

ABSTRACT
Tobacco, alcohol, and marijuana consumption is an important public health problem because of their high use worldwide and their association with the risk of mortality and many health conditions, such as hypertension, which is the commonest risk factor for death throughout the world. A likely pathway of action of substance consumption leading to persistent hypertension is DNA methylation. Here, we evaluated the effects of tobacco, alcohol, and marijuana on DNA methylation in the same cohort (N = 3,424). Three epigenome-wide association studies (EWAS) were assessed in whole blood using the InfiniumHumanMethylationEPIC BeadChip. We also evaluated the mediation of the top CpG sites in the association between substance consumption and hypertension. Our analyses showed 2,569 CpG sites differentially methylated by alcohol drinking and 528 by tobacco smoking. We did not find significant associations with marijuana consumption after correcting for multiple comparisons. We found 61 genes overlapping between alcohol and tobacco that were enriched in biological processes involved in the nervous and cardiovascular systems. In the mediation analysis, we found 66 CpG sites that significantly mediated the effect of alcohol consumption on hypertension. The top alcohol-related CpG site (cg06690548, P-value = 5.9·10−83) mapped to SLC7A11 strongly mediated 70.5% of the effect of alcohol consumption on hypertension (P-value = 0.006). Our findings suggest that DNA methylation should be considered for new targets in hypertension prevention and management, particularly concerning alcohol consumption. Our data also encourage further research into the use of methylation in blood to study the neurological and cardiovascular effects of substance consumption.
KEYWORDS: Tobacco, Alcohol, Marijuana, DNA methylation, Epigenome-wide association study, Hypertension
Key policy highlights
The consumption of tobacco, alcohol, and marijuana is very high worldwide and is associated with common diseases, like cardiovascular and neurological disorders.
This study found that tobacco and alcohol have large effects on genome wide DNA methylation while marijuana consumption has nonsignificant effects.
The genes differentially methylated were enriched in pathways related to neurodevelopment, suggesting the mediation between recreational drug consumption and neurological disorders.
More remarkably, 66 alcohol related CpG sites significantly mediated the association between heavy drinking and hypertension.
Our findings suggest that DNA methylation changes should be considered for new targets in disease prevention for recreational drug consumers.
Introduction
Tobacco, alcohol, and marijuana are the most commonly used drugs of abuse in the United States [1]. While tobacco and alcohol consumption is legal, marijuana is the most commonly used illicit drug globally [2]. The consumption of these substances is increasing, mainly among adolescents, and the health and social problems associated with them are an important public health concern.
Addiction is a major consequence of consuming tobacco, alcohol, and marijuana, which results in the strengthening of all the other health problems associated with them [3].
Cigarette smoking is the leading cause of preventable death and disease in the US and is responsible for approximately 8 million worldwide deaths every year [4]. Most of these deaths arise from cancers (mainly lung cancer), respiratory disease, and cardiovascular disease [5]. Cigarette smoke contains more than 7000 chemical compounds. Among them, 1,3-butadiene is highly associated with cancer risk, acrolein, and acetaldehyde are potential respiratory irritants, cyanide, arsenic, and cresols are the primary sources of cardiovascular risk, and nicotine is the additive component [6]. In addition, nicotine exposure causes well-characterized neurotoxic effects, which are highly important in early development [7].
Light to moderate alcohol intake is associated with reduced risks for total mortality, cardiovascular disease, and diabetes. However, excessive alcohol is the third leading cause of premature death in the US [8]. Heavy alcohol use is associated with a higher risk of cardiovascular disease, diabetes, cirrhosis of the liver, pancreatitis, and cancer [9]. One of the causes of these health consequences is the toxic effect of acetaldehyde, which comes from the metabolization of ethanol [10]. As an example, alcohol and acetaldehyde act as direct toxins to the cardiac myocytes, resulting in contractile dysfunction [11]. Furthermore, excessive exposure to alcohol can lead to severe debilitating diseases of the central and peripheral nervous systems [12].
Among marijuana health impacts, disturbances in the level of consciousness, cognition, perception, affect or behaviour, and other psychophysiological functions and responses are known as short-term effects. Additionally, long-term marijuana consumption can increase the risk of dependence, cognitive impairment, mental disorders (psychoses, depression, anxiety, and suicidal behaviour), and adverse physical health effects such as cardiovascular disease, chronic obstructive pulmonary disease, and cancer [2]. The main components of the cannabis plant are delta-9-tetrahydrocannabinol (THC), which is psychoactive, and cannabidiol (CBD), which is non-psychoactive. These compounds participate in the endocannabinoid system [13].
In summary, the consumption of tobacco, alcohol, and marijuana can lead to similar adverse effects on health, with the neurological and cardiovascular systems being particularly affected. Recent research suggests that epigenetics is a potential mediator between the consumption of toxic substances and the increase in common disease risk [14–17]. DNA methylation, the most studied epigenetic modulation, consists of the addition of a methyl group (−CH3) in the cytosine nucleotide without changing the DNA sequence. It occurs in the context of CpG sites, which are defined as adjacent cytosine and guanine nucleotides by a phosphate group. DNA methylation is dynamic and can be modified by genetic factors, disease, environmental exposures, and lifestyle [17–19]. Moreover, DNA methylation can change during the lifetime and across tissues and cell types [20,21].
Although genetic mechanisms have been the focus of understanding human diseases, the disruption of the epigenetic balance can result in the modulation of gene expression. Consequently, epigenetic disruption can cause several major pathologies, including cancer and cardiovascular disease [14]. Tobacco smoking is one of the exposures with a higher impact on the DNA methylation of smokers [22–25]. Its effect has also been observed in the newborns’ cord blood of mothers who smoked during pregnancy [26–28]. Many studies also demonstrate that alcohol consumption produces methylation changes at the CpG site level [29–34]. Conversely, only a few studies have demonstrated the effects of marijuana consumption on DNA methylation, all of which have shown small effects [35,36].
Hypertension, also known as high blood pressure, is a medical condition in which the blood pressure in the arteries is persistently elevated. It affects one billion people and is the most common risk factor for death worldwide [37]. There are many factors associated with a higher risk of hypertension, including body mass index, tobacco use, physical activity, and alcohol consumption, among others [38]. Light to moderate alcohol consumption seems to protect against hypertension because it decreases systolic and diastolic blood pressure. However, excessive intake accounts for about 16% of cases of hypertension worldwide [39,40]. Cigarette smoking enhances hypertension by inducing cardiovascular mitochondrial oxidative stress [41,42]. On the other hand, some studies evaluating the effect of marijuana consumption on blood pressure have revealed different results. For instance, Abuhasira et al. demonstrated the therapeutic effect of marijuana in reducing blood pressure in hypertensive patients [43]. However, other studies revealed an increase in blood pressure after marijuana consumption [44,45]. In light of the above, we hypothesized that changes in DNA methylation produced by substance consumption may partially explain its relationship with hypertension.
To this end, in this study we aimed to: i) perform a genome-wide association study of DNA methylation with tobacco, alcohol, and marijuana consumption; ii) identify the physiological pathways whose methylation is affected by those recreational drugs; iii) evaluate the mediation between substance consumption and hypertension by methylation changes at the CpG site level.
Materials and methods
The study cohort
Our study sample included 3,590 individuals from the TruDiagnostic DNA biobank recruited between October 2020 and February 2022. Those individuals have taken the commercial TruDiagnostic TruAge test and methylation data was generated from them. This is an EEUU population-based cohort aged between 13 and 97 years old. Among them, 58.7% are male. Demographic and substance use characteristics of the samples that met the QC requirements (N = 3,424) are displayed in Table 1. As this testing is priced to the consumer at approximately $500, this study cohort is relatively more affluent than random sampling or traditional banked cohorts. Additionally, these individuals may experience a healthy donor effect whereby they seek preventative medicine and have fewer comorbidities than normal patient populations, as is the case with blood donors [46,47]. The study involving human participants was reviewed and approved by the IRCM IRB. The patients/participants provided their written informed consent to participate in this study.
Table 1.
Characteristics of participants in the TruDiagnostic DNA Biobank.
| N = 3424 | |
|---|---|
| Sex, male | 2010 (58.7%) |
| Age in years, mean (range) | 52.9 (13.3–97.8) |
| Ethnicity | |
| European | 2584 (75.5%) |
| African American or Black | 70 (2.0%) |
| Asian | 41 (1.2%) |
| Latino or Hispanic | 276 (8.1%) |
| Middle Eastern or North African | 76 (2.2%) |
| Native American or Alaska Native | 26 (0.8%) |
| Pacific Islander or Oceanian | 23 (0.7%) |
| Sub-Saharan African | 7 (0.2%) |
| Other | 321 (9.4%) |
| BMI (kg/m2), median (range) | 25.4 (10.1–58.2) |
| Tobacco consumption | |
| None | 3275 (95.6%) |
| Less than 1 cigarette per week | 48 (1.4%) |
| Less than 1 cigarette per day | 25 (0.7%) |
| 1–5 cigarettes per day | 27 (0.8%) |
| 6–10 cigarettes per day | 21 (0.6%) |
| 11–20 cigarettes per day | 20 (0.6%) |
| More than 20 cigarettes per day | 8 (0.2%) |
| Alcohol consumption | |
| Never | 634 (18.5%) |
| On special occasions | 976 (28.5%) |
| Once per week | 578 (16.9%) |
| 3–5 times per week | 794 (23.2%) |
| Regularly | 442 (12.9%) |
| Marijuana consumption | |
| Missing | 149 |
| Never | 2908 (88.8%) |
| On special occasions | 180 (5.5%) |
| Once per week | 46 (1.4%) |
| 3–5 times per week | 73 (2.2%) |
| Regularly | 68 (2.1%) |
Note: All the continuous variables are shown as mean (range) and the categorical variables as n (%).
BMI: body mass index.
DNA methylation Assessment
Peripheral whole blood was collected by the lancet and capillary method into lysis buffer and DNA extract, and 500 ng of DNA of bisulphite were converted using the EZ DNA Methylation kit (Zymo Research) according to the manufacturer’s instructions. Bisulphite-converted DNA samples were randomly assigned to a chip well on the Infinium HumanMethylationEPIC BeadChip, amplified, hybridized onto the array, stained, washed, and imaged with the Illumina iScan SQ instrument to obtain raw image intensities.
