

Abstract
Brain region-specific degeneration and somatic expansions of the mutant Huntingtin (mHTT) CAG tract are key features of Huntington’s disease (HD). However, the relationships among CAG expansions, death of specific cell types and molecular events associated with these processes are not established. Here, we used fluorescence-activated nuclear sorting (FANS) and deep molecular profiling to gain insight into the properties of cell types of the human striatum and cerebellum in HD and control donors. CAG expansions arise at mHTT in striatal medium spiny neurons (MSNs), cholinergic interneurons and cerebellar Purkinje neurons, and at mutant ATXN3 in MSNs from SCA3 donors. CAG expansions in MSNs are associated with higher levels of MSH2 and MSH3 (forming MutSβ), which can inhibit nucleolytic excision of CAG slip-outs by FAN1. Our data support a model in which CAG expansions are necessary but may not be sufficient for cell death and identify transcriptional changes associated with somatic CAG expansions and striatal toxicity.
Subject terms: Movement disorders, Neurodegenerative diseases
Fluorescence-activated nuclear sorting combined with deep profiling shows that Huntington’s disease repeat expansions arise in specific cell types and are associated with elevated MSH2 and MSH3, which promote expansions in vitro by inhibiting excision of CAG slip-outs by FAN1.
Main
Huntington’s disease (HD) is a fatal late-onset neurodegenerative disease caused by an abnormally long CAG tract in exon 1 of the Huntingtin gene (HTT)1. HD age at onset is most often defined as the onset of motor symptoms, which are thought to arise as a consequence of early degeneration of the caudate nucleus and putamen, primarily due to loss of projection neurons of these structures, known as medium-sized spiny neurons of the direct and indirect pathways (dMSNs and iMSNs)2. Remarkably, other neuron types within these same structures are largely spared from cell death3–5. However, HTT is ubiquitously expressed, and the reason for selective vulnerability of specific cell types in HD is largely unknown6.
Tissue-specific ongoing CAG repeat expansions of the mutant allele are a central feature of HD and other repeat expansion disorders7–11. Expansion of the inherited mHTT allele to very long CAG tracts has been observed sporadically in various brain structures, including the caudate nucleus and putamen, but not in the cerebellum12. A causal role for somatic expansions of the CAG repeat in HD pathogenesis is supported by findings from a genome-wide association study looking for genetic modifiers of HD motor symptom onset other than CAG tract length itself13,14. Although analysis of individual cells captured from HD striatum and cortex by laser-microdissection capture has indicated that somatic expansion occurs more frequently in neurons15, it is not known whether CAG expansions occur in specific types of neuronal and glial cells in these regions. Therefore, it is unclear if CAG expansions are sufficient to explain selective cellular vulnerability in HD and what cell-specific factors in addition to somatically expanded mHTT CAG tract are required for toxicity.
To gain further insight into somatic CAG expansion and toxicity in HD, we developed fluorescence-activated nuclear sorting (FANS) methods for isolation of large numbers of nuclei from human striatal cell types, and we examined the relationships among selective cellular vulnerability, somatic CAG expansion and transcriptional responses in HD. We find that extensive somatic expansion of the mHTT CAG tract occurs in both medium spiny neuron (MSN) populations that are selectively vulnerable in HD, as well as in cholinergic interneurons that are not lost in the HD striatum, although we cannot rule out cell-type-specific somatic CAG expansions beyond the length limit of our assay. CAG expansion is observed also at the mutant ATXN3 locus in MSN nuclei isolated from the post-mortem brains of spinocerebellar ataxia 3 (SCA3) donors, indicating that MSNs are intrinsically prone to somatic expansion of CAG tracts. We demonstrate that the levels of DNA mismatch repair (MMR) proteins MSH2 and MSH3 are elevated in MSN nuclei, suggesting that these proteins may contribute to preferential somatic CAG expansions in MSNs. We offer mechanistic insight into how MutSβ could be promoting somatic CAG expansions by showing that increased concentrations of MutSβ inhibit excision rates of excess slipped-CAG repeats, putative intermediates of expansion mutations, by FAN1 nuclease. Our findings support models in which somatic expansion of the mHTT CAG tract is a critical first step in HD pathogenesis16, and they identify specific genes whose altered expression may modulate toxicity in HD.
Results
FANS-seq profiling of human striatal cell types
To characterize the expression profile, somatic mutations and chromatin accessibility of human striatal cell types, we further developed the FANS method17 (Fig. 1a). Nuclei were purified from samples dissected from human post-mortem caudate nucleus and putamen, stained with either antibody or RNA-binding probes specific for the nuclei of cell types of interest and resolved by passage through a fluorescence-activated cell sorter (Fig. 1a,b and Supplementary Table 1). The specificity of labeling probes and purity of isolated populations of nuclei were verified by generating RNA-sequencing (RNA-seq) libraries from their nuclear transcriptomes (FANS-seq) and comparing their gene expression profile to well-known cell-type markers and previously published data18,19 (Figs. 1c, d and 2, Extended Data Fig. 1 and Supplementary Table 2). This approach was used to generate comprehensive high-quality FANS-seq and ATAC-seq datasets for each neural cell type present in the human caudate nucleus and putamen (Extended Data Fig. 2 and Supplementary Note 1).
Fig. 1. FANS-based isolation of nuclei of striatal cell types from human post-mortem caudate nucleus and putamen.

a, Schematic representation of the procedure used to extract cell-type-specific genomic DNA and nuclear RNA from cell nuclei labeled with cell-type-specific probes. Created with BioRender.com. b, Representative FANS plots showing the labeling of striatal cell nuclei with PrimeFlow probes specific for dopamine receptor D1-expressing (DRD1+) medium spiny projection neurons of the direct pathway (dMSNs), dopamine receptor D2-expressing (DRD2+) medium spiny projection neurons of the indirect pathway (iMSNs), somatostatin-expressing interneurons (SST+ INs, SST+ nuclei), fast-spiking interneurons expressing parvalbumin (PVALB+ INs, ETV1+ TAC3− nuclei), primate-specific tachykinin precursor 3-expressing interneurons (TAC3+ INs, ETV1+ TAC3+ nuclei) and cholinergic interneurons expressing choline acetyltransferase (CHAT+ INs, TRPC3+ COL6A6+ nuclei). The probe against PPP1R1B labels all MSN nuclei. The detailed strategy used for sorting is described in Methods. c,d, Representative distribution of human FANS-seq (c) and ATAC-seq (d) reads mapped to genes expressed specifically in each of the striatal cell types studied. In panel d, arrows mark the position of annotated transcriptional start sites. The data are from a 41-year-old male control donor.
Fig. 2. Purity and reproducibility of the isolation of striatal cell-type nuclei across the two striatal brain regions studied.

a, Heatmaps depict log2-transformed relative expression level of cell-type-specific marker genes in each cell type, calculated based on the mean of DESeq2-normalized counts from six to eight control donors. b–e, Principal-component analysis (PCA) of control donor (n = 6–8 individuals) FANS-seq datasets from all putamen cell types (b,c) indicated that the first three principal components (P.C.) separated neuronal datasets from glial ones (P.C. 1), MSN datasets from those of interneurons (P.C. 2) and datasets of different glial cell types from each other (P.C. 3). For FANS-seq datasets from putamen interneurons (d,e), the major principal components separated the datasets according to interneuron subtype.
Extended Data Fig. 1. Comparison of interneuron populations collected using FANS to cell types defined from single-nucleus RNA sequencing of human striatum.

Relative expression in each cell type was calculated based on DESeq2-normalized counts from 6-8 control donors, and was log2-transformed for visualization (see Methods). The marker genes specific for each interneuron subtype were selected based on single-nucleus RNA sequencing (snRNA-seq) data19. The FANS-isolated ETV1 + TAC3- population of Parvalbumin-expressing interneuron nuclei most likely captured both the major PVALB+ interneuron population and the related smaller PVALB + TH+ interneuron population defined as a separate subtype in snRNA-seq analysis19.
Extended Data Fig. 2. Characterization of FANS-seq datasets from control donors.

a, Principal-component analysis of FANS-seq data from MSNs of caudate nucleus and putamen from 8 control donors. The main principal components separated datasets according to MSN subtype (P.C.1) and donor sex (P.C.2). Notably, none of the top principal components related to the brain region of origin (that is caudate nucleus or putamen). b, Number of genes with expression level above an arbitrary cutoff value of 5 transcripts per million (TPM) in individual FANS-seq datasets from control donors (mean is plotted). c, The number of genes with accessible transcriptional start sites (acc.), defined as having a consensus ATAC-seq peak at the transcriptional start sites (TSS), in striatal cell types from control donors. ATAC-seq datasets could not be produced from CHAT+ INs due to their low abundance in striatal tissue. Genes have been grouped according to average expression level in FANS-seq datasets from the specified cell type in control donors (n = 6-8 individuals). Excluding genes with inaccessible promoters removed a large proportion of genes with very low expression levels (<1 TPM) while affecting fewer genes with moderate to high expression (>10 TPM). See also Supplementary Note 1.
mHTT CAG expands in vulnerable MSNs and resilient CHAT+ INs
Large somatic expansions of the mHTT exon 1 CAG tract in the striatum and cerebral cortex have been demonstrated by small-pool PCR12, and laser capture studies have suggested that somatic expansion can occur in both neurons and glia, albeit more frequently in neurons15. To address the specificity of somatic expansion of mHTT CAG tract quantitatively, and to understand whether it is correlated with cell loss in HD, we isolated nuclei of each striatal cell type by FANS (Supplementary Fig. 1), verified the purity of isolated populations of nuclei by analysis of marker gene expression in the nuclear transcriptomes (Extended Data Fig. 3a) and measured the length of HTT exon 1 CAG tract in genomic DNA isolated from these populations by Illumina-sequencing of amplicons derived from HTT exon 120. Although the limited length of sequencing reads prevented the detection of CAG repeat lengths larger than 113 repeat units, reported to be present in a small minority of striatal cells12, the deep sequencing coverage over HTT CAG tract this method allows is expected to capture highly quantitative information from the vast majority of cells.
Extended Data Fig. 3. Further characterization of samples used for mHTT CAG repeat tract stability analysis.

a, Relative expression level of marker genes of striatal cell types in the nuclear transcriptome of nuclei collected for mHTT CAG repeat tract analysis. Heatmap depicts log2-transformed relative expression in each sample (calculated based on DESeq2-normalized counts). The striatal region of origin is indicated by colored letters (blue P, putamen; red C, caudate nucleus). b, Comparison of the calculated ratio of somatic expansions of mHTT CAG tract in striatal cell types other than MSNs (n = 5 individual donors). The table presents adjusted P-values as calculated by Holm–Sidak’s multiple comparisons test post one-way ANOVA (P < 0.0001). See also Supplementary Note 2. c, Ratio of somatic expansions of normal HTT allele CAG tract in striatal cell types isolated from n = 3 HD donors with 21-25 uninterrupted repeats in normal HTT allele. P = 0.599 according to one-way ANOVA. A different symbol is used for each donor. d, (Left) Relative expression level of MSN and CHAT + IN marker genes in the nuclear transcriptome of nuclei collected for mHTT CAG repeat tract analysis (for description see a). (Right) Length distribution of mHTT CAG tract in CHAT + IN samples. Blue bar marks sequencing reads derived from the initial unexpanded CAG tract. y axes denote normalized number of sequencing reads mapped to reference sequences with different CAG tract lengths (normalized by scaling to 1,000 reads). Reads derived from the normal HTT allele are not shown.
