

Graphical abstract
Keywords: Cigarette, E-cigarette, Reactive oxygen species, Oxidative stress, Cardiovascular toxicity, Adverse outcome pathway
Highlights
-
•
Reactive oxygen species and subsequent oxidative stresses are vital players in cigarettes and e-cigarettes-related cardiovascular toxicity.
-
•
The release of inflammatory cytokines and activation of nicotinic acetylcholine receptor (nAChR) is also involved in cigarettes and e-cigarettes-induced cardiovascular injury.
-
•
The toxic effects are summarized based on the adverse outcome pathway framework, which builds a comprehensive association from the molecular initiating events induced by cigarette and e-cigarette exposure to the cardiovascular adverse outcome.
-
•
The detrimental impacts on the cardiovascular system are described at different levels (from the molecular level to the population level).
Abstract
Background
Nowadays, cigarette smoking remains the leading cause of chronic disease and premature death, especially cardiovascular disease. As an emerging tobacco product, e-cigarettes have been advocated as alternatives to canonical cigarettes, and thus may be an aid to promote smoking cessation. However, recent studies indicated that e-cigarettes should not be completely harmless to the cardiovascular system.
Aim of Review
This review aimed to build up an integral perspective of cigarettes and e-cigarettes-related cardiovascular toxicity.
Key Scientific Concepts of Review
This review adopted the adverse outcome pathway (AOP) framework as a pivotal tool and aimed to elucidate the association between the molecular initiating events (MIEs) induced by cigarette and e-cigarette exposure to the cardiovascular adverse outcome. Since the excessive generation of reactive oxygen species (ROS) has been widely approved to play a critical role in cigarette smoke-related CVD and may also be involved in e-cigarette-induced toxic effects, the ROS overproduction and subsequent oxidative stress are regarded as essential parts of this framework. As far as we know, this should be the first AOP framework focusing on cigarette and e-cigarette-related cardiovascular toxicity, and we hope our work to be a guide in exploring the biomarkers and novel therapies for cardiovascular injury.
Introduction
Tobacco use has been proved to be a leading cause of morbidity and mortality, with nearly 6 million people killed each year [1]. Although cigarette smoking prevalence seemed to decline in most developed countries, it remains one of the most indispensable health risks of various chronic diseases [2]. Indeed, there are almost 13 billion smokers around the world, with nearly 80 % of them living in developing countries [3]. Thus, cigarette smoking continues to increase the health burden, especially among the poor population. Currently, epidemiologic studies emphasized that cigarette smoke should be a major cause of morbidity and mortality, especially resulting in the initiation and progression of cardiovascular disease [4]. Cigarette smoking significantly increases the incidence of both chronic and acute cardiovascular events and has been considered an independent risk factor for myocardial infarction and stroke [5], [6], [7]. Smoking is also correlated with the development and progression of atherosclerosis, which is regarded as a prevalent pathological basis for coronary artery and peripheral vascular disease [8]. Besides, tobacco use is associated with exacerbation of stable angina, inducing angina and vasospasm, increasing the risk and severity of heart failure, causing coronary or peripheral artery thrombolysis, and inducing restenosis after angioplasty [9], [10], [11], [12], [13]. As the extensive health risk induced by tobacco products has received due attention, the World Health Organization (WHO) has urged countries to invest in helping more people quit smoking. In a new press report published in November 2021, WHO noted that 60 countries are on track to meet the global target of reducing tobacco use by 30 % between 2010 and 2025. The report also highlighted the WHO's implementation of the WHO Framework Convention on Tobacco Control (WHO FCTC), which has saved millions of lives through effective and comprehensive tobacco control policies, as a great achievement in the fight against the tobacco epidemic [14]. As an alternative aid for smoking cessation, electronic cigarettes (e-cigarettes), which contain nicotine but no combustion byproducts, started to gain in popularity and become widely used worldwide.
E-cigarettes, also known as electronic nicotine delivery systems (ENDS), are one of the newest products in the tobacco industry. With sales increasing exponentially year by year, potential profits of e-cigarette retailing are supposed to surpass canonical cigarette sales margins shortly [15]. There are a wide variety of e-cigarettes available currently on sale, but they all have three basic components: a battery, a cartridge containing the e-liquid, and a nebulizer [16]. Most e-cigarettes are battery-powered and use a nebulizer to convert the liquid in the projectile into aerosols, which can mainly enter the body through the respiratory tract [17]. The toxic impact of e-cigarettes on the human body depends on the composition of the aerosol produced by the nebulizer and is mainly influenced by the formulation of e-liquid and the design of the nebulizer [18]. The e-liquid commonly contains nicotine, flavoring, glycerin, and propylene glycol, while metals, silicon, rubber, and ceramics can also be vaporized and inhaled [19]. It’s well recognized that most of the harmful ingredients of cigarette smoke come from the combustion procedure. Due to the simplifying and improvement of the aerosol formation mode, the nature and the concentration of e-cigarette aerosol may be completely different from those of tobacco smoke, consistent with it, recent studies also suggest that e-cigarette smoke is much less toxic than cigarette smoke [20]. Although the toxicity of e-cigarettes may be lower than that of cigarettes, the studies also suggest that they were not completely harmless to the organism [21]. Toxicological studies on e-liquids and aerosols have confirmed their toxic effects on numerous types of cultured cell lines, such as human and mouse fibroblasts, human embryonic stem cells, mouse neural stem cells, and cardiomyocytes [22], [23], [24]. In animal models, short-term exposure to e-cigarette smoke also induced lung inflammatory cell infiltration and increased inflammatory marker expressions such as IL-6, IL-1β, and TNFα. On the other hand, exposure to e-cigarette smoke with or without nicotine also exhibits neurotoxicity [25], [26]. Epidemiologic studies indicated that exposure to e-cigarette smoke-induced airway inflammation, oxidative stress, vascular endothelial damage, endothelial dysfunction, and vascular tone alteration [27], [28], [29], [30], [31]. Among these findings, accumulated evidence has pointed to e-cigarette use as a potential cardiovascular disease risk behavior [32]. Furthermore, current knowledge also suggests that the toxic mechanism of e-cigarettes may be similar to that of cigarettes, especially those that contain nicotine [33]. Therefore, the discussion of molecular mechanisms of their toxic effects should be combined, and recent studies tend to assume the molecular effects of e-cigarettes based on evidence achieved from the toxicity assessment of cigarette smoke.
Free radicals have been identified as a crucial activator responsible for cigarette smoke-related cardiovascular dysfunction. It is believed that free radicals are derived directly from the harmful components of cigarette smoke and can also be produced by circulating monocytes and macrophages stimulated by cigarette smoke. Meanwhile, uncoupled eNOS, xanthine oxidase, NADPH oxidase, and mitochondrial electron transport chain can also produce a large amount of endogenous ROS after exposure to cigarette smoke [34]. The excessive generation of ROS triggered a well-known cellular adverse effect named oxidative stress, which has been linked to a myriad of pathologies. Oxidative stress is a term used to describe the REDOX imbalance of the organisms, which is tending to oxidative damage. Although intracellular ROS are regulators of normal physiological functions, oxidative damage occurs when ROS levels exceed the scavenging capacity of the body's antioxidants [35]. Oxidative damage caused by ROS can directly lead to lipid peroxidation and excessive consumption of antioxidants, which may be an important factor contributing to the development of coronary artery disease [36]. In addition to directly inducing oxidative damage, ROS also serves as an important signaling molecule and regulates the death, survival, and proliferation of cells by triggering multiple types of cellular signaling pathways. The activation of ROS-related signaling pathways has been proved to trigger various cellular dysregulation including endothelial cell dysfunction, differentiation of vascular smooth cells, myocardial apoptosis, and proliferation of fibroblast, which are critical in all phases of CVD development [37]. The elevation of ROS and biomarkers of oxidative stress after exposure to e-cigarettes have been observed in both in vitro and in vivo trials, and even population research [38]. Therefore, ROS overproduction and subsequent oxidative stress should be regarded as the essential players in cigarette and e-cigarette-induced cardiovascular effects.
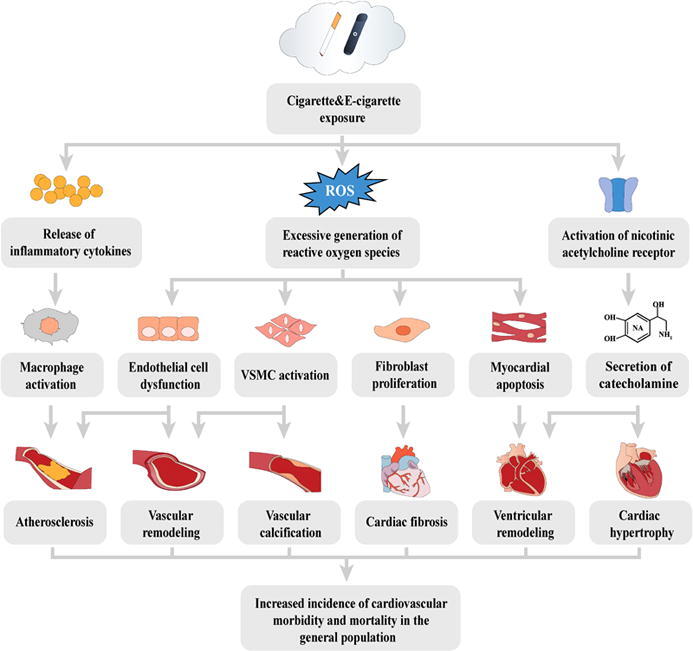
Although numerous studies have documented oxidative stress caused by cigarettes and e-cigarettes, due to the limitation of research type, the studies could not fully include the various toxic endpoints, and thus would not completely overlap the gap between the ROS overproduction and the consequent adverse effects on each level. Hence, a framework needs to be established to link the molecular/cellular events to the adverse cardiovascular effects in individuals or among the population and to provide a more comprehensive perspective on the cardiovascular toxicity of both cigarettes and e-cigarettes. Based on the published articles on PubMed and the established works on the AOP wiki, we summarize the toxic effects and underlying mechanisms of both cigarettes and e-cigarettes on the cardiovascular system. In addition to the ROS overproduction as we mentioned above, the evidence supporting the release of inflammatory cytokines after cigarette smoke exposure and the pharmacological effects of nicotine on the cardiovascular system has also been widely recognized. Therefore, the toxic effects of cigarettes and e-cigarettes are assumed to be mainly attributed to three molecular initiating events (MIEs): excessive generation of ROS, the release of inflammatory cytokines, and activation of nicotinic acetylcholine receptor (nAChR). Since ROS overproduction and subsequent oxidative stress have been identified as the most common toxic mechanism in both cigarette and e-cigarette-induced cardiovascular effects, this AOP framework is initiated with ‘excessive ROS generation’ as the essential MIE that triggers the most cellular and tissue/organ effects (summarized in Table 1 and Fig. 1). On the other hand, considering that current evidence indicated that the cardiovascular effects induced by e-cigarettes should be similar to canonical cigarettes, we tended to assess the toxic effects of both cigarettes and e-cigarettes together by using this AOP framework [33], [39], [40]. By adopting the criteria and guidelines of the Bradford-Hill weight of evidence consideration, we also summarized and evaluated the relationship of the key event (KER) (summarized in Table 2) [41]. As far as we conducted this work, there is no complete AOPs on the cardiovascular toxicity associated with either cigarettes or e-cigarettes. Therefore, this should be the first review to elucidate the cardiovascular toxicity of cigarettes and e-cigarettes based on the AOP framework. The main purpose of this review is to use the AOP framework as an effective tool to build up the association between tobacco products (both cigarettes and e-cigarettes) and the adverse effects (key events) they caused on different levels (from molecular to individual/population), thus provide guidance and assistance for toxic assessment of tobacco products.
Table 1.
Summary of the AOP.
| Sequence | Type | Event ID | Title | Short name |
|---|---|---|---|---|
| 1 | MIE | 1115 | Excessive generation of reactive oxygen species | Excessive generation of ROS |
| 2 | MIE | 151 | Release of inflammatory cytokines | Release of inflammatory cytokines |
| 3 | MIE | 559 | Activation of nicotinic acetylcholine receptor | Activation of nicotinic acetylcholine receptor |
| 4 | KE | 1392 | Oxidative stress | Oxidative stress |
| 5 | KE | 1198 | Macrophage activation | Macrophage activation |
| 6 | KE | 1913 | Endothelial cell dysfunction | Endothelial cell dysfunction |
| 7 | KE | 1925 | Vascular smooth muscle cell activation | Vascular smooth muscle cell activation |
| 8 | KE | 1500 | Fibroblast proliferation and myofibroblast differentiation | Cellular proliferation and differentiation |
| 9 | KE | 1918 | Myocardial apoptosis | Myocardial apoptosis |
| 10 | KE | 2004 | Secretion of catecholamine | Secretion of catecholamine |
| 11 | KE | 1443 | Atherosclerosis | Atherosclerosis |
| 12 | KE | 2003 | Vascular remodeling | Vascular remodeling |
| 13 | KE | 2000 | Vascular calcification | Vascular calcification |
| 14 | KE | 1924 | Cardiac fibrosis | Cardiac fibrosis |
| 15 | KE | 2002 | Ventricular remodeling | Ventricular remodeling |
| 16 | KE | 2001 | Cardiac hypertrophy | Cardiac hypertrophy |
| 18 | AO | 1929 | Increased incidence of cardiovascular morbidity and mortality in the general population | Increased incidence of cardiovascular morbidity and mortality |
Molecular Initiating Event (MIE), Key Event (KE), Adverse outcome (AO).
Fig. 1.

Adverse Outcome Pathways diagram related to cigarette and e-cigarette smoke-induced cardiovascular toxicity initiated with the excessive generation of reactive oxygen species (ROS), the release of inflammatory cytokines, and the activation of nicotinic acetylcholine receptor (nAChR). Yellow cube: Molecular Initiating Events (MIE); Blue cube: Key Events (KE); Grey cube: Adverse Outcome (AO). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
Summary and evaluation of Key Event Relationships (KER) from the AOP.
| Upstream event | Relationship Type | Downstream event | Weight of evidence |
||
|---|---|---|---|---|---|
| Biological Plausibility | Essentiality | Empirical Evidence | |||
| Excessive generation of reactive oxygen species | adjacent | Oxidative stress | Strong | Strong | Strong |
| Release of inflammatory cytokines | adjacent | Macrophage activation | Strong | Strong | Strong |
| Activation of nicotinic acetylcholine receptor | adjacent | Secretion of catecholamine | Strong | Strong | Strong |
| Oxidative stress | adjacent | Endothelial cell dysfunction | Strong | Strong | Strong |
| Oxidative stress | adjacent | Vascular smooth muscle cell activation | Strong | Strong | Strong |
| Oxidative stress | adjacent | Fibroblast proliferation and myofibroblast differentiation | Strong | Strong | Strong |
| Oxidative stress | adjacent | Myocardial apoptosis | Strong | Strong | Strong |
| Endothelial cell dysfunction | adjacent | Vascular remodeling | Strong | Moderate | Strong |
| Endothelial cell dysfunction | adjacent | Atherosclerosis | Strong | Strong | Strong |
| Vascular smooth muscle cell activation | adjacent | Vascular remodeling | Strong | Strong | Strong |
| Vascular smooth muscle cell activation | adjacent | Vascular calcification | Strong | Strong | Strong |
| Macrophage activation | adjacent | Atherosclerosis | Strong | Strong | Strong |
| Fibroblast proliferation and myofibroblast differentiation | adjacent | Cardiac fibrosis | Strong | Strong | Strong |
| Myocardial apoptosis | adjacent | Ventricular remodeling | Strong | Strong | Strong |
| Secretion of catecholamine | adjacent | Ventricular remodeling | Strong | Moderate | Strong |
| Secretion of catecholamine | adjacent | Cardiac hypertrophy | Strong | Moderate | Strong |
Toxic components in cigarette and e-cigarette smoke
Although the link between e-cigarettes and cardiovascular disease remained controversial, the existing evidence suggested that e-cigarettes should be more harmless to the cardiovascular system than canonical cigarettes. The reasons for the differences in the toxicity between them can be attributed to their components (summarized in Table 3). The canonical cigarette smoke contains over 7000 toxicants including carbon monoxide, aldehydes, nicotine, N-nitrosamines, volatile organic compounds (VOCs), polycyclic aromatic hydrocarbons (PAHs), solid particulate matter, and transition metals [33], [42]. Carbon monoxide (CO) is an important toxic component in cigarette smoke, which has been proved to bind to hemoglobin and thus inhibited its ability of oxygen transportation [43]. Biomarkers such as carboxyhemoglobin protein have been adopted to reflected the CO levels. Meanwhile, a recent study also reported that chronic CO exposure could enhance arrhythmia via increased ROS generation, which supported that CO may contribute to cigarette smoke-induced oxidative stress [44]. Aldehydes are a series of high reactive species that especially contribute to cigarette smoke-induced oxidative stress and inflammation. As a long living compounds, once being absorbed into the body, aldehydes can remain in the blood stream and thus can be transported to various body tissues. Particularly, α, β-unsaturated aldehyde from cigarette smoke can directly combine with nucleophilic amino acids to produce covalent compounds [45]. Pei et al. reported that α, β-unsaturated aldehyde could induce contractile dysfunction in cardiomyocytes via ROS elevation and mitochondrial damage [46]. Meanwhile, Facchinetti et al. also reported that two α, β-unsaturated aldehydes (acrolein and crotonaldehyde) could enhance ROS generation and secretion of inflammatory cytokines in macrophages at micromolar concentrations [47]. Therefore, aldehydes should be responsible for cigarette smoke-induced oxidative stress and inflammation. N-nitrosamines are by-products formed by nicotine and tobacco alkaloids during tobacco processing and smoking, which have been extensively proved to be one of the major carcinogens that involved in cigarette smoke-related tumorigenesis [48]. VOCs mainly contain benzene, benzopyrene, and toluene that are released from the incomplete combustion of cigarettes [49]. Occupational exposure to VOCs has been proved to induce oxidative stress, inflammation and DNA damage in patients [50], [51]. PAHs are common environmental pollutants that derived from tobacco combustion procedure [52]. PAHs have been proved to induce oxidative stress and DNA damage via ROS overproduction [53]. Meanwhile, a recent study conducted by Dai et al. has suggested that exposure to PAHs could result in low-grade inflammation in children [54]. Particulate matter is an important solid component of cigarette smoke, and particulate matter produced by tobacco combustion may be the most important source of indoor particulate matter pollution [55]. Particulate matter-induced cardiovascular toxicity has been widely elucidated in previous studies, while oxidative stress and inflammation serve as the essential players [56], [57]. The hazards of metals in cigarette smoke have often been neglected, but a recent study indicated that smokers had significantly higher levels of metals in their lungs than non-smokers [58]. In addition, another study has proved that transition metals contained in cigarette smoke were much higher than that of incense smoke, traffic-influenced aerosols, and urban aerosols, while the elevation of metal contents were associated with an increase in ROS levels [59].
Table 3.
Comparison of cigarettes and e-cigarettes-related cardiovascular toxicity.
| Canonical cigarette | Electronic cigarette | |
|---|---|---|
| Main toxicants | Carbon monoxide, aldehydes, nicotine, N-nitrosamines, VOCs, PAHs, solid particulate matter, and transition metals | Aldehydes, nicotine, N-nitrosamines, VOCs, and transition metals |
| Major mechanisms | Oxidative stress, inflammation, activation of the sympathetic nervous system | Oxidative stress, inflammation, activation of the sympathetic nervous system |
| ROS generation | Cigarette smoke induced a more significant increase in ROS levels | E-cigarette smoke induced an increase in ROS levels |
| Inflammatory cytokines | Cigarette smoke induced a more significant increase in inflammatory cytokines | E-cigarette smoke induced an increase in inflammatory cytokines |
| Activation of nAChR | Both cigarette and e-cigarette smoke induced nAChR activation but lacked evidence to compare | |
| Oxidative stress | Cigarette smoke induced a more significant increase in oxidative stress-related biomarkers | E-cigarette smoke induced an increase in oxidative stress-related biomarkers |
| Endothelial cell dysfunction | Cigarette smoke induced a more significant effect on endothelial cell dysfunction | E-cigarette smoke induced endothelial cell dysfunction |
| VSMC activation | Cigarette smoke induced VSMC activation | Lacked evidence about e-cigarette smoke-induced VSMC activation |
| Macrophage activation | Both cigarette and e-cigarette smoke induced macrophage activation but lacked evidence to compare | |
| Vascular remodeling | Cigarette smoke induced vascular remodeling | Lacked evidence about e-cigarette-induced vascular remodeling |
| Vascular calcification | Cigarette smoke induced vascular calcification | Lacked evidence about e-cigarette-induced vascular calcification |
| Atherosclerosis | Cigarette smoke induced a more significant increase in plaque progression | E-cigarette smoke induced progression in atherosclerotic plaque |
| Fibroblast activation | Cigarette smoke induced fibroblast activation | Lacked evidence about e-cigarette-induced fibroblast activation |
| Myocardial apoptosis | Both cigarette and e-cigarette smoke induced myocardial apoptosis but lacked evidence to compare | |
| Catecholamine secretion | No significant differences were observed between cigarette and e-cigarette-induced elevation of urinary catecholamine | |
| Cardiac fibrosis | Both cigarette and e-cigarette smoke induced cardiac fibrosis but lacked evidence to compare | |
| Ventricular remodeling | Both cigarette and e-cigarette smoke induced ventricular remodeling but lacked evidence to compare | |
| Cardiac hypertrophy | Both cigarette and e-cigarette smoke induced cardiac hypertrophy but lacked evidence to compare | |
| Individual and populational adverse effects | Cigarette smoke induced a significant increase in cardiovascular morbidity and mortality | Lacked evidence about e-cigarette-induced cardiovascular risks |
Due to the simplifying of the tobacco combustion procedure, the components of the e-cigarette liquid are much simpler, it contains only nicotine, propylene glycol, and glycerine. As a result, most of the toxicants detected in e-cigarette smoke were derived from all three ingredients [60]. The heating procedure of propylene glycol can produce a large number of thermal dehydration products, mainly including acetaldehyde, formaldehyde, propylene oxide, etc [61]. While glycerol produces acrolein, formaldehyde, and dehydrated glycerol. Aldehydes including formaldehyde and acrolein are responsible for inducing high blood pressure, myocardial dysfunction, and arrhythmia via oxidative stress and inflammation [62], [63]. Evidence emphasized that they may be the major toxicants responsible for cardiovascular toxic effects induced by e-cigarette emission, and was released by the heating procedure in a temperature and power-dependent manner [61]. Acrolein exposure from cigarettes has been widely documented and associated with increased cardiovascular risk. The measurement of acrolein content mainly depends on the determination of the major metabolite—3-hydroxypropylmercapturic acid (3-HPMA) in urine [64]. Exposure assessments of e-cigarettes indicated that urine 3-HPMA levels after e-cigarette use were much lower than those of cigarettes and were not significantly different from non-smokers [65]. N-nitrosamines and VOCs have also been detected in e-cigarette smoke, although the levels may be much lower than in cigarette smoke [66]. In addition, since e-cigarettes typically generate aerosols by heating metal coils, studies have suggested that e-smoke could be a potential source of exposure to toxic metals [67]. On the other hand, although it seems to be safer than canonical cigarettes, the cardiovascular toxicity of e-cigarettes can vary significantly depending on the components of e-liquids and the heating procedure. Indeed, e-cigarettes generally contain less harmful substances than cigarettes, while those substances can vary widely depending on the brand of e-cigarette [66], [68].
Nicotine should be the most well-studied component of cigarette smoke and an important contributor to the addictive properties of tobacco products. Numerous studies have proved that nicotine could induce ROS overproduction and inflammatory response in cardiovascular system [69], [70], [71]. Meanwhile, nicotine could also regulate the sympathetic nervous system and the release of catecholamine via activation of nAChR [72], [73]. These effects of nicotine will be elaborated in the discussion of MIEs and KEs. However, recent studies indicated that while second-hand smoke can accelerate atherogenesis and impair vascular function, nicotine exposure alone may cause no change to arterial lipid lesions, thus the toxic effect caused by cigarette smoke may be attributed to other combustion products [74]. Similarly, researchers also found that although high doses of nicotine exposure may favor the progress of atherosclerosis, at concentrations similar to a smoker’s blood level, it may not affect the formation or progression of the atherosclerotic plaque [34]. Therefore, Nicotine and other components in cigarettes and e-cigarettes should be studied as a whole, with particular attention to the impact of nicotine exposure dose on the toxic effects. Exposure to nicotine can be reflected in various ways, including measuring plasma nicotine levels and urine levels of the nicotine metabolite (cotinine). Although most of the commercially available e-cigarettes contain nicotine content and numerous reports indicated that potential nicotine concentration in plasma after e-cigarette exposure may be much higher than in cigarettes, the detection of mean blood nicotine levels for cigarette and e-cigarette users indicated that compared to cigarettes, e-cigarettes were associated with a lower boost in blood nicotine levels [155], [156]. Based on this evidence, we suggested that the amount of effective nicotine delivered by e-cigarettes may be lower than that delivered by cigarettes, while the nicotine content may increase with the upgrade of e-cigarettes. Therefore, due attention should be paid to nAChR-related effects induced by e-cigarette use [157]. Taken together, although the cardiovascular toxicity of cigarettes and e-cigarettes may vary from their components, their underline mechanisms should be mainly attributed to oxidative stress, inflammation, and the activation of the sympathetic nervous system.
Cardiovascular-related molecular initiating events triggered by cigarette and e-cigarette exposure
Molecular initiating event (KE 1115): Excessive generation of reactive oxygen species
Reactive oxygen species (ROS) are a general term for a class of substances that contain oxygen and are chemically active, which mainly include superoxide radical O2.−, hydroxyl radical OH., and the freely diffusible H2O2. Normal metabolism in the body can produce ROS, of which mitochondria are the main source of reactive oxygen species. Aerobic metabolism in mitochondria consumes a large amount of oxygen, part of which is converted into ROS in the mitochondrial intima and matrix [75]. ROS is an important intracellular signal molecule in the physiological state, participating in the regulation of many important physiological processes such as cell metabolism, proliferation, and apoptosis [76]. Sufficient evidence indicates that ROS and oxidative stress are common features of most cardiovascular diseases, including atherosclerosis, arrhythmia, and myocardial ischemia–reperfusion injury. While elevated ROS levels resulted in cell dysfunction, cell death, and even tissue injury [77]. ROS elevation is mainly associated with several common ways of cell death such as apoptosis and programmed necrosis. Apoptosis is a programmed cell death that is usually characterized by nuclear pyknosis and the formation of apoptotic corpuscles. Excessive generation of ROS could induce peroxidation of membrane lipid and mitochondrial damage, which eventually contribute to apoptosis. Programmed necrosis can be further classified as necroptosis, pyroptosis, etc. Both necroptosis and pyroptosis involve cell swelling and rupture of the plasma membrane, so they are also related to the release of inflammatory mediators [78]. ROS-induced necroptosis could be triggered via mPTP opening and calcium dysregulation, and the RIPK3 signaling pathway played the essential role in this process [79]. On the other hand, pyroptosis depends on inflammasome formation, and ROS is a key mechanism that triggers NLRP3 inflammasome formation and activation [80]. Fluorescence probes and electron spin resonance probes have been widely adopted to evaluate the ROS levels in cell and tissue samples [81], [82]. Numerous studies have reported the excessive ROS generation induced by cigarette, e-cigarette, or nicotine exposure (summarized in Table 4).
Table 4.
