher Scientific | Cat# 88–8824-00 |
| Deposited data | ||
| Processed imaging data | Synapse.org | doi.org/10.7303/syn30715952 |
| Experimental models: Cell lines | ||
| 293FS* viral packaging cell line | This paper | N/A |
| GreenGo 3TZ for lentiviral titering | This paper | N/A |
| 1233 KP lung adenocarcinoma | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J Jackson Laboratories | Jackson Laboratories | Stock No: 008179 |
| Mouse: B6.129P2-Trp53tm1Brn/J Jackson Laboratories | Jackson Laboratories | Stock No: 008462 |
| Mouse: Rosa26LSL-Cas9-GFP-Csy4 | Ng et al., 2020 | N/A |
| Oligonucleotides | ||
| Cxcl10 dRNA Oligo 1: CACCGACAAGCAATGCCCT | Sigma-Aldrich | N/A |
| Cxcl10 dRNA Oligo 2: AAACAGGGCATTGCTTGTC | Sigma-Aldrich | N/A |
| Tomato dRNA Oligo 1: CACCCGAGTTCGAGATCGA | Sigma-Aldrich | N/A |
| Tomato dRNA Oligo 2: AAACTCGATCTCGAACTCG | Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| Plasmid: Lenti-Cre | DuPage et al., 2011 | Addgene Cat# 198712 |
| Plasmid: Lenti-LucOS | DuPage et al., 2011 | Addgene Cat# 22777 |
| Plasmid: Lenti-SAM-Puro | This paper | Addgene Cat# 198713 |
| Plasmid: Lenti-SAM-Cre | This paper | Addgene Cat# 198714 |
| Software and algorithms | ||
| Aperio ImageScope | Leica Biosystems | Version 12 https://www.leicabiosystems.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| ImageJ BaSiC Plugin | Peng et al., 2017 | https://www.helmholtz-muenchen.de/icb/research/groups/marr-lab/software/basic/index.html |
| ASHLAR | The Python Package Index; Muhlich et al. 2021 | https://pypi.org/project/ashlar/ |
| ilastik | Berg et al., 2019 | https://www.ilastik.org/download.html |
| Visinity | Warchol et al. 2022 | https://github.com/labsyspharm/visinity. |
| Code | doi.org/10.5281/zenodo.7670911 | |
| Other | ||
Data and code availability
Data:
The imaging data reported in this study cannot be deposited in a public repository because a repository for imaging data is not yet available. To request access, contact the lead contact. Multiplexed images of a mouse lung specimen (KP LucOS can be viewed in Minerva Story 44,45 an interpretive guide for interacting with multiplexed tissue imaging data) are available at Zenodo. Summary statistics describing processed datasets derived from these data have been deposited at Synapse.org repository (doi.org/10.7303/syn30715952) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Code:
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Tissue
Formalin fixed paraffin embedded (FFPE) tissue samples of human lung adenocarcinoma were retrieved from the archives of the Brigham and Women’s Hospital Department of Pathology following approval of the research study by the Partners Healthcare Institutional Review Board at Brigham Health, Boston, MA, USA (Excess tissue, discarded tissue protocol number 2018P001627). All appropriate ethical guidelines were followed for this study.
Mice
Lung adenocarcinomas were initiated in KrasLSL-G12D/+; Trp53fl/fl (KP) on a C57BL/6 background through intratracheal installation of lentiviruses expressing Cre recombinase12. KP mice crossed to Rosa26LSL-Cas9-GFP-Csy4 46 and the Rosa26LSL-tdTomato were used for CRISPR-Cas9-mediated gene activation of Cxcl10. Mice were between 8 and 14 weeks of age at the time of lentiviral infection. Males and females were used equally across all experimental arms. All studies were performed under an animal protocol approved by the Massachusetts Institute of Technology (MIT) Committee on Animal Care. Mice were assessed for morbidity according to guidelines set by the MIT Division of Comparative Medicine and were humanely sacrificed prior to natural expiration. Information about each mouse experiment is provided in Table S2.
METHOD DETAILS
Lentiviral Tumor Induction
To initiate lung tumors, KP mice were injected intratracheally (i.t.) with 2.5 × 104 PFU of lentivirus containing Cre recombinase and model CD8 T cell antigens as previously described9,12. Details of the lentivirus production can be found below. Mice were randomized post-infection for immunotherapy trials.
Lentiviral Constructs
Lentiviral constructs containing Cre recombinase with or without LucOS antigens (Lenti-Cre and Lenti-LucOS) were previously described12. The Lenti-Cre design was modified by Gibson cloning to create Lenti-SAM-Cre for CRISPR/Cas9-mediated gene activation. A U6 promoter and an activator guide RNA cloning cassette were added upstream and inverted from the Pgk promoter driving Cre. The cloning cassette contains BsmBI restriction sites for the addition of a 15-nucleotide “dead” guide RNA (dRNA) to mediate gene activation rather than cutting by catalytically active Cas947. The cassette appends the dRNA with stem-loops containing MS2-binding aptamers as previously described48. “SAM” transcriptional activation components from p65 (NFkB) and Hsf1 were fused with the MS2 RNA binding protein47,48 and cloned in tandem with Cre, separated by a P2A self-cleaving peptide. For in vitro validation of dRNA activity, Lenti-SAM-Cre was modified to replace Cre with a Puromycin selection gene (Lenti-SAM-Puro). In the LucOS LucOS variant of the KP model, two model CD8 T cell antigens, the SIINFEKL (SIIN) epitope from chicken ovalbumin and the synthetic peptide SIYRYYGL (SIY), are expressed as a fusion to luciferase in tumor cells12. Immunogenic neoantigens isolated from MCA-induced sarcomas (i.e., mutant Alg8 and mutant Lama4)49 expressed in KP lung and pancreatic tumors15,50 have been shown to generate T cell responses of similar magnitude and functionality to the SIIN-specific response in both models (the more immunogenic between SIIN and SIY).
