

Abstract
Background and Aims
Anti-tumour necrosis factor [anti-TNF] therapy is widely used for the treatment of inflammatory bowel disease, yet many patients are primary non-responders, failing to respond to induction therapy. We aimed to identify blood gene expression differences between primary responders and primary non-responders to anti-TNF monoclonal antibodies [infliximab and adalimumab], and to predict response status from blood gene expression and clinical data.
Methods
The Personalised Anti-TNF Therapy in Crohn’s Disease [PANTS] study is a UK-wide prospective observational cohort study of anti-TNF therapy outcome in anti-TNF-naive Crohn’s disease patients [ClinicalTrials.gov identifier: NCT03088449]. Blood gene expression in 324 unique patients was measured by RNA-sequencing at baseline [week 0], and at weeks 14, 30, and 54 after treatment initiation [total sample size = 814].
Results
After adjusting for clinical covariates and estimated blood cell composition, baseline expression of major histocompatibility complex, antigen presentation, myeloid cell enriched receptor, and other innate immune gene modules was significantly higher in anti-TNF responders vs non-responders. Expression changes from baseline to week 14 were generally of consistent direction but greater magnitude [i.e. amplified] in responders, but interferon-related genes were upregulated uniquely in non-responders. Expression differences between responders and non-responders observed at week 14 were maintained at weeks 30 and 54. Prediction of response status from baseline clinical data, cell composition, and module expression was poor.
Conclusions
Baseline gene module expression was associated with primary response to anti-TNF therapy in PANTS patients. However, these baseline expression differences did not predict response with sufficient sensitivity for clinical use.
Keywords: Anti-TNF, Crohn’s disease, transcriptomic biomarkers
1. Introduction
Crohn’s disease [CD] is a chronic immune-mediated inflammatory disease [IMID] of the gastrointestinal tract. Along with ulcerative colitis [UC], it is one of the two main forms of inflammatory bowel disease [IBD]. The development of anti-tumour necrosis factor [TNF] biological therapies has revolutionized patient care for CD and a number of other IMIDs over the last two decades. Two major anti-TNF drugs, infliximab and adalimumab, are IgG1 monoclonal antibodies that bind both soluble and transmembrane TNF, inhibiting their interactions with TNF receptors.1,2 Two main mechanisms of action have been proposed: induction of CD4+ T cell apoptosis in the gut mucosa by inhibiting the TNF–TNFR2 interaction; and binding of the antibody tail [Fc region] of the drug to Fc receptors on monocytes, inducing their differentiation into wound-healing M2 macrophages.3,4
Unfortunately, anti-TNF therapy is not always effective at treating IBD. Various types of treatment failure can occur: primary non-response [PNR] within the induction period [the first 12–14 weeks for infliximab and adalimumab], secondary loss of response [LOR] during maintenance therapy after an initial response, failure to achieve remission after the treatment course, or adverse events that lead to treatment discontinuation.5 For IBD patients, the incidence of PNR is 10–40%, and the incidence of secondary LOR among initial responders is 24–46% in the first year of treatment.6–8 The ability to predict PNR and LOR could help guide changes in treatment regimens, such as dose intensification or switching to a drug class with a different mechanism of action.2,6 Reliable baseline prediction would be especially valuable, allowing stratification of patients to effective therapies from treatment initiation, minimizing healthcare costs and patient burden.
Clinical variables reported to be associated with anti-TNF response include age, disease duration, body mass index [BMI], smoking, C-reactive protein [CRP] levels, faecal calprotectin levels, serum drug concentrations, and anti-drug antibody concentrations.7,9–13 In the Personalised Anti-TNF Therapy in Crohn’s Disease [PANTS] study, the largest study of infliximab and adalimumab response in CD patients to date [enrolment n = 1610], baseline obesity, smoking, and greater disease activity were associated with low serum drug concentration after induction. A low drug concentration was in turn associated with PNR and non-remission, suggesting immunogenicity may be mediating treatment failure by increasing the drug clearance rate.8
Multiple studies have also reported transcriptomic predictors for anti-TNF response.12–20 One such example is TREM1 expression, identified as a marker of anti-TNF response in different studies with inconsistent directions of effect. Gaujoux et al.16 found TREM1 expression was lower in gut biopsies from infliximab responders than in non-responders [total cohort size n = 72], but higher in responders in a separate cohort measuring baseline whole blood expression [n = 22]. By contrast, Verstockt et al.18 reported lower TREM1 expression in responders to infliximab and adalimumab in both baseline gut biopsies [n = 44] and baseline whole blood [n = 54]. Proposed reasons for the discrepancy include false positives due to small sample sizes, differences in patient ethnicity, and differing definitions of response.12,21 In general, small sample sizes, and variation among studies in analysis methods, anti-TNF drug, response definition, tissues sampled, and disease subtype make a consensus hard to establish. Few markers for anti-TNF response of any type, clinical or transcriptomic, have been validated in independent studies, and none has yet been translated to routine clinical practice.13
To identify novel transcriptomic associations with primary response to anti-TNF therapy, we generated longitudinal RNA-sequencing [RNA-seq] data from peripheral blood samples taken from a subset of the PANTS cohort (182 primary response [PR], 142 PNR) during the first year of follow-up. Differential gene expression [DGE] between primary responders and non-responders was performed at baseline [week 0], post-induction [week 14], and during maintenance [weeks 30 and 54]. We detected differences in gene module expression that may reflect differences in disease characteristics or severity that influence risk of primary non-response. As this is one of the largest datasets currently available for assessing transcriptomic associations with anti-TNF response in IBD, we also examined the significance of previously reported transcriptomic markers from the literature. Finally, we evaluated the utility of measuring module expression for prediction of primary response status.
2. Materials and Methods
2.1. Study design
PANTS is a UK-wide, multicentre, prospective observational cohort study reporting the treatment failure rates of the anti-TNF drugs infliximab (originator, Remicade [Merck Sharp & Dohme] and biosimilar, CT-P13 [Celltrion]), and adalimumab (Humira [AbbVie]) in 1610 anti-TNF naive CD patients. The study design has been described in detail previously.8,22 In brief, patients were recruited at the time of first anti-TNF exposure between February 2013 and June 2016, and evaluated for 12 months or until drug withdrawal. Eligible patients were aged ≥6 years with evidence of active luminal CD involving the colon and/or small intestine. Four major study visits were scheduled at week 0 [baseline], week 14 [post-induction], week 30, and week 54. Additional visits were scheduled at treatment failure or study exit. At baseline, clinical and demographic data were recorded, including sex, ethnicity, BMI, smoking status, age at diagnosis, disease duration, Montreal classification, prior medical and drug history, and previous CD-related surgeries. At every visit, disease activity score, weight, current therapy, and adverse events were recorded.8
2.2. RNA-seq sample selection
A subset of PANTS patients was selected for RNA-seq, with the inclusion criteria: age ≥ 16 years; and baseline CRP ≥ 4 mg/L and/or baseline calprotectin > 100 µg/g. The target sample size was 200 patients on infliximab and 200 patients on adalimumab, with an even split between PR and PNR within each drug group. PR and PNR were defined based on patient outcome criteria from Kennedy et al.:8
Primary non-response [assessed at week 14]: exit before week 14 because of treatment failure [including resectional IBD surgery] or corticosteroid use at week 14 [new prescriptions or if previous dose had not been stopped]. Patients whose CRP did not decrease to ≤3 mg/L or by ≥50% from baseline [week 0], and whose Harvey–Bradshaw index [HBI] score did not decrease to ≤4 points or by ≥3 points from baseline were also classified as having a primary non-response.
Primary response [assessed at week 14]: decrease in CRP to ≤3 mg/L or by ≥50% from baseline [week 0], and a decrease in HBI to ≤4 points or by ≥3 points from baseline.
Remission [assessed at weeks 14, 30, and 54; implies primary response]: CRP of ≤3 mg/L and HBI score of ≤4 points, no ongoing steroid therapy, and no exit due to treatment failure.
Steroid use was defined as any systemic therapy, either oral or intravenous [including use of steroids for other conditions], but excluding single pre-infusion dosing with hydrocortisone.
PNR were required to exhibit primary non-response at week 14 and non-remission at week 54. PR were required to exhibit primary response or remission at week 14, and be in remission at week 54 [or week 30 if week 54 status was unknown]. Furthermore, within infliximab-treated patients, PNR and non-PNR were matched based on baseline immunomodulator use, baseline steroid use, age at first dose, baseline albumin, sex, and weight at study entry.
2.3. Whole blood RNA-seq
Whole blood was collected in RNA Tempus tubes [Applied Biosystems] and stored at −80°C until extraction [QIAsymphony PAXgene Blood RNA Kit, Qiagen]. RNA was quantified using the QuBit BR RNA [ThermoFisher], and RNA integrity was assessed with the 4200 TapeStation [Agilent]. RNA-seq libraries were prepared using the Kapa mRNA HyperPrep Kit, with depletion of rRNA and globin mRNA using the QIAseq FastSelect RNA Removal Kit, and adapter ligation with IDT xGEN Dual Index UMI adapters. A total of 1141 samples from 396 patients were sequenced. Raw sequencing data were demultiplexed with Picard23 and aligned to the reference genome [GRCh38] using STAR [v2.6.1d].24 Reads were deduplicated using UMI-tools25 and quantified against the Ensembl 96 gene annotation with featureCounts [v1.6.4].26
Outlier samples were excluded, defined as >2 standard deviations from the mean based on percentage of aligned reads in coding regions reported by Picard, percentage of unique reads, and number of unique reads. Samples with a sex mismatch against the documented sex were removed. As gene expression measured from bulk tissue is heavily dependent on cell composition,27 cell proportions of six common cell types in whole blood [CD4+ T cells, CD8+ T cells, B cells, NK cells, monocytes, and granulocytes] were estimated using the Houseman method28 from paired DNA methylation data.29 Samples missing clinical data and/or cell proportion estimates were removed. A total of 814 samples remained after filtering. To accommodate variability in sampling day, samples were mapped to timepoints based on Kennedy et al.’s8 windows around major visits: week 0 [week −4 to 0], week 14 [week 10–20], week 30 [week 22–38], and week 54 [week 42–66]. Samples taken at additional visits [LOR or exit] falling within one of the windows were mapped to that timepoint, unless the patient also had a major visit sample inside that window. The mapping of samples to timepoints is shown in Supplementary Figure S1a. The number of samples per patient ranged from one to four, with a median of three [Supplementary Figure S1b].