Meffil R package [48] was used for the pre-processing of DNA methylation data. In the sample quality control, we removed the sex detection mismatches and the sex detection outliers (based on the difference between median chromosome Y and chromosome X probe intensities). We also discarded those samples whose predicted median methylated signal was more than 3 standard deviations from the expected. We excluded the outliers based on deviations from mean values for control probes (dye bias, bisulphite 1, and bisulphite 2). Finally, we removed those samples with more than 5% of undetected probes (detection P-value larger than 0.01) or with a low number of beads (less than 3). This quality control resulted in 3,424 individuals, indicating that 90,3% of the samples met our QC standards. In the feature quality control, we removed those probes undetected or with low bead numbers in more than 5% of the samples. We used InfiniumAnnotation [49] to filter probes where the 30bp 3’-subsequence of the probe is non-unique, probes with INDELs, probes with extension base inconsistent with specified colour channel (type-I) or CpG (type-II) based on mapping, probes with a SNP in the extension base that causes a colour channel switch from the official annotation, and probes where 5bp 3’-subsequence overlap with any of the SNPs with global population frequency higher than 1%. The functional normalization method was further applied based on the first 10 principal components of the control probes. Consequently, the number of CpG probes kept was 745,150, which represents 86% of the total EPIC array manifest. CpG sites were annotated to genes using EPIC Illumina annotation ilm10b4.hg19. Blood cell type proportions were estimated using the blood gse35069 complete cell type methylation profile references from the meffil package. We then performed a surrogate variable analysis (SVA) to remove the batch effects using the SmartSVA package [50]. We estimated the number of surrogate variables (SVs) using the isva package [51]. Methylation levels were expressed as residual values after adjusting beta values for the first 60 SVs.
Exposure and clinical history assessment
During the recruitment of participants, they were asked to complete a survey that included questions about personal information, medical history, social history, lifestyle, and family history. Alcohol and marijuana consumption was assessed on a 5-point scale (‘never’ to ‘regularly’). Participants also reported their level of smoking according to 7 possible answers (‘none’ to ‘more than 20 cigarettes per day’). Regarding the medical history, the survey covered information about the blood type, medications and supplements, and diagnosis of any type of disease (cardiovascular, respiratory, skin and hair, endocrine, gastrointestinal, genitourinary, musculoskeletal, neuropsychological, reproductive, immune, and cancer). The main clinical outcome of this study was hypertension, assessed as a dichotomic variable (affected and unaffected.
Statistical analyses and reproducibility
Epigenome-wide association analysis
The epigenome-wide association study (EWAS) was performed using the MEAL Bioconductor package [52]. We performed a differential mean analysis on different substance consumption (tobacco, alcohol, and marijuana) using the function runPipeline that calls limma [53]. Based on previous analyses [32], we adjusted all the regression models by sex, age, ethnicity, body mass index (BMI), level of education, alcohol and tobacco consumption (except when they were the variable of interest), slide, cell type, and surrogate variables. For each substance, we fitted models
| (1) |
where Ej denotes the methylation level vector across individuals at probe j (j = 1, … 745150), S is the individuals’ consumption (separated models for alcohol, smoking, and marijuana where fitted) with its associated effect, βj, Cr is the r adjusting covariate and its effect γr, and εj is the noise that follows the distribution of methylation levels with mean 0. Adjusted P-values were calculated using Bonferroni’s correction for considering multiple comparisons. The inflation or deflation of P-values across the methylome was assessed with Q-Q plots and lambda values [54].
Enrichment analysis
We performed an enrichment in Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) pathways [55,56] using the enrichKEGG and enrichGO functions from the clusterProfiler Bioconductor package [57], respectively. Using the same package, we also evaluated the over-representation of diseases using the DisGeNET platform which contains 1,134,942 gene-disease associations [58]. Associations were corrected for multiple comparisons using Bonferroni adjustment, as computed by clusterProfiler.
Comparison of the results with previous studies
For the validation of our results, we used publicly available data from the EWAS catalogue. This catalogue includes published epigenome-wide association studies from PubMed using the search terms ‘epigenome-wide’ OR ‘epigenome wide’ OR ‘EWAS’ OR ‘genome-wide AND methylation’ OR ‘genome wide AND methylation.’ Studies are selected according to the year of publication (>2010), the use of human samples, and the number of genome-wide CpG sites (>100,000) and individuals (>100). Reported CpG sites have nominal P-values lower than 1·10−4. The data for the EWAS Catalogue is then manually curated. We obtained separate datasets for each drug of abuse containing information on the associations between DNA methylation and tobacco, alcohol, and marijuana consumption. The dataset for tobacco smoking included 30 publications, while the dataset for alcohol consumption had 6 publications, and the dataset for marijuana consumption contained 2 publications. After excluding associations that lacked information on the beta value, we obtained 26 publications for smoking, no publications for alcohol consumption, and 1 publication for marijuana consumption. For CpG sites that were reported in more than one study, we performed a metanalysis using the meta package [59]. However, due to the high heterogeneity between studies, we reported the proportion of studies that showed the same direction of effect as our study.
Estimation of epigenetic clocks
We further evaluated whether the three behaviours had an impact on epigenetic age. To calculate the principal component-based epigenetic clock for the Horvath multi-tissue clock, Hannum clock, PhenoAge clock, GrimAge clock, and telomere length we used the custom R script available via GitHub (https://github.com/MorganLevineLab/PC-Clocks). Non-principal component-based (non-PC) Horvath and Hannum epigenetic estimates were calculated using the agep function available in the wateRmelon R package [60], and non-PC PhenoAge was calculated using the methyAge function in the ENMix R package [61]. Finally, the pace of ageing clock, DunedinPACE, was calculated using the PACEProjector package available via GitHub (https://github.com/danbelsky/DunedinPACE).
Mediation analysis between recreational drug use and hypertension
To investigate the relationship between tobacco, alcohol, and marijuana consumption and hypertension, we conducted logistic regression analyses. Next, we performed a mediation analysis to explore whether methylation played a role in the relationship between recreational drug use and hypertension. We first identified CpG sites that were significantly associated with those behaviours and hypertension after correcting for multiple comparisons using Bonferroni. Then, we conducted a univariate mediation analysis for each significant CpG site using the mediate function from the mediation package [62]. This analysis allowed us to estimate the total effect, the effect of substance consumption on CpG methylation, and the effect of the mediator and substance consumption on hypertension. Finally, we performed a causal mediation analysis and estimated the average causal mediation effects, average direct effects, total effect of the independent variable on the dependent variable, and the proportion of the effect of the independent variable on the dependent variable that goes through the mediator. We adjusted all models for the same covariates as in the EWAS.
Results
We analysed 3,424 individuals from the TruDiagnostic DNA Biobank recruited from the general population in EEUU. Table 1 presents demographic and substance use characteristics. The mean age was 52.9 years (range: 13.3–97.8) and 58.7% were male. The participants were classified according to 7 ethnic groups and ‘other’ for those who had a mixed ethnicity. Most participants were Europeans (75.5%), and Latino American was the second most common ethnicity (8.1%). There were 149 current tobacco smokers, classified into seven groups according to the number of cigarettes smoked, and 3,275 non-smokers. Regarding alcohol consumption, there were 2,790 drinkers grouped by consumption frequency and 634 non-drinkers. Marijuana consumption was also classified according to consumption frequency. In total, 2,908 did not smoke marijuana, and 367 smoke marijuana at least on special occasions. It is worth noting that out of the participants, 465 individuals reported consuming more than one drug, at least on special occasions. On the other hand, 584 participants reported not consuming any of the drugs included in this study. Among those who reported using recreational drugs, the largest overlap was observed between alcohol and marijuana, with 299 individuals indicating consumption of both substances. Notably, 98 participants reported consuming both tobacco and alcohol, highlighting the co-occurring use of different substances (Additional File 2: Fig. S1).
Genome-wide effect of tobacco smoking on DNA methylation
We tested the association between the level of smoking (codified as a 7-point scale from ‘none’ to ‘more than 20 cigarettes per day;’ see Table 1) with each CpG site using linear regression models run in the MEAL R package [52]. We found 528 CpG sites associated with smoking levels after correcting for multiple comparisons and genomic inflation was not observed (λ = 1.031). Table 2 lists the top 15 CpG sites for tobacco smoking (see Additional File 1: Table S1 for all the significant CpG sites). Figure 1a shows how the CpG sites are distributed in the genome using a Manhattan plot. Among tobacco-related methylation sites, 68.2% were hypomethylated (that is, lower DNA methylation associated with higher tobacco consumption). From the 528 probes differentially methylated, 374 CpG sites were mapped to 344 unique genes. AHRR, GFI1, PRSS23, and IMMP2L had 10, 6, 4, and 4 probes differentially methylated, respectively. Moreover, these 344 genes were enriched in morphine addiction (P-value = 4.3 · 10−6), dopaminergic synapse (P-value = 1.1 · 10−4), and cholinergic synapse (P-value = 1.7 · 10−4) KEGG pathways (Additional File 2: Fig. S2). Consistent with previous studies, cg05575921 was the top-ranked CpG with a P-value = 1.3·10−226. We further demonstrated that the effect of tobacco in this CpG site was proportional to the number of cigarettes smoked (Figure 2a). In addition, we cross-referenced our findings with those previously documented in the EWAS catalogue. Out of the 528 CpG sites that displayed differential methylation in our investigation, 183 had already been documented in the EWAS catalogue, with 181 showing the same direction of change and 2 exhibiting an opposing direction. Among the 345 not-reported, 6 of them were in the top 15 CpG sites in our data, evidencing that tobacco may have important effects on them (Table 1).
Table 2.