Analysis of genomic DNA of different cell types from five HD donors carrying most prevalent disease-causing CAG tract lengths (from 42 to 45 uninterrupted CAGs; see Extended Data Fig. 3a) revealed that mHTT CAG tract was relatively stable in glial cell types and SST+, TAC3+ and PVALB+ INs, having expanded by less than 5 repeat units in great majority of these cells (Fig. 3a–c, Supplementary Table 4, Supplementary Note 2 and Extended Data Fig. 3b). In contrast, only a small fraction of dMSNs and iMSNs had mHTT copies with the original inherited CAG tract length, and approximately half of these neurons had CAG tracts that were expanded by more than 20 repeat units (mean somatic length gain (MSLG) approximately 22 repeat units, Fig. 3a–c; Methods, ‘HTT and ATXN3 CAG tract sizing’).
Fig. 3. mHTT CAG tract undergoes somatic expansion in selected striatal neuron types.

a, Length distribution of mHTT CAG tract in studied cell types of caudate nucleus and putamen of a 54-year-old female donor that carried a tract of 44 uninterrupted CAG units. Blue bar marks sequencing reads derived from the initial unexpanded CAG tract. y axes denote normalized number of sequencing reads mapped to reference sequences with different CAG tract lengths (normalized by scaling to 1,000 reads). Reads derived from the normal HTT allele are not shown. b, Frequency distribution of mHTT copies with different CAG tract length increases. Data are shown for striatal cell types of two donors that carried the most common HD-causing CAG tract lengths. c,d, Comparison of mean somatic length gain (measured in repeat units (RUs)). c, Although the mean somatic length gain of mHTT CAG tract was not different between dMSN and iMSN, comparison of each of these to any other striatal cell type showed a statistically significant difference (n = 5 individuals, P < 0.0001 by one-way analysis of variance (ANOVA), adjusted P < 0.0001 in Holm–Sidak’s multiple comparisons test). d, The mean somatic length gain of mHTT CAG tract was not different between dMSNs and CHAT+ interneurons, but comparison of each of these to unsorted nuclei showed a statistically significant difference (n = 4 individuals, P = 0.0004 by one-way ANOVA, adjusted P < 0.001 in Holm–Sidak’s multiple comparisons test). Different symbols are used for each of the four donors. e, Length distribution of mHTT CAG tract in MSNs of caudate nucleus and putamen in donors carrying mHTT alleles of reduced and full penetrance. Arrowhead indicates the initial unexpanded size of the CAG tract. f, Comparison of the mean somatic length gain of mHTT CAG tract. n = 13 individuals, P = 0.3422 between cell types, in ratio paired t-test (two sided). caud., caudate nucleus; put., putamen; y., year-old.
Because the scarcity of tissue available prevented us from isolating striatal CHAT+ IN nuclei from all but one of these five initially characterized HD donors, we isolated nuclei of this very rare cell type from three additional HD donors. Analysis of these samples revealed that mHTT CAG tract undergoes large expansions also in CHAT+ INs (MSLG approximately 18 repeat units; Fig. 3a,d, Extended Data Fig. 3d and Supplementary Table 4).
Pairwise comparisons of the two MSN types from donors carrying a wider range of initial repeat lengths revealed that the extent of repeat expansion was dependent on initial repeat length. Although the comparison is confounded by differences in age at death, more modest expansion was observed for mHTT alleles with reduced penetrance (CAG tract lengths <40 repeats) relative to longer, fully penetrant alleles (Fig. 3e,f). Interestingly, although iMSNs have been reported to be more vulnerable of the two MSN subtypes21, there was no significant difference in MSLG between dMSNs and iMSNs (Fig. 3f; P = 0.3422 by ratio paired t-test).
Taken together, these data support the hypothesis that extensive somatic expansion of the mHTT CAG tract is required for the vulnerability of MSNs in HD. However, given previous studies demonstrating that CHAT+ INs in the striatum are not lost in HD3,4, our results suggest that expansion of the mHTT CAG tract is not sufficient to cause neuronal loss in HD.
Instability of the mHTT CAG tract in the HD cerebellum
The loss of cerebellar Purkinje cells (PCs) in several spinocerebellar ataxias (SCA1, SCA2, SCA6 and SCA7) where the causal elongated CAG tracts undergo germline expansion has suggested that, in these disorders, somatic CAG expansion may occur in PCs, but this has not been documented in the cerebellum of HD donors22. Although the viability of PCs in HD has been a matter of debate, ataxia is not an uncommon symptom in HD patients23, and recent stereological studies have demonstrated that PC loss occurs in the cerebellum in HD cases with predominant motor symptoms24. Given these data, we measured mHTT CAG instability in cerebellar cell types in several HD donors (Supplementary Table 1). Although the degree of mHTT CAG expansion in PCs (MSLG approximately 5 repeat units) was relatively modest compared to MSNs, the tract had expanded more in PCs than in other cerebellar cell types except oligodendrocytes (Fig. 4a–c and Supplementary Table 4; P < 0.0001 by one-way ANOVA, adjusted P < 0.05 in Holm–Šidak’s multiple comparisons test in comparisons of PCs to other cell types except oligodendrocytes [P = 0.0617]). The instability of mHTT CAG repeat in PCs relative to cerebellar granule cells is similar to that seen for the mutant ATN1 CAG repeat causing dentatorubral pallidoluysian atrophy25,26. Our data indicate that both in striatum and cerebellum the mHTT CAG tract is somatically unstable in selected neuron types and much more stable in other neuron types and glial cells.
Fig. 4. Expansion of mHTT CAG tract in cerebellar PCs.

a,b, Length distribution of mHTT CAG tract (a) and cell-type marker gene expression (b) in cerebellar (Cb) cell nuclei isolated from a 54-year-old female donor that carried a tract of 44 uninterrupted CAG units. Blue bar marks sequencing reads derived from the initial, unexpanded CAG tract. Reads derived from the shorter normal HTT allele are not shown. b, Heatmap depicts log2-transformed relative expression in each sample (calculated based on DESeq2-normalized counts). c, Comparison of the mean somatic length gain of mHTT CAG tract in cerebellar cell types from four to seven HD donors (n = 4 individuals for PCs, n = 5 for granule cells, n = 7 for astrocytes, microglia and oligodendrocytes, and n = 5 for oligodendrocyte progenitor cells (OPCs)). The table presents adjusted P values as calculated by Holm–Sidak’s multiple comparisons test following one-way ANOVA (P < 0.0001). The variability in somatic CAG expansion observed for PC samples from different donors can most likely be attributed to the rarity of this cell type (<0.01% of all nuclei), which made it extremely difficult to collect samples entirely free of contamination with nuclei of ‘non-expanding’ cell types. Alternatively, the extent of somatic CAG expansion in PCs could be variable in the donors we analyzed, given the reported variability of PC loss between HD patients24.
Striatal MSNs are prone to somatic CAG expansion
Preferential somatic expansion of the mHTT CAG tract in selected striatal neuron types could be due to cell-type-specific properties of the HTT locus (that is, MSN and CHAT+ IN-specific factors acting in cis), putative cell-type-specific factors acting in trans (for example DNA repair proteins) or a combination of the two. Because the expansion-promoting effect of putative trans-factors would not necessarily be limited to the mHTT locus, we asked whether MSNs have a propensity to expand long and pure CAG or CTG tracts at other genomic loci as well. Because transcription through the repeat seems to be a prerequisite for somatic expansions27, we chose to analyze the CAG repeat in ATXN3 gene because, as is the case for HTT, its transcription is relatively uniform across striatal cell types (Supplementary Fig. 2a).
We isolated glial cell and MSN nuclei from striatal tissue of five donors with spinocerebellar ataxia 3 (SCA3) and striatal interneuron nuclei from two SCA3 donors, all carrying a long CAG repeat in the mutant ATXN3 allele (mATXN3) (Supplementary Table 1). Although there were no clear signs of MSN loss even in the oldest SCA3 donors analyzed, as judged by the abundance of large NeuN+ nuclei in striatal homogenates (Supplementary Fig. 2b), the mATXN3 CAG tract was clearly more unstable in the MSNs relative to glial cells and interneurons (Fig. 5a,b and Supplementary Table 4; MSLG approximately 5 repeat units in MSNs). These data indicate that MSNs have a propensity to expand long CAG tracts at other genomic loci and support the hypothesis that mHTT exon 1 CAG tract instability is modulated by rate-limiting trans-acting factors expressed at different levels in striatal cell types.
Fig. 5. MSNs are prone to somatic expansion of mATXN3 CAG tract and have elevated expression of nuclear MSH2 and MSH3 proteins.

a, Length distribution of mATXN3 CAG tract in cell types of caudate nucleus of an 84-year-old female donor that carried a tract of 64 uninterrupted CAG units. Size of the initial unexpanded CAG tract is marked with a blue bar. Reads derived from the normal ATXN3 allele are not shown. b, Comparison of the mean somatic length gain of mATXN3 CAG tract in striatal cell types from SCA3 donors (n = 5 individuals for MSNs, astrocytes, microglia and oligodendrocytes, n = 2 TAC3+ INs and PVALB+ INs, and n = 1 for SST+ INs). The table presents adjusted P values as calculated by Holm–Sidak’s multiple comparisons test following one-way ANOVA (P < 0.0001). c, Heatmaps depict log2-transformed relative expression of MMR and BER genes in cell types of putamen, calculated based on DESeq2-normalized counts from six to eight control donors. Genes identified as HD age at onset-modifying candidates or known to influence CAG tract instability in HD mouse models are marked with an asterisk or arrowhead, respectively. d, Representative immunoblots and quantification of anti-MSH3/anti-H3 (left) and anti-MSH2/anti-H3 signal ratio (right) in unfixed nuclei isolated from the putamen of control donors. These ratios were higher for MSNs compared to other cell types analyzed (P < 0.0001 by one-way ANOVA, adjusted P ≤ 0.0005 in Tukey’s multiple comparisons test). For anti-MSH3, n = 4 individuals for MSNs, astrocytes, and oligodendrocytes, and n = 2 individuals for microglia. For anti-MSH2, n = 6 individuals for MSNs, astrocytes and oligodendrocytes and n = 3 individuals for microglia. Data are presented as mean ± standard error of the mean (s.e.m.). Full-length blots are provided as Source Data.
To identify these trans-acting factors that may explain preferential CAG expansion in MSNs, we compared the FANS-seq expression profiles of striatal cell types in control donors. We focused on genes coding for DNA MMR and base-excision repair (BER) proteins, as several of these proteins have been shown to affect repeat instability in model systems28, and because several MMR genes are represented among candidate genes identified as age of motor symptom onset modifiers in HD mutation carriers14,29. We found that transcript levels of MSH2 and MSH3, encoding MMR proteins that form the MutSβ complex, were more than twofold higher in both dMSNs and iMSNs compared to other striatal neurons, including CHAT+ INs, and this difference was consistent across neuron types in both putamen and caudate nucleus (Fig. 5c, Extended Data Fig. 4a–c and Supplementary Note 3).
Extended Data Fig. 4. Relative expression level of MMR and BER genes in cell types of caudate nucleus.

