

Summary
DExD/H-box RNA helicases (DDX/DHX) are encoded by a large paralogous gene family; in a subset of these human helicase genes, pathogenic variation causes neurodevelopmental disorder (NDD) traits and cancer. DHX9 encodes a BRCA1-interacting nuclear helicase regulating transcription, R-loops, and homologous recombination and exhibits the highest mutational constraint of all DDX/DHX paralogs but remains unassociated with disease traits in OMIM. Using exome sequencing and family-based rare-variant analyses, we identified 20 individuals with de novo, ultra-rare, heterozygous missense or loss-of-function (LoF) DHX9 variant alleles. Phenotypes ranged from NDDs to the distal symmetric polyneuropathy axonal Charcot-Marie-Tooth disease (CMT2). Quantitative Human Phenotype Ontology (HPO) analysis demonstrated genotype-phenotype correlations with LoF variants causing mild NDD phenotypes and nuclear localization signal (NLS) missense variants causing severe NDD. We investigated DHX9 variant-associated cellular phenotypes in human cell lines. Whereas wild-type DHX9 was restricted to the nucleus, NLS missense variants abnormally accumulated in the cytoplasm. Fibroblasts from an individual with an NLS variant also showed abnormal cytoplasmic DHX9 accumulation. CMT2-associated missense variants caused aberrant nucleolar DHX9 accumulation, a phenomenon previously associated with cellular stress. Two NDD-associated variants, p.Gly411Glu and p.Arg761Gln, altered DHX9 ATPase activity. The severe NDD-associated variant p.Arg141Gln did not affect DHX9 localization but instead increased R-loop levels and double-stranded DNA breaks. Dhx9−/− mice exhibited hypoactivity in novel environments, tremor, and sensorineural hearing loss. All together, these results establish DHX9 as a critical regulator of mammalian neurodevelopment and neuronal homeostasis.
Keywords: DHX9, DExD/H-box RNA helicases, neurodevelopmental disorders, Charcot-Marie-Tooth disease, R-loops, DNA damage repair, homologous recombination, epilepsy, neuropathy, allelic series
Graphical abstract
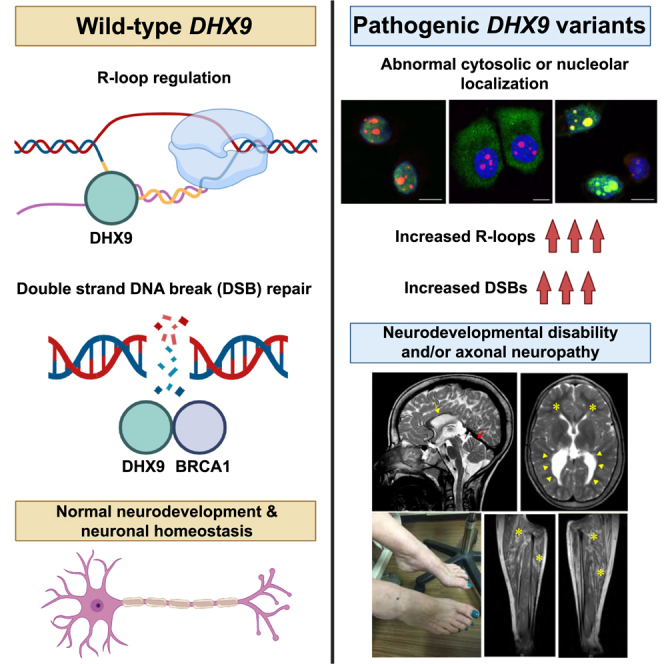
Calame et al. describe 20 individuals with monoallelic rare damaging DHX9 variants and neurologic phenotypes ranging from severe neurodevelopmental disorders to adult-onset Charcot-Marie-Tooth disease. Functional studies show that DHX9 variants disrupt DHX9 cellular localization and helicase activity and increase R-loops and double-stranded DNA breaks.
Introduction
The DExD/H-box (DDX/DHX) gene family consists of 58 paralogs encoding RNA helicases in mammals.1 Conserved from bacteria to humans, the encoded proteins share helicase core domains with a consensus DExD or DExH amino acid sequence within the Walker B motif. Despite their evolutionary origin from whole-genome duplication and tandem amplification, DDX/DHX genes are nonredundant and are often essential in model organisms and human cell lines. Although they are broadly implicated in RNA metabolism, the precise function of most DDX/DHX helicases remains unknown, and most lack associations with human disease traits.1 The genes encoding these helicases are also mutated or dysregulated in cancer and can have oncogenic or tumor-suppressive effects.2
A growing body of evidence links germline pathogenic variation in DDX/DHX genes to neurodevelopmental disorders (NDDs). The X-linked gene DDX3X is a leading cause of developmental delay and/or intellectual disability (DD/ID) in females (MIM: 300958).3,4,5 Other NDD-associated DDX/DHX genes include DHX30 (MIM: 617805),6 DDX6 (MIM: 600326),7 and DDX11 (MIM: 613398).8 A large-scale paralog study subsequently provided further evidence for helicase involvement in human brain development by identifying four additional DDX/DHX NDD genes (DHX37 [MIM: 618731], DHX16 [MIM: 618733], DDX54 [MIM: 611665], and DHX34 [MIM: 615475]) and three candidate NDD genes (DDX47 [MIM: 615428], DHX58 [MIM: 608588], and DHX8 [MIM: 600396]).1
Among DDX/DHX helicases without disease associations, DExH-box helicase 9 (DHX9) is particularly intriguing.9 DHX9 primarily localizes to the nucleus, where it regulates transcription and unwinds nucleic acid structures, such as R-loops, three-stranded structures consisting of a DNA-RNA hybrid and displaced single-stranded DNA.9,10 R-loops have a complex role in cell biology in that they regulate DNA methylation, gene expression, and transcription yet also cause single- and double-stranded DNA breaks (SSBs and DSBs, respectively) with resultant genomic instability.11 In addition, DHX9 plays an integral role in repairing DNA breaks through homologous recombination (HR) via its recruitment of BRCA1 to DSBs.12 Dysregulation of transcription, R-loops, and SSB and DSB repair is a recognized disease mechanism in neurodevelopmental and neurodegenerative disorders.13,14,15,16 We therefore hypothesized that DHX9 dysfunction underlies at least one or more neurologic rare-disease traits.
Here, we describe 20 unrelated individuals affected by sporadic neurologic diseases and found to have heterozygous, ultra-rare, de novo missense or predicted loss-of-function (pLoF) variant alleles in DHX9 (MIM: 603115). Molecular, clinical, and quantitative phenotypic analyses characterized two DHX9-associated disease traits: DD/ID and axonal Charcot-Marie-Tooth disease (i.e., the distal symmetric polyneuropathy Charcot-Marie-Tooth type 2 [CMT2]). Cell-based functional studies demonstrated that pathogenic DHX9 variants cause abnormal cellular distribution of DHX9 and in some cases alter helicase ATPase activity. We studied the impact of DHX9’s loss on organismal biology by generating and phenotyping Dhx9−/− mice, which exhibit multiple behavioral, neurological, and growth abnormalities. Mechanisms by which DHX9 variant alleles disrupt neurodevelopment and neuron axonal integrity are explored.
Subjects and methods
Participant identification and recruitment
This study adhered to the principles of the Declaration of Helsinki. The study was approved by the institutional review board (IRB) at Baylor College of Medicine (BCM) (H-29697). All individuals or their guardians provided written informed consent under BCM protocol H-29697 or through other collaborative IRBs. Participants were identified through the Baylor-Hopkins Center for Mendelian Genomics (BHCMG)/BCM Genomics Research Elucidates the Genetics of Rare Disease (BCM-GREGoR) database, the Baylor Genetics (BG) clinical diagnostic laboratory database, GeneMatcher,17,18 other research and clinical diagnostic laboratories, or a literature search.19,20 All individuals were examined by a clinical geneticist and/or neurologist. Pedigrees and deep phenotypic data for each individual were collected from collaborating clinicians with a standardized template. Brain magnetic resonance imaging (MRI) was collected whenever possible and reviewed by a board-certified neuroradiologist (J.V.H.).
Exome and genome sequencing
Exome sequencing (ES) was performed at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) with an Illumina dual indexed, paired-end pre-capture library per the manufacturer’s protocol with previously described modifications for individuals 15 (BAB12399), 16 (BAB14692), 17 (BAB704), and BAB4646 and M42-1 (https://www.hgsc.bcm.edu/content/protocols-sequencing-library-construction).21,22 Libraries were pooled and hybridized to the HGSC VCRome 2.1 plus custom Spike-In design according to the manufacturer’s (NimbleGen) protocol with minor revisions.23 Paired-end sequencing was performed with the Illumina NovaSeq6000 platform. Samples achieved 98% of the targeted exome bases covered to a depth of 20× or greater and had a sequencing yield of 13.2 Gb. Illumina sequence analysis was performed with the HGSC HgV analysis pipeline, which moves data through various analysis tools from the initial sequence generation on the instrument to annotated variant calls (SNPs and intra-read in/dels).24,25 In parallel with the exome workflow, a SNP Trace panel was generated for a final quality assessment. This included orthogonal confirmation of sample identity and purity via the Error Rate in Sequencing (ERIS) pipeline, developed at the BCM-HGSC.26 Using an “e-GenoTyping” approach, ERIS screens all sequence reads for exact matches to probe sequences defined by the variant and position of interest. A successfully sequenced sample must meet quality-control metrics of ERIS SNP array concordance (>90%) and ERIS average contamination rate (<5%).
Individual 4 underwent ES at the Genetics Institute and Genomics Center of the Tel Aviv Sourasky Medical Center with the NovaSeq 6000 platform with IDT xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA) for library preparation. Reads aligned to GRCh37/hg19. The Franklin by Genoox data analysis platform was used for bioinformatic pipeline and variant analysis.
Individuals 6, 7, and 11 underwent ES at GeneDx (Gaithersburg, MD, USA). Using genomic DNA from the proband and (when available) the parents, we captured the exonic regions and flanking splice junctions of the genome by using the SureSelect Human All Exon V4 (50 Mb) or the IDT xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Coralville, IA). Massively parallel (NextGen) sequencing was done on an Illumina system with 100 bp or greater paired-end reads. Reads were aligned to human genome build GRCh37/UCSC hg19 and analyzed for sequence variants with a custom-developed analysis tool. Reported variants were confirmed, if necessary, by an appropriate orthogonal method in the proband and, if submitted, in selected relatives.27
Genome sequencing (GS) was performed for individual 13 and her parents with the Nextera DNA Flex library preparation kit, and the libraries were 150 bp paired-end sequenced on a Novaseq 6000 (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. The generated sequencing data had a minimum of 10× coverage in at least 98% of mappable positions and an average coverage of 30×. Sequenced reads were trimmed and aligned to the human reference genome GRCh38. Data were processed in accordance with the GATK best practice through GATK v.4.1. For further variant filtering, prioritization, and interpretation, VarSeq v.2.2.3 software was used (Golden Helix Inc., Bozeman, MT, USA).
