

Abstract
Neuroendocrine carcinoma (NEC) is a highly aggressive subtype of the neuroendocrine tumor with an extremely poor prognosis. We have previously conducted a comprehensive genomic analysis of over 100 cases of NEC of the gastrointestinal system (GIS‐NEC) and unraveled its unique and organ‐specific genomic drivers. However, the epigenomic features of GIS‐NEC remain unexplored. In this study, we have described the epigenomic landscape of GIS‐NEC and small cell lung carcinoma (SCLC) by integrating motif enrichment analysis from the assay of transposase‐accessible chromatin sequencing (ATAC‐seq) and enhancer profiling from a novel cleavage under targets and tagmentation (CUT&Tag) assay for H3K27ac and identified ELF3 as one of the super‐enhancer–related transcriptional factors in NEC. By combining CUT&Tag and knockdown RNA sequencing for ELF3, we uncovered the transcriptional network regulated by ELF3 and defined its distinctive gene signature, including AURKA, CDC25B, CLDN4, ITGB6, and YWAHB. Furthermore, a loss‐of‐function assay revealed that ELF3 depletion led to poor cell viability. Finally, using gene expression of clinical samples, we successfully divided GIS‐NEC patients into two subgroups according to the ELF3 signature and demonstrated that tumor‐promoting pathways were activated in the ELF3 signature–high group. Our findings highlight the transcriptional regulation of ELF3 as an oncogenic transcription factor and its tumor‐promoting properties in NEC.
Keywords: ATAC‐seq, CUT&Tag, ELF3, neuroendocrine carcinoma, super‐enhancer
Multiorgan comprehensive epigenomic analysis identified ELF3 as a super‐enhancer–associated transcription factor and revealed its oncogenic properties in neuroendocrine carcinoma.
Abbreviations
- ATAC‐seq
assay of transposase‐accessible chromatin sequencing
- CUT&Tag
cleavage under targets and tagmentation
- GIS‐NEC
neuroendocrine carcinoma of the gastrointestinal system
- SCLC
small cell lung carcinoma
- SE
super‐enhancer
1. INTRODUCTION
Neuroendocrine neoplasm (NEN) is a relatively rare type of tumor, comprising approximately 2% of all malignancies, and is mainly found in the gastroentero‐pancreatic (GEP) tract and bronchopulmonary tree. 1 , 2 , 3 , 4 Among GEP‐NENs, neuroendocrine carcinoma of the gastrointestinal system (GIS‐NEC) is an aggressive subtype with an extremely poor prognosis. As only a few GIS‐NEC specimens have been available, their molecular characteristics have remained undefined for a long time. We have previously conducted a comprehensive genomic analysis of 115 patients with GIS‐NEC. As a result, we unraveled the unique and organ‐specific genomic drivers of GIS‐NEC, such as CCNE1 amplification in gastric NEC, inactivation mutation of APC in colonic NEC, inactivation of NOTCH family genes, SOX2 overexpression in nonpancreatic NEC, and a PTF1A‐high acinar‐type subgroup in pancreatic NEC, 5 as well as inactivation of TP53 and RB1, which was previously reported. 6
In addition to genomic alterations, growing evidence emphasizes the importance of dysregulated transcriptional programs and addiction in the development and maintenance of tumors. 7 For example, small cell lung carcinoma (SCLC) is an aggressive high‐grade neuroendocrine tumor of the lung, which accounts for over 90% of lung NENs and 10%‐20% of all lung cancers, 8 and is also characterized by the loss of TP53 and RB1, like GIS‐NEC. 9 The transcriptional regulation of SCLC is becoming clear, and gene expression profiling has identified four discrete subtypes divided by the critical transcriptional regulators, that is, ASCL1, NEUROD1, POU2F3, and YAP1. 10 , 11 , 12 , 13 Each subtype has a specific drug susceptibility (eg, ROVA‐T in the ASCL1 subtype), indicating that subgrouping by key transcriptional factors can provide novel therapeutic interventions. 14 However, the perspective of the transcriptional network and epigenomics of GIS‐NEC remains unclear.
E74‐like factor 3 (ELF3) is an epithelium‐specific ETS transcription factor (TF) that is strongly expressed in various organs such as the digestive tract and bronchus. 15 , 16 Recently, ELF3 was identified as a driver gene in ampullary carcinoma, and loss of ELF3 in normal human epithelial cells enhanced their motility and invasion. 17 More recently, we reported that ELF3 regulates epithelial integrity and host immune responses and functions in biliary tract cancer, 18 indicating that ELF3 functions as a tumor suppressor gene in the normal epithelium of the bile duct and ampulla. In contrast, emerging evidence shows its tumor‐promoting role as a master regulator in other types of cancers, such as esophageal carcinoma, gastrointestinal adenocarcinoma, and lung adenocarcinoma. 19 , 20 , 21 These findings suggest that ELF3 is context dependent and functions as both a lineage‐addicted oncogene and a tumor‐suppressive gene in cancer development and progression, similar to the role of NKX2‐1 in lung adenocarcinoma. 22 , 23 , 24 We previously found that the gene expression of ELF3 was upregulated in GIS‐NEC. 5 However, little is known about the functional role of ELF3, its transcriptional landscape, and whether it functions as a tumor suppressor or oncogene in GIS‐NEC.
In this study, we describe the epigenomic landscape of GIS‐NEC and SCLC using an assay for transposase‐accessible chromatin using sequencing (ATAC‐seq), and cleavage under targets and tagmentation (CUT&Tag), which is a recently developed technology for epigenetic profiling, 25 , 26 and identified ELF3 as one of the super‐enhancer (SE)‐related TFs. To identify the genome‐wide ELF3 binding regions, we have adopted CUT&Tag using anti‐ELF3 specific antibody and uncovered its regulatory transcriptional network and novel targets, including AURKA, CDC25B, CLDN4, ITGB6, and YWAHB. An in vitro loss‐of‐function assay and transcriptome analysis of ELF3 revealed its tumor‐promoting role in GIS‐NEC and SCLC. Finally, using clinical samples, we have successfully divided GIS‐NEC patients into two subgroups, ELF3‐sig high and ELF3‐sig low, and found that tumor‐promoting signal transduction pathways, including the cell cycle, were activated in the ELF3‐sig–high group, supporting the oncogenic role of ELF3 in GIS‐NEC.
2. METHODS
2.1. Cell cultures and reagents
Six GIS‐NEC cell lines (A99, TYUC‐1, TCC‐NECT‐2, ECC4, ECC10, and ECC12), four SCLC cell lines (DMS53, DMS454, WA‐hT, and Lu134A), and a lung adenocarcinoma cell line (A549) were used for analysis. Detailed information regarding the cell lines is provided in Table S1. (+)‐JQ1 was purchased from Sigma‐Aldrich.