Counts were normalized for library size using edgeR [v3.28.1].30 Globin genes and short non-coding RNAs were removed. Genes with low expression were filtered, requiring genes to have at least 1.25 counts per million in >10% of samples and non-zero expression in >90% of samples. Expression data from 15 511 genes remained after filtering. Finally, log2 expression values were computed using variancePartition/voom.31,32
2.4. Statistical analyses
A full description of the statistical analyses can be found in the Supplementary Methods. In brief, DGE analyses were performed in R [v3.6.2],33 with the significance threshold set at a false discovery rate [FDR] of <0.05. Variance components analysis was used to identify influential variables for inclusion in DGE models [Supplementary Figure S2]. Cell proportion estimates were found to explain large fractions of expression variance, and adjusting for cell proportions reduced the number of significant associations but improved consistency between drug subgroups, with fewer highly significant modules showing significant drug-by-response interaction effects compared to the unadjusted models [Supplementary Figure S3]. As this study was not designed to compare between drug subgroups, we focused on models adjusted for cell composition, where the improved consistency allowed us to pool expression data from both subgroups for greater statistical power. For all DGE models, cell proportions, sequencing batch, age of onset [the patient’s age at disease diagnosis], disease duration, BMI, anti-TNF drug type, prior surgery, and smoking were included as fixed effects; and patient was included as a random effect.
Per-gene linear mixed-effects models fit using DREAM [variancePartition v1.16.1]34 were used to detect pairwise DGE between study groups. Additionally, natural cubic splines [splines::ns]33 were fit to explore non-linear expression trajectories over all four timepoints, modelling expression as a function of study day in each group [day 0 = day of first drug dose]. Different expression trajectories were detected by testing for differences in spline parameters between the PR and PNR groups. Significant genes from the spline analysis were hierarchically clustered by their mean expression in PR and PNR at each timepoint, and the gap statistic35 was used to define clusters of genes with distinct trajectories. The spline analyses were only performed with drug subgroups pooled, as relatively small sample sizes at weeks 30 and 54 precluded stratification by drug.
Rank-based gene set enrichment analyses [tmod::tmodCERNOtest, v0.46.2],36 using blood transcriptomic modules [BTMs] were performed to identify coordinately up- or downregulated gene sets. These modules represent sets of genes that are coexpressed in whole blood, derived by Li et al.37 [module names prefixed with ‘LI’] and Chaussabel et al.38 [prefixed ‘DC’] from publicly available expression datasets. Gene set overrepresentation analyses were run for BTMs [tmod::tmodHGtest] and other publicly available gene sets [gprofiler2::gost, v0.2.0].39
Single-sample gene set enrichment scores [ssGSEAs, https://github.com/broadinstitute/ssGSEA2.0/] were computed as a summary measure of module expression in a sample, both at baseline and at week 14. Predictive models using clinical variables, cell proportions, and module expression scores [baseline or week 14] to predict response status were constructed using caret [v6.0-86].40 Multiple predictive algorithms were evaluated: penalized and regularized logistic regression methods, parallel random forest, eXtreme Gradient Boosting, support vector machines with a radial basis, k-nearest neighbours, naive Bayes, and Gaussian process models. Bootstrapping [50 replicates] with the area under the curve [AUC] metric was used to tune models, evaluate internal performance, and perform model selection. Pairwise tests for the difference in AUCs were performed with pROC.41
2.5. Ethical statement
The South West Research Ethics committee approved the study [Research Ethics Committee reference: 12/SW/0323] in January 2013. Patients were included after providing informed, written consent. The study is registered with ClinicalTrials.gov identifier NCT03088449 and the protocol is available at https://www.ibdresearch.co.uk/pants/.
3. Results
3.1. Baseline module expression associated with post-induction primary non-response
After RNA-seq quantification and quality control, expression data were available for 15 511 genes and 814 samples. These samples were from 324 patients, whose characteristics are shown in Table 1. We tested for associations between primary non-response and week 0 expression of genes [Figure 1a] and gene modules37 [Figure 1b], adjusting for cell composition and other influential covariates. Although no single gene was differentially expressed in the infliximab subgroup [86 PR, 59 PNR], expression of NK cell [LI.M7.2] and T cell [LI.M7.1, LI.M7.0] modules was significantly lower in responders. In the adalimumab subgroup [66 PR, 57 PNR], PDIA5 (log2 fold change [FC] = −0.3512, FDR = 0.006777), KCNN3 [log2 FC = −0.8798, FDR = 0.006777], and IGKV1-9 [log2 FC = −1.223, FDR = 0.04518] expression was significantly lower in responders, accompanied by lower expression of plasma cell/immunoglobulin [LI.M156.0, LI.M156.1] and cell cycle [LI.M4.0, LI.M4.1] modules. This heterogeneity between drug subgroups was robust to model form [Figure S4] and differences in sample size between subgroups [Figure S5]. A pooled analysis was performed to identify modules consistently differentially expressed in both drug subgroups [152 PR, 116 PNR]. The MHC-TLR7-TLR8 cluster [LI.M146], antigen presentation [LI.M71, LI.M95.0], and myeloid cell enriched receptor and transporter [LI.M4.3] modules had higher expression at baseline in responders. Several of these module associations were largely or partially driven by major histocompatibility complex [MHC] class I and class II genes [Figure S6].
Table 1.
Baseline patient characteristics for the PANTS RNA-seq subcohort.
| Adalimumab [ADA] | Infliximab [IFX] | Drugs pooled | p-value | |
|---|---|---|---|---|
| Sex [Col %] | ||||
| Female | 78 [48.4%] | 89 [54.6%] | 167 [51.5%] | |
| Male | 83 [51.6%] | 74 [45.4%] | 157 [48.5%] | |
| Age of onset [years] | 0.774 | |||
| Mean [SD] | 33.3 [15.4] | 32.8 [15.3] | 33.1 [15.3] | Wilcoxon rank-sum |
| Missing | 0 | 0 | 0 | |
| Disease duration [years] | 0.546 | |||
| Mean [SD] | 6.1 [8.1] | 5.9 [7.7] | 6.0 [7.9] | Wilcoxon rank-sum |
| Missing | 0 | 0 | 0 | |
| Smoking status [Col %] | 0.263 | |||
| Current | 28 [17.4%] | 36 [22.1%] | 64 [19.8%] | Fisher exact |
| Ex | 55 [34.2%] | 43 [26.4%] | 98 [30.2%] | |
| Never | 78 [48.4%] | 84 [51.5%] | 162 [50.0%] | |
| Crohn’s-related surgery [Col %] | 0.549 | |||
| No | 114 [70.8%] | 110 [67.5%] | 224 [69.1%] | Fisher exact |
| Yes | 47 [29.2%] | 53 [32.5%] | 100 [30.9%] | |
| On immunomodulator ever [Col %] | 0.543 | |||
| No | 23 [14.3%] | 28 [17.2%] | 51 [15.7%] | Fisher exact |
| Yes | 138 [85.7%] | 135 [82.8%] | 273 [84.3%] | |
| On immunomodulator at baseline [Col %] | 0.912 | |||
| No | 79 [49.1%] | 81 [49.7%] | 160 [49.4%] | Fisher exact |
| Yes | 82 [50.9%] | 82 [50.3%] | 164 [50.6%] | |
| On corticosteroids at baseline [Col %] | 0.011 | |||
| No | 113 [70.2%] | 92 [56.4%] | 205 [63.3%] | Fisher exact |
| Yes | 48 [29.8%] | 71 [43.6%] | 119 [36.7%] | |
| Baseline BMI | 0.237 | |||
| Mean [SD] | 25.2 [6.2] | 24.3 [5.5] | 24.8 [5.9] | Wilcoxon rank-sum |
| Missing | 0 | 0 | 0 | |
| Primary response status [Col %] | 0.263 | |||
| Primary non-response | 76 [47.2%] | 66 [40.5%] | 142 [43.8%] | Fisher exact |
| Primary response | 85 [52.8%] | 97 [59.5%] | 182 [56.2%] | |
| CD8+ T cell [%] | 0.380 | |||
| Mean [SD] | 2.8 [4.2] | 2.8 [5.2] | 2.8 [4.7] | Wilcoxon rank-sum |
| Missing | 38 | 18 | 56 | |
| CD4+ T cell [%] | 0.752 | |||
| Mean [SD] | 9.2 [6.3] | 9.2 [6.8] | 9.2 [6.5] | Wilcoxon rank-sum |
| Missing | 38 | 18 | 56 | |
| B cell [%] | 0.094 | |||
| Mean [SD] | 1.9 [2.0] | 1.5 [1.9] | 1.7 [1.9] | Wilcoxon rank-sum |
| Missing | 38 | 18 | 56 | |
| Monocyte [%] | 0.497 | |||
| Mean [SD] | 8.9 [3.5] | 9.2 [3.7] | 9.0 [3.6] | Wilcoxon rank-sum |
| Missing | 38 | 18 | 56 | |
| NK cell [%] | 0.683 | |||
| Mean [SD] | 1.9 [3.2] | 1.9 [3.8] | 1.9 [3.5] | Wilcoxon rank-sum |
| Missing | 38 | 18 | 56 | |
| Granulocyte [%] | 0.911 | |||
| Mean [SD] | 74.3 [9.7] | 74.3 [10.8] | 74.3 [10.3] | Wilcoxon rank-sum |
| Missing | 38 | 18 | 56 |
Patient characteristics are stratified by drug subgroup. Values shown are count and percentage for categorical variables, and mean and standard deviation for continuous variables. Nominal p-values are reported for the comparison between drug subgroups.