Summary of evidence supporting MIE (KE 1115): excessive generation of reactive oxygen species.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [85] | CSE | In vitro | HUVECs were pretreated (or not) with melatonin (100 μM) for 3 h and then treated with CSE for 24 h | Excessive ROS generation, pyroptosis |
| [86] | CSE | In vitro | RASMCs were treated with CSE for 5–10 h | Excessive ROS generation, DNA damage, apoptosis, and inflammation |
| [87] | CSE | In vitro | human myocardial cells (AC16) were pretreated (or not) with EGCG (10 μM) for 30 min and then treated with CSM for 24 h | Excessive ROS generation, inflammation, apoptosis |
| [88] | Cigarette smoke | In vivo | Swiss mice were exposed to cigarette smoke for 7, 15, 30, 45, and 60 days | Excessive ROS generation, autophagy |
| [89] | Cigarette smoke | In vivo | Sprague-Dawley rats were exposed to cigarette smoke for 7 days | Excessive ROS generation |
| [90] | Cigarette smoke | Clinical trial | In this study, 20 healthy subjects and 20 smokers were treated with dark chocolate or milk chocolate | Excessive ROS generation, NOX2 activation in platelets |
| [91] | Cigarette smoke | Cross-sectional study | A total of 252 healthy subjects were examined, while 212 subjects of them were smokers | Excessive ROS generation (reflected by evaluation of d-ROM) |
| [92] | E-cigarette smoke | In vitro | HPMVEC were exposed to serum from human subjects exposed to e-cigarette smoke for 2 h | Excessive ROS generation, NOX2 activation |
| [93] | E-liquid, e-cigarette smoke | In vitro | iPSC-ECs were exposed to e-liquids or serum from human subjects exposed to e-cigarette smoke for 48 h, | Excessive ROS generation, endothelial dysfunction |
| [94] | E-cigarette smoke | In vivo | Sprague Dawley rats were exposed to e-cigarette smoke for 28 days | Excessive ROS generation, inflammation |
| [95] | E-cigarette smoke | In vivo | C57BL6J mice were exposed to e-cigarette smoke for 1, 2, 4, 8 h | Excessive ROS generation, alteration of metabolites |
| [96] | Nicotine | In vivo and in vitro | ApoE-/- mice were exposed to nicotine (100 μg/mL) for 12 weeks, while HAECs were treated with nicotine (1 μM) for 24 h | Excessive ROS generation, pyroptosis, atherosclerosis |
| [97] | Nicotine | In vivo | Sprague-Dawly rats were exposed to nicotine (3 mg/kg/day) for 6 weeks | Excessive ROS generation, mitophagy |
| [102] | CSE, E-cigarette smoke extract | In vitro | HUVECs were treated with various doses of CSE and E-cigarette smoke extract for 48 h | Excessive ROS generation, inflammation |
| [103] | Cigarette smoke, e-cigarette smoke | In vitro | Immune cells isolated from Wistar rats were treated with fetal bovine serum containing cigarette smoke or e-cigarette smoke | An increase in superoxide anion (a type of ROS) |
Dysregulation of ROS generation and metabolism mainly links cigarette smoke exposure to the development of various cardiovascular diseases [83]. The ROS elevation and downstream adverse effects induced by cigarette smoke have been widely reported. Studies have concluded that stable substances in cigarette smoke, such as methyl vinyl ketone (MVK) and acrolein, are responsible for NOX activation and subsequent increase in ROS generation [84]. Wang et al. reported cigarette smoke extract (CSE)-induced excessive ROS production and pyroptosis in endothelial cells, and melatonin (an indoleamine that functioned as an antioxidant) could alleviate the toxic effects by inhibiting ROS/NLRP3 axis [85]. While Yang et al. reported that CSE exposure was associated with ROS elevation and subsequent DNA damage, apoptosis, and inflammation in rat aortic smooth muscle cells (RASMCs) [86]. ROS was also reported to involve in cigarette smoke-induced inflammation in human myocardial cells (AC16) by mediating NF-κB and p38 MAPK pathways, while EGCG (an antioxidant property in green tea) exhibited a cardioprotective effect against ROS-mediated cardiac injury [87]. ROS overproduction was also observed in vivo tests after cigarette smoke exposure. Morsch et al. reported increasing in ROS content and activation of autophagy-related pathways in mice exposed to cigarette smoke for 7, 15, 30, 45, and 60 days [88]. Another study conducted by Yang et al. illustrated that cigarette smoke was able to induce ROS generation in the carotid arteries of rats [89]. An epidemiologic study also indicated elevated ROS formation and NOX2 activation in platelets from smokers [90]. Hayashi et al. applied the derivative of reactive oxygen metabolites (d-ROM) as a biomarker of ROS, and they observed the serum ROS levels of smokers increased with the number of cigarettes consumed per day [91].
E-cigarette smoke has been proved to induce ROS overproduction and activation of NOX2 in human pulmonary microvascular endothelial cells (HPMVECs) and serum of non-smoking healthy subjects after acute e-cigarette exposure [92]. Lee et al. also observed increased ROS levels and activity of caspase 3/7 in human-induced pluripotent stem cell-derived endothelial cells (iPSC-ECs) after treatment with e-liquids, while ROS-associated endothelial dysfunction was also observed in iPSC-ECs after exposure to serum from e-cigarette users [93]. Cirillo et al. reported that e-cigarette exposure for 28 days could induce excessive ROS generation in the lungs of Sprague Dawley rats [94]. In a recent study conducted by Ren et al., acute exposure to e-cigarette smoke was reported to induce ROS elevation in the hearts of C57BL/6J mice [95]. Nicotine was also documented to mediate endothelial cell pyroptosis and promote atherosclerosis via the ROS-NLRP3 pathway [96]. Furthermore, nicotine has been proved to cause cardiac toxicity via triggering ROS burst in young adult rats [97]. On the other hand, the activation of the renin-angiotensin-aldosterone system has been regarded as an essential pathway for nicotine-related cardiovascular effects. Aldosterone is a mineralocorticoid hormone that induced direct adverse effects on cardiomyocytes, especially including the excessive generation of ROS. Cardiac dysfunction induced by aldosterone has been attributed primarily to mitochondrial damage caused by elevated ROS levels, such as oxidative damage to mitochondrial DNA [98]. Aldosterone and subsequent ROS elevation have also been proved to be associated with systolic and diastolic dysfunction, and ROS overproduction could induce deterioration of systolic and diastolic function through disturbance of cardiac mechanics [99]. Michael et al. reported that nicotine treatment for 8 weeks significantly increased the circulating levels of aldosterone in rats [100]. Cora et al. reported that nicotine promoted aldosteronism by upregulating βarrestin1, which ultimately induced cardiac dysfunction in rats [101]. An in vitro study compared the ROS levels in human umbilical vein endothelial cells (HUVECs) after exposure to either e-cigarette smoke extracts or canonical tobacco smoke extracts, and the results indicated that ROS levels induced by e-cigarette smoke extracts were much lower than that of commercially available tobacco cigarette extracts [102]. Di Biase et al. found that the medium containing tobacco cigarette smoke induced a more significant increase in superoxide anion (a type of ROS) than that of e-cigarette smoke, while the nicotine-free e-cigarette smoke didn’t induce a significant change. [103]. In summary, these results indicated that increased ROS levels should be a major cause and common feature of cardiovascular toxicity of cigarettes and e-cigarettes. Moreover, current evidence provided by various studies suggested that the lower ROS levels after exposure to e-cigarettes may be an explicable reason for their lower toxicity compared with canonical cigarettes.
Molecular initiating event (KE 151): Release of inflammatory cytokines
Inflammation refers to an organism’s defensive response to external stimuli, which is beneficial in most cases. However, inflammation can also result in tissue injury and has been regarded as a pathological basis for a sort of disease. This is a complex process that involves inflammatory cells first recognizing the affected tissue, white blood cells recruiting to the tissue, eliminating harmful substances, and repairing the site of damage. Inflammation requires interactions between the cell surface, extracellular matrix, and pro-inflammatory mediators [104]. The initiation and progression of inflammation require the participation of multiple inflammatory signaling pathways, such as NF-κB, p38 MAPK, PI3K, and JAK/STAT [105]. Activation of these signaling pathways leads to the production of downstream inflammatory mediators, including chemokines, cytokines, vasoactive amines, eicosanoids, and products of proteolytic cascades [106]. The role of inflammation in cardiovascular disease has been extensively studied. The inflammatory pathways have been proved to involve in the development of atherosclerotic plaques. As described in numerous studies, the causes of atherosclerosis are endothelial injury, abnormal lipid metabolism, and hemodynamic injury. The activation of inflammatory pathways is mainly involved in endothelial dysfunction, and atherosclerosis is thought to be accompanied by flow-mediated inflammatory changes in endothelial cells (ECs) [107]. When ECs are activated by an exogenous stimulus, they express monocyte chemotactic protein-1 (MCP-1), interleukin-8 (IL-8), intercellular adhesion molecule-1 (ICAM-1), vascular adhesion molecule-1 (VCAM-1), e-selectin, p-selectin, and other inflammatory mediators. The inflammation begins by attracting lymphocytes and monocytes that bind to the endothelium and infiltrate the artery wall [108]. Mounting studies have proved the activation of inflammatory signaling pathway induced by cigarette, e-cigarette, or nicotine exposure (summarized in Table 5).
Table 5.
Summary of evidence supporting MIE (KE 151): release of inflammatory cytokines.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [111] | Cigarette smoke, CSE | In vivo and in vitro | The macaques were exposed to environmental tobacco smoke for 6 months, while various cell lines were treated with CSE | Activation of the NF-κB signaling pathway, the release of inflammatory cytokines |
| [112] | Cigarette smoke, CSE | In vivo and in vitro | The Wistar rats were exposed to cigarette smoke for 1 week, while CAECs were treated with CSE for 24 h | Activation of the NF-κB signaling pathway, an increase in inflammatory gene expression |
| [116] | CSE | In vitro | HUVECs were treated with CSE for 24 h with (or not) MitoQ (100 nmol/L) | Activation of the NF-κB signaling pathway, activation of NLRP3 inflammasome |
| [117] | CSE | In vitro | HUVECs were pretreated (or not) with atorvastatin for 4 h, and then treated with CSE for 24 h | Activation of the NF-κB signaling pathway, the release of inflammatory cytokines |
| [118] | CSE | In vivo and in vitro | C57BL6J mice were exposed to cigarette smoke (2 h per day, 5 days per week) for 1 months, bone marrow-derived macrophages were treated with CSE for 1, 4, 8 h | Activation of the NF-κB signaling pathway, necroptosis |
| [120] | CSE | In vitro | H9c2 myocytes were pretreated with cerium oxide nanoparticles (1, 10, or 100 nM) for 24 h, and then treated with CSE for 24 h | Activation of the NF-κB signaling pathway, an increase in inflammatory gene expression |
| [87] | CSE | In vitro | human myocardial cells (AC16) were pretreated (or not) with EGCG (10 μM) for 30 min and then treated with CSM for 24 h | Activation of the p38 MAPK signaling pathway, the release of inflammatory cytokines, apoptosis |
| [124] | Cigarette total particulate matter | In vitro | BASE-2B cells were exposed to cigarette total particulate matter for 24 h | Activation of the p38 MAPK signaling pathway, the release of inflammatory cytokines, autophagy |
| [125] | CSE | In vitro | human bronchial epithelial cells were treated with CSE for 24, 48, and 72 h | Activation of the p38 MAPK signaling pathway, the release of inflammatory cytokines, apoptosis |
| [126] | CSE | In vitro | Bronchial-epithelial NCI-H292 cells were treated with CSE for 24 h | Activation of the p38 MAPK signaling pathway, the release of inflammatory cytokines, apoptosis |
| [127] | CSE | In vitro | HUVECs were treated with CSE for 6 h | Activation of the p38 MAPK signaling pathway, upregulation of cell adhesion molecules, actin filament reorganization |
| [128] | CSE | In vitro | Human pulmonary artery endothelial cells were treated with sidestream cigarette smoke extract for 5, 15, 30, and 60 min | Activation of the p38 MAPK signaling pathway, an increase in endothelial permeability |
| [129] | CSE | In vitro | HAECs were pretreated with pretreated (or not) with l-Arginine (200 μM) or l-NAME (400 μM) for 30 min, and then treated with CSE for 6 h | Activation of the p38 MAPK signaling pathway, apoptosis |
| [130] | Cigarette smoke and other nicotine products | In vitro | HUVECs were exposed to different test substances for 24 h | Activation of the PI3K/Akt/eNOS signaling pathway, activation of pro-inflammatory endothelial phenotype |
| [131] | CSE | In vitro | RASMCs or RAW cells were treated with CSE for 24 h | Activation of the Jak/Stat signaling pathway, the release of MMP-2 and MMP-9 |
| [134] | Cigarette smoke | Cross-sectional study | The study enrolled 97 smokers and 62 non-smokers | Upregulation of TLR4 mRNA |
| [135] | E-cigarette smoke extract, CSE | In vitro | Human neutrophils were isolated from the peripheral blood of healthy nonsmokers. The cells were treated with e-cigarette smoke extract or CSE for 2, 4, 6 h | Activation of the p38 MAPK signaling pathway, the release of MMP-9 |
| [136] | Nicotine | In vivo and in vitro | ApoE-/- mice fed a high-fat diet were exposed to nicotine (100 μg/mL) for 12 months, while RAW 264.7 cells were treated with nicotine at concentrations of 10, 100, and 1000 μM for 24 h | Activation of the HDAC6/NF-κB/NLRP3 signaling pathway, pyroptosis, atherosclerosis |
| [137] | Nicotine | In vitro | HUVECs were treated with nicotine at concentrations of 10-8, 10-7, 10-6 mol/L for 12 h | Activation of the NF-κB signaling pathway, apoptosis |
| [138] | Nicotine | In vitro | SMCs were monocultured or co-cultured with ECs, and then treated with 1 μM for 24 h | Activation of the NF-κB signaling pathway, |
| [139] | Cigarette smoke, e-cigarette smoke | Cross-sectional study | The study enrolled 7130 subjects, including non-smokers, e-cigarette users, cigarette users, and dual users | Elevation of inflammatory biomarkers |
The endothelial NF-κB signaling pathway plays a key role in early atherogenesis, and it has been proved to be activated in prepathological atherogenic regions of the mouse aorta [109]. As a core transcription factor of inflammation and cell death in the pathogenesis of atherosclerotic lesions, activation of NF-κB in endothelial cells increases the susceptibility to local proximal aortic atherosclerosis [110]. Activation of the NF-κB signaling pathway and downstream inflammatory mediators have been fully elucidated in cigarette smoke-induced pulmonary dysregulation, both in vivo and in vitro [111]. Recent studies also started to focus on the role of cigarette smoke-induced NF-κB activation in the cardiovascular system. Cigarette smoke exposure is reported to induce the activation of NF-κB and inflammatory gene expression in rat arteries and cultured coronary arterial endothelial cells (CAECs), accompanied by upregulation of ICAM-1, inducible nitric oxide synthase, IL-6, and tumor necrosis factor-α (TNF-α) [112]. MitoQ is a ubiquinone derivative, which can be partially inserted into the lipid bilayer and reduced by the mitochondrial respiratory chain. The subsequent product—panthenol derivative is an effective mitochondrial targeted antioxidant, which prevents lipid peroxidation and protects mitochondria from oxidative damage [113]. Atorvastatin is a widely used lipid-lowering agent that reduces total blood cholesterol and ldl-cholesterol, thereby providing a prevention of cardiovascular events [114], [115]. NF-κB activation was also observed in HUVECs exposed to cigarette smoke, along with activation of NLRP3 inflammasome and endothelial barrier dysfunction, while treatment with either MitoQ or atorvastatin can significantly alleviate the adverse effects [116], [117]. Furthermore, existing evidence suggested that cigarette smoke exposure induced necroptosis in macrophages via NF-κB activation, which may also be involved in the deterioration of atherosclerosis [118], [119]. Inflammation signals like NF-κB and its downstream genes such as TNF-α, IL-1β, and IL-6 have also been proved to be triggered by cigarette smoke-induced ROS elevation and thus played an important role in pathophysiological processes of smoking-induced cardiac injury [120], [121].
As mentioned above, p38 MAPK is another pivotal inflammatory signaling pathway involved in cardiovascular disease. The activation of p38 MAPK was associated with myocardial cell dysfunction, development of injury during myocardial ischemia, and P38 MAPK-dependent uptake of oxidized low-density lipoprotein (LDL) which leads to foam cell formation [122], [123]. Numerous studies have illustrated cigarette smoke-related p38 MAPK activation in a variety of cell lines, including BEAS-2B cells, human bronchial epithelial (HBE) cells, bronchial-epithelial NCI-H292 cells, and human AC16 cardiomyocytes [87], [124], [125], [126]. CSE also induced upregulation of cell adhesion molecules in HUVECs via activation of p38 MAPK and actin filament reorganization [127]. Meanwhile, sidestream cigarette smoke activated the p38 MAPK signaling pathway to induce endothelial permeability [128]. It was also found that the p38 MAPK signaling pathway may be involved in cigarette smoke-induced human aortic endothelial cells (HAECs) apoptosis, while endogenous NO may have a protective effect on inflammatory injury induced by cigarette smoke [129]. Although PI3K and JAK/STAT also mediate cardiovascular inflammation, there are rarely studies that reported their involvement in cigarette smoke-induced cardiovascular damage. Evidence indicated that cigarette smoke and other nicotine products reduced endothelial cell viability via activation of the PI3K/Akt/eNOS signaling pathway [130]. Cigarette smoke exposure also induced MMP2 and MMP9 secretion in aortic vascular smooth cells via activation of the JAK/STAT pathway [131]. Activation of the toll-like receptor family is also considered an initiating step of cigarette smoke-induced inflammation of the cardiovascular system [132]. Researchers suggested that cigarette smoke can induce inflammation and promote atherosclerosis via activation of the H1R-TLR2/4-COX2 signaling pathway in HUVECs [133]. Moreover, an epidemiologic study also observed upregulation of TLR4 mRNA in current smokers [134].
Although there is a lack of evidence on the molecular mechanisms of cardiovascular inflammation induced by e-cigarettes, animal data suggested a similar extent of endothelial dysfunction either by cigarette or e-cigarette smoke exposure, with inflammation as a central player in this process [33]. E-cigarette smoke exposure has been proved to induce a p38 MAPK-related inflammatory response in human neutrophils [135]. Besides, nicotine, as a major component of toxicants in most available e-cigarette aerosols, has been reported to induce macrophage pyroptosis in atherosclerosis via activation of the HDAC6/NF-κB/NLRP3 signaling pathway [136]. Nicotine also induced endothelial cell injury and mediated vascular smooth muscle cells (VSMCs) migration via activation of the NF-κB signaling pathway [137], [138]. Moreover, a recent cross-sectional study conducted by Stokes et al. has proved that the inflammatory biomarkers such as IL-6 and slCAM in e-cigarette users was significantly lower than cigarette users, which indicated that cigarette smoke may induce a more significant inflammatory response than that of e-cigarettes [139]. Taken together, all these results supported the activation of inflammatory pathways after either cigarette or e-cigarette exposure, and it’s well recognized that the release of inflammatory cytokines should be a pivotal initiating event linking cigarettes and e-cigarettes to cardiovascular injury, especially in adverse effects on the vasculature. In addition, based on current evidence, we suggested that canonical cigarette exposure induced a more severe inflammatory response than e-cigarettes, which also supported that the e-cigarettes should be harmless than cigarettes to the cardiovascular system.
Molecular initiating event (KE 559): Activation of nicotinic acetylcholine receptor
As mentioned above, nicotine has been recognized as a major toxic component of both canonical cigarettes and many commercially available e-cigarettes. As a typical cardiovascular toxicant, the toxic effects and underlying mechanisms of nicotine exposure have been extensively studied [140]. Nicotine can act on various types of nAChR in the nervous system and non-neural tissues. Activation of nAChR can regulate the nervous system by promoting the release of various types of neurotransmitters including catecholamines. Meanwhile, in non-neural tissues, nAChR can interact with other intracellular signaling molecules to regulate the physiological functions of cells [141]. Among nicotine-responded nAChR, α4 and β2 receptors are thought to mediate nicotine addiction, while α3 and β4 nAChR regulate cardiovascular function by regulating the autonomic ganglion and adrenal medulla systems [142], [143]. Recent studies indicated that α7 homologous receptors, which are present in non-neural tissues such as endothelial cells, airway epithelial cells, inflammatory cells (lymphocytes and macrophages), and keratinocytes, may also involve in nicotine-induced cardiovascular effects [144]. Activation of α7 nAChRs induced by nicotine significantly enhanced angiogenesis and promoted endothelial cell migration, proliferation, and survival, thus resulting in angiogenic responses to inflammation, ischemia, and atherosclerosis [145]. The nAChR activation mainly increases the release of catecholamines such as epinephrine and norepinephrine through sympathetic nerve stimulation, thus causing an increase in heart rate and blood pressure [146]. The β adrenalin receptor activated by catecholamine stimulation also enhanced cardiac remodeling and hypertrophy [147]. Numerous studies have documented the activation of nAChR induced by cigarette, e-cigarette, or nicotine exposure (summarized in Table 6).
Table 6.
Summary of evidence supporting MIE (KE 559): activation of nicotinic acetylcholine receptor.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [148] | Cigarette smoke | In vivo and in vitro | C57BL/6 mice were exposed to cigarette smoke (twice per day, 5 days per week for 24 weeks), while MH-S macrophages were treated with CSE for 15 min | Activation of nAChR, upregulation of HMGB1, autophagy |
| [149] | CSE | In vitro | HBE cells were treated with CSE for 48 h | Activation of nAChR, activation of STAT3/NRF2 signaling pathway |
| [150] | Cigarette smoke | In vivo | Sprague-Dawley rats were exposed to 1 h per day, 5 days per week for 13 weeks | An increase in nAChR density |
| [151] | CSE and nicotine | In vivo | Sprague-Dawley rats were injected with nicotine (0.5 mg/kg) or CSE thrice per day for 10 days | Upregulation of nAChR binding |
| [152] | Cigarette smoke | Cross-sectional study | The study included 16 women, while 8 of them were smokers | Regulation of nAChR in the placenta, increased vasoconstriction, decreased re-epithelialization |
| [153] | E-cigarette smoke | In vivo | C57BL/6J mice and nAChR α7 KO mice were exposed to e-cigarette smoke (2 h/daily, 5 days/week for 30 days) | Activation of α7 nAChR, inflammation |
| [152] | E-cigarette smoke | In vivo | C57BL/6 mice were exposed to e-cigarette smoke (1 h/daily, 5 days/week for 3 months) | Increased nAChR expression in the brain regions |
| [155] | E-cigarette smoke | Cross-sectional study | The study enrolled 7 nicotine users, including 4 e-cigarette users | Increased β2 nAChR occupancy |
| [156] | Nicotine | In vitro | VSMCs were pretreated (or not) with α-Bungarotoxin (1 μM) for 1 h, and then treated with nicotine (1 mM) for 24 h | Activation of α7 and α3 nAChR, increased intracellular Ca2+, calcification |
| [157] | Nicotine | In vivo and in vitro | ApoE-/- mice were injected with nicotine (2 mg/kg daily) for 12 weeks, while mouse VSMCs were treated with nicotine (10 μM) for 36 h | Activation of the nAChR/ROS/NF-κB signaling pathway, autophagy, atherosclerosis |
| [158] | Nicotine | In vitro | HUVECs were treated with nicotine (1 mM) for 48 h | Activation of α7 nAChR, activation of the DDAH/ADMA/NOS signaling pathway, endothelial dysfunction |
The activation of nAChR by cigarette smoke has been widely studied. Yan et al. reported cigarette smoke-induced nAChR activation in macrophages and was thought to participate in cigarette smoke-related upregulation of HMGB1 [148]. Guo et al. demonstrated α7 nAChRs dependent activation of STAT3/NRF2 in pulmonary epithelial cells after treatment with CSE [149]. In vivo studies also reported significant increases in nAChR density in the cortex, striatum, and cerebellum of Sprague-Dawley rats after cigarette smoke exposure [150]. Chronic exposure to CSE also upregulated nAChR binding in many brain regions in adult and adolescent rats [151]. An epidemiologic study even indicated that cigarette smoke exposure during pregnancy could regulate the nAChR subunits expression in the placenta. Therefore, smoking during pregnancy could contribute to increased vasoconstriction and decreased re-epithelialization, eventually resulting in calcification and apoptosis in the smoker’s placenta [152].
Evidence also suggested nAChR activation and related adverse effects induced by e-cigarette smoke exposure. Wang et al. illustrated α7 nAChR mediated inflammation and dysregulated repair in mice. Their results supported that subchronic e-cigarette exposure resulted in lung inflammation and extracellular matrix (ECM) remodeling via α7 nAChR activation [153]. Another study also found that long-term e-cigarette inhalation increased α4/β2 nAChR expression in all brain regions of C57BL/6 mice and increased α7 nAChR expression in FC and STR [154]. Epidemiologic studies also found β2 nAChR occupancy in the brains of both cigarette and e-cigarette users by PET neuroimaging, and the results indicated that compared with cigarettes, e-cigarettes even induced higher nAChR occupancy [155]. Since nicotine may be the main component that activates nAChR, more studies have focused on the cardiovascular effects of nAChR directly mediated by nicotine. In vitro study supported that activation of α7 and α3 nAChR by nicotine increased intracellular Ca2+ and promoted calcification of VSMCs via upregulation of Nox5 activity, which may serve as a novel mechanism of nicotine-induced atherosclerosis [156]. Nicotine could also mediate autophagy via activation of the nAChRs/ROS/NF-κB signaling pathway, which resulted in phenotypic switching and increased the migratory capacity of VSMCs [157]. Jiang et al. suggested that nicotine may mediate DDAH/ADMA/NOS pathway via activation of α7 nAChR, which was thought to contribute to endothelial dysfunction [158].
In conclusion, current evidence indicated that nAChR activation should be involved in nicotine product-related cardiovascular effects. Since the nAChR activation is mainly induced by nicotine, nicotine levels can directly affect the activation of nAChR. As we discussed above, several studies have suggested that e-cigarette smoke could induce a lower nicotine boost than canonical cigarette. Therefore, we suggested that the effect of e-cigarette smoke on nAChR may be lower than that of cigarette smoke, while further studies should be conducted to confirm the difference. On the other hand, although the role of nicotine in nAChR activation has been elucidated, most existing results did not manifest the direct relationship between cardiovascular effects caused by nAChR activation and cigarette exposure. After all, nicotine acts on receptors that are normally activated by endogenous acetylcholine, while cholinergic nerves regulate physiological functions through a variety of complex feedback pathways. Thus, the nicotine-induced effects observed in vitro may not be completely representative of the actual toxicity of cigarette smoke in humans [159].
Interaction between the molecular initiating events
Numerous evidence has indicated that the three types of MIEs were not completely independent but interacted with each other to jointly promote the development of cardiovascular disease. The cross-talk between ROS and inflammatory signaling pathways has been extensively elucidated. For example, NF-κB could not only be activated by proinflammatory receptors but also regulated by ROS generation [160]. ROS generally enhanced the activation and nuclear translocation of NF-κB in the cytoplasm but inhibited the tuberculous interaction of NF-κB with DNA in the nucleus [161]. By contrast, increased NF-κB activity could induce increased expression of antioxidant proteins, such as MnSOD and SOD2, which could protect cells from damage caused by excessive ROS generation [162]. On the other hand, the activation of the p38 MAPK signaling pathway in HL-1 cardiomyocytes has been proved to induce ROS elevation during hypoxia/reoxygenation [163]. As the major participants of inflammatory response, macrophages were proved to be activated via ROS-triggered activation of Jak/Stat and Toll signaling pathway [164]. Moreover, ROS was also reported to trigger the production and secretion of proinflammatory cytokines IL-1β and IL-18 via activation of NLRP3 inflammasome, which was also responsible for ROS-related pro-inflammatory programmed cell death (pyroptosis) [165]. In addition, the activation of NLRP3 inflammasome and subsequent pyroptosis were accompanied by the release of a mass of proinflammatory media, which could aggravate local inflammatory response and endothelial dysfunction [166]. Therefore, ROS and inflammation can act together and pose a synergistic threat in cardiovascular injury.
The interaction between ROS and nAChR activation remains unclear but several studies indicated that the activation of nAChR may inhibit ROS production in the neuro system. Moon et al. reported that nicotine-induced activation of α7 nAChR in microglia [167]. A study conducted by Parada et al. also indicated that melatonin could attenuate ROS elevation via activation of nAChR [168]. However, other studies have suggested that the activation of nAChR in the cardiovascular system may be associated with elevated ROS levels. Chang et al. reported that the treatment with Garcinol (an α7 nAChR antagonist) could attenuate lipoprotein (a) induced ROS elevation and inflammatory cytokine generation in cardiomyocytes [169]. Meanwhile, the treatment with another nAChR antagonist (hexamethonium) could also reduce nicotine-induced elevation of ROS and activation of the NF-κB signaling pathway in VSMCs [157]. Moreover, Ko et al. reported that hexamethonium could also attenuate cigarette smoke-induced ROS elevation in macrophages [170]. Taken together, the interaction between nAChR and ROS can vary from the acting organs/tissues, while they may play a synergistic role in the nicotine-induced cardiovascular toxicity.
Current evidence suggested that the interaction between nAChR activation and inflammation is controversial in the cardiovascular system. Saeed et al. reported that the activation of α7 nAChR induced by nicotine could reduce the production of chemokines and the nuclear translocation of NF-κB in endothelial cells [171]. Meanwhile, Yang et al. reported that the treatment with α-conotoxin MII (an α3-nAChR antagonist) could increase the inflammatory cell infiltration in the aorta of ApoE-/- mice [172]. Moreover, Li et al. reported that nicotine exhibited an anti-apoptotic effect in CVB3-induced myocarditis via the activation of α3 and β4 nAChR [173]. However, Hung et al. reported that the activation of α7 nAChR in monocytes could promote the inflammation-related development of coronary artery spasm (CAS) via activation of the p38 MAPK signaling pathway [174]. Besides, Lin et al. also reported that lipoprotein (a) could induce inflammation and macrophage polarization in CAS patients via activation of the α7 nAChR/p38 MAPK signaling pathway [175]. In summary, although the interaction between the MIEs is complex, and there may even be partial antagonism, it is clear that they should all participate in cigarette and e-cigarette-related cardiovascular toxicity via triggering specific cellular and organic effects that will be discussed in the subsequent KE section.