2.2. Small interfering RNA
Small interfering RNA (siRNA) against human ELF3 (siELF3 #1: D‐016080‐01, siELF3 #2: D‐016080‐17, siELF3 pool: M‐016080‐01) and nontargeting siRNA pool (siNC: D‐001206‐14) were purchased from Dharmacon. Cells were transfected with 5‐100 nM siRNA using lipofectamine RNAiMAX (Thermo Fisher Scientific) for DMS53 and ECC4 cells according to the manufacturer's instructions or via electroporation using NEPA21 type II (NEPA GENE Co. Ltd.) for A99 cells with a poring pulse voltage of 150 V and poring pulse width of 5 ms.
2.3. Establishment of gene knockout clone of ELF3
An ELF3 gene knockout clone was obtained using the Alt‐R CRISPR/Cas9‐system (Integrated DNA Technologies) according to the manufacturer's instructions. The complex comprising two‐part guide RNA, crRNA, and transactivating crRNA (tracrRNA) targeting the ELF3 sequence (GGACTGGATCAGCTACCAAGTGG) and Cas9 nuclease were transfected into cells using lipofectamine for ECC4 cells or electroporation for A99 cells, as described in the siRNA section. The limiting dilution cloning method was applied for the establishment of gene knockout clones. DNA of each clone was extracted using the QIAamp DNA Mini Kit (Qiagen), and genome editing was confirmed using the Alt‐R® Genome Editing Detection Kit (Integrated DNA Technologies) and Sanger sequencing.
2.4. Viability and soft agar assays
Cell viability was assessed using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay kit (Promega) as previously described. 27 Colony formation by cells in soft agar assay was examined using CytoSelect 96‐Well Cell Transformation Assay (Cell Biolabs) in accordance with the manufacturer's instructions. Cells (2 × 103) in 0.4% soft agar were plated into 96‐well plates on 0.6% base agar and incubated for 7 days.
2.5. Quantitative RT‐PCR
Total RNA was extracted using the RNeasy Mini kit (Qiagen), and the cDNA was synthesized using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) following the manufacturer's protocol. Quantification of mRNA levels was performed using an Mx‐3000P (Stratagene) and iTaq Universal SYBR Green Supermix (Bio‐Rad Laboratories Inc.). Expression levels were normalized to those of GAPDH. Sequences of specific primers for GAPDH, ELF3, ASCL1, ITGB6, AURKA, CLDN4, CDC25B, and YWHAB are shown in Table S2.
2.6. Immunoblotting
The detailed procedures have been described previously. 18 Information on the primary antibodies used is provided in Table S3.
2.7. RNA‐sequencing
Library preparation, sequencing, alignment, and read counting were performed as previously described. 28 Mutations at TP53/RB1 locus were detected using GATK4 (version 4.1.9.0). Pathway analysis was performed using Enrichr web tools, 29 and gene set enrichment analysis (GSEA) was performed as in the previous report. 30 Master TFs was determined by the gene expression of ASCL1, NEUROD1, POU2F3, and YAP and cutoff values of transcripts per kilobase million (TPM) for each TF were set to 100. The transcriptome dataset of 50 SCLC cell lines in the Cancer Cell Line Encyclopedia (CCLE) was obtained from the DepMap portal (https://depmap.org/portal/).
2.8. ATAC‐seq
Library preparation, sequencing, alignment, peak calling, quantification of read counts, and quantile normalization between samples were performed as previously described. 5 Transcription start site (TSS) enrichment and deviation scores (DSs) for motif enrichment were calculated using ChrAccR (version 0.9.11). Footprinting was performed using TOBIAS, 31 and the ClusterMotifs function was used to create consensus motifs based on similarity with the following parameter: threshold, 0.5. A heatmap was generated using deepTools (version 3.3.2), 32 and the peaks were visualized using IGV tools.
2.9. CUT&Tag
Library preparation, sequencing, alignment, and peak calling were performed as previously described. 33 The details of the antibodies used are given in Table S3. Annotation of the peaks was performed using ChIPseeker, 34 and a heatmap was generated using deepTools. The findMotifsGenome.pl function in HOMER was used for motif discovery. SEs were identified using a method described previously. 35 , 36 , 37 , 38
2.10. ChIP‐seq
Library preparation, sequencing, alignment, and peak calling were carried out as previously described. 17 The details of the antibodies used are given in Table S3. Overlapping peaks between the ChIP‐seq and CUT&Tag data were identified using bedtools.
2.11. Statistical analysis
Statistical analyses were performed using GraphPad Prism (version 5.0; GraphPad Software Inc.). The statistical significance of the differences was assessed using one‐way ANOVA, followed by the Bonferroni or Dunnett test. Data from at least three independent experiments are presented as mean ± SEM. Statistical significance was set at p < 0.05. Pearson's correlation coefficient (r) was calculated for the correlation analysis.
3. RESULTS
3.1. Chromatin status is maintained in cultured cells compared with the original frozen tissue
No reports have evaluated whether chromatin status is maintained after culture on plastic plates. Using ATAC‐seq, we first compared the chromatin status of A99, a pancreatic NEC cell line, and matched the original frozen tissue. 39 Peaks were matched between the cell line and the original tissue (Figure 1A), and almost all peaks of the original tissue were covered by those of the cell line (cell line: 59,630 peaks, frozen tissue: 23,469 peaks, shared: 21,682 peaks; Figure 1B). A strong correlation between the signal intensity of shared peaks was observed between cell lines and tissues (r = 0.88, p < 2.2 × 10−16, Figure 1C). As expected, the TSS score was 17.5 in the A99 cell line and 9.1 in frozen tissue, showing that peaks of the cell line had less signal noise. The distribution of peaks revealed that the cell line could identify more peaks in gene regulatory regions such as introns and distal intergenic regions (Figure 1D). These data suggest that the chromatin status of cell lines is retained even after being cultured on a plastic dish, and that cultured cancer cell lines are convenient and ideal tools for assessing the open chromatin status of actual tumor tissue.
FIGURE 1.