Figure 1.
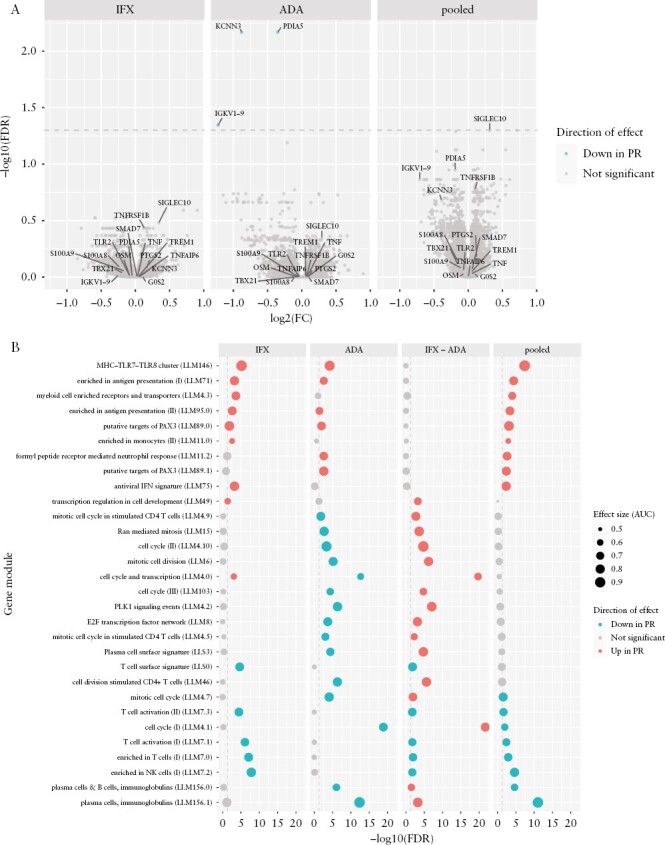
Baseline expression associated with primary response. [A] Volcano plots of differential gene expression between responders [PR] and non-responders [PNR] at week 0: for infliximab [IFX], adalimumab [ADA], or with drug subgroups pooled. Annotated genes show significant associations from this study and previously reported associations from the literature in both blood and gut biopsies. Dashed line shows significance threshold at FDR = 0.05. [B] Top gene modules differentially expressed between PR and PNR at week 0. Columns correspond to results for IFX, ADA, difference between IFX and ADA [IFX − ADA, i.e. the drug-by-response interaction], and pooled drug analyses. The top 30 modules ranked by minimum FDR in any column are shown. Dashed lines show significance thresholds at FDR = 0.05.
In addition, we collated baseline markers of anti-TNF response in gut mucosal biopsies and blood from the literature.14,15,18,42 These were not significant in our per-gene DGE analyses [Figure 1a].
3.2. Expression changes from baseline to post-induction are largely amplified in primary responders
To characterize the changes in gene expression induced by anti-TNF therapy, we compared expression at baseline to expression post-induction, and also estimated the difference between expression changes in responders and non-responders [the timepoint-by-response interaction]. As expression changes from week 0 to week 14 were relatively consistent between patients on infliximab and adalimumab after adjusting for cell composition [Figure S7], we pooled drug subgroups for these models. We found that 5572 and 626 genes were differentially expressed between week 14 and week 0 in responders and non-responders respectively, with 179 genes having a significant timepoint-by-response interaction. Of the genes differentially expressed in both responders and non-responders, and with a significant timepoint-by-response interaction, nearly all [31/32 genes] had an expression change that was amplified in responders [Figure 2a]. For example, CD177, a neutrophil marker upregulated during inflammation, was downregulated at week 14 in both responders [log2 FC = −2.225, FDR = 4.104 × 10−17] and non-responders [log2 FC = −0.8981, FDR = 0.004598], with significantly greater downregulation in responders [interaction FDR = 0.008247]. Modules differentially expressed between week 0 and week 14 included upregulation of B cell [LI.M47.0], plasma cell [LI.M156.0], and T cell activation [LI.M7.1] modules; and downregulation of immune activation [LI.M37.0], monocyte [LI.M11.0], neutrophil [LI.M37.1], and Toll-like receptor [TLR] and inflammatory signalling [LI.M16] modules [Figure 2b]. Statistically significant amplification of expression changes in responders was also observed at the module level, with nearly all module expression changes aligned in the same direction in responders and non-responders, but with larger effect sizes in responders.
Figure 2.
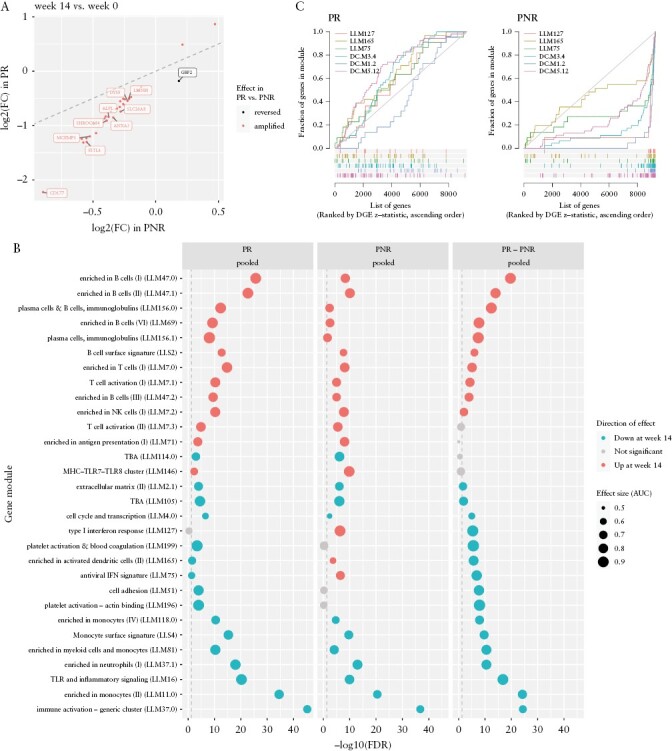
Expression changes from baseline to post-induction in responders and non-responders. [A] Expression log2 fold changes from week 0 to week 14 in primary responders [PR] and non-responders [PNR], for genes that were differentially expressed from week 0 to week 14 in both responders and non-responders, with a significantly different effect size between responders and non-responders [top ten labelled]. The identity line is shown by the dashed line. [B] Top modules differentially expressed between week 14 and week 0. Columns show effects in PR, PNR, and the PR minus PNR difference [the timepoint-by-response interaction]. The top 30 modules ranked by minimum FDR in any column are shown. Vertical dashed line shows significance threshold at FDR = 0.05. [C] Barcode plots showing interferon modules upregulated from week 0 to week 14 in PNR, but not in PR. Genes are ranked in ascending order by week 14 vs week 0 DGE z-statistic, with coloured bars indicating the rank of genes in a module. Curves show the cumulative fraction of genes in a module at a particular rank threshold. The area under the curve [AUC] reflects the effect size of the module association. Diagonal line shows the null of randomly distributed ranks. Modules sourced from Li et al.37 [prefixed ‘LI’] and Chaussabel et al.38 [prefixed ‘DC’].
In contrast, GBP2 [a member of a family of guanylate-binding proteins induced by interferons43] was downregulated from week 0 to week 14 in responders [log2 FC = −0.1783, FDR = 0.004878], but upregulated in non-responders [log2 FC = 0.1849, FDR = 0.04502; interaction FDR = 0.005977]. At the module level, upregulation of the type I interferon response [LI.M127], activated dendritic cell [LI.M165], and antiviral IFN signature [LI.M75] modules was observed in non-responders but not in responders [Figure 2b]. Extended gene set enrichment analyses including additional modules from Chaussabel et al.38 also identified interferon modules significantly upregulated at week 14 in non-responders: DC.M3.4, containing STAT2, GBP5, and PARP14 [FDR = 3.447 × 10−21]; and two modules containing IFIT3 and GBP2, DC.M1.2 [FDR = 9.492 × 10−16] and DC.M5.12 [FDR = 1.355 × 10−13]. None of these modules were differentially expressed from week 0 to week 14 in responders [Figure 2c], suggesting upregulation of interferon pathways post-induction occurs uniquely in primary non-responders.
3.3. Sustained expression differences between responders and non-responders during maintenance
As PANTS was an observational study, it was possible to retain patients who continued with anti-TNF therapy even after meeting the study definition of PNR at week 14, enabling us to sample the blood transcriptome at weeks 30 and 54 during the maintenance period. Utilizing all 814 samples over the four study timepoints, we tested for general differences in expression trajectory over time, detecting 210 differentially expressed genes between responders and non-responders after adjustment for cell composition. To visualize the expression of these genes and identify common patterns of expression change during anti-TNF therapy, significant genes were hierarchically clustered by their expression. Six clusters were identified [Figure 3a], each with distinct expression trajectories for responders and non-responders [Figure 3b]. Cluster 1 largely comprised genes previously found to have a significant difference in expression change from week 0 to week 14 between responders and non-responders [97/132 genes in the cluster]. The most significant gene was KREMEN1 [FDR = 4.287 × 10−4], part of an inflammatory apoptotic pathway in the gut epithelium.44 Cluster 1 genes were enriched in modules associated with myeloid cells and monocytes [LI.M81, hypergeometric test, FDR = 2.115 × 10−6], platelet activation [LI.M196, 1.348 × 10−5], immune activation [LI.M37.0, 1.436 × 10−4], and TLR and inflammatory signalling [LI.M16, FDR = 2.365 × 10−3] [Figure 3c]. Expression trajectories showed cluster 1 genes were more downregulated from baseline in responders than in non-responders, probably representing a lower inflammatory state in responders by week 14 that is sustained at weeks 30 and 54. An opposing trend was observed in cluster 5, which contained genes enriched for B cell development/activation [LI.M58, FDR = 0.01653] that were more upregulated from baseline in responders than non-responders.
