

Abstract
Combinatorial protein engineering for selection of proteins with novel functions, such as enzymes and affinity reagents, is an important tool in biotechnology, drug discovery, and other biochemical fields. Bacterial display is an emerging technology for isolation of new affinity proteins from such combinatorial libraries. Cells have certain properties that are attractive for directed evolution purposes, in particular the option to use quantitative flow-cytometric cell sorting for selection of binders. Here, an immune library of around 107 camelid single-domain antibody fragments (Nanobodies) was displayed on both the Gram-positive bacterium Staphylococcus carnosus and on phage. As demonstrated for the first time, the antibody repertoire was found to be well expressed on the bacterial surface and flow-cytometric sorting yielded a number of Nanobodies with subnanomolar affinity for the target protein, green fluorescent protein (GFP). Interestingly, the staphylococcal output repertoire and the binders from the phage display selection contained two slightly different sets of clones, containing both unique as well as several similar variants. All of the Nanobodies from the staphylococcal selection were also shown to enhance the fluorescence of GFP upon binding, potentially due to the fluorescence-based sorting principle. Our study highlights the impact of the chosen display technology on the variety of selected binders and thus the value of having alternative methods available, and demonstrates in addition that the staphylococcal system is suitable for generation of high-affinity antibody fragments.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1179-y) contains supplementary material, which is available to authorized users.
Keywords: Bacterial display, Combinatorial protein engineering, Nanobodies, Phage display, Recombinant antibodies
Introduction
Display technologies (e.g., phage display) are widely used for selection of protein function. Phage display has revolutionized the field of protein engineering, enabling generation of fully human antibodies, efficient affinity maturation, improvement of other properties such as stability and solubility [1] as well as the isolation of specific affinity proteins based on non-immunoglobulin scaffolds [2].
In addition to phage display, alternative display systems such as cell surface display [3, 4] and ribosome display [5] have been developed and investigated for similar purposes. The main difference between ribosome and phage display is that transcription and translation in ribosome display is performed in vitro, and the library size is hence not limited by the transformation of DNA into cells. However, while the means of producing the actual libraries is differing, the procedure for selecting binders with ribosome and mRNA display is basically identical to the capture and elution principle that is generally employed when using phage display.
In contrast, directed evolution involving cell surface display is more different compared to phage display, typically relying on screening of combinatorial libraries for identification of variants with desired function rather than true selection. This is because cells, as opposed to phages, are large enough to be detected in a flow cytometer, enabling fluorescence-based analysis and sorting of displayed libraries [6, 7]. Another important property of cell display is that recombinant proteins are expressed multivalently on the surface, and together with the flow-cytometric analysis, this provides means for quantification of the relative affinity of each cell-displayed protein variant for the target, which is the parameter that sorting is generally based upon. The use of flow-cytometric cell sorting also provides excellent control and monitoring of the selection process, as compared to other existing selection techniques. In most flow cytometers it is also possible to analyze multiple parameters simultaneously, providing means for normalization of expression level [8, 9] and engineering of crossreactivity [10, 11]. Cell display technologies have been developed and investigated using a variety of different hosts and anchoring mechanisms, and although mammalian cell-based systems have been reported [12–14], the most commonly used methods are employing bacteria [3, 7, 15–17] or yeast [4, 18]. However, although bacterial display is becoming increasingly popular, no direct comparison to phage display has been reported so far.
We have developed a bacterial display system using Gram-positive staphylococci for library purposes [17, 19]. The technology is based on the cell-wall anchoring domain from staphylococcal protein A and surface expression on the non-pathogenic strain Staphylococcus carnosus [20]. Gram-positive bacteria are well suited for recombinant display and combinatorial protein engineering applications. A single cell membrane and C-terminal anchoring mechanism results in straightforward translocation of recombinant proteins to the cell surface as well as functional display of large proteins. The thick layer of peptidoglycan that is surrounding the single cell membrane protects the cells in the harsh milieu of the flow cytometer, leading to a remarkable cell viability after cell sorting [19]. Previously, libraries based on Affibody molecules [21], bispecific albumin-binding domains (ABDs) [10] as well as peptide libraries derived from a wide variety of human proteins [19, 22, 23] have been successfully displayed on S. carnosus and used for the generation of new affinity reagents [17], affinity maturation [10, 24] as well as for epitope mapping efforts [19, 22, 23]. In this study, using an immune Nanobody library, we demonstrate for the first time that the staphylococcal system is also an excellent alternative for display of libraries derived from antibody fragments and can hence complement phage display and other methods in the protein engineering toolbox.
Nanobodies are the variable domains (VHH) from camelid heavy-chain only antibodies (HCAb). Nanobodies have several attractive properties, such as small size (15 kDa), a strict monomeric behavior and a generally excellent stability, and since they are expressed naturally without association to a light chain, the solubility is—on average—higher compared to for example single-chain variable domains from human antibodies [25]. These properties facilitate for example the construction of multivalent formats (e.g., bispecific Nanobodies) as well as production of large amounts of recombinant protein in microbial systems [26], and Nanobodies have even been reported to work intracellularly [27]. Due to the lack of pairing of a VHH with a VL domain, the natural single-domain format results in full functional diversity when working with immune Nanobody libraries in display applications [28]. The immune repertoire is hence covered with a relatively moderate complexity library and subcloning to most display formats is straightforward. Working with immune Nanobody libraries also has the advantage that an affinity maturation of the HCAb occurs during the immunization of the camelid. It is therefore expected that Nanobodies retrieved from such libraries will have high affinity and specificity for the antigen, resulting in that subsequent in vitro affinity maturation steps often can be avoided. Isolation of high-affinity Nanobodies from immune libraries has previously been performed using phage display [29], ribosome display [30], yeast display [31] and a bacterial-two-hybrid system [32], however, no bacterial display system has been investigated so far for this purpose. It has also been reported that display of other antibody fragments (e.g., scFvs) on the outer membrane of Escherichia coli is challenging and the staphylococcal system might thus be a valuable bacterial complement to phage and yeast display [15].
Here we report on the first antibody fragment library displayed on Gram-positive bacteria. Using the same immune Nanobody library generated towards green fluorescent protein (GFP) on both the staphylococcal and phage display platforms, we have investigated the outcome from each selection strategy, revealing both similarities as well as differences. Taken together, the results demonstrate that the new Gram-positive selection system is a powerful complement to phage display for selection of high-affinity antibody fragments. The fluorescence-based sorting of the bacterial library also enriched Nanobodies with fluorescence-enhancing properties. Even though naturally fluorescent targets are an exception, this indicates that cell display in combination with fluorescence-activated cell sorting (FACS) is a suitable technology for generating binders that can modulate the spectral characteristics of such targets.