All ES and GS data generated by the BHCMG/BCM-GREGoR for which informed consent for deposition into controlled-access databases was provided were deposited into either dbGaP under BHCMG dbGaP study accession number phs000711.v7.p2 or the AnVIL repository under study name “Baylor-Hopkins Center for Mendelian Genomics” (https://anvilproject.org/).
Primary analysis
The personal genome variation from 12,266 individuals within the BHCMG/BCM-GREGoR database and from 17,500 individuals in the BG clinical diagnostic laboratory database was analyzed for rare DHX9 variants predicted to be damaging. Variant prioritization utilized minor allele frequencies in the Genome Aggregation Database (gnomAD) and the BHCMG/BCM-GREGoR database, conservation (phylop100way and GERP), and functional predictions (MutationTaster, Sorting Intolerant from Tolerant [SIFT], likelihood ratio test [LRT], Combined Annotation Dependent Depletion [CADD], Rare Exome Variant Ensemble Learner [REVEL], SpliceAI, and the Human Splice Finder). De novo variants were identified from trio ES with DNMFinder.21 Copy-number variant (CNV) analysis of ES data was performed with XHMM and HMZDelFinder.28,29 Candidate variants from ES were orthogonally confirmed by Sanger di-deoxy sequencing.
Human Phenotype Ontology (HPO) analysis
A detailed description of the methods used for calculating phenotypic similarity scores has been published previously.30 In brief, proband phenotypes were annotated with Human Phenotype Ontology (HPO) terms according to their clinically observed findings.31,32,33 HPO term sets for probands were then compared by the Lin method with the OntologyX suite of packages in R.34 Phenotype similarity scores represent the symmetric Lin score (e.g., the average of the Lin score of the HPO term set for proband 1 compared with proband 2 and for proband 2 compared with proband 1). Pairwise calculations of a symmetric Lin score between each set of probands allowed for the generation of a matrix of phenotype similarity scores. We used the pairwise similarity-score matrix to generate a distance matrix. We then clustered probands according to their phenotype similarity scores on the basis of the distance matrix by using hierarchical agglomerative clustering (HAC) with the Ward method. We defined the numbers of clusters for consideration by using a gap statistic curve and chose k = 4 on the basis of the change in slope at that point. We generated heatmaps with the ComplexHeatmap package in R and organized probands on the x and y axes with clusters generated by HAC.35
Expression plasmids
We PCR amplified the EGFP coding sequence and cloned it into a pCMV5-FLAG-DHX9 vector (Sino Biological) by using restriction endonuclease KpnI to generate the EGFP-tagged DHX9 construct, which produces wild-type (WT) DHX9. We subsequently generated the EGFP empty vector by using restriction enzymes KpnI and NotI. Plasmids with DHX9 variants were generated on the basis of EGFP-tagged WT DHX9 vector via recombination using the ClonExpress Ultra One Step Cloning Kit (Vazyme). All plasmid constructs were verified by Sanger di-deoxynucleotide sequencing.
Cell culture and transfection
MCF-7 (human breast cancer), PC-3 (human prostate cancer), and HEK293T (human embryonic kidney) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were transfected with expression plasmids with Lipofectamine 3000 (Invitrogen) or Lipo 8000 (Beyotime) according to the manufacturer’s instructions. Cells were routinely tested for confirming the absence of mycoplasma contamination. Fibroblasts were generated from skin biopsies and were cultured in DMEM supplemented with 20% FBS.
Immunocytochemistry
MCF-7 cells, PC-3 cells, and fibroblasts were fixed for 10 min at room temperature with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and blocked with the blocking buffer (10% goat serum, 1% BSA, and 0.1% Tween 20). Cells were then incubated overnight with primary antibodies at 4°C according to the manufacturers’ recommendations; primary antibodies included anti-Fibrillarin/U3 RNP Rabbit pAb (ABclonal, A13490), anti-DNA-RNA Hybrid Antibody clone S9.6 (Merckmillipore, MABE 1095), anti-RNA helicase A antibody (EPR13521) (Abcam, ab238985), and anti-gamma H2A.X (phospho S139) antibody (EP854(2)Y) (Abcam, ab81299). After primary antibody incubation, cells were washed in PBS and then incubated with mouse Alexa Fluor 647 secondary antibody (Cell Signaling, 4410S) or rabbit Alexa Fluor 555 secondary antibody (Invitrogen, A-21428) for 1 h at 37°C. Nuclei were stained with 1 mg/mL DAPI (Solarbio, C0060). Images were acquired with a confocal microscope (Leica TCS SP8).
ATPase assay
HEK293T cells expressing EGFP-tagged DHX9 were purified with the GFP-Trap Agarose Kit (Chromotek) according to the manufacturer’s instructions. Transfected HEK293T cells were lysed in 200 μL ice-cold radioimmunoprecipitation assay (RIPA) buffer supplemented with DNaseI (75–150 Kunitz U/mL), MgCl2 (2.5 mM), protease inhibitor cocktail, and phenylmethylsulfonyl fluoride (PMSF) (1 mM), and cell lysates were purified by centrifugation at 17,000 × g for 10 min at 4°C. Proteins were extracted from the supernatant by being bound to 20 μL agarose bead slurry for 1 h at 4°C. Precipitates were first washed twice in washing buffer (10 mM Tris/Cl [pH 7.5], 150 mM NaCl, 0.05% NonidetTM P40 Substitute, 0.5 mM EDTA, and 0.018% sodium azide) and then washed twice in a phosphate-free buffer (40 mM KCl, 35 mM HEPES [pH 7.5], and 5 mM MgCl2). Next, precipitates were incubated in a 50 μL phosphate-free reaction mixture containing 2 mM ATP, 2 mM DTT, and 100 μg/mL yeast RNA for 30 min at 30°C. The amount of free phosphate released by ATP hydrolysis in the ATPase assay was determined by the BIOMOL Green reagent (Enzo Life Sciences). For each independent ATPase assay, we calculated ATPase activity by subtracting absorbance values of the EGFP backbone control from values of the EGFP-tagged WT or variant DHX9 proteins. Subsequently, the amount of EGFP-tagged DHX9 was determined by SDS-PAGE followed by Coomassie blue staining intensity, and ATPase activity was normalized to the amount of purified protein.
Generation of Dhx9 mouse model
The Dhx9−/− mouse line (Dhx9Tm1b (EUCOMM) Hmgu) was constructed by the IMPC “knockout first” targeting strategy at Helmholtz Zentrum München in Germany as follows. Dhx9−/− mice were generated by allele conversion of the C57BL/6NCrl-Dhx9Tm1a (EUCOMM) Hmgu mouse line originating from EUCOMM ES clone HEPD0554_5_E05 (see an overview of the clone construction here: https://www.mousephenotype.org/data/genes/MGI:108177#order). The tm1b allele was produced by deletion of exon 4 of Dhx9 and the neomycin cassette by a cell-permeable Cre recombinase. The allele is a knockout given that skipping over of the LacZ cassette does not produce a functional protein. The cassette produces LacZ under the control of the Dhx9 promoter as a fusion protein with exon 3. We genotyped the mice to verify that the mutation and heterozygous mice were intercrossed to generate Dhx9−/− mice with Dhx9+/+ controls for experimental analysis. We used mice from five cohorts in the analysis to have the following number of mutant mice per group: n = 6 male mutants and n = 9 female mutants. There was no evidence of subviability in the line, and the Dhx9 mice could be ordered; all related information is available through the IMPC website (https://www.mousephenotype.org/data/genes/MGI:108177#order). RNA quality-control analysis using RT-PCR of heterozygous brain tissue revealed that exon 3 fused with the synthetic cassette, resulting in a null allele. There was one annotated domain left in the protein. Mice were housed in individually ventilated cages in which water and standard mouse chow were available ad libitum according to European Union directive 2010/63/EU and German Mouse Clinic (GMC) housing conditions (http://www.mouseclinic.de). Moreover, all animal care and use in this study met approval by and complied with the rules of the district government of Upper Bavaria (Regierung von Oberbayern), Germany.
Mouse phenotyping
From the age of 8 to 16 weeks, the Dhx9−/− mice were phenotyped systematically in the GMC as described previously36 and in accordance with the standardized phenotyping pipeline of the IMPC (IMPReSS: https://www.mousephenotype.org/impress/index; see Figure S1 for an overview). The testing details described here are for those assays in which we identified alterations relevant for DHX9 function. Homozygous mutant (“−/−”) and WT controls (“+/+”) were compared, and the number of animals per group and the age of testing for the different assays are shown in Table S1. Body weight was measured in the different cohorts.
Data generated by the open field (OF) test, SHIRPA (SmithKline Beecham, Harwell, Imperial College, Royal London Hospital, phenotype assessment), and grip strength were obtained at 8 and 9 weeks of age. The 20-min OF test was carried out with the ActiMot system (TSE, Germany) as described previously.37 The arena was made of transparent and infrared-light-permeable acrylic with a smooth floor (internal measurements: 45.5 × 45.5 × 39.5 cm; 200 lux in middle). For neurological analysis, we applied a modified SHIRPA protocol36,38,39 covering general neurobehavioral aspects that were rated with defined rating scales. During observation in the arena, a trained observer categorized gait (normal or abnormal) and recorded the occurrence of tremor during observation. Grip strength was also measured according to our standard protocol.36,38,39
Sensorimotor gating and recruitment were measured via assessment of the acoustic startle reflex (ASR) and its pre-pulse (PP) inhibition (PPI) at 10 weeks of age with modification to the previously described protocol,40 and further details can be found here: https://www.mousephenotype.org/impress/ProcedureInfo?action=list&procID=746&pipeID=14. In brief, we used Med Associates (St. Albans, USA) startle equipment and set the background noise (no stimulus [NS]) to 65 dB. We determined the basal startle response (S; startle pulse of 110 dB/40 ms white noise) and percent PPI (%PPI) to four different PP intensities (67, 69, 73, and 81 dB [2, 4, 8, and 16 dB above background, respectively]; 50-ms interval between S and PP).