Figure 3.
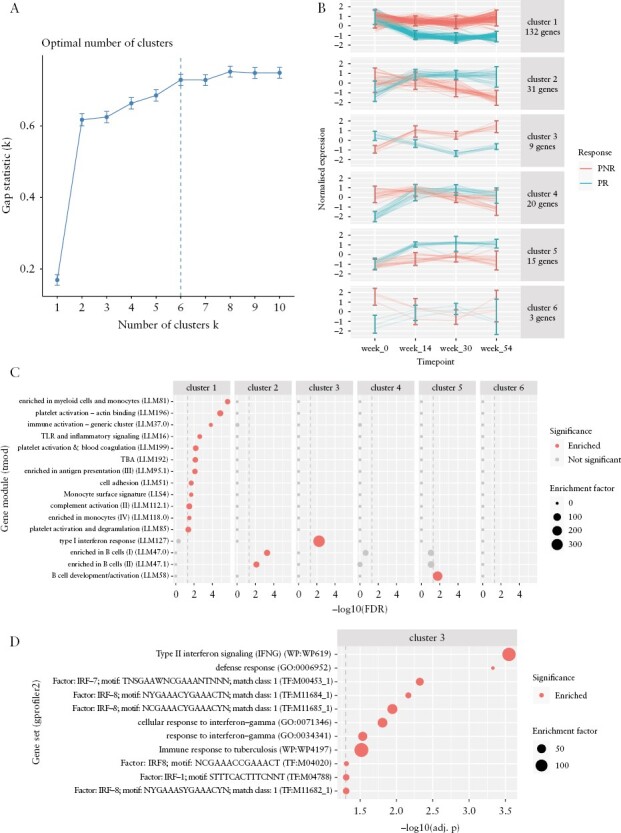
Expression differences between responders [PR] and non-responders [PNR] during maintenance. [A] Gap statistic vs cluster number k from hierarchical clustering of genes with significant expression differences between PR and PNR over all timepoints. Error bars derived from 500 bootstraps. The optimal cluster number was selected to be k = 6 by the factoextra::fviz_nbclust firstSEmax criteria [https://rpkgs.datanovia.com/factoextra/index.html]. [B] Normalized expression over study timepoints for genes in each cluster; 95% confidence intervals for expression are shown for each group at each timepoint. [C] Gene modules enriched in each cluster from gene set overrepresentation analyses. Modules significantly enriched in any cluster are shown. Vertical dashed line shows significance threshold at FDR = 0.05. [D] Gene sets enriched in cluster 3 from gene set overrepresentation analyses using gprofiler2::gost.39 Vertical dashed line shows significance threshold at an adjusted p-value = 0.05 [gost g:SCS multiple testing correction method].
Cluster 3 was uniquely enriched for the type I interferon response [LI.M127, FDR = 0.005681] [Figure 3c]. Subsequent enrichment analyses using publicly available gene sets39 revealed enrichments for type II interferon signalling [WP:WP619, adj. p = 2.826 × 10−4], and for genes containing putative transcription factor [TF] binding motifs for interferon regulatory factors IRF7 [TF:M00453_1, adj. p = 0.004768] and IRF8 [TF:M11684_1, adj. p = 0.006853; TF:M11685_1, adj. p = 0.01136] [Figure 3d]. The genes in cluster 3 showed opposing directions of expression change from week 0 to week 14 in responders vs non-responders, generating expression differences at week 14 that were also sustained at weeks 30 and 54. A significant interaction between week 0 to week 14 expression change and response status from the per-gene differential expression analyses was observed for eight of the nine genes in the cluster [STAT1, BATF2, GBP1, GBP5, IRF1, TAP1, APOL1, APOL2], many of which are key interferon signalling genes.43,45,46 Unlike the majority of genes that followed trajectories of greater expression change in responders, genes in interferon response pathways were uniquely upregulated in non-responders after anti-TNF therapy.
3.4. Prediction of primary non-response from gene expression and clinical variables
Given there were numerous module-level associations with primary response status, we evaluated the ability to predict response from module expression. For predictive models using only baseline data, the median resampling AUCs over all combinations of algorithms and predictor sets evaluated ranged from 0.5541 to 0.6686 [Supplementary Figure S8]. The best-performing model was regularized logistic regression [caret regLogistic model40; hyperparameters: cost = 0.25, loss = L1, epsilon = 0.01] using clinical variables, cell proportions, and module scores from ssGSEA as predictors, giving a median resampling AUC of 0.6686, a median sensitivity of 0.5392, and a median specificity of 0.6852, where non-response was the positive class, with a prevalence of 43% [116/268]. Including cell proportions and module scores did improve predictive performance compared to using only clinical variables [bootstrap p = 0.02629], but the increase in AUC was only 2.5% [Figure 4a]. This suggests that clinical variables provided the greatest contributions to baseline prediction, especially those variables with consistently high importance scores [high absolute t-statistics]: smoking history, BMI, and baseline steroid usage [Figure 4b].
Figure 4.
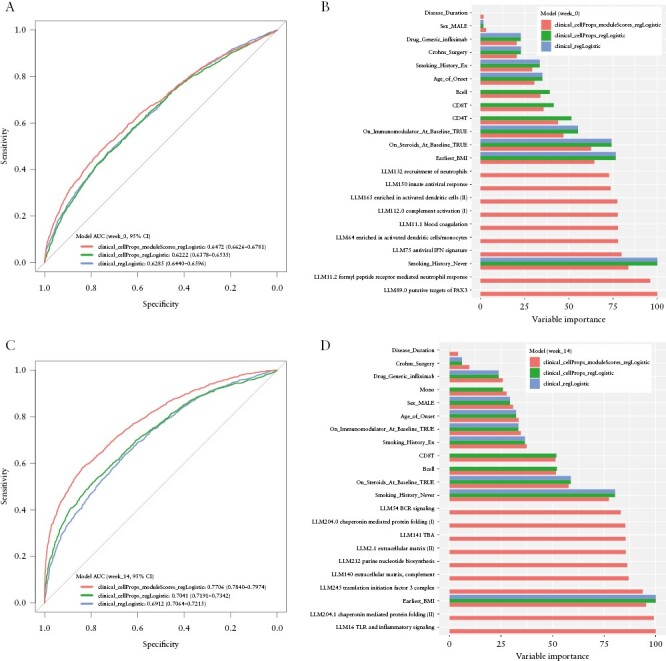
Prediction of response status from clinical variables, cell proportions, and expression data. Receiver operating characteristic [ROC] curves for the caret regLogistic method40 trained on each predictor dataset are shown at baseline [A] and week 14 [C]. ROC curves were plotted after merging all 50 resamples. Primary non-response was used as the positive class. DeLong 95% confidence intervals for the AUC are shown. The ten most important variables from models trained on each predictor dataset are shown for baseline [B] and week 14 [D] models. The overall variable importance score is computed from the absolute value of the t-statistic for each predictor from the final tuned models. Missing bars denote variables that were not in the predictor dataset for that model.
Model performance was improved by utilizing week 14 cell proportions and module scores instead of baseline [median resampling AUC range 0.6216–0.7957] [Supplementary Figure S9]. The prevalence of non-response at week 14 was 42% [104/246]. Again, the best performing model was regularized logistic regression incorporating clinical variables, cell proportions, and module scores as predictors [median resampling AUC = 0.7957, sensitivity = 0.6248, specificity = 0.7961]. Adding week 14 module scores to the predictor dataset had a larger benefit, with a 6.5% increase in AUC comparing the full model to the model including only clinical variables and cell proportions [bootstrap p = 2.306 × 10−10] [Figure 4c]. The modules with the highest variable importance included TLR and inflammatory signalling [LI.M16], chaperonin-mediated protein folding [LI.M204.0, LI.M204.1], and translation initiation factor 3 complex [LI.M245] modules [Figure 4d]. Greater predictive performance at week 14 than baseline probably reflects the larger expression differences observed between responders and non-responders after the induction period.
4. Discussion
We found substantial differences in whole blood gene expression between anti-TNF primary responders and non-responders in the PANTS cohort. At baseline, three single-gene associations detected in the adalimumab subgroup implicated similar cell types; IGKV1-9 encodes the immunoglobulin light chain variable region that forms part of antibodies produced by plasma cells, KCNN3 is annotated to a plasma cell surface signature module from Li et al.37 [LI.S3], and the expression of both KCNN3 and PDIA5 is high in plasma cells [www.proteinatlas.org/humanproteome/immune+cell, v21.1] and positively correlated with plasmablast frequencies in blood.47 These genes were downregulated in responders, as was the expression of plasma cell and immunoglobulin modules. In keeping with our observations, Martin et al.17 identified plasma cells as part of a correlated module of cell populations, where lower module expression in gut biopsies was associated with better response to anti-TNFs. Baseline plasma cell abundances in gut biopsies have also been reported to be lower in responders, albeit in relatively small cohorts of infliximab patients.16 Our findings lend credence that associations driven by immune cells observed in gut biopsies may also be observable in blood, a more accessible tissue.