Cardiovascular-related key events triggered by cigarette and e-cigarette exposure
Key event (KE 1392): Oxidative stress
Oxidative stress is a consequent result induced by excessive ROS generation, which is considered to be the initiating factor of aging and the development of various diseases. Oxidative stress is an imbalance state induced by oxidation that exceeds the antioxidant capacity of the organism, which tends to result in oxidative damage. The occurrence and progression of oxidative stress result in the activation of the antioxidant defense system, release of inflammatory cytokines, lipid peroxidation, and production of oxidative mediators [176]. Oxidative stress and its downstream effects have also been proved to play a critical role in cardiovascular disease, especially in the occurrence and development of atherosclerosis. Oxidative stress has been proved to interfere with the nitric oxide synthase via inhibition of the Nrf2 pathway and up-regulation of ADMA content. The dysregulation of nitric oxide synthase and uncoupling of eNOS eventually resulted in endothelial dysfunction, which is identified as the pathological basis of atherosclerosis, hypertension, and subsequent cardiomyopathy [177]. Meanwhile, oxidative stress also drives the activation of vascular smooth cells by promoting their proliferation and migration, which are essential for vascular calcification and remodeling [178]. On the other hand, oxidative stress can stimulate the proliferation of cardiac fibroblasts and eventually lead to extracellular matrix remodeling, which is responsible for consequent cardiac fibrosis [179]. Furthermore, oxidative stress is also considered to be directly involved in myocardial senescence and consequent apoptosis, which are believed to play a crucial role in ventricular remodeling [180]. In addition to measuring ROS levels to reflect oxidative stress levels, numerous biomarkers have been adopted to identify and assess oxidative stress in organisms, including NOX, GSH, SOD, CAT, MDA, HNE, ALE, 8-oxodG, 3-NO-Tyr, and AGE. During the progression of oxidative stress, NOX may be one of the main sources of ROS. Excessive ROS first activate the antioxidant defense system composed of GSH, SOD, and CAT. When the ROS content exceeds the antioxidant defense ability, it will cause the peroxidation of lipids, proteins, and nucleic acids, and produce corresponding oxidation products. Among them: The MDA, HNE, and ALE are lipid oxidation end products. The 8-oxodG is the most commonly used DNA oxidation biomarker. The 3-NO-Tyr is the main product of tyrosine oxidation. The AGE is the end product of glycation [181]. Current evidence strongly supported oxidative stress induced by cigarette, e-cigarette, or nicotine exposure (summarized in Table 7).
Table 7.
Summary of evidence supporting KE 1392: oxidative stress.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [183] | CSE | In vitro | CSCs were pretreated (or not) with ascorbic acid (1 mM or 2 mM) for 2 h, and then treated with CSE for 24 h | Oxidative stress, apoptosis |
| [120] | CSE | In vitro | H9c2 myocytes were pretreated with cerium oxide nanoparticles (1, 10, or 100 nM) for 24 h, and then treated with CSE for 24 h | Oxidative stress, activation of the NF-κB signaling pathway |
| [186] | CSE | In vitro | HUVECs were pretreated with resveratrol for 1 h, and then treated with CSE for 4 h | Oxidative stress, eNOS acetylation |
| [187] | Cigarette smoke | In vivo | Wistar Albinoure rats were exposed to cigarette smoke 8 h/daily for 7 days (2, 4, 8, and 24 cigarettes per day) | Oxidative stress, upregulation of hypertrophic gene |
| [188] | Cigarette smoke | In vivo | Guinea pigs were treated with 15 mg or 0.5 mg of vitamin C per day and exposed to cigarette smoke for 8 weeks | Oxidative stress, apoptosis, inflammation |
| [189] | Cigarette smoke | In vivo | C57BL/6J mice were treated with (0.2 mg·kg -1 ·day) Ang-II and exposed to cigarette smoke for 2 weeks | Oxidative stress, endothelial dysfunction, hypertension |
| [190] | Cigarette smoke | Cohort study | The study enrolled 3614 subjects and followed-up for 15 years | Oxidative stress, endothelial dysfunction, inflammation |
| [191] | Cigarette smoke | Randomized control trial | The study enrolled 577 smokers and randomized to 4 intervention groups, while 434 of smokers participated in blood sample during the 12 months-follow up | Alteration of oxidative stress biomarkers |
| [39] | E-cigarette smoke | Cross-sectional study and in vivo study | The study enrolled 20 healthy subjects, while C57BL/6 mice were exposed to e-cigarette smoke for 1, 3, or 5 days (2 h/daily) | Oxidative stress, endothelial dysfunction, inflammation, increased blood pressure |
| [192] | E-cigarette smoke | Cross-sectional study | The study enrolled 42 subjects, including 23 self-identified e-cigarette users and 19 non-tobacco users | Alteration of oxidative stress biomarkers, activation of sympathetic system |
| [193] | E-cigarette smoke | Randomized control trial | The study enrolled 25 healthy occasional tobacco users and randomized to 3 period crossover design (e-cigarette without nicotine, e-cigarette with nicotine, and sham e-cigarette) | Oxidative stress, altered endothelial dysfunction |
| [194] | Cigarette smoke, e-cigarette smoke | In vivo | ApoE-/- mice were exposed to cigarette smoke or e-cigarette smoke 3 h/daily, 5 days/week for 6 months | Oxidative stress, endothelial dysfunction, ventricle dysfunction |
| [195] | Cigarette smoke, e-cigarette smoke | Cross-sectional study | The study enrolled 33 subjects, including 12 non-smokers, 12 e-cigarette users, and 9 cigarette users | Oxidative stress |
The association between cigarette smoke and oxidative stress in the cardiovascular system has been amply documented by in vitro studies. Ascorbic acid is a non-toxic and unlimited free radical scavenger that can directly reduce ROS content by donating a single reducing equivalent [182]. Sumanasekera et al. reported that cigarette smoke extracts (CSE) exposure induced impairment of c-kit-positive cardiac stem cells (CSCs) via oxidative stress, and treatment with ascorbic acid could significantly mitigate the CSE-related malfunction [183]. Cerium Oxide nanoparticles are a recently discovered antioxidant nanomaterial that mimics the antioxidant activities of SOD and CAT by donating Ce ions [184]. CSE could induce oxidative stress and activation of NF-κB in H9c2 cardiomyocytes, and cerium oxide nanoparticles have been proved to protect the cells by utilizing their antioxidant capacity [120]. Resveratrol is a polyphenolic natural plant-derived chemical with powerful antioxidant capabilities and has been proved to reduce LDL and platelet aggregation, thereby exerting a cardiovascular protective function [185]. Arunachalam et al. suggested CSE-induced endothelial dysfunction via oxidative stress-mediated downregulation of SIRT1, while pretreatment with resveratrol on HUVECs significantly attenuated CSE-mediated down-regulation of SIRT1 levels and eNOS acetylation [186]. The casual relationship between cigarette smoke and oxidative stress has also been confirmed by in vivo studies. AI-Arifi et al. observed alteration of mRNA expression of oxidative stress-related biomarkers and cardiac hypertrophy gene in Wistar albino rats exposed to increasing doses of passive cigarette smoke for 7 days [187]. In another study, oxidative stress-related myocardial damage, inflammation, and myocardial apoptosis were also observed in guinea pigs exposed to cigarette smoke [188]. Dikalov et al. reported that cigarette smoke-induced cardiovascular mitochondrial oxidative stress in mice, and thus leads to endothelial dysfunction and hypertension [189]. Evidence obtained from epidemiologic studies also correlated with these results. Caroll et al. found that cigarette smoke exposure was associated with oxidative stress, inflammation, and endothelial dysfunction by assessment of biomarkers among 3614 adults [190]. In another randomized controlled trial, Mons et al. clarified that smoking cessation reduced levels of oxidative stress in current smokers [191].
Oxidative stress was also observed in organisms exposed to e-cigarette smoke. In a comprehensive study, Kuntic et al. manifested the correlation between short-term e-cigarette vapor exposure and vascular oxidative stress by assessing the effects of e-cigarette vapor on vascular function in smokers and experimental animals. The results indicated that acute e-cigarette vapor exposure caused endothelial dysfunction in chronic smokers, induced inflammation and oxidative stress in vessels and brain tissue, as well as increased blood pressure in experimental animals [39]. In case-control study, Moheimani et al. also suggested that habitual e-cigarette use was associated with the alteration of oxidative stress-related biomarkers and the activation of the sympathetic system [192]. A randomized crossover trial conducted by Chaumont et al. demonstrated that e-cigarettes that contained nicotine altered the vascular function and induced oxidative stress, while nicotine-free e-cigarettes did not cause changes in cardiovascular parameters [193].
Several studies have compared the oxidative stress levels induced by cigarette and e-cigarette exposure. Szostak et al. reported that compared with each type of e-cigarette aerosols, cigarette smoke could induce more significant changes in oxidative stress, cardiac function, and endothelial function in ApoE-/- mice [194]. In a cross-sectional study, Kelesidis et al. observed that e-cigarette users had lower levels of oxidative stress than cigarette users, but still higher than non-smokers [195]. Considering that oxidative stress is mainly caused by overproduction of ROS, as we mentioned above, current evidence also suggested that cigarette smoke exposure induced higher ROS production than e-cigarettes, thus we concluded that the oxidative stress induced by cigarette smoke should be more severe than e-cigarettes. Given that oxidative stress should be one of the most typical cardiovascular effects induced by tobacco products that triggered various subsequent events, we suggested that differences in oxidative stress levels may largely explain the differences in toxicity between e-cigarettes and cigarettes. As we described in the previous text, canonical antioxidants such as MitoQ and ascorbic acid have been proved to mitigate oxidative damage induced by cigarette smoke. On the other hand, emerging nanomaterials such as cerium oxide nanoparticles may also contribute to the therapy of cigarette and e-cigarette-related cardiovascular disease by directly reducing ROS or loading antioxidant drugs [196], [197].
Vascular morbidity induced by cigarette and e-cigarette exposure
Key event (KE 1913): Endothelial cell dysfunction
Both oxidative stress and inflammatory response serve as key players in endothelial cell dysfunction [198]. The vascular endothelium is the continuous inner cell layer of the vasculature, which plays an irreplaceable role in maintaining the homeostasis of the cardiovascular system. Endothelial cell dysfunction usually refers to the abnormal production or bioavailability of endothelial nitric oxide and the resulting harmful changes in vascular responsiveness. In terms of the effect of endothelial cells on atherosclerosis, this concept has now been extended to include all maladaptive changes in endothelial functional phenotypes that are closely associated with atherosclerotic cardiovascular disease [199]. In addition, endothelial dysfunction has also been associated with systemic hypertension, vascular inflammation, cardiomyopathy, and type 2 diabetes mellitus [200], [201]. Several studies have confirmed endothelial cell dysfunction induced by cigarette or e-cigarette exposure (summarized in Table 8).
Table 8.
Summary of evidence supporting KE 1913: endothelial cell dysfunction.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [202] | CSE | In vitro | HUVECs were pretreated with resveratrol for 2 h and then treated with CSE for 24 h | Autophagy, apoptosis, activation of the Notch 1 signaling pathway |
| [203] | CSE | In vitro | HUVECs were pretreated with CSE for 16 h and then treated with VEGF (10 ng/mL) for 24 h | Inhibition of NO release and Akt/eNOS phosphorylation |
| [204] | Cigarette smoke | In vivo and in vitro | Sprague-Dawley rats were exposed to cigarette smoke for 7 days (20 cigarettes per day) and injected (or not) with melatonin (10 mg/kg/day), while HAECs were treated with CSE for 24 h | Pyroptosis, upregulation of the Nrf2 signaling pathway |
| [205] | CSE, e-cigarette smoke extract | In vitro | HUVECs were treated with CSE or e-cigarette smoke extract for 4–72 h | Apoptosis, programmed necrosis |
| [206] | CSE, e-cigarette smoke extract | In vitro | HUVECs were treated with CSE, e-cigarette smoke extract for 48 h | Inhibition of proliferation, cell death |
| [130] | Cigarette smoke and other nicotine products | In vitro | HUVECs were exposed to different test substances for 24 h | Decreased of cell viability and wound-healing ability |
Endothelial cell dysfunction induced by cigarette smoke has been extensively elucidated. Zong et al. reported that treatment with CSE induced apoptosis of HUVECs, while resveratrol could attenuate the adverse effects via activating the Notch1 signaling pathway [202]. While Michaud et al. found that CSE could inhibit Akt/eNOS phosphorylation and NO release in VEGF-stimulated HUVECs, which are essential for maintaining endothelial homeostasis. Treatment with antioxidants (NAC, vitamin C) reduced ROS formation and attenuated the toxic effects [203]. CSE treatment could also induce NLRP3-related pyroptosis in human aortic endothelial cells (HAEC) via the generation of ROS and upregulation of Nrf2. Intervention with melatonin could prevent smoking-induced vascular injury and atherosclerosis by regulating Nrf2/ROS/NLRP3 signaling pathway [204].
E-cigarette aerosol was also reported to impair the vascular endothelial cells. Anderson et al. reported that e-cigarette exposure should also involve in inducing apoptosis and programmed necrosis via ROS generation and DNA damage [205]. The toxicity assessment of cigarettes and e-cigarettes to vascular endothelial cells revealed that despite e-cigarettes are cytotoxic for HUVECs via cell death induction and ROS overproduction, CSE had even more severe effects on endothelial cells [206]. Sindy Giebe et al. evaluated and compared the potential effects of various tobacco products on endothelial function. In this study, primary cultured human endothelial cells were exposed to a variety of cigarette products, including cigarette smoke and e-cigarette smoke. The results indicated that only CSE (3R4F) decreased endothelial cell viability and wound-healing ability in all subjects, while e-cigarette smoke extract (E-CIG) had almost no impact [130]. Since the oxidative damage and inflammatory response caused by cigarettes and e-cigarettes have been compared in endothelial cells, we believe that canonical cigarettes may induce more severe endothelial dysfunction than e-cigarettes. On the other hand, the severity of endothelial damage is often associated with cardiovascular risk, while endothelial dysfunction is considered as the pathological basis of severe CVD, including atherosclerosis and myocardial infarction. Therefore, differences at the endothelial dysfunction may help explain the differences in cigarette and e-cigarette related-cardiovascular toxicity.
Key event (KE 1925): Vascular smooth cell activation
Oxidative stress has been identified as a critical player in modifying the differentiation of VSMC [207]. Vascular smooth muscle cells (VSMCs) are non-striated, involuntary, and contractile cells that regulate blood pressure by contracting and relaxing in opposition to the heart which is essential for maintaining the normal function of blood vessels. VSMC has a contractile phenotype in normal conditions, in this phenotype, it proliferates slowly and mainly exhibits contractile function. VSMC changes its phenotype when it receives a local signal stimulus, such as injury, and started to migrate by downregulating contractile proteins, increasing proliferation and remodeling ECM. Abnormal activation of VSMCs has been proved to be an important cause of vascular calcification and remodeling [208]. Several studies have reported VSMC activation induced by cigarette or nicotine exposure (summarized in Table 9).
Table 9.
Summary of evidence supporting KE 1925: vascular smooth cell activation.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [209] | CSE | In vitro | VSMCs were treated with CSE and hydrogen peroxides for 24 h | Enhancement of the mitogenic effect induced by hydrogen peroxides |
| [210] | CSE | In vitro | Cerebral VSMCs were isolated from Sprague-Dawley rats and then treated with CSE for 24 h or 72 h | Phenotypic modulation, upregulation of inflammatory genes, downregulation of contractile genes |
| [211] | CSE | In vitro | HASMCs were treated with CSE for 12, 24, 36, and 48 h | Increased proliferation of VSMCs |
| [212] | CSE | In vitro | VSMCs were treated with CSE for 48 h | Increased migration of VSMCs |
| [214] | Nicotine | In vitro | VSMCs were treated with nicotine (6 × 10-4, 6 × 10-6, 6 × 10-8 mol/L) for 72 h | Increased proliferation and migration of VSMCs |
| [215] | Nicotine | In vitro | VSMCs were treated with nicotine (10-8 M) for 48 h | Increased migration of VSMCs |
| [216] | Nicotine | In vitro | VSMCs were treated with nicotine (0.1 μM) for 48 h | Phenotype transformation |
Nishio et al. reported that CSE treatment reduced the activities of superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase in VSMC in a time-dependent manner, and CSE enhances the mitogenic effect of hydrogen peroxide via dysregulation of SOD activity [209]. CSE was also reported to induce phenotypic modulation of VSMCs via oxidative stress, and the results indicated that the inflammatory genes were upregulated while contractile genes were downregulated in cerebral VSMCs after CSE exposure [210]. Guo et al. found CSE treatment significantly promoted the proliferation of human aortic smooth muscle cells (HASMCs) and induced down-regulation of P16 expression in a concentration- and time-dependent manner [211]. CSE and nicotine were also supposed to promote VSMC migration, and thus should be a contributor to the accumulation of VSMCs in atherosclerotic plaques [212]. Currently, there is still a blank in the response of VSMC to e-cigarette exposure, but nicotine has been proved to induce vascular calcification by inducing osteogenic transdifferentiation of VSMC [213]. Nicotine was also proved to induce proliferation and migration of VSMC via modulation of bFGF and TGF-β [214]. Meanwhile, nicotine treatment directly induced the transformation of VSMC from contractile type to synthetic-like type through the activation of nicotinic acetylcholine receptors and G protein-coupled receptors [215], [216]. Taken together, we hold the opinion that VSMC activation plays a key role in cigarette smoke-related vascular disease, while nicotine should be responsible for such effects to some extent. However, there is still a lack of evidence to support e-cigarette-related VSMC activation.
Key event (KE 1198): Macrophage activation
It has been well documented that systematic oxidative stress resulted in lipid modification, which serves as a harbinger of macrophage reprogramming, while inflammatory response mainly triggered the activation of macrophages [217], [218]. Monocytes and macrophages can respond to stimuli from pathogens and danger signals in rapid and specific ways. These cells can respond to the microenvironment and intercellular signaling while regulating their function by triggering complex transcription factors. Macrophages could be phenotypic dysregulated by inflammatory cytokines and oxidative lipids, which drive the development of diseases such as atherosclerosis and type 2 diabetes [119]. Macrophages are the main immune cell population in arterial plaque and have been considered to play a critical role in immune response and progression of atherosclerosis. They are mainly derived from circulating monocytes and resident tissues and recruited to the lesion site at the initial stage of atherosclerosis in which they gradually transform into foam cells through proliferation and absorption of lipid deposition particles [219]. Several studies have reported macrophage activation induced by cigarette, e-cigarette or nicotine exposure (summarized in Table 10).
Table 10.
Summary of evidence supporting KE 1198: macrophages activation.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [220] | CSE | In vitro | ANA-1 macrophages were treated with CSE for 3, 6, 12, 24 h, while HUVECs were treated with CSE or ANA-1 supernatants for 24 h | Generation of TNF-α and slCAM-1 |
| [221] | CSE | In vitro | RASMCs, RAECs, and RAW cells were treated with CSE for 24 h, and the conditioned media was collected, while fresh cells were treated with CSE-CM | Generation of MMP2 and MMP9, activation of the Jak2/Stat3 signaling pathway |
| [223] | CSE | In vitro | THP-1 monocytes were treated with CSE for 24 h | Increased the expression of CD9, CD36, and CD68 |
| [93] | E-liquid, e-cigarette smoke | In vitro | iPSC-ECs were exposed to e-liquids or serum from human subjects exposed to e-cigarette smoke for 48 h, while macrophages were exposed to iPSC-EC conditioned medium for 48 h | Increased expression of CD40 and CD163, Generation of inflammatory cytokines |
| [224] | E-cigarette smoke | In vivo and in vitro | ApoE-/- were exposed to e-cigarette smoke for 2 h/daily, 5 days/week for 16 weeks, while RAW 246.7 cells were treated with e-cigarette smoke extract for 48 h | Increased the frequency of macrophages expressing Mac2 and monocytes expressing CCR2 |
| [225] | Nicotine | In vitro | THP-1 monocytes were treated with nicotine (1, 100, 1000 nmol/L) for 24 h | Foam cell formation, activation of the CD36 signaling pathway |
Cigarette smoke has been shown to trigger multiple patterns of macrophage activation. Zhang et al. observed that CSE could indirectly induce the generation of soluble intercellular adhesion molecule-1 (slCAM-1) in HUVECs by activating macrophages and stimulating the production of TNF-α [220]. Gosh et al. analyzed the cross-talk between macrophages, smooth muscle cells, and endothelial cells in response to cigarette smoke. In this study, CSE was applied to rat aortic smooth muscle cells (RASMCs), rat aortic endothelial cells (RAECs), and RAW cells (a transformed macrophage cell line isolated from BALB/c mice), and the cigarette smoke conditioned medium (CSE-CM) was collected. They found that CSE-CM from RASMCs and RAECs significantly induced excessive production of matrix metalloproteinase-2 (MMP-2), matrix metalloproteinase-9 (MMP-9), phosphorylated Jak2, and phosphorylated Stat3 (pStat3) in RAW cells, which were previously proved to contribute to the development of atherosclerosis [221], [222]. Furthermore, CSE treatment significantly increased the expression of CD9, CD36, and CD68 in THP-1 monocytes (a transformed monocyte cell line isolated from a patient with acute monocytic leukemia), and thus may contribute to the progression of coronary artery disease (CAD) [223].
E-cigarette exposure was also supposed to induce macrophage activation. Lee et al. reported the macrophage dual-polarization induced by the e-liquid-conditioned medium. Macrophages expressing CD40 (M1 marker) and CD163 (M2 marker) were significantly increased after treatment with e-liquid-conditioned medium. Meanwhile, the generation of inflammatory cytokines such as IL-1β, IL-6 in M1 macrophages, and IL-10 in M2 macrophages was also enhanced [93]. In another study, Li et al. reported that e-cigarette vapor (ECV) exposure could remarkably enhance the accumulation of Mac2 + macrophages (macrophages expressing Mac2) in atherosclerotic plaques, which could be attenuated by TLR9 inhibitor administration. Meanwhile, ECV treatment also increased the frequency of CCR2 + cells (monocytes expressing CCR2) in classical monocytes [224]. The nicotine stimulation was also proved to enhance oxLDL-related proatherogenic effects via upregulation of the CD36 signaling pathway in macrophages [225]. In summary, these results indicated that either cigarettes or e-cigarettes could induce the activation of macrophages. However, there is still a lack of evidence to compare the effects on macrophage activation induced by canonical cigarettes and e-cigarettes.
Key event (KE 2003): Vascular remodeling
Both endothelial dysfunction and VSMC activation contribute to vascular remodeling [226], [227]. Vascular remodeling is an adaptive response to vascular disease and aging. This process mainly involves the changes in the ECM dynamic microenvironment, including the changes in ECM components and posttranslational modifications of matrix proteins, all of which affect the resident cells of the vascular wall [228]. Endothelial cells, smooth muscle cells, fibroblasts, and macrophages on the arterial wall are very sensitive to the stimulation of the external environment and they alter gene expression according to mechanical stimulation. Regulation of nitric oxide synthase (NOS) activity mediated by vascular endothelial cells altered local hemodynamics while angiotensin II (Ang II) and transforming growth factor-β (TGF-β) synthesized by VSMCs enhances extracellular matrix synthesis and promotes vascular wall thickening [229]. Indeed, evidence supported that these processes can develop into interaction, thus both endothelial cell dysfunction and VSMC activation should play a critical role in response to external stress-induced vascular remodeling [230]. Vascular remodeling induced by cigarette or nicotine exposure has been observed in several studies (summarized in Table 11).
Table 11.
Summary of evidence supporting KE 2003: vascular remodeling.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [231] | Cigarette smoke | In vivo | ApoE-/- mice were exposed to cigarette smoke for 56 min/daily, 5 days/week for 24 weeks | Medial thickening of the aorta |
| [232] | Cigarette smoke | In vivo and in vitro | ApoE-/- mice were exposed to cigarette smoke for 10 ± 1 week (1, 2, 3, or 4 h/daily, 5 days/week) and 10 % iron chloride were adopted to induce arterial thrombosis and neointima formation, while HAECs were treated with CSE for 24 h | Increased the ratio of intima/media, increased the proliferation of VSMCs |
| [233] | Cigarette smoke | In vivo and in vitro | Sprague-Dawley rats were exposed to cigarette smoke for 3 months (2 h/daily), while HPASMCs were treated with CSE for 48 h | Thickness of the pulmonary arteriolar wall, increased proliferation and migration of HPASMCs |
| [234] | Cigarette smoke | Case-control study | The study included 178 subjects in the case group and 180 subjects in the control group | Coronary artery-positive remodeling |
| [235] | Nicotine | In vivo | C57BL/6 mice were treated with nicotine (5 mg/kg/day) and Ang-II (21.6 µg per day) for 4 weeks | Thickening of the vascular wall |
Cigarette smoking has been proved to cause structural and functional remodeling of the aorta. Farra et al. reported that long-termed exposure to cigarette smoke-induced a significant thickening of the aortic wall, decreasing axial tension, and softening of vascular circumferential structure in ApoE-/- mice. The increased content of smooth muscle and extracellular matrix in the ascending aorta caused the thickness of the media, while collagen deposition increased the thickness of the descending and abdominal aorta [231]. Schroeter et al. reported that cigarette smoke exposure increased the number of α-actin positive VSMCs and increased the ratio of intima/media in ApoE-/- mice in a dose-dependent manner. Meanwhile, the in vitro trials indicated that CSE exposure promoted the proliferation of human aortic smooth muscle cells (HASMCs) when the total particulate matter was at a low concentration [232]. To evaluate the influence of cigarette smoke on the aortic and carotid artery, studies have also focused on the toxic effects on pulmonary artery remodeling. Li et al.’s reported that compared with the control group, the thickness of the pulmonary arteriolar wall was significantly increased in the Sprague-Dawley rats exposed to cigarette smoke. While the in vitro trials found that CSE treatment could increase the cell viability of human pulmonary artery smooth muscle cells (HPASMCs), accompanied by promoting their proliferation and migration [233]. Furthermore, a retrospective case-control study conducted by Alani et al. reported the association between cigarette smoke exposure and coronary artery-positive remodeling (CAPR) based on cardiac computed tomography angiography (CCTA) [234]. Currently, there is a lack of evidence on the vascular remodeling effect of e-cigarette exposure, but Colombo et al. have reported the effect of nicotine exposure on cardiovascular remodeling in mice with systemic hypertension. In this study, combined exposure to nicotine and Ang II resulted in significant thickening of vascular wall, but neither nicotine nor Ang II alone caused aortic wall thickening [235]. Taken together, cigarette smoke has been extensively proved to induce vascular remodeling, while there was still a lack of knowledge about e-cigarette-related remodeling effects. Since nicotine-induced cardiovascular remodeling has been revealed, we suggested that future studies should focus on whether these similar effects can be induced by e-cigarette smoke.
Key event (KE 2000): Vascular calcification
VSMC activation plays the essential role in vascular calcification [236]. Vascular calcification is defined as the deposit of minerals in the vascular system in the form of calcium phosphate complexes. Although vascular calcification may be part of normal aging, certain pathological processes, such as hypertension and diabetes, can also accelerate its progression. Vascular calcification is proved to be closely related to phenotypic differentiation of VSMC and ECM remodeling. In a pathological state, VSMC transforms from contractile type to synthetic-like type, promotes the secretion of stromal vesicles, and transforms target cells into a calcified state [237]. It has been found that stromal vesicles secreted by vascular smooth muscle cells may be the focus of calcium phosphate deposition on the vascular wall and play an indispensable role in mediating vascular calcification. Vascular calcification is a very complex process, which is regulated by multiple signals from both inside and outside the cell that involves a combination of multiple genetic factors and hormones [238]. Since vascular calcifications are prevalent in cardiovascular disease, they should be closely associated with the majority of cardiovascular events. Therefore, the appearance of vascular calcification may be a precursor to other cardiovascular diseases. Cigarette or e-cigarette-related vascular calcification or arterial stiffness have been observed in several studies (summarized in Table 12).
Table 12.
Summary of evidence supporting KE 2000: vascular calcification.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [239] | Cigarette smoke | In vivo | Sprague-Dawley rats were exposed to cigarette smoke for a month (2 h/daily, 5 days/week) | Increased aortic calcification |
| [240] | Cigarette smoke | Cross-sectional study | The study enrolled 3218 never smokers, 2607 former smokers, and 971 current smokers | Increased coronary artery calcification |
| [241] | Cigarette smoke | Cohort study | The study enrolled 4432 subjects and two follow-up visits were conducted in 2005–2008 and 2009–2013 | Increased coronary artery calcification |
| [242] | Cigarette smoke | Cross-sectional study | The study enrolled 66 patients with primary hypertension, including 19 active smokers, 20 passive smokers, and 27 non-smokers | Increased calcification index of carotid arteries |
| [193] | E-cigarette smoke | Randomized control trial | The study enrolled 25 healthy occasional tobacco users and randomized to 3 period crossover design (e-cigarette without nicotine, e-cigarette with nicotine, and sham e-cigarette) | Increased arterial stiffness |
| [245] | E-cigarette smoke | Randomized control trial | The study enrolled 70 current smokers, and 35 of them were assigned to use nicotine-free e-cigarette, while other 35 smokers were assigned to use e-cigarette containing nicotine | Increased arterial stiffness |
Only very few laboratory studies have focused on the link between smoking and vascular calcification. In Hariri et al.’s study, Von Cossa staining was adopted to determine the effect of cigarette smoke exposure on aortic calcification in rats. The results indicated that compared with the control group, the level of aortic calcification was significantly increased in the cigarette smoke exposure group, while antioxidant intervention could inhibit the calcific effects [239]. On the other hand, numerous epidemiologic studies have concluded that cigarette smoking has an obvious association with arterial calcification. McEvoy et al. reported that compared with never smokers, both current smokers and former smokers have a stronger association with coronary artery calcification [240]. In a large prospective cohort of black adults, current smokers were also found to have greater odds of coronary artery calcification compared with never smokers [241]. Meanwhile, Gac et al. reported that both active and passive smoking in patients with essential hypertension was associated with a higher calcification index of carotid arteries according to computed tomography angiography [242].
