

Abstract
Post-transcriptional RNA modifications have been recognized as key regulators of neuronal differentiation and synapse development in the mammalian brain. While distinct sets of 5-methylcytosine (m5C) modified mRNAs have been detected in neuronal cells and brain tissues, no study has been performed to characterize methylated mRNA profiles in the developing brain. Here, together with regular RNA-seq, we performed transcriptome-wide bisulfite sequencing to compare RNA cytosine methylation patterns in neural stem cells (NSCs), cortical neuronal cultures, and brain tissues at three postnatal stages. Among 501 m5C sites identified, approximately 6% are consistently methylated across all five conditions. Compared to m5C sites identified in NSCs, 96% of them were hypermethylated in neurons and enriched for genes involved in positive transcriptional regulation and axon extension. In addition, brains at the early postnatal stage demonstrated substantial changes in both RNA cytosine methylation and gene expression of RNA cytosine methylation readers, writers, and erasers. Furthermore, differentially methylated transcripts were significantly enriched for genes regulating synaptic plasticity. Altogether, this study provides a brain epitranscriptomic dataset as a new resource and lays the foundation for further investigations into the role of RNA cytosine methylation during brain development.
Keywords: Brain development, Neuron, Neural stem cell, RNA cytosine methylation, RNA bisulfite sequencing, RNA-seq
1. Introduction
Over the past decade, epitranscriptomics has emerged as a new field to study the post-transcriptional modifications of RNA bases on a transcriptome-wide scale [1]. Among the over 170 kinds of RNA modifications, RNA cytosine methylation (m5C) has gradually been recognized as an important form regulating RNA metabolism. The m5C modification on tRNAs regulates tRNA stability and protein translation [2], while cytosine methylation on rRNAs coordinates mitochondrial assembly and translation activity [3,4]. Despite their lower abundance, maternally derived m5C-mRNAs are essential for embryonic development during the maternal-to-zygotic transition [5] and ablation of maternal m5C-mRNAs results in developmental delay and embryonic defects [6]. Cytosine methylation on mammalian mRNAs is primarily catalyzed by NOP2/Sun RNA Methyltransferase 2 (NSUN2) [7,8] and is removed via m5C oxidation by the Ten Eleven Translocation enzymes [9,10]. At DNA damage sites, the RNA methyltransferase, TRNA Aspartic Acid Methyltransferase 1 (TRDMT1), is recruited to introduce m5C to mRNA in the form of DNA:RNA hybrids [11]. The methylated RNA in such hybrids was recognized by the m5C readers RAD51/52 [11]. Another m5C binding protein, Aly/REF Export Factor (ALYREF), mediates the export of methylated mRNAs from the nucleus to the cytoplasm [7]. mRNAs carrying m5C sites can also be recognized by the Y-box binding protein 1 (YBX1), which recruits the poly(A) binding protein cytoplasmic 1a (PABPC1A) to achieve stabilization of methylated mRNAs [12].
Accumulating literature emphasizes the important roles of RNA post-transcriptional modifications in brain development and function. In neuronal stems cells, the loss of RNA methyltransferase NSUN2 results in decreased m5C-tRNA levels, leading to aggregation of tRNA fragments, activation of a cellular stress response, and impairment of neuronal stem cell differentiation [13,14]. In mouse models, NSUN2 loss inhibits the neurogenesis of upper-layer cortical neurons inducing microcephaly and motor defects [13,15]. Later, targeted knockout of NSUN2 in the mouse prefrontal cortex was shown to have reduced levels of m5C-tRNA and resulted in the disruption of neuronal synaptic signaling patterns and behavior [16]. Although the influence of NSUN2 loss on mRNA methylation remains unclear, methylated mRNAs in brain tissue were found to be enriched for genes involved in ion transport and synapse function [17]. In neuronal oxidative stress models, an elevation in mRNA methylation level was associated with the stress response and regulation of apoptosis [18]. In glioblastoma cells, methylated transcripts mediated by the m5C writer NSUN6 were linked to the regulation of transcriptional and translational processes after alkylation treatment [19]. These findings indicate that m5C-modified RNAs may influence brain cell differentiation and function. However, no previous study has attempted to characterize m5C dynamics during mammalian brain development.
Brain development is driven by drastic transcriptome changes accompanied by an increase in cellular diversity and neuronal connectivity [20]. The expansion and migration of neuronal lineages occur at the pre-natal stage while post-natal brain development is characterized by active gliogenesis and rapid neuronal diversification [21,22]. Around postnatal day 17 (P17), the mouse brain enters a critical period of cortical plasticity that shapes neuronal circuits in response to early life experiences [23]. In this study, we performed RNA BS-seq to survey the landscape of mRNAs with methylated cytosines in the mouse brain at critical developmental time points, as well as neural stem cells and E16.5 cortical neuron cultures (Fig. 1A). In combination with bulk RNA-seq and single-cell RNA-seq data, we aimed to explore the links between the patterns of mRNA cytosine methylation and gene expression during brain development.
Fig. 1.
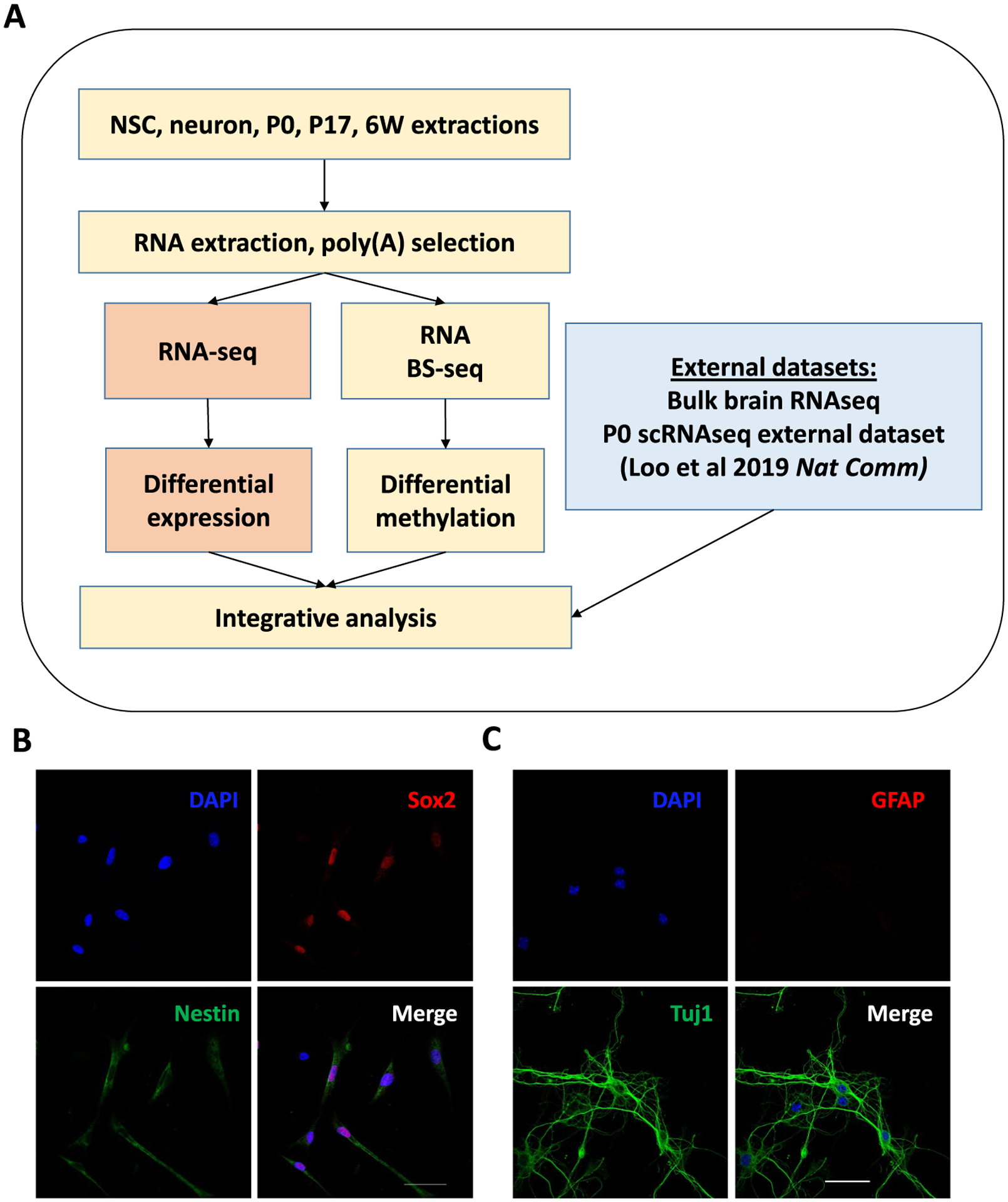
Experimental Design. (A) Overall experimental design of this study. (B) Mouse neural stem cells (NSCs) were double stained with the neural progenitor markers Nestin (cytoplasmic, green) and Sox2 (nuclear, red); nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (C) E16.5 mouse cortical neuronal culture was double stained with the neuronal marker Tuj1 (green) and glial marker GFAP (red); nuclei were counterstained with DAPI (blue). Scale bar: 50 μm.
