

Abstract
Purpose:
Despite limited genetic and histologic heterogeneity, Ewing sarcoma (EwS) tumor cells are transcriptionally heterogeneous and display varying degrees of mesenchymal lineage specification in vitro. In this study, we investigated if and how transcriptional heterogeneity of EwS cells contributes to heterogeneity of tumor phenotypes in vivo.
Experimental Design:
Single cell proteogenomic-sequencing of EwS cell lines was performed and integrated with patient tumor transcriptomic data. Cell subpopulations were isolated by FACS for assessment of gene expression and phenotype. Digital spatial profiling and human whole transcriptome analysis interrogated transcriptomic heterogeneity in EwS xenografts. Tumor cell subpopulations and matrix protein deposition were evaluated in xenografts and patient tumors using multiplex immunofluorescence staining.
Results:
We identified CD73 as a biomarker of highly mesenchymal EwS cell subpopulations in tumor models and patient biopsies. CD73+ tumor cells displayed distinct transcriptional and phenotypic properties, including selective upregulation of genes that are repressed by EWS::FLI1, and increased migratory potential. CD73+ cells were distinguished in vitro and in vivo by increased expression of matrisomal genes and abundant deposition of extracellular matrix (ECM) proteins. In epithelial-derived malignancies, ECM is largely deposited by cancer-associated fibroblasts (CAFs) and we thus labeled CD73+ EwS cells, CAF-like tumor cells. Marked heterogeneity of CD73+ EwS cell frequency and distribution were detected in tumors in situ, and CAF-like tumor cells and associated ECM were observed in peri-necrotic regions and invasive foci.
Conclusions:
EwS tumor cells can adopt CAF-like properties and these distinct cell subpopulations contribute to tumor heterogeneity by remodeling the tumor microenvironment.
INTRODUCTION
Intratumoral heterogeneity promotes cancer growth, metastasis, therapeutic failure, and drug resistance (1, 2). Cancer-associated fibroblasts (CAFs) contribute to heterogeneity of epithelial-derived tumors by depositing extracellular matrix (ECM) proteins that dynamically remodel the tumor microenvironment (TME) (3). In addition, genomic instability of tumor cells leads to clonal evolution that contributes to mutational heterogeneity of the tumor cell compartment (2). While genomic instability can drive tumor cell heterogeneity, more recent studies have highlighted the role of non-mutational epigenetic heterogeneity (4). Single-cell sequencing and spatial analyses demonstrate that tumor ecosystems are comprised of heterogeneous tumor cells with distinct epigenetic and phenotypic states (5, 6). Non-mutational tumor cell heterogeneity is particularly relevant to pediatric tumors which typically have low mutation rates but high transcriptional and phenotypic heterogeneity due to hijacking of developmental and epigenetic regulators (7). Phenotypically distinct tumor cell subpopulations differentially contribute to tumor growth, metastatic potential, and therapy response, highlighting the critical importance of studying tumor cell subpopulations in the context of contextually relevant ecosystems (8–10).
Ewing sarcomas (EwS) are histologically primitive, monomorphous bone and soft tissue tumors that present across the lifespan, with a peak incidence in adolescence (11). Intensive cytotoxic therapy has improved prognosis for patients with localized tumors, but outcomes for patients with metastastic and relapsed EwS remain dismal (12). EwS tumors are initiated by chromosomal translocations between FET and ETS gene family loci, that generate chimeric in-frame fusion proteins, most commonly EWS::FLI1 (11). These fusions function as oncogenic transcription factors that cause widespread epigenomic reprogramming and aberrant activation and repression of hundreds of target genes (11, 13, 14). Recurrent mutations in other genes are uncommon and propagation of EwS tumors depends on continued expression of the fusion (11). Although intratumoral genomic instability is not a known feature of EwS, rare subpopulations of tumor cells that express low levels of EWS::FLI1 have been identified and these cells display enhanced metastatic properties in tumor models (15). In addition, modulation of EWS::FLI1-dependent gene signatures, without altering expression of the fusion itself, influences tumor cell state (reviewed in Ref. (16)). Genomic loss of STAG2 (17, 18), expression of tissue-specific transcription factors (19–21), activation of the Wnt/β-catenin pathway (22), and other cell-intrinsic and cell-extrinsic mechanisms (16), all influence transcriptional activity of EWS::FLI1 and affect the tumorigenic and metastatic properties of EwS cells. Importantly, despite different underlying molecular origins, EWS::FLI1-depleted cells universally show increased expression of mesenchymal lineage genes that are normally repressed by the fusion. The contribution of EWS::FLI1-low, mesenchymal-high state tumor cells to EwS pathogenesis has yet to be defined in the context of tumors in vivo.
Here, we set out to identify and characterize EWS::FLI1-low cells in EwS tumors in situ. Transcriptomic profiling of EwS cell lines and patient tumors identified NT5E (CD73) as a cell surface marker that consistently marks EWS::FLI1-low tumor cells in vitro and in vivo. Phenotypic, proteogenomic, and immunofluorescence profiling confirmed that CD73+ cells exist as spatially distributed cell subpopulations that are enriched along tumor borders and invasive fronts. Further, our studies showed that CD73+ cells deposit protumorigenic ECM proteins such as tenascin-C (TNC), collagens, and proteoglycans, proteins that are largely deposited by CAFs in carcinomas (23). This matrisomal gene program is normally repressed by EWS::FLI1, validating that CD73+ tumor cells have lost EWS::FLI1 activity. Despite this, we found that expression of the EWS::FLI1-activated gene signature is largely retained by CAF-like tumor cells, revealing the novel discovery that the activating and repressive properties of the fusion can be dissociated in individual tumor cells, creating cells with hybrid transcriptional states. Together these studies identify the existence of transcriptionally and phenotypically distinct subpopulations of EwS tumor cells that contribute to tumor heterogeneity and remodel the TME via deposition of pro-tumorigenic ECM.
METHODS
Cell lines
EwS cell lines A673 (RRID:CVCL_0080), SKNMC (RRID:CVCL_0530), CHLA10 (RRID:CVCL_6583), CHLA9 (RRID:CVCL_M150), A4573 (RRID:CVCL_6245), TC32 (RRID:CVCL_7151), TC71 (RRID:CVCL_2213), RDES (RRID:CVCL_2169) as well as U2OS (RRID:CVCL_0042, osteosarcoma cell line) were obtained from ATCC and COG (https://www.childrensoncologygroup.org/) cell line repositories. A673, TC32, TC71, A4573, and SKNMC cell lines were maintained in RPMI 1640 media (Gibco) supplemented with 10% FBS (Atlas Biologicals) and 2mmol/L-glutamine (Life Technologies). RDES was cultured in RPMI 1640 media supplemented with 15% FBS and 2mmol/L-glutamine. CHLA10 and CHLA9 were maintained in IMDM media (Fisher) supplemented with 20% FBS, 2mmol/L-glutamine, and 1X Insulin-Transferrin-Selenium-Ethanolamine (Gibco). U2OS was maintained in McCoy’s 5A media (Gibco) supplemented with 10% FBS and 2mmol/L-glutamine. H7-MSCs (kind gift from Dr. Sweet-Cordero) were maintained in alpha-MEM supplemented with 10% FBS, 2 mM L-glutamine, 1% antibiotic-antimycotic (Gibco). Cells were cultured at 37°C with 5% CO2. Cells were all confirmed to be mycoplasma free and identities subject to STR-confirmation every 6 months.
PDX305 generation and characterization
PDX305 was generated from an abdominal soft tissue metastasis from 17-year-old male Ewing sarcoma patient. Cells were passaged as a patient-derived xenograft in NSG mice before cell line generation. PDX305 early passage cells were maintained in RPMI 1640 media (Gibco) supplemented with 10% FBS (Atlas Biologicals), 2mM L-glutamine (Life Technologies), and 1% Antibiotic-Antimycotic. Poly(A)-capture RNA-seq was performed on PDX305 cells as described in the RNA-seq sections.
Flow cytometry and FACS
EwS cells were trypsinized, washed with PBS, resuspended in 2% FBS in PBS, and stained with fluorophore-conjugated antibodies (Supplemental Table 1) for 30 min. on ice in the dark. Fluorophore-conjugated isotype controls and unstained controls were used for each experiment. Flow cytometry was performed on a BD Accuri C6 (10,000 events), with single cell identification by FSC and SSC. For FACS, cells were sorted on a BD FACSAria Il. Analysis was conducted in FCS Express (De Novo Software).
RT-qPCR
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen) and cDNA was generated using the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. qPCR was performed using either Taqman Fast Universal PCR Master Mix (Applied Biosystems) assays or iTaq Universal SYBR-Green Supermix (Bio-Rad) on a Roche Light-Cycler 480 instrument (Roche Applied Science). Samples were run in technical triplicates and average Ct values were normalized to the geometric mean of two reference genes. The relative mRNA expression was calculated by the ddCt method. Primers and TaqMan assays.
Tumor microarray preparation and immunohistochemistry
A tumor tissue microarray (TMA) of Ewing sarcoma tumors was prepared by Seattle Children’s Hospital Pathology. For IHC, TMA sections were pretreated with CC1 Tris buffer (pH 8) for 32 minutes, primary antibodies (see Supplemental Table 1) were incubated 1:100 for 32 minutes at 36°C. Staining was detected using OptiView DAB (Roche).
Immunofluorescence
Cells
For immunofluorescence, cells were fixed with 4% PFA in PBS for 10 min then washed with PBS. Sorted CD73−/CD73+ cells were plated at 60,000 cells/well in 8-well glass bottomed chambers overnight. Cells were permeabilized with 10 min. 0.3% Triton-X then blocked with 0.2% BSA in PBS for 1hr at room temperature. Primary antibodies were incubated for 1 hr at room temperature in 0.2% BSA (Supplemental Table 1). Alexa-fluor conjugated secondary antibodies were incubated for 1hr at room temperature in 0.2% BSA with 5% host serum (unless goat and donkey-derived secondary antibodies were used simultaneously, in which case host serum was not included). Signal was compared to matched species-specific IgG controls incubated at the same concentrations. For F-actin staining ActinGreen (ThermoFisher Scientific #R37110) was used at 2 drops/mL in the secondary antibody solution. DAPI was included during secondary antibody incubations to mark nuclei at 1 ug/mL. Coverslips were mounted with ProLong Gold and allowed to cure overnight at room temperature in the dark. Slides were imaged on a Leica SPE confocal at 40X magnification or scanned using an Olympus slide scanner (BX61) at 10X and 20X magnification.
