

Combinatorial levels of BCL2 family members in acute myeloid leukemia stem cells are a key denominator of response to BCL2 inhibitor therapy, and flow cytometry–based measurements predict response in patients both reliably and quickly.
Abstract
The BCL2 inhibitor venetoclax (VEN) in combination with azacitidine (5-AZA) is currently transforming acute myeloid leukemia (AML) therapy. However, there is a lack of clinically relevant biomarkers that predict response to 5-AZA/VEN. Here, we integrated transcriptomic, proteomic, functional, and clinical data to identify predictors of 5-AZA/VEN response. Although cultured monocytic AML cells displayed upfront resistance, monocytic differentiation was not clinically predictive in our patient cohort. We identified leukemic stem cells (LSC) as primary targets of 5-AZA/VEN whose elimination determined the therapy outcome. LSCs of 5-AZA/VEN-refractory patients displayed perturbed apoptotic dependencies. We developed and validated a flow cytometry-based “Mediators of apoptosis combinatorial score” (MAC-Score) linking the ratio of protein expression of BCL2, BCL-xL, and MCL1 in LSCs. MAC scoring predicts initial response with a positive predictive value of more than 97% associated with increased event-free survival. In summary, combinatorial levels of BCL2 family members in AML-LSCs are a key denominator of response, and MAC scoring reliably predicts patient response to 5-AZA/VEN.
Significance:
Venetoclax/azacitidine treatment has become an alternative to standard chemotherapy for patients with AML. However, prediction of response to treatment is hampered by the lack of clinically useful biomarkers. Here, we present easy-to-implement MAC scoring in LSCs as a novel strategy to predict treatment response and facilitate clinical decision-making.
This article is highlighted in the In This Issue feature, p. 1275
INTRODUCTION
Acute myeloid leukemia (AML) remains a cancer with dismal prognosis, particularly in elderly or frail patients ineligible for high-dose chemotherapy as well as patients with high-risk disease (1). The survival of AML cells is dependent on the expression of antiapoptotic factors such as BCL2 (2–4). In recent years, venetoclax (VEN), a potent BCL2 inhibitor, in combination with hypomethylating agents (HMA), such as azacitidine (5-AZA), has replaced HMAs alone as standard-of-care treatment for patients with AML unsuitable for intensive induction chemotherapy, comprising around 50% of AML cases (1, 5).
Owing to its effectiveness and good tolerability, VEN-based therapies are now under investigation as first-line treatment for adult patients with AML eligible for intensive induction chemotherapy such as cytarabine and daunorubicin (NCT04801797 and NCT05177731). Therefore, longitudinal studies linking treatment response to molecular and cytogenetic aberrations are essential to identify the most suitable therapy for each patient with AML. Moreover, biomarkers are required to predict upfront resistance or relapse following the initial response (6). The European LeukemiaNet (ELN) 2017 risk classification currently used to guide treatment decisions for patients with AML, as well as the new ELN 2022, were established prior to VEN-based treatment and might therefore not precisely predict response to 5-AZA/VEN (6, 7). Several denominators for VEN sensitivity have been proposed, such as cell of origin (8), apoptotic priming (9), and monocytic differentiation of blasts (6, 10, 11). The latter has gained particular attention in several studies, and refers to AMLs previously classified as myelomonocytic (M4) or monocytic (M5) based on the French–American–British (FAB) classification and/or that contain blast cells with high levels of CD11b+, CD64+, or CD68+ expression as detected by flow cytometry (12). Ex vivo treatment and transcriptome data have suggested that monocytic AMLs may represent a separate entity of AMLs associated with primary resistance to 5-AZA/VEN treatment (10). Others have shown that reactive oxygen species (ROS)–low, leukemic stem cell (LSC)–enriched AML fractions of monocytic but not primitive AMLs are resistant to 5-AZA/VEN due to dependence on MCL1 rather than BCL2 for survival (10). In contrast, analysis of two recent clinical AML cohorts did not identify monocytic differentiation to be associated with inferior outcomes due to impaired response to HMA/VEN treatment, suggesting more complex scenarios in clinical settings (1, 13).
To mechanistically explore the basis for these clinically relevant discrepancies, we investigated the impact of monocytic differentiation on therapy response. We report that monocytic blasts do not harbor significant functional LSC potential. However, a population of immature, GPR56+ leukemic stem cell–like (LSC-like) cells are BCL2-dependent and are rapidly cleared in patients with AML responsive to 5-AZA/VEN therapy, demonstrating the effectiveness of VEN on LSC-like cells. We further report a clinically translatable biomarker to predict 5-AZA/VEN response. We developed a flow cytometry–based “Mediators of apoptosis combinatorial score” (MAC-Score) that integrates BCL2, BCL-xL, and MCL1 protein expression levels specifically in GPR56+ LSC-like cells using standard flow cytometry. MAC-Score can predict individual patient response as well as duration of 5-AZA/VEN therapy. It outperforms genetic predictors of response and may enable rational patient selection for first-line 5-AZA/VEN treatment for a significant fraction of patients with AML.
RESULTS
Ex Vivo Resistance of AML to 5-AZA/VEN Is Associated with Myeloid Differentiation
To identify predictive parameters for the response to 5-AZA/VEN treatment, we evaluated the sensitivity of 19 AML cell lines treated for 72 hours in vitro. After stratifying these based on the mean fluorescent intensity (MFI) of the monocytic marker CD64 into monocyte-like AMLs (Mono-AML) and primitive-like AMLs (Prim-AML), we observed that Mono-AML cell lines were resistant to 5-AZA/VEN. The majority of Prim-AML cell lines were sensitive even at low concentrations of VEN (Fig. 1A). Overall, the mean IC50 of Mono-AML cell lines was 155-fold higher compared with Prim-AML cell lines (1,901 nmol/L vs. 12 nmol/L VEN in the presence of 1.5 µmol/L 5-AZA; Supplementary Fig. S1A; Supplementary Table S1). To validate the 5-AZA/VEN resistance of Mono-AMLs observed in cell lines, we treated primary cells from 12 patients with AML with increasing concentrations of 5-AZA and VEN. Unsupervised clustering on the cell viability measured after 72 hours of drug exposure revealed two independent clusters. The cluster associated with treatment resistance contained exclusively samples with more than 40% CD64+CD11b+ cells pretreatment (Fig. 1B), whereas specimens with less than 20% CD64+CD11b+ cells were highly sensitive to VEN-based treatment and clustered separately (Fig. 1B). These data further support the notion that myelomonocytic differentiation of bulk AML cells is associated with ex vivo resistance to 5-AZA/VEN treatment. The percentage of CD64+CD11b+ cells obtained from the unsupervised clustering is used from now on to stratify AML patient samples into Mono-AML (>40%) and Prim-AML (<20%).
Figure 1.

Monocytic characteristics indicate poor response to 5-AZA/VEN in cultured AML cells, but fail to predict clinical response in patients. A, Nineteen AML cell lines classified as primitive (Prim-AML, n = 11) or monocytic (Mono-AML, n = 8) based on CD64 surface expression (Mono-AML: MFI > 3,500, Prim-AML: MFI < 1,000) were treated ex vivo with 1.5 µmol/L of 5-AZA and increasing concentrations of VEN for 72 hours. Representative data of two independent replicates. Mean ± SEM of technical replicates. B, Mononuclear cells of patients with AML (N = 12) were treated ex vivo for 72 hours on a drug matrix with increasing 5-AZA and VEN concentrations. Unsupervised clustering was performed based on viability. Each quadrant represents one well with a specific 5-AZA/VEN combination on the drug matrix. C–F, Fifty-four untreated naive patients with AML treated with 5-AZA/VEN as first-line therapy were retrospectively assessed for risk factors of response/refractoriness to therapy. C, Univariate logistic regression was performed for every parameter. Multivariate logistic regression was performed on parameters with P < 0.1 in the univariate analysis. D, Percentages of CD64+ cells in pretreatment AML samples. E, Number of responding or refractory patients associated with differentiation state (based on the percentage of CD64+) or ELN classification groups. F, Abundance of different mutations within responding and refractory patients. ORR, objective response rate. Parts of the figure were created with BioRender.com.
Clinical Response of AML 5-AZA/VEN Is Independent of Myeloid Differentiation
To determine the clinical relevance of our findings, we analyzed a cohort of 54 newly diagnosed patients with AML who received 5-AZA/VEN at Heidelberg University Hospital between 2019 and 2022 for parameters associated with refractoriness to that treatment. Fourteen patients (26%) did not achieve remission. Importantly, the frequency of CD64+ blasts did not predict therapy resistance (Fig. 1C–E; Supplementary Table S2). Using univariate logistic regression analysis, we identified (i) previous myelodysplastic syndrome or myeloproliferative neoplasm (MDS/MPN), (ii) adverse risk AMLs according to the 2017 ELN classification, and (iii) complex karyotype (CK) as the only statistically significant factors predicting the risk of refractory disease (Fig. 1C). CK and MDS/MPN also trended toward independent predictors in the multivariate analysis, with 50% of these patients being refractory. ELN “adverse” was not confirmed as an independent predictor due to the high response rates of RUNX1 and ASXL1 mutated AMLs (Fig. 1E and F). Additional previously proposed predictors of response in AML, including frequency of CD34+ blasts, bone marrow blast at diagnosis, peripheral blood leukocyte count, or age, were not associated with poor response in our analysis (Fig. 1C; Supplementary Fig. S1B). In addition, we investigated the potential outgrowth of monocytic clones by studying longitudinal flow cytometry reports available for 13 patients with primary refractory disease. CD64 surface expression in bone marrow (BM) AML blasts on day 15 and/or day 30 post-treatment was not associated with therapy resistance (Supplementary Fig. S1C). Taken together, these data show that myelomonocytic differentiation based on CD64 staining is not a reliable predictor of response to 5-AZA/VEN in patients.