a, Heatmaps depict log2-transformed relative expression in each cell type, calculated based on the mean of DESeq2-normalized counts from 7-8 control donors. Genes identified as HD age at onset-modifying candidates are marked with an asterisk. Genes known to influence CAG tract instability in HD mouse models are marked with an arrowhead. b-d, Relative levels of MSH2 (b), MSH3 (c) and FAN1 (d) transcripts in MSNs compared to interneurons, calculated with DESeq2 using cell type-specific FANS-seq data from control donors. The largest P adj. value of all MSN vs. interneuron comparisons (by DESeq2, P adjusted for multiple comparisons) is indicated for each gene. For caudate nucleus samples: n = 8 individuals for dMSNs, iMSNs, TAC3 + IN, PVALB + IN and SST + IN. For putamen samples: n = 8 individuals for dMSNs, iMSNs, TAC3 + IN, n = 7 individuals for PVALB + IN and n = 6 individuals for SST + IN.
To determine whether the FANS-seq data accurately reflected nuclear protein levels for these factors in abundant striatal cell types, we measured MSH2 and MSH3 levels by western blotting of nuclear lysates from MSNs, microglia, astrocytes and oligodendrocytes (Fig. 5d). The ratio of both MSH2 and MSH3 to chromatin, assessed by anti-H3 signal, was significantly higher in MSN nuclei compared to glial cells (Fig. 5d; P < 0.0001 by one-way ANOVA, adjusted P ≤ 0.0005 in Tukey’s multiple comparisons test in comparisons involving MSNs). It is well established that the level of Msh2 and Msh3 modulates the extent of somatic CAG expansions seen in the striatum of HD mouse models carrying expanded CAG tracts30–34. Therefore, our observations suggest that elevated levels of the two components of MutSβ may explain the enhanced CAG expansions in human MSNs.
MutSβ suppresses FAN1’s excision of excess slipped-CAG DNA
FAN1 was identified as a modifier of HD disease13, and the nuclease activity of FAN1 has been shown to suppress CAG expansions in the central nervous system of HD mice and in cells derived from HD patients35–40. Unlike the levels of MSH2 and MSH3 transcripts, FAN1 transcript levels are not higher in MSNs compared to other striatal neuron types (Extended Data Fig. 4d). We asked whether a higher MutSβ to FAN1 ratio, as predicted based on elevated MSH2 and MSH3 expression in MSNs, might affect the excision rates of putative expansion intermediate DNA structure by the FAN1 nuclease. To answer this, we used purified recombinant human proteins (Extended Data Fig. 5a) and slipped-(CAG)20 DNA substrates, previously demonstrated to be cleaved by both endo- and 5′→3′ exonucleolytic activities of FAN1 (detailed in Extended Data Fig. 5b)40. Addition of increasing concentrations of MutSβ lead to progressive and substantial inhibition of endo-nucleolytic excision by FAN1 (Fig. 6a, compare lane 2 with lanes 6–8). In contrast, addition of increasing concentrations of MutSα, a dimer of MSH2 and MSH6, did not inhibit cleavage significantly (Fig. 6a, compare lane 2 with lanes 3–5). Slipped-(CTG)20 DNA substrate could also be excised by FAN1, and this was inhibited significantly by MutSβ, but not MutSα (Extended Data Fig. 5c panel i). Next, we tested the effect of the MutS complexes on FAN1s exo-nucleolytic digestion of slip-out DNA substrates, where ‘nibbling-like’ cleavage occurred throughout the repeat tract (Fig. 6b and Extended Data Fig. 5c panel ii, compare lane 1 with lane 2). Exo-nucleolytic cleavage of both slipped-CAG(20) and slipped-(CTG)20 was inhibited by MutSβ, but not MutSα (Fig. 6b and Extended Data Fig. 5c panel ii, compare lane 2 with lanes 6–8 and lanes 3–5). Thus, unlike MutSα, MutSβ inhibits FAN1’s exo- and endonucleolytic excision of excess CAG and CTG slip-outs. The inhibition of FAN1 excision by MutSβ was also significant for slip-outs in the context of anchored duplex flanks, and this effect was evident for varying slip-out sizes, with 2, 4, 8 and 14 excess repeats (Supplementary Fig. 4).
Extended Data Fig. 5. MSH2, MSH3, MSH6, and FAN1 proteins, nuclease assay, (CTG)20 digestion.

a, A representative Coomassie brilliant blue-stained SDS-PAGE of purified human MutSα (MSH2-MSH6), MutSβ (MSH2-MSH3), FAN1 and nuclease-dead FAN1p.D960A (purified in parallel to ensure an absence of contaminating nuclease activity in preparations), expressed in Sf9 cells using baculoviral overexpression. Both MutSα and MutSβ preparations were free of nuclease contamination (see c panels i and ii, lanes 9 and 10). Purity and activity were assessed for ~10 preparations, with consistent results. b, Schematic of FAN1 exo-nucleolytic and endo-nucleolytic cleavage sites on slipped-(CAG)20 DNA substrates labeled at the 3’ or 5’ ends, respectively. FAN1’s exo-nucleolytic ‘nibbling’ of excess repeats parallels the ‘inchworm’ expansions in patient brains, suggesting a role for FAN1 in regulating repeat instability46. Endo- and exo-nucleolytic activities can be distinguished by fluorescein amidite (FAM)-labeling at 5’ or 3’ ends of the (CAG)20 strand, respectively (asterisk); in this manner, only the labeled strand and its digestion products are tracked. c, Slipped-(CTG)20 DNAs are digested by endo- and exo-nucleolytic activities of FAN1, which can be inhibited by MutSβ, but not by MutSα. FAM-labelled CTG slip-out oligonucleotides (schematic, arrowhead indicated center of repeat tract) mimic putative intermediates of expansion mutations. Exo- and endo-nuclease activities were determined by labeling either 3′- or 5′-end of (CTG)20 strand, respectively (asterisk). ‘E’ is elbow at the dsDNA-ssDNA junction. Slip-out DNA substrates (100 nM) were preincubated with increasing amounts of purified human MutSα or MutSβ (0-200 nM), and nuclease digestion was initiated by addition purified human FAN1 (50 nM). Vertical schematic to the right of each gel indicates location of cleavage sites along the FAM-labeled DNA strand. Percentage cleavage (densitometry) for each reaction was then normalized to cleavage levels for FAN1 alone, and these levels were graphed (n = 3 experiments, mean ± s.d. plotted). Results of two-sided unpaired t-test are indicated (vs. ‘FAN1 alone’ in panels i and ii). d, Excess FAN1 overcomes MutSβ-mediated inhibition. (i) Flowchart outlining the competition experiment. Human MutSβ (200 nM), FAN1 (50 nM) and (ii) 5’- or (iii) 3′-FAM-labeled CAG slip-outs (100 nM), that is conditions established to be completely inhibited by MutSβ, were preincubated in non-catalytic conditions (on ice). Reactions were split at the 10-min time point and challenged with 5× BSA, to control for macromolecular crowding (lanes 2–6) or an excess (5×) of FAN1 (lanes 7–11). Aliquots were taken for analysis at different timepoints (0, 5, 10, 20 and 40 minutes) and cleavage products were quantified (n = 2 experiments).
Fig. 6. MutSβ and FAN1 levels competitively affect FAN1’s nuclease excision of excess slipped-CAG DNAs.
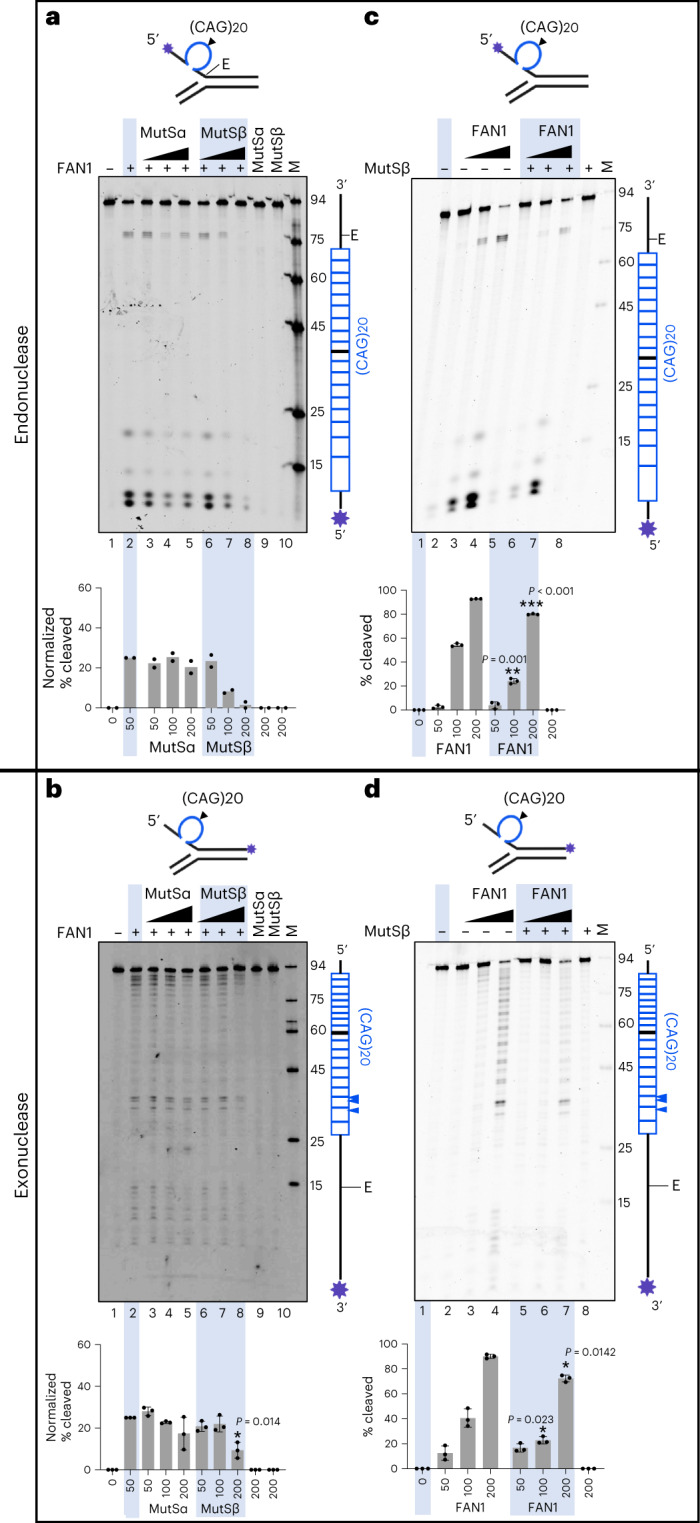
(CAG)20-slip-out DNA substrates (schematics) mimic putative intermediates of expansion mutations40. Endo- and exonucleolytic activities can be distinguished by fluorescein amidite (FAM)-labeling at 5′ or 3′ ends of the (CAG)20 strand, respectively (indicated by an asterisk); in this manner, only the labeled strand and its digestion products are tracked. ‘E’ is elbow at the dsDNA-ssDNA junction. a,b, MutSβ not MutSα inhibits FAN1. Protein-free undigested slip-out DNA substrate (100 nM), lane 1. Slip-outs were preincubated with buffer, lane 2, or increasing concentrations of purified human MutSα (50, 100 or 200 nM), lanes 3–5; or MutSβ (50, 100 or 200 nM), lanes 6–8. Nuclease digestions were initiated by addition purified human FAN1 (50 nM). Lanes 9 and 10 have slip-out DNA and only MutSα (50 nM) or only MutSβ (50 nM), ensuring these purified proteins are nuclease-free. c,d, Increasing FAN1 concentration can overcome MutSβ-mediated inhibition of cleavage. Protein-free undigested slip-out (100 nM), lane 1. Slip-outs were preincubated with buffer, lanes 1–4 or with MutSβ (200 nM), lanes 5–7. Nuclease digestions were initiated by adding increasing amounts of FAN1 (50, 100 or 200 nM), lanes 2–4 and 5–7. Lane 8 has slip-out and only MutSβ (50 nM). For gels in panels a and b, the percentage cleavage for each reaction was normalized to cleavage levels with FAN1 alone (lane 2), and these levels were graphed (GraphPad prism 9.1). For gels in panels c and d, the percentage cleavage for each reaction were graphed. The vertical schematic to the right of each gel indicates the location of cleavage sites along the FAM-labeled DNA strand. ‘E’ is elbow at the dsDNA-ssDNA junction, blue arrowheads represent cleavage hotspots. Results of two-sided unpaired t-test are indicated (versus FAN1 alone in panel b, or versus ‘no FAN1’ in panels c and d). n = 2 experiments for panel a, n = 3 experiments for the other panels and mean ± standard deviation (s.d.) are plotted.