Cxcl10 Dead Guide RNA Screening
Short guide RNA (sgRNA) sequences targeting the promoter region of Cxcl10 (up to 200 nucleotides upstream of the TSS) were selected using the Feng Zhang lab (Broad Institute of MIT and Harvard) online SAM Cas9 activator design tool (no longer operational). The 20 nucleotide sgRNA sequences were shortened to 15-nucleotide dead RNAs (dRNAs) to recruit Cas9 to the promoter region but prevent DNA cleavage by Cas9. The first nucleotide was amended to a G if it did not occur naturally to optimize expression from the U6 promoter. The dRNAs were screened for their relative ability to activate Cxcl10 expression in the 1233 KP lung adenocarcinoma cell line. Briefly, oligonucleotides were generated with BsmBI restriction site overhangs (see Key Resources Table) and annealed to create the double-stranded dRNAs for cloning into Lenti-SAM-Puro. 293FS* viral packaging cells were transfected in a 6-well plate format with the dRNA-containing Lenti-SAM-Puro constructs (1.5 μg) and psPAX2 (0.75 μg) and VSV-G (0.25 μg) helper plasmids to generate lentivirus. The lentiviral supernatant was collected through a 0.45 μm filter 48 hrs post-transfection and added 1:1 to 1233 KP Cas9 cells plated at 25,000 cells/well the day before. Polybrene was added to improve transduction efficiency at 4 μg/ml. Puromycin was added 48 hrs later to select for cells expressing the construct. Cells were expanded (under Puromycin selection) and plated in triplicate in 12-well plates at 200,000 cells/well to generate supernatant containing secreted Cxcl10. The supernatant was collected 72 hrs later and Cxcl10 protein was quantified using a Cxcl10 ELISA (R&D systems) according to the manufacturer’s protocol. The dRNA that resulted in the greatest production of Cxcl10 (GACAAGCAATGCCCT) was cloned into Lenti-SAM-Cre and used to generate large-scale lentivirus for in vivo studies. A non-targeting dRNA shortened from an sgRNA targeting tdTomato (CGAGTTCGAGATCGA;51 was used a negative control. dRNA sequences and oligonucleotides are listed in the key resources table.
Lentivirus Production for In Vivo Instillation
Lentivirus was produced by transfection of 293FS* viral packaging cells in 15 cm plates with lentiviral constructs (10 μg), VSV-G (2.5 μg) and psPAX2 (7.5 μg) viral packaging plasmids, and Mirus TransIT LT1 (MirusBio; 60 μl). Lentiviral supernatant was harvested, passed through a 0.45 um filter, and concentrated by ultracentrifugation at 25,000 rpm for 2 hrs at 4°C 48- and 72-hrs post-transfection. Viral titers were determined by measuring Cre activation of GFP expression in GreenGo 3TZ cells as previously described 51.
Anti-PD-1/Anti-CTLA-4 Therapy
KP LucOS mice were treated for one week starting at 8 wks post-tumor initiation with InvivomAb anti-PD-1 (29F.1A12; BioXCell) and InvivomAb anti-CTLA-4 (9H10; BioXCell) or isotype controls (Rag IgG2a, 2A3; Syrian Hamster, polyclonal; BioXCell). Mice received 200 μg of each antibody i.p. at day 0, followed by 200 μg anti-PD-1 and 100 μg anti-CTLA-4 (or isotype controls at the same concentrations) on days 3 and 6. Mice were sacrificed for endpoint analysis on day 7.
Antigen-targeted Vaccination
KP LucOS mice were vaccinated s.c. at the tail-base with 30 amino acid long peptides containing SIINFEKL and SIYRYYGL (10 nmol; New England Peptide) and cyclic-di-GMP adjuvant (0.25 mg/ml; Invitrogen) at 6 wks post-tumor initiation. An equivalent booster dose was given 2 wks later, and the mice were sacrificed at 9 wks post-tumor initiation for endpoint analysis. All doses were delivered in two 50 μL boluses and control mice received PBS. The long peptide sequences used were: SMLVLLPDEVSGLEQLESIINFEKLTEWTS and GRCVGSEQLESIYRYYGLLLKERSEQKLIS (New England Peptide).