LSCs Are Enriched in Immature GPR56+ Blasts in Monocytic and Primitive AML
We hypothesized that the discrepancy between clinical and preclinical data was caused by analyzing bulk AML patient samples rather than focusing on the subpopulation of disease-driving LSCs with clonal outgrowth capacity that might determine initial response to 5-AZA/VEN treatment. AML is a highly heterogeneous disease, and reliable detection of LSCs across genetic subclasses has proven difficult. Therefore, we aimed to functionally and transcriptionally characterize LSCs specifically in Mono-AML and Prim-AML samples. Cell-surface expression of CD64 and CD11b readily distinguished two predominant cell populations in our cohort of 72 diagnostic AML samples: Mature CD64+CD11b+ blasts were the predominant population in Mono-AMLs, but were present in Prim-AMLs as well, although at much lower frequencies (40%–97.6% and 0.1%–20% of leukemic blasts, respectively; Fig. 2A and B). To study subpopulations within immature blasts, we included GPR56 expression, a marker for LSCs and adverse outcome in AML (14, 15), to further enrich for functional LSCs in both Mono-AML and Prim-AML samples. We identified an immature GPR56+ population ranging from 0.4% to 92.6% of all AML cells in all examined samples including Prim-AMLs and Mono-AMLs as well as NPM1 wild-type and mutated patient samples for which classic LSC surface markers, such as CD34, often fail to define leukemia initiating cells (Fig. 2A and B; Supplementary Fig. S2A; refs. 14, 16–S19). To validate the association between GPR56 expression and leukemogenicity in Mono- and Prim-AML, FACS-sorted CD64+CD11b+ mature, immature GPR56− (non-LSCs), and immature GPR56+ (LSC-like) populations from 14 patient samples were injected into NSG mice and assessed for leukemogenic potential (Fig. 2C). In 14 of 14 AML specimens, leukemic engraftment (CD45+CD33+) was initiated by the LSC-like population while in only 2 of 14 patients non-LSC-like and mature fractions generated relevant leukemic engraftment (Fig. 2D and E). We observed no differences in the leukemogenic potential of LSC-like cells derived from Mono-AMLs or Prim-AMLs as both AML subclasses showed superior engraftment of the LSC-like population.
Figure 2.

LSC-like cells as defined by functional and transcriptomic parameters are predominantly located in the immature GPR56+ but not in the CD64+CD11b+ mature subpopulation. A, FACS gating strategy for mature, non-LSC, and LSC-like subpopulations. Displayed are AML bulk cells from primitive CD34+ (NPM1-wild-type), CD34− (NPM1-mutated), and monocytic (NPM1-mutated) samples. B, Percentages of mature, non-LSC-like, and LSC-like subpopulations among bulk AML cells in 72 diagnostic AML samples sorted by the frequency of the mature population. C, Schematic overview of the experimental setup for xenotransplantation experiments and RNA sequencing (RNA-seq) of FACS-sorted subpopulations. D, Percentage of human leukemic engraftment obtained from mature, non-LSC, and LSC-like subpopulations of 14 AML samples at endpoints in the bone marrow of NSG mice. Each dot represents an individual mouse with the line marking mean engraftment levels. E, Mean percentage of human engraftment per mouse obtained from mature, non-LSC, and LSC-like subpopulations of 14 AML samples at endpoints in the bone marrow of NSG mice. Each dot represents an individual patient with AML with the line marking mean engraftment levels. Friedmann test was used to compare LSC-like with non–LSC-like and mature subpopulations. F, PCA plot of bulk RNA-seq data from LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 14) and Mono-AML (n = 9) annotated based on subpopulation and AML subclass. Each dot represents a population from one AML sample. G, LSC17 score in LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 14) or Mono-AML (n = 9) patient samples. LSC17 score was calculated for each AML sample as the mean expression of the 17 LSC signature genes. H–J, Normalized counts of BCL2 (H), MCL1 (I), and BCL2L1 (J) expression in LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 14) or Mono-AML (n = 9) patient samples. K, Schematic representation of the experimental setup used in L–O to measure intracellular BCL2, MCL1, and BCL-xL protein expression by flow cytometry. L and M, Mean fluorescence intensity (MFI) of BCL2 in (L) AML bulk and (M) LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 11) or Mono-AML (n = 7) patient samples. N and O, MFI of MCL1 in (N) AML bulk and (O) LSC-like, non-LSC, and mature subpopulations from Prim-AML (N = 11) or Mono-AML (N = 7) patient samples. P and Q, Representative tSNE plots of (P) AML26 (Prim-AML) and (Q) AML50 (Mono-AML) showing expression of CD64, CD34, GPR56, MCL1, BCL2, and BCL-xL. AML bulk is defined as mononuclear cells from patients with AML after the exclusion of dead cells, doublets, lymphocytes, and nucleated erythrocytes. Two-way ANOVA with Tukey correction for multiple comparisons test was used to compare groups of four, and Mann–Whitney test was used to compare groups of two unless specified otherwise. Each dot represents an AML patient sample with the line marking the mean unless specified otherwise. Fix, fixation; IC-staining, intracellular staining; Mut, mutated; NL, non-LSC; NSG, NOD.Prkdcscid.Il2rgnull; Perm, permeabilization; WT, wild-type. Parts of the figure were created with BioRender.com.
The Immature GPR56+ Fraction Is Enriched for Stemness-Associated Molecular Programs
Due to the similarities in the leukemogenic potential observed in LSC-like cells from both Mono-AMLs and Prim-AMLs, we sought to further characterize LSC-like and mature cells by performing RNA sequencing on sorted cell populations from 23 patients with AML. Intriguingly, dimensionality reduction using principal component analysis (PCA) revealed clear clustering of the samples based on population (LSC-like and mature) but not based on the two AML subclasses (Fig. 2F). For selected samples, we also sequenced the non-LSCs that clustered either with LSC-like or mature cells. Differential gene-expression analysis between LSC-like and mature cells showed that the upregulated genes in LSC-like cells contained known cancer stem cell markers, including KIT, ERG, GPR56, and PROM1 (14, 15, 20–23), while upregulated genes in mature cells included monocytic markers such as S100A9, S100A8, and CD14 and were enriched for pathways associated with myeloid differentiation (refs. 24–27; Supplementary Table S3; Supplementary Fig. S2B and S2C). Moreover, several stemness scores, including the LSC17 score, were significantly higher in LSC-like compared with mature cells, irrespective of the AML subclass. As expected, non-LSCs scored in between these two populations (Fig. 2G; Supplementary Fig. S2D; ref. 15). As we observed no striking differences between LSC-like cells from AML samples stratified based on their monocytic-like surface expression on blasts, we also assessed whether genetics would explain the subtle differences observed between different LSC-like samples in the PCA. Indeed, RUNX1-mutated LSC-like cells and NPM1-mutated LSC-like cells clustered separately (Supplementary Fig. S2E). These findings highlight the transcriptomic stemness characteristics of functionally defined LSC-like cells and indicate that the transcriptomic clustering of LSCs is largely determined by the underlying mutational profile rather than the differentiation state of their blast progeny.
LSC-like Cells Predominantly Express BCL2, whereas MCL1 Is Highly Expressed in Mature Blasts
Intrigued by the similar clinical responses of Mono-AMLs and Prim-AMLs to 5-AZA/VEN treatment, we analyzed the gene expression of BCL2 family members, such as BCL2, MCL1, and BCL2L1, within the LSC-like population of either AML subclass. In line with previous studies (4), BCL2 was 4.7-fold more highly expressed in the LSC-like populations compared with the mature populations (FDR < 0.01; Fig. 2H), whereas MCL1 expression was 2.3-fold higher in the mature populations compared with LSC-like cells (FDR < 0.01; Fig. 2H and I). When we compared the expression of BCL2 and MCL1 within either LSC-like or mature cells, we did not find any significant differences between Mono-AMLs and Prim-AMLs. In contrast, expression of BCL2L1, encoding for BCL-xL, was not skewed toward a particular population (Fig. 2J). Furthermore, transcriptomic analysis of the extended BCL2 family also did not cluster based on AML subclass (Supplementary Fig. S2F). These results suggest that LSC-like cells express higher levels of BCL2 and lower levels of MCL1 compared with more mature cells, irrespective of the overall AML differentiation status.
To confirm expression levels of the BCL2 family members on the protein level, we established an intracellular staining protocol for flow cytometry combining our surface marker panel with antibodies specific for BCL2, MCL1, and BCL-xL (Fig. 2K). Without subgating on LSC-like, non-LSC, or mature populations, bulk AML cells derived from Prim-AMLs showed substantial heterogeneity and a 1.7-fold higher expression of BCL2 proteins compared with the overall more homogeneous expression in Mono-AMLs (Fig. 2L). The analysis of pregated populations, however, revealed that BCL2 was 2.7-fold more highly expressed in LSC-like cells compared with mature cells, but this was irrespective of AML subclass, abrogating the AML subclass–associated differences observed in the bulk analysis (Fig. 2M). Vice versa, MCL1 protein assessment in bulk revealed 2.1-fold higher levels in Mono-AMLs compared with Prim-AMLs (Fig. 2N). After gating on the LSC-like and mature populations, MCL1 protein expression did not differ between Mono-AML and Prim-AML samples, with MCL1 being 2.1-fold more highly expressed in the mature cells compared with LSC-like cells (Fig. 2O). In contrast to the opposing behavior of BCL2 and MCL1, BCL-xL showed no clear population nor AML subclass–specific expression differences (Supplementary Fig. S2G and H).