Addition of increasing concentrations of FAN1 led to increased endo- and exonucleolytic digestion of slip-out DNA even in the presence of MutSβ (Fig. 6c,d, compare lane 5 with lanes 6 and 7), and excess FAN1 could also overcome inhibition of slip-out DNA cleavage by pre-bound MutSβ in a competition experiment (Extended Data Fig. 5d, compare lanes 7–11 with lanes 2–6, in both panels ii and iii). As FAN1 does not interact with MutSβ38, our results support a model where CAG and CTG slip-out DNA excision rates are determined by competitive binding to either MutSβ or FAN1, thereby offering an explanation to how differences in the relative level of MutSβ to FAN1 could result in CAG expansion or stabilization in different cell types.
Altered gene expression in HD progression
To gain further insight into the molecular events that may play a role in somatic expansion or contribute to mHTT toxicity, we sequenced the nuclear transcriptomes of striatal MSNs and TAC3+, SST+ and PVALB+ INs from the putamen or caudate nucleus of six or seven HD donors (Supplementary Tables 1 and 2). We limited the comparative analysis of HD and control donors’ (n = 8) FANS-seq data to genes that had accessible promoters in cell type of interest (as indicated by ATAC-seq), reasoning that this would allow us to minimize the number of possible false-positive differences in gene expression that might have resulted from contaminating ambient transcripts, FACS sorting impurities or contamination with genomic DNA (Supplementary Note 1 and Extended Data Fig. 6a). As expected, disease-associated changes (Fig. 7a and Supplementary Tables 5 and 6) were well correlated between dMSNs and iMSNs, but the correlation was poor in comparisons involving other neuron types (Fig. 7b and Extended Data Fig. 6b). This result shows that the majority of disease-associated transcript-level changes are not common to all striatal neuron types.
Extended Data Fig. 6. Further characterization of HD-associated gene expression changes in striatal neurons.

a, The number of genes with accessible transcriptional start sites (acc.), defined based on the overlap of TSS and consensus peaks in ATAC-seq data from HD and control donor MSNs, and control donor interneurons. Genes have been grouped according to their average expression level in FANS-seq datasets from HD donors (TPM, transcripts per million). More than 98% of genes with accessible promoters in HD MSNs had accessible promoters also in MSNs from control donors. Due to the low abundance of striatal interneurons and limited amounts of striatal tissue available from HD donors, ATAC-seq datasets could not be produced from the interneurons of HD donors. Therefore, striatal interneuron FANS-seq datasets were filtered to include only those genes that had accessible promoters in control donors. b, Principal-component analysis of FANS-seq data from putamen and caudate nucleus MSNs from HD and control donors, performed after the exclusion of genes with inaccessible TSSs. The calculated CAP100 scores are shown for each HD donor. Note that disease status (P.C.1) and MSN subtype (P.C.2) were the main sources of variance in these datasets. c, Selected nonredundant Gene Ontology Cellular Component terms from enrichment analysis of genes with disease-associated expression changes (Padj < 0.05 by DESeq2 after adjusting for multiple comparisons) in each striatal neuron type HD (n = 7 individuals for HD dMSNs and HD iMSNs, n = 6 individuals for HD interneurons, n = 8 individuals for all cell types from control donors). Significance threshold for enrichment analysis: q value < 0.05, Padj < 0.05, adjusted for multiple comparisons after hypergeometric test with clusterProfiler. d, Genes essential for MSN viability in the zQ175 and R6/2 mouse models of HD43 that are also downregulated in HD iMSNs or dMSNs by more than a third (log2FC < −0.6, Padj < 0.01 by DESeq2, adjusted for multiple comparisons). See also Supplementary Note 5.
Fig. 7. Disease-associated gene expression changes in striatal neuron types.

a, Number of differentially expressed (DE) genes (Padj < 0.05 by DESeq2, adjusted for multiple comparisons) in the comparison of HD (n = 7 individuals for dMSNs and iMSNs, n = 6 individuals for all interneuron types) and control donor (n = 8 individuals) FANS-seq datasets from putamen or caudate nucleus. b, Correlation analysis of disease-associated expression changes of genes expressed in all striatal neuronal types studied. c, Selected nonredundant GOCC terms from enrichment analysis of genes with disease-associated expression changes in iMSN or dMSN (Padj < 0.05 by DESeq2), but not in any interneuron type (Padj > 0.05 in all HD versus control interneuron comparisons). d,e, Heatmaps depicting disease-associated changes in transcript levels of selected genes regulating autophagosome formation and transport (d), and transcript levels of MMR and BER genes (e). Statistically significant differences are marked with an asterisk (Padj < 0.05 by DESeq2, adjusted for multiple comparisons). CTRL, control. f, GOCC terms enriched for genes that were identified as essential for MSN viability in wild-type mice43 and are also downregulated in HD dMSNs (440 genes) or iMSNs (365 genes). GOCC terms enriched with less than 10 downregulated genes are omitted from the plot. For panels c and f, the significance threshold for enrichment analysis was: q value < 0.05, Padj < 0.05, adjusted for multiple comparisons after hypergeometric test with clusterProfiler.
Gene ontology analysis
To identify cellular processes that are affected by disease-associated gene expression changes that take place only in MSNs, thereby correlating with the presence of more toxic mHTT species in these neurons, we analyzed which gene ontology cellular component (GOCC) terms were enriched for genes that were up- or downregulated in MSNs but did not display these changes in expression in any of the interneuron populations studied. The results indicated that many genes downregulated specifically in MSNs are involved in ribosomal biogenesis (GOCC terms ‘cytosolic ribosome’ and ‘nucleolus’), pre-mRNA maturation (GOCC terms ‘nuclear body’ and ‘spliceosomal complex’) and other nuclear functions (Fig. 7c). Although transcripts of mitochondrial oxidative phosphorylation pathway genes have been reported to be downregulated in HD MSNs41, we noticed that this disease-associated change is much more evident in the nuclear transcriptome of PVALB+ INs (Extended Data Fig. 6c and Supplementary Fig. 5a). The full lists of GOCC terms enriched for MSN-specific changes and overall changes in all neuron types can be found in Supplementary Tables 7 and 8, respectively.
The GOCC terms enriched for genes that have increased expression in MSNs in the HD donor data include many terms that indicate alterations in membrane protein function. ‘Neuron projection’, ‘Synapse’ and ‘Lysosome’ were among GOCC terms enriched for genes upregulated specifically in both MSN subtypes (Fig. 7c, Extended Data Fig. 6c and Supplementary Fig. 5b). We observed also that genes central to the regulation of lysosomal biogenesis and autophagy were among the top upregulated genes in MSNs. For example, the transcripts of transcription factor TFEB, which has been shown to be essential for regulation of many genes in these pathways42, are strongly elevated in HD MSNs (Fig. 7d). The observations that TFEB is essential for MSN survival in mice in the presence of mHTT43 and can lower striatal mHTT levels44 point to the relevance of its induction for MSN survival. Several genes encoding proteins essential for autophagy are also induced in HD, including ATG9B, ATG9A, the gene encoding HTT-interacting protein ULK1 involved in autophagosome formation45, MAPK8IP1 and MAPK8IP3, which encode proteins involved in retrograde transport of autophagosomes46, and SQSTM1, the gene encoding HTT-interacting autophagy cargo receptor p62 (ref. 45) (Fig. 7d and Supplementary Note 4).
DNA repair pathways
Given the expansions of mHTT CAG tract in MSNs, we investigated whether there are disease-associated changes in the transcript levels of genes encoding MMR and BER proteins. Notably, we found that, in HD MSNs, MSH2 and MSH3 expression levels are significantly reduced relative to MSNs from control donors, whereas POLD1, coding for the large catalytic subunit of the DNA polymerase delta complex, undergoes a disease-associated upregulation that is not entirely specific to MSNs (Fig. 7e). Notably, POLD1 was recently identified as a candidate modifier of HD47. Although further validation will be required to confirm these changes, our data point to clear distinctions in the regulation of DNA repair pathways in MSNs compared to interneurons.
Genes required for MSN viability and functionality
To predict which cellular functions would be affected negatively by HD-associated transcript-level changes, we identified genes downregulated in HD MSNs that have also been shown to be required for the viability of MSNs in mice43 (Supplementary Table 10). Analysis of GOCC terms enriched for these genes revealed that many of the HD-associated expression changes could be affecting MSN viability through their effect on nuclear functions and RNA metabolism (Fig. 7f and Supplementary Note 5). We also noted that the transcript levels of MSN-enriched ANO3 and PDE10A undergo large disease-associated decreases equivalent in magnitude to complete silencing of these genes in >45% and >60% of the remaining MSNs, respectively (Supplementary Fig. 7, ANO3 transcript log2 fold change −1.35 and −0.86 for dMSNs and iMSNs, PDE10A transcript log2 fold change −1.44 and −1.33 for dMSNs and iMSNs). As missense mutations in ANO3 are known to cause dystonia (https://omim.org/entry/615034) and PDE10A mutations are known to cause childhood-onset hyperkinetic movement disorders (https://omim.org/entry/616921, in some cases with striatal degeneration: https://omim.org/entry/616922), it is likely that these transcript-level changes have a substantial effect on the function of a large fraction of remaining MSNs in HD.
Discussion
Here, we have used FANS17 to isolate thousands of nuclei of each neural cell type of human caudate nucleus and putamen to generate deep, high-resolution, cell-type-specific transcriptional and HTT CAG repeat tract length-measurement data from control and HD donors. Our data reveal that somatic expansion of mHTT CAG tract occurs in select striatal neuron types. Our findings are consistent with models of HD pathogenesis in which somatic CAG expansion is a critical first step in pathogenesis, followed by a second step in which the expanded mHTT allele has a toxic effect that eventually leads to degeneration and death of the cell16. In addition, our data indicate that somatic mHTT CAG expansions alone may not be sufficient to explain cell-type vulnerability and reveal several cell-type-specific molecular features of the disease.