2. Materials and methods
2.1. Mice
C57BL/6 mice were maintained and bred in a 12-h light/dark cycle under standard pathogen-free conditions. Mouse brains were harvested at postnatal day 0, 17, and 6wk for total RNA extraction. Adult female mice were used for setting up timed pregnancy. Embryos were timed by checking virginal plugs daily in the morning. Positive plugs were designated as E0.5. The experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Tech.
2.2. Mouse neural stem cell (NSC) culture
Mouse NSCs were isolated from the subventricular zone (SVZ) of the lateral ventricles as described previously [24]. NSCs were seeded on poly-ornithine and laminin-coated plates. The culture media was prepared by mixing Dulbecco’s Modified Eagle Medium (DMEM) and Ham’s F12 media at 1:1 volume ratio, supplemented with 2% B27, 2 mmol/L l-glutamine, 1× penicillin-streptomycin, 20 ng/ml epidermal growth factor (EGF, PeproTech), and 20 ng/ml basic fibroblast growth factor (bFGF, PeproTech).
2.3. Primary mouse cortical neuronal culture
Primary mouse cortical neurons were prepared as previously described [25] with some modifications. Briefly, C57BL/6 E16.5 mouse embryos were micro-dissected for cortex tissues and the cortex tissues were dissociated into single-cell suspensions using the neural tissue dissociation kit (Cat# 130–092-628) according to manufacturer’s instructions. After dissociation, neuronal cells were filtered through a 70-μm strainer (Falcon) and spun at 300 g for 10 min. The cell pellet was resuspended in neuronal culture medium (Neurobasal medium containing 2% B27 supplement (Invitrogen), 1% Glutamax (ThermoFisher), and 1% penicillin-streptomycin (ThermoFisher) and seeded on laminin and poly-ornithine coated 10-cm dishes. Neurons were grown in vitro for 7 days with fresh medium changed on Day 3 and Day 6.
2.4. Immunostaining
Immunostaining was performed as previously described [26]. Briefly, NSCs or E16.5 mouse cortical neurons were seeded onto an 8-well chamber. The neurons were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 15min and permeabilized with 0.2% TritonX-100 in PBS for 10min. After being blocked with 5% Normal Goat Serum (Thermo Fisher) at room temperature (RT) for 1h, the cells were incubated with mouse anti-Nestin antibody (Millipore, MAB353) and rabbit anti-Sox2 antibody (Abcam, ab97959) for NSCs, or with mouse anti-Tuj1 antibody (Biolegend, 801,201) and rabbit anti-GFAP antibody (Sigma, HPA056030) for E16.5 cortical neurons at 4°C overnight. Then, the cells were incubated with Cy3 conjugated anti-rabbit IgG (A10520, Invitrogen) and Alexa Fluor 488 conjugated anti-mouse IgG (A10680, Invitrogen) secondary antibodies at RT in darkness for 1h. After washing 3 × 5min with 1 × PBS, cells were mounted with DAPI-Fluoromount-G™ Clear Mounting Media (Southern Biotech, 010020). Fluorescent images were acquired using a confocal microscope.
2.5. RNA BS-seq library construction
RNA bisulfite conversion was performed as previously described [27] with minor modifications. Poly(A) RNA was first mixed with spiked-in Xef1 unmethylated RNA at a ratio of 0.5%. The spiked-in unmethylated mRNA was transcribed from the pTRI-Xef plasmid supplied by the MEGAscript™ T7 Transcription Kit (Invitorgen) according to manufacturer’s instructions. RNA bisulfite conversion was performed with an initial denaturation at 95 °C for 1 min, followed by three cycles of 70 °C for 10 min and 64 °C for 45 min using the EZ RNA methylation Kit (Zymo Research). The bisulfite converted RNA was subjected to the stranded RNA-seq library construction procedure using the TruSeq Stranded mRNA Library Preparation Kit (Illumina). We modified the procedure to skip the RNA fragmentation step and supply both random and ACT random hexamers during the first strand cDNA synthesis.
2.6. RNA BS-seq data analysis
RNA BS-seq data analysis was performed as previously described [27]. Raw sequencing reads were trimmed at the 5′ and 3′ ends by 6 bp to account for methylation bias and then filtered for low quality bases and adaptor sequences. After the removal of reads with short lengths, clean reads were mapped to the mm10 genome (Ensembl v.79) using meRanGh [28]. To exclude partially unconverted reads, mapped reads were further filtered by removing reads with >3 “C”s (“G”s on the cDNA strand). Methylation calling was performed using meRanCall and subjected to a series of filters to reduce false positive signals [27]. In addition to the thresholds applied on the methylation level (≥ 0.1) and read coverage (≥ 20) of m5C sites, additional filters were adopted including signal-to-noise ratio and the maximum number of m5C sites detected in a read to reduce false positive methylation calling [27]. The C-coverage threshold ranging from 6 to 10 was determined using Gini Index [8]. High-confidence sites were defined as the m5C sites passed all filters in both biological replicates.
2.7. Differential methylation analysis
Differential methylation analysis was performed using Fisher’s exact test on high-confidence sites present in at least one sample. Benjamini-Hochberg p-value correction was used to correct for multiple comparisons, and sites with p-adjusted <0.05 were reported.
2.8. RNA-seq library construction
Stranded RNA-seq libraries were constructed using the TruSeq Stranded mRNA Library Preparation Kit (Illumina) following manufacturer’s instructions. Briefly, after two rounds of poly(A) selection, the mRNA samples were fragmented and primed to synthesize first strand cDNA, followed by synthesis of the second strand cDNA. After Ampure XP bead purification, dA tailing was performed, and indexed adapters were ligated to both ends of the double-stranded cDNA. Adapter-ligated DNA fragments were enriched by PCR amplification for 12 cycles. After Ampure XP bead purification, the PCR products were size-selected with a range from 350 bp to 550 bp on 2% dye-free agarose gel using the pippin recovery system (Sage Science). The recovered libraries were sequenced on a Hiseq 4000 platform in the 150 bp paired end mode (Illumina).
2.9. RNA-seq data analysis
Trim Galore (version 0.6.5) was used to filter short reads, low quality reads, and trim adapter sequences from raw reads (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Clean reads were mapped to the mm10 genome and expression quantified using STAR (version 2.7.3a) [29]. Differentially expressed genes were identified using DESeq2 [30] using a fold change >1.5 and an adjusted p-value of 0.01 as cutoffs.
2.10. Gene ontology (GO) analysis
GO analysis was performed using DAVID (Database for Annotation, Visualization, and Integrated Discovery) [31]. The GO Direct terms, Biological Process (BP), cellular component (CC), and molecular function (MF) were identified. Significantly enriched terms were identified using Benjamini-Hochberg p-adjusted <0.05.
2.11. Clustering analysis
The z-score of bulk RNA-seq expression of transcript per million (TPM) and differentially methylated site methylation levels were used as input for clustering analysis. Clustering analysis was performed with the Python package sklearn’s Agglometric Clustering. The ‘complete’ method was used with the number of clusters set to 5 for RNA-seq and RNA BS-seq datasets. Error bars represent the standard deviation of the z-score.
2.12. Availability of data and software
Data generated in this study were submitted to the NCBI Gene Expression Omnibus under accession number GSE207092. Analyses in this study was performed using the R v4.1.1, and Python 3.9.4 packages Biopython v1.78, matplotlib v3.3.4, Seaborn v0.11, and Pysam v0.16. The software package developed in this study is available in the GitHub repository (https://github.com/zaustinj33/BrainDev).