Previously reported single-gene baseline markers in gut biopsies and blood were non-significant in this study. For example, TREM116,18 was not significantly differentially expressed between responders and non-responders in blood samples from PANTS patients. Our observation is consistent with two recent trials of comparable sample size, SERENE-CD and SERENE-UC, where baseline blood TREM1 expression was not predictive of response in either CD or UC patients.48 A variety of factors could explain failures to replicate reported markers from study to study. Many existing studies pool heterogeneous cohorts of patients taking different anti-TNF drugs due to the scarcity of large datasets, but even between arms of the PANTS study, we observed within-study differences in expression. Additional between-study variation can arise from differences in clinical setting, tissues sampled [e.g. blood vs gut biopsies], and definition of primary response [e.g. endoscopic vs clinical parameters]. Any two studies are unlikely to have adjusted for the same combinations of covariates in modelling, including covariates such as cell composition that heavily influence bulk expression data. Finally, small sample sizes have considerable sampling error. We recommend the use of set-based methods over single-gene association tests for identification of biomarkers, as drawing on differences in multiple genes improves statistical power, and may also improve reproducibility between studies. Despite the small number of single-gene associations in PANTS, we detected module-level associations that were consistent between drug subgroups, revealing a higher baseline expression of MHC and antigen presentation modules in primary responders. Genetic variation in the MHC region has been previously linked to immunogenicity rates in PANTS, where carriage of HLA-DQA1*05 was associated with a 2-fold increase in the risk for developing anti-drug antibodies, although the mechanism is yet unknown.22
Utilizing the longitudinal design of PANTS, we characterized changes in blood gene expression post-induction. Reduced expression of immune activation, monocyte, and neutrophil modules in responders at week 14 is consistent with successful drug inhibition of TNF-mediated inflammation, which correlates with reduced neutrophil activation and reduced monocyte recruitment.49 Apoptosis of monocytes induced by anti-TNF in CD patients has also been previously described.44 Certain B cell subsets are reduced in the blood of IBD patients compared to controls,50 so upregulation of B cell modules at week 14 may also represent a shift towards health. Similar expression changes were observed in responders and non-responders, but with greater magnitude in responders, potentially suggesting a continuum of response. Gaujoux et al.16 found that changes in cell proportions after anti-TNF treatment were amplified in responders; here we demonstrate a similar trend at the transcriptional level in PANTS. Post-induction expression differences between responders and non-responders were sustained at weeks 30 and 54 during the anti-TNF maintenance period. Kennedy et al.8 found that ‘continuing standard dosing regimens after primary non-response was rarely helpful’ for inducing remission by week 54. This phenomenon may also be reflected in the blood transcriptome, although non-responders in this study were selected to exclude patients in remission by week 54, so expression trajectories for non-responders at week 14 who eventually achieved remission could not be observed, and differences in trajectory between PANTS responders and non-responders may be exaggerated.
Unlike the majority of baseline vs post-induction associations, expression changes in genes and modules in the interferon pathway were uniquely upregulated in PANTS non-responders. Previous studies in IBD are conflicting, with Samie et al.51 reporting elevated expression of interferon pathway genes in colonic biopsies from non-responders compared to responders, with no significant change pre- vs post-treatment [n ≈ 40]; and Mavragani et al.20 reporting a post-treatment reduction in blood interferon expression only in non-responders [n = 30]. In studies of rheumatoid arthritis [RA], another IMID with licensed anti-TNF therapies, increases in type I interferon-regulated gene expression in blood after infliximab treatment were associated with poor clinical response [discovery n = 15, validation n = 18].52 A systematic review of our study with other studies reporting similar associations between interferon pathway genes and anti-TNF response would not only help resolve the direction of effect, if any, but also provide an opportunity to consider the shared biology of anti-TNF response in IBD, RA, and other IMIDs.
We were unable to build clinically useful predictive models of response incorporating expression data. Using only baseline clinical variables, Kennedy et al.8 used logistic regression with stepwise variable selection based on Akaike’s information criterion [AIC] to predict response in the full PANTS cohort, achieving AUCs of 0.53 (95% confidence interval [CI] 0.46–0.59) for infliximab patients and 0.54 [0.46–0.62] for adalimumab patients. Whilst our best-performing baseline model achieved a 13% improvement in AUC, expression data only contributed a small amount of predictive power on top of clinical variables and cell composition. Unsurprisingly, models had greater predictive power when provided with week 14 expression and cell composition data, and adding expression data also provided a comparatively large increase in AUC. This suggests that when expression differences between responders and non-responders are sufficiently large, transcriptomic markers do provide unique information, and are not simply proxies for clinical variables or coarse estimates of blood cell composition. A potential route to more effective prediction is to consider whether expression differences arising early in the induction period can discriminate between responders and non-responders. For example, Mesko et al.53 found that week 2 blood gene expression was predictive of infliximab response in CD [discovery n = 20, validation n = 20] and RA [discovery n = 19, validation n = 15] patients. More recently, Mishra et al.54 trained random forest models using blood DNA methylation and gene expression measured in IBD patients receiving infliximab [n = 37]. They did not find consistent baseline-only predictive signatures, but a model combining baseline with week 2 measurements predicted response in the Mesko et al. cohort with 85% accuracy [95% CI: 62–97%]. As we observed in PANTS, expression differences between responders and non-responders were far greater by week 14 than at baseline. Post-induction associations were also more consistent between drug subgroups, as baseline differences are diluted by the large transcriptomic perturbation from taking an anti-TNF. Expression changes in the innate immune system are observable within hours of treatment initiation,54 and robust prediction of non-response within that timeframe may be more valuable than a less reliable prediction at baseline.
An important limitation of our analyses is that PANTS was not designed to directly compare between drug subgroups. Differences between patient groups taking different anti-TNF drugs can arise from patient and physician preferences, influenced by cost, disease severity, location, and comorbidities. Unsurprisingly, many associations with response had significantly different effect sizes in the infliximab and adalimumab patient subgroups. We found that adjusting DGE models for estimated proportions of major cell types as a proxy for these uncontrolled factors alleviated heterogeneity between subgroups. However, the adjustment is unlikely to work well for rare cell types, and thus the associations we report may reflect differences in cell proportions rather than per-cell expression. Given the myriad of factors that could drive the observed heterogeneity, we strongly caution against interpreting associations with different effects in the PANTS infliximab and adalimumab subgroups as drug-driven differences with biological significance, and recommend that future transcriptomic studies consider influential factors such as cell composition. Additionally, PANTS defined response pragmatically as a composite, clinical outcome incorporating physician evaluation, disease severity scores, and serum CRP. Although this clinical outcome is significantly associated with faecal calprotectin, which correlates with endoscopic outcomes,8 our results would have been strengthened if paired endoscopic data were available.
In conclusion, we observed significant differences in gene module expression between responders and non-responders to anti-TNF therapy in the whole blood of PANTS CD patients at baseline and post-treatment timepoints. Interferon-induced genes were uniquely upregulated post-induction in non-responders, going against the general trend of amplified transcriptomic change in responders vs non-responders. We were unable to robustly predict response from baseline data with our current sample size. To obtain more accurate predictions, utilizing large upcoming datasets with paired drug response phenotypes and transcriptomic data such as the 1000IBD project will be essential.55
Supplementary Material
Acknowledgments
We would like to acknowledge Jim Butler, Areej Ammar, Erin Murphy, Stephen Abel, Elizabeth Asque, and Justin Wade Davis from AbbVie Inc., Chicago, IL, USA, for supporting this work. Laboratory tests were undertaken by the Exeter Blood Sciences Laboratory at the Royal Devon & Exeter [RD&E] NHS Trust [https://www.exeterlaboratory.com/]. The Exeter NIHR Clinical Research Facility coordinated sample storage and management. The sponsor of the study was the Royal Devon and Exeter NHS Foundation Trust. Simeng Lin is supported by a Wellcome GW4-CAT fellowship [222850/Z/21/Z]. Neil Chanchlani acknowledges support from Crohn’s & Colitis UK. We thank the patients who participated in the PANTS study, the Inflammatory Bowel Disease Pharmacogenetic Study Group, and research nurses who collected clinical data and biological samples at each study visit. We acknowledge the study co-ordinators of the Exeter IBD Research Group, UK: Marian Parkinson and Helen Gardner-Thorpe for their ongoing administrative support to the study.
Contributor Information
Benjamin Y H Bai, Genomics of Inflammation and Immunity Group, Wellcome Sanger Institute, Hinxton, UK; Postgraduate School of Life Sciences, University of Cambridge, Cambridge, UK.
Mark Reppell, AbbVie Inc., Chicago, IL, USA.
Nizar Smaoui, AbbVie Inc., Chicago, IL, USA.
Jeffrey F Waring, AbbVie Inc., Chicago, IL, USA.
Valerie Pivorunas, AbbVie Inc., Chicago, IL, USA.
Heath Guay, AbbVie Inc., Chicago, IL, USA.
Simeng Lin, Department of Gastroenterology, Royal Devon University Healthcare NHS Foundation Trust, Exeter, UK; Exeter Inflammatory Bowel Disease and Pharmacogenetics Research Group, University of Exeter, Exeter, UK.
Neil Chanchlani, Department of Gastroenterology, Royal Devon University Healthcare NHS Foundation Trust, Exeter, UK; Exeter Inflammatory Bowel Disease and Pharmacogenetics Research Group, University of Exeter, Exeter, UK.
Claire Bewshea, Exeter Inflammatory Bowel Disease and Pharmacogenetics Research Group, University of Exeter, Exeter, UK.
James R Goodhand, Department of Gastroenterology, Royal Devon University Healthcare NHS Foundation Trust, Exeter, UK; Exeter Inflammatory Bowel Disease and Pharmacogenetics Research Group, University of Exeter, Exeter, UK.
Nicholas A Kennedy, Department of Gastroenterology, Royal Devon University Healthcare NHS Foundation Trust, Exeter, UK; Exeter Inflammatory Bowel Disease and Pharmacogenetics Research Group, University of Exeter, Exeter, UK.
Tariq Ahmad, Department of Gastroenterology, Royal Devon University Healthcare NHS Foundation Trust, Exeter, UK; Exeter Inflammatory Bowel Disease and Pharmacogenetics Research Group, University of Exeter, Exeter, UK.