The most vulnerable cell types in the HD striatum are MSNs2. Although both dMSNs and iMSNs are progressively lost during the progression of the disease, iMSNs that express dopamine receptor D2 and enkephalin are most vulnerable in early stages21. Striatal interneurons are relatively spared early in the disease3,5. In particular, although CHAT+ INs are clearly affected, as indicated by reduced CHAT activity in histological sections, the persistent expression of acetylcholinesterase in these cells indicates that they do not die during the disease4,48.
Our data showing somatic mHTT CAG expansion in both MSNs and cerebellar PCs24,49,50 support the hypothesis that CAG expansion is an early step in disease progression that is necessary for the loss of neurons in HD. Accordingly, the stability of the mHTT CAG tract we see in SST+, TAC3+ and PVALB+ INs can explain their relative resilience in HD. However, our data also demonstrate that large expansions of the mHTT CAG tract are not sufficient for loss of CHAT+ INs in HD. Furthermore, data we collected from dMSN and iMSN nuclei isolated from HD donors, especially from carriers of reduced-penetrance mHTT alleles where the loss of MSNs is minor, establish that differences in the rate of mHTT CAG expansion are an unlikely explanation for the reportedly greater vulnerability of iMSNs than dMSNs in this disease21. The conclusion that substantial somatic expansion is required but may not be sufficient for neuronal loss in the HD brain is supported by studies of the human cerebral cortex in HD, demonstrating that extensive expansion of the mHTT CAG tract occurs in many deep layer pyramidal cell types despite selective loss of L5a corticostriatal projection neurons51.
It is important to note that the assay we have employed for determining CAG repeat lengths is limited to a tract length of 113 CAG repeats and thus cannot be used to detect the very long CAG expansions that have been reported to occur in some HD donors’ brains12. It remains possible, therefore, that cell loss from the HD striatum is due to CAG repeats that have undergone extremely large somatic expansion, and that the differential resilience of CHAT+ INs relative to MSNs during HD progression, as well as the resilience of MSNs with somatically expanded mATXN3 repeat (Supplementary Note 6), is explained by a difference in the frequency of very long CAG repeats that are undetectable by our assay. A threshold of 115 CAGs has been postulated for striatal cell loss based on computational models of somatic expansion that include acceleration of expansion as the length of the CAG repeat increases52. Analysis of the genomes of cells that have already died in the HD brain, for example by recovery of MSN DNA from cerebrospinal fluid, would provide data directly addressing the threshold of CAG expansion required for cell death.
The preferential expansion of the mATXN3 CAG tract we detect in MSN nuclei isolated from SCA3 donor samples indicates that these neurons have a general propensity to expand long CAG tracts, perhaps as a consequence of the high level of MutSβ (Supplementary Note 7). We offer mechanistic insight to how elevated MutSβ could be promoting somatic CAG expansions by showing that an excess of MutSβ inhibits FAN1 nucleolytic excision of excess CAG slip-outs, thereby allowing slip-outs to be retained as somatic expansions (Supplementary Note 8).
Our data show that HD-associated gene expression changes in human MSNs are distinct from those of other striatal neurons and have only a partial overlap to gene expression changes documented in published datasets from HD mouse models53,54 (Supplementary Note 9). The large magnitude of HD-associated downregulation seen for many genes (median log2 fold change −0.67 and −0.73 for genes downregulated in dMSNs and iMSN, respectively) indicates that extensive transcriptional disturbances occur in the majority of MSNs prior to their demise. The strong induction of genes involved in autophagic clearance argues that human MSNs mount an important defense against the mHTT misfolding and extranuclear aggregation in HD. Other changes may be more detrimental, including the strong transcriptional downregulation of genes involved in nuclear functions, perhaps as a direct effect of the presence of mHTT in the nucleus. Moreover, there are large expression changes in several genes which have been shown to be required for MSN viability in mouse models of HD43, and in genes with a clearly established link to human MSN function (ANO3) and survival (TAF1 and PDE10A).
The data we have reported here strongly support previous proposals16 that somatic CAG expansion is a necessary first step in the pathophysiological cascade that unfolds in HD. Our data suggest also that striatal MSNs are prone to somatic CAG expansion, perhaps as a consequence of the high level of MutSβ that we have documented in MSN nuclei. It remains possible that differences in the frequency of very long mHTT repeats in vulnerable and more resilient cell types is sufficient to explain their differential death in the HD brain. However, given the number and magnitude of MSN-specific transcriptional responses evident in the human striatum in HD, precedent from mouse models54, and the nature of the specific genes dysregulated, we favor the proposal that some of these gene expression changes are an indicator and likely also a cause of compromised human MSN function well before the eventual loss of these cells in HD. We hope that further analyses of the comprehensive datasets we have provided will stimulate others to interrogate them in the context of detailed mechanistic studies to clarify the degree to which these transcriptional changes perturb the implicated biological processes in human brain, and whether these changes are detrimental or compensatory.
Methods
Human samples
Deidentified tissue samples analyzed in this study were determined to be exempt from Institutional Review Board review according to 45 CFR 46.102 (f). For this work, fresh frozen brain samples were obtained from Miami’s Brain Endowment Bank, University of Washington BioRepository and Integrated Neuropathology Laboratory, Columbia University Alzheimer’s Disease Research Center, The University of Michigan Brain Bank and Netherlands Brain Bank or through the National Institutes of Health (NIH) NeuroBioBank and sourced from either the Harvard Brain Tissue Resource Center or the NIH Brain & Tissue Repository-California, Human Brain & Spinal Fluid Resource Center, VA West LA Medical Center (Los Angeles, CA). Drug addiction and schizophrenia as well as clinical evidence of brain cancers were reasons for sample exclusion, whereas samples from donors with a history of other non-brain cancers and diabetes were accepted. Caudate nucleus, putamen and cerebellar vermis were used for isolation of nuclei. The brain regions used from each donor and their age, race, sex and post-mortem interval are noted in Supplementary Table 1. The table includes information about the Vonsattel grade, calculated CAP100 score55, the number of uninterrupted CAG repeats in their HTT alleles and the sequence of the CAG tract and the adjacent CCG tract, as determined from CAG tract length measurement data.
Isolation, labeling and sorting of glial cell nuclei
Nuclei were isolated as described previously17. For the labeling of glial cell nuclei and cerebellar granule cells, the isolated nuclei were washed once with homogenization buffer (0.25 M sucrose, 150 mM KCl, 5 mM MgCl2, 20 mM Tricine pH 7.8, 0.15 mM spermine, 0.5 mM spermidine, EDTA-free protease inhibitor cocktail, 1 mM DTT, 20 U ml−1 SUPERase-In RNase inhibitor (ThermoFisher, #AM2696), 40 U ml−1 RNasin ribonuclease inhibitor (Promega, #N2515)). Each washing step constituted of resuspension of nuclei pellet followed by centrifugation (1,000 × g, 4 min, 4 °C). Resuspended nuclei were fixed in Homogenization buffer with 1% formaldehyde for 8 min at room temperature followed by quenching with 0.125 M glycine for 5 min. Following centrifugation, the nuclei were washed once with wash buffer (PBS, 0.05% TritonX-100, 0.5% BSA, 20 U ml−1 Superase-In RNase Inhibitor and 40 U ml−1 RNasin ribonuclease inhibitor) and incubated at room temperature on a shaker in wash buffer for permeabilization and blocking of unspecific binding. Nuclei were washed twice in wash buffer without TritonX-100 and resuspended in 100 µl 40% ethanol containing TrueBlack Lipofuscin Autofluorescence Quencher (Biotium, #23007) for 40–50 seconds. Nuclei were washed twice with wash buffer (w/o TritonX-100) and incubated overnight at 4 °C with the following antibodies: Rb x NeuN-Alexa-647 (1:400, Abcam, #ab190565), Rb x NeuN-Alexa594 (1:400, Abcam, #ab207279), Mm x EAAT1 (1:2,000, Santa Cruz Biotechnology, #sc-515839), Mm x IRF8-PE (1:65, ThermoFisher, #12-9852-82) and Goat x Olig2 (1:300, R&D Systems, #AF2418). After three washes with wash buffer (w/o TritonX-100), the nuclei were incubated for 30–45 min at room temperature with Donkey × Mm-Alexa-488 (1:1,000, ThermoFisher, #A-21202) and Donkey x Goat-Alexa-647 (1:300, ThermoFisher, # A-21447). After three washes with wash buffer (w/o TritonX-100), the nuclei were resuspended in Sorting buffer (PBS, 0.2% BSA, 40 U ml−1 RNasin ribonuclease inhibitor, 0.5 µg ml−1 DAPI) and separated with SONY MA900 Cell Sorter (software ver. 3.0.5). Aggregates of nuclei were excluded based on higher DAPI signal and the following gating strategies were used: neuronal nuclei (647+, 594+, 488−, large), oligodendrocyte nuclei (647+, 594−, 488−, small), microglia nuclei (647−, 594+, 488−, small) and astrocyte nuclei (647−, 594−, 488+, small). A separate sorting experiment was performed for collecting cerebellar granule cell nuclei. For this purpose, nuclei were labeled with Rb x NeuN-Alexa594 (1:400, Abcam, #ab207279) and Mm x ITPR1-Alexa-488 (Santa Cruz Biotechnology, #sc-271197 AF488), and granule cell nuclei were collected (488−, 594+).
For labeling neuronal nuclei, PrimeFlow labeling kit (ThermoFisher, #88-18005-210) was used and fixation and permeabilization were carried out according to manufacturer’s instructions but with 200 U ml−1 Superase-In RNase inhibitor and 400 U ml−1 RNasin ribonuclease inhibitor present at every incubation step. For sorting, the nuclei were resuspended in sorting buffer (PBS, 0.2% BSA, 40 U ml−1 RNasin ribonuclease inhibitor, 0.5 µg ml−1 DAPI). Probes specific to DRD1 (Alexa-647, #VA1-3002351-PF), DRD2 (Alexa-488, #VA4-3083767-PF) and PPP1R1B (Alexa-568, #VA10-3266354-PF) were used to label dMSN (647+, 568+, 488−, large) and iMSN nuclei (647−, 568+, 488+, large). In a separate set of experiments, probes specific to TAC3 (Alexa-647, #VA1-16603-PF), ETV1 (Alexa-488, # VA4-3083818-PF), SST (Alexa-568, # VA10-3252595-PF) and PPP1R1B (Alexa-568, # VA10-3266354-PF) were used to label the nuclei of TAC3+ interneurons (647+, 568−, 488+), PVALB+ interneurons (647−, 568−, 488+), SST+ interneurons (647−, 568+++, 488−) and MSNs (647−, 568+, 488−, large). Probes specific to TRPC3 (Alexa-647, # VA1-3004835-PF), COL6A6 (Alexa-647, #VA1-3014134-PF) and PPP1R1B (Alexa-568, # VA10-3266354-PF) were used in another set of experiments to label cholinergic interneuron nuclei (647+, 568−, large) and MSN nuclei (647−, 568+, large). CA8 probe (Alexa-647, #VA1-3001892-PF) was used for sorting Purkinje neuron nuclei (647+, large). Aggregates of nuclei were always excluded based on higher intensity of DAPI staining. All PrimeFlow target probes were used at a dilution of 1:40.