Materials and methods
Construction of Nanobody library
The GFPplus antigen was expressed in bacteria and purified as described by Deschamps et al. [33]. A dromedary (Camelus dromedarius) was immunized by six injections of ~0.3 mg GFPplus in an equal volume of Gerbu adjuvant (GERBU Biochemicals, Germany) at weekly intervals. Three days after the last injection, anti coagulated peripheral blood (50 ml) was collected and treated as explained by Conrath et al. [29]. A phage display Nanobody library was made in the vector pHEN4 as previously described [29]. Briefly, lymphocytes were prepared on Lymphoprep from 50 ml of blood, the total mRNA was extracted from these lymphocytes (yielding between 100 and 250 μg) of which 50 μg was converted to cDNA using dN6 as primer and Superscript II Reverse transcriptase in a total volume of 100 μl. An aliquot of 3 μl was used to amplify the Nanobody genes by standard PCR. The amplified material was used to ligate into pHEN4 vector and to transform house-made electrocompetent TG1 cells.
Selection of GFP-specific Nanobodies using phage display
The Nanobodies from the library in pHEN4 were displayed on phage and enriched for antigen binders in three rounds on GFPplus coated in wells of microtiter plates as previously described [29]. After the second and third round of panning, up to 144 individual clones were randomly selected and analyzed in a standard ELISA and the Nanobody gene from the positive clones in the ELISA were amplified and subjected to an RFLP analysis with HinfI. The nucleotide sequence of clones with a different RFLP pattern were determined and converted to amino acid sequence.
Protein production and purification of GFP-specific Nanobodies from the phage display selection
The phage display-derived Nanobodies (P-Nbs) were amplified by PCR, digested with PstI and BstEII (New England Biolabs), and ligated into the pHEN6 vector [29] for periplasmic expression. Plasmids were transformed into E. coli RR1dM15 cells, prepared using QiaPrep kit (Qiagen, Valencia, CA, USA) according to the supplier’s recommendations and transformed into E. coli WK6 for expression. A single colony from each Nanobody was inoculated to Tryptic Soy Broth (TSB) with 10 μg/ml ampicillin and cultivated at 200 rpm overnight. An aliquot of the overnight culture was inoculated to fresh TSB medium with 10 μg/ml ampicillin and grown to OD600 = 0.6–0.9. Nanobody expression was induced by addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, and cultures were grown at 30 °C and 200 rpm overnight. Cells were harvested by centrifugation for 8 min at 8,000 rpm and 14 °C, resuspended in TES buffer (0.5 M sucrose, 0.2 M Tris–HCl, 0.5 mM EDTA) and incubated on ice at 200 rpm for 1 h. Periplasmic content was released using osmotic shock by addition of TES/4 buffer (TES buffer diluted 1:4) and incubation on ice for 2 h. After centrifugation for 30 min at 8,000 rpm and 4 °C, the periplasmic extract was purified by immobilized metal ion affinity chromatography (IMAC) using PD-10 columns containing 1 ml of HisPur™ Cobalt Resin (ThermoScientific, Rockford, USA). The Nanobodies were eluted from the columns using PBS containing 0.5 M imidazole. Finally, potential multimers and salts were separated from monomers by size exclusion chromatography (SEC) using a Superdex™75 FPLC column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and an ÄKTA explorer (GE Healthcare Bio-Sciences AB, Uppsala, Sweden).
Characterization of GFP-specific Nanobodies from the phage display selection
Purified Nanobodies were immobilized onto GLM chips (Bio-Rad Laboratories, Hercules, CA, USA) by amine coupling using a ProteOn XPR36 instrument (Bio-Rad Laboratories) according to the manufacturer’s suggestions. On each chip, one surface was used as a reference surface, with no immobilized protein. For determination of kinetic constants of the Nanobodies, SPR-based biosensor analysis was performed using a ProteOn XPR36 instrument (Bio-Rad Laboratories). Three different concentrations of eGFP (MBL International) (10, 25, and 50 nM) were injected over each chip. In all experiments PBST [PBS (10 mM sodium phosphate and 150 mM sodium chloride) + 0.1 % Tween 20] was used as a running buffer and 33 mM HCl was used for regeneration of the chip surface. Response signals were double referenced by subtraction of simultaneous responses from reference surface and a buffer injection and obtained sensorgrams were thereafter fitted to a Langmuir 1:1 binding model using the ProteOn Manager Software (Bio-Rad Laboratories). The GFP enhancing effect for P-Nbs was determined as described by Kirchhofer et al. [27].
Cloning and bacterial surface expression of phage display-derived Nanobodies
Three of the GFP-specific Nanobodies selected by phage display (P-Nb1, P-Nb3, and P-Nb6) were subcloned into the staphylococcal display vector pSCZ1 [17] using the restriction sites BamHI and SalI. The Nanobody sequences were amplified by PCR using primers containing BamHI and SalI sites, and digested with the same restriction enzymes (New England Biolabs, Beverly, MA, USA). The sequences were ligated into the staphylococcal display vector pSCZ1 and transformed to E. coli RRIdM15 cells [34]. The plasmids were prepared using a JETSTAR kit (Genomed, Bad Oeynhausen, Germany), according to the supplier’s recommendations and transformed in electrocompetent S. carnosus TM300 [20] as previously described [35] and stored in 15 % glycerol at −80 °C.
Flow-cytometric analysis
Staphylococcal cells expressing the three different Nanobodies were cultured in TSB medium (Merck, Darmstadt, Germany) with 10 μg/ml chloramphenicol for 16 h at 37 °C and 150 rpm. Cells were washed in 800 μl PBS (pH 7.4) with 0.1 % Pluronic F108 NF surfactant (PBSP; BASF Corporation, Mount Olive, NJ, USA). Cells were pelleted by centrifugation (3,500 × g, 4 °C, 6 min) and re-suspended in 150 μl 200 nM eGFP (MBL International, Woburn, MA, USA) and subsequently incubated at room temperature with gentle mixing for 1 h. After the incubation, cells were washed twice in 200 μl PBSP and incubated with 225 nM Alexa Fluor 647-human serum albumin (HSA) conjugate (prepared as described previously [17]) for 30 min on ice under foil. The cells were then washed in 200 μl PBSP and re-suspended in 200 μl PBSP, and analyzed by flow cytometry using a FACS Vantage SE (BD Biosciences, San Jose, CA, USA).