Top 15 differentially methylated CpG sites by tobacco consumption.
| CpG | chr | position | Gene Symbol | Gene Group | Less than 1 per week | Less than 1 per day | 1–5 per day | 6–10 per day | 11–20 per day | More than 20 per day | F | P-Value | Adjusted P-Value | EWAS catalogue (same direction/total studies) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cg05575921 | chr5 | 373378 | AHRR | Body | −0.028 | −0.054 | −0.097 | −0.216 | −0.183 | −0.217 | 208.2 | 1.3E–226 | 9.7E–221 | 26/26 |
| cg21566642 | chr2 | 233284661 | −0.030 | −0.058 | −0.066 | −0.109 | −0.111 | −0.160 | 59.7 | 1.6E–70 | 5.9E–65 | 22/22 | ||
| cg01940273 | chr2 | 233284934 | −0.016 | −0.034 | −0.045 | −0.066 | −0.069 | −0.109 | 53.6 | 2.4E–63 | 5.9E–58 | 22/22 | ||
| cg03636183 | chr19 | 17000585 | F2RL3 | Body | −0.007 | −0.017 | −0.029 | −0.064 | −0.053 | −0.094 | 50.9 | 2.8E–60 | 5.2E–55 | 21/21 |
| ch.6.2768623F | chr6 | 144037285 | PHACTR2 | Body | −0.002 | −0.001 | 0.000 | 0.009 | −0.005 | 0.350 | 49.1 | 4.7E–58 | 7.0E–53 | Not reported |
| cg21161138 | chr5 | 399360 | AHRR | Body | −0.008 | −0.016 | −0.015 | −0.071 | −0.043 | −0.069 | 40.6 | 5.7E–48 | 7.1E–43 | 16/16 |
| cg17739917 | chr17 | 38477572 | RARA | 5‘UTR | −0.008 | −0.053 | −0.034 | −0.069 | −0.082 | −0.116 | 38.2 | 4.5E–45 | 4.8E–40 | 7/7 |
| cg15928392 | chr2 | 81694446 | 0.000 | 0.000 | 0.001 | −0.001 | −0.002 | −0.106 | 34.1 | 3.4E–40 | 3.2E–35 | Not reported | ||
| cg16774290 | chr7 | 4824628 | AP5Z1 | Body | 0.000 | 0.000 | 0.000 | −0.001 | 0.000 | −0.100 | 33.2 | 4.1E–39 | 3.4E–34 | Not reported |
| cg11173636 | chr10 | 65632259 | −0.008 | 0.017 | 0.024 | 0.018 | 0.339 | −0.020 | 30.5 | 8.5E–36 | 6.3E–31 | Not reported | ||
| cg09935388 | chr1 | 92947588 | GFI1 | Body | −0.018 | −0.030 | −0.029 | −0.100 | −0.069 | −0.134 | 30.0 | 3.5E–35 | 2.4E–30 | 20/20 |
| cg26703534 | chr5 | 377358 | AHRR | Body | −0.004 | −0.013 | −0.016 | −0.052 | −0.034 | −0.049 | 29.9 | 4.0E–35 | 2.4E–30 | 14/14 |
| cg14990808 | chr6 | 28493651 | GPX5 | TSS200 | −0.002 | −0.022 | 0.005 | 0.002 | 0.001 | 0.246 | 26.3 | 1.1E–30 | 6.2E–26 | Not reported |
| cg25648203 | chr5 | 395444 | AHRR | Body | −0.006 | −0.005 | −0.011 | −0.047 | −0.037 | −0.057 | 24.8 | 6.2E–29 | 3.3E–24 | 18/18 |
| cg04176674 | chr14 | 21121564 | −0.004 | −0.002 | 0.001 | 0.005 | 0.000 | 0.343 | 24.7 | 7.8E–29 | 3.9E–24 | Not reported |
Note: The CpG sites are annotated based on the chromosome (chr), the position (pos), the gene symbol from HGNC, and the gene group (based on the position of the CpG regarding the nearest gene). Each CpG site has a beta value for each consumer group vs non-consumers, a F-statistic (F), a nominal p-value, and an adjusted p-value by Bonferroni. The last column represents the number of studies in the EWAS catalogue that reported the same effect direction compared to the total studies that reported the beta value for the specific CpG site.
Figure 1.
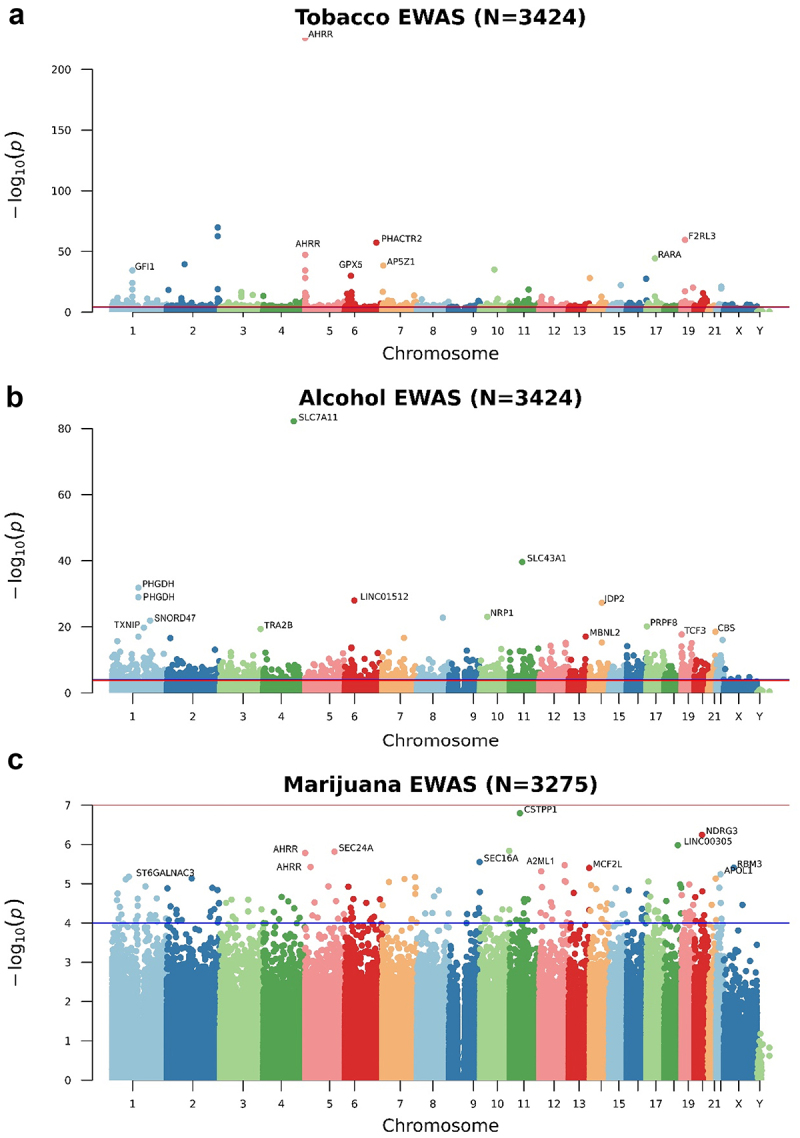
Manhattan plots of the epigenome-wide association study (EWAS) of tobacco (a), alcohol (b), and marijuana (c) consumption. The Y-axis represents the -log10(p) values and the X-axis the position of the CpG sites within the chromosome. The blue line is the suggestive nominal p-value threshold (0.0001) and the red line is the p-value adjusted threshold lower than 0.05.
Figure 2.
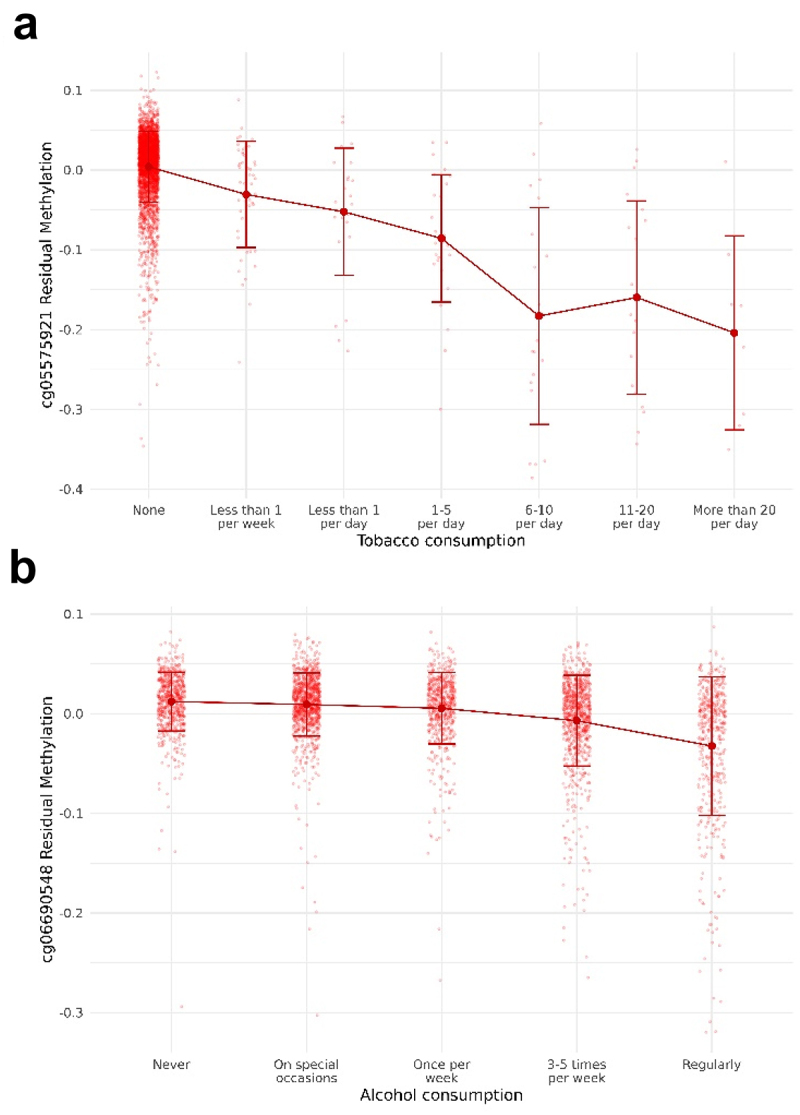
Boxplots showing the association between CpG methylation and substance consumption. (a) Association between cg05575921 methylation (AHRR) and tobacco consumption. (b) Association between cg06690548 methylation (SLC7A11) and alcohol consumption. The Y-axis represents the residuals for beta values after adjusting by covariates. The X-axis represents the number of cigarettes smoked and the frequency of drinking, respectively. Methylation means for each tobacco consumption level are represented with their 95% confidence intervals.