At the age of 11 weeks, the mice were housed individually in metabolic home cages (MHCs) for indirect calorimetry analysis. Forward (distance traveled) and vertical (rearing) locomotor activity, food intake, and bodyweight loss were measured (TSE, Germany) (for a detailed protocol, see https://www.mousephenotype.org/impress/ProcedureInfo?action=list&procID=855&pipeID=14). The measurement commenced 5 h before lights were turned off and finished 4 h after lights were turned on the next morning (21 h in total).
Altered glucose metabolism was determined with the intraperitoneal glucose tolerance test (ipGTT) at the age of 13 weeks. Glucose (2 g/kg) was administered intraperitoneally (i.p.) after a 16-h withdrawal of food, and glucose levels were measured before and 15, 30, 60, and 120 min after glucose injection. Blood glucose levels were assessed in blood collected from the tail vein with the Accu-Chek Aviva Connect glucose analyzer (Roche/Mannheim).
Auditory brainstem response (ABR) was measured at the age of 14 weeks in anesthetized mice as described here: https://www.mousephenotype.org/impress/ProcedureInfo?action=list&procID=665&pipeID=7.
At the age of 16 weeks, the final blood samples were collected from the retrobulbar vein plexus under isoflurane anesthesia in Li-heparin coated tubes (Li1000A, Kabe Labortechnik). The samples were centrifuged at 5,000 × g for 10 min at 8°C and plasma separated within 1 h of blood collection. Clinical chemistry parameters were measured immediately with an AU480 analyzer (Beckman-Coulter) and adapted reagent kits from Beckman-Coulter according to the manufacturer’s instructions, as described previously.41 The hematology was analyzed with a Sysmex XT-2000iV device using 1:5 diluted samples in the capillary mode as previously described.42
Statistics
We analyzed data by two-way ANOVA with post-hoc Tukey’s test to test genotype × sex interaction effects. For the analysis of rearing activity in the MHC over 21 h and ABR, a three-way ANOVA was used with genotype, sex, and time (for rearing) or frequency (for ABR) as independent variables. Linear regression analysis determined how body weight predicted grip strength. Data were statistically analyzed with GraphPad Prism version 8 for Windows (GraphPad Software, La Jolla, CA, USA). For all tests, a p value < 0.05 was the level of significance, and data are presented as mean ± SD or ± SEM. No correction for multiple testing was performed.
Results
Missense and loss-of-function constraint among DDX/DHX genes
The DDX/DHX superfamily consists of 58 paralogous genes underlying at least 15 rare disease traits. Despite their shared domains and overlapping functions, DDX and DHX vary in their tolerance of missense and pLoF variation (Figures 1A and 1B).43 A negative correlation between the missense and pLoF constraints is observed within the superfamily (genes with disease-phenotype associations, r = −0.82; all genes, r = −0.76). Strikingly, DHX9 has the greatest missense and pLoF intolerance of all DDX/DHX genes (missense Z score = 5.84; pLI = 1; LOEUF = 0.1) (Figure 1B). Dominant-trait-associated DDX/DHX genes tend to have higher pLoF and missense constraints than genes associated with recessive disease traits (Figure 1A). Known haploinsufficient genes DDX3X and DHX30 are also highly intolerant of pLoF (LOEUF = 0.12). Haploinsufficiency as a disease mechanism is highly enriched in genes in the highest LOEUF decile (≤0.268, dashed line in Figures 1A and 1B).43 Similarly, pHaplo and pTriplo scores for DHX9 (0.99 and 1.00, respectively) suggest that DHX9 is highly sensitive to both copy-number loss and gain.44 For comparison, DHX30 has pHaplo and pTriplo scores of 0.97 and 1.00, respectively.
Figure 1.

Missense and LoF tolerance in the DDX/DHX superfamily and DHX9 expression
(A) Relationship between the pLoF and missense constraints among known or candidate DDX/DHX genes associated with disease phenotypes. Genes linked to dominant disease traits are shown in red, those linked to recessive traits are shown in blue, and those linked to mixed dominant and recessive traits are shown in orange. The solid line indicates linear regression. The dashed line shows the top LOEUF decile. The y axis shows LOEUF, and the x axis shows missense Z scores (gnomAD v2.1.1).
(B) Relationship between pLoF and missense constraint among all DDX/DHX genes. DHX9 is indicated by a red dot. Paralogs DHX30 and DDX3X are indicated by blue dots. The solid line indicates linear regression. The dashed line shows the top LOEUF decile. The y axis shows LOEUF, and the x axis shows missense Z scores (gnomAD v2.1.1).
(C) DHX9 mRNA expression in human adult tissues from the GTEx project. Nervous system tissues are highlighted in gray. The y axis shows transcripts per million (TPM).
(D) Average mRNA expression of known or candidate DDX/DHX genes associated with disease phenotypes in the developing nervous system from BrainSpan. DHX9 is indicated by red dots. Paralogs DHX30 and DDX3X are indicated by blue dots. The y axis shows log2 reads per kilobase million (RPKM), and the x axis shows the developmental stage. Abbreviations: pcw, post-conception weeks; mo, months; yrs, years.
Tissue expression and protein-protein interactions of DHX9
Genes underlying a neurological disease trait should be well expressed within the adult and/or developing nervous system. We investigated DHX9 expression levels through human development by using Genotype-Tissue Expression (GTEx) and the BrainSpan Atlas of the Developing Human Brain. DHX9 is robustly expressed in all tissues during adulthood, but its highest neuronal expression is within the cerebellum and tibial nerve. We hypothesized that the large Purkinje cells of the cerebellum and long-track peripheral nerves, such as the tibial nerve, might be most perturbed for biological homeostasis in postmitotic neurons (Figure 1C). Similarly, DHX9 is among the most abundantly expressed DDX/DHX genes associated with disease phenotypes in the developing brain: its expression levels are comparable to those of DDX3X and DHX30 (Figure 1D). Moreover, single-cell expression data from the developing human primary cortex in the UCSC Cell Browser (https://cells.ucsc.edu/) show that DHX9 transcripts are observed in all cell types.
Protein-protein interactions between a gene and other genes with shared disease-phenotype associations provide supporting evidence for disease functional biology related to the gene. Therefore, we examined the DHX9 interactome by using the STRING database (Figure S2). DHX9 directly interacts with multiple known neurological-disease-trait-associated genes, including EWSR1 (amyotrophic lateral sclerosis [ALS] [MIM: 133450]), AGO2 (Lessel-Kreienkamp syndrome [MIM: 619149]), and HNRNPU (developmental and epileptic encephalopathy 54 [MIM: 617391]). A literature review captured other relevant protein-protein interactions involving genes or variant alleles underlying “axonopathies,” including SMN1 (spinal muscular atrophy 1 [MIM: 253300]), FUS (ALS 6 with or without frontotemporal dementia [MIM: 608030]), TAF15 (ALS [MIM: 601574]), and MATR3 (ALS 21 [MIM: 606070]).
Identification of individuals with candidate disease-causing DHX9 variants
DDX3X- and DHX30-associated NDDs result from de novo missense or pLoF variant alleles.3,4,5,6,45 The observations that DHX9 shares high missense and pLoF intolerance with DDX3X and DHX30 and that all three genes exhibit similar expression profiles in the nervous system implicate a DHX9-associated neurological disease trait potentially resulting from de novo, ultra-rare missense or pLoF variants. Therefore, we analyzed ultra-rare, potentially damaging heterozygous missense or pLoF variants in the 29,766 individuals with ES or GS data within the BHCMG/GREGoR and BG databases and subsequently identified additional individuals with DHX9 variants through the online matchmaker GeneMatcher,17,18 DECIPHER,46 and published NDD cohorts.19,20 These efforts uncovered 20 unrelated individuals with candidate disease-causing DHX9 variants and either NDDs (n = 17) or axonal CMT (n = 3) (Figure 2 and Tables S2). We also searched for individuals with biallelic DHX9 variants consistent with an autosomal recessive (AR) neurological disease trait but failed to find any compelling biallelic candidate variants. Finally, we also examined the BHCMG/GREGoR and BG databases for ultra-rare, potentially damaging heterozygous missense or pLoF variants in other highly constrained, brain-expressed paralogs without gene-disease associations—including DDX5 (MIM: 180630), DDX17 (MIM: 608469), and DDX46 (MIM: 617848) (Figure 1D)—but failed to identify any compelling additional candidate variants.
Figure 2.

Pedigrees, photographs, and brain imaging of individuals with candidate disease-causing DHX9 variants
Pedigrees, DHX9 genotypes, and representative brain MRIs of individuals with NDDs are shown above the black line. Note that family 3 and family 12 share the recurrent allele c.3497G>C (p.Arg1166Pro). Individuals, genotypes, and representative clinical images and leg-muscle MRIs of individuals with CMT are shown below the black line. The yellow arrow indicates a thin corpus callosum. Red arrows indicate cerebellar atrophy. Yellow arrowheads show enlargement of the ventricles, and yellow asterisks (brain MRI, second image from the left) show reduced white-matter volume. The photograph shows the pes cavus and hammer-toe deformity in family 15, affected by CMT. Yellow asterisks (leg MRI) highlight fatty infiltration of the lower-leg musculature, consistent with CMT. Black pedigree symbols indicate NDDs, whereas gray pedigree symbols indicate CMT. Individuals for whom limited clinical details were available were excluded from the figure (see supplemental notes).
Molecular findings
DHX9 (RefSeq: NM_001357.5) is located in chromosomal region 1q25.3, contains 28 exons, and encodes a 1,270 amino acid (aa) protein (Figure 3). Eight domains of DHX9 have been delineated: two double-stranded RNA binding domains (dsRBDs), a minimal transactivation domain (MTAD) involved in RNA polymerase II interaction, two helicase domains, a helicase-associated domain 2 (HA2) required for unwinding activity, an oligonucleotide/oligosaccharide-binding (OB) fold, and a glycine-rich RGG box that binds single-stranded nucleic acids.9 Additionally, there are two nuclear localization signals (NLSs): a 9 aa NLS between the helicase domains and a 19 aa NLS within the RGG-box domain. The C-terminal NLS is required for nuclear import via the importin-α3/importin-β pathway.47,48
Figure 3.

Location of DHX9 variants
(A) Diagram of DHX9 mRNA shows the location of NDD-associated (white) and CMT-associated (black) variants.