Cloning and sorting of the cell-displayed Nanobody library
A representative fraction of the immune Nanobody library cloned in pHEN4 was used as template to amplify the Nanobody genes. The amplicons were inserted into the staphylococcal vector pSCNb1. Plasmid pSCNb1 is a modified variant of the staphylococcal display vector pSCZ1 [17], with a NheI site introduced for insertion of Nanobody-encoding gene fragments. The Nanobody sequences were amplified by PCR using primers containing XhoI and NheI sites, and digested with the same restriction enzymes (New England Biolabs, Beverly, MA, USA). The sequences were ligated into the staphylococcal display vector pSCNb1 and transformed using electroporation to E. coli SS320 [36]. Plasmids were prepared using a JETSTAR kit (Genomed) and transformed to S. carnosus as described previously [35]. The library was stored in 15 % glycerol at −80 °C.
An aliquot of the library (corresponding to a number of cells ten times the size of the library) was inoculated to TSB with 10 μg/ml chloramphenicol and cultivated for 16 h at 37 °C and 150 rpm. A part of the overnight culture was washed with 800 μl PBSP and cells were pelleted by centrifugation (3,500 × g, 4 °C, 6 min) and resuspended in 1.4 ml of eGFP (MBL International) (starting with 200 nM in the first sorting round and increasing the stringency down to 10 nM in round 4) and incubated at room temperature with gentle mixing for 2 h. Cells were thereafter washed twice in 180 μl PBSP and incubated with 225 nM Alexa Fluor 647-HSA conjugate for 45 min on ice under foil. Finally, the cells were washed three times in 180 μl PBSP and resuspended in 1.4 ml PBSP. The library was then sorted using a FACS Vantage SE (BD Biosciences). Cells were sorted into TSB medium and incubated for 1 h with gentle mixing at 37 °C and then inoculated to 25 ml TSB with 10 μg/ml chloramphenicol and cultured for 36 h at 37 °C and 150 rpm. After the last round of sorting, sorted cells were spread on agar plates containing 10 μg/ml chloramphenicol and incubated for 24 h at 37 °C. Colonies from the sorting were screened by PCR, and sequenced using BigDye thermo cycle sequencing reactions and an ABI Prism 3700 instrument (Applied Biosystems, Foster City, CA, USA).
Subcloning, protein production, and purification
The Nanobody sequences selected for further characterization were subcloned into the pHEN6 vector and proteins were produced and purified as described for the Nanobodies from the phage selection.
Biosensor analysis
The affinities of the purified GFP-specific Nanobodies from the staphylococcal selection were determined using an SPR-based biosensor assay, as described above for the Nanobodies from the phage selection. HSA, human IgG and transferrin were immobilized on different surfaces of a GLM chip (Bio-Rad Laboratories) using amine coupling. All seven staphylococcal display-selected Nanobodies were injected over each surface in two concentrations (80 and 240 nM). As a positive control, an ABD was injected over the HSA surface, Z wt (Affibody AB, Stockholm, Sweden) over the IgG surface and a transferrin-binding Affibody molecule (Affibody AB) over the transferrin surface.
GFP fluorescence enhancement analysis
In order to investigate the fluorescence modulating effects of the selected Nanobodies, 300 nM of each Nanobody was added to 100 nM of wtGFP (Millipore, Billerica, MA, USA) in a NUNC F96 black microwell plate (ThermoScientific, Rockford, USA), and the fluorescence intensity was measured by scanning the plate using an LS400 Laser Scanner (Tecan Group Ltd, Männedorf, Switzerland) instrument, and analyzing the intensity from each well using the software Array-Pro Analyzer (version 4.5) (Media Cybernetics, Bethesda, MD, USA). As a negative control the BcII10 Nanobody with a non-related specificity was used.
Results
Rationale
In this study, an immune Nanobody library, targeting GFP, was displayed on the surface of the Gram-positive bacterium S. carnosus as well as on phage particles for isolation of binders. The aim was to investigate if bacterial display could be a complement to phage display since the selections are based on widely different principles. The rationale for the selection of GFP-specific Nanobodies using phage display and staphylococcal display is illustrated in Fig. 1. Although the two strategies are clearly different (e.g., using target in solution for bacterial display and immobilized target for phage display), in this study we preferred to use the standard procedures for selection that are currently employed in our lab.
Fig. 1.
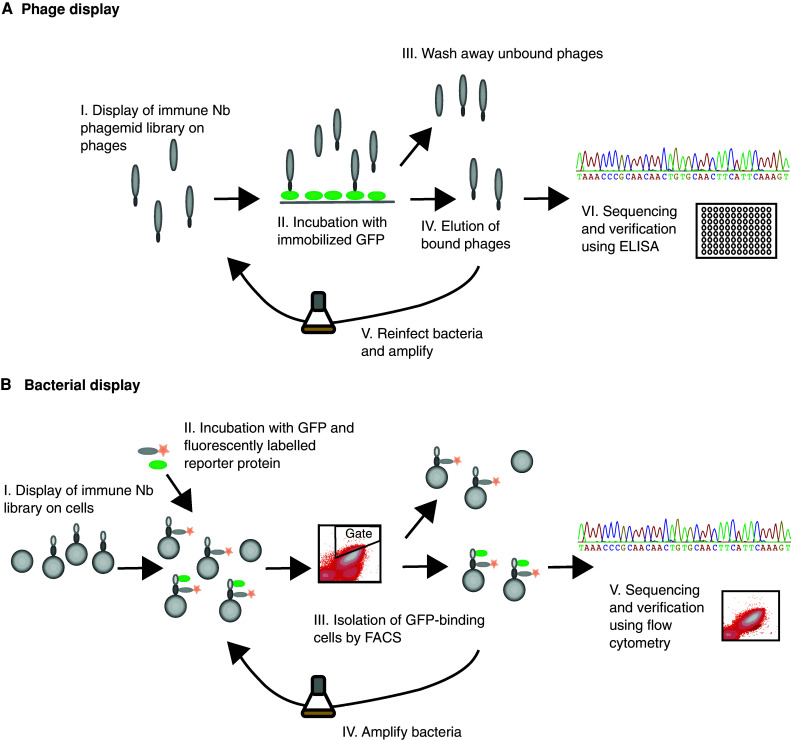
Schematic illustrations of the display procedures used for generation of GFP-binding Nanobodies. a Phage display. (I) The immune Nanobody phagemid library is displayed on the surface of bacteriophages. (II–III) After incubation with immobilized GFP, unbound phages are washed away. (IV) Bound phages are then eluted, and (V) used to re-infect bacteria in order to perform additional selection rounds, or (VI) isolated for characterization of individual clones. b Bacterial display. (I–II) The Nanobody library is displayed on the surface of S. carnosus cells, which are incubated with soluble GFP and fluorescently labeled reporter protein. (III) Cells are then sorted using flow cytometry with normalized gating. (IV) Positive cells are amplified for additional sorting rounds or (V) isolated for characterization of individual clones
Phage display selection of GFP-specific Nanobodies
After raising an immune response against GFP in the dromedary HCAb, we amplified the Nanobody gene fragments by PCR and ligated the amplicon in the phage display vector pHEN4 according to established protocols [29]. Transformation resulted in a library of 4 × 107 individual transformants and colony PCR on randomly chosen clones revealed that ~85 % of the transformants contained an insert with the size of a VHH. After three rounds of panning on GFP coated in wells of microtiter plates, and testing 144 individual clones in ELISA and RFLP analysis we retrieved five groups of GFP-specific Nanobodies (named P-Nb1, P-Nb2, P-Nb3, P-Nb5, and P-Nb6, respectively) (Online Resource 1). Within one group, all binders share the same amino acid sequence in their CDR3, and it is know that such binders originate from the same B cell lineage and target the same epitope on the antigen [37]. The amino acid sequences of all binders revealed the framework-2 hallmark residues of a dromedary VHH. It seems that all Nanobodies except for P-Nb5 were derived from germline VHH belonging to the subfamily 2a, the most frequently used VHH germline [38]. The P-Nb3 contained an interloop disulfide bond as occurs regularly in VHH; whereas the P-Nb5 of VHH germline 4b had an extra disulfide bond between the CDR3 and the framework-2 (position 50, IMGT numbering) (Online Resource 1).