3. Results
3.1. Identification of high-confidence m5C sites in mouse NSCs, neurons, and brain tissues
To obtain RNA expression and m5C epitranscriptome profiles during brain development, we performed RNA-seq and RNA BS-seq for NSCs (Fig. 1B), neurons in culture (Fig. 1C), and brain tissues isolated from postnatal day 0 (P0), postnatal day 17 (P17), and 6-week-old (6 W) mice. Two biological replicates were generated for each condition with a total of 10 libraries obtained for RNA-seq and RNA BS-seq (Table S1). Approximately 20 million paired-end reads were generated for each RNA-seq library and aligned to the mouse reference genome (mm10). For these libraries, the average percentage of sequences uniquely mapped to the reference was around 92%. The normalized gene expression level of the samples was provided (Table S2). Since methylation calling requires a much higher read depth compared with those of RNA-seq libraries, five times more sequences were generated for RNA BS-seq libraries (~100 million pair-end reads for each library). Sequencing reads derived from RNA BS-seq libraries were processed following the procedure described in our recent study [27]. In brief, bisulfite sequencing reads were first trimmed to remove adaptors and bases with low quality scores. After quality control, reads were mapped to the C2T converted mm10 genome. Around 85% of bisulfite sequencing reads were uniquely aligned (Table S1). Methylation calling was performed using meRanCall [28] and putative m5C sites were subjected to a series of filters to reduce false positive signals [27].
A total of 2259 m5C sites were identified from the ten RNA BS-seq libraries. Among these m5C sites, 1758 sites passed all filters in one biological replicate but failed in the other; thus, these were denoted as “low-confidence” m5C sites. The remaining 501 m5C sites shared by both biological replicates were considered to be “high-confidence” in this study (Fig. 2A). The methylation levels of the high-confidence m5C sites were highly correlated between the biological replicates and had Spearman R values between 0.82 and 0.92 (Fig. 2B). Notably, 76.4% of these sites had a methylation level lower than 0.3 (Fig. S1). This result was consistent with previous reports that only a small percentage of RNA copies are methylated for a given gene [8,27,32].
Fig. 2.
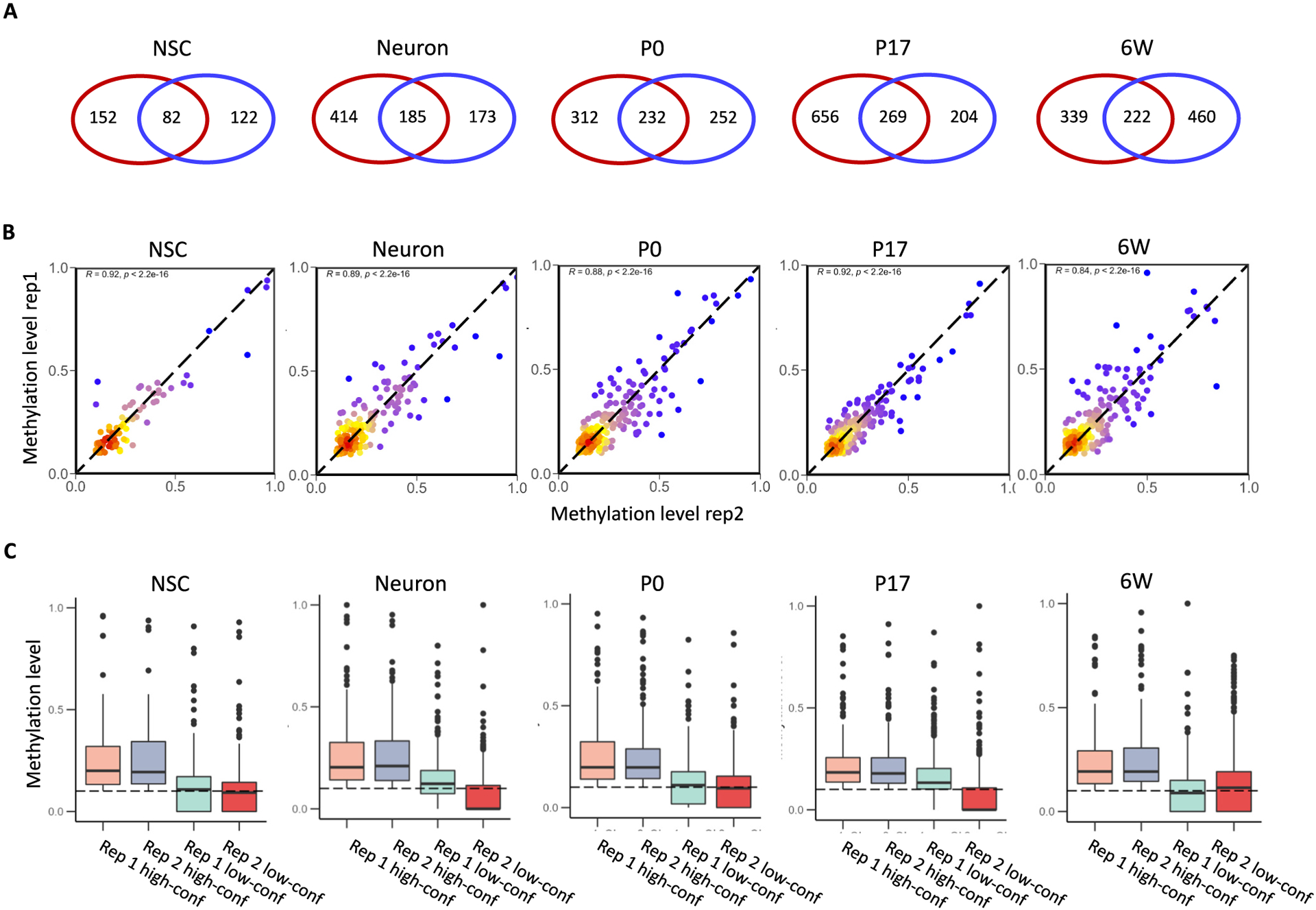
Reproducibility of replicates among RNA BS-seq samples. (A) Overlap of m5C sites identified between biological replicates. The red circle indicates replicate 1 and the blue circle indicates replicate 2. (B) Methylation correlation (Spearman’s R) of the overlapping m5C sites between biological replicates. Color code indicates the density of sites, ranging from red (high density) to blue (low density). (C) Distribution of methylation levels for the overlapping and non-overlapping m5C sites identified between biological replicates.
To explore the factors contributing to the level of confidence in methylation calling, we first compared methylation levels and read coverages of the high-confidence and low-confidence sites. Across five conditions, the high-confidence sites showed average methylation levels in the range of 0.18–0.21, while the low-confidence sites had average methylation levels in the range of 0.06–0.12 (Fig. 2C). The average read coverages for the high-confidence sites were between 109 and 227 reads per site, while the average for the low-confidence sites was only 24–48 reads per site (Fig. S2A&B). We next examined the relationship between read coverage and methylation level (Fig. S2C). A very weak positive correlation was observed between the methylation level and the read coverage for both high-confidence (Spearman R < 0.19) and low-confidence sites (Spearman R < 0.08). Altogether, the high-confidence m5C sites tended to be found with a higher methylation level and more read coverage to enable them to survive all the stringent filters in both biological replicates.
Previous studies suggested that the process of library construction, the bisulfite conversion step in particular, could have a significant impact on methylation calling [8,27,32]. In this study, we generated both RNA-seq and RNA BS-seq data for the same pool of RNA samples.
This enabled us to perform correlation analyses for the read coverage of each methylated transcript in the paired libraries for RNA-seq and RNA BS-seq. Interestingly, compared with the low-confidence sites, the high-confidence sites showed higher correlations in read coverage with or without bisulfite conversion, except for P17 brain samples in which the two correlations are similar (Fig. S3A). We further examined the average length of methylated transcripts and the distribution of m5C sites in transcripts. No significant difference in transcript length was detected for transcripts containing either high or low-confidence sites (Fig. S3B). The distribution of high-confidence sites was generally biased towards the 3’UTR of transcripts, in particular for the P17 brain samples (Fig. S3C). This result indicates that low-confidence sites tend to reside in a transcript sensitive to bisulfite treatment, but such sensitivity may not have a link with transcript length.
3.2. High-confidence m5C sites differentially methylated in mouse NSCs, neurons, and brain tissues
We next focused on the 501 high-confidence sites to examine their sequence features and distribution across five conditions. Two common features of m5C sites on mRNAs have been reported: 1) they often localize upstream of the translation initiation sites (TISs), and 2) a “GGG” motif is frequently observed downstream from the m5C sites [8,27,32,33]. To determine the distribution of high-confidence m5C sites along the transcripts, we binned the lengths of methylated transcripts into the 5′- untranslated region (5’UTR: bins 1–5), coding sequence (CDS: bins 6–28), and 3′- untranslated region (3’UTR: bins 19–46). Two density peaks surrounding TISs and an additional strong peak right before the termination of the coding region were observed for m5C sites in all five conditions (Fig. S4A). A downstream “GGG” motif was determined for the high-confidence m5C sites in all five conditions (Fig. S4B), which may serve as potential targets of the NSUN2 enzyme [8].