Further dissecting the relative distributions of LSC-like and mature cells in both subclasses, representative t-distributed stochastic neighbor embedding (tSNE) plots of two representative AML cases analyzed by flow cytometry demonstrate that Prim-AMLs contained more immature LSC-like cells, expressing high levels of GPR56 together with BCL2, but also contained a smaller fraction of mature cells expressing CD64 and MCL1 (Fig. 2P). On the contrary, Mono-AMLs were enriched for more mature cells expressing high levels of CD64 and MCL1, and contained only a small immature LSC-like population expressing CD34, GPR56, and BCL2 (Fig. 2Q). Taken together, assessment of BCL2, MCL1, and BCL-xL protein levels extends the findings of the transcriptional data and highlights the similarities of the LSC-like populations of Prim-AMLs and Mono-AMLs despite the fact that their relative frequencies vary extensively in the two different AML subclasses.
BH3 Profiling Confirms That LSC-like Cells of Both AML Subclasses Are BCL2-Dependent
Next, we studied whether the LSC-like and mature population–specific expression differences of BCL2 family members translate into functional consequences. To address this, we first performed BH3 profiling to measure the activity of apoptotic pathways in the same AML samples. Similar to the intracellular staining of BCL2 family members, we combined our surface markers with BH3 profiling to assess mitochondrial apoptotic priming and dependence on prosurvival BCL2 family proteins in bulk AML as well as in pregated LSC-like, non-LSC, and mature cells. Briefly, we exposed unperturbed cells to proapoptotic BH3 peptides plus different mimetics to assess the release of mitochondrial cytochrome C, which irreversibly drives cells into apoptosis (ref. 2; Fig. 3A and B). In unsorted, bulk AML cells, dependence on BCL2 was higher in Prim-AMLs compared with Mono-AMLs upon exposure to BH3 mimetic VEN (Fig. 3C). However, differential analysis on pregated LSC-like, non-LSC, and mature cells abrogated this difference while revealing that LSC-like cells were more dependent on BCL2 compared with the non-LSC and mature cells in both AML subclasses (Fig. 3D). This was supported by increased sensitivity of LSC-like cells to BAD, a BH3 peptide interacting with BCL2 (Supplementary Fig. S2I and S2J). The exact opposite effect was found after assessing the dependency on MCL1 via cytochrome C release upon exposure to the BH3 peptide MS-1 (Fig. 3E and F). Here, mature cells showed a 1.7-fold higher MS-1–mediated apoptotic priming compared with LSC-like cells, irrespective of the AML subclass. In agreement with the BCL-xL protein expression pattern, exposure to the BH3 peptide HRK showed neither clear population nor AML subclass–specific dependency for BCL-xL (Supplementary Fig. S2K and S2L).
Figure 3.

LSC-like and mature subpopulations show distinct dependencies on antiapoptotic proteins and response to 5-AZA/VEN therapy independent of AML subclass. A, Workflow for C–F. Mononuclear cells of diagnostic AML patient samples were stained with surface antibodies, followed by BH3 profiling and quantification of AUC to assess apoptotic susceptibility in bulk and pregated subpopulations. B, Overview of assessed BH3 mimetics and their target proteins. C and D, AUC of VEN mediated cytochrome C release in (C) AML bulk and (D) LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 11) or Mono-AML (n = 7) patient samples. E and F, AUC of MS1 mediated cytochrome C release in (E) AML bulk and (F) LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 11) or Mono-AML (n = 7) patient samples. Each dot represents an individual AML patient sample with the line marking the mean. Mann–Whitney test was used to compare groups of two. Two-way ANOVA with Tukey correction for multiple comparisons test was used to compare groups of four. G, Schematic representation of ex vivo treatment strategy for H–J. Mononuclear cells of diagnostic AML patient samples (N = 18) were treated ex vivo for 24 hours at 1.5 µmol/L 5-AZA and 100 nmol/L VEN. H, Relative viability of LSC-like, non-LSC, and mature subpopulations from Prim-AML (n = 11) or Mono-AML (n = 7) patient samples was compared using two-way ANOVA with Tukey correction for multiple comparisons test. I and J, Representative tSNE plots of (I) AML26 (Prim-AML) and (J) AML50 (Mono-AML) highlighting expression of CD64 and GPR56 in 5-AZA/VEN-treated and untreated controls. K, Schematic representation of PBMC collection strategy of patients with AML undergoing 5-AZA/VEN therapy. L, Quantification of mature, non-LSC, and LSC-like cell counts from PBMCs relative to pretherapy in the first week of 5-AZA/VEN treatment in 3 patients undergoing therapy initiation. Each line represents an individual patient with each dot on the line representing an individual timepoint of the patient. M, Representative gating strategy highlighting population dynamics of LSC-like and mature AML cell frequencies during the first week of 5-AZA/VEN treatment. All percentages represent the fraction of total live-singlet AML cells. N, Schematic representation of ex vivo treatment strategy for O. Mononuclear cells of 5-AZA/VEN first-line–treated patients with AML (N = 24) were treated ex vivo for 24 hours at 1.5 µmol/L 5-AZA and 100 nmol/L VEN. O, Relative viability of LSC-like, non-LSC, and mature subpopulations was compared using the Mann–Whitney test. Each dot represents an AML patient sample with the line marking the mean unless specified otherwise. Ab, antibody. Parts of the figure were created with BioRender.com.
Taken together, our data suggest that the differences in the mRNA and protein expression of BCL2 and MCL1 predict cellular population-specific dependencies (LSC-like/mature), which are independent of AML subclass. In general, LSC-like cells were most dependent on BCL2.
Ex Vivo 5-AZA/VEN Exposure Eradicates LSC-like but Spares Mature Cells in Both AML Subclasses
Because BH3 profiling indicated that LSC-like cells depend on BCL2 and mature cells on MCL1, we hypothesized that differential ex vivo treatment response of bulk Prim/Mono-AMLs to the combination of 5-AZA/VEN is mainly driven by these subpopulations. Therefore, we exposed bulk AML cells from 18 newly diagnosed patients to 5-AZA/VEN for 24 hours and analyzed the viability of bulk cells and subpopulations in comparison with untreated controls by flow cytometry (Fig. 3G). As expected, bulk cells from Mono-AML patient samples exhibited significantly higher resistance to the treatment compared with Prim-AMLs (Supplementary Fig. S2M). The analysis of subpopulation viability post-5-AZA/VEN revealed that in both AML subclasses 5-AZA/VEN only marginally reduced the mature population by 40% ± 32% and 22.5% ± 11.5% in Prim-AMLs and Mono-AMLs, respectively (Fig. 3H). In comparison, LSC-like cells derived from both Prim-AML and Mono-AML subclasses were efficiently eliminated by 90% ± 10.6% or 79% ± 19%, respectively (Fig. 3H). Representative tSNE plots of viable cells from two AML samples were overlaid with heat maps of CD64 and GPR56 expression to identify mature and LSC-like fractions, respectively (Fig. 3I and J). The differential response between mature and LSC-like cells was further corroborated in the remaining viable cells as LSC-like cells were replaced by an enrichment of mature cells independent of AML subclasses (Fig. 3I and J). In line with the results from BH3 profiling and of BCL2 expression, these data show that the differences in bulk AML sensitivity are driven mainly by the initial difference in proportions of the treatment-resistant but not disease-propagating mature subpopulation between Prim-AML and Mono-AML samples. Importantly, LSC-like cells were effectively eliminated by 5-AZA/VEN in both AML subclasses.
5-AZA/VEN Rapidly Clears LSC-like Cells in Responsive Patients
We next evaluated the potential of 5-AZA/VEN to target LSC-like cells in patients. For this purpose, peripheral blood mononuclear cells (PBMC) were collected from three patients with AML with peripheral blasts before treatment initiation (day 0) and during 5-AZA/VEN therapy between days 1 and 6 (Fig. 3K). All three patients responded to therapy and achieved a significant reduction of the peripheral blast count within the first 24 hours of treatment. Although the relative number of LSC-like cells decreased and remained low, a significant fraction of mature and non-LSC cells persisted in all three patients during the first days of treatment (Fig. 3L and M). Based on this, we assessed the ability of ex vivo 5-AZA/VEN treatment to predict clinical response. We compared the viability of LSC-like, non-LSC, and mature fractions after 24 hours ex vivo 5-AZA/VEN treatment in 24 patients with known clinical response to first-line 5-AZA/VEN (Fig. 3N). Despite considerable heterogeneity, LSC-like and to a lesser extent non-LSC cells from refractory patients were more resistant to treatment ex vivo, whereas mature cells remained unaffected by the ex vivo drug treatment and their ex vivo response was independent of the patient's clinical response (Fig. 3O). These data further highlight the need to study disease-driving LSC-like subpopulations in AML to assess therapy response and raise the possibility that functional properties of LSC-like cells could be predictive of the response to 5-AZA/VEN treatment.