Tissue
FFPE tumor samples from xenografts or primary patient samples were deparaffinized, then antigen retrieval was carried out using Diva Decloaker in a Biocare Medical decloaking chamber (120°C for 30 sec.) and slides were allowed to cool to room temperature. Slides were blocked with 0.2% BSA for 1 hour at room temperature. Primary and secondary antibodies (Supplemental Table 1) were incubated in 0.2% BSA for 1 hour at room temperature. When both goat and donkey derived antibodies were used, donkey-anti-goat secondary (RRID:AB_2535853) was applied first, washed 3×10 min. with PBS, then additional secondaries were added to prevent cross-species reactivity. DAPI was included during secondary antibody incubations to mark nuclei at 1 ug/mL. Coverslips were mounted with Prolong Gold and allowed to cure overnight at room temperature in the dark, then slides were stored at 4°C until imaging. Slides were imaged on a Leica SPE confocal at 40X magnification or scanned on an Olympus slide scanner at 10X and 20X magnification.
Migration and proliferation assays
Real-time cell analysis (RTCA) of cell migration and proliferation was monitored using a CIM-plate 16 or E-plate 16, respectively, on the xCELLigence DP system (Acea Bioscience). For proliferation, wells were coated with 0.2% gelatin and 100 μL complete media was added (5 × 103 cells/well), and plates equilibrated for 1 hour at 37°C. For migration, before cell seeding, electrodes were coated with 0.2% gelatin and 50 μL complete media was placed in the upper chambers and 160 μL serum-free media was added to the lower chambers. 5 × 104 cells/well were plated in the upper chamber in 100 μL media, then plates were equilibrated for 30 minutes at room temperature. Migration was evaluated up to 30 hours. Proliferation was evaluated up to 96 hours.
3D collagen invasion assays
Spheroids were formed by plating EwS single cells at a density of 50,000 viable cells/mL in complete media in non-adherent 6 well plates overnight. Debris and single cells were removed by centrifuging samples for 10 sec. at 300g. Rat tail collagen (Gibco) was prepared at 1.8–2mg/mL in 0.08N acetic acid, supplemented with 10X DMEM (final concentration 1X), neutralized with 1M NaOH and allowed to polymerize on ice for 45–60 minutes until fibers formed (24). Spheroids and collagen gels were mixed and plated as 80 μL droplets and allowed to polymerize for 30 minutes at 37°C, at which point 1mL of complete IMDM media (20% FBS, 1X L-glutamine, ITS-X) was added. 40 μL underlays of polymerized rat tail collagen were first applied to the bottom of TC treated 24wp plates to prevent spheroids from attaching to or spreading along the plate surface. Spheroids were cultured 4–6 days then assessed by phase contrast microscopy and fixed for 15 minutes with 4% PFA in PBS. Invasion was scored as protrusive cells or multicellular strands extending from the spheroid border.
Subcutaneous injections of CD73+ vs. CD73− cells
Cells were sorted based on CD73 positivity and 540,000 (CHLA10) or 720,000 (A673) CD73− or CD73+ single cells were resuspended in PBS, diluted 1:1 in Matrigel (Corning), and injected subcutaneously into NSG mice (005557, Jackson Laboratory). Tumor formation and growth was measured by calipers every other day. All mice were euthanized at end point when the tumor of one animal reach 2cm diameter in any direction.
Digital Spatial Profiling
GeoMx Digital Spatial Profiling (NanoString) was carried out according to manufacturer instructions. Tumor specimens were generated as follows: subcutaneous injection of 4×105 CHLA10 cells in 50% Matrigel, primary tumors collected 54 days post-injection; tail vein injection of 7×105 CHLA10 cells injected in PBS, tumors collected 28 days post-injection. Tumors were fixed for 24 hours in 10% non-buffered formalin before paraffin embedding. 3 sections of FFPE tumor tissues (1 from subcutaneous tumor, 2 from 2 different tail vein-injected mice) were sectioned and adhered to a single slide. Hybridization of the Human NGS Whole Transcriptome Atlas probe library was performed according to manufacturer instructions overnight at 37°C. Each probe is tagged with a photocleavable unique molecular identifier (UMI) for NGS-based readout. Slides were blocked for 30’ before staining with morphology markers (Supplemental Table 1), and SYTO DNA dye. After slide scanning, 12 ROIs comprising >1000 SYTO+ nuclei each were selected, UV-cleaved, and collected for NGS sequencing to quantify probe counts. Library preparation and Illumina NGS were carried out by NanoString. The NanoString GeoMx NGS data analysis pipeline was used (version 2.3.4) to assess probe counts, including probe and count-level QC steps using the GeoMxTools R package (RRID:SCR_023424). Gene level count data were normalized using the quartile 3 (Q3) method comparing the upper quartile of counts in each segment with the geometric mean of negative control probes. Highly variable genes were identified by ranking genes by coefficient of variation across all 12 ROIs (CV = standard deviation/mean).
Western blots
Cells were lysed with RIPA buffer (Fisher Scientific) supplemented with protease and phosphatase inhibitors (Sigma). Western blot was performed using the Bio-Rad Mini-PROTEAN Tetra System. Following transfer, nitrocellulose membranes were blocked in Odyssey Blocking Buffer (LI-COR) for 1 hour. Membranes were washed and incubated rotating overnight at 4°C with a primary antibody. Membranes were then washed three times in TBST for 5 min each and incubated with a secondary antibodies (LI-COR IRDye 700CW or 800CW; 1:10,000) for 1 hour. Membranes were imaged on a LI-COR Odyssey scanner.
Proteogenomic single-cell sequencing (CITE-seq)
CITE-seq performed as described previously (25). 500,000 cells were resuspended in PBS + 1% FBS and spun at 300g for 10 minutes at 4°C and then resuspended in 25 μL staining buffer (BioLegend #420201). 2.5 μL human TruStain FcX (BioLegend #42230) was added per sample and incubated at 4°C for 10 minutes. Hash-Tag Antibodies, used to label the samples and minimize batch effects, were added at 250 ng/sample (0.5 μL/sample) and incubated at 4°C for 30 minutes. Cells were washed twice in 1 mL staining buffer and spun at 300g for 5 minutes at 4°C. Samples were pooled 1:1 at 1000 cells/μL and libraries were generated using the 3’ V3 10X Genomics Chromium Controller following the manufacturer’s protocol (CG000183). Final library quality was assessed using the Tapestation 4200 (Agilent) and libraries were quantified by Kapa qPCR (Roche). Pooled libraries were then subjected to paired-end sequencing according to the manufacturer’s protocol (Illumina NovaSeq 6000). Bcl2fastq2 Conversion Software (Illumina) was used to generate de-multiplexed Fastq files and the CellRanger (3.1) Pipeline (10X Genomics) was used to align reads and generate count matrices. Seurat’s FindMarkers function was used to identify differentially expressed genes in NT5E expressing vs. NT5E negative cells, which were ranked by fold change. Further quality control and analyses were performed in R (see Lawlor Lab Github for code).
Pathway analysis
Gene ontology and GSEA for selected gene lists was performed using MSigDB, Enrichr (26), Metascape (27), escape and dittoSeq (Bioconductor, see supplementary R notebooks on GitHub).
RNA-sequencing
Poly(A)-capture RNA-seq was performed on PDX305 and FACS sorted CD73−/+ A673 and CHLA10 cells (RNA isolated from cell pellets after sorting). Libraries were prepared with NEBNext Ultra II RNA Library Prep kit and Paired end 150 bp sequencing was performed on a Novaseq600 by Novogene. Adapter trimming was performed using Trim Galore (RRID:SCR_011847, Babraham Institute). Trimmer reads were aligned to GRCh38 using the STAR aligner (RRID:SCR_004463) (29). Counts and differential expression analysis were calculated by DESeq2 v1.18.1 (RRID:SCR_015687) using FDR-adjusted p-values <0.05.
Public data sources
Log2 expression values for tumor microarray datasets (GSE34620, GSE142162, GSE17679) were downloaded from the R2 Genomics Analysis and Visualization Platform. Only Ewing sarcoma patient tumor samples were included.
Statistics and software
Statistical tests were conducted in GraphPad Prism 9 (RRID:SCR_002798). Error bars = SEM unless otherwise specified. Unpaired t-tests were used unless otherwise specified. ns=p>0.05, *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001. BioRender.com was used to prepare experimental schematics.
Study approval
All experiments conducted in mice were approved by the Institutional Animal Care and Use Committee of Seattle Children’s Research Institute.
Data and code availability
Sequencing data generated by this study have been deposited to NCBI Gene Expression Omnibus (RRID:SCR_005012) (GSE236289) database. Analyses in R will be available upon publication at https://github.com/LawlorLab/CD73_CAFlike.
RESULTS
CD73 marks mesenchymal-high state EwS cell subpopulations
To study EWS::FLI1-low tumor cells in patient tumors and tumor xenografts in vivo, we first sought to define a cell surface marker that would permit their identification and isolation from bulk tumor cells. Given that EWS::FLI1 normally represses mesenchymal lineage genes (28, 29), we reasoned that expression of mesenchymal genes could be used to discriminate EWS::FLI1-low cells. Single cell transcriptomic studies recently showed that EwS cells exist along a transcriptional axis that partially reflects a mesenchyme development trajectory (19, 30). Using these data, we confirmed that activation of a mesenchymal development signature highly correlates with loss of EWS::FLI1-dependent gene repression (Figure 1A). Next, we intersected the two gene signatures to identify transcripts that are normally repressed by EWS::FLI1 (31) and expressed by mesenchymal-high state tumor cells (19) Fifty-two transcripts were included in the overlap, including NT5E which encodes the cell surface protein CD73 (Figure 1B, Supplemental Table 2). NT5E/CD73 (5’-nucleotidase ecto) is a GPI-anchored enzyme that is highly expressed by normal mesenchymal stem cells (MSC) (32), and we and others have previously shown that its expression is transcriptionally inhibited by EWS::FLI1 (19, 29).
Figure 1. CD73 marks mesenchymal tumor cells that express EWS::FLI1-repressed genes.