Out of 501 high-confidence m5C sites, a total of 31 sites were identified as methylated in all conditions and 253 sites (50.8%) were shared by at least two conditions (Fig. 3A). Such a distribution indicates that at least some mRNAs are consistently methylated across multiple time points throughout brain development. Only 82 m5C sites were identified in NSCs while the libraries derived from the three-stage brain tissues reported a similar number of high-confidence sites ranging from 222 to 269. In addition, m5C sites in undifferentiated NSCs were largely over-lapped with those in neurons and brain tissues. To determine the effect of sequencing depth on methylation calling, we calculated the yield of m5C sites after normalizing to the number of total mapped reads for each library (Table S1). Despite more reads generated for NSC libraries, both NSC replicates yielded the fewest m5C sites regardless of high-confidence or low-confidence. The methylation level of high-confidence sites reported in the P17 brain were significantly lower than sites reported in the P0 brain (Fig. 3B). These results suggested that the diversity of mRNA cytosine methylation may increase during brain cell specification.
Fig. 3.
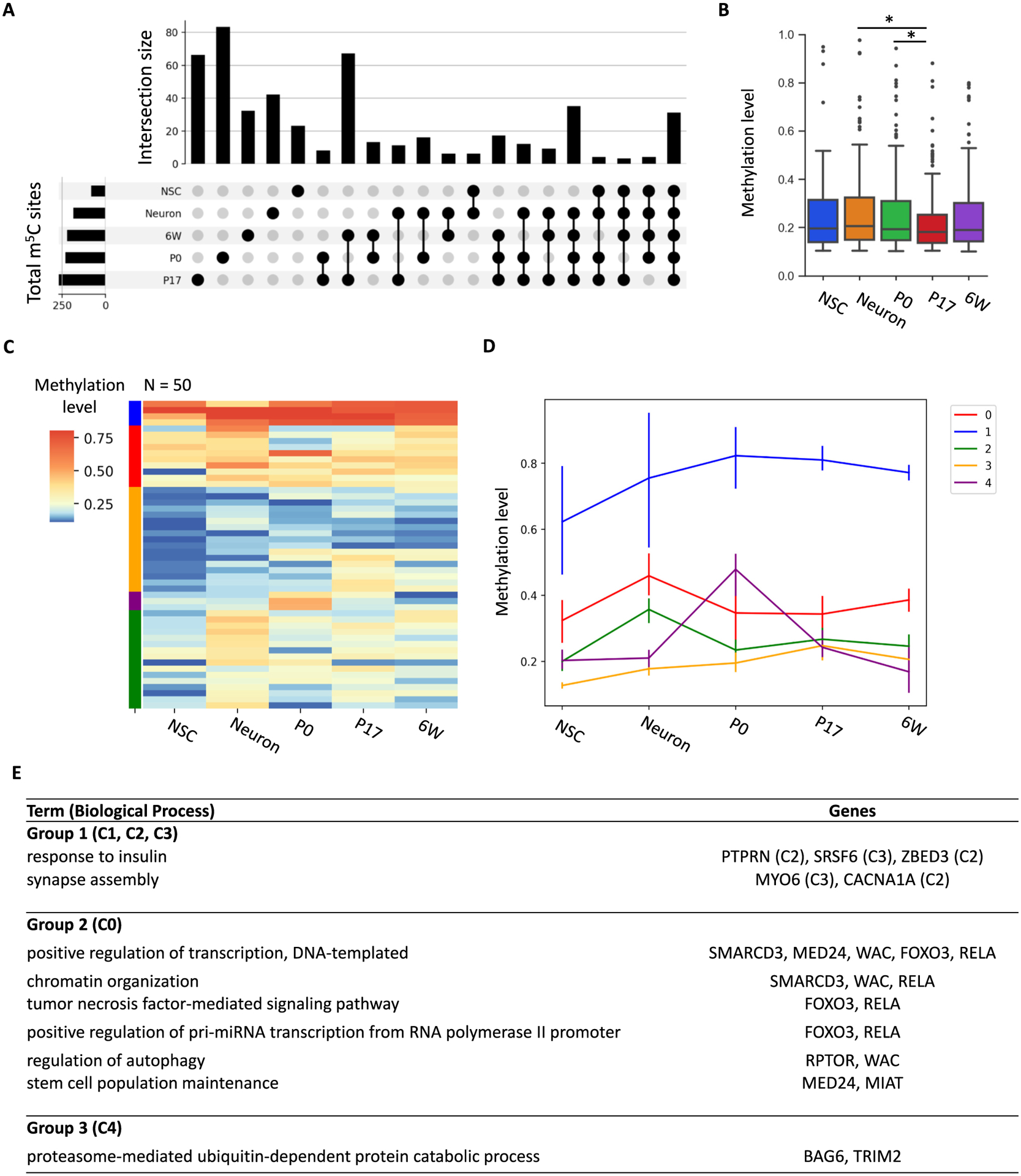
Characteristics of high-confidence m5C sites identified in NSCs, neuron, and brain samples. (A) High-confidence m5C sites shared among conditions. (B) Methylation distribution of the high-confidence m5C sites. Wilcoxon rank-sum test was performed to assess significance (*p < 0.05). (C) Clustering of m5C sites according to methylation changes. (D) Average methylation levels of clusters identified in Fig. 3C. Bars indicate standard deviation. (C) and (D) share the same color code. (E) Gene ontology analysis of methylated transcripts.
Since brain development is accompanied with substantial changes in gene expression, we selected a set of 50 high-confidence m5C sites that possessed at least 20× read coverage in all conditions. According to their average methylation levels, five clusters were identified with unique methylation patterns (Fig. 3C). The m5C sites in three clusters had relatively consistent methylation levels throughout all conditions (at high, low, and medium methylation levels, respectively). Such stable methylation patterns suggest that cells from neural lineage may share a regulatory mechanism to control the methylation of a small set of transcripts. Cluster C0 and C4 showed high methylation levels in neurons and P0 brain samples, respectively (Fig. 3D). We further determined their associated transcripts for GO enrichment analysis. Interestingly, transcripts highly methylated in neuronal culture (cluster C0) were associated with transcription, chromatin organization, and stem cell maintenance (Fig. 3E). For instance, genes including Foxo3, RelA, and Rptor are important mediators of neuronal cell functions including reprogramming and differentiation [34] and synaptic formation [35].
3.3. Gene expression dynamics of methylated mRNAs, m5C readers, writers, and erasers during brain development
We extended the analysis to identify differential methylation sites (DMSs) across five conditions. DMSs were defined as m5C sites with a methylation difference ≥ 0.05 between two conditions and a p-adjusted value ≤0.05. Pairwise comparisons determined 83, 176, 62, and 172 DMS sites for NSCs vs neurons, P0 vs P17, P17 vs 6 W, and P0 vs 6 W, respectively (Fig. 4A). Interestingly, of the 83 DMSs identified between NSCs vs neurons, 96% showed increased methylation in neurons. Gene Ontology analyses indicated that differentially methylated sites in neurons compared to NSCs are significantly enriched for positive regulation of DNA-templated transcription, including RNA polymerase II transcription (Fig. 4B). The differentially methylated sites identified in postnatal brain tissues were significantly enriched for the regulation of GTPase activity and synaptic plasticity associated with brain development. For instance, the transcripts Atxn1, Fgfr1, and Itsn1, which are known to be critical for brain development, were found to be hypomethylated in the P0 brain.
Fig. 4.