Mouse Lung Tissue Processing for Flow Cytometry
Lung tissue-resident immune cells were distinguished from circulating immune cells by retroorbital injection of a fluorescently-conjugated CD45 antibody (AlexaFluor780; 30-F11; BD Bioscience) 3 minutes prior to euthanasia52. Only tissue-resident cells were included in downstream analyses. Lung tissue was collected into RPMI 1640 media with 1% heat-inactivated fetal bovine serum, minced with spring scissors and incubated in 125 U/mL collagenase IV (Worthington Biochemical) and 40 U/mL DNase I (Sigma-Aldrich) for 30 minutes at 37°C. The tissue was then dissociated using the m_lung_2.0.1 protocol on a gentleMACS Dissociator using gentleMACS C tubes (Miltenyi Biotec) and filtered with a 70 μm cell strainer. Cell suspensions were centrifuged at 1200 rpm for 5 minutes and red blood cell lysis was performed using 1X RBC Lysis Buffer (eBioscience) per the manufacturer’s protocol. Cells were then stained with a fixable viability dye to exclude dead cells (20 minutes on ice; Zombie Dye; Invitrogen; Tonbo Ghost Dye; Tonbo Biosciences) and resuspended in FACS buffer (1% heat-inactivated FBS in PBS) and stained with the following surface antibodies for 15–30 minutes on ice: CD3e (145–2C11), CD8ɑ (53–6.7), CD4 (RM4–5), CD44 (IM7), PD-1 (RMP1–30), TIM-3 (RMT3–23), purchased from ThermoFisher Scientific, BD Biosciences or Biolegend (see Key Resources Table). In some cases, the cells were simultaneously stained with H-2Kb peptide-MHC tetramers specific to SIINFEKL and SIYRYYGL (monomer, NIH Tetramer Core Facility; PE and APC streptavidin, Invitrogen). For intracellular staining, cells were fixed for 1 hour at room temperature using the eBioscience Fixation/Permeabilization Kit (ThermoFisher Scientific). Cells were then stained overnight at 4°C with the following antibodies: TCF1/TCF7 (C63D9), Granzyme B (GB11), Ki67 (B56), Foxp3 (FJK-16s) purchased from Cell Signaling Technology, ThermoFisher Scientific, BD Biosciences (see Key Resources Table). Samples were analyzed on a BD Biosciences LSR Fortessa.
Mouse Lung Tissue Processing for Histology and H&E Staining
Tumor-bearing lung lobes were collected into 4% paraformaldehyde in PBS and incubated overnight with shaking at 4°C. Tissue was transferred into 70% ethanol and subsequently paraffin embedded and sectioned (4 μm) onto Fisherbrand Superfrost Plus Microscope Slides (ThermoFisher Scientific). After drying, slides for RNAScope™ were stored at 4°C until use. Hematoxylin and eosin (H&E) stain was performed with a standard method by the Hope Babette Tang Histology Facility at the Koch Institute at MIT.
Pathology Annotation of Mouse and Human H&E-Stained Sections
H&E and CyCIF images were reviewed and annotated by a board-certified anatomic pathologist (S.C.), blind to the underlying genotype and diagnosis (e.g., KP Cre vs. KP LucOS). For KP Cre and KP LucOS mouse tissues, all identifiable anatomic structures were delineated, including medium-large caliber airways (bronchioles, bronchi; ~50–300 μm in diameter) and medium-large caliber vascular structures (~20–300 μm in diameter). Vascular structures were further grouped into arterial (arterioles and arteries) and venous (venules and veins) categories based on typical histologic features. Smaller capillary structures were not discretely annotated. Large branches of the pulmonary artery and vein were noted when present. All tumors were identified and delineated according to morphologic features (nuclear atypia, architectural disorganization, hypercellularity, etc.). Regions of epithelial cytologic atypia that did not form discrete invasive tumors were also annotated. For human tissues, all tumors, regions of atypia, and lymphoid aggregates were annotated. In human tumor specimens, all aggregates of lymphoid cells were identified and delineated in each tissue section according to typical morphologic features. Tertiary lymphoid structures (TLS) were further defined by identifying aggregates of lymphoid cells associated with germinal center formation on H&E, or the presence of discrete aggregates of B cells (PAX5+) with surrounding T cell (CD3, CD4, and/or CD8 positive) populations on CyCIF imaging. Annotation was crosschecked between H&E and CyCIF images for all tissue sections.
Tissue-Based Cyclic Immunofluorescence (t-CyCIF) Staining and Imaging
FFPE sections were prepared and stained with a 24-plex antibody panel according to the previously described t-CyCIF protocols14,15,53 (see Table S1). This CyCIF panel has been validated across many different sample types in accordance with standards defined by our group54. The number of mice, number of lobes, and number of tumor nodules analyzed from whole slide CyCIF imaging are indicated in Table S2. As noted in Table S2, all tumor nodules were analyzed from two or three lung lobes per mouse for each experiment
Baking and Dewaxing
To prepare samples for antibody staining, slides were automatically baked at 60°C for 30 min, dewaxed at 72°C in BOND Dewax Solution, and antigen retrieval was performed at 100°C for 20 min in BOND Epitope Retrieval Solution 2 (ER2) by the Leica Bond RX machine.
Pre-Staining Background Reduction
After slides were baked and dewaxed, they were photobleached by immersing them in bleaching solution (4.5% H2O2, 20 mM NaOH in PBS) with LED light exposure for 2 × 45 min to reduce autofluorescence.
To mitigate non-specific antibody binding, slides were washed for 3 × 5 min with 1X PBS and then incubated overnight with secondary antibodies (anti-rat, anti-mouse, and anti-rabbit) diluted in 150 μL of Odyssey Blocking Buffer (1:1000) at 4°C in the dark. Slides were subsequently washed 3x with 1X PBS before photobleaching them again for 2 × 45 min.