At present, there is a lack of evidence of the link between e-cigarette exposure and vascular calcification, but coronary artery calcification (CAC) was identified as a useful biomarker of cardiovascular risk caused by e-cigarette exposure. Compared with the canonical clinical markers, CAC may be more sensitive to e-cigarette exposure-induced arterial calcification, thus it has better clinical predictive power and has been considered a reliable indicator of subclinical cardiovascular effects [243]. Since vascular calcification and ECM remodeling are direct causes of arterial stiffness, we also summarized a possible link between e-cigarette exposure and arterial stiffness reported by several epidemiologic studies [244]. Chaumont et al. reported that e-cigarettes containing nicotine could induce increased arterial stiffness index in a randomized crossover study, but nicotine-free e-cigarettes did not induce significant adverse effects [193]. Another randomized controlled trial conducted by Ikonomidis et al. found that e-cigarette smoking increased arterial stiffness and oxidative stress less than a conventional cigarette, regardless of acute or chronic exposure [245]. Taken together, current evidence supported the vascular calcification induced by cigarette smoke, while epidemiologic studies have revealed the arterial stiffness induced by e-cigarette smoke. However, there is still a lack of concrete evidence, especially the evidence obtained from laboratory studies to elucidate the vascular calcification induced by e-cigarette exposure.
Key event (KE 1443): Atherosclerosis
Macrophage activation, endothelial cell dysfunction, and VSMC activation all participate in the development and progression of atherosclerosis (AS). AS is a chronic inflammatory disease caused by the accumulation of lipids in the artery wall and is mainly characterized by the formation of atherosclerotic plaque. The plaque progression can be briefly described as follow: Initially, the stimulation of ROS, inflammatory cytokines and alteration of hemodynamics induced endothelial dysfunction, which is regarded as a pathological basis for the plaque formation. On the other hand, the abnormal lipid metabolism result in lipid accumulation in the intima, while the elevation of oxLDL can induce the activation of macrophages [246], [247]. The macrophages are subsequently turned into foam cells through engulfing lipids. Then, foam cells will undergo apoptosis or necrosis after excessive intake of modified lipids and eventually participate in forming lipid core [248]. Formation of lipid core can subsequently promote the accumulation of VSMCs and T cells in the intima, while the ECM secretion by VSMCs can result in the formation of fibrous cap that covers the plaque. Finally, cell death promotes the accumulation of cell debris and extracellular lipids, which eventually forms a lipid-rich pool called necrotic core [249]. Numerous studies have reported the pro-atherosclerotic effects induced by cigarette or e-cigarette exposure (summarized in Table 13).
Table 13.
Summary of evidence supporting KE 1443: Atherosclerosis.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [251] | Cigarette smoke | In vivo | ApoE-/- mice were fed either a normal diet or a high-fat diet and exposed to diluted cigarette smoke for 12 months (6 h/daily, 5 days/week) | Progression in atherosclerosis |
| [252] | Cigarette smoke | In vivo | ApoE-/- mice were exposed to sidestream cigarette smoke for 6, 7, 10, and 14 weeks (6 h/daily, 5 days/week) | Increased sizes of atherosclerotic lesions |
| [253] | Cigarette smoke | Cohort study | The study enrolled 1209 subjects, including 450 smokers | Increased carotid intima-media thickness |
| [254] | Cigarette smoke | Cohort study | The study enrolled 16,000 subjects aged 45 to 64 ages | Increased the progression of atherosclerosis |
| [255] | E-cigarette smoke | In vivo | ApoE-/- mice were exposed to e-cigarettes intermittently for 12 weeks (12 h/daily) | Increased sizes of arterial plaque |
| [224] | E-cigarette smoke | In vivo and in vitro | ApoE-/- were exposed to e-cigarette smoke for 2 h/daily, 5 days/week for 16 weeks, while RAW 246.7 cells were treated with e-cigarette smoke extract for 48 h | Increased sizes of atherosclerotic lesions |
| [194] | Cigarette smoke, e-cigarette smoke | In vivo | ApoE-/- mice were exposed to cigarette smoke or e-cigarette smoke 3 h/daily, 5 days/week for 6 months | Increased sizes of atherosclerotic lesions |
| [256] | Cigarette smoke, e-cigarette smoke | Randomized control trial | The study enrolled 20 cigarette smoke users | Pro-atherosclerotic effects on vascular |
Cigarette smoking has been proved to be one of the most important factors causing atherosclerosis [250]. Various toxicological studies have concluded that cigarette smoking can induce oxidative stress, vascular inflammation, platelet coagulation, vascular dysfunction, and modification of the lipid profile, all contributing to the progression of atherosclerotic plaque [8]. ApoE-/- mice, a canonical model for inhalation toxicology studies, have been adopted in numerous studies to manifest the association between cigarette exposure and atherosclerosis. K von Holt et al. reported that long-term exposure to mainstream cigarette smoke significantly accelerated the progression of atherosclerosis in ApoE-/- mice, and a high-fat diet may promote the adverse effect [251]. In another study, Gairola et al. supported that exposure to sidestream cigarette smoke could also increase the size of atherosclerotic lesions in ApoE-/- mice, especially in the thoracic region [252]. Therefore, both mainstream and sidestream cigarette smoke exposure is thought to increase the severity of atherosclerosis. Besides, numerous epidemiologic studies have also confirmed an association between cigarette smoking and atherosclerotic lesions. In a Japanese community-based cohort study, Kiriyama et al. reported that carotid intima-media thickness (cIMT) values and the incidence of high-risk atheroma were significantly higher in smokers than in non-smokers in the subgroup of participants aged ≥ 60 years [253]. Howard et al. supported that cigarette smoke contributed to the progression of atherosclerosis based on a cohort of middle-aged adults in the United States. They found that both active and passive smoking was associated with an increase in the progression of atherosclerosis, and the impact of smoking could be even more severe in subjects with diabetes and hypertension [254].
Chronic and intermittent electronic smoke exposure was also proved to aggravate atherosclerosis progression in mice. Derout et al. reported that exposure to e-cigarette smoke induced a significant increase in arterial plaque in the aortic root ApoE-/- mice [255]. Li et al. also revealed that e-cigarette smoke exposure significantly induced atherosclerotic lesions and up-regulated TLR9 expression levels in monocytes and atherosclerotic plaques in ApoE-/- mice [224]. Szostak et al. found that although e-cigarette exposure also induced the development of atherosclerotic plaques in ApoE-/- mice, they were significantly less severe than cigarette exposure [194]. Although there is still a lack of epidemiologic evidence supporting e-cigarette-related atherosclerosis, a randomized control trial has compared the pro-atherosclerotic effects induced by acute exposure to e-cigarettes and traditional tobacco combustion cigarettes. The results indicated that alteration of oxidative stress, antioxidant reserve, platelet function, flow-mediated dilation, and blood pressure induced by acute e-cigarette exposure was less severe than that of tobacco combustion cigarettes [256]. These studies suggested that either e-cigarette or cigarette exposure may promote the development and progression of atherosclerosis, and cigarette exposure may cause more severe pro-atherosclerotic effects, which was also correlated with the evidence of the differences in cigarette and e-cigarette related-oxidative stress, inflammatory response, and endothelial dysfunction. Nevertheless, considering that atherosclerosis is a serious cardiovascular disease that can develop chronically and accumulatively, long-term population studies should be conducted to confirm the e-cigarette-related atherosclerotic effects in humans.
Cardiac dysregulation induced by cigarette and e-cigarette exposure
Key event (KE 1500): Fibroblast proliferation and myofibroblast differentiation
Oxidative stress has been generally proved to play a critical role in promoting fibroblast proliferation and differentiation via upregulating TGF-β expression [257]. Cardiac fibroblast (CF) is a basic cell type, mainly derived from embryonic epicardial and endothelial cells. Physiologically, CF is responsible for ECM homeostasis, providing a structural scaffold for cardiomyocytes, distributing mechanical force through cardiac tissue, and mediating electrical conduction. In response to mechanical stimulation and injury, fibroblasts are activated and begin to proliferate and transform into myofibroblasts. Fibroblasts and their related myofibroblasts are major producers of ECM and therefore significantly contribute to cardiac fibrosis. Activated fibroblasts can also directly lead to cardiac hypertrophy through a paracrine mechanism, further leading to impaired cardiac function [258], [259]. Fibroblast activation induced by cigarette or nicotine exposure has been observed in several studies (summarized in Table 14).
Table 14.
Summary of evidence supporting KE 1500: fibroblast proliferation and myofibroblast differentiation.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [260] | Cigarette smoke | In vivo and in vitro | C57BL/6 and BALB/c mice were exposed to cigarette smoke (1 h/time, 2 times/day, 5 day/week) for 20 weeks, while MLg and MCR5 cells were treated with CSE for 4, 12, 24, 48 h | Increased the proliferation and protein production of fibroblasts |
| [262] | Cigarette smoke | In vivo and in vitro | AKR mice were exposed to cigarette smoke for 6 weeks, while primary isolated CFs were treated with CSE for 24 h | Increased collagen deposition, Increased the proliferation of fibroblasts |
| [263] | Cigarette smoke | Cross-sectional study and in vitro study | The study included samples from 86 patients undergoing valve replacement surgery and 17 control patients, while fibroblasts were treated with cigarette smoke-treated media for 24 h | Increased the ratio of collagen/elastin in stenotic valves, increased TGF-β expression |
| [264] | Nicotine | In vivo | Prenatal Sprague-Dawley rats were treated with nicotine (1 mg/kg) from the 6th day of gestation until day 22, while isolated fibroblasts were treated with nicotine (10-9 M) for 96 h | ECM deposition |
Huang et al. have reported CSE-induced proliferation and protein production in fibroblasts. They also observed cigarette smoke-induced elevated expression of hallmarks of tissue remodeling in the lungs and hearts of mice [260]. Although the profibrotic effects on the respiratory system induced by cigarette smoke have been widely elucidated, the cellular alteration on cardiac fibroblasts remains unclear. Vang et al. suggested that α7 nAChR activation led to the proliferation and increased collagen content by mediating the Ca2+/epidermal growth factor receptor (EGFR) signaling pathway in adult ventricular fibroblasts [261]. A recent study conducted by Vang et al. has directly revealed the cigarette smoke-related effects on cardiac fibroblasts. Sircol assay indicated that cigarette smoke exposure increased collagen content in the right ventricle of AKR mice, while Picrosirius staining also suggested that collagen deposition was significantly enhanced in the right ventricle. The evaluation of primary isolated rat cardiac fibroblasts (CFs) indicated that with the increase in cigarette smoke concentration, the cell viability increased in a dose-dependent manner, and the expression of proliferation marker PCNA was also enhanced. These results suggested that cigarette smoke exposure may induce a pro-fibrotic effect on CFs by activating α7 nAChR, which may negatively affect right heart function [262]. Meanwhile, Helske et al. reported that nicotine in cigarette smoke could induce the expression of TGF-β in fibroblasts, which may contribute to adverse remodeling that resulted in aortic stenosis [263]. Moreover, prenatal nicotine exposure was proved to induce ECM deposition in the hearts of offspring rats, which may be attributed to nicotine-induced activation of MIAT and miR-29 family in fibroblasts [264]. In conclusion, these studies have indicated that cigarette smoke could induce proliferation and differentiation of fibroblasts, which also provides cellular evidence for smoking-related cardiac fibrosis. Unfortunately, there is still a lack of knowledge on fibroblasts exposed to e-cigarettes.
Key event (KE 1918): Myocardial apoptosis
As we discussed above, oxidative stress induced by ROS elevation should be a major cause that triggers apoptosis. Apoptosis is a spontaneous programmed cell death process regulated by genes, which plays an irreplaceable role in maintaining the growth and development of the body and homeostasis of the internal environment. Apoptosis mainly involves the alteration of genetic and molecular programming, proapoptotic protein expression, and transformation of cell phenotypes. Apoptosis caused by pathological factors may result in a sort of cardiac disease, including ischemic heart failure, myocardial infarction, and arrhythmias [265]. Myocardial apoptosis can be mainly induced by the overproduction of ROS, which resulted in oxidative stress and mitochondrial damage. ROS can promote Ca2+ entry into the cytoplasm and mitochondria, which results in mPTP opening and mitochondrial membrane potential collapse. Therefore, ROS can promote cell apoptosis through the release of cytochrome C and apoptosis-inducing factors. Oxidative stress also contributes to the translocation of apoptotic proteins Bax and Bad into mitochondria, which bind to Bcl-2 to form heterodimers, thereby reducing Bcl-2 content. Indeed, a lower Bcl-2/Bax ratio also promotes mPTP opening. Furthermore, ROS can also trigger apoptosis by activating of MAPK cascade and inducing ER stress [266]. Various studies have confirmed myocardial apoptosis induced by cigarette, e-cigarette, or nicotine exposure (summarized in Table 15).
Table 15.
Summary of evidence supporting KE 1918: myocardial apoptosis.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [267] | Cigarette smoke | In vivo | Wistar Albino rats were exposed to cigarette smoke for 12 weeks with (or not) administration of EGCG (20 mg/kg/day) | Myocardial apoptosis, mitochondria damage, the release of cytochrome C |
| [268] | Cigarette smoke | In vivo | MIFKO mice were exposed to cigarette smoke 1 h per day for 60 days | Myocardial apoptosis, contractile dysfunction |
| [269] | CSE, E-cigarette smoke extract | In vitro | iPS-derived human cardiomyocytes were treated with cigarette and e-cigarette smoke extracts for 48 h or every other day for 14 days | Upregulation of apoptosis-related gene expression |
| [270] | E-cigarette smoke | In vivo | Pregnant mice on a high-fat diet were exposed to ECV containing 2.4 % nicotine for 16 days (12 h/daily from 9:00p.m. to 9:00 a.m.) | Fetal myocardial apoptosis |
| [271] | Nicotine | In vitro | H9c2 cardiomyocytes were treated with nicotine (10 or 100 µM) for 48 h | Myocardial apoptosis |
| [272] | Nicotine | In vitro | Neonatal rat cardiomyocytes were treated with nicotine (0.1–100 μM) for 48 h | Myocardial apoptosis, oxidative stress, alteration of apoptosis-related gene expression |
As we discussed above, oxidative stress serves as a critical player in cigarette smoke-related myocardial apoptosis [188]. Adikesavan et al. reported that cigarette smoke exposure induced cardiac apoptosis by triggering mitochondria damage and subsequent release of cytochrome C into the cytoplasm, while treatment with EGCG could reverse the pro-apoptotic effects [267]. Meanwhile, Wang et al. reported that excessive formation of autophagolysosome and elevated TFEB was involved in sidestream cigarette smoke-induced myocardial apoptosis and contractile dysfunction, while macrophage migration inhibitory factor exhibited a protective effect by facilitating mitophagy [268]. Cigarette smoke-induced myocardial apoptosis should be mainly involved in cardiac remodeling, which will be elaborated in the following discussion.
E-cigarette smoke exposure could also induce cytotoxicity by enhancing apoptosis in cardiomyocytes. Basma et al. reported that both cigarette and e-cigarette exposure resulted in a typical elevation of ROS levels in iPS-derived human cardiomyocytes. With the increase in exposure time, cell activity and pulsatile function also decreased significantly. Meanwhile, IPA analysis of differential gene expression revealed that the expression of a variety of genes involved in inflammation, cell proliferation, regeneration, and apoptosis was significantly changed in the cells exposed to both cigarettes and e-cigarettes [269]. Furthermore, Hasan et al. reported that exposure to e-cigarette smoke and a high-fat diet during pregnancy resulted in apoptosis of cardiomyocytes in offspring mice. The results indicated that intrauterine e-cigarette and HFD exposure can trigger fetal myocardial apoptosis and lead to ventricular ultrastructural abnormalities in newborn infants [270]. Nicotine treatment was also reported to reduce cell viability and induce apoptosis in H9C2 myocytes in a dose-dependent manner [271]. Meanwhile, Xiang et al. revealed that nicotine-induced myocardial apoptosis was also dependent on oxidative stress and the alteration of apoptosis-related genes [272]. Taken together, current evidence strongly supported that either cigarette or e-cigarette exposure could induce myocardial apoptosis, while the nicotine content has been proved to participate in this adverse effect. However, there is still a lack of comparison of myocardial apoptosis induced by cigarettes and e-cigarettes, and relevant studies still need to be improved.
Key event (KE 2004): Secretion of catecholamine
Since the pharmacological effects of nicotine have been extensively studied, the activation of nAChR has been proved to increase catecholamine secretion, which is widely involved in a series of nicotine-related cardiovascular effects, basically as the elevation of heart rate and blood pressure [273]. Catecholamines are hormones secreted by the adrenal medulla, mainly including epinephrine, norepinephrine, and dopamine. When the sympathetic-adrenal medulla system is excited, catecholamines are released into the blood in large quantities, causing contraction of arterioles and venules, thereby increasing blood pressure. At the same time, the stimulation of catecholamines also increases cardiac excitation and metabolites, thus improving the perfusion pressure of coronary arteries. Catecholamine acts on the heart through β-1 receptors, which results in accelerating heart rate, enhancing contractile force, increasing conduction velocity, and increasing cardiac output. Differences in catecholamine levels are also potentially associated with sudden cardiac death and coronary heart disease. It has been widely recognized that the catecholamine beta-adrenergic system facilitates both cardiac hypertrophy and ventricular remodeling [274], [275]. Various studies have confirmed myocardial apoptosis induced by cigarette or e-cigarette exposure (summarized in Table 16).
Table 16.
Summary of evidence supporting KE 2004: secretion of catecholamine.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [277] | Cigarette smoke | Cross-sectional study | The study enrolled 35 subjects and divided them into 2 groups, including 15 non-smokers and 10 smokers | Increased plasma catecholamine concentration |
| [278] | Cigarette smoke | Cross-sectional study | The study enrolled 10 monozygotic male twin-pairs with an average smoking time for 23 years | Increased total plasma catecholamine levels |
| [279] | Cigarette smoke | Clinical trial | The study enrolled 12 subjects with normal smoking habits and they were oral administrated with oxprenolol (40 mg) or placebo | Increased plasma norepinephrine levels |
| [280] | Cigarette smoke, e-cigarette smoke | Cross-sectional study | The study enrolled 36 healthy dual users of cigarettes and e-cigarettes | Increased plasma nicotine levels but without significant changes in urinary epinephrine, norepinephrine, and dopamine excretion |
Nicotine delivery by cigarette smoke can be rapidly absorbed into the blood through the lungs, and cross the blood–brain barrier to the central nervous system, where it stimulates nAChR to activate the sympathetic nervous system and promote the release of catecholamine [276]. Several epidemiologic studies have reported that smoking is associated with an increase in plasma catecholamine substances. Lowery 3rd et al. evaluated blood samples from both healthy non-smokers and smokers (15 min after smoking). The results indicated that compared with non-smokers, smoking increased plasma catecholamine concentration and platelet aggregation percentage [277]. Laustiola et al. also reported that total plasma catecholamine levels were significantly higher in smokers than in non-smokers based on a population study that enrolled 10 monozygotic male twins with inconsistent smoking [278]. In a clinical trial, the elevation of plasma norepinephrine was observed in 12 smoking subjects, while changes in blood pressure, pulse, and plasma free fatty acid content induced by norepinephrine activation could be significantly inhibited by a beta-blocker (propranolol) [279]. In another clinical trial, Benowitz et al. observed that compared with e-cigarettes, canonical cigarettes induced higher mean 24-hour plasma nicotine levels. However, there were no significant differences in urinary epinephrine, norepinephrine, and dopamine excretion between cigarette and e-cigarette users [280]. Since the secretion of catecholamine may be directly attributed to nicotine content in cigarette and e-cigarette smoke, the difference in this effect is likely to change with the update in nicotine delivery of e-cigarettes. It is also worth noting that the nicotine content of e-cigarettes is showing an increasing trend in recent years [281]. Therefore, future studies should attach due attention to the secretion of catecholamines and the activation of sympathetic nervous system induced by e-cigarette use.
Key event (KE 1924): Cardiac fibrosis
One of the major consequences of fibroblast proliferation and myofibroblast differentiation is cardiac fibrosis [282]. Cardiac fibrosis is a pathological change characterized by continuous accumulation of extracellular matrix, increased heart mass, and enlarged heart lumen. In severe cases, it may cause endocardium thickening and cardiac scarring, which may be accompanied by white fiber strips. The key event of cardiac fibrosis is the transformation of cardiac fibroblasts (CF) into myofibroblasts, which participate in the production of ECM and accelerate the fibrosis process after cardiac injury. Increased cardiac fibroblast activity during the pathological process of cardiac fibrosis leads to the accumulation of ECM proteins, such as collagen I and III. The deposition of ECM eventually results in increased myocardial hardness, increased risk of heart failure, and sudden cardiac death [283]. Therefore, the quantification of collagen content should be an important objective in cardiac fibrosis research. Besides, Masson staining, which is derived from Mallory trichromatic dyeing, is also been adopted to directly detect the fibers in the tissue and can be used for semi-quantitative analysis of cardiac fibrosis [284]. Sufficient evidence has strongly supported cardiac fibrosis induced by cigarette or e-cigarette exposure (summarized in Table 17).
Table 17.
Summary of evidence supporting KE 1924: cardiac fibrosis.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [262] | Cigarette smoke | In vivo and in vitro | C57BL/6 and BALB/c mice were exposed to cigarette smoke (1 h/time, 2 times/day, 5 day/week) for 20 weeks, while MLg and MCR5 cells were treated with CSE for 4, 12, 24, 48 h | Right ventricular fibrosis |
| [285] | Cigarette smoke | In vivo | adult male wild-type VFB and metallothionein transgenic mice were exposed to sidestream cigarette smoke for 40 days (1 h/daily) | Cardiac fibrosis, myocardial apoptosis, mitochondrial damage |
| [286] | Cigarette smoke | In vivo | Both young and old rats were exposed to exposed to secondhand smoke (SHS) for 1 month (twice a day for 30 min, 5 days/week) | Left ventricular fibrosis |
| [287] | Cigarette smoke | Case-control study | The study enrolled 95 patients, including 46 smokers and 49 non-smokers | Right atrial fibrosis |
| [288] | E-cigarette smoke | In vivo | C57BL/6 mice were exposed to e-cigarette smoke (3 h/daily) for 14 days | Increased collagen levels in cardiac tissue |
| [289] | E-cigarette smoke | In vivo | C57BL/6 mice were exposed to e-cigarette smoke (1 h/daily, 5 days/week) for 3–6 months | Cardiac fibrosis |
| [290] | Cigarette smoke, e-cigarette smoke, waterpipe smoke | In vivo | Wistar rats were exposed to either cigarettes, e-cigarettes, or water pipes for 4 weeks (1 h/daily, 5 days/week) | Cardiac fibrosis |
At present, numerous laboratory evidence supported the cardiac fibrosis caused by cigarette exposure. As we mentioned above, cigarette smoke was reported to induce right ventricular fibrosis in AKR mice [262]. Hu et al. reported that sidestream cigarette smoke-induced myocardial dysfunction and cardiac fibrosis in mice via apoptosis, intracellular Ca2+ dysregulation, and mitochondrial damage. The adverse impact could be attenuated by cardiac-specific overexpression of metallothionein and antioxidant intervention [285]. Wu et al. revealed that secondhand smoke (SHS) exposure induced a significant increase in left ventricular fibrosis in aging rats, with the elevation of TGFβ1, p-Smad2, p-Smad3, CTGF, MMP2, and MMP9 protein expression levels [286]. In a case-control study, Goette et al. concluded that cigarette smoke exposure was significantly associated with the degree of fibrosis in the right atrium, and nicotine was suspected to promote cigarette smoke-related fibrosis by upregulating mRNA expression of collagen III in a dose-dependent manner [287].
To evaluate the myocardial fibrotic effects after e-cigarette exposure, Shi et al. reported that although short-term e-cigarette exposure can slightly increase collagen levels in cardiac tissue of C57BL/6 mice, it did not lead to significant fibrotic effects [288]. Alexander et al. also reported that cardiac fibrosis was observed in C57BL/6 mice after exposure to nicotine-contained ECV for several months [289]. Mayyas et al. reported that compared with the control group, exposure to either cigarettes, e-cigarettes, or water pipes could result in a typical increase in cardiac fibrosis, while the elevation of TGF-β was significant only for the e-cigarette exposure group [290]. Taken together, the evidence suggests that e-cigarettes may promote cardiac fibrosis in a similar way to cigarettes. However, as we mentioned above, due to the lack of evidence about e-cigarette-induced fibroblast activation, it remains difficult to compare cigarette and e-cigarette smoke-related fibrotic effects.
Key event (KE 2002): Ventricular remodeling
Both myocardial apoptosis and fibroblast activation should be involved in ventricular remodeling [291], [292]. Ventricular remodeling is defined as compensatory changes in ventricular morphology and structure, often accompanied by a consequent change in systolic function. Ventricular remodeling is a complex process that mainly involves myocardial overgrowth and apoptosis, fibrosis, angiogenesis, and electrophysiological alteration [293]. It has been reported that a variety of cardiovascular diseases including myocardial infarction, hypertension, and heart failure can lead to an increase in pathological ventricular remodeling [294]. Ventricular remodeling can be divided into left ventricular remodeling and right ventricular remodeling according to the location of occurrence. Left ventricular remodeling may be a direct consequence of myocardial infarction, while right ventricular remodeling is closely related to pulmonary hypertension [295]. Currently, the remodeling effects can be mainly assessed by Doppler echocardiography with the measurement of ventricular wall parameters and diastolic function [296]. Mounting studies have reported ventricular remodeling induced by cigarette, e-cigarette, or nicotine exposure (summarized in Table 18).
Table 18.
Summary of evidence supporting KE 2002: ventricular remodeling.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [297] | Cigarette smoke | In vivo | Sprague-Dawley rats were exposed to 45 mins of cigarette smoke twice per day, accumulatively for 5 weeks | Left ventricular remodeling and systolic dysfunction |
| [298] | Cigarette smoke | In vivo | Wistar rats were exposed to cigarette smoke twice a day (once in the morning and once in the afternoon) for 8 weeks | Increased thickness of left ventricular posterior wall, decreased left ventricular diameter during both diastole and systole |
| [299] | Cigarette smoke | In vivo | Sprague-Dawley was exposed to cigarette smoke for 56 days (1 h/daily) | Increased end-systolic size of the left ventricle, decreased posterior wall size of the end-systolic left ventricle |
| [300] | Cigarette smoke | In vivo | Wistar rats were exposed to cigarette smoke for 2 months | Increased left atrial area, left ventricular mass index, and posterior wall thickness |
| [301] | Cigarette smoke | Cross-sectional study | The study included 4580 healthy participants with no significant coronary artery disease, heart failure, or severe valvular disease | Increased left ventricular (LV) mass index, LV mass/volume ratio, and the prevalence of LV hypertrophy |
| [302] | E-cigarette smoke | In vivo | C57BL/6J on fed a high-fat diet were exposed to e-cigarettes containing 0 % or 2.4 % nicotine for 12 weeks (12 h/daily) | Alteration in left ventricular systolic function but without significant effect on chamber wall thickness |
| [303] | Nicotine | In vivo | C57BL/6J mice were exposed to nicotine vapor for 8 weeks (12 h/daily) | Increased the thickness of the right ventricle |
| [304] | Nicotine | In vivo | C57BL/6J mice were exposed to nicotine vapor for 8 weeks (12 h/daily) | Increased the thickness of the right ventricle, upregulation of the right ventricular ACE |
Several studies have reported the ventricular remodeling caused by cigarette exposure. Gu et al. reported that cigarette smoke induced left ventricular remodeling and systolic dysfunction in Sprague-Dawley rats, which coordinated a significant increase in urine norepinephrine. Meanwhile, the Western-blot analysis indicated that MAPK signaling pathway was involved in cigarette smoke-related cardiac dysfunction [297]. Besides, Danilo et al.’s observed significant changes in the structure of the heart of Wistar rats, especially the left ventricle. Compared with the control group, left ventricular diameter decreased during both diastole and systole, while left ventricular posterior wall thickness increased, but there were no abnormalities in cardiac function [298]. In another study focusing on smoke-induced cardiac remodeling and dysfunction, echocardiography showed that compared with the control group, the end-systolic size of the left ventricle was significantly increased in Sprague-Dawley rats exposed to cigarette smoke, while the posterior wall size of the end-systolic left ventricle was significantly decreased. Meanwhile, cardiac systolic dysfunction was also observed in the smoking group [299]. Azevedo et al. also reported that compared with the control group, the left atrial area, left ventricular mass index, and posterior wall thickness were significantly increased in Wistar rats exposed to cigarette smoke. In addition, the left ventricular diastolic index was decreased in the cigarette smoke exposure group, accompanied by systolic and diastolic dysfunction [300]. In an observational study, Nadruz Jr et al. reported the smoking-related changes in cardiac structure and function. The chest echocardiography revealed that compared with non-smokers, current smokers had a significant increase in left ventricular (LV) mass index, LV mass/volume ratio, and the prevalence of LV hypertrophy. Meanwhile, their heart systolic function was also observed to be typically decreased [301].