Characterization of GFP-specific Nanobodies from the phage display selection
After recloning the Nanobody genes in an expression vector (pHEN6 [29]), the Nanobody proteins were expressed in bacteria, extracted from the periplasm and purified by IMAC and gel filtration (Online Resource 2 and 3). The affinities for GFP were determined using an SPR-based biosensor assay. The Nanobodies were immobilized by amine coupling to an alginate chip surface using a ProteOn XPR36 instrument, and three different concentrations of eGFP (MBL International) were injected over the surface for determination of the kinetic parameters of the binding reaction. The equilibrium dissociation constants (K D) for GFP ranged from 0.5 to 42.5 nM (Table 1, Online Resource 4). In concordance with the findings by Kirchhofer and colleagues [27], P-Nb5 (denoted “minimizer” or “GBP4” in the publication by Kirchhofer et al., affinity of 0.5 nM) reduced the fluorescence of the antigen (Table 1).
Table 1.
Affinities and effect on GFP fluorescence
| Nanobody | K D (nM) | GFP enhancing effect (%) |
|---|---|---|
| S-Nb1 | 0.5 ± 0.2 | 20 ± 8 |
| S-Nb2 | 0.25 ± 0.15 | 47 ± 11 |
| S-Nb3 | 0.14 ± 0.04 | 43 ± 7 |
| S-Nb4 | 10.1 ± 1.2 | 20 ± 4 |
| S-Nb5 | 0.21 ± 0.05 | 45 ± 8 |
| S-Nb6 | 13.8 ± 2.2 | 45 ± 21 |
| S-Nb7 | 3.1 ± 0.3 | 86 ± 16 |
| P-Nb1 | 3.2 ± 0.05 | 40 |
| P-Nb2 | 42.5 ± 1.3 | 8 |
| P-Nb3 | 20.5 ± 1.0 | 1 |
| P-Nb5 | 0.53 ± 0.05 | −49 |
| P-Nb6 | 42.2 ± 5.4 | 5 |
Equilibrium dissociation constants (K D) from SPR measurements were determined in duplicates. GFP enhancing effects were determined in triplicates. The data is shown as mean ± SD
The GFP enhancing effect for P-Nbs was determined as described by Kirchhofer et al. [27]
Cloning and expression of GFP-specific Nanobodies on the surface of S. carnosus
In order to investigate whether Nanobodies could be functionally displayed on the surface of S. carnosus, the genes encoding three GFP-binding Nanobodies selected by phage display (termed P-Nb1, P-Nb3, and P-Nb6) were subcloned into the staphylococcal display vector pSCNb1. The pSCNb1 vector is based on the previously described staphylococcal display vector pSCZ1 [17]. The Nanobody gene fragments were ligated into pSCNb1 in fusion to an albumin-binding protein (ABP), which is used for monitoring of the surface expression level and subsequent normalization during FACS [9]. A schematic representation of a surface-displayed Nanobody is shown in Fig. 2a. After transformation to electrocompetent staphylococci, bacteria displaying the three different GFP-binding Nanobodies were analyzed by flow cytometry for verification of functional display of the Nanobodies on the cell surface. Cells were incubated with eGFP as well as with fluorescently labeled HSA, and the target-binding signal and the surface expression level were measured using flow cytometry. All three of the phage-display derived Nanobodies were well expressed on the surface of S. carnosus and showed a strong signal in the channel corresponding to GFP fluorescence (Fig. 2b). The results hence suggest that staphylococcal display might be an alternative for isolation of Nanobodies from immune repertoires and encouraged the construction of a bacterial-displayed Nanobody library.
Fig. 2.