(B) Diagram of DHX9 (UniProt: Q08211) shows functional domains, including dsRBD1 and dsRBD2, the MTAD, helicase domains, HA2, the OB fold, and the RGG box. Protein domains were obtained from UniProt. The sequence of the nuclear localization signal is magnified, and the two key residues (Lys1163 and Arg1166) are underlined. DHX9’s protein-tolerance landscape, calculated by Metadome, is shown at the bottom.
Variant details, including de novo status, CADD and REVEL scores, conservation, and allele frequency, are shown in Table 1. We identified a total of nineteen different DHX9 variants, including 12 missense variants and 5 pLoF variants. All are absent from gnomAD except for c.1417G>A (p.Val473Ile) (one heterozygote; allele frequency = 4.01e−6). DHX9 variants occurred de novo in 14 individuals: 13 NDD and 1 CMT; in the remaining five individuals, parental samples were not available for genotyping. A recurrent de novo variant, c.3497G>C (p.Arg1166Pro), was seen in two unrelated individuals (from USA and Poland) each with sporadic NDDs. Ten missense variants map within functional domains: four within helicase domains, two within the HA2 domain, one in the OB fold, and three within the RGG box (Figure 3B). Site-directed mutagenesis previously demonstrated that Arg1166 and Lys1163, aa residues affected by the NLS variants c.3497G>C and c.3488A>G, respectively, are required for DHX9 nuclear import.47,48 Consistent with this, cNLS Mapper, a program that predicts import in α-dependent NLSs,49 detects the known NLS within DHX9’s C-terminal reference sequence but does not recognize an NLS in either variant sequence (Figure S3). Two of the three CMT-associated DHX9 variants cluster within the HA2 domain. All DHX9 missense variants, with the exception of the CMT-associated variant c.3763G>A (p.Ala1255Thr), lie in regions with low tolerance of missense variation (as determined by MetaDome50) (Figure 3B). DHX9 missense variants were also visualized within the DHX9 AlphaFold model (Figure S4).
Table 1.
Summary of DHX9 variant alleles
| Individual(s) | Phenotype | Position (hg19) | Nucleotide and protein | De novo | Allele count and frequency (gnomAD) | CADD score | REVEL score | Conservation (phylop100way) |
|---|---|---|---|---|---|---|---|---|
| 1 | NDD | chr1: 182822498G>A | c.422G>A (p.Arg141Gln) | yes | absent | 22.3 | 0.20 | 6.67 |
| 2 (BAB15412) | NDD | chr1: 182845191T>G | c.1822T>G (p.Cys608Gly) | yes | absent | 24.3 | 0.31 | 7.24 |
| 3 and 12 | NDD | chr1: 182856253G>C | c.3497G>C (p.Arg1166Pro) | yes | absent | 23.8 | 0.67 | 6.74 |
| 4 | NDD | chr1: 182825663A>AA | c.627−4dupA (p.?) | yes | absent | – | – | – |
| 5 | NDD | chr1: 182835663G>A | c.1417G>A (p.Val473Ile) | yes | 1 htz; 1 in 141,456 | 26.3 | 0.34 | 9.14 |
| 6 | NDD | chr1: 182856244A>G | c.3488A>G (p.Lys1163Arg) | yes | absent | 22.0 | 0.27 | 6.17 |
| 7 | NDD | chr1: 182850561G>T | c.2786+1G>T (p.?) | unknown | absent | 35.0 | – | 9.05 |
| 8 | NDD | chr1: 182827250C>T | c.685C>T (p.Arg229Ter) | yes | absent | 38 | – | 4.29 |
| 9 | NDD | chr1: 182829219G>A | c.1232G>A (p.Gly411Glu) | yes | absent | 25.1 | 0.73 | 9.09 |
| 10 | NDD | chr1: 182845626CGA>C | c.2075_2076delGA (p.Glu693GlyfsTer7) | yes | absent | – | – | – |
| 11 | NDD | chr1: 182847239G>A | c.2282G>A (p.Arg761Gln) | yes | absent | 32 | 0.90 | 7.33 |
| 13 | NDD | chr1: 182847238C>T | c.2281C>T (p.Arg761Trp) | yes | absent | 29 | 0.91 | 2.36 |
| 14 | NDD | chr1: 182856543C>T | c.3787C>T (p.Gln1263Ter) | unknown | absent | 37 | – | 0.83 |
| Iossifov et al.19,a | NDD | chr1: 182852665G>A | c.3155G>A (p.Arg1052Gln) | yes | absent | 27.2 | 0.21 | 9.10 |
| BAB4646a | NDD | chr1: 182827238G>A | c.674−1G>A (p.?) | unknown | absent | 35 | – | 8.95 |
| M42-1a | NDD | chr1: 182847247C>T | c.2290C>T (p.Arg764Ter) | unknown | absent | 36 | – | 1.75 |
| 15 (BAB12399) | CMT | chr1: 182849656A>G | c.2537A>G (p.Asp846Gly) | yes | absent | 26.7 | 0.21 | 6.84 |
| 16 (BAB14692) | CMT | chr1: 182848543G>C | c.2510G>C (p.Arg837Thr) | unknown | absent | 31 | 0.15 | 5.39 |
| 17 (BAB704) | CMT | chr1: 182856519G>A | c.3763G>A (p.Ala1255Thr) | unknown | absent | 16.19 | 0.14 | 2.18 |
Abbreviations: NDD, neurodevelopmental disorder; CMT, Charcot-Marie-Tooth disease; CADD, Combined Annotation Dependent Depletion; gnomAD, Genome Aggregation Database; htz, heterozygote; REVEL, Rare Exome Variant Ensemble Learner.
Limited clinical details are available, or there is evidence that multilocus pathogenic variation contributes to a blended phenotype. See the supplemental notes for additional details.
Five pLoF variants were identified in individuals with NDDs: c.2786+1G>T, c.2290C>T (p.Arg764Ter), c.685C>T (p.Arg229Ter), c.674−1G>A, and c.2075_2076delGA (p.Glu693GlyfsTer7). All are absent from gnomAD. c.685C>T, c.2290C>T, and c.2075_2076delGA create premature termination codons within exons 8, 18, and 20, respectively, of this 28-exon gene and therefore are predicted to undergo nonsense-mediated decay (NMD).51 c.674−1G>A and c.2786+1G>T lie within introns 7 and 23, respectively, and are predicted to alter splicing by SpliceAI. Other DHX9 candidate variants include the de novo splice variant c.627−4dupA and c.3787C>T (p.Gln1263Ter). Both are absent from gnomAD. Splicing predictors are divided for c.627−4dupA: Human Splice Finder predicts possible activation of a cryptic acceptor site, whereas SpliceAI does not (Figure S5). c.3787C>T (p.Gln1263Ter) has a CADD score of 37 and is expected to escape NMD given that it lies within the final exon. It should result in a truncated protein lacking the final eight amino acids within the RGG domain.
Because DHX9 is highly intolerant to pLoF, we also searched our ES database for CNVs encompassing DHX9 by using XHMM, as well as the BG clinical chromosomal microarray database (CMA) and DECIPHER. No small deletion CNVs spanning DHX9 were detected in the BHCMG/GREGoR database. Similarly, the smallest reported deletion encompassing DHX9 in the BG CMA database (which contains ∼90,000 personal genomes) was >20 Mb. A 191.52 kb deletion (GRCh37, chr1:182,790,120–182,981,637) encompassing DHX9 and two other coding genes (NPL, pLI = 0; SHCBP1L, pLI = 0) was reported in DECIPHER in an individual (ID 288646) with hepatic fibrosis, abnormality of the kidney, global developmental delay, and oculomotor apraxia.46 The deletion was maternally inherited and classified as “likely benign.” No maternal phenotypic data were provided for review.
Defining the DHX9-associated disease trait
Clinical records for robust organismal phenotyping of 17 individuals with candidate disease-causing DHX9 variants were available. These included data from 14 individuals with NDDs and three individuals with CMT2, and we used these data for deep clinical and phenotypic analyses (Figure 2, Table S2, and supplemental notes). All individuals with DHX9-related NDD had DD/ID. The degree of cognitive impairment ranged from autism spectrum disorder with speech delay and learning disabilities but neurotypical intelligence (individual 4) to severe DD/ID (individuals 1, 3, 5, and 12). Other common developmental or neuropsychiatric disorders included anxiety, obsessive-compulsive disorder, autistic spectrum disorders, and neurobehavioral issues (8/14). Other common clinically observed features included axial hypotonia (7/14) and dysmorphic features (8/14). Although neither a consistent facial gestalt nor a recognizable pattern of human malformation was appreciated, facial dysmorphology included ear abnormalities (low-set, posteriorly rotated ears; small ears with overfolded superior helices; and thick helices), hypertelorism, micrognathia, short downslanted palpebral fissures, a thick upper lip, a short philtrum, midface hypoplasia, and macrodontia of upper-jaw central incisors. Digital anomalies, including fifth-finger clinodactyly, brachydactyly, long tapered fingers, postaxial polydactyly, and short hypoplastic fifth fingernails, were also reported in multiple individuals. Six individuals had either congenital or postnatal microcephaly (Z scores = −2.14 to −3.39). Seizures were reported in six individuals, and drug-resistant epilepsy occurred in three individuals. Appendicular hypertonia was reported in two individuals. Brain magnetic resonance imaging (MRI) was abnormal in five individuals studied. Imaging findings included white-matter volume loss with enlargement of the ventricles, thinning of the corpus callosum, and cerebral and cerebellar atrophy (Figure 2). Other features seen in two or more individuals were cardiac abnormalities (4/14), hyperreflexia (3/14), failure to thrive (2/14), short stature (3/14), and a history of recurrent infections (2/14).
Three individuals were identified to have candidate disease-causing DHX9 variants and CMT2, also known as hereditary motor and sensory neuropathy (HMSN). Disease onset occurred in adulthood (2/3) or adolescence (1/3). All individuals had typical features of CMT, a distal symmetric polyneuropathy (DSP), including distal weakness, sensory deficits, and/or variable muscle wasting or foot deformities (Figure 2).52 Two had painful sensory neuropathy.
Electrophysiological features of long-tract nerve function were investigated by nerve conduction studies (NCSs). These studies showed either diminished compound muscle action potential (CMAP) and sensory nerve action potential (SNAP) amplitudes (2/3) or normal nerve conduction velocities (NCVs) and amplitudes (1/3). Electromyography (EMG) and the NCS in the individual with normal NCVs showed neurogenic changes in the distal lower extremities and upper extremities and active denervation in the lower extremities. EMG and NCSs were generally not performed in individuals with DHX9-related NDD; however, a moderate axonal sensorimotor polyneuropathy was detected by EMG and NCS in one individual with severe NDD (family 1, individual 1; Figure 2). Thus, electrodiagnostic studies supported an axonal neuropathic process in all individuals with CMT2.