Carl A Anderson, Genomics of Inflammation and Immunity Group, Wellcome Sanger Institute, Hinxton, UK.
UK Inflammatory Bowel Disease Pharmacogenetics Study Group:
Vinod Patel, Zia Mazhar, Rebecca Saich, Ben Colleypriest, Tony C Tham, Tariq H Iqbal, Vishal Kaushik, Senthil Murugesan, Salil Singhi, Sean Weaver, Cathryn Preston, Assad Butt, Melissa Smith, Dharamveer Basude, Amanda Beale, Sarah Langlands, Natalie Direkze, Miles Parkes, Franco Torrente, Juan De La Revella Negro, Chris Ewen MacDonald, Stephen M Evans, Anton V J Gunasekera, Alka Thakur, David Elphick, Achuth Shenoy, Chuka U Nwokolo, Anjan Dhar, Andrew T Cole, Anurag Agrawal, Stephen Bridger, Julie Doherty, Sheldon C Cooper, Shanika de Silva, Craig Mowat, Phillip Mayhead, Charlie Lees, Gareth Jones, Tariq Ahmad, James W Hart, Daniel R Gaya, Richard K Russell, Lisa Gervais, Paul Dunckley, Tariq Mahmood, Paul J R Banim, Sunil Sonwalkar, Deb Ghosh, Rosemary H Phillips, Amer Azaz, Shaji Sebastian, Richard Shenderey, Lawrence Armstrong, Claire Bell, Radhakrishnan Hariraj, Helen Matthews, Hasnain Jafferbhoy, Christian P Selinger, Veena Zamvar, John S De Caestecker, Anne Willmott, Richard Miller, Palani Sathish Babu, Christos Tzivinikos, Stuart L Bloom, Guy Chung-Faye, Nicholas M Croft, John M E Fell, Marcus Harbord, Ailsa Hart, Ben Hope, Peter M Irving, James O Lindsay, Joel E Mawdsley, Alistair McNair, Kevin J Monahan, Charles D Murray, Timothy Orchard, Thankam Paul, Richard Pollok, Neil Shah, Sonia Bouri, Matt W Johnson, Anita Modi, Kasamu Dawa Kabiru, B K Baburajan, Bim Bhaduri, Andrew Adebayo Fagbemi, Scott Levison, Jimmy K Limdi, Gill Watts, Stephen Foley, Arvind Ramadas, George MacFaul, John Mansfield, Leonie Grellier, Mary-Anne Morris, Mark Tremelling, Chris Hawkey, Sian Kirkham, Charles P J Charlton, Astor Rodrigues, Alison Simmons, Stephen J Lewis, Jonathon Snook, Mark Tighe, Patrick M Goggin, Aminda N De Silva, Simon Lal, Mark S Smith, Simon Panter, Fraser Cummings, Suranga Dharmisari, Martyn Carter, David Watts, Zahid Mahmood, Bruce McLain, Sandip Sen, Anna J Pigott, David Hobday, Emma Wesley, Richard Johnston, Cathryn Edwards, John Beckly, Deven Vani, Subramaniam Ramakrishnan, Rakesh Chaudhary, Nigel J Trudgill, Rachel Cooney, Andy Bell, Neeraj Prasad, John N Gordon, Matthew J Brookes, Andy Li, and Stephen Gore
Funding
PANTS is an investigator-led study funded by CORE [renamed Guts UK in 2018], the research charity of the British Society of Gastroenterology, and by unrestricted educational grants from AbbVie Inc, USA; Merck Sharp & Dohme Ltd, UK; NAPP Pharmaceuticals Ltd, UK; Pfizer Ltd, USA; Celltrion Healthcare, South Korea; and Cure Crohn’s Colitis [Scottish IBD Charity]. The sponsor of the PANTS study is the Royal Devon and Exeter National Health Service Foundation Trust. Data generation, sequencing, and sample analysis were funded by AbbVie. This research was funded in whole, or in part, by the Wellcome Trust [grant numbers 206194, 108413/A/15/D, and 222850/Z/21/Z]. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Conflicts of Interest
Mark Reppell, Nizar Smaoui, Jeffrey F. Waring, Valerie Pivorunas, and Heath Guay are employees of AbbVie and may own stock and/or options. Simeng Lin reports non-financial support from Pfizer outside the submitted work. James R. Goodhand reports grants from F. Hoffmann-La Roche AG, grants from Biogen Inc, grants from Celltrion Healthcare, grants from Galapagos NV, and non-financial support from Immundiagnostik outside the conduct of the study. Nicholas A. Kennedy reports grants from F. Hoffmann-La Roche AG, grants from Biogen Inc, grants from Celltrion Healthcare, grants from Galapagos NV, and non-financial support from Immundiagnostik; grants and non-financial support from AbbVie, grants and personal fees from Celltrion, personal fees and non-financial support from Janssen, personal fees from Takeda, and personal fees and non-financial support from Dr Falk, outside the submitted work. Tariq Ahmad reports grants and non-financial support from F. Hoffmann-La Roche AG, grants from Biogen Inc, grants from Celltrion Healthcare, grants from Galapagos NV, and non-financial support from Immundiagnostik; personal fees from Biogen Inc, grants and personal fees from Celltrion Healthcare, personal fees and non-financial support from Immundiagnostik, personal fees from Takeda, personal fees from ARENA, personal fees from Gilead, personal fees from Adcock Ingram Healthcare, personal fees from Pfizer, personal fees from Genentech, and non-financial support from Tillotts, outside the submitted work. Carl A. Anderson has received consultancy or lectureship fees from Genomics plc, BridgeBio, and GSK. The remaining authors have no conflicts of interest to report.
UK Inflammatory Bowel Disease Pharmacogenetics Study Group
Dr Vinod Patela, Dr Zia Mazharb, Dr Rebecca Saichc, Dr Ben Colleypriestd, Dr Tony C Thame, Dr Tariq H Iqbalf, Dr Vishal Kaushikg, Dr Senthil Murugesanh, Dr Salil Singhi, Dr Sean Weaverj, Dr Cathryn Prestonk, Dr Assad Buttl, Dr Melissa Smithl, Dr Dharamveer Basudem, Dr Amanda Bealem, Dr Sarah Langlandsn, Dr Natalie Direkzen, Dr Miles Parkeso, Dr Franco Torrenteo, Dr Juan De La Revella Negroo, Dr Chris Ewen MacDonaldp, Dr Stephen M Evansq, Dr Anton V J Gunasekerar, Dr Alka Thakurq, Dr David Elphicks, Dr Achuth Shenoyt, Prof Chuka U Nwokolou, Dr Anjan Dharv, Dr Andrew T Colew, Dr Anurag Agrawalx, Dr Stephen Bridgery, Dr Julie Dohertyz, Dr Sheldon C Cooperaa, Dr Shanika de Silvaaa, Dr Craig Mowatcc, Dr Phillip Mayheaddd, Dr Charlie Leesee, Dr Gareth Jonesee, Dr Tariq Ahmadff, Dr James W Hartff, Dr Daniel R Gayagg, Prof Richard K Russellhh, Dr Lisa Gervaishh, Dr Paul Dunckleyii, Dr Tariq Mahmoodjj, Dr Paul J R Banimkk, Dr Sunil Sonwalkarll, Dr Deb Ghoshmm, Dr Rosemary H Phillipsmm, Dr Amer Azaznn, Dr Shaji Sebastiannn, Dr Richard Shendereyoo, Dr Lawrence Armstrongpp, Dr Claire Bellpp, Dr Radhakrishnan Harirajqq, Dr Helen Matthewsrr, Dr Hasnain Jafferbhoyss, Dr Christian P Selingertt, Dr Veena Zamvartt, Prof John S De Caesteckeruu, Dr Anne Willmottuu, Mr Richard Millervv, Dr Palani Sathish Babuww, Dr Christos Tzivinikosxx, Dr Stuart L Bloomyy, Dr Guy Chung-Fayezz, Prof Nicholas M Croftaaa, Dr John ME Fellbbb, Dr Marcus Harbordccc, Dr Ailsa Hartddd, Dr Ben Hopezz, Dr Peter M Irvingeee, Prof James O Lindsayfff, Dr Joel E Mawdsleyggg, Dr Alistair McNairhhh, Dr Kevin J Monahanccc, Dr Charles D Murrayiii, Prof Timothy Orchardjjj, Dr Thankam Paulkkk, Dr Richard Pollokkkk, Dr Neil Shahlll, Dr Sonia Bouriddd, Dr Matt W Johnsonmmm, Dr Anita Modinnn, Dr Kasamu Dawa Kabiruooo, Dr B K Baburajanppp, Prof Bim Bhadurippp, Dr Andrew Adebayo Fagbemiqqq, Dr Scott Levisonrrr, Dr Jimmy K Limdisss, Dr Gill Wattsttt, Dr Stephen Foleyuuu, Dr Arvind Ramadasvvv, Dr George MacFaulwww, Dr John Mansfieldxxx, Dr Leonie Grellieryyy, Dr Mary-Anne Morriszzz, Dr Mark Tremellingzzz, Prof Chris Hawkeyaaaa, Dr Sian Kirkhamaaaa, Dr Charles PJ Charltonaaaa, Dr Astor Rodriguesbbbb, Prof Alison Simmonscccc, Dr Stephen J Lewisdddd, Dr Jonathon Snookeeee, Dr Mark Tigheeeee, Dr Patrick M Gogginffff, Dr Aminda N De Silvagggg, Prof Simon Lalhhhh, Dr Mark S Smithiiii, Dr Simon Panterjjjj, Dr Fraser Cummingskkkk, Dr Suranga Dharmisarikkkk, Dr Martyn Carterllll, Dr David Wattsmmmm, Dr Zahid Mahmoodnnnn, Dr Bruce McLainoooo, Dr Sandip Senpppp, Dr Anna J Pigottqqqq, Dr David Hobdayrrrr, Dr Emma Wesleyssss, Dr Richard Johnstontttt, Dr Cathryn Edwardstttt, Dr John Becklyuuuu, Dr Deven Vanivvvv, Dr Subramaniam Ramakrishnanwwww, Dr Rakesh Chaudharyxxxx, Dr Nigel J Trudgillyyyy, Dr Rachel Cooneyyyyy, Dr Andy Bellzzzz, Dr Neeraj Prasadaaaaa, Dr John N Gordonbbbbb, Prof Matthew J Brookesccccc, Dr Andy Liddddd, Dr Stephen Goreeeeee