ATAC-seq library preparation
For generating ATAC-seq data, the nuclei were treated with Tagment DNA TDE1 Enzyme (Illumina, #15027865) before fixation and labeling. The exact number of nuclei processed depended on the abundance of the population labeled and collected. Briefly, 800,000 nuclei were pelleted by centrifugation (5 min at 950 × g) and resuspended in 10 mM Tris-HCl pH 7.6, 10 mM NaCl, 3 mM MgCl2, 0.01% NP-40 followed by centrifugation (500 × g for 10 min at 4 °C). The pellet was resuspended in 200 μl Transposition Mix (1× TD buffer containing 20 U ml−1 Superase-In RNase Inhibitor, 40 U ml−1 RNasin ribonuclease inhibitor and 1.25 µl Illumina Tagment DNA TDE1 Enzyme per every 100,000 nuclei) and incubated at 37 °C for 30 min. The reaction was stopped and nuclei fixed by adding 1 ml homogenization buffer with 1 mM EDTA and 1% formaldehyde. After 8 min of incubation on a shaker, the fixative was quenched by adding glycine (0.125 M) for 5 min. After washing the nuclei once in homogenization buffer and once in wash buffer (without Triton X-100), the sample was processed like described above, proceeding with the steps that follow permeabilization of nuclei. After sorting, the collected nuclei were centrifuged at 1,600 × g for 10 min 4 °C and resuspended in 200 μl RC solution (50 mM Tris-HCl pH 7.6, 200 mM NaCl, 1 mM EDTA, 1% SDS and 5 µg ml−1 Proteinase K) and incubated overnight at 55 °C. Genomic DNA was isolated with using MinElute Reaction Cleanup Kit (Qiagen, #28206) and used for PCR amplification (72 °C, 5 min; 98 °C, 30 s; 12-14× (98 °C, 10 s; 63 °C, 30 s; 72 °C, 1 min)) with NEBNext High-Fidelity 2X PCR Master Mix (New England Biolabs, #M0541S) and barcoded Nextera primers (1.25 μM each)56. Following double-sided size selection by bead-purification the libraries were quantified with Qubit dsDNA HS assay kit (ThermoFisher #Q32851) and pooled for sequencing on NovaSeq6000 (SP 2 × 100 bp).
FANS-seq library preparation and sequencing
RNA extraction was carried out with AllPrep DNA/RNA FFPE Kit (Qiagen, #80234) with modifications described previously17. RNA-seq libraries were prepared with Trio RNA-Seq library preparation kit (Tecan, #0506-A01), quantified with Qubit dsDNA HS assay kit (ThermoFisher #Q32851) and pooled for sequencing on NovaSeq6000 (SP 2 x 150 bp).
RNA-seq data processing
Sequence and transcript coordinates for human hg38 UCSC genome and gene models were retrieved from the BSgenome.Hsapiens.UCSC.hg38 Bioconductor package (version 1.4.1) and TxDb.Haspiens.UCSC.hg38.knownGene (version 3.4.0) Bioconductor libraries, respectively. FANS-seq reads were aligned to the genome using Rsubread’s subjunc method (version 1.30.6)57 and exported as bigWigs normalized to reads per million using the rtracklayer package (version 1.40.6). Reads in genes were counted using the featurecounts function within the Rsubread package against the full gene bodies (Genebody.Counts) and gene exons (Gene.Counts).
ATAC-seq data processing
The ATAC-seq reads were aligned with the hg38 genome from the BSgenome.Hsapiens.UCSC.hg38 Bioconductor package (version 1.4.1) with Rsubread’s align method in paired-end mode. Fragments between 1 and 5,000 bp long were considered correctly paired. Normalized, fragment signal bigWigs were created with the rtracklayer package. Peak calls were made with MACS2 software in BAMPE mode58,59. For each striatal interneuron type except cholinergic interneurons, the ATAC-seq consensus peaks were called from four ATAC-seq datasets generated from four different control donors. For MSNs, ATAC-seq consensus peaks were called from 8 dMSNs datasets from 7 different HD donors, from 9 iMSN datasets from 8 different HD donors, and 31 dMSNs datasets and 32 iMSN datasets from 8 control donors (up to four datasets from each donor). High-confidence consensus peaks were derived by creating a nonredundant peak set for each cell type and disease state and then filtering down to peaks that were present in the majority of samples. These were then annotated to TSS based on proximity using the ChIPseeker package (version 1.28.3)60. NCBI Refseq hg38 gene annotation was used (version 109.20211119).
Differential gene expression analysis and principal-component analysis
For comparison of transcript abundance data between different cell types from control donors, the comparisons of data from caudate nucleus and putamen were done independently. For control donors from whom there were data available from both posterior and anterior parts of the same structure, a single table of average raw read counts per gene was generated for each cell type. For comparison of control donor data to HD donor data, up to four separate datasets for a given cell type (anterior putamen, posterior putamen, anterior caudate nucleus and posterior caudate nucleus) were combined by calculating the average raw read counts per gene (rounding up to integer), so that each donor was represented by a single FANS-seq dataset for each cell type. Principal-component analysis plots for 500 most variant genes were generated with pcaExplorer61 using average raw read ‘Genebody.Counts’ tables as input data (one table for each donor per cell type). Average raw read ‘Gene.Counts’ tables (that is, derived from FANS-seq reads mapped to exons), one for each donor per cell type, were converted into normalized counts by DESeq2, thereby accounting for sequencing depth differences, and used for differential gene expression analysis by DESeq2 (refs. 62,63) (version 1.36.0) (Supplementary Table 5; adjusted P < 0.05 is considered as a significant difference). Differential gene expression analysis performed based on ‘Genebody.Counts’ (that is FANS-seq reads mapped to full gene bodies) is also provided (Supplementary Table 6). Differential gene expression analysis results were filtered to exclude genes for which none of their annotated TSS positions in NCBI Refseq hg38 (version 109.20211119) overlapped with ATAC-seq consensus peaks defined separately for the cell types compared. These lists were augmented with a small number of genes (<110) for which visual inspection of mapped FANS-seq and ATAC-seq reads in Integrative Genomics Viewer64 suggested that these genes were in fact expressed (marked as ‘Visual inspection of mapped FANS-seq and ATAC-seq reads‘ in Supplementary Table 3). The subset of genes inspected visually was selected based on whether they were differentially expressed (that is, DEGs) between HD and control donors, the logic being that the exclusion of genes that have accessible TSS but are not DEGs would not have any effect on Gene Ontology analysis results. For the visualization of gene expression differences across cell types, ‘expression in cell type A’ was calculated as the mean of DESeq2-normalized ‘Gene.Counts’ from each donor. ‘Expression in a cell type A’ was then turned into relative expression (‘relative expression in cell type A’ = ‘expression in a cell type A’ divided by ‘mean of expression in all cell types compared’) and the resulting values were log2-transformed for visualization by Pheatmap R package (version 1.0.12). Relative expression was calculated in the same manner when comparing expression across individual samples instead of cell types.
Motif analysis and annotation
High-confidence consensus peaks for MSNs were annotated to genes using the ChIPseeker package60 and filtered to remove ‘Distal Intergenic’ peaks. The remaining peaks were overlapped with the list of genes either up- or downregulated in HD dMSNs and iMSNs (Supplementary Table 5) to generate three peak sets for each cell type: ‘peaks in HD-upregulated genes,’ ‘peaks in HD-downregulated genes,’ and ‘peaks in genes with no expression change’. 200-bp wide DNA sequence at the center of each peak was retrieved using the BioStrings R package (2.66.0). These DNA sequences were then used as input for MEA65 with the following comparisons: ‘HD-Upregulated vs. No Expression Change’, ‘HD-Upregulated vs. Randomized GC-content Matched Background’ and ‘HD-Upregulated vs. HD-Downregulated’ (peaks in HD-Downregulated’ genes were similarly compared to peaks in ‘HD-Upregulated’, ‘No expression change’ and ‘Randomized GC-content Matched Background’ genes). Overrepresentation of motifs in eukaryote in vivo and in vitro databases was then calculated using a Fisher’s exact test and average odds score, where matches must have a log-odds score ≥ 0.25 times the maximum possible log-odds score. Motifs with Evalue < 10 and Padj < 0.005 were considered to have significant overrepresentation over the sequences in control peak sets (Supplementary Table 9). The results were visualized by memes R package (1.6.0, using plot_ame_heatmap function), displaying those motifs that were significantly overrepresented across all three comparisons (either in dMSN or iMSNs), excluding genes for which none of their annotated TSS positions overlapped with ATAC-seq consensus peaks.
Pathway enrichment analysis
The filtered lists of DEGs (Padj < 0.05) with accessible TSS regions were analyzed for overrepresentation of GOCC terms with enrichGO function of clusterProfiler package66 (version 4.4.4, GOSOURCEDATE: 2022-03-10). The augmented list of all genes with accessible TSS regions was used as the ‘background’ list for comparison (‘universe’), and the following parameters were used: qvalueCutoff = 0.05, minGSSize = 5, maxGSSize = 2000. For identifying GOCC pathways overrepresented among genes that showed disease-associated upregulation or downregulation only in dMSNs, the list of DEGs from ‘HD_dMSN vs ctrl_dMSN’ comparison (Padj < 0.05 with DESeq2) was filtered to exclude genes that had changed expression (Padj < 0.05 with DESeq2) in the same direction in any of the three ‘HD_interneuron vs. ctrl_interneuron’ comparisons (TAC3+ INs, PVALB+ INs or SST+ INs). Significance threshold for enrichment analysis: q value < 0.05, Padj < 0.05, adjusted for multiple comparisons after hypergeometric test with clusterProfiler. GOCC term enrichment analysis (with parameters and ‘background’ list specified above) was also performed for genes downregulated in HD MSNs that are essential for MSN survival in wild-type mice (based on shRNA screening)43.