Compared with cigarettes, there has been less evidence of the structural changes in ventricular induced by e-cigarette exposure. Hasan et al. reported that nicotine-contained e-cigarette smoke induced left ventricular ultrastructural abnormalities and an increase in myocyte apoptosis in C57BL/6J mice. Echocardiographic measurements also verified that nicotine-contained e-cigarette smoke induced alteration in left ventricular systolic function but had no significant effect on chamber wall thickness, while nicotine-free e-cigarette smoke did not cause typical changes in cardiac function [302]. Besides, Oakes et al. have reported the effects of nicotine on cardiovascular remodeling in hypertensive mice. Echocardiography indicated that nicotine inhalation induced right ventricle hypertrophy, increased the thickness of the free wall of the right ventricle, the inner diameter of the right ventricle, and the systolic pressure of the right ventricle. However, no significant changes were observed in the left ventricle [303]. In another study, nicotine exposure on C57BL/6J mice also led to an increase in right ventricular systolic pressure and a thickening of the free wall, which the researchers suggest may be caused by the upregulation of the right ventricular angiotensin-converting enzyme (ACE) [304]. In summary, either cigarette or e-cigarette exposure has been confirmed to induce ventricular remodeling, which is partially related to nicotine content. Therefore, more studies should be conducted to explore the difference between cigarette and e-cigarette-induced ventricular remodeling.
Key event (KE 2001): Cardiac hypertrophy
Continuous exposure to catecholamines such as epinephrine and norepinephrine has been proved to cause cardiac hypertrophy [305]. Cardiac hypertrophy refers to an increase in the size of cardiomyocytes in the absence of cell division, which can be also defined as increased cardiac mass, accompanied by alteration in cardiac function. Hypertrophic growth of cardiomyocytes is thought to be an adaptive result of hemodynamic changes and can increase cardiac function by reducing ventricular wall tension and oxygen consumption. Hypertension and valvular disease can also induce pathological cardiac hypertrophy, which may be accompanied by cardiac fibrosis and myocardial dysfunction. Indeed, the development of cardiac hypertrophy ultimately promotes the occurrence and progression of arrhythmia and heart failure [306]. Cardiac hypertrophy can be roughly characterized by the heart/body weight ratio. Meanwhile, hematoxylin-eosin or wheat germ agglutinin (WGA) staining can be also used to determine myocardial cell diameter on paraffin sections [307]. Cardiac hypertrophy induced by cigarette or e-cigarette exposure have been observed in several studies (summarized in Table 19).
Table 19.
Summary of evidence supporting KE 2001: cardiac hypertrophy.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [239] | Cigarette smoke | In vivo | Sprague-Dawley rats were exposed to cigarette smoke for a month (2 h/daily, 5 days/week) | Increased heart/body weight ratio |
| [308] | Cigarette smoke | In vivo | SHR rats and Wistar rats were exposed to 30, 60, and 90 days (2 h/daily, 5 days/week) | Increased heart/body weight ratio |
| [309] | Cigarette smoke | In vivo | C57BL/6J mice were exposed to either 16 or 32 weeks (48 min/daily, 5 days/week) | Increased left ventricular mass and heart/body weight ratio |
| [310] | Cigarette smoke | In vivo | C57BL/6JRj mice were fed either a low-fat diet or a high-fat diet for 4 weeks, and exposed to cigarette smoke for 21 weeks (5 cigarettes/daily, 5 days/week) | Cardiac hypertrophy |
| [311] | Cigarette smoke | Clinical trial | The study enrolled 4129 subjects, including 2884 non-smokers, 503 current smokers, and 742 former smokers | Increased mean LV mass index, decreased mean LV circumferential strain |
| [312] | Cigarette smoke, e-cigarette smoke | In vivo | C57/BL6 mice were exposed to either a cigarette or e-cigarette smoke for 60 months (2 h/daily, 5 days/week) | Cardiac hypertrophy |
Cigarette smoke has been widely reported to cause typical cardiac hyperplasia. Compared with the control group, Hariri et al. observed a significant increase in heart/body weight ratio and aortic calcification in Sprague-Dawley rats exposed to cigarette smoke [239]. In another study, the SHR rats and Wistar rats were exposed to cigarette smoke, respectively for 30, 60, and 90 days. The results indicated that compared with the control group, the SHR rats exposed to cigarette smoke had a significant increase in heart/body weight ratio, while the expression of ANF, TGF-β, and ODC was significantly elevated, which has been recognized to be closely related to cardiac hypertrophy and fibrosis. However, Wistar rats exposed to cigarette smoke showed no such effect [308]. Talukder et al. found that cigarette smoke exposure caused an increase in blood pressure, left ventricular mass, and heart/body weight ratio in C57BL/6J mice, accompanied by a reduction in weight gain. Meanwhile, the in vitro trials proved that cardiac function is impaired by high load [309]. In another study focusing on the toxic effects of cigarette smoke on obese mice, Dubois-Deruy et al. reported that chronic cigarette smoke exposure increased the area of cardiomyocytes in C57BL/6JRj mice on a low-fat diet, but not in mice on a high-fat diet [310]. Epidemiologic studies have also reported cigarette smoke-induced cardiac hypertrophy. In the Jackson heart study, Kamimura et al. reported that current smoking was associated with a higher mean LV mass index and a lower mean LV circumferential strain than never-smoking [311].
Laboratory studies have also proved the association between e-cigarette smoke exposure and cardiac hypertrophy. EI-Mahdy et al. reported that long-term exposure to both cigarette and e-cigarette were observed to induce a significant increase in cardiac hypertrophy, and the degree of cardiac hypertrophy increased with exposure time. Meanwhile, although adverse effects were also observed in the absence of nicotine, they appeared earlier and more severe as nicotine content increased, suggesting that nicotine may be involved in the cardiac effects induced by e-cigarettes [312]. In conclusion, exposure to cigarette smoke could result in cardiac hypertrophy, while nicotine content should play the essential role in this pathological process. Considering that cardiac hypertrophy can be mainly induced by activation of the sympathetic adrenal system, we believe that long-term exposure to e-cigarette smoke can result in cardiac hypertrophy similar to that of canonical cigarettes.
Adverse outcome (KE 1929): Increased incidence of cardiovascular morbidity and mortality in the general population
Numerous studies have been conducted to elucidate the cardiovascular risks induced by cigarette smoke, while current researches also focused on confirming whether e-cigarettes should be safer for the cardiovascular system (summarized in Table 20). Cigarette smoke has been widely declared to be a major cause of cardiovascular morbidity and mortality in the general population. Both active and passive smoking have been fully elucidated to accelerate all phases of atherosclerosis from endothelial dysfunction and lipid profile modification to acute clinical events [35]. Indeed, compared to nonsmokers, even the use of smokeless tobacco significantly increased cardiovascular mortality [313]. Epidemiologic studies strongly supported the assertion that cigarette smoke exposure increases the rate of myocardial infarction and coronary artery disease regardless of sex or age among subjects. Meanwhile, cohort studies have concluded that smoking significantly increased the risk of cardiovascular disease in a dose-dependent manner [314], [315]. Clinical trials have also provided reliable evidence for cigarette smoke-induced cardiovascular disease. In a randomized controlled trial, G Rose et al. found that intervention in smoking behavior significantly reduced the mortality from coronary heart disease (18% lower than the control group) [316]. In a cohort study conducted by Price et al., 1592 men and women aged 55–74 years old were selected randomly and followed-up for 5 years. The results indicated that moderate and heavy smokers have a higher incidence of peripheral artery disease and coronary artery disease compared to nonsmokers [317].
Table 20.
Summary of evidence supporting AO (1929): increased incidence of cardiovascular morbidity and mortality in the general population.
| Reference | Test substance | Study type | Study design | Effects |
|---|---|---|---|---|
| [314] | Cigarette smoke | Cohort study | The study enrolled 19,782 men and 21,500 women and followed-up from 1990 to 1992 to 2001 | Increased incidence of coronary heart disease |
| [315] | Cigarette smoke | Cohort study | The study enrolled 20,000 subjects and followed-up from 1976 to 1978 to 1981–1983 | Increased risks of first acute myocardial infarction |
| [316] | Cigarette smoke | Clinical trial | The study enrolled 1445 male smokers at high risk of heart and lung disease | Smoking cessation reduced the mortality of coronary heart disease |
| [317] | Cigarette smoke | Clinical trial | The study enrolled 1592 subjects at random and followed-up for 5 years | Increased incidences of peripheral arterial disease and coronary artery disease |
| [318] | Cigarette smoke | Clinical trial | The study enrolled 12 long-term smoker and 12 non-smokers | Increased heart rate and blood pressure |
| [319] | Cigarette smoke | Clinical trial | The study enrolled 39 smokers with normal blood pressure | Smoking cessation reduced the ambulatory blood pressure |
| [320] | Cigarette smoke | Cohort study | The study enrolled 5301 subjects and followed-up for 5 years | Low levels of smoking were associated with a lower prevalence of masked hypertension |
| [321] | Cigarette smoke | Cross-sectional study | The study enrolled 1646 men, including 494 current smokers | Increased prevalence of masked hypertension |
| [322] | Cigarette smoke | Randomized control trial | The study enrolled 28,236 healthy women with no hypertension, cardiovascular disease, or cancer | Increased risk of hypertension |
| [323] | Cigarette smoke | Case-control study | The study included 115 smokers and 460 non-smokers with essential hypertension | Increased whole-day pressure |
| [324] | Cigarette smoke | Case-control study | The study included 564 male patients with primary myocardial infarction | Increased prevalence of myocardial infarction |
| [325] | Cigarette smoke | Case-control study | The study included 55 women survived myocardial infarction and 220 control | Increased prevalence of myocardial infarction |
| [326] | Cigarette smoke | Case-control study | The study included 13,926 survivors of myocardial infarction | Increased prevalence of myocardial infarction |
| [327] | Cigarette smoke | Cohort study | The study enrolled 11,472 women and 12,391 men followed for 12.3 years | Increased relative risk of myocardial infarction |
| [328] | Cigarette smoke | Cross-sectional study | The study investigated the hospitalization of myocardial infarction from December 1997 to November 2003 in Helena, Montana | Public smoking ban reduced hospitalization of myocardial infarction |
| [329] | Cigarette smoke | Cross-sectional study | The study investigated the hospitalization of myocardial infarction from January 2011 to December 2014 in urban areas of Chile | Legislation of smoking ban reduced hospitalization of myocardial infarction |
| [330] | Cigarette smoke | Cohort study | The study enrolled 11,762 men and 13,206 and followed-up for 11 years | Increased incidence of coronary heart disease |
| [73] | E-cigarette smoke | Randomized control trial | The study enrolled 12 habitual cigarette users in three experimental stages (fake smoking, TC smoking, and EC smoking) | Increased heart rate and blood pressure |
| [332] | E-cigarette smoke | Randomized control trial | The study enrolled 30 habitual e-cigarette users to investigate the effects induced by short halt in e-cigarette use | Increased heart rate and blood pressure |
| [333] | E-cigarette smoke | Randomized control trial | The study enrolled 15 healthy non-smokers | Increased arterial pressure |
| [334] | E-cigarette smoke | Cross-sectional study | The study used data from 2014 and 2016 NHIS | Increased risk of myocardial infarction |
| [335] | E-cigarette smoke | Cross-sectional study | The study used data from 2016 and 2017 NHIS | No significant association between e-cigarette use and myocardial infarction |
| [336] | Cigarette smoke, e-cigarette smoke | Randomized control trial | The study enrolled 114 smokers and randomized them into TC or EC groups for 4-week period of trial | Switching from TC to EC improved the endothelial function and vascular stiffness |
| [337] | Cigarette smoke, e-cigarette smoke | Cross-sectional study | The study included 449,092 subjects from a large cross-sectional telephone survey | No significant association between e-cigarette use and myocardial infarction, increased cardiovascular risk in dual users |
The hypertension induced by cigarette smoke has also been extensively reported as a comprehensive adverse effect induced by both vascular and cardiac pathological changes. In a clinical trial conducted in 1983, Nadler et al. reported that cigarette smoke could induce elevation of blood pressure and heart rate in non-smokers [318]. Minami et al. investigated the effects of smoking cessation (1 week) on blood pressure of smokers. The results indicated that the 24-hour ambulatory blood pressure and heart rate in non-smoking period was lower than that in smoking period [319]. In a cohort study conducted by Bromfield et al., low smoking levels were associated with a lower prevalence of masked hypertension [320]. Meanwhile, Zhang et al. reported that smoking behavior was associated with increased prevalence of masked hypertension [321]. A prospective cohort study that included 28,236 healthy women conducted by Bowman et al. aimed to investigate the association between cigarette smoke and the development of hypertension. The results indicated that smoking was moderately associated with an increased risk of hypertension while the adverse effects were much more severe in those women who consumed at least 15 cigarettes per day [322]. Besides, in a case-control study conducted by Verdecchia et al., heavy smoking (>or = 20 cigarettes/daily) was reported to be associated with an increase in whole-day blood pressure [323].
Myocardial infarction is another well-recognized adverse effect associated with smoking behavior. In 1975, an epidemiologic study conducted by Wilhelmsson has reported that smoking may be associated with a higher risk of myocardial infarction, while smoking cessation could reduce the non-fatal recurrences in myocardial infarction patients [324]. Slone et al. reported that in women under the age of 50, smoking behavior was associated a higher rate of myocardial infarction, while women who consumed 35 cigarettes/daily or more have a much higher (20-fold) rate of myocardial infarction than non-smoking women [325]. Parish et al. reported that the rate of myocardial infarction in smokers was significantly higher than that in non-smokers, and higher tar content in cigarettes was also associated with the increased rate of myocardial infarction [326]. In another cohort study conducted by Prescott et al., the relative risk of myocardial infarction increased with the consumption of cigarettes in both sexes, while women were more sensitive than men to the smoking-related increased risks [327]. Interestingly, Sargent et al. reported that public smoking ban could significantly reduce the hospitalization rates for myocardial infarction [328]. Meanwhile, in a cross-sectional study conducted by Nazzal, et al, the legislation of smoking ban in Chile also contributed to the decreased incidence of myocardial infraction [329]. Moreover, Iversen et al. confirmed that both active and passive smoking were risk factors of myocardial infraction in a large cohort study that enrolled 24,968 subjects [330].
As we have described, little is known about the cardiovascular risk induced by e-cigarettes, particularly the consequences of long-term exposure. Due to the lack of evidence-based population research and the puzzle of underlying molecular mechanisms of e-cigarette-related cardiovascular morbidity, the correlation between e-cigarette smoke and cardiovascular disease remains unclear. But one thing is certain: e-cigarette use among teens and adults is rising year by year, and tend to replace cigarettes as the dominant product in the tobacco market [32]. Several epidemiologic studies have tried to analyze the cardiovascular hazards caused by e-cigarettes. Recent studies have suggested that e-cigarette use may induce a short-term increase in blood pressure, but there is still a lack of prospective studies to determine long-term effects [331]. Dimitriadis et al. reported that acute exposure to cigarette smoke could induce an increase in heart rate and mean arterial pressure that were similar to cigarette-induced effects on healthy smokers [73]. Chaumont et al. also reported that the use of e-cigarettes containing nicotine could induce an increase in heart rate and blood pressure compared to the use of nicotine-free e-cigarettes or sham-vaping [332]. Moreover, Gonzalez et al. reported that exposure to e-cigarette smoke could induce elevation of mean arterial pressure in young non-smokers [333]. A cross-sectional study based on the 2014 and 2016 National Health Interview Survey (NHIS) indicated that daily e-cigarette use was independently associated with an increased risk of myocardial infarction, consistent with daily use of regular cigarettes [334]. However, in another pooled analysis based on NHIS in 2016 and 2017, no significant association was found between e-cigarette use and myocardial infarction or coronary heart disease [335]. To investigate the early vascular effects of tobacco cigarette (TC) conversion to e-cigarette (EC) in chronic smokers, George et al. conducted a randomized controlled trial that enrolled smokers over 18 years of age without cardiovascular disease. The results indicated that endothelial function and vascular stiffness were significantly improved within one month of switching from TC to EC, and those female users benefited more from the switch than males [336]. A large cross-sectional study focusing on the relationship between e-cigarettes and cardiovascular disease also found that e-cigarette exposure did not seem to be associated with the incidence of cardiovascular disease. However, dual use of e-cigarettes and cigarettes were associated with 36 % higher odds of cardiovascular disease compared to current cigarette users [337].
Although in vitro and in vivo studies could partially represent the cardiovascular induced by e-cigarette smoke, neither exposure time nor specific dose can accurately simulate intermittent human puffing. Therefore, the evidence obtained from population studies and clinical trials should be crucial in manifesting the precise effects. Benowitz et al. have summarized current evidence of e-cigarette-induced cardiovascular effects in a comprehensive review. Although the acute effects, including increased heart rate and blood pressure have been confirmed, the authors suggested that these acute effects should not be regarded as valuable predictors for CVD. Meanwhile, they also summarized the studies related to the alteration of cardiovascular structure and function, while the results were inconsistent [338]. Meanwhile, Farsalinos also suggested that e-cigarettes induced-acute adverse effects on endothelial function should not be considered as solid evidence supporting the cardiovascular risks since the adoption of nicotine replacement therapies and intake of caffeine could also induce similar effects on vascular function, while neither of them was considered as risk factors for CVD [339]. Besides, Callahan-Lyon suggested that there was still a lack of data to determine the long-term safety of e-cigarettes [340]. Taken together, it is now well established that e-cigarette use causes some acute cardiovascular effects, possibly with changes in biomarkers of oxidative stress and inflammation. However, the long-term effects remain unclear due to the lack of population data. On the other hand, e-cigarette use is still on the rise, with studies suggesting that perhaps more than 50 percent of e-cigarette users are under the age of 35 [341]. Although some smokers successfully quitted smoking by using e-cigarettes, in a recent systematic review conducted by Kalkhoran et al., it was surprisingly revealed that e-cigarette use was associated with a decrease in smoking cessation in the real world [342], [343]. Therefore, although current evidence supports that e-cigarette should be safer for the cardiovascular system at the population level, further research is needed to determine whether e-cigarettes are beneficial for smoking cessation.
Conclusion
This review eventually built up an AOP framework based on summarizing current evidence of cigarette and e-cigarette-related cardiovascular toxicity. We found that only a few studies have compared the cardiovascular effects induced by cigarette and e-cigarette exposure, and most of them indicated that cigarette smoke could induce more severe toxic effects (summarized in Table 3). On the other hand, cigarette smoke-induced risks of CVD have been extensively confirmed by epidemiologic evidence, while acute exposure to cigarette smoke has also been proved to be more harmful than that of e-cigarettes. Based on this complete review, we concluded that e-cigarette use should be quite safer for the cardiovascular system than canonical cigarettes. Moreover, this review also demonstrated that the ROS overproduction, the release of inflammatory cytokines, and the activation of nAChR should be responsible for both cigarette and e-cigarette induced cardiovascular injury by triggering various cellular and organic adverse effects.
Although the cardiovascular toxicity of cigarettes has been extensively elucidated, we have revealed some unavoidable difficulties and imperfections in studies focusing on e-cigarettes. First of all, although the cardiovascular toxicity of e-cigarettes is lower than that of cigarettes, there is still a lack of toxicity comparison at different levels, especially the differences in the molecular and cellular effects caused by e-cigarette exposure remain unclear. Meanwhile, most of the existing studies all focused on nicotine, but the toxicity caused by trace components such as heavy metals present in e-cigarette smoke has been rarely reported. Furthermore, since humans are usually exposed to e-cigarette smoke in an intermittent and long-term manner, most current preclinical studies may not accurately replicate the actual exposure procedure, which also limits the extrapolation of this evidence to some extent. Therefore, cohort studies and clinical trials remain the best way to verify the toxicity of e-cigarette smoke. Nevertheless, there is still a lack of population data to confirm the long-term cardiovascular effects induced by e-cigarettes. Finally, since the use of e-cigarettes may reduce smoking cessation rates and even lead to the dual use of e-cigarettes and cigarettes, we suggest that the promotion of e-cigarettes should still be treated with caution. Simultaneously, unlike canonical cigarettes, the e-cigarette device and formulation of e-liquid will be constantly updated, therefore, future studies should continue to focus on the novel modifications of e-cigarette and subsequent alterations in their cardiovascular toxicity.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Biographies

Junchao Duan received her PhD and served as a professor at School of Public Health, Capital Medical University, China. Dr. Duan is currently mainly engaged in the research fields of cardiovascular toxicology and environmental toxicology. She conducts multilateral research on the adverse health effects and toxic mechanisms of environmental pollutants.

Zhiwei Sun, Professor of School of Public Health, Capital Medical University, China. Dr. Sun received his PhD from Johannes Gutenberg University of Mainz, Germany, and is currently mainly engaged in the research fields of environmental toxicology, particulate toxicology and environmental epidemiology. He conducts multilateral research on the adverse health effects and toxic mechanisms of environmental pollutants, such as air pollution, e-cigarettes, nanomaterials and microplastics.

Ruiyang Ding is conducting his MS at the School of Public Health, Capital Medical University, China. He has conducted both in vivo and in vitro studies on microplastics-related gastrointestinal toxicity. He is currently focusing on the cardiovascular toxicity induced by cigarette and e-cigarette exposure.

Xiaoke Ren is conducting his MS at the School of Public Health, Capital Medical University, China. His research interests are focused on environmental toxicology and particulate matter toxicology. He conducts in vivo studies on e-cigarette aerosols to assess the damaging effects on the organism. He has also conducted studies in the cardiovascular toxicity of ambient particulate matter PM2.5 and its molecular mechanisms at the cellular level using different cell lines. He has contributed to several research articles on environmental pollutants.

Qinglin Sun is conducting her MS at the School of Public Health, Capital Medical University, China. Currently, she is particularly interested in cardiovascular biomolecular stress induced by e-cigarette exposure. In addition, her research interests also include the effects of particulate matter on cardiovascular toxicity at the biological and molecular levels. She has published 3 scientific articles on toxicological issues in reputable journals.
Footnotes
Peer review under responsibility of Cairo University.
Contributor Information
Zhiwei Sun, Email: zwsun@ccmu.edu.cn.
Junchao Duan, Email: jcduan@ccmu.edu.cn.