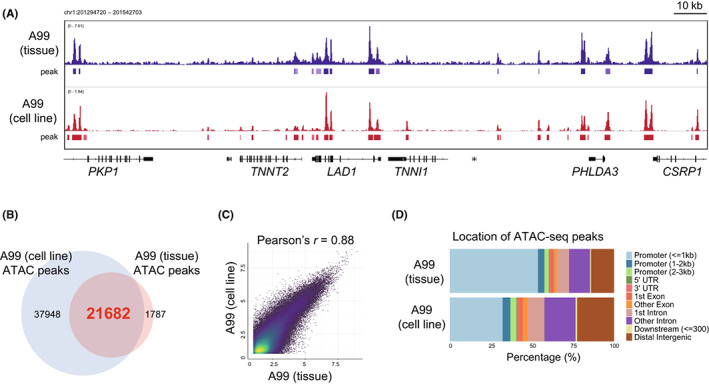
Comparison of open chromatin status between A99 frozen tissue and A99 cell line. A, IGV visualization of assay of transposase‐accessible chromatin sequencing (ATAC‐seq) signals of A99 frozen tissue (upper) and A99 cell lines (lower), which we previously established from surgically resected tumor tissue specimens. B, Venn diagram showing overlapped ATAC‐seq peaks between A99 frozen tissue and A99 cell line. C, Correlation plot showing ATAC‐seq signals in the shared peaks between A99 frozen tissue and A99 cell line. The x‐axis and y‐axis indicate log2‐transformed normalized counts. D, Bar chart showing the location of ATAC‐seq peaks in A99 frozen tissue and A99 cell lines
3.2. Molecular characterization of GIS‐NEC and SCLC cell lines
p53 and Rb are frequently lost in GIS‐NEC and SCLC. 5 , 9 Therefore, we first evaluated the status of p53 and Rb in our cell line panel, which contained six GIS‐NECs and four SCLCs, by RNA‐seq and Western blotting. Western blotting for p53 revealed the abnormal accumulation of this protein in the cell lines with a TP53 missense mutation (Figure 2A). Rb protein was not detected in seven cell lines, and among the other three cell lines that expressed Rb protein, ECC4 had a missense mutation in the RB1 gene. In contrast, the RB1 gene was intact in TYUC‐1 (esophageal NEC; POU2F3‐high tuft cell type) 5 and TCC‐NECT‐2 (duodenal NEC harboring BRAF mutation [V600E]). 40
FIGURE 2.
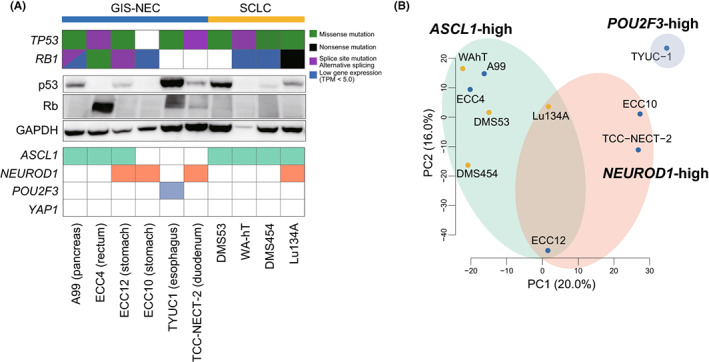
Molecular characterization of six neuroendocrine carcinoma of the gastrointestinal system (GIS‐NEC) cell lines and four small cell lung carcinoma (SCLC) cell lines. A, Mutations/abnormal splicing/gene expression of TP53 and RB1 genes, immunoblotting showing protein expression of p53 and Rb, and gene expression of master transcription factors in six GIS‐NEC and four SCLC cell lines. GAPDH was used as a loading control. B, Principal component analysis of six GIS‐NEC and four SCLC cell lines using 2500 highly variant genes
Next, we subclustered GIS‐NEC and SCLC cell lines based on the expression of master TFs, which are currently used for subtyping SCLC. 9 , 10 , 11 , 12 By setting TPM cutoff values to 100, three GIS‐NECs (A99, ECC4 [rectal NEC], and ECC12 [gastric NEC]) and all SCLC cell lines expressed ASCL1, and three GIS‐NEC cell lines (ECC12, ECC10 [gastric NEC], and TCC‐NECT2) and Lu134A (SCLC) expressed NEUROD1. Of note, ECC12 and Lu134A cells were determined to be ASCL1/NEUROD1 double‐positive NEC, which were recently reported as one of the subtypes in SCLC. 33 , 41 , 42 None of the cell lines expressed YAP1 (Figure 2A). Principal component analysis (PCA) with 2500 highly variant genes revealed that GIS‐NEC and SCLC were clustered according to the expression of TFs rather than their origin (Figure 2B), indicating that GIS‐NEC and SCLC have similar transcriptional regulation in each subgroup. For further experiments, typical GIS‐NEC and SCLC cell lines that harbor loss/mutation of TP53 and/or RB1 and high expression of ASCL1, A99, and ECC4 from GIS‐NEC and DMS53 from SCLC were chosen.
3.3. Motif enrichment analysis identified candidates for core TFs in GIS‐NEC and SCLC
We performed ATAC‐seq of the three selected cell lines and obtained 49,505, 59,630, and 63,144 peaks for A99, ECC4, and DMS53, respectively. DSs for motifs registered in the JASPAR 2022 database were calculated for each cell line (Table S4). A total of 61 motifs with highly enriched motifs (DS > 10) in at least two cell lines were extracted (Figure 3A,B) and grouped into five clusters according to motif sequence similarity (Figure 3C). Based on the TF family classification, the bHLH family, including ASCL1, ETS family, including ELF3, and Forkhead family, including FOXA1, were identified as commonly enriched TF families in GIS‐NEC and SCLC. Furthermore, footprint analysis confirmed that a clear footprint was formed at the predicted genome‐wide binding regions of ELF3 (Figure 3D), ASCL1, and FOXA1 (Figure S1).
FIGURE 3.
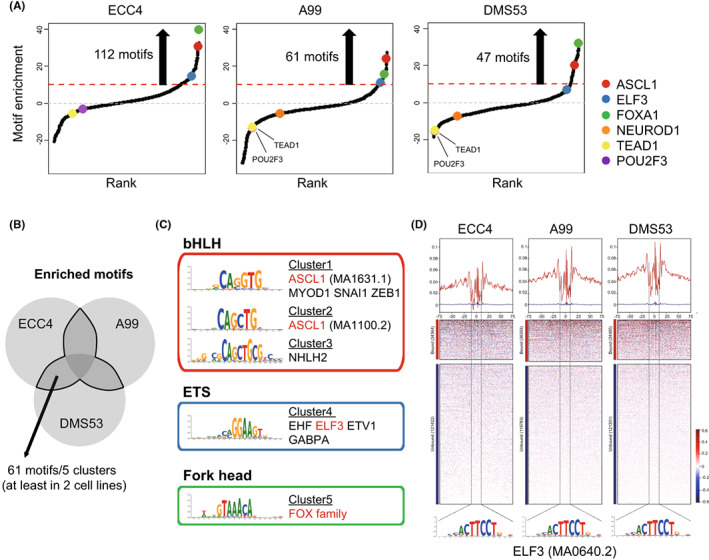
Motif enrichment in ECC4, A99, and DMS53. A, Motif distributions in ECC4, A99, and DMS53. Motifs were plotted in increasing order based on their deviation scores. B, Venn diagram showing overlapped enriched motifs in ECC4, A99, and DMS53. C, Motifs were clustered according to motif sequence similarity. Representative transcription factors for each cluster are presented. D, Chromatin accessibility footprints of ELF3 within all accessible loci for ECC4, A99, and DMS53
3.4. CUT&Tag provides low‐cost, low‐background, and simplified genome‐wide chromatin mapping
Chromatin regulates the function of enhancers by allowing TF to bind to their target motifs within an enhancer. Therefore, we evaluated the genome‐wide enhancer profiles of GIS‐NECs and SCLC. First, we performed ChIP‐seq, a traditional approach to histone modification, and then CUT&Tag, a recently developed technology for epigenetic analysis. 25 , 26 To assess the utility of this novel technology, we first compared its resolution for enhancer/promoter profiling using the A99 cell line. We found that the ChIP‐seq and CUT&Tag data had similar peak patterns (Figure S2A). Furthermore, almost all H3K4me3‐defined promoters and most H3K27ac‐defined enhancers were shared between the two approaches (Figure S2B,C), confirming that CUT&Tag can substitute ChIP‐seq. Therefore, we adopted the CUT&Tag assay, which provides low‐cost, low‐background, and simplified genome‐wide chromatin mapping for further epigenomic profiling.