Antibody Staining, Slide Mounting, and Imaging
For each round of t-CyCIF, samples were incubated overnight at 4°C in the dark with Hoechst 33342 (Dilution: 1:10,000; Thermo Fisher Scientific, cat# 62249) for nuclear staining along with either primary conjugated antibodies or primary unconjugated antibodies diluted (see Table S1 for antibody information) in 150 μL of Odyssey Blocking Buffer (LI-Cor, Cat# P/N 927–40003). Incubation with primary unconjugated antibodies was followed by secondary antibody incubation at room temperature for 2 hrs in the dark. For CyCIF antibodies that were only available from vendors as primary unconjugated antibodies, custom conjugates were requested from Cell Signaling Technology or we performed in-house conjugation of antibodies formulated without BSA and sodium azide using Invitrogen Alexa Fluor™ Antibody Labeling Kits in accordance with the manufacture’s guidelines. 100μg of antibody was labelled at a 1mg/mL dilution in an appropriate buffer (i.e., Phosphate Buffer Saline). Custom ordered antibodies from Cell Signaling Technology were generated for CD8a CST [D4W2Z], Cat# 98941 (AF 647); CD11c CST [D1V9Y] Cat# 97585 (AF 555); CD3e CST [D4V8L] Cat# 99940 (AF 555); PD-L1 [D5V3B] Cat# 64988 (AF 488); Granzyme B [E5V2L] Cat# 44153 (AF 488); Perforin [E3W4] Cat# 31647 (AF 555); TIM-3 [D3M9R] Cat# 83882 (AF 488); F4/80 [D2S9R] Cat# 70076 (AF 555). The remaining antibody was conjugated ‘in-house’: CD103 R&D [Polyclonal], Cat# AF1990 (AF 488). Key Resources Table lists all antibodies used.
Post staining, slides were washed for 3 × 5 min, mounted with 24 × 50 mm coverslips using 200 μL of 70% glycerol, and then dried. Once coverslipped, slides were manually imaged on the IN Cell Analyzer 6000 or automatically on the RareCyte Cytefinder II HT using the following channels: UV, cy3, cy5, and cy7 (Binning: 1 × 1; Objective: 20x; Numerical Aperture: 0.75; Resolution: 0.325 μm/pixel). Image exposures were optimized for each channel to avoid signal saturation and kept constant for each sample.
To demount, slides were placed in containers of 1X PBS and heated in a water bath for 1 hr. Before additional antibody staining, slides are photobleached for 2 × 45 min to deactivate the fluorophores and washed 3 × 5 min in 1X PBS.
RNA In Situ Hybridization
RNAScope™ was performed as per manufacture’s suggested protocol (Advanced Cell Diagnostics, Inc.) using the LS Multiplex Reagent Kit (cat# 322800) and probes RNAscope® 2.5 LS Probe- Mm-Cxcl9 (cat #: 489348) and RNAscope® 2.5 LS Probe- Mm-Cxcl10-C3 (cat #: 408928-C3).
QUANTIFICATION AND STATISTICAL ANALYSIS
STATISTICAL ANALYSIS
Information on the sample size (Table S2) and the statistics are included in the figure legends. Statistical tests used are Pearson correlation, two-sided t-test, and non-parametric Kolmogorov–Smirnov (KS) two-sided test as specified in the figure legends and are performed with MATLAB built-in functions. Significance was defined as a p-value of less than 0.05. For figures where mice are represented as individual data points, the data represents the average of all tumor nodules for each mouse. For figures where data is shown for groups rather than individual mice (e.g., LucOS versus Cre, Vax versus Ctrl), the average was calculated for all tumor nodules from each individual mouse prior to averaging the data from all mice in each group. In this way, we avoided skewing the data toward mice with a greater number of tumors analyzed.
QUANTIFICATION
Image Processing and Single-Cell Quantification
The image processing of tissue cyclic immunofluorescence was organized in the following steps, each of which is described in detail below:
the software ASHLAR is used to stitch, register, and correct for image acquisition artifacts (using the BaSiC algorithm). The output of ASHLAR is a single pyramid ome.tiff file for each region imaged;
the ome.tiff file is re-cut into tiles (typically 5000 × 5000 pixels) containing only the highest resolution image for all channels. One random cropped image (250 × 250 pixels) per tile is outputted for segmentation training (using Fiji);
the ilastik software is trained on the cropped images to label, nuclear, cytoplasmic, and background areas. The output of the Ilastik processing is a 3-color RGB image with label probabilities;
the RBG probability images are thresholded and watershed in MATLAB to segment the nuclear area. The cytoplasmic measurements are derived by dilating the nuclear mask;
single-cell measurements are extracted for each channel (cell pixel median and mean for both nuclear and cytoplasmic area) as well as morphological measurements of area, solidity, and cell coordinates location.
BaSiC
The BaSiC ImageJ plugin tool was used to perform background and shading correction of the original images55. The BaSiC algorithm calculates the flatfield, the change in effective illumination across an image, and the darkfield, which captures the camera offset and thermal noise. The dark field correction image is subtracted from the original image, and the result is divided by the flatfield image correction to obtain the final image.
ASHLAR
Alignment by Simultaneous Harmonization of Layer/Adjacency Registration (ASHLAR) is used to stitch together image tiles and register image tiles in subsequent layers to those in the first layer56. For the first image layer, neighboring image tiles are aligned to one another via a phase correlation algorithm that corrected for local state positioning error. A similar method is applied for subsequent layers to align tiles to their corresponding tile in the first layer. ASHLAR outputs an OME-TIFF file containing a multi-channel mosaic of the full image across all imaging cycles. Full codes available at: https://github.com/labsyspharm/ashlar.
ilastik
ilastik is a machine learning based bioimage analysis tool that is used to obtain nuclear and cytoplasmic segmentation masks from OME-TIFF files57. For increased processing speed, randomly selected 250 × 250 pixel regions from the original OME-TIFF are used as training data. ilastik’s interactive user interface allows the user to provide training annotations on the cropped regions. Users are presented with a subset of the channels stacked images and label pixels as either nuclear area, cytoplasmic area, or background area. The annotations are used to train non-linear classifiers that are applied to the entire image to obtain probability masks describing the probabilities of each pixel belonging to the nuclear, cytoplasmic, or background area. A MATLAB (version 2018a) script uses these masks to construct binary masks for nuclear and cytoplasmic area.