Genome-wide effect of alcohol consumption on DNA methylation
We identified 2,569 CpG sites differentially methylated according to alcohol consumption frequency (5 levels from ‘never’ to ‘regularly;’ see Table 1). Model fitting showed no indication of genomic inflation (λ = 1.044). The top 15 CpG sites are shown in Table 3 and all the epigenome-wide significant CpG sites are listed in Additional File 1: Table S3 and represented as a Manhattan Plot in Figure 1b. Among them, 36.9% were hypomethylated for regular consumers compared with non-consumers. However, the percentage of hypomethylated probes was increased for the most significant probes. Among the 2,569 alcohol-related methylation sites, 609 were intergenic and 1,960 were annotated to 1,670 unique genes. Five genes had seven or more significant probes mapping to their locus, including RPTOR (11 probes), JARID2 [8], and ABCG1 [8]. The enrichment revealed an over-representation of autistic disorder (P-value = 4.2 · 10−7), acquired scoliosis and curvature of the spine (P-value = 2.7 · 10−4), small nose (P-value = 2.5 · 10−4), small midface (P-value = 6.1 · 10−5), and self-injurious behaviour (P-value = 1.5 · 10−4) (Additional File 2: Fig. S3A). These genes were also enriched in the PI3k-Akt signalling pathway (P-value = 4.0 · 10−4), which is involved in the cell cycle, cholinergic synapse (P-value = 2.7 · 10−5), and longevity regulating pathway (P-value = 6.7 · 10−5) (Additional File 2: Fig. S3B). The GO enrichment revealed regulation of Wnt signalling pathway (P-value = 3.2 · 10−8) and heart growth (P-value = 2.6 · 10−5) (Additional File 2: Fig. S3C). The top CpG site (cg06690548, P-value = 5.9·10−83) mapped to the SLC7A11 gene and its methylation was significantly reduced proportionally to the alcohol consumption (Figure 2b). Due to the absence of beta value information in the studies included in the EWAS catalogue, we compared our results with the largest single-cohort EWAS of alcohol consumption performed by Lohoff et al [31]. In this study, they found 2,504 CpG sites and 909 were overlapping with our study. All the CpG sites overlapping had the same direction of the effect.
Table 3.
Top 15 differentially methylated CpG sites by alcohol consumption.
| CpG | chr | position | Gene Symbol | Gene Group | On special occasions | Once per week | 3–5 times per week | Regularly | F | P-Value | Adjusted P-Value | Lohof et al (direction) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cg06690548 | chr4 | 139162808 | SLC7A11 | Body | −0.003 | −0.005 | −0.015 | −0.031 | 103.1 | 5.9E–83 | 4.4E–77 | – |
| cg11376147 | chr11 | 57261198 | SLC43A1 | Body | −0.001 | −0.002 | −0.006 | −0.010 | 49.3 | 2.4E–40 | 8.9E–35 | – |
| cg26457483 | chr1 | 120256112 | PHGDH | Body | −0.001 | −0.004 | −0.017 | −0.028 | 39.7 | 1.5E–32 | 3.8E–27 | – |
| cg14476101 | chr1 | 120255992 | PHGDH | Body | −0.002 | −0.005 | −0.016 | −0.031 | 36.2 | 1.1E–29 | 2.0E–24 | – |
| cg18120259 | chr6 | 43894639 | LINC01512 | Body | −0.002 | −0.004 | −0.009 | −0.019 | 35.0 | 1.1E–28 | 1.6E–23 | – |
| cg06088069 | chr14 | 75895604 | JDP2 | 5‘UTR | −0.002 | −0.003 | −0.007 | −0.013 | 34.2 | 5.0E–28 | 6.1E–23 | – |
| cg21998542 | chr10 | 33605101 | NRP1 | Body | −0.002 | −0.005 | −0.009 | −0.019 | 29.0 | 9.2E–24 | 9.8E–19 | – |
| cg15837522 | chr8 | 117892654 | −0.003 | −0.011 | −0.022 | −0.031 | 28.7 | 1.7E–23 | 1.6E–18 | – | ||
| cg06644515 | chr1 | 173834831 |
SNORD47;GAS5; SNORD81;SNORD80; SNORD78;SNORD79 |
TSS1500;Body; TSS1500;TSS1500; TSS200;TSS1500 |
−0.001 | −0.001 | −0.007 | −0.013 | 27.6 | 1.3E–22 | 1.1E–17 | – |
| cg12116137 | chr17 | 1576449 | PRPF8 | Body | 0.001 | 0.001 | 0.008 | 0.018 | 25.5 | 7.5E–21 | 5.5E–16 | ++ |
| cg19693031 | chr1 | 145441552 | TXNIP | 3‘UTR | −0.004 | −0.003 | −0.011 | −0.026 | 25.1 | 1.8E–20 | 1.2E–15 | – |
| cg12825509 | chr3 | 185648568 | TRA2B | Body | 0.000 | −0.004 | −0.010 | −0.016 | 24.6 | 4.5E–20 | 2.8E–15 | – |
| cg14346162 | chr21 | 44490229 | CBS | Body | 0.001 | 0.004 | 0.009 | 0.014 | 23.5 | 3.1E–19 | 1.8E–14 | Not reported |
| cg12973487 | chr19 | 1623075 | TCF3 | Body | 0.004 | 0.005 | 0.007 | 0.014 | 22.6 | 2.0E–18 | 1.0E–13 | ++ |
| cg05713943 | chr13 | 97912352 | MBNL2 | 5‘UTR | 0.001 | 0.000 | −0.005 | −0.008 | 21.8 | 8.7E–18 | 4.2E–13 | – |
Note: The CpG sites are annotated based on the chromosome (chr), the position (pos), the gene symbol from HGNC, and the gene group (based on the position of the CpG regarding the nearest gene). Each CpG site has a beta value for each consumer group vs non-consumers, a F-statistic (F), a nominal p-value, and an adjusted p-value by Bonferroni. We also included one column with the direction of the effect in a previous study (Lohoff et al (31)) for those that were significant in their analysis.
Genome-wide effect of marijuana consumption on DNA methylation
In the EWAS for the frequency of marijuana use (codified as a 5-point scale from ‘never’ to ‘regularly;’ see Table 1), we did not find any CpG site with a P-value adjusted lower than 0.05 (Figure 1c). However, we used the P-value threshold from the EWAS catalogue (P-value < 1·10−4) and we found 195 CpG sites at a suggestive significant level (Additional File 1: Table S3). From them, almost 50%were hypomethylated for regular consumption compared to no consumption (Table 4). Gene symbols for the 195 CpG sites were tested for enrichment in KEGG pathways and Gene Ontology (GO) and we found an enrichment of myelin assembly (P-value = 8.0·10−6). We did not find CpG sites overlapping between our results and the ones reported in the EWAS catalogue. This may be in part due to the differences in the variable of interest and the study population, such as the evaluation of the effect of cannabis use on non-Hispanic white women and the risk of breast cancer [36].
Table 4.
Top 15 differentially methylated CpG sites by marijuana consumption.
| CpG | chr | position | Gene Symbol | Gene Group | On special occasions | Once per week | 3–5 times per week | Regularly | F | P-Value | Adjusted P-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| cg05107281 | chr11 | 47072710 | CSTPP1 | Body | −0.004 | −0.010 | −0.014 | 0.001 | 9.4 | 1.6E–07 | 0.12 |
| cg06499565 | chr20 | 35374747 | NDRG3 | TSS1500 | 0.001 | 0.006 | 0.000 | −0.001 | 8.7 | 5.7E–07 | 0.17 |
| cg10054857 | chr18 | 61816543 | LINC00305 | TSS1500 | −0.004 | −0.007 | −0.002 | −0.002 | 8.4 | 1.0E–06 | 0.17 |
| cg24344693 | chr10 | 133273964 | −0.004 | 0.003 | −0.001 | −0.002 | 8.2 | 1.5E–06 | 0.17 | ||
| cg20226924 | chr5 | 133985272 | SEC24A | Body | 0.001 | 0.003 | 0.003 | 0.001 | 8.2 | 1.5E–06 | 0.17 |
| cg05575921 | chr5 | 373378 | AHRR | Body | −0.003 | −0.011 | −0.012 | −0.020 | 8.1 | 1.7E–06 | 0.17 |
| cg21161138 | chr5 | 399360 | AHRR | Body | −0.004 | −0.011 | −0.009 | −0.012 | 8.1 | 1.7E–06 | 0.17 |
| cg19730404 | chr9 | 139361517 |
SEC16A; SEC16A |
ExonBnd;Body | −0.003 | −0.001 | 0.001 | −0.004 | 7.8 | 2.8E–06 | 0.25 |
| cg19308363 | chr12 | 116290566 | 0.006 | 0.005 | −0.005 | −0.015 | 7.7 | 3.4E–06 | 0.25 | ||
| cg08258765 | chr5 | 24841586 | LOC340107 | TSS1500 | 0.005 | 0.002 | 0.004 | −0.002 | 7.7 | 3.8E–06 | 0.25 |
| cg03838168 | chrX | 48433876 | RBM3 | Body | −0.003 | −0.004 | 0.015 | 0.005 | 7.7 | 3.9E–06 | 0.25 |
| cg16822035 | chr13 | 113633379 | MCF2L;MCF2L | Body;TSS1500 | 0.001 | 0.004 | 0.009 | 0.012 | 7.6 | 4.0E–06 | 0.25 |
| cg11756734 | chr12 | 9028945 | A2ML1 | 3‘UTR | −0.005 | 0.002 | −0.007 | 0.003 | 7.5 | 4.8E–06 | 0.26 |
| cg08415592 | chr22 | 36648973 | APOL1 | TSS200 | 0.008 | 0.016 | −0.004 | 0.007 | 7.4 | 5.8E–06 | 0.26 |
| cg17325792 | chr1 | 77042560 |
ST6GALNAC3; ST6GALNAC3 |
3‘UTR;Body | 0.003 | 0.001 | 0.003 | −0.011 | 7.4 | 6.6E–06 | 0.26 |
Note: The CpG sites are annotated based on the chromosome (chr), the position (pos), the gene symbol from HGNC, and the gene group (based on the position of the CpG regarding the nearest gene). Each CpG site has a beta value for each consumer group vs non-consumers, a F-statistic (F), a nominal p-value, and an adjusted p-value by Bonferroni.
Comparison between recreational drugs
We compared the differentially methylated CpG sites for the different drugs of abuse. Since no CpG sites were significant for marijuana consumption after correcting by multiple comparisons, we focused on comparing the results between tobacco and alcohol. We found that 12 CpG sites were overlapping between tobacco and alcohol, and nine of them had the same direction effect in both analyses (Figure 3a). Next, we extracted the genes where all the differentially methylated CpG sites were annotated and compared the alcohol and tobacco related genes. We observed a larger overlap and found that 61 genes were differentially methylated by both substances (Figure 3b). To determine whether these genes were involved in similar pathways, we performed an enrichment analysis, and the results revealed that these genes were implicated in various biological processes, including positive regulation of heart rate (P-value = 5.3·10−5), inositol lipid-mediated signal (P-value = 1.7·10−4), and positive regulation of blood circulation (P-value = 1.9·10−4), among others (Additional File 2: Fig. S4).