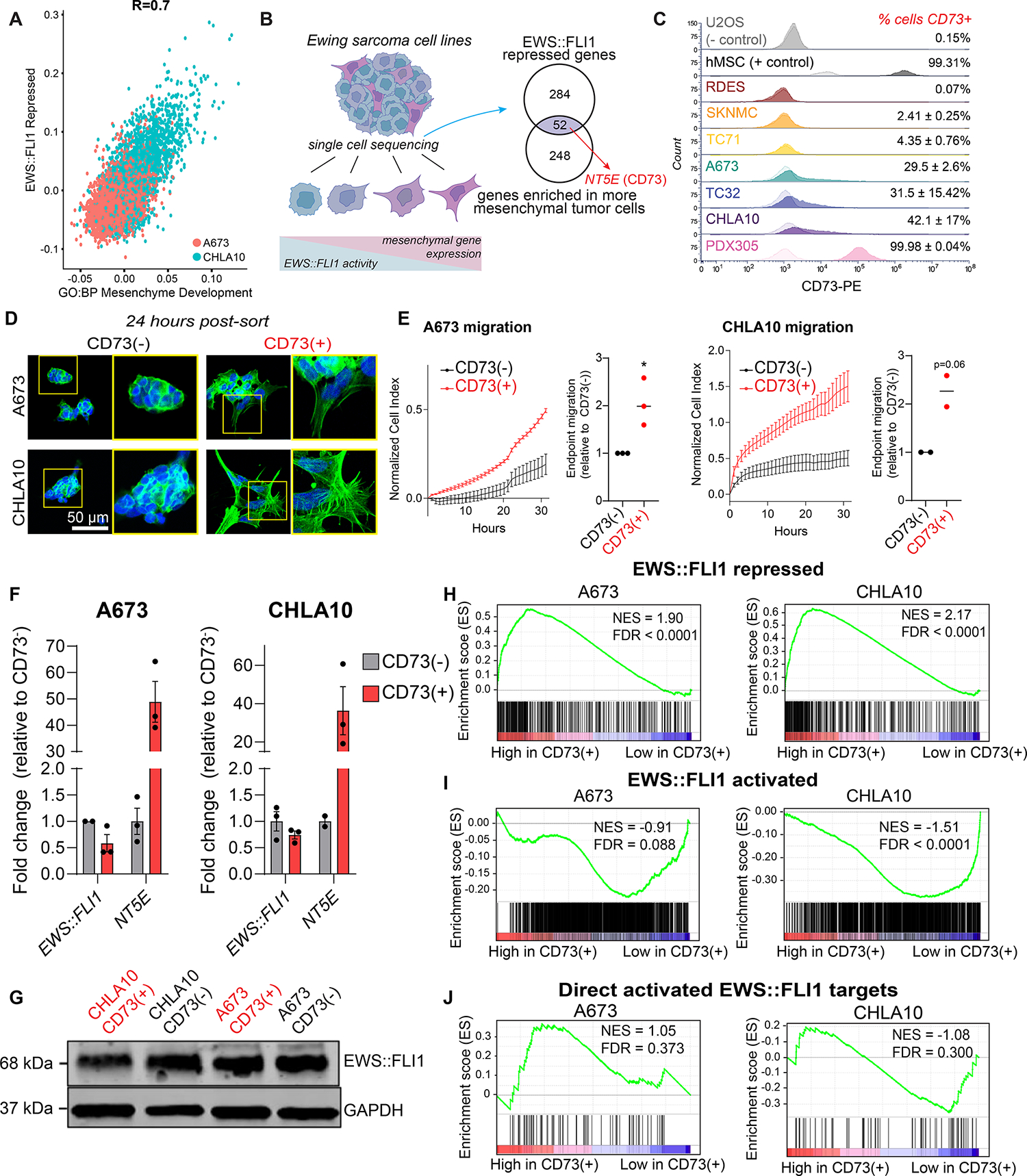
A) Scatter plot depicting expression of an EWS::FLI1 repressed gene set (31) and mesenchyme development genes (GO:0060485) in A673 and CHLA10 single-cell sequencing data (19). B) Left, Ewing sarcoma cells exist along a spectrum of mesenchymal identity. Genes associated with mesenchymal identity are repressed by EWS::FLI1. Right, Venn diagram depicting genes enriched in mesenchymal state EwS cells that are in both GO:BP Mesenchyme Development and EWS::FLI1-repressed (Kinsey et al. 2006) gene sets. C) Flow cytometry of CD73 cell surface expression in EwS and non-EwS (U2OS, hMSC) cell lines (CD73 = dark, isotype control = light). D) DAPI and F-actin staining of FACS-sorted CD73− and CD73+ EwS cells in 2D culture, 24 hours after sorting (representative images of n=3). E) Left, real-time transwell cell migration assay of isogenic CD73− and CD73+ cells (representative biological replicate shown, see Supplemental Figure 1 for additional replicates, error bars=SEM). Right, relative quantification of migration at endpoint for each replicate (A673 n=3, CHLA10 n=2). F) RT-qPCR of EWS::FLI1 and NT5E expression in CD73-sorted cell populations (n=2–3, error bars=SEM). G) Immunoblot of EWS::FLI1 protein expression in CD73-sorted cells. GAPDH is loading control. Representative of n=2. H-J) Gene set enrichment analysis (GSEA) of RNA-seq data generated from CD73-sorted cell populations (n=3) H) genes repressed by EWS::FLI1 expression (31), I) genes activated by EWS:FLI1 expression (31), and J) 78 genes directly bound and upregulated by EWS::FLI1 (30).
Having identified CD73 as a potential marker of EWS::FLI1-low cells, we next performed flow cytometry on a panel of eight established and one early passage PDX-derived EwS cell lines. Human MSC and osteosarcoma cell line U2OS were used as positive and negative controls, respectively. All cells were grown in standard tissue culture conditions (see Methods section). Surface expression of CD73 by EwS cells was highly heterogeneous both within and between cell lines (Figure 1C). Cell lines ranged from fewer than 1% (RDES) to nearly 100% (PDX305) CD73+ cells. CD73+ fractions in the remaining EwS lines ranged from 2–42% and we observed remarkable reproducibility among biologic replicates demonstrating that each model grows in a preferred homeostatic and heterogeneous state. CD73+ cells were always present in large numbers in A673, CHLA10, and TC32 cultures (ranging from ~15–50%), whereas CD73+ cells rarely comprised more than 5% of RDES, SK-N-MC, and TC71 (Figure 1C).
Next, we compared mesenchymal properties of CD73-high (>30% CD73+) and CD73-low (<5% CD73+) cell lines. As shown (Supplemental Figure S1A), CD73-low cell lines grew as rounded cells and formed multicellular clusters with few membrane protrusions. In contrast, cell lines with a high proportion of CD73+ cells were flattened and spindle-shaped with F-actin rich membrane protrusions (Supplemental Figure S1A). Cell lines with rare CD73+ cells were almost entirely non-invasive in 3D collagen I matrices, whereas CD73-high cell lines were robustly invasive (Supplemental Figure S1B). Fluorescence-activated cell sorting (FACS) was then used to isolate CD73+ and CD73− cell fractions from the same A673 and CHLA10 cell cultures (Supplemental Figure S1C). Isolated CD73− cell fractions grew as round cells that formed multicellular clusters with extensive cell-cell contacts and generated few membrane protrusions (Figure 1D). Conversely, syngeneic CD73+ cells displayed mesenchymal morphologies and generated extensive F-actin rich membrane protrusions (Figure 1D). In addition, sorted CD73+ cells migrated faster than corresponding CD73− cells from the same culture (Figure 1E, Supplemental Figure S1D). Significantly, sorted cell populations reproducibly shifted towards their baseline mixed populations over one week, demonstrating that EwS cells are plastic and interconvert between CD73+ and CD73− states (Supplemental Figure S1E). Thus, CD73 expression is remarkably plastic and heterogeneous both within and between EwS models and surface expression of CD73 reproducibly marks subpopulations of EwS cells that harbor mesenchymal properties.
CD73+ EwS cells retain tumorigenicity and EWS::FLI1 expression
Having established that CD73 surface expression marks EwS cells that have lost EWS::FLI1-mediated repression of mesenchymal development genes, we next measured EWS::FLI1 to affirm that CD73+ cells are, in fact, EWS::FLI1-low cells. Unexpectedly, we detected no reproducible depletion of EWS::FLI1 transcript (Figure 1F) or protein (Figure 1G) in CD73+ vs. CD73− cells. In addition, proliferation (Supplemental Figure S1F) and in vivo tumorigenic capacities (Supplemental Figure S1G) of FACS-sorted cells were equivalent or more pronounced in CD73+ than syngeneic CD73− cells, corroborating the observation that CD73+ cells had not downregulated expression of the oncogenic fusion. To determine if the relative transcriptional activity of EWS::FLI1 was altered in CD73+ cells we performed RNA-seq on FACS-sorted cell populations of both A673 and CHLA10 models (Supplemental Table 3). This transcriptomic profiling confirmed that CD73+ EwS cells express high levels of the EWS::FLI1-repressed gene signature (Figure 1H). Surprisingly, however, this loss of EWS::FLI1-mediated gene repression was not accompanied by a corresponding loss of fusion-dependent gene activation (Figure 1I, J). CD73+ EwS cells displayed only modest loss of the EWS::FLI1-activation signature, no significant difference in expression of 78 direct EWS::FLI1 activated targets (30) (Figure 1I, J), and no loss of expression of the cell cycle program that is positively regulated by the fusion (Supplemental Figure S1H). Consistent with the observed mesenchymal phenotypes of CD73+ EwS cells, differential gene expression analysis identified the epithelial mesenchymal transition (EMT) signature as their most enriched hallmark signature compared to CD73− cells (Supplemental Figure S1I). Thus, these studies together demonstrated that CD73 surface expression identifies subpopulations of tumor cells that have de-repressed the EWS::FLI1-repressed signature but that apparently retain expression of the fusion and much of its tumorigenic activation signature.
CD73+ EwS tumor cells selectively upregulate expression of a pro-tumorigenic matrisomal gene program
The observation of potential dissociation between EWS::FLI1-activated and -repressed signatures in FACS-sorted populations of CD73+ EwS cells led us to more deeply investigate the relationship between phenotype and gene expression in individual cells. To achieve this, we exploited single cell proteogenomic profiling using CITE-seq (25). Transcriptomes and cell surface CD73 protein expression were measured in over 12,000 cells from eight established EwS cell lines and one early passage PDX-derived line (Figure 2A and Supplemental Figure S2A). Expression of NT5E/CD73 was profoundly heterogenous within and between models and a positive correlation between mRNA and surface protein expression suggested that the CD73 status of EwS cells is largely transcriptionally determined (Figure 2B, C). The transcriptomes of 1,760 NT5E+ cells were next compared to 10,466 NT5E− cells across all cell lines and then in each cell line independently to identify markers of NT5E+ cells (Supplemental Table 4). As shown (Supplemental Figure S2B and Figure 2D), and consistent with data from RNA-seq of FACS-sorted cells, gene ontology analysis of the 200 most differentially upregulated genes in NT5E+ cells identified the EMT gene signature across all nine EwS models.
Figure 2. CD73+ EwS tumor cells selectively upregulate expression of a pro-tumorigenic matrisomal gene program.