Quantitative dissection of genotype-phenotype relationships at the DHX9 locus
HPO, a structured ontology of medical terms and a standardized terminology, is computationally accessible for informatic similarity comparisons of human phenotypic data and rare-disease traits, the latter by searchable queries of OMIM clinical synopses. HPO approaches can quantitatively dissect complex disease phenotypes resulting from multilocus pathogenic variation (MPV) and reveal previously unrecognized genotype-phenotype correlations within disease cohorts.30,53 The DHX9 variants encompassing multiple protein functional domains, variant types, and phenotypes were observed to spread over a neurological disease spectrum; therefore, we performed a quantitative phenotypic similarity analysis of the first 14 individuals identified without evidence of MPV and for whom detailed phenotypic data were available (Figure 4 and supplemental notes). Phenotypic similarity scores for each proband were calculated and visualized in a cluster heatmap where cluster number was determined by the gap statistic curve (Figure 4A).
Figure 4.

HPO analysis of the DHX9 cohort
(A) Gap statistic curve for the DHX9 cohort. The gap statistic is displayed on the y axis, and the number of clusters tested is on the x axis. The point on the curve where the slope changes from a trend of higher to lower (i.e., additional clusters do not add as much to the gap statistic) was chosen as the optimal number of clusters (k = 4).
(B) HAC and visualization of quantitative phenotypic similarity allow refinement of genotype-phenotype correlations in the DHX9 cohort. The dendrogram shown at the top and to the left of the heatmap is based on HAC analysis of the dissimilarity matrix produced from Resnik semantic similarity scores and with k = 4. Unique clusters are represented by different colors, and variants found in individual probands are labeled on top of and to the right of the heatmap. Within the heatmap, dark red indicates a higher similarity, whereas dark blue indicates a lower similarity. A key is provided on the right.
(C) Magnified dendrogram showing unique clusters and group characteristics.
HPO analysis identified four distinct phenotypic clusters (Figures 4B, 4C, and S6). Groups 1 and 3 consisted of individuals with mild NDD phenotypes (i.e., mild deviations from normotypical behaviors, such as mild DD/ID, autism spectrum disorders, and speech delay) without microcephaly or brain abnormalities, whereas group 2 contained the most severe phenotypes (severe DD/ID, microcephaly, and brain abnormalities). Group 4 consisted of all individuals with CMT. pLoF variants were exclusively found within groups 1 and 3, and NLS variants were exclusively seen in group 2. The distinction between groups 1 and 3 is not immediately apparent but could reflect phenotypic depth (average number of HPO terms per group: group 1, 13.00 ± 5.16; group 2, 22.33 ± 5.51; group 3, 3.25 ± 2.06; group 4, 9.67 ± 2.52).
Functional characterization of DHX9 variant alleles
We next investigated potential cellular phenotypes and studied whether DHX9 variants identified in individuals affect protein subcellular localization in human cells. WT DHX9 exhibits diffuse nuclear localization (Human Protein Atlas, https://www.proteinatlas.org/ENSG00000135829-DHX9). Consistently, we observed EGFP-tagged WT DHX9 diffusely distributed in the nucleus of MCF-7 cells (Figure 5A). In contrast, the EGFP-tagged NLS variant p.Lys1163Arg resulted in protein localization solely within the cytoplasm (Figure 5A). This disruption of DHX9 nuclear localization was also observed in fibroblasts derived from individual 6 (with the p.Lys1163Arg variant) but not in his unaffected father (Figure 5B). A comparable cytoplasmic distribution was also observed for the NLS variant p.Arg1166Pro (Figure S7). In contrast, nonsense or frameshift variants identified in mild NDD resulted in EGFP-tagged protein in both the nucleus and the cytoplasm (Figures 5A and S7). Missense variants identified in CMT2 instead demonstrated prominent and uniform nucleolar localization, as evidenced by their co-staining with the nucleolus marker fibrillarin (FBL) (Figures 5A and S7). Curiously, cells transfected with mild NDD-associated variants p.Gly411Glu, p.Val473Ile, p.Cys608Gly, and p.Arg761Gln showed either diffuse nuclear or nucleolar patterns (Figure S8 and Table 2). We further confirmed the changes in localization patterns in another human cell line (PC-3) for representative DHX9 proteins (Figure 5A).
Figure 5.

Subcellular localization of WT DHX9 and representative variant proteins
(A) Subcellular localization of EGFP-tagged WT DHX9, NLS p.Lys1163Arg (severe NDD), truncating p.Arg229Ter (mild NDD), and CMT p.Ala1255Thr proteins in MCF-7 and PC-3 human cells. Nucleolar loci were co-stained by the FBL marker, and DNA was stained by DAPI. Scale bar: 10 μm.
(B) Endogenous localization of DHX9 in fibroblasts from the proband with severe NDD and the p.Lys1163Arg NLS variant as well as the unaffected father. Scale bar: 40 μm.
(C) Staining of R-loop formation by the S9.6 marker and of DSBs by the γ-H2AX marker in MCF-7 cells producing the WT or p.Arg141Gln protein. Scale bar: 10 μm. Also see Figures S7 and S8.
Table 2.
Summary of disease type and severity, subcellular localization, R-loops, and DNA damage levels associated with WT DHX9 and DHX9 variants
| Variant type (individual IDs) | Disease | NDD severity | DHX9 location | DSB pattern and level | R-loop pattern and level |
|---|---|---|---|---|---|
| WT | – | – | nuclear | scattered, low | gathered, + |
| Truncating (8, 10, M42-1) | NDD | mild | nuclear and cytoplasmic | ubiquitous, + | gathered, + |
| NLS missense (3, 6, 12) | NDD | severe | cytoplasmic | ubiquitous, + | gathered, + |
| Missense (1) | NDD | severe | nuclear | ubiquitous, ++ | ubiquitous, ++ |
| Missense (2, 5, 9, 11) | NDD | mild | nuclear or nucleolar | ubiquitous, low | gathered, low |
| Missense (15, 16, 17) | CMT | – | nucleolar | ubiquitous, low | gathered, low |
Abbreviations: WT, wild type; NLS, nuclear localization signal; NDD, neurodevelopmental disease; CMT, Charcot-Marie-Tooth disease; DSB, double-stranded break; +, increased; ++, highly increased.
We also examined the R-loop and DSB levels in cells producing DHX9 variant proteins by immunofluorescence (IF) staining of the S9.6 and γ-H2AX markers, respectively. Cells transfected with an empty vector showed low levels of R-loops and DSBs (Figure S7). The DHX9 p.Arg141Gln variant associated with severe NDD induced high levels of R-loops and DSBs with ubiquitous staining throughout the nucleus; in contrast, cells transfected with the WT DHX9 vector exhibited low levels of DSBs and moderate levels of R-loops (Figure 5C). Truncated DHX9 and NLS variant proteins induced moderate levels of R-loop and DSB, whereas cells producing mild NDD- and CMT-associated variants had low levels of R-loops and DSB (Figures S7 and S8 and Table 2). Given that transcripts containing protein-truncating DHX9 variants are predicted to undergo NMD, the physiologic significance of the observed increase in DSBs is unclear.
Given that DHX9 relies on ATP hydrolysis to unwind nucleic acid structures, we inspected whether variants located within helicase domains affect DHX9 ATPase activity. Six variants fall within the helicase ATP-binding or helicase C-terminal domain; these functional domains contain eight conserved motifs (Figure S9A). Among the six variants, the pLoF variants p.Glu693GlyfsTer7 and p.Arg764Ter caused protein truncation and generated baseline ATPase activities comparable to those of the negative controls, as expected (Figure S9B and Table S3). Similar results were obtained for the p.Arg229Ter protein, which is predicted to truncate ATP-binding domains (Figure S9B and Table S3). Two missense changes located within conserved ATP binding and hydrolysis motifs, p.Gly411Glu in motif I and p.Arg761Gln in motif VI, significantly altered ATPase activity relative to that of WT DHX9. In contrast, two missense variants located outside of conserved motifs, p.Val473Ile and p.Cys608Gly, demonstrated ATPase activity comparable to that of WT DHX9 (Figure S9B).
Dhx9−/− mice are viable but have behavioral and neurological abnormalities
To further explore the role of DHX9 in the mammalian nervous system, we generated Dhx9−/− mice. Biallelic disruption of Dhx9 did not affect viability but clearly altered behavioral and neurological function in young adult mice. On exposure to a novel, mildly stressful environment (20-min open field; Figures 6A and 6B), introduction to a new home cage (Figure 6C)), Dhx9−/− mice exhibited decreased locomotor activity (both forward and vertical) and locomotor speed. Conversely, in a familiar home-cage environment, Dhx9−/− mice showed increases in vertical locomotor activity during the active dark period of the light/dark cycle (Figure 6C). These abnormal behavioral reactions to different environmental conditions could indicate altered sensory information processing. Although both 2-paw and 4-paw grip strength was clearly decreased in Dhx9−/− mice (Figure 6D), this was significantly predicted by body weight in linear regression analysis (Figure 6E). In addition, 20% of Dhx9−/− mice (3 of 15 mutants, 2 of 6 males, and 1 of 9 females) showed tremors in SHIRPA analysis (Figure 6F). The constitutive loss of Dhx9 also caused deafness in mice, as evidenced by absent hearing curves in ABR analysis (Figure 6G) and further reflected by clearly impaired acoustic startle reactivity (Figure 6H). Dhx9−/− mice not only had lower body weight than WT mice (Figure 6I) but also had reduced food intake and more weight loss than WT mice in the 21 h period after transfer to a new home cage (Figures 6J and 6K).
Figure 6.

Disruption of Dhx9 in mice causes behavioral and neurological abnormalities
(A–C) Dhx9−/− mice were hypoactive in response to a novel environment (A: decreased distance traveled; B: decreased speed) in an open field and to decreased rearing (C) upon the first introduction to a novel home cage. However, male Dhx9−/− mice showed more rearing activity with lights off during the active phase in the home cage. This increased vertical activity was evident in both male and female Dhx9−/− mice in the latter half of the active phase. Shaded areas encompass the period between lights off and lights on (18:00–06:00) and represent the mean ± SEM.