Schematic picture over a cell-displayed Nanobody and flow-cytometric analysis of cell-displayed Nanobodies selected by phage display. a The Nanobody is displayed in fusion to a cell-wall anchoring domain (XM) from staphylococcal protein A, and an albumin-binding protein (ABP), which serves as a linker as well as for binding of a reporter protein for expression normalization of the signal in the flow cytometer. b Flow-cytometric analysis of cell-displayed Nanobodies selected by phage display. Histogram showing the surface expression normalized GFP fluorescence intensity on the y-axis. All three Nanobody variants showed a high normalized signal (GFP fluorescence/expression level) compared to the negative control (Z wt)
Cloning and sorting of the Nanobody library
To evaluate the staphylococcal display system for selection of binders from Nanobody libraries, the immune VHH repertoire from a GFP-immunized dromedary was amplified by PCR, using the pHEN4 Nb library as a template, and subcloned into the staphylococcal display vector pSCNb1. After electroporation of the Nanobody library into S. carnosus, the obtained library size was estimated to approximately 107 transformants, covering a representative fraction of the VHH repertoire of B lymphocytes from a 50-ml dromedary blood sample [39]. To verify that the library was functionally expressed on the staphylococcal surface, an aliquot of the library was grown overnight, incubated with Alexa Fluor 647-HSA conjugate, and analyzed by flow cytometry. The results showed that around 60 % of the population had functional ABP expressed on the surface, which was in accordance to the proportion of in-frame clones that were identified by sequencing of the unsorted library (data not shown). The library was thereafter sorted for four rounds using eGFP as target and fluorescently labeled HSA for surface expression monitoring and normalized gating (Fig. 3). Visualizing the cell-displayed library in the flow cytometer showed a significant enrichment of fluorescent cells after each round, indicating that the isolation of eGFP-binding Nanobodies was successful. The unsorted library contained around 0.02 % binders and after the second sorting, approximately 50 % of the cells in library were binding to GFP (corresponding to an enrichment of around 2,500-fold) (Fig. 3). In the last sorting round the population appeared to consist of only GFP-binders, since the population of highly expressed clones with low or absent target binding had disappeared (Fig. 3).
Fig. 3.

Sorting of the cell-displayed Nanobody library. Density plots from the flow-cytometric sorting showing mean fluorescence intensity (MFI) corresponding to GFP binding on the y-axis and MFI corresponding to HSA binding (surface expression level) on the x-axis. Four rounds of flow-cytometric sorting were performed using normalized gating, with an enrichment of GFP-binding clones after each round. In the first three rounds 200 nM GFP was used, and in the last round 10 nM (shown in the figure), 50 nM (data not shown) or 200 nM GFP (data not shown) was used
DNA sequencing
Bacterial cells expressing isolated Nanobody clones from the last sorting round were grown into colonies and the Nanobody genes were amplified by PCR and sequenced. Out of 178 sequenced clones, 27 different variants were identified, of which 16 occurred more than once (Fig. 4, Online Resource 1). All clones were derived from the VHH germline 2a [38]. The sequences were grouped in a phylogenetic tree using Geneious software (Biomatters Ltd, Auckland, New Zealand), using the Jukes–Cantor genetic distance model and neighbor-joining and six different clusters of binders were identified (Fig. 4). Two of the clusters were homologues to phage display-selected Nanobodies (P-Nb1 and P-Nb2). One cluster was dominating the isolated repertoire, containing 19 unique variants and differed from the homologous phage display-selected variant (P-Nb2) in 2–9 amino acid positions. The other homologous cluster differed in 1–3 positions from the phage display-selected variant (P-Nb1). Notably, four clusters were unique and had no similar counterpart in the repertoire isolated by phage display (Fig. 4). Likewise, three of the phage-display selected Nanobodies were not found among the variants from the staphylococcal display selection (P-Nb3, P-Nb5, and P-Nb6).
Fig. 4.
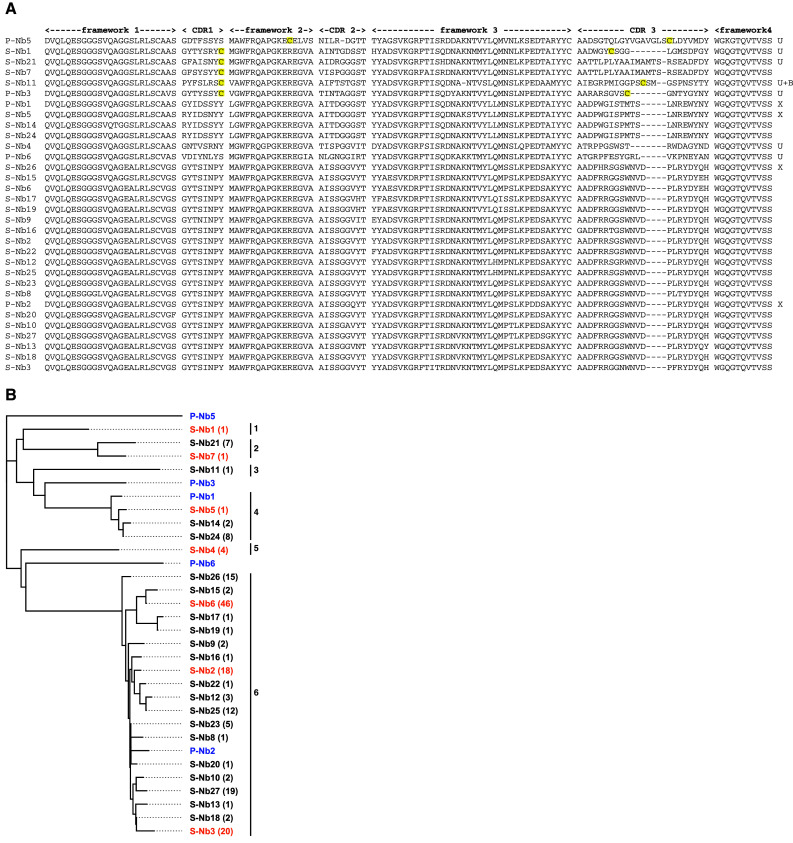
a Amino acid sequence alignment of the Nanobodies retrieved after phage display (P-Nb) and after staphylococcal display (S-Nb). All clones are derived from VHH germline sub family 2a except for P-Nb5 that is from subfamily 4b [38]. After VDJ recombination, the genes have evolved by somatic hypermutation (and possible PCR artifacts). The clones that were unique for either phage display or bacterial display are denoted U, those that were retrieved after both selections are denoted X. Clone S-Nb11 was also retrieved in a bacterial two hybrid (intracellular) selection, denoted ‘B’. The cysteines in CDR1 and CDR3 (or position 50 and CDR3) are highlighted in yellow. Remarkably, clones S-Nb21 and S-Nb7 have only one cysteine in their CDR1. b Phylogenetic tree showing genetic distances between different Nanobodies. S-Nb# denotes a Nanobody selected by staphylococcal display and P-Nb# denotes a Nanobody selected by phage display (indicated in blue). The Nanobodies that were selected for further characterization are indicated in orange. The number of identical clones is indicated after each name. The clusters are denoted cluster 1–6. The cluster number is indicated next to each cluster. Two of the clusters were homologues to phage display-selected Nanobodies (P-Nb1 and P-Nb2). One cluster was dominating the isolated repertoire, containing 19 unique variants. Four clusters were unique and had no similar counterpart in the repertoire isolated by phage display. Three of the phage-display selected Nanobodies were not found among the variants from the staphylococcal display selection (P-Nb3, P-Nb5, and P-Nb6)
Flow-cytometric analysis of isolated binders
Seven of the variants from the flow-cytometric sorting (S-Nb1, S-Nb2, S-Nb3, S-Nb4, S-Nb5, S-Nb6, and S-Nb7) were selected for further characterization. This selection was made in order to get a representation of sequences evenly distributed in the tree. Before producing the antibody fragments in a soluble format, binding to eGFP was verified by flow-cytometric analysis. Cells were labeled with eGFP and HSA-Alexa Fluor 647 conjugate as described above, and the mean fluorescence intensity was measured using flow cytometry. All variants showed a high GFP-binding signal, demonstrating that the flow-cytometric sorting for isolation of GFP-specific Nanobodies had been successful (data not shown).