Figure 3.
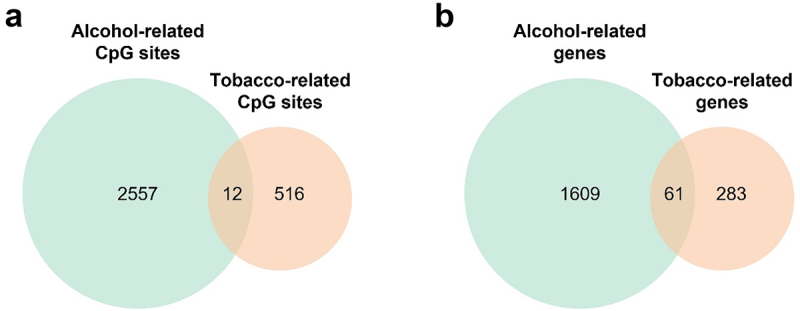
Venn diagrams comparing CpG sites and genes differentially methylated for tobacco and alcohol use. (a) Comparison between differentially methylated CpG sites with a P-value adjusted lower than 0.05. (b) Comparison between genes where the CpG sites differentially methylated are annotated.
Then, we evaluated whether recreational drug consumption was associated with multiple epigenetic clocks. For tobacco smoking, we found a significant association with telomere length (β = −0.01, P-value = 1.2·10−3), Grim Age Principal Component (β = 0.72, P-value = 1.6·10−17), and DunedinPACE (β = 0.01, P-value = 9.4·10−5). Alcohol consumption was associated also with a lower telomere length (β = −0.004, P-value = 7.5·10−3) and Grim Age Principal Component (β = 0.13, P-value = 7.7·10−4). Finally, marijuana consumption was not associated with any epigenetic clock.
Mediation between substance consumption and hypertension by CpG methylation
We evaluated whether the changes at the CpG methylation level mediated the effect of recreational drugs on hypertension. We first tested the association between smoking and hypertension. We considered that the group with the highest levels of smoking were those who smoked more than 11 cigarettes per day, joining the categories 11–20 and >20 cigarettes due to their low number of participants in each. We evaluated the association between smoking codified as numeric and high blood pressure as a binary variable and we did not detect a significant tendency (P-value = 0.26). The forest plot shows that the risk of hypertension increases with a higher number of cigarettes smoked except for the last group (more than 11 cigarettes per day) (Additional File 2: Fig. S5). Although we expected this group to be the one at the highest risk, we also observed that those participants were also the youngest (average of 5.75 years less, P-value = 0.005), suggesting a particularly strong healthy donor effect for this group. We tested the association after removing this group and we found a significant association between tobacco smoking and hypertension (P-value = 0.009, OR = 1.28). The forest plot in Figure 4a revealed a clear dose-response relationship where individuals who consume 6 to 10 cigarettes per day have 3.19 times of high blood pressure risk compared with non-smokers (P-value = 0.023). For the mediation analysis using CpG sites as mediators, we selected those CpG sites that were significantly associated with hypertension and tobacco smoking. The analysis revealed 10 CpG sites that were potential mediators. However, the univariate mediation analyses showed that none of those CpG sites was significantly mediating the effect of tobacco on hypertension with a FDR lower than 0.05 (Additional File 1: Table S4).
Figure 4.
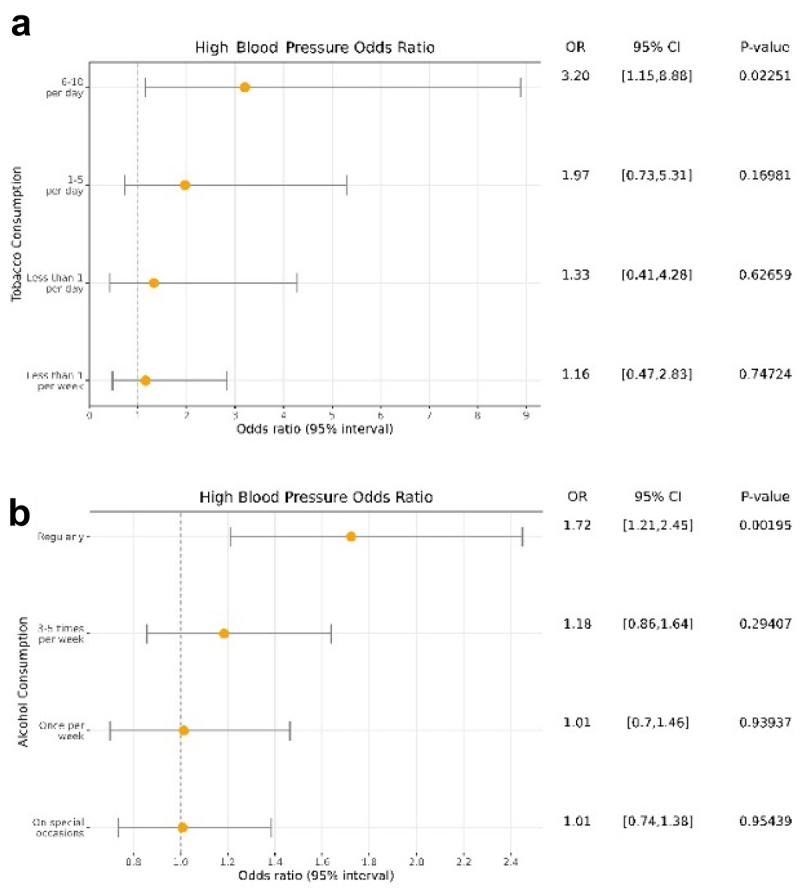
Forest plot of the association between tobacco (a) and alcohol (b) consumption with hypertension. OR: Odds Ratio.
We also tested the association between marijuana consumption and high blood pressure. We did not find any significant association when comparing the 4 levels of consumption (from ‘on special occasions’ to ‘regularly’) with non-consumers (Additional File 2: Fig. S6).
As for alcohol use, we found a significant association between alcohol and higher blood pressure (P-value = 0.001, OR = 1.13). In addition, the forest plot in Figure 4b revealed a significant association between daily consumption with high blood pressure (P-value = 0.014, OR = 1.39) and a non-significant association between light to moderate consumption with the phenotype, as expected. Thus, we evaluated whether DNA methylation was mediating this association. We first performed logistic regression between the alcohol-related CpG sites and hypertension and found 76 significant CpG sites after correcting by multiple comparisons. To see the effect of each CpG site, we performed a univariate mediation analysis for each of these probes. Among them, 66 CpG sites significantly mediated the association between alcohol and hypertension with estimated proportions ranged between 3 and 71%. The most differentially methylated CpG site by alcohol consumption, cg06690548, was also the most significant mediator between alcohol consumption and high blood pressure after adjusting by covariates. This CpG site mediated 71% of the total effect of alcohol on the phenotype (FDR = 0.019). Furthermore, we observed that 12.8% of the hypertension variance was explained by alcohol consumption after adjusting by covariates. The variance explained was increased to 13.6% after including the cg06690548 methylation in the model, and to 20.5% after including the 66 significant CpG sites. In these models, the association between hypertension and alcohol was no longer significant, further suggesting that methylation has an important mediation role in hypertension risk.
Discussion
The current study evaluated the effect of tobacco, alcohol, and marijuana consumption on genome-wide DNA methylation in 3,424 individuals from an EEUU population-based cohort. We identified 528 CpG sites differentially methylated according to tobacco smoking, 2,569 according to alcohol consumption, and no significant associations for marijuana consumption. Second, we detected a large overlapping between the differentially methylated genes by alcohol and tobacco consumption. Third, we found a significant mediation between alcohol consumption and hypertension by many alcohol-related methylation sites.
A considerable amount of literature has been published on DNA methylation changes due to smoking. The first studies evaluating these changes were carried out in single genes or small panels of genes [63–65]. It was not until 2012 that the first epigenome-wide association study on tobacco was reported by Wan et al. [66]. Since that time, several studies have demonstrated the huge impact of tobacco on DNA methylation across the human genome, even in newborns when the tobacco exposure was during pregnancy [22,23,25–27,67,68]. Our findings were in line with previous research, demonstrating a large number of CpG sites differentially methylated along the genome. We were able to compare our results with the EWAS catalogue, and this revealed a high overlap in the CpG sites differentially methylated. Moreover, 98.9% of these CpG sites had the same direction of the effect. Among the probes that were not previously reported in the EWAS catalogue, it is remarkable the CpG site at PHACTR2 gene, because it is involved in actin cytoskeleton organization and implicated in Parkinson’s disease [69], and the CpG site at GPX5 gene, since it protects cells and enzymes from oxidative damage. Additionally, our results confirm previous observations where the cg05575921 mapped to AHRR (P-value = 9.7·10−221) and the cg21566642 in the 2q37.1 region (P-value = 5.9·10−65) were the most significantly associated CpG sites to tobacco consumption [26–28]. Besides, the enrichment of the CpG sites differentially methylated revealed remarkable findings. Dopaminergic and cholinergic synapses are two systems that are affected by nicotine, the psychoactive substance found in tobacco. On one hand, nicotine activates the release of dopamine, inducing feelings of pleasure and reward [70]. This activation is one of the main reasons for which nicotine is so addictive. On the other hand, nicotine binds to receptors in the cholinergic synapse, enhancing the release of acetylcholine [71]. This neurotransmitter can improve cognition, attention, and memory. However, chronic exposure to nicotine can lead to a decrease of the numbers of receptors, which can produce cognitive impairment and other negative effects in the brain. Thus, the CpG sites differentially methylated by tobacco may be used as biomarkers to monitor addiction and neurotoxic effects on tobacco smokers.