aTameside Hospital NHS Foundation Trust, Ashton U Lyne
bBasildon and Thurrock University Hospitals NHS Foundation Trust, Basildon
cHampshire Hospitals NHS Foundation Trust, Basingstoke
dRoyal United Hospital, Bath
eUlster Hospital, Belfast
fUniversity Hospital's Birmingham NHS Foundation Trust, Birmingham
gEast Lancashire NHS Teaching Trust, Blackburn
hBlackpool Teaching Hospitals NHS Foundation Trust, Blackpool
iBolton NHS Trust, Bolton
jRoyal Bournemouth Hospital, Bournemouth
kBradford Teaching Hospitals Foundation Trust - (St Lukes Hospital &Bradford Royal Infirmary), Bradford
lBrighton and Sussex University Hospitals NHS Trust, Brighton
mUniversity Hospitals Bristol NHS Foundation Trust, Bristol
nFrimley Park Hospital NHS Foundation Trust, Camberley
oCambridge University Hospitals NHS Foundation Trust, Cambridge
pNorth Cumbria University Hospitals NHS Trust, Carlisle
qAshford & St Peter's Hospitals NHS Foundation Trust, Chertsey
rSt Peter's Hospital, Chertsey
sChesterfield Royal NHS Foundation Trust, Chesterfield
tColchester Hospital University NHS Foundation Trust, Colchester
uUniversity Hospitals Coventry and Warwickshire NHS Trust, Coventry
vCounty Durham and Darlington NHS Foundation Trust, Darlington
wDerby Hospital NHS Foundation NHS Trust, Derby
xDoncaster and Bassetlaw Hospitals NHS Foundation Trust, Doncaster
yDorset County Hospital NHS Foundation Trust, Dorchester
zDorset County Hospitals Foundation Trust, Dorchester
aaDudley Group NHS Foundation Trust, Dudley
bbRussells Hall Hospital, The Dudley Group NHS Foundation Trust, Dudley
ccNinewells Hospital & Medical School, Dundee
ddEast Sussex Healthcare Trust, Eastborne
eeNHS Lothian, Edinburgh
ffRoyal Devon and Exeter NHS Foundation Trust, Exeter
ggGlasgow Royal Infirmary, Glasgow
hhRoyal Hospital for Children, Glasgow
iiGloucestershire Hospitals NHS Trust, Gloucester
jjUnited Lincolnshire Hospitals NHS Trust, Grantham
kkJames Paget University Hospitals NHS Foundation Trust, Great Yarmouth
llCalderdale and Huddersfield NHS Trust, Halifax
mmPrincess Alexandra Hospital NHS Trust, Harlow
nnHull and East Yorkshire NHS Trust, Hull
ooAiredale NHS Foundation Trust, Keighley
ppCrosshouse Hospital, Kilmarnock
qqThe Queen Elizabeth Hospital NHS Foundation Trust, Kings Lynn
rrKingston Hospital NHS Trust, Kingston upon Thames
ssNHS Fife, Kirkcaldy
ttLeeds Teaching Hospitals NHS Trust, Leeds
uuUniversity Hospitals of Leicester NHS Trust, Leicester
vvMid Cheshire Hospitals NHS Foundation Trust, Leighton
wwUnited Lincolnshire Hospitals NHS Trust, Lincoln
xxAlder Hey Childrens Hospital, Liverpool
yyUniversity College London Hospitals NHS Foundation Trust, London
zzKings College Hospital NHS Foundation Trust, London
aaaRoyal London Childrens Hospital, Barts Health NHS Trust, London
bbbChelsea & Westminster Hospital, London
cccChelsea and Westminster Hospital NHS Foundation, London
dddNorth West London Hospitals NHS Trust, London
eeeGuys & St Thomas' NHS Foundation Trust, London
fffBarts and The London NHS Trust, London
gggGuy's and St Thomas' NHS trust, London
hhhLewisham and Greenwich Healthcare NHS Trust, London
iiiRoyal Free London NHS Foundation Trust, London
jjjImperial College Healthcare NHS Trust, London
kkkSt George's Healthcare NHS Trust, London
lllGreat Ormond Street Hospital for Children NHS Foundation Trust, London
mmmThe Luton & Dunstable University Hospital, Luton
nnnLuton and Dunstable Hospital Foundation Trust, Luton
oooThe Luton & Dunstable University Hospital, Luton
pppMaidstone and Tunbridge Wells NHS Trust, Maidstone
qqqManchester University Hospitals NHS Foundation Trust, Manchester
rrrCentral Manchester University Hospitals NHS Foundation Trust, Manchester
sssThe Pennine Acute Hospitals NHS Trust, Manchester
tttManchester University NHS Foundation Trust, Wythenshawe Hospital, Manchester
uuuSherwood Forest Hospitals NHS Foundation Trust, Mansfield
vvvSouth Tees Hospital NHS Foundation Trust, Middlesbrough
wwwMilton Keynes Hospital NHS Foundation Trust, Milton Keynes
xxxNewcastle Upon Tyne Hospital Trust, Newcastle
yyyIsle of Wight NHS Foundation Trust, Newport
zzzNorfolk & Norwich University Hospital NHS Foundation Trust, Norwich
aaaaNottingham University Hospitals NHS Trust, Nottingham
bbbbOxford University Hospitals NHS Foundation Trust, Oxford
ccccOxford University Hospitals NHS Trust, Oxford
ddddPlymouth Hospitals NHS Trust, Plymouth
eeeePoole Hospital NHS Foundation Trust, Poole
ffffPortsmouth Hospitals NHS Trust, Portsmouth
ggggRoyal Berkshire NHS Foundation Trust, Reading
hhhhSalford Royal NHS Foundation Trust, Salford
iiiiShrewsbury and Telford Hospital NHS Trust, Shrewsbury
jjjjSouth Tyneside NHS Foundation Trust, South Shields
kkkkkSouthampton University Hospitals NHS Trust, Southampton
llllEast and North Herts NHS Trust, Stevenage
mmmmNHS Forth Valley, Stirling
nnnnStockport NHS foundation Trust, Stockport
ooooNorth Tees and Hartlepool NHS Foundation Trust, Stockton
ppppUniversity Hospitals of North Staffordshire, Stoke-on Trent
qqqqUniversity Hospitals of North Midlands NHS Trust, Stoke-on Trent
rrrrCity Hospitals Sunderland NHS Foundation Trust, Sunderland
ssssTaunton and Somerset NHS Foundation Trust, Taunton
ttttSouth Devon Healthcare NHS Foundation Trust, Torquay
uuuuRoyal Cornwall Hospitals NHS Trust, Truro
vvvvMid Yorkshire Hospitals NHS Trust, Wakefield
wwwwWarrington& Halton NHS Foundation, Warrington
xxxxWest Hertfordshire Hospitals NHS Trust, Watford
yyyySandwell and West Birmingham Hospitals NHS Trust, West Bromwich
zzzzWeston Area Health NHS Trust, Weston-Super-Mare
aaaaaRoyal Albert Edward Infirmary, Wrightington, Wigan & Leigh NHS Foundation Trust, Wigan
bbbbbHampshire Hospitals NHS Foundation Trust, Winchester
cccccRoyal Wolverhampton Hospitals NHS Trust, Wolverhampton
dddddWestern Sussex Hospitals NHS Trust, Worthing
eeeeeYeovil District Hospital NHS Foundation Trust, Yeovil
Author Contributions
SL, NC, JRG, NAK, TA, and CAA participated in the conception and design of the study. CB was the project manager and coordinated recruitment. BYHB, MR, NS, JFW, VP, HG, SL, NC, CB, JRG, NAK, TA, and CAA were involved in the acquisition, analysis, or interpretation of the data. Data analysis was performed by BYHB, MR, SL, and NAK. Drafting of the manuscript was conducted by BYHB. All authors contributed to the revision of the manuscript for critically important intellectual content. TA obtained the funding for the study. CAA is the guarantor of the manuscript. All authors contributed to the final approval of the manuscript.
Data Availability
Pre-processed gene expression count data and anonymized patient metadata underlying the DGE analyses, as well as other summary-level data associated with the Figures in this article, can be found in the online Supplementary Data files. Individual participant de-identified raw data that underlie the results reported in this article will be available immediately after publication for a period of 5 years. The data will be made available to investigators whose proposed use of the data has been approved by an independent review committee. Analyses will be restricted to the aims in the approved proposal. Proposals should be directed to Tariq Ahmad [tariq.ahmad1@nhs.net]. To gain access data requestors will need to sign a data access agreement.