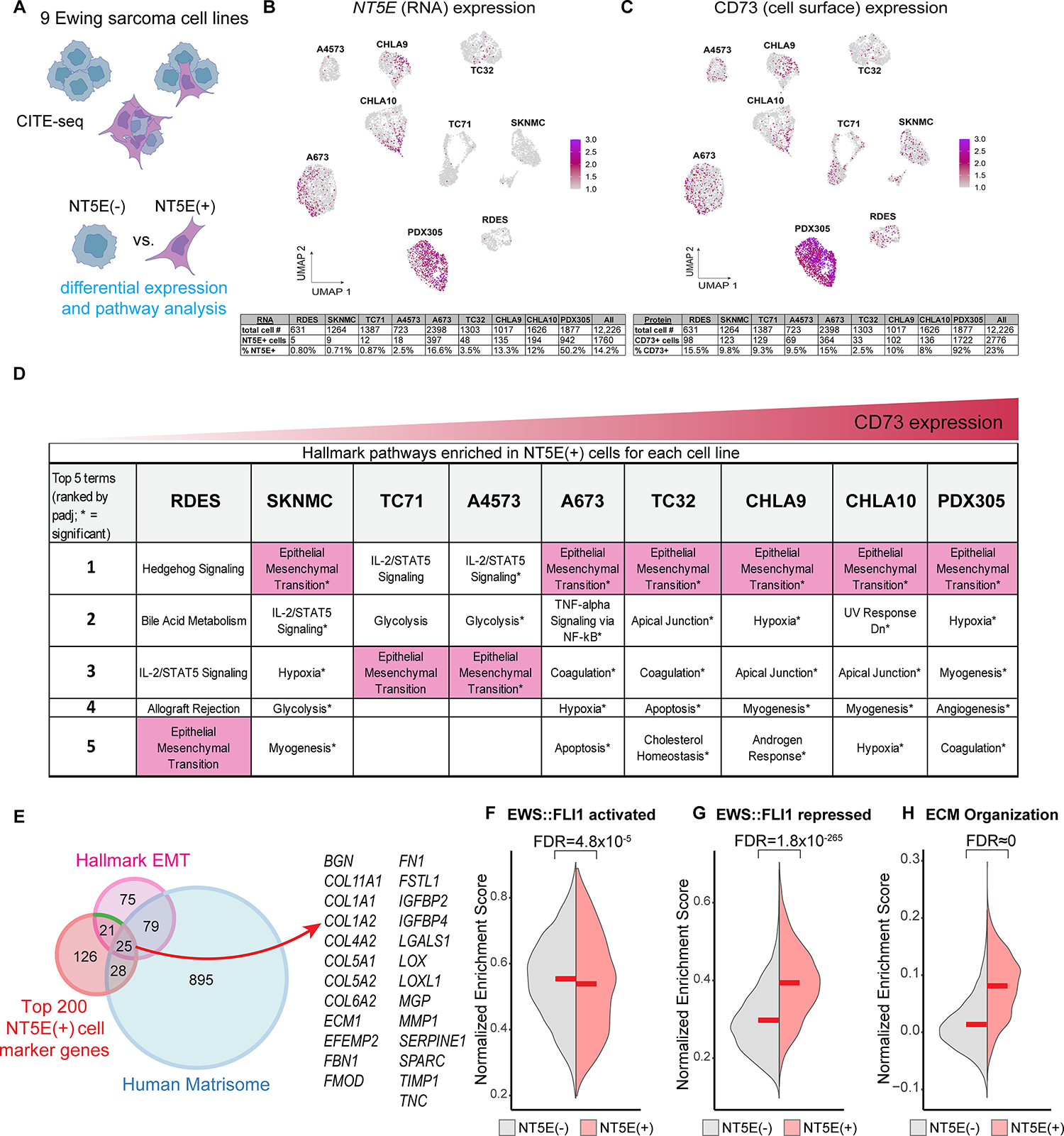
A) Workflow of CD73 CITE-seq and downstream differential expression analyses. B-C) UMAPs of CITE-seq data for NT5E (RNA) or CD73 (cell surface) expression in 9 EwS cell lines. Below, number of NT5E/CD73+ cells by cell line. D) Hallmark gene sets enriched in NT5E+ cells from each cell line. E) Venn diagram overlap of Hallmark EMT genes, human matrisome genes (35), and the 200 top markers (by log fold change) of NT5E+ cells as determined by single cell CITE-seq. Right, the 25 genes present in all 3 gene sets. F-H) Split violin plots depicting a normalized enrichment score for expression of (F) EWS::FLI1 activated (31) (G) EWS::FLI1 repressed (31), and (H) GO:BP ECM Organization (GO:0030198) gene sets in NT5E+ and NT5E– cells (CITE-seq data inclusive of 9 EwS cell lines).
EMT is a complex and highly conserved developmental process that is frequently hijacked during carcinoma pathogenesis (33). To better understand how an EMT signature could be differentially activated in sarcoma cells that are, by definition, mesenchymal (34), we explored the identity of the EMT signature genes that were upregulated in CD73+ cells. Forty-six Hallmark EMT signature genes were more highly expressed by NT5E+ cells and we noted that many of them encode for ECM and matrisome-associated proteins (35) (Figure 2E, Supplemental Figure S2C). These included numerous collagens, integrins, fibrillin 1 (FBN1), fibronectin (FN), secreted protein acidic and cysteine rich (SPARC), and tenascin-C (TNC) (Supplemental Figure S2D). The ECM of carcinomas is normally remodeled by CAFs, and we noted that 36 of the 200 most upregulated genes in NT5E+ EwS cells were recently identified as markers of diverse CAF subsets across multiple tumor types (e.g. ACTA2, COL1A1, and THY1). In addition, 24 NT5E-marker genes are included among 69 genes that specifically marked ECM-rich CAFs (dCAFs) that are associated with poor prognosis (36). Quantitative RT-PCR of FACS-sorted cells validated higher expression of ECM-related genes in CD73+ cell populations (Supplemental Figure S2E), and immunofluorescence confirmed increased deposition of TNC protein (Supplemental Figure S2F). Thus, these findings suggested that the transcriptional and phenotypic properties of CD73+ EwS tumor cells are similar to ECM-producing CAFs (3, 37).
Having established that the mesenchymal-high state of CD73+ EwS cells is characterized by significant and reproducible upregulation of matrisomal gene programs, we next evaluated expression of EWS::FLI1-activated and -repressed gene signatures in these cells. Consistent with our observations in CD73+ vs. CD73− sorted cell populations (Figure 1H–J), single NT5E+ cells showed only a modest decrease in expression of the EWS::FLI1-activated signature (Figure 2F), but a marked upregulation of EWS::FLI1-repressed and ECM genes (Figure 2G–H). These findings demonstrate a partial decoupling of EWS::FLI1-dependent gene activation and gene repression in NT5E+ cells, with a significant and reproducible re-activation of mesenchymal and ECM genes that are normally repressed by the fusion.
Spatial and transcriptional heterogeneity of EwS tumor cells exists in vivo
Given our finding of extensive intratumoral heterogeneity in vitro, we next investigated transcriptional and phenotypic profiles of EwS tumor cell states in vivo. Local and disseminated CHLA10 tumor xenografts were generated in immunocompromised mice by subcutaneous and tail vein injections, respectively. Tumors were harvested at humane endpoints and subjected to H&E staining and immunofluorescence (Figure 3A–F). As shown, CD73+ tumor cells were detected in all tumors with a notable increase in frequency in tail vein-derived tumors compared to subcutaneous masses (Figure 3B–F). Spatial heterogeneity of CD73+ cell subpopulations was also apparent within each tumor (Figure 3). In the tail vein-derived tumors, we observed the highest CD73 expression in tumor cells in peri-necrotic regions, tumor borders, and in invasive foci that had infiltrated the retroperitoneal cavity (Figure 3E–F, Supplemental Figure S3A). In keeping with our in vitro findings, tumor cell-derived ECM deposition was evident in regions adjacent to the CD73+ tumor cells (Figure 3B–F). Similar spatial heterogeneity in CD73 expression and proximity to ECM deposition was observed in additional A673 and PDX305 subcutaneous xenografts (Supplemental Figure S3B, C).
Figure 3. Spatial heterogeneity of CD73+ tumor cells and ECM deposition in vivo.
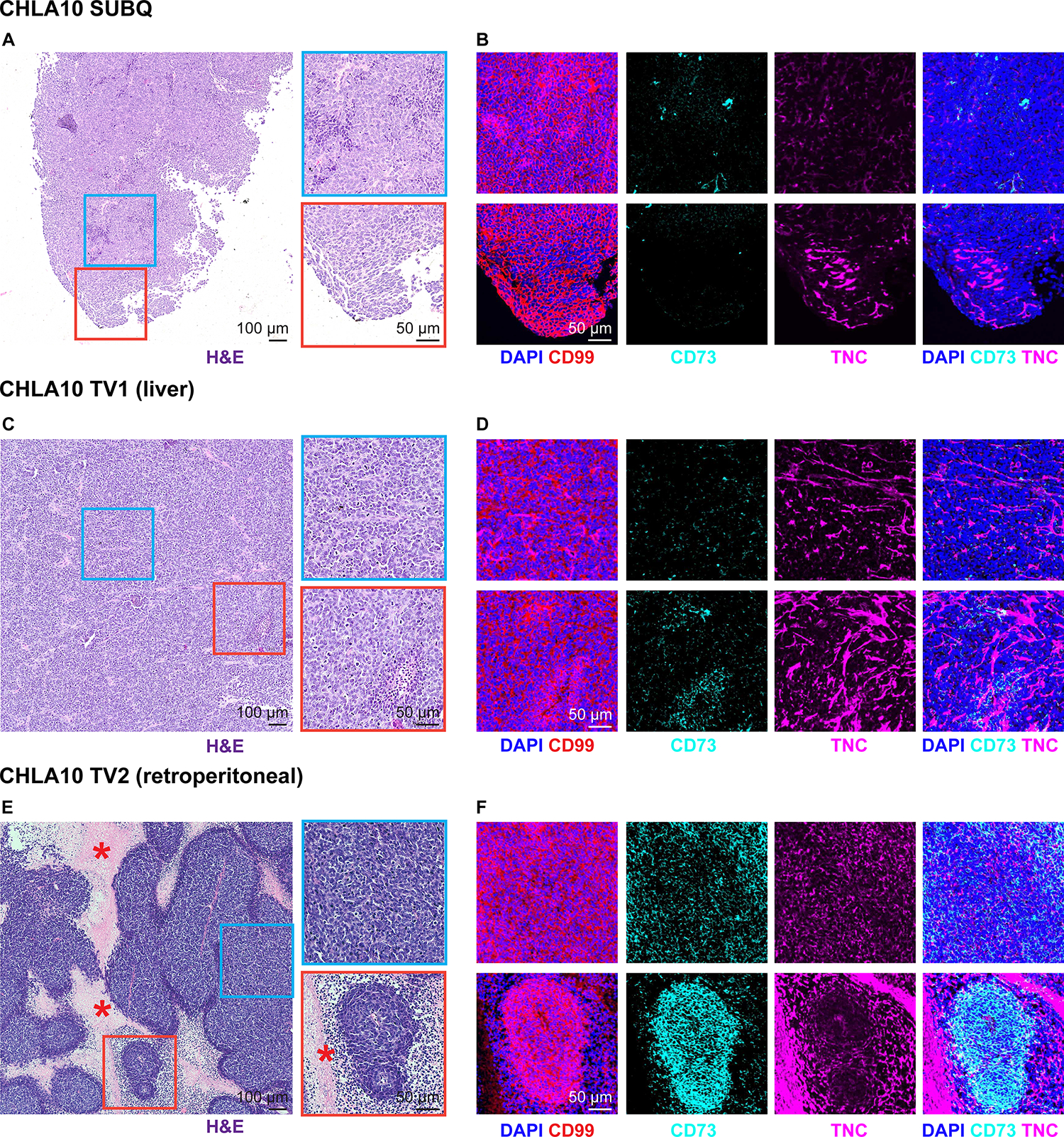
FFPE sections were generated from 3 CHLA10 tumor xenografts from 3 separate mice: one subcutaneous tumor (SUBQ), a liver tumor (TV1), and a retroperitoneal tumor (TV2) formed after tail vein injection. A) H&E staining of the SUBQ xenograft tumor. Right, zoomed insets. B) Immunofluorescence of the same regions in adjacent SUBQ FFPE sections for DAPI (nuclei), CD99 (tumor membrane marker), CD73, and human TNC. C) H&E staining of the TV1 xenograft liver tumor. Right, zoomed insets. D) Immunofluorescence of the same regions in adjacent TV1 FFPE sections for DAPI (nuclei), CD99 (tumor membrane marker), CD73, and human TNC. E) H&E staining of the TV2 xenograft retroperitoneal tumor. Right, zoomed insets. Asterisk marks regional necrosis adjacent to viable tumor borders. F) Immunofluorescence of the same regions in adjacent TV2 FFPE sections for DAPI (nuclei), CD99 (tumor membrane marker), CD73, and human TNC.