(D and E) Both 2-paw and 4-paw grip strength were reduced in Dhx9−/− mice (D). Lower grip strength was predicted by the lower body weight in linear regression analysis (E).
(F) 20% of Dhx9−/− mice showed tremors in the SHIRPA analysis.
(G and H) Both an absent auditory response curve in the auditory brainstem response (G) and decreased acoustic startle reactivity (H) indicated deafness in Dhx9−/− mice.
(I–K) The body weight of Dhx9−/− mice was lower than that of WT mice (I). Dhx9−/− mice had lower cumulative food intake and lost more weight over a 21 h period than WT mice after transferring to a new home cage. #p < 0.05, male +/+ vs. −/−; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, +/+ vs. −/−. Data represent the mean ± SD.
In addition to noticing differences in body weight, we observed further indications that altered metabolism and organ function are associated with DHX9 loss. Impaired glucose clearance in the ipGTT (Figure S10A) and decreased ad libitum fed glucose (Figure S10B) and cholesterol levels (Figure S10C) suggest that glucose and fat metabolism are possibly affected by altered liver metabolism. Females showed increased creatinine and urea levels (Figures S10D and S10E), suggesting possible altered renal function. Hematologic analysis provided evidence of abnormal erythropoiesis and thrombopoiesis. This included a mild hypochromic microcytosis of erythrocytes, as indexed by increased red blood cell counts with decreased mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH). Moreover, lowered platelet counts in males were associated with higher mean platelet volumes and increased anisocytosis of platelets (Figure S11).
Discussion
Despite the rapid increase in variant identification and gene-disease associations in Mendelian diseases over the last decade and the importance of RNA metabolism in health and disease, most DDX/DHX paralogs still lack gene-disease association. Here, we provide evidence for the existence of at least two autosomal-dominant (AD) rare-disease traits associated with variants in DHX9: DD/ID and axonal CMT2. Like DDX3X and DHX30, the best characterized DDX/DHX paralogs,3,4,5,6,45 de novo DHX9 missense and pLoF variant alleles, associate with a wide phenotypic spectrum. Among individuals with NDDs, the spectrum ranges from neurobehavioral differences of isolated ASD without ID on one end to severe DD/ID with microcephaly, brain anomalies, refractory epilepsy, and movement disorders on the other. Other parallels with DDX3X- and DHX30- associated rare-disease traits include the association between structural brain abnormalities with severe phenotypes and variable but frequent extra-neurological findings.
Quantitative HPO analysis of the DHX9 cohort demonstrated four distinct phenotypic clusters: two associated with mild DD/ID (groups 1 and 3), one associated with severe DD/ID (group 2), and another associated with CMT (group 4). Although the total number of cases was modest (n = 14), these analyses provide evidence consistent with the contention of emerging genotype-phenotype correlations reminiscent of DDX3X and DHX30: an association between pLoF variant alleles and mild NDD phenotypes and between specific missense variants and severe phenotypes.3,4,5,6,45 The sole instance of a maternally inherited 191.52 kb CNV deletion encompassing DHX9 in DECIPHER in an individual with a neurodevelopmental phenotype could corroborate this genotype-phenotype schema; alternatively, it could indicate reduced penetrance of DHX9 LoF. Exploring these hypotheses will require additional individual ascertainment and variant studies.
The association between variants in a single gene and distinct phenotypes such as NDDs and CMT2 might be surprising but is not without precedence. The allelic series of PNKP, a gene encoding another SSB/DSB repair enzyme, polynucleotide kinase 3-prime phosphatase, is relevant because it ranges from CMT type 2B2 (MIM: 605589) to ataxia-oculomotor apraxia 4 (MIM: 616267) and microcephaly, seizures, and developmental delay (MIM: 613402).54 Other examples include SURF1 (CMT type 4K [MIM: 616684] and mitochondrial complex IV deficiency [MIM: 220110]), YARS1 (CMT, dominant intermediate C [MIM: 608323] and infantile-onset multisystem neurologic, endocrine, and pancreatic disease 2 [MIM: 619418]), ATP1A1 (CMT, axonal, type 2DD [MIM: 618036]; and hypomagnesemia, seizures, and impaired intellectual development 2 [MIM: 618314]), MORC2 (CMT, axonal, type 2Z [MIM: 616688]; and developmental delay, impaired growth, dysmorphic facies, and axonal neuropathy [MIM: 619090]), and AIFM1 (combined oxidative phosphorylation deficiency 6 [MIM: 300816] and Cowchock syndrome [MIM: 310490]). Because PNKP, SURF1, YARS1, ATP1A1, MORC2, AIFM1, and DHX9 all play integral roles in neuronal homeostasis and survival, each gene’s phenotypic spectrum most likely reflects distinct mutational mechanisms within the allelic series (e.g., amorphs versus hypomorphs) and a phenotypic gradient caused by a range of residual gene function or gene dosage.55
Rapid repair of SSBs and DSBs is essential for neurodevelopment and neuronal homeostasis.56,57,58,59,60 SSB and DSB repair limits the pathologic consequence of DNA damage—genomic instability, compromised genomic integrity, and apoptosis—and has essential physiologic functions in the brain. Consequently, pathogenic variation in SSB/DSB repair genes results in a spectrum of neurodevelopmental and neurodegenerative disorders.14,15,56,57,61,62,63 Here, we show that a subset of pathogenic variants in DHX9, a BRCA1-interacting R-loop helicase required for homologous recombination (HR),10,12,64 cause aberrant increases in the levels of R-loops and/or DSBs. Thus, DHX9 joins the expanding list of genes associated with Mendelian disorders of SSB and DSB repair.
The mechanisms underlying DHX9-related neurologic disorders are likely to be complex, and dissecting them will take a great deal of experimental biology, human genomics, and time. The paucity of pLoF variants in gnomAD and the exclusive identification of high-confidence pLoF variants in affected individuals in research and clinical genomic databases suggest haploinsufficiency as a disease mechanism, as in DDX3X and DHX30. However, the increased R-loops and DSBs induced by DHX9 variants c.422G>A (p.Arg141Gln), c.3497G>C (p.Arg1166Pro), and c.3488A>G (p.Lys1163Arg) and their association with severe phenotypes point toward a gain-of-function (GoF), perhaps dominant-negative mechanism. Similarly, the nucleolar DHX9 accumulation seen with CMT- and mild-NDD-associated DHX9 variants might indicate a separate, perhaps toxic GoF mechanism. Given that cellular stressors, including RNA PolII-mediated transcriptional inhibition, growth arrest, viral replication, and hypothermia, all induce nucleolar DHX9 translocation,65,66 it is possible that pathogenic DHX9 variants might disrupt neurodevelopment or cause neurodegeneration by inducing cellular stress responses. Furthermore, because some mild-NDD-associated DHX9 variants cause a mixed nuclear-nucleolar distribution and NDD-affected individuals are young (average age of 10.5 years), it is possible that individuals with DHX9-associated NDD could develop axonal neuropathy in adulthood and that adults with DHX9-associated CMT2 could have had unreported mild NDD traits (e.g., learning disabilities or speech delay). It is also possible that the prevalence of axonal neuropathy in DHX9-associated NDD might be higher given that axonal neuropathy was identified in the single individual who underwent EMG and a NCS. Moreover, nucleolar accumulation of DHX9 variants could alter phase separation, enhance partitioning into the nucleolus, and cause nucleolar dysfunction, such as last-exon frameshifts in HMGB1.67
Consistent with the association between pathogenic DHX9 variant alleles and DD/ID and CMT in humans, we have found that homozygous deletion of the mouse ortholog Dhx9 results in multiple behavioral and neurological abnormalities. Although further phenotyping of aged mice is needed, the observation of tremor in a subset of young adult mice is intriguing given that Trembler and TremblerJ mice, both Pmp22 CMT models, also exhibit tremor.68,69 Curiously, the homozygous-null line described here was viable, whereas the previously reported knockout mouse line exhibited embryonic lethality.70 Although the explanation for the variation in viability is not currently clear, differences in genetic construct and background could play a role. An important construct distinction in Lee et al.’s 1998 model was the insertion of a neomycin resistance (neo) cassette in the opposite reading frame in the middle of exon 2. On the C57BL/6 (unspecified substrain) × Sv129 mixed background, the neo cassette might have been toxic. This also illustrates how genetic modifiers and epigenetic variation between different genetic backgrounds could modify how mutagenesis approaches influence the essentialome.71,72 Nonetheless, our findings are consistent with those of prior studies of the Drosophila ortholog mle (maleless) in demonstrating the importance of DHX9 in development and nervous system function at the organismal level.73,74,75 Additional phenotyping of Dhx9+/− mice, including cognitive testing, epileptic susceptibility, and peripheral nerve studies, will provide important insights as a model of human haploinsufficiency.
In summary, we have provided evidence that variants in DHX9 are a cause of two AD human neurogenetic disease traits: DD/ID and axonal CMT. Our allelic affinity studies, like those performed on SOX10, where nonsense variants and premature termination codons (PTCs) conveyed different neurological disease traits depending upon whether they escaped NMD (PCWH syndrome [MIM: 609136]) or not (Waardenburg syndrome type 4C [MIM: 613266]), provide insight into human nervous system development.55,76 Our allelic series provides a starting point for the dissection of the role of DHX9 in human neurodevelopment and neurodegeneration. As for DDX3X and DHX30, expansion of the DHX9 allelic series and functional studies will facilitate genotype-phenotype correlations and provide greater insights into the contribution of DHX9 to human health and disease.
Acknowledgments
This study was supported in part by the US National Human Genome Research Institute (NHGRI) and National Heart, Lung, and Blood Institute (NHBLI) via the Baylor-Hopkins Center for Mendelian Genomics (BHCMG; grant UM1 HG006542 to J.R.L), by the NHGRI via the GREGoR Consortium (grant U01 HG011758 to J.E.P., J.R.L., and R.A.G.) and the Baylor College of Medicine Human Genome Sequencing Center (grant U54HG003273 to R.A.G.), by the US National Institute of Neurological Disorders and Stroke (NINDS; grant R35NS105078 to J.R.L), by the Muscular Dystrophy Association (MDA; grant 512848 to J.R.L.), and by Spastic Paraplegia Foundation Research (grant to J.R.L.). This study was also supported by the General Research Fund of the Research Grants Council of Hong Kong (grants 24101921 to S.G. and 14104321 to H.H.C.) and the National Natural Science Foundation of China (grant 82202045 to S.G.). D.M. was supported by a Medical Genetics Research Fellowship Program through the US National Institutes of Health (NIH; grant T32 GM007526-42). T.M. was supported by the Uehara Memorial Foundation. D.P. was supported by a NINDS grant (1K23 NS125126-01A1) and a Rett Syndrome Research Trust fellowship award from the International Rett Syndrome Foundation (grant 3701-1). J.E.P. was supported by NHGRI K08 HG008986. D.G.C. was supported by an NIH Brain Disorders and Development Training Grant (T32 NS043124), the NIH Medical Genetics Research Fellowship Program (grant T32 GM007526), a Chao Physician Scientist Award, and an MDA Development Grant (873841). Additional acknowledgments can be found within the supplemental information. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Declaration of interests
J.R.L. has stock ownership in 23andMe, is a paid consultant for Genome International, and is a co-inventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at the Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories. F.M. and T.S.-S. are employees of GeneDx.