3.5. SE‐related genes play essential roles in transcriptional regulation in NEC
Super‐enhancers can drive the expression of genes essential for cell lineage specification and are characterized by the enrichment of master TFs and mediators or high levels of H3K27ac. First, we explored if SE formation is actually involved in the progression of GIS‐NEC and SCLC by using JQ1, a BET bromodomain inhibitor, which is known to specifically inhibit SE activity. 43 In ECC4 and DMS53 cells, gene expression of ASCL1, which is known to encode a SE‐related master TF, was suppressed by JQ1 (Figure S3A). Furthermore, cell viability was significantly decreased in a dose‐dependent manner in both cell lines (Figure S3B), indicating that the specific suppression of SEs leads to the inhibition of tumor growth in GIS‐NEC and SCLC.
To define SEs in GIS‐NEC and SCLC, we performed H3K27ac CUT&Tag and 11,776, 20,693, and 14,863 enhancer regions were identified in ECC4, A99, and DMS53, respectively. Then, we defined SE using the ROSE algorithm, and 536, 882, and 610 SEs were defined, respectively (Figure 4A). SE genes were extracted from each cell line (Table S5), and 798 genes were overlapped in at least two cell lines (Figure 4B). KEGG pathway analysis revealed that 798 SE genes regulated a variety of cancer‐related signaling pathways, including cell cycle (p = 0.004; CDKN1C, CDKN1B, CDKN2C, MCM7, PRKDC, GADD45A, E2F1, E2F4, YWHAZ, YWHAG, ANAPC2, and GADD45G) and SCLC (p = 0.011; LAMA5, CDKN1B, GADD45A, E2F1, PIK3R3, PTK2, CKS1B, GADD45G, and BCL2L1). Of note, Notch signaling, which we previously showed is inactivated in GIS‐NEC, 5 was also listed as an SE gene–related pathway (p = 0.03; LFNG, NCOR2, JAG2, CTBP2, HES1, and DLL1) (Table S6), and this is in line with previous studies showing that the expression levels of these negative regulators of the Notch pathway are upregulated in SCLC. 44 , 45 Finally, we extracted SE‐related TFs that overlapped in at least two cell lines and found that ASCL1, ELF3, and FOXA1, which were also identified as core TFs by ATAC‐seq, were listed (Figure 4D), indicating that these SE‐related TFs play important roles in oncogenic pathway regulation, determination of the neuroendocrine lineage, or maintaining its phenotype by regulating many genes and pathways.
FIGURE 4.
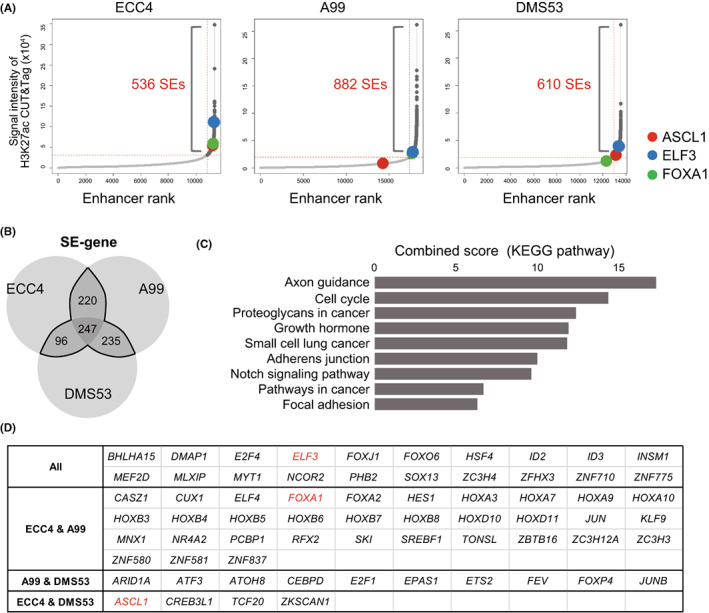
Super‐enhancers in ECC4, A99, and DMS53. A, Enhancer distributions in ECC4, A99, and DMS53. Enhancer regions are plotted in increasing order based on their H3K27Ac cleavage under targets and tagmentation (CUT&Tag) signals. B, Venn diagram showing overlapping super‐enhancer (SE) genes in ECC4, A99, and DMS53. C, Bar chart showing the combined score calculated by EnrichR pathway analysis (KEGG) of 798 SE genes. D, List of SE‐related transcription factors that overlapped in ECC4, A99, and DMS53
3.6. CUT&Tag revealed genome‐wide ELF3 binding locus in GIS‐NEC and SCLC
Next, we integrated core TFs defined by ATAC‐seq, SE‐related TFs defined by H3K27ac CUT&Tag, and highly expressed TFs in GIS‐NEC defined by RNA‐seq, 5 and ELF3 was identified as a novel candidate master TF in GIS‐NEC, in addition to ASCL1 and FOXA1, which were previously reported as key master TFs of neuroendocrine differentiation in SCLC or other types of NEC (Figure 5A). SE regions near the ELF3 gene locus were ranked at 15, 436, and 72 among 11,776, 20,693, and 14,863 enhancers in ECC4, A99, and DMS53, respectively (Figure 4A), and broad SE was observed around the ELF3 genomic locus in all three cell lines (Figure 5B). SE regions near the ASCL1 and FOXA1 gene loci are also shown in Figure S4.
FIGURE 5.