We next performed integrated digital spatial profiling and whole human transcriptome analysis to define the transcriptional states of phenotypically distinct tumor cells in these in vivo tumor microenvironments (Figure 4A). Ki67 and cytoskeletal protein vimentin were used as morphology markers to identify differentially proliferative and mesenchymal regions, respectively, and CD31 was used to localize vasculature (Figure 4B). ROIs were chosen to represent a broad range of proliferation (by Ki67), mesenchymal identity (by Vimentin expression), and nearby microenvironment structures (e.g. tumor-stromal borders, vasculature, and necrosis). We then selected four spatially distinct regions of interest (ROIs) from each tumor for capture and next generation sequencing (NGS). Between 1400 and 6500 cells were captured from each of the 12 ROIs which were distributed in tumor interiors (e.g. ROI1, ROI5, ROI11), along tumor-stromal borders (e.g. ROI2, ROI7), and in invasive tumor foci that were surrounded by necrotic tissue (e.g. ROI9, ROI10) (Figure 4B). Normalized MKI67 and VIM gene expression in the captured ROIs (Supplemental Table 5) corroborated the observed spatial differences in Ki67 and vimentin immunofluorescence (Supplemental Figure S4A, B). As shown, tumor cells that were captured from subcutaneous and tail vein-derived focal liver tumors generally expressed higher levels of MKI67 than cells captured from an infiltrating retroperitoneal tumor (Supplemental Figure S4A, B). In contrast, expression of VIM was generally higher in disseminated tumors and heterogeneity among ROIs was apparent in all three tumors (Supplemental Figure S4A, B).
Figure 4. Digital spatial profiling reveals transcript and protein-level spatial heterogeneity in EwS tumors.
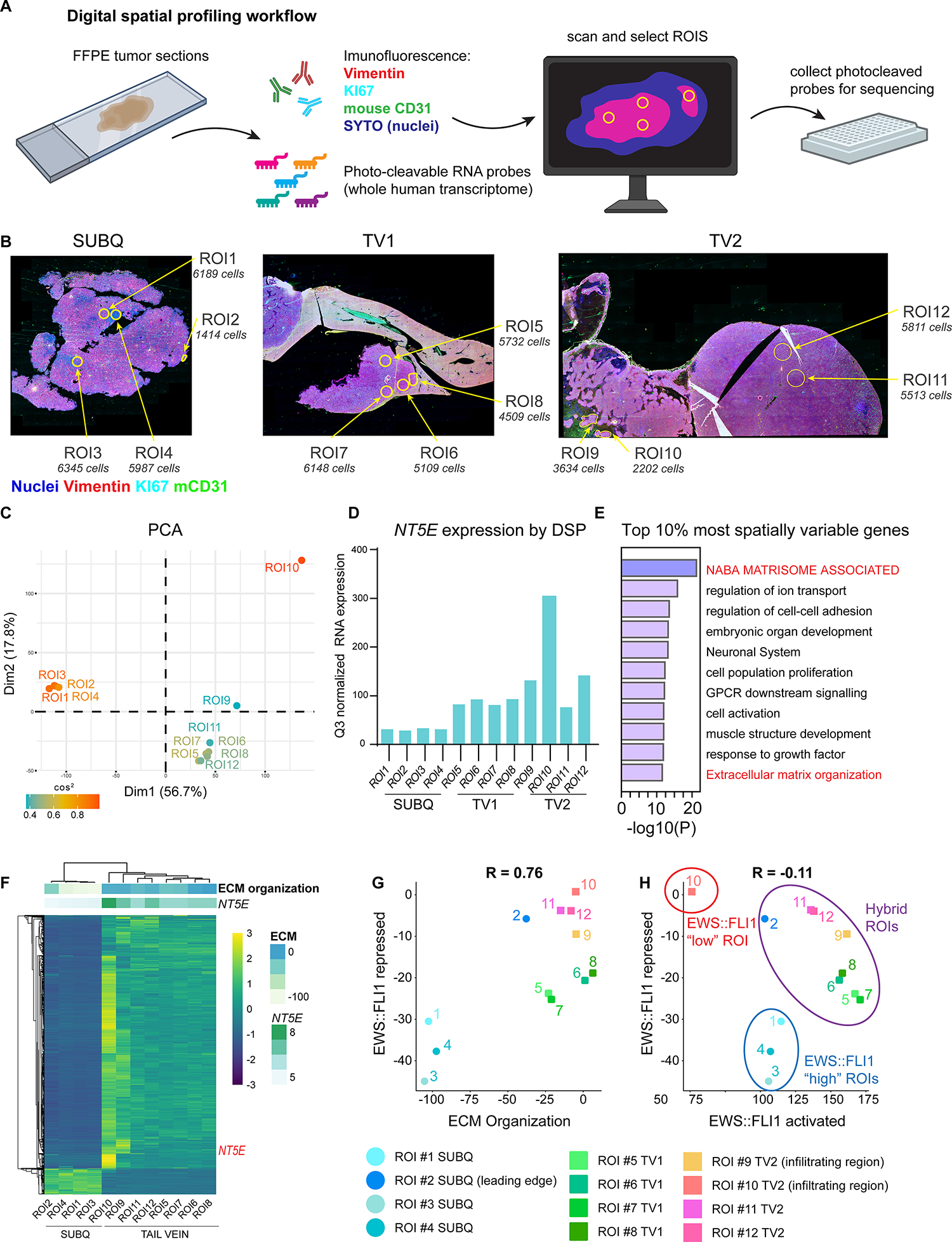
A) Workflow of the Digital Spatial Profiling (DSP) method used. Briefly, immunofluorescence of Vimentin (mesenchymal cytoskeletal marker), KI67 (proliferative nuclei), SYTO (all nuclei), anti-mouse CD31 (murine blood vessels) were used to select regions of interest (ROIs) on xenograft sections hybridized with a whole human transcriptome probe library. ROIs were captured and gene expression quantified by next-generation sequencing after UV cleavage of probes from selected ROIs. B) Vimentin, KI67, mCD31, SYTO (nuclei) immunofluorescence and selected ROIs indicated for each of 3 tumors subjected to DSP: CHLA10 (SUBQ) and tail vein-derived liver (TV1) and retroperitoneal (TV2) tumors as in Figure 3. Approximate DAPI+ cell count is listed for each ROI. C) PCA plot of normalized whole human transcriptome gene expression for each ROI. Colored by cos2 value (indicates quality of representation by the principal components). D) Bar graph depicting Q3 normalized mRNA expression of NT5E within each ROI as determined by DSP and next generation sequencing. E) Gene ontology (Metascape) of the top 10% most spatially variable genes across 12 ROIs (highest coefficient of variation between ROIs, n=1339 genes). E) Unsupervised heatmap of the top 10% most spatially variable genes across all ROIs. Top panels show expression of NT5E (log2) and the GO:BP ECM Organization gene set. G) Scatter plot of expression of EWS::FLI1-repressed gene signature (31) vs. ECM Organization gene set expression. Individual ROIs are labeled. H) Scatter plot of RNA expression of EWS::FLI1 repressed signature vs. EWS::FLI1 activated signature in each ROI (31). EWS::FLI1-high, EWS::FLI1-low, and EWS::FLI1-hybrid ROIs are indicated.
Having confirmed that CHLA10 cells display heterogeneity of VIM expression in vivo, we next performed unsupervised analyses of whole transcriptomic data to interrogate spatial and biologic heterogeneity in an unbiased fashion. First, principal components analysis (PCA) of the 12 ROI-transcriptome dataset showed that subcutaneous tumor ROIs clustered tightly and separately from ROIs captured from disseminated tumors despite the fact that all tumors were generated from CHLA10 cells (Figure 4C). Transcriptomes of ROIs captured from tail vein-derived tumors also demonstrated considerably more heterogeneity than subcutaneous ROIs. This was especially notable when comparing tumor cells captured from tumor interiors to those captured from invasive tumor foci (ROI9 and ROI10) (Figure 4C). The transcriptomic profile of tumor cells in ROI10 was distinct from all other ROIs and, significantly, targeted analysis of the data reveals that these tumor cells expressed the highest level of NT5E (Figure 4D). Next, we sought to identify genes with the most spatial heterogeneity. To achieve this, we again took an unbiased approach and identified the most variably expressed genes across all 12 ROIs as determined by coefficient of variance (38). Limiting analysis to the top 10% most differentially expressed transcripts generated a list 1,339 genes and gene ontology analysis revealed these to be highly enriched for ECM-related gene programs (Figure 4E). Unsupervised hierarchical clustering of these 1,339 genes clearly separated ROIs captured from subcutaneous and disseminated tumors and showed that NT5E and an ECM organization gene signature were markedly upregulated in tumor cells captured from peri-necrotic foci (Figure 4F; ROI9 & ROI10). One notable exception to this trend was ROI2, a region along the tumor-stromal interface of the subcutaneous tumor that highly expressed ECM genes but not NT5E/CD73. Thus, intratumoral spatial heterogeneity of EwS cell phenotypes and transcriptional profiles is evident in vivo. NT5E and matrisomal gene programs are, in general, more highly expressed by tumor cells in distant sites than those that are growing in localized masses and expression of ECM-encoding genes is particularly enriched along tumor borders and invasive foci.