References
- 1.D'Angelo D., Ahluwalia I.B., Pun E., Yin S., Palipudi K., Mbulo L. Current Cigarette Smoking, Access, and Purchases from Retail Outlets Among Students Aged 13–15 Years - Global Youth Tobacco Survey, 45 Countries, 2013 and 2014. MMWR Morb Mortal Wkly Rep. 2016;65:898–901. doi: 10.15585/mmwr.mm6534a3. [DOI] [PubMed] [Google Scholar]
- 2.Lubin J.H., Couper D., Lutsey P.L., Woodward M., Yatsuya H., Huxley R.R. Risk of Cardiovascular Disease from Cumulative Cigarette Use and the Impact of Smoking Intensity, Epidemiology (Cambridge. Mass) 2016;27:395–404. doi: 10.1097/EDE.0000000000000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nansseu J.R., Bigna J.J. Electronic Cigarettes for Curbing the Tobacco-Induced Burden of Noncommunicable Diseases: Evidence Revisited with Emphasis on Challenges in Sub-Saharan Africa. Pulmonary medicine. 2016;2016:4894352. doi: 10.1155/2016/4894352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jha P., Ramasundarahettige C., Landsman V., Rostron B., Thun M., Anderson R.N., et al. 21st-century hazards of smoking and benefits of cessation in the United States. The New England journal of medicine. 2013;368:341–350. doi: 10.1056/NEJMsa1211128. [DOI] [PubMed] [Google Scholar]
- 5.Benowitz N.L., Burbank A.D. Cardiovascular toxicity of nicotine: Implications for electronic cigarette use. Trends Cardiovasc Med. 2016;26:515–523. doi: 10.1016/j.tcm.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haig C., Carrick D., Carberry J., Mangion K., Maznyczka A., Wetherall K., et al. Current Smoking and Prognosis After Acute ST-Segment Elevation Myocardial Infarction: New Pathophysiological Insights. JACC Cardiovascular imaging. 2019;12:993–1003. doi: 10.1016/j.jcmg.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan B., Jin X., Jun L., Qiu S., Zheng Q., Pan M. The relationship between smoking and stroke: A meta-analysis. Medicine. 2019;98 doi: 10.1097/MD.0000000000014872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siasos G., Tsigkou V., Kokkou E., Oikonomou E., Vavuranakis M., Vlachopoulos C., et al. Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Curr Med Chem. 2014;21:3936–3948. doi: 10.2174/092986732134141015161539. [DOI] [PubMed] [Google Scholar]
- 9.Shinozaki N., Yuasa T., Takata S. Cigarette smoking augments sympathetic nerve activity in patients with coronary heart disease. International heart journal. 2008;49:261–272. doi: 10.1536/ihj.49.261. [DOI] [PubMed] [Google Scholar]
- 10.Choi B.G., Rha S.W., Park T., Choi S.Y., Byun J.K., Shim M.S., et al. Impact of Cigarette Smoking: a 3-Year Clinical Outcome of Vasospastic Angina Patients. Korean circulation journal. 2016;46:632–638. doi: 10.4070/kcj.2016.46.5.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Y., Xu Z., Georgakis M.K., Wang Z., Lin H., Zheng L. Smoking and heart failure: a Mendelian randomization and mediation analysis. ESC heart failure. 2021;8:1954–1965. doi: 10.1002/ehf2.13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong X., Wang C., Liao X., Pan Y., Yan H., Cao Y., et al. Smoking-Thrombolysis Relationship Depends on Ischemic Stroke Subtype. Stroke. 2016;47:1811–1816. doi: 10.1161/STROKEAHA.116.013124. [DOI] [PubMed] [Google Scholar]
- 13.Yang G., Zeng R., Song X., Li T., Liu C., Ni L. Sophocarpine prevents cigarette smoke-induced restenosis in rat carotid arteries after angioplasty. Annals of palliative medicine. 2020;9:1622–1630. doi: 10.21037/apm-19-568. [DOI] [PubMed] [Google Scholar]
- 14.WHO, Tobacco use falling: WHO urges countries to invest in helping more people to quit tobacco, 2021, https://www.who.int/news/item/16-11-2021-tobacco-use-falling-who-urges-countries-to-invest-in-helping-more-people-to-quit-tobacco. [PMC free article] [PubMed]
- 15.Heidenreich P.A., Trogdon J.G., Khavjou O.A., Butler J., Dracup K., Ezekowitz M.D., et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 16.Bertholon J.F., Becquemin M.H., Annesi-Maesano I., Dautzenberg B. Electronic cigarettes: a short review. Respiration; international review of thoracic diseases. 2013;86:433–438. doi: 10.1159/000353253. [DOI] [PubMed] [Google Scholar]
- 17.C.J. Brown, J.M. Cheng, Electronic cigarettes: product characterisation and design considerations, Tobacco control, 23 Suppl 2 (2014) ii4-10. [DOI] [PMC free article] [PubMed]
- 18.M.S. Orr, Electronic cigarettes in the USA: a summary of available toxicology data and suggestions for the future, Tobacco control, 23 Suppl 2 (2014) ii18-22. [DOI] [PMC free article] [PubMed]
- 19.Williams M., Villarreal A., Bozhilov K., Lin S., Talbot P. Metal and silicate particles including nanoparticles are present in electronic cigarette cartomizer fluid and aerosol. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0057987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flouris A.D., Chorti M.S., Poulianiti K.P., Jamurtas A.Z., Kostikas K., Tzatzarakis M.N., et al. Acute impact of active and passive electronic cigarette smoking on serum cotinine and lung function. Inhalation Toxicol. 2013;25:91–101. doi: 10.3109/08958378.2012.758197. [DOI] [PubMed] [Google Scholar]
- 21.Marques P., Piqueras L., Sanz M.J. An updated overview of e-cigarette impact on human health. Respir Res. 2021;22:151. doi: 10.1186/s12931-021-01737-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romagna G., Allifranchini E., Bocchietto E., Todeschi S., Esposito M., Farsalinos K.E. Cytotoxicity evaluation of electronic cigarette vapor extract on cultured mammalian fibroblasts (ClearStream-LIFE): comparison with tobacco cigarette smoke extract. Inhalation Toxicol. 2013;25:354–361. doi: 10.3109/08958378.2013.793439. [DOI] [PubMed] [Google Scholar]
- 23.Bahl V., Lin S., Xu N., Davis B., Wang Y.H., Talbot P. Comparison of electronic cigarette refill fluid cytotoxicity using embryonic and adult models, Reproductive toxicology (Elmsford. NY) 2012;34:529–537. doi: 10.1016/j.reprotox.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Farsalinos K.E., Romagna G., Tsiapras D., Kyrzopoulos S., Spyrou A., Voudris V. Impact of flavour variability on electronic cigarette use experience: an internet survey. Int J Environ Res Public Health. 2013;10:7272–7282. doi: 10.3390/ijerph10127272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Husari A., Shihadeh A., Talih S., Hashem Y., El Sabban M., Zaatari G. Acute Exposure to Electronic and Combustible Cigarette Aerosols: Effects in an Animal Model and in Human Alveolar Cells. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco. 2016;18:613–619. doi: 10.1093/ntr/ntv169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zelikoff J.T., Parmalee N.L., Corbett K., Gordon T., Klein C.B., Aschner M. Microglia Activation and Gene Expression Alteration of Neurotrophins in the Hippocampus Following Early-Life Exposure to E-Cigarette Aerosols in a Murine Model, Toxicological sciences : an official journal of the Society of. Toxicology. 2018;162:276–286. doi: 10.1093/toxsci/kfx257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Marco L.Y., Farkas E., Martin C., Venneri A., Frangi A.F. Is Vasomotion in Cerebral Arteries Impaired in Alzheimer's Disease? Journal of Alzheimer's disease : JAD. 2015;46:35–53. doi: 10.3233/JAD-142976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carnevale R., Sciarretta S., Violi F., Nocella C., Loffredo L., Perri L., et al. Acute Impact of Tobacco vs Electronic Cigarette Smoking on Oxidative Stress and Vascular Function. Chest. 2016;150:606–612. doi: 10.1016/j.chest.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 29.Vlachopoulos C., Ioakeimidis N., Abdelrasoul M., Terentes-Printzios D., Georgakopoulos C., Pietri P., et al. Electronic Cigarette Smoking Increases Aortic Stiffness and Blood Pressure in Young Smokers. J Am Coll Cardiol. 2016;67:2802–2803. doi: 10.1016/j.jacc.2016.03.569. [DOI] [PubMed] [Google Scholar]
- 30.Franzen K.F., Willig J., Cayo Talavera S., Meusel M., Sayk F., Reppel M., et al. E-cigarettes and cigarettes worsen peripheral and central hemodynamics as well as arterial stiffness: A randomized, double-blinded pilot study, Vascular medicine. 2018;23:419–425. doi: 10.1177/1358863X18779694. [DOI] [PubMed] [Google Scholar]
- 31.Caporale A., Langham M.C., Guo W., Johncola A., Chatterjee S., Wehrli F.W. Acute Effects of Electronic Cigarette Aerosol Inhalation on Vascular Function Detected at Quantitative MRI. Radiology. 2019;293:97–106. doi: 10.1148/radiol.2019190562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bold K.W., Krishnan-Sarin S., Stoney C.M. E-cigarette use as a potential cardiovascular disease risk behavior. The American psychologist. 2018;73:955–967. doi: 10.1037/amp0000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Münzel T., Hahad O., Kuntic M., Keaney J.F., Deanfield J.E., Daiber A. Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes. Eur Heart J. 2020;41:4057–4070. doi: 10.1093/eurheartj/ehaa460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ambrose J.A., Barua R.S. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 35.Zhao M.J., Yuan S., Zi H., Gu J.M., Fang C., Zeng X.T. Oxidative Stress Links Aging-Associated Cardiovascular Diseases and Prostatic Diseases. Oxid Med Cell Longevity. 2021;2021:5896136. doi: 10.1155/2021/5896136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang P.Y., Xu X., Li X.C. Cardiovascular diseases: oxidative damage and antioxidant protection. Eur Rev Med Pharmacol Sci. 2014;18:3091–3096. [PubMed] [Google Scholar]
- 37.E. Dubois-Deruy, V. Peugnet, A. Turkieh, F. Pinet, Oxidative Stress in Cardiovascular Diseases, Antioxidants (Basel, Switzerland), 9 (2020). [DOI] [PMC free article] [PubMed]
- 38.Snoderly H.T., Nurkiewicz T.R., Bowdridge E.C., Bennewitz M.F. E-Cigarette Use: Device Market, Study Design, and Emerging Evidence of Biological Consequences. Int J Mol Sci. 2021;22 doi: 10.3390/ijms222212452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuntic M., Oelze M., Steven S., Kröller-Schön S., Stamm P., Kalinovic S., et al. Short-term e-cigarette vapour exposure causes vascular oxidative stress and dysfunction: evidence for a close connection to brain damage and a key role of the phagocytic NADPH oxidase. NOX-2Eur Heart J. 2020;41:2472–2483. doi: 10.1093/eurheartj/ehz772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai H., Wang C. Graphical review: The redox dark side of e-cigarettes; exposure to oxidants and public health concerns. Redox Biol. 2017;13:402–406. doi: 10.1016/j.redox.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Becker R.A., Ankley G.T., Edwards S.W., Kennedy S.W., Linkov I., Meek B., et al. Increasing Scientific Confidence in Adverse Outcome Pathways: Application of Tailored Bradford-Hill Considerations for Evaluating Weight of Evidence. Regulatory toxicology and pharmacology : RTP. 2015;72:514–537. doi: 10.1016/j.yrtph.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Soleimani F., Dobaradaran S., De-la-Torre G.E., Schmidt T.C., Saeedi R. Content of toxic components of cigarette, cigarette smoke vs cigarette butts: A comprehensive systematic review. The Science of the total environment. 2022;813 doi: 10.1016/j.scitotenv.2021.152667. [DOI] [PubMed] [Google Scholar]
- 43.Kim H.H., Choi S. Therapeutic Aspects of Carbon Monoxide in Cardiovascular Disease. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19082381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.André L., Gouzi F., Thireau J., Meyer G., Boissiere J., Delage M., et al. Carbon monoxide exposure enhances arrhythmia after cardiac stress: involvement of oxidative stress. Basic Res Cardiol. 2011;106:1235–1246. doi: 10.1007/s00395-011-0211-y. [DOI] [PubMed] [Google Scholar]
- 45.Colombo G., Aldini G., Orioli M., Giustarini D., Gornati R., Rossi R., et al. Water-Soluble alpha, beta-unsaturated aldehydes of cigarette smoke induce carbonylation of human serum albumin. Antioxid Redox Signal. 2010;12:349–364. doi: 10.1089/ars.2009.2806. [DOI] [PubMed] [Google Scholar]
- 46.Pei Z., Zhuang Z., Sang H., Wu Z., Meng R., He E.Y., et al. α, β-Unsaturated aldehyde crotonaldehyde triggers cardiomyocyte contractile dysfunction: role of TRPV1 and mitochondrial function. Pharmacol Res. 2014;82:40–50. doi: 10.1016/j.phrs.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 47.Facchinetti F., Amadei F., Geppetti P., Tarantini F., Di Serio C., Dragotto A., et al. Alpha, beta-unsaturated aldehydes in cigarette smoke release inflammatory mediators from human macrophages. Am J Respir Cell Mol Biol. 2007;37:617–623. doi: 10.1165/rcmb.2007-0130OC. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann D., Djordjevic M.V., Rivenson A., Zang E., Desai D., Amin S. A study of tobacco carcinogenesis. LI. Relative potencies of tobacco-specific N-nitrosamines as inducers of lung tumours in A/J mice. Cancer Lett. 1993;71:25–30. doi: 10.1016/0304-3835(93)90092-n. [DOI] [PubMed] [Google Scholar]
- 49.D.M. Chambers, J.M. Ocariz, M.F. McGuirk, B.C. Blount, Impact of cigarette smoking on volatile organic compound (VOC) blood levels in the U.S. population: NHANES 2003-2004, Environment international, 37 (2011) 1321-1328. [DOI] [PubMed]
- 50.Sisto R., Cavallo D., Ursini C.L., Fresegna A.M., Ciervo A., Maiello R., et al. Direct and Oxidative DNA Damage in a Group of Painters Exposed to VOCs: Dose - Response Relationship. Front Public Health. 2020;8:445. doi: 10.3389/fpubh.2020.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwon J.W., Park H.W., Kim W.J., Kim M.G., Lee S.J. Exposure to volatile organic compounds and airway inflammation. Environmental health : a global access science source. 2018;17:65. doi: 10.1186/s12940-018-0410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.S. Dobaradaran, T.C. Schmidt, N. Lorenzo-Parodi, M.A. Jochmann, I. Nabipour, A. Raeisi, et al., Cigarette butts: An overlooked source of PAHs in the environment?, Environmental pollution (Barking, Essex : 1987), 249 (2019) 932-939. [DOI] [PubMed]
- 53.Zhang Y.J., Huang C., Lv Y.S., Ma S.X., Guo Y., Zeng E.Y. Polycyclic aromatic hydrocarbon exposure, oxidative potential in dust, and their relationships to oxidative stress in human body: A case study in the indoor environment of Guangzhou. South China, Environment international. 2021;149 doi: 10.1016/j.envint.2021.106405. [DOI] [PubMed] [Google Scholar]
- 54.Dai Y., Huo X., Cheng Z., Wang Q., Zhang Y., Xu X. Alterations in platelet indices link polycyclic aromatic hydrocarbons toxicity to low-grade inflammation in preschool children. Environ Int. 2019;131 doi: 10.1016/j.envint.2019.105043. [DOI] [PubMed] [Google Scholar]
- 55.Dobson R., Semple S. “How do you know those particles are from cigarettes?”: An algorithm to help differentiate second-hand tobacco smoke from background sources of household fine particulate matter. Environ Res. 2018;166:344–347. doi: 10.1016/j.envres.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 56.Grahame T.J., Schlesinger R.B. Oxidative stress-induced telomeric erosion as a mechanism underlying airborne particulate matter-related cardiovascular disease. Part Fibre Toxicol. 2012;9:21. doi: 10.1186/1743-8977-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pope C.A., 3rd, Bhatnagar A., McCracken J.P., Abplanalp W., Conklin D.J., O'Toole T. Exposure to Fine Particulate Air Pollution Is Associated With Endothelial Injury and Systemic Inflammation. Circ Res. 2016;119:1204–1214. doi: 10.1161/CIRCRESAHA.116.309279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pinto E., Cruz M., Ramos P., Santos A., Almeida A. Metals transfer from tobacco to cigarette smoke: Evidences in smokers' lung tissue. J Hazard Mater. 2017;325:31–35. doi: 10.1016/j.jhazmat.2016.11.069. [DOI] [PubMed] [Google Scholar]
- 59.See S.W., Wang Y.H., Balasubramanian R. Contrasting reactive oxygen species and transition metal concentrations in combustion aerosols. Environ Res. 2007;103:317–324. doi: 10.1016/j.envres.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 60.Stefaniak A.B., LeBouf R.F., Ranpara A.C., Leonard S.S. Toxicology of flavoring- and cannabis-containing e-liquids used in electronic delivery systems. Pharmacol Ther. 2021;224 doi: 10.1016/j.pharmthera.2021.107838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sleiman M., Logue J.M., Montesinos V.N., Russell M.L., Litter M.I., Gundel L.A., et al. Emissions from Electronic Cigarettes: Key Parameters Affecting the Release of Harmful Chemicals. Environ Sci Technol. 2016;50:9644–9651. doi: 10.1021/acs.est.6b01741. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y., Yang Y., He X., Yang P., Zong T., Sun P., et al. The cellular function and molecular mechanism of formaldehyde in cardiovascular disease and heart development. J Cell Mol Med. 2021;25:5358–5371. doi: 10.1111/jcmm.16602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava S., Sithu S.D., Vladykovskaya E., Haberzettl P., Hoetker D.J., Siddiqui M.A., et al. Oral exposure to acrolein exacerbates atherosclerosis in apoE-null mice. Atherosclerosis. 2011;215:301–308. doi: 10.1016/j.atherosclerosis.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeJarnett N., Conklin D.J., Riggs D.W., Myers J.A., O'Toole T.E., Hamzeh I., et al. Acrolein exposure is associated with increased cardiovascular disease risk. Journal of the American Heart Association. 2014;3 doi: 10.1161/JAHA.114.000934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McRobbie H., Phillips A., Goniewicz M.L., Smith K.M., Knight-West O., Przulj D., et al. Effects of Switching to Electronic Cigarettes with and without Concurrent Smoking on Exposure to Nicotine, Carbon Monoxide, and Acrolein, Cancer prevention research (Philadelphia. Pa) 2015;8:873–878. doi: 10.1158/1940-6207.CAPR-15-0058. [DOI] [PubMed] [Google Scholar]
- 66.Goniewicz M.L., Knysak J., Gawron M., Kosmider L., Sobczak A., Kurek J., et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tobacco control. 2014;23:133–139. doi: 10.1136/tobaccocontrol-2012-050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olmedo P., Goessler W., Tanda S., Grau-Perez M., Jarmul S., Aherrera A., et al. Metal Concentrations in e-Cigarette Liquid and Aerosol Samples: The Contribution of Metallic Coils. Environ Health Perspect. 2018;126 doi: 10.1289/EHP2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng T. Chemical evaluation of electronic cigarettes. Tobacco control. 2014;23 Suppl 2:ii11-17. doi: 10.1136/tobaccocontrol-2013-051482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mao C., Li D., Zhou E., Zhang J., Wang C., Xue C. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging. 2021;13:7627–7643. doi: 10.18632/aging.202660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fu X., Zong T., Yang P., Li L., Wang S., Wang Z., et al. Nicotine: Regulatory roles and mechanisms in atherosclerosis progression. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2021;151 doi: 10.1016/j.fct.2021.112154. [DOI] [PubMed] [Google Scholar]
- 71.Ramalingam A., Budin S.B., Mohd Fauzi N., Ritchie R.H., Zainalabidin S. Targeting mitochondrial reactive oxygen species-mediated oxidative stress attenuates nicotine-induced cardiac remodeling and dysfunction. Sci Rep. 2021;11:13845. doi: 10.1038/s41598-021-93234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Menshov V.A., Trofimov A.V., Zagurskaya A.V., Berdnikova N.G., Yablonskaya O.I., Platonova A.G. Influence of Nicotine from Diverse Delivery Tools on the Autonomic Nervous and Hormonal Systems. Biomedicines. 2022;10 doi: 10.3390/biomedicines10010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dimitriadis K., Narkiewicz K., Leontsinis I., Konstantinidis D., Mihas C., Andrikou I., et al. Acute Effects of Electronic and Tobacco Cigarette Smoking on Sympathetic Nerve Activity and Blood Pressure in Humans. Int J Environ Res Public Health. 2022;19 doi: 10.3390/ijerph19063237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun Y.P., Zhu B.Q., Browne A.E., Sievers R.E., Bekker J.M., Chatterjee K., et al. Nicotine does not influence arterial lipid deposits in rabbits exposed to second-hand smoke. Circulation. 2001;104:810–814. doi: 10.1161/hc3301.092788. [DOI] [PubMed] [Google Scholar]
- 75.Gough D.R., Cotter T.G. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2 doi: 10.1038/cddis.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.D'Autréaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 77.Sugamura K., Keaney J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radical Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang D., Kang R., Berghe T.V., Vandenabeele P., Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu P., Hu S., Jin Q., Li D., Tian F., Toan S., et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abais J.M., Xia M., Zhang Y., Boini K.M., Li P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111–1129. doi: 10.1089/ars.2014.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kenmoku S., Urano Y., Kojima H., Nagano T. Development of a highly specific rhodamine-based fluorescence probe for hypochlorous acid and its application to real-time imaging of phagocytosis. J Am Chem Soc. 2007;129:7313–7318. doi: 10.1021/ja068740g. [DOI] [PubMed] [Google Scholar]
- 82.Utsumi H., Muto E., Masuda S., Hamada A. In vivo ESR measurement of free radicals in whole mice. Biochem Biophys Res Commun. 1990;172:1342–1348. doi: 10.1016/0006-291x(90)91597-l. [DOI] [PubMed] [Google Scholar]
- 83.Niemann B., Rohrbach S., Miller M.R., Newby D.E., Fuster V., Kovacic J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J Am Coll Cardiol. 2017;70:230–251. doi: 10.1016/j.jacc.2017.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim M., Han C.H., Lee M.Y. NADPH oxidase and the cardiovascular toxicity associated with smoking. Toxicological research. 2014;30:149–157. doi: 10.5487/TR.2014.30.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang X., Bian Y., Zhang R., Liu X., Ni L., Ma B., et al. Melatonin alleviates cigarette smoke-induced endothelial cell pyroptosis through inhibiting ROS/NLRP3 axis. Biochem Biophys Res Commun. 2019;519:402–408. doi: 10.1016/j.bbrc.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 86.Yang H.L., Korivi M., Chen C.H., Peng W.J., Chen C.S., Li M.L., et al. Antrodia camphorata attenuates cigarette smoke-induced ROS production, DNA damage, apoptosis, and inflammation in vascular smooth muscle cells, and atherosclerosis in ApoE-deficient mice. Environ Toxicol. 2017;32:2070–2084. doi: 10.1002/tox.22422. [DOI] [PubMed] [Google Scholar]
- 87.Liang Y., Ip M.S.M., Mak J.C.W. (-)-Epigallocatechin-3-gallate suppresses cigarette smoke-induced inflammation in human cardiomyocytes via ROS-mediated MAPK and NF-κB pathways. Phytomedicine : international journal of phytotherapy and phytopharmacology. 2019;58 doi: 10.1016/j.phymed.2018.11.028. [DOI] [PubMed] [Google Scholar]
- 88.Morsch A., Wisniewski E., Luciano T.F., Comin V.H., Silveira G.B., Marques S.O., et al. Cigarette smoke exposure induces ROS-mediated autophagy by regulating sestrin. AMPK, and mTOR level in mice, Redox report : communications in free radical research. 2019;24:27–33. doi: 10.1080/13510002.2019.1601448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang G., Li Y., Wu W., Liu B., Ni L., Wang Z., et al. Anti-oxidant effect of heme oxygenase-1 on cigarette smoke-induced vascular injury. Mol Med Rep. 2015;12:2481–2486. doi: 10.3892/mmr.2015.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carnevale R., Loffredo L., Pignatelli P., Nocella C., Bartimoccia S., Di Santo S., et al. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. Journal of thrombosis and haemostasis : JTH. 2012;10:125–132. doi: 10.1111/j.1538-7836.2011.04558.x. [DOI] [PubMed] [Google Scholar]
- 91.Hayashi I., Morishita Y., Imai K., Nakamura M., Nakachi K., Hayashi T. High-throughput spectrophotometric assay of reactive oxygen species in serum. Mutat Res. 2007;631:55–61. doi: 10.1016/j.mrgentox.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 92.Chatterjee S., Tao J.Q., Johncola A., Guo W., Caporale A., Langham M.C., et al. Acute exposure to e-cigarettes causes inflammation and pulmonary endothelial oxidative stress in nonsmoking, healthy young subjects, American journal of physiology. Lung cellular and molecular physiology. 2019;317:L155–L166. doi: 10.1152/ajplung.00110.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee W.H., Ong S.G., Zhou Y., Tian L., Bae H.R., Baker N., et al. Modeling Cardiovascular Risks of E-Cigarettes With Human-Induced Pluripotent Stem Cell-Derived Endothelial Cells. J Am Coll Cardiol. 2019;73:2722–2737. doi: 10.1016/j.jacc.2019.03.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.S. Cirillo, F. Vivarelli, E. Turrini, C. Fimognari, S. Burattini, E. Falcieri, et al., The customizable e-cigarette resistance influences toxicological outcomes: lung degeneration, inflammation and oxidative stress-induced in a rat model, Toxicological sciences : an official journal of the Society of Toxicology, (2019). [DOI] [PubMed]
- 95.Ren X., Lin L., Sun Q., Li T., Sun M., Sun Z., et al. Metabolomics-based safety evaluation of acute exposure to electronic cigarettes in mice. The Science of the total environment. 2022;839 doi: 10.1016/j.scitotenv.2022.156392. [DOI] [PubMed] [Google Scholar]
- 96.Wu X., Zhang H., Qi W., Zhang Y., Li J., Li Z., et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9:171. doi: 10.1038/s41419-017-0257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jia G., Meng Z., Liu C., Ma X., Gao J., Liu J., et al. Nicotine induces cardiac toxicity through blocking mitophagic clearance in young adult rat. Life Sci. 2020;257 doi: 10.1016/j.lfs.2020.118084. [DOI] [PubMed] [Google Scholar]
- 98.Solesio M.E., Mitaishvili E., Lymperopoulos A. Adrenal βarrestin1 targeting for tobacco-associated cardiac dysfunction treatment: Aldosterone production as the mechanistic link. Pharmacol Res Perspect. 2019;7 doi: 10.1002/prp2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Münzel T., Gori T., Keaney J.F., Jr., Maack C., Daiber A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J. 2015;36:2555–2564. doi: 10.1093/eurheartj/ehv305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Michael O.S., Olatunji L.A. Ameliorative effect of nicotine exposure on insulin resistance is accompanied by decreased cardiac glycogen synthase kinase-3 and plasminogen activator inhibitor-1 during oral oestrogen-progestin therapy. Arch Physiol Biochem. 2018;124:139–148. doi: 10.1080/13813455.2017.1369549. [DOI] [PubMed] [Google Scholar]
- 101.Cora N., Ghandour J., Pollard C.M., Desimine V.L., Ferraino K.E., Pereyra J.M., et al. Nicotine-induced adrenal beta-arrestin1 upregulation mediates tobacco-related hyperaldosteronism leading to cardiac dysfunction. World journal of cardiology. 2020;12:192–202. doi: 10.4330/wjc.v12.i5.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.L. Su, M. Zhao, F. Ma, Z. An, Q. Yue, C. Zhao, et al., A comparative assessment of e-cigarette aerosol extracts and tobacco cigarette smoke extracts on in vitro endothelial cell inflammation response, Human & experimental toxicology, 41 (2022) 9603271221088996. [DOI] [PubMed]
- 103.Di Biase A., Attorri L., Di Benedetto R., Sanchez M. Comparative effects between electronic cigarette and tobacco cigarette smoke on oxidative markers in cultured immune cells isolated from rats. Annali dell'Istituto superiore di sanita. 2018;54:300–307. doi: 10.4415/ANN_18_04_06. [DOI] [PubMed] [Google Scholar]
- 104.Dinh Q.N., Drummond G.R., Sobey C.G., Chrissobolis S. Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int. 2014;2014 doi: 10.1155/2014/406960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yeung Y.T., Aziz F., Guerrero-Castilla A., Arguelles S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr Pharm Des. 2018;24:1449–1484. doi: 10.2174/1381612824666180327165604. [DOI] [PubMed] [Google Scholar]
- 106.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 107.Tabas I., García-Cardeña G., Owens G.K. Recent insights into the cellular biology of atherosclerosis. The Journal of cell biology. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chistiakov D.A., Melnichenko A.A., Grechko A.V., Myasoedova V.A., Orekhov A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp Mol Pathol. 2018;104:114–124. doi: 10.1016/j.yexmp.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 109.Karunakaran D., Nguyen M.A., Geoffrion M., Vreeken D., Lister Z., Cheng H.S., et al. RIPK1 Expression Associates With Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation. 2021;143:163–177. doi: 10.1161/CIRCULATIONAHA.118.038379. [DOI] [PubMed] [Google Scholar]
- 110.Hajra L., Evans A.I., Chen M., Hyduk S.J., Collins T., Cybulsky M.I. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. PNAS. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu J., Chen S.J., Hsu S.W., Zhang J., Li J.M., Yang D.C., et al. MARCKS cooperates with NKAP to activate NF-kB signaling in smoke-related lung cancer. Theranostics. 2021;11:4122–4136. doi: 10.7150/thno.53558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Csiszar A., Labinskyy N., Podlutsky A., Kaminski P.M., Wolin M.S., Zhang C., et al. Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations, American journal of physiology. Heart and circulatory physiology. 2008;294:H2721–H2735. doi: 10.1152/ajpheart.00235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. The Journal of biological chemistry. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 114.S.P. Adams, M. Tsang, J.M. Wright, Lipid-lowering efficacy of atorvastatin, The Cochrane database of systematic reviews, 2015 (2015) Cd008226. [DOI] [PMC free article] [PubMed]
- 115.Arca M., Gaspardone A. Atorvastatin efficacy in the primary and secondary prevention of cardiovascular events. Drugs. 2007;67(Suppl 1):29–42. doi: 10.2165/00003495-200767001-00004. [DOI] [PubMed] [Google Scholar]
- 116.Chen S., Wang Y., Zhang H., Chen R., Lv F., Li Z., et al. The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. International journal of biological sciences. 2019;15:1440–1451. doi: 10.7150/ijbs.30193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang H., Yin J., Guo Y. Atorvastatin might resist tobacco smoking-induced endothelial inflammation through the inhibition of NF-κB signal pathway, Clinical and experimental hypertension (New York. NY. 1993;41(2019):1–4. doi: 10.1080/10641963.2018.1433196. [DOI] [PubMed] [Google Scholar]