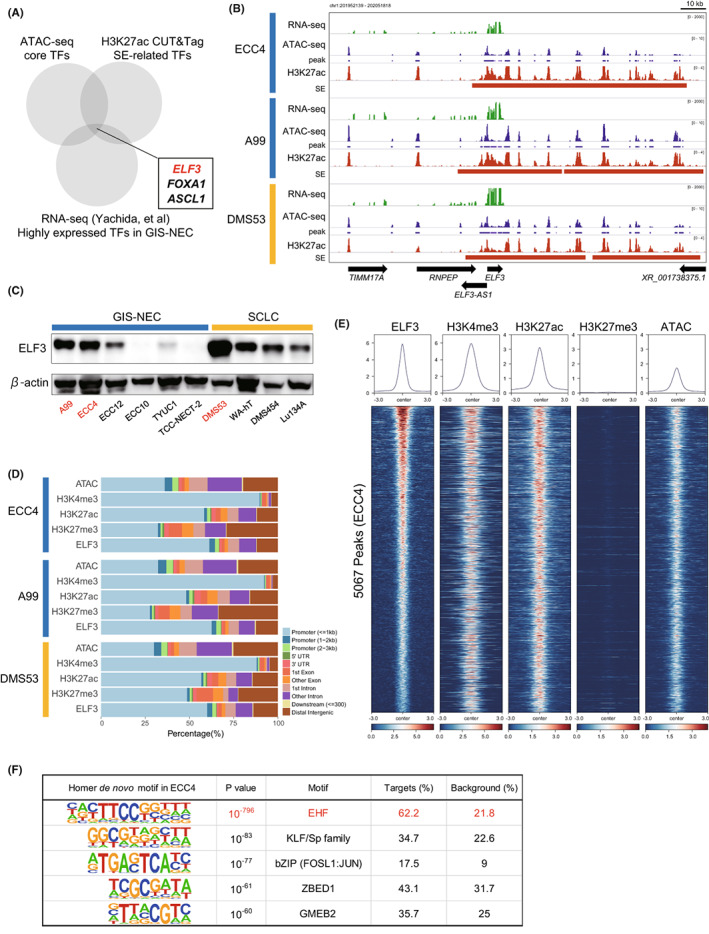
Cleavage under targets and tagmentation (CUT&Tag) assay for ELF3 and histone markers in ECC4, A99, and DMS53. A, Venn diagram showing overlapping core transcription factors (TFs) identified by assay of transposase‐accessible chromatin sequencing (ATAC‐seq), super‐enhancer (SE)‐related TFs, and highly expressed TFs. B, IGV visualization of RNA‐seq signals, ATAC‐seq signals and peaks, and H3K27ac CUT&Tag signals and SEs around the ELF3 genomic locus in ECC4, A99, and DMS53. C, Immunoblotting for ELF3 in six neuroendocrine carcinoma of the gastrointestinal system (GIS‐NEC) and four small cell lung carcinoma (SCLC) cell lines. β‐actin was used as the loading control. D, Bar chart showing annotation of ATAC‐seq and CUT&Tag (H3K4me3, H3K27ac, H3K27me3, and ELF3) peaks in ECC4, A99, and DMS53. E, Heatmaps of ATAC‐seq and CUT&Tag (H3K4me3, H3K27ac, H3K27me3, and ELF3) signals around 5067 ELF3 genomic binding regions in ECC4. Regions are shown in descending order of signal intensity of the ELF3 CUT&Tag. F, Top five motifs in the ELF3 genomic binding regions identified by the ELF3 CUT&Tag assay in ECC4
We confirmed the abundant expression of ELF3 protein in ASCL1‐high NEC cell lines, including ECC4, A99, and DMS53 (Figure 5C), and weak expression in the POU2F3‐high NEC cell line (TYUC‐1). This result was validated based on the CCLE transcriptome dataset, which showed that ELF3 was expressed in SCLC‐A and SCLC‐P clusters (Figure S5). Next, we explored the mechanisms through which ELF3 expression is induced in ASCL1‐positive NEC. By integrating copy number variation via whole‐genome sequencing and gene expression based on RNA‐seq analysis of 34 GIS‐NEC samples, 28 no correlation between focal amplification of the ELF3 gene locus and ELF3 expression was observed (r = −0.07, p = 0.69, Figure S6). However, clear negative correlations between DNA methylation of the SE regions, especially in the gene body region, and ELF3 expression were observed (Figure S7A,B). Furthermore, silencing ASCL1 using siRNA did not affect ELF3 expression in DMS53 and ECC4 cells (data not shown), suggesting that the activation of ELF3 is independent of ASCL1 and that DNA methylation of SE regions is one of the causes of ELF3 overexpression in ASCL1‐positive GIS‐NEC and SCLC.
To explore transcriptional regulation by ELF3, we performed CUT&Tag using an anti‐ELF3 antibody, as well as other histone modification markers. We successfully obtained clear peaks from the ELF3 CUT&Tag (5067, 3837, and 6415 peaks in ECC4, A99, and DMS53, respectively). These peaks were mostly located in promoter regions (Figure 5D) and were shared by ATAC‐seq and H3K27ac peaks but not H3K27me3 peaks (Figure 5E, Figures S8 and S9), indicating that ELF3 binds to promoter/enhancer/open chromatin regions. HOMER de novo motif analysis of ELF3 peak regions revealed that significant ETS motif enrichment was observed in all cell lines (p = 1.0 × 10−796 in ECC4, p = 1.0 × 10−717 in A99, and p = 1.0 × 10−722 in DMS53), supporting the validity of ELF3 CUT&Tag data. Interestingly, KLF5 and AP1 motifs were also identified in the ELF3 peaks (Figure 5F, Figure S10), indicating that these transcriptional factors may interact with ELF3. To explore whether the context‐dependent roles of ELF3 could be explained by the context‐dependent interactions with other TFs, 21 CUT&Tag sequencing of A549 cells, a non‐neuroendocrine lung adenocarcinoma line, was performed, and enriched motifs in A549 cells were compared with those of NEC cell lines. The top five enriched motifs in A549 cells in the ELF3 binding regions were shared with those in NEC (Figure S10), indicating that transcriptional network formation and oncogenic properties mediated by ELF3 in GIS‐NEC, SCLC, and NSCLC are similar to each other. Collectively, we successfully defined ELF3‐binding genomic regions in GIS‐NECs and SCLC using ELF3 CUT&Tag.