Finally, we compared expression of EWS::FLI1-activated and -repressed genes in tumor cells captured from the 12 spatially distributed ROIs. As predicted from our in vitro data, EwS cells that highly expressed the ECM gene signature showed loss of EWS::FLI1-dependent gene repression (Figure 4G). However, loss of the repressive signature was not always accompanied by loss of EWS::FLI1-mediated gene activation, and hybrid transcriptional states were uncovered (Figure 4H). Transcriptomes of EwS cells isolated from the core of the subcutaneous tumor (ROIs 1, 3, & 4) were typical of EWS::FLI1-high cells (activated genes ON/repressed genes OFF). Likewise, the ROI10 transcriptional signature was fully compatible with EWS::FLI1-low cells (activated genes OFF/repressed genes ON). In contrast, transcriptomes of tumor cells captured from the remaining regions displayed features of both EWS::FLI1-high and -low cells. This is particularly evident for ROIs 9,11, and 12 wherein EwS cells were found to express high levels of both EWS::FLI1-activated and -repressed genes. Thus, these studies demonstrate dissociation of EWS::FLI1 activating and repressive transcriptional activity in EwS tumor regions in vivo and suggest that tumor subpopulations can upregulate ECM signatures without downregulating expression of EWS::FLI1-activated genes. Although it is possible that hybrid ROIs were comprised of mixed populations of EWS::FLI1-high and EWS::FLI1-low cells, when combined with our identification of transcriptionally hybrid states in vitro (Fig 2), these data lead us to conclude that hybrid EwS cells are present in tumors in vivo in and these cells acquire mesenchymal properties while retaining EWS::FLI1-dependent gene activation.
Heterogeneity of NT5E/CD73+ tumor cells and tumor-derived ECM is evident in patient tumors
There is growing evidence that cells in transitional EMT cell states, rather than fully epithelial or mesenchymal cells, drive growth and metastatic progression of carcinomas (39, 40). Having identified NT5E/CD73 as a marker of transcriptionally hybrid and phenotypically distinct CAF-like tumor cell subpopulations in EwS models, we sought to investigate the presence of these cells in primary patient tumors. Analysis of three EwS gene expression datasets (GSE34620, GSE142162, GSE17679) (18, 41, 42) revealed marked inter-tumor variability in NT5E expression (Figure 5A). Next, we identified genes that were strongly correlated with NT5E (R>0.6) across patient tumors and discovered a striking overlap among the independent cohorts (Figure 5B). Hallmark EMT and ECM-related gene ontology signatures were highly enriched in these 217 shared NT5E-correlated genes (Supplemental Table 6), corroborating the enrichment of these signatures in NT5E+ EwS cells in tumor models (Figure 5C). Integrating the top 200 markers of NT5E+ EwS cells in vitro (Figure 2E, Supplemental Table 3) with the 217 genes NT5E-correlated genes in patient tumors in vivo, generated a list of 27 genes that were highly reproducibly upregulated alongside NT5E in EwS tumor cells and these include numerous EMT and ECM genes, most of which are normally repressed by EWS::FLI1 (31, 43, 44) (Figure 5D). Consistent with this observation, expression of the 28 gene signature by individual patient tumors correlated positively with expression of both ECM organization and EWS::FLI1-repressed gene signatures (Figure 5E and Supplemental Figure S5A–B).
Figure 5. Heterogeneity of NT5E and related mesenchymal transcriptomic signatures in EwS patient tumor biopsies.
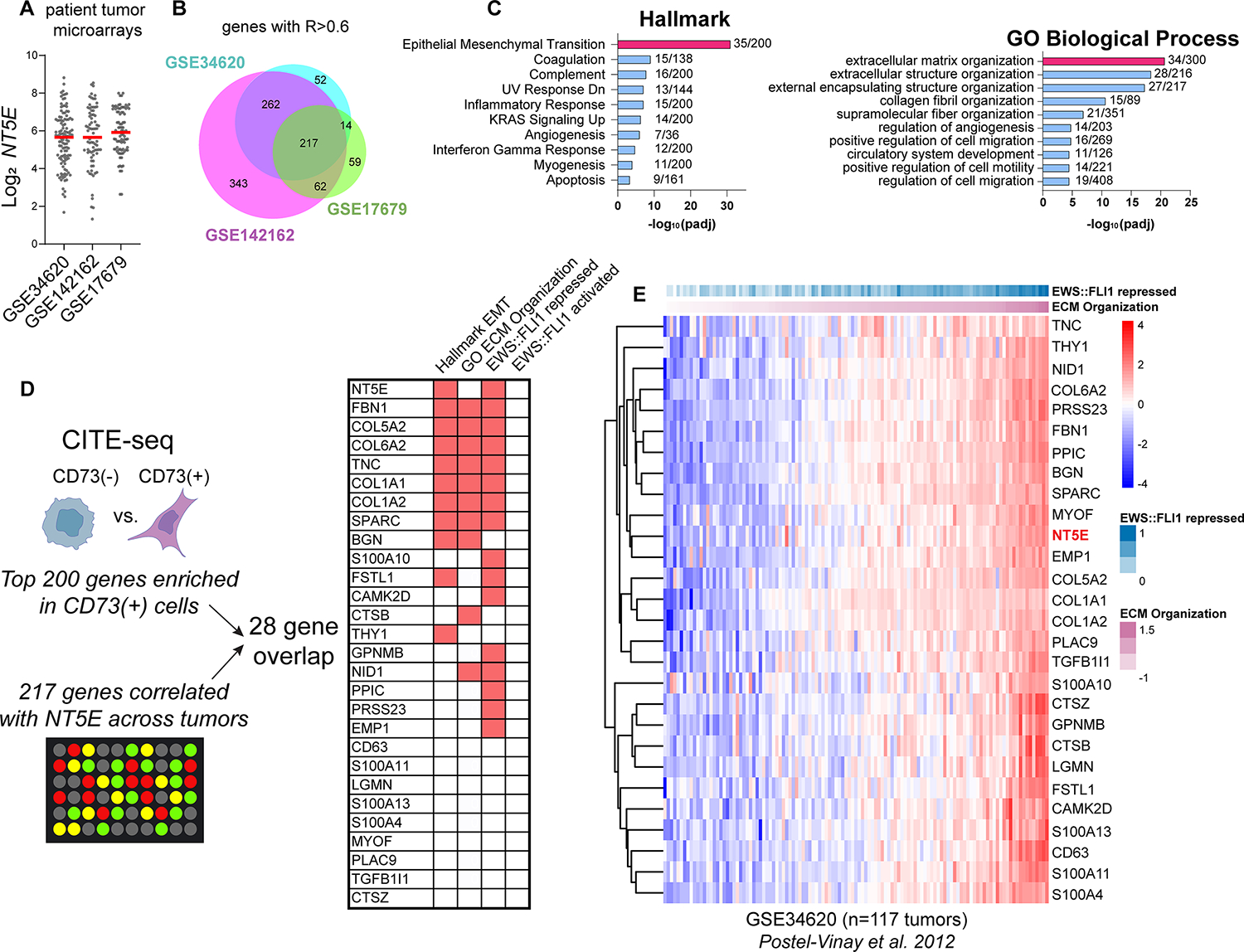
A) Log2 NT5E expression derived from three independent gene expression microarray studies of patient tumors (18, 41, 42). B) Venn diagram of genes positively correlated with NT5E (R>0.6) in each dataset and the intersection of these genes. C) Hallmark and GO:BP gene ontology (Enrichr) of the 217 NT5E-correlated genes (and NT5E itself) shared across three studies. D) Workflow and characterization of the overlap between genes marking NT5E+ cells (from Figure 2) and genes highly correlated with NT5E in patient tumor biopsies. Right, membership of overlapping 28 genes in Hallmark EMT, GO:BP ECM Organization, EWS::FLI1 repressed (31, 43, 44), and EWS::FLI1 activated gene sets (30, 31, 43, 44). E) Heatmap of NT5E and 27 associated marker gene expression vs. EWS::FLI1 repressed genes (31), ranked by ECM organization expression in Ewing sarcoma patient tumor microarray GSE34620 (41).
This analysis of bulk tumor RNA profiling confirmed inter-tumor heterogeneity of NT5E expression and the relationship between high NT5E and expression of EWS::FLI1-repressed matrisomal genes. To assess intratumoral heterogeneity and the potential existence of distinct CAF-like tumor cell subpopulations we evaluated CD73 and ECM protein expression in tumor sections from archived patient biopsies. We first stained a tumor tissue microarray (TMA) with an anti-CD73 antibody using the EWS::FLI1-induced protein NKX2–2 as a tumor marker. CD73+ cells were detected in only 7 of 24 tumor cores (20 unique patients; Supplemental Table 7) and the frequency of CD73+ tumor cells varied from rare to extensive (Supplemental Figure S6A–C). Staining of adjacent sections of the TMA with an antibody against TNC confirmed inter-and intra-tumoral heterogeneity of TNC and deposition of this protumorigenic ECM protein was most robust in regions adjacent to CD73+ tumor cells (Supplemental Figure S6D).
Given the observed spatial heterogeneity of CD73+ tumor cells and tumor-derived ECM in EwS xenografts (Figure 3, Supplemental Figure S3) and TMA cores (Supplemental Figure S6B, C), we next stained full biopsy sections from two tumors that were included in the TMA. One of these (#20) showed extensive CD73+ cells on the TMA core while the other (#35) was scored negative (Supplemental Table 7). Whole sections of both biopsies showed the presence of large acellular regions that denoted areas where core punches had been previously harvested for inclusion on TMAs (Figure 6A,B). Staining for the CD99 tumor marker showed that biopsy #20 included EwS tumor embedded within adjacent non-tumor tissue (Figure 6A) while biopsy #35 was predominantly comprised of viable tumor (Figure 6B). Importantly, CD73+ tumor cells within CD99+ regions were detected in both tumors, including biopsy #35 which was devoid of CD73+ cells on the TMA (Figure 6A,B). Spatial heterogeneity of ECM proteins TNC and biglycan (BGN) was also evident between and within the tumors and both proteins were more highly expressed adjacent to CD73+ tumor cells (Figure 6A,B). Thus, EwS tumor cells are both transcriptionally and phenotypically heterogeneous in vitro and in vivo and spatial co-localization of CD73+ tumor cells with pro-tumorigenic ECM suggests that EWS::FLI1-low and EWS::FLI1-high/low hybrid tumor cells function as CAF-like cells (Figure 6C).
Figure 6. Intra-tumoral heterogeneity of CD73+ tumor cells and ECM deposition in patient biopsies.