Alcohol is known to affect DNA methylation. To date, several EWAS have detected CpG sites associated with alcohol consumption [29,31–33,72,73]. Here, we identified 2569 probes associated with drinking. As we found, many studies detected cg06690548 mapped at the SLC7A11 promoter as the most alcohol-related methylation site [29,31,32]. Furthermore, Lohoff et al. demonstrated that various liver biomarkers were robustly associated with SLC7A11 methylation status [31], suggesting an implication of this gene in the disturbance of the gastrointestinal system when consuming alcohol. In addition, we compared our results with those reported in study where they also performed an EWAS for alcohol consumption in 8,161 individuals. We found that 909 probes overlapped, all showing the same direction of effect. We observed that certain probes did not overlap, which could be attributed to differences in study design. Specifically, Lohoff et al used information on alcohol consumption during the previous week, while our study assessed general alcohol consumption habits. As a result, these discrepancies suggest that the timing and specificity of the alcohol consumption measure used in different studies can influence epigenetic changes. Besides, the genes differentially methylated in our study were highly enriched in autistic disorder, acquired scoliosis, curvature of the spine, small nose, and small midface. Engagingly, all these features are symptoms of the foetal alcohol spectrum disorder (FASD), which encompasses the range of adverse effects associated with alcohol exposure during pregnancy [74–77]. In addition, previous studies have already investigated the epigenetic mechanism linking autism and FASD [78]. Our results suggest that DNA methylation changes are important contributors to the relationship between alcohol consumption and FASD. Future studies should examine whether pregnant women with alcohol disorders could be monitored using epigenetic changes to prevent disorders in children.
The studies that have investigated DNA methylation modifications after marijuana consumption are limited. In 2015, Watson et al. evaluated in rats the effect of cannabis parental exposure on the epigenome of the nucleus accumbens [79] and they identified 1027 differentially methylated regions. Five years later, Osborne et al. carried out the first EWAS on heavy cannabis consumption with and without tobacco comparing 48 consumers with 48 controls [35]. They found five differentially methylated sites in cannabis and tobacco users that replicated previous studies on the effects of tobacco. However, cannabis-only users had no evidence of significant differential methylation in any gene. Markunas et al. performed another EWAS with a larger sample size (1,247 ever users) consisting of women at risk of developing breast cancer [36]. They identified a unique significant CpG mapped to CEMIP 5’ region. However, they designed a biomarker for lifetime cannabis use based on the top 50 EWAS CpG sites. In our study, 367 individuals smoked marijuana from occasionally to daily. The EWAS did not reveal significant CpG sites at the Bonferroni adjustment. Nonetheless, the genes where the 195 CpG sites with a P-value lower than 1·10−4 were annotated were enriched in myelin assembly, essential for the proper functioning of the nervous system, as it enables the rapid and efficient transmission of nerve impulses between neurons. This suggests a possible implication of DNA methylation changes on the effects of THC, the active substance in marijuana, in cognitive and behavioural impairments. In addition, we detected cg05575921 (AHRR), the most significant tobacco-associated CpG site, differentially methylated according to marijuana use with a nominal P-value equal to 1.7·10−6. Allen et al. already found that the link between marijuana use and epigenetic age acceleration was statistically mediated via hypomethylation at site cg05575921 [80]. This is consistent with the association of the AHRR gene with exposure to tobacco and fine particulate matter (PM2.5) which suggests that marijuana inhalation can produce similar effects [80,81].
In the current study, we aimed to investigate whether the adverse health effects commonly associated with the use of multiple substances, such as cardiovascular disease, could be attributed to epigenetic alterations. Our findings revealed a significant overlap of 12 CpG sites that displayed differential methylation levels in response to both tobacco and alcohol. Moreover, 61 genes overlapped between these two substances and were enriched in biological processes involved in the proper functioning of the nervous and cardiovascular systems. Therefore, similar epigenetic changes may explain the shared consequences of drug abuse. Targeting these specific pathways could be a strategy for preventing addiction, as well as neurological and cardiovascular disorders. In line with this, we found that tobacco and alcohol consumption were associated with shorter telomere lengths, as reported previously [82–84].
In our data, hypertension was partially associated with tobacco and highly associated with alcohol consumption, as demonstrated previously [39–42]. Marijuana was not associated with hypertension, in line with previous studies that have revealed ambiguous results [43–45]. In the case of tobacco exposure, we found unexpected results because the individuals who smoked the most were the ones who had less risk to develop hypertension. These results may be explained due to the lower age of the individuals in that group and also by the healthy donor effect of the data. This means that participants are volunteers who have paid for the TruAge test and may have healthy habits that protect them against hypertension although they are heavy smokers.
Our data replicated prior studies where light to moderate drinking was not associated with high blood pressure and heavy drinking increased the risk of the disease [8,40]. Another important finding was that 66 CpG sites significantly mediated the effect of alcohol consumption on hypertension. Importantly, the methylation levels of these probes increased the variance explained in hypertension by alcohol from 13.6% to 20.5%. More interestingly, lower methylation levels of cg06690548 at SLC7A11, cg18120259 at LINC01512, and cg19693031 at TXNIP have been seen previously associated with higher systolic and diastolic blood pressure [85–87]. Additionally, hypomethylation of cg06690548 was associated with higher expression of SLC7A11 [87]. SLC7A11 enhances antioxidant defence and protects against endothelial dysfunction and vascular inflammation. This increases vascular tone and rigidity, and consequently blood pressure. Also, Richard et al. evidenced triangular associations between methylation, gene expression, and blood pressure [86]. The univariate mediation based on cg06690548 methylation revealed that 70.5% of the effect of alcohol on high blood pressure was mediated by the CpG methylation level (P-value = 0.006). In essence, we have demonstrated that the effect of heavy drinking on high blood pressure is partially mediated by the methylation of CpG sites that are significantly associated with the disease. This finding provides new insights on targets to prevent and manage hypertension in individuals with regular alcohol consumption.
The generalizability of these results is subject to certain limitations. First, DNA methylation was obtained from blood samples, thus, further research is required to understand the implication of the identified markers in each tissue. Second, genetics has an important role in substance use predisposition. In our analysis, we were not able to remove the genetic factor because of the lack of data. Some of the differentially methylated probes may be a consequence of the genetic differences and not the exposure itself. Notwithstanding this limitation, we filtered all the probes with a SNP in the extension base and all probes where 5bp 3’-subsequence overlapped with any of the SNPs with a global population frequency higher than 1%. Third, the consumption assessment was self-reported and not specific for a time period, limiting credibility and enhancing misclassification. In addition, we did not have information on whether marijuana was smoked mixed or not with tobacco. This information could benefit future studies on removing the tobacco effect. Fourth, we have compared our results with the EWAS catalogue for smoking and with a unique paper for alcohol consumption. It would be useful to compare our results with other published papers.
Our study also had notable strengths, including a large number of drinkers and a high variability in drinking frequency. This allowed us to test the mediation analysis between alcohol consumption and hypertension. Moreover, most studies are focused on evaluating the effects of one substance in drug-specific cohorts. Our data provided information on tobacco, alcohol, and marijuana consumption in the same individuals, along with clinical data. While marijuana consumption may be reported or suffer from healthy donor effect, we observed that fewer individuals smoked tobacco compared with marijuana, yet the effect of tobacco and alcohol on DNA methylation was strong. This suggests that marijuana may have a lower effect on blood DNA methylation compared to other substances. However, further studies with better consumption assessment are needed to confirm this observation.
Conclusions
To the best of our knowledge, this is the first study to assess simultaneously the effect of tobacco, alcohol, and marijuana on DNA methylation. We have shown that tobacco and alcohol have large effects on genome-wide DNA methylation, while marijuana consumption has small effects. Most importantly, many genes differentially methylated by smoking are also affected by alcohol consumption, suggesting a similar epigenetic impact after the consumption of recreational drugs. The results of this research also have significant implications for the understanding of how alcohol consumption increases hypertension. We demonstrated that 66 CpG sites were partially mediating the association between heavy drinking and hypertension and the most alcohol-related CpG site mediated 70.5% of this association. Finally, the current data highlight the importance of using blood methylation biomarkers in clinical practice to detect and monitor the adverse effects, such as addiction, derived from substance consumption.
Supplementary Material
Acknowledgments
We are grateful to all participants and researchers who took part in this study.
Funding Statement
This research has received funding from the Spanish Ministry of Science and Innovation through the “Centro de Excelencia Severo Ochoa 2019-2023 (CEX2018-000806-S) program, and support from the Generalitat de Catalunya through the CERCA Program. NC is supported by the Spanish regional program PERIS (Ref.: SLT017/20/000061), granted by Departament de Salut de la Generalitat de Catalunya. TruDiagnostics also provided funding for data analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are available upon reasonable request due to ensure the privacy of the participants. Please email varun@trudiagnostic.com for data requests. Any custom code or software used in our analysis is available at DOI: 10.5281/zenodo.6417926 (URL: https://zenodo.org/badge/latestdoi/296552532).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2214392
References
- [1].Substance Abuse and Mental Health Services Administration (SAMHSA) . Results from the 2015 national survey on drug use and health: Summary of National Findings [Internet]. 2017. [cited 2022 Jun 27]. Available from: https://www.samhsa.gov/
- [2].World Health Organization . Management of substance abuse team. The health and social effects of nonmedical cannabis use. World Health Organization; 2016. [Google Scholar]
- [3].Substance Abuse and Mental Health Services Administration . Key substance use and mental health indicators in the United States: results from the 2017 national survey on drug use and health. Rockville, MD: Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental HealthServices Administration; 2018. https://www.samhsa.gov/data/ [Google Scholar]
- [4].Geneva: World Health Organization . WHO global report on trends in prevalence of tobacco use 2000-2025, fourth edition. 2021.
- [5].West R. Tobacco smoking: health impact, prevalence, correlates and interventions. Psychol Health Internet. 2017 Aug 3[cited 2022 Jun 20];32(8):1018–20. https://pubmed.ncbi.nlm.nih.gov/28553727/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].(US) C for DC and P, (US) NC for CDP and HP, (US) O on S and H . Chemistry and Toxicology of Cigarette Smoke and Biomarkers of Exposure and Harm. 2010. [cited 2023 Feb 14]; Available from: https://www.ncbi.nlm.nih.gov/books/NBK53014/
- [7].Levin ED. Neurotoxicology of nicotine and tobacco. Adv Neurotoxicology. 2022 Jan 1;8:93–103. [Google Scholar]
- [8].O’Keefe JH, Bhatti SK, Bajwa A, et al. Alcohol and cardiovascular health: the dose makes the poison … or the remedy. Mayo Clin Proc. [cited 2022 Jun 27] 2014;89:382–393. https://pubmed.ncbi.nlm.nih.gov/24582196/ [DOI] [PubMed] [Google Scholar]
- [9].Geneva: World Health Organization . Global status report on alcohol and health 2018. 2018.