Single Cell Segmentation and Quantification
Using ilastik’s Pixel Classification workflow, a random forest classifier is trained for each experimental dataset based on manual annotations of nuclear, cytoplasmic, and background regions within the CroppedData. Batch processing is subsequently performed by the classifier on the FullStacks, generating .tif probability maps for nuclei, background, and cytoplasm.
Cell nuclei are segmented through thresholding maps based on nuclear, cytoplasm, and background probabilities and performing water shedding on them using MATLAB. Cytoplasmic segmentation masks are produced by dilating nuclear segmentation masks radially by 3 pixels and then excluding the segmented nuclear area.
Median nuclear and cytoplasmic marker expression, centroid coordinates, area (nuclear and cytoplasmic), and solidity are quantified for each segmented cell using MATLAB’s regionprops function and outputted as a single “Results.mat” file for each FFPE slide. All MATLAB scripts used for segmentation and quantification can be found here: https://github.com/santagatalab.
Data analysis workflow
The data analysis is divided in a set of pre-processing steps in which data from different tissues is i) log2-transformed and aggregated together, ii) filtered for image analysis errors, and iii) normalized on a channel-by-channel basis across the entire data from a single experiment. All the steps are performed in MATLAB.
Data aggregation
The image processing workflow outputs one ome.tiff image and one data file (.mat) for each tissue area imaged. The data matrices from each .mat file are concatenated into a single matrix for each metric measured (median/mean, nuclear/cytoplasmic) into a single structure (“AggrResults”). The morphological data (i.e., area, solidity, and centroid coordinates) is concatenated into a single structure (“MorpResults”), which also contains the indexing vector to keep track of the tissue of origin within the dataset.
Data filtering
Single cells are filtered to identify and potentially exclude from subsequent analysis errors in segmentation and cells lost through the rounds of imaging. Two types of criteria are used to filter cells: morphological criteria based on cell object segmented area, which are applied to all the rounds for the cell object, and DAPI-based criteria which are applied to the DAPI measurement for each imaging round. The latter corrects for cell loss during cycling and computational misalignment, which are both round specific.
Morphological filtering criteria are:
nuclear area within a user-input range;
cytoplasmic area within a user-input range;
nuclear object solidity above a user-input threshold.
DAPI-based criteria are:
nuclear DAPI measurement above a user-input threshold;
ratio between nuclear and cytoplasmic DAPI measurement above a user-input threshold;
The filter information for the criteria is allocated to a logical (0–1) structure ‘Filter’, which is used to select the cells to analyze in the further analysis by indexing. The threshold selection is dataset dependent and is performed by data inspection. The values used in each dataset are available with the codes used for data analysis in the Synapse.org repository syn30715952.
Data normalization
Each channel distribution is normalized by probability density function (pdf) centering and rescaling. The aim is to center the distribution of the log2 fluorescent signal at 0 and rescale the width of the distribution to be able to compare across channels. The data is first log-transformed (base 2). The standard normalization is performed using a 2-component Gaussian mixture model, each model capturing the negative and the positive cell population. If the 2-component model fails to approximate the channel distribution, two other strategies are attempted: i) a 3-component model is used assuming the components with the two highest means are the negative and positive distribution (i.e., discarding the lowest component) or ii) the user selects a percentage ‘x’ of assumed positive cells and a single Gaussian distribution fit is performed on the remainder of the data to capture the negative distribution. The single Gaussian fit is then used as the lower component in a 2-component model to estimate the distribution of the positive population. The strategy chosen for each channel in each dataset is available in the code section of the Synapse.org repository syn30715952.
The “add_coeff” is defined as the intersection of the negative and positive distributions. The “mult_coeff” is defined as the difference between the mean of the negative and positive distributions. The full distribution is normalized by subtracting the add_coeff and dividing by the mult_coeff. The normalization is performed on the nuclear and cytoplasmic single-cell, single-channel distributions individually.
The data preprocessing workflow is performed on all datasets. The individual analyses used in the paper are performed only in selected datasets as follows.
Cell type classification
Cell type classification is performed hierarchically on the filtered, normalized expression data. Each cell is evaluated based on marker expression and then assigned to cell types in a layered fashion according to the dendrogram schematic in Figure S1C, with each successive layer being more specific than the previous one. A cell is considered to be positive for a marker if its median expression is above 0. Cell types are defined in the dendrogram by the presence or exclusion of multiple markers using “&&” and “||” operators representing “AND” and “OR” logic respectively. If multiple marker conditions must be met to assign a cell type, these marker conditions are grouped using parentheses. If a cell is “positive” for two markers that are expected to be mutually exclusive, the marker that is expressed at a higher value takes precedence as long as the difference in expression surpasses a user-defined threshold.
Multimodal Data Integration
H&E, RNAScope™ and CyCIF images are rescale and registered using the open-source software elastix58 using non-shearing global transformation. The CyCIF images are used as the fixed images in elastix. To integrate the CyCIF and histological data, H&Es are annotated for tumors, blood vessels, and airways by a trained pathologist. The elastix registration is used to overlay the pathology annotation onto the CyCIF single cell coordinates and then to calculate the distance from tumor boundaries and blood vessels.