HTT and ATXN3 CAG tract sizing
Genomic DNA was purified using AllPrep DNA/RNA FFPE Kit (Qiagen, #80234) and concentrated in a vacuum concentrator if required. HTT CAG tract sizing was done by next generation sequencing of PCR amplicons of HTT exon 1 using a modified version of a previously published protocol20. Up to 10 ng gDNA was amplified in a 20 μl volume using NEBNext High-Fidelity 2X PCR Master Mix (New England Biolabs, #M0541S) supplemented with 5% dimethyl sulfoxide and barcoded primers specific to HTT exon 1 (0.5 μM each)20 or ATXN3 exon 10 (primer sequences in Supplementary Table 12): 1 cycle 96 °C, 5 min; 30× [96 °C, 45 s; 61 °C, 45 s; 72 °C, 3 min]; 72 °C, 10 min. The number of amplification cycles was raised to 32 cycles if the amount of gDNA input was below 4 ng. After PCR, the samples were combined into small pool of two to six samples and size selection was carried out by adding 0.55× volume of AMPure XP beads (Beckman Coulter, #A68831). The concentrations of purified library pools were quantified with Collibri Library Quantification Kit (ThermoFisher, #A38524100), combined into a sequencing library and sequenced on MiSeq sequencer using a 500 cycle MiSeq Reagent Nano Kit v2 with both index reads, but with 400-nt long read 1 and no read 2. Demultiplexed sequencing read data was aligned using Burrows-Wheeler Aligner (https://github.com/lh3/bwa, using BWA MEM default settings except: -O 6,6 -E 4,4) to a set of HTT exon 1 or ATXN3 exon 10 reference sequences (Supplementary Data 1 and 2) that differed by the number of CAG repeat units in the repeat tract. The number of reads uniquely mapped to each of the reference sequences in the set was considered to reflect the distribution of CAG tract lengths in the two HTT or ATXN3 alleles in the cell population analyzed. HTT read mapping data from each donor was inspected manually for determining the nucleotide sequence of the adjacent polyproline tract and the presence/absence of interruptions in CAG tract. If mHTT exon 1 structure of the donor was atypical, then sequencing reads were realigned to a set of reference sequences matching that mHTT exon 1 structure. The length of CAG repeat tracts reliably mapped was limited to 113 repeat units. Uninterrupted CAG tract lengths of progenitor/unexpanded mHTT allele (M repeat units) and normal HTT allele (N repeat units) were defined from the two modes of mapped read length-distribution in CAG-sizing data from non-expanding cell types (usually striatal microglia and astrocytes, or, if available, cerebellar granule cells). R is the number of reads mapped to a reference sequence with the specified CAG tract length. The ratio of somatic expansions (RoSE)20 and mean somatic length gain (MSLG, measured in repeat units (RUs)) were calculated as follows:
Mean somatic length gain is the average uninterrupted CAG repeat length in sequencing reads from which the progenitor allele CAG repeat length (M) has been subtracted. It is important to note that the term is not meant to reflect the size of incremental change per mutation event. Quantification of CAG tract length changes for mATXN3 was done in the same way. Statistical analysis of differences between cell types was carried out by comparing their ratio of somatic expansions or mean somatic length gains with one-way ANOVA, followed by Holm–Sidak’s multiple comparisons test.
Western blotting
When isolating nuclei for western blotting the tissue homogenization and ultracentrifugation steps were carried out as described by Xiao et al.17. After washing the nuclei once in homogenization buffer the nuclei were resuspended in 1 ml 1× PBS, 0.05% Triton X-100, 2% BSA and incubated at room temperature on a shaker for ~15–20 min. The nuclei were labeled by adding the following antibodies: Rb x NeuN-Alexa-647 (1:300, Abcam #ab190565), Rb x NeuN-Alexa594 (1:300, Abcam, #ab207279), Mm x EAAT1-Alexa-488 (1:200, Santa Cruz Biotechnology, #sc-515839 AF488), Mm x IRF8-PE (1:65, ThermoFisher, #12-9852-82) and Goat x Olig2 (1:200, R&D Systems, #AF2418). After two washes with WB wash buffer (1× PBS, 0.05% Triton X-100, 0.2% BSA), the nuclei were incubated for 30 min at room temperature with Donkey x Goat-Alexa-647 (1:400, ThermoFisher, # A-21447). Nuclei were washed twice with WB wash buffer and resuspended in Sorting buffer (w/o RNase inhibitors). After sorting, the collected nuclei were centrifuged at 1,600 × g for 10 min 4 °C and the residual volume was kept to a minimum. Nuclei were treated with DNase I (in the presence of 0.5 mM MgCl2) at 37 °C for 10 min, mixed with NuPAGE Sample Reducing Agent (ThermoFisher #NP0004) and β-mercaptoethanol (final concentration 4%, Sigma, #M3148), and heat-denatured at 96 °C for 3 min. Material from 25,000 to 50,000 nuclei were loaded on NuPAGE 4% to 12% Bis-Tris Mini gels (ThermoFisher, #NP0322BOX), aiming for equal loading in each well. After blotting the samples onto nitrocellulose membrane and blocking unspecific binding by incubating the membrane in a 5% solution of non-fat dry milk, the membranes were probed with Rb × Histone H3 antibody (1:5,000, Abcam, #ab1791) and Mm × Human MSH2 (1:300 BD Biosciences, #556349) or Mm x Human MSH3 (1:300 BD Biosciences, #611390) by incubating overnight at 4 °C. After three washes with TBS-T (1× TBS, 0.1% Tween-20), the membranes were probed with IRDye 680LT Donkey anti-Rabbit IgG Secondary Antibody (1:10,000, LICOR, #926-68023) and IRDye 800CW Goat anti-Mouse IgG Secondary Antibody (1:10,000, LICOR, #926-32210) by 1 h at room temperature. After three washes with TBS-T, the membranes were imaged with Odyssey CLx Imaging System.
Protein purification
Recombinant human FAN1 protein was expressed from a Baculovirus and purified from Sf9 insect cells as described previously40,67. Recombinant human MutSα (MSH2-MSH6) and MutSβ (MSH2-MSH3) were generated from Sf9 insect cells using Baculoviruses expressing his-tagged hMSH2, hMSH3 and hMSH6, and a purification procedure described previously68–70.
FAN1 nuclease assay
FAN1 nuclease assays were performed as described40 in nuclease assay buffer (50 mM Tris-HCl pH 8.0, 25 mM NaCl, 1 mM MnCl2, 1 mM dithiothreitol, 200 mg ml−1 BSA) with 100 nM of fluorescently labeled DNA incubated with 50 nM of recombinant human FAN1 protein. Reactions were initiated by the addition of FAN1 protein, incubated at 37 °C for 20 min and then stopped with formamide loading buffer (95% formamide, 10 mM EDTA). Reaction products were electrophoretically resolved on 6% denaturing sequencing gel for 1 h at 2,000 V and detected at fluorescence filter in the Typhoon FLA (GE Healthcare). Nuclease activity quantification compared the densitometric intensity of cleaved versus uncleaved DNA (ImageQuant). In some of the experiments, different incubation time and concentration of proteins are used and are mentioned in respective figure legends. Sequences of oligonucleotides used to generate slipped-DNA substrates with anchored flanks can be found in Supplementary Note 10.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41588-024-01653-6.
Supplementary information
Supplementary Notes 1–10 and Supplementary Figures 1–9.
HTT exon 1 reference sequences.
ATXN3 exon 10 reference sequences.
Supplementary Tables 1–12.
Source data
Unprocessed images of western blots.
Acknowledgements
This study was supported by funding from the CHDI Foundation, and we are thankful to T.F. Vogt and J. Chen for helpful discussions throughout the project. We thank N. Didkovsky, C. Wang, H. Chetia and Y. Coulombe for technical assistance, M. Ciosi for his advice on CAG tract sizing assay and K. Mätlik for advice on data visualization and for critical comments on the paper. We are also grateful to the Rockefeller University Genomics Resource Center for advice and support. We thank J.P.G. Vonsattel and A.F. Teich from Columbia University Alzheimer’s Disease Research Center (funded by NIH grant P30AG066462), D. Keene from University of Washington BioRepository and Integrated Neuropathology Laboratory (supported by Alzheimer’s Disease Research Center grants AG066567 and AG066509), S. Berretta and the Harvard Brain Tissue Resource Center, The University of Michigan Brain Bank (P30AG053760/ P30AG072931 University of Michigan Alzheimer’s Disease Core Center), Netherlands Brain Bank, and UCLA Human Brain & Spinal Fluid Resource Center (supported by the NIH and the US Department of Veterans Affairs) for assisting and providing post-mortem brain tissues. K.M. was supported by a fellowship from the Sigrid Jusélius Foundation. A.L.D. is supported by a Postdoctoral Researcher Fellowship from the Hereditary Disease Foundation and the Fox Family Foundation. J.-Y.M. is supported by the Canadian Institutes of Health Research (FRN-388879). C.E.P. is supported by the Canadian Institutes of Health Research (FRN-148910; FRN-173282), the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-08355 RGPIN-2016-06355/498835), The Petroff Family Foundation, The Marigold Foundation, Tribute Communities and the Fox Family Foundation. J.-Y.M. holds a Tier 1 Canada Research Chair in DNA Repair and Cancer Therapeutics. C.E.P. holds a Tier 1 Canada Research Chair in Disease-Associated Genome Instability.
Extended data
Author contributions
Conceptualization: K.M., C.E.P., N.H. Formal analysis: K.M., M.B., M.R.P., T.S.C., A.L.D. Investigation: K.M., M.B., A.L.D. Resources: L.K., D.A.D., M.-C.C., J.-Y.M., N.H. Visualization: K.M., M.B. Writing of original draft: K.M., N.H. Writing, review and editing: M.R.P., M.B., L.K., C.E.P. Supervision: C.E.P., J.-Y.M., N.H. Funding acquisition: C.E.P., N.H.
Peer review
Peer review information
Nature Genetics thanks Darren Monckton and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
All sequencing datasets generated as part of this study are publicly available in NCBI GEO under accession GSE227729 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227729). Further information and requests for resources and reagents should be directed to the lead contact, N. Heintz (heintz@rockefeller.edu). Altered expression of mouse genes in the striatum of BAC-CAG mice54 (10.1016/j.neuron.2022.01.006), the Str266R gene set71 (10.1101/2022.02.04.479180), list of genes essential for MSN survival in wild-type mice43 (10.1016/j.neuron.2020.01.004) and TRAP data from zQ175 and R6/2 mice41 (GEO dataset GSE152058, 10.1016/j.neuron.2020.06.021) have been published before. Sequence and transcript coordinates for human hg38 UCSC genome and gene models were retrieved from the BSgenome.Hsapiens.UCSC.hg38 Bioconductor package (version 1.4.1) and TxDb.Haspiens.UCSC.hg38.knownGene (version 3.4.0) Bioconductor libraries (https://bioconductor.org/packages/release/data/annotation/html/BSgenome.Hsapiens.UCSC.hg38.html). NCBI Refseq hg38 gene annotation (version 109.20211119, https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Homo_sapiens/109.20211119/) was used for annotating ATAC-seq consensus peaks to transcriptional start sites. Gene Ontology Cellular Compartment (GOCC) terms for enrichment analysis were derived through enrichGO function of clusterProfiler package (version 4.4.4, GOSOURCEDATE: 2022-03-10, https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html). Eukaryote in vivo and in vitro databases were accessed through MEME Suite 5.5.4 (https://meme-suite.org/meme/tools/ame). Source data are provided with this paper.
Code availability
Only publicly available tools were used in data analysis. The analysis parameters used have been described in Methods.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41588-024-01653-6.
Supplementary information
The online version contains supplementary material available at 10.1038/s41588-024-01653-6.