Nanobody production, affinity determination and specificity analysis
The seven variants selected for further characterization were expressed in fusion to a C-terminal His-tag in the periplasm of E. coli WK6 cells and purified by immobilized metal IMAC followed by SEC (Online Resource 2). Analysis of the eluted fractions using SDS-PAGE demonstrated proteins of the correct size with no detectable contamination (Online Resource 3).
The affinities of the Nanobodies for eGFP were determined using an SPR-based biosensor assay. The Nanobodies were immobilized by amine coupling to an alginate chip surface using a ProteOn XPR36 instrument, and three different concentrations of eGFP (MBL International) were injected over the surface for determination of the kinetic parameters of the binding reaction. The obtained sensorgrams were fitted using non-linear regression to a monovalent binding equation using the ProteOn software and the equilibrium dissociation constants (K D) were calculated from the rate constants (k on and k off). The analysis revealed that four of the seven Nanobodies from the staphylococcal selection had subnanomolar affinities for eGFP, and the strongest binder a K D of around 140 pM (Fig. 5a; Table 1, Online Resource 4). On average, the binders from the staphylococcal selection had a K D of around 3 nM.
Fig. 5.

Biosensor affinity analysis and analysis of enhancement effect on GFP fluorescence. a Representative sensorgram from SPR-analysis of immobilized S-Nb2 binding to GFP, showing the response signal as well as the fitted data. GFP was injected at concentrations ranging from around 10–50 nM. Data is double referenced by subtraction of simultaneous responses from reference surface and a buffer injection. b Histogram showing the GFP fluorescence intensity when incubated with respective Nanobody. Each Nanobody was incubated with a threefold molar excess of wtGFP, and the fluorescence intensity was measured by scanning a 96-well microplate using an LS400 Laser Scanner (Tecan). The experiment was performed in triplicates. The fluorescence enhancement effect was calculated as the mean fluorescence intensity obtained from wtGFP incubated with GFP-binding Nanobody divided by the mean fluorescence intensity from wtGFP incubated with the negative control Nanobody. All seven Nanobodies enhanced the intensity of GFP fluorescence, with effects ranging from 20 up to 82 % compared to the control Nanobody
To investigate the specificity of the Nanobodies that were isolated in the sorting, potential binding to three different human proteins was analyzed in a biosensor assay. Transferrin, HSA and IgG were immobilized on three surfaces of a biosensor chip followed by injection of the seven binders at two concentrations each (80 and 240 nM). To ensure functional immobilization of the serum proteins, an ABD, the IgG-binding Z domain [40] and a transferrin-specific Affibody molecule [41] were included as positive controls. The results from the biosensor experiment showed no binding of any of the seven Nanobodies to the serum proteins (data not shown), whereas the positive controls all bound to their respective surface (Online Resource 4), hence strongly indicating that the Nanobodies selected using staphylococcal surface display are specific for GFP.
GFP fluorescence enhancement analysis
In a previous article by Kirchhofer et al. [27], it was reported that some GFP-specific Nanobodies modulate the fluorescence properties of GFP upon binding. It was also demonstrated that in particular the enhancing effect on the fluorescence could be exploited in a variety of assays [27]. Since the staphylococcal display system selects GFP-binding Nanobodies based on the intensity of the fluorescence signal, we wanted to investigate if this was reflected in their spectral-modulating properties. In order to determine the effect on the GFP fluorescence by the binding of the Nanobodies, a threefold molar excess of each binder was added to 100 nM of wtGFP (MBL International). Fluorescence intensity was measured by scanning a 96-well microplate using an LS400 Laser Scanner (Tecan). Interestingly, the results revealed that all of the seven Nanobodies indeed enhanced the intensity of GFP fluorescence, with effects ranging from 20 up to 82 % compared to the negative control Nanobody (Fig. 5b; Table 1).
Discussion
Display of antibody fragment libraries have been reported using a number of different systems, including several that are based on microbial hosts such as phage, yeast and E. coli. In this work, we have for the first time displayed an antibody fragment library on the surface of a Gram-positive bacterium, followed by isolation of GFP-specific binders using flow-cytometric sorting. The immune Nanobody library, which comprises the single variable domains (VHH) from heavy-chain only camelid antibodies was found to be well expressed on the staphylococcal surface and several subnanomolar affinity binders for GFP were isolated by FACS. Phage display and staphylococcal display selections of GFP binders from the same library generated five and six different repertoires of binders, respectively. Several variants with high sequence similarities belonging to two clusters were isolated from both selections. Interestingly, both systems also generated unique clones that were not found using the other technology. Four out of six clusters were unique for the staphylococcal display and had no counterpart in the repertoire generated by phage display (Fig. 4). A Nanobody from the third staphylococcal-derived cluster, S-Nb11, was also selected using the bacterial-two-hybrid system [32]. Correspondingly, three of the five phage-display selected Nanobodies (P-Nb3, P-Nb5, and P-Nb6) were not found after the staphylococcal display selection. Notably, the strongest Nanobody from the phage display selection (P-Nb5, denoted ‘GBP4’ in Kirchhofer et al. [27]) has a minimizing effect on the fluorescence of GFP [27], and was potentially therefore not isolated by FACS. Comparison of the affinities of homologous Nanobodies selected by staphylococcal and phage display shows that a small number of amino acid differences can have significant effects on biochemical parameters. For example, the phage display selected variant P-Nb2 had an affinity of 42.5 nM and a GFP-enhancing effect of 8 %, whereas one of the staphylococcal-display derived homologues (S-Nb3, belonging to the same sequence cluster as P-Nb2 but differing in six amino acid positions) had an affinity of around 0.14 nM and a GFP-enhancing effect of 43 %. The other staphylococcal-display derived homologue (S-Nb2, which differs in four amino acid positions) had an affinity of around 0.25 nM and a GFP-enhancing effect of 47 %. Similarly, the phage-display selected variant P-Nb1 had an affinity of 3.2 nM, whereas the staphylococcal-display derived homologue (S-Nb5, which differs in three amino acid positions) had an affinity of 0.21 nM (but in this case the GFP fluorescence enhancement was not drastically affected). Since the staphylococcal display system selects GFP-binding Nanobodies by the intensity of the fluorescence signal, it is likely that this system favors to select binders with a GFP-enhancing effect. Indeed, all seven analyzed Nanobodies from the staphylococcal selection demonstrated an enhancing effect on the GFP fluorescence (higher than 20 %), whereas only two Nanobodies from the phage selection showed a fluorescence enhancing effect of more than 20 %. Furthermore, no Nanobodies with a minimizing effect on the GFP fluorescence were selected using staphylococcal display. The bias in the FACS-based selection might thus have an impact on the observed differences in output. In addition, even though the staphylococcal system appears to select for GFP-enhancing Nanobodies, the affinities of the selected clones were also higher on average compared to the Nanobodies derived from phage display. It should be noted that if more clones from each selection were to be sequenced, additional rare candidates might probably be identified, thereby potentially increasing the overlap between the repertoires. Nevertheless, the study demonstrates that the dominating candidates from the two selections are slightly different.