3.7. ELF3 regulates oncogenic signaling pathways
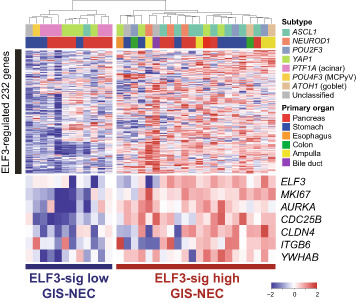
Next, we explored the downstream targets of ELF3 by integrating ELF3 CUT&Tag and ELF3 knockdown RNA‐seq. ELF3 was silenced using siRNAs, and the high knockdown efficiency of ELF3 was confirmed at the protein level in all three cell lines (Figure 6A). By merging the downregulated 1121 genes (fold change < 0.75) in at least two cell lines and 3288 genes near ELF3 CUT&Tag peaks in at least two cell lines, we obtained 232 ELF3‐regulated genes (Figure 6B, Tables S7–S9). Pathway analysis revealed that oncogenic pathways such as the G2‐M checkpoint and E2F targets were enriched in ELF3 signature genes (Figure 6C, Table S10). Clear ELF3 peaks of representative genes were observed in AURKA (cell growth and death), CDC25B (cell cycle), CLDN4 (tight junction), ITGB6 (focal adhesion), and YWHAB (Hippo signaling) in at least two cell lines (Figure 6D). ELF3 knockdown mediated by siRNA significantly decreased cell viability in all cell lines (Figure 6E, Figure S11), indicating that ELF3 has a tumor‐promoting function in vitro. Furthermore, we established ELF3 knockout clones of ECC4 and A99 cells (Figure S12A), and soft agar assays showed decreased colony formation in the knockout clones, suggesting that ELF3 is involved in anchorage‐independent growth in NEC (Figure 6F, Figure S12B). To explore the clinical relevance of ELF3 in GIS‐NECs, we clustered 34 GIS‐NEC samples by gene expression using ELF3‐regulated 232 genes. 5 GIS‐NEC was divided into two clusters: ELF3‐sig high (N = 23) and ELF3‐sig low (N = 11). In the ELF3‐sig–high group, MKI67 (Ki‐67), a well‐established tumor proliferation marker, and ELF3‐regulated genes such as AURKA, CDC25B, CLDN4, ITGB6, and YWAHB were highly expressed (Figure 6G). Downregulation of the expression of these genes mediated by siELF3 was confirmed based on quantitative RT‐PCR (Figure S13). GSEA revealed that the ELF3‐sig–high group was enriched in various gene signatures, including tight junctions, cell cycles, adherent junctions, and regulation of Hippo signaling in clinical samples (Figure 6H), indicating that ELF3 can play a tumor‐promoting role by regulating various oncogenic signaling pathways in GIS‐NEC and SCLC.
FIGURE 6.
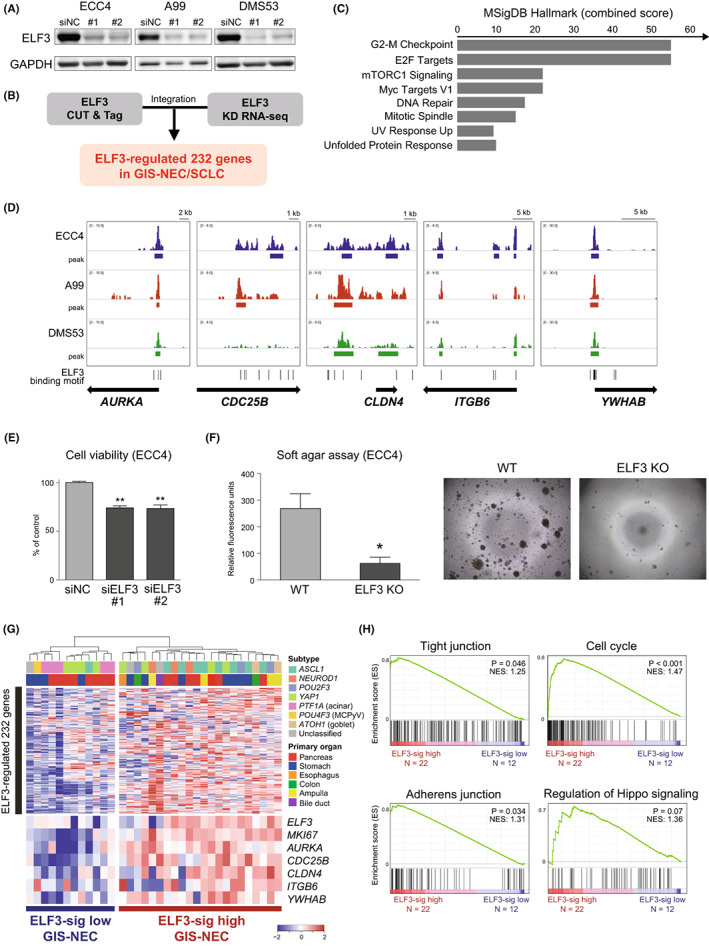
Identification of ELF3‐regulated gene signature and clinical relevance. A, Immunoblotting for ELF3 in ECC4, A99, and DMS53 cells treated with the negative control (siNC) or siELF3. GAPDH was used as a loading control. B, Flowchart for extracting ELF3‐regulated genes by combining cleavage under targets and tagmentation (CUT&Tag) for ELF3 and ELF3 knockdown RNA‐seq. C, Bar chart showing the combined scores calculated by EnrichR pathway analysis (MSIgDB Hallmark) of 232 ELF3‐regulated genes. D, IGV visualization of ELF3 CUT&Tag signals for representative five ELF3‐regulated genes (AURKA, CDC25B, CLDN4, ITGB6, and YWAHB). E, Bar chart showing cell viability at 96 h after ELF3 knockdown by siRNA in ECC4, A99, and DMS53 cells. Data are representative of three independent experiments. **p < 0.01. F, Bar chart showing colony formation in soft agar at day 7 in the ELF3 knockout clone of ECC4 cells. Data are representative of three independent experiments. *p < 0.05. G, Heatmap showing ELF3‐regulated 232 genes in 34 neuroendocrine carcinomas of the gastrointestinal system (GIS‐NECs). Unsupervised clustering identified two clusters: ELF3‐sig–high GIS‐NEC and ELF3‐sig–low GIS‐NEC. H, Enrichment plot of GSEA with 34 GIS‐NEC gene expression data
4. DISCUSSION
The present study performed an integrative epigenomic analysis of ATAC‐seq and CUT&Tag in GIS‐NEC and SCLC cell lines. By integrating open chromatin status from ATAC‐seq and enhancer profiling from H3K27ac CUT&Tag, we identified ELF3 as one of the SE‐related TFs in ASCL1‐positive GIS‐NEC and SCLC. Furthermore, we confirmed that the protein expression of ELF3 was abundant in most GIS‐NECs and SCLC cell lines. Using an anti‐ELF3 specific antibody, we successfully performed ELF3 CUT&Tag, integrated ELF3 CUT&Tag with knockdown RNA‐seq, and uncovered the transcriptional network and distinctive tumor‐promoting pathways regulated by ELF3. In vitro, the functional assay showed that loss of function of ELF3 decreased cell viability. Finally, using clinical samples, we successfully divided GIS‐NEC patients into two subgroups (ELF3‐sig high and ELF3‐sig low), and GSEA revealed regulations of a variety of pathways by ELF3 in these patients.
As mutation of the ELF3 gene is frequently observed in many epithelial tumors, especially in bladder urothelial carcinoma and ampullary carcinoma, ELF3 is assumed to function as a tumor suppressor in these cancers 17 , 46 , 47 , 48 ; however, amplification and/or overexpression of ELF3 was also observed in other types of cancer and ELF3 was shown to function as an oncogene by regulating cell metastasis and cell growth through PI3K/AKR and ERK signaling, 19 , 20 , 21 , 49 suggesting that ELF3 functions in a context‐dependent manner. Interestingly, our data suggested that DNA demethylation in the SE regions might cause ELF3 overexpression in ASCL1‐positive GIS‐NEC, but not the genomic amplification or direct regulation of ASCL1, indicating that the mechanism of ELF3 activation in ASCL1‐positive GIS‐NEC is distinct from that in other cancer types.