Rapid and Robust Prediction of Ex Vivo Response by a BCL2 Family–Based LSC-like Response Score
Flow cytometry is a routine diagnostic tool for the diagnosis and clinical monitoring of AML and can support clinical decision-making within several hours after sampling. We hypothesized that BCL2 family protein expression levels in LSC-like cells might predict patient response to 5-AZA/VEN treatment at diagnosis. To address this, we performed ex vivo 5-AZA/VEN treatment with simultaneous intracellular staining of BCL2, MCL1, and BCL-xL on 54 diagnostic AML patient samples (Supplementary Fig. S3A). Stratification based on the viability of LSC-like cells (<5%, 5%–20%, or > 20%) after 24 hours ex vivo 5-AZA/VEN treatment showed higher intracellular BCL2 expression scores in AML specimens sensitive to ex vivo treatment (Supplementary Fig. S3B).
Although BCL2 conveys sensitivity to VEN, MCL1 and BCL-xL can promote survival independent of BCL2 (5). Thus, to additionally account for these factors contributing to resistance, we incorporated all three proteins into a singular response score and termed it the MAC-Score, which can be calculated for subpopulations defined by flow cytometry. MAC-Score calculates the ratio between the normalized MFI of the drug target (BCL2) and the normalized MFI of the resistance factors (sum of MCL1 and BCL-xL) as follows: BCL2Norm. MFI/(MCL1Norm. MFI + BCL-xLNorm. MFI; for details, see Methods).
Accounting for the alternative inhibitors of apoptosis and potential mediators of VEN resistance, MAC-Score further improved separation between ex vivo 5-AZA/VEN sensitivity AML samples compared with individual protein levels (Supplementary Fig. S3B–S3E). These data highlight the benefit of combinatorial assessment of BCL2 family proteins specifically within LSC-like cells.
MAC-Score in LSC-like Cells Predicts Clinical Response and Remission Duration of 5-AZA/VEN
We next assessed whether individual BCL2 family protein levels or MAC-Score predict clinical response to 5-AZA/VEN treatment. We analyzed the expression of BCL2 family proteins BCL2, MCL1, and BCL-xL together with cell-surface expression profiling in 35 diagnostic samples from two independently processed multicenter cohorts (Fig. 4A, cohorts 1+2). Here, BCL2 was significantly more highly expressed in LSC-like cells of patients achieving CR, CRi, or morphologic leukemia-free state (MLFS; responder), whereas MCL1 and BCL-xL were more highly expressed in patients with stable disease (SD), partial response (PR), or progressive disease (PD; nonresponder; Fig. 4B). Similar trends were observed in non-LSC cells and total blasts, whereas BCL2 family expression in mature cells did not differ between responders and nonresponders (Supplementary Fig. S4A–S4C). However, expression of neither BCL2 family protein alone in LSC-like cells nor in other subpopulations provided a clear separation between responders and nonresponders, and the levels of all three proteins showed high interpatient variability (Fig. 4B; Supplementary Fig. S4A–S4C).
Figure 4.

Response to 5-AZA/VEN therapy in patients with AML can be predicted by MAC scoring in LSC-like cells. A, Schematic representation of the experimental design for B–G. Mononuclear cells of AML patient samples treated first-line with 5-AZA/VEN from three independently processed cohorts (cohort 1: n = 17, cohort 2: n = 18, and vohort 3: n = 24) were stained with surface antibodies, followed by intracellular staining of three BCL2 family proteins. MAC-Score was calculated based on normalized BCL2 family protein expression levels in LSC-like, non-LSC, mature, and total blast cells. B, Expression of BCL2, MCL1, and BCL-xL in LSC-like cells of patients with AML from cohorts 1 and 2 combined and associated 5-AZA/VEN therapy outcome. Protein expression is shown as MFI z-scores. C, MAC-Score in LSC-like cells of patients with AML from cohorts 1 and 2 combined and association to 5-AZA/VEN therapy outcome. D, Comparison of MAC-Score in LSC-like, non-LSC, mature, and total blast cells of patients with AML from cohorts 1 and 2 and association to 5-AZA/VEN therapy outcome. E, EFS of first-line 5-AZA/VEN AML patients from cohorts 1 and 2 combined with above and below median MAC-Score, BCL2 expression, MCL1 expression, or BCL-xL expression in LSC-like cells. F, MAC-Score in LSC-like cells of patients with AML from cohort 3 and associated 5-AZA/VEN therapy outcome. G, EFS of first-line 5-AZA/VEN AML patients from cohort 3 with above (>0.4) and below (<0.4) median MAC-Score in LSC-like cells. H, Schematic representation of the experimental design for I–J. Mononuclear cells of patients with relapsed/refractory AML who received 5-AZA/VEN as a salvage therapy (cohort 4: n = 23) were stained with surface antibodies, followed by intracellular staining of BCL2 family proteins. I, MAC-Score in LSC-like cells of patients with AML from cohort 4 and associated 5-AZA/VEN therapy outcome. J, EFS of salvage-treated 5-AZA/VEN AML patients from cohort 4 with above (>0.4) and below (<0.4) median MAC-Score determined in LSC-like cells. Each dot represents an AML patient sample with the line marking the mean unless specified otherwise. Mann–Whitney test was used to compare groups and log-rank test to compare therapy durations of AML patients. R/R, relapsed/refractory to standard induction. Parts of the figure were created with BioRender.com.
As MAC-Score outperformed the individual BCL2 family proteins in predicting 5-AZA/VEN response ex vivo, we calculated MAC-Scores in LSC-like, non-LSC, mature, and total blast cells also in these two patient cohorts (Fig. 4C and D; Supplementary Table S4). Both cohorts individually and together showed significantly higher MAC-Scores in LSC-like cells derived from patients who responded to 5-AZA/VEN treatment compared with the LSC-like cells analyzed from nonresponder patients (Fig. 4C and D; Supplementary Fig. S5A and S5B). This phenomenon was also detected in total blasts but to a lower extent and without a clear separation between responders and nonresponders (Fig. 4D). These findings indicate that MAC-Score in LSC-like cells outperforms expression of individual BCL2 family proteins as a binary predictor of clinical response to 5-AZA/VEN.
To study response duration, we assessed event-free survival (EFS) of these two cohorts. Patients who discontinued treatment due to reasons other than disease progression were censored. For both cohorts, we observed a significantly longer 5-AZA/VEN response in patients with median MAC-Scores greater than 0.4 compared with patients with median MAC-Scores less than 0.4 in LSC-like cells (Fig. 4E; Supplementary Fig. S5C–S5E). BCL2 family proteins alone did not reach the same predictive value compared with MAC scoring, even though high BCL2 levels alone predicted longer and high MCL1 shorter EFS if analyzed in LSC-like cells (Fig. 4E; Supplementary Fig. S5F–S5H). Moreover, neither MAC-Score nor individual protein levels of the three BCL2 members determined from the other subpopulations provided reasonable predictive power and all were outperformed by MAC-Score in LSC-like cells (Supplementary Fig. S5E–S5H). We then validated these findings in a third, independently processed cohort (n = 24), in which MAC-Scores in LSC-like cells were again significantly higher in patients who responded to 5-AZA/VEN treatment and were low in patients who did not (Fig. 4F). EFS was also longer for patients with above- median MAC-Scores, suggesting improved therapy response (Fig. 4G). Collectively, these findings support the concept that MAC-Score in LSC-like cells can accurately predict response to first-line treatment with 5-AZA/VEN in older, frail patients, outperforming prediction based on individual BCL2 family proteins alone.
5-AZA/VEN is currently under investigation for first-line treatment of younger patients and has shown efficacy as a salvage therapy option for relapsed and refractory patients. Therefore, we tested if response to 5-AZA/VEN can be predicted in young, relapsed/refractory patients by assessing MAC-Scores in 23 AML samples receiving 5-AZA/VEN as salvage therapy (Fig. 4H). The samples were biobanked either at diagnosis or before the start of 5-AZA/VEN treatment. In this salvage setting, MAC-Scores above median were again highly predictive of binary clinical response and longer EFS, opening the path for the prospective assessment of biomarker-based choice of induction regimen (Fig. 4I and J). Together, MAC-Score enabled a robust identification of patients who benefit from 5-AZA/VEN as first-line and salvage therapy and is a step toward selecting the best therapy for each patient on an individual basis.
MAC-Score in LSC-like Cells Outperforms BH3 Profiling
Measurement of apoptotic dependence by BH3 profiling has previously been proposed as a predictor of response to 5-AZA/VEN (9). To compare MAC-Score with BH3 profiling, we selected 15 samples (7 nonresponders vs. 8 responders) from the three cohorts above treated first-line with 5-AZA/VEN and assessed both readouts. As expected, MAC-Score was highly predictive of binary clinical response (Supplementary Fig. S6A). Moreover, EFS showed robust differences when stratified by either the median MAC-Score of the selected 15 samples or the previously established cutoff of 0.4 (Supplementary Fig. S6B and S6C). In contrast, BH3 profiling in LSC-like cells based on VEN-, HRK-, or MS1-induced cytochrome C release did not predict clinical response (Supplementary Fig. S6D–S6F). Assessing the sum of the HRK- and MS1-induced cytochrome C release, as previously reported, did not improve the prediction (Supplementary Fig. S6G; ref. 9). BH3 profiling in LSC-like cells also did not correlate with EFS in these patients (Supplementary Fig. S6H). When total blasts were assessed, only VEN-induced cytochrome C release predicted binary clinical response but showed only a trend toward longer EFS (Supplementary Fig. S6I–S6M). Importantly, the observed separation was weaker compared with MAC-Score in this patient cohort. In summary, MAC-Score is a robust alternative to BH3 profiling for VEN response prediction.