RNAscope foci detection
Custom spot detection scripts (https://github.com/Yu-AnChen/wsi-fish) are used to identify RNAScope™ foci and quantify their intensity. Each RNAScope™ dot is assigned as belonging to the closest cell based on the segmented area. A cell is considered Cxcl positive if it is assigned at least two RNA foci and if the cumulative RNAScope™ dot intensity of all the dots assigned to the cell exceed a preset threshold (based on the positive tail of the single cell distribution).
Lymphonet definition
The single cell centroids are tessellated using the Delaunay Triangulation using a custom script in MATLAB (https://github.com/santagatalab) to obtain a 2D graph, setting a maximum edge length of 16.25 microns (50 pixels). Using conventional graph operations, the graph edges are then filtered to include only connection between lymphocytes (Lv3 of cell type dendogram), after which connected subgraphs of length greater than 5 are than defined as “lymphonets”.
Palantir algorithm and CD8 T cell state definition
The algorithm Palantir25 was adapted to CyCIF data by bypassing the initial dimensionality reduction applied to single-cell RNA-seq data and using the CyCIF channel information as the dimensionality reduction output. The Python Jupiter Notebooks used to run the Palantir analyses can be found at https://github.com/santagatalab. The CD8 T cell phenotypic states S1-S3 and T1-T3 were obtained using a flow cytometry manual gating approach combining Palantir point density and marker intensity. The gating was performed in MATLAB using the “Flow Cytometry GUI for Matlab” by Nitai Steinberg (2022) available at https://www.mathworks.com/matlabcentral/fileexchange/38080-flow-cytometry-gui-for-matlab, MATLAB Central File Exchange.
Spatial and Phenotypic Correlation Analysis
Spatial correlations Cxy(r) were computed as the Pearson correlation between two groups of spatially defined objects; (1) a cell of group X and (2) its kth nearest neighbor of group Y, for their respective variables x and y. A value of Cxy (r) is computed for each k up to 100, and a distance r was assigned to each k as the average distance between kth nearest neighbors. More detail can be found in Gaglia et al.53. In Figure 7F and S7D, spatial correlation between direct neighbors k = 2 was calculated between the likelihood of lymphocytes belonging to lymphonets and non-lymphocyte cells’ likelihood of being positive for the indicated markers. Hence in the Cxy formula above: X = lymphocytes, x = {0 not part of a lymphonet, or 1 part of a lymphonet}, Y=non-lymphocyte cell, y = {0 negative or, 1 positive for marker}. For each marker the analysis is repeated within each of 14 human lung cancer tissues independently. The phenotypic correlation (in Figure 7H) is calculated by comparing the 2D probability density function in Palantir space, by correlating the likelihood of CD8 Tc belonging to a lymphonet (binned by size) and the likelihood of CD8 Tc being TCF1+ PD-1+ double positive.
Visinity - Visual Spatial Neighborhood Analysis
To visually explore the spatial neighborhoods within these data, we use the Visinity18, a scalable system for visual analysis in whole-slide multiplexed tissue imaging data. This system supports the analysis of recurrent cellular spatial neighborhoods across cohorts of specimens. Visinity is an open-source project (https://github.com/labsyspharm/visinity), with a JavaScript client for browser-based visualization and a Python server for efficient and scalable backend computation.
Quantifying Cellular Neighborhoods
Visinity quantifies the spatial neighborhood for each cell in terms of the types of cells that surround it (for Visinity the information contained in level 4 (Lv4) was used as the cell type information). More specifically, this process is as follows:
A ball-tree index structure is constructed using nuclei centroids of each segmented cell in a specimen, which allows for O(n + k) range queries, where n is the number of cells and k is the number of points within this range. We use the scikit-learn59 implementation of this data structure.
With the ball-tree, we identify neighboring cells within a 50 μm radius of each cell.
We create feature vectors representing the neighborhood of each cell. Vectors are 1 × n, where n is the number of cell types. Columns in this vector correspond to the presence of a specific cell type. We linearly weight each cell in a neighborhood by its distance from the center so that cells just at the edge of the neighborhood radius contribute the least and sum these weights by cell type.
We repeat this process for every cell across all specimens, L1 normalizing the vectors. Each vector, which represents the neighborhood of an individual cell, is a row in a matrix representing all cells across all specimens.
We create a 2D embedding of this matrix using UMAP60 with the parameters n_neighbors = 50, min_dist = 0.1. Points close to each other in this embedding space represent cells with similar spatial neighborhoods
We display this embedding as an interactive scatterplot. Selecting regions in this embedding highlights the corresponding cells within the tissue image and we visualize the cell types that compose the selected neighborhood with a parallel coordinates plot.
Visinity supports both confirmatory and exploratory analysis, allowing users to detect spatial neighborhood patterns in a semi-automated manner and visually query across specimens for specific cellular neighborhoods. This workflow and the system as a whole are described in detail in ref 18.
Supplementary Material
Highlights.
Interacting networks of lymphocytes (lymphonets) from in the KP GEMM of lung cancer
Small lymphonets have mostly T cells, and B cell fraction rises as networks enlarge
A key feature of lymphonets is that they contain TCF1+PD-1+CD8+ T cell progenitors
Lymphonets gain cytotoxic CD8+ T cells after immunotherapy
ACKNOWLEDGEMENTS
Work was supported by the Bridge Project (partnership between Koch Institute for Integrative Cancer Research at MIT and Dana-Farber/Harvard Cancer Center (P.K.S., S.S., T.J.)), Ludwig Center at Harvard (P.K.S., S.S.), Ludwig Center for Molecular Oncology at MIT (M.L.B.), Gray Foundation, David Liposarcoma Research Initiative, U2C-CA233262 (P.K.S., S.S.), R01-CA194005 (S.S.), R41-CA224503 (P.K.S.), U54-CA225088 (P.K.S., S.S.), T32-GM007748 (S.C.), T32-HL007627 (G.G.), HHMI (T.J.), American-Italian Cancer Foundation (G.G.), K99-CA256497 (A.J.N), Jane Coffin Childs Memorial Fund for Medical Research, and BWH President’s Scholar Award (S.S.). We thank DF/HCC for Specialized Histopathology Core use (P30-CA06516). This work was supported by Koch Institute Support Grant P30-CA014051. T.J. is a Daniel K. Ludwig Scholar.