The downstream analysis of ELF3 by CUT&Tag revealed absence of peaks near ASCL1 or INSM1, master TFs of NE differentiation, or neuroendocrine markers, such as CHGA, NCAM1, SYP, or CALCA suggesting that ELF3 does not directly regulate neuroendocrine differentiation in GIS‐NEC/SCLC. These results are in line with those of a previous report by Wooten et al, in which systems‐level network modeling identified ELF3 as one of the master regulators of the ASCL1‐positive subgroup of SCLC and suggested that ELF3 might be involved in a variety of pathway, such as the immune response, drug catabolism, ion transport, homeostasis, epithelial cell differentiation, and glycosylation, but not in neuron differentiation. 50 In addition to regulating tight‐junction proteins and focal adhesion molecules, which have been previously reported in other cancer cell types, 18 our data suggest that ELF3 is involved in tumor promotion by regulating cell growth and cell cycle genes such as AURKA, which is identified as a direct target of ELF3. In SCLC, RB1‐mutant SCLC cells are highly sensitive to AURKA inhibitors, 51 and AURKA inhibitors such as alisertib are attracting attention as promising therapies in subsets of SCLC. 14 Considering that most GIS‐NECs are ELF3‐sig high (Figure 6G) and ELF3‐sig genes are involved in E2F signaling (Figure 6C), which is closely related to RB1, AURKA inhibitors could also be a potential therapy for ELF3‐sig–high GIS‐NEC patients.
Instead of ChIP‐seq, we applied CUT&Tag for histone modification and transcriptional factor binding. CUT&Tag was recently developed to assess the enrichment of modified histones. For profile TF binding, cleavage under targets and release using nuclease (CUT&RUN) has been used primarily instead of CUT&Tag because of the size of pA‐Tn5, which is bulkier than MNase and requires stringent washing to avoid binding and tagmentation to the accessible DNA in unfixed cells. 25 We have successfully performed CUT&Tag for ELF3 and confirmed the specific enrichment of the ELF3 binding motif in the peak regions (Figure 5F, Figure S10). However, only a few reports have successfully performed CUT&Tag for TFs, such as CTCF and Sox2. 25 Further application of the CUT&Tag methods to TFs is expected.
We used six GIS‐NEC cell line panels covering most GIS‐NEC subtypes (ASCL1‐type, NEUROD1‐type, and POU2F3‐type). Nonetheless, our previous paper had identified rarer variants of GIS‐NEC, such as PTF1A‐high acinar‐type pancreatic NEC or virus‐induced GIS‐NEC (human papilloma virus or Merkel cell polyomavirus), which usually do not harbor TP53/RB1 aberrations. 5 Therefore, although these subtypes of GIS‐NEC are rare and cell lines are not available to date, further epigenetic examination should be performed to understand the entire perspective of GIS‐NEC transcriptional and epigenomic regulatory networks. In conclusion, we identified ELF3 as one of the SE‐related TFs and identified its oncogenic properties in NEC using an integrative epigenomic approach. This finding might help to understand the transcriptional network and establish novel therapies for GIS‐NEC and SCLC.
CONFLICT OF INTEREST STATEMENT
S.Y. is an Associate Editor of Cancer Science.
ETHICAL APPROVAL
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal studies: N/A.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Figure S10
Figure S11
Figure S12
Figure S13
Tables S1
ACKNOWLEDGEMENTS
This work was supported by JSPS KAKENHI (20K16440 [PAGS] to H.M., 21K16830 to H.T., 21K06800 to M.S., and 20H03662 to S.Y.); the Takeda Science Foundation to M.H and S.Y.; Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development (AMED) (JP21ck0106558 to S.Y., JP21ck0106693 to S.Y., JP21ck0106690 to S.Y., and JP21ck0106547 to S.Y.); Project for Cancer Research and Therapeutic Evolution (P‐CREATE) from AMED (JP17cm0106612 to S.Y.); United States‐Japan Cooperative Medical Science Program from AMED (JP20jk0210009 to S.Y.); Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives, Osaka University (to S.Y.); Joint Research Project of the Institute of Medical Science, the University of Tokyo (to S.Y.); the Yasuda Medical Foundation (to S.Y.); the Mitsubishi Foundation (to S.Y.); and the Princess Takamatsu Cancer Research Fund (to S.Y.).
Horie M, Tanaka H, Suzuki M, et al. An integrative epigenomic approach identifies ELF3 as an oncogenic regulator in ASCL1‐positive neuroendocrine carcinoma. Cancer Sci. 2023;114:2596‐2608. doi: 10.1111/cas.15764
Masafumi Horie and Hidenori Tanaka contributed equally to this work.
Contributor Information
Masafumi Horie, Email: mhorie@med.kanazawa-u.ac.jp.
Shinichi Yachida, Email: syachida@cgi.med.osaka-u.ac.jp.
DATA AVAILABILITY STATEMENT
The data generated in this study were deposited in GSE190618.