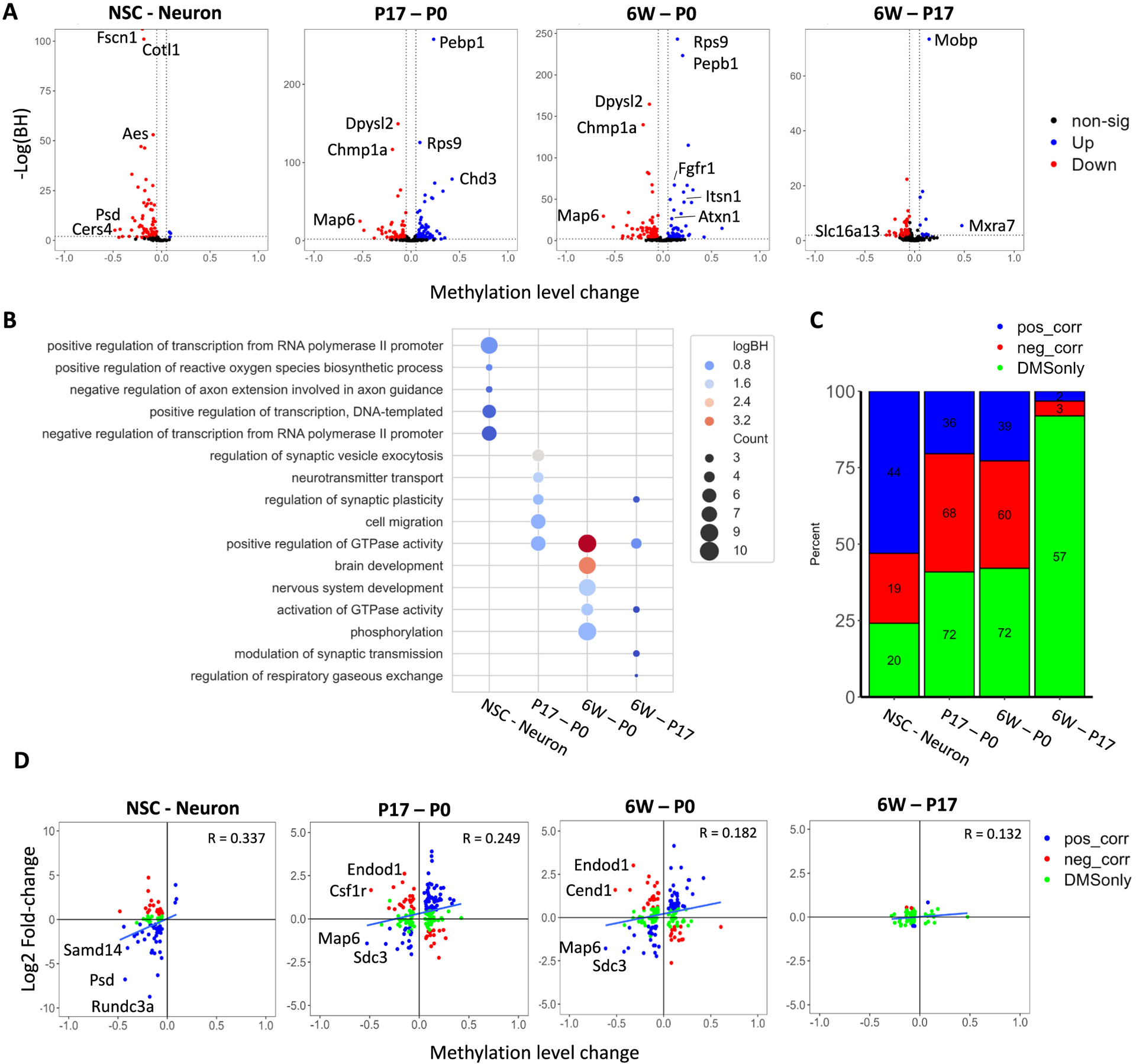
Methylation dynamics throughout brain development. (A) Differential methylation analysis of neuron and brain samples. Blue and red points represent significantly hyper and hypo methylated transcripts, respectively. Significance was determined using Fisher exact test followed by Benjamini-Hochberg correction for multiple comparison (p < 0.05), requiring a minimum difference of 0.05 in methylation. (B) Gene Ontology analysis of differentially methylated transcripts identified in (A). (C) Correlation analysis of changes in RNA expression and methylation for transcripts carrying differentially methylated sites. Blue and red bars represent positive and negative correlation between methylation level difference and expression fold-change, respectively. Green bars represent transcripts without expression fold-changes above 2 and p-adjusted <0.01. (D) Dot plot representation of transcripts to indicate the changes in methylation and expression. Overall correlation was determined using Spearman’s R. Transcripts with methylation difference over 0.3 are notated.
To examine the relationship between RNA cytosine methylation and gene expression, we performed differential expression analysis of sample-matched RNA-seq libraries. These differentially expressed genes (DEGs) were defined as transcripts with an expression difference ≥ 1.5-fold between two conditions and a p-adjusted value ≤0.01. The majority of transcripts carrying DMSs were not differentially expressed between P17 and 6 W (Fig. 4C), while a number of genes showed significant changes in both gene expression and RNA methylation during early brain development. A moderate positive correlation (R = 0.337 in NSCs vs neurons) was observed between the changes in gene expression and RNA methylation (Fig. 4D). As aforementioned, transcripts carrying m5C sites tend to have a higher methylation level in neurons compared to those in NSCs. Of those differentially methylated transcripts, 52.3% exhibited an increased expression level in neurons. Notably in neuronal samples, the transcript Psd, which codes for a Plekstrin homology and SEC7 domain-containing protein, was over-expressed and hyper-methylated. SEC7 domains were conserved throughout many proteins and served to catalyze the guanine nucleotide exchange factor initiating the formation of vesicle coating [36]. However, both positive and negative correlations between gene expression and RNA cytosine methylation were observed for pairwise comparisons of NSCs vs neurons, P0 vs P17, and P0 vs 6 W. For instance, the differentially methylated transcripts Csf1r and Endod1, which were hyper-methylated and under-expressed in the P0 brain, are markers for microglia and endothelial cell types, suggesting cell-type specific methylation of transcripts. Conversely, two transcripts, Map6 and Sdc3, were consistently hyper-methylated and over-expressed in P0 samples compared to P17 and 6 W samples. Map6 infers structural stability in developing neuronal microtubules, promoting interconnectivity and signaling [37,38], while Sdc3 is a well-known mediator of neuronal lineage, promoting neuronal migration and synapse formation [39,40]. This result indicated that the regulation of RNA cytosine methylation and gene expression may be cell-type specific and is positively correlated for some transcripts.
Recently, significantly increased understanding has been gained about the writers, readers, and erasers of m5C in mRNAs [41–44]. To examine the expression patterns of these factors, we first identified the top 1000 variably expressed genes in our RNA-seq dataset and performed clustering analysis (Fig. 5A). Not surprisingly, NSCs showed distinct gene expression profiles from other samples. We further checked the expression profiles of the factors regulating RNA cytosine methylation. It showed highly dynamic expression patterns during neural cell specification and postnatal brain development (Fig. 5B). In the comparison between NSCs and neurons, it showed that NSUN2 and YBX1 are highly expressed in both, while NSUN4, ALYREF, and FMR1 are highly expressed in NSCs, TET1 and TET3 are highly expressed in neurons (Fig. 5B). In the comparison among the three postnatal stages (P0, P17 and 6 W), it underwent clear downregulation trend, with the P0 brain displayed the highest expression for most m5C readers/writers/erasers. Except that NSUN4 and RAD52 showed increasing upregulation during postnatal brain development (Fig. 5B). We further performed pairwise comparisons to determine the transitions in gene expression (Fig. 5C). With the cutoff of fold change >1.5 and an adjusted p-value <0.01, 9501 DEGs were identified in between NSCs and neurons, 6672 DEGs and 6997 DEGs were identified in the comparison P17 vs P0, and 6 W vs P0, respectively, while only 430 DEGs were identified in the comparison between 6 W and P17 (Fig. 5C). Consistent with the transcriptome-wide differences among the four comparisons, the 6 W vs P17 comparison yielded the fewest differentially expressed factors related to RNA m5C regulation (Fig. 5D). This suggests that m5C regulation is highly dynamic during early postnatal brain development and may reach homeostasis throughout the maturation process.
Fig. 5.