- [10].Zakhari S. Director is. Overview: how is alcohol metabolized by the body? Alcohol Res Heal. 2006. [cited 2023 Feb 14];29(4):245. https://pmc/articles/PMC6527027/ [PMC free article] [PubMed] [Google Scholar]
- [11].Gardner JD, Mouton AJ. Alcohol effects on cardiac function. Compr Physiol. 2015. Apr 1 [cited 2023 Feb 14];5(2):791–802. https://pubmed.ncbi.nlm.nih.gov/25880513/ [DOI] [PubMed] [Google Scholar]
- [12].La Monte SM D, Kril JJ. Human alcohol-related neuropathology. Acta Neuropathol. 2014. Jan-Mar [[cited 2023 6]];127(1):71–90. https://pubmed.ncbi.nlm.nih.gov/24370929/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Atakan Z. Cannabis, a complex plant: different compounds and different effects on individuals. Ther Adv Psychopharmacol. [cited 2023 Feb 14] 2012;2:241. https://pmc/articles/PMC3736954/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004. May 27 [cited 2022 May 3];429(6990):457–463. https://pubmed.ncbi.nlm.nih.gov/15164071/. [DOI] [PubMed] [Google Scholar]
- [15].Szutorisz H, Hurd YL. Epigenetic Effects of Cannabis Exposure. Biol Psychiatry. [cited 2022 May 4] 2016;79(7):586. https://pmc/articles/PMC4789113/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cecil CAM, Walton E, Methylation VED. Substance Use and Addiction: a Systematic Review of Recent Animal and Human Research from a Developmental Perspective. Curr Addict Reports. 2015. Dec 1 [cited 2022 Jul 8];2(4):331–346. DOI: 10.1007/s40429-015-0072-9. [DOI] [Google Scholar]
- [17].Moosavi A, Ardekani AM. Role of Epigenetics in Biology and Human Diseases. Iran Biomed J. 2016. Nov 1 [cited 2022 Jul 13];20(5):246. https://pmc/articles/PMC5075137/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Leenen FAD, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clin Epigenetics. 2016. Sep 6 [cited 2022 May 2]. 8(1). https://pubmed.ncbi.nlm.nih.gov/27602172/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Carreras-Gallo N, Cáceres A, Balagué-Dobón L, et al. The early-life exposome modulates the effect of polymorphic inversions on DNA methylation. Commun Biol. 2022. DecJul. [cited 2022 20];5(1). https://pubmed.ncbi.nlm.nih.gov/35550596/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li E, Zhang Y. DNA Methylation in Mammals. Cold Spring Harb Perspect Biol. [cited 2022 Jul 19] 2014;6:a019133. https://pmc/articles/PMC3996472/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Laurent L, Wong E, Li G, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010. MarJul [cited 2022 20];20(3):320–331. https://pubmed.ncbi.nlm.nih.gov/20133333/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dugué PA, Jung CH, Joo JE, et al. Smoking and blood DNA methylation: an epigenome-wide association study and assessment of reversibility. Epigenetics. 2020. Apr 2 [cited 2022 Jun 2];15(4):358–368. https://pubmed.ncbi.nlm.nih.gov/31552803/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zeilinger S, Kühnel B, Klopp N, et al.Tobacco smoking leads to extensive genome-wide changes in DNA methylation PLoS ONE. 2013. May 17 [cited 2022 Jun 2];8(5). https://pubmed.ncbi.nlm.nih.gov/23691101/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dogan MV, Beach SRH, Philibert RA. Genetically contextual effects of smoking on genome wide DNA methylation. Am J Med Genet B Neuropsychiatr Genet. 2017. Sep 1 [cited 2022 Jun 2];174(6):595–607. https://pubmed.ncbi.nlm.nih.gov/28686328/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Christiansen C, Castillo-Fernandez JE, Domingo-Relloso A, et al.Novel DNA methylation signatures of tobacco smoking with trans-ethnic effects Clin Epigenetics. 2021. Dec 1 [cited 2022 Jun 2];13(1). https://pubmed.ncbi.nlm.nih.gov/33593402/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sikdar S, Joehanes R, Joubert BR, et al. Comparison of smoking-related DNA methylation between newborns from prenatal exposure and adults from personal smoking. Epigenomics. 2019. Oct 1 [cited 2022 Jun 2];11(13):1487–1500. https://pubmed.ncbi.nlm.nih.gov/31536415/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Joubert BR, Håberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012. Oct;120(10):1425–1431. [cited 2022 Jul 7] https://pubmed.ncbi.nlm.nih.gov/22851337/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the life course: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet. 2015. Apr 15 [cited 2022 Jul 7];24(8):2201–2217. https://pubmed.ncbi.nlm.nih.gov/25552657/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu K, Montalvo-Ortiz JL, Zhang X, et al. Epigenome-Wide DNA Methylation Association Analysis Identified Novel Loci in Peripheral Cells for Alcohol Consumption Among European American Male Veterans. Alcohol Clin Exp Res. 2019. Oct 1 [cited 2022 May 12];43(10):2111–2121. https://pubmed.ncbi.nlm.nih.gov/31386212/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Toinét Cronjé H, Elliott HR, Nienaber-Rousseau C, et al. Replication and expansion of epigenome-wide association literature in a black South African population. Clin Epigenetics. 2020. Jan 7 [cited 2022 May 12];12(1). https://pubmed.ncbi.nlm.nih.gov/31910897/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lohoff FW, Clarke TK, Kaminsky ZA, et al. Epigenome-wide association study of alcohol consumption in N = 8161 individuals and relevance to alcohol use disorder pathophysiology: identification of the cystine/glutamate transporter SLC7A11 as a top target. Mol Psychiatry. [cited 2022 May 18]. 2022;27:1754–1764. https://pubmed.ncbi.nlm.nih.gov/34857913/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dugué PA, Wilson R, Lehne B, et al.Alcohol consumption is associated with widespread changes in blood DNA methylation: analysis of cross-sectional and longitudinal data Addict Biol. 2021. Jan 1 [cited 2022 May 5];26(1). https://pubmed.ncbi.nlm.nih.gov/31789449/ [DOI] [PubMed] [Google Scholar]
- [33].Wilson LE, Xu Z, Harlid S, et al. Alcohol and DNA Methylation: an Epigenome-Wide Association Study in Blood and Normal Breast Tissue. Am J Epidemiol. 2019. Jun 1 [cited 2022 May 12];188(6):1055–1065. https://pubmed.ncbi.nlm.nih.gov/30938765/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stephenson M, Bollepalli S, Cazaly E, et al. Associations of Alcohol Consumption with Epigenome-Wide DNA Methylation and Epigenetic Age Acceleration: individual-Level and Co-twin Comparison Analyses. Alcohol Clin Exp Res. 2021. Feb 1 [cited 2022 May 12];45(2):318–328. https://pubmed.ncbi.nlm.nih.gov/33277923/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Osborne AJ, Pearson JF, Noble AJ, et al. Genome-wide DNA methylation analysis of heavy cannabis exposure in a New Zealand longitudinal cohort. Transl Psychiatry. 2020. Apr 22 [cited 2022 Apr 5];10(1):1–10. https://www.nature.com/articles/s41398-020-0800-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Markunas CA, Hancock DB, Xu Z, et al. Epigenome-wide analysis uncovers a blood-based DNA methylation biomarker of lifetime cannabis use. Am J Med Genet B Neuropsychiatr Genet. 2021. Apr 1 [cited 2022 May 5];186(3):173–182. https://pubmed.ncbi.nlm.nih.gov/32803843/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kumar J. Epidemiology of hypertension. Clin Queries Nephrol. 2013. Apr 1 [cited 2022 Jul 25];2(2):56–61. https://linkinghub.elsevier.com/retrieve/pii/S2211947713000162. [Google Scholar]
- [38].Singh S, Shankar R, Singh GP. Prevalence and Associated Risk Factors of Hypertension: a Cross-Sectional Study in Urban Varanasi. Int J Hypertens. 2017. [cited 2022 Jul 25];2017:1–10. https://pubmed.ncbi.nlm.nih.gov/29348933/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Minzer S, Losno RA, Casas R. The Effect of Alcohol on Cardiovascular Risk Factors: is There New Information? Nutrients. 2020. Apr 1 [cited 2022 Jun 20];12(4):1–22. https://pmc/articles/PMC7230699/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tasnim S, Tang C, Musini VM, et al. Effect of alcohol on blood pressure. Cochrane Database Syst Rev. 2020. Jul 1 [cited 2022 Jul 18]. 2020(7). https://pmc/articles/PMC8130994/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Virdis A, Giannarelli C, Fritsch Neves M, et al. Cigarette smoking and hypertension. Curr Pharm Des. 2010. Aug 6 [cited 2022 Jun 20];16(23):2518–2525. https://pubmed.ncbi.nlm.nih.gov/20550499/. [DOI] [PubMed] [Google Scholar]
- [42].Dikalov S, Itani H, Richmond B, et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol. 2019. Mar 1 [cited 2022 Jun 20];316(3):H639–46. https://pubmed.ncbi.nlm.nih.gov/30608177/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Abuhasira R, Haviv YS, Leiba M, et al., Cannabis is associated with blood pressure reduction in older adults - a 24-hours ambulatory blood pressure monitoring study. Eur J Intern Med. 2021. Apr 1 [cited 2022 Jul 25];86:79–85. https://pubmed.ncbi.nlm.nih.gov/33483174/ [DOI] [PubMed] [Google Scholar]
- [44].Alshaarawy O, Elbaz HA. Cannabis use and blood pressure levels: United States National Health and Nutrition Examination Survey, 2005-2012. J Hypertens. 2016. Aug 1 [cited 2022 Jul 25];34(8):1507–1512. https://pubmed.ncbi.nlm.nih.gov/27270185/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jones RT. Cardiovascular system effects of marijuana. J Clin Pharmacol. 2002. Nov 1 [cited 2022 Jul 25];42(S1):58S–63S. https://pubmed.ncbi.nlm.nih.gov/12412837/. [DOI] [PubMed] [Google Scholar]
- [46].Atsma F, De Vegt F. The healthy donor effect: a matter of selection bias and confounding. Transfusion. 2011. Sep [cited 2023 6];51(9):1883–1885. https://pubmed.ncbi.nlm.nih.gov/21790637/ [DOI] [PubMed] [Google Scholar]
- [47].Brodersen T, Rostgaard K, Lau CJ, et al.The healthy donor effect and survey participation, becoming a donor and donor career Transfusion. 2023. Dec 8 [cited 2023 Feb 6];631https://pubmed.ncbi.nlm.nih.gov/36479702/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Min JL, Hemani G, Smith GD, et al. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018. Dec 1 [cited 2022 Jun 23];34(23):3983–3989. https://pubmed.ncbi.nlm.nih.gov/29931280/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhou W Infinium Annotation. https://zwdzwd.github.io/InfiniumAnnotation. Accessed 14 June 2022.