Combined Analysis of MAC-Scores in LSC-like Cells Identifies Drivers of VEN Resistance
To further assess the prognostic accuracy of MAC-Score, we combined the MAC-Scores in LSC-like cells from all three first-line–treated cohorts (cohorts 1–3), extending the analysis to 59 patients treated first-line with 5-AZA/VEN. As expected, MAC-Scores were significantly higher in responders compared with nonresponders (Fig. 5A). We assessed the prognostic accuracy of MAC-Score in the combined cohorts with ROC analysis and observed a ROC value of 0.95, revealing high accuracy to predict 5-AZA/VEN treatment response in patients (Fig. 5B). The combined EFS analysis of all 59 5-AZA/VEN first-line–treated patients showed a 4-fold prolongation of EFS in patients with above-median scores with an EFS of 3 versus 12 months (Fig. 5C). As some patients with MAC-Scores less than 0.4 responded to 5-AZA/VEN, we next assessed whether MAC-Scores within responders would discriminate with regard to response duration. Indeed, responders with greater than 0.4 MAC-Scores had a longer EFS with 5-AZA/VEN treatment compared with responders with MAC-Scores less than 0.4, with a median EFS of 6 versus 12 months (Fig. 5D). This shows that MAC-Score can be used to distinguish patients with long-lasting response to 5-AZA/VEN.
Figure 5.

MAC-Score in LSC-like cells predicts response to 5-AZA/VEN with high accuracy. A, MAC-Score in LSC-like cells of patients with AML from all first-line 5-AZA/VEN AML patients combined (cohorts 1–3) and association to 5-AZA/VEN therapy outcome. B, Receiver operating characteristic (ROC) curve of MAC-Score and therapy outcomes of all first-line 5-AZA/VEN AML patients combined (cohorts 1–3). C, EFS of all first-line 5-AZA/VEN AML patients combined (cohorts 1–3) with above and below median MAC-Score. D, EFS of all first-line 5-AZA/VEN AML patients who achieved complete remission from combined cohorts (cohorts 1–3) with above (>0.4) and below (<0.4) median MAC-Score. E, Patient characteristics of first-line 5-AZA/VEN cohorts with retrospectively assessed risk factors of refractoriness to therapy. Univariate logistic regression was performed for every parameter. Multivariate logistic regression was performed on parameters with P < 0.15 in the univariate analysis. F, EFS from combined cohorts (cohorts 1–3) with above (>0.4) and below (<0.4) median MAC-Score based in patients with complex karyotype, RUNX1, or NPM1 mutation. G, MAC-Score in LSC-like cells from diagnostic AML patients receiving first-line standard induction chemotherapy and association to therapy outcome. H, Schematic representation of the experimental design for I–J. MAC-Score was calculated based on normalized BCL2 family protein expression levels in LSC-like cells from diagnostic AML patients independent of received therapy (n = 95). I, MAC-Score of patients with AML differentiated by the number of structural variants. J, MAC-Score of patients with AML with different recurrent AML mutations. K, Schematic model outlining the MAC-Score concept for predicting clinical response to 5-AZA/VEN. Each dot represents an AML patient sample with the line marking the mean unless specified otherwise. Mann–Whitney test was used to compare groups and the log-rank test to compare therapy durations of patients with AML. Resp, responder. Parts of the figure were created with BioRender.com.
Finally, to identify predictors of response to 5-AZA/VEN, we performed logistic regression analysis on all 59 first-line–treated patients (Supplementary Table S4) and included MAC-Score as one of the assessed variables. Using univariate analyses, MAC-Score and CK were identified as statistically significant parameters in this set of samples (Fig. 5E). We then performed multivariate analysis on all parameters with P < 0.15 and observed that MAC-Score remained as the sole predictor of response with an odds ratio of 5.1 per 0.1 points (Fig. 5E). MAC-Score predicted EFS even within genetic subgroups such as CK, RUNX1, or NPM1-mutated patients, highlighting its potential role in patient stratification beyond genetics (Fig. 5F; Supplementary Fig. S7A). Of note, MAC-Score showed no difference between patients who did or did not respond to standard induction therapy, highlighting its specificity to predict response to VEN-based therapies (Fig. 5G).
Taken together, MAC-Score predicts patients’ initial response to 5-AZA/VEN with an accuracy superior to mutational profiling and can identify patients with long response durations.
MAC-Score Deconvolutes Response Heterogeneity within Mutational Patterns
To gain insights into the genetic profiles of patients with AML with different MAC-Scores, we correlated clinical structural variant (SV) analysis and targeted mutational profiling with MAC-Scores for 95 diagnostic AML patient samples independent of the received treatment (Fig. 5H; Supplementary Fig. S7B; Supplementary Table S5). We observed that patients with low MAC-Scores harbored a higher number of SVs compared with patients with high MAC-Scores (Fig. 5I). Moreover, patients with AML with mutations in TET2 or JAK2 and CALR had exclusively low MAC-Scores (Fig. 5J). This is in line with the observed association of prior MDS/MPN diagnosis with clinical refractoriness to 5-AZA/VEN, although the sample size remains small (Fig. 1C). In contrast, patients with high MAC-Scores were enriched for IDH1/2, DNMT3A, and splicing mutations (Fig. 5J), suggesting that specific genetic alterations affect the differential dependency of LSC-like cells on BCL2 family proteins. Importantly, the heterogeneity of MAC-Scores within complex karyotypic or IDH1/2-mutant AMLs and the associated 5-AZA/VEN response highlights the value of patient individualized assessment. Taken together, MAC scoring in LSC-like cells aggregates the impact of the correlatively associated mutational background and other still-elusive nongenetic factors by providing causality-based information that predicts patient response to 5-AZA/VEN therapy (Fig. 5K).
DISCUSSION
Treatment with 5-AZA in combination with the BCL2 inhibitor VEN is a highly effective and reasonably well-tolerated regimen that has transformed the standard of care for unfit patients with AML. Due to its efficacy, VEN-based therapies are now evaluated as treatment for adult patients with AML eligible for intensive induction chemotherapy, highlighting the need to identify patients with good and poor response to 5-AZA/VEN, respectively, for tailored AML treatment. Here, we present the MAC-Score that allows the prediction of response to 5-AZA/VEN. It is based on the combinatorial calculation of the individual protein expression levels of BCL2, BCL-xL, and MCL1 specifically in disease-driving LSC-like cells. MAC scoring predicts therapy response and duration of first-line as well as salvage treatment with 5-AZA/VEN. MAC-Score–based prediction of response would enable patient selection for clinical trials that go beyond genetic or phenotypic risk factors and enable a true personalized medicine approach in AML (Fig. 5K).
Targeting the disease-driving LSC population is key to eradicating AML, making correct identification of LSCs and a deeper understanding of their biology of paramount importance (28–30). LSCs have been implicated with adverse outcomes in AML, which entails both refractory and relapse cases (14, 15). Initially, LSCs were treated as a therapy-resilient population that upon completion of therapy would reinitiate the disease, much like in murine transplantation recipients of LSCs. In refractory disease, the focus has shifted from studying classic stemness features to dissecting metabolic rewiring and functional changes such as senescence and dormancy in response to chemotherapy (31–34). Meanwhile, the role of LSCs for initial response to different induction therapies in patients remains elusive. Here, we identify specific ratios of BCL2 family proteins as a dominant mechanism of a priori VEN resistance.
Importantly, they show a markedly improved prediction when measured in LSC-like cells compared with more differentiated cells and total blasts. Because clinical response relies on features specifically present in these disease-driving cells, LSC-specific assessment is essential to deconvolute resistant mechanisms or drug vulnerabilities, as shown previously (4, 34). Assessment of LSC-like cells was not part of otherwise powerful datasets such as BEAT-AML or The Cancer Genome Atlas, which combine bulk-level, unfractionated RNA-sequencing data with unfractionated drug response data, both confounded by unknown size of the typically highly variable presence of more differentiated cells that blur the results (35, 36). Here, we show that gating on immature GPR56+ blasts serves as a robust LSC marker set for clinical stratification. Our findings extend the observations, in which LSCs of Mono-AML defined by ROS levels showed VEN resistance (10). In our cohort, gating on the GPR56+ LSC population revealed highly variable LSC frequencies between 0.5% and 90% of AML cells, whereas ROS-low–based default gating identifies cells displaying the 15% to 20% lowest ROS within all AML blasts, irrespective of the vastly differing actual size of the LSC population in each patient. Samples with low LSC frequencies are thereby likely contaminated by more mature, MCL1-expressing cell types influencing results and complicating the interpretation.
In AML, mutational profiles are commonly correlated with clinical outcomes. However, responses to 5-AZA/VEN treatment based on genetic alterations are distinct from high-dose induction therapies and results are inconsistent across different studies (1, 6, 7, 37). Indeed, 5-AZA/VEN clinical trials and preclinical research reported that AML patients with mutations in NPM1, IDH1/2, and RUNX1 experience a high rate of durable responses (1, 6, 38). In contrast, CK, activating mutations of FLT3 or RAS have been associated with resistance to 5-AZA/VEN, but are never dichotomous predictors of response, highlighting the need for personalized response prediction (1, 6, 37). Interestingly, we observed that the mutational background influences BCL2 family expression, explaining the association of specific genomic features with 5-AZA/VEN response. Our MAC-Score may be particularly useful for situations with contradicting genetic predictors such as IDH mutations in CK AMLs or NPM1 mutations in MPN-transformed AMLs without clear risk classification.