References
- 1. Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P.. Anti-TNF therapy in Crohn’s disease. Int J Mol Sci 2018;19:2244. doi: 10.3390/ijms19082244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lichtenstein GR. Comprehensive review: antitumor necrosis factor agents in inflammatory bowel disease and factors implicated in treatment response. Therap Adv Gastroenterol 2013;6:269–93. doi: 10.1177/1756283X13479826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol 2014;7:6–19. doi: 10.1038/mi.2013.73. [DOI] [PubMed] [Google Scholar]
- 4. Levin AD, Wildenberg ME, van den Brink GR.. Mechanism of action of anti-TNF therapy in inflammatory bowel disease. J Crohns Colitis 2016;10:989–97. doi: 10.1093/ecco-jcc/jjw053. [DOI] [PubMed] [Google Scholar]
- 5. Roda G, Jharap B, Neeraj N, Colombel J-F.. Loss of response to anti-TNFs: Definition, epidemiology, and management. Clin Transl Gastroenterol 2016;7:e135. doi: 10.1038/ctg.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ben-Horin S, Kopylov U, Chowers Y.. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev 2014;13:24–30. doi: 10.1016/j.autrev.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 7. Flamant M, Roblin X.. Inflammatory bowel disease: towards a personalized medicine. Therap Adv Gastroenterol 2018;11:1756283X1774502. doi: 10.1177/1756283X17745029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kennedy NA, Heap GA, Green HD, et al. ; UK Inflammatory Bowel Disease Pharmacogenetics Study Group. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: a prospective, multicentre, cohort study. Lancet Gastroenterol Hepatol 2019;4:341–53. doi: 10.1016/S2468-1253(19)30012-3. [DOI] [PubMed] [Google Scholar]
- 9. D’Haens GR, Panaccione R, Higgins PDR, et al. The London position statement of the World Congress of Gastroenterology on Biological Therapy for IBD With the European Crohn’s and Colitis Organization: When to start, when to stop, which drug to choose, and how to predict response? Am J Gastroenterol 2011;106:199–212; quiz 213. doi: 10.1038/ajg.2010.392. [DOI] [PubMed] [Google Scholar]
- 10. Ding NS, Hart A, De Cruz P.. Systematic review: predicting and optimising response to anti-TNF therapy in Crohn’s disease - algorithm for practical management. Aliment Pharmacol Ther 2016;43:30–51. doi: 10.1111/apt.13445. [DOI] [PubMed] [Google Scholar]
- 11. Kopylov U, Seidman E.. Predicting durable response or resistance to antitumor necrosis factor therapy in inflammatory bowel disease. Therap Adv Gastroenterol 2016;9:513–26. doi: 10.1177/1756283X16638833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Digby-Bell JL, Atreya R, Monteleone G, Powell N.. Interrogating host immunity to predict treatment response in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol 2019;17:9–20. doi: 10.1038/s41575-019-0228-5. [DOI] [PubMed] [Google Scholar]
- 13. Noor NM, Verstockt B, Parkes M, Lee JC.. Personalised medicine in Crohn’s disease. Lancet Gastroenterol Hepatol 2020;5:80–92. doi: 10.1016/S2468-1253(19)30340-1. [DOI] [PubMed] [Google Scholar]
- 14. Arijs I, Li K, Toedter G, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009;58:1612–9. doi: 10.1136/gut.2009.178665. [DOI] [PubMed] [Google Scholar]
- 15. Arijs I, Quintens R, Van Lommel L, et al. Predictive value of epithelial gene expression profiles for response to infliximab in Crohn’s disease. Inflamm Bowel Dis 2010;16:2090–8. doi: 10.1002/ibd.21301. [DOI] [PubMed] [Google Scholar]
- 16. Gaujoux R, Starosvetsky E, Maimon N, et al. ; Israeli IBD research Network (IIRN). Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 2019;68:604–14. doi: 10.1136/gutjnl-2017-315494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin JC, Chang C, Boschetti G, et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 2019;178:1493–508.e20. doi: 10.1016/j.cell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verstockt B, Verstockt S, Dehairs J, et al. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019;40:733–42. doi: 10.1016/j.ebiom.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. West NR, Hegazy AN, Owens BMJ, et al. ; Oxford IBD Cohort Investigators. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med 2017;23:579–89. doi: 10.1038/nm.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mavragani CP, Nezos A, Dovrolis N, et al. Type I and II interferon signatures can predict the response to anti-TNF agents in inflammatory bowel disease patients: Involvement of the microbiota. Inflamm Bowel Dis 2020;26:1543–53. doi: 10.1093/ibd/izaa216. [DOI] [PubMed] [Google Scholar]
- 21. Verstockt B, Verstockt S, Blevi H, et al. TREM-1, the ideal predictive biomarker for endoscopic healing in anti-TNF-treated Crohn’s disease patients? Gut 2019;68:1531–3. doi: 10.1136/gutjnl-2018-316845. [DOI] [PubMed] [Google Scholar]
- 22. Sazonovs A, Kennedy NA, Moutsianas L, et al. ; PANTS Consortium. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn’s disease. Gastroenterology 2019;158:189–99. doi: 10.1053/j.gastro.2019.09.041. [DOI] [PubMed] [Google Scholar]
- 23. Broad Institute. Picard toolkit. 2019. https://broadinstitute.github.io/picard/.
- 24. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith T, Heger A, Sudbery I.. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 2017;27:491–9. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao Y, Smyth GK, Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxford, England) 2014;30:923–30. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 27. Piasecka B, Duffy D, Urrutia A, et al. ; Milieu Intérieur Consortium. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc Natl Acad Sci USA 2018;115:E488–97. doi: 10.1073/pnas.1714765115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin S, Hannon E, Reppell M, et al. ; PANTS Consortium. Whole blood DNA methylation changes are associated with anti-TNF drug concentration in patients with Crohn’s disease. MedRxiv 2023. doi: 10.1093/ecco-jcc/jjad133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robinson MD, McCarthy DJ, Smyth GK.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoffman GE, Schadt EE.. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinf 2016;17:17. doi: 10.1186/s12859-016-1323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Law CW, Chen Y, Shi W, Smyth GK.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing, 2019. [Google Scholar]
- 34. Hoffman GE, Roussos P.. dream: Powerful differential expression analysis for repeated measures designs. Bioinformatics 2020;37:192–201. doi:0.1093/bioinformatics/btaa687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tibshirani R, Walther G, Hastie T.. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc Series B: Stat Methodol 2001;63:411–23. doi: 10.1111/1467-9868.00293. [DOI] [Google Scholar]
- 36. Weiner J 3rd, Domaszewska T.. tmod: an R package for general and multivariate enrichment analysis. 2016. doi: 10.7287/peerj.preprints.2420v1. [DOI] [Google Scholar]
- 37. Li S, Rouphael N, Duraisingham S, et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 2013;15:195–204. doi: 10.1038/ni.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chaussabel D, Quinn C, Shen J, et al. A modular analysis framework for blood genomics studies: Application to systemic lupus erythematosus. Immunity 2008;29:150–64. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raudvere U, Kolberg L, Kuzmin I, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019;47:W191–8. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuhn M. caret: Classification and regression training. 2020. https://github.com/topepo/caret/ [Google Scholar]
- 41. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salvador-Martín S, Raposo-Gutiérrez I, Navas-López VM, et al. Gene signatures of early response to anti-TNF drugs in pediatric inflammatory bowel disease. Int J Mol Sci 2020;21:3364. doi: 10.3390/ijms21093364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tretina K, Park E-S, Maminska A, MacMicking JD.. Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J Exp Med 2019;216:482–500. doi: 10.1084/jem.20182031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lügering A, Schmidt M, Lügering N, Pauels H-G, Domschke W, Kucharzik T.. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology 2001;121:1145–57. doi: 10.1053/gast.2001.28702. [DOI] [PubMed] [Google Scholar]
- 45. Liu S-Y, Sanchez DJ, Aliyari R, Lu S, Cheng G.. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA 2012;109:4239–44. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schneider WM, Chevillotte MD, Rice CM.. Interferon-stimulated genes: A complex web of host defenses. Annu Rev Immunol 2014;32:513–45. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsang JS, Schwartzberg PL, Kotliarov Y, et al. ; Baylor HIPC Center. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell 2014;157:499–513. doi: 10.1016/j.cell.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Verstockt B, Pivorunas V, Al Mahi N, et al. Baseline whole blood gene expression of TREM-1 does not predict response to adalimumab treatment in patients with ulcerative colitis or Crohn’s disease in the SERENE studies. 2022. doi: 10.1093/ecco-jcc/jjad170. [DOI] [PMC free article] [PubMed]
- 49. Prame Kumar K, Nicholls AJ, Wong CHY.. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res 2018;371:551–65. doi: 10.1007/s00441-017-2753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pararasa C, Zhang N, Tull TJ, et al. Reduced CD27-IgD- B cells in blood and raised CD27-IgD- B cells in gut-associated lymphoid tissue in inflammatory bowel disease. Front Immunol 2019;10:10. doi: 10.3389/fimmu.2019.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Samie M, Lim J, Verschueren E, et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol 2018;19:246–54. doi: 10.1038/s41590-017-0042-6. [DOI] [PubMed] [Google Scholar]
- 52. van Baarsen LG, Wijbrandts CA, Rustenburg F, et al. Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res Ther 2010;12:R11. doi: 10.1186/ar2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mesko B, Poliska S, Váncsa A, et al. Peripheral blood derived gene panels predict response to infliximab in rheumatoid arthritis and Crohn’s disease. Genome Med 2013;5:59. doi: 10.1186/gm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mishra N, Aden K, Blase JI, et al. ; SYSCID Consortium. Longitudinal multi-omics analysis identifies early blood-based predictors of anti-TNF therapy response in inflammatory bowel disease. Genome Med 2022;14:110. doi: 10.1186/s13073-022-01112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Imhann F, Van der Velde KJ, Barbieri R, et al. The 1000IBD project: multi-omics data of 1000 inflammatory bowel disease patients; data release 1. BMC Gastroenterol 2019;19:5. doi: 10.1186/s12876-018-0917-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Pre-processed gene expression count data and anonymized patient metadata underlying the DGE analyses, as well as other summary-level data associated with the Figures in this article, can be found in the online Supplementary Data files. Individual participant de-identified raw data that underlie the results reported in this article will be available immediately after publication for a period of 5 years. The data will be made available to investigators whose proposed use of the data has been approved by an independent review committee. Analyses will be restricted to the aims in the approved proposal. Proposals should be directed to Tariq Ahmad [tariq.ahmad1@nhs.net]. To gain access data requestors will need to sign a data access agreement.