References
- 1.Gusella JF, Lee JM, MacDonald ME. Huntington’s disease: nearly four decades of human molecular genetics. Hum. Mol. Genet. 2021;30:R254–R263. doi: 10.1093/hmg/ddab170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heinsen H, et al. Cortical and striatal neurone number in Huntington’s disease. Acta Neuropathol. 1994;88:320–333. doi: 10.1007/BF00310376. [DOI] [PubMed] [Google Scholar]
- 3.Ferrante RJ, et al. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1987;46:12–27. doi: 10.1097/00005072-198701000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Massouh M, Wallman MJ, Pourcher E, Parent A. The fate of the large striatal interneurons expressing calretinin in Huntington’s disease. Neurosci. Res. 2008;62:216–224. doi: 10.1016/j.neures.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington’s disease. Brain Res Brain Res Rev. 2000;34:80–101. doi: 10.1016/S0165-0173(00)00039-4. [DOI] [PubMed] [Google Scholar]
- 6.Landwehrmeyer GB, et al. Huntington’s disease gene: regional and cellular expression in brain of normal and affected individuals. Ann. Neurol. 1995;37:218–230. doi: 10.1002/ana.410370213. [DOI] [PubMed] [Google Scholar]
- 7.Telenius H, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet. 1994;6:409–414. doi: 10.1038/ng0494-409. [DOI] [PubMed] [Google Scholar]
- 8.Thornton CA, Johnson K, Moxley RT., 3rd Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol. 1994;35:104–107. doi: 10.1002/ana.410350116. [DOI] [PubMed] [Google Scholar]
- 9.Long A, et al. Somatic instability of the expanded GAA repeats in Friedreich’s ataxia. PLoS ONE. 2017;12:e0189990. doi: 10.1371/journal.pone.0189990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouro Pinto R, et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020;29:2551–2567. doi: 10.1093/hmg/ddaa139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lokanga RA, et al. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 2013;34:157–166. doi: 10.1002/humu.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy L, et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003;12:3359–3367. doi: 10.1093/hmg/ddg352. [DOI] [PubMed] [Google Scholar]
- 13.Genetic Modifiers of Huntington’s Disease Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genetic Modifiers of Huntington’s Disease Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. 2019;178:887–900. doi: 10.1016/j.cell.2019.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shelbourne PF, et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum. Mol. Genet. 2007;16:1133–1142. doi: 10.1093/hmg/ddm054. [DOI] [PubMed] [Google Scholar]
- 16.Hong EP, et al. Huntington’s disease pathogenesis: two sequential components. J. Huntingtons Dis. 2021;10:35–51. doi: 10.3233/JHD-200427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, et al. Species and cell-type properties of classically defined human and rodent neurons and glia. Elife. 2018;7:e37551. doi: 10.7554/eLife.37551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle JP, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krienen FM, et al. Innovations present in the primate interneuron repertoire. Nature. 2020;586:262–269. doi: 10.1038/s41586-020-2781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciosi M, et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. 2019;48:568–580. doi: 10.1016/j.ebiom.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiner A, et al. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl Acad. Sci. USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulson H. Repeat expansion diseases. Handb. Clin. Neurol. 2018;147:105–123. doi: 10.1016/B978-0-444-63233-3.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franklin GL, et al. Is ataxia an underestimated symptom of Huntington’s disease? Front. Neurol. 2020;11:571843. doi: 10.3389/fneur.2020.571843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh-Bains MK, et al. Cerebellar degeneration correlates with motor symptoms in Huntington disease. Ann. Neurol. 2019;85:396–405. doi: 10.1002/ana.25413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe H, et al. Differential somatic CAG repeat instability in variable brain cell lineage in dentatorubral pallidoluysian atrophy (DRPLA): a laser-captured microdissection (LCM)-based analysis. Hum. Genet. 2000;107:452–457. doi: 10.1007/s004390000400. [DOI] [PubMed] [Google Scholar]
- 26.Hashida H, et al. Single cell analysis of CAG repeat in brains of dentatorubral-pallidoluysian atrophy (DRPLA) J. Neurol. Sci. 2001;190:87–93. doi: 10.1016/S0022-510X(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 27.Nakamori M, Pearson CE, Thornton CA. Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum. Mol. Genet. 2011;20:580–588. doi: 10.1093/hmg/ddq501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheeler VC, Dion V. Modifiers of CAG/CTG repeat instability: insights from mammalian models. J. Huntingtons Dis. 2021;10:123–148. doi: 10.3233/JHD-200426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flower M, et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain. 2019;142:1876–1886. doi: 10.1093/brain/awz115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 31.Wheeler VC, et al. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum. Mol. Genet. 2003;12:273–281. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- 32.Dragileva E, et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 2009;33:37–47. doi: 10.1016/j.nbd.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovalenko M, et al. Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington’s disease knock-in mice. PLoS ONE. 2012;7:e44273. doi: 10.1371/journal.pone.0044273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tome S, et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 2013;9:e1003280. doi: 10.1371/journal.pgen.1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim KH, et al. Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am. J. Hum. Genet. 2020;107:96–110. doi: 10.1016/j.ajhg.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshmukh AL, et al. FAN1, a DNA repair nuclease, as a modifier of repeat expansion disorders. J. Huntingtons Dis. 2021;10:95–122. doi: 10.3233/JHD-200448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loupe JM, et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum. Mol. Genet. 2020;29:3044–3053. doi: 10.1093/hmg/ddaa196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goold R, et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 2019;28:650–661. doi: 10.1093/hmg/ddy375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McAllister B, et al. Exome sequencing of individuals with Huntington’s disease implicates FAN1 nuclease activity in slowing CAG expansion and disease onset. Nat. Neurosci. 2022;25:446–457. doi: 10.1038/s41593-022-01033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deshmukh AL, et al. FAN1 exo- not endo-nuclease pausing on disease-associated slipped-DNA repeats: a mechanism of repeat instability. Cell Rep. 2021;37:110078. doi: 10.1016/j.celrep.2021.110078. [DOI] [PubMed] [Google Scholar]
- 41.Lee H, et al. Cell type-specific transcriptomics reveals that mutant Huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron. 2020;107:891–908. doi: 10.1016/j.neuron.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 43.Wertz MH, et al. Genome-wide in vivo CNS screening identifies genes that modify CNS neuronal survival and mHTT toxicity. Neuron. 2020;106:76–89. doi: 10.1016/j.neuron.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vodicka P, et al. Autophagy activation by transcription factor EB (TFEB) in striatum of HDQ175/Q7 mice. J. Huntingtons Dis. 2016;5:249–260. doi: 10.3233/JHD-160211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rui YN, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015;17:262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cason SE, et al. Sequential dynein effectors regulate axonal autophagosome motility in a maturation-dependent pathway. J. Cell Biol. 2021;220:e202010179. doi: 10.1083/jcb.202010179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JM, et al. Genetic modifiers of Huntington disease differentially influence motor and cognitive domains. Am. J. Hum. Genet. 2022;109:885–899. doi: 10.1016/j.ajhg.2022.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki M, et al. Vesicular neurotransmitter transporters in Huntington’s disease: initial observations and comparison with traditional synaptic markers. Synapse. 2001;41:326–336. doi: 10.1002/syn.1089. [DOI] [PubMed] [Google Scholar]
- 49.Rub U, et al. Degeneration of the cerebellum in Huntington’s disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol. 2013;23:165–177. doi: 10.1111/j.1750-3639.2012.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeste DV, Barban L, Parisi J. Reduced Purkinje cell density in Huntington’s disease. Exp. Neurol. 1984;85:78–86. doi: 10.1016/0014-4886(84)90162-6. [DOI] [PubMed] [Google Scholar]
- 51.Pressl, C. et al. Layer 5a corticostriatal projection neurons are selectively vulnerable in Huntington’s disease. Preprint at bioRxiv10.1101/2023.04.24.538096 (2023).
- 52.Kaplan S, Itzkovitz S, Shapiro E. A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput. Biol. 2007;3:e235. doi: 10.1371/journal.pcbi.0030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langfelder P, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 2016;19:623–633. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu X, et al. Uninterrupted CAG repeat drives striatum-selective transcriptionopathy and nuclear pathogenesis in human Huntingtin BAC mice. Neuron. 2022;110:1173–1192 e7. doi: 10.1016/j.neuron.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warner JH, et al. Standardizing the CAP score in Huntington’s disease by predicting age-at-onset. J. Huntingt. Dis. 2022;11:153–171. doi: 10.3233/JHD-210475. [DOI] [PubMed] [Google Scholar]
- 56.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Q, et al. Exploring epigenomic datasets by ChIPseeker. Curr. Protoc. 2022;2:e585. doi: 10.1002/cpz1.585. [DOI] [PubMed] [Google Scholar]
- 61.Marini F, Binder H. pcaExplorer: an R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinform. 2019;20:331. doi: 10.1186/s12859-019-2879-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Hogenesch JB, Irizarry RA. Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. Nat. Biotechnol. 2016;34:1287–1291. doi: 10.1038/nbt.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love MI, Soneson C, Patro R. Swimming downstream: statistical analysis of differential transcript usage following Salmon quantification. F1000Res. 2018;7:952. doi: 10.12688/f1000research.15398.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLeay RC, Bailey TL. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinform. 2010;11:165. doi: 10.1186/1471-2105-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maity R, et al. GST-His purification: a two-step affinity purification protocol yielding full-length purified proteins. J. Vis. Exp. 2013;80:e50320. doi: 10.3791/50320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pearson CE, Ewel A, Acharya S, Fishel RA, Sinden RR. Human MSH2 binds to trinucleotide repeat DNA structures associated with neurodegenerative diseases. Hum. Mol. Genet. 1997;6:1117–1123. doi: 10.1093/hmg/6.7.1117. [DOI] [PubMed] [Google Scholar]
- 69.Panigrahi GB, Slean MM, Simard JP, Gileadi O, Pearson CE. Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSbeta, but clustered slip-outs are poorly repaired. Proc. Natl Acad. Sci. USA. 2010;107:12593–12598. doi: 10.1073/pnas.0909087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Panigrahi GB, Lau R, Montgomery SE, Leonard MR, Pearson CE. Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol. 2005;12:654–662. doi: 10.1038/nsmb959. [DOI] [PubMed] [Google Scholar]
- 71.Obenauer, J.C. et al. Expression analysis of Huntington disease mouse models reveals robust striatum disease signatures. Preprint at bioRxiv10.1101/2022.02.04.479180 (2023).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Notes 1–10 and Supplementary Figures 1–9.
HTT exon 1 reference sequences.
ATXN3 exon 10 reference sequences.
Supplementary Tables 1–12.
Unprocessed images of western blots.
Data Availability Statement
All sequencing datasets generated as part of this study are publicly available in NCBI GEO under accession GSE227729 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227729). Further information and requests for resources and reagents should be directed to the lead contact, N. Heintz (heintz@rockefeller.edu). Altered expression of mouse genes in the striatum of BAC-CAG mice54 (10.1016/j.neuron.2022.01.006), the Str266R gene set71 (10.1101/2022.02.04.479180), list of genes essential for MSN survival in wild-type mice43 (10.1016/j.neuron.2020.01.004) and TRAP data from zQ175 and R6/2 mice41 (GEO dataset GSE152058, 10.1016/j.neuron.2020.06.021) have been published before. Sequence and transcript coordinates for human hg38 UCSC genome and gene models were retrieved from the BSgenome.Hsapiens.UCSC.hg38 Bioconductor package (version 1.4.1) and TxDb.Haspiens.UCSC.hg38.knownGene (version 3.4.0) Bioconductor libraries (https://bioconductor.org/packages/release/data/annotation/html/BSgenome.Hsapiens.UCSC.hg38.html). NCBI Refseq hg38 gene annotation (version 109.20211119, https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Homo_sapiens/109.20211119/) was used for annotating ATAC-seq consensus peaks to transcriptional start sites. Gene Ontology Cellular Compartment (GOCC) terms for enrichment analysis were derived through enrichGO function of clusterProfiler package (version 4.4.4, GOSOURCEDATE: 2022-03-10, https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html). Eukaryote in vivo and in vitro databases were accessed through MEME Suite 5.5.4 (https://meme-suite.org/meme/tools/ame). Source data are provided with this paper.
Only publicly available tools were used in data analysis. The analysis parameters used have been described in Methods.