Footnotes
DECLARATION OF INTERESTS
P.K.S. is a BOD member of Applied Biomath and Glencoe Software (cofounder), SAB member for RareCyte, NanoString, and Montai Health, and consultant for Merck. T.J. is a BOD member of Amgen and ThermoFisher, co-founder of Dragonfly Therapeutics and T2-Biosystems, SAB member of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics, and President of Break-Through-Cancer. Jacks lab receives funding from J&J Lung Cancer Initiative and Lustgarten Foundation (not supporting research in this manuscript). These affiliations do not represent a COI in this manuscript with respect to design/execution/interpretation. Other authors declare no COI.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hanahan D (2022). Hallmarks of Cancer: New Dimensions. Cancer Discovery 12, 31–46. 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 2.Nirmal AJ, Maliga Z, Vallius T, Quattrochi B, Chen AA, Jacobson CA, Pelletier RJ, Yapp C, Arias-Camison R, Chen Y-A, et al. (2022). The spatial landscape of progression and immunoediting in primary melanoma at single cell resolution. Cancer Discov, candisc.1357.2021. 10.1158/2159-8290.CD-21-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey C, Black JRM, Reading JL, Litchfield K, Turajlic S, McGranahan N, Jamal-Hanjani M, and Swanton C (2021). Tracking Cancer Evolution through the Disease Course. Cancer Discov 11, 916–932. 10.1158/2159-8290.CD-20-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20. 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin J-R, Wang S, Coy S, Chen Y-A, Yapp C, Tyler M, Nariya MK, Heiser CN, Lau KS, Santagata S, et al. (2023). Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell 186, 363–381.e19. 10.1016/j.cell.2022.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baertsch M-A, Nolan GP, and Hickey JW (2022). Multicellular modules as clinical diagnostic and therapeutic targets. Trends Cancer 8, 164–173. 10.1016/j.trecan.2021.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodenmiller B (2016). Multiplexed Epitope-Based Tissue Imaging for Discovery and Healthcare Applications. Cell Syst 2, 225–238. 10.1016/j.cels.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Lewis SM, Asselin-Labat M-L, Nguyen Q, Berthelet J, Tan X, Wimmer VC, Merino D, Rogers KL, and Naik SH (2021). Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods 18, 997–1012. 10.1038/s41592-021-01203-6. [DOI] [PubMed] [Google Scholar]
- 9.DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072. 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, and Jacks T (2001). Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116. 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 11.McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, et al. (2016). Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A 113, E6409–E6417. 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85. 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Qu S, Zhang T, Gao Y, Shi H, Song K, Chen W, and Yin W (2021). Applications of Single-Cell Omics in Tumor Immunology. Frontiers in Immunology 12. 10.3389/fimmu.2021.697412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin J-R, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, and Sorger PK (2018). Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 7, e31657. 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burger ML, Cruz AM, Crossland GE, Gaglia G, Ritch CC, Blatt SE, Bhutkar A, Canner D, Kienka T, Tavana SZ, et al. (2021). Antigen dominance hierarchies shape TCF1+ progenitor CD8 T cell phenotypes in tumors. Cell 184, 4996–5014.e26. 10.1016/j.cell.2021.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan S-L, Gibbons G, Smith OC, Kim JY, Westcott P, et al. (2021). Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 54, 2338–2353.e6. 10.1016/j.immuni.2021.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Facciabene A, Motz GT, and Coukos G (2012). T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 72, 2162–2171. 10.1158/0008-5472.CAN-11-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warchol S, Krueger R, Nirmal AJ, Gaglia G, Jessup J, Ritch CC, Hoffer J, Muhlich J, Burger ML, Jacks T, et al. (2022). Visinity: Visual Spatial Neighborhood Analysis for Multiplexed Tissue Imaging Data. IEEE Trans Vis Comput Graph PP. 10.1109/TVCG.2022.3209378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delaunay BN (1934). Sur la sphère vide. Bull. Acad. Sci. URSS 1934, 793–800. [Google Scholar]
- 20.Liebling TM, and Pournin L (2012). Voronoi Diagrams and Delaunay Triangulations: Ubiquitous Siamese Twins. In.