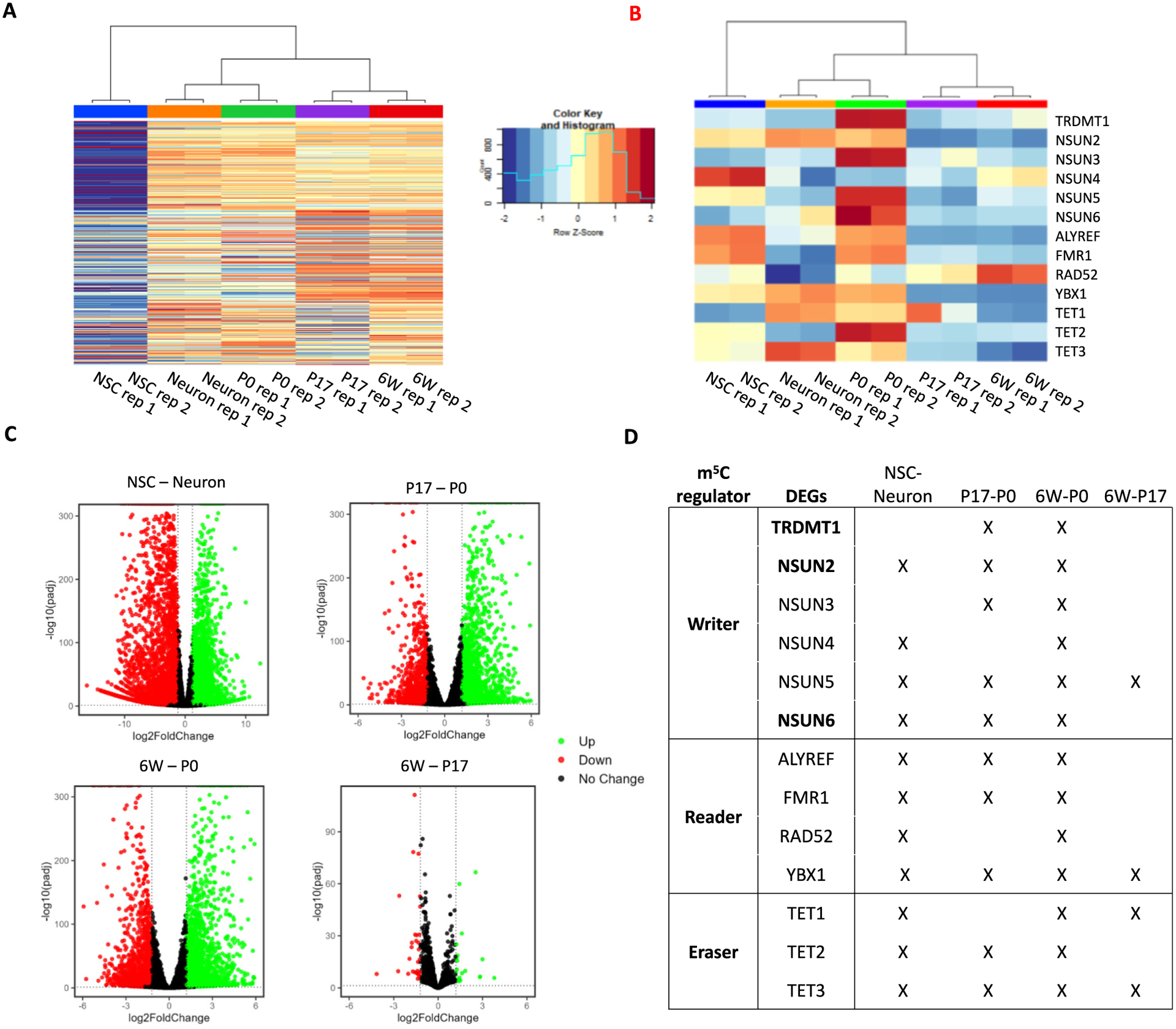
Differentially expressed transcripts among NSCs, neurons, and brain tissues. (A) TPM expression values of the top 200 differentially expressed genes identified in each comparison (NSC vs Neuron, P0 vs P17, P0 vs 6 W, and P17 vs 6 W). TPM values and density are displayed in the figure legend. (B) TPM expression values of m5C reader, writer, and eraser enzymes. (C) Volcano plots showing differentially expressed transcripts identified in each comparison (NSC vs Neuron, P0 vs P17, P0 vs 6 W, and P17 vs 6 W). Overexpressed genes are highlighted in green while under expressed genes are highlighted in red (fold-change ≥2, p-adjusted ≤0.01). (D) Dynamic expression of m5C-RNA readers, writers, and erasers. In pair-wise comparisons, differentially expressed transcripts are denoted with an “X”.
3.4. Differentially methylated transcripts have cell type-specific expression patterns and are temporally regulated in developing brains
During brain cell specification, more m5C sites (Fig. 3A) and increased methylation (Fig. 3B) were observed in differentiated neurons and maturing brains. This inspired us to further examine the temporal expression patterns of differentially methylated transcripts using bulk RNA-seq data publicly available for mouse embryonic/postnatal brain tissues including different neuronal subsets. We focused on 42 differentially methylated transcripts identified in the RNA BS-seq datasets, which also showed expression fold-change >2 across the five RNA-seq conditions (Fig. 6A). According to their expression profiles in bulk RNA-seq, four distinct clusters were identified (Fig. 6B). Cluster 1 and cluster 3 were characterized with high expression levels in the adult brain and newborn brain, respectively. Of the Cluster 1 transcripts, Endod1, Wbp2, and Rapgef4 were highly expressed in the mature 22-month-old brain samples and were found to be both over-expressed and hyper-methylated in our 6 W brain samples compared to the P0 brain. ENDOD1, an endothelial marker, was found in a previous study to have high expression in the cerebral cortex and hippocampus, while WW domain binding protein 2 (WBP2) and Rap guanine nucleotide exchange factor 4 (RAPGEF4) were mainly localized to the cerebral cortex [45,46]. WBP2 is a transcriptional co-activator of estrogen receptor α (ESR1), of which mutations induce abnormal glutamatergic synapse development [47].
Fig. 6.
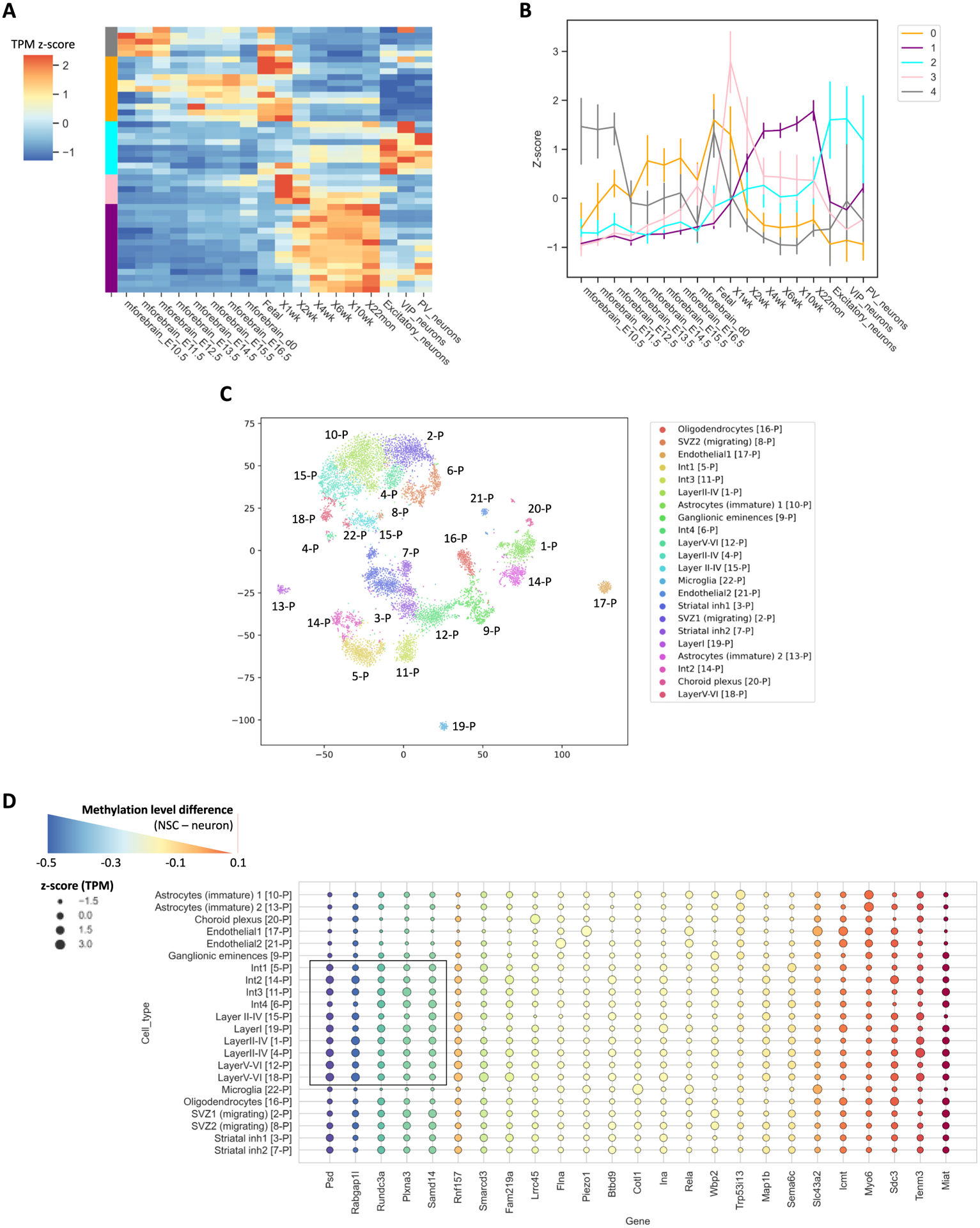
Temporal and spatial expression of differentially methylated transcripts. (A) Clustering analysis of expression profiles for differentially methylated transcripts. Z-score of TPM was used to perform clustering. (B) Linear representation of clusters identified in (A). Error bars represent standard deviation. (A) and (B) share the same color code. (C) tSNE clustering of scRNAseq P0 brain tissue from Loo et al., 2019 [22]. p is denoted as postnatal. (D) TPM z-score of differentially methylated transcripts using scRNA-seq data generated for P0 brain. Color of bubble plot indicates methylation level differences between NSC and neuron RNA BS-seq samples.