Although the correlation between high BCL2 levels and response to 5-AZA/VEN treatment seems clear in theory, we and others observed BCL2 levels alone to poorly predict response to 5-AZA/VEN therapy in AML when unfractionated bulk cells were assessed (5). Indeed, combinatorial reliance on other BCL2 family members plays a pivotal role in controlling the balance between survival and apoptosis (3, 5, 9, 39). Moreover, disease- and tissue-specific dependencies on these proteins have been proposed. For example, primary myelofibrosis seems to be sensitive to cotargeting of BCL2 and BCL-xL, whereas chronic lymphocytic leukemia is highly dependent on BCL2 alone (40, 41). Selective dependencies such as BCL-xL in the megakaryocytic lineage or myocardial MCL1 dependency are key determinants of drug toxicities and have to be considered in the design of treatment regimens (42, 43). These dependencies are usually not absolute and can plastically change during disease course (44). Our approach enables direct quantification of BCL2 family protein ratios between AML subpopulations and allows focusing on disease-driving LSC-like cells, which outperformed the predictive power of ex vivo drug testing or BH3 profiling. This critically distinguishes MAC scoring (Fig. 5K) from previous attempts to predict the efficacy of VEN-based treatment based on gene expression of BCL2 family ratios in unfractioned bulk samples or cell lines (5, 45–47). Furthermore, responders with low MAC-Scores are prone to early relapse, implying that treatment duration is already determined at a pretreatment, naive stage and arguing that clonal outgrowth is only one of many mechanisms of 5-AZA/VEN resistance.
Turnaround time for RNA analysis, BH3 profiling, or ex vivo culture assays spans from days to weeks and is resource-intensive, especially when subpopulations such as LSC-like cells are assessed. Most importantly, they also have inferior predictive accuracy (44). In contrast, MAC scoring by flow cytometry can easily be incorporated into clinical routine diagnosis and monitoring of patients with AML and can support clinical decision-making within several hours after sampling. These attributes make MAC scoring a simple, globally accessible, and affordable tool with low turnover time that can support therapeutic decision-making regarding 5-AZA/VEN versus intensive induction as first-line therapy for selected fit patients. Prospective studies to further validate MAC scoring will provide more insights into its applicability to guide therapy in first-line and relapsed/refractory disease. Importantly, the MAC-Score concept could be easily adapted to predict apoptotic dependencies beyond BCL2 and beyond AML. Adapting MAC-scoring strategies to other inhibitors of the BCL2 family may identify patients particularly sensitive to inhibition of MCL1 or BCL-xL and thus extend biomarker-guided personalized medicine.
METHODS
Primary AML Patient Samples and Clinical Data
AML samples were collected from diagnostic peripheral blood (PB) or BM aspirations at the University Hospitals in Heidelberg or Hannover in accordance with the Declaration of Helsinki and based on institutional approvals after obtaining written informed consent from each patient. The project was approved by the Ethics Committee of the Medical Faculty of Heidelberg (S-169/2017 and S-648/2021) and the local Ethics Review Committee of Hannover Medical School (ethical vote No.7972_BO_K_2018). PBMCs were isolated by density gradient centrifugation using Ficoll Paque Plus (GE Healthcare, cat #GE17-1440-03), and stored in liquid nitrogen until further use. Detailed patient and specimen characteristics are listed in the Supplementary Tables and are based on the clinical diagnostic reports. An experienced hematologist categorized all patients by retrospectively reviewing all pathology and flow cytometry reports in accordance with ELN 2017 (7). Patients achieving CR, CRi, and MLFS were defined as responders, and patients achieving only PR, SD, or PD as nonresponders. For EFS calculations, time to proven molecular, flow cytometry–based, or cytologic relapse from start of therapy was calculated.
Processing of Primary AML Cells
Viable cryopreserved AML PB samples were thawed at 37°C in Iscove's Modified Dulbecco's Medium (IMDM) containing 10% FBS and treated with DNase I for 15 minutes (100 µg/mL).
Ex Vivo Drug Screening with Primary Leukemia Cells
Recovered cells were cultured using previously established protocols (14, 48) using IMDM, 15% BIT (bovine serum albumin, insulin, transferrin; Stem Cell Technologies, cat #09500), 100 ng/mL SCF (PeproTech, cat #300-07), 50 ng/mL FLT3L (PeproTech, cat #300-19), 20 ng/mL IL3 (PeproTech, cat #200-03), 20 ng/mL G-CSF (PeproTech, cat #300-23), 100 µmol/L β-mercaptoethanol (Thermo Fisher, cat #31350010), 500 nmol/L SR1 (StemRegenin 1, STEMCELL Technologies, cat #72342), 1 µmol/L UM729 (STEMCELL Technologies, cat #72332), and 1% penicillin–streptomycin (Sigma, cat #P4458-100ML). For drug assay in Fig. 1, 0.5 × 105 AML cells/well were seeded in flat-bottom 96-well plates, and cells were treated with increasing concentration of 5-AZA (0.5, 1.5, 4.5, 13.5, and 40.5 µmol/L) and VEN (0.3, 0.9, 2.7, 8.1, 24.3, 72.9, 218.7, 656.1, and 1968.3 nmol/L) alone and in combination for 72 hours. After 72 hours, viability was assessed using CellTiter-Glo (Promega, cat #G7571) Luminescent Cell Viability Assay. For drug assay in Fig. 3, 0.5 × 105 AML cells/well were seeded in flat-bottom 96-well plates, and cells were treated with 1.5 µmol/L of 5-AZA and 100 nmol/L of VEN for 24 hours. After 24 hours, nonadherent cell fraction was collected, and any adherent cells were trypsinized and scraped off the well bottom using a pipette tip. Lastly, both fractions were combined before the cells were stained with cell-surface antibodies (see Supplementary Table S6). The same amount of CountBright Absolute Counting Beads (Thermo Fisher Scientific, cat #C36950) together with 7-AAD (BD Biosciences, cat #559925) was added to each sample prior to analysis with BD LSRFortessa Cell Analyzer.
Intracellular Staining for BCL2 Family Members
Thawed cells were stained with Zombie NIR Fixable Viability stain in PBS (BioLegend, cat #423105), followed by staining with cell-surface antibodies (see Supplementary Table S6). Stained cells were fixed and permeabilized using the Fixation/Permeabilization Solution Kit (BD Biosciences, cat #554714) according to the manufacturer's instructions, followed by a secondary permeabilization step for enhanced intracellular staining using Permeabilization Buffer Plus (BD Biosciences, cat #561651). Fixed and permeabilized cells were stained separately for anti-human-BCL2-PE (clone 124, Cell Signaling Technology, cat #26295S), anti-human-MCL1-PE (clone D2W9E, Cell Signaling Technology, cat #65617S), and anti-human-BCL-xL-PE (clone 54H6, Cell Signaling Technology, cat #13835S), or together for anti-human-BCL2-AF647 (clone 124, Cell Signaling Technology, cat #82655), anti-human-MCL1-AF488 (clone D2W9E, Cell Signaling Technology, cat #58326) and anti-human-BCL-xL-PE (clone 54H6, Cell Signaling Technology, cat #13835S). Samples were analyzed with BD LSRFortessa Cell Analyzer.
MAC-Score Calculation
To ensure consistent and comparable MFI measurements of samples processed and analyzed on separate days, a reference AML sample was processed and run along with each cohort. Detector voltages were adjusted to keep MFI for each BCL2, MCL1, and BCL-xL of the LSC-like population in the reference sample constant. Small fluctuations of reference sample MFIs were adjusted by normalizing the measurement day's reference sample to match previous reference sample measurements. For each sample, normalized MFI for each BCL2, MCL1, and BCL-xL of the LSC-like population were divided by the respective median MFI of patients with AML classified as responders within the cohort to obtain relative MFI values (rel.MFI). Afterward, MAC-Score was calculated using the following formula:
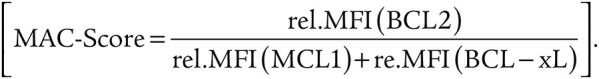 |
BH3 Profiling of Primary AML Samples
Thawed cells were stained with Zombie NIR Fixable Viability stain in PBS (BioLegend, cat #423105), followed by staining with cell-surface antibodies (see Supplementary Table S6). BH3 profiling was performed as previously described (49). Cells were exposed to increasing concentrations of synthetic BH3 peptides as well as BH mimetics in MEB buffer (150 mmol/L mannitol, 10 mmol/L HEPES-KOH pH 7.5, 50 mmol/L KCl, 0.02 mmol/L EGTA, 0.02 mmol/L EDTA, 0.1% BSA, and 5 mmol/L succinate) for 60 minutes and plasma membrane permeabilized with digitonin (0.002%). After 60 minutes, peptide/mimetic exposure at room temperature cells were fixed using 4% formaldehyde for 15 minutes, followed by neutralization for 10 minutes using N2 buffer (1.7M Tris, 1.25M glycine pH 9.1). Sensitivity to BH3 peptides/mimetics was measured as cytochrome C release using anti-cytochrome C FITC antibody (clone 6H2.B4, BioLegend, cat #612302) via BD FACSymphony A3 Cell Analyzer. DMSO was used as a negative control for cytochrome C retention, whereas Alamethicin (ALM) was used as a positive control for 100% cytochrome C release. Cytochrome C loss was calculated using the following equation: 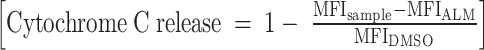 . Cytochrome C release was assessed in each FACS-gated population. To assess the variation of cytochrome C release as a function of BH3 peptide/mimetic concentration, area under the curve (AUC) for each BH3 peptide/mimetic was calculated using Prism v.9.
. Cytochrome C release was assessed in each FACS-gated population. To assess the variation of cytochrome C release as a function of BH3 peptide/mimetic concentration, area under the curve (AUC) for each BH3 peptide/mimetic was calculated using Prism v.9.