- 118.Wang Y., Wang X.K., Wu P.P., Wang Y., Ren L.Y., Xu A.H. Necroptosis Mediates Cigarette Smoke-Induced Inflammatory Responses in Macrophages. International journal of chronic obstructive pulmonary disease. 2020;15:1093–1101. doi: 10.2147/COPD.S233506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tabas I., Bornfeldt K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Niu J., Wang K., Kolattukudy P.E. Cerium oxide nanoparticles inhibit oxidative stress and nuclear factor-κB activation in H9c2 cardiomyocytes exposed to cigarette smoke extract. The Journal of pharmacology and experimental therapeutics. 2011;338:53–61. doi: 10.1124/jpet.111.179978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gokulakrisnan A., Jayachandran Dare B., Thirunavukkarasu C. Attenuation of the cardiac inflammatory changes and lipid anomalies by (-)-epigallocatechin-gallate in cigarette smoke-exposed rats. Mol Cell Biochem. 2011;354:1–10. doi: 10.1007/s11010-011-0785-6. [DOI] [PubMed] [Google Scholar]
- 122.Bassi R., Heads R., Marber M.S., Clark J.E. Targeting p38-MAPK in the ischaemic heart: kill or cure? Curr Opin Pharmacol. 2008;8:141–146. doi: 10.1016/j.coph.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 123.Rahaman S.O., Lennon D.J., Febbraio M., Podrez E.A., Hazen S.L., Silverstein R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–221. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu L., Li X., Wang H., Xie F., Liu H., Xie J. Cigarette smoke triggers inflammation mediated by autophagy in BEAS-2B cells. Ecotoxicol Environ Saf. 2019;184 doi: 10.1016/j.ecoenv.2019.109617. [DOI] [PubMed] [Google Scholar]
- 125.Jing X., Luan Z., Liu B. miR-558 Reduces the Damage of HBE Cells Exposed to Cigarette Smoke Extract by Targeting TNFRSF1A and Inactivating TAK1/MAPK/NF-κB Pathway. Immunol Invest. 2022;51:787–801. doi: 10.1080/08820139.2021.1874977. [DOI] [PubMed] [Google Scholar]
- 126.Hulina-Tomašković A., Rajković M.G., Somborac-Bačura A., Čeri A., Dabelić S., Rumora L. Extracellular Hsp70 modulates the inflammatory response of cigarette smoke extract in NCI-H292 cells. Exp Physiol. 2018;103:1704–1716. doi: 10.1113/EP087180. [DOI] [PubMed] [Google Scholar]
- 127.Chen H.W., Lii C.K., Ku H.J., Wang T.S. Cigarette smoke extract induces expression of cell adhesion molecules in HUVEC via actin filament reorganization. Environ Mol Mutagen. 2009;50:96–104. doi: 10.1002/em.20441. [DOI] [PubMed] [Google Scholar]
- 128.Low B., Liang M., Fu J. p38 mitogen-activated protein kinase mediates sidestream cigarette smoke-induced endothelial permeability. J Pharmacol Sci. 2007;104:225–231. doi: 10.1254/jphs.fp0070385. [DOI] [PubMed] [Google Scholar]
- 129.Raveendran M., Wang J., Senthil D., Wang J., Utama B., Shen Y., et al. Endogenous nitric oxide activation protects against cigarette smoking induced apoptosis in endothelial cells. FEBS Lett. 2005;579:733–740. doi: 10.1016/j.febslet.2004.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Giebe S., Hofmann A., Brux M., Lowe F., Breheny D., Morawietz H., et al. Comparative study of the effects of cigarette smoke versus next generation tobacco and nicotine product extracts on endothelial function. Redox Biol. 2021;47 doi: 10.1016/j.redox.2021.102150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ghosh A., Pechota A., Coleman D., Upchurch G.R., Jr., Eliason J.L. Cigarette smoke-induced MMP2 and MMP9 secretion from aortic vascular smooth cells is mediated via the Jak/Stat pathway. Hum Pathol. 2015;46:284–294. doi: 10.1016/j.humpath.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 132.Sharma S., Garg I., Ashraf M.Z. TLR signalling and association of TLR polymorphism with cardiovascular diseases. VascPharmacol. 2016;87:30–37. doi: 10.1016/j.vph.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 133.Barua R.S., Sharma M., Dileepan K.N. Cigarette Smoke Amplifies Inflammatory Response and Atherosclerosis Progression Through Activation of the H1R-TLR2/4-COX2 Axis. Front Immunol. 2015;6:572. doi: 10.3389/fimmu.2015.00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yeh H.Y., Hung S.H., Chen S.C., Guo F.R., Huang H.L., Peng J.K., et al. The Expression of Toll-Like Receptor 4 mRNA in PBMCs Is Upregulated in Smokers and Decreases Upon Smoking Cessation. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.667460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Higham A., Rattray N.J., Dewhurst J.A., Trivedi D.K., Fowler S.J., Goodacre R., et al. Electronic cigarette exposure triggers neutrophil inflammatory responses. Respir Res. 2016;17:56. doi: 10.1186/s12931-016-0368-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xu S., Chen H., Ni H., Dai Q. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway. Atherosclerosis. 2021;317:1–9. doi: 10.1016/j.atherosclerosis.2020.11.021. [DOI] [PubMed] [Google Scholar]
- 137.Liu X., Wang C.N., Qiu C.Y., Song W., Wang L.F., Liu B. Adipocytes promote nicotine-induced injury of endothelial cells via the NF-κB pathway. Exp Cell Res. 2017;359:251–256. doi: 10.1016/j.yexcr.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 138.Wang Z., Wu W., Tang M., Zhou Y., Wang L., Xu W., et al. NF-κB pathway mediates vascular smooth muscle response to nicotine. The international journal of biochemistry & cell biology. 2013;45:375–383. doi: 10.1016/j.biocel.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 139.Stokes A.C., Xie W., Wilson A.E., Yang H., Orimoloye O.A., Harlow A.F., et al. Association of Cigarette and Electronic Cigarette Use Patterns With Levels of Inflammatory and Oxidative Stress Biomarkers Among US Adults: Population Assessment of Tobacco and Health Study. Circulation. 2021;143:869–871. doi: 10.1161/CIRCULATIONAHA.120.051551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Herman M., Tarran R. E-cigarettes, nicotine, the lung and the brain: multi-level cascading pathophysiology. The Journal of physiology. 2020;598:5063–5071. doi: 10.1113/JP278388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.C.D. Fowler, J.R. Turner, M. Imad Damaj, Molecular Mechanisms Associated with Nicotine Pharmacology and Dependence, Handbook of experimental pharmacology, 258 (2020) 373-393. [DOI] [PubMed]
- 142.Picciotto M.R., Kenny P.J. Mechanisms of Nicotine Addiction. Cold Spring Harbor perspectives in medicine. 2021;11 doi: 10.1101/cshperspect.a039610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Deng J., Jiang H. Role of Nicotinic Acetylcholine Receptors in Cardiovascular Physiology and Pathophysiology: Current Trends and Perspectives. Curr Vasc Pharmacol. 2021;19:370–378. doi: 10.2174/1386207323666200917104920. [DOI] [PubMed] [Google Scholar]
- 144.Bertrand D., Lee C.H., Flood D., Marger F., Donnelly-Roberts D. Therapeutic Potential of α7 Nicotinic Acetylcholine Receptors. Pharmacol Rev. 2015;67:1025–1073. doi: 10.1124/pr.113.008581. [DOI] [PubMed] [Google Scholar]
- 145.Lee J., Cooke J.P. Nicotine and pathological angiogenesis. Life Sci. 2012;91:1058–1064. doi: 10.1016/j.lfs.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Scholze P., Orr-Urtreger A., Changeux J.P., McIntosh J.M., Huck S. Catecholamine outflow from mouse and rat brain slice preparations evoked by nicotinic acetylcholine receptor activation and electrical field stimulation. Br J Pharmacol. 2007;151:414–422. doi: 10.1038/sj.bjp.0707236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.van Berlo J.H., Maillet M., Molkentin J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Investig. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Le Y., Wang Y., Zhou L., Xiong J., Tian J., Yang X., et al. Cigarette smoke-induced HMGB1 translocation and release contribute to migration and NF-κB activation through inducing autophagy in lung macrophages. J Cell Mol Med. 2020;24:1319–1331. doi: 10.1111/jcmm.14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kwok H.H., Gao B., Chan K.H., Ip M.S., Minna J.D., Lam D.C. Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells. Cancers. 2021;13 doi: 10.3390/cancers13215345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yates S.L., Bencherif M., Fluhler E.N., Lippiello P.M. Up-regulation of nicotinic acetylcholine receptors following chronic exposure of rats to mainstream cigarette smoke or alpha 4 beta 2 receptors to nicotine. Biochem Pharmacol. 1995;50:2001–2008. doi: 10.1016/0006-2952(95)02100-0. [DOI] [PubMed] [Google Scholar]
- 151.Cano M., Reynaga D.D., Belluzzi J.D., Loughlin S.E., Leslie F. Chronic exposure to cigarette smoke extract upregulates nicotinic receptor binding in adult and adolescent rats. Neuropharmacology. 2020;181 doi: 10.1016/j.neuropharm.2020.108308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Machaalani R., Ghazavi E., Hinton T., Waters K.A., Hennessy A. Cigarette smoking during pregnancy regulates the expression of specific nicotinic acetylcholine receptor (nAChR) subunits in the human placenta. Toxicol Appl Pharmacol. 2014;276:204–212. doi: 10.1016/j.taap.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 153.Wang Q., Sundar I.K., Li D., Lucas J.H., Muthumalage T., McDonough S.R., et al. E-cigarette-induced pulmonary inflammation and dysregulated repair are mediated by nAChR α7 receptor: role of nAChR α7 in SARS-CoV-2 Covid-19 ACE2 receptor regulation. Respir Res. 2020;21:154. doi: 10.1186/s12931-020-01396-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alasmari F., Crotty Alexander L.E., Hammad A.M., Horton A., Alhaddad H., Schiefer I.T., et al. E-cigarette aerosols containing nicotine modulate nicotinic acetylcholine receptors and astroglial glutamate transporters in mesocorticolimbic brain regions of chronically exposed mice. Chem Biol Interact. 2021;333 doi: 10.1016/j.cbi.2020.109308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Baldassarri S.R., Hillmer A.T., Anderson J.M., Jatlow P., Nabulsi N., Labaree D., et al. Use of Electronic Cigarettes Leads to Significant Beta2-Nicotinic Acetylcholine Receptor Occupancy: Evidence From a PET Imaging Study. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco. 2018;20:425–433. doi: 10.1093/ntr/ntx091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Petsophonsakul P., Burgmaier M., Willems B., Heeneman S., Stadler N., Gremse F., et al. Nicotine promotes vascular calcification via intracellular Ca2+-mediated, Nox5-induced oxidative stress and extracellular vesicle release in vascular smooth muscle cells. Cardiovasc Res. 2021 doi: 10.1093/cvr/cvab244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang Z., Liu B., Zhu J., Wang D., Wang Y. Nicotine-mediated autophagy of vascular smooth muscle cell accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway. Atherosclerosis. 2019;284:1–10. doi: 10.1016/j.atherosclerosis.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 158.Jiang D.J., Jia S.J., Yan J., Zhou Z., Yuan Q., Li Y.J. Involvement of DDAH/ADMA/NOS pathway in nicotine-induced endothelial dysfunction. Biochem Biophys Res Commun. 2006;349:683–693. doi: 10.1016/j.bbrc.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 159.Dani J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int Rev Neurobiol. 2015;124:3–19. doi: 10.1016/bs.irn.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kabe Y., Ando K., Hirao S., Yoshida M., Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 162.Kairisalo M., Korhonen L., Blomgren K., Lindholm D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-kappaB activation. Biochem Biophys Res Commun. 2007;364:138–144. doi: 10.1016/j.bbrc.2007.09.115. [DOI] [PubMed] [Google Scholar]
- 163.Ashraf M.I., Ebner M., Wallner C., Haller M., Khalid S., Schwelberger H., et al. A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury. Cell communication and signaling : CCS. 2014;12:6. doi: 10.1186/1478-811X-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chakrabarti S., Visweswariah S.S. Intramacrophage ROS Primes the Innate Immune System via JAK/STAT and Toll Activation. Cell reports. 2020;33 doi: 10.1016/j.celrep.2020.108368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tschopp J., Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 166.Bai B., Yang Y., Wang Q., Li M., Tian C., Liu Y., et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11:776. doi: 10.1038/s41419-020-02985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Moon J.H., Kim S.Y., Lee H.G., Kim S.U., Lee Y.B. Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar beta amyloid peptide (1–42)-stimulated microglia. Exp Mol Med. 2008;40:11–18. doi: 10.3858/emm.2008.40.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Parada E., Buendia I., León R., Negredo P., Romero A., Cuadrado A., et al. Neuroprotective effect of melatonin against ischemia is partially mediated by alpha-7 nicotinic receptor modulation and HO-1 overexpression. J Pineal Res. 2014;56:204–212. doi: 10.1111/jpi.12113. [DOI] [PubMed] [Google Scholar]
- 169.Chang N.C., Yeh C.T., Lin Y.K., Kuo K.T., Fong I.H., Kounis N.G., et al. Garcinol Attenuates Lipoprotein(a)-Induced Oxidative Stress and Inflammatory Cytokine Production in Ventricular Cardiomyocyte through α7-Nicotinic Acetylcholine Receptor-Mediated Inhibition of the p38 MAPK and NF-κB Signaling Pathways, Antioxidants (Basel. Switzerland) 2021;10 doi: 10.3390/antiox10030461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ko H.K., Lee H.F., Lin A.H., Liu M.H., Liu C.I., Lee T.S., et al. Regulation of Cigarette Smoke Induction of IL-8 in Macrophages by AMP-activated Protein Kinase Signaling. J Cell Physiol. 2015;230:1781–1793. doi: 10.1002/jcp.24881. [DOI] [PubMed] [Google Scholar]
- 171.Saeed R.W., Varma S., Peng-Nemeroff T., Sherry B., Balakhaneh D., Huston J., et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201:1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yang C., Li Z., Yan S., He Y., Dai R., Leung G.P., et al. Role of the nicotinic acetylcholine receptor α3 subtype in vascular inflammation. Br J Pharmacol. 2016;173:3235–3247. doi: 10.1111/bph.13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li P., Yan Y., Shi Y., Cheng B., Zhan Y., Wang Q., et al. Nicotinic Agonist Inhibits Cardiomyocyte Apoptosis in CVB3-Induced Myocarditis via α3β4-nAChR/PI3K/Akt-Dependent Survivin Upregulation. Oxid Med Cell Longevity. 2019;2019:9496419. doi: 10.1155/2019/9496419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hung M.Y., Wu Y.H., Bamodu O.A., Chen X., Lin Y.K., Hu P., et al. Activation of the monocytic α7 nicotinic acetylcholine receptor modulates oxidative stress and inflammation-associated development of coronary artery spasm via a p38 MAP-kinase signaling-dependent pathway. Free Radical Biol Med. 2018;120:266–276. doi: 10.1016/j.freeradbiomed.2018.03.050. [DOI] [PubMed] [Google Scholar]
- 175.Lin Y.K., Yeh C.T., Kuo K.T., Fong I.H., Yadav V.K., Kounis N.G., et al. Apolipoprotein (a)/Lipoprotein(a)-Induced Oxidative-Inflammatory α7-nAChR/p38 MAPK/IL-6/RhoA-GTP Signaling Axis and M1 Macrophage Polarization Modulate Inflammation-Associated Development of Coronary Artery Spasm. Oxid Med Cell Longevity. 2022;2022:9964689. doi: 10.1155/2022/9964689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Apel K., Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol. 2004;55:373–399. doi: 10.1146/annurev.arplant.55.031903.141701. [DOI] [PubMed] [Google Scholar]
- 177.Nair N., Gongora E. Oxidative Stress and Cardiovascular Aging: Interaction Between NRF-2 and ADMA. Current cardiology reviews. 2017;13:183–188. doi: 10.2174/1573403X13666170216150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhou Y., Zhang M.J., Li B.H., Chen L., Pi Y., Yin Y.W., et al. PPARγ Inhibits VSMC Proliferation and Migration via Attenuating Oxidative Stress through Upregulating UCP2. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0154720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tsutsui H., Kinugawa S., Matsushima S. Oxidative stress and heart failure, American journal of physiology. Heart and circulatory physiology. 2011;301:H2181–H2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 180.Sheydina A., Riordon D.R., Boheler K.R. Molecular mechanisms of cardiomyocyte aging, Clinical science (London. England. 1979;121(2011):315–329. doi: 10.1042/CS20110115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Marrocco I., Altieri F., Peluso I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid Med Cell Longevity. 2017;2017:6501046. doi: 10.1155/2017/6501046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Njus D., Kelley P.M., Tu Y.J., Schlegel H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radical Biol Med. 2020;159:37–43. doi: 10.1016/j.freeradbiomed.2020.07.013. [DOI] [PubMed] [Google Scholar]
- 183.Sumanasekera W.K., Dao H.T., Shekhovtsova V., Schultz K., Jani M., Gyamfi F., et al. The mechanistic role of oxidative stress in cigarette smoke-induced cardiac stem cell dysfunction and prevention by ascorbic acid. Cell Biol Toxicol. 2019;35:111–127. doi: 10.1007/s10565-018-9437-x. [DOI] [PubMed] [Google Scholar]
- 184.Thakur N., Manna P., Das J. Synthesis and biomedical applications of nanoceria, a redox active nanoparticle. Journal of nanobiotechnology. 2019;17:84. doi: 10.1186/s12951-019-0516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Das M., Das D.K. Resveratrol and cardiovascular health. Mol Aspects Med. 2010;31:503–512. doi: 10.1016/j.mam.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 186.Arunachalam G., Yao H., Sundar I.K., Caito S., Rahman I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: Role of resveratrol. Biochem Biophys Res Commun. 2010;393:66–72. doi: 10.1016/j.bbrc.2010.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Al-Arifi M.N., Maayah Z.H., Alshamrani A.A., Korashy H.M. Impact of cigarette smoke exposure on the expression of cardiac hypertrophic genes, cytochrome P450 enzymes, and oxidative stress markers in rats. The Journal of toxicological sciences. 2012;37:1083–1090. doi: 10.2131/jts.37.1083. [DOI] [PubMed] [Google Scholar]
- 188.Das A., Dey N., Ghosh A., Das S., Chattopadhyay D.J., Chatterjee I.B. Molecular and cellular mechanisms of cigarette smoke-induced myocardial injury: prevention by vitamin C. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0044151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dikalov S., Itani H., Richmond B., Vergeade A., Rahman S.M.J., Boutaud O., et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension, American journal of physiology. Heart and circulatory physiology. 2019;316:H639–H646. doi: 10.1152/ajpheart.00595.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Carroll A.J., Huffman M.D., Zhao L., Jacobs D.R., Jr., Stewart J.C., Kiefe C.I., et al. Evaluating Longitudinal Associations Between Depressive Symptoms, Smoking, and Biomarkers of Cardiovascular Disease in the CARDIA Study. Psychosom Med. 2019;81:372–379. doi: 10.1097/PSY.0000000000000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mons U., Muscat J.E., Modesto J., Richie J.P., Jr., Brenner H. Effect of smoking reduction and cessation on the plasma levels of the oxidative stress biomarker glutathione–Post-hoc analysis of data from a smoking cessation trial. Free Radical Biol Med. 2016;91:172–177. doi: 10.1016/j.freeradbiomed.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Moheimani R.S., Bhetraratana M., Yin F., Peters K.M., Gornbein J., Araujo J.A., et al. Increased Cardiac Sympathetic Activity and Oxidative Stress in Habitual Electronic Cigarette Users: Implications for Cardiovascular Risk. JAMA cardiology. 2017;2:278–284. doi: 10.1001/jamacardio.2016.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chaumont M., de Becker B., Zaher W., Culié A., Deprez G., Mélot C., et al. Differential Effects of E-Cigarette on Microvascular Endothelial Function. Arterial Stiffness and Oxidative Stress: A Randomized Crossover Trial, Scientific reports. 2018;8:10378. doi: 10.1038/s41598-018-28723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Szostak J., Wong E.T., Titz B., Lee T., Wong S.K., Low T., et al. A 6-month systems toxicology inhalation study in ApoE(-/-) mice demonstrates reduced cardiovascular effects of E-vapor aerosols compared with cigarette smoke, American journal of physiology. Heart and circulatory physiology. 2020;318:H604–H631. doi: 10.1152/ajpheart.00613.2019. [DOI] [PubMed] [Google Scholar]
- 195.Kelesidis T., Tran E., Arastoo S., Lakhani K., Heymans R., Gornbein J., et al. Elevated Cellular Oxidative Stress in Circulating Immune Cells in Otherwise Healthy Young People Who Use Electronic Cigarettes in a Cross-Sectional Single-Center Study: Implications for Future Cardiovascular Risk. Journal of the American Heart Association. 2020;9 doi: 10.1161/JAHA.120.016983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhao T., Wu W., Sui L., Huang Q., Nan Y., Liu J., et al. Reactive oxygen species-based nanomaterials for the treatment of myocardial ischemia reperfusion injuries. Bioact Mater. 2022;7:47–72. doi: 10.1016/j.bioactmat.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chen L., Huang Q., Zhao T., Sui L., Wang S., Xiao Z., et al. Nanotherapies for sepsis by regulating inflammatory signals and reactive oxygen and nitrogen species: New insight for treating COVID-19. Redox Biol. 2021;45 doi: 10.1016/j.redox.2021.102046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.El Assar M., Angulo J., Rodríguez-Mañas L. Oxidative stress and vascular inflammation in aging. Free Radical Biol Med. 2013;65:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 199.Gimbrone M.A., Jr., García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Konukoglu D., Uzun H. Endothelial Dysfunction and Hypertension. Adv Exp Med Biol. 2017;956:511–540. doi: 10.1007/5584_2016_90. [DOI] [PubMed] [Google Scholar]
- 201.Shi Y., Vanhoutte P.M. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes. 2017;9:434–449. doi: 10.1111/1753-0407.12521. [DOI] [PubMed] [Google Scholar]
- 202.Zong D.D., Liu X.M., Li J.H., Ouyang R.Y., Long Y.J., Chen P., et al. Resveratrol attenuates cigarette smoke induced endothelial apoptosis by activating Notch1 signaling mediated autophagy. Respir Res. 2021;22:22. doi: 10.1186/s12931-021-01620-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Michaud S.E., Dussault S., Groleau J., Haddad P., Rivard A. Cigarette smoke exposure impairs VEGF-induced endothelial cell migration: role of NO and reactive oxygen species. J Mol Cell Cardiol. 2006;41:275–284. doi: 10.1016/j.yjmcc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 204.Zhao Z., Wang X., Zhang R., Ma B., Niu S., Di X., et al. Melatonin attenuates smoking-induced atherosclerosis by activating the Nrf2 pathway via NLRP3 inflammasomes in endothelial cells. Aging. 2021;13:11363–11380. doi: 10.18632/aging.202829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Anderson C., Majeste A., Hanus J., Wang S. E-Cigarette Aerosol Exposure Induces Reactive Oxygen Species. DNA Damage, and Cell Death in Vascular Endothelial Cells, Toxicological sciences : an official journal of the Society of Toxicology. 2016;154:332–340. doi: 10.1093/toxsci/kfw166. [DOI] [PubMed] [Google Scholar]
- 206.Putzhammer R., Doppler C., Jakschitz T., Heinz K., Förste J., Danzl K., et al. Vapours of US and EU Market Leader Electronic Cigarette Brands and Liquids Are Cytotoxic for Human Vascular Endothelial Cells. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0157337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hu C.T., Shao Y.D., Liu Y.Z., Xiao X., Cheng Z.B., Qu S.L., et al. Oxidative stress in vascular calcification. Clinica chimica acta; international journal of clinical chemistry. 2021;519:101–110. doi: 10.1016/j.cca.2021.04.012. [DOI] [PubMed] [Google Scholar]
- 208.Durham A.L., Speer M.Y., Scatena M., Giachelli C.M., Shanahan C.M. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590–600. doi: 10.1093/cvr/cvy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nishio E., Watanabe Y. Cigarette smoke extract is a modulator of mitogenic action in vascular smooth muscle cells. Life Sci. 1998;62:1339–1347. doi: 10.1016/s0024-3205(98)00068-x. [DOI] [PubMed] [Google Scholar]
- 210.Starke R.M., Thompson J.W., Ali M.S., Pascale C.L., Martinez Lege A., Ding D., et al. Cigarette Smoke Initiates Oxidative Stress-Induced Cellular Phenotypic Modulation Leading to Cerebral Aneurysm Pathogenesis. Arterioscler Thromb Vasc Biol. 2018;38:610–621. doi: 10.1161/ATVBAHA.117.310478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Guo T., Chai X., Liu Q., Peng W., Peng Z., Cai Y. Downregulation of P16 promotes cigarette smoke extract-induced vascular smooth muscle cell proliferation via preventing G1/S phase transition. Experimental and therapeutic medicine. 2017;14:214–220. doi: 10.3892/etm.2017.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yoshiyama S., Horinouchi T., Miwa S., Wang H.H., Kohama K., Nakamura A. Effect of cigarette smoke components on vascular smooth muscle cell migration toward platelet-derived growth factor BB. J Pharmacol Sci. 2011;115:532–535. doi: 10.1254/jphs.10283sc. [DOI] [PubMed] [Google Scholar]
- 213.Babic M., Schuchardt M., Tölle M., van der Giet M. In times of tobacco-free nicotine consumption: The influence of nicotine on vascular calcification. Eur J Clin Invest. 2019;49 doi: 10.1111/eci.13077. [DOI] [PubMed] [Google Scholar]
- 214.Cucina A., Sapienza P., Corvino V., Borrelli V., Mariani V., Randone B., et al. Nicotine-induced smooth muscle cell proliferation is mediated through bFGF and TGF-beta 1. Surgery. 2000;127:316–322. doi: 10.1067/msy.2000.104249. [DOI] [PubMed] [Google Scholar]
- 215.Di Luozzo G., Pradhan S., Dhadwal A.K., Chen A., Ueno H., Sumpio B.E. Nicotine induces mitogen-activated protein kinase dependent vascular smooth muscle cell migration. Atherosclerosis. 2005;178:271–277. doi: 10.1016/j.atherosclerosis.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 216.Yoshiyama S., Chen Z., Okagaki T., Kohama K., Nasu-Kawaharada R., Izumi T., et al. Nicotine exposure alters human vascular smooth muscle cell phenotype from a contractile to a synthetic type. Atherosclerosis. 2014;237:464–470. doi: 10.1016/j.atherosclerosis.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 217.Cuschieri J., Maier R.V. Oxidative stress, lipid rafts, and macrophage reprogramming. Antioxid Redox Signal. 2007;9:1485–1497. doi: 10.1089/ars.2007.1670. [DOI] [PubMed] [Google Scholar]
- 218.Yunna C., Mengru H., Lei W., Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877 doi: 10.1016/j.ejphar.2020.173090. [DOI] [PubMed] [Google Scholar]
- 219.Xu H., Jiang J., Chen W., Li W., Chen Z. Vascular Macrophages in Atherosclerosis. Journal of immunology research. 2019;2019:4354786. doi: 10.1155/2019/4354786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhang X., Wang L., Zhang H., Guo D., Zhao J., Qiao Z., et al. The effects of cigarette smoke extract on the endothelial production of soluble intercellular adhesion molecule-1 are mediated through macrophages, possibly by inducing TNF-alpha release. Methods Find Exp Clin Pharmacol. 2002;24:261–265. doi: 10.1358/mf.2002.24.5.802302. [DOI] [PubMed] [Google Scholar]
- 221.Ghosh A., Pechota L.V., Upchurch G.R., Jr., Eliason J.L. Cross-talk between macrophages, smooth muscle cells, and endothelial cells in response to cigarette smoke: the effects on MMP2 and 9. Mol Cell Biochem. 2015;410:75–84. doi: 10.1007/s11010-015-2539-3. [DOI] [PubMed] [Google Scholar]
- 222.Lombardi M., Mantione M.E., Baccellieri D., Ferrara D., Castellano R., Chiesa R., et al. P2X7 receptor antagonism modulates IL-1β and MMP9 in human atherosclerotic vessels. Sci Rep. 2017;7:4872. doi: 10.1038/s41598-017-05137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Momeni-Moghaddam M.A., Asadikaram G., Nematollahi M.H., Esmaeili Tarzi M., Faramarz-Gaznagh S., Mohammadpour-Gharehbagh A., et al. Effects of Cigarette Smoke and Opium on the Expression of CD9, CD36, and CD68 at mRNA and Protein Levels in Human Macrophage Cell Line THP-1, Iranian journal of allergy, asthma. and immunology. 2020;19:45–55. doi: 10.18502/ijaai.v19i1.2417. [DOI] [PubMed] [Google Scholar]
- 224.Li J., Huynh L., Cornwell W.D., Tang M.S., Simborio H., Huang J., et al. Electronic Cigarettes Induce Mitochondrial DNA Damage and Trigger TLR9 (Toll-Like Receptor 9)-Mediated Atherosclerosis. Arterioscler Thromb Vasc Biol. 2021;41:839–853. doi: 10.1161/ATVBAHA.120.315556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhou M.S., Chadipiralla K., Mendez A.J., Jaimes E.A., Silverstein R.L., Webster K., et al. Nicotine potentiates proatherogenic effects of oxLDL by stimulating and upregulating macrophage CD36 signaling, American journal of physiology. Heart and circulatory physiology. 2013;305:H563–H574. doi: 10.1152/ajpheart.00042.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Langenickel T.H., Olive M., Boehm M., San H., Crook M.F., Nabel E.G. KIS protects against adverse vascular remodeling by opposing stathmin-mediated VSMC migration in mice. J Clin Investig. 2008;118:3848–3859. doi: 10.1172/JCI33206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.V.J. Dzau, G.H. Gibbons, Endothelium and growth factors in vascular remodeling of hypertension, Hypertension (Dallas, Tex. : 1979), 18 (1991) Iii115-121. [DOI] [PubMed]
- 228.Ma Z., Mao C., Jia Y., Fu Y., Kong W. Extracellular matrix dynamics in vascular remodeling. Am J Physiol Cell Physiol. 2020;319:C481–C499. doi: 10.1152/ajpcell.00147.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Humphrey J.D. Mechanisms of Vascular Remodeling in Hypertension. Am J Hypertens. 2021;34:432–441. doi: 10.1093/ajh/hpaa195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Dai Z., Zhu M.M., Peng Y., Jin H., Machireddy N., Qian Z., et al. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am J Respir Crit Care Med. 2018;198:788–802. doi: 10.1164/rccm.201709-1835OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Farra Y.M., Matz J., Ramkhelawon B., Oakes J.M., Bellini C. Structural and functional remodeling of the female Apoe(-/-) mouse aorta due to chronic cigarette smoke exposure, American journal of physiology. Heart and circulatory physiology. 2021;320:H2270–H2282. doi: 10.1152/ajpheart.00893.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schroeter M.R., Sawalich M., Humboldt T., Leifheit M., Meurrens K., Berges A., et al. Cigarette smoke exposure promotes arterial thrombosis and vessel remodeling after vascular injury in apolipoprotein E-deficient mice. J Vasc Res. 2008;45:480–492. doi: 10.1159/000127439. [DOI] [PubMed] [Google Scholar]