Published: July 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.06.013.
Contributor Information
Shen Gu, Email: shengu@cuhk.edu.hk.
James R. Lupski, Email: jlupski@bcm.edu.
Web resources
AlphaFold, https://alphafold.ebi.ac.uk/
Baylor College of Medicine Human Genome Sequencing Center, https://www.hgsc.bcm.edu
Baylor College of Medicine Lupski lab, https://github.com/BCM-Lupskilab
BrainSpan, https://www.brainspan.org/
Genotype-Tissue Expression (GTEx), https://gtexportal.org/home/
gnomAD Browser, https://gnomad.broadinstitute.org/
Human Splice Finder, https://www.genomnis.com/access-hsf
Online Mendelian Inheritance in Man, http://www.omim.org
phylop100way, GERP, MutationTaster, SIFT, and REVEL scores, https://varsome.com/
STRING, https://string-db.org/
UCSC Cell Browser, https://cells.ucsc.edu/
VarSeq, https://www.goldenhelix.com/products/VarSeq/index.html
Supplemental information
Data and code availability
All data described in this study are provided within the article and supplemental information. Raw sequencing data are available in dbGaP under BHCMG dbGaP study accession phs000711.v7.p2 or in the AnVIL repository under study name Baylor-Hopkins Center for Mendelian Genomics (https://anvilproject.org/).
References
- 1.Paine I., Posey J.E., Grochowski C.M., Jhangiani S.N., Rosenheck S., Kleyner R., Marmorale T., Yoon M., Wang K., Robison R., et al. Paralog studies augment gene discovery: DDX and DHX genes. Am. J. Hum. Genet. 2019;105:302–316. doi: 10.1016/j.ajhg.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cargill M., Venkataraman R., Lee S. DEAD-box RNA helicases and genome stability. Genes. 2021;12:1471. doi: 10.3390/genes12101471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snijders Blok L., Madsen E., Juusola J., Gilissen C., Baralle D., Reijnders M.R.F., Venselaar H., Helsmoortel C., Cho M.T., Hoischen A., et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am. J. Hum. Genet. 2015;97:343–352. doi: 10.1016/j.ajhg.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X., Posey J.E., Rosenfeld J.A., Bacino C.A., Scaglia F., Immken L., Harris J.M., Hickey S.E., Mosher T.M., Slavotinek A., et al. Phenotypic expansion in DDX3X – a common cause of intellectual disability in females. Ann. Clin. Transl. Neurol. 2018;5:1277–1285. doi: 10.1002/acn3.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lennox A.L., Hoye M.L., Jiang R., Johnson-Kerner B.L., Suit L.A., Venkataramanan S., Sheehan C.J., Alsina F.C., Fregeau B., Aldinger K.A., et al. Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development. Neuron. 2020;106:404–420.e8. doi: 10.1016/j.neuron.2020.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lessel D., Schob C., Küry S., Reijnders M.R.F., Harel T., Eldomery M.K., Coban-Akdemir Z., Denecke J., Edvardson S., Colin E., et al. De novo missense mutations in DHX30 impair global translation and cause a neurodevelopmental disorder. Am. J. Hum. Genet. 2017;101:716–724. doi: 10.1016/j.ajhg.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balak C., Benard M., Schaefer E., Iqbal S., Ramsey K., Ernoult-Lange M., Mattioli F., Llaci L., Geoffroy V., Courel M., et al. Rare de novo missense variants in RNA helicase DDX6 cause intellectual disability and dysmorphic features and lead to P-body defects and RNA dysregulation. Am. J. Hum. Genet. 2019;105:509–525. doi: 10.1016/j.ajhg.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Lelij P., Chrzanowska K.H., Godthelp B.C., Rooimans M.A., Oostra A.B., Stumm M., Zdzienicka M.Z., Joenje H., de Winter J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee T., Pelletier J. The biology of DHX9 and its potential as a therapeutic target. Oncotarget. 2016;7:42716–42739. doi: 10.18632/oncotarget.8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cristini A., Groh M., Kristiansen M.S., Gromak N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 2018;23:1891–1905. doi: 10.1016/j.celrep.2018.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hegazy Y.A., Fernando C.M., Tran E.J. The balancing act of R-loop biology: the good, the bad, and the ugly. J. Biol. Chem. 2020;295:905–913. doi: 10.1074/jbc.REV119.011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty P., Hiom K. DHX9-dependent recruitment of BRCA1 to RNA promotes DNA end resection in homologous recombination. Nat. Commun. 2021;12:4126. doi: 10.1038/s41467-021-24341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groh M., Gromak N. Out of balance: R-loops in human disease. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takashima H., Boerkoel C.F., John J., Saifi G.M., Salih M.A.M., Armstrong D., Mao Y., Quiocho F.A., Roa B.B., Nakagawa M., et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002;32:267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 15.El-Khamisy S.F., Saifi G.M., Weinfeld M., Johansson F., Helleday T., Lupski J.R., Caldecott K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 16.Ahel I., Rass U., El-Khamisy S.F., Katyal S., Clements P.M., McKinnon P.J., Caldecott K.W., West S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 17.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wohler E., Martin R., Griffith S., Rodrigues E.d.S., Antonescu C., Posey J.E., Coban-Akdemir Z., Jhangiani S.N., Doheny K.F., Lupski J.R., et al. PhenoDB, GeneMatcher and VariantMatcher, tools for analysis and sharing of sequence data. Orphanet J. Rare Dis. 2021;16:365. doi: 10.1186/s13023-021-01916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dawidziuk M., Gambin T., Bukowska-Olech E., Antczak-Marach D., Badura-Stronka M., Buda P., Budzynska E., Castaneda J., Chilarska T., Czyzyk E., et al. Exome sequencing reveals novel variants and expands the genetic landscape for congenital microcephaly. Genes. 2021;12:2014. doi: 10.3390/genes12122014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eldomery M.K., Coban-Akdemir Z., Harel T., Rosenfeld J.A., Gambin T., Stray-Pedersen A., Küry S., Mercier S., Lessel D., Denecke J., et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26. doi: 10.1186/s13073-017-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calame D.G., Bakhtiari S., Logan R., Coban-Akdemir Z., Du H., Mitani T., Fatih J.M., Hunter J.V., Herman I., Pehlivan D., et al. Biallelic loss-of-function variants in the splicing regulator NSRP1 cause a severe neurodevelopmental disorder with spastic cerebral palsy and epilepsy. Genet. Med. 2021;23:2455–2460. doi: 10.1038/s41436-021-01291-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bainbridge M.N., Wang M., Wu Y., Newsham I., Muzny D.M., Jefferies J.L., Albert T.J., Burgess D.L., Gibbs R.A. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12 doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Challis D., Yu J., Evani U.S., Jackson A.R., Paithankar S., Coarfa C., Milosavljevic A., Gibbs R.A., Yu F. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinf. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid J.G., Carroll A., Veeraraghavan N., Dahdouli M., Sundquist A., English A., Bainbridge M., White S., Salerno W., Buhay C., et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinf. 2014;15:30. doi: 10.1186/1471-2105-15-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansen A.W., Murugan M., Li H., Khayat M.M., Wang L., Rosenfeld J., Andrews B.K., Jhangiani S.N., Coban Akdemir Z.H., Sedlazeck F.J., et al. A genocentric approach to discovery of Mendelian disorders. Am. J. Hum. Genet. 2019;105:974–986. doi: 10.1016/j.ajhg.2019.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Retterer K., Juusola J., Cho M.T., Vitazka P., Millan F., Gibellini F., Vertino-Bell A., Smaoui N., Neidich J., Monaghan K.G., et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016;18:696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 28.Fromer M., Moran J.L., Chambert K., Banks E., Bergen S.E., Ruderfer D.M., Handsaker R.E., McCarroll S.A., O’Donovan M.C., Owen M.J., et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012;91:597–607. doi: 10.1016/j.ajhg.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gambin T., Akdemir Z.C., Yuan B., Gu S., Chiang T., Carvalho C.M.B., Shaw C., Jhangiani S., Boone P.M., Eldomery M.K., et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45:1633–1648. doi: 10.1093/nar/gkw1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C., Jolly A., Shayota B.J., Mazzeu J.F., Du H., Dawood M., Soper P.C., Ramalho de Lima A., Ferreira B.M., Coban-Akdemir Z., et al. Novel pathogenic variants and quantitative phenotypic analyses of Robinow syndrome: WNT signaling perturbation and phenotypic variability. HGG Adv. 2022;3 doi: 10.1016/j.xhgg.2021.100074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Posey J.E., Harel T., Liu P., Rosenfeld J.A., James R.A., Coban Akdemir Z.H., Walkiewicz M., Bi W., Xiao R., Ding Y., et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017;376:21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu P., Meng L., Normand E.A., Xia F., Song X., Ghazi A., Rosenfeld J., Magoulas P.L., Braxton A., Ward P., et al. Reanalysis of clinical exome sequencing data. N. Engl. J. Med. 2019;380:2478–2480. doi: 10.1056/NEJMc1812033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson P.N., Köhler S., Bauer S., Seelow D., Horn D., Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greene D., Richardson S., Turro E. ontologyX: a suite of R packages for working with ontological data. Bioinforma. Oxf. Engl. 2017;33:1104–1106. doi: 10.1093/bioinformatics/btw763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinforma. Oxf. Engl. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 36.Fuchs H., Aguilar-Pimentel J.A., Amarie O.V., Becker L., Calzada-Wack J., Cho Y.-L., Garrett L., Hölter S.M., Irmler M., Kistler M., et al. Understanding gene functions and disease mechanisms: phenotyping pipelines in the German Mouse Clinic. Behav. Brain Res. 2018;352:187–196. doi: 10.1016/j.bbr.2017.09.048. [DOI] [PubMed] [Google Scholar]