- 21.Metzemaekers M, Vanheule V, Janssens R, Struyf S, and Proost P (2017). Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front Immunol 8, 1970. 10.3389/fimmu.2017.01970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6, 7458. 10.1038/ncomms8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellis SL, Gysbers V, Manders PM, Li W, Hofer MJ, Müller M, and Campbell IL (2010). The cell-specific induction of CXC chemokine ligand 9 mediated by IFN-gamma in microglia of the central nervous system is determined by the myeloid transcription factor PU.1. J Immunol 185, 1864–1877. 10.4049/jimmunol.1000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Setty M, Kiseliovas V, Levine J, Gayoso A, Mazutis L, and Pe’er D (2019). Characterization of cell fate probabilities in single-cell data with Palantir. Nat Biotechnol 37, 451–460. 10.1038/s41587-019-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1−CD8+ Tumor-Infiltrating T Cells. Immunity 50, 181–194.e6. 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 30.Jaeger AM, Stopfer LE, Ahn R, Sanders EA, Sandel DA, Freed-Pastor WA, Rideout WM, Naranjo S, Fessenden T, Nguyen KB, et al. (2022). Deciphering the immunopeptidome in vivo reveals new tumour antigens. Nature 607, 149–155. 10.1038/s41586-022-04839-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 32.Shenoy AT, Lyon De Ana C, Arafa EI, Salwig I, Barker KA, Korkmaz FT, Ramanujan A, Etesami NS, Soucy AM, Martin IMC, et al. (2021). Antigen presentation by lung epithelial cells directs CD4+ TRM cell function and regulates barrier immunity. Nat Commun 12, 5834. 10.1038/s41467-021-26045-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang S-R, Kurian A, Valen DV, West R, et al. (2018). A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174, 1373–1387.e19. 10.1016/j.cell.2018.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colbeck EJ, Ager A, Gallimore A, and Jones GW (2017). Tertiary Lymphoid Structures in Cancer: Drivers of Antitumor Immunity, Immunosuppression, or Bystander Sentinels in Disease? Front Immunol 8, 1830. 10.3389/fimmu.2017.01830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher TN, and Thommen DS (2022). Tertiary lymphoid structures in cancer. Science 375, eabf9419. 10.1126/science.abf9419. [DOI] [PubMed] [Google Scholar]
- 36.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. (2020). Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 37.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fridman WH, Meylan M, Petitprez F, Sun C-M, Italiano A, and Sautès-Fridman C (2022). B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol, 1–17. 10.1038/s41571-022-00619-z. [DOI] [PubMed] [Google Scholar]
- 39.Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43, 579–590. 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philip M, and Schietinger A (2022). CD8+ T cell differentiation and dysfunction in cancer. Nat Rev Immunol 22, 209–223. 10.1038/s41577-021-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, Singhal S, Mitchell JD, Franklin WA, Merrick DT, et al. (2017). Antigen-Presenting Intratumoral B Cells Affect CD4+ TIL Phenotypes in Non-Small Cell Lung Cancer Patients. Cancer Immunol Res 5, 898–907. 10.1158/2326-6066.CIR-17-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, Chang C, Walker L, Lansky A, Le Berichel J, et al. (2021). Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 39, 1594–1609.e12. 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffer J, Rashid R, Muhlich JL, Chen Y-A, Russell DPW, Ruokonen J, Krueger R, Pfister H, Santagata S, and Sorger PK (2020). Minerva: a light-weight, narrative image browser for multiplexed tissue images. J Open Source Softw 5, 2579. 10.21105/joss.02579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rashid R, Chen Y-A, Hoffer J, Muhlich JL, Lin J-R, Krueger R, Pfister H, Mitchell R, Santagata S, and Sorger PK (2022). Narrative online guides for the interpretation of digital-pathology images and tissue-atlas data. Nat Biomed Eng 6, 515–526. 10.1038/s41551-021-00789-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng SR, Rideout WM, Akama-Garren EH, Bhutkar A, Mercer KL, Schenkel JM, Bronson RT, and Jacks T (2020). CRISPR-mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proceedings of the National Academy of Sciences 117, 513–521. 10.1073/pnas.1821893117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, and Konermann S (2015). Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol 33, 1159–1161. 10.1038/nbt.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, Mercer KL, Garcia AP, Lin L, Rideout WM, et al. (2021). The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell 39, 1342–1360.e14. 10.1016/j.ccell.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, et al. (2014). Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431. 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, et al. (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9, 209–222. 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaglia G, Kabraji S, Rammos D, Dai Y, Verma A, Wang S, Mills CE, Chung M, Bergholz JS, Coy S, et al. (2022). Temporal and spatial topography of cell proliferation in cancer. Nature Cell Biology 24, 316–326. 10.1038/s41556-022-00860-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du Z, Lin J-R, Rashid R, Maliga Z, Wang S, Aster JC, Izar B, Sorger PK, and Santagata S (2019). Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging. Nat Protoc 14, 2900–2930. 10.1038/s41596-019-0206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng T, Thorn K, Schroeder T, Wang L, Theis FJ, Marr C, and Navab N (2017). A BaSiC tool for background and shading correction of optical microscopy images. Nat Commun 8, 14836. 10.1038/ncomms14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muhlich J, Chen Y-A, Russell D, and Sorger PK (2021). Stitching and registering highly multiplexed whole slide images of tissues and tumors using ASHLAR software. bioRxiv, 2021.04.20.440625. 10.1101/2021.04.20.440625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nature Methods 16, 1226–1232. 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- 58.Klein S, Staring M, Murphy K, Viergever MA, and Pluim JPW (2010). elastix: A Toolbox for Intensity-Based Medical Image Registration. IEEE Transactions on Medical Imaging 29, 196–205. 10.1109/TMI.2009.2035616. [DOI] [PubMed] [Google Scholar]
- 59.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- 60.McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861. 10.21105/joss.00861. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data:
The imaging data reported in this study cannot be deposited in a public repository because a repository for imaging data is not yet available. To request access, contact the lead contact. Multiplexed images of a mouse lung specimen (KP LucOS can be viewed in Minerva Story 44,45 an interpretive guide for interacting with multiplexed tissue imaging data) are available at Zenodo. Summary statistics describing processed datasets derived from these data have been deposited at Synapse.org repository (doi.org/10.7303/syn30715952) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Code:
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.