- [50].Chen J, Behnam E, Huang J, et al. Fast and robust adjustment of cell mixtures in epigenome-wide association studies with SmartSVA. BMC Genomics. 2017. May 26 [cited 2021 Mar 9];18(1):413. DOI: 10.1186/s12864-017-3808-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011. Mar-Jun [cited 2021 9];27(11):1496–1505. https://pubmed.ncbi.nlm.nih.gov/21471010/ [DOI] [PubMed] [Google Scholar]
- [52].Ruiz-Arenas C, JR G. MEAL: perform methylation analysis. R package version 1.22.0. 2019. [cited 2020 Apr 9]; Available from: https://bioconductor.org/packages/release/bioc/html/MEAL.html
- [53].Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for {RNA}-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. DOI: 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Guintivano J, Shabalin AA, Chan RF, et al. Test-statistic inflation in methylome-wide association studies. Epigenetics. 2020. Nov 1 [cited 2022 Jul 15];15(11):1163–1166. DOI: 10.1080/15592294.2020.1758382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ogata H, Goto S, Sato K, et al. Kyoto Encyclopedia of Genes and Genomes [Internet]. Nucleic Acids Res. 1999. [cited 2020 Apr 9];27(1):29–34. https://academic.oup.com/nar/article-abstract/27/1/29/1238108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nature genet. 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omi A J Integr Biol. 2012;16(5):284–287. DOI: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Piñero J, Piñero P, Bravo A, et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016. [cited 2020 Apr 9];45:833–839. http://www.disgenet.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schwarzer G, Carpenter JR, Rücker G. Meta-Analysis with R. 2015. [cited 2023 Mar 6]. DOI: 10.1007/978-3-319-21416-0. [DOI]
- [60].Pidsley R, Wong CC Y, Volta M, et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14(1):293. DOI: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xu Z, Niu L, Li L, et al. Enmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016. Feb 18;44(3):e20. DOI: 10.1093/nar/gkv907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tingley D, Yamamoto T, Hirose K, et al. Mediation: r Package for Causal Mediation Analysis. J Stat Softw. 2014;59(5):1–38. http://www.jstatsoft.org/v59/i05/26917999 [Google Scholar]
- [63].Enokida H, Shiina H, Urakami S, et al. Smoking influences aberrant CpG hypermethylation of multiple genes in human prostate carcinoma. Cancer. 2006. Jan 1 [cited 2022 Jul 8];106(1):79–86. https://pubmed.ncbi.nlm.nih.gov/16323173/ [DOI] [PubMed] [Google Scholar]
- [64].Ostrow KL, Michalidi C, Guerrero-Preston R, et al. Cigarette smoke induces methylation of the tumor suppressor gene NISCH. Epigenetics. 2013. Apr 4 [cited 2023 Feb 9];8(4):383. https://pmc/articles/PMC3674047/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Breitling LP, Salzmann K, Rothenbacher D, et al. F2RL3 methylation, and prognosis in stable coronary heart disease. Eur Heart J. 2012. Feb-Nov [cited 2023 9];33(22):2841–2848. https://pubmed.ncbi.nlm.nih.gov/22511653/ [DOI] [PubMed] [Google Scholar]
- [66].Wan ES, Qiu W, Baccarelli A, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012. Jul [cited 2022 8];21(13):3073–3082. https://pubmed.ncbi.nlm.nih.gov/22492999/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shenker NS, Polidoro S, van Veldhoven K, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013. Mar-Jul;22(5):843–851. [cited 2022 8] https://pubmed.ncbi.nlm.nih.gov/23175441/ [DOI] [PubMed] [Google Scholar]
- [68].Monick MM, Beach SRH, Plume J, et al. Coordinated Changes in AHRR Methylation in Lymphoblasts and Pulmonary Macrophages from Smokers. Am J Med Genet. [cited 2022 Jul 8] 2012;159B:141. https://pmc/articles/PMC3318996/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wider C, Lincoln SJ, Heckman MG, et al. Phactr2 and Parkinson’s disease. Neurosci Lett. 2009. Mar 3 [cited 2022 Jul 26];453(1):9. https://pmc/articles/PMC2684848/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wise RA, Robble MA. Dopamine and Addiction. Annu Rev Psychol. 2020. Jan 4 [cited 2023 Mar 9];71(1):79–106. https://pubmed.ncbi.nlm.nih.gov/31905114/. [DOI] [PubMed] [Google Scholar]
- [71].Subramaniyan M, Dani JA. Dopaminergic and cholinergic learning mechanisms in nicotine addiction. Ann N Y Acad Sci. 2015. Sep 1 [cited 2023 Mar 9];1349(1):46–63. https://pubmed.ncbi.nlm.nih.gov/26301866/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Liang X, Justice AC, So-Armah K, et al. DNA methylation signature on phosphatidylethanol, not on self-reported alcohol consumption, predicts hazardous alcohol consumption in two distinct populations. Mol Psychiatry. 2021. Jun 1 [cited 2022 May 12];26(6):2238–2253. https://pubmed.ncbi.nlm.nih.gov/32034291/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu C, Marioni RE, Hedman AK, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2016 Nov 15 [cited 2022 May 13] 2018;23:422–433. https://www.nature.com/articles/mp2016192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Aragona J, Lee CK. Scoliosis in fetal alcohol syndrome: a case report. Orthopedics. 1981;4(10):1141–1143. [DOI] [PubMed] [Google Scholar]
- [75].Gallagher C, McCarthy FP, Ryan RM, et al. Maternal Alcohol Consumption During Pregnancy and the Risk of Autism Spectrum Disorders in Offspring: a Retrospective Analysis of the Millennium Cohort Study. J Autism Dev Disord. 2018. Nov 1 [cited 2022 Jul 26];48(11):3773. https://pmc/articles/PMC6182718/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Singer AB, Aylsworth AS, Cordero C, et al. Prenatal Alcohol Exposure in Relation to Autism Spectrum Disorder: findings from the Study to Explore Early Development (SEED). Paediatr Perinat Epidemiol. 2017. Nov 1 [cited 2022 Jul 26];31(6):573. https://pmc/articles/PMC5690833/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wilhoit LF, Scott DA, Simecka BA. Fetal Alcohol Spectrum Disorders: characteristics, Complications, and Treatment. Community Ment Health J. 2017. Aug 1 [cited 2023 Feb 17];53(6):711–718. https://pubmed.ncbi.nlm.nih.gov/28168434/. [DOI] [PubMed] [Google Scholar]
- [78].Varadinova M, Boyadjieva N. Epigenetic mechanisms: a possible link between autism spectrum disorders and fetal alcohol spectrum disorders. Pharmacol Res. 2015. Dec 1;102:71–80. [DOI] [PubMed] [Google Scholar]
- [79].Watson CT, Szutorisz H, Garg P, et al. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated with Cross-Generational Effects of Adolescent THC Exposure. Neuropsycho pharmacology. 2015. Dec 1 [cited 2022 May 13];40(13):2993–3005. https://pubmed.ncbi.nlm.nih.gov/26044905/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Allen JP, Danoff JS, Costello MA, et al.Lifetime marijuana use and epigenetic age acceleration: a 17-year prospective examination. Drug Alcohol Depend. [cited 2022 Apr 5];233109363.https://pubmed.ncbi.nlm.nih.gov/35231715/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tantoh DM, Lee KJ, Nfor ON, et al.Methylation at cg05575921 of a smoking-related gene (AHRR) in non-smoking Taiwanese adults residing in areas with different PM 2.5 concentrations. Clin Epigenetics. 2019. May 6 [cited 2022 Jul 8];111. https://pubmed.ncbi.nlm.nih.gov/31060609/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Astuti Y, Wardhana A, Watkins J, et al. Cigarette smoking and telomere length: a systematic review of 84 studies and meta-analysis. Environ Res. 2017. [cited 2023 Mar 9];158:480–489. https://pubmed.ncbi.nlm.nih.gov/28704792/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Topiwala A, Taschler B, Ebmeier KP, et al. Alcohol consumption and telomere length: Mendelian randomization clarifies alcohol’s effects. Mol Psychiatry. 2022. Jul 26 [cited 2023 Mar 9];27:4001–4008. https://www.nature.com/articles/s41380-022-01690-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Maugeri A, Barchitta M, Lio RMS, et al. The effect of alcohol on telomere length: a systematic review of epidemiological evidence and a pilot study during pregnancy. Int J Environ Res Public Health. 2021. May 1 [cited 2023 Mar 9];18(9):5038. https://pmc/articles/PMC8126216/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gonzalez-Jaramillo V, Portilla-Fernandez E, Glisic M, et al. The role of DNA methylation and histone modifications in blood pressure: a systematic review. J Hum Hypertens. 2019. Oct 1 [cited 2022 Jul 11];33(10):703–715. https://pubmed.ncbi.nlm.nih.gov/31346255/ [DOI] [PubMed] [Google Scholar]
- [86].Richard MA, Huan T, Ligthart S, et al. DNA Methylation Analysis Identifies Loci for Blood Pressure Regulation. Am J Hum Genet. 2017. Dec 7 [cited 2022 Jul 11];101(6):888–902. https://pubmed.ncbi.nlm.nih.gov/29198723/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Syme C, Shin J, Richer L, et al. Epigenetic Loci of Blood Pressure. Circ Genomic Precis Med. 2019. Jan 1 [cited 2022 Jul 11];12(1):E002341. https://pubmed.ncbi.nlm.nih.gov/30645168/ [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available upon reasonable request due to ensure the privacy of the participants. Please email varun@trudiagnostic.com for data requests. Any custom code or software used in our analysis is available at DOI: 10.5281/zenodo.6417926 (URL: https://zenodo.org/badge/latestdoi/296552532).