Methylated transcripts in cluster 2 showed highest expression level in neurons. To further examine the expression patterns of these methylated transcripts, we integrated the epitranscriptome profiles generated in this study with an scRNA-seq dataset recently published for the P0 cortex [22] (Fig. 6C). Previous studies reported that the methylation of tRNAs is essential for proper development of cortical layers [13,15]. Intriguingly, scRNA-seq data also suggested that some genes with abundant neuronal expression were associated with a relatively high level of mRNA methylation (Fig. 6D). In neurons, the Psd, Rundc3a, and Smarcd3 transcripts were hyper-methylated, and their expressions were also broadly enriched for many neuronal subpopulations in cortical layers I-VI, interneurons 1–3, and striatal inhibitor neurons 1 and 2. For these genes, such a cell-type predominant expression was also reported in previous studies. For example, localized expression of Rundc3a was previously found to be concentrated within noradrenergic populations of multiple brain regions [45,46]. Taken together, these results indicate that highly methylated transcripts in mature neuron populations have temporal and cell-type predominant expression patterns.
4. Discussion
Cytosine methylation of mRNA has emerged as a critical regulator of mRNA transportation, stability and translation [5,7,33,48,49]. Despite distinct mRNA methylation profiles reported in brain and NSCs [17], no attempt has been made to characterize mRNA m5C methylation patterns in the developing mammalian brain. In this study, we provide both gene expression and mRNA cytosine methylation profiles for NSCs, mature neurons, and postnatal brain tissues.
RNA cytosine methylation profiling can be achieved with m5C-specific antibodies to enrich methylated transcripts followed by deep-sequencing [50,51]. Such an approach has resolution limitations and is highly dependent on the quality of antibody used. Despite the fact that bisulfite sequencing remains the gold standard for RNA cytosine methylation detection, our recent experiments [27], together with reports from other labs [8,52], indicated that both experimental procedure and methylation calling have a significant impact on the detection of RNA methylation. Thus, inconsistent methylation sites may be observed between biological replicates. In this study, we found that high-confidence m5C sites shared by two biological replicates tend to have a higher methylation level and more read coverage than those of low-confidence m5C sites identified in one biological replicate only. In addition, low-confidence sites frequently reside in a transcript sensitive to bisulfite treatment.
Notwithstanding current technical challenges in the determination of RNA cytosine methylation, our analyses on high-confidence m5C sites led to a few interesting findings. Consistent with previous studies [7,8,17], methylated cytosines are preferentially enriched around the translation initiation sites of mRNAs and usually have a methylation level between 20 and 30% with a downstream “GGG” motif. Approximately 6% of high-confidence m5C sites identified in this study were methylated across all samples. However, the diversity of mRNA methylation increases in differentiated neurons. In addition, increased methylation in neurons were observed in 96% of the DMSs identified between NSCs vs neurons. Previous studies linked the methylation of both tRNA and mRNA to synapse formation [13,15]. Intriguingly, the DMSs identified in P17 vs P0 and 6 W brain tissues were significantly enriched for synaptic plasticity. This suggests that mRNA cytosine methylation may also play important roles during brain development.
In this study, we examined the expression profiles of RNA cytosine methylation readers, writers, and erasers. As a result of tRNA degradation, neuronal synapse function was impaired in the NSUN2 knockout mouse model [16]. Interestingly, most of the factors regulating m5C methylation were downregulated during development in the comparison between P17 and P0 but showed no significant change in the comparison between 6 W and P17. This result suggests the highly dynamic regulation of mRNA m5C methylation during early postnatal brain development, and it may reach homeostasis in the maturing brain. The integration of RNA-seq data with RNA BS-seq data allowed us to explore the correlation between the levels of gene expression and RNA methylation. A weak to moderate positive correlation was observed in general, but such a trend may not be true for all transcripts. This suggests that the regulation of RNA expression and methylation might be positively correlated and the methylated transcripts could be cell-type predominant, spatially, and/or temporally regulated. Future studies are needed to explore how cell-type predominant mRNA methylation may contribute to neuronal differentiation. Collectively, our study provided insight into the RNA m5C methylation dynamics of the developing brain and is a rich resource to facilitate further investigation on the role of brain mRNA cytosine methylation.
Supplementary Material
Funding
This study was supported by NIH grant ES031521, NS094574, MH120498, NSF1922428, the Center for One Health Research at the Virginia-Maryland College of Veterinary Medicine and the Edward Via College of Osteopathic Medicine, the Center for Engineered Health, the Virginia-Maryland College of Veterinary Medicine at Virginia Tech, and the Fralin Life Sciences Institute faculty development fund for H.X.
Acknowledgments
We thank Dr. Janet Webster for English language editing.
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygeno.2023.110604.
Data availability
Data will be made available on request.