Longitudinally Collected Primary AML Samples
From three newly diagnosed patients with AML (AML55, AML61, and AML62), PB was drawn prior to treatment with 5-AZA/VEN (day 0), followed by blood draws during therapy (days 1–6). PBMCs were isolated as explained above. 0.2 × 106 cells were stained with cell-surface antibodies (see Supplementary Table S6) and analyzed with BD LSRFortessa Cell Analyzer.
Processing of AML Cell Lines
Twenty-four AML cell lines were cultivated at 37°C in a humidified incubator with 5% CO2 following the German Collection of Microorganisms and Cell Cultures (DSMZ) culture recommendations. Authenticated and Mycoplasma-screened cell lines were received from DSMZ and Cell Services at Francis Crick Institute (courtesy of Dr. Dominque Bonnet).
In Vitro Drug Screening in Leukemia Cell Lines
0.1 × 105 cells/well from each cell line were seeded in flat-bottom 96-well plates, and cells were treated with increasing concentrations of VEN (1, 3, 9, 27, 81, 243, 729, and 2187 nmol/L) in combination with a single dose of 5-AZA (1.5 µmol/L) for 72 hours. After 72 hours, viability was assessed using CellTiter-Glo (Promega, cat #G7571) Luminescent Cell Viability Assay.
FACS
Primary AML cells were stained with cell-surface antibodies (see Supplementary Table S6). Cells were sorted into four populations according to CD11b, CD64, and GPR56 expression within the lineage-negative gate. Cells from each population and lineage-negative bulk were sorted directly into RNA extraction buffer (Thermo Fisher, cat #KIT0214), snap-frozen, and stored at −80°C until RNA extraction. For xenotransplantations, cells from each population were sorted into PBS.
Mouse Experiments
NOD.Prkdcscid.Il2rgnull (NSG) mice were bred and housed under specific pathogen-free conditions at the central animal facility of the German Cancer Research Center (DKFZ). Animal experiments were conducted in compliance with all relevant ethical regulations. All animal experiments were approved and performed in accordance with all regulatory guidelines of the official committee at the Regierungspräsidium Karlsruhe (G-140-21 and G42/18).
Xenotransplantations and Analysis of Leukemic Engraftment
Female mice 8 to 12 weeks of age were sublethally irradiated (175 cGy) 24 hours before xenotransplantation assays. Up to 1 × 106 cells from FACS-sorted primary AML samples (see above) were injected into the femoral BM cavity of sublethally irradiated mice. Mice were monitored daily and at the endpoint, when mice reached defined termination criteria, BM cells were harvested from tibiae, femurs, and iliac crests by bone crushing. Spleen cells were harvested by mincing the spleen with a plunger. Following red blood cell lysis, cells were resuspended in FBS + 10% DMSO (Sigma, cat #D2650-100) and stored in liquid nitrogen until further use. Human leukemic engraftment in mouse BM was evaluated by flow cytometry (maximum 45 weeks unless endpoint criteria were reached earlier) using anti-human-CD45-FITC (clone HI30), anti-human-CD34-BUV395 (clone 581), anti-human-GPR56-PE (clone CG4), anti-human-CD19-FITC (clone HIB19), anti-human-CD33-PE-Cy7 (clone WM53), CD64-APC, CD11b-BV711, and anti-mouse-CD45-Alexa700 (clone 30-F11).
RNA-Sequencing AML Populations
RNA extraction and purification of FACS-sorted cells were done using the PicoPure RNA Isolation Kit according to the manufacturer's instructions (Thermo Fisher, cat #KIT0214). RNA quality assessment and quantification were performed with Bioanalyzer using Agilent RNA 6000 Pico Kit (Agilent, cat #5067-1513). Whole-transcriptome amplification was performed using a modified smart-seq2 protocol (50), with 5 µL of a modified RT buffer containing 1 × SMART First Strand Buffer (Takara Bio Clontech, cat #639538), 1 mmol/L dithiothreitol (Takara Bio Clontech), 1 µmol/L template switching oligo (IDT), 10 U/µL SMARTScribe (Takara Bio Clontech, cat #639538), and 1 U/µL RNasin Plus RNase Inhibitor (Promega, cat #N2615). Tagmentation of cDNA was done using Nextera XT DNA Library Preparation Kit (Illumina, cat #FC-121-1030). All RNA libraries were pooled and sequenced together on an Illumina NextSeq 550 high-output sequencer (1.4 pmol/L with 1% PhiX loading concentration, single-end 75 bp read configuration).
Raw Data Processing and Quality Control of RNA-Sequencing Data
Reads were demultiplexed, and STAR aligner v. 2.5.3a was used to align FASTQ files containing reads for individual samples by two-pass alignment (51). Reads were aligned to a STAR index generated from the 1000 Genomes Project human genomes assembly (hs37d5), using GENCODE v.19 gene models. Default alignment call parameters were used with the following modifications: –outSAMtype BAM Unsorted SortedByCoordinate –limitBAMsortRAM 100000000000 –outBAMsortingThreadN = 1 –outSAMstrandField intronMotif –outSAMunmapped Within KeepPairs –outFilterMultimapNmax 1 –outFilterMismatchNmax 5 –outFilterMismatchNoverLmax 0.3 –chimSegmentMin 15 –chimScoreMin 1 –chimScoreJunctionNonGTAG 0 –chimJunctionOverhangMin 15 –chimSegmentReadGapMax 3 –alignSJstitchMismatchNmax 5 -1 5 5 –alignIntronMax 1100000 –alignMatesGapMax 1100000 –alignSJDBoverhangMin 3 –alignIntronMin 20.
Sambamba v.0.6.5 was used for the alignment file sorting, duplicate marking, and BAM index generation using eight threads (52). Quality control analysis was performed using the sambamba flagstat command and rnaseqc v.1.1.8 with the hs37d5 assembly and GENCODE v.19 gene models. Depth of coverage analysis for rnaseqc was turned off. Gene-specific gene counting over exon features based on GENCODE v.19 gene models was performed using featureCounts v.1.5.1 (53). Quality threshold was set to 255, which indicates that STAR found a unique alignment. Strand-unspecific counting was used.
DESeq2 (54) was used for statistical analysis of the read counts to identify differentially expressed genes between the LSC-like and mature populations in Prim-AMLs and Mono-AMLs. Genes with an FDR-corrected P value d 0.05 and at least a 2-fold change in expression (|log2FC| e 1) were considered as differentially expressed. Gene-expression estimates for PCA visualization were adjusted by variance stabilization. Gene set enrichment analysis for Hallmark gene sets between LSC-like and mature cells was performed based on a log fold change order-ranked gene list. LSC17 scores were calculated for each AML sample as the mean expression of the normalized log2-transformed gene counts of the 17 LSC signature genes from Ng and colleagues (15) as follows:
 |
Quantification and Statistical Analysis
Overview of all assays performed in which patient sample is shown in Supplementary Table S7. Flow cytometry data analysis was done using FlowJo v.10.5.3. TSNE plots for BCL2 family members (Fig. 2P and Q), and 5-AZA/VEN sensitivities (Fig. 3I and J) were done using the FlowJo TSne plugin v.2.0.0. All statistical analyses excluding RNA-sequencing data were done using Prism v.9.
Data Availability
RNA-sequencing data from this study have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) and can be retrieved under the accession number E-MTAB-11976.
Supplementary Material
Cell line sensitivity and additional patient data
Transcriptomic and functional characterization of LSC-like cells
Bcl-2 family gene expression in LSCs predicts in vitro response to HMA/VEN
Subpopulation specific Bcl-2 family levels and response to HMA/Venetoclax
Subpopulation specific Bcl-2 family levels and time on HMA/Venetoclax
Comparison of MAC-Score and BH-3 profiling based response prediction
MAC-Score predict response within mutational subgroups
7 Tables showing: molecular and clinical patient data, cell line info, antibody panels and overview of assays used
Acknowledgments
The authors thank all technicians and in particular Markus Sohn of the Trumpp laboratory for technical assistance; Steffen Schmitt, Marcus Eich, Klaus Hexel, Tobias Rubner, and Florian Blum from the DKFZ Flow Cytometry Core Facility for their assistance; and K. Reifenberg, P. Prückl, M. Durst, A. Rathgeb, and animal caretaker of the DKFZ Central Animal Laboratory for excellent animal welfare and husbandry. The authors also thank the DKFZ Genomics and Proteomics Core Facility for their assistance, as well as the DKFZ ODCF System Administration. This work was in part supported by the SPP2036, FOR2674, and SFB873 funded by the Deutsche Forschungsgemeinschaft (DFG); the DKTK joint funding project “RiskY-AML,” the “Integrate-TN” Consortium funded by the Deutsche Krebshilfe, the ERC Advanced Grant SHATTER-AML (AdG-101055270), and the Dietmar Hopp Foundation (all to A. Trumpp), Emmy Noether DFG RA 3166/2-1, and LeukoSyStem BMBF 01ZX1911D (to S. Raffel), Deutsche Krebshilfe BW70113908, DFG MU1328/18-1, DFG MU1328/23-1 BMBF/Projektträger Jülich 031L0212A, Wilhelm Sander-Stiftung Nr. 2021.145.1, Deutsche José Carreras Leukämie-Stiftung DJCLS 04 R/2022 (to C. Muller-Tidow). A. Waclawiczek was supported by the EMBO Postdoctoral Fellowship and the Marie Curie Individual Fellowship. Graphical illustrations were created using a BioRender.com license. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101028612.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
A. Waclawiczek reports personal fees from the European Molecular Biology Organization and Horizon 2020 during the conduct of the study, as well as a patent for 22177579.4 pending. A.-M. Leppä reports a patent for 22177579.4 pending. S. Renders reports other support from Gilead outside the submitted work, as well as a patent for 22177579.4 pending. B. Betz reports personal fees from Helmholtz International Graduate School for Cancer Research (HIGS) during the conduct of the study. J.M. Unglaub reports other support from AbbVie, Jazz Pharmaceuticals, and Novartis outside the submitted work. C. Rollig reports grants and personal fees from AbbVie and Pfizer, personal fees from Bristol Myers Squibb, Astellas, Jazz Pharmaceuticals, and Servier, and grants from Novartis outside the submitted work. M. Heuser reports grants from AbbVie during the conduct of the study. C. Muller-Tidow reports personal fees from MorphoSys AG, Exeter Pharma Consultancy, Wilhelm Sander Stiftung, and DKFZ outside the submitted work. T. Sauer reports other support from AbbVie outside the submitted work. A. Trumpp reports a patent for therapy assessment for hematopoietic cancer pending. No disclosures were reported by the other authors.