- 233.Li Q., Wu J., Xu Y., Liu L., Xie J. Role of RASEF hypermethylation in cigarette smoke-induced pulmonary arterial smooth muscle remodeling. Respir Res. 2019;20:52. doi: 10.1186/s12931-019-1014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Alani A., Luo Y., Nakanishim R., Matsumoto S., Budoff M.J. Coronary artery-positive remodeling in current smokers. Coron Artery Dis. 2018;29:17–22. doi: 10.1097/MCA.0000000000000555. [DOI] [PubMed] [Google Scholar]
- 235.Colombo E.S., Davis J., Makvandi M., Aragon M., Lucas S.N., Paffett M.L., et al. Effects of nicotine on cardiovascular remodeling in a mouse model of systemic hypertension. Cardiovasc Toxicol. 2013;13:364–369. doi: 10.1007/s12012-013-9217-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Furmanik M., Chatrou M., van Gorp R., Akbulut A., Willems B., Schmidt H., et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ Res. 2020;127:911–927. doi: 10.1161/CIRCRESAHA.119.316159. [DOI] [PubMed] [Google Scholar]
- 237.Lee S.J., Lee I.K., Jeon J.H. Vascular Calcification-New Insights Into Its Mechanism. Int J Mol Sci. 2020;21 doi: 10.3390/ijms21082685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Leopold J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. 2015;25:267–274. doi: 10.1016/j.tcm.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Al Hariri M., Zibara K., Farhat W., Hashem Y., Soudani N., Al Ibrahim F., et al. Cigarette Smoking-Induced Cardiac Hypertrophy, Vascular Inflammation and Injury Are Attenuated by Antioxidant Supplementation in an Animal Model. Front Pharmacol. 2016;7:397. doi: 10.3389/fphar.2016.00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.McEvoy J.W., Nasir K., DeFilippis A.P., Lima J.A., Bluemke D.A., Hundley W.G., et al. Relationship of cigarette smoking with inflammation and subclinical vascular disease: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:1002–1010. doi: 10.1161/ATVBAHA.114.304960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Oshunbade A.A., Kassahun-Yimer W., Valle K.A., Hamid A., Kipchumba R.K., Kamimura D., et al. Cigarette Smoking. Incident Coronary Heart Disease, and Coronary Artery Calcification in Black Adults: The Jackson Heart Study, Journal of the American Heart Association. 2021;10 doi: 10.1161/JAHA.120.017320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gać P., Jaźwiec P., Mazur G., Poręba R. Exposure to Cigarette Smoke and the Carotid Arteries Calcification Index in Patients with Essential Hypertension. Cardiovasc Toxicol. 2017;17:335–343. doi: 10.1007/s12012-016-9391-x. [DOI] [PubMed] [Google Scholar]
- 243.Navas-Acien A., Martinez-Morata I., Hilpert M., Rule A., Shimbo D., LoIacono N.J. Early Cardiovascular Risk in E-cigarette Users: the Potential Role of Metals. Current environmental health reports. 2020;7:353–361. doi: 10.1007/s40572-020-00297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chen Y., Zhao X., Wu H. Arterial Stiffness: A Focus on Vascular Calcification and Its Link to Bone Mineralization. Arterioscler Thromb Vasc Biol. 2020;40:1078–1093. doi: 10.1161/ATVBAHA.120.313131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ikonomidis I., Vlastos D., Kourea K., Kostelli G., Varoudi M., Pavlidis G., et al. Electronic Cigarette Smoking Increases Arterial Stiffness and Oxidative Stress to a Lesser Extent Than a Single Conventional Cigarette: An Acute and Chronic Study. Circulation. 2018;137:303–306. doi: 10.1161/CIRCULATIONAHA.117.029153. [DOI] [PubMed] [Google Scholar]
- 246.Puddu P., Puddu G.M., Cravero E., De Pascalis S., Muscari A. The emerging role of cardiovascular risk factor-induced mitochondrial dysfunction in atherogenesis. J Biomed Sci. 2009;16:112. doi: 10.1186/1423-0127-16-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Di Francesco V., Gurgone D., Palomba R., Ferreira M., Catelani T., Cervadoro A., et al. Modulating Lipoprotein Transcellular Transport and Atherosclerotic Plaque Formation in ApoE(-/-) Mice via Nanoformulated Lipid-Methotrexate Conjugates. ACS Appl Mater Interfaces. 2020;12:37943–37956. doi: 10.1021/acsami.0c12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Liu Z., Zhu H., Dai X., Wang C., Ding Y., Song P., et al. Macrophage Liver Kinase B1 Inhibits Foam Cell Formation and Atherosclerosis. Circ Res. 2017;121:1047–1057. doi: 10.1161/CIRCRESAHA.117.311546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Libby P., Ridker P.M., Hansson G.K. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 250.Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–533. doi: 10.1038/s41586-021-03392-8. [DOI] [PubMed] [Google Scholar]
- 251.von Holt K., Lebrun S., Stinn W., Conroy L., Wallerath T., Schleef R. Progression of atherosclerosis in the Apo E-/- model: 12-month exposure to cigarette mainstream smoke combined with high-cholesterol/fat diet. Atherosclerosis. 2009;205:135–143. doi: 10.1016/j.atherosclerosis.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 252.Gairola C.G., Drawdy M.L., Block A.E., Daugherty A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein E-/- mice. Atherosclerosis. 2001;156:49–55. doi: 10.1016/s0021-9150(00)00621-3. [DOI] [PubMed] [Google Scholar]
- 253.Kiriyama H., Kaneko H., Itoh H., Yoshida Y., Nakanishi K., Mizuno Y., et al. Effect of cigarette smoking on carotid artery atherosclerosis: a community-based cohort study. Heart Vessels. 2020;35:22–29. doi: 10.1007/s00380-019-01455-5. [DOI] [PubMed] [Google Scholar]
- 254.Howard G., Wagenknecht L.E., Burke G.L., Diez-Roux A., Evans G.W., McGovern P., et al. Cigarette smoking and progression of atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Study. JAMA. 1998;279:119–124. doi: 10.1001/jama.279.2.119. [DOI] [PubMed] [Google Scholar]
- 255.Espinoza-Derout J., Hasan K.M., Shao X.M., Jordan M.C., Sims C., Lee D.L., et al. Chronic intermittent electronic cigarette exposure induces cardiac dysfunction and atherosclerosis in apolipoprotein-E knockout mice, American journal of physiology. Heart and circulatory physiology. 2019;317:H445–H459. doi: 10.1152/ajpheart.00738.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Biondi-Zoccai G., Sciarretta S., Bullen C., Nocella C., Violi F., Loffredo L., et al. Acute Effects of Heat-Not-Burn, Electronic Vaping, and Traditional Tobacco Combustion Cigarettes: The Sapienza University of Rome-Vascular Assessment of Proatherosclerotic Effects of Smoking (SUR - VAPES) 2 Randomized Trial. Journal of the American Heart Association. 2019;8 doi: 10.1161/JAHA.118.010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Purnomo Y., Piccart Y., Coenen T., Prihadi J.S., Lijnen P.J. Oxidative stress and transforming growth factor-β1-induced cardiac fibrosis. Cardiovasc Hematol Disord: Drug Targets. 2013;13:165–172. doi: 10.2174/1871529x11313020010. [DOI] [PubMed] [Google Scholar]
- 258.Travers J.G., Kamal F.A., Robbins J., Yutzey K.E., Blaxall B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Krenning G., Zeisberg E.M., Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Huang Q., Li C.D., Yang Y.R., Qin X.F., Wang J.J., Zhang X., et al. Role of the IL-33/ST2 axis in cigarette smoke-induced airways remodelling in chronic obstructive pulmonary disease. Thorax. 2021 doi: 10.1136/thoraxjnl-2020-214712. [DOI] [PubMed] [Google Scholar]
- 261.Vang A., da Silva Gonçalves D., Bos A., Fernandez-Nicolas P., Zhang A.R., Morrison T.J.M., et al. α7 Nicotinic acetylcholine receptor mediates right ventricular fibrosis and diastolic dysfunction in pulmonary hypertension. JCI insight. 2021;6 doi: 10.1172/jci.insight.142945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Vang A., Clements R.T., Chichger H., Kue N., Allawzi A., O'Connell K., et al. Effect of α7 nicotinic acetylcholine receptor activation on cardiac fibroblasts: a mechanism underlying RV fibrosis associated with cigarette smoke exposure, American journal of physiology. Lung cellular and molecular physiology. 2017;312:L748–L759. doi: 10.1152/ajplung.00393.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Helske S., Syväranta S., Kupari M., Lappalainen J., Laine M., Lommi J., et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–1504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- 264.Chuang T.D., Ansari A., Yu C., Sakurai R., Harb A., Liu J., et al. Mechanism underlying increased cardiac extracellular matrix deposition in perinatal nicotine-exposed offspring, American journal of physiology. Heart and circulatory physiology. 2020;319:H651–H660. doi: 10.1152/ajpheart.00021.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Feuerstein G.Z., Young P.R. Apoptosis in cardiac diseases: stress- and mitogen-activated signaling pathways. Cardiovasc Res. 2000;45:560–569. doi: 10.1016/s0008-6363(99)00372-7. [DOI] [PubMed] [Google Scholar]
- 266.Xiang M., Lu Y., Xin L., Gao J., Shang C., Jiang Z., et al. Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments. Oxid Med Cell Longevity. 2021;2021:6614009. doi: 10.1155/2021/6614009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Adikesavan G., Vinayagam M.M., Abdulrahman L.A., Chinnasamy T. (-)-Epigallocatechin-gallate (EGCG) stabilize the mitochondrial enzymes and inhibits the apoptosis in cigarette smoke-induced myocardial dysfunction in rats. Mol Biol Rep. 2013;40:6533–6545. doi: 10.1007/s11033-013-2673-5. [DOI] [PubMed] [Google Scholar]
- 268.Wang S., Chen X., Zeng B., Xu X., Chen H., Zhao P., et al. Knockout of macrophage migration inhibitory factor accentuates side-stream smoke exposure-induced myocardial contractile dysfunction through dysregulated mitophagy. Pharmacol Res. 2020;157 doi: 10.1016/j.phrs.2020.104828. [DOI] [PubMed] [Google Scholar]
- 269.Basma H., Tatineni S., Dhar K., Qiu F., Rennard S., Lowes B.D. Electronic cigarette extract induced toxic effect in iPS-derived cardiomyocytes. BMC cardiovascular disorders. 2020;20:357. doi: 10.1186/s12872-020-01629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Hasan K.M., Munoz A., Tumoyan H., Parveen M., Espinoza-Derout J., Shao X.M., et al. Adverse effects of fetal exposure of electronic-cigarettes and high-fat diet on male neonatal hearts. Exp Mol Pathol. 2021;118 doi: 10.1016/j.yexmp.2020.104573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Huang C., Guo X., Zhao H., An R., Lian K., Zhang X., et al. Nicotine induces H9C2 cell apoptosis via Akt protein degradation. Mol Med Rep. 2017;16:6269–6275. doi: 10.3892/mmr.2017.7331. [DOI] [PubMed] [Google Scholar]
- 272.Zhou X., Sheng Y., Yang R., Kong X. Nicotine promotes cardiomyocyte apoptosis via oxidative stress and altered apoptosis-related gene expression. Cardiology. 2010;115:243–250. doi: 10.1159/000301278. [DOI] [PubMed] [Google Scholar]
- 273.Bourlaud I., Underner M., Perault M.C., Patte F. Pharmacology of nicotine. Rev Mal Respir. 1992;9:367–374. [PubMed] [Google Scholar]
- 274.Zimmer H.G. Catecholamine-induced cardiac hypertrophy: significance of proto-oncogene expression. Journal of molecular medicine (Berlin, Germany) 1997;75:849–859. doi: 10.1007/s001090050176. [DOI] [PubMed] [Google Scholar]
- 275.Kayki Mutlu G., Arioglu Inan E., Karaomerlioglu I., Altan V.M., Yersal N., Korkusuz P., et al. Role of the β(3)-adrenergic receptor subtype in catecholamine-induced myocardial remodeling. Mol Cell Biochem. 2018;446:149–160. doi: 10.1007/s11010-018-3282-3. [DOI] [PubMed] [Google Scholar]
- 276.Lowery C.L., 3rd, Woulfe D., Kilic F. Responses of Plasma Catecholamine. Serotonin, and the Platelet Serotonin Transporter to Cigarette Smoking, Frontiers in neuroscience. 2019;13:32. doi: 10.3389/fnins.2019.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Lowery C.L., 3rd, Elliott C., Cooper A., Hadden C., Sonon R.N., Azadi P., et al. Cigarette Smoking-Associated Alterations in Serotonin/Adrenalin Signaling Pathways of Platelets. Journal of the American Heart Association. 2017;6 doi: 10.1161/JAHA.116.005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Laustiola K.E., Lassila R., Kaprio J., Koskenvuo M. Decreased beta-adrenergic receptor density and catecholamine response in male cigarette smokers. A study of monozygotic twin pairs discordant for smoking, Circulation. 1988;78:1234–1240. doi: 10.1161/01.cir.78.5.1234. [DOI] [PubMed] [Google Scholar]
- 279.Carruthers M. Modification of the noradrenaline related effects of smoking by beta-blockade. Psychol Med. 1976;6:251–256. doi: 10.1017/s0033291700013799. [DOI] [PubMed] [Google Scholar]
- 280.Benowitz N.L., St Helen G., Nardone N., Addo N., Zhang J.J., Harvanko A.M., et al. Twenty-Four-Hour Cardiovascular Effects of Electronic Cigarettes Compared With Cigarette Smoking in Dual Users. Journal of the American Heart Association. 2020;9 doi: 10.1161/JAHA.120.017317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Jackler R.K., Ramamurthi D. Nicotine arms race: JUUL and the high-nicotine product market. Tobacco control. 2019;28:623–628. doi: 10.1136/tobaccocontrol-2018-054796. [DOI] [PubMed] [Google Scholar]
- 282.Nagpal V., Rai R., Place A.T., Murphy S.B., Verma S.K., Ghosh A.K., et al. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation. 2016;133:291–301. doi: 10.1161/CIRCULATIONAHA.115.018174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Baudino T.A., Carver W., Giles W., Borg T.K. Cardiac fibroblasts: friend or foe?, American journal of physiology. Heart and circulatory physiology. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 284.Schipke J., Brandenberger C., Rajces A., Manninger M., Alogna A., Post H., et al. Assessment of cardiac fibrosis: a morphometric method comparison for collagen quantification, Journal of applied physiology (Bethesda. Md. 1985;122(2017):1019–1030. doi: 10.1152/japplphysiol.00987.2016. [DOI] [PubMed] [Google Scholar]
- 285.Hu N., Han X., Lane E.K., Gao F., Zhang Y., Ren J. Cardiac-specific overexpression of metallothionein rescues against cigarette smoking exposure-induced myocardial contractile and mitochondrial damage. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0057151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wu J.P., Chang-Lee S.N., Day C.H., Ho T.J., Viswanadha V.P., Chung L.C., et al. Secondhand Smoke Exposure Enhances Cardiac Fibrosis Effects on the Aging Rat Hearts. Acta Cardiologica Sinica. 2016;32:594–603. doi: 10.6515/ACS20150824C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Goette A., Lendeckel U., Kuchenbecker A., Bukowska A., Peters B., Klein H.U., et al. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart (British Cardiac Society) 2007;93:1056–1063. doi: 10.1136/hrt.2005.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Shi H., Fan X., Horton A., Haller S.T., Kennedy D.J., Schiefer I.T., et al. The Effect of Electronic-Cigarette Vaping on Cardiac Function and Angiogenesis in Mice. Sci Rep. 2019;9:4085. doi: 10.1038/s41598-019-40847-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Crotty Alexander L.E., Drummond C.A., Hepokoski M., Mathew D., Moshensky A., Willeford A., et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am J Physiol Regul Integr Comp Physiol. 2018;314:R834–R847. doi: 10.1152/ajpregu.00270.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Mayyas F., Aldawod H., Alzoubi K.H., Khabour O., Shihadeh A., Eissenberg T. Comparison of the cardiac effects of electronic cigarette aerosol exposure with waterpipe and combustible cigarette smoke exposure in rats. Life Sci. 2020;251 doi: 10.1016/j.lfs.2020.117644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Konstam M.A., Kramer D.G., Patel A.R., Maron M.S., Udelson J.E. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovascular imaging. 2011;4:98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 292.Zeisberg E.M., Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107:1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Burchfield J.S., Xie M., Hill J.A. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Bhatt A.S., Ambrosy A.P., Velazquez E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Current cardiology reports. 2017;19:71. doi: 10.1007/s11886-017-0876-4. [DOI] [PubMed] [Google Scholar]
- 295.Franco V. Right ventricular remodeling in pulmonary hypertension. Heart failure clinics. 2012;8:403–412. doi: 10.1016/j.hfc.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 296.Scherrer-Crosbie M., Kurtz B. Ventricular remodeling and function: insights using murine echocardiography. J Mol Cell Cardiol. 2010;48:512–517. doi: 10.1016/j.yjmcc.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Gu L., Pandey V., Geenen D.L., Chowdhury S.A., Piano M.R. Cigarette smoke-induced left ventricular remodelling is associated with activation of mitogen-activated protein kinases. Eur J Heart Fail. 2008;10:1057–1064. doi: 10.1016/j.ejheart.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Bocalini D.S., da Silva Luiz R., Silva K.A.S., Serra A.J., Avila R.A., Leopoldo A.S., et al. Short-Term Cigarette Smoking in Rats Impairs Physical Capacity and Induces Cardiac Remodeling. Biomed Res Int. 2020, 2020,:2589892. doi: 10.1155/2020/2589892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Liang Y., Li X., Zhang Y., Yeung S.C., Zhen Z., Ip M.S.M., et al. Induced Pluripotent Stem Cells-Derived Mesenchymal Stem Cells Attenuate Cigarette Smoke-Induced Cardiac Remodeling and Dysfunction. Front Pharmacol. 2017;8:501. doi: 10.3389/fphar.2017.00501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Azevedo P.S., Polegato B.F., Paiva S., Costa N., Santos P., Bazan S., et al. The role of glucose metabolism and insulin resistance in cardiac remodelling induced by cigarette smoke exposure. J Cell Mol Med. 2021;25:1314–1318. doi: 10.1111/jcmm.16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Nadruz W., Jr., Claggett B., Gonçalves A., Querejeta-Roca G., Fernandes-Silva M.M., Shah A.M., et al. Smoking and Cardiac Structure and Function in the Elderly: The ARIC Study (Atherosclerosis Risk in Communities) Circulation Cardiovascular imaging. 2016;9 doi: 10.1161/CIRCIMAGING.116.004950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hasan K.M., Friedman T.C., Parveen M., Espinoza-Derout J., Bautista F., Razipour M.M., et al. Electronic cigarettes cause alteration in cardiac structure and function in diet-induced obese mice. PLoS ONE. 2020;15 doi: 10.1371/journal.pone.0239671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.J.M. Oakes, J. Xu, T.M. Morris, N.D. Fried, C.S. Pearson, T.D. Lobell, et al., Effects of Chronic Nicotine Inhalation on Systemic and Pulmonary Blood Pressure and Right Ventricular Remodeling in Mice, Hypertension (Dallas, Tex. : 1979), 75 (2020) 1305-1314. [DOI] [PMC free article] [PubMed]
- 304.Fried N.D., Morris T.M., Whitehead A., Lazartigues E., Yue X., Gardner J.D. Angiotensin II type 1 receptor mediates pulmonary hypertension and right ventricular remodeling induced by inhaled nicotine, American journal of physiology. Heart and circulatory physiology. 2021;320:H1526–H1534. doi: 10.1152/ajpheart.00883.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Zierhut W., Zimmer H.G. Significance of myocardial alpha- and beta-adrenoceptors in catecholamine-induced cardiac hypertrophy. Circ Res. 1989;65:1417–1425. doi: 10.1161/01.res.65.5.1417. [DOI] [PubMed] [Google Scholar]
- 306.Samak M., Fatullayev J., Sabashnikov A., Zeriouh M., Schmack B., Farag M., et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med Sci Monit Basic Res. 2016;22:75–79. doi: 10.12659/MSMBR.900437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Baudouy D., Michiels J.F., Vukolic A., Wagner K.D., Wagner N. Echocardiographic and Histological Examination of Cardiac Morphology in the Mouse. Journal of visualized experiments : JoVE. 2017 doi: 10.3791/55843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Meurrens K., Ruf S., Ross G., Schleef R., von Holt K., Schlüter K.D. Smoking accelerates the progression of hypertension-induced myocardial hypertrophy to heart failure in spontaneously hypertensive rats. Cardiovasc Res. 2007;76:311–322. doi: 10.1016/j.cardiores.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 309.Talukder M.A., Johnson W.M., Varadharaj S., Lian J., Kearns P.N., El-Mahdy M.A., et al. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice, American journal of physiology. Heart and circulatory physiology. 2011;300:H388–H396. doi: 10.1152/ajpheart.00868.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Dubois-Deruy E., Rémy G., Alard J., Kervoaze G., Chwastyniak M., Baron M., et al. Modelling the Impact of Chronic Cigarette Smoke Exposure in Obese Mice: Metabolic, Pulmonary, Intestinal, and Cardiac Issues. Nutrients. 2020;12 doi: 10.3390/nu12030827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Kamimura D., Cain L.R., Mentz R.J., White W.B., Blaha M.J., DeFilippis A.P., et al. Cigarette Smoking and Incident Heart Failure: Insights From the Jackson Heart Study. Circulation. 2018;137:2572–2582. doi: 10.1161/CIRCULATIONAHA.117.031912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.El-Mahdy M.A., Mahgoup E.M., Ewees M.G., Eid M.S., Abdelghany T.M., Zweier J.L. Long-term electronic cigarette exposure induces cardiovascular dysfunction similar to tobacco cigarettes: role of nicotine and exposure duration, American journal of physiology. Heart and circulatory physiology. 2021;320:H2112–H2129. doi: 10.1152/ajpheart.00997.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Bolinder G., Alfredsson L., Englund A., de Faire U. Smokeless tobacco use and increased cardiovascular mortality among Swedish construction workers. Am J Public Health. 1994;84:399–404. doi: 10.2105/ajph.84.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Baba S., Iso H., Mannami T., Sasaki S., Okada K., Konishi M. Cigarette smoking and risk of coronary heart disease incidence among middle-aged Japanese men and women: the JPHC Study Cohort I, European journal of cardiovascular prevention and rehabilitation : official journal of the European Society of Cardiology, Working Groups on Epidemiology & Prevention and Cardiac Rehabilitation and Exercise. Physiology. 2006;13:207–213. doi: 10.1097/01.hjr.0000194417.16638.3d. [DOI] [PubMed] [Google Scholar]
- 315.Nyboe J., Jensen G., Appleyard M., Schnohr P. Smoking and the risk of first acute myocardial infarction. Am Heart J. 1991;122:438–447. doi: 10.1016/0002-8703(91)90997-v. [DOI] [PubMed] [Google Scholar]
- 316.Rose G., Hamilton P.J., Colwell L., Shipley M.J. A randomised controlled trial of anti-smoking advice: 10-year results. J Epidemiol Community Health. 1982;36:102–108. doi: 10.1136/jech.36.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Price J.F., Mowbray P.I., Lee A.J., Rumley A., Lowe G.D., Fowkes F.G. Relationship between smoking and cardiovascular risk factors in the development of peripheral arterial disease and coronary artery disease: Edinburgh Artery Study. Eur Heart J. 1999;20:344–353. doi: 10.1053/euhj.1998.1194. [DOI] [PubMed] [Google Scholar]
- 318.Nadler J.L., Velasco J.S., Horton R. Cigarette smoking inhibits prostacyclin formation. Lancet (London, England) 1983;1:1248–1250. doi: 10.1016/s0140-6736(83)92698-3. [DOI] [PubMed] [Google Scholar]
- 319.Minami J., Ishimitsu T., Matsuoka H. Effects of smoking cessation on blood pressure and heart rate variability in habitual smokers, Hypertension (Dallas. Tex. 1979;33(1999):586–590. doi: 10.1161/01.hyp.33.1.586. [DOI] [PubMed] [Google Scholar]
- 320.Bromfield S.G., Shimbo D., Booth J.N., 3rd, Correa A., Ogedegbe G., Carson A.P., et al. Cardiovascular Risk Factors and Masked Hypertension: The Jackson Heart Study, Hypertension (Dallas. Tex. 1979;68(2016):1475–1482. doi: 10.1161/HYPERTENSIONAHA.116.08308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Zhang D.Y., Huang J.F., Kang Y.Y., Dou Y., Su Y.L., Zhang L.J., et al. The prevalence of masked hypertension in relation to cigarette smoking in a Chinese male population. J Hypertens. 2020;38:1056–1063. doi: 10.1097/HJH.0000000000002392. [DOI] [PubMed] [Google Scholar]
- 322.Bowman T.S., Gaziano J.M., Buring J.E., Sesso H.D. A prospective study of cigarette smoking and risk of incident hypertension in women. J Am Coll Cardiol. 2007;50:2085–2092. doi: 10.1016/j.jacc.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 323.Verdecchia P., Schillaci G., Borgioni C., Ciucci A., Zampi I., Battistelli M., et al. Cigarette smoking, ambulatory blood pressure and cardiac hypertrophy in essential hypertension. J Hypertens. 1995;13:1209–1215. doi: 10.1097/00004872-199510000-00016. [DOI] [PubMed] [Google Scholar]
- 324.Wilhelmsson C., Vedin J.A., Elmfeldt D., Tibblin G., Wilhelmsen L. Smoking and myocardial infarction. Lancet (London, England) 1975;1:415–420. doi: 10.1016/s0140-6736(75)91488-9. [DOI] [PubMed] [Google Scholar]
- 325.Slone D., Shapiro S., Rosenberg L., Kaufman D.W., Hartz S.C., Rossi A.C., et al. Relation of cigarette smoking to myocardial infarction in young women. The New England journal of medicine. 1978;298:1273–1276. doi: 10.1056/NEJM197806082982302. [DOI] [PubMed] [Google Scholar]
- 326.Parish S., Collins R., Peto R., Youngman L., Barton J., Jayne K., et al. Cigarette smoking, tar yields, and non-fatal myocardial infarction: 14,000 cases and 32,000 controls in the United Kingdom. The International Studies of Infarct Survival (ISIS) Collaborators. BMJ (Clinical research ed) 1995;311:471–477. doi: 10.1136/bmj.311.7003.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Prescott E., Hippe M., Schnohr P., Hein H.O., Vestbo J. Smoking and risk of myocardial infarction in women and men: longitudinal population study. BMJ (Clinical research ed) 1998;316:1043–1047. doi: 10.1136/bmj.316.7137.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Sargent R.P., Shepard R.M., Glantz S.A. Reduced incidence of admissions for myocardial infarction associated with public smoking ban: before and after study. BMJ (Clinical research ed) 2004;328:977–980. doi: 10.1136/bmj.38055.715683.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Nazzal C., Harris J.E. Lower incidence of myocardial infarction after smoke-free legislation enforcement in Chile. Bull World Health Organ. 2017;95:674–682. doi: 10.2471/BLT.16.189894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Iversen B., Jacobsen B.K., Løchen M.L. Active and passive smoking and the risk of myocardial infarction in 24,968 men and women during 11 year of follow-up: the Tromsø Study. Eur J Epidemiol. 2013;28:659–667. doi: 10.1007/s10654-013-9785-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Martinez-Morata I., Sanchez T.R., Shimbo D., Navas-Acien A. Electronic Cigarette Use and Blood Pressure Endpoints: a Systematic Review. Curr Hypertens Rep. 2020;23:2. doi: 10.1007/s11906-020-01119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Chaumont M., Tagliatti V., Channan E.M., Colet J.M., Bernard A., Morra S., et al. Short halt in vaping modifies cardiorespiratory parameters and urine metabolome: a randomized trial, American journal of physiology. Lung cellular and molecular physiology. 2020;318:L331–L344. doi: 10.1152/ajplung.00268.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Gonzalez J.E., Cooke W.H. Acute effects of electronic cigarettes on arterial pressure and peripheral sympathetic activity in young nonsmokers, American journal of physiology. Heart and circulatory physiology. 2021;320:H248–H255. doi: 10.1152/ajpheart.00448.2020. [DOI] [PubMed] [Google Scholar]
- 334.Alzahrani T., Pena I., Temesgen N., Glantz S.A. Association Between Electronic Cigarette Use and Myocardial Infarction. Am J Prev Med. 2018;55:455–461. doi: 10.1016/j.amepre.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.K.E. Farsalinos, R. Polosa, F. Cibella, R. Niaura, Is e-cigarette use associated with coronary heart disease and myocardial infarction? Insights from the 2016 and 2017 National Health Interview Surveys, Therapeutic advances in chronic disease, 10 (2019) 2040622319877741. [DOI] [PMC free article] [PubMed]
- 336.George J., Hussain M., Vadiveloo T., Ireland S., Hopkinson P., Struthers A.D., et al. Cardiovascular Effects of Switching From Tobacco Cigarettes to Electronic Cigarettes. J Am Coll Cardiol. 2019;74:3112–3120. doi: 10.1016/j.jacc.2019.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Osei A.D., Mirbolouk M., Orimoloye O.A., Dzaye O., Uddin S.M.I., Benjamin E.J., et al. Association Between E-Cigarette Use and Cardiovascular Disease Among Never and Current Combustible-Cigarette Smokers. The American journal of medicine. 2019;132:949–954.e942. doi: 10.1016/j.amjmed.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 338.Benowitz N.L., Fraiman J.B. Cardiovascular effects of electronic cigarettes, Nature reviews. Cardiology. 2017;14:447–456. doi: 10.1038/nrcardio.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Farsalinos K.E. Acute vs. chronic effects of e-cigarettes on vascular function. Eur Heart J. 2020;41:1525. doi: 10.1093/eurheartj/ehaa073. [DOI] [PubMed] [Google Scholar]
- 340.Callahan-Lyon P. Electronic cigarettes: human health effects. Tobacco control. 2014;23 Suppl 2:ii36-40. doi: 10.1136/tobaccocontrol-2013-051470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.M. Mirbolouk, P. Charkhchi, S. Kianoush, S.M.I. Uddin, O.A. Orimoloye, R. Jaber, et al., Prevalence and Distribution of E-Cigarette Use Among U.S. Adults: Behavioral Risk Factor Surveillance System, 2016, Annals of internal medicine, 169 (2018) 429-438. [DOI] [PMC free article] [PubMed]
- 342.Kalkhoran S., Glantz S.A. E-cigarettes and smoking cessation in real-world and clinical settings: a systematic review and meta-analysis, The Lancet. Respir Med. 2016;4:116–128. doi: 10.1016/S2213-2600(15)00521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Brown J., Beard E., Kotz D., Michie S., West R. Real-world effectiveness of e-cigarettes when used to aid smoking cessation: a cross-sectional population study. Addiction (Abingdon, England) 2014;109:1531–1540. doi: 10.1111/add.12623. [DOI] [PMC free article] [PubMed] [Google Scholar]