- 37.Garrett L., Lie D.C., Hrabé de Angelis M., Wurst W., Hölter S.M. Voluntary wheel running in mice increases the rate of neurogenesis without affecting anxiety-related behaviour in single tests. BMC Neurosci. 2012;13:61. doi: 10.1186/1471-2202-13-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuchs H., Gailus-Durner V., Neschen S., Adler T., Afonso L.C., Aguilar-Pimentel J.A., Becker L., Bohla A., Calzada-Wack J., Cohrs C., et al. Innovations in phenotyping of mouse models in the German Mouse Clinic. Mamm. Genome. 2012;23:611–622. doi: 10.1007/s00335-012-9415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gailus-Durner V., Fuchs H., Becker L., Bolle I., Brielmeier M., Calzada-Wack J., Elvert R., Ehrhardt N., Dalke C., Franz T.J., et al. Introducing the German Mouse Clinic: open access platform for standardized phenotyping. Nat. Methods. 2005;2:403–404. doi: 10.1038/nmeth0605-403. [DOI] [PubMed] [Google Scholar]
- 40.Heermann T., Garrett L., Wurst W., Fuchs H., Gailus-Durner V., Hrabě de Angelis M., Graw J., Hölter S.M. Crybb2 mutations consistently affect schizophrenia endophenotypes in mice. Mol. Neurobiol. 2019;56:4215–4230. doi: 10.1007/s12035-018-1365-5. [DOI] [PubMed] [Google Scholar]
- 41.Rathkolb B., Hans W., Prehn C., Fuchs H., Gailus-Durner V., Aigner B., Adamski J., Wolf E., Hrabě de Angelis M. Clinical chemistry and other laboratory tests on mouse plasma or serum. Curr. Protoc. Mouse Biol. 2013;3:69–100. doi: 10.1002/9780470942390.mo130043. [DOI] [PubMed] [Google Scholar]
- 42.Rathkolb B., Fuchs H., Gailus-Durner V., Aigner B., Wolf E., Hrabě de Angelis M. Blood collection from mice and hematological analyses on mouse blood. Curr. Protoc. Mouse Biol. 2013;3:101–119. doi: 10.1002/9780470942390.mo130054. [DOI] [PubMed] [Google Scholar]
- 43.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins R.L., Glessner J.T., Porcu E., Lepamets M., Brandon R., Lauricella C., Han L., Morley T., Niestroj L.-M., Ulirsch J., et al. A cross-disorder dosage sensitivity map of the human genome. Cell. 2022;185:3041–3055.e25. doi: 10.1016/j.cell.2022.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mannucci I., Dang N.D.P., Huber H., Murry J.B., Abramson J., Althoff T., Banka S., Baynam G., Bearden D., Beleza-Meireles A., et al. Genotype–phenotype correlations and novel molecular insights into the DHX30-associated neurodevelopmental disorders. Genome Med. 2021;13:90. doi: 10.1186/s13073-021-00900-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aratani S., Oishi T., Fujita H., Nakazawa M., Fujii R., Imamoto N., Yoneda Y., Fukamizu A., Nakajima T. The nuclear import of RNA helicase A is mediated by importin-alpha3. Biochem. Biophys. Res. Commun. 2006;340:125–133. doi: 10.1016/j.bbrc.2005.11.161. [DOI] [PubMed] [Google Scholar]
- 48.Tang H., McDonald D., Middlesworth T., Hope T.J., Wong-Staal F. The carboxyl terminus of RNA helicase A contains a bidirectional nuclear transport domain. Mol. Cell Biol. 1999;19:3540–3550. doi: 10.1128/MCB.19.5.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kosugi S., Hasebe M., Tomita M., Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiel L., Baakman C., Gilissen D., Veltman J.A., Vriend G., Gilissen C. MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 2019;40:1030–1038. doi: 10.1002/humu.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coban-Akdemir Z., White J.J., Song X., Jhangiani S.N., Fatih J.M., Gambin T., Bayram Y., Chinn I.K., Karaca E., Punetha J., et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am. J. Hum. Genet. 2018;103:171–187. doi: 10.1016/j.ajhg.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.England J.D., Gronseth G.S., Franklin G., Carter G.T., Kinsella L.J., Cohen J.A., Asbury A.K., Szigeti K., Lupski J.R., Latov N., et al. Vol. 72. Neurology; 2008. Practice parameter: evaluation of distal symmetric polyneuropathy: role of autonomic testing, nerve biopsy, and skin biopsy (an evidence-based review) pp. 177–184. [DOI] [PubMed] [Google Scholar]
- 53.Herman I., Jolly A., Du H., Dawood M., Abdel-Salam G.M.H., Marafi D., Mitani T., Calame D.G., Coban-Akdemir Z., Fatih J.M., et al. Quantitative dissection of multilocus pathogenic variation in an Egyptian infant with severe neurodevelopmental disorder resulting from multiple molecular diagnoses. Am. J. Med. Genet. 2022;188:735–750. doi: 10.1002/ajmg.a.62565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garrelfs M.R., Takada S., Kamsteeg E.-J., Pegge S., Mancini G., Engelen M., van de Warrenburg B., Rennings A., van Gaalen J., Peters I., et al. The phenotypic spectrum of PNKP-associated disease and the absence of immunodeficiency and cancer predisposition in a Dutch cohort. Pediatr. Neurol. 2020;113:26–32. doi: 10.1016/j.pediatrneurol.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 55.Lupski J.R. Biology in balance: human diploid genome integrity, gene dosage, and genomic medicine. Trends Genet. 2022;38:554–571. doi: 10.1016/j.tig.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caldecott K.W. DNA single-strand break repair and human genetic disease. Trends Cell Biol. 2022;32:733–745. doi: 10.1016/j.tcb.2022.04.010. [DOI] [PubMed] [Google Scholar]
- 57.Alt F.W., Schwer B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair. 2018;71:158–163. doi: 10.1016/j.dnarep.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madabhushi R., Gao F., Pfenning A.R., Pan L., Yamakawa S., Seo J., Rueda R., Phan T.X., Yamakawa H., Pao P.-C., et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suberbielle E., Sanchez P.E., Kravitz A.V., Wang X., Ho K., Eilertson K., Devidze N., Kreitzer A.C., Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013;16:613–621. doi: 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McConnell M.J., Lindberg M.R., Brennand K.J., Piper J.C., Voet T., Cowing-Zitron C., Shumilina S., Lasken R.S., Vermeesch J.R., Hall I.M., Gage F.H. Mosaic copy number variation in human neurons. Science. 2013;342:632–637. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou T., Lee J.W., Tatavarthi H., Lupski J.R., Valerie K., Povirk L.F. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1) Nucleic Acids Res. 2005;33:289–297. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Date H., Onodera O., Tanaka H., Iwabuchi K., Uekawa K., Igarashi S., Koike R., Hiroi T., Yuasa T., Awaya Y., et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 2001;29:184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- 63.Grunseich C., Wang I.X., Watts J.A., Burdick J.T., Guber R.D., Zhu Z., Bruzel A., Lanman T., Chen K., Schindler A.B., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Mol. Cell. 2018;69:426–437.e7. doi: 10.1016/j.molcel.2017.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chakraborty P., Huang J.T.J., Hiom K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018;9:4346. doi: 10.1038/s41467-018-06677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fuchsová B., Hozák P. The localization of nuclear DNA helicase II in different nuclear compartments is linked to transcription. Exp. Cell Res. 2002;279:260–270. doi: 10.1006/excr.2002.5617. [DOI] [PubMed] [Google Scholar]
- 66.Zhang S., Herrmann C., Grosse F. Nucleolar localization of murine nuclear DNA helicase II (RNA helicase A) J. Cell Sci. 1999;112(Pt 16):2693–2703. doi: 10.1242/jcs.112.16.2693. [DOI] [PubMed] [Google Scholar]
- 67.Mensah M.A., Niskanen H., Magalhaes A.P., Basu S., Kircher M., Sczakiel H.L., Reiter A.M.V., Elsner J., Meinecke P., Biskup S., et al. Aberrant phase separation and nucleolar dysfunction in rare genetic diseases. Nature. 2023;614:564–571. doi: 10.1038/s41586-022-05682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suter U., Welcher A.A., Ozcelik T., Snipes G.J., Kosaras B., Francke U., Billings-Gagliardi S., Sidman R.L., Shooter E.M. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 69.Suter U., Moskow J.J., Welcher A.A., Snipes G.J., Kosaras B., Sidman R.L., Buchberg A.M., Shooter E.M. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc. Natl. Acad. Sci. USA. 1992;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee C.-G., da Costa Soares V., Newberger C., Manova K., Lacy E., Hurwitz J. RNA helicase A is essential for normal gastrulation. Proc. Natl. Acad. Sci. USA. 1998;95:13709–13713. doi: 10.1073/pnas.95.23.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bertomeu T., Coulombe-Huntington J., Chatr-Aryamontri A., Bourdages K.G., Coyaud E., Raught B., Xia Y., Tyers M. A high-resolution genome-wide CRISPR/Cas9 viability screen reveals structural features and contextual diversity of the human cell-essential proteome. Mol. Cell Biol. 2018;38 doi: 10.1128/MCB.00302-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Belote J.M., Lucchesi J.C. Control of X chromosome transcription by the maleless gene in Drosophila. Nature. 1980;285:573–575. doi: 10.1038/285573a0. [DOI] [PubMed] [Google Scholar]
- 74.Kernan M.J., Kuroda M.I., Kreber R., Baker B.S., Ganetzky B. napts, a mutation affecting sodium channel activity in Drosophila, is an allele of mle, a regulator of X chromosome transcription. Cell. 1991;66:949–959. doi: 10.1016/0092-8674(91)90440-a. [DOI] [PubMed] [Google Scholar]
- 75.Garber G., Smith L.A., Reenan R.A., Rogina B. Effect of sodium channel abundance on Drosophila development, reproductive capacity and aging. Fly (Austin) 2012;6:57–67. doi: 10.4161/fly.18570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Inoue K., Khajavi M., Ohyama T., Hirabayashi S.i., Wilson J., Reggin J.D., Mancias P., Butler I.J., Wilkinson M.F., Wegner M., Lupski J.R. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described in this study are provided within the article and supplemental information. Raw sequencing data are available in dbGaP under BHCMG dbGaP study accession phs000711.v7.p2 or in the AnVIL repository under study name Baylor-Hopkins Center for Mendelian Genomics (https://anvilproject.org/).