Authors’ Contributions
A. Waclawiczek: Conceptualization, resources, data curation, formal analysis, validation, investigation, methodology, writing–original draft, writing–review and editing. A.-M. Leppä: Conceptualization, resources, data curation, software, formal analysis, methodology, writing–original draft, writing–review and editing. S. Renders: Conceptualization, resources, formal analysis, validation, investigation, methodology, writing–review and editing. K. Stumpf: Resources, investigation. C. Reyneri: Resources. B. Betz: Resources, investigation. M. Janssen: Resources. R. Shahswar: Resources. E. Donato: Formal analysis, investigation, methodology. D. Karpova: Methodology. V. Thiel: Resources. J.M. Unglaub: Resources. S. Grabowski: Resources. S. Gryzik: Resources. L. Vierbaum: Resources. R.F. Schlenk: Resources. C. Rollig: Resources. M. Hundemer: Resources. C. Pabst: Resources. M. Heuser: Re-sources. S. Raffel: Resources. C. Muller-Tidow: Conceptualization, resources, supervision, writing–original draft, writing–review and editing. T. Sauer: Resources, supervision, writing–review and editing. A. Trumpp: Conceptualization, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020;383:617–29. [DOI] [PubMed] [Google Scholar]
- 2. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012;151:344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014;4:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 2016;6:1106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cherry EM, Abbott D, Amaya M, McMahon C, Schwartz M, Rosser J, et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv 2021;5:5565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai SF, Chu SH, Goldberg AD, Parvin S, Koche RP, Glass JL, et al. Leukemia cell of origin influences apoptotic priming and sensitivity to LSD1 inhibition. Cancer Discov 2020;10:1500–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhatt S, Pioso MS, Olesinski EA, Yilma B, Ryan JA, Mashaka T, et al. Reduced mitochondrial apoptotic priming drives resistance to BH3 mimetics in acute myeloid leukemia. Cancer Cell 2020;38:872–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov 2020;10:536–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuusanmaki H, Leppa AM, Polonen P, Kontro M, Dufva O, Deb D, et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 2020;105:708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gorczyca W. Flow cytometry immunophenotypic characteristics of monocytic population in acute monocytic leukemia (AML-M5), acute myelomonocytic leukemia (AML-M4), and chronic myelomonocytic leukemia (CMML). Methods Cell Biol 2004;75:665–77. [DOI] [PubMed] [Google Scholar]
- 13. Stahl M, Menghrajani K, Derkach A, Chan A, Xiao W, Glass J, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv 2021;5:1552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pabst C, Bergeron A, Lavallee VP, Yeh J, Gendron P, Norddahl GL, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016;127:2018–27. [DOI] [PubMed] [Google Scholar]
- 15. Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016;540:433–7. [DOI] [PubMed] [Google Scholar]
- 16. Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008;112:568–75. [DOI] [PubMed] [Google Scholar]
- 17. Quek L, Otto GW, Garnett C, Lhermitte L, Karamitros D, Stoilova B, et al. Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med 2016;213:1513–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He L, Arnold C, Thoma J, Rohde C, Kholmatov M, Garg S, et al. CDK7/12/13 inhibition targets an oscillating leukemia stem cell network and synergizes with venetoclax in acute myeloid leukemia. EMBO Mol Med 2022;14:e14990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Donato E, Correia NC, Andresen C, Karpova D, Wurth R, Klein C, et al. Retained functional normal and preleukemic HSCs at diagnosis are associated to good prognosis in DNMT3Amut NPM1mut AMLs. Blood Adv 2022. doi 10.1182/bloodadvances.2022008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Florek M, Haase M, Marzesco AM, Freund D, Ehninger G, Huttner WB, et al. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res 2005;319:15–26. [DOI] [PubMed] [Google Scholar]
- 21. Tume L, Paco K, Ubidia-Incio R, Moya J. CD133 in breast cancer cells and in breast cancer stem cells as another target for immunotherapy. Gaceta Mexicana de Oncología 2016;15:22–30. [Google Scholar]
- 22. Loughran SJ, Kruse EA, Hacking DF, de Graaf CA, Hyland CD, Willson TA, et al. The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat Immunol 2008;9:810–9. [DOI] [PubMed] [Google Scholar]
- 23. Klonisch T, Wiechec E, Hombach-Klonisch S, Ande SR, Wesselborg S, Schulze-Osthoff K, et al. Cancer stem cell markers in common cancers: therapeutic implications. Trends Mol Med 2008;14:450–60. [DOI] [PubMed] [Google Scholar]
- 24. Lagasse E, Clerc RG. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol 1988;8:2402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem 1991;266:7706–13. [PubMed] [Google Scholar]
- 26. Koike T, Harada N, Yoshida T, Morikawa M. Regulation of myeloid-specific calcium binding protein synthesis by cytosolic protein kinase C. The Journal of Biochemistry 1992;112:624–30. [DOI] [PubMed] [Google Scholar]
- 27. Ziegler-Heitbrock HWL, Ulevitch RJ. CD14: Cell surface receptor and differentiation marker. Immunol Today 1993;14:121–5. [DOI] [PubMed] [Google Scholar]
- 28. Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017;551:384–8. [DOI] [PubMed] [Google Scholar]
- 29. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017;547:104–8. [DOI] [PubMed] [Google Scholar]
- 30. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645–8. [DOI] [PubMed] [Google Scholar]
- 31. Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov 2021;11:1542–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones CL, Stevens BM, D'Alessandro A, Culp-Hill R, Reisz JA, Pei S, et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019;134:389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones CL, Stevens BM, D'Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018;34:724–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018;24:1859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DiNardo CD, Maiti A, Rausch CR, Pemmaraju N, Naqvi K, Daver NG, et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: a single-centre, phase 2 trial. Lancet Haematol 2020;7:e724–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang H, Nakauchi Y, Kohnke T, Stafford M, Bottomly D, Thomas R, et al. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer 2020;1:826–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 2018;25:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harrison CN, Garcia JS, Somervaille TCP, Foran JM, Verstovsek S, Jamieson C, et al. Addition of navitoclax to ongoing ruxolitinib therapy for patients with myelofibrosis with progression or suboptimal response: phase II safety and efficacy. J Clin Oncol 2022;40:1671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood 1998;91:3379–89. [PubMed] [Google Scholar]
- 42. Debrincat MA, Josefsson EC, James C, Henley KJ, Ellis S, Lebois M, et al. Mcl-1 and Bcl-x(L) coordinately regulate megakaryocyte survival. Blood 2012;119:5850–8. [DOI] [PubMed] [Google Scholar]
- 43. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 2013;27:1365–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Montero J, Haq R. Adapted to survive: targeting cancer cells with BH3 mimetics. Cancer Discov 2022;12:1217–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tessoulin B, Papin A, Gomez-Bougie P, Bellanger C, Amiot M, Pellat-Deceunynck C, et al. BCL2-family dysregulation in B-cell malignancies: from gene expression regulation to a targeted therapy biomarker. Front Oncol 2018;8:645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiron D, Dousset C, Brosseau C, Touzeau C, Maiga S, Moreau P, et al. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget 2015;6:8750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez MS, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood 2018;132:2656–69. [DOI] [PubMed] [Google Scholar]
- 48. Pabst C, Krosl J, Fares I, Boucher G, Ruel R, Marinier A, et al. Identification of small molecules that support human leukemia stem cell activity ex vivo. Nat Methods 2014;11:436–42. [DOI] [PubMed] [Google Scholar]
- 49. Ryan J, Montero J, Rocco J, Letai A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol Chem 2016;397:671–8. [DOI] [PubMed] [Google Scholar]
- 50. Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014;9:171–81. [DOI] [PubMed] [Google Scholar]
- 51. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015;31:2032–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 54. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell line sensitivity and additional patient data
Transcriptomic and functional characterization of LSC-like cells
Bcl-2 family gene expression in LSCs predicts in vitro response to HMA/VEN
Subpopulation specific Bcl-2 family levels and response to HMA/Venetoclax
Subpopulation specific Bcl-2 family levels and time on HMA/Venetoclax
Comparison of MAC-Score and BH-3 profiling based response prediction
MAC-Score predict response within mutational subgroups
7 Tables showing: molecular and clinical patient data, cell line info, antibody panels and overview of assays used
Data Availability Statement
RNA-sequencing data from this study have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) and can be retrieved under the accession number E-MTAB-11976.