References
- [1].Xu X, Wei X, Xie H, Advances in methods and software for RNA cytosine methylation analysis, Genomics 112 (2020) 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F, RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis, Nat. Struct. Mol. Biol 19 (2012) 900–905. [DOI] [PubMed] [Google Scholar]
- [3].Metodiev MD, Spahr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson NG, Ruzzenente B, NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly, PLoS Genet. 10 (2014), e1004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cámara Y, Asin-Cayuela J, Metodi Chan Y Shi B Ruzzenente C Kukat B Habermann R Wibom K Hultenby, et al. , MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome, Cell Metab. 13 (2011) 527–539. [DOI] [PubMed] [Google Scholar]
- [5].Yang Y, Wang L, Han X, Yang WL, Zhang M, Ma HL, Sun BF, Li A, Xia J, Chen J, et al. , RNA 5-Methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay, Mol. Cell 75 (6) (2019) 1188–1202.e11. [DOI] [PubMed] [Google Scholar]
- [6].Liu J, Huang T, Chen W, Ding C, Zhao T, Zhao X, Cai B, Zhang Y, Li S, Zhang L, et al. , Developmental mRNA m5C landscape and regulatory innovations of massive m5C modification of maternal mRNAs in animals, Nat. Commun (2022) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, Li A, Wang X, Bhattarai DP, Xiao W, et al. , 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader, Cell Res. 27 (2017) 606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang T, Chen W, Liu J, Gu N, Zhang R, Genome-wide identification of mRNA 5-methylcytosine in mammals, Nat. Struct. Mol. Biol 26 (2019) 380–388. [DOI] [PubMed] [Google Scholar]
- [9].Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, Cai Q, Ji D, Jin S-G, Niedernhofer LJ, et al. , Tet-mediated formation of 5-Hydroxymethylcytosine in RNA, J. Am. Chem. Soc 136 (2014) 11582–11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lan J, Rajan N, Bizet M, Penning A, Singh NK, Guallar D, Calonne E, Li Greci A, Bonvin E, Deplus R, et al. , Functional role of Tet-mediated RNA hydroxymethylcytosine in mouse ES cells and during differentiation, Nat. Commun (2020) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen H, Yang H, Zhu X, Yadav T, Ouyang J, Truesdell SS, Tan J, Wang Y, Duan M, Wei L, et al. , m5C modification of mRNA serves a DNA damage code to promote homologous recombination, Nat. Commun (2020) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang Y, Wang L, Han X, Yang WL, Zhang M, Ma HL, Sun BF, Li A, Xia J, Chen J, et al. , RNA 5-Methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay, Mol. Cell 75 (1188–1202) (2019), e1111. [DOI] [PubMed] [Google Scholar]
- [13].Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M, et al. , Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders, EMBO J 33 (2014) 2020–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Blanco S, Frye M, Role of RNA methyltransferases in tissue renewal and pathology, Curr. Opin. Cell Biol 31 (2014) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Flores JV, Cordero-Espinoza L, Oeztuerk-Winder F, Andersson-Rolf A, Selmi T, Blanco S, Tailor J, Dietmann S, Frye M, Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility, Stem Cell Reports 8 (2017) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Blaze J, Navickas A, Phillips HL, Heissel S, Plaza-Jennings A, Miglani S, Asgharian H, Foo M, Katanski CD, Watkins CP, et al. , Neuronal Nsun2 deficiency produces tRNA epitranscriptomic alterations and proteomic shifts impacting synaptic signaling and behavior, Nat. Commun (2021) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Amort T, Rieder D, Wille A, Khokhlova-Cubberley D, Riml C, Trixl L, Jia XY, Micura R, Lusser A, Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain, Genome Biol. 18 (2017) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jian H, Zhang C, Qi Z, Li X, Lou Y, Kang Y, Deng W, Lv Y, Wang C, Wang W, et al. , Alteration of mRNA 5-Methylcytosine modification in neurons after OGD/R and potential roles in cell stress response and apoptosis, Front. Genet 12 (2021), 633681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Awah CU, Winter J, Mazdoom CM, Ogunwobi OO, NSUN6, an RNA methyltransferase of 5-mC controls glioblastoma response to temozolomide (TMZ) via NELFB and RPS6KB2 interaction, Cancer Biol. Therapy 22 (2021) 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dillman AA, Cookson MR, Transcriptomic Changes in Brain Development, Elsevier, 2014, pp. 233–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tan L, Ma W, Wu H, Zheng Y, Xing D, Chen R, Li X, Daley N, Deisseroth K, Xie XS, Changes in genome architecture and transcriptional dynamics progress independently of sensory experience during post-natal brain development, Cell 184 (2021) 741–758.e717. [DOI] [PubMed] [Google Scholar]
- [22].Loo L, Simon JM, Xing L, McCoy ES, Niehaus JK, Guo J, Anton ES, Zylka MJ, Single-cell transcriptomic analysis of mouse neocortical development, Nat. Commun (2019) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hensch TK, Critical period plasticity in local cortical circuits, Nat. Rev. Neurosci 6 (2005) 877–888. [DOI] [PubMed] [Google Scholar]
- [24].Theus MH, Ricard J, Liebl DJ, Reproducible expansion and characterization of mouse neural stem/progenitor cells in adherent cultures derived from the adult subventricular zone, Curr. Protoc. Stem Cell Biol. Chapter 2 (2012). Unit 2D.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, Greenberg ME, Genome-wide identification and characterization of functional neuronal activity-dependent enhancers, Nat. Neurosci 17 (2014) 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sun Z, Xu X, He J, Murray A, Sun MA, Wei X, Wang X, McCoig E, Xie E, Jiang X, et al. , EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity, Nat. Commun 10 (2019) 3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Johnson Z, Xu X, Pacholec C, Xie H, Systematic evaluation of parameters in RNA bisulfite sequencing data generation and analysis, NAR Genom Bioinform 4 (2022) lqac045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rieder D, Amort T, Kugler E, Lusser A, Trajanoski Z, meRanTK: methylated RNA analysis ToolKit, Bioinformatics 32 (2016) 782–785. [DOI] [PubMed] [Google Scholar]
- [29].Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome, BMC Bioinform. 12 (2011) 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15 (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA, The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists, Genome Biol. 8 (2007) R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Z, Chen T, Chen HX, Xie YY, Chen LQ, Zhao YL, Liu BD, Jin L, Zhang W, Liu C, et al. , Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library, Nat. Methods 18 (2021) 1213–1222. [DOI] [PubMed] [Google Scholar]
- [33].Schumann U, Zhang HN, Sibbritt T, Pan A, Horvath A, Gross S, Clark SJ, Yang L, Preiss T, Multiple links between 5-methylcytosine content of mRNA and translation, BMC Biol. 18 (2020) 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ahlenius H, Chanda S, Webb AE, Yousif I, Karmazin J, Prusiner SB, Brunet A, Südhof TC, Wernig M, FoxO3 regulates neuronal reprogramming of cells from postnatal and aging mice, Proc. Natl. Acad. Sci 113 (2016) 8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Boersma MCH, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK, A requirement for nuclear factor- B in developmental and plasticity-associated synaptogenesis, J. Neurosci 31 (2011) 5414–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wright J, Kahn RA, Sztul E, Regulating the large Sec7 ARF guanine nucleotide exchange factors: the when, where and how of activation, Cell. Mol. Life Sci 71 (2014) 3419–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Deloulme J-C, Gory-Fauŕe S, Mauconduit F, Chauvet S, Jonckheere J, Boulan B, Mire E, Xue J, Jany M, Maucler C, et al. , Microtubule-associated protein 6 mediates neuronal connectivity through Semaphorin 3E-dependent signalling for axonal growth, Nat. Commun 6 (2015) 7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cuveillier C, Delaroche J, Seggio M, Gory-Fauŕe S, Bosc C, Denarier E, Bacia M, Schoehn G, Mohrbach H, Kulíc I, et al. , MAP6 is an intraluminal protein that induces neuronal microtubules to coil, Sci. Adv 6 (2020) eaaz4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hienola A, Tumova S, Kulesskiy E, Rauvala H, N-syndecan deficiency impairs neural migration in brain, J. Cell Biol 174 (2006) 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hsueh Y-P, Sheng M, Regulated expression and subcellular localization of Syndecan Heparan sulfate proteoglycans and the Syndecan-binding protein CASK/LIN-2 during rat brain development, J. Neurosci 19 (1999) 7415–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Trixl L, Lusser A, The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark, Wiley Interdiscip. Rev. RNA 10 (2019), e1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bohnsack KE, Hobartner C, Bohnsack MT, Eukaryotic 5-methylcytosine (m(5)C) RNA methyltransferases: mechanisms, cellular functions, and links to disease, Genes (Basel) (2019) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Motorin Y, Lyko F, Helm M, 5-methylcytosine in RNA: detection, enzymatic formation and biological functions, Nucleic Acids Res. 38 (2010) 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Garcia-Vilchez R, Sevilla A, Blanco S, Post-transcriptional regulation by cytosine-5 methylation of RNA, Biochim. Biophys. Acta Gene Regul. Mech 1862 (2019) 240–252. [DOI] [PubMed] [Google Scholar]
- [45].Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, Oksvold P, Edfors F, Limiszewska A, Hikmet F, et al. , An atlas of the protein-coding genes in the human, pig, and mouse brain, Science 367 (2020) eaay5947. [DOI] [PubMed] [Google Scholar]
- [46].Uhĺen M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. , Tissue-based map of the human proteome, Science 347 (2015) 1260419. [DOI] [PubMed] [Google Scholar]
- [47].Buniello A, Ingham NJ, Lewis MA, Huma AC, Martinez-Vega R, Varela-Nieto I, Vizcay-Barrena G, Fleck RA, Houston O, Bardhan T, et al. , Wbp2 is required for normal glutamatergic synapses in the cochlea and is crucial for hearing, EMBO Mol. Med 8 (2016) 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen X, Li A, Sun BF, Yang Y, Han YN, Yuan X, Chen RX, Wei WS, Liu Y, Gao CC, et al. , 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs, Nat. Cell Biol 21 (2019) 978–990. [DOI] [PubMed] [Google Scholar]
- [49].Xu X, Johnson Z, Wang A, Padget RL, Smyth JW, Xie H, Folate regulates RNA m(5)C modification and translation in neural stem cells, BMC Biol. 20 (2022) 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cui X, Liang Z, Shen L, Zhang Q, Bao S, Geng Y, Zhang B, Leo V, Vardy LA, Lu T, et al. , 5-methylcytosine RNA methylation in Arabidopsis Thaliana, Mol. Plant 10 (2017) 1387–1399. [DOI] [PubMed] [Google Scholar]
- [51].Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, Sorek R, Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs, PLoS Genet. 9 (2013), e1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Legrand C, Tuorto F, Hartmann M, Liebers R, Jacob D, Helm M, Lyko F, Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs, Genome Res. 27 (2017) 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.