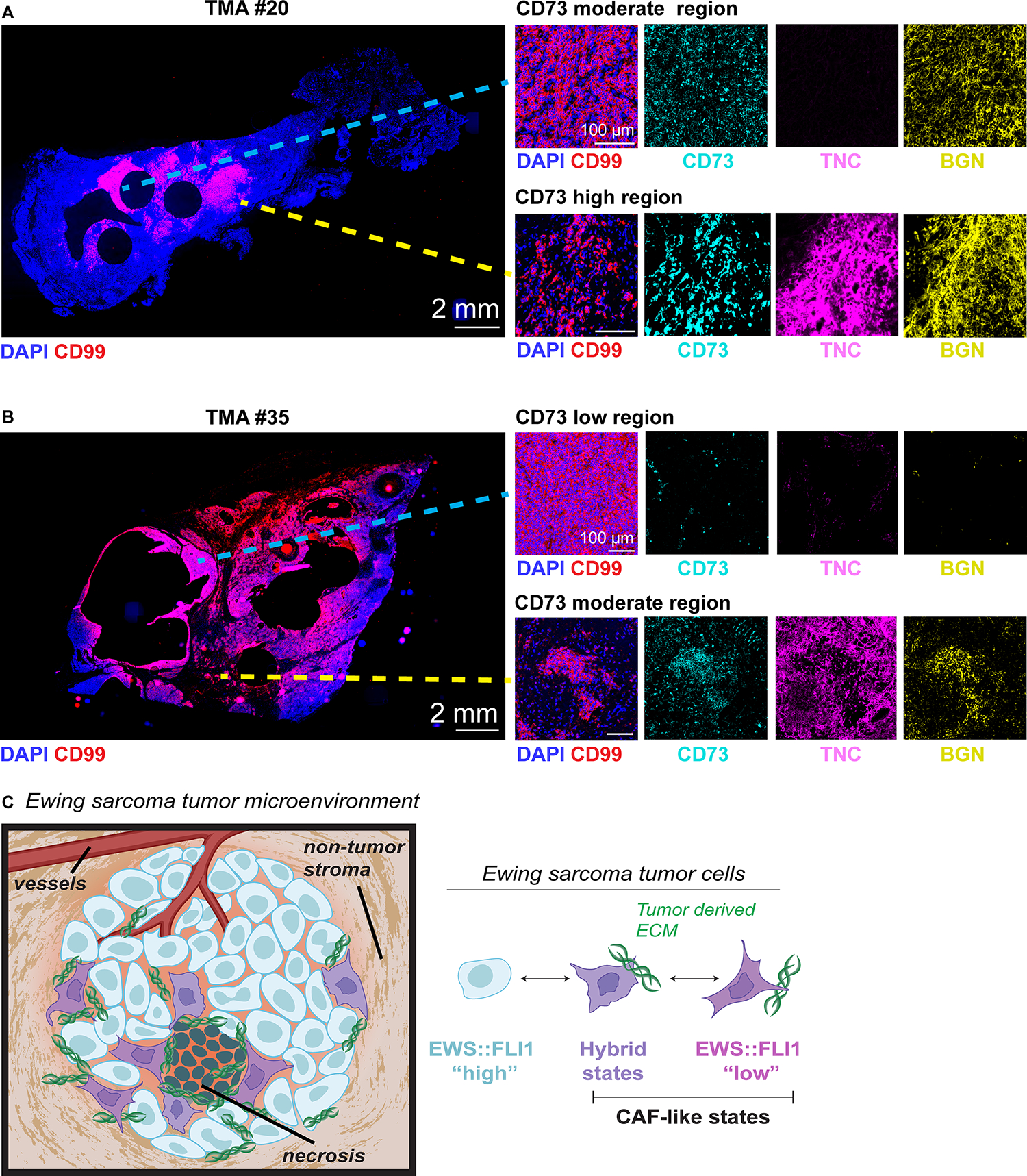
Immunofluorescence analysis of full tumor biopsy sections: (A) TMA ID #20 (extensive CD73+ scoring on TMA) and (B) TMA ID #35 (negative CD73 scoring on TMA). DAPI (nuclei), CD99 (tumor membrane marker), CD73, Tenascin-C, and biglycan. Right panels, zoomed insets show regions from the same tumor section with disparate CD73 expression and ECM deposition. C) Model of cell state heterogeneity in EwS wherein tumors contains spatially and transcriptionally heterogeneous subpopulations of tumor cells. Selective derepression of EWS::FLI1-suppressed mesenchymal target genes leads to acquisition of mesenchymal identity without loss of EWS::FLI1-dependent gene activation and tumorigenicity. These transcriptionally hybrid cells display properties of CAFs and promote remodeling of the TME by depositing pro-tumorigenic ECM.
DISCUSSION
Accruing evidence suggests that tumor cell heterogeneity is a universal feature of all cancers and that even genetically homogeneous tumors develop extensive cell diversity through epigenetic processes (4, 5). Phenotypically distinct tumor cell subpopulations can contribute in different ways to tumor progression as they may have very different proliferation rates, metastatic potential, and differential responses to treatment (5). EwS are genetically quiet tumors that are driven by a single fusion oncoprotein (11). They show little evidence of clonal genetic diversity or evolution and they are histologically classified as homogeneous, small round blue cell tumors (12). Here, we have identified that EwS tumor cells exist in a previously uncharacterized spectrum of cell states that differentially express mesenchymal gene programs. In particular, our findings show that transcriptionally distinct subpopulations of highly mesenchymal tumor cells are present in EwS cell lines in vitro, xenograft models in vivo, and in patient tumors in situ. These cells can be identified by cell surface expression of CD73 and they are distinguished by their upregulation of matrisomal protein-encoding genes and their deposition of ECM. The distinct capacity of these cells to remodel the local TME through deposition of pro-tumorigenic ECM leads us to label them “CAF-like” EwS cells (Figure 6C).
The critical importance of ECM remodeling to tumor growth, invasion, and metastatic progression is well established (45–47). In epithelial-derived tumors ECM is largely deposited by CAFs, non-malignant stromal cells that most often arise from resident tissue fibroblasts or other mesenchymal cells and that are activated to secrete pro-tumorigenic matrisomal proteins in response to cues from the tumor cell compartment (3). More rarely, subpopulations of carcinoma cells themselves acquire the capacity to deposit ECM and promote pro-tumorigenic remodeling of the local TME and there is evidence that tumor cell-derived ECM confers a more aggressive tumor state (47, 48). The critical contribution of ECM and non-malignant stromal cells to sarcomas is less well studied but post-translational remodeling of collagen by tumor cells plays an essential role in mediating metastatic progression of soft tissue sarcomas (49). The contribution of ECM remodeling to growth and progression of EwS has not been deeply investigated, though clinico-pathologic correlative studies suggested that a stroma-rich TME influences EwS gene expression and patient prognosis (50). EwS cells and tumors can deposit fibrillar and non-fibrillar proteins (51–53) and we previously showed that activation of canonical Wnt/beta-catenin and TGF-beta signaling upregulates secretion of pro-tumorigenic and -angiogenic matrisomal proteins by EwS cells, including collagens and TNC (54, 55). In the current work, we have identified discrete subpopulations of EwS cells that deposit pro-tumorigenic ECM proteins in vivo. These cells are largely CD73+ and they are enriched on tumor borders, peri-necrotic regions, and in invasive foci. Importantly, these cells were rarely found on our examination of small TMA tumor cores. Diagnostic EwS tumor specimens are normally obtained from needle or small open biopsies of viable tumor interiors (56). Thus, these CD73+ tumor cells and tumor cell-derived ECM can be missed when histologic and molecular analyses are limited to small tumor specimens. As our understanding of the critical importance of tumor heterogeneity and tumor: TME crosstalk grows, so too does our appreciation that TMA cores may not be adequate for studies of tumor biology that aim to dissect tumors as spatially dynamic ecosystems (57).
Our single cell studies of EwS cell transcriptomes and spatial profiling studies of tumors in vivo generated the unanticipated discovery that, although they expressed high levels of matrisomal genes that are normally repressed by EWS::FLI1, CD73+ EwS cells retained expression of the fusion and were largely not “EWS::FLI1-low”. While we did identify invasive cell clusters that were consistent with a fully EWS::FLI1-low state (e.g., Figure 4H, ROI10), CAF-like EwS cells most often existed in hybrid EWS::FLI1 transcriptional states. These cells retained expression of the fusion-activated gene signature alongside high expression of genes that are normally repressed by EWS::FLI1. Thus, these EwS cells gained mesenchymal properties, including the capacity to deposit ECM, without losing proliferative or tumorigenic potential. Notably, we also observed transcriptional heterogeneity among CAF-like populations in vivo, including cells that expressed NT5E but not TNC (Supplemental Figure S2D), and deposition of TNC protein in xenograft regions without detectable CD73 expression (Figure 3B). Bone fide CAFs exist as transcriptionally distinct subsets and dynamically switch between these different states (58, 59). Further studies will be needed to determine if CAF-like subpopulations of EwS cells can likewise be further subdivided into multiple transcriptional states with different functions in TME remodeling.
These observations lead us to conclude that EwS cell states are more nuanced than first appreciated. While more migratory and metastatic EWS::FLI1-low cells were first presumed to express lower levels of the fusion (15), it is now evident that the activity of the fusion can be influenced by cell intrinsic and extrinsic factors that modulate its transcriptional output (16) including context-dependent transcription factors (19–21), chromatin remodelers (17, 18), and nuclear condensates (14, 60). Our current work adds further complexity, revealing that the activating and repressing properties of the fusion can be decoupled in individual tumor cells. This decoupling may be possible because EWS::FLI1 gene activation and repression occur through distinct mechanisms requiring the activity of different partners. Gene repression can occur by multiple means including direct EWS::FLI1 binding, upregulation of transcriptional repressors, and recruitment of HDACs (16). Further, recent studies have identified mechanisms that antagonize this repression, including Wnt-induced sensitization to TGF-beta signaling (22, 55), chromatin reorganization after STAG2 loss (17, 18), and activity of developmental transcription factors such as HOXD13 (19). Whether these or other mechanisms facilitate de-repression of NT5E and ECM genes and generation of a CAF-like state remain to be elucidated, and this is a key gap in knowledge that the field must now fill.
Tumor cell heterogeneity and cell plasticity are key factors in tumor evolution (4) and metastatic progression of carcinoma models is, at least in part, driven by transitional or hybrid cells that exist along a continuum between epithelial and mesenchymal states (39, 40). In addition, interactions between dynamic tumor cells and an equally dynamic TME create complex tumor ecosystems that support tumor growth, enable metastatic progression, and promote resistance to current treatment approaches (61–63). Given this, the tumor: TME interface and ECM remodeling processes are increasingly being investigated as novel targets on which to focus basic and translational cancer research efforts (23, 64–67). Here we have reported the existence of CAF-like EwS cells that contribute to ECM remodeling, and we propose that these cells are likely to play critical roles in mediating tumor progression and influencing treatment response. A deeper understanding of CAF-like EwS cells is now needed, including determination of their prevalence, origins, fundamental biology, and impact on neighboring tumor cells as well as the immune and non-immune TME. Elucidating the dynamic and highly heterogeneous nature of EwS ecosystems will afford novel insights into tumor biology that will guide development and rational integration of TME-targeted therapeutics into EwS treatment regimens.
Supplementary Material
Statement of Translational Relevance.
Remodeling of the tumor microenvironment (TME) is essential for propagation of solid tumors. In epithelial-derived cancers, non-malignant cancer-associated fibroblasts (CAFs) deposit pro-tumorigenic extracellular matrix (ECM) proteins that support local growth, metastatic progression, and treatment resistance. Ewing sarcomas are aggressive mesenchymal tumors that are generally stroma-poor and the contributions of ECM and TME remodeling to disease progression are largely unknown. We have identified subpopulations of Ewing sarcoma cells that upregulate expression and deposition of ECM proteins in a manner reminiscent of CAFs. Importantly, these cells are both transcriptionally and phenotypically distinct from the tumor bulk and they are enriched along tumor borders and invasive foci. These findings demonstrate that pro-tumorigenic remodeling of the in situ Ewing sarcoma TME is achieved, at least in part, by subpopulations of CAF-like tumor cells and we propose that these cells are likely to be critical for tumor progression.