The data from this work show that both systems have biases that result in different sets of binders depending on which technology that is employed for the selection. In a previously reported study, Burton and colleagues performed a comparison of yeast and phage display for selection of scFvs [42]. The authors discovered that several binders that were isolated from the yeast library could not be retrieved after phage display biopanning, and speculated that this was mainly due to incorrect folding of scFvs in E. coli [42]. It is possible that similar reasons prevent some library members from being correctly expressed on the surface of the staphylococci as well as on phages, hence resulting in the observed differences in output repertoires. However, although it is likely that the choice of host influences the final functional library, it should also be noted that the strategy and basics for isolating binders is different in cell display and phage display, which probably also have an effect on the outcome of the selection. Nevertheless, whatever the underlying origin of the observed differences is, the results suggest that parallel use of a panel of display systems is valuable in order to isolate the highest possible number of binders from a combinatorial library. It also highlights the importance of choosing a display system that is suitable for the target protein and the purpose of the selection.
In summary, here we report on the first successful display of an antibody fragment library on Gram-positive bacteria and the results demonstrate that the staphylococcal system is a powerful technology for selection of high-affinity Nanobodies from an immune library, and potentially other antibody fragments as well. It should also be noted that this is the first library based on a protein scaffold that is dependent on correct disulfide pairing to be displayed on Gram-positive bacteria. Including the results from this study, Nanobodies have now been displayed using a number of different systems [29–32], indicating that the format is relatively easy to express in a functional manner in fusion to various surface-anchored proteins. This is an attractive feature of a protein scaffold that is used in combinatorial protein engineering, enabling an array of technologies for generation of new high-affinity binders and increasing the possibility of finding a candidate with optimal properties.
Surprisingly few new microbial hosts for display of antibody fragment libraries have been investigated during the last decade and the work presented in this paper will hopefully encourage other groups to investigate new species for such purposes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by the Swedish Research Council (VR) [2009-5758], Affinomics (EU-collaborative Project) and the VINNOVA excellence center for protein technology (ProNova). The VIB laboratory was financially supported by Affinomics (EU-collaborative Project, 241481).
References
- 1.Bradbury ARM, Marks JD. Antibodies from phage antibody libraries. J Immunol Methods. 2004;290(1–2):29–49. doi: 10.1016/j.jim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Lofblom J, Frejd FY, Stahl S. Non-immunoglobulin based protein scaffolds. Curr Opin Biotechnol. 2011;22(6):843–848. doi: 10.1016/j.copbio.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Lofblom J. Bacterial display in combinatorial protein engineering. Biotechnol J. 2011;6(9):1115–1129. doi: 10.1002/biot.201100129. [DOI] [PubMed] [Google Scholar]
- 4.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17(4):467–473. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipovsek D, Pluckthun A. In vitro protein evolution by ribosome display and mRNA display. J Immunol Methods. 2004;290(1–2):51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15(6):553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 7.Daugherty PS, Chen G, Olsen MJ, Iverson BL, Georgiou G. Antibody affinity maturation using bacterial surface display. Protein Eng. 1998;11(9):825–832. doi: 10.1093/protein/11.9.825. [DOI] [PubMed] [Google Scholar]
- 8.VanAntwerp JJ, Wittrup KD. Fine affinity discrimination by yeast surface display and flow cytometry. Biotechnol Prog. 2000;16(1):31–37. doi: 10.1021/bp990133s. [DOI] [PubMed] [Google Scholar]
- 9.Lofblom J, Wernerus H, Stahl S. Fine affinity discrimination by normalized fluorescence activated cell sorting in staphylococcal surface display. FEMS Microbiol Lett. 2005;248(2):189–198. doi: 10.1016/j.femsle.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 10.Nilvebrant J, Alm T, Hober S, Lofblom J. Engineering bispecificity into a single albumin-binding domain. PLoS ONE. 2011;6(10):e25791. doi: 10.1371/journal.pone.0025791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, Geren I, Stevens RC, Marks JD. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat Biotechnol. 2007;25(1):107–116. doi: 10.1038/nbt1269. [DOI] [PubMed] [Google Scholar]
- 12.Ho M, Nagata S, Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. P Natl Acad Sci USA. 2006;103(25):9637–9642. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beerli RR, Bauer M, Buser RB, Gwerder M, Muntwiler S, Maurer P, Saudan P, Bachmann MF. Isolation of human monoclonal antibodies by mammalian cell display. Proc Natl Acad Sci USA. 2008;105(38):14336–14341. doi: 10.1073/pnas.0805942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowers PM, Horlick RA, Neben TY, Toobian RM, Tomlinson GL, Dalton JL, Jones HA, Chen A, Altobell L, 3rd, Zhang X, Macomber JL, Krapf IP, Wu BF, McConnell A, Chau B, Holland T, Berkebile AD, Neben SS, Boyle WJ, King DJ. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc Natl Acad Sci USA. 2011;108(51):20455–20460. doi: 10.1073/pnas.1114010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daugherty PS. Protein engineering with bacterial display. Curr Opin Struct Biol. 2007;17(4):474–480. doi: 10.1016/j.sbi.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, Rogers GK. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Natl Acad Sci USA. 2004;101(25):9193–9198. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kronqvist N, Lofblom J, Jonsson A, Wernerus H, Stahl S. A novel affinity protein selection system based on staphylococcal cell surface display and flow cytometry. Protein Eng Des Sel. 2008;21(4):247–255. doi: 10.1093/protein/gzm090. [DOI] [PubMed] [Google Scholar]