REFERENCES
- 1. Basu B, Sirohi B, Corrie P. Systemic therapy for neuroendocrine tumours of gastroenteropancreatic origin. Endocr Relat Cancer. 2010;17:R75‐R90. [DOI] [PubMed] [Google Scholar]
- 2. Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing but NET: a review of neuroendocrine tumors and carcinomas. Neoplasia. 2017;19:991‐1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008;113:2655‐2664. [DOI] [PubMed] [Google Scholar]
- 4. Tsai HJ, Wu CC, Tsai CR, Lin SF, Chen LT, Chang JS. The epidemiology of neuroendocrine tumors in Taiwan: a nation‐wide cancer registry‐based study. PLoS One. 2013;8:e62487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yachida S, Totoki Y, Noe M, et al. Comprehensive genomic profiling of neuroendocrine carcinomas of the gastrointestinal system. Cancer Discov. 2022;12:692‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yachida S, Vakiani E, White CM, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well‐differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36:173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bradner JE. Hnisz D Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168:629‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alwan H, La Rosa S, Andreas Kopp P, et al. Incidence trends of lung and gastroenteropancreatic neuroendocrine neoplasms in Switzerland. Cancer Med. 2020;9:9454‐9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horie M, Saito A, Ohshima M, Suzuki HI, Nagase T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016;107:1755‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pozo K, Minna JD, Johnson JE. Identifying a missing lineage driver in a subset of lung neuroendocrine tumors. Genes Dev. 2018;32:865‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito T, Matsubara D, Tanaka I, et al. Loss of YAP1 defines neuroendocrine differentiation of lung tumors. Cancer Sci. 2016;107:1527‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rudin CM, Poirier JT, Byers LA, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer. 2019;19:289‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Poirier JT, George J, Owonikoko TK, et al. New approaches to SCLC therapy: from the laboratory to the clinic. J Thorac Oncol. 2020;15:520‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oliver JR, Kushwah R, Hu J. Multiple roles of the epithelium‐specific ETS transcription factor, ESE‐1, in development and disease. Lab Invest. 2012;92:320‐330. [DOI] [PubMed] [Google Scholar]
- 16. Ng AY, Waring P, Ristevski S, et al. Inactivation of the transcription factor Elf3 in mice results in dysmorphogenesis and altered differentiation of intestinal epithelium. Gastroenterology. 2002;122:1455‐1466. [DOI] [PubMed] [Google Scholar]
- 17. Yachida S, Wood LD, Suzuki M, et al. Genomic sequencing identifies ELF3 as a driver of Ampullary carcinoma. Cancer Cell. 2016;29:229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki M, Saito‐Adachi M, Arai Y, et al. E74‐like factor 3 is a key regulator of epithelial integrity and immune response genes in biliary tract cancer. Cancer Res. 2021;81:489‐500. [DOI] [PubMed] [Google Scholar]
- 19. Chen L, Huang M, Plummer J, et al. Master transcription factors form interconnected circuitry and orchestrate transcriptional networks in oesophageal adenocarcinoma. Gut. 2020;69:630‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan J, Silva TC, Gull N, et al. Lineage‐specific Epigenomic and genomic activation of oncogene HNF4A promotes gastrointestinal adenocarcinomas. Cancer Res. 2020;80:2722‐2736. [DOI] [PubMed] [Google Scholar]
- 21. Enfield KSS, Marshall EA, Anderson C, et al. Epithelial tumor suppressor ELF3 is a lineage‐specific amplified oncogene in lung adenocarcinoma. Nat Commun. 2019;10:5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanaka H, Yanagisawa K, Shinjo K, et al. Lineage‐specific dependency of lung adenocarcinomas on the lung development regulator TTF‐1. Cancer Res. 2007;67:6007‐6011. [DOI] [PubMed] [Google Scholar]
- 23. Saito RA, Watabe T, Horiguchi K, et al. Thyroid transcription factor‐1 inhibits transforming growth factor‐beta‐mediated epithelial‐to‐mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 2009;69:2783‐2791. [DOI] [PubMed] [Google Scholar]
- 24. Winslow MM, Dayton TL, Verhaak RG, et al. Suppression of lung adenocarcinoma progression by Nkx2‐1. Nature. 2011;473:101‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaya‐Okur HS, Wu SJ, Codomo CA, et al. CUT&tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaya‐Okur HS, Janssens DH, Henikoff JG, Ahmad K, Henikoff S. Efficient low‐cost chromatin profiling with CUT&tag. Nat Protoc. 2020;15:3264‐3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohmoto A, Suzuki M, Takai E, et al. Establishment of preclinical chemotherapy models for gastroenteropancreatic neuroendocrine carcinoma. Oncotarget. 2018;9:21086‐21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yachida S, Totoki Y, Noe M, et al. Comprehensive genomic profiling of neuroendocrine carcinomas of the gastrointestinal system. Cancer Discov. 2022;12:692‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie Z, Bailey A, Kuleshov MV, et al. Gene set knowledge discovery with Enrichr. Curr Protoc. 2021;1:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horie M, Saito A, Noguchi S, et al. Differential knockdown of TGF‐beta ligands in a three‐dimensional co‐culture tumor–stromal interaction model of lung cancer. BMC Cancer. 2014;14:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bentsen M, Goymann P, Schultheis H, et al. ATAC‐seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun. 2020;11:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramirez F, Ryan DP, Gruning B, et al. deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res. 2016;44:W160‐W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyakawa K, Miyashita N, Horie M, et al. ASCL1 regulates super‐enhancer‐associated miRNAs to define molecular subtypes of small cell lung cancer. Cancer Sci. 2022;113:3932‐3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382‐2383. [DOI] [PubMed] [Google Scholar]
- 35. Suzuki HI, Young RA, Sharp PA. Super‐enhancer‐mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017;168:e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell. 2013;153:307‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horie M, Miyashita N, Mikami Y, et al. TBX4 is involved in the super‐enhancer‐driven transcriptional programs underlying features specific to lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2018;314:L177‐L191. [DOI] [PubMed] [Google Scholar]
- 38. Miyashita N, Horie M, Suzuki HI, et al. An integrative analysis of transcriptome and Epigenome features of ASCL1‐positive lung adenocarcinomas. J Thorac Oncol. 2018;13:1676‐1691. [DOI] [PubMed] [Google Scholar]
- 39. Yachida S, Zhong Y, Patrascu R, et al. Establishment and characterization of a new cell line, A99, from a primary small cell carcinoma of the pancreas. Pancreas. 2011;40:905‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yanagihara K, Kubo T, Mihara K, et al. Establishment of a novel cell line from a rare human duodenal poorly differentiated neuroendocrine carcinoma. Oncotarget. 2018;9:36503‐36514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baine MK, Hsieh MS, Lai WV, et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive Immunohistochemical and histopathologic characterization. J Thorac Oncol. 2020;15:1823‐1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato Y, Okamoto I, Kameyama H, et al. Integrated Immunohistochemical study on small‐cell carcinoma of the lung focusing on transcription and Co‐transcription factors. Diagnostics (Basel). 2020;10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super‐enhancers. Cell. 2013;153:320‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tendler S, Kanter L, Lewensohn R, Ortiz‐Villalon C, Viktoersson K, De Petreis L. The prognostic implications of Notch1, Hes1, Ascl1, and DLL3 protein expression in SCLC patients receiving platinum‐based chemotherapy. PLoS One. 2020;15:e0240973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pozo K, Kollipara RK, Kelenis DP, et al. ASCL1, NKX2‐1, and PROX1 co‐regulate subtype‐specific genes in small‐cell lung cancer. iScience. 2021;24:102953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pandey A, Stawiski EW, Durinck S, et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat Commun. 2020;11:4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47:1003‐1010. [DOI] [PubMed] [Google Scholar]
- 49. Zhang T, Song X, Zhang Z, et al. Aberrant super‐enhancer landscape reveals core transcriptional regulatory circuitry in lung adenocarcinoma. Oncogenesis. 2020;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wooten DJ, Groves SM, Tyson DR, et al. Systems‐level network modeling of small cell lung cancer subtypes identifies master regulators and destabilizers. PLoS Comput Biol. 2019;15:e1007343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gong X, Du J, Parsons SH, et al. Aurora a kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2019;9:248‐263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Figure S10
Figure S11
Figure S12
Figure S13
Tables S1
Data Availability Statement
The data generated in this study were deposited in GSE190618.