Acknowledgments
The authors thank members of the Lawlor lab for helpful discussion, and the staff at the Fred Hutchinson and Seattle Children’s Research Institute flow cytometry, comparative medicine, and bioinformatics cores. Grant and gift support for this work is gratefully acknowledged and was provided by the following sources: NIH/NCI R01 CA215981 (ERL), F31CA247104 (AA); research supported by the 2022 AACR-QuadW Foundation Sarcoma Research Fellowship in Memory of Willie Tichenor, Grant Number 22-40-37-WREN (EDW); Sam Day Foundation (ERL); the 1M4Anna Foundation (ERL, SNF); Lafontaine U-Can-Cer-Vive Foundation (ERL). This research was supported by the Flow Cytometry Shared Resource of the Fred Hutch/University of Washington/Seattle Children’s Cancer Consortium (P30 CA015704).
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Greaves M Evolutionary determinants of cancer. Cancer discovery. 2015;5(8):806–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGranahan N, and Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613–28. [DOI] [PubMed] [Google Scholar]
- 3.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nature Reviews Cancer. 2020;20(3):174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discovery. 2022;12(1):31–46. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Seehawer M, and Polyak K. Untangling the web of intratumour heterogeneity. Nature cell biology. 2022;24(8):1192–201. [DOI] [PubMed] [Google Scholar]
- 6.Tabassum DP, and Polyak K. Tumorigenesis: it takes a village. Nature Reviews Cancer. 2015;15(8):473–83. [DOI] [PubMed] [Google Scholar]
- 7.Filbin M, and Monje M. Developmental origins and emerging therapeutic opportunities for childhood cancer. Nature medicine. 2019;25(3):367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178(4):835–49.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danielli SG, Porpiglia E, De Micheli AJ, Navarro N, Zellinger MJ, Bechtold I, et al. Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance. Science advances. 2023;9(6):eade9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nature Genetics. 2017;49(8):1261–6. [DOI] [PubMed] [Google Scholar]
- 11.Riggi N, Suvà ML, and Stamenkovic I. Ewing’s Sarcoma. The New England journal of medicine. 2021;384(2):154–64. [DOI] [PubMed] [Google Scholar]
- 12.Grünewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H, et al. Ewing sarcoma. Nature Reviews Disease Primers. 2018;4(1):5. [DOI] [PubMed] [Google Scholar]
- 13.Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schönegger A, Datlinger P, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell reports. 2015;10(7):1082–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 2017;171(1):163–78 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene. 2017;36(25):3505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apfelbaum AA, Wrenn ED, and Lawlor ER. The importance of fusion protein activity in Ewing sarcoma and the cell intrinsic and extrinsic factors that regulate it: A review. Frontiers in oncology. 2022;12:1044707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adane B, Alexe G, Seong BKA, Lu D, Hwang EE, Hnisz D, et al. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell. 2021;39(6):827–44. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Surdez D, Zaidi S, Grossetête S, Laud-Duval K, Ferre AS, Mous L, et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer cell. 2021. [DOI] [PubMed] [Google Scholar]
- 19.Apfelbaum AA, Wu F, Hawkins AG, Magnuson B, Jimenez JA, Taylor SD, et al. EWS-FLI1 and HOXD13 control tumor cell plasticity in Ewing sarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, He XY, Wu XS, Huang YH, Toneyan S, Ha T, et al. ETV6 dependency in Ewing sarcoma by antagonism of EWS-FLI1-mediated enhancer activation. Nat Cell Biol. 2023;25(2):298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu DY, Ellegast JM, Ross KN, Malone CF, Lin S, Mabe NW, et al. The ETS transcription factor ETV6 constrains the transcriptional activity of EWS-FLI to promote Ewing sarcoma. Nat Cell Biol. 2023;25(2):285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen EA, Menon R, Bailey KM, Thomas DG, Van Noord RA, Tran J, et al. Activation of Wnt/β-Catenin in Ewing Sarcoma Cells Antagonizes EWS/ETS Function and Promotes Phenotypic Transition to More Metastatic Cell States. Cancer research. 2016;76(17):5040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, and Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11(1):5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padmanaban V, Grasset EM, Neumann NM, Fraser AK, Henriet E, Matsui W, et al. Organotypic culture assays for murine and human primary and metastatic-site tumors. Nat Protoc. 2020;15(8):2413–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery with Enrichr. Current Protocols. 2021;1(3):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fadul J, Bell R, Hoffman LM, Beckerle MC, Engel ME, and Lessnick SL. EWS/FLI utilizes NKX2–2 to repress mesenchymal features of Ewing sarcoma. Genes & cancer. 2015;6(3–4):129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, and Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer cell. 2007;11(5):421–9. [DOI] [PubMed] [Google Scholar]
- 30.Aynaud M-M, Mirabeau O, Gruel N, Grossetête S, Boeva V, Durand S, et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell reports. 2020;30(6):1767–79.e6. [DOI] [PubMed] [Google Scholar]
- 31.Kinsey M, Smith R, and Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Molecular cancer research : MCR. 2006;4(11):851–9. [DOI] [PubMed] [Google Scholar]
- 32.Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, and Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. npj Regenerative Medicine. 2019;4(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sannino G, Marchetto A, Kirchner T, and Grünewald TGP. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas? Cancer research. 2017;77(17):4556–61. [DOI] [PubMed] [Google Scholar]
- 35.Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, and Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016;49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galbo PM Jr, ., Zang X, and Zheng D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clinical cancer research : an official journal of the American Association for Cancer Research. 2021;27(9):2636–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biffi G, and Tuveson DA. Diversity and Biology of Cancer-Associated Fibroblasts. Physiological Reviews. 2021;101(1):147–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salem F, Perin L, Sedrakyan S, Angeletti A, Ghiggeri GM, Coccia MC, et al. The spatially resolved transcriptional profile of acute T cell–mediated rejection in a kidney allograft. Kidney International. 2022;101(1):131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pastushenko I, and Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends in Cell Biology. 2019;29(3):212–26. [DOI] [PubMed] [Google Scholar]
- 40.Simeonov KP, Byrns CN, Clark ML, Norgard RJ, Martin B, Stanger BZ, et al. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer cell. 2021;39(8):1150–62.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Postel-Vinay S, Véron AS, Tirode F, Pierron G, Reynaud S, Kovar H, et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet. 2012;44(3):323–7. [DOI] [PubMed] [Google Scholar]
- 42.Savola S, Klami A, Myllykangas S, Manara C, Scotlandi K, Picci P, et al. High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing’s Sarcoma Family of Tumour Patients. ISRN Oncol. 2011;2011:168712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hancock JD, and Lessnick SL. A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle. 2008;7(2):250–6. [DOI] [PubMed] [Google Scholar]
- 44.Kauer M, Ban J, Kofler R, Walker B, Davis S, Meltzer P, et al. A Molecular Function Map of Ewing’s Sarcoma. PloS one. 2009;4(4):e5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kok SY, Oshima H, Takahashi K, Nakayama M, Murakami K, Ueda HR, et al. Malignant subclone drives metastasis of genetically and phenotypically heterogenous cell clusters through fibrotic niche generation. Nature communications. 2021;12(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox TR. The matrix in cancer. Nature Reviews Cancer. 2021;21(4):217–38. [DOI] [PubMed] [Google Scholar]
- 47.Oskarsson T, Acharyya S, Zhang XHF, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature Medicine. 2011;17(7):867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian C, Öhlund D, Rickelt S, Lidström T, Huang Y, Hao L, et al. Cancer Cell–Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer research. 2020;80(7):1461–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisinger-Mathason TS, Zhang M, Qiu Q, Skuli N, Nakazawa MS, Karakasheva T, et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013;3(10):1190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Volchenboum SL, Andrade J, Huang L, Barkauskas DA, Krailo M, Womer RB, et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. The journal of pathology Clinical research. 2015;1(2):83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harvey W, Squier MV, Duance VC, and Pritchard J. A biochemical and immunohistological study of collagen synthesis in Ewing’s tumour. Br J Cancer. 1982;46(6):848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scarpa S, Modesti A, and Triche TJ. Extracellular matrix synthesis by undifferentiated childhood tumor cell lines. Am J Pathol. 1987;129(1):74–85. [PMC free article] [PubMed] [Google Scholar]
- 53.Stracca-Pansa V, Dickman PS, Zamboni G, Bevilacqua PA, and Ninfo V. Extracellular matrix of small round cell tumors of childhood: an immunohistochemical study of 67 cases. Pediatr Pathol. 1994;14(1):111–25. [DOI] [PubMed] [Google Scholar]
- 54.Hawkins AG, Basrur V, da Veiga Leprevost F, Pedersen E, Sperring C, Nesvizhskii AI, et al. The Ewing Sarcoma Secretome and Its Response to Activation of Wnt/beta-catenin Signaling. Molecular & cellular proteomics : MCP. 2018;17(5):901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hawkins AG, Pedersen EA, Treichel S, Temprine K, Sperring C, Read JA, et al. Wnt/β-catenin–activated Ewing sarcoma cells promote the angiogenic switch. JCI Insight. 2020;5(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zöllner SK, Amatruda JF, Bauer S, Collaud S, de Álava E, DuBois SG, et al. Ewing sarcoma—diagnosis, treatment, clinical challenges and future perspectives. Journal of clinical medicine. 2021;10(8):1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin J-R, Wang S, Coy S, Chen Y-A, Yapp C, Tyler M, et al. Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell. 2023;186(2):363–81.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lavie D, Ben-Shmuel A, Erez N, and Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer. 2022;3(7):793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nurmik M, Ullmann P, Rodriguez F, Haan S, and Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer. 2020;146(4):895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chong S, Graham TGW, Dugast-Darzacq C, Dailey GM, Darzacq X, and Tjian R. Tuning levels of low-complexity domain interactions to modulate endogenous oncogenic transcription. Mol Cell. 2022;82(11):2084–97 e5. [DOI] [PubMed] [Google Scholar]
- 61.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine. 2018;24(5):541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin M-Z, and Jin W-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduction and Targeted Therapy. 2020;5(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brabletz T, Lyden D, Steeg PS, and Werb Z. Roadblocks to translational advances on metastasis research. Nat Med. 2013;19(9):1104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiao Y, and Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacology & Therapeutics. 2021;221:107753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hauge A, and Rofstad EK. Antifibrotic therapy to normalize the tumor microenvironment. Journal of Translational Medicine. 2020;18(1):207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foster DS, Jones RE, Ransom RC, Longaker MT, and Norton JA. The evolving relationship of wound healing and tumor stroma. JCI insight. 2018;3(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Visser KE, and Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41(3):374–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data generated by this study have been deposited to NCBI Gene Expression Omnibus (RRID:SCR_005012) (GSE236289) database. Analyses in R will be available upon publication at https://github.com/LawlorLab/CD73_CAFlike.