- 18.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JMW, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, Graff C, Wiley HS, Wittrup KD. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21(2):163–170. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 19.Kronqvist N, Malm M, Rockberg J, Hjelm B, Uhlen M, Stahl S, Lofblom J. Staphylococcal surface display in combinatorial protein engineering and epitope mapping of antibodies. Recent Pat Biotechnol. 2010;4(3):171–182. doi: 10.2174/187220810793611536. [DOI] [PubMed] [Google Scholar]
- 20.Gotz F. Staphylococcus carnosus: a new host organism for gene cloning and protein production. Soc Appl Bacteriol Symp Ser. 1990;19:49S–53S. doi: 10.1111/j.1365-2672.1990.tb01797.x. [DOI] [PubMed] [Google Scholar]
- 21.Lofblom J, Feldwisch J, Tolmachev V, Carlsson J, Stahl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584(12):2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Hjelm B, Fernandez CD, Lofblom J, Stahl S, Johannesson H, Rockberg J, Uhlen M. Exploring epitopes of antibodies toward the human tryptophanyl-tRNA synthetase. N Biotechnol. 2010;27(2):129–137. doi: 10.1016/j.nbt.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Rockberg J, Lofblom J, Hjelm B, Uhlén M, Stahl S. Epitope mapping of antibodies using bacterial surface display. Nat Methods. 2008;5(12):1039–1045. doi: 10.1038/nmeth.1272. [DOI] [PubMed] [Google Scholar]
- 24.Kronqvist N, Malm M, Gostring L, Gunneriusson E, Nilsson M, Hoiden Guthenberg I, Gedda L, Frejd FY, Stahl S, Lofblom J. Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng Des Sel. 2011;24(4):385–396. doi: 10.1093/protein/gzq118. [DOI] [PubMed] [Google Scholar]
- 25.Conrath KE, Wernery U, Muyldermans S, Nguyen VK. Emergence and evolution of functional heavy-chain antibodies in Camelidae. Dev Comp Immunol. 2003;27(2):87–103. doi: 10.1016/S0145-305X(02)00071-X. [DOI] [PubMed] [Google Scholar]
- 26.van der Linden RH, de Geus B, Frenken GJ, Peters H, Verrips CT. Improved production and function of llama heavy-chain antibody fragments by molecular evolution. J Biotechnol. 2000;80(3):261–270. doi: 10.1016/S0168-1656(00)00274-1. [DOI] [PubMed] [Google Scholar]
- 27.Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, Leonhardt H, Hopfner KP, Rothbauer U. Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol. 2010;17(1):133–138. doi: 10.1038/nsmb.1727. [DOI] [PubMed] [Google Scholar]
- 28.Harmsen MM, Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biot. 2007;77(1):13–22. doi: 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conrath KE, Lauwereys M, Galleni M, Matagne A, Frere JM, Kinne J, Wyns L, Muyldermans S. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the Camelidae. Antimicrob Agents Chemother. 2001;45(10):2807–2812. doi: 10.1128/AAC.45.10.2807-2812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yau KYF, Groves MAT, Li SH, Sheedy C, Lee H, Tanha J, MacKenzie CR, Jermutus L, Hall JC. Selection of hapten-specific single-domain antibodies from a non-immunized llama ribosome display library. J Immunol Methods. 2003;281(1–2):161–175. doi: 10.1016/j.jim.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Ryckaert S, Pardon E, Steyaert J, Callewaert N. Isolation of antigen-binding camelid heavy chain antibody fragments (nanobodies) from an immune library displayed on the surface of Pichia pastoris . J Biotechnol. 2010;145(2):93–98. doi: 10.1016/j.jbiotec.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Pellis M, Pardon E, Zolghadr K, Rothbauer U, Vincke C, Kinne J, Dierynck I, Hertogs K, Leonhardt H, Messens J, Muyldermans S, Conrath K (2012) A bacterial-two-hybrid selection system for one-step isolation of intracellularly functional Nanobodies. Arch Biochem Biophys. doi:10.1016/j.abb.2012.04.023 [DOI] [PubMed]
- 33.Deschamps JR, Miller CE, Ward KB. Rapid purification of recombinant green fluorescent protein using the hydrophobic properties of an HPLC size-exclusion column. Protein Expr Purif. 1995;6(4):555–558. doi: 10.1006/prep.1995.1073. [DOI] [PubMed] [Google Scholar]
- 34.Ruther U. pUR 250 allows rapid chemical sequencing of both DNA strands of its inserts. Nucleic Acids Res. 1982;10(19):5765–5772. doi: 10.1093/nar/10.19.5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lofblom J, Kronqvist N, Uhlén M, Stahl S, Wernerus H. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J Appl Microbiol. 2007;102(3):736–747. doi: 10.1111/j.1365-2672.2006.03127.x. [DOI] [PubMed] [Google Scholar]
- 36.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol. 2000;328:333–363. doi: 10.1016/S0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 37.de Genst E. Strong in vivo maturation compensates for structurally restricted H3 loops in antibody repertoires. J Biol Chem. 2005;280(14):14114–14121. doi: 10.1074/jbc.M413011200. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen VK, Hamers R, Wyns L, Muyldermans S. Camel heavy-chain antibodies: diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000;19(5):921–930. doi: 10.1093/emboj/19.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riechmann L, Muyldermans S. Single-domain antibodies: comparison of camel VH and camelised human VH domains. J Immunol Methods. 1999;231(1–2):25–38. doi: 10.1016/S0022-1759(99)00138-6. [DOI] [PubMed] [Google Scholar]
- 40.Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlen M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1(2):107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 41.Gronwall C, Sjoberg A, Ramstrom M, Hoidén Guthenberg I, Hober S, Jonasson P, Stahl S. Affibody-mediated transferrin depletion for proteomics applications. Biotechnol J. 2007;2(11):1389–1398. doi: 10.1002/biot.200700053. [DOI] [PubMed] [Google Scholar]
- 42.Bowley DR, Labrijn AF, Zwick MB, Burton DR. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng Des Sel. 2007;20(2):81–90. doi: 10.1093/protein/gzl057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.